Изобретение относится к соединениям и их фармацевтически приемлемым солям, обладающим способностью ингибировать матричные металлопротеазы, в частности интерстициальные коллагеназы, и, следовательно, пригодным для лечения болезненных состояний у млекопитающих, облегчаемых ингибированием таких матричных металлопротеаз.
Матричные металлопротеазы представляют собой семейство протеаз, ответственных за разложение и реконструирование соединительных тканей. Представители этого семейства ферментов обладают многочисленными свойствами, в том числе цинк- и кальций-зависимостью, секрецией в виде проферментов и 40-50% гомологичностью последовательностей аминокислот.
Семейство матричных металлопротеаз включает интерстициальные коллагеназы, полученные из фибробластов/макрофагов и нейтрофилов, которые катализируют начальное расщепление и расщепление с ограниченной скоростью нативного коллагена типов I, II, III и X.
Коллаген, главный структурный протеин у млекопитающих, является основным компонентом межклеточного вещества многих тканей, например хрящевой, костной, ткани сухожилий и кожи. Интерстициальные коллагеназы являются очень специфичными матричными металлопротеазами, которые расщепляют коллаген с получением двух фрагментов, которые спонтанно денатурируют при физиологических температурах и поэтому становятся чувствительными к расщеплению менее специфическими ферментами. Поскольку расщепление с помощью коллагеназ приводит к потере структурной целостности ткани-мишени, то вследствие этого по существу возможен и обратный процесс, и, следовательно, речь идет о хорошей мишени для терапевтического воздействия.
Помимо интерстициальных коллагеназ, семейство матричных металлопротеаз из ферментов включает две различные, но очень близкие желатиназы: фермент 72 кДа, секретируемый фибробластами, и фермент 92 кДа, высвобождаемый из одноядерных фагоцитов. Эти желатиназы обладают способностью разлагать желатины (денатурированные коллагены), нативный коллаген типов IV и V, фибронектин и нерастворимый эластин.
Семейство матричных металлопротеаз также включает стромелизины 1 и 2, обладающие способностью расщеплять широкий спектр матричных субстратов, в том числе ламинин, фибронектин, протеогликаны и коллаген типов IV и IX в их негеликоидальной области.
Матрилизин (мнимая металлопротеаза или ММП) является недавно открытым представителем семейства матричных металлопротеаз. Матрилизин обладает способностью разлагать широкий спектр матричных субстратов, в том числе протеогликаны, желатины, фибронектин, эластин и ламинин. Его экспрессия была зафиксирована в одноядерных фагоцитах, эксплантатах матки крыс и в единичных случаях в опухолях.
Считается, что ингибиторы матричных металлопротеаз пригодны для лечения артритов, болезней, связанных с резорбцией кости (таких, как остеопорозы), увеличения деструкции коллагена, связанного с диабетами, болезней периодонта, язв на роговице, язв на коже и метастазов опухолей. Предназначение и потенциальные возможности использования ингибиторов коллагеназ описаны, например, в J. Enzyme Inhibition (1987), т. 8, стр. 1-22, и в Drug News &. Prospectives (1990), т. 3, N 8, стр. 453-458. Ингибиторы матричных металлопротеаз также являются предметом различных патентов и заявок на патент, например, патентов US 5189178 (Galardy) и US 5183900 (Galardy), опубликованных заявок на Европейские патенты ЕР 0438223 (Beecham) и ЕР 0276436 (F.Hoffmann-La Roche), Международных заявок WO 92/21360 (Merck), WO 92/06966 (Beecham) и WO 92/09563 (Glycomed).
Объектом настоящего изобретения являются новые соединения, которые пригодны в качестве ингибиторов матричных металлопротеаз, в частности интерстициальных коллагеназ, которые эффективны при лечении болезненных состояний, характеризующихся повышенной активностью матричных металлопротеаз.
Таким образом, настоящее изобретение касается производных амидов общей формулы (I):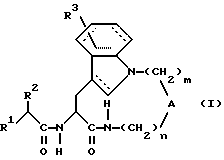
где штриховые линии обозначают необязательные двойные связи; и, если n равно 1, 2 или 3; m равно 3 или 4; то
А обозначает - CH2-;
R1 обозначает;
a) -CH2-R4, где R4 обозначает меркапто, ацетилтио, карбокси, гидроксиаминокарбонил, N-гидроксиформиламино, алкоксикарбонил, морфолино(С1-С4)алкоксикарбонил, арилоксикарбонил, бензилоксиаминокарбонил или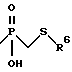
где R6 обозначает хинол-2-ил;
б) -CH(R7)-R8, где R7 обозначает алкил, гидрокси, амино, алкоксикарбонил, аминокарбонил или карбокси; и R8 обозначает карбокси, гидроксиаминокарбонил, алкоксикарбонил или аралкоксикарбонил;
в) -NH-CH(R9)-R10, где R9 обозначает водород или алкил; и R10 обозначает карбокси или аралкоксикарбонил;
R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил;
R3 обозначает водород;
или, если n равно 2 или 3; m равно 3 или 4; то А обозначает -N(R11)-, где R11 обозначает водород или алкил; и
R1, R2 и R3 имеют указанные выше значения;
в виде отдельных стереоизомеров или в виде их смеси; или их фармацевтически приемлемые соли.
Кроме вышеопределенных обозначений для R3, R4, R6, R9 и R10 в соединениях формулы (I) указанные радикалы могут иметь следующие дополнительные обозначения:
R3 - галоген, алкил или алкокси;
R4 - аралкоксикарбонил;
R6 - необязательно замещенный арил, где арильная группа обозначает хинолил-2-ил, нафт-1-ил, нафт-2-ил, пиридил или фенил;
R9 - аралкил;
R10 - алкоксикарбонил, фосфонил, диалкилфосфонил, метоксифосфонил.
В настоящем изобретении описывается также и способ ингибирования активности матричных металлопротеаз у млекопитающих, включающий назначение млекопитающему, в случае необходимости, терапевтически эффективного количества соединения формулы (I), как определено выше, в виде отдельного стереоизомера или в виде их смеси; или его фармацевтически приемлемой соли.
Другим объектом настоящего изобретения является фармацевтическая композиция, обладающая ингибирующей активностью в отношении матричных металлопротеаз у млекопитающих, включающая в качестве действующего вещества терапевтически эффективное количество соединения формулы (I), как определено выше, в виде отдельного стереоизомера или в виде их смеси; или его фармацевтически приемлемой соли; и фармацевтически приемлемый эксципиент.
В описании изобретения и в формуле изобретения, если не указано иное, применяются следующие определения и термины, которые имеют указанные ниже значения.
"ВОС" обозначает трет-бутоксикарбонил.
"КБЗ" обозначает бензилоксикарбонил (карбобензилокси).
"ДМФ" обозначает N,N-диметилформамид.
"ЭДКИ" обозначает N-этил-N'-(3-диметиламинопропил)карбодиимид.
"ГОБТ" обозначает 1-гидроксибензтриазол.
"Ацетилтио" обозначает радикал -SC(O)CH3.
"Галоген" обозначает бром, хлор или фтор.
"Алкил" обозначает одновалентный радикал с прямой или разветвленной цепью, состоящий только из углерода и водорода, не содержащий ненасыщенных связей и имеющий от одного до четырех атомов углерода, например метил, этил, н-пропил, 2-метилпропил (изобутил), 1-метилэтил (изопропил), н-бутил и 1,1-диметилэтил (трет.-бутил).
"Алкокси" обозначает радикал формулы -ORa, где Ra обозначает алкил, как определено выше, например метокси, этокси, н-пропокси, изопропокси, 1-метилэтокси, н-бутокси, трет.-бутокси и т.п.
"Арил" обозначает фенильный или нафтильный радикал.
"Арилокси" обозначает радикал формулы -ORb, где Rb обозначает арил, как определено выше, например фенокси, хинол-2-илокси, нафт-1-илокси или нафт-2-илокси.
"Аралкил" обозначает радикал формулы -RaRb, где Ra обозначает алкил, как определено выше, и Rb обозначает арил, как определено выше, например бензил, фенилэтилен, 3-фенилпропил и т.п.
"Аралкокси" обозначает радикал формулы -ORaRb, где Ra обозначает алкил, как определено выше, и Rb обозначает арил, как определено выше, например бензилокси или 3-нафт-2-илпропокси и т.п.
"Алкоксикарбонил" обозначает радикал формулы -C(O)Rb, где Rb обозначает алкокси, как определено выше, например метоксикарбонил, этоксикарбонил, трет.-бутоксикарбонил и т.п.
"Аралкоксикарбонил" обозначает радикал формулы -C(O)Rc, где Rc обозначает аралкокси, как определено выше, например бензилоксикарбонил, нафт-2-илэтоксикарбонил и т.п.
"Бензилоксиаминокарбонил" обозначает радикал формулы - C(O)NHCH2Rd, где Rd обозначает фенил.
"Карбамоил" обозначает радикал -C(O)NH2.
"Карбокси" обозначает радикал -С(O)ОН.
"Гидроксиамино" обозначает радикал -NHOH.
"Гидроксиаминокарбонил" обозначает радикал -С(О)NHOH.
"Меркапто" обозначает радикал -SH.
"Сульфонил" обозначает радикал =S(O)2.
"Фосфонил" обозначает радикал -PO(ОН)2.
"Необязательный" или "необязательно" обозначает, что последовательно описанные действия или обстоятельства могут иметь место или их может не быть и что определение включает ситуации, когда указанные действия или обстоятельства имеют место, и ситуации, когда они отсутствуют. Например, "необязательно замещенный хинол-2-ил" обозначает, что радикал хинол-2-ила может быть замещенным или может быть незамещенным и что определение включает как радикалы замещенного хинол-2-ила, так и радикалы хинол-2-ила, не имеющие замещения.
"Необязательно замещенный арил" обозначает радикал хинол-2-ила, нафт-1-ила, нафт-2-ила, пиридила или фенила, необязательно замещенный одним или более заместителями, например, такими, как галоген, алкил, алкокси, гидрокси и нитро, например 6-нитрохинол-2-ил, 6-фторхинол-2-ил, 6-гидроксихинол-2-ил, 6-метоксихинол-2-ил, 6-нитронафт-1-ил, 6-хлорнафт-1-ил, 6-гидроксинафт-1-ил, 6-метоксинафт-1-ил, 6-нитронафт-2-ил, 6-хлорнафт-2-ил, 6-гидроксинафт-2-ил, 6-метоксинафт-2-ил, 6-нитрофенил, 6-хлорфенил, 6-гидроксифенил, 6-метоксифенил, 3- метилпиридил, 4-этилпиридил и т.п.
"Необязательно замещенный карбамоил" обозначает радикал карбамоила, необязательно замещенный по атому азота одним или более заместителями, выбранными из группы, включающей алкил или аралкил.
"Аминозащитная группа", как это используется в настоящем описании, обозначает органические группы, предназначенные для защиты атомов азота от нежелательных реакций во время процессов синтеза, и включает, но не ограничена ими, бензил, ацил, ацетил, бензилоксикарбонил (карбобензилокси), пара-метоксибензилоксикарбонил, пара-нитробензилоксикарбонил, трет.-бутоксикарбонил и т.п.
"Фармацевтически приемлемая соль" включает как фармацевтически приемлемые кислотно-аддитивные соли, так и фармацевтически приемлемые соли присоединения оснований.
"Фармацевтически приемлемая кислотно-аддитивная соль" обозначает такие соли, которые сохраняют биологическую эффективность и свойства свободных оснований, которые не являются нежелательными по биологическим или иным причинам и которые образуются с помощью неорганических кислот, таких как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., и органических кислот, таких как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, пара-толуолсульфоновая кислота, салициловая кислота и т.п.
"Фармацевтически приемлемая соль присоединения оснований" обозначает такие соли, которые сохраняют биологическую эффективность и свойства свободных кислот, которые не являются нежелательными по биологическим или иным причинам. Эти соли получают при добавлении неорганического основания или органического основания к свободной кислоте. Соли, образованные из неорганических оснований, включают, но не ограничены ими, соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и т.п. Предпочтительными неорганическими солями являются соли аммония, натрия, калия, кальция и магния. Соли, образованные из органических оснований, включают, но не ограничены ими, соли первичных, вторичных и третичных аминов, замещенных аминов, включающих встречающиеся в естественных условиях замещенные амины, циклические амины и основные ионообменные смолы, такие как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диметиламиноэтанол, 2-диэтиламиноэтанол, триметамин (trimethamin), дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин (hydrabamine), холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, N-этилпиперидин, полиаминные смолы и т.п. Особенно предпочтительными органическими основаниями являются изопропиламин, диэтиламин, этаноламин, триметамин, дициклогексиламин, холин и кофеин.
"Млекопитающие" включают людей и всех домашних и диких животных, в том числе, но не ограничиваясь ими, крупный рогатый скот, лошадей, свиней, овец, коз, собак, кошек и т.п.
"Терапевтически эффективное количество" относится к такому количеству соединения формулы (1), которое, будучи назначенным млекопитающему в случае необходимости, является достаточным для осуществления лечения, как определено ниже, состояний болезни, облегчаемых путем ингибирования активности матричной металлопротеазы, в частности, активности интерстициальной коллагеназы. Количество соединения формулы (I), которое составляет "терапевтически эффективное количество", в значительной степени варьируется в зависимости от соединения, состояния болезни и ее серьезности, а также от млекопитающего, подвергающегося лечению, однако оно может быть определено принятым образом обычным специалистом в данной области техники, полагаясь на его собственные знания и данное описание.
"Лечение" или "лечить", как это используется в настоящем описании, охватывают любое лечение состояния болезни у млекопитающего, прежде всего у человека, которое облегчается путем ингибирования активности матричной металлопротеазы, в частности активности интерстициальной коллагеназы и т.п.; и включает:
(I) предупреждение наступления болезненного состояния у млекопитающего, в частности, если указанное млекопитающее предрасположено к болезненному состоянию, но еще не установлено, что таковое наступило;
(II) ингибирование болезненного состояния, т.е. прекращение развития болезни; или
(III) облегчение болезненного состояния, т.е. регрессию болезненного состояния.
"Стереоизомеры" относятся к соединениям, имеющим одинаковую молекулярную формулу и природу или последовательность связей, но отличаются по ориентации их атомов в пространстве. Используемая в описании номенклатура в основном представляет собой модифицированную форму номенклатуры I.U.P.A.C. (Международного союза теоретической и прикладной химии), согласно которой соединения по изобретению названы как производные фосфиновой или алкановой кислот, имеющих трициклоалкильный заместитель. Соединения формулы (I) или их фармацевтически приемлемые соли имеют в своей структуре по крайней мере два асимметричных атома углерода; один углерод является углеродом, к которому присоединен заместитель R2, а другой углерод является углеродом, к которому присоединена индолилметильная группа. Соединения формулы (I) и их фармацевтически приемлемые соли могут также существовать в виде отдельных стереоизомеров, рацематов и в виде смесей энантиомеров и диастереомеров. Все указанные отдельные стереоизомеры, рацематы и их смеси включены в объем данного изобретения.
При обозначении отдельных стереоизомеров соединений формулы (I) R или S может быть отнесен к хиральным атомам углерода в соответствии с методикой "Правила последовательности" Кана, Ингольда и Прелога.
Например, следующее соединение формулы (I), где n равно 2; m равно 3; А обозначает -CH2-; R1 обозначает -CH2-R4, где R4 обозначает -C(O)NHOH; R2 обозначает 2-метилпропил и R3 обозначает водород, т.е. соединение, имеющее следующую формулу: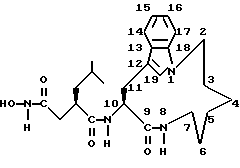
названо в настоящем описании как (3R,10S)-N-гидрокси-5- метил-3-(9-оксо-1,8-диазатрицикло-[10.6.1.013,18] нонадека- 12(19),13(18),14,16-тетраен-10-илкарбамоил) гексанамид.
Соединения формулы (I) пригодны в качестве ингибиторов матричных металлопротеаз млекопитающих, в частности интерстициальных коллагеназ млекопитающих, предотвращая тем самым разложение коллагена в организме млекопитающего. Эти соединения, кроме того, пригодны для лечения болезненных состояний, связанных с увеличением активности матричных металлопротеаз, прежде всего с увеличением активности интерстициальной коллагеназы, таких, как артриты и остеоартриты, метастазы опухолей, болезни периодонта и язвы роговицы. См., например, Arthritis and Rheumatism (1993), т. 36, N 2, стр. 181-189; Arthritis and Rheumatism (1991), т. 34, N 9, стр. 1073-1075; Seminars in Arthritis and Rheumatism (1990), т. 19, N 4, Supplement I, (February), стр. 16-20; Drugs of the Future (1990), т. 15, N 5, стр. 495-508; и J. Enzyme Inhibition (1987), т. 2, стр. 1-22.
Способность соединений формулы (I) ингибировать активность матричной металлопротеазы, в частности активность интерстициальной коллагеназы, может быть продемонстрирована различными опытами in vitro и ex vivo, известными специалистам в данной области техники. Например, активность отдельной металлопротеазы может быть продемонстрирована в опытах in vitro, описанных в Anal.Biochem. (1985), т. 147, стр. 437, или их модификациях. Физиологический эффект ингибирования матричных металлопротеаз может быть продемонстрирован в опытах ex vivo на эксплантанте бычьего хряща, описанных в Methods of Enzymology (1987), т. 144, стр. 412-419, или их модификациях; или в опытах ex vivo на длинной кости эмбриона крыс, описанных в Proc. Natl. Acad. Sci. USA (1988), т. 85, стр. 8761-8765, или их модификациях, или в J.Clin. Invest. (1965), т. 44, стр. 103-116, или их модификациях.
Введение соединений формулы (I) или их фармацевтически приемлемых солей в чистом виде или в виде приемлемой фармацевтической композиции может быть осуществлено с помощью любых приемлемых способов назначения или агентов, служащих подобным целям. Таким образом, введение может осуществляться, например, оральным, назальным, парентеральным, локальным, трансдермальным или ректальным путем в виде твердых, полутвердых или жидких дозируемых форм или дозируемых форм в виде лиофилизированного порошка, например, таких, как таблетки, суппозитории, пилюли, мягкие эластичные или твердые желатиновые капсулы, порошки, растворы, суспензии или аэрозоли и т.п., предпочтительно в виде унифицированных доз, пригодных для простого введения точных доз. Композиции могут включать обычный фармацевтический носитель или эксципиент и соединение формулы (I) в качестве действующего вещества и, кроме того, могут включать другие лекарственные средства, фармацевтические средства, носители, адъюванты и т.д.
Обычно в зависимости от назначаемого способа введения фармацевтически приемлемые композиции будут содержать от примерно 1 до примерно 99 вес.% соединения(ий) формулы (I) или его фармацевтически приемлемой соли и от 99 до 1 вес. % фармацевтически пригодного эксципиента. Предпочтительно композиция должна содержать от примерно 5 до примерно 75 вес.% соединения(ий) формулы (I) или его фармацевтически приемлемой соли, при этом остальное приходится на долю фармацевтически приемлемых эксципиентов.
Предпочтительным путем введения является оральный с использованием обычной суточной схемы приема лекарственного средства, которая может корректироваться в зависимости от степени сложности заболевания, которое подлежит лечению. Для такого орального введения фармацевтически приемлемую композицию, включающую соединение(я) формулы (I) или его фармацевтически приемлемую соль, получают путем добавления любых обычно используемых эксципиентов, таких как, например, фармацевтически чистые маннитол, лактоза, крахмал, прежелатинизированный крахмал, стеарат магния, натрийсахарин, тальк, эфирные производные целлюлозы, глюкоза, желатин, сахароза, цитрат, пропилгаллат и т.п.. Такие композиции имеют форму растворов, суспензий, таблеток, пилюль, капсул, порошков, композиций с непрерывным высвобождением лекарства и т.п.
Предпочтительно такие композиции должны иметь форму капсул, капель или таблеток и, кроме того, должны включать разбавитель, такой как лактоза, сахароза, дифосфат кальция и т.п., дезинтегратор, такой как натрийкроскармелоза или ее производные; замасливатель, такой как стеарат магния и т.п.; и связующее вещество, такое как крахмал, смола акации, поливинилпирролидон, желатин, эфирные производные целлюлозы и т.п.
Соединения формулы (I) или их фармацевтически приемлемые соли могут быть также использованы в форме суппозитория, который включает от примерно 0,5% до примерно 50% действующего вещества, распределенного в медленно растворяющемся внутри организма носителе, например, полиоксиэтиленгликолях и полиэтиленгликолях (ПЭГ), например ПЭГ 1000 (96%) и ПЭГ 4000 (4%).
Жидкие композиции для фармацевтического назначения могут, например, быть приготовлены путем растворения, диспергирования и т.д. соединения(ий) формулы (I) (от примерно 0,5% до примерно 20%) или его фармацевтически приемлемой соли и необязательно фармацевтических адъювантов в носителе, таком как, например, вода, физиологический раствор, водная декстроза, глицерин, этанол и т.п., для получения раствора или суспензии.
При необходимости фармацевтическая композиция по изобретению может также содержать небольшие количества добавочных веществ, таких как смачивающие или эмульгирующие агенты, pH буферные агенты, антиокислители и т.п., такие как, например, лимонная кислота, сорбитанмонолаурат, триэтаноламинолеат, бутилированный гидрокситолуол и т.д.
Практические способы получения таких форм дозировки известны или очевидны для специалистов в данной области техники; например, см. в Remington's Pharmaceutical Sciences, 18-е издание, (Mack Publishing Company, Easton, Pennsylvania, 1990). Назначаемые композиции в любом случае должны содержать терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли для лечения заболевания, которое может быть облегчено путем ингибирования матричной металлопротеазы в соответствии с рекомендациями данного изобретения.
Соединения формулы (I) или их фармацевтически приемлемые соли должны назначаться в терапевтически эффективном количестве, которое в значительной степени зависит от различных факторов, включающих активность конкретного применяемого соединения, метаболическую стабильность и продолжительность действия соединения, возраст, вес тела, общее состояние здоровья, пол, диету, режим и время введения, скорость экскреции, комбинацию лекарств, серьезность конкретного состояния болезни и терапию, которой подвергается пациент. Обычно терапевтически эффективная суточная доза составляет от приблизительно 0,14 мг до приблизительно 14,3 мг/кг веса тела в день для соединения формулы (I) или его фармацевтически приемлемой соли; предпочтительно от приблизительно 0,7 мг до приблизительно 10 мг/кг веса тела в день; и наиболее предпочтительно от приблизительно 1,4 мг до приблизительно 7,2 мг/кг веса тела в день. Например, при назначении человеку весом 70 кг диапазон доз может быть от приблизительно 10 мг до приблизительно 1,0 г соединения формулы (I) или его фармацевтически приемлемой соли в день, предпочтительно от приблизительно 50 мг до приблизительно 700 мг в день, и наиболее предпочтительно от приблизительно 100 мг до приблизительно 500 мг в день.
Предпочтительной группой соединений формулы (I) согласно изобретению являются те соединения, в которых n равно 2 или 3; m равно 3; А обозначает - CH2-; R2 обозначает алкил или аралкил.
Предпочтительным классом соединений этой группы являются те соединения, где R1 обозначает -CH2-R4, где R4 обозначает карбокси, гидроксиаминокарбонил, N-гидроксиформиламино, алкоксикарбонил, арилоксикарбонил или бензилоксиаминокарбонил; и R2 обозначает 2-метилпропил.
Предпочтительным подклассом соединений этого класса являются те соединения, где n равно 2 и R1 обозначает -CH2-C(O)ОН или -CH2-C(O)NHOH.
Из указанного подкласса соединений предпочтителен такой отдельный стереоизомер, как: (3R, 10S)-5-метил-3-(9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16-тетраен-10-илкарбамоил)гексановая кислота или (3R, 10S)-N-гидрокси-5-метил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18]нонадека-12(19),13(18),14,16-тетраен-10- илкарбамоил)гексанамид.
Другим предпочтительным подклассом указанного выше класса соединений являются те соединения, где n равно 3 и R1 обозначает - CH2-C(O)NHOH.
При этом предпочтителен такой отдельный стереоизомер, как: (3R,11S)-N-гидрокси-5-метил-3-(10-оксо-1,9-диазатрицикло [11.6.1.014,19]эйкоза-13(20), 14(19),15,17-тетраен-11- илкарбамоил)гексанамид.
Кроме того, предпочтительным подклассом указанного выше класса являются соединения, где n равно 1 и R1 обозначает -CH2-C(O)ОН или -CH2-C(O)NHOH.
При этом предпочтителен отдельный стереоизомер, а именно: (3R,9S)-5-метил-3-(8-оксо-1,7-диазатрицикло[9.6.1.012,17] октадека-11(18), 12(17), 13,15-тетраен-9-илкарбамоил)гексановая кислота; или (3R,9S)-N-гидрокси-5-метил-3-(8-оксо-1,7-диазатрицикло [9.6.1.012,17] октадека-11(18), 12(17), 13,15-тетраен-9- илкарбамоил)гексанамид.
Другим предпочтительным классом соединений группы являются те соединения, где R1 обозначает -CH2-R4, где R4 обозначает меркапто или ацетилтио.
При этом предпочтителен подкласс соединений, где n равно 2 и R1 обозначает -CH2SH или -CH2C(O)CH3.
Из указанного подкласса соединений предпочтителен: (10S)-2- меркаптометил-4-метил-N-(9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16-тетраен-10-илкарбамоил)пентанамид; или (10S)-2-ацетилтиометил-4-метил-N-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18),14,16-тетраен-10- илкарбамоил)пентанамид.
Другим предпочтительным классом соединений указанной группы являются также соединения, где R6 обозначает хинол-2-ил.
При этом предпочтительно соединение, где n равно 2.
В частности предпочтительно следующее соединение: (10S)-[4-метил-2-(9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19),13(18),14,16- тетраен-10-илкарбамоил)пентил]-(хинолин-2-илтиометил)фосфиновая кислота.
Другим предпочтительным классом соединений группы являются те соединения, где R1 обозначает -CH(R7)-R8, где R7 обозначает алкил, алкоксикарбонил или карбокси; и R8 обозначает карбокси, гидроксиаминокарбонил, алкоксикарбонил или аралкоксикарбонил.
Предпочтительным подклассом соединений этого класса являются те соединения, где R7 обозначает метоксикарбонил или метил.
Особенно предпочтительными в этом подклассе являются те соединения, где R8 обозначает гидроксиаминокарбонил.
В этом подклассе особенно предпочтителен отдельный стереоизомер, где n равно 2, а именно: (3R,10S)-N-гидрокси-5-метил-2-метоксикарбонил-3-(9-оксо-1,8- диазатрицикло[10.6.1.013,18]нонадека-12(19),13(18),14,16- тетраен-10-илкарбамоил)гексанамид.
Другим предпочтительным классом соединений группы являются те соединения, где R1 обозначает -NH-CH(R9)-R10, где R9 обозначает водород или алкил; и R10 обозначает карбокси или аралкоксикарбонил.
При этом предпочтительны те соединения, где R9 обозначает алкил и R10 обозначает карбокси.
Другой предпочтительной группой соединений формулы (I) являются те соединения, где n равно 2 или 3; m равно 4; А обозначает - N(R11)-, где R11 обозначает водород или алкил; R2 обозначает алкил.
Предпочтительным классом этой группы являются те соединения, где R2 обозначает 2-метилпропил; и R11 обозначает метил.
Предпочтительным подклассом соединений этого класса являются те соединения, где R1 обозначает -CH2-R4, где R4 обозначает карбокси, гидроксиаминокарбонил, N-гидроксиформиламино, алкоксикарбонил, арилоксикарбонил или бензилоксиаминокарбонил.
Особенно предпочтительны в этом подклассе те соединения, где n равно 2 и R' обозначает -CH2-C(O)NHOH.
Соединения формулы (I) в виде отдельных стереоизомеров или в виде их смесей, а также их фармацевтически приемлемые соли являются производными пептидов, которые могут быть получены из производных составляющих альфа-аминокислот. Стандартные способы образования пептидных связей описаны в M.Bodanszky и др. The Practice of Peptide Synthesis (1984), Springer-Verlag; M. Bodanszky, Principles of Peptide Synthesis (1984), Springer-Verlag; J.P. Greenstein и др. , Chemistry of the Amino Acids (1961), т. 1-3, John Wiley and Sons Inc.; G.R.Pettit, Synthetic Peptides (1970), т. 1-2, Van Nostrand Reinhold Company.
Амидные сочетания, используемые для образования соединений формулы (I), обычно выполняют с помощью карбодиимидного метода с такими реагентами, как дициклогексилкарбодиимид или N'-этил-N'-(3- диметиламинопропил)карбодиимид (ЭДКИ) в присутствии 1-гидроксибензтриазола (ГОБТ) в инертном растворителе, таком как диметилформамид (ДМФ). Другие способы образования амидной или пептидной связи включают, но не ограничены ими, способы синтеза с использованием хлорангидрида кислоты, ацилазида, смешанного ангидрида или активированного эфира, такого как нитрофениловый эфир. Обычно проводят амидные сочетания в фазе раствора с пептидными фрагментами или без них.
Выбор защитных групп для концевых амино- или карбоксильных групп соединений, используемых для получения соединений формулы (I), определяется отчасти конкретными условиями амидного или пептидного сочетания, а отчасти компонентами аминокислоты и/или пептида, включенными в сочетание. Обычно используемые аминозащитные группы включают те, которые хорошо известны в данной области техники, например бензилоксикарбонил (карбобензилокси), пара-метоксибензилоксикарбонил, пара-нитробензилоксикарбонил, трет.-бутоксикарбонил (ВОС) и т.п. Предпочтительно использовать либо ВОС, либо бензилоксикарбонил (КБЗ) в качестве защитной группы для α-аминогруппы из-за относительной простоты ее удаления слабыми кислотами, например трифторуксусной кислотой (ТФК) или соляной кислотой в этилацетате; или с помощью каталитического гидрирования.
Индивидуальные стереоизомеры соединений формулы (I) можно разделять друг от друга способами, известными специалистам в данной области техники, например путем селективной кристаллизации или хроматографии, и/или способами, приведенными в описании.
Комбинации заместителей и/или переменных в соединениях формулы (I) допустимы только в тех случаях, если такие комбинации приводят к получению стабильных соединений.
Способ получения соединений формулы I включает нижеописанные стадии.
А. Получение промежуточных продуктов: соединения формулы (J)
Соединения следующей формулы (J):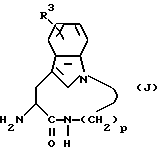
где R3 обозначает водород, галоген, алкил или алкокси; а p равно 5, 6, 7 или 8; используют при получении соединений формулы (I) и получают в соответствии с реакционной схемой 1 (см. в конце описания), где R3 обозначает водород, галоген, алкил или алкокси; p равно 5, 6, 7 или 8; ВОС обозначает трет-бутоксикарбонил, и R13 обозначает водород, мезил или тозил.
Соединения формул (В) и (F) являются коммерчески доступными, например, выпускаются фирмами Karl Industries, Inc. или Sigma соответственно, или могут быть получены способами, известными специалистам в данной области техники.
Обычно соединения формулы (J) получают способом, который включает сначала этерификацию спирта формулы (В) уксусным ангидридом в присутствии основания, предпочтительно пиридина, для образования соединения формулы (С), которое затем восстанавливают в присутствии уксусного ангидрида для образования соединения формулы (D). Соединение формулы (D) подвергают гидролизу в кислотных условиях, предпочтительно соляной кислотой, для образования соединения формулы (Е), которое затем подвергают сочетанию с соединением формулы (F) в стандартных условиях пептидного сочетания, например, с ЭДКИ в присутствии ГОБТ в ДМФ, для образования соединения формулы (G), где R13 обозначает гидроксил. Это соединение затем обрабатывают либо тозилхлоридом, либо мезилхлоридом для образования соединения формулы (G), где R13 обозначает мезил или тозил. Путем циклизации полученных таким образом тозилатов с избытком NaH в инертном растворителе, предпочтительно ТГФ, при сильном разбавлении и при комнатной температуре получают соединения формулы (Н). Защитную группу в соединениях формулы (Н) удаляют в слабокислой среде предпочтительно в присутствии трифторуксусной кислоты (ТФК), получая соединения формулы (J).
Б. Получение соединений формул (Ia), (Ib), (Ic) и (Id)
Соединения формулы (Ia) представляют собой соединения формулы (I), где n равно 1, 2 или 3; m равно 3 или 4; А обозначает -CH2-; R1 обозначает -CH2-R4, где R4 обозначает трет-бутоксикарбонил; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R3 обозначает водород, галоген, алкил или алкокси.
Соединения формулы (Ib) представляют собой соединения формулы (I), где n равно 1, 2 или 3; m равно 3 или 4; А обозначает -CH2-; R1 обозначает -CH2-R4, где R4 обозначает карбокси; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R3 обозначает водород, галоген, алкил или алкокси.
Соединения формулы (Ic) представляют собой соединения формулы (I), где n равно 1, 2 или 3; m равно 3 или 4; А обозначает -CH2-; R1 обозначает -CH2-R4, где R4 обозначает бензилоксиаминокарбонил; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R3 обозначает водород, галоген, алкил или алкокси.
Соединения формулы (Id) представляют собой соединения формулы (I), где n равно 1, 2 или 3; m равно 3 или 4; А обозначает -CH2-; R1 обозначает -CH2-R4, где R4 обозначает гидроксиаминокарбонил; R обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R3 обозначает водород, галоген, алкил или алкокси.
Соединения формул (Ia), (Ib), (Ic) и (Id) получают по приведенной ниже реакционной схеме 2, где p равно 5, 6, 7 или 8; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси, R14 обозначает трет.-бутил или бензил и R7a обозначает водород или алкоксикарбонил.
Соединения формулы (К) получают способами, приведенными в описании, или они могут быть получены способами, известными специалистам в данной области техники.
Обычно соединения формул (Ia), (Ib) и (Ic) получают сначала сочетанием соединения формулы (J) с соединением формулы (К) в стандартных условиях пептидного сочетания для образования соединения формулы (Ia). Защитную группу в соединении формулы (Ia) затем удаляют в слабо кислой среде, получая соединение формулы (Ib).
Соединение формулы (Ib) затем подвергают сочетанию с О-бензилгидроксиламином в стандартных условиях пептидного сочетания для образования соединения формулы (Ic). Бензильную защитную группу в соединении формулы (Ic) затем удаляют в условиях каталитического гидрирования, получая соединение формулы (Id).
В. Получение соединений формул (Ie) и (If)
Соединения формулы (Ie) представляют собой соединения формулы (I), где n равно 1, 2 или 3; m равно 3 или 4; А обозначает -CH2-; R1 обозначает - CH2-R4, где R4 обозначает ацетилтио; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R3 обозначает водород.
Соединения формулы (If) представляют собой соединения формулы (I), где n равно 1, 2 или 3; m равно 3 или 4; А обозначает -CH2-; R1 обозначает -CH2-R4, где R4 обозначает меркапто; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R3 обозначает водород.
Соединения формул (Ie) и (If) получают в соответствии с приведенной ниже реакционной схемой 3, где R2 и R3 имеют вышеуказанные значения и p равно 5, 6, 7 или 8.
Соединения формулы (M) являются коммерчески доступными или могут быть получены способами, известными специалистам в данной области техники.
Обычно соединения формул (Ie) и (If) получают сначала сочетанием соединения формулы (M) с соединением формулы (J) в стандартных условиях пептидного сочетания для образования соединения формулы (Ie). Путем обработки соединений формулы (Ie) концентрированным NH4OH в метаноле получают соответствующие соединения формулы (If).
Г. Получение индивидуальных стереоизомеров соединений формулы (К)
Соединения формулы (К):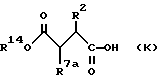
где R14 обозначает трет-бутил или бензил и R7a обозначает водород, алкоксикарбонил, гидроксикарбамоил, карбокси или необязательно замещенный карбамоил, используют для получения соединений формулы (I)
Индивидуальные стереоизомеры соединений формулы (К) используют для получения индивидуальных стереоизомеров соединений формулы (I). В частности, соединения нижеприведенной формулы (Ka):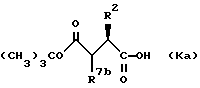
где R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R7b обозначает водород, являются стереоизомерами соединений формулы (К), которые имеют R-конфигурацию вокруг атома углерода, к которому присоединен заместитель R2.
Соединения формулы (Ka) получают в соответствии с приведенной ниже реакционной схемой 4, где R2 и R7b имеют значения, указанные выше.
Аналогичным способом, но заменяя сультам D-(-)-2,10-камфоры на сультам L-(+)-2,10-камфоры, получали соответствующие индивидуальные изомеры с S-конфигурацией.
Соединения формулы (HH) являются коммерчески доступными или могут быть получены в соответствии со способами, известными специалистам в данной области техники, например способом, описанным ниже в примере 11. Сультам L-(+)-2,10-камфоры и сультам D-(-)-2,10-камфоры являются коммерчески доступными, например, выпускаются фирмой Aldrich.
Обычно соединения формулы (Ka) получают, конденсируя сначала соединение формулы (HH) с сультамом L-(+)-2,10-камфоры и получая соединение формулы (N). Используя NaHMDS для образования аниона в течение часа, реакцию затем останавливают добавлением трет. -бутилбромацетата для получения соответствующего эфира формулы (Q). Затем удаляют камфорную группу в щелочной среде, получая индивидуальный стереоизомер соединения формулы (Ka), где углерод, к которому присоединен заместитель R2, находится в (R)-конфигурации.
Соединения формулы (Kb)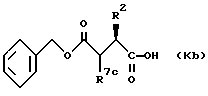
где R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R7c обозначает алкоксикарбонил,
также являются индивидуальными стереоизомерами соединений формулы (К) и их получают в соответствии с приведенной ниже реакционной схемой 5, где R2 и R7c имеют значения, указанные выше.
Соединения формул (R) и (Т) являются коммерчески доступными или могут быть получены в соответствии со способами, известными специалистам в данной области техники.
Обычно соединения формулы (Kb) получают, сначала обрабатывая соединение формулы (R) изобутеном и каталитическим количеством концентрированной H2SO4 в метиленхлориде с последующей перегонкой для получения соединения формулы (S). Соединение формулы (S) затем подвергают взаимодействию с соединением формулы (Т) в присутствии трет.-бутоксида калия для получения соединения формулы (U). Путем гидролиза соединения формулы (U) в кислой среде, предпочтительно с трифторуксусной кислотой при комнатной температуре, получают соединение формулы (Kb), где R7c обозначает алкоксикарбонил.
Соединения формулы (К), где R7a обозначает карбокси, могут быть получены из соединений формулы (Kb), где R7c обозначает алкоксикарбонил, способами, известными специалистам в данной области техники.
В дополнение к вышеописанным способам получения индивидуальных стереоизомеров соединений формулы (К), соединения формулы (К), где R7a обозначает алкил, могут быть получены обработкой соединения формулы (К), где R7a обозначает водород, в апротонном растворителе, например ТГФ, в присутствии NaN(тетраметилсилана)2 с галоалканом, предпочтительно йодметаном, для получения соединения формулы (К), где R7a обозначает алкил.
Д. Получение соединений формулы (Ig)
Соединения формулы (Ig) представляют собой соединения формулы (I), где n равно 1, 2 или 3; m равно 3 или 4; А обозначает -CH2-; R1 обозначает -CH2-R4, где R4 обозначает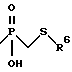
где R6 обозначает необязательно замещенный арил, причем арильная группа обозначает хинол-2-ил, нафт-1-ил, нафт-2-ил, пиридил или фенил; R2 обозначает алкил и R3 обозначает водород.
Соединения формулы (Ig) могут быть получены в соответствии с приведенной ниже реакционной схемой 6, где p равно 5, 6, 7 или 8; R2, R3 и R6 имеют указанные выше значения и R12a обозначает мезил или тозил.
Соединения формулы (W) могут быть получены в соответствии со способами, известными специалистам в данной области техники, или могут быть получены в соответствии со способом, описанным ниже в примере 19. Соединения формулы (Z) являются коммерчески доступными или могут быть получены в соответствии со способами, известными специалистам в данной области техники.
Обычно соединения формулы (Ig) получают, сначала обрабатывая соединение формулы (W) формамидом для получения соединения формулы (X). Соединение формулы (X) затем обрабатывают хлоридом тозила или мезила в щелочной среде для получения соединения формулы (Y). Соединение формулы (Y) затем подвергают взаимодействию с солью соединения формулы (Z) (предпочтительно натриевой солью, полученной взаимодействием соединения формулы (Z) с гидридом натрия) для получения соединения формулы (AA). Соединение формулы (AA) затем подвергают гидролизу в щелочной среде для получения соединения формулы (BB). Соединение формулы (BB) подвергают сочетанию с соединением формулы (J) в стандартных для пептидов условиях, предпочтительно с 1,1'-карбонилдиимидазолом, для получения соединения формулы (Ig).
Е. Получение соединений формул (Ih), (Ii) и (Ij)
Соединения формул (Ih), (Ii) и (Ij) являются соединениями формулы (Ib), формулы (Ic) и формулы (Id) соответственно, как описано выше в разделе Б, где индольное кольцо является полностью насыщенным. Их получают в соответствии с приведенной ниже реакционной схемой 7, где R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси; R7a обозначает водород и p равно 5, 6, 7 или 8.
Обычно соединения формул (Ih), (Ii) и (Ij) получают, восстанавливая сначала соединение формулы (Ib) в условиях каталитического гидрирования для образования соединения формулы (Ih). Соединение формулы (Ih) затем подвергают взаимодействию с О-бензилгидроксиламином в условиях стандартного пептидного сочетания для получения соединения формулы (Ii). После этого из соединения формулы (Ii) удаляют бензильную защитную группу в условиях каталитического гидрирования для получения соединения формулы (Ij).
Ж. Получение соединений формул (Ik), (Il), (Im) и (In)
Соединения формулы (Ik) представляют собой соединения формулы (I) с аллильной связью, где n равно 2 или 3; m равно 4; А обозначает - NR11, где R11 обозначает водород или алкил; R1 обозначает -CH2-R4, где R4 обозначает трет-бутоксикарбонил; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси.
Соединения формулы (II) представляют собой соединения формулы (I) с аллильной связью, где n равно 2 или 3; m равно 4; А обозначает -NR11, где R11 обозначает водород или алкил; R1 обозначает -CH2-R4, где R4 обозначает карбокси; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси.
Соединения формулы (Im) представляют собой соединения формулы (I) с аллильной связью, где n равно 2 или 3; m равно 4; А обозначает -NR11, где R11 обозначает водород или алкил; R1 обозначает -CH2-R4, где R4 обозначает бензилоксиаминокарбонил; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси.
Соединения формулы (In) представляют собой соединения формулы (I), где n равно 2 или 3; m равно 4; А обозначает -NR11, где R11 обозначает водород или алкил; R1 обозначает -CH2-R4 где R4 обозначает гидроксиаминокарбонил; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси.
Соединения формул (Ik), (Il), (Im) и (In) получают в соответствии с реакционной схемой 8 (см. в конце описания), где n равно 2 или 3; R2 и R11 имеют указанные выше значения; R14 обозначает трет.-бутил; R7a обозначает водород; и ВОС обозначает трет.-бутоксикарбонил.
Соединения формулы (К) получают способами, известными специалистам в данной области техники, или представленными в описании способами.
Обычно соединения формул (Ik), (II), (Im) и (In) получают сначала взаимодействием соединений формулы (F) с диаминоалканом или моноалкилзамещенным диаминоалканом в стандартных условиях пептидного сочетания, например с ГОБТ и ЭДКИ, в инертном растворителе, например ДМФ, для образования соединения формулы (DD). Соединение формулы (DD) затем подвергают взаимодействию с транс-1,4-дихлорбут-2-еном в щелочной среде для образования соединения формулы (ЕЕ). Аминозащитную группу соединения формулы (ЕЕ) удаляют в слабокислой среде, предпочтительно с трифторуксусной кислотой, для образования соединения формулы (FF).
Соединение формулы (FF) затем подвергают сочетанию с соединением формулы (К) в стандартных условиях пептидного сочетания, например с ГОБТ и ЭДКИ, для образования соединения формулы (Ik). Защитную группу соединения формулы (Ik) удаляют в слабокислой среде, предпочтительно с трифторуксусной кислотой, для образования соединения формулы (Il). Соединение формулы (II) затем обрабатывают О-бензилгидроксиламином в стандартных условиях пептидного сочетания для получения соединения формулы (Im). Защитную группу соединения формулы (Im) удаляют в условиях каталитического гидрирования для получения соединения формулы (In).
З. Получение соединений формул (Io) и (Ip)
Соединения формулы (Io) представляют собой соединения формулы (I), где n равно 1, 2 или 3; m равно 3 или 4; А обозначает -CH2-, R1 обозначает -NH-CH(R9)-R10, где R9 обозначает водород, алкил или аралкил и R10 обозначает аралкоксикарбонил; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси.
Соединения формулы (Ip) представляют собой соединения формулы (I), где n равно 1, 2 или 3; m равно 3 или 4; А обозначает -CH2-, R1 обозначает -NH-CH(R9)-R10, R9 обозначает водород, алкил или аралкил и R10 обозначает карбокси; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси.
Соединения формул (Io) и (Ip) получают в соответствии с приведенной ниже реакционной схемой 9, где p равно 5, 6, 7 или 8; R2, R3 и R9 имеют указанные выше значения.
Соединения формулы (JJ) получают способами, известными специалистам в данной области техники, или способом, описанным ниже в примере 36.
Обычно соединения формулы (Il) и (Im) получают сначала обработкой соединения формулы (JJ) ангидридом трифторметансульфокислоты, а затем обработкой соединением формулы (KK) в щелочной среде для образования соединения формулы (LL). Затем соединение формулы (LL) подвергают гидролизу в слабокислой среде, предпочтительно с трифторуксусной кислотой, для образования соединения формулы (ММ). После чего соединение формулы (ММ) подвергают сочетанию с соединением формулы (J) в стандартных условиях пептидного сочетания для образования соединения формулы (Io). Затем в соединении (Io) удаляют защитную группу для образования соединения формулы (Ip).
Кроме того, все соединения формулы (I), существующие в виде свободного основания, могут быть превращены в их фармацевтически приемлемые соли путем обработки подходящей неорганической или органической кислотой. Соли соединений формулы (I) могут также быть превращены в форму свободного основания или другую соль.
В целом, соединения формул (Ia), (Ib), (Ic), (Id), (Ie), (If), (Ig), (Ih), (Ii), (Ij), (Ik), (Il), (Im), (In), (Io) и (Ip), которые все являются соединениями формулы (I), получают путем:
1. взаимодействия соединения формулы (К), где R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R7a обозначает водород или алкоксикарбонил; и R14 обозначает трет.-бутил или бензил; с соединением формулы (J), где p равно 5, 6, 7 или 8; и R3 обозначает водород, галоген, алкил или алкокси; для получения соединения формулы (Ia), где p, R2, R3, R7a и R14 имеют значения, указанные для соединений формул (К) и (J);
2. обработки соединения формулы (Ia), где p равно 5, 6, 7 или 8; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси; R7a обозначает водород или алкоксикарбонил; R14 обозначает трет. -бутил или бензил; для получения соединения формулы (Ib), где p, R2, R3 и R7a имеют значения, указанные для соединения формулы (Ia);
3. обработки соединения формулы (Ib), где p равно 5, 6, 7 или 8; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси; и R7a обозначает водород или алкоксикарбонил; с О-бензилгидроксиламином для получения соединения формулы (Ic), где p, R2, R3 и R7а имеют значения, указанные для соединения формулы (Ib);
4. обработки соединения формулы (Ic), где p равно 5, 6, 7 или 8; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси; и R7a обозначает водород или алкоксикарбонил; для получения соединения формулы (Id), где p, R2, R3 и R7a имеют значения, указанные для соединения формулы (Ic);
5. взаимодействия соединения формулы (М), где R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; с соединением формулы (J), где p равно 5, 6, 7 или 8; и R3 обозначает водород, для получения соединения формулы (Ie), где p, R2 и R3 имеют значения, указанные для соединений формул (М) и (J);
6. обработки соединения формулы (Ie), где p равно 5, 6, 7 или 8; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R3 обозначает водород; для получения соединения формулы (If), где p, R2, R3 имеют значения, указанные для соединения формулы (Ie);
7. взаимодействия соединения формулы (BB), где R2 обозначает алкил; и R6 обозначает необязательно замещенный арил, причем арильная группа обозначает хинол-2-ил, нафт-1-ил, нафт-2-ил и фенил; с соединением формулы (J), где p равно 5, 6, 7 или 8; и R3 обозначает водород, для получения соединения формулы (Ig), где p, R2, R3 и R6 имеют значения, указанные для соединений формул (BB) и (J);
8. обработки соединения формулы (Ib), где p равно 5, 6, 7 или 8; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R3 обозначает водород, галоген, алкил или алкокси; для получения соединения формулы (Ih), где p, R2 и R3 имеют значения, указанные для соединения формулы (Ib);
9. обработки соединения формулы (Ih), где p равно 5, 6, 7 или 8; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R3 обозначает водород, галоген, алкил или алкокси; для получения соединения формулы (Ii), где p, R2 и R3 имеют значения, указанные для соединения формулы (Ih);
10. обработки соединения формулы (Ii), где p равно 5, 6, 7 или 8; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R3 обозначает водород, галоген, алкил или алкокси; для получения соединения формулы (Ij), где p, R2 и R3 имеют значения, указанные для соединения формулы (Ii);
11. взаимодействия соединения формулы (FF), где n равно 2 или 3; R3 обозначает водород, галоген, алкил или алкокси; и R11 обозначает водород или алкил; с соединением формулы (К), где R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R7a обозначает водород; и R14 обозначает трет. -бутил; для получения соединения формулы (Ik), где n, R2, R3, R7a, R11 и R14 имеют значения, указанные для соединений формул (FF) и (К);
12. обработки соединения формулы (Ik), где n равно 2 или 3; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси; R7a обозначает водород; R11 обозначает водород или алкил и R14 обозначает трет.-бутил; для получения соединения формулы (Il), где n, R2, R3, R7a и R11 имеют значения, указанные для соединения формулы (Ik);
13. обработки соединения формулы (Il), где n равно 2 или 3; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R3 обозначает водород, галоген, алкил или алкокси; R7a обозначает водород; R11 обозначает водород или алкил; О-бензилгидроксиламином для получения соединения формулы (Im), где n, R2, R3, R7a и R11 имеют значения, указанные для соединения формулы (Il);
14. обработки соединения формулы (Im), где n равно 2 или 3; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R3 обозначает водород, галоген, алкил или алкокси; R7a обозначает водород; R11 обозначает водород или алкил; для получения соединения формулы (In), где n, R2, R3, R7a и R11 имеют значения, указанные для соединения формулы (GG);
15. взаимодействия соединения формулы (ММ), где R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; и R9 обозначает водород, алкил или аралкил; с соединением формулы (J), где p равно 5, 6, 7 или 8; и R3 обозначает водород, галоген, алкил или алкокси, для получения соединения формулы (Io), где p, R2, R3 и R9 имеют значения, указанные для соединений формул (ММ) и (J); и
16. обработки соединения формулы (Io), где p равно 5, 6, 7 или 8; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил; R3 обозначает водород, галоген, алкил или алкокси; и R9 обозначает водород, алкил или аралкил; для получения соединения формулы (Ip), где p, R2, R3 и R9 имеют значения, указанные для соединения формулы (Io).
Следующие примеры приведены в качестве рекомендаций для осуществления настоящего изобретения на практике и не ограничивают объем изобретения.
Пример 1
Соединение формулы (Е)
А. 6-Циано-1-гексанол (7,1 г, 55,8 ммоля) растворяли в 30 мл уксусного ангидрида в атмосфере аргона. К этому материалу добавляли по каплям 5,3 мл (65,4 ммоля) пиридина и смесь оставляли перемешиваться в течение 2 часов. Содержимое колбы затем сливали в химический стакан, содержащий 50 мл ледяной воды, и материал перемешивали в течение 15 минут. Затем смесь перемещали в делительную воронку объемом 250 мл и добавляли простой эфир (100 мл). После встряхивания эфирную фазу выделяли, а водную фазу дважды промывали простым эфиром (2х100 мл). Объединенную эфирную фазу промывали соляным раствором, сушили (MgSO4) и фильтровали. Путем выпаривания (роторный испаритель и вакуумный насос) получали 6-циано-1-ацетоксигексан (соединение формулы (С)), которое непосредственно использовали на следующей стадии.
Б. 6-Циано-1-ацетоксигексан (55,8 ммоля) растворяли в приблизительно 100 мл уксусного ангидрида в реакционном сосуде Парра (объемом 500 мл). Сюда же добавляли уксусную кислоту (0,5 мл), а затем оксид платины (100 мг). Сосуд помещали в гидрогенизатор Парра и пропускали газообразный водород при давлении 40 фунт/дюйм2 (2,8 кг/см3). Материал встряхивали в течение 12 часов, фильтровали через целит (для удаления катализатора), загружали свежий оксид платины (100 мг) и водород (при давлении 40 фунт/дюйм2) и встряхивали еще в течение 24 часов. Материал фильтровали через целит и все летучие компоненты удаляли при пониженном давлении (роторный испаритель). Целевой 1-ацетокси-7-ацетамидогептан был достаточно чистым для использования на следующей стадии (выход 11,8 г).
В. 1-Ацетокси-7-ацетамидогептан (11,8 г, 54,3 ммоля) растворяли в 20 мл метанола в круглодонной колбе объемом 200 мл. Сюда же добавляли 50 мл 40%-ной водной соляной кислоты и смесь нагревали с обратным холодильником в течение 60 часов. Все летучие компоненты удаляли при пониженном давлении. Целевой 7-амино-1- гептанол получали в виде кристаллической гидрохлоридной соли, температура плавления 74-81oC, МС: 131 (MH+).
Пример 2
Соединение формулы (G)
А. N-метилморфолин (2,2 мл, 19,7 ммоля) добавляли по каплям при комнатной температуре к гидрохлоридной соли 7-амино-1-гептанола (3,3 г, 19,7 ммоля) в 50 мл сухого ДМФ в атмосфере аргона при перемешивании. После перемешивания в течение 5 минут добавляли следующую смесь: N-трет.-бутоксикарбонил-L-триптофан (5 г, 16,45 ммоля), 1-гидроксибензотриазол (2,52 г, 16,45 ммоля) и гидрохлорид ЭДКИ (4,73 г, 24,7 ммоля). Смесь перемешивали в течение 2 часов и затем ДМФ удаляли при пониженном давлении. Остаток экстрагировали холодной 2,5%-ной HCl (100 мл) и этилацетатом (3х100 мл) и перемещали в делительную воронку. Органическую фазу выделяли и промывали последовательно холодной 2,5%-ной HCl (100 мл), а затем соляным раствором (100 мл). Этилацетатную фазу сушили (MgSO4), фильтровали и концентрировали, получая N-трет.-бутоксикарбонил-L- триптофан-N'-(7-гидроксигептил)амид.
ИК (чистый): 3300, 2921, 1685, 1645, 1490, 1356, 1157 см-1.
1H-ЯМР (80 МГц, CDCl3): δ 0,98-1,62 (m, 10Н, -(CH2)5-), 1,45 (s, 9H, трет. -бутил), 2,86-3,32 (m, 4H, CH-CH2, HN-CH2), 3,68 (t, 2Н, J=5,6 Гц, -CH2ОН), 4,22-4,55 (m, 1H, CH), 5,12-5,32 (широкий d, 1H, NH-CH), 5,65-5,9 (широкий t, 1H, NH-CH2), 6,98-7,92 (m, 5H, ArH), 8,63 (широкий s, индол NH).
Б. Раствор N-трет. -бутоксикарбонил-L-триптофан-N'-(7- гидроксигептил)амида (8,2 г) в 150 мл безводного пиридина охлаждали до 0oC (ледяная баня). Пара-толуолсульфонилхлорид (4,7 г) добавляли к раствору в виде одной порции и охлажденную смесь оставляли перемешиваться в течение 7 часов. Реакцию останавливали, добавляя 50 мл ледяной воды и удаляя все летучие компоненты при пониженном давлении. Продукт N-трет.-бутоксикарбонил-L-триптофан-N'-[7-(4'-метилфен-1- илсульфонилокси)гептил]амид выделяли с помощью колоночной хроматографии на силикагеле, используя в качестве растворителей для элюирования смесь 10-40% этилацетат/гексан. Этот материал кристаллизовался при стоянии, МС: 572 (MH+).
В. В другом варианте к раствору, содержащему N-трет.- бутоксикарбонил-L-триптофан (5,0 г, 16,45 ммоля), 6-амино-1- гексанол (2,31 г, 19,74 ммоля) и 1-гидроксибензотриазол H2О (2,52 г, 16,45 ммоля) в сухом ДМФ (50 мл), при комнатной температуре в атмосфере аргона добавляли ЭДКИ (4,73 г, 24,68 ммоля). После перемешивания в течение ночи ДМФ удаляли в высоком вакууме. Осадок распределяли между этилацетатом (150 мл) и 1Н HCl (75 мл). Органический слой затем промывали 1Н HCl (75 мл), насыщенным раствором бикарбоната натрия (2х75 мл) и в завершение соляным раствором (50 мл). Органический слой сушили (MgSO4) и выпаривали досуха, получая 6,45 г (97%) N-трет.-бутоксикарбонил-L-триптофан- N'-(6-гидроксигексил)амида в виде белой пены, МС: 404,3 (М+Н)+. Чистота продукта подтверждена анализом с помощью высокоэффективной жидкостной хроматографии.
Г. Продолжая далее процесс, к N-трет.-бутоксикарбонил-L- триптофан-N'-(6-гидроксигексил)амиду (5,5 г, 13,64 ммоля) в 150 мл сухого пиридина при 0oC в атмосфере аргона добавляли 3,9 г (20,46 ммолей) пара-толуолсульфонилхлорида. Гомогенный раствор перемешивали при этой температуре в течение ночи. Реакцию останавливали добавлением 25 мл воды и избыток пиридина удаляли при пониженном давлении. Остаток растворяли в этилацетате (120 мл) и промывали 1Н HCl (2х50 мл), насыщенным раствором NaHCO3 (50 мл) и соляным раствором (50 мл). Органический слой сушили (MgSO4) и выпаривали для получения N-трет-бутоксикарбонил-L-триптофан-N'-[6- (4'-метилфен-1-илсульфонилокси)гексил]амида в виде бледно-желтого масла (5,77 г, 76%), МС: 558,3 (М+Н)+.
Д. К 5-гидрокситриптофану (3,5 г, поставляется фирмой Sigma) и триэтиламину (5,6 мл) в воде (25 мл) и тетрагидрофуране (50 мл) добавляли BOC-ON [2-(трет. -бутоксикарбонилоксиимино)-2- фенилацетонитрил] . Спустя 2,5 часа тетрагидрофуран удаляли, добавляли 10% Na2CO3 (20 мл) и смесь разделяли с помощью простого эфира (50 мл). Водную фракцию далее экстрагировали простым эфиром (20 мл) и затем подкисляли холодной 10% HCl в двухфазной системе, содержащей этилацетат (100 мл). Этилацетатную фракцию отделяли и промывали водой (30 мл), соляным раствором, сушили над безводным MgSO4 и концентрировали до получения сиропа. Сироп подвергали взаимодействию с 6-амино-1-гексанолом способом, аналогичным описанному выше в примере 1В для получения N-трет. - бутоксикарбонил-L-(5-гидрокси)триптофан-N'-(6- гидроксигексил)амида. Половину этого продукта экстрагировали 40 мл ДМФ и обрабатывали K2CO3 (5 г) и йодметаном (1,2 г) при комнатной температуре в течение ночи. Реакционную смесь затем распределяли между водой (50 мл) и этилацетатом (80 мл), органическую фракцию затем промывали водой (2х20 мл), соляным раствором, сушили над безводным MgSO4 и концентрировали до получения масла. Затем производили очистку продукта, N-трет.-бутоксикарбонил-L-(5- метокси)триптофан-N'-(6-гидроксигексил)амида, с помощью хроматографии на силикагеле; 1H-ЯМР: δ CDCl3): 0,9-1,6 (m, CH2, 8Н); 1,45 (s, 9Н); 2,7-3,3 (m, 5H); 3,6 (t, 2Н); 13,85 (s, 3H); 4,35 (m, 1H); 5,3 (широкий d, 1H); 5,85 (широкий t, 1H); 6,75-8,3 (m, 4H); 8,73 (широкий s, 1H).
E. Аналогичным способом были получены следующие соединения:
N-трет. -бутоксикарбонил-L-(5-этокси)триптофан-N'-(6- гидроксигексил)амид;
N-трет. -бутоксикарбонил-L-(5-пропокси)триптофан-N'-(6- гидроксигексил)амид;
N-трет. -бутоксикарбонил-L-(5-этил)триптофан-N'-(6- гидроксигексил)амид; и
N-трет.-бутоксикарбонил-L-(4-метил)триптофан-N'-(6- гидроксигексил)амид.
Пример 3
Соединения формулы (Н)
А. N-трет.-бутоксикарбонил-L-триптофан-N'-[7-(4'-метилфен- 1-илсульфонилокси)гептил] амид (6,78 г) порциями добавляли к раствору NaH (60% в масле, 1,9 г) в 1,1 литр безводного тетрагидрофурана и оставляли перемешиваться в течение ночи. Реакционную смесь концентрировали и экстрагировали водой (150 мл) и CH2Cl2 (150 мл). Водную фазу слегка подкисляли 2,5%-ной HCl (pH 3-4) и органическую фазу выделяли (3х150 мл) и промывали последовательно холодной 2,5% HCl (150 мл), 5% NaHCO3 (150 мл) и соляным раствором (150 мл). Органическую фазу сушили (MgSO4), фильтровали и концентрировали, получая желто-зеленый полутвердый материал. Путем очистки с помощью хроматографии на силикагеле получали (11S)-11-N'-(бензилоксикарбонил)амино-10-оксо-1,9- диазатрицикло[11.6.1.014,19] эйкоза-13(20), 14(19),15,17-тетраен с температурой плавления 208-209oC, МС: 400 (М+Н)+.
Б. В другом варианте к N-трет.-бутоксикарбонил-L-триптофан-N'- [6-(4'-метилфен-1-илсульфонилокси)гексил] амиду (5,0 г, 8,97 ммолей) в одном литре сухого ТГФ при 0oC в атмосфере аргона добавляли 4 эквивалента 60% NaH (1,44 г, 36 ммолей) небольшими порциями в течение 10 минут. Затем смесь перемешивали в течение ночи при комнатной температуре. Полученную желтую смесь упаривали до ~ 200 мл и затем добавляли 1 литр дистиллированной воды. Затем смесь подкисляли 1Н HCl при интенсивном перемешивании. Желтый осадок собирали путем фильтрации и сушили над P2О5 в высоком вакууме в течение ночи. Сухой неочищенный продукт (8 г) хроматографировали на силикагеле 60, элюируя 30% этилацетатом в CH2Cl2 для получения 1,2 г (35%) (10S)-10-N'- (бензилоксикарбонил)амино-9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18), 14,16-тетраена в виде белого порошка, МС: 386 (М+Н)+, tпл 222-223oC.
В. В другом варианте к раствору N-трет.-бутоксикарбонил-L- триптофан-N'-[6-(4'-метилфен-1-илсульфонилокси)гексил]амиду (1,21 г, 2,17 ммолей) в 45 мл химически чистого метиленхлорида добавляли 15 мл 40% водного КОН и 0,3 эквивалента хлорида бензилтриэтиламмония (0,65 ммоля, 148 мг). Двухфазную смесь интенсивно перемешивали при комнатной температуре в течение ночи. Органический слой отделяли и водный слой экстрагировали 25 мл метиленхлорида. Объединенный органический слой промывали водой (25 мл), сушили (MgSO4) и выпаривали досуха. Остаток перемешивали в 10% эфире в петролейном эфире при 0oC в течение 15 минут и фильтровали для получения 792 мг (93%) (10S)-10-N'- (бензилоксикарбонил)амино-9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19),13(18),14,16- тетраена в виде белого порошка.
Пример 4
Соединения формулы (J)
А. (11S)-10-N'-(бензилоксикарбонил)амино-10-оксо-1,9- диазатрицикло[11.6.1.014,19]эйкоза-13(20),14(19),15,17-тетраена (850 мг) растворяли в 5 мл раствора 10% трифторуксусной кислоты в метиленхлориде и перемешивали в течение 1 часа. Летучие компоненты удаляли при пониженном давлении. Остаток экстрагировали CH2Cl2 (40 мл) и 1H NaOH (40 мл) и помещали в делительную воронку. Органическую фазу выделяли и промывали соляным раствором, сушили (MgSO4), фильтровали и концентрировали для получения 654 мг (11S)-10-амино-10-оксо-1,9-диазатрицикло[11.6.1.014,19]эйкоза- 13(20),14(19),15,17-тетраена.
Б. В другом варианте (10S)-10-N'-(бензилоксикарбонил)амино-9- оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13 (18),14,16-тетраен (0,5 ммоля, 193 мг) перемешивали в 20% ТФК/CH2Cl2 (10 мл) при комнатной температуре в течение 2 часов. Избыток ТФК и растворителя удаляли при пониженном давлении. Остаток растворяли в этилацетате (30 мл) и промывали 1H HCl (25 мл), соляным раствором (10 мл) и сушили (MgSO4). Путем выпаривания досуха получали 140 мг (кол.) (10S)-10-амино-9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18),14,16- тетраена в виде белой пены, tпл 157-160oC, МС: 286,2 (М+Н)+.
Пример 5
Соединения формулы (Ia)
А. К перемешиваемому раствору, содержащему (11S)-10-амино-10- оксо-1,9-диазатрицикло[11.6.1.014,19]эйкоза-13(20),14(19),15,17- тетраен (654 мг) и рацемическую 4-метил-2-(трет. - бутоксикарбонилметил)пентановую кислоту (800 мг) в 30 мл безводного ДМФ, добавляли в атмосфере аргона 1-гидроксибензтриазол (360 мг), а затем ЭДКИ (940 мг). Смесь оставляли перемешиваться в течение ночи и затем удаляли ДМФ при пониженном давлении. Остаток экстрагировали смесью, содержащей CH2Cl2 (100 мл) и 1,5%-ную холодную HCl (100 мл), и помещали в делительную воронку. Органическую фазу выделяли и промывали последовательно 1,5% HCl (100 мл), 5% NaHCO3 (100 мл) и соляным раствором (100 мл). Фазу CH2Cl2 сушили (MgSO4), фильтровали и концентрировали, получая полукристаллический продукт: трет.-бутиловый эфир (11S)-5-метил-3-(10-оксо-1,9-диазатрицикло [11.6.1.014,19]эйкоза-13 (20),14 (19),15,17-тетраен-11- илкарбамоил)гексановой кислоты. Два отдельных стереоизомера этого соединения разделяли с помощью хроматографии на силикагеле с этилацетатом/гексаном в качестве растворителя для элюирования. Менее полярный стереоизомер имел температуру плавления 154-157oC, [α]
Б. В другом варианте к раствору, содержащему (2R)-4-метил-2- (трет.-бутоксикарбонилметил)пентановую кислоту, полученную описанным выше способом, (2,39 г, 10,4 ммолей), ГОБТ•H2О (2,5 г, 1 экв.), N-метилморфолин (2,3 мл, 2 экв. ) и (10S)-10-амино-9-оксо- 1,8-диазатрицикло[10.6.1.013,18]нонадека-12(19), 13(18),14,16- тетраен (2,96 г, 1 экв.) в сухом ДМФ (200 мл), в атмосфере аргона добавляли ЭДКИ (3,96 г, 2,0 экв.). Полученную смесь перемешивали в течение ночи, затем на следующее утро удаляли ДМФ при 35oC в высоком вакууме. Остаток распределяли между CH2Cl2 (150 мл)/водой (75 мл), затем органический слой промывали 0,5Н HCl (2х75 мл), насыщенным NaHCO3 (2х75 мл) и в завершение соляным раствором (1х75 мл). После сушки слоя CH2Cl2 над Na2SO4 его фильтровали и упаривали досуха. Путем очистки с помощью колоночной хроматографии (петролейный эфир или смесь до 30% этилацетат/петролейный эфир) получали трет. -бутиловый эфир (3R,10S)-5-метил-3-(9-оксо-1,8- диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18),14,16- тетраен-10-илкарбамоил)гексановой кислоты (3,24 г, 62,7%).
В. Аналогичным способом были получены следующие соединения формулы (Ia):
трет. -бутиловый эфир (3R, 10S)-4-фенил-3-(9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16- тетраен-10-илкарбамоил)бутановой кислоты, МС: 532 (М+Н)+;
трет. -бутиловый эфир (3R, 10S)-4-циклогексил-3-(9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13 (18), 14,16- тетраен-10-илкарбамоил)бутановой кислоты, МС: 538 (М+Н)+;
трет. -бутиловый эфир (3R, 10S)-6-фенил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19), 13 (18), 14,16-тетраен-10- илкарбамоил)гексановой кислоты, МС: 560 (М+Н)+;
трет.-бутиловый эфир (3R,10S)-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18]нонадека-12(19),13(18),14,16-тетраен-10- илкарбамоил)гексановой кислоты, МС: 484 (М+Н)+; и
бензиловый эфир (3R,10S)-2-метоксикарбонил-5-метил-3-(9- оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13 (18),14,16-тетраен-10-илкарбамоил)гексановой кислоты, МС: 590 (М+Н)+.
Г. В другом варианте к (2R)-4-метил-2-(трет. - бутоксикарбонилметил)пентановой кислоте (1 г, 4,34 ммоля) в сухом ТГФ (100 мл) при -78oC в атмосфере аргона добавляли по каплям NaN(тетраметилсилан)2 (1,0 М в ТГФ, 10,9 мл, 2,5 экв.) и смесь перемешивали в течение 1 часа. Добавляли йодметан (0,33 мл, 1,2 экв.) и образовавшуюся смесь перемешивали в течение ночи при температуре от -78oC до комнатной. На следующий день реакцию останавливали путем добавления воды (100 мл). После экстрагирования эфиром (3х100 мл) водный слой объединяли с этилацетатом и при перемешивании добавляли 4Н HCl до pH 2. Также добавляли до насыщения хлорид натрия и водный слой экстрагировали этилацетатом (3х100 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали, получая (2R)-4-метил-2- [(метил)(трет.-бутоксикарбонил)метил]пентановую кислоту в виде темно-коричневого масла (1 г). К этому неочищенному продукту реакции (500 мг) и (10S)-10-амино-9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18), 14,16-тетраену (399 мг, 0,7 экв.) в сухом ДМФ при 0oC в атмосфере аргона добавляли ГОБТ•H2О (1,1 экв., 234 мг), а затем ЭДКИ (663 мг, 2,5 экв). Образовавшуюся смесь перемешивали в течение ночи при температуре от 0oC до комнатной. Большую часть ДМФ удаляли с помощью вакуумной отгонки при 65oC. Затем остаток распределяли между CH2Cl2 (150 мл)/водой. После промывки 0,5Н HCl (2х75 мл), насыщенным NaHCO3 (2х75 мл) и соляным раствором (1х75 мл) органический слой сушили над Na2SO4, фильтровали и упаривали досуха. Неочищенный материал очищали с помощью мгновенной колоночной хроматографии на кремнеземе, элюируя 30% этилацетатом в петролейном эфире, получая на выходе смесь трех соединений, двух индивидуальных стереоизомеров трет.-бутилового эфира (3R,10S)-2-метил-5-метил-3-(9-оксо-1,8 -диазатрицикло [10.6.1.013,18]нонадека-12(19), 13(18),14,16-тетраен-10- илкарбамоил)гексановой кислоты и трет.-бутилового эфира (3R,10S)-5- метил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18]нонадека-12(19), 13(18),14,16-тетраен-10-илкарбамоил)гексановой кислоты. Дальнейшая очистка предусматривает выделение трех соединений: трет. -бутилового эфира (3R,10S)-5-метил-3-(9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18),14,16-тетраен-10-илкарбамоил)гексановой кислоты (13 мг) в виде белого твердого вещества; смеси 1:1 стереоизомеров (5 мг) в виде белого твердого вещества и менее полярного стереоизомера трет. -бутилового эфира (3R,10S)-2-метил-5- метил-3-(9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека- 12(19),13(18),14,16-тетраен-10-илкарбамоил)гексановой кислоты (15 мг); 300 МГц, 1H-ЯМР в CDCl3 (менее полярный диастереомер): δ (-0,2)-(-0,05) (m, 1Н); 0,5-0,7 (m, 1Н); 0,9 (dd, J= 4 Гц, J=6,7 Гц, 6Н); 1,15 (d, J=8,4 Гц, 3H); 1,18-1,4 (m, 3Н); 1,41 (s, 9Н); 1,45-1,73 (m, 4Н); 1,75-1,8 (m, 2Н); 2,5-2,7 (m, 3Н); 2,89 (dd, J=10,9 Гц, J= 15 Гц, 1Н); 3,34-3,5 (m, 2Н); 3,95- 4,1 (m, 1Н); 4,25-4,4 (m, 1Н); 4,72-4,82 (m, 1Н); 5,22-5,3 (m, 1Н); 6,52 (d, J=7,5 Гц, 1Н); 6,91 (s, 1H); 7,13 (dd, J= 6,7 Гц, J=8,4 Гц, 1Н); 7,22 (dd, J=5 Гц, J=7,1 Гц, 1H); 7,34 (d, J= 8,4 Гц, 1H); 7,84 (d,J=8,4 Гц, 1H).
Пример 6
Соединения формулы (Ib)
А. В менее полярный стереоизомер трет.-бутилового эфира (11S)- 5-метил-3-(10-оксо-1,9-диазатрицикло[11.6.1.014,19] эйкоза- 13(20), 14(19), 15,17-тетраен-11-илкарбамоил)гексановой кислоты (300 мг) добавляли 5 мл 10%-ного раствора трифторуксусной кислоты в метиленхлориде таким образом, чтобы покрыть его, и оставляли перемешиваться. Через 2,5 часа тонкослойная хроматография показала, что реакция закончена. Все летучие компоненты удаляли при пониженном давлении. Остаток экстрагировали CH2Cl2 (40 мл), помещали в делительную воронку и постепенно промывали 0,5%-ной HCl (40 мл) и соляным раствором (40 мл). Органическую фазу сушили (MgSO4), фильтровали и концентрировали для получения менее полярного стереоизомера (11S)-5-метил-3-(10-оксо-1,9- диазатрицикло[11.6.1.014,19]эйкоза-13(20),14(19),15,17-тетраен-11- илкарбамоил)гексановой кислоты.
Б. Аналогичным способом более полярный стереоизомер трет.- бутилового эфира (11S)-5-метил-3-(10-оксо-1,9- диазатрицикло[11.6.1.014,19] эйкоза-13(20), 14(19), 15,17-тетраен-11- илкарбамоил)гексановой кислоты подвергали гидролизу, получая более полярный стереоизомер (11S)-5-метил-3-(10-оксо-1,9-диазатрицикло [11.6.1.014,19] эйкоза-13(20), 14(19), 15,17-тетраен-11- илкарбамоил)гексановой кислоты.
В. В другом варианте трет.-бутиловый эфир (3R,10S)-5-метил-3- (9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13 (18),14,16-тетраен-10-илкарбамоил)гексановой кислоты (3,24 г, 6,5 ммоля) экстрагировали 95% ТФК (водной) (30 мл) при 0oC, затем перемешивали в течение 20 минут, удаляли ледяную баню и смесь перемешивали еще один час. После концентрирования до масла остаток экстрагировали этилацетатом (250 мл) и промывали водой (7х150 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали досуха, получая отдельный стереоизомер (3R, 10S)-5-метил-3-(9-оксо- 1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16- тетраен-10-илкарбамоил)гексановой кислоты в виде белого порошка, 2,83 г (выход: 98,4%); МС: 442 (М+Н)+ (соединение 1).
Г. Аналогичным способом, но заменяя трет.-бутиловый эфир (3R,10S)-5-метил-3-(9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16-тетраен-10-илкарбамоил)гексановой кислоты на соответствующее соединение формулы (Ia), были получены следующие соединения формулы (Ib):
(3R, 10S)-4-фенил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18]нонадека-12(19), 13(18), 14,16-тeтpaeн-10- илкapбaмoил)бутaнoвaя кислота, МС: 474(M-H)-;
(3R, 10S)-4-циклогексил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18), 14,16-тетраен-10- илкарбамоил)бутановая кислота, МС: 482 (М+Н)+;
(3R, 10S)-3-циклогексил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18),14,16-тетраен-10- илкарбамоил)пропионовая кислота, МС: 468 (M+H)+;
(3R, 10S)-6-фенил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18]нонадека-12(19), 13(18),14,16-тетраен-10- илкарбамоил)гексановая кислота, МС: 502(M-H)-;
(3R, 10S)-3-(9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18),14,16-тетраен-10-илкарбамоил)гексановая кислота, МС: 426(M-H)-;
(3R, 10S)-2-aминo-5-мeтил-3-(9-oкco-l, 8-диaзaтpицикло [10.6.1.013,18] нонадека-12(19),13(18),14,16-тетраен-10- илкарбамоил)гексановая кислота, МС: 457 (М+Н)+;
(3R, 10S)-2-гидрокси-5-метил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19),13(18),14,16-тетраен-10- илкарбамоил)гексановая кислота, МС: 458 (М+Н)+; и
(3R, 9S)-5-метил-3-(8-оксо-1,7-диазатрицикло[9.6.1.012,17] октадека-11(18),12(17),13,15-тетраен-9-илкарбамоил)гексановая кислота.
Д. (3R, 10S)-5-мeтил-3-(9-oкco-1,8-диaзaтpициклo [10.6.1.013,18]нонадека-12(19), 13(18), 14,16-тетраен-10- илкарбамоил)гексановую кислоту (183 г) вносили в 40 мл сухого CH2Cl2 и при 0oC добавляли этанол (0,5 мл, 5 экв.), а затем N, N-диметиламинопиридин (0,1 экв., 5 мг) и, наконец, ЭДКИ (209 мг, 5 экв. ). Образовавшийся раствор перемешивали в течение ночи при температуре в интервале от 0oC до комнатной. Дополнительно добавляли CH2Cl2 (100 мл) и смесь промывали 0,5Н HCl (2х50 мл), насыщенным NaHCO3 (2х50 мл) и в завершение соляным раствором (1х50 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали досуха. Путем перекристаллизации из этилацетата и петролейного эфира получали этиловый эфир (3R,10S)-5-метил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18]нонадека-12(19),13(18),14,16-тетраен-10- илкарбамоил)гексановой кислоты в виде белого твердого вещества (Выход: 108 мг, 55%), МС: 470 (М+Н)+.
Е. В другом варианте в менее полярный стереоизомер трет.- бутилового эфира (3R,10S)-2-метил-5-метил-3- (9-оксо-1,8- диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18), 14,16- тетраен-10-илкарбамоил)гексановой кислоты, полученного выше в примере 5Г (15 мг), добавляли CH2Cl2 (2,4 мл) и ТФК (0,6 мл) и образовавшуюся смесь перемешивали при комнатной температуре в течение 4 часов. Растворитель удаляли при пониженном давлении при 35oC. Затем добавляли этилацетат и раствор промывали водой (3х10 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали досуха. Путем перекристаллизации из этилацетата/петролейного эфира получали (3R,10S)-2-метил-5-метил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18]нонадека-12(19),13(18),14,16-тетраен-10- илкарбамоил)гексановую кислоту в виде белого твердого вещества (7 мг), МС: 456,3 (М+Н)+.
Ж. Аналогичным способом смесь 1:1 стереоизомеров трет.- бутилового эфира (3R, 10S)-2-метил-5-метил-3-(9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16- тетраен-10-илкарбамоил)гексановой кислоты (полученную выше в примере 5Г) (5 мг) гидролизовали, получая 3 мг белого твердого вещества;
300 МГц, 1H-ЯМР в CDCl3: δ (-0,5)-(-0,3) (m, 1Н); 0,6-0,8 (m, 1Н); 0,8-1,05 (m, 6Н); 1,05-1,22 (m, 2Н); 1,35 (3H, dd, J=9 Гц); 1,4-1,7 (m, 3H); 1,7-1,95 (m, 3H); 2,3-2,48 (m, 1Н); 2,54-2,73 (m, 1Н); 2,8-3,0 (m, 2Н); 3,38-3,5 (m, 1Н); 3,52-3,72 (m, 1Н); 3,8-3,98 (m, 1Н); 4,34-4,45 (m, 1Н); 4,7-4,84 (m, 1Н); 5,0-5,08 (m, 1Н); 6,8 (d, 1Н); 7,15-7,25 (m, 1Н); 7,25-7,32 (m, 1Н); 7,35 (d, J=8,4 Гц, 1Н); 7,88 (d, J=8,4 Гц).
З. В другом варианте бензиловый эфир (3R,10S)-2- метоксикарбонил-5-метил-3-(9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16-тетраен-10-илкарбамоил)гексановой кислоты, экстрагировали этанолом (35 мл, требовалось некоторое нагревание) и добавляли формиат аммония (1642 мг, 3 экв.), а затем добавляли 10% Pd на активированном древесном угле (100 мг). После перемешивания в атмосфере аргона при комнатной температуре в течение 3 часов реакция была закончена. Смесь фильтровали под вакуумом через слой целита толщиной 1 см, затем концентрировали, добавляли MeOH и полученный продукт фильтровали через ватную пробку. После концентрирования к остатку добавляли CH2Cl2 и полученный продукт интенсивно перемешивали и затем фильтровали. Фильтрат концентрировали и перекристаллизовывали из этилацетата/петролейного эфира, получая (3R,10S)-2-метоксикарбонил-5-метил-3-(9-оксо-1,8- диазатрицикло[10.6.1.013,18]нонадека-12(19),13(18),14,16- тетраен-10-илкарбамоил)гексановую кислоту в виде белого твердого вещества (выход: 140 мг), МС: 500,3 (М+Н)+.
И. (3R, 10S)-2-метоксикарбонил-5-метил-3-(9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16- тетраен-10-илкарбамоил)гексановую кислоту экстрагировали этанолом (25 мл) и затем добавляли по каплям 1H LiOH (0,3 мл, 3 экв.). Полученный гомогенный раствор перемешивали при комнатной температуре в течение 3 часов. Большую часть этанола удаляли при пониженном давлении при 30oC. Затем добавляли при перемешивании воду (5 мл) и этилацетат (30 мл), добавляли 4Н HCl до получения pH 2. Этилацетатный слой далее промывали соляным раствором, сушили над Na2SO4, фильтровали и упаривали досуха. Очищали с помощью высокоэффективной жидкостной хроматографии с обращенной фазой, получая 47 мг (3R,10S)-2-карбокси-5-метил-3-(9-оксо-1,8- диазатрицикло[10.6.1.013,18]нонадека-12(19),13(18),14,16- тетраен-10-илкарбамоил)гексановую кислоту в виде белого твердого вещества, МС: 484,5 (M-H)-.
Пример 7
Соединения формулы (Ic)
А. Раствор менее полярного стереоизомера (11S)-5-метил-3-(10- оксо-1,9-диазатрицикло[11.6.1.014,19] эйкоза-13(20), 14(19),15,17- тетраен-11-илкарбамоил)гексановой кислоты (210 мг) и моногидрата 5-гидроксибензтриазола (109 мг) в безводном ДМФ (20 мл) охлаждали в атмосфере аргона до 0oC (в ледяной бане). К этой смеси добавляли ЭДКИ (282 мг) и перемешивание продолжали в течение 0,5 часа. Затем к раствору добавляли O-бензилгидроксиламин (0,27 мл) и оставляли реакционную смесь нагреваться до комнатной температуры в течение ночи. Все летучие компоненты удаляли при пониженном давлении. Остаток экстрагировали CH2Cl2 (100 мл) и 20% HCl (100 мл) и помещали в делительную воронку. Органическую фазу выделяли и водную фазу промывали (2х100 мл) CH2Cl2. Органический материал затем промывали последовательно 5% NaHCO3, соляным раствором, сушили (MgSO4), фильтровали и концентрировали для получения (11S)-N- бензилокси-5-метил-3-(10-оксо-1,9- диазатрицикло[11.6.1.014,19]эйкоза-13(20),14(19),15,17-тетраен-11- илкарбамоил)гексанамида в виде кристаллического вещества. Продукт далее очищали с помощью колоночной хроматографии на силикагеле, после чего путем кристаллизации из горячей смеси этилацетат/CH2Cl2 получали более полярный стереоизомер соединения, имеющий температуру плавления 232-233oC, и менее полярный стереоизомер соединения, имеющий температуру плавления 251-253oC.
Б. В другом варианте к раствору, содержащему (3R,10S)-5-метил- 3-(9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13 (18),14,16-тетраен-10-илкарбамоил)гексановую кислоту (2,5 г, 5,82 ммолей), ГОБТ•H2О (0,89 г, 1 экв.) и О-бензилгидроксиламин (2,2 мл, 3 экв.) в 200 мл ДМФ, при 0oC добавляли ЭДКИ (2,77 г, 2,5 экв.). Образовавшуюся смесь затем перемешивали в течение ночи. ДМФ удаляли с помощью вакуумной отгонки при 65oC. К остатку затем добавляли метанол (14 мл), затем эфир (140 мл). При 0oC и перемешивании добавляли 0,5Н HCl (140 мл), а после этого петролейный эфир (140 мл). Смесь перемешивали при 0oC в течение 15 минут, затем белое твердое вещество фильтровали под вакуумом и последовательно промывали водой (100 мл), а затем смесью 1:1 простой эфир/петролейный эфир (100 мл). Путем сушки под вакуумом (P2О5) в течение 3 часов получали (3R,10S)-N-бензилокси-5- метил-3-(9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13 (18),14,16-тетраен-10-илкарбамоил)гексанамид в виде белого твердого вещества (2,7 г, 84,9%).
В. Аналогичным способом, но заменяя (3R,10S)-5-метил-3-(9- оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13 (18),14,16-тетраен-10-илкарбамоил)гексановую кислоту на соответствующе замещенное соединение формулы (Ib), получали следующие соединения формулы (Ic):
(3R, 105)-N-бензилокси-2-метоксикарбонил-5-метил-3-(9-оксо- 1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16- тетраен-10-илкарбамоил)гексанамид, МС: 605,3 (М+Н)+;
(3R,10S)-N-бензилокси-6-фенил-3-(9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16- тетраен-10-илкарбамоил)гексанамид, МС: 609 (М+Н)+;
(3R, 10S)-N-бензилокси-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18]нонадека-12(19),13(18),14,16-тетраен-10- илкарбамоил)гексанамид, МС: 533 (М+Н)+.
Пример 8
Получение соединений формулы (Id)
А. К раствору более полярного стереоизомера (11S)-N- бензилокси-5-метил-3-(10-оксо-1,9-диазатрицикло[11.6.1.014,19] эйкоза-13(20), 14(19), 15,17-тетраен-11-илкарбамоил)гексанамида (90 мг) в смеси этанол/тетрагидрофуран (350 мл; 2:1) добавляли 10% палладий на активированном древесном угле (30 мг). Материал перемешивали, пропуская через него постоянно барботирующий поток газообразного водорода. Через 3 часа тонкослойная хроматография (10% CH3OH/CH2Cl2) показала, что реакция завершена. Материал фильтровали через слой целита (3 раза) и концентрировали при пониженном давлении до почти сухого остатка. Добавляли метиленхлорид (15 мл) и материал вновь концентрировали при пониженном давлении до почти сухого остатка и затем повторяли процедуру вновь. К остатку добавляли 3-4 капли метанола, вслед за этим добавляли метиленхлорид (15 мл). Материал перемешивали с охлаждением (в ледяной бане) и добавляли простой эфир (5 мл), а затем гексан (2 мл). Медленно образующийся кристаллический материал собирали путем фильтрации, получая 50 мг более полярного стереоизомера (11S)-N-гидрокси-5-метил-3-(10-оксо-1,9- диазатрицикло[11.6.1.014,19] эйкоза-13(20), 14(19),15,17-тетраен-11- илкарбамоил)гексанамида с температурой плавления 197-201oC, [α]
Б. Аналогичным способом, но заменяя более полярный стереоизомер (11S)-N-бензилокси-5-метил-3-(10-оксо-1,9- диазатрицикло[11.6.1.014,19] эйкоза-13(20), 14(19),15,17-тетраен- 11-илкарбамоил)гексанамида менее полярным, был получен менее полярный стереоизомер (11S)-N-гидрокси-5-метил-3-(10-оксо-1,9- диазатрицикло[11.6.1.014,19] эйкоза-13(20),14(19),15,17-тетраен- 11-илкарбамоил)гексанамида, температура плавления 212-216oC, [α]
В. В другом варианте (3R,10S)-N-бензилокси-5-метил-3-(9-оксо- 1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16- тетраен-10-илкарбамоил)гексанамид (1,0 г, 1,83 ммоля) экстрагировали раствором 20% ТГФ в этаноле (500 мл) и затем порциями добавляли Pd на активированном древесном угле (200 мг). Образовавшуюся суспензию перемешивали, пропуская через раствор слегка барботирующий его газообразный H2. Спустя 4 часа реакционную смесь фильтровали под вакуумом через слой целита (1,5 см) и фильтрат концентрировали и затем экстрагировали метанолом (30 мл) и фильтровали через ватную пробку. Путем перекристаллизации из смеси метанол/этилацетат/простой эфир/петролейный эфир получали (3R,10S)-N- гидрокси-5-метил-3-(9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16-тетраен-10-илкарбамоил)гексанамид (768 мг, 92%), МС: 455 (M-H)+.
Г. Аналогичным способом, но заменяя (3R,10S)-N-бензилокси-5- метил-3-(9-оксо-1,8-диазатрицикло[10.6.1.013,18]нонадека-12(19), 13(18),14,16-тетраен-10-илкарбамоил)гексанамид на соответствующее соединение формулы (Ic), получали следующие соединения формулы (Id):
(3R, 10S)-N-гидрокси-2-метоксикарбонил-5-метил-3-(9- оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека- 12(19), 13(18), 14,16-тетраен-10-илкарбамоил)гексанамид, МС: 515(М+Н)+;
(3R, 10S)-N-гидрокси-6-фенил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18), 14,16-тетраен-10- илкарбамоил)гексанамид, МС: 517(M-H)-;
(3R, 10S)-N-гидрокси-2-аминокарбонил-5-метил-3-(9- оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13 (18), 14,16-тетраен-10-илкарбамоил)гексанамид, MC: 483 (MH)+-H2O;
(3R, 10S)-N-гидрокси-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18]нонадека-12(19),13(18),14,16-тетраен-10- илкарбамоил)гексанамид, MC: 443(MH)+; и
(3R,9S)-N-гидрокси-5-метил-3-(8-оксо-1,7-диазатрицикло [9.6.1.012,17]октадека-11 (18),12(17),13,15-тетраен-9-илкарбамоил) гексанамид.
Пример 9
Соединения формулы (Ie)
А. К раствору, содержащему 4-метил-2-ацетилтиометилпентановую кислоту (612 мг, 3 ммоля), (10S)-10-амино-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18), 14,16-тетраен (427 мг, 1,5 ммоля) и ГОБТ•H2О, (230 мг, 1,5 ммоля) в сухом ДМФ (30 мл) в атмосфере аргона при комнатной температуре добавляли ЭДКИ (863 мг, 4,5 ммоля) в виде одной порции. После перемешивания в течение ночи ДМФ удаляли при 30oC в высоком вакууме, получая желтоватое полутвердое вещество. Его растворяли в этилацетате (50 мл), промывали 1Н HCl (30 мл), 5% раствором NaHCO3 (30 мл) и, наконец, соляным раствором (30 мл). Органический слой сушили (MgSO4) и упаривали досуха. Образовавшееся светло-желтое масло перемешивали в смеси 50% простой эфир/петролейный эфир (40 мл), получая 600 мг (85%) (10S)-2-ацетилтиометил-4-метил-N-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18),14,16-тетраен-10- ил)пентанамида в виде смеси стереоизомеров 1:1. Смесь стереоизомеров разделяли с помощью быстрой колоночной хроматографии (LPS-2), элюировали 20% этилацетатом в петролейном эфире, получая менее полярный стереоизомер с температурой плавления 226oC и более полярный стереоизомер с температурой плавления 220oC.
Пример 10
Соединения формулы (If)
А. К раствору менее полярного стереоизомера (10S)- 2-ацетилтиометил-4-метил-N-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18), 14,16- тетраен-10-ил)пентанамида (50 мг, 0,106 ммоля) в 10 мл метанола при 0oC в атмосфере аргона добавляли 0,5 мл концентрированного NH4OH. Реакционную смесь перемешивали при температуре от 0oC (ледяную баню удаляли после добавления NH4OH) до комнатной и далее выдерживали при комнатной температуре в течение 1 часа. Избыток метанола удаляли при пониженном давлении, получая белый твердый остаток. Остаток распределяли между этилацетатом (30 мл) и 0.1Н HCl (15 мл). Органический слой промывали соляным раствором (15 мл), сушили (MgSO4) и упаривали досуха. Твердый остаток перемешивали в смеси 50% простой эфир/петролейный эфир и фильтровали, получая более полярный стереоизомер (10S)-2-меркаптометил-4-метил-N-(9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18),14,16- тетраен-10-ил)пентанамида в виде белого порошка, 41 мг (90%), температура плавления 224oC (соединение 3).
Пример 11
Соединения формулы (HH)
А. К 4-метилпентановой кислоте (25 г, 0,215 ммоля) в водяной бане с температурой 25oC медленно добавляли тионилхлорид (20,4 мл, 1,3 г). Затем смесь выдерживали при 50oC в атмосфере аргона в течение 3 часов (пока не прекращалось выделение газа). Неочищенную реакционную смесь перегоняли при атмосферном давлении, получая 4-метилпентаноилхлорид (25,3 г, 87,3%) с температурой кипения 143oC.
Б. Аналогичным способом, но заменяя 4-метилпентановую кислоту на 5-фенилпентановую кислоту (5 г), получали 5-фенилпентаноилхлорид (4,4 г) в виде бесцветной жидкости, температура кипения 91-93oC.
Пример 12
Соединения формулы (N)
А. К суспензии 60% NaH (836 мг, 1,5 экв.) в толуоле (200 мл) при комнатной температуре в атмосфере аргона добавляли порциями сультам L-(+)-2,10-камфоры (3,0 г, 3,9 ммоля). Смесь интенсивно перемешивали при комнатной температуре в течение 1 часа. Затем к раствору осторожно по каплям добавляли 4-метилпентаноилхлорид при 0oC. После перемешивания реакционной смеси при комнатной температуре в течение 3 часов реакцию останавливали путем смешения с 10 мл воды и добавляли 70 мл простого эфира. Реакционную смесь сначала промывали 0,5Н HCl (2х50 мл), затем 5% К2СО3 (3х50 мл) и, наконец, соляным раствором (1х50 мл). Органический слой сушили над MgSO4, фильтровали и упаривали досуха. Путем очистки с помощью колоночной хроматографии (смесь 1: 6 этилацетат/петролейный эфир в качестве растворителя для элюирования) получали сультам N-4-метилпентаноил-L-(+)-2,10-камфоры (3,39 г, 78%).
Б. Аналогичным способом, но заменяя 4-метилпентаноилхлорид соответствующим хлоридом, получали следующие соединения формулы (N):
сультам N-3-фенилпропаноил-L-(+)-2,10-камфоры, МС: 347 (М+);
сультам N-5-фенилпентаноил-L-(+)-2,10-камфоры, МС: 375 М+;
сультам N-пентаноил-L-(+)-2,10-камфоры, МС: 300 (М+Н)+.
Пример 13
Соединения формулы (О)
А. К раствору сультама N-4-метилпентаноил-L-(+)-2,10-камфоры (3,39 г, 10,8 ммоля) в 75 мл сухого ТГФ при -78oC в атмосфере аргона добавляли по каплям в течение примерно 5 минут NaN(тетраметилсилан)2 (1,0 М в ТГФ, 11,34 мл, 1,05 экв.). После перемешивания при -78oC в течение 1 часа к смеси добавляли гексаметилфосфорамид (5 мл), далее трет.-бутилбромацетат (5,2 мл, 3 экв. ), а затем в виде одной порции добавляли 400 мг йодида тетра-н- бутиламмония. Полученный раствор выдерживали при -78oC в атмосфере аргона в течение ночи. На следующее утро реакцию останавливали путем смешения с водой (100 мл) и затем экстрагировали простым эфиром (3х100 мл). Объединенные эфирные слои промывали соляным раствором, затем сушили над Na2SO4, фильтровали и концентрировали. Путем очистки с помощью колоночной хроматографии (при соотношении этилацетат/петролейный эфир от 5:95 до 10:90 в качестве элюента) получали сультам N-(4-метил-2-трет.- бутоксикарбонилметил)пентаноил-L-(+)-2,10-камфоры (4 г, 86,5%).
Б. Аналогичным способом, но заменяя сультам N-4-метилпентаноил-L-(+)-2,10-камфоры соответствующим соединением формулы (N), получали следующие соединения формулы (Q):
сультам N-(3-фенил-2-трет.- бутоксикарбонилметил)пропаноил-L-(+)-2,10-камфоры, МС: 461 М+;
сультам N-(5-фенил-2-трет.- бутоксикарбонилметил)пентаноил-L-(+)-2,10-камфоры, МС: 490 (М+Н)+;
сультам N-(2-трет. -бутоксикарбонилметил)пентаноил-L- (+)-2,10-камфоры, МС: 414 (М+Н)+.
Пример 14
Соединения формулы (Ka)
А. К перемешиваемому раствору сультама N-(4-метил-2-трет- бутоксикарбонилметил)пентаноил-L-(+)-2,10-камфоры (5,45 г, 12,7 ммоля) в 50% водном ТГФ (150 мл) при 0oC в атмосфере аргона добавляли кристаллы LiOH•H2O (2,14 г, 4 экв.), затем 30% H2О2 (11,5 мл). Затем ледяную баню удаляли и образовавшуюся эмульсию перемешивали в течение 3 часов до тех пор, пока она не стала прозрачной. Большую часть ТГФ удаляли при пониженном давлении при 35oC. Затем добавляли CH2Cl2 (150 мл) и при перемешивании добавляли 4Н HCl до pH = 2. После добавления NaCl водный слой снова экстрагировали CH2Cl2 (3х150 мл). CH2Cl2 удаляли при пониженном давлении при 35oC и затем остаток экстрагировали этилацетатом (150 мл). Этот раствор затем экстрагировали 5% K2CO3 (3х50 мл) и объединенные экстракты промывали простым эфиром (50 мл). Затем CH2Cl2 добавляли к водному слою и при перемешивании с NaCl водный слой экстрагировали CH2Cl2 (3х70 мл) и объединенные экстракты сушили над Na2SO4, фильтровали и концентрировали, получая (2R)-4-метил-2-трет.-бутоксикарбонилметилпентановую кислоту в виде бесцветного масла (2,95 г, выход количественный).
Б. Аналогичным способом, но заменяя сультам N-(4-метил-2- трет.-бутоксикарбонилметил)пентаноил-L-(+)-2,10-камфоры соответствующим соединением формулы (Q), получали следующие соединения формулы (Ka):
(2R)-3-фенил-2-трет. - бутоксикарбонилметилпропановую кислоту, МС: 265 (М+Н)+;
(2R)-5-фенил-2-трет. -бутоксикарбонилметилпентановую кислоту, МС: 293 (M+H)+;
(2R)-2-трет. -бутоксикарбонилметилпентановую кислоту (бесцветное масло, 1,09 г).
В. (2R)-3-фенил-2-трет. -бутоксикарбонилметилпропановую кислоту (55 мг) экстрагировали ледяной уксусной кислотой (20 мл) и добавляли PtO2 (25 мг) в уксусной кислоте. Затем химический стакан помещали в сосуд высокого давления Парра, создавали вакуум и заряжали H2 под давлением 100 фунт/дюйм2 (7,03 кг/см2). После перемешивания в течение 3 дней смесь фильтровали под вакуумом через слой целита толщиной 1 см. Фильтрат затем концентрировали, получая (2R)-3-циклогексил-2- трет.-бутоксикарбонилметилпропановую кислоту (56 мг) в виде желтого масла, МС: 269 (M-H)-.
Пример 15
Соединения формулы (R)
К раствору D-лейцина (50 г, 0,381 моля) в 570 мл 3H HBr (вод.) при 0oC добавляли нитрит натрия (42 г, 1,6 экв.) порциями в течение 1 часа 15 минут. Далее реакционную смесь перемешивали в течение 3 часов при 0oC и затем экстрагировали простым эфиром (1000 мл). После промывки эфирного слоя водой (2х500 мл) его сушили над MgSO4 и концентрировали. Красный сироп затем выпаривали вместе с хлороформом (3х200 мл) до удаления окраски и затем отсасывали, получая (2R)-2-бром-4-метилпетановую кислоту в виде бесцветного масла с постоянным весом 71,3 г.
Пример 16
Соединения формулы (S)
В дихлорметане (80 мл) конденсировали изобутен до двойного объема (при -50oC CHCl3/сухой лед). К этому раствору добавляли (2R)-2-бром-4-метилпетановую кислоту (28 г, 143,6 ммоля) и, поддерживая температуру на уровне между -40o и -50oC, по каплям добавляли концентрированную серную кислоту (1 мл). Реакционной смеси далее давали нагреться до комнатной температуры в течение 20 часов. Затем раствор концентрировали перед добавлением дополнительного количества метиленхлорида (300 мл), после чего последовательно промывали насыщенным NaHCO3 (2х100 мл), а затем водой (2х100 мл). После сушки над Na2SO4 органический слой отфильтровывали и концентрировали, получая желтое масло. Материал перегоняли, получая 23 г трет.-бутилового эфира (2R)-2-бром-4-метилпетановой кислоты в виде прозрачного бесцветного масла.
Пример 17
Соединения формулы (U)
К бензилметилмалонату (2,13 мл, 1 экв.) и трет.-бутоксиду калия (1,36 г, 1 экв. ) в сухом ДМФ (100 мл) при 0oC добавляли по каплям в течение 1 часа трет. -бутиловый эфир (2R)-2-бром-4- метилпентановой кислоты (2,89 г, 11,5 ммоля) в 50 мл ДМФ. Полученный раствор затем перемешивали при 0oC в течение 3 дней. Затем реакционную смесь распределяли между простым эфиром (150 мл) и насыщенным хлоридом аммония (80 мл). Образовавшуюся смесь фильтровали под вакуумом через целит и отделяли две фазы. Затем водный слой дополнительно экстрагировали простым эфиром (3х100 мл) и объединенные эфирные экстракты промывали водой (6х100 мл). После сушки над MgSO4 органический слой фильтровали и упаривали досуха. Путем очистки с помощью мгновенной колоночной хроматографии (с элюированием смесью 4:96 этилацетат/петролейный эфир) получали трет. -бутиловый эфир (2R)-2[(1-метоксикарбонил-1- бензилоксикарбонил)метил]-4-метилпентановой кислоты (2,55 г) в виде прозрачного бесцветного масла, МС: 322 (М-ацетон)+.
Пример 18
Соединения формулы (Kb)
Трет. -бутиловый эфир (2R)-2[(1-метоксикарбонил-1- бензилоксикарбонил)метил]-4-метилпентановой кислоты помещали в 5 мл 80% ТФК (вод.) при комнатной температуре и перемешивали в течение 1,5 часов, контролируя с помощью тонкослойной хроматографии. Реакция была завершена только на 30%, поэтому добавляли дополнительное количество ТФК (10 мл). Через 0,5 часа реакция была завершена. ТФК удаляли в высоком вакууме при 45oC и остаток экстрагировали этилацетатом и промывали водой (5х30 мл). После сушки над Na2SO4 слой этилацетата фильтровали, концентрировали и отсасывали, получая (2R)-2[(1-метоксикарбонил-1- бензилоксикарбонил)метил]-4-метилпентановую кислоту в виде твердого вещества (1,68 г), МС: 322 (М+).
Пример 19
Соединения формулы (W)
Кристаллическую фосфиновую кислоту (8,4 г, 0,13 моля) перемешивали в чистом триэтил-орто-формиате (22 мл, 0,20 ммоля) в течение 90 минут при комнатной температуре. Затем эту смесь вводили через канюлю в перемешиваемый раствор, содержащий этилизобутилакрилат (8 г, 0,036 моля) и тетраметилгуанидин (4,5 мл, 0,036 моля), который был охлажден до 0oC в течение 10 минут. Ледяную баню удаляли и реакционную смесь перемешивали в течение 4 часов. Смесь разбавляли 200 мл диэтилового эфира и раствор промывали 1Н HCl (100 мл), водой (4х100мл) и соляным раствором (100 мл) и сушили над сульфатом магния. Путем выпаривания при пониженном давлении получали 8,15 г этилового эфира 2-(этоксифосфиноилметил)- 4-метилпентановой кислоты в виде масла бледно-желтого цвета, МС: 349 (M-H2О)+.
Пример 20
Соединения формулы (X)
Неочищенный этиловый эфир 2-(этоксифосфиноилметил)-4- метилпентановой кислоты (26 г) растворяли в 600 мл ТГФ/CH2Cl2 (50/50) и охлаждали до 0oC. Затем к раствору добавляли диизопропилэтиламин (32 мл) и 90,8 мл бис-(триметилсилил)ацетамида и образовавшуюся смесь перемешивали в течение 20 минут перед добавлением пара-формальдегида (5,5 г). Раствор доводили до комнатной температуры и после нагрева до 37oC выдерживали при этой температуре в течение 18 часов. Растворитель удаляли выпариванием и образовавшееся масло растворяли в 200 мл этилацетата. Раствор промывали 50 мл 1Н HCl (2 раза), 50 мл соляного раствора (2 раза), сушили над MgSO4, фильтровали и выпаривали, получая 19,3 г этилового эфира 2-[(этокси)(гидроксиметил)фосфиноилметил] -4- метилпентановой кислоты в виде желтоватого масла, МС: 281 (MH+).
Пример 21
Соединения формулы (Y)
А. Этиловый эфир 2-[(этокси)(гидроксиметил)фосфиноилметил]-4- метилпентановой кислоты (5 г) растворяли в 20 мл CH2Cl2 и охлаждали до -20oC (с дублированием). Метансульфонилхлорид (1,5 мл и триэтиламин (3,0 мл) добавляли по каплям к раствору. После 15 минут баню удаляли и реакционную смесь оставляли при комнатной температуре в течение 3,5 часов. Затем каждый раствор промывали 10 мл холодной 2% HCl, 10 мл NaHCO3 (насыщенного), 10 мл соляного раствора, сушили над MgSO4, фильтровали и выпаривали, получая 12,8 г (объединенный выход) этилового эфира 2-[(этокси) (метансульфонилоксиметил)фосфиноилметил]-4-метилпентановой кислоты.
Б. Аналогичным способом, но заменяя метансульфонилхлорид на пара-толуолсульфонилхлорид, получали этиловый эфир 2-[(этокси) (пара-толуолсульфонилоксиметил) фосфиноилметил] -4-метилпентановой кислоты.
Пример 22
Соединения формулы (AA)
Гидрид натрия (1,52 г, (60%)) и 6 г 2-хинолинтиола смешивали вместе при 0oC в 50 мл ДМФ. После прекращения начального выделения H2 смесь перемешивали при комнатной температуре в течение 2,5 ч. Затем смесь охлаждали до 0oC и через канюлю добавляли этиловый эфир 2-[(этокси)(метансульфонилоксиметил)фосфиноилметил]-4- метилпентановой кислоты (12,8 г) в 10 мл ДМФ и образовавшуюся смесь затем перемешивали в течение 18 часов, медленно нагревая до комнатной температуры. ДМФ удаляли с помощью выпаривания, остаток растворяли в 50 мл этилацетата и промывали 50 мл H2О (2 раза), соляным раствором (50 мл), сушили над MgSO4 и выпаривали до желтого полутвердого продукта. Путем очистки с помощью мгновенной хроматографии с использованием смеси 10%-80% этилацетат/гексан для элюирования получали 10 г этилового эфира 2-[(этокси)(хинолин-2- илтиометил)фосфиноилметил]-4-метилпентановой кислоты (Rf=0,35 в 80% этилацетате/гексане), МС: 424 (MH+).
Б. Аналогичным способом, но заменяя 2-хинолинтиол на 1-нафталинтиол, 2-нафталинтиол или тиофенол, получают следующие соединения формулы (AA):
этиловый эфир 2-[(этокси)(нафт-1- илтиометил)фосфиноилметил]-4-метилпентановой кислоты;
этиловый эфир 2-[(этокси)(нафт-2- илтиометил)фосфиноилметил]-4-метилпентановой кислоты; и
этиловый эфир 2-[(этокси)(фенилтиометил)фосфиноилметил]-4- метилпентановой кислоты.
Пример 23
Соединения формулы (BB)
А. Этиловый эфир 2-[(этокси)(хинолин-2- илтиометил)фосфиноилметил]-4-метилпентановой кислоты (4,5 г) растворяли в 100 мл ТГФ и добавляли 12,5 мл 2Н NaOH вместе с достаточным количеством метанола для получения гомогенного раствора. Через 18 часов ТГФ удаляли выпариванием, остаток растворяли в 50 мл H2О и промывали 50 мл этилацетата. Водную фазу затем подкисляли до pH 4 и продукт экстрагировали 50 мл этилацетата (2 раза). Этилацетатную фазу промывали 20 мл соляного раствора, сушили над MgSO4 и выпаривали, получая 3,8 г 2-[(гидрокси)(хинолин-2-илтиометил)фосфиноилметил]-4- метилпентановой кислоты в виде желтого масла, МС: 368 (MH+).
Б. Аналогичным способом получали следующие соединения формулы (BB):
2-[(гидрокси)(нафт-1-илтиометил)фосфиноилметил] -4- метилпентановую кислоту;
2-[(гидрокси)(нафт-2-илтиометил)фосфиноилметил] -4- метилпентановую кислоту; и
2-[(гидрокси)(фенилтиометил)фосфиноилметил]-4- метилпентановую кислоту.
Пример 24
Пептизация соединения формулы (BB)
2-[(Гидрокси)(хинолин-2-илтиометил)фосфиноилметил] -4- метилпентановую кислоту (5,3 г) растворяли в 50 мл теплого этанола (абсолютного) и добавляли 4,2 г (-)-цинхонидина. По истечении 30 минут выдержки при комнатной температуре начинала осаждаться соль. Колбу покрывали фольгой и оставляли стоять в течение 2 дней. Затем соль удаляли фильтрацией под вакуумом и фильтрат упаривали до состояния желтой пены. Соль и фильтрат соответственно растворяли в 100 мл этилацетата и промывали последовательно 1% HCl для удаления цинхонидина, поддерживая pH выше 4. Оба раствора сушили по отдельности над MgSO4 и выпаривали, получая 2,4 г отдельного стереоизомера [α]
Пример 25
Соединение формулы (Ig)
Отдельный стереоизомер 2-[(гидрокси)(хинолин-2- илтиометил)фосфиноилметил] -4-метилпентановой кислоты (300 мг, 0,81 ммоля) и 1,1'-карбонилдиимидазол (174 мг, 1,0 ммоль) перемешивали при 0oC в 6 мл ТГФ в течение 1,5 ч. К раствору добавляли (10S)-10- амино-9-оксо-1,8-диазатрицикло[10.6.1.013,18]нонадека-12(19),13 (18),14,16-тетраен (270 мг, 0,95 ммоля) и образовавшейся смеси давали нагреться до комнатной температуры и затем перемешивали в течение 18 часов. ТГФ удаляли с помощью выпаривания и остаток растворяли в 60 мл этилацетата. Этилацетатный раствор промывали 10 мл H2О, 10 мл соляного раствора, сушили над MgSO4 и выпаривали, получая (10S)-[4метил-2-(9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19),13(18), 14,16-тетраен-10-илкарбамоил)пентил] - (хинолин-2-илтиометил)фосфиновую кислоту в виде желтого масла. Очистку проводили с помощью высокоэффективной жидкостной хроматографии с обращенной фазой, используя в качестве растворителей для элюирования градиент ацетонитрила и 50 мл буфера NH4OAc. Более полярный стереоизомер (30 мг) выделяли при 41% ацетонитрила, а менее полярный стереоизомер (10 мг) - при 43% ацетонитрила. Фракции лиофилизировали до получения беловатого порошка, МС: 635 (MH+) (соединение 4).
Пример 26
Соединение формулы (Ih)
(3R, 10S)-5-метил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18),14,16-тетраен-10-илкарбамоил)гексановую кислоту (200 мг, 0,45 ммоля) растворяли в 10 мл ледяной уксусной кислоты и гидрировали при давлении водорода 100 фунт/дюйм2 в присутствии Pt2O (60 мг) в реакторе Парра при комнатной температуре в течение 15 часов. Газообразным аргоном барботировали реакционную смесь в течение 15 минут, катализатор (Pt2O) отфильтровывали (через воронку с целитом). Прозрачный фильтрат упаривали досуха и далее дважды проводили совместное выпаривание с толуолом, получая количественный выход (3R, 10S)-5-метил-3-(9-оксо-1,8- диазатрицикло[10.6.1.013,18]нонадекан-10-илкарбамоил) гексановой кислоты в виде белого твердого вещества, МС: 448 (M-H)+.
Пример 27
Соединение формулы (Ii)
К (3R, 10S)-5-метил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18]нонадеканилкарбамоил)гексановой кислоте и О-бензилгидроксиламину (5 экв., 2,25 ммоля, 277 мг) в 10 мл сухого ДМФ при комнатной температуре добавляли 1-гидроксибензтриазол•H2О (2 экв. , 0,9 ммоля, 122 мг) и ЭДКИ (5 экв., 2,25 ммоля, 431 мг). Образовавшуюся прозрачную реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли в высоком вакууме при комнатной температуре и остаток распределяли между этилацетатом (15 мл) и 1Н HCl (15 мл). Этилацетатный слой затем экстрагировали 1Н HCl (15 мл). Объединенные водные экстракты подщелачивали с помощью 4Н NaOH до pH 10 и насыщали NaCl и экстрагировали CH2Cl2 (3х15 мл). Объединенный метиленхлоридный экстракт CH2Cl2 сушили (MgSO4), фильтровали и выпаривали досуха, получая полутвердый продукт. Этот полутвердый продукт перемешивали в простом эфире (10 мл) при 0oC в течение 30 минут и фильтровали, получая 85 мг (34%) (3R, 10S)-N-бензилокси-5-метил-3-(9-оксо-1,8- диазатрицикло[10.6.1.013,18]нонадеканилкарбамоил)гексанамида в виде белого порошка, МС: 555 (M+H)+
Пример 28
Соединение формулы (Ij)
(3R,10S)-N-бензилокси-5-метил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадекан-10-илкарбамоил)гексанамид (75 мг, 0,135 ммоля) гидрировали при давлении водорода 1 атмосфера в абсолютном этаноле (5 мл) в присутствии 10% Pd/C (35 мг) в течение 2 часов. Газообразным аргоном барботировали реакционную смесь в течение 10 минут и реакционную смесь отфильтровывали через воронку с целитом. Далее катализатор на целите промывали 5 мл этанола. Объединенный фильтрат упаривали досуха до образования твердого остатка. Твердый остаток перемешивали в 10 мл 5% MeOH в простом эфире при 0oC в течение 30 минут и после фильтрации получали 57 мг (91%) (3R,10S)-N-гидрокси-5-метил-3-(9-оксо- 1,8-диазатрицикло[10.6.1.013,18] нонадекан-10-илкарбамоил)гексанамида в виде беловатого порошка, МС: 465 (М+Н)+.
Пример 29
Соединение формулы (DD)
К N-трет. -бутоксикарбонилтриптофану (3 ммоля, 914 мг) и N-метилэтандиамину (3,6 ммоля, 0,27 г, 0,32 мл) в сухом ДМФ (15 мл) добавляли 1-гидроксибензтриазол•H2О (3 ммоля, 459 мг) и ЭДКИ (4,5 ммоля, 863 мг). Смесь перемешивали при комнатной температуре в течение ночи в атмосфере аргона и избыток растворителя (ДМФ) удаляли в высоком вакууме при 35oC. Остаток растворяли в этилацетате (40 мл) и аддукт экстрагировали 1Н HCl (3х25 мл). Объединенный водный экстракт подщелачивали твердым K2CO3 и экстрагировали этилацетатом (3х25 мл). Объединенный органический экстракт промывали соляным раствором (30 мл) и сушили (MgSO4). Путем выпаривания досуха получали 920 мг (85%) очищенного с помощью высокоэффективной жидкостной хроматографии продукта, N'-трет. - бутоксикарбонилтриптофан-N-[(метил)аминоэтил]амида в виде белой пены.
Пример 30
Соединение формулы (ЕЕ)
К интенсивно перемешиваемому раствору N'-трет- бутоксикарбонилтриптофан-N-[(метил)аминоэтил] амида (2 г, 5,55 ммоля) и транс-1,4-дихлорбут-2-ена (8,32 ммоля, 1,04 г, 0,88 мл) в метиленхлориде (75 мл) и 40% КОН (50 мл) добавляли 0,3 экв. хлорида бензилтриэтиламмония (1,66 ммоля, 378 мг). После перемешивания при комнатной температуре в течение ночи желтоватый органический слой отделяли и водный слой затем экстрагировали 30 мл CH2Cl2. Объединенный метиленхлоридный экстракт промывали соляным раствором и сушили (MgSO4). Остаток растворяли в 10 мл MeOH и при 0oC добавляли при перемешивании 50 мл простого эфира. Образовавшийся желтый твердый продукт отфильтровывали. Фильтрат упаривали досуха, получая 1,12 г очищенного с помощью высокоэффективной жидкостной хроматографии продукта 11-N'-(трет.-бутоксикарбонил)амино-10-оксо- 1,6,9-триаза-6-метил-трицикло[11.6.1.014,19]эйкоза-3(4), 13(20), 14(19),15,17-пентаена в виде светло-желтого порошка, МС: 413 (М+Н)+.
Пример 31
Соединение формулы (FF)
11-N'-(трет. -бутоксикарбонил)амино-10-оксо-1,6,9-триаза-6- метилтрицикло[11.6.1.014,19] эйкоза-3(4),13(20),14(19),15,17- пентаен (414 мг, 1 ммоль) перемешивали в 40% ТФК в CH2Cl2 (10 мл) при комнатной температуре в течение 1 часа. Избыток ТФК и растворителя удаляли при пониженном давлении. Остаток растворяли в метиленхлориде (30 мл) и промывали 5% раствором K2CO3 (2х15 мл) и соляным раствором (15 мл). Органический слой сушили (MgSO4) и выпаривали для получения 240 мг (76%) в виде светло-желтой пены 11- амино-10-оксо-1,6,9-триаза-6-метилтрицикло[11.6.1.014,19] эйкоза-3(4), 13(20), 14(19), 15,17-пентаена, МС: 313 (М+Н)+.
Пример 32
Соединения формулы (Ik)
11-Амино-10-оксо-1,6,9-триаза-6-метилтрицикло[11.6.1.014,19] эйкоза-3(4), 13(20),14(19),15,17-пентаен (161 мг, 0,7 ммоля) и (2R)-4-метил-2-трет. -бутоксикарбонилметилпентановую кислоту (220 мг, 0,7 ммоля) перемешивали в сухом ДМФ (15 мл) в присутствии ГОБТ (0,7 ммоля), ЭДКИ (1,4 ммоля, 268 мг) и N-метилморфолина (1,4 ммоля, 0,15 мл) в атмосфере аргона при комнатной температуре в течение ночи. Избыток ДМФ удаляли при пониженном давлении. Остаток растворяли в метиленхлориде (30 мл) и промывали водой (30 мл). Водную фракцию экстрагировали CH2Cl2 (30 мл). Объединенный метиленовый экстракт сушили (MgSO4) и выпаривали до получения светло-коричневого масла. Светло-коричневое масло очищали с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (C18-колонка; градиент CH3CN-50 MM NH4OH). Целевые фракции лиофилизировали для получения 148 мг (40%) трет.-бутилового эфира (3R, 11S)-5-метил-3-(10-оксо-1,6,9-триаза-6- метилтрицикло[11.6.1.014,19] эйкоза-3(4),13(20),14(19),15,17-пентаен -11-илкарбамоил)гексановой кислоты в виде светло-желтого порошка, МС: 525,2 (M+Н)+.
Пример 33
Соединения формулы (Il)
Трет. -бутиловый эфир (3R, 11S)-5-метил-3-(10-оксо-1,6,9-триаза- 6-метил-трицикло[11.6.1.014,19] эйкоза-3(4), 13(20), 14(19),15,17- пентаен-11-илкарбамоил)гексановой кислоты (0,228 ммоля, 120 мг) перемешивали в 20% ТФК в CH2Cl2 (5 мл) при комнатной температуре в течение 1 часа. Избыток растворителя удаляли при пониженном давлении (ротационный испаритель при 30oC) для получения неочищенной кислоты, (3R,11S)-5-метил-3-(10-оксо-1,6,9-триаза-6- метилтрицикло [11.6.1.014,19]эйкоза-3(4),13(20),14(19),15,17- пентаен-11-илкарбамоил)гексановой кислоты в виде светло-коричневого масла. Путем очистки с помощью высокоэффективной жидкостной хроматографии с обращенной фазой при элюировании буфером CH3CN- NH4OAc в градиентных условиях получали 90 мг (75%) кислоты в виде светло-желтого порошка, МС: 469,1 (М+Н)+.
Пример 34
Соединение формулы (Im)
(3R,11S)-5-метил-3-(10-оксо-1,6,9-триаза-6-метилтрицикло [11.6.1.014,19] эйкоза-3(4),13(20),14(19),15,17-пентаен-11- илкарбамоил)гексановую кислоту и О-бензилгидроксиламин (5 экв., 2,5 ммоля, 308 мг) перемешивали в сухом ДМФ (30 мл) в присутствии ГОБТ (2 экв., 1 ммоль, 135 мг), ЭДКИ (5 экв., 2,5 ммоля, 479 мг) и N-метилморфолина (10 экв., 5 ммолей, 0,55 мл) при комнатной температуре в атмосфере аргона в течение 15 часов. Растворитель удаляли в высоком вакууме при комнатной температуре. Остаток растворяли в дистиллированной воде (35 мл) и экстрагировали 10% этилацетатом в петролейном эфире для удаления менее полярных примесей. Целевой продукт затем экстрагировали из водного слоя с помощью CH2Cl2 (2х35 мл). Органический экстракт промывали соляным раствором (25 мл), сушили (MgSO4) и выпаривали, получая желтое масло (330 мг). Неочищенный продукт очищали с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (буфер CH3CN-NH4OAc; градиент) и лиофилизировали, получая 175 мг (61%) (3R,11S)-N-бензилокси-5-метил-3-(10-оксо-1,6,9-триаза-6- метилтрицикло[11.6.1.014,19]эйкоза-3(4),13(20),14(19),15,17- пентаен-11-илкарбамоил)гексанамида в виде беловатого порошка, МС: 572 (M-H)-.
Пример 35
Соединение формулы (In)
Смесь (3R,11S)-N-бензилокси-5-метил-3-(10-оксо-1,6,9-триаза-6- метилтрицикло[11.6.1.014,19] эйкоза-3(4), 13(20),14(19),15,17- пентаен-11-илкарбамоил)гексанамида (40 мг, 0,07 ммоля) и 10% Pd/C (10 мг) перемешивали в растворе 3% HCOOH в этаноле (5 мл) при комнатной температуре в течение 1 часа. Смесь фильтровали через воронку с целитом и концентрировали в вакууме. К твердому остатку в 1 мл 50% AcOH/MeOH при перемешивании добавляли в виде одной порции 5 мл простого эфира. Беловатый порошок затем собирали с помощью фильтрации, получая 26 мг (68%) (3R,11S)-N-гидрокси-5-метил-3-(10- оксо-1,6,9-триаза-6-метил-трицикло[11.6.1.014,19] эйкоза-3 (4), 13(20), 14(19), 15,17-пентаен-11-илкарбамоил)гексанамида, МС: 488,5 (MH+).
Пример 36
Соединения формулы (JJ)
А. К раствору натриевой соли (±)-2-гидроксибутановой кислоты (2,54 г, 20,1 ммоля) в сухом ДМФ (30 мл) добавляли бензилбромид (2,9 мл, 1,2 экв.) и безводный KI (330 мг, 0,1 экв.). Суспензию выдерживали при 100oC в течение 24 часов и ДМФ отгоняли при пониженном давлении. Остаток экстрагировали простым эфиром, промывали водой и насыщенным Na2S2O3, сушили (MgSO4) и концентрировали. Путем перегонки оставшегося масла получали 3,7 г (95%) бензил-(±)-2-гидроксибутаноата в виде бесцветного масла, температура кипения 95oC (0,45 торр).
Б. В другом варианте к холодной (0oC) суспензии NaH (3,8 г 60%-ной (по массе) дисперсии в минеральном масле, 95,0 ммолей) в ТГФ (50 мл) добавляли по каплям через канюлю раствор гликолевой кислоты (7,2 г, 95 ммолей) в ТГФ (50 мл). Образовавшийся раствор нагревали до 25oC и концентрировали в вакууме. Образовавшуюся соль суспендировали в ДМФ (100 мл) и обрабатывали KI (1,57 г, 0,1 экв.) и бензилбромидом (12,3 мл, 1,1 экв). Смесь выдерживали при 100oC в течение 23 часов в атмосфере аргона и выпаривали ДМФ. Остаток растворяли в простом эфире и промывали водой, насыщенным Na2S2O3 и соляным раствором и сушили над MgSO4. Путем перегонки получали бензилгликолят (8,5 г, 54%) в виде бесцветного масла, температура кипения 85-87oC (0,5 торр).
Пример 37
Соединения формулы (LL)
А. К холодному (0oC) раствору бензил-(±)-2-гидроксибутаноата (1,75 г, 9,01 ммоля) в CH2Cl2 (50 мл) добавляли 2,6-лутидин (1,2 мл, 1,1 экв.), после чего по каплям добавляли ангидрид трифторметансульфокислоты (1,7 мл, 1,1 экв.). Через 10 мин по каплям при 0oC добавляли раствор трет.-бутилового эфира L-лейцина (1,7 г, 1 экв.) и диизопропилэтиламина (1,7 мл, 1,1 экв.) в CH2Cl2 (30 мл). Раствор выдерживали при 25oC в течение 36 часов, разбавляли CH2Cl2 и промывали насыщенным NaHCO3 (50 мл). После сушки (Na2SO4) и концентрации в вакууме оставшееся масло подвергали мгновенной хроматографии (кремнезем, 5% этилацетат/гексаны) для разделения диастереомеров. Менее полярный диастереомер трет. -бутиловый эфир (1R)-N-(2- бензилоксикарбонилпропил)-L-лейцина (Rf 0,22; 5% этилацетат/гексаны) и более полярный диастереомер трет. -бутиловый эфир (1S)-N-(2-бензилоксикарбонилпропил)-L-лейцина (Rf 0,13), в дальнейшем очищали по-отдельности с помощью высокоэффективной жидкостной хроматографии (5% этилацетат/гексаны) для получения 1,1 г более полярного диастереомера и 0,78 г менее полярного диастереомера, МС(БТЯ): 364 (MH+).
Б. Аналогичным способом из бензилгликолята (379 мг, 2,7 ммоля), трет.-бутилового эфира L-лейцина (435 мг, 2,7 ммоля), 2,6 лутидина (0,35 мл, 2,8 ммоля), диизопропилэтиламина (0,53 мл, 0,28 ммоля) и ангидрида трифторметансульфокислоты (0,51 мл, 2,8 ммоля) получали 383 мг (50%) трет.-бутилового эфира N-бензилоксикарбонилметил-L-лейцина в виде бесцветного масла, МС(БТЯ): 336(MH+).
Пример 38
Соединения формулы (Io)
А. К раствору трет.-бутилового эфира (1S)-N-(2- бензилоксикарбонилпропил)-L-лейцина (143 мг, 0,393 ммоля) в CH2Cl2 (3 мл) при 0oC добавляли ТФК (0,5 мл). Раствор перемешивали при 25oC в течение 4 часов и затем концентрировали в вакууме для получения соли (1S)-N-(2-бензилоксикарбонилпропил)-L-лейцина (соединение формулы (MM)), которую затем растворяли в ДМФ (3 мл) с (10S)-10-амино-9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13 (18), 14,16- тетраеном (124 мг, 1,1 экв.) и ГОБТ (80 мг, 1,5 экв.). После охлаждения до 0oC добавляли N-метилморфолин (60 мл, 1,5 экв.) и ЭДКИ (113 мг, 1,5 экв.). После выдержки при 25oC в течение 18 часов реакционную смесь разбавляли 10 мл этилацетата, промывали насыщенным NaHCO3 (3х10 мл) и водой (2х10 мл), сушили (Na2SO4) и концентрировали. Остаток хроматографировали с помощью мгновенной хроматографии (кремнезем, 1% MeOH/CH2Cl2) и собирали фракции с Rf 0,5 (5% MeOH/CH2Cl2). Путем высокоэффективной жидкостной хроматографии с обращенной фазой получали (2R,1'S,10S)-2-[N-(1- бензилоксикарбонилпропил)амино] -4-метил-N-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19),13(18),14,16-тетраен-10- ил)пентанамид (46 мг), МС(БТЯ): 575 (М+).
Б. Аналогичным способом, но заменяя трет.-бутиловый эфир (1S)- N-(2-бензилоксикарбонилпропил)-L-лейцина (270 мг) на трет.- бутиловый эфир (1R)-N-(2-бензилоксикарбонилпропил)-L-лейцина, получали (2R,1'R,10S)-2-[N-(1-бензилоксикарбонилпропил)амино]-4- метил-N-(9-оксо-1,8-диазатрицикло[10.6.1.013,18]нонадека- 12(19),13(18),14,16-тетраен-10-ил)пентанамид (175 мг), МС(БТЯ): 575 (MH+).
Пример 39
Соединения формулы (Ip)
А. К раствору (2R,1'S,10S)-2-[N-(1- бензилоксикарбонилпропил)амино]-4-метил-N-(9-оксо-1,8- диазатрицикло [10.6.1.013,18] нонадека-12(19),13(18), 14,16- тетраен-10-ил)пентанамида (46 мг) в ТГФ/MeOН (1:1, 2 мл) добавляли в атмосфере аргона 1М гидроксид бария (0,3 мл). После выдержки при 25oC в течение 24 часов через раствор пропускали СО2 и образовавшийся осадок карбоната бария отфильтровывали. Растворитель удаляли при пониженном давлении и водный остаток доводили до pH 5,5 с помощью 1М HCl. После удаления воды с помощью высокоэффективной жидкостной хроматографии с обращенной фазой получали (2R, 1'S,10S)- 2-[N'-(1-карбоксипропил)амино]-4-метил-N-(9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18)14,16-тетраен-10- ил)пентанамид в виде белого твердого продукта, МС(БТЯ): 483 (M-H)- (соединение 5).
Аналогичным способом получали:
(2RS, 10S)-2-(карбоксиметиламино)-2-циклогексил-N-(9- оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19),13(18),14,16- тетраен-10-ил)ацетамид, МС: 497 (М+Н)+
(2RS, 10S)-2-(карбоксиметиламино)-3-метил-N-(9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18),14,16- тетраен-10-ил)пентамид, МС: 471 (М+Н)+;
(2RS, 10S)-2-(фосфонилметиламино)-4-метил-N-(9-оксо-1,8- диазатрицикло[10.6.1.013,18]нонадека-12(19),13(18),14,16- тетраен-10-ил)пентамид.
Б. Аналогичным способом из (2R,1'R,10S)-2-[N'-(1- бензилоксикарбонилпропил)амино] -4-метил-N-(9-оксо-1,8- диазатрицикло[10.6.1.013,18]нонадека-12(19), 13(18), 14,16-тетраен- 10-ил)пентанамида получали (2R,1'S,10S)-2-[N'-(1- карбоксипропил)амино]-4-метил-N-(9-оксо-1,8-диазатрицикло [10.6.1.013,18] нонадека-12(19), 13(18), 14,16-тетраен-10- ил)пентанамид (16 мг), МС(БТЯ): 483 (M-H)-.
В. В соответствии с описанной выше процедурой получения соединения формулы (Io) из трет. -бутилового эфира N-[(бензилоксикарбонил)метил]-L-лейцина (156 мг, 0,465 ммоля), ГОБТ (94 мг, 1,5 экв.), ЭДКИ (134 мг, 1,5 экв. ), N-метилморфолина (77 мкл) и (10S)-10-амино-9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16-тетраена (133 мг, 1 экв.) после мгновенной хроматографии (3% MeOH/CH2Cl2) получали неочищенный бензиловый эфир. Неочищенный бензиловый эфир растворяли в ТГФ/MeOH (1:1, 6 мл) и гидролизовали с помощью 1М гидроксида бария (0,9 мл) в течение ночи. Через раствор пропускали двуокись углерода и образовавшийся осадок отфильтровывали. Фильтрат концентрировали и водный остаток доводили до pH 5,5 с помощью 1М HCl. Путем высокоэффективной жидкостной хроматографии с обращенной фазой получали (2R, 10S)-N'-(карбоксиметил)амино-4-метил-N-(9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18),14,16-тетраен- 10-ил)пентанамид (17 мг) в виде белого твердого продукта, МС(БТЯ): 457 (MH+).
Пример 40
4-Метил-2-метиленпентановая кислота
А. К чистому этилизобутилмалонату (25 г, 0,13 моля) при 0oC медленно добавляли охлажденный на льду диэтиламин (15,1 мл, 0,15 моля). После перемешивания в течение 15 минут добавляли по каплям формалин (11,1 мл, 37% водный формальдегид). Смесь перемешивали при 25oC в течение 3 дней. Затем реакционную смесь обрабатывали раствором, содержащим 20 г К2СО3 в 40 мл воды, и экстрагировали простым эфиром (2х100 мл). Эфирные экстракты объединяли, промывали соляным раствором, сушили над MgSO4 и концентрировали при пониженном давлении при 20oC. Неочищенный продукт, этил-4-метил-2- метиленпентаноат, содержащий некоторое количество эфира, растворяли в абсолютном этаноле (250 мл) и обрабатывали ацетонитрилом (250 мл), 1M LiOH (9,7 г в 250 мл воды, 0,23 моля). После перемешивания в течение ночи органические растворители выпаривали при пониженном давлении и водный остаток экстрагировали этилацетатом (2х150 мл). Объединенные экстракты промывали соляным раствором, сушили над MgSO4 и концентрировали при пониженном давлении для получения 10,5 г 4-метил-2-метиленпентановой кислоты в виде бесцветного масла.
Б. Аналогичным способом получали следующие соединения:
4-фенил-2-метиленбутановую кислоту;
3-циклогексил-2-метиленпропановую кислоту;
5-фенил-2-метиленпентановую кислоту;
2-метиленпентановую кислоту; и
3,3-диметил-2-метиленбутановую кислоту.
Пример 41
Соединения формулы (Id)
(Обращенные гидроксаматы)
А. Раствор 4-метил-2-метиленпентановой кислоты (3,5 г) и О-бензилгидроксиламина выдерживали при 120oC в течение 8 часов. Реакционную смесь затем разделяли между 50 мл 1,0 Н NaOH и 50 мл диэтилового эфира. Водную фракцию отделяли, подкисляли до pH 3 с помощью 10% HCl и промывали 50 мл простого эфира. Путем ионообменной хроматографии (Dowex-50W) при элюировании смесью 20% пиридин/вода получали 2-(бензилоксиаминометил)-4-]метилпентановую кислоту.
300 МГц 1H-ЯМР в CDCl3: δ 0,9-1,0 (dd, 6Н, CH3); 1,25-1,35 (m, 1H, CH); 1,6-1,75 (m, 2H, CH2); 2,8-2.9 (m, 1H, Caльфа-H); 3,0-3,2 (ABq, 2H, CH2N); 4,7-4,75 (ABq, 2H, CH2O); 7,3-7,4 (m, 5H, Ph).
Б. Формилирование 2-(бензилоксиаминометил)-4-метилпентановой кислоты проводили в дихлорметане смесью муравьиная кислота/уксусный ангидрид для получения N-формил-2-(бензилоксиаминометил)-4- метилпентановой кислоты.
Сочетание с соединением формулы (J): к N-формил-2- (бензилоксиаминометил)-4-метилпентановой кислоте (175 мг) и соединению формулы (J) (230 мг) в смеси 5% пиридин/дихлорметан (30 мл) добавляли 4-диметиламинопиридин (ДМАП) (200 мг) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (ЭДКИ) при комнатной температуре. Реакционную смесь перемешивали в течение 8 часов и затем концентрировали и разделяли между 30 мл этилацетата и 30 мл 20% HCl. Органическую фракцию промывали водой (20 мл), 5% NaHCO3 (20 мл) и соляным раствором, сушили над MgSO4 и концентрировали. Путем очистки на силикагеле с элюированием смесью 50% этилацетат/гексан получали продукт в виде смеси двух изомеров. Путем гидрогенолиза в метаноле над 10% Pd/C получали (2R,10S)-2- (N-формил-N-гидроксиаминометил)-4-метил-N-(9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18), 14,16- тетраен-10-ил)пентамид в виде смеси двух изомеров, МС: 455 (M-H)+, (соединение 6).
Аналогичным способом получали:
(2RS, 10S)-2-(изопропоксикарбонилметил)-4-метил-N-(9- оксо-1,8-диазатрицикло [10.6.1.013,18]нонадека-12(19),13 (18),14,16-тетраен-10-ил)пентамид, МС: 455 (М+Н)+.
(2RS, 10S)-2-(морфолинокарбэтоксиметил)-4-метил-N-(9- оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19),13 (18),14,16-тетраен-10-ил)пентамид, МС: 455 (M+H)+.
Пример 42
Данный пример иллюстрирует получение характерных фармацевтических композиций для орального введения, содержащих соединение формулы (I) или его фармацевтически приемлемую соль, например (3R,10S)-2-гидрокси-5-метил-3-(9-оксо-1,8-диазатрицикло [10.6.1.013,18]нонадека-12(19),13(18),14,16-тетраен-10- илкарбамоил)гексанамид:
А. Ингредиенты - Процентное соотношение массы
Соединение формулы (I) - 20,0%
Лактоза - 79,5%
Стеарат магния - 0,5%
Вышеуказанные ингредиенты смешивают и помещают в желатиновые капсулы с твердой оболочкой, содержащие по 100 мг каждая, одна капсула содержит приблизительно суточную полную дозу.
Б. Ингредиенты - Процентное соотношение массы
Соединение формулы (I) - 20,0%
Стеарат магния - 0,9%
Крахмал - 8,6%
Лактоза - 79,6%
ПВП (поливинилпирролидин) - 0,9%
Вышеуказанные ингредиенты, за исключением стеарата магния, смешивают и гранулируют с использованием воды в качестве жидкости для гранулирования. Затем композицию сушат, смешивают со стеаратом магния и таблетируют с помощью соответствующей таблетирующей машины.
В. Ингредиенты
Соединение формулы (I) - 0,1 г
Пропиленгликоль - 20,0 г
Полиэтиленгликоль 400 - 20,0 г
Полисорбат 80 - 1,0 г
Вода - q.s. до 100 мл
Соединение формулы (I) растворяют в пропиленгликоле, полиэтиленгликоле 400 и полисорбате 80. Затем при перемешивании добавляют воду в количестве, необходимом для получения общего объема раствора 100 мл, который фильтруют и разливают по флаконам.
Г. Ингредиенты - Процентное соотношение массы
Соединение формулы (I) - 20,0%
Арахисовое масло - 78,0%
Спан 60 - 2,0%
Вышеуказанные ингредиенты расплавляют, смешивают и заполняют ими мягкие эластичные капсулы.
Пример 43
Данный пример иллюстрирует получение характерной фармацевтической композиции для парентерального назначения, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, например (3R,11S)-N-гидрокси-5-метил-3-(10-оксо-1,9-диазатрицикло[11.6.1.014,19] эйкоза-13(20), 14(19), 15,17-тетраен-11- илкарбамоил)гексанамид:
Ингредиенты
Соединение формулы (I) - 0,02 г
Пропиленгликоль - 20,0 г
Полиэтиленгликоль 400 - 20,0 г
Полисорбат 80 - 1,0 г
0,9% Физиологический раствор - q.s. 100 мл
Соединение формулы (I) растворяют в пропиленгликоле, полиэтиленгликоле 400 и полисорбате 80. Затем добавляют при перемешивании 0,9%-ный физиологический раствор в количестве, достаточном для получения 100 мл раствора для внутривенного вливания, который фильтруют через мембранный фильтр с размером ячеек 0,2 микрона и упаковывают в стерильных условиях.
Пример 44
Данный пример иллюстрирует получение характерной фармацевтической композиции в форме суппозитория, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, например (10S)-2-меркаптометил-4-метил-1-4-(9-оксо-1,8- диазатрицикло[10.6.1.013,18] нонадека-12(19), 13(18),14,16-тетраен-10- илкарбамоил)пентанамид:
Ингредиенты - Процентное соотношение массы
Соединение формулы (I) - 1,0%
Полиэтиленгликоль 1000 - 74,5%
Полиэтиленгликоль 4000 - 24,5%
Ингредиенты расплавляют вместе и смешивают в паровой бане и разливают в формы, вмещающие 2,5 г общего веса.
Пример 45
Данный пример иллюстрирует получение характерной фармацевтической композиции для инсуффляции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, например (10S)-4-метил-2-(9-оксо-1,8-диазатрицикло[10.6.1.013,18] нонадека- 12(19), 13(18), 14,16-тетраен-10-илкарбамоил)пентил-(хинолин-2- илтиометил)фосфиновую кислоту:
Ингредиенты - Процентное соотношение массы
Тонкоизмельченное соединение формулы (I) - 1,0%
Тонкоизмельченная лактоза - 99,0%
Ингредиенты измельчают, смешивают и упаковывают в инсуффлятор, снабженный дозирующим насосом.
Пример 46
Данный пример иллюстрирует получение характерной фармацевтической композиции в беспропеллентной распылительной форме, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, например (3R,10S)-N-гидрокси-5-метил-3-(9-оксо- 1,8-диазатрицикло[10.6.1.013,18] нонадека-12(19),13(18), 14,16- тетраен-10-илкарбамоил)гексанамид:
Ингредиенты - Процентное соотношение массы
Соединение формулы (I) - 0,005%
Вода - 89,995%
Этанол - 10,000%
Соединение формулы (I) растворяют в этаноле и смешивают с водой. Затем композицию упаковывают в распылитель, снабженный дозирующим насосом.
Пример 47
Данный пример иллюстрирует получение характерной фармацевтической композиции в аэрозольной форме, содержащей соединение формулы (I) или его фармацевтически приемлемую соль, например (3R,11S)-N-гидрокси-5-метил- 3-(10-оксо-1,9-диазатрицикло[11.6.1.014,19] эйкоза- 13(20), 14(19),15,17-тетраен-11-илкарбамоил)гексанамид:
Ингредиенты - Процентное соотношение массы
Соединение формулы (I) - 0,10%
Пропеллент 11/12 - 98,90%
Олеиновая кислота - 1,00%
Соединение формулы (I) диспергируют в олеиновой кислоте и вводят пропелленты. Затем полученную смесь заливают в аэрозольную емкость, снабженную дозирующим клапаном.
Пример 48
Анализ in vitro
Коллагеназу типа 1 фибробласта очищали от лишенной сыворотки среды для культивирования клеток GMOO10A, стимулированной РМА, с помощью гепариновой и цинк-хелатирующей сефарозных колонок, а затем с помощью жидкостной хроматографии быстрого разрешения (колонка MONO S). Фермент активировали с помощью инкубации трипсином.
Коллагеназу типа IV очищали от содержащей сыворотку среды для культивирования клеток фибробласта (GMOO10A) с помощью цинк-хелатирующей и желатин-сефарозной колонок, а затем с помощью жидкостной хроматографии быстрого разрешения (колонка MONO S). Электрофорез в полиакриламидном геле с додецилсульфатом натрия показал, что фермент является гомогенным. Фермент активировали с помощью инкубации 1 ммолем АРМА в течение 1 часа при 35-37oC.
Соединение формулы (I) растворяли в ДМСО и добавляли в кювету, содержащую 0,2 мкг коллагеназы типа I или 0,03 мкг коллагеназы типа IV в ТК-буфере (20 мМ Трис, 5 мМ CaCl2, pH 7,5) (конечная концентрация ДМСО 2%). Концентрации соединений формулы (I) выбирали таким образом, чтобы для каждых 20% изменения активности иметь по крайней мере одну экспериментальную точку. Ферменту и соединению давали возможность пройти предварительную 3-минутную инкубацию при 37oC. Для того чтобы инициировать реакцию, к каждым 20 мкМ добавляли N-(7-диметиламино-4-метил)кумаринил ("DACM") (фирма Sigma) и тиопептид (Ac-Pro-Leu-Gly-S- "Leu"-Leu-Gly-OEt, фирма Bachem Bioscience Inc.). Флуоресценцию регистрировали при длинах волн возбуждения и эмиссии 395 и 485 нм соответственно. Каждую экспериментальную точку получали осреднением по двукратному эксперименту. Для определения значения IC50 с помощью программы Enzfitter использовали по крайней мере шесть экспериментальных точек, показывающих зависимость изменения флуоресценции в минуту от концентрации соединения.
Этот анализ подтверждает, что соединения формулы (I) обладают способностью ингибировать коллагеназы (см. таблицу).
Пример 49
Анализ in vitro
Этот анализ позволяет определить, обладают ли соединения формулы (I) с способностью ингибировать выход меченных по 35S гликозаминогликанов (GAG) из хрящевых эксплантатов.
Небольшие хрящевые эксплантаты (3 мм в диаметре) получали из коленных суставов свежеумерщвленных быков и метили с помощью 35SO4. Меченные по 35S гликозаминогликаны (GAG) выходили в среду для культивирования в ответ на добавление rhIL-1-альфа, индуцирующего экспрессию хондроцитных матричных металлопротеаз (ММП), включающих стромелизин и коллагеназу. При определении процента ингибирования меченных GAG вводили поправку на спонтанный выход в отсутствии rhIL-1-альфа. Результаты для каждой группы представляют собой средние значения ± среднеквадратичная ошибка по пяти эксплантантам.
Соединения формулы (I) при тестировании этим методом проявили способность ингибировать выход меченных по 35S (GAG) из хрящевых эксплантантов.
Соединение 2 (пример 8) IC50 = 40 мкМ
Соединение 4 (пример 25) IC50 = 50 мкМ
Пример 50
Анализ in vitro
Для изучения in vitro влияния соединений формулы (I) на резорбцию кости использовали в качестве модели длинную кость крысиных эмбрионов. Для индуцирования резорбции кости in vitro использовали бычий РТН. Влияние на резорбцию кости выражали с помощью количества 45Ca, вышедшего в среду для культивирования из предварительно меченных по 45Ca длинных костей крысиных эмбрионов. Ингибирующий эффект соединений формулы (I) в отношении индуцированной бычьим РТН резорбции кости выражали в виде среднего процента ингибирования ± среднеквадратичная ошибка.
Предварительно меченные по 45Ca длинные кости крысиных эмбрионов (из предплечья) рассекали и культивировали в чашках Linbro при 37oC в течение ночи в среде BGJb, дополненной 1 мг/мл бычьим сывороточным альбумином (БСА). В каждой группе использовали по пять пар костей. Соединения формулы (I) сначала растворяли в этаноле, затем разбавляли до различных концентраций и добавляли одновременно с бычьим РТН (1-34) в концентрации 1•10-8 М в первый день. Концентрации этанола в растворах соединений составляли менее 0,05%, что не оказывало влияния на анализ. Анализ заканчивали на шестой день с однократной заменой среды на третий день.
В конце каждой замены среды подсчитывали количество 45Ca, присутствующего в среде для культивирования. Оставшиеся кости разлагали с помощью 0,1Н HCl и также подсчитывали количество 45Ca, присутствующего в продукте разложения кости. Результаты выражали в виде % по отношению к общему количеству 45Ca, вышедшему из каждой пары костей. Бычий РТН в концентрации 1•10-8 М индуцирует резорбцию кости до максимального уровня, который принимается за 100%, и эту концентрацию использовали в качестве стандарта. Уровень резорбции кости в присутствии только среды принимали за базовую линию (за 0%). Все группы, обработанные соединениями, сравнивали с бычьим РТН (1-34) в концентрации 1•10-8 М. Концентрация, при которой соединение ингибирует резорбцию кости на 50%, обозначали как IC50.
Соединения формулы (I) при тестировании этим методом проявили способность ингибировать индуцируемую бычьим РТН резорбцию кости.
Соединение 3 (пример 10) - IC50 = 0,1 мкМ
Соединение 4 (пример 25) - IC50 = 5 мкМ
Токсикология
В вышеописанных анализах не было обнаружено никаких серьезных токсикологических эффектов.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОСТИКОВЫЕ ИНДОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ МАТРИЧНЫХ МЕТАЛЛОПРОТЕАЗ | 1996 |
|
RU2191779C2 |
| ЗАМЕЩЕННЫЕ КАРБОКСАМИДЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2132327C1 |
| КАРБОКСАМИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2163232C2 |
| Способ получения (4,2,0)бициклооктановых производных, или их фармацевтически приемлемых нетоксичных солей, или фармацевтически приемлемых нетоксичных сложных эфиров | 1986 |
|
SU1500153A3 |
| Способ получения (4,2,0)-бициклооктановых производных или их фармацевтически приемлемых нетоксичных солей | 1987 |
|
SU1588275A3 |
| ПРОИЗВОДНЫЕ БЕНЗОЦИКЛОАЛКИЛАЗОЛОТИОНА | 1995 |
|
RU2145321C1 |
| ПРОИЗВОДНЫЕ 10,11-МЕТАНДИБЕНЗОСУБЕРАНА, ХЕМОСЕНСИБИЛИЗИРУЮЩАЯ КОМПОЗИЦИЯ, СПОСОБ ПРЕОДОЛЕНИЯ РЕЗИСТЕНТНОСТИ К ПРОТИВООПУХОЛЕВЫМ ЛЕКАРСТВАМ И СПОСОБ ЛЕЧЕНИЯ ОПУХОЛЕВЫХ ЗАБОЛЕВАНИЙ | 1994 |
|
RU2167154C2 |
| Способ получения (2-оксо-1,2,3,5-тетрагидроимидазо-/2,1- @ /хиназолинил)оксиалкиламидов,их оптических изомеров или фармакологически приемлемых солей с кислотами | 1984 |
|
SU1349700A3 |
| Фармацевтически эффективные соединения, селективно ингибирующие изоформы миозина 2 | 2019 |
|
RU2817643C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛО[1,5-а]ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ ТРК КИНАЗЫ | 2014 |
|
RU2666367C2 |
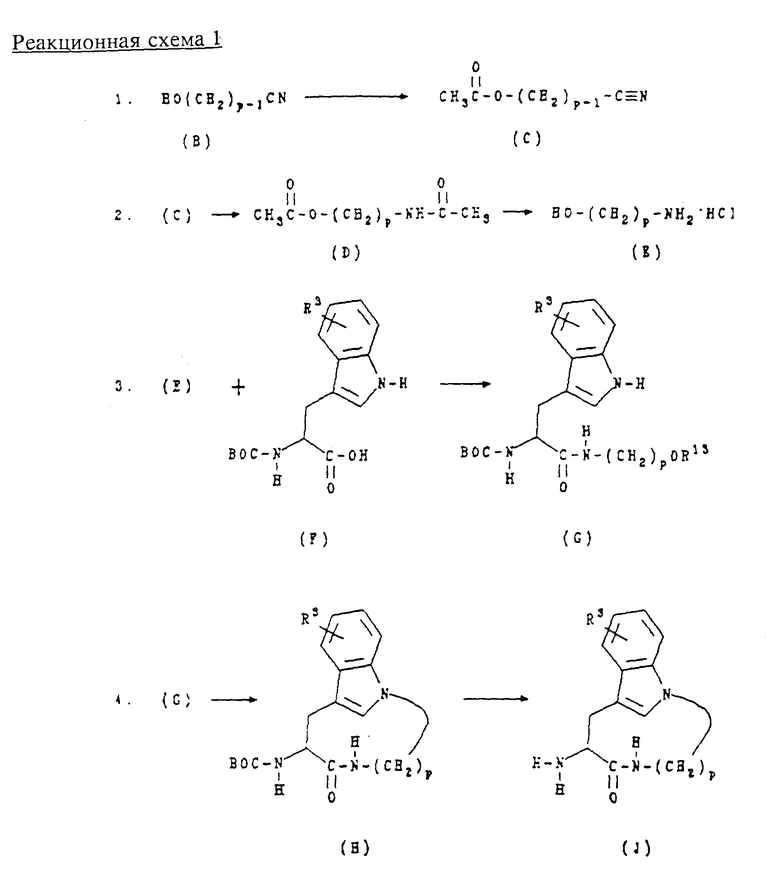
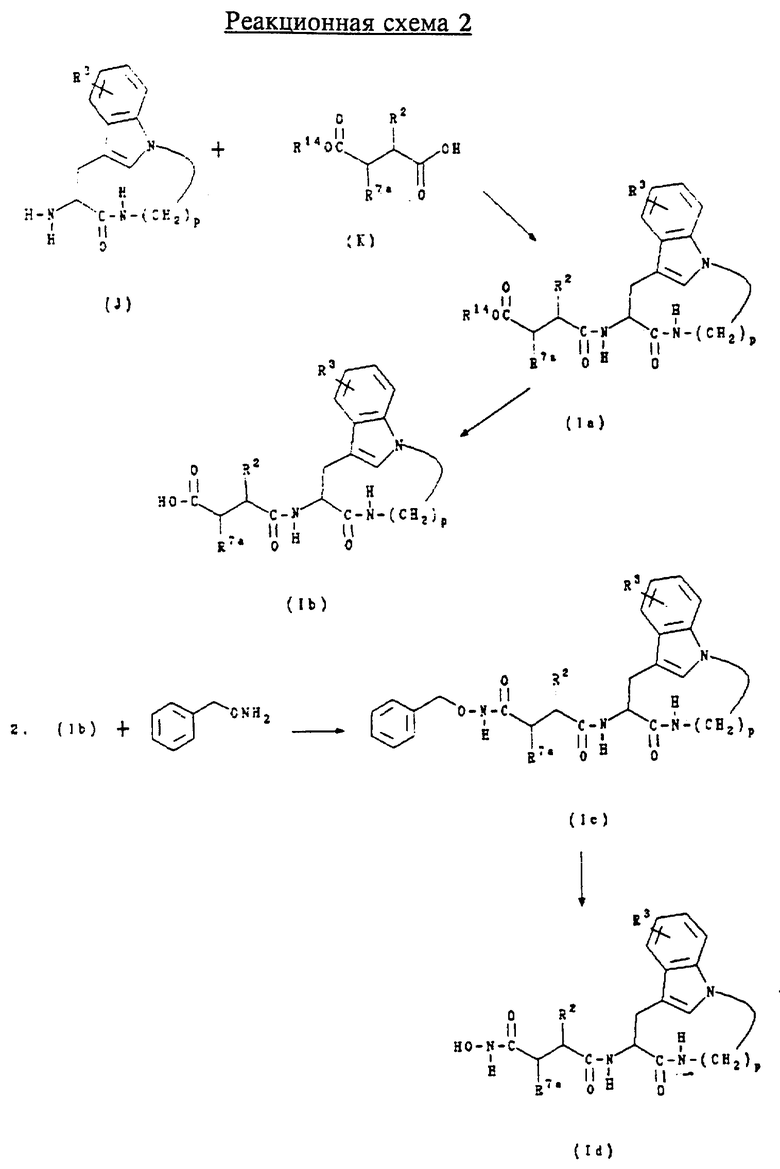
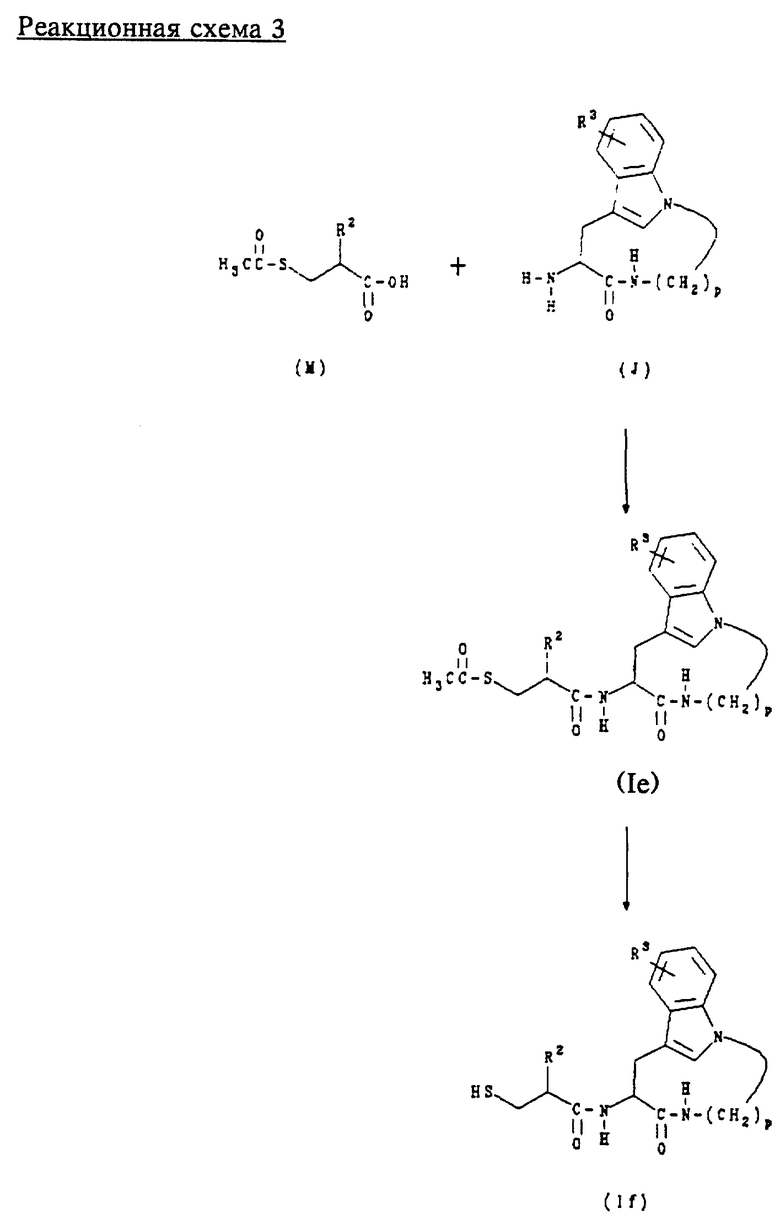
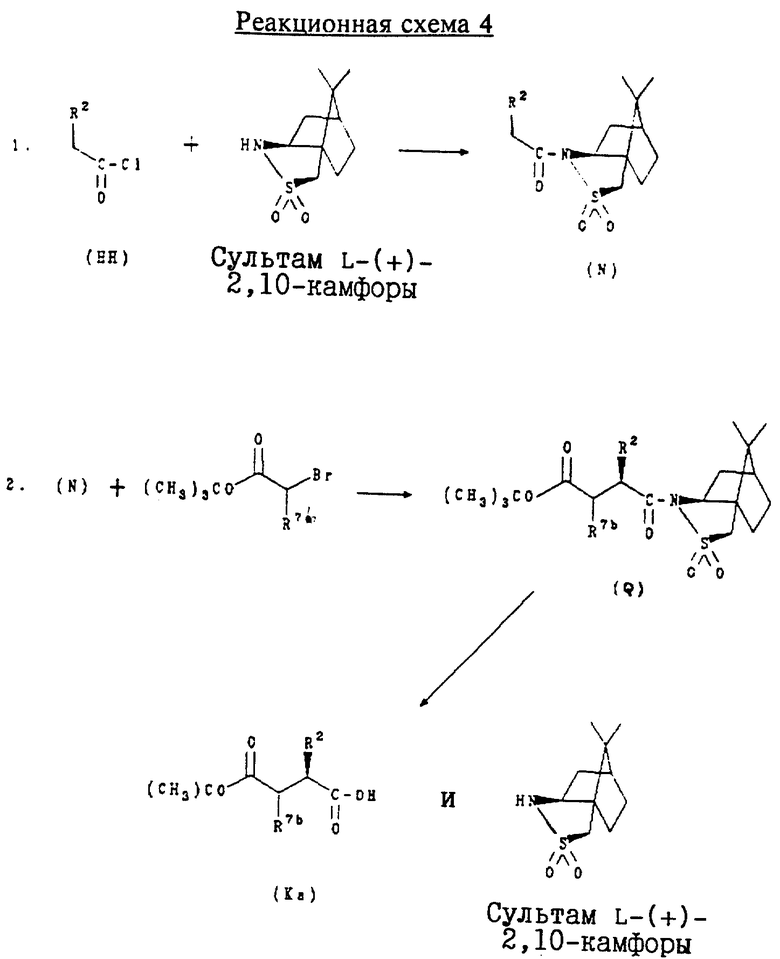
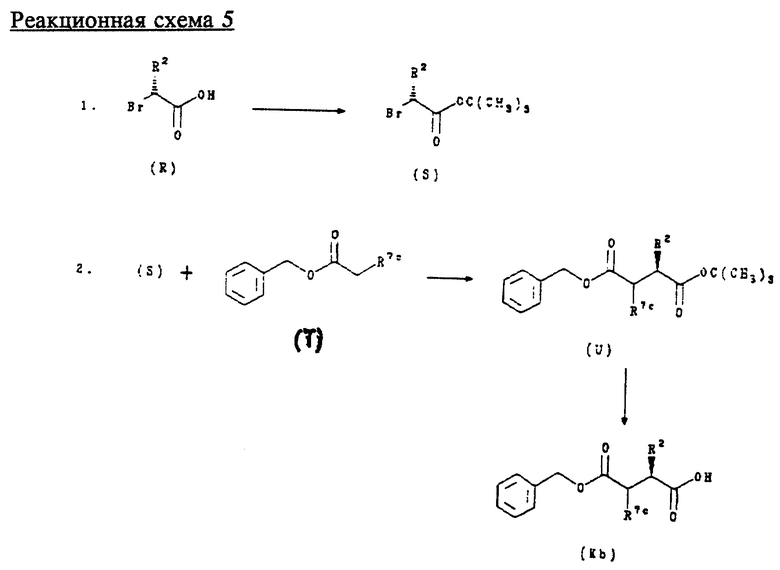
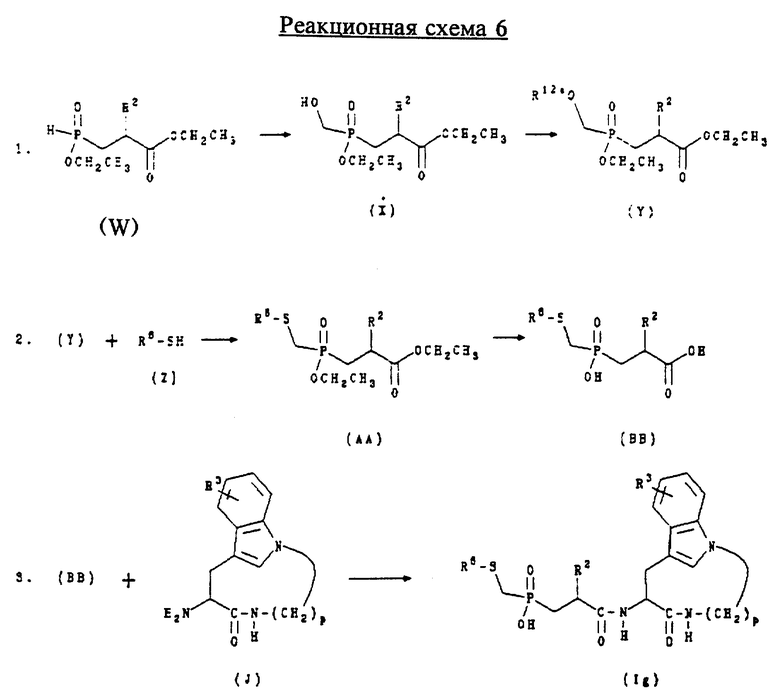
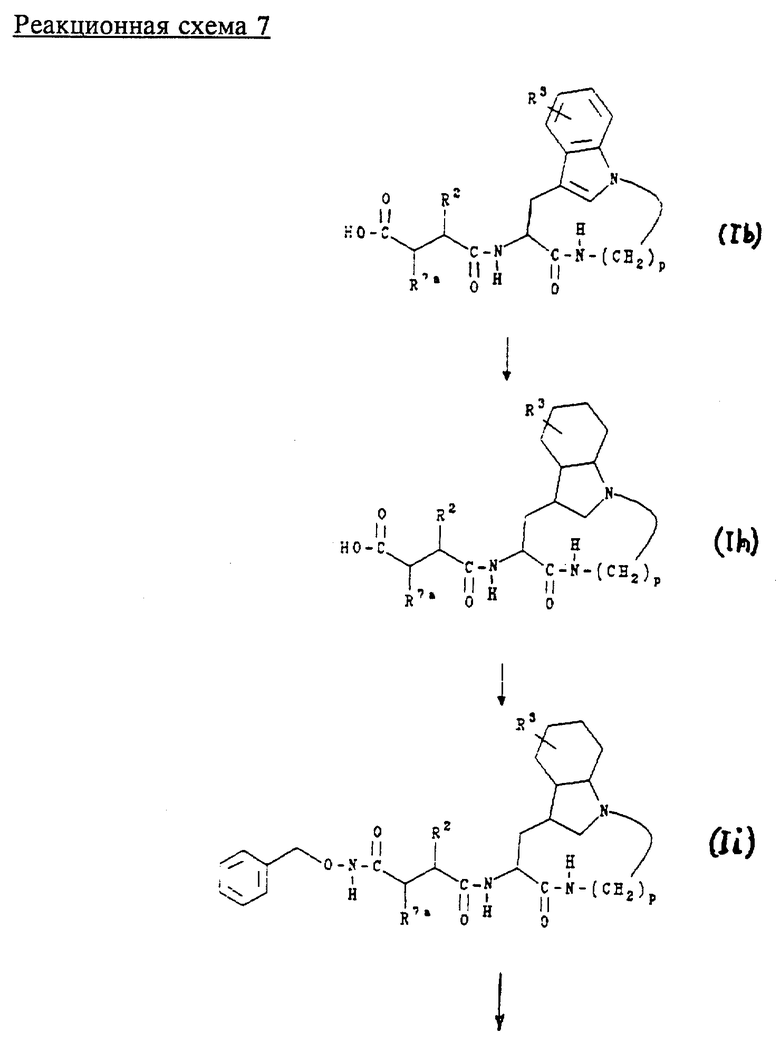
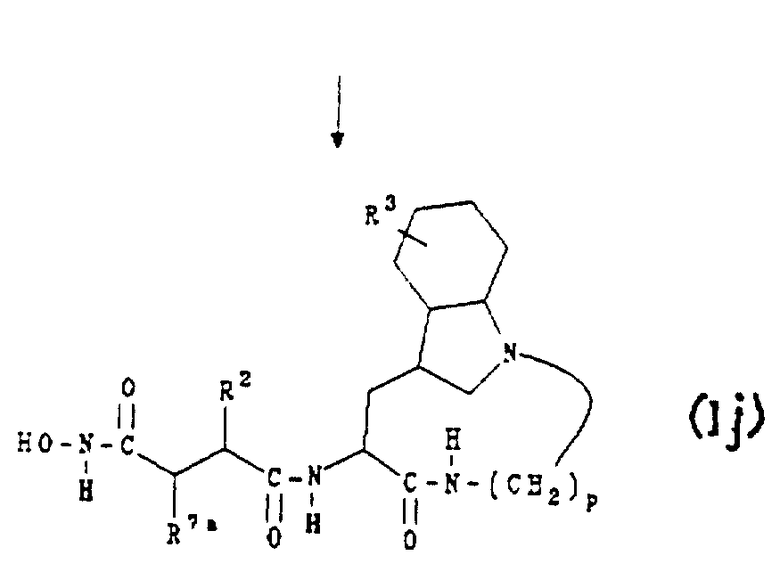
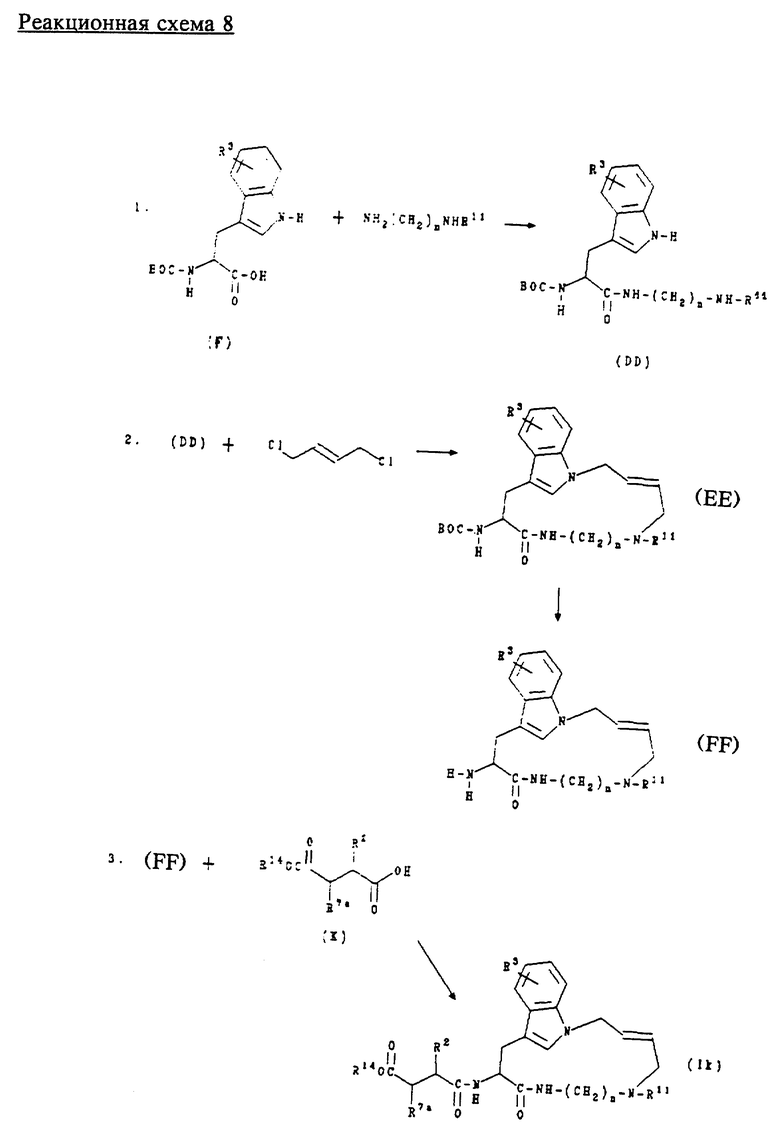
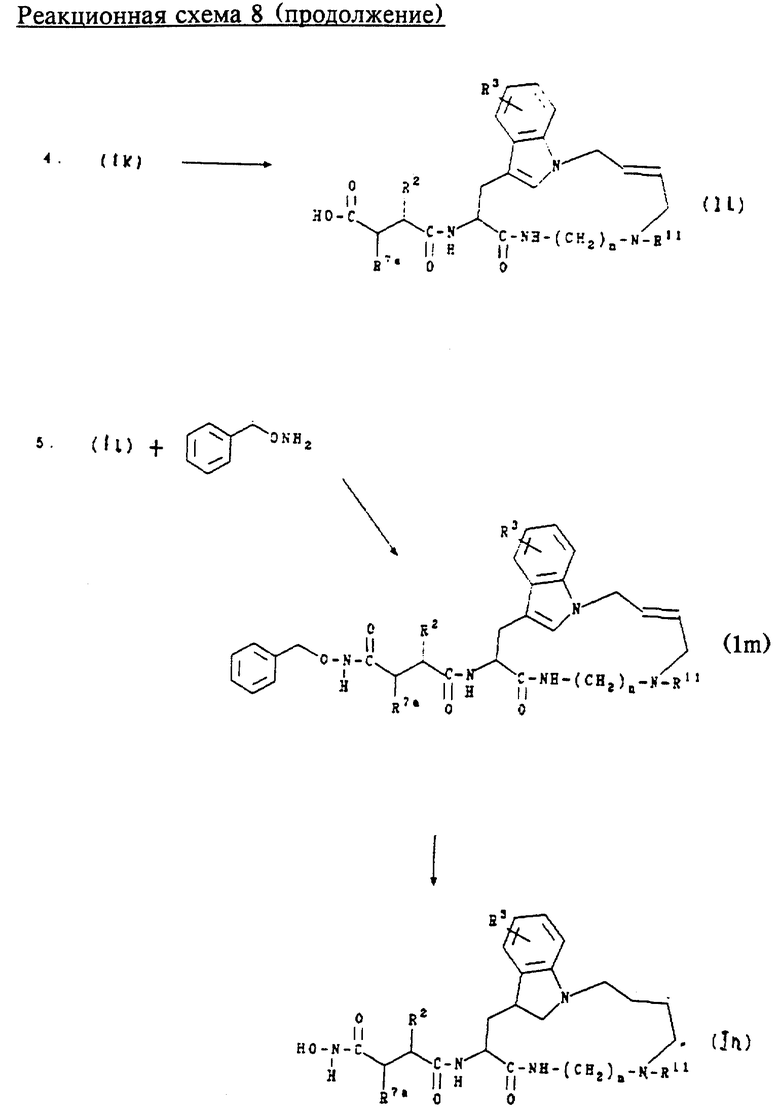
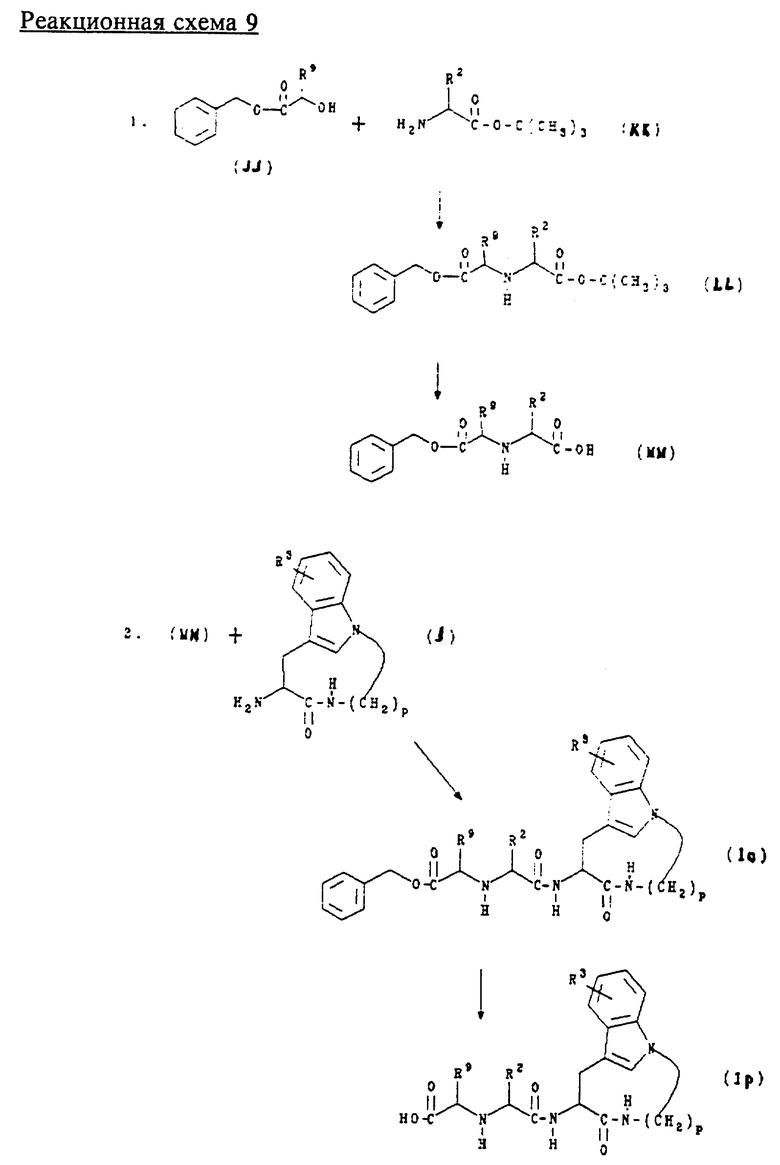

Соединения формулы 1, где штриховые линии обозначают необязательные двойные связи, и, если n = 1, 2 или 3; m = 3 или 4, то А обозначает -СН2-; R1 обозначает а) -CH2-R4, где R4 обозначает меркапто, ацетилтио, карбокси, гидроксиаминокарбонил, N -гидроксиформиламино, алкоксикарбонил, морфолино (С1-С4)алкоксикарбонил, арилоксикарбонил, бензилоксиаминокарбонил или остаток формулы (i), где R6 обозначает хинол-2-ил; б) -СН(R7)-R8, где R7, обозначает алкил, гидрокси, амино, алкоксикарбонил, аминокарбонил или карбокси и R8 обозначает карбокси, гидроксиаминокарбонил, алкоксикарбонил или аралкоксикарбонил, в) -NН-СН(R9)-R10, где R9 обозначает водород или алкил и R10 обозначает карбокси, аралкоксикарбонил; R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил и R3 обозначает водород, или, если n = 2 или 3; m = 3 или 4, то А обозначает - N(R11)-, где R11 обозначает водород или алкил; и R1, R2 и R3 имеют указанные выше значения, в виде отдельных стереоизомеров или в виде их смеси или их фармацевтически приемлемые соли; пригодны для ингибирования активности матричной металлопротеазы у млекопитающих. 2 с. и 24 з.п. ф-лы, 1 табл.
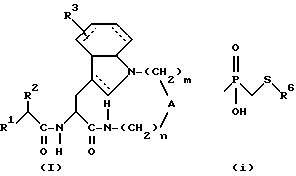
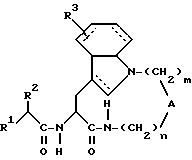
где штриховые линии обозначают необязательные двойные связи и если n = 1, 2 или 3; m = 3 или 4, то А обозначает -CH2-;
R1 обозначает а) -CH2-R4, где R4 обозначает меркапто, ацетилтио, карбокси, гидроксиаминокарбонил, N-гидроксиформиламино, алкоксикарбонил, морфолино(C1-C4)алкоксикарбонил, арилоксикарбонил, бензилоксиаминокарбонил или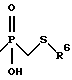
где R6 обозначает хинол-2-ил;
б) -CH(R7)-R8, где R7 обозначает алкил, гидрокси, амино, алкоксикарбонил, аминокарбонил или карбокси и R8 обозначает карбокси, гидроксиаминокарбонил, алкоксикарбонил или аралкоксикарбонил;
в) -NH-CH(R9)-R10, где R9 обозначает водород или алкил и R10 обозначает карбокси или аралкоксикарбонил;
R2 обозначает алкил, циклоалкил, циклоалкилалкил или аралкил;
R3 обозначает водород;
или, если n = 2 или 3,; m = 3 или 4, то А обозначает -N(R11)-, где R11 обозначает водород или алкил;
и R1, R2 и R3 имеют указанные выше значения;
в виде отдельных стереоизомеров или в виде их смеси или их фармацевтически приемлемые соли.
| WO 9221360 A, 10.12.92 | |||
| RU 94005002 A1, 27.09.95 | |||
| WO 9309136 A, 13.05.93 | |||
| WO 9206966 A, 30.04.92 | |||
| EP 0438223 A, 24.07.91. |

