Изобретение относится к новым тропановым производным, которые представляют собой сильные ингибиторы обратного захвата нейромедиаторных моноаминов, то есть дофамина, серотонина и норадреналина, и к применению новых тропановых производных для лечения расстройств или заболеваний, на которые влияет ингибирование обратного захвата нейромедиаторных моноаминов, таких как болезнь Паркинсона, депрессия, обсессивно-компульсивные расстройства, панические состояния, деменция, нарушения памяти, гиперактивность при дефиците внимания, ожирение, состояние тревоги, нарушения аппетита и токсикомания или злоупотребление лекарственными средствами, включая злоупотребление кокаином.
Мозг состоит из множества нейронов, которые контактируют друг с другом через химические переносчики. Каждый нейрон генерирует нейрохимические вещества, или нейромедиаторы, которые действуют по сайтам на клеточной мембране нейрона, относящимся к рецепторам. Одна из групп нейромедиаторов, которая относится к нейромедиаторным моноаминам, включает в себя серотонин, дофамин и норадреналин.
Нейромедиаторные моноамины высвобождаются в синаптическую щель между нейронами, чтобы стимулировать активность постсинаптического рецептора. Удаление (или инактивация) нейромедиаторных моноаминов осуществляется главным образом по механизму обратного захвата в пресинаптические окончания. При ингибировании обратного захвата имеет место усиление физиологической активности нейромедиаторных моноаминов.
Показано, что серотонэргическая невральная система мозга влияет на разнообразные физиологические функции, и предсказывают, что соединения, обладающие ингибирующей обратный захват серотонина активностью, способны лечить у млекопитающих, включая человека, разнообразные расстройства, связанные с этой невральной системой, например нарушения аппетита, депрессию, обсессивно-компульсивные расстройства, панические состояния, алкоголизм, боль, нарушения памяти и состояние тревоги. В эту группу заболеваний входят расстройства, связанные с депрессией, такие как псевдодеменция или синдром Ганзера, мигрень, булимия, ожирение, предменструальный синдром или синдром поздней лютеиновой фазы, табачная зависимость, паническое состояние, посттравматический синдром, потеря памяти, деменция старения, комплекс деменции синдрома приобретенного иммунодефицита, нарушение памяти при старении, социальная фобия, гиперактивность при дефиците внимания, синдром хронического утомления, преждевременная эякуляция, затруднение эрекции, нервная анорексия, нарушения сна, аутизм, мутизм или трихотилломания.
Патофизиология основного эмоционального нарушения недостаточно понята, и предполагалось, что в патогенез основной депрессии вовлечено несколько нейромедиаторов.
Смешанные ингибиторы обратного захвата норадреналина и серотонина, такие как Имипрамин и Амитриптилин, и ингибиторы обратного захвата норадреналина, такие как Дезипрамин, Нортриптилин и Протриптилин, представляют собой современные фармацевтические препараты, применяемые в антидепрессантной терапии. Более того, несколько серий предклинических и клинических данных указывают на то, что повышение серотонин-опосредованной нейротрансмиссии, возможно, лежит в основе терапевтического эффекта большинства лекарственных средств прошлого и современных лекарственных средств, применяемых в антидепрессантной терапии: Флуоксетина, Циталопрама и Пароксетина.
Парадоксальные современные ингибиторы обратного захвата серотонина ингибируют переносчик серотонина в течение минут, тогда как их антидепрессантный эффект в полной мере наблюдается лишь через три-четыре недели лечения, что указывает на то, что ингибирование обратного захвата per se не ответственно за антидепрессантный ответ, а скорее дальнейшие адаптивные изменения лежат в основе терапевтического эффекта и/или вносят вклад в него. Считается, что запоздалое проявление антидепрессантного эффекта является серьезным недостатком современных ингибиторов обратного захвата моноаминов.
Считают, что сильная ингибиторная активность обратного захвата дофамина связана с риском нежелательных центрально-стимулирующих эффектов. С другой стороны, полагают, что активирующее воздействие на мезолимбическую дофаминовую систему лежит в основе общего механизма современного антидепрессантного лечения посредством усиления эндогенной компенсаторной системы. Соединения с сильной ингибиторной активностью обратного захвата серотонина в сочетании с хорошо сбалансированной умеренной ингибиторной активностью обратного захвата дофамина могут, следовательно, продуцировать агенты с быстрым появлением антидепрессантного эффекта.
Соединения по настоящему изобретению являются также сильными ингибиторами обратного захвата дофамина и, таким образом, применимы для лечения паркинсонизма, депрессии, ожирения, нарколепсии, лекарственной зависимости или злоупотребления лекарственными средствами, включая злоупотребление кокаином, гиперактивности при дефиците внимания, болезни Жилль де ля Туретта и сенильной деменции. Ингибиторы обратного захвата дофамина косвенно усиливают через дофаминовые нейроны высвобождение ацетилхолина и, следовательно, также применимы для лечения нарушения памяти, например при болезни Альцгеймера, пресенильной деменции, нарушения памяти при старении и синдрома хронического утомления. Ингибиторы обратного захвата норадреналина считают применимыми для повышения внимания, улучшения состояний тревоги, возбуждения, бессонницы и для лечения депрессии.
Задачей настоящего изобретения является создание новых тропановых производных, которые представляют собой ингибиторы обратного захвата нейромедиаторных моноаминов и, следовательно, применимы для лечения таких расстройств, как болезнь Паркинсона, депрессия и родственные ей заболевания, обсессивно-компульсивные расстройства, панические состояния, деменция, нарушения памяти, гиперактивность при дефиците внимания, ожирение, состояние тревоги, нарушения аппетита, лекарственная зависимость или злоупотребление лекарственными средствами, включая злоупотребеление кокаином.
Другой задачей настоящего изобретения является создание новых фармацевтических композиций, содержащих новые тропановые производные.
Еще одной задачей изобретения является создание способа лечения заболеваний или расстройств, на которые влияет ингибирование обратного захвата нейромедиаторных моноаминов, таких как болезнь Паркинсона, депрессия и родственные ей заболевания, обсессивно-компульсивные расстройства, панические состояния, деменция, нарушения памяти, гиперактивность при дефиците внимания, ожирение, состояние тревоги, нарушения аппетита, лекарственная зависимость или злоупотребление лекарственными средствами, включая злоупотребление кокаином.
Другие задачи станут очевидны специалисту далее.
Таким образом, изобретение, inter alia, включает в себя следующее, по отдельности или в сочетании:
Соединение формулы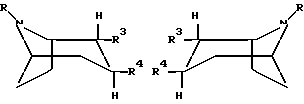

или любая их смесь, или их фармацевтически приемлемая соль;
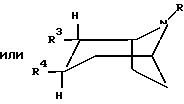
где R представляет собой водород, алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил или 2-гидроксиэтил;
R3 представляет собой -CH2-X-R', где X представляет собой O, S или NR'', где R'' является водородом или алкилом, а R' представляет собой алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил или -CO-алкил; и
R4 представляет собой
фенил, который может быть замещен один или более чем один раз заместителями, выбранными из группы, состоящей из галогена, CF3, CN, алкокси, циклоалкокси, алкила, циклоалкила, алкенила, алкинила, амино, нитро, гетероарила и арила;
3,4-метилендиоксифенил;
бензил, который может быть замещен один или более чем один раз заместителями, выбранными из группы, состоящей из галогена, CF3, CN, алкокси, циклоалкокси, алкила, циклоалкила, алкенила, алкинила, амино, нитро, гетероарила и арила;
гетероарил, который может быть замещен один или более чем один раз заместителями, выбранными из группы, состоящей из галогена, CF3, CN, алкокси, циклоалкокси, алкила, циклоалкила, алкенила, алкинила, амино, нитро, гетероарила и арила; или
нафтил, который может быть замещен один или более чем один раз заместителями, выбранными из группы, состоящей из галогена, CF3, CN, алкокси, циклоалкокси, алкила, циклоалкила, алкенила, алкинила, амино, нитро, гетероарила и арила;
соединение по п.1, которое представляет собой
2-метоксиметил-3-(3,4-дихлорфенил)-тропан,
2-изопропоксиметил-3-(3,4-дихлорфенил)-тропан,
2-этоксиметил-3-(3,4-дихлорфенил)-тропан,
2-циклопропилметилоксиметил-3-(3,4-дихлорфенил)-тропан,
2-метоксиметил-3-(4-хлорфенил)-тропан,
N-норметил-2-метоксиметил-3-(4-хлорфенил)-тропан,
2-этоксиметил-3-(4-хлорфенил)-тропан,
N-норметил-2-метоксиметил-3-(3,4-дихлорфенил)-тропан,
N-норметил-2-этоксиметил-3-(3,4-дихлорфенил)-тропан,
N-норметил-2-этоксиметил-3-(4-хлорфенил)-тропан,
2-этилтиометил-3-(3,4-дихлорфенил)-тропан,
2-циклопропилметилоксиметил-3-(4-хлорфенил)-тропан, или
N-норметил-2-циклопропилметилоксиметил-3-(4-хлорфенил)-тропан,
или его фармацевтически приемлемую соль присоединения;
соединение по п.1, которое представляет собой
(1R,2R,3S)-2-метоксиметил-3-(3,4-дихлорфенил)-тропан,
(1R,2R,3S)-2-изопропоксиметил-3-(3,4-дихлорфенил)-тропан,
(1R,2R,3S)-2-этоксиметил-3-(3,4-дихлорфенил)-тропан,
(1R,2R,3S)-2-циклопропилметилоксиметил-3-(3,4-дихлорфенил)-тропан,
(1R,2R,3S)-2-метоксиметил-3-(4-хлорфенил)-тропан,
(1R,2R,3S)-N-норметил-2-метоксиметил-3-(4-хлорфенил)-тропан,
(1R,2R,3S)-2-этоксиметил-3-(4-хлорфенил)-тропан,
(1R,2R,3S)-N-норметил-2-метоксиметил-3-(3,4-дихлорфенил)-тропан,
(1R,2R,3S)-N-норметил-2-этоксиметил-3-(3,4-дихлорфенил)-тропан,
(1R,2R,3S)-N-норметил-2-этоксиметил-3-(4-хлорфенил)-тропан,
(1R, 2R, 3S)-N-норметил-2-циклопропилметилоксиметил-3-(4- хлорфенил)-тропан,
(1R,2R,3S)-2-циклопропилметилоксиметил-3-(4-хлорофенил)-тропан, или
(1R,2R,3S)-2-этилтиометил-3-(3,4-дихлорофенил)-тропан
или его фармацевтически приемлемую соль присоединения;
фармацевтическая композиция, содержащая терапевтически эффективное количество соединения, такого как любое из указанных выше, или его фармацевтически приемлемой соли присоединения, вместе с по меньшей мере одним фармацевтически приемлемым носителем или разбавителем;
применение соединения, такого как любое из указанных выше, для изготовления лекарственного средства для лечения расстройства или заболевания у живого организма, включая человека, причем расстройства или заболевания, реагирующего на ингибирование обратного захвата нейромедиаторных моноаминов в центральной нервной системе;
применение соединения, такого как любое из указанных выше, где расстройство или заболевание представляет собой паркинсонизм, депрессию, псевдодеменцию, ожирение, нарколепсию, лекарственную зависимость и/или злоупотребление лекарственными средствами, гиперактивность при дефиците внимания, сенильную деменцию или нарушение памяти;
способ получения соединений, таких как указано выше, включающий в себя стадию, на которой соединение формулы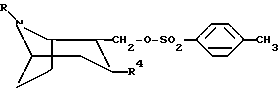
или любой из его энантиомеров или любую их смесь, где R и R4 такие, как определено в п. 1, подвергают взаимодействию с алкоголятом R'-Z-Na, где R' такой, как определено в п.1, а Z представляет собой O или S, с образованием соединения по изобретению, в котором X представляет собой O или S;
соединение формулы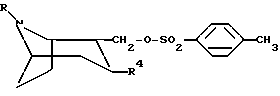
или любой из его энантиомеров или любую их смесь, где R и R4 такие, как определено в п.1, подвергают взаимодействию с амином NHR''-R' с образованием соединения по изобретению, в котором X представляет собой NR''; или
соединение формулы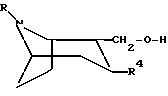
или любой из его энантиомеров или любую их смесь, где R и R4 такие, как определено в п.1, подвергают взаимодействию с гидридом натрия и соединением формулы R'-SO2, где R' такой, как определено в п.1, с образованием соединения по изобретению, в котором X представляет собой O;
способ лечения расстройства или заболевания у живого организма, включая человека, причем расстройства или заболевания, реагирующего на ингибирование обратного захвата нейромедиаторных моноаминов, содержащий стадию, на которой нуждающемуся в таком лечении живому организму, включая человека, вводят терапевтически эффективное количество соединения по пп. 1-3; и
способ, как указано выше, отличающийся тем, что лечат паркинсонизм, депрессию, псевдодеменцию, ожирение, нарколепсию, лекарственную зависимость и/или злоупотребление лекарственными средствами, гиперактивность при дефиците внимания, нарушение познавательной способности, сенильную деменцию или нарушение памяти.
Примеры фармацевтически приемлемых солей присоединения включают в себя соли присоединения неорганических и органических кислот, такие как гидрохлорид, гидробромид, фосфат, нитрат, перхлорат, сульфат, цитрат, лактат, тартрат, малеат, фумарат, манделат, бензоат, аскорбат, циннамат, бензолсульфонат, метансульфонат, стеарат, сукцинат, глутамат, гликолат, толуол-п-сульфонат, формат, малонат, нафталин-2-сульфонат, салицилат и ацетат. Такие соли получают по методикам, хорошо известным специалисту.
Другие кислоты, такие как щавелевая кислота, хотя сами по себе не являются фармацевтически приемлемыми, можно использовать при получении солей, применимых в качестве промежуточных соединений для получения соединений по изобретению и их солей присоединения фармацевтически приемлемых кислот.
Галоген представляет собой фтор, хлор, бром или иод.
Алкил означает нормальную или разветвленную цепь, содержащую от одного до шести атомов углерода, включая в себя, но не ограничиваясь ими, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил и гексил; предпочтительными группами являются метил, этил, пропил и изопропил.
Циклоалкил означает циклический алкил, содержащий от трех до семи атомов углерода, включая в себя, но не ограничиваясь ими, циклопропил, циклобутил, циклопентил и циклогексил.
Алкенил означает группу, содержащую от двух до шести атомов углерода, содержащую по меньшей мере одну двойную связь, например этенил, 1,2- или 2,3-пропенил, 1,2-, 2,3- или 3,4-бутенил, но не ограничиваясь этими примерами.
Алкинил означает группу, содержащую от двух до шести атомов углерода, содержащую по меньшей мере одну тройную связь, например этинил, 2,3-пропинил, 2,3- или 3,4-бутинил.
Циклоалкилалкил означает циклоалкил, как указано выше, и алкил, как указано выше, например, циклопропилметил.
Алкоксигруппа представляет собой O-алкил, где алкил такой, как определено выше.
Циклоалкоксигруппа представляет собой O-циклоалкил, где циклоалкил такой, как определено выше.
Аминогруппа представляет собой NH2, или NH-алкил, или N-(алкил)2, где алкил такой, как определено выше.
Гетероарил представляет собой 5- или 6-членную гетероциклическую моноциклическую группу. Такая гетероарильная группа включает в себя, например, оксазол-2-ил, оксазол-4-ил, оксазол-5-ил, изооксазол-3-ил, изооксазол-4-ил, изооксазол-5-ил, тиазол-2-ил, тиазол-4-ил, тиазол-5-ил, изотиазол-3-ил, изотиазол-4-ил, изотиазол-5-ил, 1,2,4-оксадиазол-3-ил, 1,2,4-оксадиазол-5-ил, 1,2,4-тиадиазол-3-ил, 1,2,4-тиадиазол-5-ил, 1,2,5-оксадиазол-3-ил, 1,2,5-оксадиазол-4-ил, 1,2,5-тиадиазол-3-ил, 1,2,5-тиадиазол-4-ил, 1-имидазолил, 2-имидазолил, 4-имидазолил, 1-пирролил, 2-пирролил, 3-пирролил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидтинил, 4-пиримидтинил, 5-пиримидтинил, 3-пиридазинил, 4-пиридазинил, 2-пиразинил и 3-пиразинил и 1-пиразолил, 3-пиразолил и 4-пиразолил.
Арил представляет собой ароматический углеводород, такой как фенил и нафтил.
Кроме того, соединения по настоящему изобретению могут существовать как в несольватированной, так и в сольватированной форме с фармацевтически приемлемыми растворителями, такими как вода, этанол и им подобные. Как правило, считается, что для назначений настоящего изобретения сольватированные формы эквивалентны несольватированным.
Специалисты должны принимать во внимание, что соединения по настоящему изобретению содержат несколько хиральных центров, и что такие соединения существуют в форме изомеров, то есть, энантиомеров. Изобретение включает в себя все такие изомеры и любые их смеси, включая рацемические смеси.
Рацемические формы могут быть разделены на оптические антиподы известными способами, например разделением их диастереомерных солей с помощью оптически активной кислоты и высвобождением оптически активного аминосоединения обработкой основанием. Другой способ разделения рацематов на оптические антиподы основан на хроматографии на оптически активной стационарной фазе. Рацемические соединения по настоящему изобретению можно, таким образом, разделить на оптические антиподы, например, путем фракционной кристаллизации d- или l-солей, например тартратов, манделатов или камфорсульфонатов. Соединения по настоящему изобретению можно также разделить путем образования диастереомерных амидов посредством взаимодействия этих соединений с оптически активной активированной карбоновой кислотой, такой как производное от (+) или (-) фенилаланина, (+) или (-) фенилглицина, (+) или (-) камфановой кислоты, или путем образования диастереомерных карбаматов взаимодействием соединений по настоящему изобретению с оптически активным хлорформатом или ему подобным веществом.
Можно использовать известные дополнительные методы разделения оптических изомеров, которые очевидны для среднего специалиста. Такие методы описаны J. Jaques, A. Collet, S.Wilen ("Enantiomers, racemates and resolutions", John Wiley and Sons, New York, 1981).
Оптически активные соединения можно также получить из исходных оптически активных материалов.
Следующая схема иллюстрирует способы, которыми можно получить соединения по изобретению: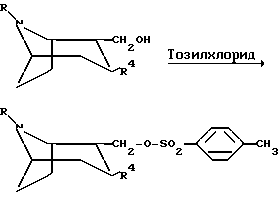
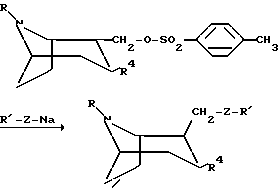
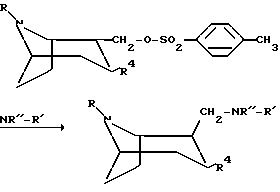
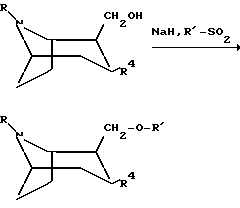
Процессы на реакционной схеме, приведенной выше, осуществляют традиционным образом. Заместитель Z в приведенной выше схеме означает O или S.
Исходные материалы для способов, описанных в настоящей патентной заявке, известны или могут быть получены известными способами из имеющихся в продаже материалов (см. WO-95/28401).
Соединение по изобретению может быть превращено в другое соединение по изобретению с использованием стандартных способов.
Продукты описанных здесь реакций выделяют стандартными способами, такими как экстракция, кристаллизация, дистилляция, хроматография и т.п.
Биология
Соединения по настоящему изобретению были исследованы на их способность связываться с медиатором дофамина с помощью следующих тестов на in vitro ингибирование 3H-WIN 35428.
Ингибирование in vitro связывания 3H-WIN 35428
Предпосылки
Дофаминовые медиаторы/сайты захвата на нервных окончаниях предположительно функционируют как терминаторы нейронного импульса путем удаления дофамина из синаптической щели. Активность или наличие транспортного интегрального белка дофамина могут быть измерены в анализах in vitro по синаптосомальному захвату или мембранному связыванию 3H-дофамина с помощью 3H-лигандов, для которых известно связывание с переносчиком.
Исследования связывания in vitro кокаина показали, что он связывается с переносчиком дофамина и ингибирует захват 3H-дофамина. Сообщалось, что многочисленные лиганды нескольких структурных типов связываются по сайту захвата дофамина, но остается под вопросом, идентичны ли эти сайты таковым для кокаина. Структурный аналог кокаина, 3H-WIN 35428, избирательно связывается с транспортным комплексом дофамина с высокой степенью сродства.
Препараты ткани: Препараты готовят при 0-4oC, если не указано иначе. Полосатое тело из самцов крыс линии Вистар (150-200 г) гомогенизируют в течение 5-10 сек в 10 мл NaH2PO4 (50 мМ, pH 7,4) в гомогенизаторе Ultra-Turrax. Суспензию центрифугируют при 27000 • g в течение 15 мин. Надосадочную жидкость удаляют, а остаток заново суспендируют в 50 мМ NaH2PO4, pH 7,4 (1000 мл на г исходной ткани) и используют для анализов на связывание.
Анализ: К 25 мл исследуемого раствора и 25 мл 3H-WIN 35428 (конечная концентрация 1 нМ) добавляют аликвоты ткани по 0,5 мл, смешивают и инкубируют в течение 60 мин при 2oC. Неспецифическое связывание определяют с использованием кокаина (конечная концентрация 30 мМ). После инкубации к образцам добавляют 5 мл охлажденного во льду буфера, наливают их прямо на фильтры из стекловолокна Whatman GF/C под отсосом и немедленно промывают 5 мл охлажденного во льду буфера. Количество радиоактивности на фильтрах определяют стандартным счетом жидкостной сцинтилляции. Специфическим связыванием является общее связывание минус неспецифическое.
Прежде чем вычислять IC50, должно быть достигнуто 25-75% ингибирование специфического связывания. Тестируемое значение приводят в виде IC50 (концентрация (мкМ) исследуемого вещества, которая ингибирует специфическое связывание 3H-WIN 35428 на 50%).
Результаты, полученные тестированием соединений по изобретению, приведены в таблице 1.
Приведенные выше результаты тестов показывают, что соединения по изобретению связываются с комплексом транспорта дофамина с высоким сродством.
Соединения по изобретению также были протестированы на их способность ингибировать обратный захват дофамина (DA), норадреналина (NA) и серотонина (5-HT) в синаптосомах.
Предпосылки
Дофаминовые медиаторы/сайты захвата на нервных окончаниях предположительно функционируют как терминаторы нейронного импульса путем удаления нейромедиаторов дофамина, норадреналина и серотонина, соответственно, из синаптической щели. Активность транспортных интегральных белков может быть измерена in vitro на синаптосомальный захват 3H-дофамина, 3H-норадреналина и 3H-серотонина, соответственно.
Ингибирование in vitro захвата 3H-дофамина (3H-DA) в стриарных синаптосомах
Препараты ткани: Препараты готовят при 0-4oC, если не указано иначе. Corpi striati из самцов крыс линии Вистар (150-200 г) гомогенизируют 5-10 сек в 100 объемах охлажденной во льду 0,32 М сахарозе, содержащей 1 мМ паргилина, в гомогенизаторе Ultra-Turrax. Активность моноаминоксидазы должна ингибироваться в присутствии паргилина. Гомогенат центрифугируют при 1000 • g в течение 10 мин. Полученную надосадочную жидкость затем центрифугируют при 27000 • g в течение 50 мин и надосадочную жидкость отбрасывают. Остаток (P2) заново суспендируют в оксигенированном (уравновешенном в атмосфере 96% O2 : 4% CO2 в течение по меньшей мере 30 мин) инкубационном буфере Кребса-Рингера (8000 мл на г исходной ткани) с pH 7,2, содержащем 122 мМ NaCl, 0,16 мМ ЭДТА, 4,8 мМ KCl, 12,7 мМ Na2HPO4, 3,0 мМ NaH2PO4, 1,2 мМ MgSO4, 1 мМ CaCl2, 10 мМ глюкозы и 1 мМ аскорбиновой кислоты.
Анализ: К 100 мкл тестируемого раствора и 100 мкл 3H-DA (конечная концентрация 1 нМ) добавляют аликвоты по 4,0 мл тканевой суспензии, смешивают и инкубируют в течение 25 мин при 37oC. Неспецифический захват определяют с использованием бензтропина (конечная концентрация 10 мкМ). После инкубации образцы наливают прямо на фильтры из стекловолокна Whatman GF/C под отсосом. Затем фильтры промывают три раза 5 мл охлажденного во льду 0,9% (мас./об.) раствора NaCl. Количество радиоактивности на фильтрах определяют стандартным счетом жидкостной сцинтилляции. Специфический захват вычисляют как разницу между общим и неспецифическим захватом.
Перед вычислением IC50 должно быть получено 25-75% ингибирование специфического связывания.
Тестируемое значение приводят в виде IC50 (концентрация (мкМ) тестируемого вещества, которая ингибирует специфическое связывание 3H-DA на 50%).
Ингибирование in vitro захвата 3H-норадреналина (3H-NA) в синаптосомах гиппокампа
Препараты ткани: Препараты готовят при 0-4oC, если не указано иначе. Hippocampi из самцов крыс линии Вистар (150-200 г) гомогенизируют 5-10 сек в 100 объемах охлажденной во льду 0,32М сахарозе, содержащей 1 мМ паргилина, в гомогенизаторе Ultra-Turrax. Активность моноаминоксидазы должна ингибироваться в присутствии паргилина. Гомогенат центрифугируют при 1000 • g 10 мин. Полученную надосадочную жидкость затем центрифугируют при 27000 • g в течение 50 мин, и надосадочную жидкость отбрасывают. Остаток (P2) заново суспендируют в оксигенированном (уравновешенном в атмосфере 96% O2 : 4% CO2 в течение по меньшей мере 30 мин) инкубационном буфере Кребса-Рингера (2000 мл на г исходной ткани) с pH 7,2, содержащем 122 мМ NaCl, 0,16 мМ ЭДТА, 4,8 мМ KCl, 12,7 мМ Na2HPO4, 3,0 мМ NaH2PO4, 1,2 мМ MgSO4, 1 мМ CaCl2, 10 мМ глюкозы и 1 мМ аскорбиновой кислоты.
Анализ: К 100 мкл тестируемого раствора и 100 мкл 3H-NA (конечная концентрация 1 нМ) добавляют аликвоты по 4,0 мл тканевой суспензии, смешивают и инкубируют в течение 90 мин при 37oC. Неспецифический захват определяют с использованием дезипрамина (конечная концентрация 1 мкМ). После инкубации образцы наливают прямо на фильтры из стекловолокна Whatman GF/C под отсосом. Затем фильтры промывают три раза 5 мл охлажденного во льду 0,9% (мас./об.) раствора NaCl. Количество радиоактивности на фильтрах определяют стандартным счетом жидкостной сцинтилляции. Специфический захват вычисляют как разницу между общим и неспецифическим захватом.
Перед вычислением IC50 должно быть получено 25-75% ингибирование специфического связывания.
Тестируемое значение приводят как IC50 (концентрация (мкМ) исследуемого вещества, которая ингибирует специфическое связывание 3H-NA на 50%).
Ингибирование in vitro захвата 3H-5-гидрокситриптамина (3H-5-HT, серотонин) в кортикальных синаптосомах
Препараты ткани: Препараты готовят при 0-4oC, если не указано иначе. Кору головного мозга из самцов крыс линии Вистар (150-200 г) гомогенизируют в течение 5-10 сек в 100 объемах охлажденной во льду 0,32М сахарозе, содержащей 1 мМ паргилина, в гомогенизаторе Ultra-Turrax. Активность моноаминоксидазы должна ингибироваться в присутствии паргилина. Гомогенат центрифугируют при 1000 • g в течение 10 мин. Полученную надосадочную жидкость затем центрифугируют при 27000 • g в течение 50 мин и надосадочную жидкость отбрасывают. Остаток (P2) заново суспендируют в оксигенированном (уравновешенном в атмосфере 96% O2 : 4% CO2, в течение по меньшей мере 30 мин) инкубационном буфере Кребса-Рингера (1000 мл на г исходной ткани) с pH 7,2, содержащем 122 мМ NaCl, 0,16 мМ ЭДТА, 4,8 мМ KCl, 12,7 мМ Na2HPO4, 3,0 мМ NaH2PO4, 1,2 мМ MgSO4, 1 мМ CaCl2, 10 мМ глюкозы и 1 мМ аскорбиновой кислоты.
Анализ: К 100 мкл тестируемого раствора и 100 мкл 3H-5-HT (конечная концентрация 1 нМ) добавляют аликвоты по 4,0 мл тканевой суспензии, смешивают и инкубируют в течение 30 мин при 37oC. Неспецифический захват определяют с использованием циталопрама (конечная концентрация 1 мкМ). После инкубации образцы наливают прямо на фильтры из стекловолокна Whatman GF/C под отсосом. Затем фильтры промывают три раза 5 мл охлажденного во льду 0,9% (мас./об.) раствора NaCl. Количество радиоактивности на фильтрах определяют стандартным счетом жидкостной сцинтилляции. Специфический захват вычисляют как разницу между общим и неспецифическим захватом.
Перед вычислением IC50 должно быть получено 25-75% ингибирование специфического связывания.
Тестируемое значение приводят в виде IC50 (концентрация (мкМ) исследуемого вещества, которая ингибирует специфическое связывание 3H-5-HT на 50%).
Результаты, полученные при тестировании соединений по настоящему изобретению, приведены в таблице 2.
Представленные выше результаты показывают, что тестируемые соединения эффективно ингибируют обратный захват дофамина, норадреналина и серотонина в синаптосомах.
Фармацевтические композиции
Хотя возможно, что для применения в терапии соединение по изобретению можно вводить в виде химического соединения как такового, предпочтительно представлять активный ингредиент в виде фармацевтического препарата.
В изобретении, таким образом, далее предложены фармацевтические препараты, содержащие соединение по изобретению или его фармацевтически приемлемую соль или производное вместе с одним или более чем одним фармацевтически приемлемым носителем для него и возможно другими терапевтическими и/или профилактическими ингредиентами. Носитель(и) должен быть "приемлемым", то есть совместимым с другими ингредиентами препарата и безвредным для реципиента.
Фармацевтические препараты включают в себя формы, пригодные для перорального, ректального, назального, местного (включая защечное и подъязычное), вагинального или парентерального введения (включая внутримышечное, подкожное и внутривенное) или формы, пригодные для введения путем ингаляции или инсуффляции.
Соединения по изобретению вместе со стандартным адъювантом, носителем или разбавителем, таким образом, могут быть уместны в форме их фармацевтических композиций и стандартных доз, и в такой форме они могут быть использованы в виде твердых форм, таких как таблетки или капсулы, или жидких форм, таких как растворы, суспензии, эмульсии, эликсиры, или заполненные ими капсулы, все для перорального применения; в форме суппозиториев для ректального введения или в форме стерильных инъекционных растворов для парентерального применения (включая подкожное). Такие формы фармацевтических композиций и их стандартных доз могут включать в себя стандартные ингредиенты в стандартных соотношениях, с дополнительными активными соединениями или элементами или без них, и такие формы стандартных доз могут содержать любое соответствующее эффективное количество активного ингредиента, соразмерное с предназначенным диапазоном суточной дозировки, которая должна применяться. Препараты, содержащие 10 мг активного ингредиента или, в более широком диапазоне, от 0,1 до 100 мг на таблетку, являются, таким образом, подходящими характерными формами стандартных доз.
Соединения по настоящему изобретению можно вводить в большом разнообразии пероральных и парентеральных дозовых форм. Специалисту должно быть очевидно, что следующие дозовые формы могут включать в себя в качестве активного компонента либо соединение по изобретению, либо его фармацевтически приемлемую соль.
Для получения фармацевтических композиций из соединений по настоящему изобретению фармацевтически приемлемые носители могут быть либо твердыми, либо жидкими. Твердые лекарственные формы включают в себя порошки, таблетки, драже, капсулы, облатки, суппозитории и диспергированные гранулы. Твердым носителем могут быть вещества, одно или более чем одно, которые могут также действовать как разбавители, корригенты, солюбилизаторы, смазки, суспендирующие агенты, связующие вещества, консерванты, разрыхляющие агенты таблеток или инкапсулирующий материал.
В порошках носитель представляет собой тонко измельченное твердое вещество, которое находится в смеси с тонко измельченным активным компонентом.
В таблетках активный компонент смешан с носителем, имеющим необходимую связывающую способность, в подходящих соотношениях и спрессован в желаемую форму желаемого размера.
Порошки и таблетки предпочтительно содержат от пяти или десяти до примерно семидесяти процентов активного соединения. Подходящими носителями являются карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлоза, натрий-карбоксиметилцеллюлоза, легкоплавкий воск, масло какао и тому подобное. Термин "препарат" должен включать в себя препарат активного соединения вместе с инкапсулирующим материалом в качестве носителя, образующего капсулу, где активный компонент, с носителями или без них, окружен носителем, который таким образом с ним связан. Сюда относятся также облатки и лепешки. Таблетки, порошки, капсулы, драже, облатки и лепешки можно применять как твердые формы, подходящие для перорального введения.
Для приготовления суппозиториев легкоплавкий воск, такой как смесь глицеридов жирных кислот или масло какао, сначала плавят, и активный компонент гомогенно диспергируют в нем путем перемешивания. Расплавленную гомогенную смесь затем заливают в формочки подходящего размера, дают остыть и затем затвердеть.
Препараты, подходящие для вагинального введения, могут быть представлены в форме пессариев, тампонов, кремов, гелей, паст, пен или аэрозолей, содержащих в дополнение к активному ингредиенту соответствующие носители, известные специалистам.
Жидкие лекарственные формы включают в себя растворы, суспензии и эмульсии, например растворы в воде или в воде с пропиленгликолем. Например, жидкие препараты для парентерального введения могут быть приготовлены в виде растворов в водном растворе полиэтиленгликоля.
Соединения по настоящему изобретению, таким образом, могут быть приготовлены в виде препарата для парентерального введения (например инъекцией, например болюсной инъекцией или непрерывной инфузией) и представлены в форме разовой дозы в ампулах, предварительно наполненных шприцах, малых объемов вливания или в контейнерах для многоразовых доз с добавлением консерванта. Композиции могут иметь такие формы, как суспензии, растворы или эмульсии в масляных или водных растворителях и могут содержать суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный ингредиент может находиться в форме порошка, полученного путем асептического выделения стерильного твердого вещества или путем лиофилизации из раствора, для приготовления состава с подходящим растворителем, например стерильной апирогенной водой, перед использованием.
Водные растворы, подходящие для перорального применения, можно приготовить путем растворения активного компонента в воде и добавления подходящих красителей, корригентов, стабилизирующих и загущающих агентов по желанию.
Водные растворы, подходящие для перорального применения, можно приготовить путем диспергирования тонко измельченного активного компонента в воде с вязким материалом, таким как натуральные и синтетические смолы, метилцеллюлоза, натрий-карбоксиметилцеллюлоза или другие хорошо известные суспендирующие агенты.
Включены также твердые лекарственные формы, которые предназначены для превращения непосредственно перед применением в жидкие лекарственные формы для перорального введения. Такие жидкие формы включают в себя растворы, суспензии и эмульсии. Эти препараты могут содержать в дополнение к активному компоненту красители, корригенты, стабилизаторы, буферы, искусственные и натуральные подсластители, диспергирующие агенты, загустители, солюбилизирующие агенты и тому подобное.
Для местного введения через кожу соединения по изобретению могут быть представлены в виде мазей, кремов или лосьонов, или в виде трансдермального пластыря. Мази и кремы могут быть приготовлены, например, на водной или масляной основе с добавлением подходящих загущающих или желирующих агентов. Лосьоны могут быть приготовлены на водной или масляной основе, и, как правило, они также должны содержать один или более чем один эмульгирующий, стабилизирующий, диспергирующий, суспендирующий, загущающий агент или краситель.
Препараты, пригодные для местного введения через рот, включают в себя лепешки, в состав которых входит активный агент на основе корригента, обычно это сахароза и аравийская камедь или трагакант; пастилки, в состав которых входит активный ингредиент на инертной основе, такой как желатин и глицерин или сахароза и аравийская камедь; и полоскания, состоящие из активного ингредиента в подходящем жидком носителе.
Растворы и суспензии вводят непосредственно в носовую полость с помощью стандартных средств, например, капельницы, пипетки или распылителя. Препараты могут быть представлены в форме разовых или многократных доз, в последнем случае в виде капельницы или пипетки, что достигается введением пациенту соответствующего определенного объема раствора или суспензии. В случае распылителя это достигается, например, с помощью дозирующего клапана.
Введение в дыхательные пути также достигается с помощью аэрозольных препаратов, в которых активный ингредиент находится в герметичной упаковке с подходящим пропеллентом, таким как хлорфторуглерод (CFC), например дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, диоксид углерода или другой подходящий газ. Аэрозоль приемлемо может также содержать поверхностно-активное вещество, такое как лецитин. Дозу лекарства можно контролировать с помощью дозирующего клапана.
Альтернативно, активные ингредиенты могут быть представлены в форме сухого порошка, например порошковой смеси соединения в подходящей порошковой основе, такой как лактоза, крахмал, производные крахмала, например, гидроксипропилметилцеллюлоза и поливинилпирролидон (PVP). Для удобства порошковый носитель должен образовывать гель в носовой полости. Порошковая композиция может быть представлена в форме стандартной дозы, например в капсулах или картриджах, например из желатина, или в блистерных упаковках, из которых порошок можно вводить с помощью ингалятора.
В препаратах, предназначенных для введения в дыхательные пути, включая интраназальные препараты, соединение должно, как правило, иметь малый размер частиц, например порядка 5 мкм или меньше. Частицы такого размера можно получить способами, известными специалистам, например микронизацией.
При желании можно применять препараты, адаптированные к постепенному высвобождению активного ингредиента.
Фармацевтические препараты находятся предпочтительно в форме стандартной дозировки. В такой форме препарат разделен на разовые стандартные дозы, содержащие соответствующие количества активного компонента. Форма разовых стандартных доз может представлять собой препарат в упаковке, содержащей дискретные количества препарата, такой как упаковка таблеток, капсул, и порошки во флаконах или ампулах. Форма стандартной дозировки может быть также сама по себе капсулой, таблеткой, облаткой или лепешкой, или соответствующим количеством любой из них в упаковке.
Таблетки или капсулы для перорального введения и жидкости для внутривенного введения являются предпочтительными композициями.
Способ лечения
Соединения по настоящему изобретению полезны для лечения расстройств и заболеваний, на которые влияет ингибирующая обратный захват нейромедиаторных моноаминов активность этих соединений моноаминов. Эта активность соединений по изобретению делает их чрезвычайно полезными для лечения паркинсонизма, депрессии, ожирения, нарколепсии, злоупотребления наркотическими средствами, например злоупотребления кокаином, гиперактивности при дефиците внимания, сенильной деменции и нарушения познавательной деятельности, а также других расстройств, чувствительных к ингибирующей обратный захват нейромедиаторных моноаминов активности этих соединений. Соединения по этому изобретению можно, соответственно, вводить живому организму, включая человека, нуждающемуся в лечении, купировании или элиминации симптома, ассоциированного с или чувствительного к ингибирующей обратный захват нейромедиаторных моноаминов активности. Это относится в особенности к паркинсонизму, депрессии, ожирению, нарколепсии, злоупотреблению кокаином, гиперактивности при нарушениях внимания, сенильной деменции и нарушению памяти при старении. Подходящим диапазоном дозировки является 0,1-500 мг в сутки и в особенности 10-70 мг в сутки, которые вводят один или два раза в день, в зависимости, как правило, от точного способа введения, формы введения, симптома, на который направлено введение, больного и его массы тела и, наконец, предпочтения и опыта лечащего врача или ветеринара.
I. p. означает "внутрибрюшинно", что представляет собой хорошо известный способ введения.
P. o. означает "перорально", что представляет собой хорошо известный способ введения.
Следующие примеры иллюстрируют изобретение, однако, не должны рассматриваться как ограничивающие.
Пример 1
Метиловый эфир (-)ангидроэкгонина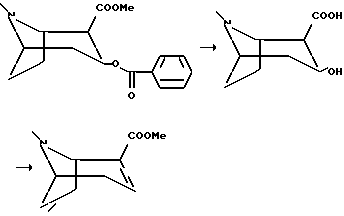
(1R, 2R, 3S)-2-Карбометокси-3-бензокситропана гидрохлорид (100 г, 0,29 моль) кипятили с обратным холодильником в 1000 мл 1М соляной кислоты в течение 18 часов, и раствор охлаждали во льду. Бензойную кислоту собирали фильтрацией, и фильтрат концентрировали in vacuo. Растиранием остатка с этанолом и фильтрацией получали (1R,2R,3S)-3-гидрокси- тропан-2- карбоксилата гидрохлорид в виде белого кристаллического соединения, которое без дальнейшей очистки сушили и кипятили с обратным холодильником в оксихлориде фосфора (50 мл) в течение двух часов. Раствор концентрировали in vacuo и медленно добавляли абсолютный метанол (150 мл) при охлаждении во льду. Раствор перемешивали при температуре окружающей среды в течение 16 часов и концентрировали in vacuo. Остаток охлаждали во льду, делали щелочным путем добавления раствора гидроксида натрия (10 М, приблизительно 100 мл) и экстрагировали 5 раз диэтиловым эфиром. Комбинированную органическую фазу сушили и концентрировали in vacuo с получением масла, которое дистиллировали in vacuo (70-74oC, 1 мбар) с получением соединения, указанного в заголовке, в виде чистого масла.
Альтернативно метиловый эфир (-)ангидроэкгонина получали следующим образом:
К 103 г (3,05 экв.) натрия в 3,25 л абсолютного этанола добавляли 3 л этилацетата (HPLC (высокоэффективная жидкостная хроматография) сорта) и 500 г гидрохлорида кокаина. Реакционную смесь кипятили с обратным холодильником 2,5 часа. Добавляли 150 мл уксусной кислоты, pH 8, затем 1,5 л толуола. 2 л растворителей выпаривали при пониженном давлении. Добавляли еще 2 л толуола и еще 2 л выпаренных растворителей. Эту обработку повторяли еще раз. Общее количество добавленного толуола составило 5,5 л, и около 6 л растворителей выпаривали. Реакционную смесь фильтровали, и соли промывали общим количеством толуола 1 л. Растворитель выпаривали при пониженном давлении, а остаток 570 г дистиллировали с использованием колонки Vigreux 15 см. Этилбензоат дистиллировали под давлением 12 мм рт. ст., Tкип 80-95oC, и соединение, указанное в заголовке, дистиллировали без колонки Vigreux при 0,2-0,4 мбар, Tкип 56-80oC. Продукт представлял собой чистую желтую жидкость.
Выход: 218 г (76%).
Пример 2
(1R, 2S, 3S)-2-Карбометокси-3-(4-фторфенил)тропан и (1R,2R,3S)-2-карбометокси-3-(4-фторфенил)тропан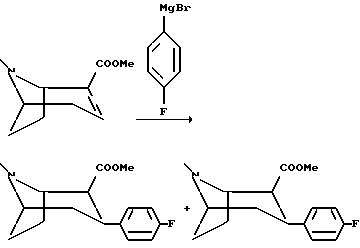
Реактив Гриньяра получали в трехгорлой реакционной колбе, оборудованной механической мешалкой, интенсивным конденсором и воронкой, уравновешивающей давление, используя 4-бромфторбензол (27,5 мл, 250 мМоль) и магниевую стружку (6,3 г, 260 мМоль) в 250 мл абсолютного диэтилового эфира. Раствор реактива Гриньяра охлаждали до -20oC и добавляли раствор метилового эфира (-)ангидроэкгонина (21,7 г, 120 мМоль) в 100 мл абсолютного диэтилового эфира в течение 1/2 часа. Реакционную смесь перемешивали один час при -20oC, и реакцию гасили одним из двух следующих способов:
1. Реакционную смесь перемешивали с 250 мл колотого льда, и водную фазу подкисляли добавлением приблизительно 100 мл 4М соляной кислоты. Органическую фазу сливали, а водную фазу промывали 100 мл диэтилового эфира. Водную фазу подщелачивали добавлением 25% раствора гидроксида аммония, а затем насыщали хлоридом натрия и, наконец, экстрагировали три раза диэтиловым эфиром. Комбинированную органическую фазу сушили и концентрировали in vacuo с получением масла, которое дистиллировали in vacuo (150-160oC, 2 мбар). Этим способом получали смесь двух стереоизомеров (2S/2R - 1/3), которую разделяли колоночной хроматографией с использованием смеси диэтилового эфира и пентана (1+1)+1% триэтиламина в качестве элюента. Неочищенные продукты растирали в пентане с получением (1R,2S,3S)-2-карбометокси-3-(4-фторфенил)тропана, белых кристаллов с Tпл 91-92oC и (1R,2R,3S)-2-карбометокси-3-(4-фторфенил)тропана, белых кристаллов с Tпл 65-66oC.
2. Реакционную смесь охлаждали до -78oC и добавляли раствор трифторуксусной кислоты (20 мл, 250 мМоль) в 50 мл диэтилового эфира в течение 10 минут. Охлаждающую баню убирали, и когда температура достигала 0oC, смесь перемешивали с 700 мл воды. pH водной фазы доводили до pH 1 добавлением концентрированной соляной кислоты, за чем следовала водная обработка и очистка таким же способом, как описано выше. Этим способом получали смесь двух стереоизомеров (2S/2R - 2/1).
Подобным способом получали следующие соединения:
(1R, 2R, 3S)-2-Карбометокси-3-бензилтропан и (1R,2S,3S)-2-карбометокси-3-бензилтропан, способ 2, только (1R,2S,3S)-2-карбометокси-3-бензилтропан получали без загрязнения другим изомером, в виде масла, которое кристаллизуется при стоянии, Tпл 53-54oC. (1R,2R,3S)-2-Карбометокси-3-бензилтропан получали изомеризацией смеси, как описано в примере 3.
(1R, 2R,3S)-2-Карбометокси-3-(4-хлорфенил)тропан и (1R,2S,3S)-2-карбометокси-3-(4-хлорфенил)тропан, способ 2. Два изомера не разделяли, но смесь изомеризовали, как описано в примере 3.
(1R, 2R, 3S)-2-Карбометокси-3-(4-хлорфенил)тропан, (1R,2S,3S)-2-карбометокси-3-(4-хлорфенил)тропан, (1S,2S,3R)-2-карбометокси-3-(4-хлорфенил)тропан и (1S, 2R, 3R)-2-карбометокси-3-(4-хлорфенил)тропан, способ 2. Две серии энантиомерных пар не разделяли, но смесь изомеризовали, как описано в примере 3.
(1R,2R,3S)-2-Карбометокси-3-(4-метилфенил)тропан и (1R,2S,3S)-2-карбометокси-3-(4-метилфенил)тропан, способ 2. Два изомера не разделяли, но смесь изомеризовали, как описано в примере 3.
(1R, 2S, 3S)-2-Карбометокси-3-(2-нафтил)тропан и (1R,2R,3S)-2-карбометокси-3-(2-нафтил)тропан, способ 2. Реактив Гриньяра получали добавлением смеси одного эквивалента 2-бромнафталина и 1,2-дибромэтана в диэтиловом эфире к кипящей с обратным холодильником суспензии двух эквивалентов магния. Оба продукта представляли собой белые кристаллические соединения с Tпл 79-80oC и 86-87oC, соответственно.
(1R, 2R, 3S)-2-Карбометокси-3-(1-нафтил)тропан и (1R,2S,3S)-2-карбометокси-3-(1-нафтил)тропана гидрохлорид, способ 2. Реактив Гриньяра получали добавлением смеси одного эквивалента 2-бромнафталина и 1,2-дибромэтана в диэтиловом эфире к кипящей с обратным холодильником суспензии двух эквивалентов магния. Соединения, указанные в заголовке, выделяли, соответственно, как белое кристаллическое соединение, Tпл 133-135oC и аморфное соединение.
(1R, 2S,3S)-2-Карбометокси-3-(3,4-дихлорфенил)тропан и (1R,2R,3S)-2-карбометокси-3-(3,4-дихлорфенил)тропан, способ 2. Оба продукта представляли собой белые кристаллические соединения с Tпл 69-70oC и 61-63oC, соответственно.
Рацемическую смесь (1R,2R,3S)-2-карбометокси-3-(3,4-дихлорфенил)тропана и его энантиомера (1S,2S,3R)-2-карбометокси-3-(3,4-дихлорфенил)тропана получали с использованием метилового эфира (±)-ангидроэкгонина в качестве исходного материала, способ 2, с последующей изомеризацией как описано в примере 3.
(1S, 2S, 3R)-2-Карбометокси-3-(3,4-дихлорфенил)тропан получали, применяя способ 2. Соединение не выделяли, но изомеризовали, как описано в примере 3.
(1R, 2S,3S)-2-Карбометокси-3-(4-фенил-фенил)тропан и (1R,2R,3S)-2-карбометокси-3-(4-фенил-фенил)тропан, способ 2. Оба продукта представляли собой белые кристаллические соединения с Tпл 130-132oC и 95-96oC, соответственно.
(1R, 2S,3S)-2-Карбометокси-3-(4-трет-бутил-фенил)тропан и (1R,2R,3S)-2-карбометокси-3-(4-трет-бутил-фенил)тропан, способ 2. Оба продукта представляли собой белые кристаллические соединения с Tпл 84-85oC и 83-84oC, соответственно.
Пример 3
(1R,2R,3S)-2-карбометокси-3-бензилтропан, гидрохлорид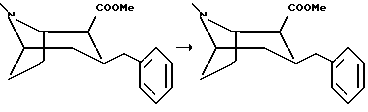
К раствору (1R,2S,3S)-2-карбометокси-3-бензилтропана (5,6 г, 20,5 мМоль) в абсолютном метаноле (100 мл) добавляли раствор метанолята в метаноле (2 М, 2 мл), и смесь кипятили с обратным холодильником в течение 16 часов. Реакционную смесь концентрировали in vacuo, и остаток растворяли в диэтиловом эфире и промывали водой. Органическую фазу сушили и концентрировали in vacuo. Сырой продукт очищали колоночной хроматографией с использованием смеси диэтилового эфира и пентана (1+1)+1% триэтиламина в качестве элюента, что давало выход (1R,2R,3S)-2-карбометокси-3-бензилтропана, гидрохлорида, в виде масла. Растворением этого продукта в диэтиловом эфире и последующим добавлением раствора соляной кислоты в диэтиловом эфире соединение, указанное в заголовке, осаждали в виде белых кристаллов, Tпл 188-190oC.
Пример 4
2-Карбометокси-3-тропанон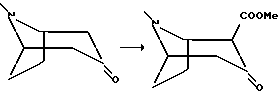
К суспензии гидрида натрия (3,2 г 80%, 107 мМоль, предварительно промытого в циклогексане) и диметилкарбоната (9,13 мл, 108 мМоль) в абсолютном циклогексане, нагретом до температуры дефлегмации, добавляли раствор (±)-3-тропанона (6,9 г, 50 мМоль) в 50 мл абсолютного циклогексана в течение 15 минут. Когда не наблюдалось образования водорода, добавляли 0,2 мл метанола. Реакционную смесь перемешивали в течение ночи при температуре дефлегмации, и после охлаждения до температуры окружающей среды осторожно добавляли 75 мл воды. К водной фазе добавляли 40 г хлорида аммония, и полученную смесь экстрагировали 8 раз метиленхлоридом. Комбинированные метиленхлоридные органические фазы сушили и концентрировали in vacuo с последующей колоночной хроматографией неочищенного продукта, используя метиленхлорид с возрастающими количествами (до 10%) метанола в качестве элюента. Фракции, содержащие продукт, концентрировали in vacuo, и полученное масло подвергали Kugelrohr дистилляции (1 мбар, 120oC) с получением соединения, указанного в заголовке, в виде оранжевых кристаллов, Tпл 104-107oC.
Пример 5
2-Карбометокси-3-гидрокситропан, гидрохлорид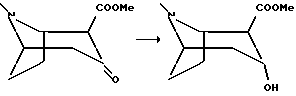
К раствору 2-карбометокси-3-тропанона, полученного в примере 4 (17 г, 85 мМоль) в 750 мл метанола, охлажденного до -35oC, добавляли боргидрид натрия (17 г, 450 мМоль), и смесь перемешивали в течение 4 часов. Охлажденный раствор гасили медленным добавлением концентрированной соляной кислоты (40 мл), и смесь концентрировали in vacuo. Добавляли воду (400 мл) и доводили pH до 3 добавлением концентрированной соляной кислоты. После того как водная фаза была промыта три раза диэтиловым эфиром, pH доводили до 11 добавлением концентрированного гидроксида аммония, и водную фазу экстрагировали три раза метиленхлоридом. Концентрированием in vacuo получали масло, которое растворяли в этаноле, и добавляли концентрированную соляную кислоту с последующим концентрированием in vacuo. Сушкой замораживанием остатка получали соединение, указанное в заголовке, в виде аморфного продукта.
(1S)-Карбометокси-3-гидрокситропан, аморфное твердое вещество, получали аналогичным способом с использованием в качестве исходного материала (1S)-2-карбометокси-3-тропанона, полученного путем разделения соединения, полученного в примере 4, как описано в J. Med. Chem., 37, 2007 (1994).
Пример 6
Метиловый эфир (1RS)-ангидроэкгонина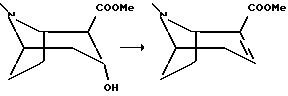
Смесь 2-карбометокси-3-гидрокси-тропана, гидрохлорида, полученного в примере 5 (0,5 г, 2,1 мМоль), и тионилхлорида (0,4 мл, 5,3 мМоль) перемешивали при 60oC в течение двух часов, в результате чего получали чистый раствор. После охлаждения до температуры окружающей среды добавляли колотый лед и доводили pH до 11 добавлением концентрированного гидроксида аммония. Смесь дважды экстрагировали метиленхлоридом и удаляли растворитель in vacuo с получением соединения, указанного в заголовке, в виде масла, которое дистиллировали при 1 мбар 70-85oC.
Метиловый эфир (1S)-ангидроэкгонина, масло, получали подобным способом с использованием (1S)-карбометокси-3-гидрокси-тропана, гидрохлорида, полученного в примере 5, в качестве исходного материала.
Пример 7
(1R,2R,3S)-N-Норметил-2-карбометокси-3-(3,4-дихлорфенил)тропан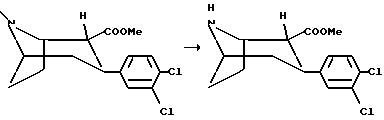
Смесь (1R, 2R, 3S)-2-карбометокси-3-(3,4-дихлорфенил)тропана (8,7 г, 27 мМоль) и 2,2,2-трихлорэтилхлорформата (14,6 мл, 106 мМоль) в сухом толуоле (100 мл) кипятили с обратным холодильником в течение 18 часов. Реакционную смесь концентрировали in vacuo, и к остатку добавляли метиленхлорид, который затем промывали водой. Органическую фазу сушили и концентрировали in vacuo. Остаток растворяли в 75% водном растворе уксусной кислоты (60 мл) и добавляли цинковую пыль (8,7 г) в реакционную смесь, которую затем
перемешивали при температуре окружающей среды в течение 18 часов. Добавляли концентрированный раствор гидроксида аммония (pH>7), и смесь дважды экстрагировали диэтиловым эфиром. Комбинированную органическую фазу сушили и концентрировали in vacuo с получением соединения, указанного в заголовке, в виде масла, которое использовали без дальнейшей очистки.
Пример 8
(1R, 2R,3S)-N-Норметил-N-(трет-бутоксикарбонил)-2-карбометокси- 3-(3,4-дихлорфенил)тропан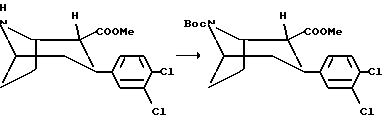
Раствор (1R,2R,3S)-N-норметил-2-карбометокси-3-(3,4-дихлорфенил)тропана (7 г, 22,3 мМоль) и ди-трет-бутил-дикарбоната (7,7 мл, 33,6 мМоль) в сухом тетрагидрофуране (50 мл) перемешивали при комнатной температуре в течение часа. Реакцию гасили добавлением льда (100 мл), и смесь дважды экстрагировали диэтилэфиром, который сушили и концентрировали in vacuo с получением соединения, указанного в заголовке, в виде масла, которое использовали без дальнейшей очистки.
Пример 9
(1R,2S,3S)-2-Гидроксиметил-3-(4-фторфенил)тропан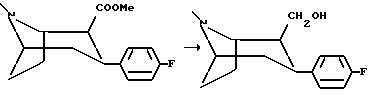
К суспензии литийалюминийгидрида (0,8 г, 21 мМоль) в диэтиловом эфире (30 мл) при комнатной температуре медленно добавляли раствор (1R,2S,3S)-2-карбометокси-3-(4-фторфенил)тропана (5 г, 18 мМоль) в 100 мл диэтилового эфира. Реакцию завершали после перемешивания в течение 10 минут и гасили добавлением 0,8 мл воды, 0,8 мл гидроксида натрия (15%) и 2 мл воды. Соли алюминия удаляли фильтрованием, а растворитель удаляли in vacuo с получением масла. Соединение, указанное в заголовке, осаждали при растирании с пентаном в виде белых кристаллов, Tпл 79-80oC.
Подобным способом получали следующие соединения:
(1R, 2R,3S)-2-гидроксиметил-3-(4-фторфенил)-тропан, белые кристаллы, Tпл 169-170oC;
(1R, 2R,3S)-2-гидроксиметил-3-(3,4-дихлорфенил)-тропан, белые кристаллы, Tпл 145-150oC;
(1R, 2R, 3S)-N-норметил-N-(трет-бутоксикарбонил)-2-гидроксиметил- 3-(3,4-дихлорфенил)-тропан, масло;
(1R, 2S,3S)-2-гидроксиметил-3-(3,4-дихлорфенил)-тропан, белые кристаллы, Tпл 83-89oC;
рацемическую смесь (1R,2R,3S)-2-гидроксиметил-3-(3,4-дихлорфенил)-тропана и его энантиомера (1S,2S,3R)-2-гидроксиметил-3-(3,4-дихлорфенил)-тропана, Tпл 186-187oC:
(1S,2S,3R)-2-гидроксиметил-3-(3,4-дихлорфенил)-тропан, Tпл 179-184oC;
(1R, 2R,3S)-2-гидроксиметил-3-(3,4-дихлорфенил)-тропан, белые кристаллы, Tпл 200-202oC.
Пример 10
(1R,2R,3S)-2-Гидроксиметил-3-(3,4-дихлорфенил)тропана тозилат
К суспензии (1R,2R,3S)-2-гидроксиметил-3-(3,4-дихлорфенил)тропана (15 г, 0,05 моль) в метиленхлориде (250 мл) добавляли триэтиламин (8 мл) и тозилхлорид (10,5 г, 0,06 моль). Реакцию перемешивали в течение ночи при комнатной температуре. Растворитель выпаривали, а остаток растворяли в эфире. Эфирную фазу промывали гидроксидом натрия (1 н.) и дважды промывали водой. Сушкой над сульфатом магния и выпариванием растворителя получали 21,1 г (93%) соответствующего тозилата.
Другим способом тозилат получали так:
К холодной (5oC) суспензии (1R,2R,3S)-2-гидроксиметил-3-(3,4-дихлорфенил)тропана (1,5 г, 5 ммоль) в пиридине (5 мл) добавляли тозилхлорид (1,15 г, 6 мМоль). Реакционную смесь перемешивали при комнатной температуре в течение часа. Добавляли воду (50 мл) при температуре < 10oC, и смесь перемешивали в течение 15 мин.
Добавляли 4 н. NaOH (2,5 мл). Продукт выделяли, промывали водой и сушили. Выход 2,12 г (93%).
Перекристаллизация из 100 мл гептана давала 1,61 г чистого тозилата. Tпл 124-125oC.
Пример 11
(1R,2R,3S)-2-Метоксиметил-3-(3,4-дихлорфенил)тропан
(1R, 2R, 3S)-2-Гидроксиметил-3-(3,4-дихлорфенил)тропана тозилат (9,2 г, 0,02 моль) растворяли в безводном метаноле (100 мл). Добавляли метоксид натрия в метаноле (15 мл, 2H, 30 мМоль), и реакционную смесь кипятили с обратным холодильником в течение 96 часов. Растворитель выпаривали, а остаток растворяли в эфире. Эфирную фазу промывали три раза водой и сушили над сульфатом магния. Выпариванием растворителя получали 5,98 г (95%) соединения, указанного в заголовке.Tпл 73-76oC.
(1R, 2R, 3S)-2-Метоксиметил-3-(3,4-дихлорфенил)тропана цитрат получали следующим образом:
К раствору (1R,2R,3S)-2-метоксиметил-3-(3,4-дихлорфенил)-тропана (16 г, 50 мМоль) в 96% этаноле (200 мл) добавляли лимонную кислоту (10,5 г, 55 мМоль). Смесь нагревали до получения чистого раствора. Раствор охлаждали, осадок отфильтровывали и промывали 2х25 мл этанола. Выход 21,0 г (83%). Tпл 159-160oC.
(1R, 2R, 3S)-2-Метоксиметил-3-(3,4-дихлорфенил)тропана сульфат получали следующим образом:
К раствору (1R,2R,3S)-2-метоксиметил-3-(3,4-дихлорфенил)тропана (2,2 г, 7 мМоль) добавляли серную кислоту в изопропаноле (2М, 3,6 мл). Сульфат кристаллизуется при охлаждении и затравливании кристаллами. Кристаллы отфильтровывали, промывали холодным изопропанолом и сушили. Выход 1,61 г. Tпл 171-172oC.
Следующие соединения получали аналогично:
(1R, 2R, 3S)-2-изопропоксиметил-3-(3,4-дихлорфенил)-тропана фумарат, Tпл 154-155oC:
(1R, 2R, 3S)-2-циклопропилметилоксиметил-3-(3,4-дихлорфенил)-тропана сульфат, Tпл 66-75oC;
(1R,2R,3S)-2-метоксиметил-3-(4-хлорфенил)-тропана цитрат, Tпл 165-166oC;
(1R,2R,3S)-2-этоксиметил-3-(4-хлорфенил)-тропана цитрат, Tпл 166-167oC;
(1R,2R,3S)-N-норметил-2-этоксиметил-3-(4-хлорфенил)-тропана фумарат, Tпл 184-186oC;
(1R,2R,3S)-N-норметил-2-метоксиметил-3-(4-хлорфенил)-тропана цитрат. Tпл 112-114oC;
(1R, 2R, 3S)-2-циклопропилметилоксиметил-3-(4-хлорфенил)-тропана цитрат, Tпл 155-157oC;
(1R, 2R, 3S)-N-норметил-2-циклопропилметилоксиметил-3-(4- хлорфенил)-тропана фумарат, Tпл 176-178oC;
Пример 13
(1R,2R,3S)-2-Этоксиметил-3-(3,4-дихлорфенил)тропан
(1R, 2R, 3S)-2-Гидроксиметил-3-(3,4-дихлорфенил)тропана тозилат (2,5 г, 5,5 мМоль) растворяли в безводном этаноле (20 мл). Добавляли этоксид натрия в этаноле (2,4 мл, 2,5 М, 6 мМоль), и реакционную смесь кипятили с обратным холодильником в течение 72 часов. Растворитель выпаривали. Остаток перемешивали с водой и эфиром, экстрагировали три раза эфиром (3х50 мл) и сушили над MgSO4. Выпариванием растворителя получали 1,75 г соединения, указанного в заголовке. Продукт очищали колоночной хроматографией на силикагеле с использованием EtOAc:Et3N (99:1). Выход 1,24 г.
Соль фумаровой кислоты вышеуказанного соединения получали следующим образом:
К раствору (1R,2R,3S)-2-этоксиметил-3-(3,4-дихлорфенил)тропана (450 мг, 1,38 ммоль) в эфире добавляли фумаровую кислоту (160 мг, 1,38 мМоль), суспендированную в MeOH, и нагревали смесь до получения чистого раствора. Раствор выпаривали, а остаток растирали в эфире, затравливали кристаллами и перемешивали в течение ночи. Осадок отфильтровывали, промывали эфиром и сушили с получением 370 мг соли фумаровой кислоты. Tпл 134-137oC.
Пример 14
(1R,2R,3S)-2-Этоксиметил-3-(3,4-дихлорфенил)тропан
К (1R, 2R, 3S)-2-гидроксиметил-3-(3,4-дихлорфенил)тропану (26,9 г, 0,09 моль) в ТГФ (200 мл) добавляли 60% гидрид натрия в масле (4,6 г, 0,12 моль) и этилсульфат (15,7 мл, 0,12 моль) и нагревали до 30-40oC на масляной бане в течение 1/2 часа. Реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Затем реакционную смесь нагревали до 30-40oC на масляной бане в течение часа и вливали в воду (500 мл). Смесь экстрагировали дважды трет-бутилметиловым эфиром, органические фазы промывали водой, сушили над MgSO4 и выпаривали с получением 32,82 г соединения, указанного в заголовке.
(1R, 2R, 3S)-2-Этоксиметил-3-(3,4-дихлорфенил)тропана цитрат получали следующим образом:
К раствору (1R, 2R, 3S)-2-этоксиметил-3-(3,4-дихлорфенил)тропана в 96% этаноле (275 мл) добавляли лимонную кислоту (19,2 г, 0,1 моль). Раствор нагревали до температуры дефлегмации. Оставляли раствор при температуре окружающей среды на 3 часа, что приводило к его кристаллизации. Смесь оставляли на ледяной бане на 1/2 часа, кристаллический продукт отфильтровывали и промывали 96% этанолом (50 мл и 25 мл). Кристаллический продукт сушили. Выход 32,85 мг (70%). Tпл 153-155,5oC.
Пример 15
(1R,2R,3S)-N-Норметил-2-метоксиметил-3-(3,4-дихлорфенил)тропана цитрат
К раствору (1R,2R,3S)-N-норметил-2-метоксиметил-3-(3,4- дихлорфенил)-тропана (5,98 г, 19 мМоль) в дихлорэтане (50 мл) добавляли хлорэтилхлорформат (2,7 мл, 25 мМоль). Реакционную смесь кипятили с обратным холодильником в течение ночи. Растворитель выпаривали, а остаток кипятили с обратным холодильником в метаноле в течение 30 минут. Растворитель выпаривали, и остаток растворяли в воде. Раствор делали щелочным с помощью водного аммиака и экстрагировали эфиром. Эфирную фазу промывали водой, сушили над сульфатом магния и выпаривали до сухости с выходом 5,4 г. Остаток очищали колоночной хроматографией на силикагеле, используя CH2Cl2/MeOH/NH3 (водн.) (40:9:1). Получали 2,64 г очищенного материала. Этот материал растворяли в этаноле (20 мл, 96%) и добавляли лимонную кислоту (1,7 г) в этаноле (20 мл, 96%). Выдержка при 5oC давала 3,82 г (41%) кристаллического твердого вещества, Tпл 118-120oC.
(1R,2R,3S)-N-Норметил-2-этоксиметил-3-(3,4-дихлорфенил)тропана цитрат
К раствору (1R, 2R, 3S)-N-норметил-2-этоксиметил-3-(3,4-дихлорфенил)-тропана (4,85 г, 14,8 мМоль) в дихлорэтане (50 мл) добавляли хлорэтилхлорформат (2,4 мл, 22 мМоль). Реакционную смесь кипятили с обратным холодильником в течение ночи. Растворитель выпаривали, и остаток кипятили с обратным холодильником в метаноле (50 мл) в течение 30 минут. Растворитель выпаривали, и остаток растворяли в воде. Раствор делали щелочным с помощью NH4OH и экстрагировали эфиром. Эфирную фазу промывали водой, сушили над MgSO4 и выпаривали с получением 4,35 г неочищенного продукта. Продукт очищали колоночной хроматографией на силикагеле, используя CH2Cl2/MeOH/NH3 (води.) (40:9: 1) в качестве элюента. Выход 2,49 г.
Соль фумаровой кислоты получали путем растворения продукта в этаноле и добавления фумаровой кислоты в этаноле (0,25М). Соль отфильтровывали, промывали этанолом и сушили. Tпл 220-222oC.
Пример 16
(1R,2R,3S)-2-этилтиометил-3-(3,4-дихлорфенил)тропан
К холодному (0oC) раствору этантиола (0,5 мл) в диметилформамиде (30 мл) добавляли гидрид натрия (60%, 0,27 г). Когда прекращалось образование водорода, добавляли (1R, 2R,3S)-2-тозилметил-3-(3,4-дихлорфенил)тропан (2,0 г, 4,4 ммоль) в диметилформамиде (20 мл). Смесь перемешивали при 0oC в течение 25 минут. Реакционную смесь нагревали при 100oC в течение 5 дней. Реакционную смесь охлаждали до температуры окружающей среды и вливали в смесь воды (500 мл) и эфира (100 мл). Фазы разделяли, и водную фазу экстрагировали еще раз эфиром (100 мл). Эфирную фазу выпаривали, а остаток растворяли в эфире (75 мл) и промывали водой (2х400 мл), сушили над MgSO4 и выпаривали до сухости. Выход: 1,4 г соединения, указанного в заголовке. Сырой продукт очищали колоночной хроматографией на силикагеле, используя смесь CH2Cl2/MeOH/NH3 (води.) (40:9:1) + 1% NH3 (води.). 0,6 г соединения, указанного в заголовке, получали в виде масла.
К суспензии (1R,2R,3S)-2-этилтиометил-3-(3,4-дихлорфенил)тропана (0,3 г) в эфире (3-4 мл) добавляли фумаровую кислоту (1,02 экв.) в теплом MeOH (4 мл). Раствор затравливали кристаллами и оставляли на ночь при температуре окружающей среды. Кристаллический продукт выделяли фильтрацией. Кристаллы суспендировали в петролейном эфире, перемешивали в течение 30 мин, выделяли фильтрацией и сушили. Выход 0,38 г. Tпл 69-71oC.
Примеры фармацевтических композиций
Химическое соединение по изобретению может быть облечено в любую желательную лекарственную форму и может быть дозировано в любом желаемом количестве. Ниже представлены стандартный препарат в форме капсулы, стандартный препарат в форме таблетки и стандартный препарат в форме раствора для инъекций, соответственно.
Стандартный препарат в форме капсулы
Капсулы, содержащие 1 мг активного фармацевтического ингредиента (АФИ) на капсулу, могут быть получены с использованием следующей композиции (см. табл. А).
Рассчитанные количества лекарственного вещества и наполнителя, соответствующие 1 мг активного лекарственного вещества и 117 мг наполнителя на одну капсулу, отвешивают и смешивают всухую. Смесью затем заполняют рассчитанное количество капсул (размер 4).
Стандартный препарат в форме таблетки
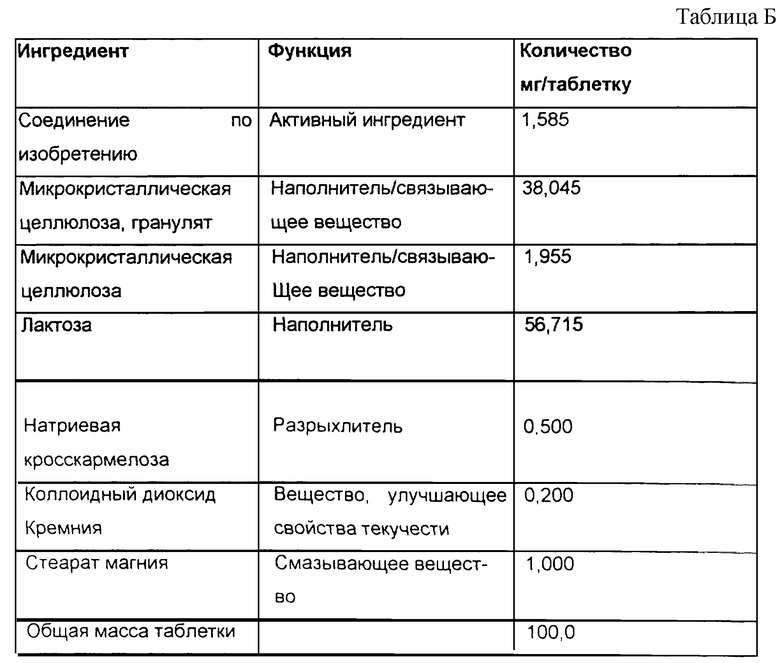
Таблетки, содержащие 1,585 мг активного ингредиента на таблетку, получают, используя следующую композицию (см табл. Б).
Активный ингредиент растворяют в растворителе для грануляции, который состоит из метилцеллюлозы и воды, и затем используют для грануляции микрокристаллической целлюлозы. Гранулят оставляют сохнуть на поддоне. Полученный гранулят, содержащий активный фармацевтический ингредиент (АФИ), микрокристаллическую целлюлозу, лактозу и натриевую кросскармелозу отвешивают, просеивают в смеситель и смешивают. Отвешивают стеарат магния и просеивают его в смеситель вместе со смесью, полученной ранее, и смешивают. Полученную смесь затем прессуют в таблетки.
Стандартный препарат в форме раствора для инъекций
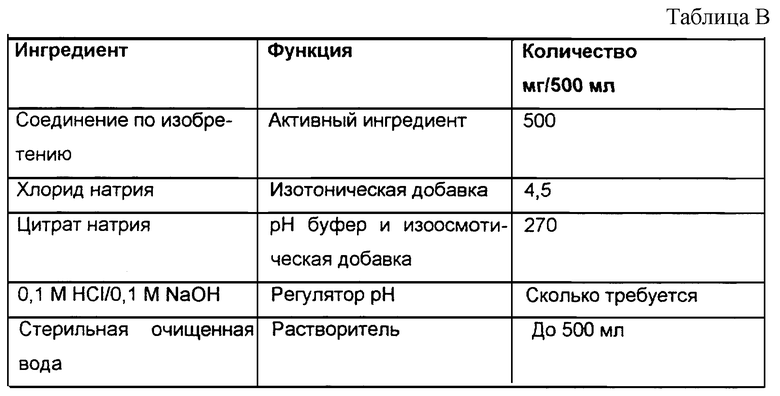
Раствор для инъекций, содержащий 1 мг/мл активного ингредиента, получают, используя следующую композицию (см. табл. В).
Рассчитанное количество активного ингредиента отвешивают, растворяют в стерильной очищенной воде, добавляют предписанное количество хлорида натрия и цитрата натрия, затем pH раствора доводят до желаемого значения, обычно в диапазоне от примерно pH 6,5 до примерно pH 8.



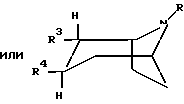
Описывается новое 2,3-трансдизамещенное тропановое производное общей формулы I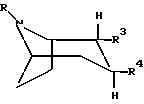
или любая их смесь, или их фармацевтически приемлемая соль, где R представляет собой водород, метил, этил или пропил; R3 представляет собой -CH2-X-R', где Х представляет собой О или S, а R' представляет собой метил, этил, пропил или циклопропилметил; и R4 представляет собой фенил, который может быть замещен один или более чем один раз заместителями, выбранными из группы, состоящей из галогена, CF3 и CN. Новые тропановые производные представляют собой сильные ингибиторы обратного захвата нейромедиаторных моноаминов, то есть дофамина, серотонина и норадреналина, и к применению новых тропановых производных для лечения расстройств или заболеваний, на которые влияет ингибирование обратного захвата нейромедиаторных моноаминов, таких как болезнь Паркинсона, депрессия, обсессивно-компульсивные расстройства, панические состояния, деменция, нарушения памяти, гиперактивность при дефиците внимания, ожирение, состояние тревоги, нарушения аппетита и токсикомания или злоупотребление кокаином. Описывается также способ получения соединений формулы I и фармацевтическая композиция на их основе. 4 с. и 7 з. п.ф-лы, 5 табл.
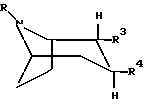
или любая их смесь, или их фармацевтически приемлемая соль,
где R представляет собой водород, метил, этил или пропил,
R3 представляет собой -CH2-X-R', где Х представляет собой О или S, а R' представляет собой метил, этил, пропил или циклопропилметил;
R4 представляет собой фенил, который может быть замещен один или более чем один раз заместителями, выбранными из группы, состоящей из галогена.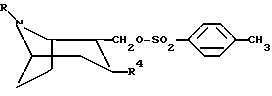
или любой из его энантиомеров или любую их смесь,
где R и R4 такие, как определено в п.1,
подвергают взаимодействию с алкоголятом R'-Z-Na, где R' такой, как определено в п.1, a Z представляет собой О или S, с образованием тропанового производного, имеющего формулу I.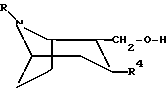
или любой из его энантиомеров или любую их смесь,
где R и R4 такие, как определено в п.1,
подвергают взаимодействию с гидридом натрия и соединением, имеющим формулу
R'-SO2,
где R' такой, как определено в п.1,
с образованием тропанового производного, имеющего формулу I, где Х представляет собой О.
| Способ приготовления связывающего вещества для минеральных наполнителей | 1928 |
|
SU12497A1 |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| US 4281130, 1981 | |||
| А.П | |||
| Орехов "Химия алкалоидов", М., 1968, с.91-116. | |||
