ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к новым производным пиперидина, полезным в качестве ингибиторов обратного захвата нейромедиаторных моноаминов.
В других аспектах данное изобретение относится к применению этих соединений в способе терапии и к фармацевтическим композициям, содержащим соединения по данному изобретению.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Petukhov et al. [Petukhov Р A, Zhang J, Kozikowski A Р, Wang С Z, Ye Y Р, Johnson К M and Tella S R]; J. Med. Chem. 2002, 45, 3161-3170] описывают SAR (structure activity relationship, зависимость «структура-активность») исследования аналогов кокаина на основе пиперидина.
В WO 00/20390 (Georgetown University) описаны мономерные и димерные гетероциклы и их терапевтическое применение.
В WO 98/51668 (NeuroSearch A/S) описаны производные 3-алкоксиимидометил-пиперидина, активные в качестве ингибиторов обратного захвата нейромедиаторов. В Примерах 1 и 2 описаны две промежуточные смеси, (±)-цис/транс-1-метил-3-метоксикарбонил-4-(3,4-дихлорфенил)-пиперидин и (±)-цис/транс-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин. Никакого фармакологического применения этих двух промежуточных смесей не описано.
Однако имеется постоянная сильная необходимость в поиске соединений с оптимизированным биохимическим профилем в отношении активности обратного захвата нейромедиаторных моноаминов - серотонина, дофамина и норадреналина, такой как соотношение обратного захвата серотонина против активности норадреналина и дофамина.
Кроме того, имеется сильная необходимость в поиске эффективных соединений, которые не родственны кокаину в структурном и синтетическом отношении.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В первом аспекте данного изобретения предложено производное пиперидина формулы I:
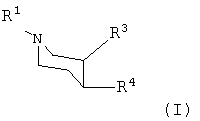
или любой из его изомеров, или любая смесь его изомеров, или его фармацевтически приемлемая соль,
где R1, R3 и R4 такие, как определено ниже.
Во втором аспекте данного изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по данному изобретению, или любого из его изомеров, или любой смеси его изомеров, или его фармацевтически приемлемой соли вместе с по меньшей мере одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
В еще одном аспекте данного изобретения предложено применение соединения по данному изобретению, или любого из его изомеров, или любой смеси его изомеров, или его фармацевтически приемлемой соли для изготовления фармацевтической композиции для лечения, предупреждения или облегчения заболевания, или расстройства, или состояния млекопитающего, включая человека, которое чувствительно к ингибированию обратного захвата нейромедиаторных моноаминов в центральной нервной системе.
В еще одном аспекте данное изобретение относится к способу лечения, предупреждения или облегчения заболевания, или расстройства, или состояния живого организма животного, включая человека, которое чувствительно к ингибированию обратного захвата нейромедиаторных моноаминов в центральной нервной системе, включающему стадию введения в указанный живой организм животного, нуждающегося в этом, терапевтически эффективного количества соединения по данному изобретению, или любого из его изомеров, или любой смеси его изомеров, или его фармацевтически приемлемой соли.
Другие задачи данного изобретения будут очевидны специалистам в данной области техники из следующего подробного описания и примеров.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Производные пиперидина
В первом аспекте данного изобретения предложено производное пиперидина формулы I:
или любой из его изомеров, или любая смесь его изомеров, или его фармацевтически приемлемая соль,
где
R1 представляет собой водород, алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил или 2-гидроксиэтил;
R3 представляет собой -С(=О)-О-Rc или -CH2-O-Rc;
где Rc представляет собой водород, алкил, алкенил, алкинил, циклоалкил или циклоалкилалкил;
R4 представляет собой
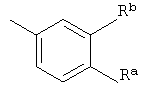
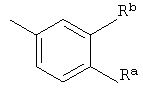
где Ra и Rb независимо друг от друга представляют собой галогено или трифторметил;
при условии, что смесь изомеров не представляет собой
(±)-цис/транс-1-метил-3-метоксикарбонил-4-(3,4-дихлорфенил)-пиперидин или
(±)-цис/транс-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин.
В одном воплощении R1 представляет собой водород или алкил.
Во втором воплощении Ra и Rb независимо друг от друга представляют собой галогено. В конкретном воплощении Ra представляет собой хлоро. В еще одном воплощении Rb представляет собой хлоро. В еще одном воплощении Ra представляет собой хлоро, и Rb представляет собой хлоро.
В еще одном воплощении R3 представляет собой -C(=O)-O-Rc. В еще одном воплощении R3 представляет собой -CH2-O-Rc.
В еще одном воплощении Rс представляет собой водород, алкил или циклоалкилалкил. В еще одном воплощении Rc представляет собой водород или алкил. В еще одном воплощении Rc представляет собой алкил или циклоалкилалкил. В конкретном воплощении Rc представляет собой водород. В еще одном воплощении Rc представляет собой алкил, такой как метил или этил. В еще одном воплощении Rc представляет собой циклоалкилалкил, такой как циклопропилметил.
В еще одном воплощении соединения формулы I
R1 представляет собой водород, алкил или циклоалкилалкил;
Rc представляет собой водород или алкил; и
Ra и Rb независимо друг от друга представляют собой галогено.
В еще одном воплощении соединения формулы I
R1 представляет собой водород или алкил;
Rc представляет собой водород или алкил; и
Ra и Rb независимо друг от друга представляют собой галогено.
В еще одном воплощении соединения формулы I
R1 представляет собой водород или алкил;
R3 представляет собой -CH2-O-Rc;
Rc представляет собой алкил или циклоалкилалкил; и
Ra и Rb независимо друг от друга представляют собой галогено.
В конкретном воплощении химическое соединение по данному изобретению представляет собой
метиловый эфир 1-метил-4-(3,4-дихлорфенил)-пиперидин-3-карбоновой кислоты;
1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин;
1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
1-метил-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидин;
3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
или любой из его изомеров, или любую смесь его изомеров, или его фармацевтически приемлемую соль.
В еще одном конкретном воплощении химическое соединение по данному изобретению представляет собой
метиловый эфир (±)-цис-1-метил-4-(3,4-дихлорфенил)-пиперидин-3-карбоновой кислоты;
метиловый эфир (±)-транс-1-метил-4-(3,4-дихлорфенил)-пиперидин-3-карбоновой кислоты;
(±)-цис-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин;
(±)-транс-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин;
(±)-цис-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(±)-цис-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(±)-транс-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(±)-транс-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-1-метил-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-1-метил-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-1-метил-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-1-метил-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидин;
или его фармацевтически приемлемую соль.
Любая комбинация двух или более чем двух воплощений, как описано выше, рассматривается в пределах объема настоящего изобретения.
Определение заместителей
В контексте этого изобретения галогено представляет собой фторо, хлоро, бромо или йодо.
В контексте этого изобретения алкильная группа означает одновалентную насыщенную прямую или разветвленную углеводородную цепь. Углеводородная цепь предпочтительно содержит от одного до шести атомов углерода (С1-6-алкил), включая пентил, изопентил, неопентил, третичный пентил, гексил и изогексил. В предпочтительном воплощении алкил представляет собой С1-4-алкильную группу, включая бутил, изобутил, вторичный бутил и третичный бутил. В другом предпочтительном воплощении этого изобретения алкил представляет собой C1-3-алкильную группу, которая в частности может быть метильной, этильной, пропильной или изопропильной.
В контексте этого изобретения алкениловая группа означает углеродную цепь, содержащую одну или более двойных связей, включая диены, триены и полиены. В предпочтительном воплощении алкениловая группа по данному изобретению содержит от двух до шести атомов углерода (С2-6-алкенил), включая по меньшей мере одну двойную связь. В наиболее предпочтительном воплощении алкениловая группа по данному изобретению представляет собой этенил; 1- или 2-пропенил; 1-, 2- или 3-бутенил или 1,3-бутдиенил; 1-, 2-, 3-, 4- или 5-гексенил, или 1,3-гексдиенил, или 1,3,5-гекстриенил.
В контексте этого изобретения алкиниловая группа означает углеродную цепь, содержащую одну или более тройных связей, включая диины, триины и полиины. В предпочтительном воплощении алкиниловая группа по данному изобретению содержит от двух до шести атомов углерода (С2-6-алкинил), включая по меньшей мере одну тройную связь. В наиболее предпочтительном воплощении алкиниловая группа по данному изобретению представляет собой этинил; 1- или 2-пропинил; 1-, 2- или 3-бутинил или 1,3-бутдиинил; 1-, 2-, 3-, 4-пентинил или 1,3-пентдиинил; 1-, 2-, 3-, 4- или 5-гексинил, или 1,3-гексдиинил, или 1,3,5-гекстриинил.
В контексте этого изобретения циклоалкильная группа означает циклическую алкильную группу, предпочтительно содержащую от трех до семи атомов углерода (С3-7-циклоалкил), включая циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Фармацевтически приемлемые соли
Химическое соединение по данному изобретению может быть предложено в любой форме, подходящей для предназначенного введения. Подходящие формы включают фармацевтически (то есть физиологически) приемлемые соли и пре- или пролекарственные формы химического соединения по данному изобретению.
Примеры фармацевтически приемлемых солей присоединения включают, без ограничения, такие нетоксичные соли присоединения неорганических и органических кислот, как гидрохлорид, полученный из соляной кислоты, бромистоводородную соль, полученную из бромистоводородной кислоты, нитрат, полученный из азотной кислоты, перхлорат, полученный их хлорной кислоты, фосфат, полученный из фосфорной кислоты, сульфат, полученный из серной кислоты, формиат, полученный из муравьиной кислоты, ацетат, полученный из уксусной кислоты, аконат, полученный из аконитовой кислоты, аскорбат, полученный из аскорбиновой кислоты, бензолсульфонат, полученный из бензолсульфоновой кислоты, бензоат, полученный из бензойной кислоты, циннамат, полученный из коричной кислоты, цитрат, полученный из лимонной кислоты, эмбонат, полученный из памовой кислоты, энантат, полученный из энантовой кислоты, фумарат, полученный из фумаровой кислоты, глутамат, полученный из глутаминовой кислоты, гликолят, полученный из гликолевой кислоты, лактат, полученный из молочной кислоты, малеат, полученный из малеиновой кислоты, малонат, полученный из малоновой кислоты, манделат, полученный из миндальной кислоты, метансульфонат, полученный из метансульфоновой кислоты, нафталин-2-сульфонат, полученный из нафталин-2-сульфоновой кислоты, фталат, полученный из фталевой кислоты, салицилат, полученный из салициловой кислоты, сорбат, полученный из сорбиновой кислоты, стеарат, полученный из стеариновой кислоты, сукцинат, полученный из янтарной кислоты, тартрат, полученный из винной кислоты, толуол-лара-сульфонат, полученный из пара-толуолсульфокислоты, и им подобные. Такие соли могут быть образованы посредством хорошо известных и описанных в данной области техники методик.
Другие кислоты, такие как щавелевая кислота, которые не могут рассматриваться как фармацевтически приемлемые, могут быть полезны в получении солей, полезных в качестве промежуточных соединений в получении химического соединения по данному изобретению и его фармацевтически приемлемой соли присоединения кислоты.
Примеры фармацевтически приемлемых катионных солей химического соединения по данному изобретению включают, без ограничения, натриевую, калиевую, кальциевую, магниевую, цинковую, алюминиевую, литиевую, холиновую, лизиновую и аммонийную соль и им подобные соли химического соединения по данному изобретению, содержащего анионную группу. Такие катионные соли могут быть образованы посредством хорошо известных и описанных в данной области техники методик.
В контексте этого изобретения «ониевые соли» N-содержащих соединений также рассматриваются в качестве фармацевтически приемлемых солей. Предпочтительные «ониевые соли» включают алкил-ониевые соли, циклоалкил-ониевые соли и циклоалкилалкил-ониевые соли.
Примеры пре- или пролекарственных форм химического соединения по данному изобретению включают примеры подходящих пролекарств веществ, которые согласно изобретению включают соединения, модифицированные по одной или более активной группе исходного соединения либо одной или более группе исходного соединения, способной к дериватизации. Особый интерес представляют соединения, модифицированные по карбоксильной группе, гидроксильной группе или аминогруппе. Примерами подходящих производных являются сложные эфиры или амиды.
Химическое соединение по данному изобретению может быть предложено в растворимой или нерастворимой форме вместе с фармацевтически приемлемым растворителем, таким как вода, этанол и им подобные. Растворимые формы также могут включать такие гидратированные формы, как моногидрат, дигидрат, полугидрат, тригидрат, тетрагидрат и им подобные. Как правило, растворимые формы считают эквивалентными нерастворимым формам для целей данного изобретения.
Стерические изомеры
Химические соединения по настоящему изобретению могут существовать в виде энантиомеров в (+) и (-) формах, а также в виде рацемических форм (±). Рацематы этих изомеров и сами индивидуальные изомеры входят в объем настоящего изобретения.
Рацемические формы можно разделить на оптические антиподы известными методами и техниками. Одним из способов разделения изомерных солей является использование оптически активной кислоты и выделение оптически активного аминного соединения посредством обработки основанием. Другой способ разделения рацематов на оптические антиподы основан на хроматографии на оптически активной матрице. Рацемические соединения по настоящему изобретению можно таким образом разделить на их оптические антиподы, например, путем фракционной кристаллизации d- или l-солей (тартратов, манделатов или камфорсульфоната).
Химические соединения по настоящему изобретению можно также разделить путем образования диастереомерных амидов посредством взаимодействия химических соединений по настоящему изобретению с оптически активной активированной карбоновой кислотой, такой как полученная из (+)- или (-)-фенилаланина, (+)- или (-)-фенилглицина, (+)- или (-)-камфановой кислоты, либо путем образования диастереомерных карбаматов посредством взаимодействия химического соединения по настоящему изобретению с оптически активным хлорформиатом или ему подобным.
Дополнительные способы разделения оптических изомеров известны из уровня техники. Такие способы включают способы, описанные Jaques J, Collet А, & Wilen S в «Enantiomers. Racemates and Resolutions». John Wiley and Sons, New York (1981).
Оптические активные соединения также могут быть получены из оптически активных исходных материалов.
Кроме того, соединения по настоящему изобретению могут существовать в цис- или транс-конфигурациях, а также в их смесях. Заместитель R3 и заместитель R4 пиперидиновой структуры формулы I могут, в частности, быть в цис- или транс-конфигурации по отношению друг к другу. В одном воплощении данного изобретения заместители R3 и R4 находятся в транс-конфигурации. В другом воплощении данного изобретения заместители R3 и R4 находятся в цис-конфигурации. Данное изобретение включает все такие изомеры и любые их смеси, включая рацемические смеси.
Меченые соединения
Соединения по данному изобретению могут быть использованы в своей меченой или немеченой форме. В контексте этого изобретения «метка» означает связывание маркера с интересующим соединением, что обеспечит легкое количественное детектирование указанного соединения.
Меченые соединения по данному изобретению могут быть полезны в качестве диагностических инструментов, радиоактивных индикаторов или контролирующих агентов в различных диагностических методах и для визуализации рецепторов in vivo.
Меченый изомер по данному изобретению предпочтительно содержит по меньшей мере один радионуклид в качестве метки. Все испускающие позитроны радионуклиды являются кандидатами для использования. В контексте этого изобретения радионуклид предпочтительно выбран из 2H (дейтерия), 3H (трития), 13С, 14С, 131I, 125I, 123I и 18F.
Физический метод детектирования меченого изомера по настоящему изобретению может быть выбран из позитронно-эмиссионной томографии (PET), однофотонной эмиссионной компьютерной томографии (SPECT), магнитно-резонансной спектроскопии (MRS), магнитно-резонансной томографии (MRI) и компьютерной аксиальной рентгеновской томографии (CAT) или их комбинаций.
Методы получения
Химические соединения по данному изобретению могут быть получены традиционными способами химического синтеза, например, описанными в рабочих примерах. Исходные материалы для способов, описанных в настоящей заявке, известны или легко могут быть получены с помощью традиционных способов из имеющихся в продаже реактивов.
Также одно соединение по данному изобретению можно превратить в другое соединение по данному изобретению, используя традиционные способы.
Конечные продукты взаимодействий, описанных здесь, могут быть выделены посредством традиционных методов, например посредством экстракции, кристаллизации, дистилляции, хроматографии и так далее.
Биологическая активность
Соединения по данному изобретению могут быть протестированы на их способность ингибировать обратный захват моноаминов - дофамина, норадреналина и серотонина в синаптосомах, например так, как описано в WO 97/30997.
Таким образом, в еще одном аспекте, основываясь на сбалансированной активности, наблюдаемой в этих тестах, соединения по данному изобретению считаются полезными в лечении, предупреждении или облегчении заболевания, или расстройства, или состояния млекопитающего, включая человека, которое чувствительно к ингибированию обратного захвата нейромедиаторных моноаминов в центральной нервной системе.
В конкретном воплощении соединения по данному изобретению считаются полезными для лечения, предупреждения или облегчения расстройства настроения, депрессии, атипичной депрессии, большого депрессивного расстройства, дистимического расстройства, биполярного расстройства, биполярного расстройства I типа, биполярного расстройства II типа, циклотимического расстройства, расстройства настроения, вызванного общим медицинским состоянием, расстройства настроения, вызванного лекарствами, псевдодеменции, синдрома Ганзера, обсессивно-компульсивного расстройства, панического расстройства, панического расстройства без агорафобии, панического расстройства с агорафобией, агорафобии без истории панического расстройства, панической атаки, дефицита памяти, потери памяти, синдрома дефицита внимания с гиперактивностью, ожирения, тревоги, генерализованного тревожного расстройства, расстройства приема пищи, болезни Паркинсона, паркинсонизма, деменции, возрастной деменции, старческой деменции, болезни Альцгеймера, комплекса СПИД (синдром приобретенного иммунодефицита) - деменция, нарушения памяти при старении, социальной фобии, посттравматического стрессового расстройства, острого стрессового расстройства, зависимости от лекарств, неправильного употребления лекарств, злоупотребления кокаином, злоупотребления никотином, злоупотребления табаком, хронического алкоголизма, алкоголизма, боли, воспалительной боли, невропатической боли, боли при мигрени, головной боли напряжения, хронической головной боли напряжения, боли, ассоциированной с депрессией, фибромиалгии, артрита, остеоартрита, ревматоидного артрита, боли в спине, боли при раке, боли при раздраженном толстом кишечнике, синдрома раздраженного толстого кишечника, послеоперационной боли, боли после удара, невропатии, вызванной лекарствами, диабетической невропатии, боли, поддерживаемой симпатической нервной системой, невралгии тройничного нерва, зубной боли, миофасциальной боли, фантомной боли, булимии, предменструального синдрома, синдрома поздней лютеиновой фазы, посттравматического синдрома, синдрома хронической усталости, недержания мочи, недержания мочи при напряжении, неудержания мочи, ночного недержания, преждевременной эякуляции, эректильной дисфункции, нервной анорексии, расстройств сна, аутизма, мутизма, трихотилломании, нарколепсии, депрессии после удара, повреждения мозга, вызванного ударом, нейронального повреждения, вызванного ударом, или болезни Жиль де ла Туретта. В предпочтительном воплощении данные соединения считаются полезными для лечения, предупреждения или облегчения депрессии.
В настоящем изобретении предусматривается, что подходящая дозировка активного фармацевтического ингредиента (API, active pharmaceutical ingredient) находится в диапазоне от примерно 0,1 до примерно 1000 мг API в сутки, более предпочтительно от примерно 10 до примерно 500 мг API в сутки, наиболее предпочтительно от примерно 30 до примерно 100 мг API в сутки, в зависимости, однако, от конкретного способа введения, формы, в которой его вводят, рассматриваемого показания, пациента и, в частности, массы тела рассматриваемого пациента и, кроме того, предпочтения и опыта ответственного врача или ветеринара.
Предпочтительные соединения по данному изобретению проявляют биологическую активность в субмикромолярном и микромолярном диапазоне, то есть от менее 1 до примерно 100 мкМ.
Фармацевтические композиции
В другом аспекте данного изобретения предложены новые фармацевтические композиции, содержащие терапевтически эффективное количество химического соединения по данному изобретению.
Несмотря на то что химическое соединение по данному изобретению для использования в терапии можно вводить в виде необработанного химического соединения, предпочтительно включать активный ингредиент, возможно в форме физиологически приемлемой соли, в фармацевтическую композицию вместе с одним или более чем одним адъювантом, эксципиентом, носителем, буфером, разбавителем и/или другими обычными фармацевтическими добавками.
В предпочтительном воплощении данного изобретения предложены фармацевтические композиции, содержащие химическое соединение по данному изобретению или его фармацевтически приемлемую соль или производное вместе с одним или более чем одним фармацевтически приемлемым носителем и, возможно, другими терапевтическими и/или профилактическими ингредиентами, известными и используемыми в данной области техники. Носитель(и) должен(ны) быть «приемлемым(и)» в смысле его совместимости с другими ингредиентами препарата и отсутствия вреда для получающего препарат.
Фармацевтическими композициями по данному изобретению могут быть композиции, подходящие для перорального, ректального, бронхиального, назального, пульмонального, местного (включая буккальное и сублингвальное), трансдермального, вагинального или парентерального (включая кожную, подкожную, внутримышечную, внутрибрюшинную, внутривенную, внутриартериальную, интрацеребральную, внутриглазную инъекцию или инфузию) введения либо композиции в форме, подходящей для введения путем ингаляции или инсуффляции, включая введение порошков и жидких аэрозолей, или посредством систем с длительным высвобождением. Подходящие примеры систем с длительным высвобождением включают полупроницаемые матрицы твердых гидрофобных полимеров, содержащие соединение по данному изобретению, которые могут быть в виде продуктов определенной формы, например пленок или микрокапсул.
Химическое соединение по данному изобретению вместе с общепринятым адъювантом, носителем или разбавителем может быть таким образом помещено в форму фармацевтических композиций и их унифицированных лекарственных форм. Такие формы включают твердые, в частности, таблетки, наполненные капсулы, порошки и формы в виде пеллет, и жидкие формы, в частности водные и неводные растворы, суспензии, эмульсии, эликсиры и капсулы, наполненные тем же, все для перорального применения, суппозитории для ректального введения и стерильные растворы для инъекций для парентерального применения. Такие фармацевтические композиции и их унифицированные лекарственные формы могут содержать традиционные ингредиенты в общепринятых соотношениях с дополнительными активными соединениями или началами либо без них, и такие унифицированные лекарственные формы могут содержать любое походящее эффективное количество активных ингредиентов соответственно с предназначенным диапазоном суточной дозировки, которую будут применять.
Химическое соединение по настоящему изобретению можно вводить в виде целого ряда пероральных и парентеральных лекарственных форм. Специалистам в данной области техники будет очевидно, что следующие лекарственные формы могут содержать в качестве активного ингредиента либо химическое соединение по данному изобретению, либо фармацевтически приемлемую соль химического соединения по данному изобретению.
Для изготовления фармацевтических композиций из химического соединения по настоящему изобретению фармацевтически приемлемые носители могут быть либо твердыми, либо жидкими. Препараты твердой формы включают порошки, таблетки, пилюли, капсулы, облатки, суппозитории и диспергируемые гранулы. Твердым носителем может быть одно или более чем одно вещество, которое может также действовать в качестве разбавителей, корригентов, растворителей, смазывающих агентов, суспендирующих агентов, связующих агентов, консервантов, агентов, способствующих распадению таблетки, или инкапсулирующего материала.
В порошках носитель представляет собой тонкоизмельченное твердое вещество, которое находится в смеси с тонкоизмельченным активным компонентом.
В таблетках активный компонент смешан с носителем, имеющим необходимую связывающую способность, в подходящих соотношениях и спрессован в виде формы желаемого размера.
Порошки и таблетки предпочтительно содержат от пяти или десяти до примерно семидесяти процентов активного соединения. Подходящими носителями являются карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, низкоплавкий воск, масло какао и им подобные. Подразумевается, что термин «препарат» включает препарат активного соединения с инкапсулирующим материалом в качестве носителя, обеспечивающего капсулу, в которой активный компонент, с носителями или без них, окружен носителем, который таким образом находится в ассоциации с ним. Аналогично включены облатки и лепешки. Таблетки, порошки, капсулы, пилюли, облатки и лепешки можно применять в виде твердых форм, подходящих для перорального введения.
Для получения суппозиториев вначале расплавляют низкоплавкий воск, такой как смесь глицерида жирной кислоты или масла какао, и в нем гомогенно диспергируют активный компонент, например, путем перемешивания. Затем расплавленную гомогенную смесь заливают в формы удобного размера, оставляют охлаждаться и тем самым затвердеть.
Композиции, подходящие для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пенок или спреев, содержащих в дополнение к активному ингредиенту такие носители, которые известны как подходящие из уровня техники.
Жидкие препараты включают растворы, суспензии и эмульсии, например водные или водно-пропиленгликолевые растворы. Например, жидкие препараты для парентерального введения могут быть изготовлены в виде растворов в водном растворе полиэтиленгликоля.
Химическое соединение по настоящему изобретению может таким образом быть изготовлено в виде препарата для парентерального введения (например, путем инъекции, например болюсной инъекции, или непрерывной инфузии) и может быть представлено в виде однодозовых лекарственных форм в ампулах, предварительно наполненных шприцах, инфузиях малого объема либо в многодозовых контейнерах с добавленным консервантом. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать такие агенты препарата, как суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный ингредиент может быть в порошковой форме, полученной посредством асептического выделения стерильного твердого вещества или посредством лиофилизации раствора, для смешения с подходящим носителем, например стерильной апирогенной водой, перед применением.
Водные растворы, подходящие для перорального применения, могут быть получены путем растворения активного компонента в воде и добавления подходящих красителей, вкусовых ароматизирующих веществ, стабилизирующих агентов и загустителей, по желанию.
Водные растворы, подходящие для перорального применения, могут быть изготовлены посредством диспергирования тонкоизмельченного активного компонента в воде с вязким материалом, таким как природные или синтетические смолы, полимеры, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы или другие общеизвестные суспендирующие агенты.
Также включены препараты твердой формы, предназначенные для превращения в препараты жидкой формы для перорального введения незадолго перед применением. Такие жидкие формы включают растворы, суспензии и эмульсии. В дополнение к активному компоненту такие препараты могут содержать красители, вкусовые ароматизирующие вещества, стабилизаторы, буферы, искусственные и природные подсластители, диспергаторы, загустители, солюбилизирующие агенты и им подобные.
Для местного применения на эпидермис химическое соединение по данному изобретению может быть изготовлено в виде мазей, кремов или лосьонов или в виде трансдермального пластыря. Мази и кремы могут, например, быть изготовлены с водной или масляной основой с добавлением подходящих загустителей и/или желирующих агентов. Лосьоны могут быть изготовлены с водной или масляной основой и также обычно содержат один или более чем один эмульгирующий агент, стабилизирующий агент, диспергирующий агент, суспендирующий агент, загуститель или корригент.
Композиции, подходящие для местного введения в ротовую полость, включают лепешки, содержащие активный агент в ароматизированной основе, обычно сахарозе и акации или трагаканте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и акация; и полоскания для рта, содержащие активный ингредиент в подходящем жидком носителе.
Растворы или суспензии применяют непосредственно в носовую полость с помощью традиционных средств, например капельницы, пипетки или спрея. Композиции могут быть предложены в однодозовой или многодозовой форме.
Введение в дыхательные пути также может быть достигнуто посредством аэрозольного препарата, в котором активный ингредиент предложен в герметичной упаковке с подходящим пропеллентом, таким как хлорфторуглерод (ХФУ), например дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, двуокись углерода или другой подходящий газ. Аэрозоль может также для удобства содержать поверхностно-активное вещество, такое как лецитин. Дозу лекарственного средства можно контролировать посредством снабжения измерительным клапаном.
Альтернативно, активные ингредиенты могут быть предложены в форме сухого порошка, например порошковой смеси соединения в подходящей порошковой основе, такой как лактоза, крахмал, производные крахмала, такие как гидроксипропилметилцеллюлоза и поливинилпирролидон (ПВП). Удобно, если порошковый носитель образует гель в носовой полости. Порошковая композиция может быть представлена в однодозовой лекарственной форме, например в капсулах или картриджах, например из желатина, или в блистерных упаковках, из которых порошок можно вводить посредством ингалятора.
В композициях, предназначенных для введения в дыхательные пути, включая интраназальные композиции, соединение обычно имеет маленький размер частиц, например порядка 5 микрон или менее. Такой размер частиц может быть получен с помощью способов, известных из уровня техники, например путем микронизации.
При желании можно применять композиции, адаптированные для получения длительного высвобождения активного ингредиента.
Фармацевтические препараты предпочтительно находятся в унифицированных лекарственных формах. В такой форме препарат подразделен на унифицированные дозы, содержащие соответствующие количества активного компонента. Унифицированная лекарственная форма может представлять собой упакованный препарат, где упаковка содержит разделенные количества препарата, такой как расфасованные таблетки, капсулы и порошки во флаконах или ампулах. Также унифицированная лекарственная форма может представлять собой капсулу, таблетку, облатку или лепешку саму по себе, либо она может представлять собой подходящее количество любой из этих упакованных форм.
Таблетки или капсулы для перорального введения и жидкости для внутривенного введения и непрерывной инфузии представляют собой предпочтительные композиции.
Дополнительные подробности, касающиеся методик для препаратов и введения, могут быть найдены в последнем издании Remington's Pharmaceutical Sciences (Maack Publishing Co., Easton, PA).
Терапевтически эффективная доза относится к такому количеству активного ингредиента, которое облегчает симптомы или состояние. Терапевтическая эффективность и токсичность, например ED50 и LD50, могут быть определены с помощью стандартных фармакологических процедур на клеточных культурах или экспериментальных животных. Соотношение доз между терапевтическим и токсическим эффектом представляет собой терапевтический индекс и может быть выражен соотношением LD50/ED50. Фармацевтические композиции, проявляющие большие терапевтические индексы, являются предпочтительными.
Вводимая доза, конечно, должна быть тщательно скорректирована в соответствии с возрастом, массой и состоянием пациента, которого лечат, а также со способом введения, лекарственной формой, режимом и желаемым результатом, и точную дозировку, конечно, должен назначать лечащий врач.
Действительная дозировка зависит от природы и тяжести заболевания, которое лечат, и находится на усмотрении лечащего врача, и ее можно варьировать путем титрования дозировки на конкретные воплощения этого изобретения для получения желаемого терапевтического эффекта. Однако в настоящем изобретении предусматривается, что фармацевтические композиции, содержащие от примерно 0,1 до примерно 500 мг активного ингредиента на индивидуальную дозу, предпочтительно от примерно 1 до примерно 100 мг, наиболее предпочтительно от примерно 1 до примерно 10 мг, подходят для терапевтического лечения.
Активный ингредиент можно вводить в одной или нескольких дозах в сутки. Удовлетворительный результат может в определенных случаях быть достигнут при такой низкой дозировке, как 0,1 мкг/кг i.v. (внутривенно) и 1 мкг/кг р.о. (перорально). Предполагается, что верхняя граница диапазона дозировки составляет около 10 мг/кг i.v. и 100 мг/кг р.о. Предпочтительными диапазонами являются диапазоны от примерно 0,1 мкг/кг до примерно 10 мг/кг/сутки i.v. и от примерно 1 мкг/кг до примерно 100 мг/кг/сутки р.о.
Способы терапии
В другом аспекте данного изобретения предложен способ лечения, предупреждения или облегчения заболевания, или расстройства, или состояния живого организма животного, включая человека, которое чувствительно к ингибированию обратного захвата нейромедиаторных моноаминов в центральной нервной системе, включающий введение в указанный живой организм животного, включая человека, нуждающегося в этом, эффективного количества химического соединения по данному изобретению.
В настоящем изобретении предусматривается, что подходящими диапазонами дозировки являются от 0,1 до 1000 миллиграмм в сутки, 10-500 миллиграмм в сутки и особенно 30-100 миллиграмм в сутки, в зависимости от конкретного способа введения, формы, в которой осуществляют введение, показания, на которое направлено введение, рассматриваемого пациента и массы тела рассматриваемого пациента и также от предпочтения и опыта ответственного лечащего врача или ветеринара.
ПРИМЕРЫ
Изобретение далее проиллюстрировано со ссылкой на следующие примеры, которые не предназначены для какого-либо ограничения объема заявленного изобретения.
Пример 1
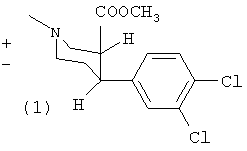
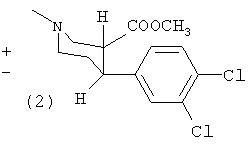
Метиловый эфир (±)-цис-1-метил-4-(3,4-дихлорфенил)-пиперидин-3-карбоновой кислоты (1) и метиловый эфир (±)-транс-1-метил-4-(3,4-дихлорфенил)-пиперидин-3-карбоновой кислоты (2)
К перемешиваемой суспензии магниевой стружки (3,4 г, 142 ммоль) в диэтиловом эфире (20 мл) добавляли раствор 1-бром-3,4-дихлорбензола (29 г, 130 ммоль) в диэтиловом эфире (150 мл). Смесь кипятили с обратным холодильником в течение 20 минут и затем охлаждали до -40°С. Раствор ареколина (10 г, 65 ммоль) в толуоле (100 мл) добавляли медленно при поддержании внутренней температуры между -40°С и -30°С. Реакционную смесь перемешивали при -20°С в течение 6 часов и затем добавляли 4 н. HCl (50 мл). Затем фазы разделяли и в водную фазу добавляли аммиак (водный) до щелочного рН и экстрагировали дихлорметаном (4×100 мл), сушили сульфатом магния и упаривали с получением масла. Изомеры (1) и (2) разделяли путем колоночной хроматографии (петролейный эфир, эфир, триэтиламин 70:25:5) с получением 5,0 г (25%) (1) (Т.пл. (точка плавления) 70-75°С) и 2,0 г (10%) (2) (масло).
Пример 2
Способ А1
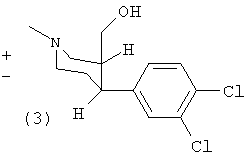
(±)-Цис-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин (3)
К раствору (1) (5,0 г, 17 ммоль) в тетрагидрофуране (50 мл) при -50°С добавляли LiAIH4 (0,50 г, 13 ммоль). Перемешивали при температуре -30°С в течение трех часов, затем гасили путем добавления воды и упаривали с получением твердого вещества. Остаток растворяли в дихлорметане, сушили сульфатом магния и упаривали до сухого состояния. Выход 4,6 г (3) (100%). Т.пл. 127-129°С.
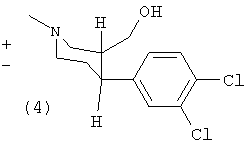
(±)-Транс-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин (4)
Раствор (2) (2,0 г, 6,6 ммоль) восстанавливали согласно способу (А1) с получением 1,9 г (100%) продукта (4). Т.пл. 109-111°С.
Процедура (а)
Рацемат может быть разделен на индивидуальные энантиомеры путем осаждения солей манделатов.
(+)-Цис-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин (5)
Смесь (3) (23,8 г, 86,8 ммоль) и (-)-миндальной кислоты (6,6 г, 43,4 ммоль) в абсолютном этаноле (60 мл) нагревали до прозрачного раствора. Реакционную смесь упаривали до сухого состояния и повторно кристаллизовали один раз из толуола (100 мл) и один раз из толуола (100 мл) и абсолютного этанола (12 мл). Осадок выделяли и помещали в воду (75 мл). Концентрированный аммиак (водный) добавляли до щелочного рН и смесь экстрагировали этилацетатом (3×75 мл). Объединенные органические фазы сушили сульфатом магния и упаривали до сухого состояния. Выход 8,2 г (69%) (5), т.пл. 94,5-96,5°С, [α]D 25=+67°C.
Соль фумарат (+)-цис-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин, т.пл. 147-149°С, [α]D 25=+68°C.
Процедура (б)
(-)-Цис-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин (6)
К толуолу из повторной кристаллизации, упомянутой в процедуре (а), добавляли воду (75 мл) и концентрированный аммиак (водный) до щелочного рН. Смесь экстрагировали этилацетатом (2×75 мл). Объединенные органические фазы сушили сульфатом магния и упаривали до сухого состояния. Остаток растворяли в абсолютном этаноле (60 мл) и добавляли (+)-миндальную кислоту (6,6 г, 43,4 ммоль). Затем процедура повторяет упомянутую выше процедуру (а), давая выход 7,4 г (62%) (6), т.пл. 95-97°С, [α]D=-65°C.
Соль фумарат (-)-цис-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидина, т.пл. 148-150°С, [α]D 25=-37°C.
Способ А2
Чистые энантиомеры, (+)- и (-)-транс-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин, могут быть получены посредством изомеризации (5) и (6).
(+)-Транс-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин (7)
Раствор (5) (10,0 г, 36 ммоль) и трет-бутилата калия (12,0 г, 1,08 ммоль) в диметилформамиде (75 мл) перемешивали в течение ночи. Добавляли CaCl2 (75 мл, 3 М) и реакционную смесь экстрагировали этилацетатом (2×200 мл). Объединенные органические фазы сушили сульфатом магния и упаривали до сухого состояния. Остаток повторно кристаллизовали из этилацетата (7 мл). Выход 6,3 г (63%) (7), т.пл. 137-139°С, [α]D 25=+39°C.
(-)-Транс-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидин (8)
(8) синтезировали из (6) (2,0 г, 7,3 ммоль) согласно способу (А2), с получением 1,4 г (70%) (8), т.пл. 137-139°С, [α]D 25=-38°C.
Соль фумарат (-)-транс-1-метил-3-гидроксиметил-4-(3,4-дихлорфенил)-пиперидина, т.пл. 138-140°С, [α]D 25=-25°C.
Пример 3
Способ В1
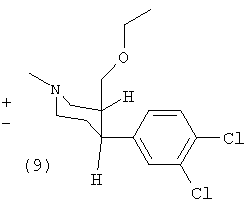
(±)-Цис-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин (9)
К раствору (3) (2,4 г, 8,6 ммоль) в тетрагидрофуране (40 мл) добавляли 60% NaH (0,69 г, 17 ммоль) и перемешивали при комнатной температуре в течение одного часа. Добавляли диэтилсульфат (1,4 мл, 11 ммоль) и перемешивали реакционную смесь в течение ночи. Добавляли воду и экстрагировали реакционную смесь диэтиловым эфиром (3×40 мл). Объединенные органические фазы сушили сульфатом магния и упаривали до сухого состояния. Колоночной хроматографией с использованием смеси дихлорметана, метанола и аммиака (водного) (9:1:1%) получили 1,5 г (56%) продукта (9) (масло).
Подобным образом получили:
(±)-Цис-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин (10) путем метилирования (3) диметилсульфатом (масло).
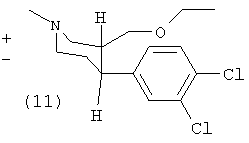
(±)-Транс-1-метил-3-эгпоксиметил-4-(3,4-дихлорфенил)-пиперидин (11)
(11) синтезировали из (4) (1,8 г, 6,6 ммоль) согласно способу (В1), с получением 0,83 г (43%) продукта (11) (масло).
Подобным образом получили:
(±)-Транс-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин (12) путем метилирования (4) диметилсульфатом (масло).
Способ B2
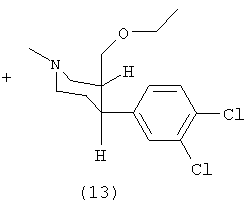
Соль фумарат (+)-цис-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидина (13)
К раствору (5) (11,0 г, 40 ммоль) в тетрагидрофуране (150 мл) добавляли трет-бутилат калия (13,3 г, 120 ммоль). Реакционную смесь перемешивали в течение одного часа и охлаждали до 5°С. Добавляли диэтилсульфат (5,7 мл, 44 ммоль) и перемешивали реакционную смесь в течение ночи. Добавляли насыщенный хлорид натрия (150 мл) и воду (50 мл) и экстрагировали реакционную смесь этилацетатом (2×80 мл), сушили сульфатом натрия и упаривали до сухого состояния. Выход 12 г (99%) (13). Т.пл. 72,5-74°С, [α]D25=+65°C.
Подобным образом получили:
(+)-Цис-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин (14) путем алкилирования (5) диметилсульфатом (масло).
(-)-Цис-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин (15) путем алкилирования (6) диметилсульфатом (масло).
Соль фумарат (+)-цис-1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидина (16) путем алкилирования (5) (бромметил)циклопропаном. Т.пл. 187-188,5°С, [α]D 25=+63°C.
Соль фумарат (-)-цис-1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидина (17) путем алкилирования (6) (бромметил)циклопропаном. Т.пл. 184,5-187,6°С, [α]D 25=-66°C.
Соль фумарат (-)-цис-1-метил-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидина (18) путем алкилирования (6) 1-бром-2-метилпропаном. Т.пл. 181-183°С, [α]D 25=-64°C.
(+)-Транс-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин (19) путем алкилирования (7) диэтилсульфатом (масло).
(-)-Транс-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин (20) путем алкилирования (8) диэтилсульфатом (масло).
(+)-Транс-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин (21) путем алкилирования (7) диметилсульфатом, (масло), [α]D 25=+44°C.
Соль фумарат (-)-транс-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидина (22) путем алкилирования (8) диметилсульфатом. Т.пл. 50-70°С (гигроскопический), [α]D 25=-23°C.
Соль фумарат (-)-транс-1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидина (23) путем алкилирования (8) (бромметил)циклопропаном. Т.пл. 180-182°С, [α]D 25=-31°C.
Соль фумарат (-)-транс-1-метил-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидина (24) путем алкилирования (8) 1-бром-2-метилпропаном. Т.пл. 162-164°С, [α]D 25=-29°C.
Подобным образом получили:
Соль фумарат (+)-цис-1-метил-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидина
Соль фумарат (+)-транс-1-метил-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидина
Соль фумарат (+)-транс-1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидина
Пример 4
Способ С
Рацемат (±)-цис-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидина (9) можно разделить на индивидуальные энантиомеры с использованием солей дибензоилтартратов.
Процедура (а)
Гидробромид (+)-цис-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидина (13)
(±)-Цис-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин (9) (1,1 г, 3,7 ммоль) и (-)-дибензоилвинную кислоту (0,48 г, 1,3 ммоль) растворяли в 99% этаноле (10 мл) и упаривали до сухого состояния. Упаренный осадок кристаллизовали из толуола (3 мл). Проводили повторную кристаллизацию из смеси толуола (10 мл) и этанола (10 мл). Осадок выделяли и растворяли в смеси 4 н. NaOH (5 мл) и диэтилового эфира (10 мл). Диэтиловый эфир отделяли и сушили сульфатом магния с получением 0,25 г (45%) продукта в виде свободного основания. Добавляли бромистоводородную кислоту (0,20 мл, 1,7 ммоль) и упаривали смесь до сухого состояния. Остаток повторно кристаллизовали из этанола (2 мл) и диэтилового эфира (10 мл) с получением 0,20 г (28%) (13), т.пл. 183-185°С, [α]D 25=+62,8°, (с=14 мг/мл в 99% этаноле).
Процедура (b)
Гидробромид (-)-цис-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидина (25)
К толуолу со стадии повторной кристаллизации, упомянутой в процедуре (а), добавляли 4 н. NaOH (5 мл) и экстрагировали диэтиловым эфиром (3×25 мл), сушили сульфатом магния и упаривали до сухого состояния. Остаток растворяли в 99% этаноле (10 мл) и добавляли (+)-дибензоилвинную кислоту (0,67 г, 1,8 ммоль). Затем процедура повторяет упомянутую выше процедуру (а), давая выход 0,14 г (20%) (25), т.пл. 183-185°С, [α]D 25=-66,1°, (с=14 мг/мл в 99% этаноле).
Гидробромид (+)-транс-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидина (19) и
Гидробромид (-)-транс-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидина (20)
(19) и (20) были выделены из (11) (0,83 г, 2,8 ммоль) согласно процедуре, используемой в способе С, давая выход 0,16 г (30%) (19), т.пл. 222-224°С, [α]D 25=+34,9°, (с=10 мг/мл в 99% этаноле); и 0,14 г (26%) (20), т.пл. 219-221°С, [α]D 25=-32,7°, (с=10 мг/мл в 99% этаноле).
Гидробромид (+)-цис-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидина (14) и
Гидробромид (-)-цис-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидина (15)
(14) и (15) были выделены из (10) согласно процедуре в способе С. [α]D 25=+65°, т.пл. 212-215°С для (14) и [α]D 25=-65°, т.пл. 212-215°С для (15).
Пример 5
(+)-Цис-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин (26)
Смесь (13) 0,70 г и 1-хлорэтилхлорформиата (2,5 мл) перемешивали при 100°С в течение 2 суток, после чего добавляли 4 н. NaOH (25 мл). Смесь перемешивали при кипячении с обратным холодильником в течение ночи. После охлаждения смесь экстрагировали толуолом. Органическую фазу сушили и упаривали с получением масла, которое подвергали колоночной хроматографии (SiO2, метиленхлорид, МеОН, аммиак 9:1:1%) с получением продукта в виде бледных кристаллов. Т.пл. 68-70°С.
Соль фумарат (+)-цис-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидина, т.пл. 149-151°С. [α]D 25=+68°.
Подобным образом получили:
Гидробромид (-)-цис-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидина (27), т.пл. 182-184°С, [α]D 25=-75°.
Соль фумарат (+)-цис-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидина (28), т.пл. 154,5-156°С, [α]D 25=+66°.
Соль фумарат (-)-цис-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидина (29), т.пл. 147-149°С, [α]D 25=+68°.
Соль фумарат (+)-цис-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидина (30), т.пл. 172-173,5°С, [α]D 25=+63°.
Соль фумарат (-)-цис-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидина (31), т.пл. 175-176,5°С, [α]D 25=-65°.
(+)-Транс-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин (32), (масло), [α]D25=+46°.
Соль фумарат (-)-транс-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидина (33), т.пл. 140-142°С, [α]D 25=-37°.
(+)-Транс-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин (34), (масло), [α]D 25=+44°.
Соль фумарат (-)-транс-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидина (35), т.пл. 140-142°С, [α]D 25=-31°.
Соль фумарат (-)-транс-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидина (36), т.пл. 153,5-155,5°С, [α]D 25=37°.
Подобным образом получили:
Соль фумарат (+)-транс-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидина
Соль фумарат (+)-цис-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидина
Соль фумарат (-)-цис-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидина
Соль фумарат (+)-транс-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидина
Соль фумарат (-)-транс-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидина
Биологические данные
Соединения по изобретению были протестированы на их способность ингибировать обратный захват нейромедиаторных моноаминов: дофамина (DA), норадреналина (NA) и серотонина (5-НТ) в синаптосомах.
Предпосылки
Дофаминовые медиаторы/сайты захвата на нервных окончаниях предположительно функционируют как терминаторы нейронного импульса путем удаления нейромедиаторов дофамина, норадреналина и серотонина, соответственно, из синаптической щели. Активность транспортных интегральных белков может быть измерена in vitro на синаптосомальный захват 3H-дофамина, 3H-норадреналина и 3H-серотонина, соответственно.
Ингибирование in vitro захвата 3Н-дофамина (3H-DA) в стриарных синаптосомах
Препараты ткани: Препараты готовят при 0-4°С, если не указано иначе. Corpi striati из самцов крыс линии Вистар (150-200 г) гомогенизируют 5-10 с в 100 объемах охлажденной во льду 0,32 М сахарозе, содержащей 1 мМ паргилина, в гомогенизаторе Ultra-Turrax. Активность моноаминоксидазы должна ингибироваться в присутствии паргилина. Гомогенат центрифугируют при 1000·g в течение 10 мин. Полученную надосадочную жидкость затем центрифугируют при 27000·g в течение 50 мин и надосадочную жидкость отбрасывают. Остаток (P2) заново суспендируют в оксигенированном (уравновешенном в атмосфере 96% O2: 4% CO2 в течение по меньшей мере 30 мин) инкубационном буфере Кребса-Рингера (8000 мл на г исходной ткани) с рН 7,2, содержащем 122 мМ NaCl, 0,16 мМ ЭДТА, 4,8 мМ KCl, 12,7 мМ Na2HPO4, 3,0 мМ NaH2PO4, 1,2 мМ MgSO4, 1 мМ CaCl2, 10 мМ глюкозы и 1 мМ аскорбиновой кислоты.
Анализ: К 100 мкл тестируемого раствора и 100 мкл 3H-DA (конечная концентрация 1 нМ) добавляют аликвоты по 4,0 мл тканевой суспензии, смешивают и инкубируют в течение 25 мин при 37°С. Неспецифический захват определяют с использованием бензтропина (конечная концентрация 10 мкМ). После инкубации образцы наливают прямо на фильтры из стекловолокна Whatman GF/C под отсосом. Затем фильтры промывают три раза 5 мл охлажденного во льду 0,9% (мас./об.) раствора NaCl. Количество радиоактивности на фильтрах определяют стандартным счетом жидкостной сцинтилляции. Специфический захват вычисляют как разницу между общим и неспецифическим захватом.
Перед вычислением IC50 должно быть получено 25-75% ингибирование специфического связывания.
Тестируемое значение приводят в виде IC50 (концентрация (мкМ) тестируемого вещества, которая ингибирует специфическое связывание 3H-DA на 50%).
Ингибирование in vitro захвата 3H-норадреналина (3H-NA) в синаптосомах гиппокампа
Препараты ткани: Препараты готовят при 0-4°С, если не указано иначе. Hippocampi из самцов крыс линии Вистар (150-200 г) гомогенизируют 5-10 с в 100 объемах охлажденной во льду 0.32М сахарозе, содержащей 1 мМ паргилина, в гомогенизаторе Ultra-Turrax. Активность моноаминоксидазы должна ингибироваться в присутствии паргилина. Гомогенат центрифугируют при 1000·g 10 мин. Полученную надосадочную жидкость затем центрифугируют при 27000·g в течение 50 мин и надосадочную жидкость отбрасывают. Остаток (P2) заново суспендируют в оксигенированном (уравновешенном в атмосфере 96% O2: 4% CO2 в течение по меньшей мере 30 мин) инкубационном буфере Кребса-Рингера (2000 мл на г исходной ткани) с рН 7,2, содержащем 122 мМ NaCl, 0,16 мМ ЭДТА, 4,8 мМ KCl, 12,7 мМ Na2HPO4, 3,0 мМ NaH2PO4, 1,2 мМ MgSO4, 1 мМ CaCl2, 10 мМ глюкозы и 1 мМ аскорбиновой кислоты.
Анализ: К 100 мкл тестируемого раствора и 100 мкл 3H-NA (конечная концентрация 1 нМ) добавляют аликвоты по 4,0 мл тканевой суспензии, смешивают и инкубируют в течение 90 мин при 37°С. Неспецифический захват определяют с использованием дезипрамина (конечная концентрация 1 мкМ). После инкубации образцы наливают прямо на фильтры из стекловолокна Whatman GF/C под отсосом. Затем фильтры промывают три раза 5 мл охлажденного во льду 0,9% (мас./об.) раствора NaCl. Количество радиоактивности на фильтрах определяют стандартным счетом жидкостной сцинтилляции. Специфический захват вычисляют как разницу между общим и неспецифическим захватом.
Перед вычислением IC50 должно быть получено 25-75% ингибирование специфического связывания.
Тестируемое значение приводят как IC50 (концентрация (мкМ) исследуемого вещества, которая ингибирует специфическое связывание 3H-NA на 50%).
Ингибирование in vitro захвата 3H-5-гидрокситриптамина (3H-5-HT, серотонин) в кортикальных синаптосомах
Препараты ткани: Препараты готовят при 0-4°С, если не указано иначе. Кору головного мозга из самцов крыс линии Вистар (150-200 г) гомогенизируют в течение 5-10 с в 100 объемах охлажденной во льду 0,32М сахарозе, содержащей 1 мМ паргилина, в гомогенизаторе Ultra-Turrax. Активность моноаминоксидазы должна ингибироваться в присутствии паргилина. Гомогенат центрифугируют при 1000·g в течение 10 мин. Полученную надосадочную жидкость затем центрифугируют при 27000·g в течение 50 мин и надосадочную жидкость отбрасывают. Остаток (P2) заново суспендируют в оксигенированном (уравновешенном в атмосфере 96% O2: 4% CO2, в течение по меньшей мере 30 мин) инкубационном буфере Кребса-Рингера (1000 мл на г исходной ткани) с рН 7,2, содержащем 122 мМ NaCl, 0,16 мМ ЭДТА, 4,8 мМ KCl, 12,7 мМ Na2HPO4, 3,0 мМ Na2HPO4, 1,2 мМ MgSO4, 1 мМ CaCl2, 10 мМ глюкозы и 1 мМ аскорбиновой кислоты.
Анализ: К 100 мкл тестируемого раствора и 100 мкл 3H-5-НТ (конечная концентрация 1 нМ) добавляют аликвоты по 4,0 мл тканевой суспензии, смешивают и инкубируют в течение 30 мин при 37°С. Неспецифический захват определяют с использованием циталопрама (конечная концентрация 1 мкМ). После инкубации образцы наливают прямо на фильтры из стекловолокна Whatman GF/C под отсосом. Затем фильтры промывают три раза 5 мл охлажденного во льду 0,9% (мас./об.) раствора NaCl. Количество радиоактивности на фильтрах определяют стандартным счетом жидкостной сцинтилляции. Специфический захват вычисляют как разницу между общим и неспецифическим захватом.
Перед вычислением IC50 должно быть получено 25-75% ингибирование специфического связывания.
Тестируемое значение приводят в виде IC50 (концентрация (мкМ) исследуемого вещества, которая ингибирует специфическое связывание 3H-5-НТ на 50%).
Результаты, полученные при тестировании соединений по изобретению, приведены в таблице 1 ниже.
Изобретение относится к новым производным пиперидина формулы I:
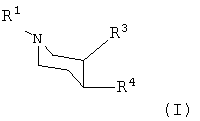
или к любым из его изомеров, или к любой смеси его изомеров, или к его фармацевтически приемлемой соли, где R1 представляет собой водород или алкил; R3 представляет собой -CH2-O-Rc; где Rc представляет собой алкил или циклоалкилалкил; R4 представляет собой
где Ra и Rb независимо друг от друга представляют собой галогено. Изобретение также относится к фармацевтической композиции и к применению соединений на основе формулы I, обладающих ингибирующей активностью обратного захвата нейромедиаторных моноаминов в центральной нервной системе. Технический результат - получение новых биологически активных соединений и фармацевтических композиций на их основе, обладающих ингибирующей активностью обратного захвата нейромедиаторных моноаминов в центральной нервной системе. 3 н. и 7 з.п. ф-лы, 1 табл.
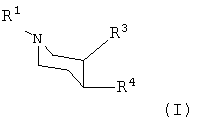
или любой из его изомеров, или любая смесь его изомеров, или его фармацевтически приемлемая соль,
где R1 представляет собой водород или алкил;
R3 представляет собой -CH2-O-Rc;
где Rc представляет собой алкил или циклоалкилалкил;
R4 представляет собой
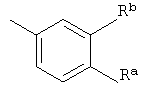
где Ra и Rb независимо друг от друга представляют собой галогено.
1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
1-метил-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидин;
3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
или любой из его изомеров, или любую смесь его изомеров, или его фармацевтически приемлемую соль.
(+)-цис-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-1-метил-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-1-метил-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-1-метил-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-1-метил-3-изобутоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-1-метил-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-цис-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-цис-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-3-этоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(+)-транс-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-3-метоксиметил-4-(3,4-дихлорфенил)-пиперидин;
(-)-транс-3-циклопропилметоксиметил-4-(3,4-дихлорфенил)-пиперидин;
или его фармацевтически приемлемую соль.
| СВЕКЛОКОПАТЕЛБ | 1928 |
|
SU20390A1 |
| WO 9851668 A1, 19.11.1998 | |||
| RU 99124412 A, 20.08.2001. | |||

