Изобретение относится к новым оксимным производным, являющимся ингибиторами обратного захвата моноаминных нейротрансмиттеров, а именно дофамина, серотонина и норадреналина. Настоящее изобретение, в частности, относится к новым оксимным производным, которые являются сильными ингибиторами обратного захвата дофамина, и в качестве таковых обладают выраженной антипаркинсонической, антидепрессивной, препятствующей ожирению, антинарколептической и препятствующей токсикомании активностью, и, в то же время, в слабой степени дают нежелательные побочные эффекты.
Дофамин высвобождается в синаптическую щель для стимуляции постсинаптических дофаминовых рецепторов. Удаление дофамина происходит в норме с помощью механизма обратного захвата в пресинаптические окончания. Путем торможения такого обратного захвата достигается усиление физиологической дофаминэргической активности. Можно предсказать, что соединения, способные тормозить обратный захват дофамина, будут полезны при лечении болезни Паркинсона, депрессии, кокаиновой наркомании, ожирения и нарколепсии.
Хорошо известным веществом, обладающим свойствами как сильного высвобождения дофамина, так и сильного торможения обратного захвата дофамина, является кокаин. Кокаин обладает различными фармакологическими свойствами, и в первую очередь действует как сильный стимулятор ЦНС и локальный анестетик. Эти эффекты сопровождаются высокой токсичностью и развитием зависимости [R. L. Clarke et al in Journal of Medicinal Chemistry 16 (11). 1261- 1267 (1973)] . Считается, что развитие зависимости связано с сочетанием сильной стимулирующей активности кокаина, короткого периода действия, быстрого проявления эффекта и сильным высвобождением дофамина. Полагают, что соединения, обладающие свойством в течение длительного времени селективно тормозить обратный захват дофамина и не обладающие свойством высвобождать дофамин, будут очень полезны в качестве антипаркинсонических, антидепрессивных, препятствующих ожирению и антинарколептических агентов нового типа. Более того, такие соединения будут очень полезны при лечении привыкания к чрезмерному употреблению лекарственных средств, особенно при лечении кокаиновой наркомании или злоупотребления.
В течение ряда лет было сделано много попыток оптимизации свойств кокаина. Было синтезировано много производных кокаина и их изомеров [R.L. Clarke et al in Journal of Medicinal Chemistry 16(11). 1261-1267 (1973), F. ivy Caroll et al in Journal of Medicinal Chemistry 34, 883-886 (1991)] . Многие из этих производных, особенно описанные в [R.L. Clarke et al in Journal of Medicinal Chemistry 16 (11I). 1261-1267 (1973)], являются сильными стимулирующими соединениями, и было обнаружено, что они являются и сильными ингибиторами обратного захвата дофамина. Однако, не было обнаружено ни одного производного кокаина, синтезированного до настоящего момента, которое не давало бы нежелательных побочных эффектов. Таким образом, необходимость в новых ингибиторах обратного захвата дофамина существует до сих пор.
Некоторые из описанных здесь соединений обладают также свойством сильного торможения обратного захвата серотонина (5-гидрокси-триптамин, 5НТ) в сочетании со свойством торможения обратного захвата дофамина.
Обычно используемые при антидепрессивной терапии фармацевтические средства являются ингибиторами обратного захвата норадреналина (Дезипрамин, Нортриптилин и Протриптилин) или смешанными ингибиторами обратного захвата серотонина и обратного захвата норадреналина (Имипрамин и Амитриптилин). Серьезным недостатком этих агентов является позднее проявление эффекта (несколько недель). Предполагается, что смешанные ингибиторы обратного захвата серотонина и обратного захвата норадреналина могли бы дать лучшее антидепрессантное действие с быстрым проявлением эффекта.
Задачей данного изобретения является создание новых оксимных производных, обладающих анти-паркинсонической, анти-депрессивной, препятствующей ожирению, анти-нарколепсической и препятствующей токсикомании активностью, способа их получения, а также фармацевтической композиции и веществ для производства лекарств на их основе и способа лечения паркинсонизма, депрессии, ожирения, нарколепсии и токсикомании с помощью предложенных соединений.
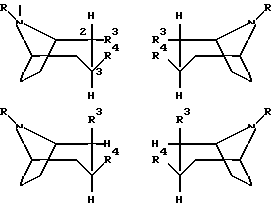
Согласно настоящему изобретению предложено соединение формулы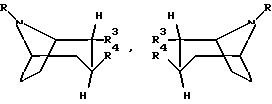
любая их смесь или их фармацевтически приемлемая соль;
где R обозначает водород или алкил;
R3 представляет собой CH=NOR',
где R' обозначает водород или алкенил, алкинил, циклоалкил, циклоалкилалкил, арил или алкил, возможно замещенный COOH, COO- алкилом или фенилом;
R4 обозначает фенил, который может быть замещен один или несколько раз заместителями, выбранными из группы, состоящей из галогена, CF3, CN, NO2 или алкила; бензил; или нафтил.
Предпочтительными являются следующие соединения:
3-(3,4-Дихлорфенил)тропан-2-альдоксим,
3-(3,4-Дихлорфенил)тропан-2-О-метил-альдоксим,
3-(3,4-Дихлорфенил)тропан-2-О-бензил-альдоксим,
3-(3,4-Дихлорфенил)тропан-2-О-этоксикарбонилметил-альдоксим,
3-(3,4-Дихлорфенил)тропан-2-О-метоксикарбонилметил-альдоксим,
3-(3,4-Дихлорфенил)тропан-2-O-(1-этоксикарбонил-1,1-диметил-метил)-альдоксим,
3-(3,4-Дихлорфенил)тропан-2-O-карбоксиметил-2-альдоксим,
N-норметил-3-(3,4-дихлорфенил)тропан-2-O-метил-альдоксим или
N-норметил-3-(3,4-дихлорфенил)тропан-2-0-бензил-альдоксим, или их фармацевтически приемлемые соли присоединения.
Также предложены следующие соединения:
(1R, 2R, 3S)-3-(3,4-Дихлорфенил)тропан-2-альдоксим,
(1R, 2R, 3S)-3-(3,4-Дихлорфенил)тропан-2-О-бензил-альдоксим,
(1R, 2R, 3S)-3-(3,4-Дихлорфенил)тропан-2-O-этоксикарбонилметил-альдоксим,
(1R, 2R, 3S)-3-(3,4-Дихлорфенил)тропан-2-О-метоксикарбонилметил-альдоксим,
(1R, 2R, 3S)-3-(3,4-Дихлорфенил)тропан-2-О-(1- этоксикарбонил-1,1-диметил-метил)-альдоксим,
(1R, 2R, 3S)-3-(3,4-Дихлорфенил)тропан-2-O-карбоксиметил-2-альдоксим,
(1R, 2R, 3S)-N-норметил-3-(3,4-дихлорфенил)тропан-2-О-метил-альдоксим или
(1R, 2R, 3S)-N-норметил-3-(3,4-дихлорфенил)тропан-2-О-бензил-альдоксим, или их фармацевтически приемлемые соли присоединения.
Предпочтительным является соединение:
(1R, 2R, 3S)-3-(3,4-Дихлорфенил)тропан-2-О-метил-альдоксим или его фармацевтически приемлемая соль присоединения.
Также предпочтительными являются следующие соединения:
анти-изомер (1R, 2R, 3S)-3-(3,4-Дихлорфенил)тропан-2-O-метил-альдоксима,
син-изомер (1R, 2R, 3S)-3-(3,4-Дихлорфенил)тропан-2-O-метил-альдоксима, их смесь или их фармацевтически приемлемая соль присоединения.
Согласно настоящему изобретению предложена также фармацевтическая композиция для ингибирования обратного захвата моноаминных нейротрансмиттеров, содержащая активный ингредиент и по меньшей мере один фармацевтически приемлемый носитель или разбавитель, которая в качестве активного ингредиента содержит эффективное количество упомянутого выше соединения или его фармацевтически приемлемой соли присоединения.
Также предложены вещества для производства лекарств для лечения расстройства или болезни организма животного, включая человека, на которые оказывает влияние торможение обратного захвата моноаминных нейротрансмиттеров и дофамина в центральной нервной системе, содержащее упомянутые выше соединения, а также вещество для производства лекарства, оказывающего влияние на торможение обратного захвата дофамина при лечении паркинсонизма, депрессии, ожирения, нарколепсии и токсикомании, которое представляет собой соединение упомянутое выше.
Согласно изобретению предложен также способ лечения расстройства или болезни организма животного, включая человека, на которые оказывает влияние торможение обратного захвата дофамина, содержащий стадию, на которой в нуждающийся в таком лечении организм вводят лекарство, при этом в организм вводят предложенное соединение в эффективном количестве.
Предпочтительно когда воздействие оказывают на торможение обратного захвата дофамина при лечении паркинсонизма, депрессии, ожирения, нарколепсии или привыкания к чрезмерному употреблению лекарственных средств и/или при их злоупотреблении.
Также предлагается способ получения предложенного выше соединения, содержащий стадию, на которой соединение формулы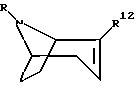
его энантиомер или их смесь;
где R обозначает водород или алкил;
R12 является эфиром карбоновой кислоты подвергают взаимодействию с соединением формулы
R4-A
где R4 обозначает фенил, который может быть замещен один или несколько раз заместителями, выбранными из группы, состоящей из галогена, CF3, CN, NO2 или алкила; бензил; или нафтил;
A - любой тип реакционноспособной группы, пригодной для генерации карбаниона как ее противоионной части, такой как Li, MgX, где X обозначает галоген, и CuLi, в реакции 1,4 присоединения, подобной реакции Михаэля,
и, если R12 является эфиром карбоновой кислоты, превращают полученное соединение в предложенное выше соединение, используя традиционные методы.
Примеры фармацевтически приемлемых аддитивных солей включают в себя соли, полученные добавлением неорганических и органических кислот, такие как гидрохлорид, гидробромид, фосфат, нитрат, перхлорат, сульфат, цитрат, лактат, тартрат, малеат, фумарат, манделат, бензоат, аскорбат, циннамат, бензенсульфонат, метансульфонат, стеарат, сукцинат, глутамат, гликоллат, толуол-p-сульфонат, формиат, малонат, нафтален-2-сульфонат, салицилат и ацетат. Такие соли образуют хорошо известными способами.
Другие кислоты, такие как щавелевая кислота, которые сами не являются фармацевтически приемлемыми, могут быть полезны при получении солей, используемых как промежуточные соединения в получении соединений по изобретению и их фармацевтически приемлемых солей с кислотами.
Галоген является фтором, хлором, бромом или иодом.
Алкил обозначает нормальную или разветвленную цепь атомов углерода в количестве от 1 до 6, включая, но не ограничиваясь метилом, этилом, пропилом, изопропилом, бутилом, изобутилом, трет-бутилом, пентилом и гексилом; причем метил, этил, пропил и изопропил являются предпочтительными группами.
Циклоалкил обозначает циклический алкил, содержащий от 3 до 7 атомов углерода, включая, но не ограничиваясь циклопропилом, циклобутилом, циклопентилом и циклогексилом.
Алкенил обозначает гриппу, содержащую от 2 до 6 атомов углерода, имеющую по меньшей мере одну двойную связь, например этенил, 1,2- или 2,3-пропенил, 1,2-, 2,3- или 3,4-бутенил.
Алкинил обозначает группу, содержащую от 2 до б атомов углерода, имеющую по меньшей мере одну тройную связь, например этинил, 2,3-пропинил, 2,3- или 3,4- бутинил.
Циклоалкилалкил обозначает определенный выше циклоалкил и определенный выше алкил, например циклопропилметил.
Алкокси обозначает O-алкил, в котором алкил определен выше.
Циклоалкокси обозначает O-циклоалкил, в котором циклоалкил определен выше.
Амино обозначает NH2 или NH-алкил, или N-(алкил)2, в котором алкил определен выше.
В качестве гетероарила подходит 5- или 6-членная гетероциклическая моноциклическая группа. Такие гетероарильные группы включают в себя, например, оксазол-2-ил, оксазол-4-ил, оксазол-5-ил, изоксазол-3-ил, изоксазол-4-ил, изоксазол-5-ил, тиазол-2-ил, тиазол-4-ил, тиазол-5-ил, изотиазол-3-ил, изотиазол-4-ил, изотиазол-5-ил, 1,2,4-оксадиазол- 3-ил, 1,2,4-оксадиазол-5-ил, 1,2,4-тиадиазол-3-ил, 1,2,4-тиадиазол-5-ил, 1,2,5-оксадиазол-3-ил, 1,2,5-оксадиазол-4-ил, 1,2,5-тиадиазол-3- ил, 1,2,5-тиадиазол-4-ил, 2-имидазолил, 4-имидазолил, 5-имидазолил, 2-пирролил, 3-пирролил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 2- пиридил, 3-пиридил, 4-пиридил.
Арил представляет собой ароматический углеводород, такой как фенил и нафтил.
Соединения по изобретению могут существовать как в форме несольватированной, так и в сольватированной фармацевтически приемлемыми растворителями, такими как вода, этанол и подобные. В общем, сольватированные формы рассматриваются как эквиваленты несольватированных для целей данного изобретения.
Предложенные соединения содержат несколько хиральных центров и существуют в форме изомеров (т.е. энантиомеров). Данное изобретение охватывает все такие изомеры и любые их смеси, включая рацемические.
Некоторые соединения по изобретению существуют в (+) и (-) формах, а также в рацемических формах. Рацемические формы могут быть разделены на оптические антиподы известными методами, например путем отделения диастереоизомерных солей с оптически активной кислотой и выделения в свободном состоянии оптически активных аминных соединений путем обработки основанием. Другой метод разделения рацематов на оптически активные антиподы основан на хроматографии на оптически активных матрицах. Рацемические соединения по изобретению, таким образом, могут быть разделены на их оптические антиподы, например путем фракционной кристаллизации d- и l-солей (тартраты, манделаты или камфор-сульфонаты). Соединения по изобретению также могут быть разделены путем образования диастереомерных амидов реакцией соединений по изобретению с оптически активными активированными карбоновыми кислотами, такими как полученные из (+) или (-) фенилаланина, (+) или (-) фенилглицина, (+) или (-) камфановой кислоты или путем образования диастереомерных карбаматов реакцией соединений по изобретению с оптически активным хлороформиатом и т.п.
Могут быть использованы дополнительные методы разделения оптических изомеров, известные и очевидные для специалистов [J.Jaques, A. Collet, and S. Wilen in "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, New York (1981)] .
Так как соединения по изобретению являются оксимами, они могут существовать в двух формах, син- и анти-форме, в зависимости от расположения заместителей относительно двойной связи -C=N-. Настоящее изобретение охватывает обе эти формы данных соединений, а также их смесь. Анти-син изомеризацию катализируют кислоты.
Соединения по изобретению могут быть получены многочисленными путями. Соединения по изобретению и их фармацевтически приемлемые производные могут, таким образом, быть получены любым известным из уровня техники способом получения соединений аналогичной структуры, в частности как показано в нижеследующих иллюстративных примерах.
Следующая схема иллюстрирует один способ, которым могут быть получены соединения по изобретению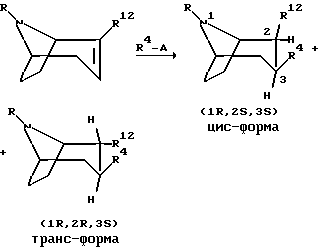
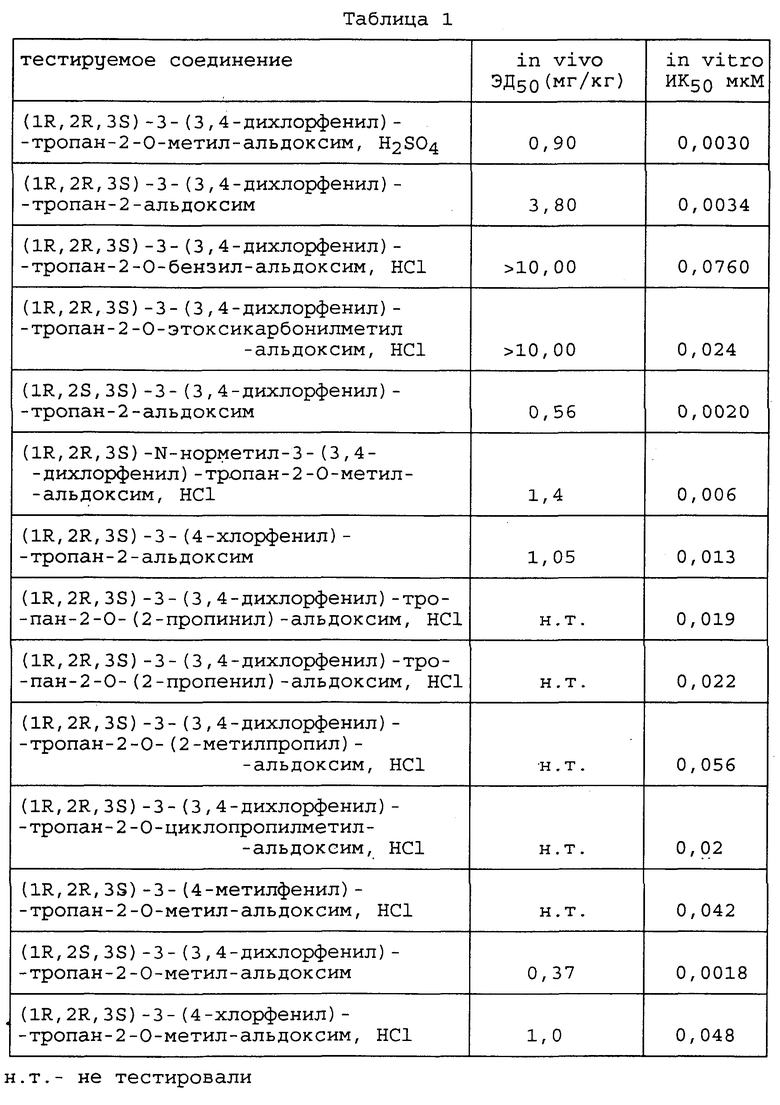
В приведенной выше реакционной схеме A обозначает любой тип реакционноспособной группы, пригодной для генерации карбаниона как ее противоионной части, например Li, MgX, где X обозначает галоген, и CuLi, в реакции 1,4 присоединения, подобной реакции Михаэля, R12 является эфиром карбоновой кислоты, таким, например, как COO-метил, COO-этил, COO-изопропил, или R12 имеет значения, определенный выше для R3, и R и R4 определены выше.
Обе формы, цис- и транс-, а также их смесь могут быть получены путем вышеописанной взаимодействия соединения формулы (i) с соединением R4-A, но почти во всех полученных соединениях заместитель R4 обнаруживается в экваториальном положении.
Изомеризация цисизомера в трансизомер может быть проведена в сильном основании, таком как алкоголят.
Энантиомеры цис- и транс- соединений на вышеприведенной схеме могут быть получены с использованием того же способа и энантиомера соединения формулы (i), имеющего формулу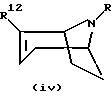
в качестве исходного вещества.
Рацемические смеси цис- и транс- соединений, соответственно, могут быть получены с использованием смеси соединений формулы (i) и (iv) в качестве исходного вещества.
Соединения, полученные описанным выше способом, где R12 обозначает эфир карбоновой кислоты, могут быть превращены в соединения по изобретению с использованием традиционных способов. Такие способы включают в себя восстановление 2-карбонового эфира в 2-гидроксиметил с последующим окислением до соответствующего 2-альдегида. Оксимы по изобретению могут затем быть получены путем взаимодействия 2-альдегидного соединения с гидроксиламин-производными NH2-OR', где R' такое, как определено выше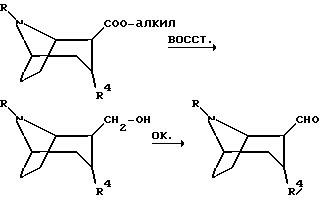
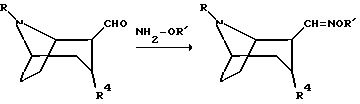
Соединение по изобретению может быть превращено в другое соединение по изобретению с использованием традиционных способов.
Исходные вещества для описанных здесь способов известны или могут быть получены известными способами из имеющихся в продаже веществ.
Исходные вещества формулы (i), где R12 является эфиром карбоновой кислоты, могут быть, таким образом, получены из кокаина с использованием традиционных способов, например как описано ниже в примерах.
Исходные вещества формулы (i), где R12 является оксимом, могут быть получены с использованием такого же способа, как описанный выше способ получения оксимов по изобретению, а также известный способ [EP-A2-316718].
Продукты описанных здесь реакций выделяют традиционными способами, такими как экстракция, кристаллизация, дистилляция, хроматография и подобные.
Фармацевтические композиции.
Хотя соединение по изобретению может быть использовано в терапевтических целях и в виде необработанного химического вещества, все же предпочтительно использовать его в составе фармацевтической композиции.
Таким образом, согласно данному изобретению предложены фармацевтические композиции, содержащие соединение по изобретению или его фармацевтически приемлемые соли или производные вместе с одним или несколькими фармацевтически приемлемыми носителями и, возможно, другими терапевтическими и/или профилактическими ингредиентами. Носитель (и) должен быть "приемлемым" в смысле совместимости с остальными ингредиентами композиции и безвредности для реципиента.
Фармацевтические композиции включают в себя композиции, подходящие для перорального, ректального, интерназального, местного (включая трансбуккальное и подъязычное), вагинального или парэнтерального (включая внутримышечное, подкожное и внутривенное) введения или формы, подходящие для введения ингаляцией или вдуванием.
Соединения по изобретению вместе с адъювантами, носителями или разбавителями могут войти в состав фармацевтических композиций и их единичных доз и в такой форме могут быть использованы в виде твердых веществ, таких как таблетки или заполненные капсулы, жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или наполненные ими капсулы, предназначенные для перорального введения, в виде суппозиториев для ректального введения или в форме стерильных растворов для инъекций для парэнтерального (в том числе подкожного) введения. Такие фармацевтические композиции и их единичные дозовые формы могут включать в себя традиционные ингредиенты в традиционных пропорциях, содержащие или не содержащие дополнительные активные соединения или действующие начала, такие единичные дозовые формы могут содержать любое подходящее эффективное количество активного ингредиента, соответствующее предполагаемой суточной дозе. Композиции, содержащие 10 мг активного ингредиента или, в более широком диапазоне, от 0,1 до 100 мг активного ингредиента на таблетку, соответственно являются подходящими представителями единичных дозовых форм.
Соединения по изобретению могут быть введены в виде большого числа пероральных и парэнтеральных дозовых форм. Специалисту очевидно, что следующие дозовые формы могут содержать в качестве активного ингредиента либо соединение по изобретению, либо его фармацевтически приемлемую соль.
Для получения фармацевтических композиций из соединений по изобретению фармацевтически приемлемые носители должны быть либо жидкостями, либо твердыми веществами. Твердые формы препаратов включают в себя порошки, таблетки, пилюли, капсулы, лепешки, суппозитории и дисперсионные гранулы. Твердым носителем может быть одно или несколько веществ, которые также могут являться разбавителями, ароматизаторами, растворителями, любрикантами, суспендирующими агентами, связующими веществами, консервантами, дезинтегрирующими агентами для таблеток или материалом для капсулирования.
В порошковых препаратах носитель является тонко измельченным твердым веществом, которое смешано с тонко измельченным активным компонентом.
В таблетках активный компонент в подходящей пропорции смешан с носителем, имеющим необходимую связывающую емкость, и смеси придан вид таблетки желаемой формы и размера.
Порошки и таблетки предпочтительно содержат от 5 или 10 до 70% активного ингредиента. Подходящими носителями являются карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, воск с низкой температурой плавления, масло какао и т.п. Термин "препарат" подразумевает композицию активного ингредиента с материалом для капсулирования, который является носителем, формирующим капсулу, в которую заключен соединенный или не соединенный с носителями активный ингредиент, окруженный носителем, который, таким образом, находится в ассоциации с ним. Аналогичный состав имеют лепешки и пастилки. Таблетки, порошки, капсулы, пилюли, пастилки и лепешки могут быть использованы как твердые формы, подходящие для перорального введения.
Для получения суппозиториев плавят воск, имеющий низкую температуру плавления, такой как смесь глицеридов жирных кислот или масло какао, и равномерно диспергируют в нем с помощью перемешивания активный ингредиент. Расплавленную гомогенную смесь затем выливают в формы традиционного размера, позволяют остыть и затвердеть.
Препараты для вагинального введения могут представлять собой пессарии, тампоны, кремы, гели, пасты, пенки или спрэи, содержащие дополнительно к активному ингредиенту известные из уровня техники подходящие носители.
Жидкие формы включают в себя растворы, суспензии, эмульсии, например водные или водо-пропилен гликолевые растворы. Например, жидкие препараты для парэнтеральных инъекций могут быть приготовлены в виде растворов в водном растворе полиэтилен гликоля.
Соединения по изобретению, таким образом, могут быть приготовлены для парэнтерального введения (например инъекций, в частности инъекций из ампул или непрерывных инфузий) и могут представлять собой в единичной дозовой форме ампулы, предварительно наполненные шприцы, инфузий небольшого объема или находиться в много дозовых контейнерах с добавкой консерванта. Композиции могут иметь форму суспензий, растворов или эмульсий в масле или водных носителях, и могут содержать такие агенты, как суспендирующие, стабилизирующие и/или диспергирующие агенты. В альтернативном случае активный ингредиент может находиться в форме порошка, полученного асептическим выделением твердого вещества или путем лиофилизации из раствора, подлежащего перед использованием соединению с подходящим носителем, например стерильной апирогенной водой.
Водные растворы, подходящие для перорального введения могут быть получены путем растворения активного компонента в воде с добавлением соответствующего красителя, ароматизатора, стабилизатора или загустителя по желанию.
Водные суспензии, подходящие для перорального введения могут быть получены путем диспергирования тонко измельченного активного ингредиента в воде с вязким материалом, таким как природные или синтетические смолы, полимеры, метилцеллюлоза, натрий карбоксиметилцеллюлоза или другие известные суспендирующие агенты.
В жидкие препараты для перорального введения также могут быть превращены, незадолго до использования, и препараты, находящиеся в твердой форме. Такие жидкие препараты представляют собой растворы, суспензии и эмульсии. Эти препараты могут содержать дополнительно к активному ингредиенту красители, ароматизаторы, стабилизаторы, буферы, искусственные или натуральные подслащивающие вещества, дисперсанты, загустители, растворяющие агенты и т.п.
Для местного применения на эпидермис соединения по изобретению могут быть приготовлены в виде мазей, кремов, лосьонов или трансдермальных пластырей. Мази и кремы могут быть, напри мер, приготовлены на водной или масляной основе с добавлением подходящих сгущающих и/или желирующих агентов. Лосьоны могут быть получены на водной или масляной основе и обычно содержат один или несколько эмульгирующих агентов, стабилизирующих агентов, диспергирующих агентов, суспендирующих агентов, загущающих агентов или красителей.
Препараты для местного применения в полости рта включают в себя пастилки, содержащие активный ингредиент на инертной основе, такой как желатин и глицерин или сахароза и гуммиарабик. Жидкости для полоскания рта содержат активный ингредиент на подходящем жидком носителе.
Растворы и суспензии вводят непосредственно в носовую полость традиционными способами, например капельницей, пипеткой или спрэем. Эти препараты могут готовиться в единичной или многодозовой форме. В последнем случае капельницу или пипетку можно использовать так, чтобы пациент вводил соответствующий заранее определенный объем раствора или суспензии. В случае спрэя можно предусмотреть насос с дозированным распылением.
Введение в респираторный тракт можно обеспечить с помощью аэрозоля, в котором активный ингредиент помещен в баллон, находящийся под давлением, снабженный подходящим пропеллантом, таким как хлорфторуглерод (CFC), например дихлорфторметан, трихлорфторметан или дихлортетрафторэтан, диоксид углерода или другой подходящий газ. Дозу лекарства можно контролировать, снабдив баллон мерным клапаном.
В альтернативном случае активный ингредиент может находится в форме сухого порошка, например порошковой смеси соединения по изобретению с подходящим порошковым носителем, таким как лактоза, крахмал, производные крахмала, такие как гидроксипропилметил целлюлоза и поливинилпирролидон (PVP). Обычно порошковый носитель превращается в носовой полости в гель. Порошковый препарат может находиться в единичной дозовой форме, например капсуле или картридже, например из желатина, пузырька, из которого порошок может быть введен при помощи ингалятора.
В препаратах, предназначенных для введения в респираторный тракт, включая интраназальные препараты, соединения обычно представлены частицами малого размера, например порядка 5 микрон или меньше. Частицы такого размера могут быть получены известными способами, например путем микронизации.
При желании могут быть использованы препараты, обеспечивающие замедленное высвобождение активного ингредиента.
Фармацевтические препараты предпочтительно находятся в единичной дозовой форме. В такой форме препарат разделен на единичные дозы, содержащие подходящее количество активного ингредиента. Единичные дозовые формы могут представлять собой упакованный препарат, упаковка содержит дискретное количество препарата, например упакованные таблетки, капсулы, порошки в виалах и ампулах. Единичная дозовая форма сама может быть капсулой, таблеткой, лепешкой или пастилкой, или может являться соответствующим числом любой из этих упаковок.
Таблетки или капсулы для перорального введения и жидкости для внутривенного введения являются предпочтительными композициями.
Способ лечения.
Соединения по изобретению очень полезны для лечения паркинсонизма, депрессии, ожирения, нарколепсии и токсикомании благодаря их свойству сильно тормозить захват дофамина и наблюдающемуся при этом низкому уровню нежелательных побочных эффектов. Эти свойства делают соединения по изобретению очень полезными для лечения паркинсонизма, депрессии, ожирения, нарколепсии и токсикомании, а также других расстройств, на которые может оказать влияние свойство соединений по изобретению тормозить захват дофамина. Соединениями по изобретению можно воздействовать на организм животного, включая человека, который нуждается в лечении, уменьшении или устранении показаний, связанных со свойством торможения захвата дофамина или на которые можно влиять путем торможения захвата дофамина. Среди них особенно важны паркинсонизм, депрессия, ожирение, нарколепсия и токсикомания. Подходящей суточной дозой является доза 0,1-500 мг, особенно 10-70 мг, которую вводят в один прием или за два раза в зависимости от конкретного способа введения, формы введения, показаний, по которым производится введение, пациента, веса его тела, а также практических навыков и опыта занимающегося лечением врача или ветеринара.
Следующие примеры иллюстрируют изобретение, не ограничивая его сферу.
Пример 1.
Метиловый эфир(-)-ангидроэкгонина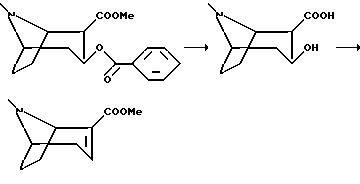
Гидрохлорид (1R, 2R, 3S)-2-карбометокси-3-бензокситропана (100 г, 0,29 моль) кипятили с обратным холодильником в 1000 мл 1М соляной кислоты в течение 18 ч и затем раствор остужали на льду. Фильтрацией собирали бензойную кислоту и концентрировали фильтрат в вакууме. Растирали остаток с этанолом и фильтровали с получением гидрохлорида (1R,2R,3S)-3-гидрокси-тропан-2-карбоксилата в виде белого кристаллического соединения, которое без дальнейшей очистки высушивали и кипятили с обратным холодильником в оксихлориде фосфора (50 мл) в течение 2 ч. Раствор концентрировали в вакууме и медленно при охлаждении на льду добавляли абсолютный метанол (150 мл). Раствор перемешивали при комнатной температуре в течение 16 ч и концентрировали в вакууме. Остаток охлаждали на льду, подщелачивали добавлением раствора гидроксида натрия (10 М, около 100 мл) и 5 раз экстрагировали диэтиловым эфиром. Объединенные органические фазы высушивали и концентрировали в вакууме с получением масла, которое подвергали дистилляции в вакууме (70-74oC, 1 мбар) с получением указанного в заголовке соединения в виде прозрачного масла.
Пример 2.
(1R,2S,3S)-2-карбометокси-3-(4-фторфенил)тропан и
(1R,2R,3S)-2-карбометокси-3-(4-фторфенил)тропан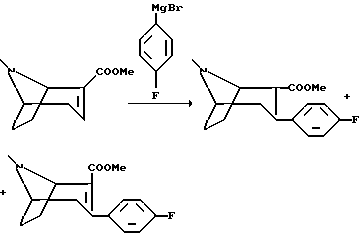
Реактив Гриньяра готовили в трехгорлой реакционной колбе, снабженной механической мешалкой, интенсивным холодильником и воронкой для уравновешивания давления, используя 4-бром-фторбензол (27,5 мл, 250 ммоль), источники магния (6,3 г, 260 мл) в 250 мл абсолютного диэтилового эфира. Раствор реактива Гриньяра охлаждали до -20oC и в течение 1/2 ч добавляли раствор метилового эфира (-)-ангидроэкгонина (21,7 г, 120 мл) в 100 мл абсолютного диэтилового эфира. Реакционную смесь перемешивали в течение 1 ч при -20oC и останавливали реакцию одним из двух способов:
1) Реакционную смесь перемешивали в 250 мл дробленого льда и подкисляли водную фазу путем добавления около 100 мл 4М соляной кислоты. Органическую фазу сливали и промывали водную фазу 100 мл диэтилового эфира. Водную фазу подщелачивали путем добавления 25%-ного раствора гидроксида аммония, затем насыщали хлоридом натрия, и, наконец, экстрагировали трижды диэтиловым эфиром. Объединенную органическую фазу высушивали и концентрировали в вакууме с получением масла, которое подвергали дистилляции в вакууме (150-160oC, 2 мбар). Этим способом получают смесь двух стереоизомеров (2S/2R - 1/3), которую разделяют колоночной хроматографией с использованием смеси диэтилового эфира и пентана (1+1) +1% триэтиламина в качестве элюента. Неочищенные продукты растирают в пентане с получением (1R,2S,3S)-2-карбометокси-3-(4-фторфенил)тропана в виде белых кристаллов, т.пл. 91- 92oC и (1R,2R,3S)-2-карбометокси-3-(4-фтор-фенил)тропана в виде белых кристаллов, т.пл. 65-66oC.
2) Реакционную смесь охлаждают до -78oC и в течение 10 мин добавляют раствор трифторуксусной кислоты (20 мл, 250 ммоль) в 50 мл диэтилового эфира. Охлаждающую баню убирают и когда температура смеси достигнет 0oC, ее выливают с перемешиванием в 700 мл воды. pH водной фазы подводят до pH 1 путем добавления концентрированной соляной кислоты, а затем проводят водную обработку и очищают как описано выше. Этим способом получают смесь двух стереоизомеров (2S/2R-2/1).
Аналогичным способом могут быть получены следующие соединения:
(1R,2R,3S)-2-карбометокси-3-бензилтропан и
(1R, 2S,3S)-2-карбометокси-3-бензилтропан, способ 2, только (1R,2S,3S)-2-карбометокси-3-бензилтропан был получен без примесей других изомеров в виде масла, которое кристаллизовалось с течением времени, т.пл. 53-54oC.
(1R, 2R,3S)-2-карбометокси-3-бензилтропан был получен путем изомеризации смеси, как описано в примере 3.
(1R,2R,3S)-2- карбометокси-3-(4-хлорфенил)тропан и
(1R, 2S, 3S)-2-карбометокси-3-(4-хлорфенил)тропан, способ 2. Эти два изомера не были выделены, но смесь была изомеризована как описано в примере 3.
(1R, 2R, 3S)-2-карбометокси-3-(4-хлорфенил)тропан,
(1R, 2S, 3S)-2-карбометокси-3-(4-хлорфенил)тропан,
(1S, 2S, 3R)-2-карбометокси-3-(4-хлорфенил)тропан и
(1S, 2R, 3R)-2-карбометокси-3-(4-хлорфенил) тропан, способ 2. Эти две пары энантиомерных пар не были выделены, но смесь была изомеризована как описано в примере 3.
(1R, 2R, 3S)-2-карбометокси-3-(4-метилфенил) тропан и
(1R, 2S, 3S)-2-карбометокси-3-(4-метилфенил) тропан, способ 2. Эти два изомера не были выделены, но смесь была изомеризована как описано в примере 3.
(1R, 2S, 3S)-2-карбометокси-3-(2-нафтил) тропан и
(1R, 2R, 3S)-2-карбометокси-3-(2-нафтил)тропан, способ 2. Реактив Гиньяра получили, добавив смесь одного эквивалента 2-бромнафталина и 1,2-дибромэтана в диэтиловом эфире к кипятящейся с обратным холодильником суспензии двух эквивалентов магния. Оба продукта были получены в виде белых кристаллов, т.пл. 79-80oC и т.пл. 86-87oC, соответственно.
(1R, 2R, 3S)-2-карбометокси-3-(1-нафтил)тропан и (1R,2S,3S)-2-карбометокси-3-(1-нафтил)тропан в виде гидрохлорида, способ 2. Реактив Гиньяра получили, добавив смесь одного эквивалента 1-бромнафталина и 1,2-дибромэтана в диэтиловом эфире к кипятящейся с обратным холодильником суспензии двух эквивалентов магния. Указанные в заголовке соединения были выделены в виде, соответственно, белых кристаллов, т.пл. 79-80oC, и аморфного вещества.
(1R, 2S, 3S)-2-карбометокси-3-(3,4-дихлорфенил)тропан и (1R, 2R, 3S)-2-карбометокси-3-(3,4-дихлорфенил)тропан, способ 2. Оба продукта были получены в виде белых кристаллов, т.пл. 69-70 С и т.пл. 61-63oC, соответственно.
Рацемическая смесь (1R, 2S, 3S)-2-карбометокси-3-(3,4-дихлорфенил)тропана и его энантиомера (1S,2S,3R)-2-карбометокси-3-(3,4-дихлорфенил)тропана была получена с использованием в качестве исходного вещества метилового эфира (+-)-ангидроэкгонина, способ 2, с последующей изомеризацией как описано в примере 3.
(1S, 2S, 3R)-2-карбометокси-3-(3,4-дихлорфенил)тропан был получен с использованием способа 2. Это соединение не было выделено, но было изомеризовано как описано в примере 3.
(1R, 2S, 3S)-2-карбометокси-3-(4-фенилфенил) тропан и (1R, 2R, 3S)-2-карбометокси-3-(4-фенилфенил)тропан, способ 2. Оба продукта были получены в виде белых кристаллов, т.пл. 130-132oC и т.пл. 95-96oC, соответственно.
(1R, 2S, 3S)-2-карбометокси-3-(4-трет-бутил-фенил)тропан и (1R, 2R, 3S)-2-карбометокси-3-(4-трет-бутил-фенил)тропан, способ 2. Оба продукта были получены в виде белых кристаллов, т.пл. 84-85oC и т.пл. 83-84oC, соответственно.
Пример 3.
Гидрохлорид (1R,2R,3S)-2-карбометокси-3-бензилтропана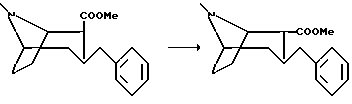
К раствору (1R,2S,3S)-2-карбометокси-3-бензилтропана (5,6 г, 20,5 ммоль) в абсолютном метаноле (100 мл) добавили раствор метанолата натрия в метаноле (2 М, 2 мл) и смесь кипятили с обратным холодильником в течение 16 ч. Концентрировали реакционную смесь в вакууме и остаток растворили в диэтиловом эфире и промыли водой. Органическую фазу высушивали и концентрировали в вакууме. Неочищенный продукт очищали с помощью колоночной хроматографии, используя смесь диэтилового эфира и пентана (1+1) + 1% триэтиламина в качестве элюента, с получением (1R, 2R, 3S)-2-карбометокси-3-бензилтропана в виде масла. После растворения этого продукта в диэтиловом эфире и последующего добавления раствора соляной кислоты в диэтиловом эфире указанное в заголовке соединение выпадало в осадок в виде белых кристаллов, т.пл. 188-190oC.
Пример 4.
2-Карбометокси-3-тропанон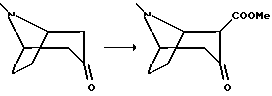
К суспензии гидрида натрия (3,2 г 80%, 107 ммоль, предварительно промытого в циклогексане) и диметилкарбоната (9,13 мл, 108 ммоль) в абсолютном циклогексане нагревали до температуры флегмообразования, в течение 15 мин добавляли раствор (+-)-3-тропанона (6,9 г, 50 ммоль) в 50 мл абсолютного циклогексана. Если выделения водорода не наблюдалось, добавляли 0,2 мл метанола. Реакционную смесь перемешивали в течение ночи при кипячении с обратным холодильником и после охлаждения до комнатной температуры осторожно добавляли 75 мл воды. К водной фазе добавляли 40 г хлорида аммония и полученную смесь 8 раз экстрагировали метилен хлоридом. Объединенные метиленхлоридные органические фазы высушивали и концентрировали в вакууме, затем неочищенный продукт подвергали колоночной хроматографии, используя метилен хлорид с повышенным (до 10%) содержанием метанола в качестве элюента. Фракции, содержащие указанный продукт концентрировли в вакууме и образующееся масло подвергали дистилляции kugelrohr (1 мбар, 120oC), с получением указанного в заголовке соединения в виде оранжевых кристаллов, т.пл. 104-107oC.
Пример 5.
Гидрохлорид 2-карбометокси-3-гидрокси-тропана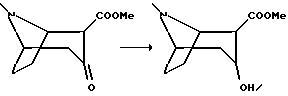
К раствору 2-карбометокси-3-тропанона, полученному в примере 4 (17 г, 85 ммоль), в 750 мл метанола, охлажденного до -35oC, добавили борогидрид натрия (17 г, 450 ммоль) и перемешивали смесь в течение 4 ч. Охлажденный раствор нейтрализовали медленным добавлением концентрированной соляной кислоты (40 мл) и смесь концентрировали в вакууме. Добавили 400 мл воды и подвели pH до 3 путем добавления концентрированной соляной кислоты. После того, как водную фазу трижды промыли диэтиловым эфиром подвели pH до 11 путем добавления концентрированного гидроксида аммония и трижды экстрагировали водную фазу метилен хлоридом. После концентрации в вакууме получили масло, которое растворили в этаноле, затем добавили концентрированную соляную кислоту, после чего концентрировали в вакууме. Остаток подвергли сушке вымораживанием с получением указанного в заголовке соединения в виде аморфного вещества.
(1S) - карбометокси-3-гидрокси-тропан в виде аморфного твердого вещества был получен аналогичным образом, при использовании в качестве исходного вещества (1S)-2-карбометокси-3-тропанона, полученного разделением известным [J.Med.Chem.,37, 2007(1994)] способом соединения, полученного в примере 4.
Пример 6.
Метиловый эфир (1R)-ангидроэкгонина.
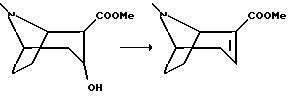
Смесь гидрохлорида 2-карбометокси-3-гидрокси-тропана, полученного в примере 5, (0,5 г, 2,1 ммоль) и тионил хлорида (0,4 мл, 5,3 ммоль) перемешивали при 60oC в течение 2 ч с получением прозрачного раствора. После охлаждения до комнатной температуры добавляли дробленый лед и подводили pH до 11 путем добавления концентрированного гидроксида аммония. Смесь экстрагировали дважды метиленхлоридом и удаляли растворитель в вакууме с получением указанного в заголовке соединения в виде масла, которое подвергали дистилляции при 1 мбар, 70-85oC.
Метиловый эфир (1S) -ангидроэкгонина в виде масла был получен аналогичным образом с использованием в качестве исходного вещества (1S)-кapбoмeтoкcи-3-гидpoкcи-тpoпaнa, полученного в примере 5.
Пример 7.
(1R, 2R, 3S)-N-норметил-2-карбометокси-3-(3,4-дихлорфенил)тропан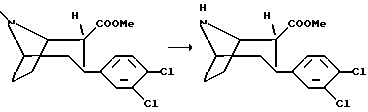
Смесь (1R, 2R, 3S)-2-карбометокси-3-(3,4-дихлорфенил) тропана (8,7 г, 27 ммоль) и 2,2,2-трихлорэтилового эфира хлормуравьиной кислоты (14,6 мл, 106 ммоль) в сухом толуоле (100 мл) кипятят с обратным холодильником в течение 18 ч. Реакционную смесь концентрируют в вакууме и к остатку добавляют метилен хлорид, который последовательно был промыт водой. Органическую фазу высушивали и концентрировали в вакууме. Остаток растворяли в 75%-ной водной уксусной кислоте (60 мл) и добавили к реакционной смеси цинковую пыль (8,7 г), после чего реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Добавили концентрированный гидроксид аммония (pH>7) и экстрагировали смесь дважды диэтиловым эфиром. Объединенную органическую фазу высушивали и концентрировали в вакууме с получением указанного в заголовке соединения в виде масла, которое использовали без дальнейшей очистки.
Пример 8.
(1R, 2R, 3S)-N-норметил-N-(трет-бутоксикарбонил)-2-карбометокси-3-(3,4-дихлорфенил)тропан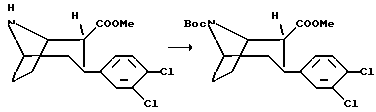
Раствор (1R, 2R, 3S)-N-норметил-2-карбометокси-3-(3,4-дихлорфенил)тропан (7 г, 22,3 ммоль) и ди-трет-бутил-дикарбонат (7,7 мл, 33,6 ммоль) в сухом тетрагидрофуране (50 мл) перемешивают при комнатной температуре в течение 1 ч. Реакцию останавливают добавлением льда (100 мл) и экстрагируют смесь дважды диэтиловым эфиром, высушивают и концентрируют в вакууме с получением указанного в заголовке соединения в виде масла, которое использовали без дальнейшей очистки.
Пример 9.
(1R, 2S, 3S)-2-гидроксиметил-3-(4-фторфенил)тропан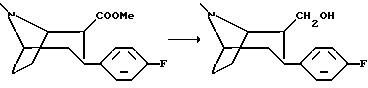
К суспензии литий алюминий гидрида (0,8 г, 21 ммоль) в диэтиловом эфире (30 мл) при комнатной температуре медленно добавляли раствор (1R, 2S, 3S)-2-карбометокси-3-(4-фторфенил) тропана (5 г, 18 ммоль) в 100 мл диэтилового эфира. Реакционную смесь перемешивали в течение 10 мин и останавливали реакцию путем добавления 0,8 мл воды, 0,8 мл гидроксида натрия (15%) и 2 мл воды. Соли алюминия удаляли путем фильтрации, а растворитель удаляли в вакууме с получением масла. Указанное в заголовке соединение в виде белых кристаллов выпадало в осадок при растирании с пентаном, т.пл. 79-80oC.
Аналогичным образом могут быть получены следующие соединения:
(1R, 2R, 3S)-2-гидроксиметил-3-(4-фторфенил)тропан, белые кристаллы, т. пл. 169-170oC.
(1R, 2R, 3S)-2-гидроксиметил-3-(3,4-дихлорфенил)тропан, белые кристаллы, т.пл. 145-150oC.
(1R, 2R,3S)-N-норметил-N-(трет-бутоксикарбонил)-2-гидроксиметил-3-(3,4-дихлорфенил)тропан, масло.
(1R, 2S, 3S)-2-гидроксиметил-3-(3,4-дихлорфенил)тропан, белые кристаллы, т.пл. 83-89oC.
Рацемическая смесь (1R,2R,3S)-2-гидроксиметил-3-(3,4-дихлорфенил) тропана и его энантиомера, (1R, 2S, 3R)-2-гидроксиметил-3-(3,4-дихлорфенил)тропана, т. пл. 186-187oC. (1S,2S,3R)-2-гидроксиметил-3-(3,4-дихлорфенил)тропан, т.пл. 179-184oC.
(1R,2R,3S)-2-гидроксиметил-3-(4-хлорфенил)тропан, белые кристаллы, т.пл. 200-202oC.
Пример 10.
(1R,2R,3S)-2-формил-3-(3,4-дихлорфенил)тропан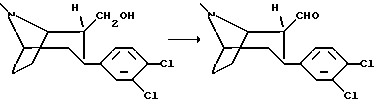
К раствору оксалилхлорида (2,3 мл) в абсолютном метилен хлориде (60 мл) при -60oC в течение 10 мин добавляли раствор диметилсульфоксида (4 мл) в абсолютном метилен хлориде (10 мл). Смесь перемешивали 10 мин и в течение 15 мин добавляли суспензию (1R, 2R, 3S)-2-гидроксиметил-3-(3,4-дихлорфенил) тропана (7 г, 23,3 ммоль) в абсолютном метилен хлориде (400 мл). Полученную смесь перемешивали 10 мин, затем добавляли триэтиламин (17 мл, 0,12 моль) и перемешивали еще 10 мин. Реакционной смеси позволяли нагреться до комнатной температуры и останавливали реакцию путем добавления воды (200 мл). Органическую фазу дважды промывали водой, высушивали и концентрировали в вакууме с получением указанного в заголовке соединения в виде белых кристаллов, т. пл. 131-135oC.
Аналогичным образом могут быть получены следующие соединения:
Рацемическая смесь (1R, 2R, 3S)-2-формил-3-(3,4-дихлорфенил)тропана и его энантиомера (1S, 2S, 3R)-2-формил-3-(3,4-дихлорфенил)тропана, которую использовали без дальнейшей очистки.
(1S,2S,3R)-2-формил-3-(3,4-дихлорфенил) тропан, который использовали без дальнейшей очистки.
(1R, 2R,3S)-2-формил-3-(4-хлорфенил)тропан, который использовали без дальнейшей очистки.
(1R, 2R, 3S)-N-норметил-N-(трет-бутоксикарбонил)-2-формил-3-(3,4-дихлорфенил)тропан, масло, которое использовали без дальнейшей очистки.
Следующие соединения были получены аналогичным образом, но промежуточный альдегид не выделяли из-за того, что могла произойти изомеризация из (1R, 2S, 3S) изомера в (1R, 2R, 3S) изомер. Вместо нагревания реакционной смеси до комнатной температуры и добавления воды для остановки реакции, был добавлен избыток (3 эквивалента) соли гидроксиламмония и оставляли смесь нагреваться до комнатной температуры, при которой ее перемешивали в течение 18 ч. Реакционную смесь дважды промывали водой и органическую фазу высушивали и концентрировали в вакууме.
(1R, 2S,3S)-3-(3,4-дихлорфенил)тропан-2-альдоксим. Был выделен путем фильтрации реакционной смеси в виде смеси син/анти изомеров 20%+80% (по результатам ЯМР) без установления идентичности изомеров. Продукт далее не очищали, белые кристаллы, т.пл. 248-251oC.
(1R, 2S, 3S)-3-(3,4-дихлорфенил) тропан-2-O-метил-альдоксим. После концентрации метиленхлоридной фазы получали масло, которое подвергали колоночной хроматографии с использованием смеси метилен хлорида, ацетона и метанола (4+1+1) в качестве элюента с получением указанного в заголовке соединения в виде белых кристаллов, т.пл. 98-100oC.
Гидрохлорид (1R,2S,3S)-3-(3,4-дихлорфенил)тропан-2-О-бензил-альдоксима. После концентрации метиленхлоридной фазы получали масло, которое подвергали колоночной хроматографии, используя этилацетат в качестве элюента и получая на выходе масло. Окончательно это масло растворяли в небольшом объеме диэтилового эфира и добавляли раствор соляной кислоты в диэтиловом эфире с получением указанного в заголовке соединения в виде белых кристаллов, т.пл. 196-197oC.
Пример 11.
(1R,2R,3S)-3-(3,4-дихлорфенил)тропан-2-альдоксим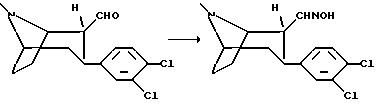
К раствору (1R, 2R, 3S)-2-формил-3-(3,4-дихлорфенил)тропана (6,9 г, 23 ммоль) в метаноле (100 мл) добавили карбонат натрия (4 г) и хлорид гидроксиламмония (2,6 г, 37 ммоль), смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь концентрировали в вакууме и остаток растирали с водой. Неочищенный продукт выделяли путем фильтрации и рекристаллизации сначала в смеси этанола и воды (1+1), а затем в 99%-ном этаноле с получением указанного в заголовке соединения (смесь син/анти изомеров, примерно 1+2) в виде белых кристаллов, т.пл. 230-235oC.
Аналогичным образом были получены следующие соединения:
(1R, 2R,3S)-3-(4-хлорфенил) тропан-2-О-альдоксим, белые кристаллы, т.пл. 220-222oC.
Гидрохлорид (1R,2R,3S)-3-(4-хлорфенил)тропан-2-О-метил-альдоксима, белые кристаллы, т.пл. 90-93oC.
(1R,2R,3S)-3-(3, 4-дихлорфенил) тропан-2-О-метил-альдоксим, масло.
Рацемическая смесь (1R,2R,3S)-3-(3,4-дихлорфенил)тропан-2-О-метил-альдоксима и его энантиомера
(1S, 2S,3R)-3-(3,4-дихлорфенил) тропан-2-О-метил-альдоксима, т.пл. 172-178oC.
(1S, 2S, 3R)-3-(3,4-дихлорфенил)тропан-2-О-метил-альдоксима, т.пл. 123-130oC.
(1R, 2R, 3S)-3-(3,4-дихлорфенил)тропан-2-О-бензил-альдоксим, белые кристаллы, т.пл. 161-163oC.
(1R, 2R, 3S)-3-(3,4-дихлорфенил)тропан-2-О-фенил-альдоксим, H2SO4, т.пл. 100-102oC.
(1R, 2R, 3S)-N-норметил-N-(трет-бутоксикарбонил)-3-(3,4-дихлорфенил)тропан-2-O-метил-альдоксим, масло, которое использовали без дальнейшей очистки.
(1R, 2R, 3S)-N-норметил-N-(трет-бутоксикарбонил)-3-(3,4-дихлорфенил)тропан-2-O-бензил-альдоксим, масло, которое использовали без дальнейшей очистки.
Гидрохлорид (1R, 2R, 3S)-3-(3,4-дихлорфенил)тропан-2-O-(2-пропинил)-альдоксима, белые кристаллы, т.пл. < 100oC.
Гидрохлорид (1R, 2R, 3S)-3-(3,4-дихлорфенил) тропан-2-O--(2-пропенил)-альдоксима, белые кристаллы, т.пл. 131-133oC.
Гидрохлорид (1R,2R,3S)-3-(3,4-дихлорфенил) тропан-2-O-(2-метилпропил)-альдоксима, белые кристаллы, т.пл. < 161- 163oC.
Гидрохлорид (1R, 2R, 3S)-3-(3,4-дихлорфенил) тропан-2-O-циклопропилметил-альдоксима, белые кристаллы, т.пл. 173-175oC.
Гидрохлорид (1R, 2R,3S)-3-(3,4-дихлорфенил) тропан-2-O-этил-альдоксима, белые кристаллы, т.пл. < 110oC.
Гидрохлорид (1R, 2R, 3S)-3-(3,4-дихлорфенил) тропан-2-O-(1,1-диметилэтил)-альдоксима, белые кристаллы, т.пл. 213-215oC.
Гидрохлорид (1R, 2R, 3S)-3-(4-метилфенил) тропан-2-O-метил-альдоксима, белые кристаллы, т.пл. < 95oC (гигроскопичный).
Соли (1R,2R,3S)-3-(3,4-дихлорфенил)тропан-2- О-метил-альдоксима получали как описано ниже:
Раствор кислоты (3,25 ммоль) добавляли к раствору (1R,2R,3S)-3-(3,4-дихлорфенил)тропан-2-O-метил-альдоксима (1 г, 3,0 ммоль) в 96%-ном этоноле (5 мл) и перемешивали смесь при комнатной температуре. Если через 18 ч не выпадал осадок, смесь концентрировали в вакууме и концентрированную смесь оставляли для выпадения осадка в холодильнике. Кристаллический продукт выделяли путем фильтрации и промывали небольшим количеством охлажденного на льду 96%-ного этанола. Соль рекристаллизировали из воды или изопропанола.
Были получены следующие соли (1R,2R,3S)-3-(3,4-дихлорфенил)тропан-2-О-метил-альдоксима:
Малеат: белые кристаллы, т.пл. (H2О) 140-142oC
Цитрат: белые кристаллы, т.пл. (изопропанол) 143-144oC
Малонат: белые кристаллы, т.пл. (изопропанол) 116-118oC
Фумарат: белые кристаллы, т.пл. (H2О) 158-159oC
H2SO4: белые кристаллы, т.пл. (H2О) 84-87oC рекристаллизация из H2О дает дисульфат и некоторое количество гидросульфатов. Осаждение соли из изопропанола дает гидросульфат, т. пл. 161-163oC.
HCl: белые кристаллы, т.пл. (H2О) 74-75oC.
Пример 12.
Гидрохлорид (1R, 2R,3S)-3-(3,4-дихлорфенил)тропан-2-O-этоксикарбонилметил-альдоксима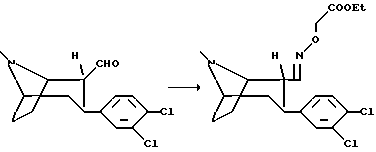
Раствор (1R, 2R, 3S)-2-формил-3-(3,4-дихлорфенил)тропана (1 г), хлорида O-(2-ацетокси)-гидроксиламмония (0,5 г) и концентрированной соляной кислоты в этаноле (1 мл) кипятят с обратным холодильником в течение 5 ч, после чего перемешивают 18 ч при комнатной температуре. Смесь концентрируют в вакууме и остаток растирают с изопропанолом. Указанное в заголовке соединение получают путем фильтрации в виде белых кристаллов, т.пл. 220-222oC.
Аналогичным образом получают следующие соединения:
Гидрохлорид (1R, 2R,3S)-3-(3,4-дихлорфенил) тропан-2-O-метоксикарбонилметил-альдоксима, белые кристаллы, т.пл. 193-195oC.
Гидрохлорид (1R, 2R, 3S)-3-(3,4-дихлорфенил) тропан-2-O-(1-этоксикарбонил-1,1-диметил-метил)-альдоксима, белые кристаллы, т.пл. 214-215oC.
Гидрохлорид (1R,2R,3S)-3-(4-хлорфенил)тропан-2-О-метоксикарбонилметил-альдоксима, белые кристаллы, т.пл. 202-203oC.
Гидрохлорид (1R, 2R,3S)-3-(3,4-дихлорфенил)тропан-2-O-карбоксиметил-2-альдоксима. Получают вместе с гидрохлоридом (1R,2R,3S)-3-(3,4-дихлорфенил)тропан-2-O-метоксикарбонилметил-альдоксима после кипячения с обратным холодильником в течение 1 ч. Белые кристаллы, т.пл. 158-160oC.
Пример 13.
Гидрохлорид (1R, 2R, 3S)-N-норметил-3-(3,4-дихлорфенил) тропан-2-О-метил-альдоксима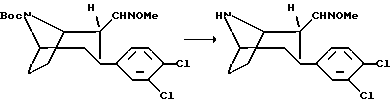
Смесь (1R, 2R, 3S)-N-норметил-N-(трет-бутоксикарбонил)-3-(3,4-дихлорфенил)тропан-2-O-метил-альдоксима (1,3 г, 3,1 ммоль) и трифторуксусной кислоты (10 мл) в абсолютном метилен хлориде (10 мл) перемешивают в течение 1 ч. Добавляют лед и метилен хлорид (50 мл) и подводят pH смеси до 10 путем добавления 4М гидроксида натрия. Органическую фазу высушивают и концентрируют в вакууме с получением масла, которое подвергали колоночной хроматографии, используя смесь метилен хлорида, метанола и 25%-ного гидроксида аммония (90+10+1). Фракции, содержащие желаемый продукт, концентрировали в вакууме, полученное масло растворяли в малом количестве диэтилового эфира и добавляли раствор соляной кислоты в диэтиловом эфире. Указанное в заголовке соединение выделяли в виде белых кристаллов, т. пл. 226-230oC.
Аналогичным образом получают следующие соединения:
Гидрохлорид (1R, 2R, 3S)-N-норметил-3-(3,4-дихлорфенил) тропан-2-О-бензил-альдоксима, т.пл. 70-72oC.
Малонат (1R,2R,3S) -N-норметил-3-(4-хлорфенил)тропан-2-альдоксима, т.пл. 70-75oC.
(1R,2R,3S)-N-норметил-3-(4-хлорфенил)тропан-2-O-метил-альдоксим, H2SO4.
Пример 14.
Син- и анти- изомеры (1R,2R,3S) и (1S,2S,3R)-3-(3,4-дихлорфенил)тропан-О-метил-альдоксима
Образование (1R, 2R, 3S) и (1S,2S,3R)-3-(3,4-дихлорфенил) тропан-О-метил-альдоксима путем взаимодействия соответствующего 2- формильного соединения и хлорида метоксил аммония, как описано в примере 12, приводит к получению смеси син- и анти-изомера, которая является результатом кинетического контроля продукта и изомеризации первоначально образовавшейся смеси. Кинетическая смесь благоприятствует получению анти-изомера (более 90%) и равновесная смесь содержит анти/син изомеры в соотношении 7/3. Равновесие син/анти катализируется кислотой и равновесная смесь может быть легко получена путем нагревания водного раствора материала при 100oC и pH 4 в течение 3 ч. Колоночная хроматография равновесной смеси с использованием толуола, этил ацетата и триэтиламина (2+1) +2% в качестве элюента дает син-изомер в виде масла, которое может быть подвергнуто дистилляции kugelrohg без изомеризации.
Биология
Соединения по изобретению были протестированы на их способность связываться с транспортером дофамина в следующих тестах на in vitro и in vivo ингибирование 3H-WIN 35428.
In vitro ингибирование связывания 3H-WIN 35428
Предполагается, что функцией транспортеров/сайтов захвата дофамина на нервных окончаниях является прекращение нервного импульса путем удаления дофамина из синаптической щели. Активность или наличие интегрального белка, являющегося транспортером дофамина, может быть измерено in vitro синаптосомальным захватом 3H-дофамина или пробой на мембранное связывание с 3H-лигандами, способными связываться с транспортером.
Изучение связывание in vitro кокаина показало, что кокаин связывается с транспортером дофамина и тормозит захват 3H-дофамина. Известно, что многочисленные лиганды нескольких структурных типов связываются с сайтом захвата дофамина, но остается непонятным идентичны ли сайты связывания кокаина и таких лигандов. Структурный аналог кокаина, 3H-WIN 35428, связывается селективно и с высоким сродством с транспортерным комплексом дофамина.
Приготовление ткани: Приготовление осуществляют при 0-4oC, если не указано иное. Полосатое тело из мужских особей крыс линии Wistar (150-200 г) гомогенизируют 5-10 с в 10 мл NaH2PO4 (50 мМ, pH 7,4), используя гомогенизатор Ultra-Turrax. Суспензию центрифугируют при 27000 g в течение 15 мин. Супернатант сливают, а осадок ресуспендируют в 50 мМ NaH2PO4, pH 7,4 (1000 мл на 1 г исходной ткани) и используют в пробе на связывание.
Проба: Аликвоты по 0,5 мл ткани добавляют к 25 мл тестируемого раствора и 25 мл 3H-WIN 35428 (1 нМ, конечная концентрация), перемешивают и инкубируют в течение 60 мин при 2oC. Неспецифическое связывание определяют, используя кокаин (30 мМ, конечная концентрация). После инкубации к образцам добавляют 5 мл охлажденного на льду буфера и выливают прямо на фильтры из стекловолокна Whatman GF/C с отсасыванием и немедленно промывают 5 мл охлажденного на льду буфера. Количество радиоактивности на фильтрах определяют традиционным подсчетом жидкостных сцинтилляций. Специфическое связывание определяют, вычитая величину неспецифического связывания из величины общего связывания.
Перед вычислением ИК50 должно быть достигнуто 25-75% ингибирования специфического связывания. Тестируемая величина дается как ИК50 (концентрация (мкМ) тестируемого вещества, при которой на 50% ингибируется специфическое связывание 3H-WIN 35428).
In vivo ингибирование связывания 3H-WIN 35428
3H-WIN 35428 может быть также использован для in vivo изучения меченых рецепторов. Аккумуляция 3H-WIN 35428 обнаруживается избирательно в областях мозга, содержащих дофаминэргические нервные окончания. В полосатом теле, которое содержит дофамин в самой высокой концентрации, в самой высокой степени аккумулируется 3H-WIN 35428. Специфическое связывание в полосатом теле достигает максимума через 30 мин после внутривенной инъекции 3H-WIN 35428, и этот максимум сохраняется на протяжении 30 мин. Такое специфическое связывание 3H-WIN 35428 может быть частично или полностью предотвращено путем одновременного или предварительного введения лекарств, способных тормозить транспорт дофамина, и лигандов, связывающихся с транспортерным комплексом дофамина, то есть GBR 12909, кокаина и номифенсина [Scheffel et al., J.Pharm.Exp.Ther. 257.954-958 (1991)].
Все тестируемые вещества использовали в виде растворов или суспензий, приготовленных в 10%-ном Tween 80. Группы из 3 самок мышей линии NMRI (25 г) получали внутрибрюшинно инъекцию тестируемого вещества. Немедленно после этой инъекции эти мыши получали внутривенную инъекцию в хвостовую вену 2,0 мКи 3H-WIN 35428 в 0,2 мл физраствора. Через 45 мин после инъекции 3H-WIN 35428 мышей убивали путем декапитации и быстро на льду извлекали полосатое тело. Ткани взвешивали и растворяли в течение 36 ч в 1 мл 2%-ного лаурилсульфата натрия. К растворенной ткани добавляли затем 2 мл сцинтилляционной смеси и подсчитывали количество радиоактивности на 1 мг ткани путем традиционной жидкостной сцинтилляции. Группы необработанных мышей служили контролем. Для определения неспецифического связывания группы мышей инъецировали внутрибрюшинно WIN 35428 (2,5 мг/кг) во время 3H-WIN 35428-инъекции. Специфическое связывание определяется разницей между количеством связывания в контроле и количеством связывания у мышей, обработанных WIN 35428.
Величину ЭД50 определяли по доза-зависимым кривым. Если вводилась только одна доза тестируемого вещества, величину ЭД50 вычисляли как указано ниже, при условии, что ингибирование специфического связывания находится в пределах от 25 до 75%:
ЭД50= (введенная доза, мг/кг) х 1/(CO/CX-1), где CO обозначает специфическое связывание в контроле и
CX обозначает специфическое связывание у мышей, обработанных тестируемым веществом.
Результаты, полученные при тестировании соединений по изобретению, даны ниже в таблице 1.
Представленные выше результаты тестов показывают, что соединения по изобретению с высоким сродством связываются с транспортерным комплексом дофамина как in vitro, так и in vivo.
Соединения по изобретению также были протестированы на их способность тормозить возврат (обратный захват) дофамина (DA), норадреналина (NA) и серотонина (5-НТ) в синаптосомы.
Предполагается, что функцией транспортеров/сайтов захвата специфических нейротрансмиттеров на нервных окончаниях является прекращение нервного импульса путем удаления нейротрансмиттеров дофамина, норадреналина и серотонина, соответственно, из синаптической щели. Активность транспортерного интегрального белка может быть измерена in vitro путем синаптосомального захвата 3H-дофамина, 3H-норадреналина и 3H-серотонина, соответственно.
Торможение in vitro захвата 3H-дофамина (3H-DA) в стриарных синаптосомах
Приготовление ткани. Приготовление осуществляют при 0-4oC, если не указано иное. Полосатое тело из мужских особей крыс линии Wistar (150-200 г) гомогенизируют 5-10 с в 100 объемах охлажденной на льду 0,32 М сахарозы, содержащей 1 мМ паргилина, используя гомогенизатор Ultra-Turrax. Активность моноамин оксидазы будет ингибироваться в присутствии паргилина. Гомогенат центрифугируют при 1000 g в течение 10 мин. Образовавшийся супернатант центрифугируют затем при 27000 g в течение 50 мин и сливают супернатант. Осадок (P2) ресуспендируют в насыщенном кислородом (приведенном в равновесие с атмосферой 96% O2:4% CO2 по меньшей мере на 30 мин) инкубационном буфере Кребса-Ринждера (8000 мл на 1 г первоначальной ткани) при pH 7,2, содержащим 122 мМ NaCl, 0,16 мМ EDTA, 4,8 мМ KCl, 12,7 мМ Na2HPO4, 3,0 мМ NaH2PO4 1.2 мМ MgSO4, 1 мМ CaCl2, 10 мМ глюкозы и 1 мМ аскорбиновой кислоты.
Проба. Аликвоты по 4,0 мл суспензии ткани добавляют к 100 мкл тестируемого раствора и 100 мкл 3H-DA (1 нМ, конечная концентрация), перемешивают и инкубируют в течение 25 мин при 37oC. Неспецифический захват определяют, используя бензтропин (10 мкМ, конечная концентрация). После инкубации образцы выливают прямо на фильтры из стекловолокна Whatman GF/C с отсасыванием. Фильтры затем промывают три раза 5 мл охлажденного на льду 9%-ного (в/о) раствора NaCl. Количество радиоактивности на фильтрах определяют традиционным подсчетом жидкостных сцинтилляций. Специфический захват определяют, вычитая величину неспецифического захвата из величины общего захвата.
Перед вычислением ИК50 должно быть достигнуто 25-75% ингибирования специфического связывания.
Тестируемая величина дается как ИК50 (концентрация (мкМ) тестируемого вещества, при которой на 50% ингибируется специфическое связывание 3H-DA).
Торможение in vitro захвата 3Н-норадреналина (3H-NA) в синаптосомах гиппокампа
Приготовление ткани: Приготовление осуществляют при 0-4oC, если не указано иное. Гиппокамп из мужских особей крыс линии Wistar (150-200 г) гомогенизируют 5-10 с в 100 объемах охлажденной на льду 0,32 М сахарозы, содержащей 1 мМ паргилина, используя гомогенизатор Ultra-Turrax. Активность моноамин оксидазы будет ингибироваться в присутствии паргилина. Гомогенат центрифугируют при 1000 g в течение 10 мин. Образовавшийся супернатант центрифугируют затем при 27000 g в течение 50 мин и сливают супернатант. Осадок (P2) ресуспендируют в насыщенном кислородом (приведенном в равновесие с атмосферой 96% O2:4% CO2 по меньшей мере на 30 мин) инкубационном буфере Кребса-Ринждера (2000 мл на 1 г первоначальной ткани) при pH 7,2, содержащим 122 мМ NaCl, 0,16 мМ EDTA, 4,8 мМ KCl, 12,7 мМ Na2HPO4, 3,0 мМ NaH2PO4 1,2 мМ MgSO4, 0,97 мМ CaCl2, 10 мМ глюкозы и 1 мМ аскорбиновой кислоты.
Проба. Аликвоты по 4,0 мл суспензии ткани добавляют к 100 мкл тестируемого раствора и 100 мкл 3H-NA (1 нМ, конечная концентрация), перемешивают и инкубируют в течение 90 мин при 37oC. Неспецифический захват определяют, используя дезипрамин (1 мкМ, конечная концентрация). После инкубации образцы выливают прямо на фильтры из стекловолокна Whatman GF/C с отсасыванием. Фильтры затем промывают три раза 5 мл охлажденного на льду 9%-ного (в/о) раствора NaCl. Количество радиоактивности на фильтрах определяют традиционным подсчетом жидкостных сцинтилляций. Специфический захват определяют, вычитая величину неспецифического захвата из величины общего захвата.
Перед вычислением ИК50 должно быть достигнуто 25-75% ингибирования специфического связывания.
Тестируемая величина дается как ИК50 (концентрация (мкМ) тестируемого вещества, при которой на 50% ингибируется специфическое связывание 3H-DA).
Торможение in vitro захвата 3Н-5-гидрокситриптамина (3Н-5-НТ, серотонина) в кортикальных синаптосомах
Приготовление ткани. Приготовление осуществляют при 0-4oC, если не указано иное. Кору головного мозга от мужских особей крыс линии Wistar (150-200 г) гомогенизируют 5-10 с в 100 объемах охлажденной на льду 0,32 М сахарозы, содержащей 1 мМ паргилина, используя гомогенизатор Ultra-Turrax. Активность моноамин оксидазы будет ингибироваться в присутствии паргилина. Гомогенат центрифугируют при 1000 g в течение 10 мин. Образовавшийся супернатант центрифугируют затем при 27000 g в течение 50 мин и сливают супернатант. Осадок (P2) ресуспендируют в насыщенном кислородом (приведенном в равновесие с атмосферой 96% O2:4% CO2 по меньшей мере на 30 мин) инкубационном буфере Кребса-Ринждера (1000 мл на 1 г первоначальной ткани) при pH 7,2, содержащем 122 мМ NaCl, 0,16 мМ EDTA, 4,8 мМ KCl, 12,7 мМ Na2HPO4, 3,0 мМ NaH2PO4, 1,2 мМ MgSO4, 1мМ CaCl2, 10 мМ глюкозы и 1 мМ аскорбиновой кислоты.
Проба. Аликвоты по 4,0 мл ткани добавляют к 100 мкл тестируемого раствора и 100 мкл 3Н-5-НТ (1 нМ, конечная концентрация), перемешивают и инкубируют в течение 30 мин при 37oC. Неспецифический захват определяют, используя циталопрам (10 мкМ, конечная концентрация). После инкубации образцы выливают прямо на фильтры из стекловолокна Whatman GF/C с отсасыванием. Фильтры затем промывают три раза 5 мл охлажденного на льду 9%-ного (в/о) раствора NaCl. Количество радиоактивности на фильтрах определяют традиционным подсчетом жидкостных сцинтилляций. Специфический захват определяют, вычитая величину неспецифического захвата из величины общего захвата.
Перед вычислением ИК50 должно быть достигнуто 25-75% ингибирования специфического связывания.
Тестируемая величина дается как ИК50 (концентрация (мкМ) тестируемого вещества, при которой на 50% ингибируется специфическое связывание 3Н-5-НТ).
Результаты, полученные при тестировании некоторых соединений по изобретению, даны ниже в таблице 2.
Представленные выше результаты тестов показывают, что соединения по изобретению эффективно тормозят обратный захват (возврат) дофамина, норадреналина и серотонина в синаптосомы.
Соединения по изобретению также были протестированы на следующих животных моделях болезни Паркинсона.
Антагонизм МТРТ-индуцированного паркинсонизма у мартышек.
Введение МРТР (гидрохлорид 1-метил-4-фенил-1,2,3,6-тетра-гидропипридина) обезьянам индуцирует поражение дофаминэргической системы в мозгу и дает паркинсонические симптомы. Путем последующего введения тестируемых соединений может быть проверена способность смягчения этими соединениями симптомов паркинсонизма.
Способ.
Были использованы обыкновенные мартышки обоих полов весом 350-400 г в возрасте 3-5 лет. Животных содержали поодиночке в стандартных условиях при температуре 25-27oC, относительной влажности 50% и 12-часовом световом дне. Животные имели свободный доступ к пище и воде.
За несколько месяцев до начала локомоторного или поведенческого теста животных обрабатывали МРТР (гидрохлоридом 1-метил-4-фенил-1,2,3,6-тетрагидропипридина), растворенным в стерильном 0,9%-ном физиологическом растворе, в дозе 2 мг/кг, вводимом подкожно ежедневно в течение 5 дней или до развития явновыраженного паркинсонизма. Введенная кумулятивная доза находилась в пределах от 8 до 12 мг/кг. Во время обработки МРТР и последующие 2-3 недели животных кормили с руки, до тех пор пока они не поправлялись настолько, чтобы быть способными питаться самостоятельно. Перед поведенческим тестом все животные показывали ослабление базальной локомоторной активности, слабую координацию, ослабленные контрольные движения головы и атипичное положение тела, в частности позвоночника и конечностей.
Тестируемые соединения растворяли в 100%-ном Tween 80 с подогревом до 40-50oC, а затем разводили водой. Тестируемые соединения давали через зонд в объеме 2 мл/кг. Дополнительно каждый экспериментальный день вводили носитель тестируемого соединения для сравнения с обработкой лекарством. Каждое животное в течение следующих недель было обработано носителем или одной из трех доз тестируемого соединения, причем между обработками делали одну неделю перерыва для восстановления.
Оценка нарушения дееспособности: Нарушение дееспособности животных было индексировано следующим образом;
живость (нормальная 0, вялая 2);
реакция на стимулы (нормальная 0, пониженная 1, медленная 2, отсутствие 3);
контрольные движения (наличие 0, ослабленные 1, отсутствие 2);
внимание и движения глаз (нормальные 0, атипичные 1);
поза (нормальная 0, атипичное туловище 1, атипичные конечности 1, атипичный хвост, или все атипично 4);
баланс/координация (нормальная 0, ослабленная 1, нестабильная 2, самопроизвольные падения 3);
издавание звуков (нормальное 0, ослабленное 1, отсутствие 2).
Измерение локомоторной активности: Локомоторную активность измеряли одновременно у четырех отдельных животных, каждое из которых было помещено в металлическую клетку (50ш х 60д х 70в см) с прозрачными пластиковыми дверями (50ш х 70в см), похожую на клетки, в которых они содержались, но снабженную восьмью горизонтально ориентированными инфракрасными фотоэлементами. Эти лучи были локализованы на уровне пола; поперек клетки и один вдоль каждого из двух насестов. Другие лучи были направлены от лицевой к задней стене клетки на уровне пола и над каждым насестом. Число движений было измерено как число прерываний световых лучей, которые происходили при движении животных. Это число движений суммировали за 30 минутные промежутки времени и записывали в течение 10 ч проведения эксперимента.
Анализ данных: Было вычислено среднее (+ -) стандартная ошибка за время течения эксперимента для суммарного числа движений или индекса нарушения поведенческой дееспособности для различно обработанных групп.
Результаты, полученные при тестировании соединения по изобретению (1R, 2R, 3S)-3-(3,4-дихлорфенил)тропан-2-О-метил-альдоксима (NS 2214) показаны на фиг. 1 и фиг. 2. Легко увидеть, что соединения по изобретению в дозе от 0,1 до 2,5 мг/кг обеспечивают дозозависимое возрастание локомоторной активности и дозозависимое уменьшение индекса нарушения дееспособности.
Соединения по изобретению также были протестированы в следующем тесте на антидепрессантную активность.
Подвешивание за хвост.
Уменьшение времени неподвижности у мышей, подвешенных за хвост, наблюдается после системного введения центральных стимуляторов и антидепрессантов [Steru. L. , Chermat. R. , Thierry,B. & Simon, P (1985) The tail suspension test: A new method for screening antidepressant in mice. Psychopharmacology 85:367-370].
Способ. Используют самок мышей линии NMRI (20-25 г), содержащихся в комнате (12-часовой световой день) по меньшей мере 16 ч по 25 особей в клетке. Через 30 мин после перорального введения носителя или лекарства мышь подвешивают за хвост клейкой лентой на шест, находящийся на расстоянии 30 см над лабораторным столом. В течение следующих б мин определяют суммарную продолжительность неподвижности как отсутствие движений тела или конечностей (движения головы не считаются). Каждую дозу проверяют на 6 мышах.
Для обработанной физиологическим раствором или носителем мыши время неподвижности составляет в среднем 160-180 с. Величина ЭД50 рассчитывается графической интерполяцией от по меньшей мере 3 доз как доза, уменьшающая неподвижность на 100 с.
В следующей таблице 3 представлены результаты, полученные при тестировании нескольких соединений по изобретению.
Результаты, приведенные в таблице 3, позволяют предполагать наличие сильной антидепрессантной активности у соединений по изобретению.
Профиль побочных эффектов, оказываемых соединениями по изобретению.
Побочные эффекты кокаина и производных амфетамина, являющихся центральными стимуляторами, включают в себя центральное возбуждение и стимуляцию животных, включая приматов, и такие же эффекты могут наблюдаться у человека. Серьезным побочным эффектом кокаина и производных амфетамина также является их способность провоцировать токсические психотические симптомы, сильно напоминающие умственное расстройство шизофрению и включающие в себя галлюцинации, паранойю и атипичную странную стереотипную умственную активность и стереотипию.
В настоящее время считается, что эти симптомы у приматов и у человека вызываются экстенсивным и массивным высвобождением дофамина в стриарном комплексе и, в частности, в мезолимбической дофаминовой системе, которая иннервирует лимбические структуры, включая прилегающие ядра.
Индукция стереотипного атипичного поведения у грызунов, таким образом, также представляет одну из наиболее часто используемых животных моделей шизофрении среди моделей, используемых для антипсихотических нейролептических лекарств (включая галоперидол и хлорпромазин).
Развитие токсического атипичного стереотипного амфетаминового синдрома, как описано ниже, может позволить предсказать токсический центрально-стимулирующий побочный эффект соединений, высвобождающих дофамин у человека.
Классификация атипичного стереотипного поведения
В общем, атипичное стереотипное поведение после введения амфетамина и подобных кокаину центральных стимулирующих лекарств может быть классифицировано на стереотипное поведение с "низким" и "высоким" индексом интенсивности. Низкий индекс интенсивности стереотипии включает в себя атипичное и непрерывное повторение движений, вставания и сопения, и эти синдромы обычно видны только после низких доз дофаминэргических центрально-стимулирующих лекарств или могут наблюдаться в начальной и конечной фазе действия высоких доз таких лекарств. Поведенческие эффекты низкой интенсивности представлены здесь двигательным синдромом и синдромом вставания. Синдром высокой интенсивности стереотипии рассматривается здесь, если поведенческий репертуар крысы четко ограничивается в вариациях и состоит из непрерывного повторения одного или малого количества элементов поведения.
Синдром стереотипичного сопения, таким образом, наблюдается непрерывно только на небольшой ограниченной площади клетки. Такая активность обычно начинается на верхней части стенки и последующие более высокие дозы лекарств приводят к увеличению интенсивности сопения с передвижением к нижней части клетки, по стенке или на проволочный пол. В течение этой стадии стереотипии высокой интенсивности все нормальные поведенческие элементы отсутствуют, включая такое поведение, как еда, питье, чистка и нормальное исследование окружающей среды.
У крыс сопение высокой интенсивности может развиться в сопение, совмещенное с лизанием и/или кусанием-грызением проволочной сетки клетки после еще более высоких доз лекарства-стимулянта. Эти крысы обычно сидят в типичной сжатой позе в углу клетки. Иногда можно заметить медленное движение.
Следующая шкала использована для стереотипии высокой интенсивности, при условии, что поведенческие синдромы такие, как описано выше:
+ = только стереотипное сопение
++ = стереотипное сопение и эпизодическое лизание
+++ = постоянное лизание и/или кусание-грызение (см. табл.4).


Описываются новые производные тропан-2-альдоксима, любая их смесь или их фармацевтически приемлемая соль, где R обозначает водород или алкил; R3 представляет собой CH = NOR1, где R1 обозначает водород или алкенил, алкинил, циклоалкил, циклоалкилалкил, арил или алкил, возможно замещенный COOH, COO-алкилом или фенилом; R4 обозначает фенил, который может быть замещен один или несколько раз заместителями, выбранными из группы, состоящей из галогена, CF3, CN, NO2 или алкила, бензил или нафтил. Такие соединения обладают ценными терапевтическими свойствами в качестве ингибиторов обратного захвата моноаминных нейротрансмиттеров, то есть дофамина, серотонина, норадреналина. Описывается также фармацевтическая композиция для ингибирования обратного захвата моноаминных нейротрансмиттеров на основе соединений предложенной формулы. Описываются способы получения соединений и способ лечения расстройства или болезни организма животного (включая человека), на которые оказывает влияние торможение обратного захвата дофамина с использованием соединений формулы. 7 с. и 5 з.п.ф-лы, 4 табл.
любая их смесь или их фармацевтически приемлемая соль,
где R обозначает водород или алкил;
R3 представляет собой CH=NOR', где R' обозначает водород или алкенил, алкинил, циклоалкил, циклоалкилалкил, арил или алкил, возможно замещенный COOH, COO-алкилом или фенилом;
R4 обозначает фенил, который может быть замещен один или несколько раз заместителями, выбранными из группы, состоящей из галогена, CF3, CN, NO2 или алкила, бензил или нафтил.
3-(3,4-дихлорфенил)тропан-2-альдоксим,
3-(3,4-дихлорфенил)тропан-2-О-метил-альдоксим,
3-(3,4-дихлорфенил)тропан-2-О-бензил-альдоксим,
3-(3,4-дихлорфенил)тропан-2-О-этоксикарбонилметил-альдоксим,
3-(3,4-дихлорфенил)тропан-2-О-метоксикарбонилметил-альдоксим,
3-(3,4-дихлорфенил)тропан-2-О(1-этоксикарбонил-1,1-диметил-метил)-альдоксим,
3-(3,4-дихлорфенил)тропан-2-О-карбоксиметил-2-альдоксим,
N-норметил-3-(3,4-дихлорфенил)тропан-2-О-метил-альдоксим
или N-норметил-3-(3,4-дихлорфенил)тропан-2-О-бензил-альдоксим,
или их фармацевтически приемлемые соли присоединения.
(1R,2R,3S)-3-(3,4-дихлорфенил)тропан-2-альдоксим,
(1R,2R,3S)-3-(3,4-дихлорфенил)тропан-2-О-бензил-альдоксим,
(1R,2R,3S)-3-(3,4-дихлорфенил)тропан-2-О-этоксикарбонилметил-альдоксим,
(1R,2R,3S)-3-(3,4-дихлорфенил)тропан-2-О-метоксикарбонилметил-альдоксим,
(1R, 2R, 3S)-3-(3,4-дихлорфенил)тропан-2-О-(1-этоксикарбонил-1,1-диметил-метил)-альдоксим,
(1R,2R,3S)-3-(3,4-дихлорфенил)тропан-2-О-карбоксиметил-2-альдоксим,
(1R,2R,3S)-N-норметил-3-(3,4-дихлорфенил)тропан-2-О-метил-альдоксим или
(1R,2R,3S)-N-норметил-3-(3,4-дихлорфенил)тропан-2-О-бензил-альдоксим,
или их фармацевтически приемлемые соли присоединения.
его энантиомер или их смесь,
где R определено в п.1 и R12 является эфиром карбоновой кислоты,
подвергают взаимодействию с соединением формулы
R4 - A
где R4 определено в п.1;
A - любой тип реакционноспособной группы, пригодной для генерации карбаниона как ее противоионной части, такой, как Li, MgX, где X обозначает галоген, и CuLi, в реакции 1,4 присоединения, подобной реакции Михаэля,
и, если R12 является эфиром карбоновой кислоты, превращают полученное соединение в соединение по п.1, используя традиционные методы.
Приоритет по пунктам:
24.11.94 - по п.1;
19.04.94 - по пп.2 - 5;
24.11.94 - по пп.6 - 11;
12.04.95 - по п.12.
| МНОГОКАНАЛЬНЫЙ АНАЛИЗАТОР ИМПУЛЬСОВ | 0 |
|
SU316718A1 |
| Способ размножения копий рисунков, текста и т.п. | 1921 |
|
SU89A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Хлоргидраты W-аминоацильных эфиров тропан-3-ола как стимуляторы центральной нервной системы | 1978 |
|
SU941369A1 |
| Способ получения азонийспиронортропанолового сложного эфира | 1986 |
|
SU1711674A3 |
| US 3813404, 1974 | |||
| Экономайзер | 0 |
|
SU94A1 |
| Карлов В.А | |||
| Терапия нервных болезней | |||
| - М.: Медицина, 1987, с.304 - 308. | |||
