Изобретение относится к области анализа материалов, а именно к анализу с помощью рентгеновского излучения, включая неразрушающий анализ химического состава сложных многокомпонентных материалов, таких как оксидные высокотемпературные сверхпроводники (ВТСП), диэлектрические материалы на основе собственных оксидов многоэлементных полупроводников типа A3B5, A2B6, тройных растворов и других. Изобретение направлено также на определение такой фундаментальной константы, как энергия связи остовного уровня атома, находящегося в определенном химическом состоянии, и на решение проблемы неразрушающей диагностики химического состава таких микроскопических объектов, как нанометровые пленки и кластеры. Условие неразрушаемости и нано-масштаб объектов диагностики выводят за рамки обсуждения аналогов чисто химические и физико-химические методы, такие, например, как хроматографические.
В качестве аналогов предлагаемого изобретения можно привести широко известные методы электронной оже-спектроскопии [1] и рентгеновской фотоэлектронной спектроскопии [2], используемые в режиме определения элементного состава материала. В первом случае образец облучается электронами и измеряются спектры оже-электронов, во втором случае облучение осуществляется рентгеновскими квантами определенной энергии и измеряются спектры фотоэлектронов. По интенсивностям линий спектров определяется содержание химических элементов. Их соотношение позволяет делать вывод о химической формуле вещества. Очевидно, что такой подход эффективен лишь в случае химически однородных веществ и в общем случае не позволяет определять содержание различных химических фаз. Аналогом способа определения энергии связи электрона остовного уровня атома Ei является адсорбционная спектроскопия, то есть метод, основанный на измерении спектров поглощения излучения непрерывного спектра, такого, например, как синхротронное излучение, и определения искомой величины по так называемому краю поглощения [3]. Точность этого метода оказывается недостаточной для надежной идентификации химического состояния атома.
Качественным скачком на пути решения проблемы неразрушающего определения химического состава вещества явилось обнаружение изменения энергии связи электрона остовного уровня Ei при изменении химического состояния атома. Для определения химического состава (содержания химических фаз) было предложено использовать энергии связи Ei или соответствующие им химические сдвиги линий фотоэлектронных спектров. Таким образом, в описываемом методе исследуемый образец облучают рентгеновскими квантами определенной энергии и измеряют спектры фотоэлектронов. Задача определения химического состава материала сводится при этом к задаче разделения экспериментального спектра (экспериментальной линии) на элементарные составляющие, соответствующие определенным химическим состояниям атомов. По вкладам этих составляющих с учетом стехиометрии химических фаз определяется искомый химический состав в молекулярных процентах. Решение задачи ищется путем минимизации функционала ошибок между экспериментальным фотоэлектронным спектром и суммарной огибающей набора элементарных линий. Свободными параметрами являются искомые интенсивности элементарной линии и, иногда, их ширины. Обычно форма элементарных линий описывается аппаратной функцией спектрометра, чаще всего гауссовой, и их ширины определяются в отдельных экспериментах. В общем случае для того, чтобы решение было единственным, необходимо заранее установить возможное число элементарных линий (число химических фаз), а также соответствующие им значения энергий связи. Энергии связи определяются по положению фотоэлектронных линий чистых эталонных материалов (химических фаз). Таким образом, описываемый метод используется как для определения химического состава, так и для определения энергий связи Ei электронов остовных уровней. В большинстве прикладных работ используются литературные данные об энергиях связи. Описанный метод получил название рентгеновской фотоэлектронной спектроскопии (РФЭС) для химического анализа, а ее автор К.Зигбан был удостоен в 1968 году Нобелевской премии по физике [4, 2]. По большинству существенных признаков данный метод ближе других к предлагаемому изобретению, поэтому он и был выбран за прототип.
Метод РФЭС для химического анализа зарекомендовал себя, как чрезвычайно эффективный, а соответствующие приборы-спектрометры имеются в настоящее время во всех лабораториях и университетах, во многих промышленных фирмах. Вместе с тем, появление новых сложных многокомпонентных материалов выявило и недостатки прототипа, а именно невозможность в случае большого числа химических фаз корректно выделить элементарные фотоэлектронные линии из экспериментальной суммарной линии. Дело в том, что химические сдвиги в таких случаях не превышают полуширины аппаратной функции спектрометра, и погрешности в определении остовных энергий связи Ei (или химических сдвигов) оказываются недостаточно малыми для надежного разделения сложных линий исследуемых материалов на составляющие. Дорогостоящая монохроматизация рентгеновского излучения далеко не всегда помогает решить проблему разделения экспериментальной линии спектра, поскольку повышает разрешение прибора всего в два раза (с 0.8-0.7 eV до 0.5-0.3 eV), но уменьшает потоки рентгеновских квантов более, чем на порядок, уменьшая тем самым чувствительность метода. Дальнейшее повышение разрешения обычно бессмысленно, поскольку ширина элементарной линии в конечном итоге определяется естественной шириной линии. Точность и, даже, достоверность определения энергий связи Ei методом РФЭС для сложных многокомпонентных материалов часто оказываются неудовлетворительными из-за сложности или невозможности синтезировать эталоны в чистом виде и сохранять их поверхность неизменной. Точность определения энергий Ei ограничивают и неконтролируемые систематические ошибки, разные в экспериментах с разными эталонами.
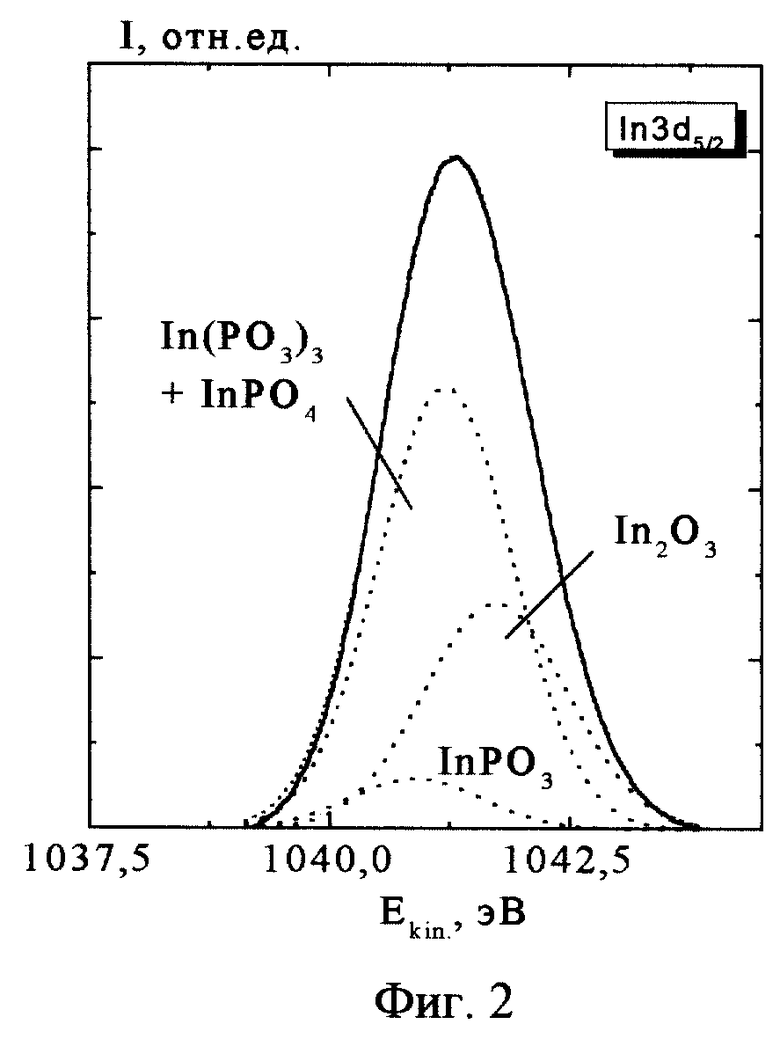
Проиллюстрируем недостатки прототипа на конкретном примере анализа фазового состава тонкого (50  ) слоя собственного оксида, полученного на поверхности фосфида индия InP. Образец этого материала облучался рентгеновскими квантами AlKα (hν = 1486.6 eV). Измерялись спектры фотоэлектронов, испускаемых из внутренних (остовных) оболочек индия и фосфора: In3d5/2 и P2р. На фиг. 1 и 2 приведены примеры таких спектров (линий) за вычетом квазинепрерывного фона. По оси x отложена кинетическая энергия Ek фотоэлектрона, однозначно связанная с энергией связи остовного электрона Ei уравнением Эйнштейна: Ek = hν-Ei-e Φ, где eΦ - работа выхода спектрометра. По оси y - количество испущенных фотоэлектронов.
) слоя собственного оксида, полученного на поверхности фосфида индия InP. Образец этого материала облучался рентгеновскими квантами AlKα (hν = 1486.6 eV). Измерялись спектры фотоэлектронов, испускаемых из внутренних (остовных) оболочек индия и фосфора: In3d5/2 и P2р. На фиг. 1 и 2 приведены примеры таких спектров (линий) за вычетом квазинепрерывного фона. По оси x отложена кинетическая энергия Ek фотоэлектрона, однозначно связанная с энергией связи остовного электрона Ei уравнением Эйнштейна: Ek = hν-Ei-e Φ, где eΦ - работа выхода спектрометра. По оси y - количество испущенных фотоэлектронов.
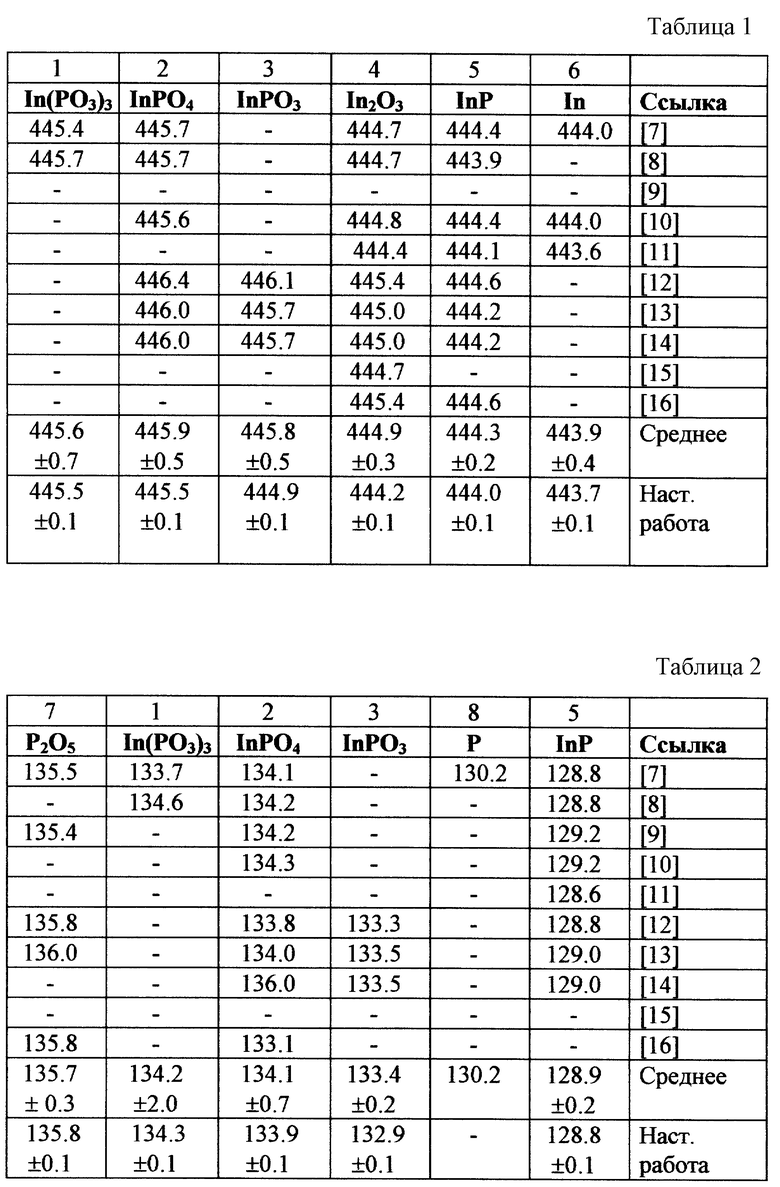
Известно, что возможно образование следующих термодинамически стабильных оксидов InP: In(PO3)3 (1), InPO4 (2), InPO3 (3), In2O3 (4), P2O5 (7). В табл. 1 и 2 для этих соединений приведены энергии связи Ei (в электронвольтах) электрона 3d5/2 оболочки индия и 2р оболочки фосфора, соответственно, и данные для исходного материала InP (5), металлического индия In (6) и элементарного фосфора P (8).
Таким образом, количество возможных химических фаз известно. Форма элементарной линии в нашем случае определяется аппаратной функцией спектрометра, близкой к гауссовой. Ее ширина была определена в экспериментах на однофазных материалах (на чистом серебре). В качестве энергий связи индия и фосфора в указанных соединениях использовались значения, полученные усреднением литературных данных (табл. 1, 2). Заметим, что определенные таким образом значения характеризуются большими погрешностями (±~0.5 eV) несмотря на то, что обычные для каждой из работ погрешности, по оценкам их авторов, как правило, не превышают ±0.1 eV. Содержание химических фаз определялось по амплитудам элементарных составляющих, которые устанавливались, исходя из минимума площадей суммарной огибающей и экспериментальной кривой. На фиг. 1 и 2 приведены результаты такого разложения, выполненного в соответствии с прототипом (амплитуда элементарной линии InP (5) на хвостах спектров пренебрежимо мала. ) Полученный вариант, однако, невозможно признать приемлемым, поскольку содержание одноименных фаз, определенное независимо по линиям индия и фосфора отличается в разы: "1+2" (P) - 40%, "1+2" (In)- 74%. Доля фазы "3", определенная относительно фаз "1+2" независимо по линиям индия и фосфора, различается почти на порядок: 41% и 5%. Вариации ширины элементарных линий не уменьшают указанные различия.
Таким образом, можно сделать вывод о том, что для сложных систем недостаточная точность определения энергий связи, погрешности измерения спектра, формы аппаратной функции и вычислений, а также погрешности, связанные с коррекцией эффектов статической зарядки диэлектрического образца под действием излучения, не позволяют решать задачу достоверного определения их химического состава.
Предлагаемый способ позволяет решать следующие задачи: повышать достоверность и точность определения химического состава вещества и энергий связи остовных электронов.
Задачи решаются тем, что в известном способе количественного определения химического состава вещества и энергий связи остовных электронов, включающем измерение линии фотоэлектронного спектра по меньшей мере одного элемента, разложение ее по известному набору элементарных составляющих, соответствующих различным химическим фазам, и известной последовательности энергий связи остовного электрона выбранного элемента в этих фазах, минимизацию функционала ошибок между измеренной линией спектра и суммарной расчетной огибающей набора элементарных составляющих с выбором их амплитуд и ширины в качестве свободных параметров и определение искомого состава по относительному вкладу этих составляющих в разлагаемую линию спектра с учетом стехиометрии химических фаз, согласно формуле изобретения, вышеупомянутые операции производят для каждой линии спектра двух или более выбранных элементов, дополнительно выбирая в качестве свободных параметров энергии связи элементов в химических фазах, и, сравнивая полученные для всех линий спектра результаты между собой, варьируют свободными параметрами до совпадения результатов с учетом заданной ошибки.
Покажем сущность изобретения, новизну и существенность признаков.
В прототипе задача разложения на элементарные составляющие становится имеющей одно решение благодаря заданности энергий связи Ei для каждой химической фазы (заданности положений элементарных составляющих). Однако погрешности этих значений, как отмечалось выше, приводят к неправильному решению, или к отсутствию решения, согласованного по экспериментальным линиям-спектрам различных элементов. Авторы установили, что получение согласованного по различным спектрам решения невозможно без варьирования энергии связи Ei. В изобретении вводятся дополнительные свободные параметры - энергии связи Ei остовных электронов в химических фазах. Единственность решения достигается разложением на элементарные составляющие спектра не одного, а двух или более элементов и сравнением результатов разложения по каждому элементу. Введение этих двух признаков позволило получить дополнительные условия для выбора свободных параметров. При этом количество условий может быть сделано большим количеством свободных параметров и задача разложения получает единственное решение. Отсутствие любого из указанных признаков делает задачу либо некорректной, либо несамосогласованной. Поэтому приведенные признаки необходимо признать существенными.
Новизна способа проявляется в том, что впервые предлагается проводить самосогласованный анализ спектров двух или более элементов, входящих в состав химического соединения, с варьированием дополнительных свободных параметров - энергий связи Ei. В результате безэталонный анализ образца сложного многокомпонентного химического состава впервые позволил получить не только информацию об этом составе, но и о таких фундаментальных константах, как энергии связи остовных электронов атомов Ei, входящих в различные химические соединения. При этом, как будет показано авторами, точность и достоверность определения этих энергий значительно повышается по сравнению с литературными данными.
Способ осуществляется следующим образом. Исследуемый образец вводят в камеру фотоэлектронного спектрометра и очищают его поверхность от адсорбированного слоя молекул атмосферы. Далее образец облучают рентгеновскими квантами и измеряют энергетические распределения испущенных образцом электронов, или фотоэлектронные спектры, в диапазонах энергий, включающих фотоэлектронные линии двух или более входящих в состав образца элементов. Из спектров по стандартным процедурам вычитается квазинепрерывный фон, в результате чего остаются экспериментальные линии-спектры, состоящие из неразрешенных элементарных линий, каждая из которых соответствует определенному химическому состоянию элемента. Далее решается задача разложения спектров на составляющие. Для этого определяется (по справочной литературе) количество химических соединений-фаз, которые могут образовывать элементы, входящие в состав исследуемого образца, и соответствующее количество химических состояний исследуемых элементов. Иными словами, определяется базис элементарных составляющих, по которым необходимо провести разложение экспериментальных линий-спектров. Каждая элементарная линия характеризуется своим энергетическим положением (химическим сдвигом), или соответствующей энергией связи Ei. Знать эти энергии в предлагаемом способе не нужно. Но необходимо для каждого элемента выстроить энергии связи по возрастанию (убыванию), то есть пронумеровать базис, и выбрать в качестве нулевого приближения любые значения в пределах экспериментальной линии. Для оксидных материалов в случае отсутствия литературных данных об энергиях связи последовательность может быть установлена из термодинамических расчетов, в частности, из значений приходящейся на атом свободной энергии. Величина этой энергии, по наблюдениям авторов, пропорциональна энергии связи. Следующим этапом решения задачи является разложение в нулевом приближении экспериментального спектра на составляющие, которое осуществляется, как и в прототипе, вариацией интенсивностей и ширин элементарных линий.
Выполненное по каждому экспериментальному спектру (элементу) разложение дает в нулевом приближении содержание каждой из химических фаз. Необходимо сравнить эти данные по каждой из фаз, оценить величины расхождений и так изменить свободные параметры, прежде всего энергии связи, чтобы в следующем итерационном цикле уменьшить эти расхождения. Итерационную процедуру необходимо повторять до достижения такого значения расхождения, которое определяется условиями задачи.
Суммируя вышеизложенное можно предложить следующий алгоритм решения задачи разложения спектра:
- определение максимально возможного количества химических фаз, выстраивание последовательности энергий связи Ei каждого выбранного для анализа элемента и выбор в нулевом приближении любых значений Ei в пределах экспериментальной линии;
- описание экспериментальных спектров набором элементарных (гауссовых) линий с центрами при энергиях Ei 1,2.. и определение по каждому из анализируемых элементов относительных долей химических фаз Ci 1, Ci 2,...(i - номер химической фазы; 1, 2 - номер исследуемого элемента, или линии-спектра). Под относительной долей понимается отношение площади соответствующей элементарной линии, скорректированной на стехиометрический коэффициент фазы, к сумме площадей элементарных линий фаз, общих для каждого из анализируемых элементов. Определение состава осуществляется вариацией интенсивностей элементарных линий (Ci 1, Ci 2, ...,) (и ширин, если ширина аппаратной функции меньше естественной ширины линии) путем минимизации разницы между экспериментальным и расчетным спектрами;
- установление разности Δ
- осуществление нового итерационного цикла при новом наборе энергий Ei 1,2.. и возвращение ко второму пункту предлагаемой методики. Окончание процедуры при достижении заданного условиями задачи значения δ: Δ
- определение химического состава в молекулярных процентах путем перенормировки относительных долей химических фаз на суммарную площадь элементарных линий всех присутствующих в образце химических фаз.
Пример. Способ был применен при решении задачи определения химического состава тонкого (50  ) диэлектрического слоя собственного оксида, полученного в ФТИ им.А.Ф.Иоффе РАН на поверхности фосфида индия InP при разработке приборной структуры "полупроводник/диэлектрик" для быстродействующего полевого транзистора и диодов [5]. Используемый в данном примере образец был тем же самым, что и в примере, иллюстрирующем недостатки прототипа.
) диэлектрического слоя собственного оксида, полученного в ФТИ им.А.Ф.Иоффе РАН на поверхности фосфида индия InP при разработке приборной структуры "полупроводник/диэлектрик" для быстродействующего полевого транзистора и диодов [5]. Используемый в данном примере образец был тем же самым, что и в примере, иллюстрирующем недостатки прототипа.
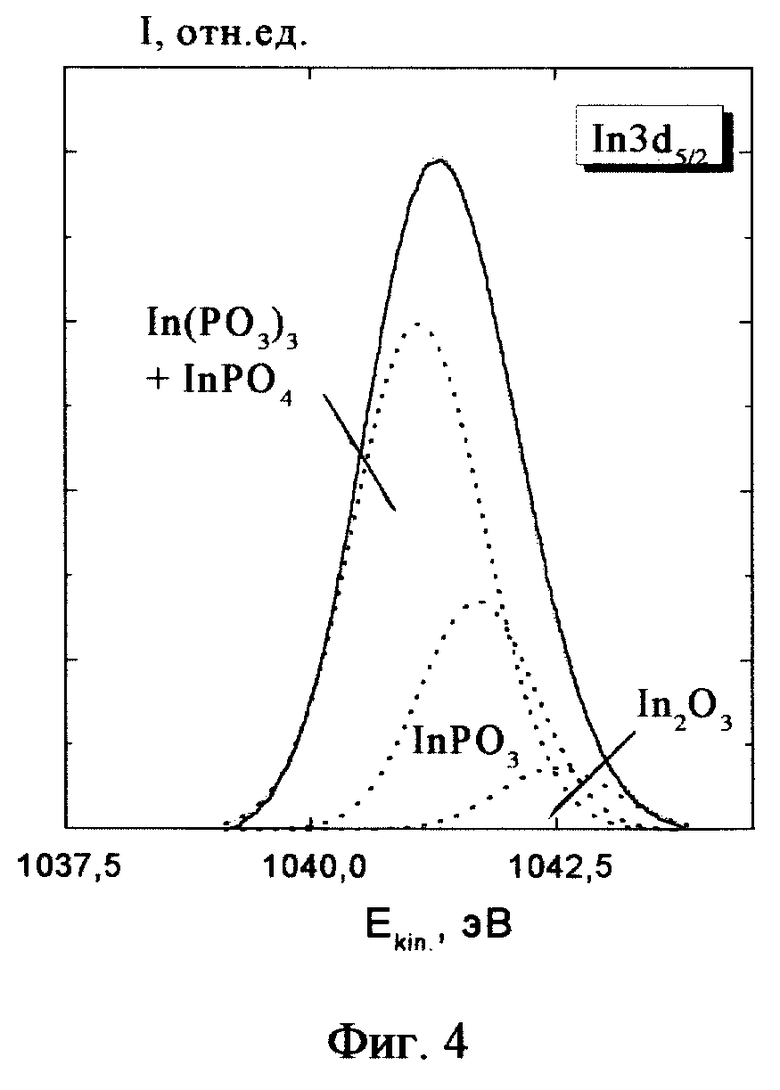
После ввода в высоковакуумную камеру спектрометра LHS-11 (Leybold AG) поверхность образца очищалась мягким ионным травлением от адсорбированного слоя молекул атмосферы. Затем в процессе облучения образца рентгеновскими квантами AlKα (1486.6 eV) измерялись спектры фотоэлектронов, испускаемых из внутренних (остовных) оболочек индия и фосфора In3d5/2 и P2p. Обработка спектров сводилась к их сглаживанию и вычитанию квазинепрерывного фона. Примеры полученных спектров приведены на фиг. 3-4.
Необходимый для анализа базис разложения хорошо известен. Как уже отмечалось выше InP образует следующие термодинамически стабильные оксиды: In(PO3)3 (1), InPO4 (2), InPO3 (3), In2O3 (4), P2O5 (7).
Как для индия, так и для фосфора известны, хотя и с большой погрешностью (± 0.3 - 0.7 eV), энергии связи Ei остовных электронов In3d5/2 и P2р (табл. 1, 2). Однако в отличие от прототипа, в изобретении эта информация не нужна. Используется только информация о последовательности этих энергий в указанных соединениях: E7 P > E1 P > E2 P > E3 P (> > E8 P > E5 P) и E1 In > E2 In > E3 In > E4 In > E5 In. Более того, для указанных оксидов эта информация была нами ранее получена независимо из термодинамических оценок. В качестве нулевого приближения для указанных энергий связи в этом примере были выбраны средние энергии, приведенные в табл. 1 и 2. Однако приводимый ниже конечный результат получается при любом выборе этих энергий из интервала экспериментальных спектров при условии соблюдения вышеприведенного неравенства.
В разложении экспериментальных линий-спектров использовалась гауссова аппроксимация аппаратной функции спектрометра, которая была определена в экспериментах на однофазных материалах (на чистом серебре). Однако варьирование ширин элементарных линий не приводило к их переоценке. Результаты нулевого и конечного разложения спектров индия и фосфора приведены соответственно на фиг. 1 - 4. Из-за близости энергий связи фаз (1) и (2) в спектре индия приводится их суммарная элементарная огибающая. В высокоэнергетический хвост этого спектра попадает также линия неокисленного InP (5). Однако ее вклад ничтожен. В спектре фосфора незначительные вклады неокисленного InP (5) и элементарного фосфора P(8) представлены отдельной малоинтенсивной линией, отстоящей от спектра оксида примерно на 6 eV. Сходимость итерационной процедуры оказалась весьма быстрой. Для получения конечного разложения потребовалось всего пять циклов. Доли химических фаз 1, 2, 3, 4 и 7 оказались равными соответственно 16, 49, 29, 4 и 2%. В последнем разложении доли химических фаз, определенные по спектрам индия и фосфора, совпадают с точностью до 3%. В пределах этой ошибки точность определения количества основных фаз не хуже 5-10%.
В результате решения задачи разложения были также получены значительно более точные и достоверные энергии связи. Из табл. 1 и 2, видно, что данные настоящей работы, как правило, близки к усредненным по многим источникам данным, хотя в некоторых случаях разница достигает 0.6 - 0.9 eV. Но при этом погрешность определения энергий связи в настоящей работе уменьшена в несколько раз, и для основных фаз она не превышает 0.1 eV. Смещение центра элементарной линии на величину большую, чем 0.1 eV, не позволяет достичь заданной ошибки разложения. Вывод о повышении достоверности определения энергий связи в описываемом примере сделан на том основании, что в противном случае, в случае недостаточной точности определения этой величины, возможны ошибки в установлении соответствия энергии Ei и вида соединения. Так, определенная в работе [12] энергия связи In3d5/2 для InPO3 (446.1 eV) на 0.2 eV превышает усредненное по многим источникам значение энергии для оксида InPO4 (табл. 1, 2) вместо того, чтобы быть ниже этого значения. Определенная в работе [14] энергия связи Р2р для InPO4 (136.0 eV) на 0.2 eV превышает усредненное значение для P2O5, также меняя последовательность указанных фаз. Напротив, энергия связи Р2р для InPO4 (136.0 eV) из работы [16] не выше на 0.7 - 1.0 eV, а на 0.3 eV ниже средней энергии для InPO3. Следовательно при использовании этих ошибочных данных технологические процессы были бы ориентированы на получение совершенно иных веществ.
Приведенный пример был частью исследования, в результате которого удалось получить рекордно большую долю термодинамически наиболее стабильных фаз - полимерной In(PO3)3 (1) и InPO4 (2) и, как следствие, рекордные параметры приборной структуры [5].
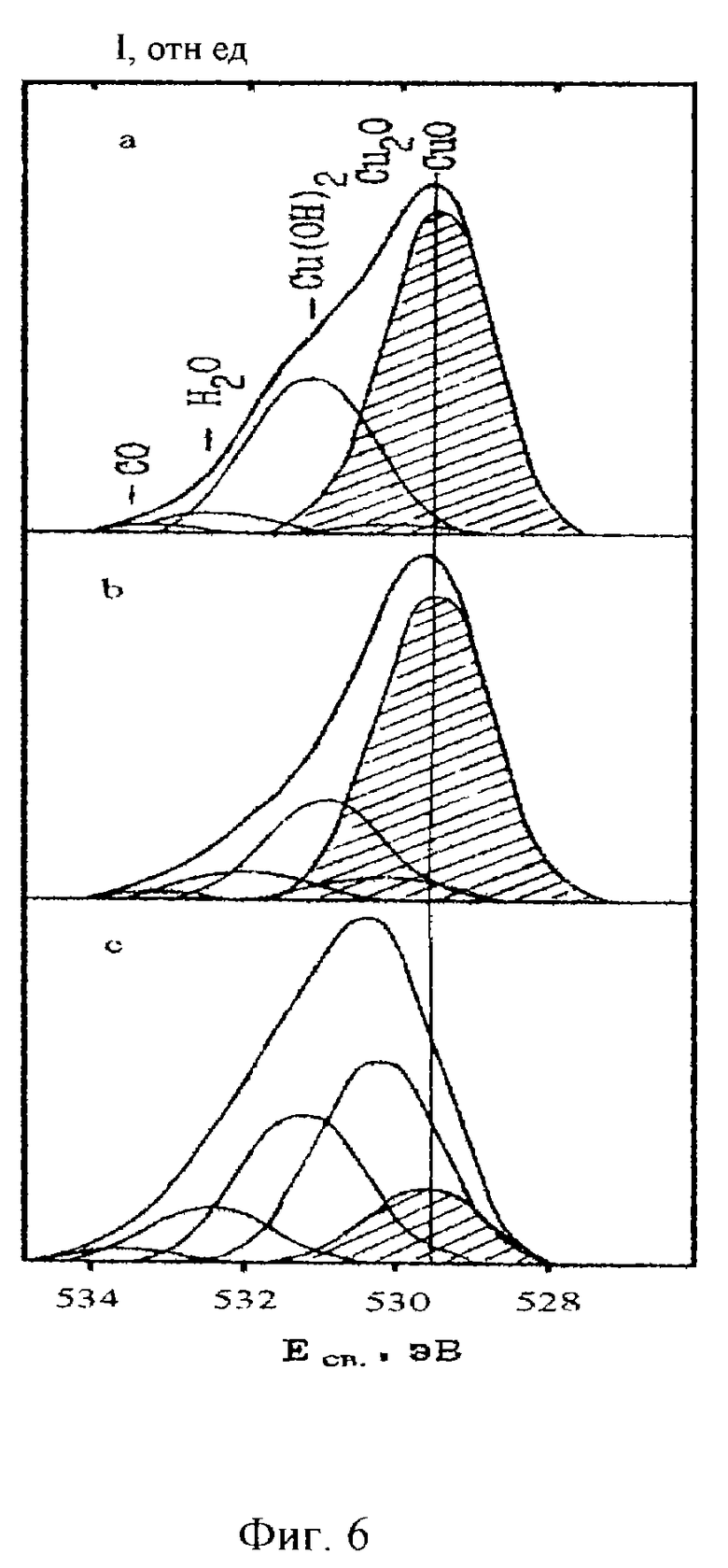
На фиг. 5 и 6 приведены результаты разложения фотоэлектронных спектров кислорода и меди, полученные в соответствии с предлагаемым способом для оксидной пленки, выращивавшейся магнетронным распылением меди в присутствии атомарного потока кислорода [6]. Заштрихованные элементарные составляющие соответствуют двухвалентному оксиду меди CuO. В работе ставилась задача получения максимальной доли этого оксида. Видно, что максимальное окисление достигается в режимах, при которых были получены спектры, приведенные в средней части фиг. 5 и 6.
Таким образом, приведенные примеры свидетельствуют об эффективности предлагаемого способа: способ повышает точность и достоверность количественного определения химического состава вещества и энергий связи электронов остовных уровней и, кроме того, является безэталонным.
Литература
1. М.П.Сих, Д.Бриггс. "Анализ поверхности методами оже- и рентгеновской фотоэлектронной спектроскопии". М.: Мир, 1987, с. 203-220, 598 с.
2. М.П.Сих, Д.Бриггс. "Анализ поверхности методами оже- и рентгеновской фотоэлектронной спектроскопии". М.: Мир, 1987, 221-241, 598 с.
3. Д.Вудраф, Т.Денчар. Современные методы исследования поверхности. М.: Мир, 1989, 540 с.
4. К. Зигбан, К.Нордлинг, А.Фальман, Р.Нордберг, К.Хамрин, Я.Хедман, Г. Иоханссон, Т. Бергмарк, С. Карлссон, И.Линдгрен, Б.Линдберг. "Электронная спектроскопия". М.: Мир, 1971, 493 с.
K. Siegban, C.Nordling, A.Fahlman, R.Nordberg, K.Hamrin, J.Hedman, G.Johansson, T. Bergmark, S.-E.Karlsson, I.Lindgren, B.Lindberg. "Atomic, Molecular and Solid State Structure Studied by Means of Electron Spectroscopy" Nova Acta Regiae Societatis Scientarium Upsalien, Ser.IV, vol.20, ESCA. Uppsala 1967.
5 Е. Д.Белякова, А.В.Габараева, А.Т.Гореленок, Р.В.Каржавин, В.М.Микушкин, С.Е.Сысоев, Н.М.Шмидт, Поверхность, 1992, N 7, с. 88-93.
6. Э.М.Шер, В.М.Микушкин, С.Е.Сысоев, Б.Т.Мелех, ЖТФ, т. 70 (2000).
7. G. Hollinger, E.Berngignat, J.Joseph, and Y.Robach, J.Vac.Sci.Tech., v.A3, p.2082, 1985.
8. Maria Faur, Mircea Faur, D.T.Jayne, M.Goradia and C.Goradia, Surface and Interface Analysis, v. 15, 641 (1990).
9. H. Ishii, H.Hasegawa, A.Ishii, H.Ohno, Appl. Surf.Sci., v. 41/42, p. 390 (1989).
10. S.M.Thurgate, N.E.Erickson, J.Vac.Sci.Tech., v. A8, p. 3669 (1990).
11. C.W.Wilmsen, R.W.Kee, J.Vac.Sci.Tech.,v. 15, p. 1513 (1978).
12. I.K.Han, E.K.Kim, J.I.Lee, S.H.Kim, K.N.Kang, Y.Kim, H.Lim, H.L.Park, J.Appl.Phys. v. 81, 6986 (1997).
13. Y. S. Lee and W.A.Anderson, J.Appl.Phys., v. 65, Issue 10, p. 4051 (1989).
14. N. Shibata and H.Ikoma, Jap. J.Appl.Phys., v. 31, part 1, N 12A, p. 3976 (1992).
15. L.L. Kazmerski, P.J.Ireland, P.Sheldon, T.L.Chu, S.S.Chu, C.L.Li, J. Vac.Sci.Tech.,v. 17, p. 1061 (1980).
16. A.Guivarc'h, H.L'Haridon and G.Pelous, J.Appl Physics, v. 55, Issue 4, p. 1139 (1984).


Изобретение относится к области анализа материалов с помощью рентгеновского излучения и может быть использовано для неразрушающего анализа химического состава многокомпонентных материалов и определения энергии связи остовного уровня атома, находящегося в определенном химическом состоянии. Для этого в известном способе, включающем измерение линии фотоэлектронного спектра по меньшей мере одного элемента, разложение ее по известному набору элементарных составляющих, соответствующих различным химическим фазам, и известной последовательности энергий связи остовного электрона выбранного элемента в этих фазах, минимизацию функционала ошибок между измеренной линией спектра и суммарной расчетной огибающей набора элементарных составляющих с выбором их амплитуд и ширины в качестве свободных параметров и определение искомого состава по относительному вкладу этих составляющих в разлагаемую линию спектра с учетом стехиометрии химических фаз согласно формуле изобретения, вышеупомянутые операции производят для каждой линии двух или более выбранных элементов, дополнительно выбирая в качестве свободных параметров энергии связи элементов в химич. фазах и, сравнивая полученные для всех линий спектра результаты между собой, варьируют свободными параметрами до совпадения результатов с учетом заданной ошибки. Техническим результатом изобретения является возможность без использования эталонов определять с высокой точностью не только химический состав сложных соединений, но и энергий связи элементов. 6 ил., 2 табл.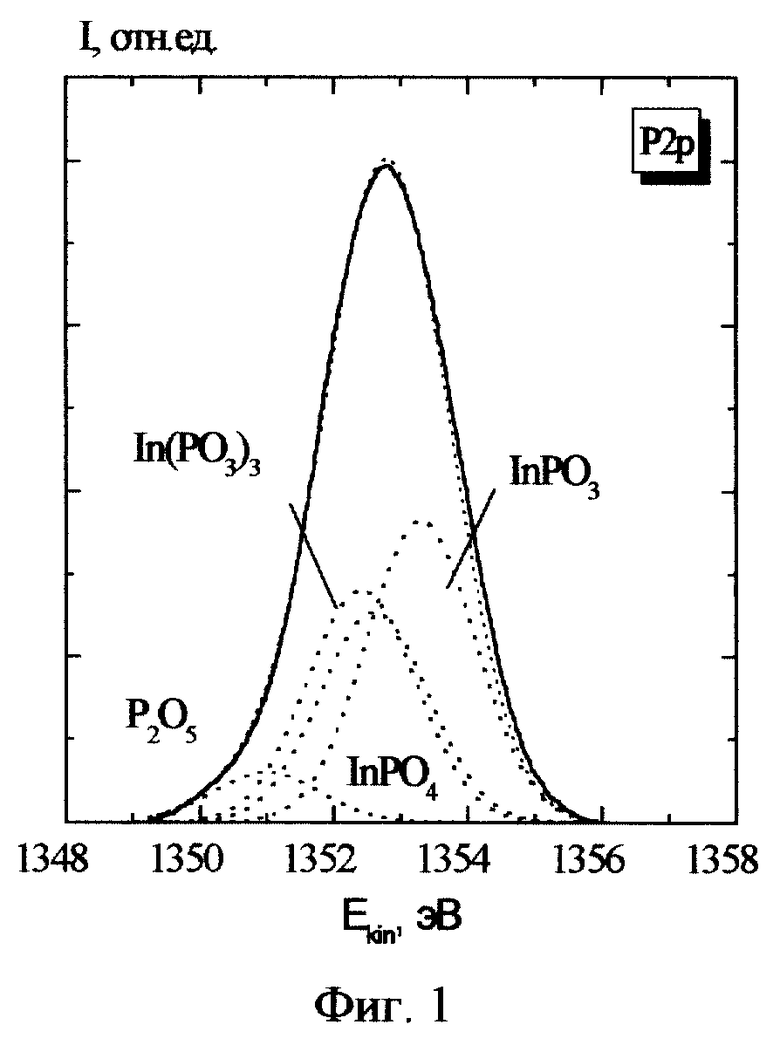
Способ количественного определения химического состава вещества и энергий связи остовных электронов, включающий измерение линии фотоэлектронного спектра по меньшей мере одного элемента, разложение ее по известному набору элементарных составляющих, соответствующих различным химическим фазам, и известной последовательности энергий связи остовного электрона выбранного элемента в этих фазах, минимизацию функционала ошибок между измеренной линией спектра и суммарной расчетной огибающей набора элементарных составляющих с выбором их амплитуд и ширины в качестве свободных параметров и определение искомого состава по относительному вкладу этих составляющих в разлагаемую линию спектра с учетом стехиометрии химических фаз, отличающийся тем, что вышеупомянутые операции производят для каждой линии спектра двух или более выбранных элементов, дополнительно выбирая в качестве свободных параметров энергии связи элементов в химических фазах и, сравнивая полученные для всех линий спектра результаты между собой, варьируют свободными параметрами до совпадения результатов с учетом заданной ошибки.
| СИХ М.П., Бриггс Д | |||
| Анализ поверхности методами оже- и рентгеновской фотоэлектронной спектроскопии | |||
| - M.: Мир, 1987, с.221-241 и 598 | |||
| ПРИБОР СИСТЕМЫ БЕЗОПАСНОСТИ ЖЕЛЕЗНОДОРОЖНОЙ ЦИСТЕРНЫ | 2013 |
|
RU2600422C1 |
| УСТРОЙСТВО ДЛЯ ОЧИСТКИ ДЛИННОМЕРНЫХ ЦИЛИНДРИЧЕСКИХ ИЗДЕЛИЙ ОТ ОКАЛИНЫ | 2004 |
|
RU2268802C1 |
| СПОСОБ ПРОИЗВОДСТВА ВОДКИ | 1995 |
|
RU2105805C1 |

