Настоящее изобретение относится к новым производным пептидных гормонов и их аналогам, обладающим пролонгированным профилем действия, а также к способам получения и использования этих производных.
Предпосылки создания изобретения
Пептидные гормоны широко используются в медицинской практике, и поскольку они могут быть продуцированы с использованием техники рекомбинантных ДНК, то можно ожидать, что с годами их значение будет возрастать. При использовании нативных пептидных гормонов или их аналогов в терапии было установлено, что они имеют высокую скорость выведения. Такая высокая скорость выведения терапевтического агента может оказаться нежелательной в случаях, когда необходимо поддерживать высокий уровень этого агента в крови в течение продолжительного периода времени, поскольку, в противном случае, его следует затем вводить повторно. Примерами пептидных гормонов с высокой скоростью выведения являются АСТН; кортикотропин-высвобождающий фактор; ангиотензин; кальцитонин; инсулин и его фрагменты и аналоги; глюкагон; глюкагон-подобный пептид и его аналоги и фрагменты; ICF-1; ICF-2; энтерогастрин; соматостатин, соматотропин; соматомедин; паратироидный гормон; тромбопоэтин; эритропоэтин; гипоталамические рилизинг-факторы; пролактин; тиреотропные гормоны; эндорфины; энкефалины; вазопрессин; окситоцин; опиаты и их аналоги; супероксид-дисмутаза; интерферон; аспарагиназа; аргиназа; аргининдеаминаза; аденозиндеаминаза; и рибонуклеаза.
Хотя в некоторых случаях на профиль высвобождения пептидных гормонов может оказать влияние применение подходящих фармацевтических композиций, этот способ имеет ряд недостатков и, в основном, не применяется. В соответствии с вышеизложенным очевидно, что необходимо разработать более эффективные способы введения пептидных гормонов.
Краткое описание изобретения
В настоящем описании, термин "пептид" используется для обозначения небольших пептидов, полипептидов и белков. Термин "пептид" и "пептидный гормон", используемые в настоящем описании, охватывают как встречающиеся в природе, так и синтетические пептидные гормоны, а также их фрагменты и аналоги. Аналоги представляют собой пептиды, в которых одна или несколько аминокислот исходного пептида были делетированы или заменены другими аминокислотами, или в которые была добавлена одна или несколько аминокислот, и которые, кроме того, в качественном (но не обязательно в количественном) отношении обладают фармакологическим действием, аналогичным действию исходного пептида.
В общих чертах настоящее изобретение относится к новым производным пептидных гормонов, которые имеют пролонгированный профиль действия.
Таким образом, в наиболее широком своем аспекте настоящее изобретение относится к фармакологически активному пептидному гормону, который был модифицирован путем введения липофильного заместителя, содержащего 8 - 40 атомов углерода, и N-концевую или C-концевую аминокислоту нативного пептидного гормона или его аналога, при условии, что, в случае, если липофильный заместитель связан с N-концевой аминогруппой, то этот заместитель содержит группу, которая может быть отрицательно заряжена, и при условии, что указанный пептидный гормон не является инсулином или его аналогом.
В одном из предпочтительных вариантов настоящего изобретения карбоксильная группа, содержащаяся в липофильной группе W, образует амидную связь вместе с α-аминогруппой N-концевой аминокислоты исходного пептида.
В другом предпочтительном варианте настоящего изобретения карбоксильная группа, содержащаяся в липофильной группе W, образует амидную связь вместе с ε-аминогруппой N-концевого лизина.
В другом предпочтительном варианте настоящего изобретения липофильная группа W состоит из спейсера и объемного липофильного заместителя. Предпочтительным спейсером является янтарная кислота, Glu или Asp. Объемным липофильным заместителем является, предпочтительно, жирная кислота с прямой цепью, которая необязательно имеет аминогруппу. Если в качестве спейсера используется янтарная кислота, то одна из ее карбоксильных групп образует амидную связь с аминогруппой, присутствующей в N-концевой аминокислоте исходного пептида, а другая карбоксильная группа образует амидную связь с аминогруппой, содержащейся в объемной липофильной группе. Если в качестве спейсера используется Glu или Asp, то одна из карбоксильных групп образует амидную связь с аминогруппой, присутствующей в N-концевой аминокислоте исходного пептида, а объемным липофильным заместителем предпочтительно является ацильная группа жирной кислоты с прямой цепью, или кислоты, которая имеет частично или полностью гидрогенизированный циклопентанофенантреновый остов, где указанная ацильная группа связана с аминогруппой спейсера.
В другом предпочтительном варианте настоящего изобретения аминогруппа, содержащаяся в липофильной группе Z, образует амидную связь вместе с карбоксильной группой C-концевой аминокислоты исходного пептида.
В другом предпочтительном варианте настоящего изобретения, Z представляет жирную кислоту с прямой цепью, имеющую аминогруппу.
В другом предпочтительном варианте настоящего изобретения Z имеет группу, которая может быть отрицательно заряжена.
В другом предпочтительном варианте настоящего изобретения Z имеет свободную карбоновокислотную группу.
В другом предпочтительном варианте настоящего изобретения липофильная группа Z состоит из спейсера и объемного липофильного заместителя. Спейсером являются предпочтительно Lys, Glu или Asp. Если в качестве спейсера используется Lys, то объемным липофильным заместителем, в одном из предпочтительных вариантов, является ацильная группа жирной кислоты, имеющей прямую цепь, или кислоты, имеющей частично или полностью гидрогенизированный циклопентанофенантреновый остов, где указанная ацильная группа связана с аминогруппой спейсерной группы. В еще одном предпочтительном варианте настоящего изобретения, в случае, если в качестве спейсера используется Lys, то между ε-аминогруппой Lys и объемным липофильным заместителем вставляется дополнительный спейсер. В одном из предпочтительных вариантов таким дополнительным спейсером является янтарная кислота, которая образует амидную связь с ε-аминогруппой Lys и аминогруппой, присутствующей в объемном липофильном заместителе. В другом предпочтительном варианте таким дополнительным спейсером является Glu или Asp, которые образуют одну амидную связь с ε-аминогруппой Lys, а другую амидную связь с карбоксильной группой, присутствующей в объемном липофильном заместителе, который является предпочтительно жирной кислотой, имеющей прямую цепь, или кислотой, имеющей частично или полностью гидрогенизированный циклопентанофенантреновый остов.
В другом своем предпочтительном варианте настоящее изобретение относится к использованию пептидных производных настоящего изобретения в качестве лекарственных средств.
В другом своем предпочтительном варианте настоящее изобретение относится к лекарственным средствам, содержащим пептидные производные настоящего изобретения.
В другом своем предпочтительном варианте настоящее изобретение относится к способу лечения остеопороза у пациента, нуждающегося в таком лечении, предусматривающему введение указанному пациенту терапевтически эффективного количества производного IGF-1 настоящего изобретения в сочетании с фармацевтически приемлемым носителем.
В качестве примеров исходных пептидных гормонов, которые могут быть использованы в целях настоящего изобретения, являются следующие пептиды: АСТН (адрено-кортикотропный гормон), кортикотропин-рилизинг-фактор, ангиотензин, кальцитонин, глюкагон, глюкагонподобный пептид и его аналоги и фрагменты, например, GLP-1 u GLP-2 и их аналоги и фрагменты, IGF-1, IGF-2, энтерогастрин, соматостатин, соматотропин, соматомедин, паратироидный гормон, тромбопоэтин, эритропоэтин, гипоталамические рилизинг-факторы, пролактин, тиреотропные гормоны, эндорфины, энкефалины, вазопрессин, окситоцин, опиаты и их аналоги, супероксид-дисмутаза, интерферон, аспарагиназа, аргиназа, аргининдеаминаза, аденозиндеаминаза и рибонуклеаза.
Примерами особенно предпочтительных производных IGF-I и аналогов IGF-I являются
Lys68 (Nε-тeтpaдeкaнoил)дeз (69,70) IGF-I человека;
Lys68 [Νε-γ-GLu ( Nα-гексадеканоил)-ОН]-ОН дез (69, 70) IGF-1 человека;
Lys69( Νε- етрадеканоил) дез (70) IGF-1 человека;
Ser69- NH(CH2)nCOOH дез (70) IGF-1 человека, где n является целым числом от 12 до 24;
Ser69- NH(CH2)nCH3 дез (70)IGF-1 человека, где n является целым числом от 12 до 24;
Lys71( Νε-тетрадеканоил) IGF-1 человека;
Ala70-NH(CH2)nCOOH IGF-1 человека, где n является целым числом от 12 до 24; и
Ala70-NH(CH2)nCH3 IGF-1 человека, где n является целым числом от 12 до 24.
Предпочтительным производным соматостатина является Ala- Gly-Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Tyr-Thr-Ser-Cys-Lys[ Νε-γ-Gly( Να-тетрадеканоил)-ОН] -ОН (два цистеиновых остатка соединены посредством дисульфидного мостика).
Предпочтительным производным GLP-1 является His-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Тyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys[Νε-γ-Glu( Να-тетрадиканоил) -OH]-OH.
Предпочтительным аналогом ANP является
Ser-Leu-Arg-Arg-Ser-Ser-Cys-Phe-Gly-Gly-Arg-Met-Asp-Arg-Ile- Gly-Ala-Gln-Ser-Gly-Leu-Gly-Cys-Asn-Ser-Phe-Arg-Tyr-Lys[ Νε-γ-Glu( Να-тетрадеканоил)-ОН]-ОН.
Предпочтительным типом производного аналога динорфина является:
Tyr-Gly-Gly-Phe-Cys-Arg-Arg-D-Ala-Arg-Pro-Cys-NH-(CH2)n-COOH, где n является целым числом от 8 до 24.
Предпочтительным производным энтерогастрина является H-Ala-Pro-Gly-Pro-Arg-Lys ( Νε -тетрадеканоил)-ОН.
Подробное описание изобретения
Фармацевтические композиции, содержащие пептидное производное настоящего изобретения, могут быть введены парентерально пациенту, нуждающемуся в соответствующем лечении. Парентеральное введение может быть осуществлено путем подкожной, внутримышечной или внутривенной инъекции с помощью шприца, необязательно, перобзного шприца. Альтернативно парентеральное введение может быть осуществлено с помощью инфузионного насоса. В качестве другой альтернативы может быть использована композиция в виде порошка или раствора для введения пептидного производного в форме интраназального аэрозоля. В качестве еще одной альтернативы пептидные производные могут быть также введены чрескожно.
Фармацевтические композиции, содержащие соединение настоящего изобретения, могут быть получены с использованием стандартной технологии, описанной, например, в Remington s Pharmaceutical Sciences, 1985.
Так, например, инъецируемые композиции, содержащие пептидные производные настоящего изобретения, могут быть получены стандартными методами, используемыми в фармацевтической промышленности, которые предусматривают растворение и смешивание соответствующих ингредиентов с получением нужного целевого продукта.
Например, в соответствии с одним из таких методов пептидное производное растворяют в некотором количестве воды, несколько меньшем, чем конечный объем изготавливаемой композиции. Если необходимо, могут быть добавлены изотонический агент, консервант и буфер, а pH раствора, если это требуется, может быть скорректирован с использованием кислоты, например соляной кислоты, или основания, например водного гидроксида натрия. И наконец, объем раствора может быть также скорректирован путем добавления воды для получения нужной концентрации ингредиентов.
Примерами изотонических агентов являются хлорид натрия, маннит и глицерин.
Примерами консервантов являются фенол, м-крезол, метил, п-гидроксибензоат и бензиловый спирт.
Примерами подходящих буферов являются ацетат натрия и фосфат натрия.
Композиция для интраназального введения некоторых пептидных гормонов может быть получена, например, как описано в Европейском патенте N 272097 (to Novo Nordisk A/S). Пептидные производные настоящего изобретения могут быть использованы для лечения различных заболеваний. Выбор конкретного пептидного производного, а также оптимальной дозы для введения конкретному пациенту зависит от заболевания пациента и от ряда других факторов, включая эффективность конкретно используемого пептидного производного, возраст, вес тела, физическое состояние и режим питания пациента, возможное использование комбинации с другими лекарственными средствами и тяжесть заболевания. При этом следует отметить, что доза пептидного производного настоящего изобретения может быть определена специалистом для каждого отдельного пациента в соответствии с существующей практикой использования известных пептидных гормонов.
Более подробно, настоящее изобретение проиллюстрировано в нижеследующих примерах, которые, однако, не должны рассматриваться как некоторое ограничение объема изобретения. Отличительные признаки изобретения, раскрытые в вышеприведенном описании и в нижеприведенных примерах, и взятые как отдельно, так и в комбинации друг с другом, могут служить основой для осуществления изобретения в его различных вариантах.
Примеры
Сокращения:
Fmoc: 9-флуоренилметилоксикарбонил;
For: формил;
Dde: 1-(4,4-диметил-2,6-диоксоциклогексилидин)-этил;
ДМФ: N,N-диметилформамид;
Tbu: трет-бутил;
Acm: ацетамидометил;
DIG: N,N' -диизопропилкарбодиимид;
НОВТ: 1-гидроксибензотриазол;
TFA: трифторуксусная кислота.
Аналитическая часть
Молекулярные массы полученных продуктов определяли с помощью плазменной десорбционной масс-спектрометрии (ПДМС) с использованием прибора Bio-Ion 20 (Bio-Ion Nordic AB, Uppsala, Sweden).
Определение липофильности:
Липофильность пептидов и производных пептидов по отношению к инсулину человека, k'rel, измеряли на ВЭЖХ-колонке LiChrosorb RP18 (5 мкм, 4 x 250 мм) путем изократного элюирования при 40oC с использованием в качестве элюентов смесей A) 0,1М фосфатно-натриевого буфера, pH 7,3, содержащего 10% ацетонитрил, и В) 50% ацетонитрил в воде. За ходом элюирования следили с помощью УФ-абсорбции элюата при 214 нм. Время пробега tо определяли путем введения 0,1 мМ нитрата натрия. Время удерживания для инсулина человека, tинсулин, доводили по крайней мере до 2tо путем изменения отношения между растворами A и В. k'rel = (tпроизв одное- tо/(tинсулин -tо).
Пример 1
Синтез For-Nle-Leu-Phe-Nle-Tyr-Lys
( Νε-тетрадеканоил)-ОН
For-Nle-Leu-Phe-Nle-Tyr-Lys-OH поставлялся от Bachem Feinchemikalien AG, Switzerland. Этот пептид является сильно действующим хемоаттрактаном для нейтрофилов человека. Целевое соединение получали путем растворения 17 мг For-Nle-Leu-Phe-Nle-Tyr-Lys-OH в 5 мл ДМФ, с после дующим добавлением 35 мкл триэтиламина, и добавлением к полученному раствору 20 мг твердого сукцинимидил-Н-гидрокси-эфира тетрадекановой кислоты. За ходом реакции наблюдали с помощью обращенно-фазовой жидкостной хроматографии высокого разрешения (ОФ-ЖХВР) с использованием обращенно-фазовой колонки с двуокисью кремния С18. Для элюирования был использован градиент смеси 30% этанола ---> 80% этанола в 0,1% водном растворе трифторуксусной кислоты. Полученный продукт очищали на обращенно-фазовой колонке (длина = 250 мм; диаметр = 20 мм) с двуокисью кремния C18. Затем, соединение растворяли в 74% этаноле/0,1% водном растворе трифторуксусной кислоты, после чего наносили на колонку и очищали при температуре 40oC путем изократного элюирования в том же буфере при скорости потока 6 мл/час. Выход составлял 20 мг. Идентичность соединения подтверждали с помощью ПДМС.
Молекулярная масса (ПДМС): Найдено, 1034, теор.:1034.
Липофильность целевого соединения по отношению к инсулину человека, составила 8,2 x 103.
Стандарт
Референс-соединение For-Nle-Leu-Phe-Nle-Tyr-Lys-OH поставлялось от Bachem Feinchemikalien AG, Switzerland и было использовано в том виде, в каком оно было получено. Липофильность референс-соединения по отношению к инсулину человека составляла 2,3.
Пример 2
Синтез H-Tyr-D-Ala-Gly-Phe-Leu-Lys (Νε-тетрадеканоил)-ОН
Энкефалиновое производное H-Tyr-D-Ala-Gly-Phe-Leu-Lys ( Νε -тетрадеканоил)-ОН получали из соединения Вос-Tyr-D- Ala-Gly-Phe-Leu-Lys-OH (A-2435 Bachem Feinchemikalien AG, Switzerland). Воc-Tyr-D-Ala-Gly-Phe-Leu-Lys-OH ацилировали с использованием сукцинимидил-N-гидроксиэфира тетрадекановой кислоты, как описано в Примере 1. Полученную реакционную смесь выпаривали досуха, и образовавшийся остаток растворяли в трифторуксусной кислоте и выпаривали досуха, а затем солюбилизировали в этаноле/воде/0,1% трифторуксусной кислоте, и очищали с помощью обращенно-фазовой жидкостной хроматографии высокого разрешения как описано в Примере 1. Выход составлял 15 мг.
Молекулярная масса (ПДМС-анализ): найдено: 909, теоретич.: 907.
Липофильность целевого соединения по отношению к инсулину человека составляла 2,3 х 103.
Стандарт
Референс-соединение H-Tyr-D-Ala-Gly-Phe-Leu-Lys-OH синтезировали из Boc-Tyr-D-Ala-Gly-Phe-Leu-Lys-OH путем растворения 20 мг этого соединения в 200 мкл трифторуксусной кислоты и выпаривали досуха. Остаток растворяли в 5% уксусной кислоте и осушали вымораживанием. Липофильность референс-соединения по отношению к инсулину человека составляла 3,0 x 10-3.
Пример 3
Синтез H-Pro-His-Pro-Phe-His-Phe-Phe-Val-Tyr-Lys(Νε- тетрадеканоил)-ОН
Fmoc-Pro-His-Pro-Phe-His-Phe-Phe-Val-Tyr- Lys-OH (поставляемый от Bachem Feinchemikalien, AG, Switzerland), который является сильнодействующим ингибитором ренина, подвергали реакции с сукцинимидил-N-гидроксиэфиром тетрадекановой кислоты, как описано в Примере 1. После реакции ацилирования группу Fmoc удаляли путем добавления пиперидина к реакционной смеси до конечной концентрации 20%. Целевое соединение выделяли с помощью обращенно-фазовой жидкостной хроматографии высокого разрешения, как описано в Примере 1. Выход: 23 мг.
Молекулярная масса (ПДМС-анализ): найдено: 1529,6, теоретич.1529.
Липофильность целевого соединения по отношению к инсулину человека составляла 5,3 x 103.
Стандарт
Референс-соединение H-Pro-His-Pro-Phe-His-Phe-Phe- Val-Tyr-Lys-OH синтезировали из Fmos-Pro-His-Pro-Phe-His-Phe-Phe-Val-Tyr-Lys-OH (от фирмы Bachem Fein-chemikalien AG, Switzerland). Таким образом, 20 мг Fmor-Pro-His-Pro-Phe-His-Phe-Phe-Val-Tyr-Lys- OH растворяли в 500 микролитрах 20%-ного пиперидина в ДМФ и оставляли на 20 минут. Референс-соединение очищали с помощью обращенно-фазовой жидкостной хроматографии высокого разрешения, как описано в Примере 1.
Липофильность сравнительного соединения по отношению к инсулину человека составляла 2,3 х 102.
Пример 4
Синтез Arg4, Arg9, Lys15 (Νε-тетрадеканоил)сомотостатина
Целевое соединение синтезировали из Fmoc-Arg4, Arg9, Lys15 - соматостатина, который поставлялся фирмой Saxon Biochemicals GMBH, Hannover, Germ. 50 мг Fmoc-Arg4, Arg9, Lys15-соматостатина растворяли в смеси 346 микролитров ДМФ и 53,9 микролитров 4-метилморфолина. Полученную смесь охлаждали до температуры 15oC и добавляли 15,9 мг сукцинимидил-N-гидроксиэфира тетрадекановой кислоты, растворенного в 100 мкл ДМФ. Полученную реакционную смесь оставляли для реакции на 3 часа 20 минут, а затем реакцию прекращали путем добавления 4140 мкл 5% уксусной кислоты в ДМФ. Целевое соединение очищали с помощью обращенно-фазовой жидкостной хроматографии высокого разрешения следующим образом: образец наносили на колонку (10 x 250 мм) Lichrosorb RP-18 (7 мкм) Merck, Germany, Art. 9394. Колонку уравновешивали смесью 90% буфера A (50 мМ Триса, 75 мМ (NH4)2SO4, доведенного до pH 7,0 путем добавления H2SO4 и 20% CH3CN) и 10% буфера B (80% CH3CN). И наконец, образец наносили на колонку и элюировали линейным градиентом 10% ---> 90% буфера В в буфере А при скорости потока 4 мл/час и при температуре 40oC. Фракции, содержащие целевое соединение, выпаривали досуха, растворяли в 50% уксусной кислоте, и обессоливали путем гель-фильтрации при 4oC на колонке (16 x 150 мм) BIO GEL P2 (BIO RAD, Калифорния, США). Фракции, содержащие нужный продукт разводили водой и осушали вымораживанием. Выход составлял 2 мг. Идентичность соединения подтверждали с помощью ПДМС. Молекулярная масса (ПДМС-анализ): найдено: 2033, теор.: 2032.
Определение пролонгированного действия пептидного производного у свиней
Целевое пептидное производное Примера 4 подвергали 125I-мечению с использованием реагента Boulton & Hunters (Bolton A.E. & Hunter, W.M. (1973) Biochem.J. 133, 529- 539) следующим образом: 50 нмоль пептида растворяли в 1 мл диметилсульфоксида, а затем добавляли 400 мкл диметилформамида и 2 мкл N-этилизопропиламина. Полученный раствор добавляли к определенному количеству реагента Boulton & Hunters, имеющему радиоактивность 500 мкКи. Реакционную смесь оставляли для прохождения реакции на 20 минут, а затем добавляли 10 мкл этаноламина в ДМФ. Полипептид выделяли и очищали с помощью обращенно-фазовой ВЭЖХ на колонке (4 x 250 мм) при скорости потока 1 мл/мин, как описано выше.
Для оценки продолжительности присутствия пептидного производного у свиней была измерена скорость выведения этого производного из организма животных и было определено T50%. T50% обозначает время, за которое из места инъекции выводится 50% 125I-меченного пептида, определяемого с помощью внешнего γ - счетчика (Ribel, U.et al., The Pig as a Model for Sugcutaneous Absorption in Man. B: M. Serra-no-Rios and P.J. Lefebre (Eds): Diabetes 1985; Proceedings of the 12th Congress of the International Diabetes Federation., Madrid, Spain, 1985 (Excerpta Medica, Amsterdam, (1986) 891-96).
Подкожные инъекции 125-I-меченного пептидного производного, введенного свиньям, показали время Т50%= 1,7 ± 0,5 ч. (n=4), а подкожная инъекция нететрадеканоилированного 125I-меченного эталонного пептида показала время T50%= 0,7 ± 0,1 ч.
Стандарт
125I-меченный эталонный пептид синтезировали из Fmoc-Arg4, Arg9, Lys15-соматостатина. Так, например, 20 мг Fmos-Arg4, Arg9, Lys15-соматостатина растворяли в 1000 мкл 20% пиперидина/ДМФ. Через 20 минут продукт очищали с помощью обращенно-фазовой ВЭЖХ, обессоливали и осушали вымораживанием, и метили реагентом Boulton & Hunters, как описано в Примере 4.
Пример 5
Синтез атриального натрийуретического Lys15 (Νε-тетрадеканоил)-пептида.
Человеческий пептид
(H-Ser-Leu-Arg-Arg-Ser-Ser-Cys-Phe-Gly-Gly-Arg-Met-Asp-Arg- Ile-Gly-Ala-Gln-Ser-Gly-Leu-Gly-Cys-Asn-Ser-Phe-Arg-Tyr-Lys(Νε- тетрадеканоил) -COOH) синтезировали с помощью стандартного твердофазного пептидного синтеза с использованием Fmoc- группы (Methods in Molecular Biology, Vol.35: Peptide Synthesis Protocols). ε - Аминогруппу C-концевого лизина лизировали с использованием сукцинимидил-N-гидрокси-эфира тетрадекановой кислоты в соответствии с процедурой, описанной ниже. Синтез осуществляли вручную в полипропиленовых шприцах, при этом в качестве полимерного носителя использовали низкомолекулярный структурированный полистироловый остов с привитым к нему полиоксиэтиленом (Tenta-Gel Resin).
Процедура:
Один грамм смолы добавляли к 3 эквивалентам кислотного лабильного линкера, 4-гидроксиметилфеноксиуксусной кислоте (НМРA). 3 Эквивалента Fmoc-Lys(Dde)-OH в качестве первой аминокислоты связывали с 0,5 эквивалентами 4-диметиламинопиридина, используемого в качестве активирующего агента. Защитную Fmoc-группу отщепляли путем воздействия 20% пиперидином/ДМФ в течение 30 минут. Все остальные аминкоислоты присоединяли в виде N-Fmoc- защищенных аминокислот с использованием смеси DIC /НОВТ ((1:1 экв.) в ДМФ в качестве активирующих реагентов. Аминокислоту Cys присоединяли в виде Fmoc-Cys(Acm)-OH.
Цистеины деблокировали и окисляли путем обработки 10 мМ иода в ДМФ в течение 2 минут. После удаления последней защитной Fmoc-группы, Να-группу последней присоединенной аминокислоты защищали Вос-группой путем связывания с 5 эквивалентами ди-трет-бутил-дикарбоната. Защитную Dde-группу Νε-Lys отщепляли путем обработки 2% гидразином/ДМФ в течение 20 минут, и свободную Νε-группу ацилировали 5 эквивалентами сукцинимидил-N-гидроксиэфира тетрадекановой кислоты. Защитные группы Bоc- и tBu- а также НМРА-линкер отщепляли путем обработки 95% TFA/5% H2О в течение полутора часов. Затем TFA/H2О выпаривали при пониженном давлении, и пептид осаждали в диэтиловом эфире в виде HCl-соли, после чего проводили осушку вымораживанием из 10 мМ бикарбоната аммония (pH 8,8). Полный выход составил 35 мг. N-концевое секвенирование продукта подтвердило получение правильной последовательности.
Молекулярная масса, определенная с помощью ПДМС, составляет 3417, что соответствует вычисленной массе плюс натрий.
Пример 6
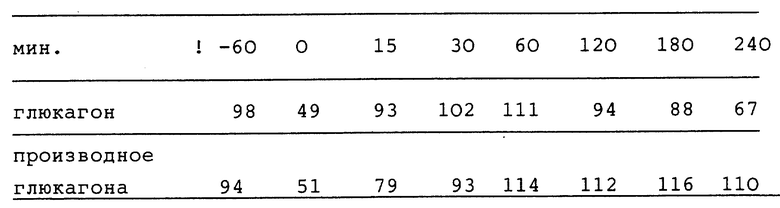
Lys30 (Νε-деканоил)-глюкагон
Целевое соединение поставлялось от фирмы Saxon Biochemicals GMBH, Ганновер, Германия, в качестве заказа для синтеза. 4,32 мг Lys30 ( Νε-деканоил), глюкагон (эквивалентные 4 мг глюкагона) растворяли в 4 мл 1,8 мМ соляной кислоты, добавляли 0,9% хлорида натрия, и pH раствора доводили до 2,7 Полученный раствор солюбилизировали путем фильтрации и переносили в сосуд.
Двум группам кроликов (по 6 кроликов в каждой) инъецировали 2 МЕ/животное инсулин-актрапида (Actrapid) во время t =-60 мин. Во время t=0, группе 1 подкожно инъецировали молярный эквивалент 0,54 мг Lys30( Νε- декаканоил) глюкагон на одного кролика, а группе 2 подкожно инъецировали 0,5 мг глюкагона на одного кролика. Пробы крови брали в интервалы времени: -60, 0, 15, 30, 60, 120, 180 и 240 мин, а концентрацию глюкозы определяли гексокиназным методом. Полученные результаты концентраций глюкозы в крови представлены в таблице и выражены в мг глюкозы/100 мл:
Как видно из таблицы, действие глюкагона, направленное на повышение уровня глюкозы в крови, сохраняется yLys30 ( Νε-деканоил)-глюкагона, но действие последнего является более продолжительным.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИДНАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ПРОПИЛЕНГЛИКОЛЬ, ЯВЛЯЮЩАЯСЯ ОПТИМАЛЬНОЙ ДЛЯ ИЗГОТОВЛЕНИЯ И ПРИМЕНЕНИЯ В ИНЪЕКЦИОННЫХ УСТРОЙСТВАХ | 2004 |
|
RU2421238C2 |
| ПЕПТИДНАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ПРОПИЛЕНГЛИКОЛЬ, ЯВЛЯЮЩАЯСЯ ОПТИМАЛЬНОЙ ДЛЯ ИЗГОТОВЛЕНИЯ И ПРИМЕНЕНИЯ В ИНЪЕКЦИОННЫХ УСТРОЙСТВАХ | 2004 |
|
RU2831321C2 |
| ПРОИЗВОДНОЕ АНАЛОГА GLP-1 ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2565536C2 |
| СТАБИЛЬНЫЕ СОВМЕСТНЫЕ АГОНИСТЫ РЕЦЕПТОРА ГЛЮКАГОНОПОДОБНОГО ПЕПТИДА-1/ГЛЮКАГОНА ПРОЛОНГИРОВАННОГО ДЕЙСТВИЯ ДЛЯ МЕДИЦИНСКОГО ПРИМЕНЕНИЯ | 2014 |
|
RU2683039C2 |
| АНАЛОГИ ЭФР(А) С ЗАМЕСТИТЕЛЯМИ - ЖИРНЫМИ КИСЛОТАМИ | 2017 |
|
RU2747877C2 |
| ПРИМЕНЕНИЕ ДОЛГОДЕЙСТВУЮЩИХ ПЕПТИДОВ GLP-1 | 2013 |
|
RU2657573C2 |
| НОВЫЕ АНАЛОГИ ГЛЮКАГОНА | 2011 |
|
RU2559320C2 |
| ТОЛЕРОГЕННАЯ ДНК-ВАКЦИНА | 2017 |
|
RU2752608C2 |
| ПРОИЗВОДНОЕ ИНСУЛИНА, РАСТВОРИМАЯ ПРОЛОНГИРОВАННАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРОЛОНГИРОВАНИЯ ГИПОГЛИКЕМИЧЕСКОГО ДЕЙСТВИЯ ПРИ ЛЕЧЕНИИ ДИАБЕТА | 1994 |
|
RU2164520C2 |
| ДВОЙНОЙ АГОНИСТ РЕЦЕПТОРОВ ГЛЮКАГОНОПОДОБНОГО ПЕПТИДА-1 И ГЛЮКАГОНА ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2021 |
|
RU2799327C1 |
В изобретении раскрывается фармакологически активное производное пептидного гормона, которое происходит от нативного пептидного гормона, модифицированного путем введения липофильного заместителя Z в C-концевую аминокислоту указанного пептидного гормона или его аналога, где указанный липофильный заместитель имеет от 8 до 40 атомов углерода. Целевые соединения относятся к лекарственным средствам для лечения остеопороза. 2 с. и 5 з.п.ф-лы, 1 табл.
-NHCH(COOH) (CH2)4NH-CO(CH2)mCH3,
где m является целым числом от 8 до 18,
то есть Z представляет собой Nε -ацилированный лизиновый остаток.
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИПЕПТИДОВ | 1990 |
|
RU2037498C1 |
| WO 9508567 A1, 30.03.1995 | |||
| WO 9514035 A1, 26.05.1995 | |||
| WO 9516703 A1, 22.06.1995. | |||

