Изобретение относится к твердым фармацевтическим препаратам на основе циклодекстрина. Более конкретно, настоящее изобретение относится к препаратам на основе сульфоалкилового эфира циклодекстрина (САЭ-ЦД), в которых основная часть терапевтического агента не образует комплекс с САЭ-ЦД.
Уровень техники
Патент США N 5134127 (патент '127) относится к производным сульфоалкилового эфира циклодекстрина (САЭ-ЦД). Производные САЭ-ЦД предложены для использования в качестве солюбилизирующих агентов для плохо растворимых в воде или не растворимых в воде лекарственных средств в различных фармацевтических лекарственных формах.
Комплексы циклодекстрин/лекарственное средство обычно получают до их использования в фармацевтических препаратах. Патент '127 относится к композициям и препаратам, содержащим лекарственные средства, образующие комплекс с производным САЭ-ЦД, т.е. комплексы клатрат/лекарственное средство или комплексы включения. Фармацевтические препараты, рассматриваемые в нем, относятся к препаратам, которые содержат клатратный комплекс и фармацевтически приемлемый носитель.
Клатратные комплексы САЭ-ЦД/лекарство готовят отдельно до получения желаемого фармацевтического препарата. Способы получения таких препаратов включают стадии, для которых необходим значительный мониторинг и контроль, что может усложнить процесс приготовления препарата. В фармацевтической промышленности упрощенные способы предпочтительнее сложных способов, и в случае композиций, содержащих циклодекстрин и в особенности САЭ-ЦД, сохраняется необходимость в упрощенных композициях и способах их получения.
Предпринимались попытки получить препараты на основе циклодекстринов с лекарственными средствами, плохо растворимыми в воде, как в виде физических смесей, так и в виде комплексов включения. Muranushi с соавторами (1988) сравнили профили растворения чистого бенексата, физической смеси бенексат/циклодекстрин и комплекса бенексат/циклодекстрин. Они обнаружили значительно более высокую растворимость комплексов бенексата по сравнению с физической смесью или чистым продуктом.
Аналогичные результаты получены J.J. Torres-Labandeira и др. (1994). Ими установлено, что биодоступность комплекса глиборнурида с β-циклодекстрином в 2-3 раза выше, чем биодоступность физической смеси глиборнурид/β-циклодекстрин. D. Peri и др. (1994) также показали, что в случае толнафата комплекс лекарственное средство/β-циклодекстрин имеет улучшенную растворимость по сравнению с растворимостью физической смеси или свободного лекарственного средства. При изучении напроксена и β-циклодекстрина было установлено, что соответствующий комплекс включения имеет в 6-9 раз более высокую растворимость через 5 минут по сравнению с растворимостью физической смеси (Otero-Espinar et al., 1991).
Дополнительное доказательство того, что комплекс включения лекарство/ β-циклодекстрин обладает значительно улучшенным профилем растворения, чем соответствующая физическая смесь, получено Lin с соавторами (1989) при изучении комплексов и физических смесей β-циклодекстрина с ацетаминофеном, индометацином, пироксикамом и варфарином. Esclusa-Diaz и др. (1996) также сообщили, что комплекс кетоконазол/β-циклодекстрин имеет значительно более высокую растворимость, чем соответствующая физическая смесь.
В патенте США N 4946686 (McClelland и др.) обсуждается другое применение физических смесей лекарственное средство/циклодекстрин, но примеры не приводятся. Эта композиция была получена только для лекарственного средства с контролируемым высвобождением, в котором модулирующие растворимость единицы представлены в виде частиц с медленным высвобождением, диспергированных в смеси лекарственных наполнителей. Все компоненты были окружены микропористой нерастворимой в воде стенкой.
Таким образом, в данной области техники известно, что комплексы лекарственное средство/циклодекстрин будут иметь значительно более высокую растворимость, профиль растворения и биодоступность, чем соответствующая физическая смесь. В фармацевтической области существует необходимость в фармацевтических препаратах, содержащих физическую смесь лекарственное средство/циклодекстрин, которая будет обладать профилем растворения, биодоступностью и растворимостью, которые приблизительно равны профилю растворения, биодоступности и растворимости соответствующего комплекса лекарственное средство/циклодекстрин.
Сущность изобретения
В настоящем описании, если не оговорено особо, термины "a" или "an" означают один или несколько.
Настоящее изобретение предполагает способ преодоления недостатков, которыми обладают известные твердые фармацевтические препараты, содержащие физическую смесь терапевтический агент/циклодекстрин. Изобретение относится к упрощенным твердым фармацевтическим композициям и препаратам, содержащим сульфоалкиловый эфир циклодекстрина и предназначенным для доставки терапевтических агентов, а также к способам их получения. Фармацевтические препараты настоящего изобретения преимущественно получают упрощенными способами, которые не требуют предварительного приготовления комплексов САЭ-ЦД с терапевтическими агентами до получения препаратов. Препараты содержат пленочное покрытие вокруг твердого ядра, которое содержит физическую смесь терапевтический агент/простой сульфоалкиловый эфир циклодекстрина, которая при воздействии воды или жидкостей тела образует комплекс терапевтический агент/сульфоалкиловый эфир циклодекстрина. Фармацевтический препарат, содержащий физическую смесь терапевтический агент/простой сульфоалкиловый эфир циклодекстрина, будет иметь растворимость, профиль растворения и/или биодоступность, которые близки к растворимости, профилю растворения и/или биодоступности соответствующего комплекса включения.
Таким образом, в соответствии с одним из аспектов, настоящее изобретение предлагает твердый фармацевтический препарат, содержащий пленочное покрытие и твердое ядро, в котором пленочное покрытие включает пленкообразующий агент и порообразующий агент, а твердое ядро содержит фармацевтически приемлемый носитель и физическую смесь терапевтически эффективного количества терапевтического агента и простого сульфоалкилового эфира циклодекстрина (САЭ-ЦД), где основная часть терапевтического агента не образует комплекс с САЭ-ЦД.
Препараты настоящего изобретения представляют собой простые композиции, полученные с помощью упрощенного способа. Настоящее изобретение также дает возможность готовить разнообразные лекарственные формы, обладающие уникальными характеристиками.
В одном из вариантов осуществления изобретения простой сульфоалкиловый эфир циклодекстрина представляет собой соединение формулы (1):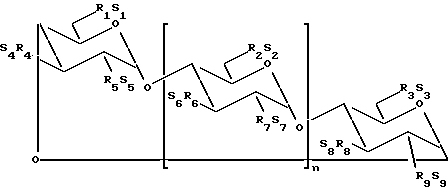
где n принимает значения 5 или 6;
каждый из заместителей R1, R2, R3, R4, R5, R6, R7, R8 и R9 независимо друг от друга представляют собой -O- или -O-(C2-C6-алкилен)-SO3-группу, где, по меньшей мере, один из заместителей R1 и R2 независимо друг от друга представляет собой -O-(C2-C6-алкилен)-SO3-группу, предпочтительно -O-(CH2)m-SO3-группу, где m принимает значение 4 (например, -OCH2CH2CH2SO3- или -OCHCH2CH2CH2SO3-); и
каждый из S1, S2, S3, S4, S5, S6, S7, S8 и S9 независимо друг от друга представляют собой фармацевтически приемлемый катион, который включает, например, H+, щелочные металлы (например, Li+, Na+, K+), щелочноземельные металлы (например, Ca2+, Mg+2), аммонийные ионы и аминные катионы, такие как катионы (C1-C6)-алкиламинов, пиперидина, пиразина, (C1-C6)-алканоламина и (C4-C8)-циклоалканоламина,
где основная часть терапевтического агента не образует комплекс с производным простого сульфоалкилового эфира циклодекстрина.
Пленочное покрытие служит для регулирования высвобождения терапевтического агента и сульфоалкилового эфира циклодекстрина (САЭ-ЦД) из твердого ядра. Пленкообразующий агент является основным компонентом пленочного покрытия и обычно служит для снижения скорости высвобождения терапевтического агента и/или САЭ-ЦД. Подразумевается использование различных пленкообразующих агентов. Порообразующий агент служит для повышения проницаемости пленочного покрытия за счет образования в пленке или пор или участков с повышенной проницаемостью для воды, образованной пленкообразующим агентом.
Фармацевтические препараты, описанные в настоящем изобретении, могут дополнительно содержать один или несколько дополнительных адъювантов и/или активных ингредиентов, которые могут быть выбраны из адъювантов, известных в данной области и включающих вещества, корригирующие вкус и запах (корригенты), разбавители, красители, связующие вещества, наполнители, поверхностно-активные вещества, диспергирующие добавки, биоадгезивы, усилители проникновения, стабилизаторы протеазных ингибиторов и уплотняющие носители.
В соответствии с еще одним аспектом настоящее изобретение предлагает упрощенный способ получения фармацевтических препаратов, содержащих производное простого сульфоалкилового эфира циклодекстрина. Таким образом, изобретение предлагает способ получения содержащей САЭ-ЦД твердой фармацевтической лекарственной формы, который включает:
получение твердого ядра, содержащего физическую смесь производного простого сульфоалкилового эфира и циклодекстрина формулы (1), фармацевтический носитель и эффективное количество терапевтического агента, основная часть которого не образует комплекс с производным сульфоалкилового эфира циклодекстрина; и
нанесение на указанное твердое ядро пленочного покрытия, содержащего пленкообразующий агент и порообразующий агент с получением фармацевтически приемлемой твердой лекарственной формы.
В способе настоящего изобретения нет необходимости в том, чтобы образовывался комплекс простой сульфоалкиловый эфир циклодекстрина/терапевтический агент. Следовательно, основная часть терапевтического агента в конечной лекарственной форме будет находиться в свободной форме, а не в виде комплекса.
В соответствии с еще одним аспектом настоящее изобретение предлагает способ модификации биодоступности и/или скорости биоабсорбции терапевтических агентов. Следовательно, в одном из вариантов настоящее изобретение предлагает способ модификации биодоступности или скорости биоабсорбции терапевтического агента, который включает стадии:
получения простого сульфоалкилового эфира циклодекстрина и терапевтического агента, основная часть которого не образует комплекс с сульфоалкиловым эфиром циклодекстрина, и
введения пациенту терапевтического агента и сульфоалкилового эфира циклодекстрина, причем указанный сульфоалкиловый эфир циклодекстрина модифицирует биодоступность или скорость биоабсорбции указанного терапевтического агента.
Подразумевается, что сульфоалкиловый эфир циклодекстрина и терапевтический агент будут находиться в одной и той же лекарственной форме. Необходимо только, чтобы САЭ-ЦД и терапевтический агент образовывали комплекс после введения их пациенту. Приемлемая лекарственная форма будет обеспечивать гидратацию физической смеси САЭ-ЦД и терапевтического агента, когда они находятся в лекарственной форме, что будет приводить к образованию соответствующего комплекса САЭ-ЦД/терапевтический агент. В рассматриваемых препаратах может быть использован широкий спектр терапевтических агентов, в том числе растворимые в воде, гидрофильные и плохо растворимые в воде гидрофобные терапевтические агенты.
Другие признаки, преимущества и варианты осуществления настоящего изобретения будут очевидны для квалифицированного в данной области специалиста из приведенных ниже описания, примеров и формулы изобретения.
Краткое описание чертежей
Следующие чертежи составляют часть настоящего описания и предназначены для дополнительного пояснения некоторых аспектов изобретения. Изобретение может быть лучше понято при рассмотрении одного или нескольких из этих чертежей и более подробного описания конкретных вариантов осуществления изобретения.
Фиг. 1. Профиль высвобождения препаратов, содержащих метилпреднизолон и СБЭ7-β-ЦД.
Фиг. 2а. Профили высвобождения препаратов, содержащих физическую смесь и лиофилизированный комплекс метилпреднизолона и СБЭ7-β-ЦД.
Фиг. 2б. Профиль высвобождения СБЭ7-β-ЦД препаратов, содержащих физическую смесь и лиофилизированный комплекс метилпреднизолона.
Фиг. 3а и 3б. Профили высвобождения метилпреднизолона (МП) и СБЭ7-β-ЦД из препаратов с 200 мкм пленочным покрытием, содержащих физическую смесь и лиофилизированный комплекс.
Фиг. 4а и 4б. Влияние толщины пленки на профиль высвобождения МП и СБЭ7-β-ЦД в препарате в виде таблетки с пленочным покрытием.
Фиг. 5. Зависимость скорости высвобождения МП и СБЭ7-β-ЦД от увеличения толщины пленки из препарата в виде таблетки, содержащего физическую смесь.
Фиг. 6. Влияние СБЭ7-β-ЦД на высвобождение МП из ядра таблетки без покрытия, содержащего или лиофилизированный комплекс или физическую смесь. Также приведен контроль, в котором СБЭ7-β-ЦД отсутствует.
Фиг. 7а и Фиг. 7б. Влияние мольного отношения МП/СБЭ7-β-ЦД на высвобождение МП из ядер таблеток с пленочным покрытием, содержащих физическую смесь или лиофилизированный комплекс.
Фиг. 7в и Фиг. 7г. Влияние мольного отношения МП/СБЭ7-β-ЦД на профиль высвобождения СБЭ7-β-ЦД из ядер таблеток с пленочным покрытием, содержащих физическую смесь или лиофилизированный комплекс.
Фиг. 8. Профиль высвобождения препаратов тестостерон/СБЭ7-β-ЦД с контролируемым высвобождением.
Фиг. 9. Профиль высвобождения препаратов с замедленным высвобождением дипиридамола (ДП) из ядра таблетки, покрытого мембраной из EUDRAGIT-L и мочевины (120 мкм толщиной) и содержащего физическую смесь ДП и СБЭ7-β-ЦД.
Фиг. 10. Влияние толщины пленки на высвобождение ДП через мембрану из EUDRAGIT-L и мочевины из ядра таблетки, содержащей физическую смесь ДП и СБЭ7-β-ЦД.
Фиг. 11. Профиль высвобождения ДП через пленочное покрытие толщиной 180 мкм из EUDRAGIT-S и мочевины, которое окружает ядро таблетки, содержащей физическую смесь ДП и СБЭ7-β-ЦД.
Фиг. 12. Профиль высвобождения ДП из ядра таблетки, содержащей физическую смесь ДП и СБЭ7-β-ЦД, которая покрыта пленкой толщиной 90 мкм из ацетата целлюлозы (АЦ) и фталата гидроксипропилметилцеллюлозы (ФГПМЦ).
Фиг. 13. Влияние толщины пленки на высвобождение ДП из таблетки, содержащей физическую смесь ДП и СБЭ7-β-ЦД и покрытую пленкой из АЦ и ФГПМЦ (50:50).
Фиг. 14. Профиль высвобождения ДП из препаратов, имеющих комбинированный профиль высвобождения - высвобождение с задержкой и контролируемое высвобождение, и влияние толщины и состава пленки на профиль высвобождения.
Подробное описание изобретения
Изобретение преодолевает недостатки, которыми отличаются известные фармацевтические препараты, содержащие физическую смесь терапевтического агента и циклодекстрина, за счет создания препарата, который легко приготовить и который имеет растворимость, профиль растворения и/или биодоступность терапевтического агента, приближающиеся к растворимости, профилю растворения и биодоступности соответствующего фармацевтического препарата, содержащего комплекс терапевтического агента и циклодекстрина. В настоящем изобретении при получении фармацевтических препаратов используются производные простого сульфоалкилового эфира циклодекстрина (САЭ-ЦД). Настоящие препараты могут быть использованы для быстрой, контролируемой, замедленной, рассчитанной по времени, пульсирующей и длительной доставки широкого спектра терапевтических агентов. Препараты могут быть также изготовлены в виде различных лекарственных форм, которые описаны ниже.
Производные простого сульфоалкилового эфира циклодекстрина
Понятия "алкилен" и "алкил", которые используются в данном описании (например, в случае -O-(C2-C6-алкилен)SO3-группы или алкиламина), включает линейные, циклические и разветвленные, насыщенные и ненасыщенные (т.е. содержащие одну двойную связь) двухвалентные алкиленовые группы и моновалентные алкильные группы соответственно. Понятие "алканол" в данном случае также включает линейные, циклические и разветвленные, насыщенные и ненасыщенные алкильные компоненты алканольных групп, в которых гидроксильные группы могут находиться в любом положении алкильного фрагмента. Определение "циклоалканол" включает незамещенные или замещенные (например, метилом или этилом) циклические спирты.
Настоящее изобретение предлагает композиции, содержащие смесь производных циклодекстрина, имеющих структуру формулы (1), где вся композиция содержит в среднем, по меньшей мере, от 1 до 3n+6 фрагментов алкилсульфокислоты на молекулу циклодекстрина. Настоящее изобретение также предлагает композиции, содержащие один тип производного циклодекстрина или, по меньшей мере, 50% производного циклодекстрина одного типа.
Рассматриваемые производные циклодекстрина замещены или, по меньшей мере, по одной первичной гидроксильной группе (т.е., по меньшей мере, один из R1-R3 представляет собой заместитель), или они замещены как по первичной гидроксильной группе, так и по гидроксильной группе в 3-ем положении (т.е., по меньшей мере, один из R1-R3 и, по меньшей мере, один из R4, R6 и R8 представляет собой заместитель). Замещение по гидроксильной группе во 2-ом положении, хотя и возможно теоретически, как оказалось, не представляет интереса для настоящего изобретения.
Производные циклодекстрина настоящего изобретения получают в виде очищенных композиций, т. е. композиций, содержащих, по меньшей мере, 95% вес. производного(ых) циклодекстрина, замещенных, по меньшей мере, по первичной гидроксильной группе молекулы циклодекстрина (т.е. R1, R2 или R3 в формуле 1). В предпочтительном варианте осуществления изобретения могут быть получены очищенные композиции, содержащие, по меньшей мере, 98% производного(ых) циклодекстрина.
В некоторых композициях изобретения непрореагировавший циклодекстрин по существу удаляется, причем оставшиеся примеси (т.е. менее 5% вес. из расчета на композицию) не оказывают существенного влияния на свойства композиции, содержащей производное циклодекстрина.
Производные циклодекстрина, используемые в данном случае, могут быть в общем случае получены в соответствии с описанием патента США N 5134127, который приведен в качестве справочного материала. Этот способ может включать растворение циклодекстрина в водном основании при подходящей температуре, например при 70-80oC, до получения возможной наиболее высокой концентрации. Например, для получения необходимых производных циклодекстрина при интенсивном перемешивании для обеспечения максимального контакта гетерогенной фазы добавляют подходящий алкилсульфон в количестве, соответствующем числу молей первичных гидроксильных групп, присутствующих в ЦД.
Проведена оценка различных производных САЭ-ЦД, в том числе СБЭ4-β, СБЭ7-β, СБЭ11-β и СБЭ4-γ, которые соответствуют производным САЭ-ЦД формулы 1, где n = 5, 5, 5 и 6; m = 4; и где присутствует 4, 7, 11 и 4 простых сульфоалкилэфирных заместителей, соответственно. Установлено, что производные САЭ-ЦД в различной степени повышают растворимость плохо растворимых в воде лекарственных средств. Например, в представленной табл. 1 обобщены данные для констант связывания и растворимости, наблюдаемых в случае некоторых производных САЭ-ЦД (0,1 М при 25oC) и метилпреднизолона.
В другом варианте осуществления изобретения используется дипиридамол (ДП), который является основным лекарственным соединением (pKa = 6,28) с плохой растворимостью в воде его свободного основания (3,6 мкг/мл при 25oC) и с низкой и непостоянной биодоступностью. СБЭ7-β, как установлено, резко повышает растворимость ДП. В приведенной табл. 2 обобщены данные по растворимости ДП в присутствии и в отсутствие СБЭ7-β-ЦД при различных значениях pH.
Хотя описанные выше варианты изобретения дают примеры некоторых производных САЭ-ЦД, предусмотренных изобретением, их не следует рассматривать в качестве ограничивающих весь объем изобретения.
Фармацевтический препарат, содержащий простой сульфоалкиловый эфир циклодекстрина
Чтобы получить циклодекстриновую фармацевтическую композицию, имеющую приемлемые характеристики растворимости, профиля растворения и биодоступности, в данной области обычно подразумевается, что клатрат или комплекс включения циклодекстрина и терапевтического агента должны быть получены отдельно до получения содержащей их фармацевтической композиции. Однако заявители настоящего изобретения установили, что получение комплекса САЭ-ЦД/терапевтический агент не является необходимым.
Содержащий САЭ-ЦД препарат настоящего изобретения будет содержать производное САЭ-ЦД формулы (1), описанной выше, фармацевтический носитель, терапевтический агент и, необязательно, дополнительные адъюванты и активные ингредиенты, где основная часть терапевтического агента не образует комплекс с производным САЭ-ЦД.
Так как подразумевается, что только основная часть терапевтического агента, содержащегося в настоящем препарате, не будет образовывать комплекс с САЭ-ЦД, возможно присутствие некоторого количества комплекса САЭ-ЦД/терапевтический агент. Присутствие комплекса САЭ-ЦД/терапевтический агент в настоящем препарате может быть намеренным или не намеренным, т.е. комплекс может быть получен отдельно в соответствии с патентом Stella и др., а затем введен в препарат, или комплекс может образовываться в процессе получения настоящего препарата.
Под "комплексом САЭ-ЦД/терапевтический агент" обычно подразумевают клатрат или комплекс включения производного сульфоалкилового эфира циклодекстрина формулы (1) и терапевтического агента. Отношение САЭ-ЦД/терапевтический агент, присутствующих в комплексе, может сильно меняться, но обычно находится в интервале приблизительно от 1:2 до 2:1 (мольное отношение) соответственно, и предпочтительно составляет 1:1. Под выражением "образует комплекс" подразумевается, что "существует часть клатрата и комплекса включения с", т. е. образующий комплекс терапевтический агент представляет собой часть клатрата или комплекса включения с производным сульфоалкилового эфира циклодекстрина. Под определением "основная часть" подразумевается, по меньшей мере, приблизительно 50% вес. терапевтического соединения. Таким образом, препарат в соответствии с настоящим изобретением будет содержать терапевтический агент, более чем приблизительно 50% вес. которого не образует комплекс с САЭ-ЦД. В различных вариантах осуществления изобретения предпочтительно более чем 60% вес., более предпочтительно более, чем 75% вес. и даже более предпочтительно более, чем 90% вес., и наиболее предпочтительно более, чем 95% вес. терапевтического агента не будет образовывать комплекс с САЭ-ЦД, когда терапевтический агент находится в фармацевтическом препарате.
Полагают, что терапевтический агент будет образовывать комплекс с САЭ-СД при введении его в виде лекарственной формы, содержащей композицию настоящего изобретения, пациенту и при воздействии на композицию жидкостей тела. Например, когда капсула, содержащая порошки терапевтического агента и САЭ-ЦД, вводится перорально пациенту, капсула будет растворяться, позволяя, таким образом, желудочному соку контактировать с терапевтическим агентом и САЭ-СД. В результате будет образовываться комплекс САЭ-СД/терапевтический агент. Подходящая лекарственная форма будет способствовать гидратации физической смеси до ее высвобождения из лекарственной формы, что гарантирует образование соответствующего комплекса.
Соотношение САЭ-ЦД: терапевтический агент в препарате будет зависеть от ряда таких факторов, как собственная растворимость агента, предполагаемая доза агента и константа связывания при образовании комплекса включения между конкретным лекарственным средством (агентом) и конкретным САЭ-СД. Эти факторы в сумме будут определять количество САЭ-СД, необходимое для лекарственной формы и, следовательно, соотношение САЭ-СД:терапевтический агент.
Молекулярный вес большинства САЭ-ЦД составляет приблизительно 2000, а большинство терапевтических агентов имеет молекулярные веса в интервале 20~ 500. Кроме того, большинство лекарственных средств образует комплексы включения с САЭ-ЦД в соотношении 1:1. Из-за таких различий в молекулярных весах необходимое количество САЭ-ЦД будет минимально в 4-10 раз больше количества агента, т.е. один моль ЦД будет солюбилизировать один моль лекарственного средства. Однако, как показано ниже, при этом подразумевается бесконечно высокая константа связывания между агентом и ЦД. Для большинства твердых лекарственных форм наилучшим вариантом являются таблетки, которые имеют массу менее одного грамма, и из-за необходимости в таблетках других наполнителей должны содержать менее чем 500 мг СД. Таким образом, исходя из этого простого предположения, можно понять, что количество лекарственного средства, которое может быть использовано с САЭ-ЦД, должно составлять менее 50 мг. Т. к. большинство лекарственных средств не имеет бесконечно высокую константу связывания с САЭ-ЦД, суммарная доза лекарственного средства, которая может находиться в препарате, будет менее 50 мг.
Если говорить более конкретно, то агенты могут образовывать комплексы включения с САЭ-ЦД от слабых до очень прочных. Очень слабый комплекс включения представляет собой комплекс, в котором константа связывания составляет менее чем приблизительно 500 М-1. Слабый комплекс будет иметь константу связывания приблизительно от 500 до 1000 М-1. При умеренном связывании комплекс имеет константу связывания приблизительно от 1000 до 5000 М-1; сильное связывание должно быть в комплексе с константой связывания приблизительно от 5000 до 20000 М-1; и очень сильное связывание будет в комплексе с константой связывания больше чем приблизительно 20000 М-1.
Относительное повышение растворимости плохо растворимого лекарственного средства в присутствии САЭ-ЦД обусловлено константой связывания и мольной концентрацией присутствующего САЭ-ЦД. Для очень слабо связанного лекарственного средства необходимо мольное соотношение САЭ-СД/агент 100:1. В этом случае количество лекарственного средства в препарате должно быть менее чем 1 мг. Если константа связывания между САЭ-СД и агентом очень велика, то может быть допустимо соотношение приблизительно 1:1. В таком случае может быть использована доза лекарственного средства до 50 мг, при условии, что собственная растворимость лекарственного средства является приемлемой. Рассмотрим лекарственное средство с константой связывания 10000 М-1, которая реальна для ряда лекарственных средств. В присутствии 0,1 М САЭ-ЦД растворимость лекарства будет повышаться приблизительно в 1000 раз в сравнении с растворимостью в отсутствие САЭ-ЦД. Если собственная растворимость лекарственного средства составляет приблизительно 1 мкг/мл, то в присутствии 0,1 М САЭ-ЦД будет возможна растворимость только приблизительно 1 мг/мл; однако, если собственная растворимость лекарственного средства составляет приблизительно 10 мкг/мл, то в присутствии 0,1 М САЭ-ЦД возможна растворимость приблизительно 10 мг/мл.
Для лекарственных форм, описанных в этом патенте, мольное соотношение САЭ-ЦД:терапевтический агент будет обычно находиться в интервале от 100:1 до приблизительно 1: 1 и предпочтительно приблизительно от 20:1 до 1:1. Таким образом, САЭ-ЦД обычно будет присутствовать в избытке по отношению к терапевтическому агенту для некоторых агентов, тогда как для других агентов - в минимальном избытке. Величина избытка будет определяться собственной растворимостью агента, ожидаемой дозы агента и константы связывания при образовании комплекса включения между конкретным лекарственным средством (агентом) и конкретным САЭ-ЦД.
В настоящем изобретении подразумеваются различные фармацевтические препараты, содержащие физическую смесь терапевтического агента и САЭ-ЦД: таблетка с осмотическим насосом, слоеная таблетка, таблетка с покрытием, пеллеты с покрытием, порошок для воссоздания, капсулы, гранулы с покрытием и экструдированные из горячего расплава пленки.
В настоящем изобретении таблетки, гранулы и пеллеты с покрытием содержат пленочное покрытие и твердое ядро. Пленочное покрытие содержит пленкообразующий агент и порообразующий агент. Пленочное покрытие может содержать большое число пленкообразующих агентов и/или порообразующих агентов, например, в конкретном пленочном покрытии могут быть использованы комбинации пленкообразующих агентов.
Термины "пленкообразующий агент" и "агент, контролирующий высвобождение" используются в данном описании взаимозаменяемо и означают полимерные соединения (природные, синтетические, полусинтетические и полученные генетическим путем), которые будут образовывать пленочное покрытие вокруг твердого ядра препарата и которые контролируют высвобождение или уменьшают скорость высвобождения терапевтического агента или САЭ-ЦД из твердого ядра. Пленкообразующие агенты, использование которых подразумевается в настоящем изобретении, описаны дополнительно, а для некоторых вариантов осуществления изобретения приведены примеры.
На фиг. 1 (процедура получения подробно описана в Примере 1) представлены профили высвобождения для двух таблеток с осмотическим насосом, содержащих метилпреднизолон (МП). Таблетки отличаются только образованием комплекса САЭ-ЦД и терапевтического агента. Из двух композиций, первая на основе физической смеси метилпреднизолон/СБЭ7-β-ЦД, и вторая, содержащая комплекс метилпреднизолон/СБЭ7-β-ЦД, в соответствии с методикой Примера 1 готовят таблетки с осмотическим насосом с контролируемым высвобождением. МП и СБЭ7-β-ЦД (в мольном отношении 1: 7) вместе с фармацевтическим носителем прессуют в твердое ядро, на которое наносят покрытие путем распыления смеси этилцеллюлозы, ПЭГ3350, ПЭГ400 и этанола с образованием вокруг твердого ядра пленки толщиной 140 мкм. Профиль растворения определяют с помощью устройства для растворения II (Фармакопея США) (100 об/мин, 37oC) и метода ВЭЖХ для метилпреднизолона (МП). Флуориметрическая оценка с использованием 2,6-толуидиннафталинсульфоната (2,6-ТНС) была разработана для количественного определения САЭ-СД. Первый препарат, обозначенный на фиг. 1 полыми кругами, содержит отдельно приготовленный лиофилизированный комплекс МП-СБЭ7-β-ЦД. Второй препарат, обозначенный с помощью закрашенных кругов, содержит основную часть МП в свободной форме в виде физической смеси с СБЭ7-β-ЦД. Третий препарат, обозначенный с помощью квадратов, содержит физическую смесь лактозы, фруктозы и МП. Из подобия кривых, соответствующих заранее полученному комплексу и физической смеси, очевидно, что последний имеет профиль высвобождения аналогичный или практически аналогичный профилю высвобождения первого. Следует отметить, что для этой конкретной лекарственной формы МП и СБЭ7-β-ЦД имеют по существу одинаковый профиль высвобождения. Результаты представлены на фиг. 2а и 2б соответственно для МП и СБЭ7-β-ЦД.
Если в качестве терапевтического агента используется тестостерон (ТСТ), препарат с физической смесью СБЭ7-β-ЦД и ТСТ имеет такой же профиль высвобождения, что и соответствующая лиофилизированная смесь (фиг. 8). Твердое ядро таблетки содержит ТСТ и СБЭ7-β-ЦД в мольном соотношении 1:1. Пленочное покрытие этой таблетки содержит сорбит, ПЭГ400 и ацетат целлюлозы. Профили высвобождения препарата с физической смесью и препарата с комплексом были сравнены с профилем высвобождения базового препарата ТСТ/фруктоза-лактоза.
При увеличении толщины пленочного покрытия или мембраны, которые окружают ядро таблетки, содержащее или физическую смесь или лиофилизированный комплекс МП и СБЭ7-β-ЦД, до 200 мкм наблюдается небольшое отличие в профилях высвобождения для физической смеси и лиофилизированного комплекса; однако СБЭ7-β-ЦД фактически имеет профиль высвобождения по существу аналогичный профилю высвобождения МП. Эти результаты изображены на фиг. 3а и 3б соответственно для МП и СБЭ7-β-ЦД. Приготовлены и изучены дополнительные примеры таблеток с пленочным покрытием, которые имеют толщину пленки 38, 89, 137, 198 и 234 мкм. Полученные результаты представлены на фиг. 4а и 4б, которые показывают, что СБЭ7-β-ЦД имеет фактически такой же профиль высвобождения, что и МП, в каждой лекарственной форме. В варианте изобретения с толщиной пленки 234 мкм лиофилизированный комплекс, как оказывается, высвобождает СБЭ7-β-ЦД быстрее, чем МП; однако при построении графика зависимости скорости высвобождения для физической смеси фиг. 4а и 4б от толщины пленки видно, что СБЭ7-β-ЦД имеет по существу такой же профиль высвобождения, что и МП (фиг. 5).
Толщина пленки не обязательно оказывает значительное влияние на профиль высвобождения данной лекарственной формы. На фиг. 10 представлено влияние, которое оказывает толщина пленки на препарат с замедленным высвобождением, содержащий пленочное покрытие из EUDRAGIT-L/мочевины и твердое ядро с физической смесью дипиридамол/СБЭ7-β-ЦД. Полученные результаты показывают, что для этого варианта осуществления изобретения профиль высвобождения ДП не зависит от толщины пленки, а зависит от pH раствора.
При замене композиции пленочного покрытия на EUDRAGIT-S и мочевину можно приготовить препарат с замедленным высвобождением, который высвобождает ДП при pH приблизительно 7,2, а не при 6,8 (фиг. 11). Более высокое щелочное значение pH соответствует значению pH в тонком или толстом кишечнике пациента. Таким образом, можно приготовить препарат с замедленным высвобождением для кишечного или ректального высвобождения терапевтического агента, который содержит твердое ядро и пленочное покрытие, состоящее из пленкообразующего агента, который представляет собой полимер с растворимостью, зависящей от pH.
Пленка, окружающая твердое ядро, будет влиять на высвобождение МП и СБЭ7-β-ЦД. В вариантах осуществления изобретения, в которых пленка, окружающая ядро отсутствует, ядро, содержащее физическую смесь СБЭ7-β-ЦД и МП, может иметь те же или почти те же характеристики высвобождения, что и ядро, содержащее комплекс этих компонентов. На фиг. 6 представлен профиль высвобождения МП из твердых ядер, содержащих лиофилизированный комплекс (закрашенные круги), физическую смесь (полые круги) и физическую смесь фруктоза-лактоза-МП (квадраты). В этом примере смесь фруктоза-лактоза выполняет функцию осмотического, а не солюбилизирующего агента. Физическая смесь по существу имеет те же профили высвобождения, что и комплекс.
Мольное отношение МП/СБЭ7-β-ЦД может влиять на профиль высвобождения данной лекарственной формы. На фиг. 7а-7г представлен профиль высвобождения МП и СБЭ7-β-ЦД из покрытых пленкой таблеток, содержащих МП и СБЭ7-β-ЦД в виде физической смеси (фиг. 7а и 7в) и лиофилизированиого комплекса (фиг. 7б и 7г), где мольные отношения МП/СБЭ7-β-ЦД составляют 10/1, 1/7 и 1/3 (вес. /вес. ). Полученные результаты показывают, что уменьшение относительного количества СБЭ7-β-ЦД понижает профиль высвобождения МП. Следовательно, лекарственные формы, имеющие различные профили высвобождения, могут быть приготовлены путем регулирования соотношения МП/СБЭ7-β-ЦД. Эти результаты также показывают, что физическая смесь и лиофилизированный комплекс имеют по существу одинаковые характеристики высвобождения.
Используемое пленочное покрытие может содержать полимер, растворимость которого зависит от величины pH. На фиг. 9 изображен профиль высвобождения для препарата с замедленным высвобождением, который состоит из ядра таблетки и пленочного покрытия. Ядро таблетки содержит физическую смесь СБЭ7-β-ЦД и дипиридамола (ДП). Пленочное покрытие (150 мкм) содержит EUDRAGIT-L, который имеет зависящую от pH растворимость. Когда pH раствора, в который погружена таблетка, повышается через два часа от 1,5 до 6,8, СБЭ7-β-ЦД и ДП имеют практически одинаковый профиль высвобождения. Двухчасовая задержка соответствует лекарственной форме, из которой основная часть ДП будет высвобождаться в подвздошной кишке и тонкой кишке пациента.
Пленочные покрытия или мембраны настоящего изобретения могут содержать сочетание пленкообразующих агентов. На фиг. 12 представлен один из вариантов осуществления изобретения, в котором пленочное покрытие содержит смесь ацетата целлюлозы (АД) и фталата гидроксипропилметилцеллюлозы (ФГПМЦ) в соотношении 1:1 и твердое ядро, состоящее из СБЭ7-β-ЦД и ДП.
Такое сочетание пленкообразующих агентов обеспечивает получение препарата, имеющего замедленное и контролируемое высвобождение терапевтического агента.
Изменение толщины пленки от 90 до 170 мкм, как оказывается, фактически не влияет на профиль высвобождения ДП при использовании пленкообразующих агентов, имеющих pH-зависимую растворимость. Следовательно, в этом варианте осуществления изобретение обеспечивает получение фармацевтического препарата с замедленным и контролируемым высвобождением, имеющего профиль высвобождения, который лишь незначительно зависит от толщины пленки (фиг. 13).
Конкретные варианты осуществления изобретения могут обладать замедленным высвобождением, комбинированным замедленным и контролируемым высвобождением и точно контролируемым высвобождением. В варианте изобретения, представленном на фиг. 14, содержащее ДП/СБЭ7-β-ЦД ядро таблетки покрыто смесью АЦ: ФГПМЦ в различных соотношениях и с различной толщиной пленки. Вариант с замедленным высвобождением, обозначенный квадратами, содержит пленочное покрытие толщиной 90 мкм, которое состоит из смеси АЦ:ФГПМЦ в соотношении 1: 1. Вариант изобретения с комбинированным замедленным и контролируемым высвобождением, обозначенный ромбами, имеет пленочное покрытие толщиной 105 мкм, которое состоит из АЦ:ФГПМЦ в соотношении 6:4. Таким образом, путем изменения соотношения АЦ:ФГПМЦ можно регулировать относительный вклад контролируемого и замедленного высвобождения в суммарный профиль высвобождения лекарственной формы.
Следует отметить, что в отсутствие САЭ-ЦД в соответствии с настоящим изобретением подходящий профиль высвобождения лекарственного средства не будет получен для терапевтических агентов, рассмотренных в данном описании. Например, ядро таблетки, содержащее ДП, лимонную кислоту и фруктозу-лактозу, окруженное пленкой толщиной 120 мкм из АЦ:ФГПМЦ (50:50), не высвобождает ДП. В еще одном примере, в котором такое же ядро таблетки окружено пленкой толщиной 120 мкм из EUDRAGIT-L и мочевины (50:50), наблюдается неполное высвобождение ДП.
Таким образом, настоящее изобретение также предлагает фармацевтический препарат, имеющий замедленное высвобождение, контролируемое высвобождение или комбинированное замедленное и контролируемое высвобождение и состоящий из ядра таблетки и пленочного покрытия вокруг ядра таблетки, причем ядро таблетки содержит физическую смесь терапевтического агента и САЭ-ЦД, а пленочное покрытие состоит из сочетания пленкообразующих агентов.
Кроме того, в соответствии с Примером 2 получены таблетки с осмотическим насосом и оценены характеристики их растворения. Эти таблетки состоят из ядра, содержащего ДП/САЭ-ЦД, окруженного пленочным покрытием, состоящим из одного или нескольких следующих компонентов: ацетат целлюлозы, этилцеллюлоза, воск, EUDRAGIT E100, EUDRAGIT RS и EUDRAGIT RL, EUDRAGIT L, EUDRAGIT S, ацетатфталат целлюлозы, фталат гидроксипропилметилцеллюлозы и ацетатсукцинат гидроксипропилметилцеллюлозы. Порообразующие агенты, которые были изучены, включают: поли(этиленгликоль) 3350 (ПЭГ 3350), сорбит, сахарозу и мочевину.
Определение "порообразующий агент", которое используется в данном описании, относится к агенту, который способствует образованию пор в пленочном покрытии настоящего изобретения или улучшает водопроницаемость пленки. Примерами порообразующих агентов являются углеводороды, такие как лактоза, декстроза, фруктоза, сахароза, манноза; α-гидроксикислоты, такие как лимонная кислота, винная кислота, фумаровая кислота, янтарная кислота, гликолевая кислота, молочная кислота, смеси этих кислот и их соли; галогеновые противоионы, такие как бромид, фторид, иодид и хлорид; катионы двухвалентных металлов, такие как ионы магния и кальция; анионные агенты, такие как фосфаты, сульфаты, сульфонаты, нитраты, бикарбонаты, их смеси и их соли; целлюлозы, такие как ГПЦ, ГПМЦ, гидроксиэтилцеллюлоза, метилцеллюлоза; поли(этиленоксид); поли(винилпирролидон); камеди и желатинизирующие агенты, такие как кизельгур, ксантановая камедь, альгиновая кислота, аравийская камедь, трагакант, их сочетания и их соли; глины, такие как монтмориллонит, бентонит, Veegum, каолин; различные компоненты, такие как кизельгур, силикат магния, бентон, гекторит, PLURONICS, гидрофильные поверхностно-активные вещества; полиолы, такие как сорбит, маннит, ксилит; протеины, такие как альбумин, коллаген, желатин; растворимые в воде аминокислоты; диспергирующие добавки, такие как крахмал, натриевая соль гликолята крахмала, кроскармелоза; и растворимые в воде органические соединения; и их сочетания. Порообразующие агенты, которые проницаемы для воды, могут быть использованы для улучшения проницаемости пленки.
Препараты настоящего изобретения предназначены для получения комплексов САЭ-ЦД, когда на них воздействуют жидкости тела. В одном из вариантов осуществления изобретения лекарственные формы настоящего изобретения будут обеспечивать гидратацию физической смеси САЭ-ЦД/терапевтический агент до высвобождения терапевтического агента, что способствует образованию комплекса.
Способ модификации биодоступности и скорости биоабсорбции
Для плохо растворимых в воде, гидрофобных лекарственных
средств с плохой биодоступностью настоящее изобретение предлагает способ улучшения растворимости в воде и модифицикации биодоступности и/или скорости биоабсорбции в организме пациента. Для растворимых в воде гидрофильных лекарственных средств с экстремально высокой биодоступностью настоящее изобретение предлагает способ модификации скорости биоабсорбции в организме пациента.
Определение "плохо растворимый в воде" и "гидрофобный" означает, что при 20oC терапевтический агент имеет растворимость в воде с нейтральным значением pH менее чем приблизительно 1 мг/мл. Под определением "растворимый в воде" или "гидрофильный" подразумевается, что при 20oC терапевтический агент имеет растворимость в нейтральной воде больше чем приблизительно 1 мг/мл.
В некоторых вариантах осуществления изобретения способ настоящего изобретения по модификации биодоступности или скорости абсорбции терапевтического агента включает стадии получения комбинации терапевтического агента и производного сульфоалкилового эфира циклодекстрина и введения полученной комбинации пациенту. Под "модификацией биодоступности и/или скорости биоабсорбции" подразумевается, что биодоступность и/или скорость биоабсорбции терапевтического агента при введении в комбинации с САЭ-ЦД будут отличаться от (или будут модифицированы по отношению к) биодоступности и/или скорости биоабсорбции при введении их по отдельности.
В других вариантах рассматриваемый способ включает стадии получения препарата производного сульфоалкилового эфира циклодекстрина и свободного (не находящегося в виде комплекса) терапевтического агента в одной фармацевтически приемлемой лекарственной форме и введения лекарственной формы пациенту.
Полагают, что САЭ-ЦД модифицирует биодоступность и/или скорость абсорбции терапевтического агента за счет образования с ним клатрата или комплекса включения после воздействия жидкостей тела пациента. На основе комбинации САЭ-ЦД/терапевтический агент может быть получен препарат различными путями, которые более подробно описаны ниже. Необходимо только, чтобы САЭ-ЦД присутствовал в количестве, достаточном для комплексообразования с терапевтическим агентом в организме пациента, который принял препарат.
Общие положения
Терапевтический агент, который используется в настоящем изобретении, может обладать широким спектром значений растворимости в воде, биодоступности и гидрофильности. Таким образом, настоящее изобретение подразумевает любой терапевтический агент, который будет образовывать клатрат или комплекс включения с производным САЭ-ЦД формулы (I). Терапевтические агенты, для которых настоящее изобретение особенно подходит, представляют собой плохо растворимые в воде гидрофобные терапевтические агенты и растворимые в воде гидрофильные терапевтические агенты. Настоящее изобретение может быть использовано для приготовления стандартных лекарственных форм, содержащих менее чем приблизительно 500 мг, в частности менее чем приблизительно 150 мг, и в особенности менее чем приблизительно 50 мг терапевтического агента.
Количество терапевтического соединения, введенного в рассматриваемые препараты, может быть выбрано в соответствии с положениями, известными в фармацевтике. Подразумевается введение терапевтически эффективного количества соединения. Под определением "терапевтически эффективное количество" понимается количество, под которым, например, в фармацевтике, подразумевается фармацевтически эффективное количество. Фармацевтически эффективное количество представляет собой количество лекарственного средства или фармацевтически активного вещества, которого достаточно для того, чтобы вызвать заметную биологическую реакцию при введении пациенту. По отношению к витаминам или минеральным веществам понятие "эффективное количество" означает количество, по меньшей мере, приблизительно 10% от рекомендуемого в США суточного потребления (РСП) этого конкретного ингредиента для пациента. Например, если ингредиент представляет собой Витамин C, то эффективное количество Витамина C должно включать количество Витамина C, достаточное для обеспечения 10% или более от РСП. Обычно, когда таблетка содержит минерал или витамин, в нее будет вводиться большее количество, предпочтительно приблизительно 100% или более от РСП.
Терапевтическое соединение обычно используется в тонко измельченной форме, т. е. в виде порошка или гранул с тем, чтобы увеличить скорость растворения. Предпочтительно использовать тонко измельченное порошкообразное соединение для повышения скорости растворения, причем более предпочтительно, чтобы не менее чем на 80% терапевтического соединения, желательно не менее чем на 90%, проходило через сито размером 100 меш (150 мкм). Количество терапевтического соединения, которое должно быть введено, обычно лежит в интервале приблизительно от 0,1 до 50%, предпочтительно приблизительно от 1 до 25% вес., из расчета на композицию, а соотношение может быть приемлемым образом модифицировано в зависимости от используемого терапевтического соединения.
В качестве терапевтического агента могут быть использованы антибактериальные агенты типа плохо растворимой в воде пиридонкарбоновой кислоты, такие как бенофлоксацин, налидиксоновая кислота, эноксацин, офлоксацин, амифлоксацин, флумеквин, тозфлоксацин, пиромидиновая кислота, пипемидиновая кислота, милоксацин, оксолиновая кислота, циноксацин, норфлоксацин, ципрофлоксацин, пефлоксацин, ломефлоксацин, энрофлоксацин, данофлоксацин, бинфлоксацин, сарафлоксацин, ибафлоксацин, дифлоксацин и их соли. Другие терапевтические агенты представляют собой пенициллин, тетрациклин, цефалоспорины и другие антибиотики, антибактерицидные вещества, антигистамины и противоотечные средства, противовоспалительные агенты, противопаразитные средства, противовирусные средства, местные анестетики, противогрибковые агенты, amoebicidal или трихомоноцидные агенты, анальгетики, противоартритные агенты, противоастматические агенты, антикоагулянты, противосудорожные средства, антидепрессанты, антидиабетические агенты, противоопухолевые агенты, антипсихотические средства, гипотензивные средства и миорелаксанты. Примерами антибактериальных веществ являются бета-лактамовые антибиотики, тетрациклины, хлорамфеникол, неомицин, грамицидин, бацитрацин, сульфонамиды, нитрофуразон, налидиксоновая кислота и их аналоги и противомикробная комбинация флудаланин/пентизидон. Примерами антигистаминов и противоотечных агентов являются периламин, хлорфенирамин, тетрагидрозолин и антазолин. Примерами противовоспалительных лекарственных средств являются кортизон, гидрокортизон, бетаметазон, дексаметазон, флуокортолон, преднизолон, триамцинолон, индометацин, сулиндак и их соли и соответствующие сульфиды. Примером противопаразитного средства является ивермектин.
Примерами противовирусных соединений являются ацикловир и интерферон. Примерами болеутоляющих лекарственных средств являются дифлунизал, аспирин или ацетаминофен. Примерами противоартритных агентов являются фенилбутазон, индометацин, силиндак, их соли и соответствующие сульфиды, дексаметазон, ибупрофен, аллопуринол, оксифенбутазон или пробенецид. Примерами противоастматических лекарственных средств являются теофиллин, эфедрин, беклометазондипропионат и эпинефрин. Примерами антикоагулянтов являются бисгидроксикумарин и варфарин. Примерами противосудорожных средств являются дифенилгидантоин и диазепам. Примерами антидепрессантов являются амитриптилин, хлордиазепоксид перфеназина, протриптилин, имипрамин и доксепин. Примерами противодиабетических агентов являются инсулин, соматостатин и их аналоги, толбутамид, толазамид, ацетогексамид и хлорпропамид. Примерами противоопухолевых агентов являются адриамицин, фторурацил, метотрексат и аспарагиназа. Примерами противопсихотических агентов являются прохлорперазин, тиоридазин, молиндон, флуфеназин, трифлуоперазин, перфеназин, амитриптилин и трифлуопромазин. Примерами гипотензивных средств являются спиронолактон, метилдопа, гидралазин, клонидин, хлортиазид, дезерпидин, тимолол, пропранолол, метопролол, празозингидрохлорид и резерпин. Примерами миорелаксантов являются сукцинилхолинхлорид, данбролен, циклобензаприн, метокарбамол и диазепам.
Некоторыми конкретными примерами терапевтических агентов, которые могут быть использованы, но которые не ограничивают изобретение, являются адифенин, аллобарбитал, аминобензойная кислота, амобарбитал, ампициллин, анетол, аспирин, азопропазон, азаленбарбитуровая кислота, беклометазон, беклометазондипропронат, бенциклан, бензальдегид, бензокаин, бензодиазепины, бензотиазид, бетаметазон, бетаметазон-17-валерат, бромбензойная кислота, бромизовалерилмочевина, бутил-п-аминобензоат, хлоральгидрат, хлорамбуцил, хлорамфеникол, хлорбензойная кислота, хлорпромазин, коричная кислота, клофибрат, коэнзим A, кортизон, кортизонацетат, циклобарбитал, циклогексилантранилат, деоксихолевая кислота, дексаметазон, дексаметазонацетат, диазепам, дигитоксин, дигоксин, эстрадиол, флуфенаминовая кислота, флуоцинолонацетонид, 5-фторурацил, флурбипрофен, гризеофульвин, гуаиазулен, гидрокортизон, гидрокортизонацетат, ибупрофен, индикан, индометацин, йод, кетопрофен, антибиотики ланкацидиновой группы, мефенаминовая кислота, менадион, мефобарбитал, метарбитал, метициллин, метронидазол, митомицин, нитразепам, нитроглицерин, нитромочевины, параметазон, пенициллин, пентобарбитал, фенобарбитал, фенобарбитон, фенилмасляная кислота, фенилвалериановая кислота, фенитоин, преднизолон, преднизолонацетат, прогестерон, пропилпарабен, просцилларидин, простагландин серии A, простагландин серии B, простагландин серии E, простагландин серии F, хинолоновые противомикробные агенты, резерпин, спиронолактон, сульфацетамид натрия, сульфонамид, тестостерон, талидомид, тиаминдилаурилсульфат, тиамфениколпальмитат, тиопентал, триамцинолон, витамин A, витамин D3, витамин E, витамин K3 и варфарин.
Терапевтические соединения, содержащиеся в фармацевтическом препарате, могут находиться в виде их фармацевтически приемлемых солей. Как подразумевается в данном описании, определение "фармацевтически приемлемая соль" относится к производным названных соединений, где терапевтическое соединение модифицировано путем получения его кислой или основной соли. Примерами фармацевтически приемлемых солей являются, но не ограничиваются только ими, соли минеральных и органических кислот по основным остаткам, таким как аминогруппы; щелочные или органические соли по кислотным остаткам, таким как остатки карбоновых кислот; и др. Фармацевтически приемлемые соли представляют собой обычные нетоксичные соли или четвертичные аммонийные соли исходного соединения, полученные, например, из нетоксичных неорганических или органических кислот. Например, такими обычными нетоксичными солями являются соли, полученные из неорганических кислот, таких как соляная, бромистоводородная, серная, сульфоновая, сульфаминовая, фосфорная, азотная и др. кислоты; и соли, полученные из таких органических кислот, как аминокислоты, уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, пальмовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изэтионовая кислоты и др.
Фармацевтически приемлемые соли настоящего изобретения могут быть синтезированы из исходного терапевтического соединения, которое содержит основный или кислотный остаток, с использованием обычных химических способов. Обычно такие соли могут быть приготовлены при взаимодействии свободной кислотной или основной форм этих соединений с предварительно рассчитанным количеством подходящего основания или кислоты в воде или в органическом растворителе или в их смеси. Обычно предпочтительна неводная среда. Список подходящих солей приведен в Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA, ch. 40 (работа приведена в качестве справочного материала).
Выражение "фармацевтически приемлемый" используется в данном описании для соединений, материалов, композиций и/или лекарственных форм, которые с медицинской точки зрения приемлемы для использования в контакте с тканями человека и животного, без чрезмерной токсической, раздражающей, аллергической реакций или без других проблем или осложнений, соизмеримых с разумным соотношением положительный эффект/опасность.
Определение "активный ингредиент", используемое в данном описании, означает корригирующий вкус или запах агент, подслащивающий агент, витамин, минерал и другие соединения, предназначенные для применения в фармацевтике. Настоящий препарат также может содержать адъюванты, такие как красящие агенты, диспергирующие агенты, смазывающие вещества, биоадгезивы и др.
К диспергирующим агентам относятся крахмалы, такие как кукурузный крахмал, картофельный крахмал, предварительно желатинизированные и модифицированные крахмалы, целлюлозные агенты, такие как Ac-di-sol, монтмориллонитовые глины, поперечно-сшитый ПВП, подслащивающие агенты, бентонит и VEEGUMTM, микрокристаллическая целлюлоза, альгинаты, натриевая соль гликолята крахмала, камеди, такие как агар, кизельгур, плоды рожкового дерева, karaya, пектин и трагакант. В одном из вариантов осуществления изобретения заявляемая таблетка не будет растворяться слишком быстро так, что будет существовать возможность гидратации в ней физической смеси САЭ-ЦД/терапевтический агент.
Ингибиторы протеазы, которые могут присутствовать в препаратах настоящего изобретения, включают, но не ограничиваются только ими, антипаин, лейпептин, химостатин, амистатин и пуромицин.
Усилители проникновения, которые могут присутствовать в препаратах настоящего изобретения, включают, но не ограничиваются только ими, хелатообразователи для ионов кальция, такие как этилендиаминтетрауксусная кислота и поликарбоновые кислоты; поверхностно-активные вещества, такие как лаурилсульфат натрия, додецилсульфат натрия и Tween; соли желчных кислот, такие как таурохолат натрия; жирные кислоты, такие как олеиновая и линолевая кислоты; а также вещества, не являющиеся ПАВами, такие как AZONE и диалкилсульфоксиды.
Вводимые в композицию вещества, корригирующие вкус и запах (корригенты), могут быть выбраны из синтетических ароматических масел и ароматических отдушек и/или минеральных масел, экстрактов растений, листьев, цветов, фруктов и т. д. и их сочетаний. Они могут включать коричное масло, масло грушанки, масло перечной мяты, голубиное масло, лавровое масло, анисовое масло, эвкалиптовое масло, тимьяновое масло, кедровое масло, масло мускатного ореха, масло шалфея, масло горького миндаля и масло кассии. Также приемлемыми корригентами являются ванилин, цитрусовое масло, в том числе лимонное масло, апельсиновое масло, виноградное масло, масло лайма и грейпфрутовое масло, и фруктовые эссенции, в том числе яблочная, грушевая, персиковая, клубничная, малиновая, вишневая, сливовая, ананасовая, абрикосовая и т.д. эссенции. Корригенты, которые, как установлено, особенно предпочтительны, представляют собой коммерчески доступные апельсиновый, виноградный, вишневый корригент и корригент жевательных резинок, а также их смеси. Количество корригента может зависеть от ряда факторов, в том числе от желаемого органолептического эффекта. Особенно предпочтительными корригентами являются виноградный и вишневый корригенты, а также цитрусовый корригент, например апельсиновый.
Материалы, которые вводятся в рассматриваемые препараты, могут быть предварительно приготовлены в виде гранул. Этот процесс известен как процесс гранулирования. Под определением "гранулирование" обычно подразумевается любой процесс увеличения размера частиц, в результате которого небольшие частицы собираются вместе в более крупные, постоянные агломераты с образованием свободно текучих композиций, имеющих подходящую консистенцию. Гранулирование может быть осуществлено при перемешивании в смесительном оборудовании или путем прессования, экструзии и агломерации.
Используемое в данном описании понятие "витамин" относится к небольшим количествам органических веществ, которые необходимы для питания. В целях настоящего изобретения понятие "витамин(ы)" включает, но не ограничивается только ими, тиамин, рибофлавин, никотиновую кислоту, пантотеновую кислоту, пиридоксин, биотин, фолиевую кислоту, витамин B12, липоевую кислоту, аскорбиновую кислоту, витамин A, витамин D, витамин E и витамин K. Также понятие "витамин" включает их коферменты. Коферменты являются специальными химическими формами витаминов. Коферменты включают тиаминпирофосфаты (ТПФ), флавинмононуклеотид (ФМН), флавинадениндинуклеотид (ФАД), никотинамидадениндинуклеотид (НАД), никотинамидадениннуклеотидфосфат (НАНФ), кофермент A (КоA, пиридоксальфосфат, биоцитин, тетрагидрофолиевая кислота, кофермент B12, липоиллизин, 11-цис-ретинал и 1,25-дигидроксихолекалциферол. Понятие "витамин(ы)" также включает холин, карнитин и альфа-, бета- и гамма-каротины.
В настоящем описании понятие "минерал" относится к неорганическим веществам, металлам и т.д., которые необходимы для питания человека. Таким образом, понятие "минерал" включает, но не ограничивается только ими, кальций, железо, цинк, селен, медь, йод, магний, фосфор, хром и т.д., а также их смеси.
Понятие "пищевая добавка", используемое в данном описании, означает вещество, которое оказывает заметный питательный эффект при введении в небольших количествах. Пищевые добавки включают, но не ограничиваются только ими, такие ингредиенты как пчелиное молочко, отруби, пшеничные зародыши, бурую водоросль, жир печени трески, женьшень и рыбий жир, аминокислоты, протеины и их смеси. Пищевые добавки могут включать витамины и минеральные вещества.
Биоадгезивы также могут входить в состав препарата настоящего изобретения. Биоадгезив определяется как материал, который прикрепляется к биологической поверхности, такой как слизистая оболочка или ткани кожи. Биоадгезив будет плотно локализовывать лекарственную форму на клеточной мембране. Предпочтительный биоадгезив представляет собой волокно или частицы, набухаемые в воде, но не растворимые в ней. Подходящее соотношение биоадгезива и других компонентов будет давать прочное биоприлипание. Биоадгезивные полимеры, которые могут быть использованы в настоящем изобретении, представляют собой гидрофильные и диспергирующиеся в воде полимеры, содержат свободные карбоксильные группы и имеют относительно высокую основную связывающую способность. Эти полимеры, а также гидрофильные целлюлозы, представляют собой поликарбоксилированные виниловые полимеры и полимеры полиакриловой кислоты. Некоторые гидрофильные полисахаридные камеди, такие как гуаровая камедь, смола плодов акации, смола семян psyllium и др. также могут быть использованы в рецептуре. Весовое отношение биоадгезива к активному ингредиенту может находиться в достаточно широком интервале. На практике весовое отношение биоадгезива к активному ингредиенту может составлять приблизительно от 1:10 до 10:1.
Для получения приемлемого продукта содержащий САЭ-ЦД фармацевтический препарат настоящего изобретения может требовать использования определенных гидрофобных или гидрофильных связующих веществ. Подходящими гидрофобными связующими веществами являются ацетатбутират целлюлозы, ацетатпропионат целлюлозы, пропионат целлюлозы с высоким молекулярным весом (200000), пропионат целлюлозы со средним молекулярным весом (75000), пропионат целлюлозы с низким молекулярным весом (25000), ацетат целлюлозы, нитрат целлюлозы, этилцеллюлоза, поливинилацетат и др. Подходящими гидрофильными связующими веществами являются поливинилпирролидон, поливиниловый спирт, полиэтиленоксид, растворимые в воде или набухающие в воде целлюлоза и производные крахмала и т.д.
Другие соединения, приемлемые в качестве пленкообразующего агента, включают ацетатбутират целлюлозы, ацетатпропионат целлюлозы, пропионат целлюлозы, ГПМЦ, каррагенан, ацетат целлюлозы, нитрат целлюлозы, метилцеллюлозу, гидроксиэтилцеллюлозу, этилцеллюлозу, поливинилацетат и дисперсии латексов, аравийскую камедь, трагакант, гуаровую камедь, желатин и их сочетания.
Примерами других связующих веществ, которые могут быть добавлены к препарату, являются, например, аравийская камедь, трагакант, желатин, крахмал, целлюлозные материалы, такие как метилцеллюлоза и натриевая соль карбоксиметилцеллюлозы, альгиновые кислоты и их соли, полиэтиленгликоль, гуаровая камедь, полисахариды, сахара, инвертированные сахара, полоксомеры (PLURONIC F68, PLURONIC F127), коллаген, альбумин, желатин, целлюлозы в неводных растворителях, предварительно желатинизированный крахмал, пастообразный крахмал, их смеси и др. Другими связующими веществами являются, например, полипропиленгликоль, полиоксиэтиленполипропиленовый сополимер, сложный полиэтиленовый эфир, сложный полиэтиленовый сорбитовый эфир, полиэтиленоксид или их смеси и т.д.
Температуры плавления и/или размягчения этих связующих веществ обычно растут с увеличением их молекулярного веса. Связующие вещества, имеющие температуру плавления или размягчения больше чем 150oC, могут требовать использования пластификаторов в процессе приготовления соответствующей лекарственной формы с тем, чтобы уменьшить температуру плавления и/или размягчения связующего вещества до температуры ниже 150oC. Связующее вещество может быть использовано в любой форме, такой как порошок, гранулы, чешуйки или терморасплавленная жидкость.
В данном описании понятие "пластификатор" включает соединения, способные пластифицировать связующее вещество, используемое в изобретении. Пластификатор должен понижать температуру плавления или температуру перехода в стеклообразное состояние (температура размягчения) связующего вещества. Пластификаторы, такие как низкомолекулярный ПЭГ, обычно расширяют интервал молекулярного веса связующего вещества, понижая за счет этого температуру стеклования или температуру размягчения. Пластификаторы также уменьшают вязкость полимера. Возможно, что пластификатор будет придавать положительные физические свойства препарату настоящего изобретения.
Пластификаторы, которые могут быть использованы в настоящем изобретении, включают, например, но не ограничиваются только ими, низкомолекулярные полимеры, олигомеры, сополимеры, масла, небольшие органические молекулы, низкомолекулярные полиолы, имеющие алифатические гидроксильные группы, пластификаторы сложноэфирного типа, гликолевые эфиры, поли(пропиленгликоль), мультиблоксополимеры, блоксополимеры, низкомолекулярный поли(этиленгликоль), эфиры лимонной кислоты, триацетин, пропиленгликольфталат, фосфатные эфиры, эфиры себациновой кислоты, производные гликоля, эфиры жирных кислот и глицерин.
Такими пластификаторами также могут быть этиленгликоль, 1,2-бутиленгликоль, 2,3-бутиленгликоль, стирол гликоль, диэтиленгликоль, дипропиленгликоль, триэтиленгликоль, тетраэтиленгликоль и другие поли(этиленгликолевые) соединения, моноизопропиловый эфир монопропиленгликоля, моноэтиловый эфир пропиленгликоля, моноэтиловый эфир этиленгликоля, моноэтиловый эфир диэтиленгликоля, лактат сорбита, этиллактат, бутиллактат, этилгликолят, дибутилсебацат, диметилсебацат, ди-2-этилгексилсебацат, трикрезилфосфат, триэтилфосфат, трифенилфосфат, ацетилированные моноглицериды, минеральное масло, касторовое масло, триацетат глицерина, бутилстеарат, моностеарат глицерина, бутоксиэтилстеарат, стеариловый спирт, циклогексилэтилфталат, циклогексилметилдибутилфталат, диэтилфталат, дибутилфталат, диизопропилфталат, диметилфталат, диоктилфталат, ацетилтрибутилцитрат, триэтилцитрат, ацетилтриэтилцитрат, трибутилцитрат и аллилгликолят. Такие пластификаторы являются коммерчески доступными продуктами, выпускаемыми фирмами Aldrich, Sigma Chemical Co. или Morflex, Inc. Также предполагается, и это входит в объем изобретения, что в настоящем препарате может быть использована комбинация пластификаторов.
Настоящие фармацевтические препараты обычно содержат твердое ядро, содержащее сульфоалкиловый эфир циклодекстрина формулы I, которая описана выше, фармацевтически приемлемый носитель и терапевтически эффективное количество терапевтического агента, основная часть которого не образует комплекс с сульфоалкиловым эфиром циклодекстрина. Твердое ядро будет окружено пленочным покрытием. Такие препараты могут быть представлены в твердых лекарственных формах, например, (но без ограничения ими) в виде жевательной пластины, капсулы волокна, пленки, геля, гранулы, жевательной резинки, имплантанта, вставки, пеллеты, порошка, таблетки, ленты, пастилки, пилюли, бруска, полоски и облатки.
Предполагаемыми способами применения являются пероральный, трансбуккальный, назальный, имплантантный, ректальный, вагинальный, подъязычный, ушной и уретральный. Препарат может быть введен с фармацевтически приемлемыми носителями или разбавителями, количество и природа которых определяются растворимостью и свойствами выбранного терапевтического агента, выбранной лекарственной формы и стандартной фармацевтической практикой. Твердые пероральные лекарственные формы могут содержать обычные наполнители, например, лактозу, сахарозу, стеарат магния, смолу и другие материалы, корригент, краситель, буферные агенты, консерванты или стабилизирующие агенты. Эти препараты могут также содержать гигроскопичные агенты, которые способны подавать воду в ядро таблетки. Такие гигроскопичные агенты могут включать растворимые в воде электролиты, небольшие органические соединения, регулирующие осмотическое давление агенты, с целью повышения осмотического давления в пределах лекарственной формы и впитывания воды.
Используемое в данном описании понятие "пациент" относится к теплокровным животным, таким как, например, кошки, собаки, морские свинки, лошади, коровы, овцы. Это понятие относится также к человеку.
Понятие "стандартная лекарственная форма" означает в данном случае единичную или многократную лекарственную форму, содержащую некоторое количество активного ингредиента и разбавителя или носителя, причем указанное количество представляет собой такое количество, что обычно требуется одна или несколько заранее определенных стандартных единиц для одного терапевтического приема. В случае многократных лекарственных форм, таких как жидкости или таблетки с зарубками, указанная заранее определенная единица будет составлять одну часть, такую как половина и четверть таблетки с зарубками многократной лекарственной формы. Следует понимать, что конкретная доза для любого пациента будет зависеть от различных факторов, в том числе от показаний для лечения, используемого терапевтического агента, активности терапевтического агента, серьезности показаний, здоровья, возраста, веса пациента, диеты и фармакологической реакции, используемой конкретной лекарственной дозы и других подобных факторов.
Различные компоненты или соединения могут быть использованы для содействия в приготовлении подходящих для настоящего изобретения лекарственных форм. Такие компоненты или соединения включают, но не ограничиваются только ими, подкисляющий агент, подщелачивающий агент, адсорбент, противогрибковый консервант, антиоксидант, буферный агент, краситель, капсулирующий агент, корригент, загуститель, основу для свечей, подслащивающий агент, антиадгезив, разбавитель для таблеток и капсул, агент для покрытия таблеток, наполнитель для прямого прессования таблеток, диспергирующий агент для таблеток, агент для скольжения, смазывающее вещество, замутняющий агент для таблеток/капсул и полирующий агент.
Понятие "подкисляющий агент" означает соединение, используемое для создания кислой среды с целью стабилизации продукта. Такие соединения включают, но не ограничиваются только ими, уксусную кислоту, лимонную кислоту, фумаровую кислоту, соляную кислоту и азотную кислоту и т.д.
Понятие "подщелачивающий агент" означает соединение, используемое для создания щелочной среды в целях стабилизации продукта. Такие соединения включают, но не ограничиваются только ими, раствор аммиака, карбонат аммония, диэтаноламин, моноэтаноламин, гидроксид калия, борат натрия, карбонат натрия, гидроксид натрия, триэтаноламин, троламин и др.
Понятие "адсорбент" означает агент, способный удерживать другие молекулы на его поверхности физическим или химическим (хемосорбция) путем. Такие соединения включают, но не ограничиваются только ими, порошкообразный и активированный древесный уголь и др.
Определение "консервант" означает соединение, используемое для предупреждения роста микроорганизмов. Такие соединения включают, но не ограничиваются только ими, бензальконийхлорид, бензетонийхлорид, бензиловый спирт, цетилпиридинийхлорид, хлорбутанол, фенол, фенилэтиловый спирт, нитрат фенилртути, тимеросал и др.
Понятие "антиоксидант" означает агент, который ингибирует окисление и, таким образом, используется для предупреждения порчи препаратов из-за процессов окисления. Такими соединениями являются, но не ограничиваются только ими, аскорбиновая кислота, аскорбилпальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол, пирофосфористая кислота, монотиоглицерин, пропилгаллат, аскорбат натрия, бисульфит натрия, сульфоксилат формальдегида натриевая соль и метабисульфит натрия и др.
Понятие "буферный агент" означает соединение, используемое для предупреждения изменения pH при разбавлении или добавлении кислоты или щелочи. Такие соединения включают, но не ограничиваются только ими, метафосфат калия, фосфат калия, моноосновный ацетет натрия, безводный ацетат натрия и др.
Понятие "краситель" означает соединение, используемое для придания окраски твердым фармацевтическим препаратам (например, таблеткам и капсулам). Такие соединения включают, но не ограничиваются только ими FD&C Красный N 3, FD& C Красный N 20, FD&C Желтый N 6, FD&C Голубой N 2, D&C Зеленый N 5, D&C Оранжевый N 5, D& C Красный N 8, карамельный краситель и оксид железа, красный и т.д. Красящие агенты могут также включать диоксид титана, природные красящие агенты, такие как экстракт кожуры винограда, порошок красной свеклы, бета-каротин, аннатто (красно-желтый растительный краситель), кармин, куркума, паприка и т.д.
Понятие "капсулирующий агент" означает соединение, используемое для формирования тонких оболочек с целью капсулирования лекарственного вещества или лекарственного препарата для облегчения его введения. Такие соединения включают, но не ограничиваются только ими, желатин, найлон, биодеградируемые сложные полиэфиры, D,L-поли(молочная)кислота, сополимер полимолочной кислоты и гликолевой кислоты, ацетатфталат целлюлозы и т.д.
Понятие "корригент" означает соединение, используемое для придания фармацевтическому препарату приятного вкуса и часто запаха. Помимо природных корригентов могут быть также использованы различные синтетические корригенты. Такие соединения включают, но не ограничиваются только ими, анисовое масло, коричное масло, какао, ментол, апельсиновое масло, масло мяты перечной, ванилин и др.
Понятие "подслащивающий агент" означает соединение, используемое для придания препарату сладкого вкуса. Такие соединения включают, но не ограничиваются только ими, аспартам, декстрозу, глицерин, маннит, натриевую соль сахарина, сорбит, сахарозу и др.
Понятие "антиадгезив для таблеток" относится к агентам, которые предупреждают прилипание ингредиентов препарата к плунжерам и головкам таблетирующей машины в процессе изготовления таблеток. Такие соединения включают, но не ограничиваются только ими, стеарат магния, кукурузный крахмал, диоксид кремния, тальк и др.
Понятие "связующие вещества для таблеток" означает вещества, используемые для того, чтобы обеспечить адгезию частиц порошка в таблетках. Такие соединения включают, но не ограничиваются только ими, аравийскую камедь, альгиновую кислоту, натрийкарбокиметилцеллюлозу, прессующийся сахар (например NuTab), этилцеллюлозу, желатин, жидкую глюкозу, метилцеллюлозу, повидон, предварительно желатинизированный крахмал и т.д.
Понятие "разбавитель для таблеток и капсул" означает инертные вещества, используемые в качестве наполнителей для получения необходимого объема, текучих свойств и характеристик прессования препарата в виде таблеток и капсул. Такие вещества включают, но не ограничиваются только ими, двухосновный фосфат кальция, каолиновую глину, фруктозу, сахарозу, декстрозу, лактозу, маннит, микрокристаллическую целлюлозу, порошкообразную целлюлозу, осажденный карбонат кальция, сорбит, сульфат кальция, крахмал и т.д.
Понятие "агент для покрытия таблеток" означает соединение, используемое для нанесения покрытия на приготовленные таблетки с целью защиты лекарства от разложения под действием атмосферного кислорода или атмосферной влаги, для обеспечения необходимого профиля высвобождения лекарственного средства после его введения, для маскировки вкуса или запаха лекарственного вещества или для эстетических целей. Покрытие может быть различного типа, в том числе покрытие из сахара, пленочное покрытие или энтеросолюбильное покрытие. Покрытие из сахара имеет водную основу и образуется в результате утолщающегося покрытия вокруг приготовленной таблетки. Покрытие из сахара обычно растворяется в кишечнике при более высоких значениях pH. Пленочное покрытие представляет собой тонкое покрытие вокруг ядра. Если пленочное покрытие не является энтеросолюбильным покрытием, оно растворяется в желудке. Таблетки или шарики с энтеросолюбильным покрытием будут проходить через желудок и будут разрушаться в кишечнике. Некоторые нерастворимые в воде покрытия (например, из этилцеллюлозы) могут быть использованы в качестве покрытия таблетки и шарика с целью замедления высвобождения лекарственного средства при прохождении через желудочно-кишечный тракт. Такие соединения покрытия включают, но не ограничиваются только ими, жидкую глюкозу и сахарозу - в качестве агентов для сахарного покрытия; гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, метилцеллюлозу (например, Methocel) и этилцеллюлозу (например Ethocel) - в качестве пленочного покрытия; и ацетатфталат целлюлозы и шеллак (35% в спирте, "фармацевтическая глазурь") - в качестве энтеросолюбильного покрытия и др.
Понятие "наполнитель для прямого прессования таблеток" означает соединение, используемое в таблетках, получаемых прямым прессованием. Такие соединения включают, но не ограничиваются только ими, двухосновный фосфат кальция (например, Ditab), микрокристаллическую целлюлозу, Avicel, декстран (EMDEX), сахарозу (NUTAB) и т.д.
Понятие "добавка для скольжения таблеток" означает агенты, используемые в таблетках и капсулах для уменьшения трения в процессе прессования. Такие соединения включают, но не ограничиваются только ими, коллоидную или мелкодисперсную двуокись кремния, стеарат магния, кукурузный крахмал, тальк и т. д.
Понятие "смазывающее вещество для таблеток" означает вещества, используемые в препаратах в виде таблеток для уменьшения трения в процессе прессования. Такие соединения включают, но не ограничиваются только ими, стеарат кальция, стеарат магния, минеральное масло, стеариновую кислоту, гидрированное растительное масло, бензойную кислоту, поли(этиленгликоль), NaCl, PRUV, стеарат цинка и др.
Понятие "затемняющий агент для таблеток/капсул" относится к соединению, используемому для получения непрозрачного покрытия таблеток или капсул. Он может быть использован отдельно или в сочетании с красителем. Такие соединения включают, но не ограничиваются только ими, диоксид титана и т.д.
Понятие "полирующий агент для таблеток" означает соединение, используемое для придания приятного блеска покрытым таблеткам. Такие соединения включают, но не ограничиваются только ими, карнаубский воск, белый воск и др.
Настоящий препарат, содержащий физическую смесь терапевтический агент/САЭ-ЦД, как установлено, особенно подходит для терапевтических агентов, в том числе симвастатина, криптофицина, яспамида, амброзина, бисульфана, пропанолола, этопозида, таксола, брефельдина A, пролекарства брефельдина A (NSC# D656202), 9-амино-20(S)-камптотецина, камптотецина, преднизолонацетата, преднизолона, панкреастатина, ризоксина, бриостатина 1, таксотер-O6-бензилгуанина, андростана, гуанина, хлорамфеникола, дапсона, сульфакона, бенклометазона, дипропионата, менадмона, тамоксифенцитрата, холестерина, эстрона, верапмила-HCl, эквилина, варфарина, индометацина, фенитоина, циннаризина, амиодарона-HCl, напроксена, пироксикама, тиабендазола, папаверина, миконазола (свободное основание), нифедипина, тестостерона, прогестерона, карбамазепина, метилпреднизолона, дексаметазона, гидрокортизона и миконазолнитрата.
Все сказанное выше можно лучше понять с помощью приведенных ниже примеров, которые подробно описывают некоторые методики приготовления препаратов в соответствии с настоящим изобретением. Все ссылки, приведенные в примерах, даны в целях иллюстрации. Их не следует рассматривать как ограничивающие объем и суть настоящего изобретения.
Пример 1
Препарат с длительным высвобождением тестостерона-(СБЭ)7-β-ЦД
Данный пример иллюстрирует применение настоящего изобретения для приготовления препаратов с длительным высвобождением фармакологически активного агента, причем примером такого агента является тестостерон.
Изучение фазовой растворимости
Избыточные количества тестостерона добавляют к растворам СБЭ)7-β-ЦД объемом 0,25 мд с концентрациями от 0,0 до 0,05 моль/л. Дисперсии оставляют для уравновешивания минимум на 24 часа во встряхиваемой водяной бане (100 встряхиваний в минуту, 25oC). Дисперсии центрифугируют в течение 10 мин при 2500 об/мин, отбирают образец надосадочной жидкости объемом 20 мкл с помощью газонепроницаемого шприца объемом 100 мкл (Hamilton Co., NV), разбавляют подвижной фазой и анализируют методом ВЭЖХ для определения концентрации тестостерона в растворе. Затем по методу Хигучи (Higuchi) и Коннорса (Connors) для диаграммы типа AL определяют константу связывания тестостерон-(СБЭ)7-β-ЦД K1:1.
Приготовление ядра таблетки
Ядро таблетки готовят при молярном соотношении тестостерон-(СБЭ)7-β-ЦД 1/1. Ядро таблетки состоит либо из комплекса тестостерон-(СБЭ)7-β-ЦД, либо из физической смеси этих двух соединений. Комплекс получают лиофилизированием раствора тестостерона-(СБЭ)7-β-ЦД (5-15% в (СБЭ)7-β-ЦД). Также готовят таблетки, не содержащие (СБЭ)7-β-ЦД. Они состоят из смеси с соотношением 1/1 тестостерона и смеси фруктоза/лактоза (50:50 вес/вес) (Fischer Scientific, NJ). Смеси растирают в ступке и просеивают через сито 200 меш (75 мкм) в условиях низкой влажности. Смеси хранят в эксикаторе. Таблетки весом приблизительно 120 мг прессуют в матрице для растворения с использованием лабораторного пресса Carver Laboratory Press (Fred S. Carver Inc., NJ) при нагрузке 1 т/мин.
Получение полупроницаемой мембраны
Покрывающую рецептуру получают путем растворения 1,0% сорбита (Sigma, MO) в 3,7% дважды перегнанной воды и 0,4% ПЭГ 400 (Sigma, MO). В растворе суспендируют 2,0% ацетата целлюлозы (CA-398-10, Eastman Chemical Co., TN). К смеси добавляют 55,7% метиленхлорида и 37,2% метанола. Дисперсию встряхивают и обрабатывают ультразвуком до полного растворения твердых компонентов. Покрывающий раствор распыляют воздухом (Airbrush, Paasche) на поверхность из нержавеющей стали при постоянном потоке воздуха (40oC), затем оставляют при комнатной температуре на 24 часа. Мембраны снимают с поверхности, проверяют на наличие трещин и повреждений под световым микроскопом (х70) и измеряют их толщину микрометром (Ames, МА). Мембраны закрепляют на матрице для растворения, содержащей таблетку, стороной, которая была распылена на сталь, обращенной к поверхности таблетки.
Изучение высвобождения in vitro
Высвобождение изучают в матрице для растворения в аппарате для растворения USP II (Vanderkamp 600, VanKel Industries Inc.), содержащий 900 мл воды при температуре 37oC и 100 об/мин. Образцы отбирают в различных временных точках. Высвобождение на 100% определяют путем удаления мембраны с матрицы и полного растворения лекарственного средства. Образцы анализируют методом ВЭЖХ, определяя концентрацию тестостерона.
Определение тестостерона метолом ВЭЖХ
Концентрацию тестостерона определяют с использованием колонки ODS Hypersil длиной 15 см с последующим УФ детектированием при 238 нм (Shimadzu scientific Instruments, Inc., Japan). Подвижная фаза состоит из 60% ацетонитрила и 40% дважды перегнанной воды.
ПРИМЕР 2
Препарат с замедленным высвобождением дипиридамола-(СБЭ)7-β-ЦД
Аналитические методики
Дипиридамол анализируют с использованием колонки ODS Hypersil длиной 15 см. Объем образца составляет 20 мкл, длина волны при УФ детектировании равна 285 нм (Shimadzu 6A, Shimadzu, Japan). Подвижную фазу, состоящую из 70% метанола и 30% аммонийфосфатного буфера (pH 5,0), пропускают через колонку со скоростью потока 1,5 мл/мин. (СБЭ)7-β-ЦД определяют с использованием флуориметрического анализа. Раствор из 0,2 мл 1 мМ раствора 2,6-толуидинонафталинсульфоната и 0,8 мл образца возбуждают при 325 нм и флуоресценцию наблюдают при 455 нм с использованием флуоресцентного детектора Perkin-Elmer (Perkin-Elmer, CT).
Опыты по фазовой растворимости
Готовят растворы (CБЭ)7-β-ЦД (0-0,1 М) в различных буферных растворах при значениях pH от 4,0 до 7,0 (цитрат для pH 4 и 5, фосфат для pH 6 и 7). К 0,25 мл этих растворов добавляют избыток дипиридамола и выдерживают для уравновешивания в течение как минимум 24 часов во встряхиваемой водяной бане при 25oC (предварительные эксперименты показали, что равновесная растворимость достигается за 24 часа). Растворы центрифугируют в течение 10 минут при 2500 об/мин, осторожно отбирают образец надосадочной жидкости объемом 20 мкл с помощью газонепроницаемого шприца Hamilton объемом 100 мкл (Hamilton Co., NV), разбавляют подвижной фазой и анализируют методом ВЭЖХ. Затем данные по растворимости используют для определения константы связывания по методу Хигучи (Higuchi) и Коннорса (Connors) для фазового поведения типа AL.
Получение физической смеси
Дипиридамол (SIGMA, MO), (СБЭ)7-β-ЦД и лимонную кислоту (SIGMA, MO) (мольное отношение 1:9:3) физически смешивают и с помощью ступки и пестика растирают вручную. Затем растертую физическую смесь просеивают через сито 200 меш (75 мкм). Эту операцию повторяют дважды. Полученную смесь всегда хранят в эксикаторе, если она не используется.
Описание матрицы для растворения и получения таблетки
Матрица для растворения состоит из цилиндрической, изготовленной из нержавеющей стали центральной части, платформы из нержавеющей стали, верхней крышки из нержавеющей стали, двух тефлоновых прокладок (верхней и нижней) и тефлоновых вставок. Центральная цилиндрическая часть имеет отверстие (радиус 7,5 мм) в центре, в котором прессуется таблетка. Верхняя крышка из нержавеющей стали и верхняя тефлоновая прокладка обе имеют в центре отверстия того же радиуса. Центральную часть переворачивают и прикручивают к
платформе. В цилиндрическое отверстие помещают приблизительно 120 мг физической смеси, содержащей лекарственное средство, (СБЭ)7-β-ЦД и лимонную кислоту и в отверстие плотно вводят плунжер. Ядро таблетки прессуют с усилием в одну тонну в течение одной минуты с использованием пресса Carver (Fred Carver Inc., NJ). Плунжер осторожно удаляют из центральной части, выбивая с обратной стороны.
Пленочное покрытие
Приготовление полимерных растворов
Покрытие из Eudragit получают путем растворения 5% (вес/вес) Eudragit R или S (Huls America, NJ), 5% мочевины (SIGMA, MO) или полиэтиленгликоля (ПЭГ 3350, SIGMA, MO) и 0,75% триэтилцитрата (ТЭЦ, SIGMA, MO) в 89,25% этанола, до тех пор пока не образуется прозрачный раствор. Полимерные растворы ацетата целлюлозы (CA-320S, Eastman Chemical Co., TN) И фталата гидроксипропилметилцеллюлозы (ФГПМЦ, Eastman Chemical Co., TN) получают растворением 5% полимеров и 1% ТЭЦ в 94% растворителя, содержащего равные количества метиленхлорида и метанола. Соотношение ацетата целлюлозы и ФГПМЦ варьируют от 50: 50 до 75:25, но общее количество полимера всегда составляет 5%. Растворение проводят до тех пор, пока не образуется прозрачный раствор.
Нанесение покрытия на таблетки
Этот покрывающий раствор затем распыляют воздухом непосредственно на поверхность таблетки при постоянной скорости потока воздуха (приблизительно 70oC). Покрытые таблетки дополнительно сушат в течение 15 мин при том же потоке воздуха. Таблетки дополнительно сушат в течение 12-16 часов при комнатной температуре. Толщину мембраны принимают равной разности толщины таблетки после и перед нанесением покрытия. Толщины измеряют с помощью микрометра Screwgauge.
Изучение высвобождения in vitro
Изучение высвобождения для таблеток, покрытых Eudragit L и смесью АЦ: ФГПМЦ проводят, помещая матрицу для растворения в аппарат для растворения USP II (Vanderkamp 600, VanKel Industries Inc.), содержащий 450 мл HCl (pH 1,5, 37oC при 100 об/мин). Через 2 часа матрицу осторожно извлекают и помещают в 450 мл фосфатного буфера (pH 1,5, 37oC при 100 об/мин) и эксперимент по растворению продолжают. Периодически отбирают образцы объемом 1,5 мл и эквивалентные количества среды для растворения возвращают в сосуд для растворения. Для таблетки, покрытой АЦ:ФГПМЦ 100%-ное высвобождение определяют путем удаления мембраны с матрицы и полного растворения лекарства. Эксперименты по высвобождению для таблеток, покрытых Eudragit S, проводят аналогичным образом в 450 мл HCl в течение первых 2 часов, затем в фосфатном буфере (pH 6,4) в течение дополнительных 5 часов, а в фосфатном буфере при pH 7,2. Условия высвобождения и методики аналогичны описанным выше.
Образец объемом 0,5 мл разбавляют наполовину подвижной фазой, и разбавленные образцы анализируют методом ВЭЖХ для определения концентрации лекарственного средства, как описано в последнем разделе. Остаток образца фильтруют через мембрану из поливинилиденфторида (Fischer Scientific, NJ) и не содержащие лекарственное средство образцы анализируют на содержание (СБЭ)7-β-ЦД с использованием флуориметрического анализа, описанного ниже.
ПРИМЕР 3
Препарат с длительным высвобождением метилпреднизолона-(СБЭ)7-β-ЦД
Изучение фазовой растворимости
Избыточные количества метилпреднизолона (МП) добавляют к растворам (СБЭ)7-β-ЦД объемом 0,25 мл с концентрациями от 0,0 до 0,2 моль/л. Дисперсии оставляют для уравновешивания как минимум на 24 часа во встряхиваемой водяной бане (100 встряхиваний в минуту, 25oC). Дисперсии центрифугируют в течение 10 мин при 2500 об/мин, отбирают образец надосадочной жидкости объемом 20 мкл с помощью газонепроницаемого шприца объемом 100 мкл (Hamilton Co., NV), разбавляют подвижной фазой и анализируют методом ВЭЖХ для определения концентрации метилпреднизолона в растворе. Затем способом Хигучи (Higuchi) и Коннорса (Connors) для диаграммы типа AL определяют константу связывания метилпреднизолон-(СБЭ)7-β-ЦД K1:1.
Приготовление ядра таблетки
Ядро таблетки готовят при молярном соотношении метилпреднизолон/(СБЭ)7-β-ЦД 1/7. Это соотношение вычисляют с использованием предварительно определенной константы связывания, чтобы иметь в ядре таблетки (СБЭ)7-β-ЦД, достаточное для солюбилизации присутствующего метилпреднизолона. Кроме того, для изучения влияния соотношения метилпреднизолон-(СБЭ)7-β-ЦД, готовят ядра таблетки с отношением 1/3 и 1/10 (ср. результаты 4). Ядро таблетки состоит либо из комплекса метилпреднизолон-(СБЭ)7-β-ЦД, либо из физической смеси этих двух соединений. Комплекс получают лиофилизированием раствора метилпреднизолон-(СБЭ)7-β-ЦД (5-15% в (СБЭ)7-β-ЦД). Также готовят таблетки, не содержащие (СБЭ)7-β-ЦД. Они состоят из смеси в соотношении 1/7 метилпреднизолона и смеси фруктозы и лактозы 50:50 (вес/вес) (Fischer Scientific, NJ). Смеси растирают в ступке и просеивают через сито 200 меш (75 мкм) в условиях низкой влажности. Смеси хранят в эксикаторе, если они не используются. Таблетки весом приблизительно 150 мг прессуют в матрице для растворения с использованием лабораторного пресса Carver (Fred S. Carver Inc., NJ) при нагрузке 1 т/мин.
Получение полупроницаемой мембраны
Покрывающую рецептуру получают путем смешения 4,5% этилцеллюлозы (Ethocel Standart 10 Premium, Dow Chemicals, MI) с эквивалентным количеством полиэтиленгликоля 3350 (ПЭГ 3350, Sigma, MO). К смеси добавляют 0,9% ПЭГ 400 (Sigma, MO) и 90,1% абсолютного этанола. Дисперсию встряхивают и обрабатывают ультразвуком до полного растворения твердых компонентов. Покрывающий раствор распыляют воздухом (Airbrush, Paasche) на тефлоновую поверхность при постоянном потоке воздуха (40oC). После распыления мембраны сушат в потоке воздуха с температурой 40oC в течение 5 мин, затем оставляют при комнатной температуре на 24 часа. Мембраны снимают с тефлоновой поверхности, проверяют на наличие трещин и повреждений под световым микроскопом (х70) и измеряют их толщину микрометром (Ames, MA). Мембраны закрепляют на матрице для растворения, содержащей таблетку, стороной, которая находилась на тефлоне, обращенной к поверхности таблетки.
Изучение высвобождения in vitro
Изучение высвобождения проводят, помещая матрицу для растворения в аппарат для растворения USP II (Vanderkamp 600, VanKel Industries Inc.), содержащий 350 мл воды при температуре 37oC при 100 об/мин. Образцы отбирают в различных временных точках. Определяют 100%-ное высвобождение путем удаления мембраны с матрицы и полного растворения лекарственного средства. Образцы анализируют методом ВЭЖХ и флуориметрическим методом для определения концентраций метилпреднизолона и (СБЭ)7-β-ЦД соответственно.
Определение метилпреднизолона методом ВЭЖХ
Концентрацию метилпреднизолона определяют с использованием колонки ODS Hypersil длиной 15 см с последующим УФ детектированием при 254 нм (LC-10AT, Shimadzu Scientific Instruments, Inc., Japan). Подвижная фаза состоит из 30% ацетонитрила и 70% ацетатного буфера с pH 4,7.
Флуориметрическое определение (СБЭ)7-β-ЦД
Концентрацию (СБЭ)7-β--ЦД определяют путем добавления 0,2 мл раствора 2,6-толуидинонафталинсульфоната с концентрацией 1E-3 моль/л к 0,8 мл образца. Раствор возбуждают при 325 нм и наблюдают флуоресценцию при 455 нм (флуоресцентный спектрофотометр, 65040 Fluorescence Spectrophotometer, Perkin-Elmer, CT).
ПРИМЕР 4
Таблетки, содержащие (СБЭ)7-β-ЦД и терапевтический агент
Лекарственная форма в виде таблеток в соответствии с настоящим изобретением в общем случае может быть приготовлена следующим образом. Терапевтический агент и (СБЭ)7-β-ЦД смешивают в сухом виде в течение приблизительно 10 минут. К смеси добавляют остальные ингредиенты и смешивают в сухом виде в течение приблизительно 10 минут. Затем таблетки прессуют до твердости приблизительно 8-10 кг. Для приготовления лекарственных форм настоящего изобретения могут использоваться рецептуры, приведенные в табл. 3.
Приведенные в табл. 3 ингредиенты используются для получения ядра таблетки с массой 600 мг, имеющего быстрый профиль высвобождения. Номер около ингредиента означает общий порядок добавления. После добавления каждой группы ингредиентов смесь перемешивают в сухом виде в течение 5-10 мин. Таким образом, PRUV и кукурузный крахмал добавляют отдельно (стадия 3) от других ингредиентов и к общей методике добавляют дополнительную стадию сухого перемешивания в течение 5 минут.
Приведенные в табл. 4 ингредиенты используются для получения ядра таблетки с массой 555 мг, имеющего быстрый профиль высвобождения. Номер около ингредиента означает общий порядок добавления. После добавления каждой группы ингредиентов смесь перемешивают в сухом виде в течение 5-10 мин. Таким образом, PRW и Ac-Di-Sol добавляют отдельно (стадия 3) от других ингредиентов и к общей методике добавляют дополнительную стадию сухого перемешивания в течение 5 минут.
Приведенные в табл. 5 ингредиенты используются для получения ядра таблетки с массой 500 мг, имеющего быстрый профиль высвобождения. Номер около ингредиента означает общий порядок добавления. После добавления каждой группы ингредиентов смесь перемешивают в сухом виде в течение 5-10 мин. Таким образом, PRUV и натрийкроскармелозу добавляют отдельно (стадия 3) от других ингредиентов и к общей методике добавляют дополнительную стадию сухого перемешивания в течение 5 минут.
Приведенные в табл. 6 ингредиенты используются для получения ядра таблетки с массой 600 мг, имеющего быстрый профиль высвобождения. Номер около ингредиента означает общий порядок добавления. После добавления каждой группы ингредиентов смесь перемешивают в сухом виде в течение 5-10 мин. Таким образом, PRUV и натриевую соль гликолята крахмала добавляют отдельно (стадия 3) от других ингредиентов и к общей методике добавляют дополнительную стадию сухого перемешивания в течение 5 минут.
Приведенные в табл. 7 ингредиенты используются для получения ядра таблетки с массой 315 мг, имеющего быстрый профиль высвобождения. Номер около ингредиента означает общий порядок добавления. После добавления каждой группы ингредиентов смесь перемешивают в сухом виде в течение 5-10 мин. Таким образом, PRUV добавляют отдельно (стадия 3) от других ингредиентов и к общей методике добавляют дополнительную стадию сухого перемешивания в течение 5 мин.
Приведенные в табл. 8 ингредиенты используются для получения ядра таблетки с массой 560 мг, имеющего быстрый профиль высвобождения. Номер около ингредиента означает общий порядок добавления. После добавления каждой группы ингредиентов смесь перемешивают в сухом виде в течение 5-10 мин. Таким образом, PRUV добавляют отдельно (стадия 3) от других ингредиентов и к общей методике добавляют дополнительную стадию сухого перемешивания в течение 5 минут.
Приведенные в табл. 9 ингредиенты используются для получения 600 миллиграммовой твердой желатиновой капсулы, состоящей из 3 х 200 мг покрытых пленкой минитаблеток в соответствии с настоящим изобретением. Непокрытые ядра минитаблеток имеют быстрый профиль высвобождения. Минитаблетки готовят следующим образом. Номер около ингредиента означает общий порядок добавления. После добавления каждой группы ингредиентов смесь перемешивают в сухом виде в течение 5-10 мин. Таким образом, PRUV и кукурузный крахмал добавляют отдельно (стадия 3) от других ингредиентов и к общей методике добавляют дополнительную стадию сухого перемешивания в течение 5 минут. Затем смесь разделяют на три равных части и каждую часть прессуют в минитаблетку. После покрытия ядра таблетки пленкообразующим агентом настоящего изобретения в соответствии с приведенным ниже примером, покрытые минитаблетки помещают внутрь твердой желатиновой капсулы.
Следует заметить, что в некоторых из приведенных выше примеров связующие вещества, такие как EMDEX и полиокс-0,4 М, можно заменить на контролирующие высвобождение агенты, такие как ГПМЦ (гидроксипропилметилцеллюлоза), ГПЦ (гидроксипропилцеллюлоза), ацетатбутират целлюлозы, ацетатпропионат целлюлозы, пропионат целлюлозы, каррагенан, ацетат целлюлозы, нитрат целлюлозы, метилцеллюлозу, гидроксиэтилцеллюлозу, этилцеллюлозу, поливинилацетат, дисперсии латексов, аравийскую камедь, трагакант, гуаровую камедь и т.д. Таким образом, могут быть приготовлены непокрытые ядра таблеток, которые имеют контролируемый или постепенный профиль высвобождения. После дополнительного покрытия пленкообразующими агентами настоящего изобретения получают препарат в виде таблетки, который имеет комбинированный замедленный и контролируемый или постепенный профиль высвобождения, т.е. при достижении заранее определенной части желудочно-кишечного тракта оболочка таблетки становится пористой и позволяет терапевтическому агенту высвобождаться из ядра таблетки с контролируемым или постепенным профилем. Ядро таблетки с контролируемым или постепенным высвобождением может применяться для получения препарата в виде таблетки, состоящей из хорошо растворимого в воде пленкообразующего агента, очень пористой пленки, большого количества осмотического или солюбилизирующего агента и других подобных условий.
Другие способы получения ядра таблетки включают, например, сухое гранулирование, мокрое гранулирование и прессование - измельчение - повторное прессование. Соответственно, способ сухого гранулирования может включать предварительное формирование таблетки или зерен из всех ингредиентов таблетки, кроме САЭ-ЦД, измельчение предварительно формованной таблетки или зерен, смешение размолотого продукта с САЭ-ЦД, и повторное прессование смеси для получения необходимого препарата в форме таблетки.
Приведенные в табл. 10 ингредиенты используются для получения ядра таблетки массой 525 мг, имеющего контролируемый или постепенный профиль высвобождения. Номер около ингредиента означает общий порядок добавления. После добавления каждой группы ингредиентов смесь перемешивают в сухом виде в течение 5-10 мин. Таким образом, PRUV и кукурузный крахмал добавляют отдельно (стадия 3) от других ингредиентов и к общей методике добавляют дополнительную стадию сухого перемешивания в течение 5 минут.
Пример 5
Ядро таблеток, получаемое из гранул, содержащих (СБЭ7)-β-ЦД и терапевтический агент
Лекарственные формы в соответствии с настоящим изобретением могут содержать гранулы, изготовленные мокрым гранулированием следующим образом. Приведенное процентное содержание означает весовые проценты по отношению к весу конечного препарата. Настоящий пример основан на дозе метилпреднизолона (МП) в 10 мг. Терапевтический агент (20%) и (СБЭ)7-β-ЦД (375) смешивают в сухом виде. С помощью водной суспензии поливинилпирролидона (ПВП) гранулируют путем мокрого гранулирования лактозу (40%) и декстрозу (8%) до получения 2% увеличения веса с образованием необходимых гранул. NaHCO3 (3,5%), PRUV (4,5%), SiO2 (0,5%) и ксилит (2%) в сухом виде смешивают с гранулами и полученную смесь прессуют в таблетки до твердости 8-10 кг.
Пример 6
Пленочные покрытия таблеток
Пленочные покрытия для таблеток в соответствии с настоящим изобретением могут быть изготовлены с использованием следующих ингредиентов и условий. Пленочные покрытия могут быть изготовлены на основе воды или растворителя, например спирта. Обычно пленкообразующий агент растворяют или суспендируют в половине объема от предполагаемого объема раствора, и добавляют остальные ингредиенты. При необходимости смесь затем доводят до конечного объема путем добавления воды или растворителя. Приготовленный раствор или суспензию используют в соответствии с Примером 7 для нанесения покрытия на ядра таблеток, полученные, как описано выше. Приведенные в табл. 11-17 рецептуры пленок относятся к объему конечного раствора или суспензии в 100 мл.
EUDRAGIT RS получают от производителя в виде 30% вес. суспензии. Пленкообразующий агент EUDRAGIT RS растворяют в воде (50 мл) при перемешивании и последовательно добавляют ТЭЦ, тальк и ГПМЦ (порообразователь). Конечный объем раствора доводят до 100 мл путем добавления воды. Можно использовать и другие порообразователи и пленкообразующие агенты.
Рецептура EUDRAGIT RL может быть приготовлена в изопропаноле (ИПА). EUDRAGIT RL растворяют в ИПА (50 мл) при перемешивании, затем последовательно добавляют ТЭЦ, тальк и ГПЦ. Конечный объем раствора доводят до 100 мл с помощью ИПА.
EUDRAGIT RL и EUDRAGIT RS при перемешивании смешивают в воде (50 мл), затем последовательно добавляют ТЭЦ, тальк и ГПЦ. Конечный объем раствора доводят до 100 мл водой. EUDRAGIT RL служит для улучшения водопроницаемости пленки из EURODRAGIT.
В воде (50 мл) при перемешивании растворяют этилцеллюлозу и последовательно добавляют дибутилсебацат, тальк и ГПМЦ E5. Конечный объем раствора доводят до 100 мл водой. Аналогичная методика может быть осуществлена с использованием ИПА вместо воды и ГПЦ (1,0 г) вместо ГПМЦ E5.
В воде (50 мл) при перемешивании растворяют ацетат целлюлозы и последовательно добавляют ТЭЦ, тальк и лактозу. Конечный объем раствора доводят до 100 мл водой. При использовании этой рецептуры пленки может быть необходимо применение аппарата Hi-Coater при температуре 45oC или выше.
Полимеры EUDRAGIT при перемешивании растворяют или суспендируют в воде (50 мл) и последовательно добавляют ТЭЦ и тальк. Конечный объем раствора доводят до 100 мл водой. В этих комбинированных рецептурах пленок можно использовать и другие пленкообразующие агенты, такие как ацетат целлюлозы и ГПМЦ.
EUDRAGIT L при перемешивании растворяют или суспендируют в воде (50 мл) и последовательно добавляют ТЭЦ и тальк. Конечный объем раствора доводят до 100 мл водой. Эта пленка может быть использована для получения препарата в виде таблетки с высвобождением в тонком кишечнике и для покрытия таблеток, уже покрытых другими пленочными покрытиями настоящего изобретения. Полученный препарат в виде таблетки будет представлять собой препарат, имеющий замедленное контролируемое или постепенное высвобождение терапевтического агента из ядра таблетки.
Пример 7
Нанесение покрытия на ядра таблеток с помощью пленкообразующих агентов
Покрытый пленкой препарат в виде таблетки обычно может быть приготовлен следующим образом. Предполагается, что другие эквивалентные условия и оборудование, известные специалисту в данной области, могут быть использованы для получения настоящих препаратов без проведения каких-либо дополнительных экспериментов.
При следующих условиях используют устройство Hi-Coater (Vector) (устройство для нанесения покрытия на таблетки с перфорированным поддоном):
Температура на входе - 65-75oC
Температура на выходе - 30-35oC
Скорость распыления - 2-3 г/мин
Загрузка таблеток - 300 г
Скорость вращения - 20 об/мин
После получения раствора или суспензии, содержащей пленкообразующий агент и другие ингредиенты (в соответствии с примером 6), ядра таблеток помещают внутрь аппарата Hi-Coater и проводят процедуру нанесения пленки до тех пор, пока не образуется покрытие толщиной 100-125 мкм. Покрытые таблетки сушат приблизительно при 40oC в течение ночи. Толщина таблетки и композиция пленки могут варьироваться. Настоящий способ может быть использован в случае композиций для нанесения покрытия на водной основе или на основе растворителя.
Выше приведено подробное описание конкретных вариантов осуществления настоящего изобретения. В свете настоящего изобретения специалист в данной области должен понимать, что могут быть проведены очевидные модификации описанных здесь вариантов без отклонения от сути и объема настоящего изобретения. Следовательно, все описанные и заявленные здесь варианты осуществления изобретения могут быть проведены без дополнительных экспериментов. Весь объем изобретения и его эквивалентов представлен в следующей формуле изобретения. Таким образом, формулу изобретения и описание не следует использовать для необоснованного сужения полного объема защиты, на которую имеет право настоящее изобретение.
Ссылки
Приведенные ниже ссылки в объеме, в котором они дают примеры методик или других деталей дополнительно к приведенным в описании, специально введены в качестве справочного материала.
1. Pharmaceutical Dosage Forms-Tablets vol. 1, 2nd edition, Herbert A. Lieberman, ed. pp. 372-376.
2. Патент США 3426011, Parmerter et al.
3. Патент США 3816393, Hayashi et al.
4. Патент США 4497803, Harada et al.
5. Патент США 4535152, Szejtli et al.
6. Патент США 4555504, Jones
7. Патент США 4582900, Brandt et al.
8. Патент США 4596795, Pitha
9. Патент США 4638058, Brandt et al.
10. Патент США 4727064, Pitha
11. Патент США 4746734, Tsuchiyama et al.
12. Патент США 4764604, Mueller
13. Патент США 4774329, Friedman
14. Патент США 4808232, Beesley
15. Патент США 4869904, Uekama et al.
16. B. W. Mueller et al. Proceedings of the Fourth International Symposium on Cyclodextrins, (1988) pp. 369-382, "Cyclodextrin Derivatives for Solubilization, Stabilization, and Absorption of Drugs".
17. Josef Pitha, Third International Symposium on Recent Advances in Drug Delivery Systems, (1987), pp. 1-12, "Amorphous Water Soluble Derivatives of Cyclodextrins: From Test Tube to Patent".
18. Патент США 5134127, Stella et al.
19. M. E. Brewster et al. , J. Pharm. Sci. (1988), 77(11), 981-985; "Improved delivery through biological membranes. XXXL: Solubilization and stabilization of an estradiol chemical delivery system by modified SBE7-β-CD-Cyclodextrins".
20. N. Muranushi et al., Nippon Yakurigaku Zasahi (Japan) (1988), 91(6), 377-383; "Studies on benexate. CD: effect of inclusion compound formation on the anti-ulcer activity of benexate, the effective ingredient in benexate. CD".
21. J.J. Torres-Labandeira et al., STP Pharma. Sci. (1994), 4(3), 235-239; "Biopharmaceutical stability of the glibomuride/β-cyclodextrin inclusion complex after one year of storage".
22. D. Peri et al. Drug Dev. Ind. Pharm. (USA) (1994), 20(4), 1401-1410; "Inclusion complexes of tolnaftate with β-cyclodextrin and hydroxypropyl β-cyclodextrin".
23. F. J. Otero-Espinar, et al. Int. J. Pharm. (Netherlands) (1991), 75(1), 3744; "Oral bioavailability of naproxen-β-cyclodextrin inclusion complex".
24. S.Y. Lin et al. Int. J. Pharm. (Netherlands) (1989), 56(3), 249-259; "Solid particulates of drug-β-cyclodextrin inclusion complexes directly prepared by a spray-drying technique".
25. M.T. Esclusa-Diaz et al. Int. J. Pharm. (Amsterdam) (1996), 142(2), 183-187; "Preparation and evaluation of ketoconazole-β-cyclodextrin multi-component complexes".
26. Патент США 5134127, Stella, et al.

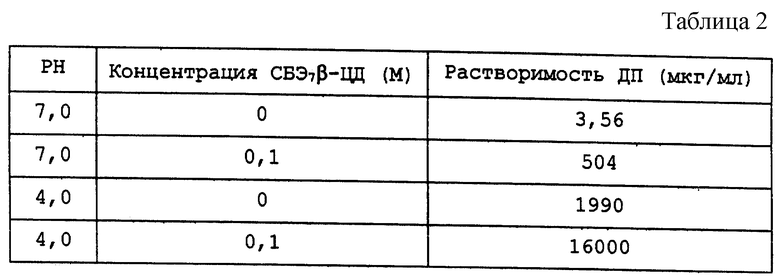
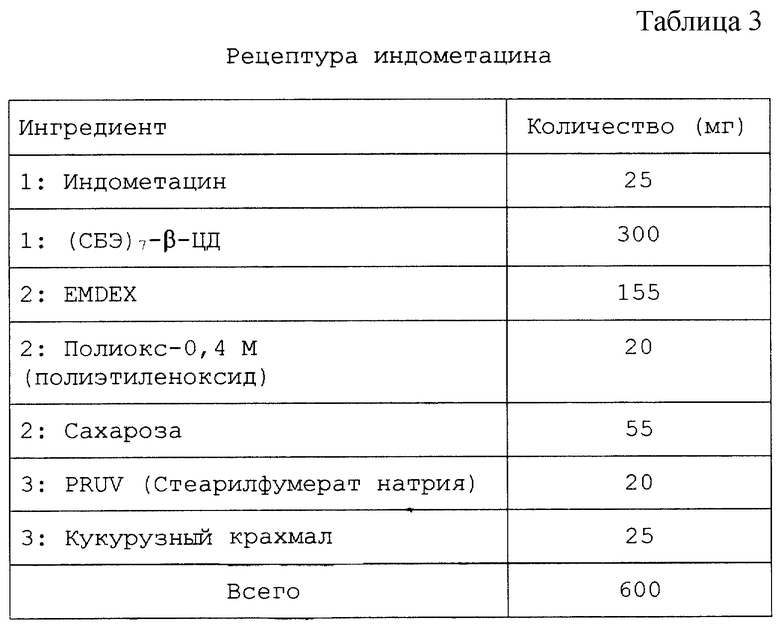
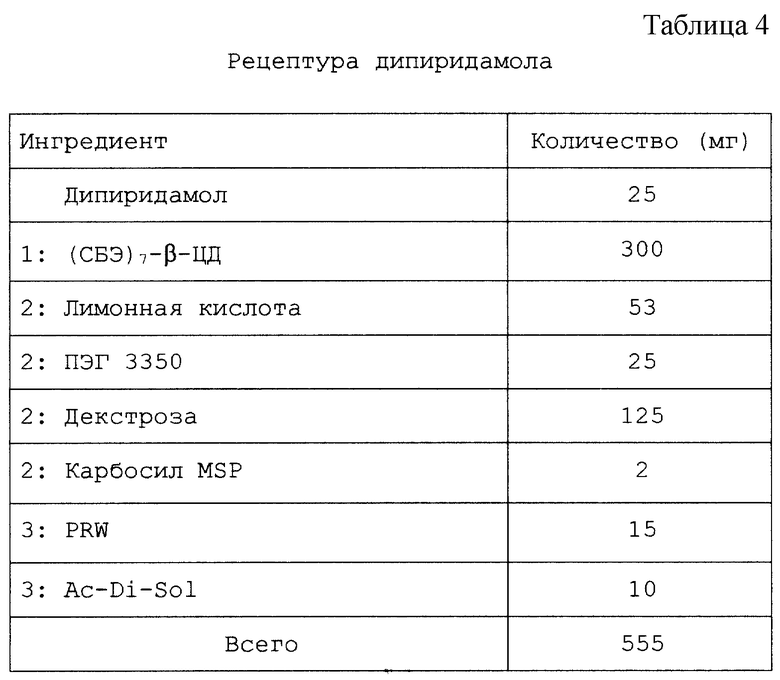
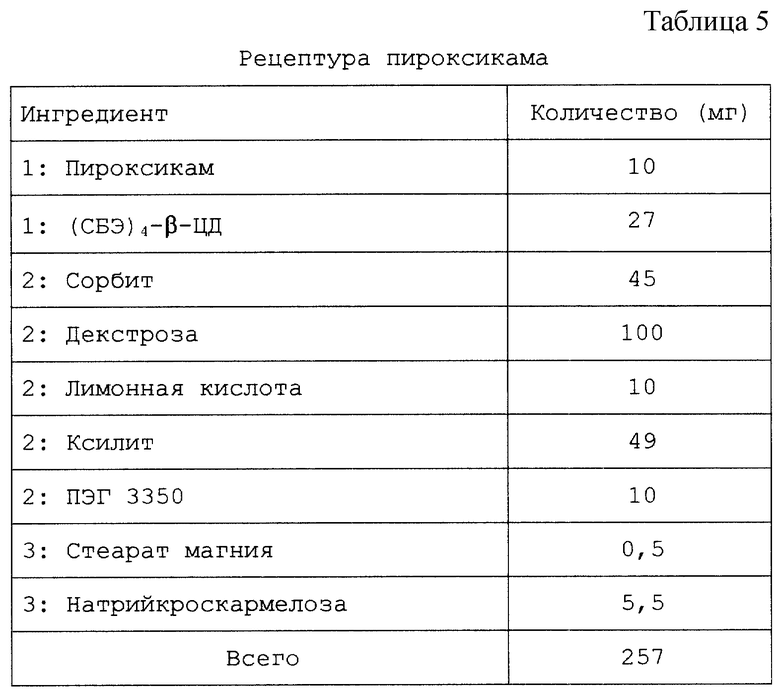
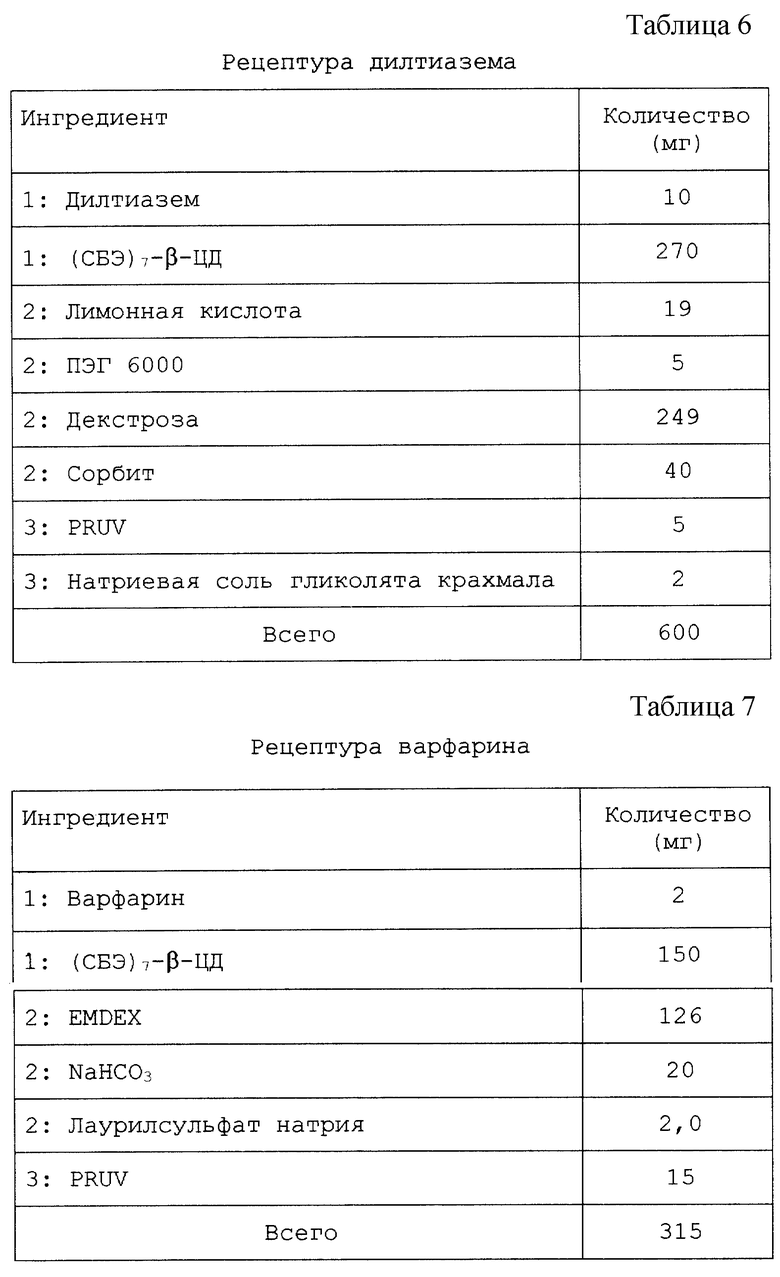
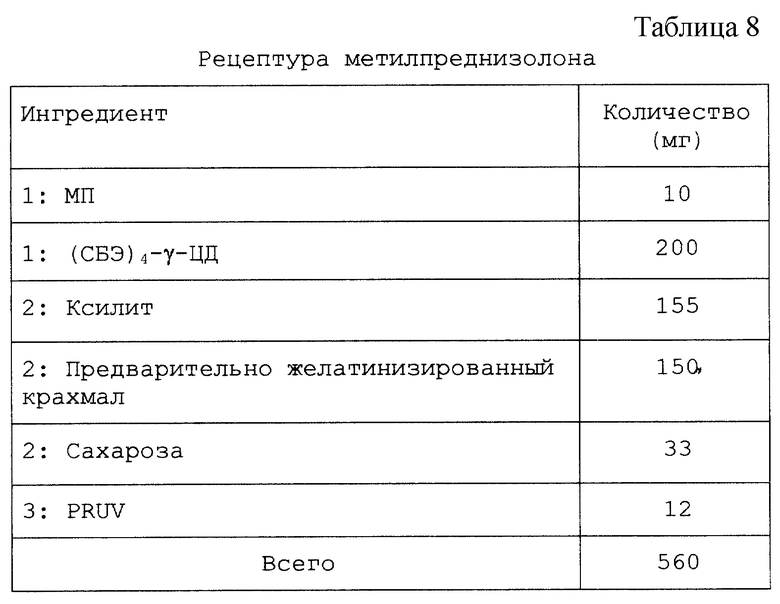
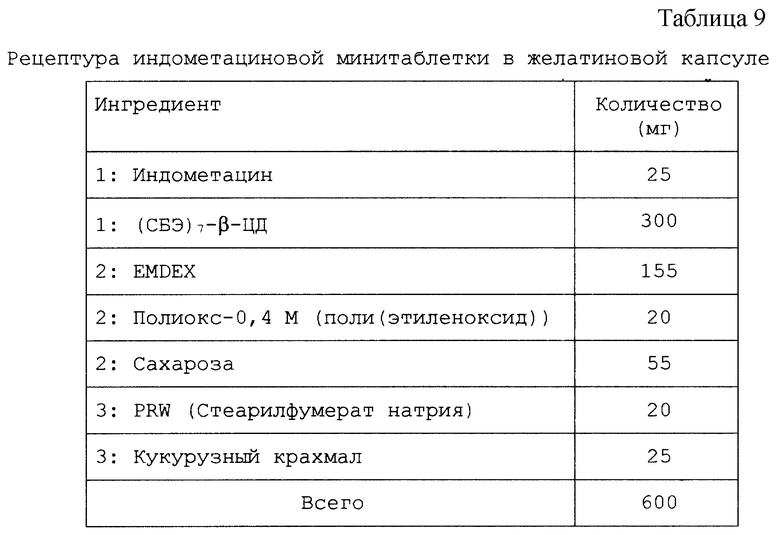
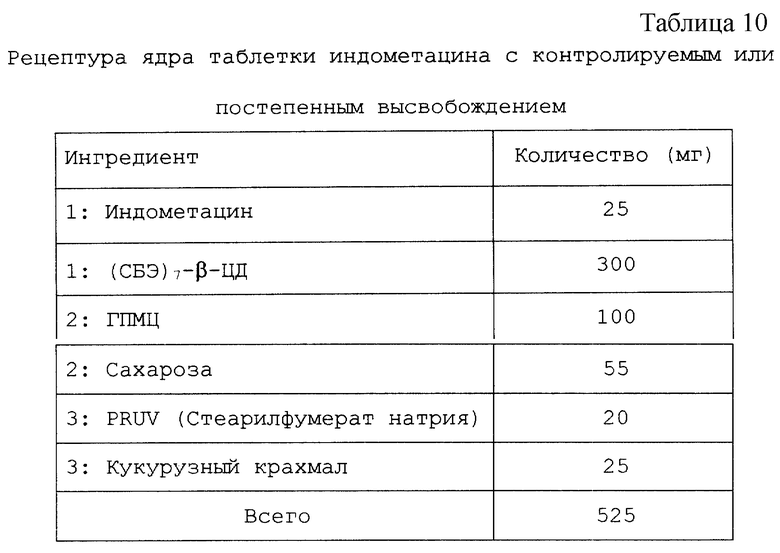

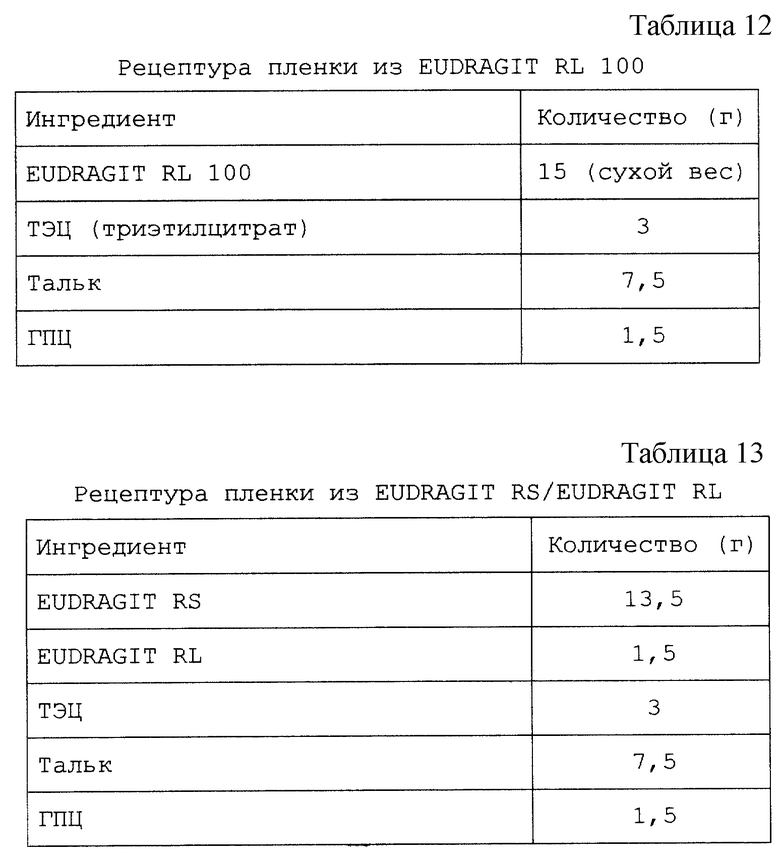
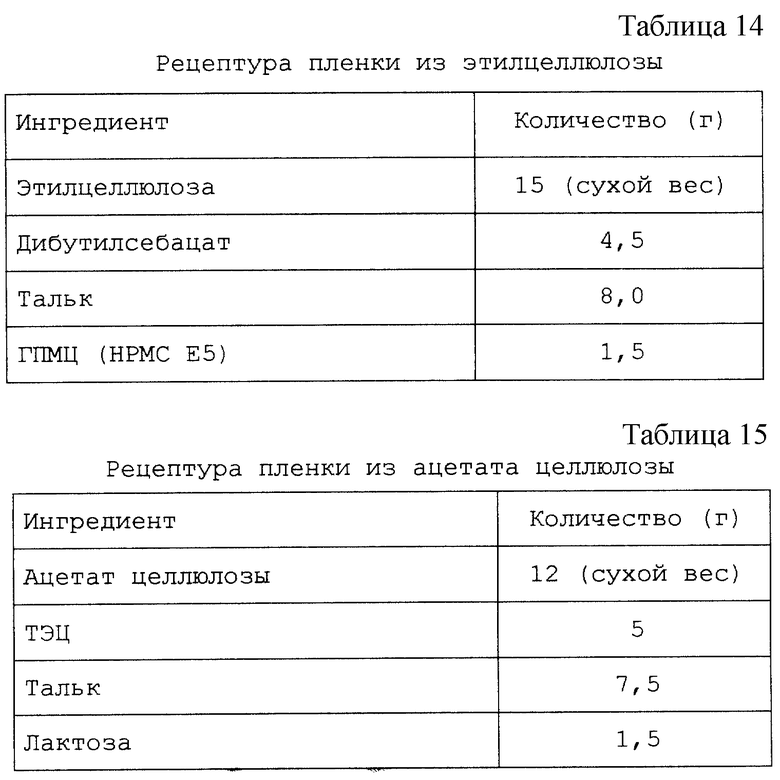
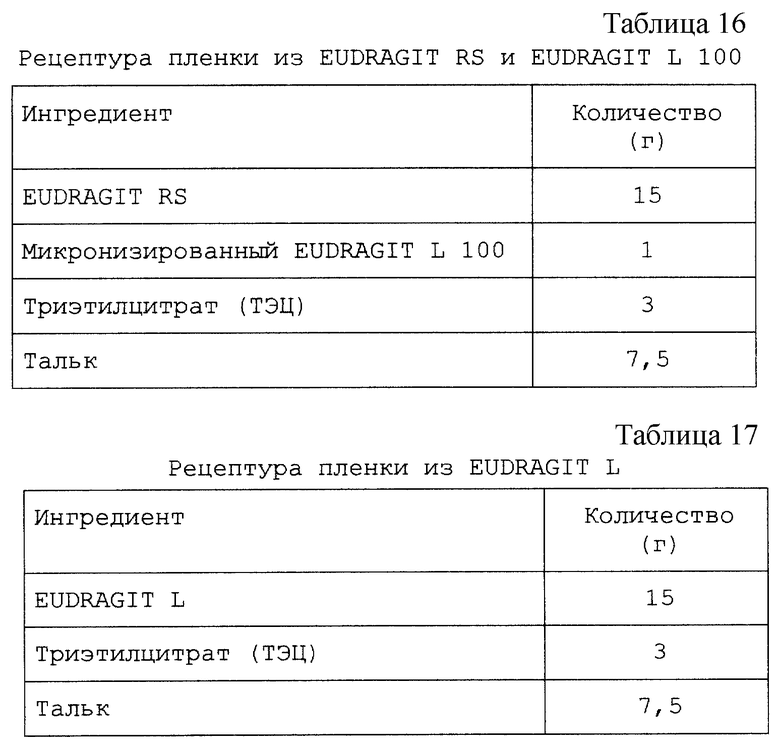

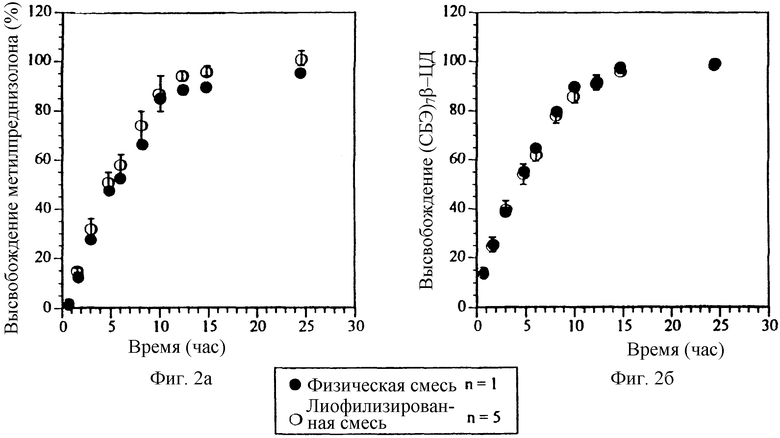
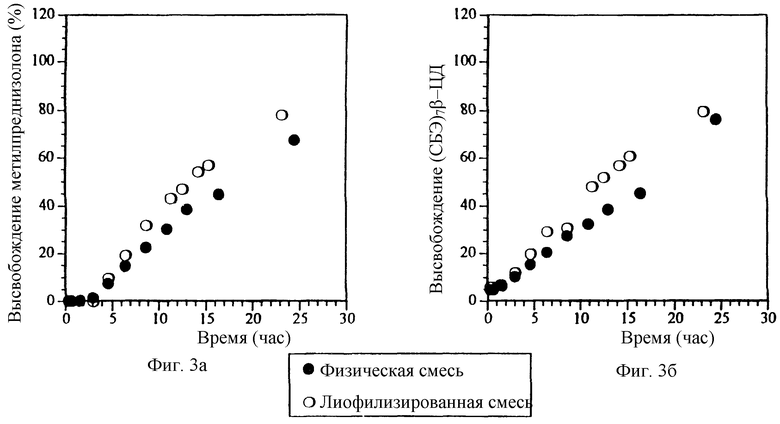
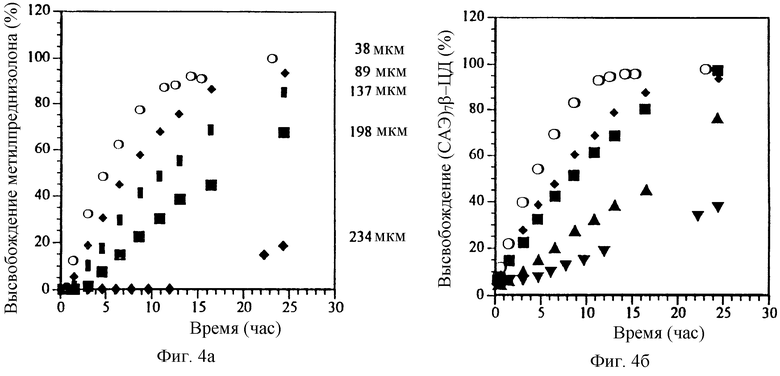
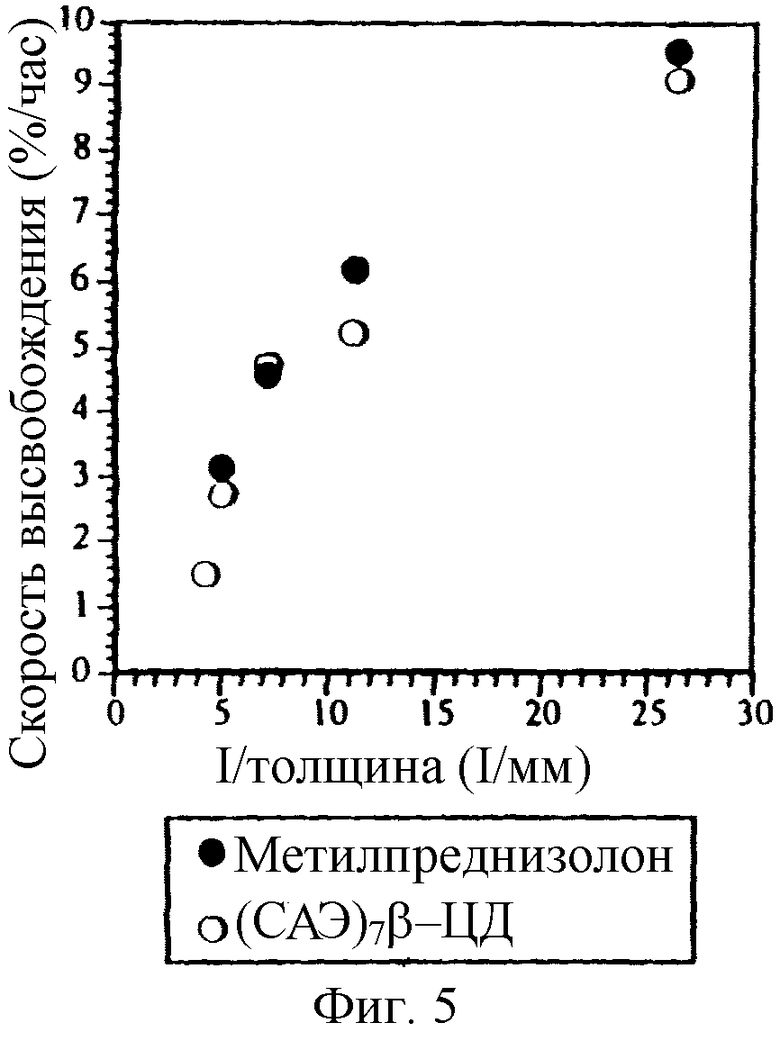
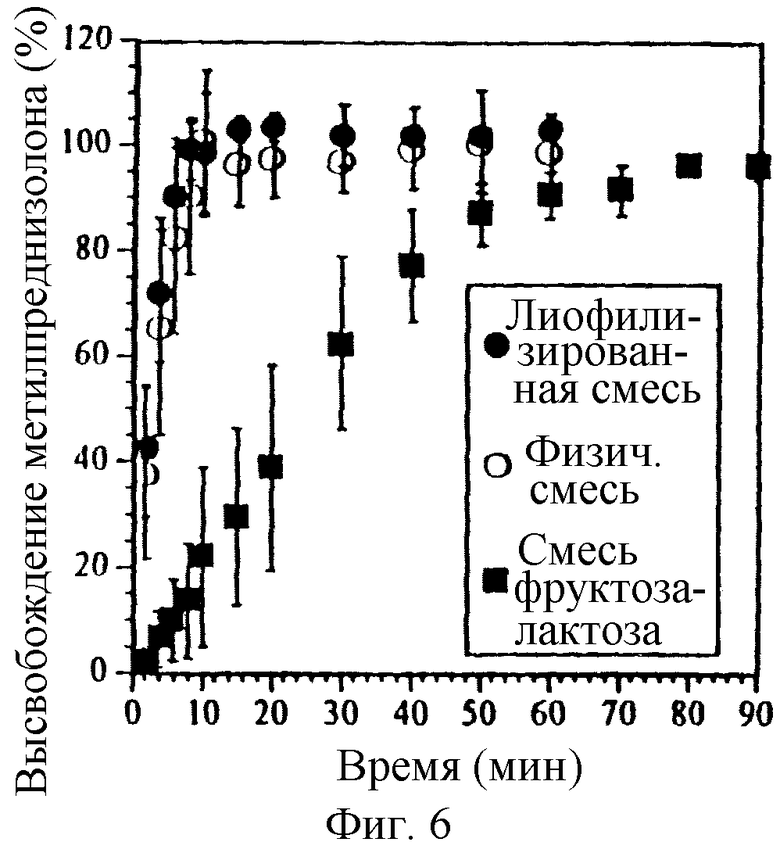
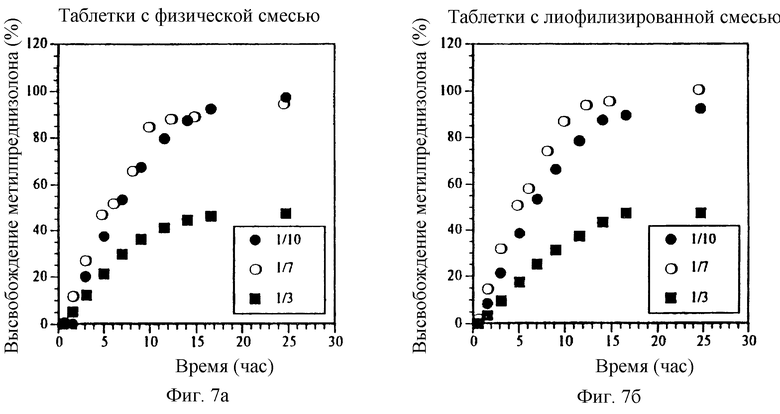
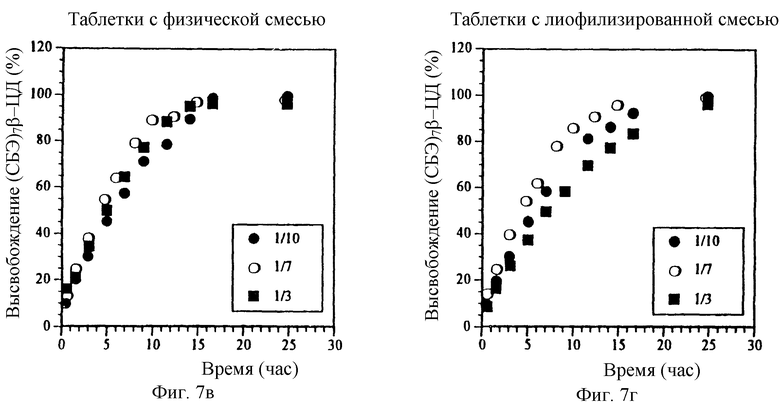
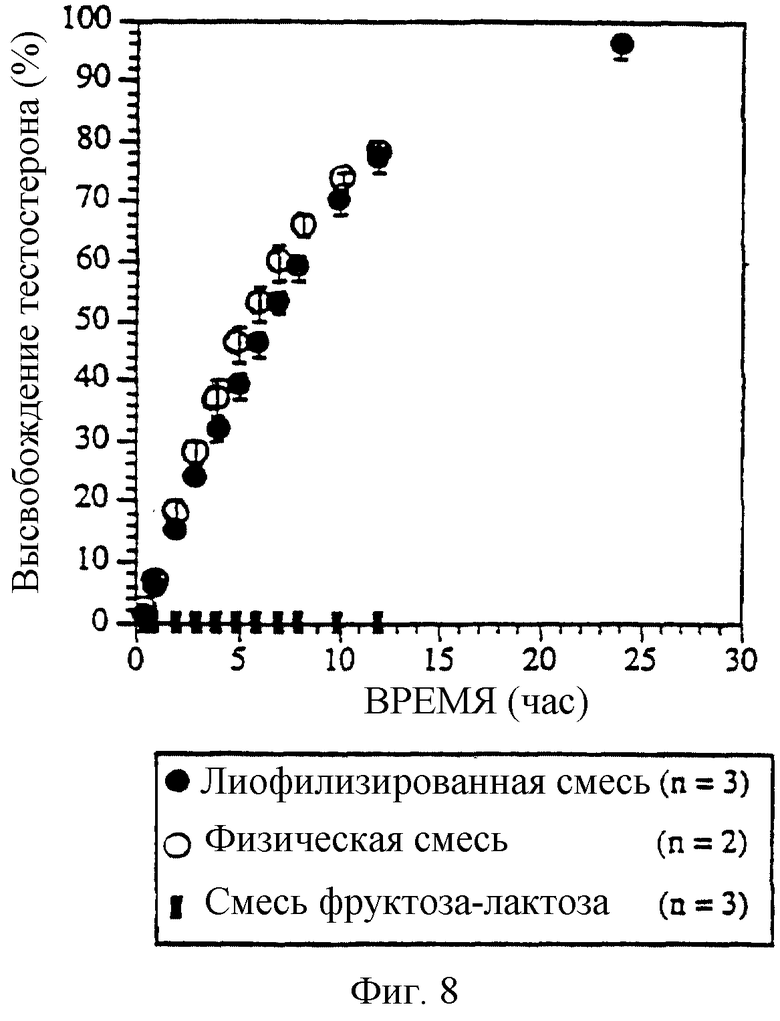
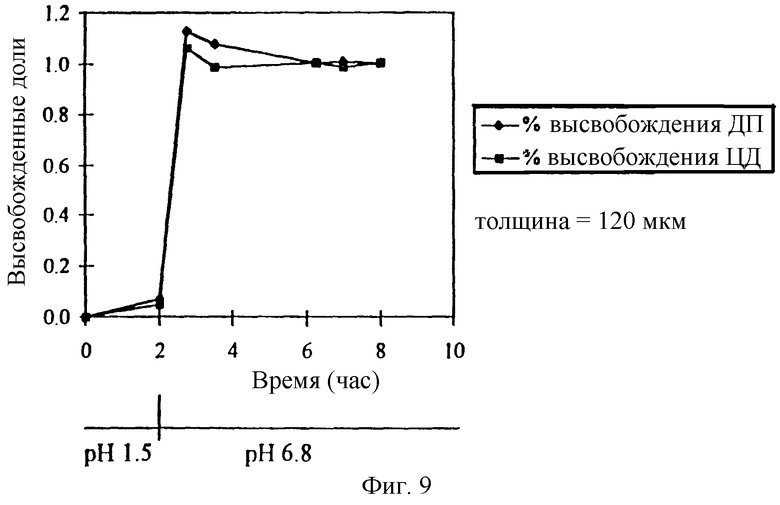

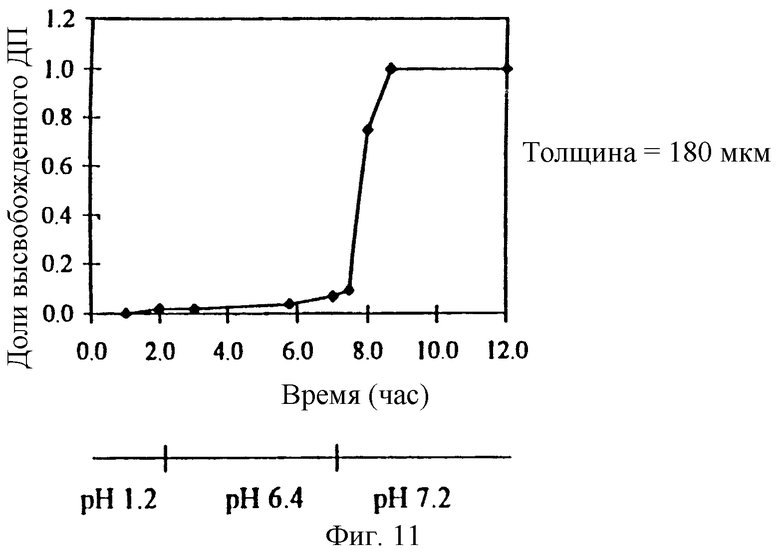
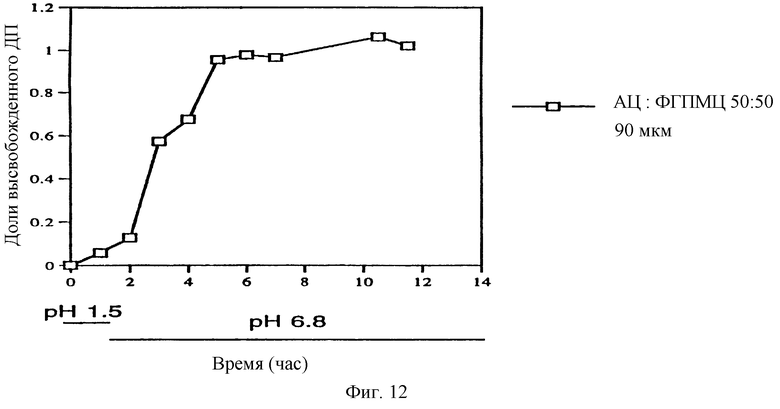
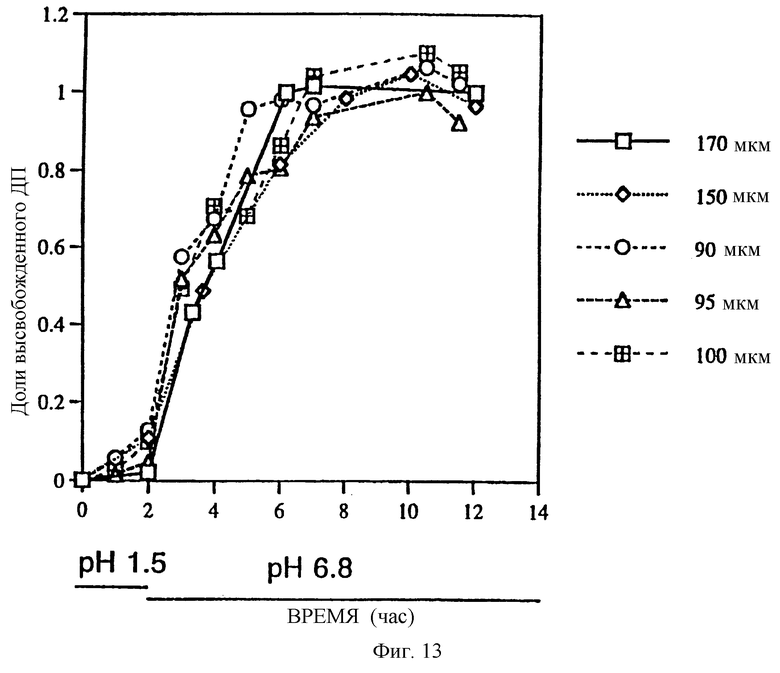
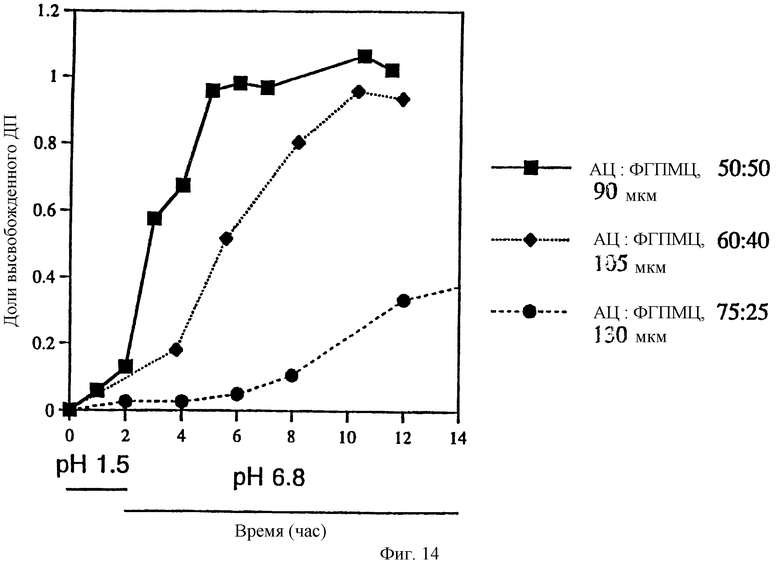
Изобретение может быть использовано в фармации и медицине. Заявлена фармацевтическая композиция, содержащая приемлемый носитель и физическую смесь производного простого сульфоалкилового эфира циклодекстрина и терапевтически эффективное количество терапевтического агента. По меньшей мере 50% терапевтического агента не образует комплекс с производным сульфоалкилового эфира циклодекстрина, который имеет формулу 1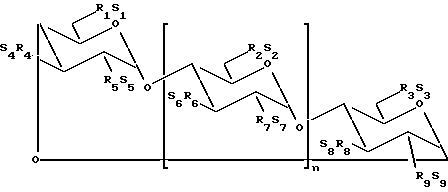
где n принимает значения 4, 5 или 6, каждый из R1, R2, R3, R4, R5, R6, R7, R8 и R9 независимо друг от друга представляет -О- или -O-(С2-С6-алкилен)-S03-, где, по меньшей мере, один из R1 и R2 независимо друг от друга представляет -O-(С2-С6-алкилен)-SO3-. Каждый из S1 - S9 независимо представляет фармацевтически приемлемый катион. Заявлен способ получения фармацевтической лекарственной формы, содержащей соединение I. Сначала получают твердое ядро, содержащее указанную выше физическую смесь, фармацевтический носитель, на ядро наносят пленочное покрытие, содержащее пленкообразующий агент и порообразующий агент. Заявлен твердый фармацетвический препарат, получаемый заявленным способом. Полученные препараты имеют повышенную растворимость и биодоступность активного агента. Способ их получения не требует предварительного приготовления комплексов активных веществ с производным циклодекстрина, что упрощает его технологию. 3 с. и 14 з.п. ф-лы, 14 ил., 17 табл.
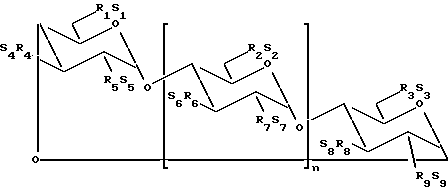
где n принимает значения 4, 5 или 6;
каждый из R1, R2, R3, R4 R5, R6, R7, R8 и R9 независимо друг от друга представляет -О- или -O-(С2-С6-алкилен)-SO3-, где, по меньшей мере, один из R1 и R2 независимо друг от друга представляет -O-(С2-С6-алкилен)-SO3-; и
каждый из заместителей S1, S2, S3, S4, S5, S6, S7, S8 и S9 независимо друг от друга представляет фармацевтически приемлемый катион.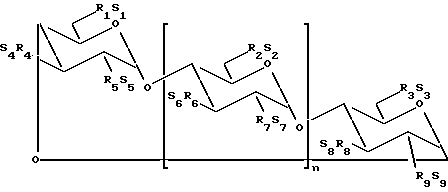
где n принимает значения 4, 5 или 6;
каждый из R1, R2, R3, R4, R5, R6 R7, R8 и R9 независимо друг от друга представляет -О- или -O-(С2-С6-алкилен)-SO3-, где, по меньшей мере, один из R1 и R2 независимо друг от друга представляет -O-(С2-С6-алкилен)-SO3-;
каждый из заместителей S1, S2, S3, S4, S5, S6, S7, S8 и S9 независимо друг от друга представляет фармацевтически приемлемый катион.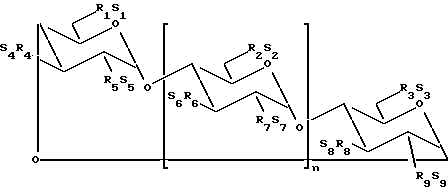
где n принимает значения 4, 5 или 6;
каждый из R1, R2, R3, R4, R5, R6, R7, R8 и R9 независимо друг от друга представляет -О- или -O-(С2-С6-алкилен)-SO3-, где, по меньшей мере, один из R1 и R2 независимо друг от друга представляет -O-(С2-С6-алкилен)-SO3-; и
каждый из заместителей S1, S2, S3, S4, S5, S6, S7, S8 и S9 независимо друг от друга представляет фармацевтически приемлемый катион.
| Огнетушитель | 0 |
|
SU91A1 |
| Экономайзер | 0 |
|
SU94A1 |
| RU 94017930 A1, 27.01.1996. | |||

