Изобретение относится, в основном, к области биологии опухоли. Более конкретно, изобретение относится к композициям и способам лечения сквамозной (чешуйчатой) клеточной карциномы. Обеспечивается также животная модель для изучения микроскопических остаточных опухолей и опухолевого обсеменения в полостях тела, а также способы их лечения.
Балансирование скоростями клеточной пролиферации и отмирания клеток является важным в поддержании гомеостаза нормальной ткани. Нарушение этого баланса может быть основным фактором в многоступенчатом процессе онкогенеза, а ингибирование апоптоза, или запрограммированного отмирания клеток, является одной из причин этого нарушения. Влияние таких дефектов является катастрофичным, вызывающим около полумиллиона смертей ежегодно в одних Соединенных Штатах.
Имеются существенные доказательства причастности мутаций p53 гена, опухолевого супрессора, в этиологии многих раковых заболеваний человека. Сообщения демонстрировали, что рост различных раковых клеточных линий человека, включающих представителей рака толстой кишки, глиобластому, рак груди, остеосаркому и рак легкого может функционально подавляться с помощью вирусно-опосредованного переноса p53 гена дикого типа. Было показано, что индуцирование экзогенной p53 экспрессии p53 дикого типа индуцирует апоптоз в раковых клеточных линиях толстой кишки и в легочных раковых сфероидах человека, наводя на мысль о роли p53 гена в запрограммированном отмирании клеток.
Пациенты со сквамозной клеточной карциномой головы и шеи (SCCHN) поражаются заболеванием, которое часто оказывает сильное влияние на речь, глотание и хирургическую операцию, затрагивающую внешность пациента. Кроме того, общая степень выживания среди этих пациентов, приблизительно 50%, остается неизменной в течение последних 30 лет, с тех пор как были основаны современная хирургия и радиационная терапия. Рецидивы являются преимущественно локальными и региональными в противоположность систематическим, указывая на то, что микроскопическая остаточная карцинома в первичном опухолевом участке является главной причиной смертности. Благодаря этим фактам, способность эффективно воздействовать на микроскопические остаточные заболевания в SCCHN является попыткой, которая может улучшить терапевтическую эффективность лечения рака.
Поэтому, задачей настоящего изобретения является создание улучшенных способов лечения сквамозной клеточной карциномы in vivo. Другой задачей настоящего изобретения является создание способа оценки развития и лечения микроскопической остаточной карциномы и микроскопического опухолевого обсеменения полостей тела.
Для решения этих задач предлагается способ лечения субъекта со сквамозной клеточной карциномой, включающий стадии (а) обеспечения экспрессионной конструкции, включающей промоторную функцию в эукариотических клетках и полинуклеотид, кодирующий p53, где полинуклеотид располагается в смысловой ориентации к промотору и находится под контролем промотора; (б) контактирования экспрессионной конструкции со сквамозной клеточной карциномой in vivo.
Сквамозная клеточная карцинома может быть карциномой головы и шеи. Эндогенный p53 сквамозной клеточной карциномы может быть, а может и не быть мутированным. Экспрессионная конструкция предпочтительно является вирусным вектором, таким как ретровирусный вектор, аденовирусный вектор и адено-ассоциированный вирусный вектор, с репликационно дефицитным аденовирусным вектором, который является наиболее предпочтительным. В конкретном варианте, p53 ген направляется таким образом, что может быть обнаружена экспрессия p53 из экспрессионного вектора. Предпочтительной мишенью является иммунологическая мишень, такая как секвенциальный эпитоп антитела.
Способ может далее включать хирургическую резекцию опухоли с дополнительным контактированием опухолевого ложа или "искусственной полости тела" с экспрессионной конструкцией после резекции. Объем, используемый для контакта с опухолевым ложем, составляет от около 3 мл до около 10 мл. Там, где применяется аденовирусный вектор, количество аденовируса, введенного в каждый процесс контактирования, составляет 107, 108, 109, 1010, 1011 или 1012 pfu.
Рассматривается также непрерывная перфузия экспрессионной конструкции. Количество конструкции, доставленное в процессе непрерывной перфузии, будет определяться исходя из количества, доставленного путем инъекций, так чтобы аппроксимировать ту же самую полную дозу в течение данного периода времени, хотя могут быть достигнуты несколько большие общие дозы с использованием непрерывной перфузии.
В другом варианте экспрессионная конструкция вводится в виде инъекции в естественную полость тела, такую как рот, глотка, трахея, плевральная полость, перитонеальная полость, или полости полых органов, включающие мочевой пузырь, толстую кишку или другие висцеральные органы.
Также заявляется способ определения эффективности терапии на микроскопический остаточный рак, включающий (а) обеспечение грызуна с рассечением в подкожной ткани; (б) обсеменение рассечения опухолевыми клетками; (с) лечение грызуна терапевтическим способом; (д) оценка воздействия режима на развитие опухоли. Рассечение может быть изолировано после стадии (б) и до стадии (с). Кроме того, режим может включать введение терапевтической композиции в рассечение, которое повторно открывается после изоляции и повторно изолируется после введения указанной терапевтической композиции.
Другие задачи, черты и преимущества настоящего изобретения будут очевидны из следующего детального описания. Должно быть, однако, понятно, что детальное описание и конкретные примеры, в то время как они указывают на предпочтительные варианты изобретения, даются исключительно с целью иллюстрации, так как различные изменения и модификации в пределах сути и объема изобретения станут очевидны специалисту в этой области из этого детального описания.
Следующие чертежи являются частью настоящего описания и включаются для дальнейшей демонстрации определенных аспектов настоящего изобретения. Изобретение может быть лучше понято со ссылкой на один или большее количество этих чертежей вместе с детальным описанием конкретных вариантов, представленных здесь.
Фиг. 1 - эффективность трансдукции SCCHN клеточных линий Tu-138 (сплошные треугольники) и Tu-177 (сплошные квадраты). Был использован рекомбинантный β-гал аденовирус для инфицирования клеток при различных MOI областях от 10 до 100. Процентное содержание β-гал-позитивных клеток было получено из подсчета 500 клеток, находящихся на каждой чашке для репликации.
Фиг. 2A и 2B - ингибирование роста SCCHN клеток in vitro. Фиг. 2A - кривая роста mock-зараженных Tu-138 клеток (сплошные кружки), d-1312-зараженных клеток (сплошные треугольники) и Ad5CMV-p53-зараженных клеток (сплошные квадраты). Фиг. 2B - кривая роста mock-зараженных Tu-177 клеток (открытые кружки), d1312-зараженных клеток (открытые треугольники), Ad5CMV-p53-зараженных клеток (открытые квадраты). В каждый указанный момент времени три чашки клеток были подвергнуты трипсинизации и подсчитаны. Значение ±SEM подсчитанных клеток на тройные лунки после заражения представляли в виде зависимости от времени (количества суток) с момента заражения.
Фиг. 3A, 3B, 3C и 3D - составная кривая роста четырех SCCHN клеточных линий. Фиг. 3A - Tu-138. Фиг. 3B - Tu-177. Фиг. 3C - MDA 686-LN. Фиг. 3D - MDA 886. Mock-зараженные клетки (сплошные кружки), d1312-зараженные клетки (сплошные треугольники) и Ad5CMV-p53 зараженные клетки (сплошные квадраты). Значение подсчитанных клеток на тройные лунки после заражения представляли в виде зависимости от количества суток с момента заражения; bars, SEM (сканирующая электронная микроскопия).
Фиг. 4 - кривая роста клеточной линии нормальных фибробластов. Mock-зараженные клетки (сплошные кружки), d1312-зараженные клетки (сплошные треугольники) и Ad5CMV-p53 зараженные клетки (сплошные квадраты).
Фиг. 5A и 5B - составная кривая роста SCCHN клеточных линий. Фиг. 5A - Tu-138. Фиг. 5B - MDA 686LN. Mock-зараженные клетки (открытые квадраты), d1312-зараженные клетки (открытые треугольники) и Ad5CMV-p53 зараженные клетки (открытые кружки). В каждый указанный момент времени три чашки клеток были подвергнуты трипсинизации и подсчитаны. Значение подсчитанных клеток на тройные лунки представляли в виде зависимости от количества часов после заражения; bars, SEM.
Фиг. 6A и 6B - мечение ДНК разрывов в апоптотических клетках биотинилированным dUTP с помощью TUNEL способа. После заражения, проточный цитометрический анализ для апоптоза проводили в процессе исследования. Фиг. 6A - Tu-138 клетки, которые заражали d1312, репликационно-дефицитным аденовирусом (панель 1 - панель 4), Tu-138 клетки, зараженные p53 аденовирусом дикого типа (панель 5 - панель 8). Фиг 6B - MDA 686LN клетки, которые заражали d1312, репликационно-дефицитным аденовирусом (A-D), MDA 686LN клетки, зараженные p53 аденовирусом дикого типа (E-H). Ap стенды (Ap stands) для апоптоза.
Фиг. 7A и 7B - составная кривая роста SCCHN клеточной линии. Фиг. 7A - Tu-138. Фиг. 7B - MDA 686LN. Mock-зараженные клетки (открытые кружки), d1312-зараженные клетки (закрытые треугольники) и Ad5CMV-p53 зараженные клетки (закрытые квадраты). В каждый указанный момент времени три чашки клеток были подвергнуты трипсинизации и подсчитаны. Значение подсчитанных клеток на тройные лунки представляли в виде зависимости от времени (количества часов) после заражения; bars, SEM.
Информация доступных данных предполагает, что одним из первичных недостатков лечения SCCHN является неспособность к полной ликвидации заболевания в первичном опухолевом участке, или в непосредственных локальных или региональных тканях, поэтому настоящее изобретение направлено на обеспечение методологий генной терапии, которые позволяют проводить более полное и эффективное лечение SCCHN, особенно за счет воздействия на микроскопическую остаточную карциному. Эта методология может быть использована сама по себе или вместе с более обычными методами лечениями, такими как химио- и радиотерапия или хирургическое вмешательство. Кроме того, использование животной модели конкретно направленной на лечение микроскопической остаточной карциномы, а также микроскопического опухолевого обсеменения полостей тела, настоящее изобретение демонстрирует эффективность этих способов. Детали изобретения описываются более полно ниже.
Обычно наблюдали, что p53 генная терапия рака может быть эффективной независимо от статуса p53 опухолевой клетки. Неожиданно терапевтические эффекты наблюдали, когда вирусный вектор, несущий p53 ген дикого типа, использовался для лечения опухолевых заболеваний, клеток, которые экспрессировали p53 молекулу. Этот результат не был предсказуемым на основании существующего понимания как функционируют опухолевые супрессоры. Неожиданным также является факт, что нормальные клетки, которые также экспрессируют функциональную p53 молекулу, явно не затрагиваются экспрессией высоких уровней p53 из вирусной конструкции. Это увеличивает возможность того, что p53 генная терапия может быть более широко применима для лечения раковых заболеваний, чем первоначально ожидали.
A. P53 белки и полинуклеотиды.
Во всем тексте этой заявки термин "p53" относится к служащим примером p53 молекулам, а также ко всем p53 гомологам из других видов. "Дикого типа" и "мутантный" p53 относится соответственно к p53 гену, экспрессирующему нормальную опухолевую супрессорную активность, и к p53 гену, испытывающему недостаток или имеющему пониженную супрессорную активность и/или обладающему трансформирующей активностью. Таким образом, "мутантные" p53's являются скорее просто вариантами последовательности, а не вариантами, показывающими измененные функциональные профили.
p53 в настоящее время признается как опухолевый супрессорный ген (Montenarh, 1992). Были найдены высокие уровни гена во многих клетках, трансформированных с помощью химического карциногенеза, ультрафиолетового облучения и некоторыми вирусами, включая SV40. p53 ген является частой мишенью мутационной инактивации в широком разнообразии опухолей человека и является уже документированным как являющийся наиболее часто-мутированным геном в обычных человеческих опухолевых раковых заболеваниях (Mercer, 1992). Ген мутируется в около 50% человеческих NSCLC (Hollestein et. al., 1991) и широком спектре других опухолей.
В то время как опухоли, содержащие мутированный p53 ген, являются предпочтительной мишенью согласно настоящему изобретению, пригодность заявленных p53 векторов экспрессии простирается на лечение опухолей, имеющих p53 дикого типа или функциональный p53. Хотя механизм является не полностью понятным, в настоящем изобретении установлено, что p53 экспрессия может ограничивать рост опухолей экспрессирующих функциональный p53 продукт и даже индуцировать апоптоз в таких клетках. Таким образом, p53 статус опухоли, хотя потенциально полезный в диагностических целях, не является необходимым для практики настоящего изобретения. Это явление не ограничивается SCCHN опухолями, но может быть применимо к широкому разнообразию злокачественного развития болезней, включающих глиомы, саркомы, карциномы, лейкемии, лимфомы и меланому, включая опухоли кожи, печенки, яичек, кости, мозга, поджелудочной железы, головы, шеи, желудка, печени, легкого, яичника, груди, слепой кишки, простаты и мочевого пузыря.
p53 Полипептиды.
p53 ген кодирует 375-аминокислотный фосфопротеин, который может образовывать комплексы с вирусными белками такими, как большой T антиген E1B. Белок находится в нормальных тканях и клетках, но при концентрациях, которые являются минимальными при сравнении с многими трансформированными клетками или опухолевой тканью. Интересно, что появляется p53 дикого типа, который является важным в регулировании клеточного роста и деления. В некоторых случаях была показана сверхэкспрессиия p53 дикого типа, которая является антипролиферативной в человеческих опухолевых линиях клеток. Таким образом, p53 может выступать в качестве отрицательного регулятора клеточного роста (Weinberg, 1991) и может непосредственно вызывать супрессию неконтролированного клеточного роста или косвенно активировать гены, которые подавляют этот рост. Таким образом, отсутствие или инактивация p53 дикого типа может способствовать трансформации. Однако некоторые исследования указывают на то, что присутствие мутантного p53 может быть необходимым для полной экспрессии, трансформирующей потенциал гена.
Хотя p53 дикого типа признается в качестве центрально важного регулятора роста во многих клеточных типах, его генетические и биохимические черты также, по-видимому, имеют значение. Мис-сенс мутации являются обычными для p53 гена и являются существенными для трансформирующей способности онкогена. Единичное генетическое изменение, вызванное за счет точечной мутации, может создать карциногенный p53. Известно, что в отличие от других онкогенов, однако, p53 точечные мутации происходят по крайней мере в 30 четких кодонах, часто создавая доминантные аллели, которые производят сдвиги в клеточном фенотипе без восстановления гомозиготности. Кроме того, многие из этих доминантных отрицательных аллелей оказываются толерантными в организме и передаются в зародышевую линию. Различные мутантные аллели проявляются в области от минимально дисфункциональных до сильно проникающих, доминантных отрицательных аллелей (Weinberg, 1991).
Casey с коллегами сообщали, что трансфекция ДНК, кодирующей p53 дикого типа, в двух раковых клеточных линиях человеческой груди восстанавливает контроль супрессии роста в таких клетках (Casey et al., 1991). Аналогичный эффект был также продемонстрирован на трансфекции дикого типа, но не мутантного p53, в раковых клеточных линиях человеческого легкого (Takahasi et al. , 1992). p53 дикого типа оказывается доминантным в пределах мутантного гена и будет селективным по отношению к пролиферации, когда трансфекцируется в клетки с мутантным геном. Экспрессия трансфекцированного p53 не влияет на рост нормальных клеток с эндогенным p53. Таким образом, такие конструкции могут поглощаться нормальными клетками без отрицательных эффектов.
Таким образом, становится возможным то, чтобы лечение p53-ассоциированных раковых заболеваний с помощью p53 дикого типа могло снижать количество злокачественных клеток. Однако исследования, такие как те, которые описаны выше, являются далеки от достижения такой цели по крайней мере, но не потому, что ДНК трансфекция не может быть применена для введения ДНК в раковые клетки внутри тела пациента.
р53-кодирующие полинуклеотиды.
Полинуклеотиды согласно настоящему изобретению могут кодировать полный p53 ген, функциональный p53 белковый домен или любой p53 полипептид. "Комплементарные" полинуклеотиды являются такими, которые являются способными к спариванию оснований согласно стандартным правилам комплементарности Watson-Crick. Таким образом, пара оснований с большим количеством пуринов будет парой оснований с меньшим количеством пиримидинов для образования комбинаций гуанина, спаренного с цитозином (G:C), и аденина, спаренного с простым эфиром тимина (A:T) в случае ДНК, или аденина, спаренного с урацилом (A: U) в случае РНК. Включение меньшего количества оснований таких, как инозин, 5-метилцитозин, 6-метиладенин, гипоксантин и других, в гибридизующие последовательности не является помехой спариванию.
Термин "комплементарные последовательности", как он использован здесь, обозначает полинуклеотидные последовательности, которые являются, по существу, комплементарными в пределах всей их длины и имеют очень небольшое количество ошибочных спариваний оснований. Например, последовательности пятнадцати оснований в длину могут быть названы комплементарными, когда они имеют комплементарный нуклеотид в тринадцати или четырнадцати положениях. Естественно, последовательности, которые являются "полностью комплементарными", будут являться последовательностями, которые являются полностью комплементарными по всей их полной длине и не содержат ошибочных спариваний оснований.
Рассматриваются также другие последовательности с более низкими степенями гомологии. Например, может быть сконструирована антисмысловая конструкция, которая содержит ограниченные области высокой гомологии, но также содержит негомологичную область (например, рибозим). Эти молекулы, хотя и имеющие менее 50% гомологии, будут связываться с последовательностями мишенями при соответствующих условиях.
Полинуклеотиды могут быть получены из геномной ДНК, т.е. клонированы непосредственно из генома или конкретного организма. В других вариантах, однако, полинуклеотиды могут быть комплементарными ДНК (кДНК). кДНК представляет ДНК, полученную с использованием информационной (матричной) РНК (мРНК) в качестве матрицы. Таким образом, кДНК не содержит каких либо нарушенных кодирующих последовательностей и содержит обычно почти исключительно кодирующую область(ти) для соответствующего белка. В других вариантах полинуклеотид может быть получен синтетически.
Может оказаться удобным комбинировать белки геномной ДНК с кДНК или синтетическими последовательностями для генерирования специфических конструкций. Например, там где желательным является интрон в последней конструкции, будет необходимо использование геномного клона. Интроны могут быть получены из других генов дополнительно к p53. кДНК или синтезированный полинуклеотид могут обеспечивать более убедительные сайты рестрикции для остающихся частей конструкции и поэтому могут быть использованы для остатка последовательности.
Человеческая или мышиная ДНК последовательность для p53 обеспечивается в SEQ ID NO:1 и SEQ ID NO:3 соответственно с соответствующими аминокислотами, которые обеспечиваются в SEQ ID NO:2 и SEQ ID NO:4 соответственно.
Рассматривается также, что существуют природные (дикие) варианты p53, которые содержат последовательности, отличающиеся от тех, которые раскрываются здесь. Таким образом, настоящее изобретение не ограничивается применением предусматриваемой полинуклеотидной последовательности p53, а скорее включает применение любых вариантов природного происхождения. Настоящее изобретение включает также химически полученные мутанты этих последовательностей.
Другой тип варианта последовательности возникает из кодоновой вариации. Так как существует несколько кодонов для большинства 20 нормальных аминокислот, многие различные ДНК могут кодировать p53. Табл. 1 позволяет идентифицировать такие варианты.
Учитывая дегенерацию генетического кода, последовательности, которые находятся между около 50% и около 75% или между около 76% и около 99% нуклеотидов, являющихся идентичными нуклеотидам, раскрытым здесь, будут предпочтительными. Последовательности, которые находятся внутри диапазона "p53 кодирующего полинуклеотида", являются такими, которые способны к спариванию оснований с сегментом полинуклеотида, представленным выше при внутриклеточных условиях.
Как установлено выше, хотя p53 кодирующие последовательности могут быть копиями полной длины или кДНК копиями, или большими фрагментами, настоящее изобретение может также применять более короткие олигонуклеотиды p53. Последовательности длиной в 17 оснований способны появляться лишь однажды в геноме человека, и поэтому являются достаточными для определения уникальной последовательности-мишени. Хотя более короткие олигомеры должны более легко получаться и повышать in vivo доступность, множество других факторов вовлекаются в определение специфичности спаривания оснований. И связывающее сродство и специфичность последовательности олигонуклеотида к его комплементарной мишени возрастают с увеличением длины. Предполагается, что олигонуклеотиды 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 пар оснований будут использоваться, например, в приготовлении p53 мутантов и в PCR реакциях.
Любая последовательность 17 оснований в длину должна иметь место только однажды в человеческом геноме и поэтому достаточна для специфичной уникальной последовательности-мишени. Хотя более короткие олигомеры легче получаются и повышают in vivo доступность, множество других факторов вовлекаются в определение специфичности гибридизации. Оба параметра, связывающее сродство и специфичность последовательности олигонуклеотида к его комплементарной мишени, возрастают с увеличением длины.
В определенных вариантах каждый может применять конструкции, которые включают другие элементы, например те, которые включают C-5 пропиновые пиримидины. Олигонуклеотиды, которые содержат C-5 пропиновые аналоги уридина и цитидина, связывают РНК с более высоким сродством (Wagner et al., 1993).
Специалисту в этой области также понятно, что неотъемлемой в определении биологически функционального эквивалентного белка или пептида является концепция, которая накладывает ограничение на количество изменений, которые могут быть проведены внутри определенной части молекулы и еще привести к молекуле с приемлемым уровнем эквивалентной биологической активности. Биологически функциональные эквивалентные пептиды определяются, таким образом, здесь как такие пептиды, в которых определенные, но наибольшая их часть или все аминокислоты могут быть замещенными. В частности, что касается N-терминуса p16 белка, рассматривается, что только около 16 или более, предпочтительно около 5 аминокислот могут быть изменены внутри данного пептида. Конечно, множество четких белков/пептидов с различными замещениями могут быть легко изготовлены и использованы в соответствии с изобретением.
Аминокислотные замещения обычно основываются на относительном подобии аминокислотных боковых заместителей, например, их гидрофобности, гидрофильности, заряде, размере и им подобных свойствах. Анализ размера, формы и типа аминокислотных боковых заместителей показывает, что аргинин, лизин и гистидин все являются положительно заряженными остатками; что аланин, глицин и серин все имеют аналогичный размер; и что фенилаланин, триптофан и тирозин все имеют обычно аналогичную форму. Поэтому на основании этих рассмотрений аргинин, лизин и гистидин; аланин, глицин и серин; фенилаланин, триптофан и тирозин; определяются здесь как биологически функциональные эквиваленты.
При проведении изменений, может быть рассмотрена степень гидропатичности (hydropathic) аминокислот. Каждой аминокислоте приписывается степень гидропатичности на основании ее гидрофобности и характеристик заряда, которые представляют: изолейцин (+4.5); валин (+4.2); лейцин (+3.8); фенилаланин (+2.8); цистеин/цистин (+2.5); метионин (+1.9); аланин (+1.8); глицин (-0.4); треонин (-0.7); серин (-0.8); триптофан (-0.9); тирозин (-1.3); пролин (-1.6); гистидин (-3.2); глутамат (-3.5); глутамин (-3.5); аспартат (-3.5); аспарагин (-3.5); лизин (-3.9); и аргинин (-4.5).
Роль степени гидропатичности аминокислоты в обсуждении биологической функции взаимодействия на белке обычно понятна специалисту (Kyte and Doolittle, 1982, которая вводится здесь ссылкой). Известно, что определенные аминокислоты могут быть замещены на другие аминокислоты, обладающие подобной степенью гидропатичности или меткой и еще сохраняют подобную биологическую активность. При проведении изменений на основании степени гидропатичности замещение аминокислот, чья гидропатичность находится в пределах +/-2, является предпочтительной, аминокислоты, которые имеют степень гидропатичности в пределах +/-1, являются особенно предпочтительными, а аминокислоты, которые имеют степень гидропатичности в пределах +/-0.5, являются даже более предпочтительными.
Понятно, что аминокислота может быть замещена на другую, обладающую подобной величиной гидрофильности, при этом еще получается биологически эквивалентный белок. Как детально описано в патенте США 4554101, следующие величины гидрофильности были приписаны аминокислотным остаткам: аргинин (+3.0); лизин (+3.0); аспартат (+3.0 +/-1); глутамат (+3.0 +/-1); серин (+0.3); аспарагин (+0.2); глутамин (+0.2); глицин (0); треонин (-0.4); пролин (-0.5 +/-1); аланин (-0.5); гистидин (-0.5); цистеин (-1.0); метионин (-1.3); валин (-1.5); лейцин (-1.8); изолейцин (-1.8); тирозин (-2.3); фенилаланин (-2.5); триптофан (-3.4).
При проведении изменений на основании подобия величин гидрофильности замещение аминокислот, чьи величины гидрофильности находятся в пределах +/-2, являются предпочтительными, и аминокислоты, которые имеют величины гидрофильности в пределах +/-1, являются особенно предпочтительными, а аминокислоты, которые имеют величины гидрофильности в пределах +/-0.5, являются даже более предпочтительными.
B. Экспрессионные векторы.
В пределах этого применения термин "экспрессионные конструкции" обозначает включение любого типа генетической конструкции, содержащей нуклеиновую кислоту, кодирующую генный продукт, в которой часть или вся кодирующая последовательность нуклеиновой кислоты является способной транскрибироваться. Транскрипт может быть транслирован в белок, но в этом нет нужды. Таким образом, в определенных вариантах, экспрессия включает и транскрипцию гена p53 и трансляцию p53 мРНК в p53 генный продукт. В других вариантах экспрессия включает только транскрипцию нуклеиновой кислоты, кодирующей p53 или его комплемент.
Для того чтобы конструкция оказывала влияние на экспрессию, по крайней мере p53 транскрипта, полинулкеотид, кодирующий p53 полинуклеотид, должен находиться под транскрипциональным контролем промотора. "Промотор" относится к последовательности ДНК, узнаваемой синтетическим механизмом клетки-хозяина, или введенным синтетическим механизмом, который требуется для инициации специфической транскрипции гена. Фраза "под транскрипциональным контролем" означает, что промотор находится в правильном положении по отношению к полинуклеотиду для контроля инициирования РНК полимеразы и экспрессии полинуклеотида.
Термин "промотор" может быть использован здесь для применения к группе модулей транскрипционального контроля, которые являются кластеризованными вокруг инициирующего сайта РНК полимеразы II. Большая часть размышлений о том, как промоторы организованы, исходит из анализов нескольких вирусных промоторов, включая HSV тримидин киназу (tk) и SV40 единицы ранней транскрипции. Эти исследования, наиболее дополненные недавней работой, показали, что промоторы составляются из дискретных функциональных модулей, каждый из которых состоит приблизительно из 7-20 bp ДНК, и содержат один или больше узнаваемых сайтов для транскрипционального активатора или репрессорных белков.
По крайней мере один модуль в каждом промоторе функционирует к позиции стартового сайта для синтеза РНК. Наиболее известным примером этого является TATA box, но в некоторых промоторах, в которых отсутствует TATA box, таких как промотор для терминального деоксинуклеотидильного трансферазного гена млекопитающего, и промотор SV40 поздних генов, дискретный элемент, покрывающий стартовый сайт, сам помогает фиксировать место инициации.
Дополнительные элементы промотора регулируют частоту транскрипциональной инициации. Обычно они располагаются в области 30-110 bp перед стартовым сайтом, хотя недавно было показано, что ряд промоторов содержит также функциональные элементы после стартового сайта. Пространство между промоторными элементами является часто гибким, так что функция промотора сохраняется, когда элементы инвертируются или передвигаются друг относительно друга. В tk промоторе пространство между промоторными элементами может увеличиваться до 50 bp независимо от того, что активность до этого начала убывать. В зависимости от промотора становится очевидным, что индивидуальные элементы могут функционировать или совместно, или независимо для активации транскрипции.
Конкретный промотор, который применяется для контролирования экспрессии полинуклеотида p53, не рассматривается как важный, поскольку он является способным экспрессировать полинуклеотид в нацеленной клетке (клетке-мишени) с достаточными уровнями. Таким образом, когда клетка человека является нацеленной, является предпочтительным располагать кодирующую область полинуклеотида рядом и под контролем промотора, который способен экспрессироваться в клетке человека. В общем, такой промотор может включать либо человеческий, либо вирусный промотор.
В различных вариантах промотор прямых ранних генов человеческого цитомегаловируса (CMV), ранний промотор SV40 и длинный терминальный повтор вируса саркомы Rous могут использоваться для получения высокоуровневой экспрессии p53 полинуклеотида. Рассматривается также применение других вирусных промоторов или клеточных промоторов млекопитающего или бактерильных фаговых промоторов, которые хорошо известны в данной области для достижения экспрессии полинуклеотидов, при условии, что уровни экспрессии являются достаточными для продуцирования ингибиторного эффекта роста.
Путем применения промотора с хорошо известными свойствами уровень и структура экспрессии p53 полинуклеотида может быть оптимизирована после трансфекции. Например, выбор промотора, который является активным в специфических клетках, таких как тирозиназа (меланома), альфа-фетопротеин и альбумин (опухоль печени), CC10 (опухоль легкого) и простато-специфический антиген (опухоль простаты), будет способствовать ткань-специфический
экспрессии p53 полинуклеотидов. Табл. 2 дает перечень нескольких элементов/промоторов, которые могут применяться в контексте настоящего изобретения, для регулирования экспрессии p53 конструкций. Этот перечень не претендует на то, чтобы быть исчерпывающим для всех возможных элементов, вовлеченных в ускорение p53 экспрессии, но лишь содержит в себе примеры.
Энхансеры были первоначально обнаружены в виде генетических элементов, которые повышали транскрипцию из промотора, расположенного на некотором расстоянии на той же молекуле ДНК. Эта способность действовать на большие расстояния имеет мало прецедентов в классических исследованиях прокариотических транскрипциональных регуляций. Последующие работы показали, что области ДНК с активностью энхансеров организованы в значительной степени подобно промоторам. То есть они состоят из многих индивидуальных элементов, каждый из которых связывается с одним или большим числом транскрипциональных белков.
Основное различие между энхансерами и промоторами заключается в способе действия. Область энхансера как целое может быть способна стимулировать транскрипцию на расстоянии; не обязательно, чтобы она точно соответствовала области промотора или его составляющим элементам. С другой стороны, промотор должен иметь один или большее количество элементов, которые непосредственно инициируют синтез РНК при конкретном сайте и в конкретной ориентации, тогда как энхансеры не имеют таких особенностей. Промоторы и энхансеры часто перекрываются и являются смежными, часто создавая видимость, что они имеют сходную модульную организацию.
Кроме того, любая комбинация промотор/энхансер (согласно банку данных эукариотического промотора EPDB) может также использоваться, чтобы привести в действие экспрессию p53 конструкции. Применение T3, T7 или SP6 цитоплазматической экспрессионной системы является другим возможным вариантом. Эукариотические клетки могут поддерживать цитоплазматическую транскрипцию от определенных бактериальных промоторов, если обеспечивается соответствующая бактериальная полимераза, либо в виде части освобождаемого комплекса, либо как дополнительный генетический экспрессионный вектор.
Кроме того, выбор промотора, который регулируется в ответ на специфические физиологические сигналы, может позволять индуцируемую экспрессию p53 конструкции. Например, с полинуклеотидом под контролем человеческого PAI-1 промотора: экспрессия индуцируется фактором опухолевого некроза. Табл. 3 иллюстрирует некоторые комбинации промотор/индуктор.
В определенных вариантах изобретения доставка экспрессионного вектора в клетку может быть идентифицирована in vitro или in vivo введением маркера в экспрессионный вектор. Маркер может приводить к идентифицируемому изменению в трансфекцированной клетке, позволяя легкую идентификацию экспрессии. Обычно введение селективного лекарственного маркера помогает в клонировании и в подборе трансформантов. Альтернативно могут использоваться ферменты, такие как (tk) тимидин киназа герпесного простого вируса (эукариотическая) или хлорамфеникольная ацетилтрансфераза (CAT) (прокариотическая). Могут использоваться также иммунологические маркеры. Применяемый селектируемый маркер, не рассматривается как имеющий важное значение, поскольку он является способным экспрессироваться вместе с полинуклеотид кодирующим p53. Другие примеры селективно способных маркеров хорошо известны специалистам в данной области.
Обычно пример будет включать сигнал полиаденилирования для осуществления соответствующего полиаденилирования транскрипта. Нет оснований верить, что природа сигнала полиаденилирования является решающей для успешного применения изобретения, и может быть использована любая такая последовательность. В изобретении был использован сигнал полиаденилирования SV40 в том смысле, что он был стандартным и известным для успешного функционирования в клетках-мишенях, которые использовались. Рассмотренным также в качестве элемента экспрессионной конструкции является терминатор. Эти элементы могут служить для повышения уровней послания и для минимизации считывания непосредственно из конструкции в других последовательностях.
В предпочтительных вариантах изобретения экспрессионная конструкция включает вирус или сконструированную конструкцию, полученную из вирусного генома. Способность определенных вирусов входить в клетки посредством рецептор-опосредованного эндоцитоза и, в некоторых случаях, интегрироваться в хромосомах клетки хозяина делает их привлекательными кандидатами для переноса генов в клетки млекопитающего. Однако из-за того, что было продемонстрировано, что прямое поглощение "оголенной" ДНК так же, как рецептор-опосредованное поглощение ДНК, осложняется (обсуждается ниже), экспрессионные векторы не должны быть вирусными, но вместо этого они могут быть любой плазмидой, космидой или фаговой конструкцией, которая является способной поддерживать экспресссию кодированных генов в клетках млекопитающего, таких как pUC или BluescriptT плазмидных серий.
Ретровирусы
Ретровирусы являются группой вирусов одноцепочечной РНК, характеризующихся способностью превращать свои РНК в двухцепочечные ДНК в инфицированных клетках способом обратной транскрипции (Coffin, 1990). Получающаяся в результате ДНК затем устойчиво интегрируется в клеточных хромосомах как провирус и направляет синтез вирусных белков. Интеграция приводит к удерживанию последовательностей вирусного гена в клетке реципиента и ее производных. Ретровирусный геном содержит три гена, gag, pol и env, которые кодируют капсидные белки, фермент полимеразы и элементы оболочки соответственно. Последовательность, которая обнаружена впереди gag гена, обозначенного Ψ, функционирует как сигнал для упаковки генома в вирионы. Две последовательности длинного терминального повтора (LTR) присутствуют на 5' и 3' концах вирусного генома. Они содержат сильные последовательности промотора и энхансера и также требуются для интеграции в геноме клетки хозяина (Coffin, 1990).
Для того чтобы конструировать ретровирусный вектор, нуклеиновая кислота кодирующая p53 инсертируется в вирусный геном в месте определенных вирусных последовательностей для продуцирования вируса, который является репликационно-дефицитным. Для того чтобы создать вирионы, конструируется упаковочная клеточная линия, содержащая гены gag, pol и env, но без LTR и Ψ компонентов (Mann et al. , 1983). Когда рекомбинантная плазмида, содержащая кДНК человека, вместе с ретровирусной LTR и Ψ последовательностями вводятся в эту клеточную линию (путем кальций фосфатного осаждения, например), Ψ последовательность позволяет РНК транскрипту рекомбинантной плазмиды быть упакованным в вирусные частицы, которые затем секретируются в культурную среду (Nicolas and Rubenstein, 1988; Temin, 1986; Mann et al., 1983). Среда, содержащая рекомбинантные ретровирусы, затем собирается, необязательно концентрируется и используется для переноса гена. Ретровирусные векторы являются способными к заражению широкого разнообразия типов клеток. Однако интеграция и устойчивая экспрессия требуют разделения клеток-хозяев (Paskind et al., 1975).
Новый подход, предназначенный способствовать специфическому нацеливанию ретровирусных векторов, был разработан недавно на основе химической модификации ретровирусов посредством химической добавки остатков ластозы к вирусной оболочке. Эта модификация может позволить проводить специфическое инфицирование гепатоцитов через сиалогликопротеиновые рецепторы.
Был разработан другой подход для нацеливания рекомбинантных ретровирусов, в которых были использованы биотинилированные антитела по отношению к ретровирусному белку оболочки и по отношению к специфическому клеточному рецептору. Антитела сочетали через биотиновые компоненты путем использования стрептавидина (Roux et al., 1989). Используя антитела по отношению к главным гистосовместимым комплексным антигенам класса I и класса II, они демонстрировали инфицирование различных клеток человека, которые порождали эти поверхностные антигены с экотропным вирусом in vitro (Roux et al., 1989).
Аденовирусы
Человеческие аденовирусы являются двутяжными ДНК опухолевыми вирусами с размерами генома приблизительно 36 пар оснований (Tooze, 1981). В качестве модельной системы для эукариотической генной экспрессии были широко изучены и хорошо охарактеризованы аденовирусы, которые делают их привлекательной системой для разработки аденовируса в качестве системы генного переноса. Эта группа вирусов является легкой для выращивания и манипулирования ими и они проявляют широкую область хозяина in vitro и in vivo. В литически зараженных клетках, аденовирусы являются способными закрывать белковый синтез хозяина, направляя клеточные механизмы на синтез больших количеств вирусных белков и продуцируя распределенные группами количества вирусов.
E1 область генома включает E1A и E1B, которые кодируют белки, ответственные за регуляцию транскрипции вирусного генома, а также небольшого числа клеточных генов. E2 экспрессия, включающая E2A и E2B области, способствует синтезу вирусных репликативных функций, например ДНК-связывающего белка, ДНК полимеразы и терминального белка, которые служат затравкой репликации. E3 генные продукты предотвращают цитолиз за счет цитотоксических T клеток и фактора опухолевого некроза и очевидно являются важными для вирусного роста. Функции, связанные с E4 белками, включают ДНК репликацию, экспрессию позднего гена и закрытие клетки хозяина. Продукты позднего гена включают наибольшую часть варион капсидных белков, и эти белки экспрессируются только после того, как происходит переработка большей части единичного первичного транскрипта из большей части позднего промотора. Большая часть позднего промотора (MLP) показывает высокую эффективность в течение поздней фазы инфекции (Stratford-Perricaudet and Perricaudet, 1991 a).
Как только малая часть вирусного генома, очевидно, требуется in cis (Tooze, 1981), векторы производные аденовируса представляют превосходный потенциал для замещения больших фрагментов ДНК, когда используются совместно с клеточными линиями, такими как 293 клетки. Клеточные линии Ad5 трансформированной человеческой эмбриональной почки (Graham, et al., 1977) были разработаны для обеспечения основных вирусных белков in trans. В изобретении, таким образом, приводятся доводы, что характеристики аденовирусов превращали их в хороших кандидатов для использования в нацеливании раковых клеток (раковых клеток в качестве клеток-мишеней) in vivo (Grunhaus and Horwitz, 1992).
Конкретные преимущества аденовирусной системы для доставки чужеродных белков в клетку включают (i) способность замещать относительно большие участки вирусной ДНК чужеродной ДНК; (ii) структурную стабильность рекомбинантных аденовирусов; (iii) безопасность аденовирусного введения людям и (iv) отсутствие любой известной ассоциации аденовирусной инфекции с раком или злокачественными образованиями; (v) способность получать высокие титры рекомбинантного вируса; и (vi) высокая инфективность аденовируса.
Дальнейшее преимущество аденовирусных векторов по сравнению с ретровирусами включает более высокие уровни генной экспрессии. Кроме того, аденовирусная репликация является независимой от генной репликации хозяина, в отличие от ретровирусных последовательностей. Из-за того, что аденовирусные трансформирующие гены в E1 области могут быть легко делетированы и еще обеспечивать эффективные экспрессионные векторы, предполагается, что онкогенный риск от аденовирусных векторов является незначительным (Grunhaus and Horwitz, 1992).
Вообще, системы аденовирусного генного переноса основываются на рекомбинанте сконструированного аденовируса, которая делается репликационно-некомпетентной за счет делеции части ее генома, такой как E1, и еще сохраняет ее компетентность для инфекции. Последовательности, кодирующие относительно большие чужеродные белки, могут быть экспрессированными, когда производятся дополнительные делеции в аденовирусном геноме. Например, аденовирусы, делетированные в E1 и в E2 областях, являются способными нести вплоть до 10Kb чужеродной ДНК и могут расти до высоких титров в 293 клетках ((Stratford-Perricaudet and Perricaudet, 1991 a). Неожиданно сообщалась также устойчивая экспрессия трансгенов после аденовирусной инфекции.
Генный перенос, опосредованный аденовирусом, недавно был исследован как средство опосредования генного переноса в эукариотические клетки и во всех животных. Например, при обработке мыши с редким рецессивным генетическим нарушением орнитин транскарбамилазной (OTC) недостаточности, было найдено, что аденовирусные конструкции могут быть применены для подачи нормального OTC фермента. К сожалению, экспрессия нормальных уровней OTC достигается только в 4 из 17 примеров (Stratford-Perricaudet et al., 1991b). Поэтому, дефект только частично корректировался у большинства мышей и не приводил к физиологическому или фенотипическому изменению. Поэтому эти результаты являются малопривлекательными для применения аденовирусных векторов в раковой терапии.
Попытки применения аденовируса в переносе гена для цитического фиброзного трансмембранного проводящего регулятора (CFTR) в легочный эпителий хлопкового хомяка также были частично успешными, хотя не было возможности оценить биологическую активность перенесенного гена в эпителии животных (Rosenfeld et al., 1992). Вновь эти исследования продемонстрировали, что генный перенос и экспрессия CFTR белка в клетки легочного пути не показывает физиологического эффекта. В статье 1991 Science, Rosenfeld et al. показали легочную экпрессию 1-антитрипсинового белка, но вновь не показали физиологического эффекта. Фактически, они установили, что уровни экспрессии, которые они наблюдали, составляли только около 2% уровней, требуемых для защиты легких у людей, т.е. значительно ниже того, который необходим для физиологического эффекта.
Ген для человеческого α1-антитрипсина был введен в легкие нормальных крыс за счет внутрипортальной инъекции, где он был экспрессирован и приводил к секреции введенного человеческого белка в плазму этих крыс (Jaffe et al., 1992). Однако, и неутешительно, уровни, которые были получены, были недостаточно высокими, чтобы представлять терапевтическое значение.
Эти результаты не продемонстрировали, что аденовирус является способным направлять экспрессию достаточного количества белка в рекомбинантные клетки для достижения физиологически соответствующего эффекта, и они поэтому не предполагают полезность аденовирусной системы для применения вместе с терапией рака. Кроме того, до настоящего изобретения считалось, что p53 не может входить в упаковывающуюся клетку, как те гены, использованные для приготовления аденовируса, так как он может быть токсичным. Так как E1B аденовируса связывается с p53, считается, что это является другой причиной, почему технология аденовируса и технология p53 не могут быть объединены.
Другие вирусные векторы в качестве экспрессионных конструкций
В качестве экспрессионных конструкций в настоящем изобретении могут использоваться другие вирусные векторы. Могут использоваться векторы, полученные из вирусов, таких как вирус коровьей оспы (Ridgeway, 1988; Baichwal and Sugden, 1986; Coupar et al., 1988), адено-ассоциированный вирус (AAV) (Ridgeway, 1988; Baichwal and Sugden, 1986; Hermonat and Muzycska, 1984) и вирусы герпеса. Они представляют несколько привлекательных качеств для различных клеток млекопитающих (Friedmann, 1986; Ridgeway, 1988; Baichwal and Sugden, 1986; Coupar et al., 1988; Horwich et al., 1990).
С недавним обнаружением дефектных вирусов гепатита B было совершено новое проникновение в структурно-функциональные отношения различных вирусных последовательностей. In vitro исследования показали, что вирус может сохранять способность для хелперзависимой упаковки и обратной транскрипции, несмотря на делецию вплоть до 80% его генома (Horwich et al., 1990). Это предполагает, что значительные части генома могут быть заменены чужеродным генетическим материалом. Гепатотропизм и персистенция (интеграция) были особенно привлекательными качествами для печеночно-направленного переноса гена. Chang et al. недавно ввели хлорамфеникол ацетилтрансферазный (CAT) ген в вирусный геном гепатита B утки вместо полимеразной, поверхностной и приповерхностной кодирующих последовательностей. Он был сотрансфекцирован с вирусом дикого типа в клеточную линию гепатомы птицы. Для инфицирования первичных гепатоцитов утенка использовалась культурная среда, содержащая высокие титры рекомбинантного вируса. Была обнаружена устойчивая экспрессия CAT гена по крайней мере в течение 24 дней после трансфекции (Chang et al. 1991).
Альтернативные способы генной доставки
Для того чтобы вызвать экспрессию p53 конструкций, экспрессионный вектор должен быть доставлен в клетку. Как описано выше, предпочтительный механизм для доставки осуществляется через вирусную инфекцию, где экспрессионный вектор инкапсулируется в инфекционную аденовирусную частицу.
Настоящим изобретением рассматриваются также некоторые невирусные способы переноса экспрессионных векторов в культивированные клетки млекопитающего. Они включают кальцийфосфатное осаждение (Graham and Van Der Eb, 1973; Chen and Okayama, 1987; Rippe et al. 1990), DEAE-декстрановый способ (Gopal, 1985), электропорацию (Tur-Kaspa et al., 1986; Potter et al., 1984), прямое микроинъецирование (Harland and Weintraub, 1985), способ ДНК-нагруженных липосом (Nicolau and Sene, 1982; Fraley et al., 1979) и способ липофектамин-ДНК комплексов, обработку клеток ультразвуком (Fechheimer et al., 1987), бомбардировку гена с использованием высокоскоростных микропроекций (Yang et al. , 1990), поликатионов (Boussif et. al., 1995) и рецептор-опосредованную трансфекцию (Wu and Wu, 1987; Wu and Wu, 1988). Некоторые из этих технических приемов могут быть успешно приспособлены для применения in vivo или ex vivo.
В одном варианте изобретения аденовирусный экспрессионный вектор может состоять просто из оголенного рекомбинантного вектора. Перенос конструкции может быть проведен с помощью любого из способов, упомянутых выше, который обеспечивает физически или химически проницаемость клеточной мембраны. Например, Dubensky et al. (1984), успешно провели инъекцию полиомавируса ДНК в форме осадков CaPO4 в печень и селезенку взрослых и новорожденных мышей, демонстрируя активную вирусную репликацию и острую инфекцию. Benvenisty and Neshif (1986) также продемонстрировали, что прямая внутрибрюшинная инъекция CaPO4 осажденных плазмид приводит к экспрессии трансфекцированных генов. Можно предвидеть, что ДНК кодирующая p53 конструкция может быть также перенесена аналогичным образом in vivo.
Другой вариант изобретения для переноса экспрессионного вектора оголенной ДНК в клетки может включать бомбардировку частицами. Этот способ зависит от способности ускорять микрочастицы, покрытые ДНК, до высокой скорости, позволяющей им пронизывать клеточные мембраны и входить в клетки, не убивая их (Klein et al., 1987). Было создано несколько устройств для ускорения маленьких частиц. Одно такое устройство основано на высоковольтном разряде для генерирования электрического тока, который, в свою очередь, обеспечивает движущую силу (Yang et al., 1990). Использованные микрочастицы, состояли из биологически инертных веществ, таких как вольфрамовый или золотой бисер.
Выделенные органы, включающие печень, кожу и мышечную ткань крыс и мышей, бомбардировались in vivo (Yang et al., 1990; Zelenin et al., 1991). Эти органы могут нуждаться в хирургическом вскрытии ткани или клеток для того, чтобы удалить любую мешающую ткань между пушкой и органом-мишенью. ДНК, кодирующая p53 конструкцию, может быть доставлена этим способом.
В дальнейшем варианте изобретения экспрессионный вектор может быть захвачен в липосому. Липосомы являются ячеистыми структурами, характеризующимися фосфолипидной двухслойной мембраной и внутренней водянистой средой. Многослойные липосомы имеют множество липидных слоев, разделенных водянистой средой. Они образуются спонтанно, когда фосфолипиды суспендируются в избытке водного раствора. Липидные компоненты претерпевают сомо-перегруппировку до образования закрытых структур и захвата воды и растворенных веществ между липидными бислоями (Ghosh and Bachhawat, 1991). Предполагается также рассмотреть липофектамин-ДНК комплексы.
Доставка липосомо-опосредованной нуклеиновой кислоты и экспрессия чужеродной ДНК in vitro оказались очень успешными. Wong et al., (1980) продемонстрировали осуществимость липосомо-опосредованной доставки и экспрессии чужеродной ДНК в культивированных клетках HeLa и гепатомах зародыша цыпленка. Nicolau et al., (1987) успешно выполнили липосомо-опосредованный перенос гена в крысах после внутривенной инъекции.
В некоторых вариантах изобретения липосома может быть комплексосвязанной с гемагглютинирующим вирусом (HVJ). Это, как показано, облегчает слияние с клеточной мембраной и ускоряет клеточное вхождение липосомо-инкапсулированной ДНК (Kaneda et al., 1989). В других вариантах липосома может комплексоваться или использоваться вместе с ядерными негистонными хромосомными белками (HGM-1) (Kato et al., 1991). Еще в других вариантах липосома может комплексоваться или применяться вместе с обоими HVJ и HMG-1. Тем, что такие экспрессионные векторы были успешно применены в переносе и экспрессии полинуклеотида in vitro и in vivo, они тем самым являются применимыми для настоящего изобретения. Там, где используется бактериофаговый промотор в конструкции ДНК, также будет желательным включить внутрь липосомы подходящую бактериофаговую полимеразу.
Другим механизмом для переноса экспрессионных векторов в клетки, является рецептор-опосредованная доставка. Этот подход обладает преимуществом селективного поглощения макромолеул за счет рецептор-опосредованного эндоцитоза почти во всех эукариотических клетках. Из-за клеточного тип-специфического распределения различных рецепторов доставка может быть высокоспецифичной (Wu and Wu, 1993).
Нацеливающие проводники рецептор-опосредованных генов обычно состоят из двух компонентов: клеточного рецептор-специфического лиганда и ДНК-связующего агента. Несколько лигандов было использовано для рецептор-опосредованного генного переноса. Наиболее широко охарактеризованными лигандами являются азиалооросомукоид (ASOR) (Wu and Wu, 1987) и трансферрин (Wagner et al., 1993). Недавно синтетический неогликопротеин, который узнает тот же рецептор, как ASOR, был использован в качестве проводника доставки гена (Ferkol et al. , 1993; Perales et al., 1994) и был также использован эпидермальный фактор роста (EGF) для доставки генов в клетки сквамозной карциномы (Myers, EPO 0273085).
В других вариантах проводник доставки может включать лиганд и липосому. Например, Nicolae et al. (1987) использовали лактозил-керамид (ceramid), галактоза-терминальный азиалганглиозид, введенный в липосомы, и наблюдали увеличение поглощения гена инсулина гепатоцитами. Таким образом, возможно, что аденовирусный экспрессионный вектор, также может специфично доставляться в тип клетки, такой как клетки легкого, эпителиальные или опухолевые клетки, посредством любого количества рецепторно-лигандных систем с липосомами или без липосом. Например, эпидермальный фактор роста (EGF) может использоваться в качестве рецептора для опосредованной доставки p53 конструкции во многие опухолевые клетки, которые показывают сверхрегуляцию EGF рецептора. Может быть использована манноза для нацеливания рецептора маннозы на клетки печени. Кроме того, антитела к CD5 (CLL), CD22 (лимфома), CD25 (T-клеточная лейкемия) и MAA (меланома) могут подобным же образом использоваться как нацеливающие половины.
В определенных вариантах перенос гена может более легко осуществляться при ex vivo условиях. Ex vivo генная терапия относится к выделению клеток из животного, доставки полинуклеотида в клетки in vitro и затем возвращению модифицированных клеток обратно в животное. Это может включать хирургическое удаление ткани/органов из животного или первичной культуры клеток и тканей. Anderson et al., патент США 5399346, раскрывают ex vivo терапевтические способы. Во время применения ex vivo культуры экспрессионный вектор может экспрессировать p53 конструкцию. Наконец, клетки могут быть повторно введены в первоначальное животное или введены в отдельное животное в фармацевтически приемлемой форме с помощью любого из приемов, описанных ниже.
D. Фармацевтические композиции и пути введения.
Когда рассматривается клиническое применение аденовирусного экспрессионного вектора согласно настоящему изобретению, будет необходимо приготовление комплекса в качестве фармацевтической композиции, соответствующей для предполагаемого применения. Обычно это будет приводить к приготовлению фармацевтической композиции, которая, по существу, является свободной от пирогенов, а также любых других примесей, которые могут быть вредными для людей или животных. К тому же обычно желательно применять соответствующие соли и буферные растворы для того, чтобы делать комплекс устойчивым, и для того, чтобы позволить комплексу поглощаться клетками-мишенями.
Водные композиции настоящего изобретения включают эффективное количество экспрессионного вектора, растворенного или диспергированного в фармацевтически приемлемом носителе или водной среде. Такие композиции могут быть также отнесены к так называемой инокуле. Фраза "фармацевтически или фармакологически приемлемый" относится к молекулярным соединениям или композициям, которые не продуцируют вредных, аллергических или других нежелательных реакций, когда вводятся животному или человеку. Термин "фармацевтический носитель", как он использован здесь, включает любые или все растворители, дисперсионные среды, покрытия, антибактериальные или противогрибковые агенты, изотонические агенты и замедляющие поглощение агенты и им подобные. Использование таких сред и агентов для фармацевтически активных веществ хорошо известно в этой области. Поскольку за исключением того, что любая обычная среда или агент является несовместимым с активным ингредиентом, рассматривается его использование в терапевтических композициях. Могут также вводиться дополнительные ингредиенты в композиции.
Растворы активных соединений в виде свободного основания или фармакологически приемлемых солей могут быть приготовлены в воде, соответственно смешанной с поверхностно-активным соединением, таким как гидроксипропилцеллюлоза. Могут быть также приготовлены дисперсии в глицерине, жидких полиэтиленгликолях и их смесях в маслах. При обычных условиях хранения и применения эти препараты содержат консерванты для предотвращения роста микроорганизмов.
Экспресионные векторы и проводники доставки настоящего изобретения могут включать классические фармацевтические препараты. Введение терапевтических композиций согласно настоящему изобретению может происходить любым обычным путем введения, пока ткань мишени является доступной этому пути введения. Этот путь включает оральное, назальное, введение в щеку, ректальное, вагинальное или точечное введение. Или же введение может быть осуществлено ортоточечной, внутрикожной, подкожной, внутримышечной, внутрибрюшинной или внутривенной инъекцией. Такие композиции будут обычно вводиться в виде фармацевтически приемлемых композиций, которые включают физиологически приемлемые носители, буферы или другие наполнители.
Терапевтические композиции настоящего изобретения преимущественно вводятся в форме композиций для инъекции либо в виде жидких растворов либо суспензий; могут быть приготовлены твердые формы, пригодные для растворения в жидкости или суспендирования в жидкости до инъекции. Эти препараты также могут быть эмульгированными. Типичная композиция для такой цели включает фармацевтически приемлемый носитель. Например, композиция может содержать 10 мг, 25 мг, 50 мг или вплоть до 100 мг человеческого сывороточного альбумина на миллилитр фосфатного забуференного солевого раствора. Другие фармацевтически приемлемые носители включают водные растворы, нетоксичные наполнители, включающие соли, консерванты, буферы и им подобные. Примерами неводных растворителей являются пропиленгликоль, полиэтиленгликоль, растительное масло и органические эфиры для инъекций, такие как этилолеат. Водные носители включают воду, водно-спиртовые растворы, солевые растворы, парентеральные наполнители, такие как хлористый натрий, декстроза Рингера и т.д. Внутривенные наполнители включают жидкие и питательные специальные наполнители. Консерванты включают антимикробные агенты, антиоксиданты, хелатирующие агенты и инертные газы. pH и точная концентрация различных компонентов фармацевтической композиции доводится согласно хорошо известным параметрам.
Дополнительные составы являются пригодными для орального введения. Оральные композиции включают такие типичные наполнители, как например маннитол, лактозу, крахмал, стеарат магния, натрий сахарин, целлюлозу, карбонат магния и им подобные реагенты фармацевтического качества. Композиции выбирают в форме растворов, суспензий, таблеток, пилюль, капсул, композиций с контролируемым высвобождением или порошков. Если путь введения является точечным, форма может быть кремом, мазью жидкостью или спрэем.
Эффективное количество терапевтического агента определяют на основании предполагаемой цели, например, (1) ингибирования пролиферации опухолевой клетки или (11) удаления опухолевых клеток. Термин "стандартная доза" относится к физически дискретным единицам, пригодным для использования субъекту, такой единице, которая содержит предварительно определенное количество терапевтической композиции, рассчитанной для получения желаемых ответов, обсуждавшихся выше, вместе с ее введением, т.е. соответствующим путем и режимом обработки. Количество, которое вводится в соответствии с количеством обработок и стандартной дозой, зависит от субъекта, который подвергается лечению, состояния субъекта и желаемой защиты. Точные количества терапевтической композиции также зависят от решения лечащего врача и являются конкретными для каждого индивидуума.
В определенных вариантах может быть желательным обеспечение непрерывной подачи терапевтических композиций пациенту. При внутривенных или внутриартериальных путях введения эта подача может сопровождаться капельными системами. Для локальных применений следует использовать повторные применения. Замедленное высвобождение композиций может быть
использовано для различных подходов, что обеспечивает ограниченные, но непрерывные количества терапевтического агента в течение определенного и продолжительного периода времени. Для внутреннего применения может быть предпочтительной непрерывная перфузия интересуемой области. Это применение может сопровождаться введением катетера, в некоторых случаях послеоперативно, с последующим непрерывным введением терапевтического агента. Продолжительность перфузии будет выбираться лечащим врачом для конкретного пациента и ситуации, но время может быть в области от около 1-2 часов до 2-6 часов, до около 6-10 часов, до около 10-24 часов, до около 1-2 недель или больше. Обычно доза терапевтической композиции при непрерывной перфузии будет эквивалентна дозе, которая дается с помощью одной или множества инъекций, отрегулированной для периода времени, в течение которого вводятся инъекции. Предполагается, однако, что могут быть достигнуты более высокие дозы при перфузии.
Клинический протокол для SCCHN
Был разработан клинический протокол для облегчения лечения SCCHN заболевания с использованием аденовирусных конструкций, обсужденных ниже в примерах. В соответствии с этим протоколом, будут выбираться пациенты у которых имеется гистологическое доказательство сквамозной клеточной карциномы головы и шеи. Пациенты могли, но не обязательно получали предварительные химио-, радио- или генные терапии. Оптимально, пациенты должны иметь адекватную функцию костного мозга (определенную в виде количества периферических абсолютных гранулоцитов >2000/мм3 и количества тромбоцитов 100000/мм3), адекватную функцию печени (билирубин ≤1.5 мг/дл) и адекватную почечную функцию (креатинин <1.5 мг/дл).
Протокол требует однократной дозы введения при интратуморальной (внутриопухолевой) инъекции фармацевтической композиции, содержащей между 106 и 109 зараженных частиц p53 аденовирусной экспрессионной конструкции. Для опухолей ≥4 см вводимый объем будет составлять 4-10 мл (предпочтительно 10 мл), в то время как для опухолей < 4 см будет использоваться объем 1-3 мл (предпочтительно 3 мл). Множество инъекций будет доставляться для однократной дозы, в 0.1-0.5 мл объемах с расстоянием приблизительно 1 см или больше).
Курс лечения состоит из шести доз, введенных в течение двух недель. При выборе с помощью клинических испытаний режим может быть продолжен, шесть доз каждые две недели или с меньшей частотой (ежемесячно, в течение двух месяцев, в течение четырех месяцев и т.д.).
Когда пациенты являются подходящими для хирургической резекции, опухоль будет лечиться как описано выше в течение по крайней мере двух последовательных двухнедельных курсов лечения. В пределах одной недели окончания второго курса (или более, например, третьего, четвертого, пятого, шестого, седьмого, восьмого и т.д.) пациенту будет проводиться хирургическая резекция. До закрытия иссечения 10 мл фармацевтической композиции, содержащей p53 аденовирсную экспрессионную конструкцию (106-109 зараженных частиц), будет доставляться в хирургический участок (оперативное ложе) и оставляться в контакте в течение по крайней мере 60 минут. Рана закрывается и в нее помещается дренаж или катетер. На третий послеоперационный день вводится дополнительно 10 мл фармацевтической композиции через дренаж и оставляется в контакте с оперативным ложем в течение по крайней мере двух часов. Затем проводится удаление композиции отсасыванием и удаление дренажа в клинически соответствующее время.
Обработка искусственных и естественных полостей тела
Одним из первичных источников возвратного SCCHN является остаточное микроскопическое заболевание, которое остается в первичном опухолевом участке, а также локально или регионально после опухолевого иссечения. Кроме того, существуют аналогичные ситуации когда естественные полости тела обсеменяются с помощью микроскопических опухолевых клеток. Эффективное лечение таких микроскопических заболеваний будет создавать значительное преимущество в терапевтических режимах.
Таким образом, в определенных вариантах рак может быть удален с помощью хирургического иссечения, создающего "полость". Во время операции и после нее (периодически или непрерывно) в полость тела вводится терапевтическая композиция настоящего изобретения. Это является, в сущности, "локальной" обработкой поверхности полости. Объем композиции должен быть достаточным для того, чтобы гарантировать, что вся поверхность полости подвергалась контактированию за счет экспрессионной конструкции.
В одном варианте введение просто будет вести за собой инъекцию терапевтической композиции в полость образованную иссечением опухоли. В другом варианте может быть желательным механическое применение через сифон, тампон или другое устройство. Может быть использован любой из этих подходов после удаления опухоли, а также в течение начальной операции. Еще в другом варианте катетер вводится в полость до закрытия описанного операционного участка. Полость может быть затем подвергнута непрерывной перфузии в течение желаемого промежутка времени.
В другой форме этой обработки "локальное" применение терапевтической композиции нацеливается в естественную полость, такую как рот, глотка, пищевод, гортань, трахея, плевральная полость, брюшная полость или полости полых органов, включающие мочевой пузырь, ободочную кишку или другие висцеральные органы. В этой ситуации может быть, а может и не быть значительной первичной опухоли в полости. Обработка нацеливается на микроскопические заболевания в полости, но случайно может затрагивать также первичную опухолевую массу, если она не была предварительно удалена, или предварительное неопластическое повреждение, которое может присутствовать внутри этой полости. Вновь могут быть применены различные способы для того, чтобы влиять на "локальное" применение в этих висцеральных органах или поверхностях полости. Например, на оральную полость в глотке можно оказать воздействие простым оральным рассечением и полосканием растворами. Однако локальная обработка внутри глотки и трахеи может требовать эндоскопической визуализации и локальной доставки терапевтической композиции. Висцеральные органы, такие как мочевой пузырь или слизистая оболочка ободочной кишки, могут требовать постоянных катетеров с инфузией или вновь прямой визуализации цитоскопом или другим эндоскопическим инструментом. Полости, такие как плевральная и брюшная полости, могут быть доступными с помощью постоянных катетеров или хирургических подходов, которые обеспечивают доступ к этим зонам.
Наблюдение p53 экспрессии после введения
Другой аспект настоящего изобретения включает наблюдение p53 экспрессии после введения терапевтической композиции. Из-за разрушения микроскопических опухолевых клеток они не могут наблюдаться, поэтому важно определить эффективно ли контактирует участок-мишень с экспрессионной конструкцией. Это явление может сопровождаться идентификацией клеток, в которых экспрессионная конструкция активно продуцирует p53 продукт. Важно, однако, быть способными провести различие между экзогенным p53 и тем, который присутствует в опухоли и здоровых клетках в обрабатываемой области. Мечение экзогенного p53 индикаторным элементом будет обеспечивать определенное доказательство экспрессии такой молекулы, а не ее эндогенной версии.
Один такой индикаторный элемент обеспечивается FLAC биосистемой (Hopp et al., 1988). FLAC полипептид представляет октапептид (AspTyrLysAspAspAspAspLys) и его небольшой размер не разрушает экспрессию доставленного генного терапевтического белка. Соэкспрессия FLAC и белка, представляющего интерес, метится за счет использования антител, возникающих против FLAC белка.
Могут быть также использованы другие иммунологические системы-маркеры, такая как 6XHis система (Qiagen). Для этой цели может быть использован любой линейный эпитоп для генерирования составного белка с p53, поскольку (i) иммунологическая целостность эпитопа не компрометируется составным белком и (ii) функциональная целостность p53 не компрометируется составным белком.
E. Объединенные терапевтические протоколы.
Сопротивление опухолевой клетки агентам повреждающим ДНК представляет большую проблему в клинической онкологии. Одной из целей текущих исследований рака является нахождение путей улучшения эффективности химио- и радиотерапии путем комбинирования ее с генной терапией. Например, симплекс-тримидин киназный (HS-tK) ген герпеса, когда он доставляется в опухоли мозга за счет системы ретровирусного вектора, успешно индуцирует чувствительность к ганцикловиру антивирусного агента (Culver, et al., 1992). В контексте настоящего изобретения предполагается, что терапия p53 может применяться вместе с химио- или радиотерапевтическим вмешательством.
Для того чтобы убить клетки, такие как злокачественные или метастатические клетки, используя способы и композиции настоящего изобретения, обычно проводится контакт клетки "мишени" с экспрессионным вектором и по крайней мере с одним ДНК повреждающим агентом. Эти композиции могут обеспечиваться в объединенном количестве, эффективном, чтобы убить или ингибировать пролиферацию клетки. Этот процесс может включать контактирование клеток с экспрессионным вектором и с ДНК повреждающим агентом(ами) или фактором(ами) в то же самое время. Этого можно достигнуть контактированием клетки с единичной композицией или фармакологическим составом, который включает оба агента, или контактированием клетки с двумя отдельными композициями или составами, в то же самое время, где одна композиция включает p53 экспрессионный вектор, а другая включает ДНК повреждающий агент.
Альтернативно, p53 обработка может предварять или следовать за обработкой ДНК повреждающим агентом с интервалами, изменяющимися от минут до недель. В вариантах, где ДНК повреждающий фактор и p53 экспрессионный вектор применяются раздельно к клетке, обычно заботятся о том, чтобы не проходил значительный период времени между временем каждой доставки, так чтобы ДНК повреждающий агент и экспрессионный вектор были бы еще способны оказывать преимущественно объединенное воздействие на клетку. В таких примерах предполагается, что будет иметь место контакт клетки с обоими агентами в пределах от 6 часов до одной недели друг с другом и более предпочтительно в пределах 24-72 часов друг с другом с задержкой времени всего лишь около 48 часов, которая является наиболее предпочтительной. Однако в некоторых ситуациях может быть желательным существенно растянуть временной период обработки, при котором между соответствующими применениями проходит от нескольких дней (2, 3, 4, 5, 6 или 7) до нескольких недель (1, 2, 3, 4, 6, 7 или 8).
Предполагается также, что более чем одно введение либо p53 конструкции, либо ДНК повреждающего агента будет желательным. Могут быть использованы различные комбинации, где p53 является "A", а ДНК повреждающий агент является "B":
A/B/A B/A/B B/B/A A/A/B B/A/A A/B/ A/B/B/B
B/B/B/A B/B/A/B A/A/B/B A/B/A/B A/B/B/A B/B/A/A B/A/B/B
B/A/B/A B/A/A/B A/A/A/B B/A/A/A A/B/A/A A/A/B/A B/A/A/B
Для достижения убийства клетки оба агента доставляются в клетку в объединенном количестве, эффективном для убийства клетки.
ДНК повреждающие агенты или факторы определяются здесь как любое химическое соединение или способ обработки, который индуцирует повреждение ДНК, когда доставляется в клетку. Такие агенты и факторы включают излучение и волны, которые индуцируют повреждение ДНК так же, как γ-облучение, рентгеновское облучение, УФ-облучение, облучение микроволнами, электронное излучение и им подобные. Разнообразие химических соединений, также описанных как "химиотерапевтические агенты", выполняющих функцию индуцирования повреждения ДНК, все из которых предназначены для применения в способах комбинированной обработки, раскрываются здесь. Химиотерапевтические агенты, которые предполагаются для применения, включают, например, адриамицин, 5-фторурасил (5FU), этопосид (VP-16), камптотецин, актиномицин-D, митомицин C, цисплатин (CDDP) и даже перекись водорода. Изобретение также включает в себя использование комбинации одного или большего числа ДНК повреждающих агентов, или основанных на излучении или реальных соединений, таких как применение X-лучей с цисплатином или применение цисплатина с этопозидом (etoposide). В конкретных вариантах применение цисплатина в комбинации с p53 экспрессионным вектором является особенно предпочтительным.
При лечении рака в соответствии с изобретением будет осуществляться контакт опухолевых клеток с ДНК повреждающим агентом в дополнение к экспрессионному вектору. Это может быть достигнуто облучением локализованного опухолевого участка ДНК повреждающим облучением, таким как X-лучи, УФ-свет, γ-лучи или даже микроволны. Альтернативно, опухолевые клетки могут подвергаться контакту с ДНК повреждающим агентом путем введения субъекту терапевтически эффективного количества фармацевтической композиции, содержащей ДНК повреждающее соединение, такое как адриамицин, 5-фторурацил, этопозид, камптотецин, актиномицин-D, митомицин C или более предпочтительно цисплатин. ДНК повреждающий агент можно приготовить и использовать как комбинированную терапевтическую композицию, или набор, путем объединения его с p53 экспрессионным вектором, как описано выше.
Агенты, которые непосредственно сшивают полинуклеотиды, конкретно ДНК, рассматриваются и показываются здесь для того, чтобы вызвать ДНК повреждение, ведущее к синергичной антинеопластичной комбинации. Могут применяться агенты, такие как цисплатин, и другие ДНК алкилирующие агенты. Цисплатин широко применяется для лечения рака, с эффективными дозами используемыми в клинических применениях 20 мг/м2 в течение 5 дней каждые три недели для общих трех курсов. Цисплатин не абсорбируется при оральном применении и должен поэтому доставляться путем инъекции внутривенно, подкожно, внутриопухолевой инъекции или внутрибрюшинно.
Агенты, которые повреждают ДНК, также включают соединения, которые являются помехой ДНК репликации, митозу и хромосомной сегрегации. Такие химиотерапевтические соединения включают адриамицин, также известный как доксорубицин, этопозид, верапамил, подофиллотоксин и им подобные. Широко используемые в клинической постановке для лечения неоплазм, эти соединения вводятся путем болюсных инъекций внутривенно при дозах, изменяющихся в пределах от 25-75 мг/м2 при 21-дневных перерывах для адриамицина до 35-50 мг/м2 для этопозида внутривенно или орально двойной внутривенной дозой.
Агенты, которые разрушают синтез и привязанность полинуклеотидных предшественников и субъединиц, также ведут к повреждению ДНК. По существу, был разработан ряд полинуклеотидных предшественников. Особенно полезными являются агенты, которые подвергались широким испытаниям и является легкодоступными. По существу, агенты, такие как 5-фторурацил (5-FU), предпочтительно используются неопластической тканью, делая этот агент особенно полезным для нацеливания на неопластические клетки. Хотя умеренно токсичный 5-FU является применимым в широком ряду носителей, включая локальные, однако обычно применяются дозы внутривенного введения, изменяющиеся в пределах от 3 до 15 мг/кг/день.
Другие факторы, которые вызывают повреждение ДНК и были интенсивно использованы, включают такие, которые общеизвестны как γ-лучи, X-лучи, и/или направленная доставка радиоизотопов в опухолевые клетки. Рассматриваются также другие формы ДНК повреждающих факторов, такие как микроволны и УФ-излучение. По всей вероятности, все эти факторы создают широкий диапазон повреждения ДНК, или предшественников ДНК, репликации и восстановления ДНК, и сборки и поддержания хромосом. Интервалы дозирования для X-лучей изменяются от дневных доз 50-200 рентген в течение длительных периодов времени (3-4 недели), до однократных доз от 2000 до 6000 рентген. Интервалы дозирования для радиоизотопов широко меняются, и зависят от периода полураспада изотопа, силы и типа радиоизлучения, и поглощения неопластическими клетками.
Специалиста в этой области отсылают к "Remington's Pharmaceutical Sciences" 15-е издание, глава 33, в частности страницы 624-652. Некоторые изменения в дозах неизбежно возникнут в зависимости от состояния субъекта, который подвергается лечению. Специалист, ответственный за введение препарата, будет, в любом случае, определять соответствующую дозу для индивидуального субъекта. Кроме того, для введения человеку препараты должны отвечать стерильности, пирогенности, общей безопасности и чистоте стандартов, как требуется FDA Office of Biologics standarts.
Авторы изобретения полагают, что региональная доставка p53 экспрессионных векторов пациентам, страдающим раковыми заболеваниями, связанными с p53, будет очень эффективным способом доставки терапевтически эффективного гена для противодействия клиническому заболеванию. Аналогично, химио- и радиотерапия могут быть направленными в конкретную, пораженную область тела субъекта. Альтернативно, системная доставка экспрессионного вектора или ДНК повреждающего агента может быть соответствующей в определенных обстоятельствах, например, где возникли обширные метастазы.
Было доказано также, что цитокиновая терапия является эффективным партнером для объединенных терапевтических режимов. В таких комбинированных подходах могут применяться различные цитокины. Примеры цитокинов включают IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-11, IL-12, IL-13, TGF-β, GM-CSF, M-CSF, G-CSF, TNF-α, TNF-β, LAF, TCGF, BCGF, TRF, BAF, BDG, MP, LIF, OSM, TMF, PDGF, IFN-α, IFN-β, IFN-γ. Цитокины вводятся согласно стандартным режимам, как описано ниже, согласуясь с клиническими указаниями, такими как состояние пациента и относительная токсичность цитокина.
В дополнение к комбинированию p53-нацеленных терапий с химио- и радио- и цитокиновыми терапиями предполагается также, что комбинация с другими генными терапиями будет преимущественной. Например, нацеливание K-ras и p53 мутаций в то же самое время может продуцировать улучшенное антираковое лечение. Любой другой связанный с опухолью ген возможно может быть нацелен этим способом, например p21, p16, p27, E2F, Dp семейство генов, Rb, APC, DCC, NF-1, NF-2, WT-1, MEN-1, MEN-II, BRCA1, VHL, FCC, MCC, другие ras молекулы, myc, neu, raf, erb, src, fms, jun, trk, ret, gsp, hst, bcl и abl. Может быть также желательным комбинировать p53 терапию с генной терапевтической обработкой, основанной на антителе, включающей использование конструкции одноцепочечного антитела, в которой антитело связывается с любыми вышеуказанными молекулами, или в комбинации с генами, кодирующими один или большее количество цитокинов, перечисленных выше. Также может быть предпочтительным комбинировать p53 с другими генами, которые были вовлечены в процессы апоптоза, например аденовирусными E1A, Bax, BclXs и т.д.
Наборы.
Все необходимые материалы и реагенты, требуемые для ингибирования пролиферации опухолевой клетки, могут быть соединены вместе в набор. Этот набор обычно может включать выбранные аденовирусные экспрессионные векторы. Кроме того, включенными могут быть различные среды для репликации экспрессионных векторов и клеток хозяев для такой репликации. Такие наборы могут включать отдельные контейнеры для каждого индивидуального реагента.
Если компоненты набора обеспечиваются в одном или большем количестве жидких растворов, жидкий раствор предпочтительно является водным раствором, со стерильным водным раствором, являющимся особенно предпочтительным. Для использования in vivo экспрессионный вектор может быть составлен в фармацевтически приемлемую композицию, способную вводиться с помощью шприца. В этом случае контейнерное средство может быть само по себе контейнером для вдыхания, шприцем, пипеткой, глазной капельницей или любым другим подобным средством, из которого композиция может быть применена к зараженному участку тела, такому как легкие, может быть введена животному в виде инъекции, или даже может быть добавлена и смешана с другими компонентами набора.
Компоненты набора могут также обеспечиваться в сухих или лиофилизованных формах. Если реагенты или компоненты обеспечиваются в виде сухой формы, их повторное приготовление осуществляется обычно путем добавления соответствующего растворителя. Совершенно очевидно, что растворитель также может быть обеспечен в другом контейнерном средстве.
Наборы настоящего изобретения также могут обычно включать средства для хранения пробирок в закрытом виде для коммерческой продажи, такие как, например, контейнеры для инъекций или формованные пластиковые контейнеры, в которых удерживаются желаемые пробирки.
Независимо от количества и типа контейнеров наборы изобретения могут также включать или упаковываться вместе с инструментом для облегчения инъекции/введения или помещения однородной комплексной композиции внутрь тела животного. Такой инструмент может быть ингалятором, шприцем, пипеткой, форсептами (forcepts), мерной ложкой, глазной капельницей или любым таким медицински принятым высвобождающим проводником.
Животная модель для микроскопического опухолевого обсеменения и остаточного заболевания.
Другой аспект настоящего изобретения включает развитие животной модели для анализа микроскопических остаточных карцином и микроскопического обсеменения полостей тела. Термин "карцинома", как он использован здесь, может относиться к единичной клетке или многоклеточной опухолевой массе. При микроскопическом заболевании "опухоль" будет состоять из одной или нескольких клеток карциномы, которые не могут наблюдаться невооруженным глазом.
Животная модель, описанная здесь, является особенно полезной для имитации (i) послеоперационной среды для пациентов с раком головы и шеи, особенно в стадиях развития заболевания и (ii) полости тела зараженного субъекта, где была установлена микроскопическая карцинома. Модель, аналогичная другим животным моделям для рака, происходит от инокуляции опухолевых клеток в животное. Различие, однако, лежит в создании, подкожно, кармана (углубления), который является физиологически эквивалентным естественной полости тела или послеоперационной полости, созданной путем иссечения опухолевой массы.
Настоящее изобретение в качестве примера использует голую мышь, как модельный организм. Фактически, однако, может быть использовано любое животное для применения согласно настоящему изобретению. Особенно предпочтительными животными будут являться маленькие млекопитающие, которые рутинно используются в лабораторных протоколах. Даже более предпочтительными животными будут животные группы грызунов, такие как мыши, крысы, морские свинки и хомячки. Кролики также являются предпочтительными видами. Критерий выбора животного будет в значительной степени зависеть от конкретного предпочтения исследователя.
Первой стадией является создание тканевого лоскута в экспериментальном животном. Термин "тканевый лоскут" обозначает любое иссечение в теле животного, которое выдерживает ткань-мишень. Обычно предпочтительным является иссечение, выполненное в заднем боку животного, так как он представляет легкодоступный участок. Однако должно быть понятно, что иссечение может быть выполнено в других участках на животном, и выбор участков ткани может зависеть от различных факторов, таких как конкретный тип терапий, которые будут исследоваться.
Как только участок ткани-мишени подвергается воздействию, клетки карциномы либо индивидуально, либо в виде микроскопических опухолей контактируют с участком ткани. Наиболее убедительным способом обсеменения раковыми клетками в участке ткани является подача суспензии культурной среды ткани, содержащей клетки для воздействия на ткань. Применение раковых клеток может быть достигнуто просто использованием стерильной пипетки или любого другого обычного аппликатора. Естественно, эта процедура будет проводиться в стерильных условиях.
В одном варианте 2.5 • 106 SCCHN клеток инокулируется в подвергаемый воздействию тканевый лоскут голой мыши. Специалист в этой области будет способен легко определить для данной цели, какое соответствующее количество клеток будет требоваться. Количество клеток будет зависеть от различных факторов, таких как размер животного, участок иссечения, репликативная способность самих опухолевых клеток, времени, предполагаемом для роста опухоли, потенциальной противоопухолевой терапии, которая будет испытываться, и им подобных факторов. Хотя установление оптимальной модельной системы для любого конкретного типа опухоли может требовать определенного регулирования количества вводимых клеток, это не создает чрезмерного количества экспериментов. Специалист в области испытания животных будет оценивать то, что требуется такая оптимизация.
Это может сопровождаться, например, проведением предварительных исследований, в которых в животное доставляются различные количества клеток и наблюдается рост клетки после высвобождения тканевого лоскута. Естественно, введение большего количества клеток будет приводить к большей популяции микроскопических остаточных опухолевых клеток.
В настоящем исследовании лоскуты были эффективно герметизированы с использованием матрацных швов. Однако является очевидным, что специалисты в этой области могут использовать любое разнообразие способов, которые обычно применяются для герметизации иссечения, таких как применение адгезивов, скобок, стежков, швов и т. д., зависящих от конкретного рассматриваемого применения.
Следующие примеры обеспечиваются больше для иллюстрации определенных конкретных вариантов и не должны, в любом случае, считаться как ограничивающие объем изобретения.
ПРИМЕР 1
Подавление роста раковых клеток головы и шеи человека введением гена p53 дикого типа посредством рекомбинантного аденовируса
Материалы и способы
Клеточные линии и условия культуры. Клеточные линии человеческих SCCHN, Tu-138 и Tu-177, обе были установлены при Департаменте хирургии головы и шеи, Раковом центре M.D. Anderson. Tu-138 и Tu-177 были выделены из десно-губной умеренно дифференцированной сквамозной карцииномы и слабо дифференцированной сквамозной карцииномы гортани соответственно. Обе клеточные линии были сконструированы с помощью основной техники эксплантации, и являются цитокератин позитивными и онкогенными у атимичных голых и SCID мышей. Эти клетки выращивали в среде DMEM/F12, дополненной 10%-й инактивированной теплом фетальной бычьей сывороткой (FBS) с пенициллином/стрептомицином.
Приготовление рекомбинантного аденовируса и заражение. Рекомбинантный аденовирус p53 (Ad5CMV-p53) (Zhang et al., 1994) содержит цитомегаловирусный (CMV) промотор, кДНК p53 дикого типа, и сигнал полиаденилирования SV40 в кассете минигена, инсертированной в E1-делетированную область модифицированного типа 5 аденовируса (Ad5). Вирусные исходные культуры были выращены в 293 клетках. Клетки собирали спустя 36-40 часов после инфекции, таблетировали, ресуспендировали в фосфат-забуференном солевом растворе, лизировали, и клеточный дебрис удаляли путем градиентной очистки за счет воздействия CsCl. Концентрированный вирус диализировали, проводили разделение на аликвоты и хранили при -80oC. Инфекцию проводили добавлением вируса в среде DMEM/F12 и 2% FBS с клеточными монослоями, и клетки инкубировали при 37oC в течение 60 мин с постоянным перемешиванием, затем добавляли полную среду (DMEM/F12/10% FBS) и клетки инкубировали при 37oC в течение желаемого промежутка времени.
Анализ Нозерн-блоттинга. Полную РНК выделяли с помощью кислотно-гуанидин-тиоцианатного способа (Chomczynsky and Sacchi, 1987). Нозерн-блоттинг анализы проводили на 20 мкг полной РНК. Мембрану гибридизовали зондом p53 кДНК, меченным с помощью способа случайного праймера в 5 х SSC/Denhardt's растворе/0.5% SDS/ДНК денатурированной спермы лосося (20 мкг/мл). Мембрану также делили на полосы и повторно зондировали GAPDH кДНК для РНК нагружающего контроля. Относительные количества экспрессированного p53 определяли с помощью денситометра (Molecular Dynamics Inc., Sunnyvale, CA).
Анализ Вестерн-блоттинга. Готовили полные клеточные лизаты путем ультразвуковой обработки клеток, после 24-часового заражения в буфере RIPA (150 мМ NaCl, 1.0% NP-40, 0.5% DOC, 0.1% SDS, 50 мМ Tris, pH 8.0), в течение 5 секунд. Пятьдесят микрограммов белка из образцов были подвергнуты воздействию 10% SDS-PAGE и перенесены на Hybond-ECL мембрану (Amersham). Мембрану блокировали Blotto/Tween (5% нежирного сухого молока, 0.2% Tween 20, 0.02% азида натрия в фосфат-забуференном солевом растворе) и зондировали первичными антителами, мышиным моноклональным антителом Pab1801 античеловеческого p53 и мышиным моноклональным антителом (Amersham) античеловеческого β-актина, и вторичным антителом пероксидаза хрена, конъюгированным овечьим антимышиным IgG (Boehringer Mannheim, Indianapolis, IN). Мембрану обрабатывали и проявляли, как предложено производителем.
Иммуногистохимический анализ. Зараженные клеточные монослои фиксировали 3.8% формалином и обрабатывали 3% H2O2 в метаноле в течение 5 мин. Иммуногистохимическое окрашивание проводили путем использования набора Vectastain Elite (Vector, Burlingame, CA). Первичное использованное антитело было Pab1801 антителом анти-p53, и вторичное антитело было авидинмеченым антимышиным IgG (Vector). Биотинилированный пероксидазой хрена комплексный реагент ABC использовался для обнаружения комплекса антиген-антитела. Применялись предварительные адсорбционные контроли в каждом эксперименте иммуноокрашивания. Затем клетки противоокрашивали Harris гематоксилином (Sigma Chemical Co., St. Louis, MO).
Анализ клеточного роста. Клетки высевали при плотности 2•104 клеток/мл в 6-луночные планшеты в трижды повторенных вариантах. Клетки заражали либо аденовирусом дикого типа (Ad5CMV-p53), либо репликативно-дефицитным аденовирусом в качестве контроля. Клетки собирали каждые 2 дня, подсчитывали и их выживаемость определяли исключением трепановым синим.
Ингибирование роста опухоли in vivo. Воздействие Ad5CMV-p53 на образование подкожных опухолевых узелков определялось в голых мышах в определенной непатогенной окружающей среде. Эксперименты повторно рассматривались и подтверждались обоими комитетами по защите и утилизации животных и для исследования рекомбинантной ДНК. После индуцирования ацепромазиновой/кетаминовой анестезии были подняты три отдельных подкожных лоскута на каждом животном и 5 • 106 клеток в 150 мл полной среды вводили подкожно в каждый лоскут, используя тупую иглу; клетки хранили в кармане с горизонтальным матрацным швом. Для каждой клеточной линии использовали четыре животных. После 4 дней животных повторно анестезировали и лоскуты повторно поднимали для доставки 100 мл 1) Ad5CMV-p53 (50 MOI) в правый передний лоскут; 2) репликативно-дефективного вируса (50 MOI) в правый задний лоскут; и 3) одной транспортной среды в левый задний бок. Все участки инъекций имели развитые подкожные видимые и пальпируемые (осязаемые) узлы до того, как проводилась обработка. Животных осматривали ежедневно и умерщвляли на 20-й день. Объем опухоли in vivo рассчитывали путем допущения сферической формы со средним диаметром опухоли, определенным как корень квадратный произведения поперечно пересекающихся диаметров. Вслед за умерщвлением проводилось трехмерное измерение опухоли с помощью микроштангенциркуля для определения объема опухоли. Применялся непараметрический Friedman's 2-way ANOVA тест для определения величины различия между значениями образцов; использовался SPSS/PC + блок программного обеспечения (SPSS Inc., Chicago, II).
Результаты
Аденовирусное заражение SCCHN клеток. Условия для оптимальной аденовирусной трансдукции Tu-138 и Tu-177 клеток были определены заражением этих клеток аденовирусом, экспрессирующим E. Colip β-gal ген. Эффективность трансдукции была оценена путем подсчета количества голубых клеток после X-gal окрашивания. Оказывается, что имеет место линейное отношение между количеством зараженных клеток и количеством использованных аденовирусных частиц. Клетки, инокулированные однократной дозой 100 MOI β-gal аденовируса, показывали 60% голубых клеток (фиг. 1), и это было доказано в 100% случаев путем множества заражений. Эффективность трансдукции этого вектора в SCCHN клетках является совершенно отличной от эффективности трансдукции другой клеточной линией, исследованной ранее: HeLa, HepG2, LM2 и человеческие клеточные линии немалых клеток рака легкого показали 97-100% эффективности заражения после инкубации 30-50 MOI β-gal аденовирусом (Zhang et al., 1994).
Экспрессия экзогенного p53 мРНК в SCCHN клетках, зараженных аденовирусом. Две человеческие SCCHN клеточные линии были выбраны для этого исследования: обе клеточные линии Tu-138 и Tu-177 обладали мутированным p53 геном. Недавно созданный рекомбинантный p53 аденовирус дикого типа Ad5CMV-p53 был использован для заражения Tu-138 и Tu-177 клеток. Через 24 часа после заражения была выделена полная РНК и был проведен анализ Нозерн блоттинга. Трансформированная первичная клеточная линия 293 человеческой эмбриональной почки была использована в качестве положительного контроля из-за ее высокого уровня экспрессии p53 генного продукта, тогда как К562 клеточная линия лимфобластомы с гомозиготной делецией p53 гена служила в качестве отрицательного контроля. Уровни 2.8-kb эндогенного p53 мРНК, обнаруженного в образцах, выделенных из mock-зараженных клеток и из клеток, зараженных аденовирусом с репликационными дефектами, d1312 были аналогичными. Вплоть до 10-крат более высокие уровни экзогенного 1.9-Kb p53 мРНК присутствовали в клетках, зараженных Ad5CMV-p53, указывая на то, что экзогенный p53 кДНК подвергался успешной трансдукции в эти клетки и эффективно транскрибировался. Интересно, что уровень эндогенного p53 мРНК в этих клетках был в 5 раз выше, чем в экспериментальных контрольных образцах. Анализ Нозерн блоттинга не подтверждал Ad5CMV-p53 (ДНК) загрязнения РНК.
Экспрессия p53 белка в SCCHN клетках, зараженных аденовирусом
Для сравнения уровней p53 мРНК по отношению к количеству продуцированного p53 белка был проведен анализ Вестерн блоттинга. p53 полосу, определенную с помощью моноспецифического анти-p53 антитела, PAb1801, наблюдали в клеточных экстрактах, выделенных из всех образцов за исключением K562 клеток. Линия клеток 293 показывала высокие уровни p53 белка. Образцы, выделенные из mock-зараженных Tu-138 и Tu-177 клеток, показывали низкие уровни p53 белка. Уровень p53 экспрессии оставался аналогичным уровню в клетках, зараженных d1312 аденовирусом. Уровни p53 антигена, обнаруженные в Ad5CMV-p-53-зараженных клетках, были значительно выше, чем уровни эндогенных мутированных белков в обеих линиях клеток. Этот результат демонстрирует, что экзогенный p53 мРНК, продуцированный из клеток, зараженных Ad5CMV-p-53, эффективно транслируется в иммунореактивный p53 белок. Кроме того, иммуногистохимический анализ клеток, зараженных Ad5CMV-p-53 показывает характерное ядерное окрашивание p53 белка, в то время как mock-зараженные клетки не в состоянии показывать аналогичное окрашивание, несмотря на присутствие p53 белка в этих клетках. Эта неспособность к обнаружению белка может приписываться к нечувствительности анализа.
Влияние экзогенного p53 на SCCHN клеточный рост in Vitro. Клетки, зараженные контрольным вирусом d1312, имели скорости роста, аналогичные скорости роста mock-инфицированных клеток (фиг. 2A и 2B), тогда как рост Ad5CMV-p53 клеток, зараженных Tu-138 (фиг. 2A) и Tu-177 (фиг. 2B) клеток был сильно подавлен. Через 24 часа после заражения происходило кажущееся морфологическое изменение с участками клеточной популяции, которые округлялись, и их внешними мембранами, которые образовывали волдыри. Это являлось частью серии гистологически предсказуемых событий, которые составляют запрограммированную гибель клетки. Эффект был более заметен для Tu-138, чем для Tu-177 клеток. Клетки, зараженные аденовирусом с дефектом репликации, d1312 демонстрировали нормальные характеристики роста с отсутствием гистологических аномалий. Анализы роста были воспроизведены в четырех повторяющихся экспериментах.
Ингибирование роста опухоли in Vivo. Для каждой линии клеток были испытаны четыре животных. Одно животное в Tu-177 группе умирало после второй операции лоскута и проведения (доставки) терапевтического вмешательства, преимущественно благодаря глубокой анестезии и последующего увечья за счет клеточного спаривания. Вскрытие трупа не показало наличия метастаз или систематических эффектов. Измеримые опухоли наблюдались на обоих задних лоскутах животных (т. е. участках, которые не получали Ad5CMV-p53). Отсутствие прогрессии опухоли является значительным в правых передних лоскутах животных, которые получали Ad5CMV-p53 (p<0.4). Такие Tu-177 клетки имеют более медленную скорость роста, чем была ранее установлена у этих животных. Двое животных в Tu-138 группе были умерщвлены раньше из-за того, что у них происходил быстрый рост и изъязвление контрольных опухолевых участков. Все хирургические участки развивали повреждения по крайней мере 9 мм3 до вмешательства. Объемы опухолей при вскрытии трупа показаны в табл. 4. Различия в объеме не были статистически значительными в Tu-177 группе, которые могли отражать ограниченный размер образца.
Пример 2
Молекулярная терапия in vivo аденовирусом p53 для микроскопической остаточной сквамозной карциномы головы и шеи.
Материалы и способы
Клеточные линии и состояния культуры. Человеческие SCCHN линии клеток Tu-138, Tu-177, MDA 686-LN и MDA 886 были установлены и предварительно охарактеризованы (Clayman et al., 1993; Sacks et al., 1988). Эти клетки были выращены в Dulbecco's среде модифицированной Eagle's (DMEM/F12) дополненной 10% термически инактивированной фетальной бычьей сывороткой (FBS) и пенициллин/стрептомицином.
Приготовление рекомбинантного аденовируса и заражение. Анализ клеточного роста. Вестерн блоттинг анализы. Все процедуры были ранее описаны в Примере 1. Анализы клеточного роста все были проведены трижды.
Трансдукция in vivo аденовирусом β-галактозидазы. X-gal окрашивание образцов ткани было проведено на O.C.T. замороженных тканевых срезах для определения эффективности трансдукции. Восьми микрометровой толщины образцы были промыты в холодном PBS и зафиксированы в 0.5% глутаровом альдегиде при комнатной температуре в течение 5 минут. Затем слайды были дважды промыты 4oC PBS и проинкубированы в течение 4 часов в X-gal растворе (1.3 mM MgCl2, 15 mM NaCl, 44 мМ Hepes буфера, pH 7.4; 3 mM феррицианида калия; 3 mM ферроцианида калия; 2% X-gal в ДМФА). Слайды были противоокрашены гематоксилином и эозином.
Иммуногистохимический анализ. Экспериментальные ткани животного in vivo, зафиксированные формалином и заключенные в парафин, разрезали на куски 4-5 мкм, сушили при 60oC, подвергали депарафинизации и гидратировали дистиллированной водой. Срезы обрабатывали 0.5% раствором сапонина в дистиллированной воде и промывали несколько раз, заменяя дистиллированную воду; эндогенную пероксидазную активность блокировали 3% раствором пероксида водорода в метаноле, с последующим промыванием дистиллированной водой. Среды подвергали микроволновому облучению в дистиллированной воде в течение 3 минут с использованием микроволновой печи Sharp Model R9H81 работающей при частоте 2450 МГц при 700 Вт. После охлаждения срезы промывали, заменяя несколько раз дистиллированную воду, и помещали в PBS; иммуногистохимическое исследование проводили с использованием способа Hsu et al., (1981), использующего авидин-биотин-пероксидазный комплекс (ABC) в следующем виде: срезы блокировали нормальной лошадиной сывороткой и инкубировали в течение ночи при 4oC кроличьим античеловеческим p53 поликлональным антителом, клон OM-1, 1:80 (Signet Laboratories, Denham, MA). Затем был использован антикроличий IgG Elite набор (Vector Laboratories, Burlingame, CA) для применения биотинилированного анти-кроличьего IgG и ABC комплексов, которые были проинкубированы в течение 45 минут каждый. Реакцию иммуноокрашивания подвергали визуализации за счет использования 0.5% DAB в PBS, содержащем 0.01% пероксида водорода (pH 7.6), противоокрашивали 0.01% толуидинового голубого, дигидратировали, просветляли и вводили в Permount. Для подтверждения специфичности реакции иммуноокрашивания было выполнено иммунопероксидазное окрашивание с использованием того же самого способа, как и в испытываемых образцах, на известном положительном cytospin культуры ткани клеточной линии сквамозной карциномы, а также на отрицательном контроле кроличьего моноклонального антитела.
Ингибирование роста опухоли in vivo. Эта процедура была выполнена как описано в Примере 1. Все хирургические участки были оценены на патологию, а также с помощью анализа вскрытия трупов на систематическую токсичность.
Результаты
Влияние экзогенного p53 на SCCHN клеточный рост in vitro. Пример 1 описывал ингибирование клеточного роста in vitro с помощью Ad5CMV-p53 в SCCHN клеточных линиях, эндогенно мутированным p53. Этот настоящий пример приводится для определения того, будут ли SCCHN клеточные линии аналогично затрагиваться эндогенным p53 дикого типа. Исследуется также влияние Ad5CMV-p53 на незлокачественные фибробласты.
Четыре человеческие SCCHN клеточные линии были выбраны для этого исследования: Tu-138 и Tu-177 обладали мутированным p53 геном, тогда как MDA 686-LN и 886 обе являются гомозиготными для p53 гена дикого типа. Клеточная линия фибробластов, полученная из нормального выросшего фибробласта, которая является кариотически нормальной и неонкогенной, была использована в качестве незлокачественной контрольной клеточной линии. Клетки, зараженные контрольным вирусом d1312, имели скорость роста, аналогичную скорости роста mock-зараженных клеток, тогда как рост опухолевых клеток, зараженных Ad5CMV-p53, был в значительной степени подавленным (фиг. 3A, фиг. 3B, фиг. 3C и фиг. 3D). Через двадцать четыре - сорок восемь часов после заражения имело место кажущееся морфологическое изменение с участками клеточной популяции, которые округлялись, и их внешними мембранами, которые образовывали волдыри. Это являлось частью серии гистологически предсказуемых событий, которые составляют запрограммированную гибель клетки. Эффект ранее имел место скорее в клетках с эндогенным мутированным p53, чем в клетках с p53 дикого типа. Клетки, зараженные аденовирусом с дефектом репликации d1312, демонстрировали нормальные характеристики роста с отсутствием гистоморфологических аномалий. Анализы роста были воспроизведены в четырех повторяющихся экспериментах.
Экспрессия экзогенного p53 белка в нормальных фибробластах, зараженных аденовирусом, и ее влияние на скорость роста. Дополнительно был также исследован эффект Ad5CMV-p53 на кариотипично нормальные и не опухолегенные линии клеток фибробластов. Эти клетки были выделены в процессе установления первичных клеточных опухолевых линий. Через двадцать четыре часа после заражения был проведен анализ Вестерн блоттинга для сравнению уровней белка, продуцированного различными зараженными клеточными типами. Полосу p53, узнаваемую моноспецифическим анти-p53 антителом, PAb1801, наблюдали в клеточных экстрактах, выделенных из всех образцов, зараженных Ad5CMV-p53. Как видно из Примера 1, клеточная линия Tu-138, зараженная p53 аденовирусом, показывала высокие уровни p53 белка после трансдукции и служила в качестве контроля. Уровень p53 экспрессии оставался подобным в обоих типах клеток в mock-зараженных и d1312 зараженных клетках. Ad5CMV-p53 зараженные фибробласты показывали более высокие уровни p53 белка, чем уровни контрольных клеток. Этот результат указывает на то, что p53 ген эффективно транслируется в нормальные фибробласты, зараженные Ad5CMV-p53, как доказано за счет продуцирования иммунореактивного p53 белка. Экспрессия белка и эффективность трансдукции цитоспинов Ad5CMV-p53 зараженных фибробластов были подтверждены с помощью иммуногистохимических анализов. Эта фибробластовая клеточная линия проявляла нормальную скорость роста и морфологию независимо от вмешательства (mock, репликационно-дефектного вируса, или Ad5CMV-p53) (фиг. 4). Эти эксперименты были повторены дважды и также подтверждены в других нормальных человеческих фибробластовых клеточных линиях.
Эффективность трандукции in vivo. Для измерения эффективности генного переноса in vivo, участок подкожного лоскута был вырезан через 72 часа после молекулярного или контрольного вмешательства. Эксперименты доза-отклик с вектором маркером аденовируса β-галлактозидазы демонстрировали эффективность трансдукции доза-отклик в этой модели. Это было подтверждено иммуногистохимическими анализами через 4 суток после заражения Ad5CMV-p53. Обе группы экспериментов показывали зависимость in vivo доза-отклик, которая была ранее описана in vitro (Пример 1). Ни в одном из примеров, которые приводились, дозы вируса, превышающие 1010 PFU, не вызывали экспрессию p53 в других системах органов, включающих мозг, печень, легкое, сердце, обдоминальные висцеральные органы и кожу. Эти эксперименты иллюстрировали отношение доза-отклик между вирусным титром и эффективностью трансдукции, а также возможность достижения обширной временной экспрессии гена, подвергнутого трансдукции внутри желаемой хирургической модельной области.
Супрессия роста опухоли in Vivo. Были спланированы исследования для определения будет ли Ad5CMV-p53 опосредованный генный перенос in vivo оказывать влияние на установление или рост SCCHN клеток, имплантированных в подкожный лоскут. Для достижения этой цели была создана модель микроскопического остаточного заболевания. В этой модели три подкожных лоскута были подняты на атимичной голой самке мыши и было проведено обсеменение 2.5 • 106 опухолевых клеток с помощью пипетки. Вместо того чтобы позволить опухолевым клеткам образовать узлы (обычно имеющие место на 4 сутки), однократная доза молекулярного вмешательства была проведена через 48 часов после обсеменения опухолевыми клетками. В этом способе, хотя большие опухоли не присутствовали, микроскопические опухолевые клетки были внутри хирургического участка, имитируя клиническую дилему хирургического иссечения всей большой опухоли. Развитие опухолей было непосредственно связано с количеством опухолевых клеток, временем, распределяемым для имплантации, и дозой Ad5CMV-p53. Из мышей, которые получили микроскопически имплантированные опухолевые клетки (2.5 • 106) и были обработаны Ad5CMV-p53 при концентрации 108 плашкообразующих единиц (PFU) или больше, только у двух развивались опухоли, у обеих мышей, имплантированных клеточной линией p53 дикого типа (MDA 886-LN). Все другие клеточные линии показывали отсутствие развития опухоли (табл. 5). Эти эксперименты ясно демонстрируют, что рост микроскопических опухолевых клеток может быть эффективно подавлен in vivo, если подвергается воздействию Ad5CMV-p53. Образование опухоли было оценено по окончании 12 недельного периода (раньше животного умерщвляли при обстоятельствах избыточной опухолевой нагрузки) с помощью суммарного и гистологического анализов хирургических участков. Данные установления опухоли суммируются в табл. 5.
Иммуногистохимический анализ был выполнен на опухолевых срезах экспериментальных животных. Эта клеточная линия обладала эндогенным p53 геном дикого типа. Имело место отсутствие значительного базального иммуноокрашивания жизнеспособной опухолью MDA 686-LN (mock-инфекция). 107 PFU Ad5CMV-p53 показывало периферийный опухолевый некроз с иммуноокрашиванием в более центральной части опухоли. 108 PFU Ad5CMV-p53 обнаруживало полный некроз опухоли с иммуноокрашиванием, найденным во всем хирургическом кармане с множеством слоев, экспрессирующих белок, включающих строму и поверхностные мышечные слои. 109 PFU Ad5CMV-p53 показывало результаты, аналогичные с результатами 108 PFU Ad5CMV-p53, однако заметными являлись увеличенная экзогенная p53 экспрессия по всему хирургическому участку и отек.
Используя животных, которые служили в качестве их собственного внутреннего контроля, имплантаты 4.0 • 106 или большего количества клеток значительно увеличивали установление подкожных имплантатов по сравнению с опухолевой имплантаций 2.5 • 106 клеток (P < 0.01), даже будучи обработанными в хирургическом участке Ad5CMV-p53 через 48 часов после инокуляции. Позволение имплантированнным клеткам укореняться в течение 72 или 96 часов до Ad5CMV-p53 вмешательства аналогично увеличивало опухолевый результат (заражения). Эксперименты доза-отклик установили, что 108 и 109 PFU Ad5CMV-p53 были в равной степени эффективны в ингибировании опухоли, нагруженной 2.5 • 106 клетками, имплантированными в течение 48 часов (фиг. 6). Эндогенный p53 статус имплантированных опухолевых клеточных линий (либо гомозиготно мутированных, либо p53 дикого типа) оказывал небольшое воздействие на эффективность Ad5CMV-p53 в прекращении развития опухоли.
Пример 3
Индукция апоптоза, опосредованная с помощью генного переноса аденовируса p53 дикого типа в сквамозной клеточной карциноме головы и шеи.
Материалы и способы
Клеточные линии и условия культуры. Приготовление и заражение рекомбинантного аденовируса. Все процедуры были выполнены и клеточные линии поддерживали как описано ранее в Примерах 1 и 2.
Анализ ДНК фрагментации. После инкубации аденовирусом p53 дикого типа, а также репликации дефектных аденовирусных контролей с различными временными интервалами клетки были собраны, ресуспендированы в 300 мкл PBS с добавлением 3 мл экстракционного буфера (10 mM Трис, pН 8.0, 0.1 М ЭДТА, 20 мкг/мл РНК, 0.5% SDS) и проинкубированы при 37oC в течение 1-2 часов. К концу инкубации была добавлена протеиназа K с конечной концентрацией 100 мкг/мл и раствор помещен в водяную баню при 50oC по крайней мере на 3 часа. ДНК была проэкстрагирована равным объемом 0.5 М Трис (pH 8.0), насыщенного фенолом, затем экстракция повторена смесью фенол/хлороформ. Осажденная ДНК была проанализирована в 1% агарозном геле.
Клеточная фиксация. Для способа TUNEL клетки были зафиксированы в 1% растворе формальдегида в PBS (pH 7.4) в течение 30 минут во льду. Затем клетки были промыты 3 мл PBS, ресуспендированы в 70% этаноле, охлажденном во льду, и хранились при -20oC до использования. Для анализа клеточного цикла клетки фиксировали только в 70% этаноле, охлажденном во льду.
Анализ терминальной деоксинуклеотидил трансферазы. Анализ был выполнен согласно Gorczyca et al. процедуры (Gorczyca et al., 1993). После фиксации и промывания, клетки были ресуспендированы в 50 мкл TdT буфера, содержащего 0.2 М какодилата натрия (pH 7.0), 2.5 mM Трис-HCl CoCl2 (Sigma Chemical Company, St. Louis, MO), 0.1 mM DTT (Sigma Chemical Company), 0.25 мг/мл BSA (Sigma Chemical Company), 5 единиц терминальной трансферазы (Boehringer Mannheim Biochemicals, Indianapolis, IN) и 0.5 нмолей биотин-16dUTP вместе с dATP, dGTP и dCTP при концентрации 20 мкМ. Контроли были приготовлены путем инкубирования отдельных аликвот каждого испытываемого образца без dUTP. Клетки были проинкубированы в растворе при 37oC в течение 30 минут, промыты в PBS и ресуспендированы в 100 мкл FITC, окрашивающего раствора, содержащего 4X SSC, 0.1% Тритона X-100 и 2.5 мкг/мл флуоресцеинового авидина (Vector Labs. Inc. , Burlingame, CA). Пробирки были проинкубированы в течение 30 минут в темноте при комнатной температуре. Клетки были промыты в PBS с 0.1% Тритона X-100 и ресуспендированы в 0.5 мл PBS, содержащего пропидиний йодид (5 мкг/мл) и 70 мкл (1 мг/мл) РНКазы. Пробирки были проинкубированы в темноте во льду в течение 30 минут до проточного цитометрического анализа.
Проточной цитометрический анализ. Все образцы были проанализированы с использованием проточного цитометрического анализа EPICS Profile II (Coulter Corp. , Healeah, FL) со стандартной оптической конфигурацией. Для каждого образца были собраны по крайней мере 5000 событий. Позитивность для TdT концевого-мечения была определена вычитанием контрольной гистограммы из гистограммы испытываемого образца с использованием рабочей программы иммуно-4 Elite (Coulter Corp., Healeah, FL).
Анализ клеточного роста. Клетки высевали и рост регистрировали, как описано в Примере 1.
Анализ на апоптоз in Vivo. Генная терапия в модели микроскопического остаточного заболевания SCCHN была описана выше в Примере 2.
Концевое мечение in Situ. Процедура была выполнена как ранее описано (Wijsman et al., 1993). Парафиновые срезы депарафинизировали растворением в ксилоле в течение 5 минут, три раза каждый и успешно гидратировали погружением слайдов в течение 3 минут каждый в 100%, 90%, 70% и 30% растворы этанола. Эндогенная пероксидаза была инактивирована погружением слайдов в течение 20 минут в 0.75% H2O2 об./об. в 100% метаноле. После этого слайды промывали в PBS, срезы гидролизовали 0.1% пепсином (Fisher Scientific, Houston, TX) вес./об. в 0.1 N HCl в течение 5 минут при 37oC и интенсивно промывали в PBS. Затем срезы были проинкубированы во влажной камере при 37oC в течение 1 часа коктейлем с концевым мечением, включающим следующие компоненты: терминальную деоксинуклеотидилтрансферазу 0.5 единиц/мкл; биотинилированный dUTP, 0.06 mM; 5X tdt буфер, 10 мкл; бидистиллат вплоть до 50 мкл. Реакцию обрывали погружением слайдов в буфер, содержащий 300 mM NaCl и 30 mM Na-цитрата в бидистиллате. После промывания слайдов в PBS срезы были проинкубированы в авидине, конъюгированном пероксидазой хрена в течение 1 часа при 37oC во влажной камере. Окрашивание проявляли с использованием 3,3'-диаминобензидина и срезы противоокрашивали метиловым зеленым.
Результаты
Супрессия роста SCCHN клеточных линий аденовирусом p53. Приведенные выше Примеры демонстрируют, что ген p53 дикого типа может быть эффективно трансдуцирован в SCCHN клеточные линии с помощью рекомбинантного аденовирусного вектора. Следовательно, поврежденные опухолевые клетки теряют их способность к пролиферации in vitro, а также к пролиферации in vivo. Эффект супрессии является независимым от эндогенного p53 статуса клеточных линий. Анализы предварительной скорости роста были выполнены в течение недельного периода времени. Настоящий пример исследует ранние эффекты p53 дикого типа на SCCHN клеточный рост (т.е. более ранние временные интервалы, в часах).
В этом исследовании были использованы две представительные клеточные линии. Клеточная линия Tu-138 обладала мутированным p53 геном, тогда как MDA 686-LN обладала p53 геном дикого типа. Клетки, зараженные репликационно-дефектным вирусом d1312, имели скорость роста, аналогичную скорости роста mock-зараженных клеток (фиг. 5A и фиг. 6B). С другой стороны, рост Ad5CMV-p53, зараженных Tu-138 (фиг. 5A) и MDA 686LN (фиг. 5B), был в значительной степени подавленным. Очевидно, что экзогенный p53 белок обладал более ранней и более глубокой супрессией роста Tu-138, по сравнению с MDA 686-LN. Наблюдали кажущееся морфологическое изменение с участками клеточных популяций, округляющихся сверху, и с их внешними мембранами, образующими волдыри, напоминающие апоптоз, которое сопутствовало с инициированием супрессии роста. Клетки, зараженные репликационно-дефектным аденовирусом d1312, демонстрировали нормальные характеристики роста с отсутствием гистоморфологических аномалий. Важно, что эти эффекты не наблюдали после заражения p53 аденовирусом кариотипично нормальных фибробластов, как детально представлено в Примере 2, приведенном выше, а также в человеческих оральных кератиноцитах (умерщвленных, но не онкогенных).
Анализ ДНК фрагментации. Одним из характерных маркеров в апоптозе, который отличает апоптоз от некроза, является биохимически наблюдаемое проявление ступенчатости ДНК фрагментов. Для подтверждения представления о том, что клетки подвергаются апоптозу после заражения p53 аденовирусом, был проведен анализ осуществления ДНК фрагментации. Хромосомальная ДНК, экстрагированная из жизнеспособных клеток после заражения репликационно-дефектным аденовирусом или аденовирусом p53 дикого типа, была подвергнута электрофорезу в агарозном геле. Вид ДНК фрагментов эквивалентен приблизительно 200 bp (пар оснований) и их многочисленность была отмечена в обеих клеточных линиях. Фрагментированная ДНК появлялась через 22 часа после заражения p53 аденовирусом в клеточной линии Tu-138, тогда как в клеточной линии MDA 686 LN, фрагментированная ДНК была видимой после 30 часов и более очевидной после 48 часов после заражения p53 аденовирусом дикого типа. Не появлялось обнаруживаемой фрагментированной ДНК из mock-зараженных клеток и d1312-зараженных клеток.
Анализ терминальной деоксинуклеотидил трансферазы in Vitro. Другим характерным маркером апоптоза является морфологическое изменение и разрушение структурной организации ядер, которое приводят к конденсации хроматина. Электронная микроскопия была широко использована для обнаружения такого ультраструктурного изменения. Однако недавно проточные цитометрические способы для идентификации апоптозных клеток достигли больших успехов благодаря способности сканирования и анализа клеточных популяций по сравнению с электронной микроскопией (Gorczyca et al., 1993). Автор изобретения применил здесь способ TUNEL (терминального деоксинуклеотидил трансфераза опосредованного-dUTP-биотин nick концевого-мечения) (Gorczyca et al., 1993), который основан на обнаружении обширного разрыва ДНК для идентификации апоптозных клеток. Через пятнадцать часов после заражения p53 аденовирусом 4.4% жизнеспособной Tu-138 клеточной популяции находились в апоптотических стадиях по сравнению с отсутствием такого состояния для MDA 686LN (фиг. 6A и фиг. 6B). Количество апоптотических клеток увеличивалось пропорционально увеличению продолжительности наблюдения после инкубации p53 аденовирусом. Около 31% Tu-138 клеток подвергались апоптозу после 22 часов. Хотя имело место замедление в начальном индуцировании апоптоза, приблизительно 60% MDA 686 LN клеток находились в апоптотических стадиях после 48 часов заражения p53 аденовирусом. Заслуживает внимания то, что процентное содержание апоптотических клеток, как оно определено с помощью способа TUNEL, может быть в значительной степени неустановленным, так как только жизнеспособные клетки подвергались анализу. Эти данные коррелируют с данными анализов скорости роста и ДНК фрагментации. Не наблюдали обнаруживаемых клеточных популяций, подвергающихся апоптозу, в контрольных экспериментах с использованием mock-заражения, а также в репликационно дефектных вирусных контролях (100 M.O.I). Следовательно, апоптоз не являлся сам по себе функцией продуктов трансдуцированного аденовирусного гена.
Анализ на апоптоз in Vivo. Примеры, приведенные выше, показывают, что p53 аденовирус подавляет образование опухоли in vivo. Этот пример приводится для того, чтобы показать имеет ли место супрессия роста опухоли in vivo после апоптоза. Был проведен анализ концевого мечения in situ для обнаружения апоптотических клеток в срезах, заключенных в парафин, которые были получены из Примера 2. Ясно, что окрашивания не наблюдается в тканевых срезах, выделенных из животных, несущих MDA 686LN, которым в качестве контроля была проведена обработка PBS. С другой стороны, тканевые срезы, выделенные из мыши, несущей MDA 686LN, обработанные аденовирусом p53 дикого типа, показывали высокоположительное окрашивание, демонстрируя, что апоптоз действительно являлся событием, включенным в супрессию роста опухоли in vivo.
Кроме того, в этих исследованиях пришли к определению того, является ли супрессия роста, частично обусловленной арестом клеточного цикла за счет индуцирования p21 белка, или главным образом, является результатом апоптоза. Анализ Вестерн блоттинга показал, что p21 белок был индуцирован зараженными аденовирусом p53 дикого типа SCCHN клетками. Однако анализы клеточного цикла указывали на то, что несмотря на повышенный уровень p21 белка в клетках, зараженных аденовирусом p53, не происходило значительного аккумулирования клеток в G1 фазе, когда сравнивали с S фазой.
Пример 4
Супрессия роста сквамозных клеток головы и шеи с помощью p53-FLAG: эффективный маркер для испытаний генной терапии.
Материалы и способы
Клеточные линии и состояния культуры. Приготовление и заражение рекомбинантным аденовирусом. Анализ Нозерн блоттинга. Анализ Вестерн блоттинга. Анализ клеточного роста. Иммуногистохимическое окрашивание клеточных слоев in vitro. Все процедуры были выполнены и клеточные линии поддерживались как описано в Примере 1.
Генерирование p53-FLAG аденовируса. p53 кДНК последовательность была вырезана из pC53-SN путем гидролиза BamHI и клонирована в BamHI сайте pGEM7Z. Рекомбинантная плазмида с соответствующей ориентацией инсерта была затем гидролизована AccI и KpnI для удаления 22 аминокислот из 3' конца p53 кДНК. Линкер с AccI-KpnI совместимыми концами, содержащий последовательность пептида FLAG, включающую стоп кодон, был затем лигирован в гидролизованную плазмиду для создания p53-FLAG составного гена. Образующийся p53-FLAG составной ген был затем клонирован в векторе экспрессии человеческим CMV промотором и SV сингалом полиаденилирования. Конечная конструкция была впоследствии инсертирована в шаттл вектор pXCJL.1 (Zhang et al., 1994) для генерирования рекомбинантного p53-FLAG аденовируса.
Эксперименты с микроскопическим остаточным заболеванием in Vivo. Исследования были проведены в свободной окружающей среде определенного патогена с использованием модельной системы атимичной голой мыши, описанной в Примере 1. Были выполнены два различных набора повторяющихся экспериментов. Первый был экспериментом доза-отклик с использованием AdCMV-p53-FLAG вируса в трех лоскутах со снижающейся концентрацией (1010 pfu, 109 pfu, 108 pfu). Четвертый лоскут служил в качестве контроля и был подвергнут методу слепого отбора либо с PBS, либо с репликационно-дефектным аденовирусом (DL312). Второе исследование было выполнено с использованием 1010 pfu AdCMV-p53-FLAG, AdCMV-p53 и репликационно-дефектного аденовируса в трех раздельных лоскутах. Четвертый лоскут был инокулирован тем же самым объемом (100 мкл) стерильного PBS. Через сорок восемь часов после обработки двое из этих животных были умерщвлены и собраны лоскуты для иммуногистохимического анализа. Оставшихся животных наблюдали в течение 21 дня и затем умерщвляли. Объемы опухоли измеряли для сравнения с использованием циркулей.
Результаты
Экспрессия мРНК после заражения AdCMV-p53 и AdCMV-p53-FLAG вирусом. Обе Tu-138 и MDA 686LN были исследованы на экспрессию p53 мРНК. Полная РНК была выделена после заражения аденовирусом. Был проведен анализ Нозерн-блоттинга. Были обнаружены аналогичные уровни экзогенной AdCMV-p53 мРНК между AdCMV-p53 и AdCMV-p53-FLAG зараженными клетками. Уровень p53 мРНК экспрессии после заражения AdCMV-p53 и AdCMV-p53-FLAG были сравнимыми для Tu-138 и для MDA 686LN. Изменение интенсивности является ощутимым для того, чтобы его можно было связать с дозой нагрузки. Эндогенная экспрессия p53 мРНК видна в линиях 2 и 3 в мутированной клеточной линии Tu-138. Не наблюдалось значительной эндогенной p53 мРНК экспрессии в MDA 686LN клеточной линии, которая является дикого типа для p53 гена. Эти данные подтверждают, что AdCMV-p53-FLAG вирус аналогично AdCMV-p53 вирусу успешно трансдукцируется и эффективно транскрибируется. Анализ Нозерн блоттинга не подтверждает загрязнения AdCMV-p53 ДНК.
Экспрессия экзогенного p53 белка в зараженные AdCMV-p53 и AdCMV-p53-FLAG клеточные линии SCCHN. Для сравнения количества белка, экспрессированного клетками, зараженными AdCMV-p53 и зараженными AdCMV-p53-FLAG, был выполнен анализ Вестерн блоттинга. Полосы белка были идентифицированы с использованием моноспецифического p53 антитела (РAb1801) и анти-FLAG M2 антитела (IB13025) на двух одновременно испытываемых гелях. Используя p53 антитела (pAB1801), получены аналогичные высокие уровни экспрессии p53 белка в обеих клеточных линиях, которые заражали AdCMV-p53 и AdCMV-p53-FLAG. Были испытаны также Tu-138 и MDA 686LN клетки, зараженные AdCMV-p53. Не было отмечено изменения экспрессии p53 белка ни в клетках, зараженных репликационно-дефектным аденовирусом, ни в группе mock-зараженных клеток, когда аналогично обработанный гель метили мышиным анти-FLAG M2 антителом. Уровень экспрессии p53-FLAG белка, очевидно, аналогичен уровню, экспрессированному после мечения p53 антителом, но не наблюдалось обнаруживаемой полосы в этих клетках, зараженных AdCMV-p53 вирусом. Mock- и DL312 зараженные клетки не показывали обнаруживаемого уровня иммунореактивного p53 или FLAG белка в любой клеточной линии.
Влияние AdCMV-p53 и AdCMV-p53-FLAG на клеточный рост SCCHN in Vitro. Цитотоксический эффект терапии p53 дикого типа в Tu-138 и MDA 686LN клеточных линиях был детально представлен выше. Tu-138 клеточная линия содержит эндогенно мутированный p53 ген и MDA 686LN клеточная линия обладает p53 геном дикого типа. Это исследование рассматривается для определения, будет ли какое-либо различие в эффективности наблюдаться после рекомбинации AdCMV-p53 вируса за счет инсертирования FLAG последовательности. Клетки, зараженные репликационно-дефектным аденовирусом, имеют аналогичную скорость роста в mock-зараженных клетках. Умеренный цитотоксический эффект можно видеть с репликационно-дефектным аденовирусом (фиг. 7A). Напротив, те клетки, которые заражены либо AdCMV-p53 либо AdCMV-p53-FLAG испытывали фактически общую гибель опухолевых клеток на третьи сутки. Гистологическое исследование подтвердило образование волдыря за счет плазменной мембраны, который является характерной чертой апоптоза и был охарактеризован в виде механизма клеточной гибели в AdCMV-p53 зараженных SCCHN клеточных линиях (Пример 1). Как отмечено выше, эффект был более заметен для Tu-138 клеточной линии (мутированной p53), чем он был заметен для MDA 686LN клеточной линии (p53 дикого типа). Анализы кривой роста были воспроизводимыми в трех повторяющихся исследованиях без значительного различия, которое было бы заметно между эффектом AdCMV-53 и AdCMV-53-FLAG вирусами, предполагая, что добавление FLAG пептида не оказывает влияния на способность p53 в супрессии клеточного роста.
Иммуногистохимическое окрашивание SCCHN клеточных линий, зараженных аденовирусом. Зараженные клеточные монослои были сравнены на экспрессию p53 и p53-FLAG белка с использованием стандартного иммуногистохимического способа. Ни p53, ни FLAG белок не могли быть четко идентифицированы в mock инфекции DL312 зараженных клеток в MDA 686LN клеточной линии. Однако в Tu-138, которая содержит мутированный p53 ген, эндогенное окрашивание для p53 было положительным. Когда клетки были заражены AdCMV-p53 вирусом, было отмечено сильное окрашивание в обеих клеточных линиях. Визуальное наблюдение этих клеток, зараженных AdCMV-p53-FLAG вирусом, показывало идентичную интенсивность окрашивания и количество положительных клеток с PAb1801 для антитела по сравнению с клетками, зараженными AdCMV-p53 вирусом. Клетки, зараженные AdCMV-p53-FLAG вирусом, также показывали сильную иммуногистохимическую положительную реакцию с M2 FLAG антителом. Хотя качество окрашивания отличалось и внутри ядра и, в меньшей степени, в цитоплазме.
Супрессия роста in Vivo. Исследование доза-отклик с использованием 108, 109 и 1010 плашкообразующих единиц (pfu) AdCMV-p53-FLAG вируса по сравнению с контрольным лоскутом, который был обработан либо PBS либо DL312, были выполнены с использованием микроскопического модельного способа, описанного в Примере 1 на Tu-138 клеточной линии. Средний размер опухоли для mock инфекции составлял 1205 +/- 205 мм3. Размер линейно уменьшался с увеличением концентрации вируса, использованного в молекулярной интервенции. Средний размер опухоли составлял 637 +/- 113 мм3, 392 +/- 109 мм3 и 193 +/- 74 мм3 для лоскутов, которые были обработаны 108, 109 и 1010 pfu AdCMV-p53-FLAG соответственно. Было проведено сравнение каждого животного относительно самого себя с использованием спаренного t теста и было отмечено значение доза-отклик при p<0.05 во всех сравнениях, за исключением сравнения между лоскутом, обработанным 109 и 1010 pfu. Ясно, что большее количество вируса в большей степени визуально ингибирует рост опухоли. В дополнительном исследовании эффекты AdCMV-p53 были сравнены с эффектами AdCMV-p53-FLAG. Не отмечали значительных различий в активности.
Иммуногистохимическая демонстрация эффекта супрессии экзогенной опухоли в животной модели микроскопического остаточного заболевания. После обеспечения сравнимой in vivo и in vitro активности AdCMV-p53 и AdCMV-p53-FLAG в изобретении были применены иммуногистохимические способы для демонстрации p53-FLAG составного белкового продукта in vivo. Используя Tu-138 и MDA 686LN клеточные линии, лоскуты микроскопического остаточного заболевания были собраны через 48 часов после обработки, зафиксированы в формалине и заключены в парафин. На соседствующих срезах опухолевых клеток, обработанных AdCMV-p53-FLAG вирусом, было применено окрашивание и для p53 и FLAG белка. Интенсивность окрашивания и количество клеток, окрашивающихся положительно, было прямо пропорционально количеству использованного в заражении вируса. Контроли были отрицательными на окрашивание и с p53 и с FLAG антителами в MDA 686LN клетках. Эндогенное окрашивание для p53 отмечали в Tu-138 опухолевых клетках. Гистологический образец окрашивал Гематоксилином и Эозином p53 антитело и FLAG антитело. Характерное цитоплазмическое окрашивание FLAG M2 антитела контрастировало с внутриядерным окрашиванием p53 антитела. Это, во-первых, представляет, что FLAG M2 антитело, как было доказано, является эффективным в парафин заключенной фиксированной ткани. Окрашивание демонстрирует, что опухолевый супрессивный эффект направляется с помощью экзогенной терапии, и так, что в модели in vivo, он может идентифицировать экзогенную терапию с использованием FLAG системы.
В заключение, ясно, что совместная доставка FLAG белка вместе с желаемой генной терапией представляет потенциальную пригодность в качестве маркера генной терапии. Изобретение ясно показывает, что он одновременно промотировал вместе с p53 геном и что экспрессия РНК мессенджера и белка не снижалась. Более важно, что биологическая активность высвобожденного опухолевого супрессорного гена не изменялась. Впервые было доказано, что FLAG антитело эффективно, когда иммуногистохимический анализ проводят на формалин фиксированной заключенной в парафин ткани. Эти факторы предполагают пригодность этого нового белка в качестве метки в дальнейших исследованиях генной терапии.
Пример 5
Обработка сквамозной клеточной карциномы головы и шеи с использованием p53 аденовируса.
Пациент A
Представлен 53-летний мужчина с неоперабельной SCCHN опухолью головы. Масса опухоли представляла приблизительно 6.5 см в диаметре. После исследования функции костного мозга, количества тромбоцитов и почечной функции пациент получил первую обработку 108 зараженными частицами экспрессионной конструкции p53 аденовируса, разбавленной в стерильном фосфатном забуференном солевом растворе, путем проведения восьми отдельных внутриопухолевых инъекций (общий объем 10 мл). Каждые три дня пациент получал идентичную обработку, пока не было проведено шесть полных обработок.
Через трое суток после шести обработок исследовалась опухоль и найдено, что она составляет больше 4.0 см в диаметре. Гистологическое исследование показывает значительную фрагментацию клеток в опухолевом крае. Был проведен второй курс из шести обработок, после которого опухоль составляла больше 2.0 см в диаметре и была некротической. Пациент продолжал получать еженедельную обработку в течение трех месяцев, время, после которого опухоль уже больше не обнаруживалась.
Пациент B
Представлена 44-летняя женщина с операбельной SCCHN опухолью шеи. Размер опухоли представляет приблизительно 2.5 см в диаметре. После исследования функции костного мозга, количества тромбоцитов и почечной функции пациентка получает первую обработку 5 • 107 зараженными частицами экспрессионной конструкции p53 аденовируса, разбавленной в стерильном фосфатном забуференном солевом растворе, путем трех отдельных внутриопухолевых инъекций (общий объем 3 мл). Каждые три дня пациентка получает идентичную обработку, пока не было проведено шесть полных обработок.
Через трое суток после шести обработок, проводится иссечение опухоли. Опухолевое ложе промывается 6 мл стерильного забуференного солевого раствора в течение 60 минут. Инокулюм удаляется, рану закрывают и в опухолевом ложе оставляют дренаж. На 4, 7, 10 и 14 сутки после операции проводят инфузию через дренаж 5 • 107 зараженных частиц экспрессионной конструкции p53 аденовируса, разбавленного в стерильном забуференном солевом растворе (общий объем 3 мл). После контактирования с опухолевым ложем в течение двух часов инокулюм удаляют отсасыванием. Через шесть месяцев после окончания обработки не наблюдается первичных локальных или региональных опухолей.
Пример 6
Генный перенос дикого типа через аденовирусный вектор в A фазе I испытаний пациентов с прогрессированной возвратной сквамозной карциномой головы и шеи.
Аденовирусный вектор, содержащий p53 ген "дикого типа", был доставлен в логарифмически повышающихся дозах пациентам с возвратной сквамозной карциномой головы и шеи, подтвержденных с помощью биопсии. Были проведены прямые опухолевые инъекции три раза еженедельно в течение двух текущих недель. Пациенты были разделены на две группы: 1) пациенты с возвратным заболеванием, которое может подвергаться резекции, 2) пациенты с возвратным заболеванием, которое не может подвергаться резекции.
Пациентов, отделенных в группу с заболеванием, которое может подвергаться резекции, направляли на полную обширную хирургическую резекцию их возвратного неоплазма через 72 часа после шестого события генного переноса в течение 2-недельного периода. Аденовирусный вектор был также доставлен внутриоперативно и через 72 часа после хирургической процедуры через регрессивную катетерную инфузию. Пациенты, которые не могли подвергаться резекции, подвергались повторной попытке переноса гена через прямые опухолевые инъекции ежемесячно в течение двухнедельных циклов до тех пор, пока не наблюдали прогрессирование заболевания или ухудшение статуса активности пациента. Надежность этой обработки контролировали путем закрытого больничного наблюдения, биопсий для оценки эффективности переноса гена, анализа содержания воды в организме для тромбоцитных (Shed) векторов и анализа некропсии.
Способы
Субъекты исследования. Двадцать один пациент с прогрессированными возвратными сквамозными карциномами верхнего аэропищеварительного тракта (aerodigestive) с Eastern Cooperative Oncology Group статусом активности у 2 пациентов поступали в одну из двух исследуемых групп (arms), состоящих из пациентов, которые могли подвергаться резекции (Группа 1) или которые не могли подвергаться резекции (Группа 2) с возвратным злокачественным развитием болезни. Характеристики субъектов исследования и дозы аденовирусного вектора показываются в табл. 6 и 7 соответственно. У всех женщин были отрицательные тесты на беременность и все пациенты использовали контрацептивные способы. Данное сведение было получено от всех пациентов до начала исследования.
Вектор генного переноса. Настоящее изучение применяло вектор репликационно-дефектного аденовирусного серотипа 5 с энхансером (цитомегаловирусом)-промотором, обозначенным Ad5CMVp53. Были продуцированы три серии аденовирусного вектора с плашкообразующими единицами (PFU) в области от 109 до 1011 за счет хорошей производственной практики Magenta, Inc and Introgen Therapeutics, Inc, и хранились замороженными (-70oC) в University of Texas, M.D. Anderson Cancer Centre. Каждая серия была эффективной для трансдукции, использующей Вестерн блоттинг, а также для анализов супрессии роста опухолевых клеток in vitro. Вектор оттаивали и разбавляли в фосфатном забуференном солевом растворе (связующее) сразу до генного переноса и направляли в комнаты пациентов при 4oC.
Дозировка. Аденовирусный вектор вводили каждой из пяти групп пациентов в логарифмически увеличивающихся дозах. Увеличение дозировки было установлено после двухнедельных наблюдений последнего пациента, обработанного предыдущей дозой. После записи первых шести пациентов в исследовании: три пациента записывали при каждом уровне дозировки независимо от того, является ли пациент способным к резекции или не является способным к резекции. Доза биологического вектора описывается в терминах полной дозы (в плашкообразующих единицах). Установленное количество векторов, введенных на злокачественную эпителиальную клетку, не аппроксимировалось. Общий объем введения показывается в табл. 7. Объем аденовирусного вектора, введенного в твердую злокачественную опухоль, определяли клиническим и радиографическим способом установления объема опухоли. Вектор непосредственно вводился в возвратные сквамозные карциномы при прямом наблюдении и с помощью ручной пальпации. Инъекции осуществляли по всему пространству через 1 см вдоль масс опухоли. После переноса гена субъект оставался под постоянным наблюдением в течение по крайней мере 1-1/2 часа. Респираторные выделения и секреторные выделения тела поддерживали в течение 72 часов после окончания генного переноса вектора.
Обнаружение вектора. Образцы мочи и сыворотки регистрировали для обсемененного аденовирусного вектора, используя вирусную культуру 293 клеток, а также реакции полимеразной цепи (PCR), используя праймеры, которые амплифицируют E1b область аденовируса и 5' конец p53 гена дикого типа, которые были специфическими к вектору. PCR продукты были затем перенесены с помощью саузерна для улучшения обнаружения вируса в 1-5 вирусных частицах, а также подтверждения специфичности PCR продукта. Были проанализированы положительные и отрицательные контрольные образцы в каждой реакции.
Безопасность. Были проведены регистрация симптомов, жизненные признаки и количество крови, и пациентов исследовали физически и ежедневно фотографически документировали. Радиография груди, химическое исследование крови и анализ статуса состояния были выполнены с начала каждого цикла обработки. Сывороточные титры аденовирусного антитела были
измерены до и после каждого цикла генного переноса. Через три дня после шестого генного переноса первого цикла были проведены опухолевые биопсии (или хирургическая резекция). Образцы в каждом примере хранили в виде замороженных-snap, патологически включенных образцов, а также в виде образцов, зафиксированных в формалине.
Экстракция нуклеиновых кислот из сыворотки и мочи. Ad5CMV-p53 аденовирусную ДНК экстрагировали из 0.5 мл аликвот сыворотки или мочи с помощью модифицированного метода Cunningham et al., (1995). Дистиллированную воду добавляли до получения 1 мл, и аликвоты осаждали 30% полиэтиленгликолем (PEG). Так как одного SDS было недостаточно для высвобождения вирусной ДНК из ее частиц, протеиназа K была добавлена к SDS при 50oC в течение 2-16 часов после осаждения полиэтиленгликолем (Norder et al., 1990). Образцы были проэкстрагированы фенолом и вирусную ДНК осаждали этанолом в присутствии гликогена (Cunningham et al., 1995). Осажденную ДНК выделяли центрифугированием при 1400 g в течение 10 минут при 4oC, ресуспендировали 0.3 мл дистиллированной воды и повторно осаждали этанолом. Гранулы ДНК промывали 70% этанолом, сушили в вакууме и растворяли в 10 мкл дистиллированной воды. Образцы анализировали либо тотчас же, либо хранили при -20o до использования. Экстракцию нуклеиновых кислот проводили в биологически безопасных камерах для предотвращения возможного перекрестного загрязнения образцов.
PCR реакции на ДНК, выделенной из образцов сыворотки. Были сконструированы праймеры для специфической амплификации p53 гена из аденовирусного вектора. Верхний праймер (5'-CACTGCCCAACAACACCA-3', SEQ ID NO:5) соответствует 3' концу p53 гена и нижний праймер (5'-GCCACGCCCACACATTT-3', SEQ ID NO: 6) соответствует E1B области аденовирусного типа 5 (нуклеотиды 3517-3533 последовательности дикого типа). Каждая пробирка PCR реакции содержала 0.2 mM каждого олигонуклотида, 0.4 mM dNTPs, 1X TaqPlus Long слабо солевого буфера (из Stratagene), 0.6 мкл TaqPlus Long (5U/мл) (из Stratagene), и 5 мкл испытываемой ДНК. Образцы помещали в MJ Research Pelter Thermal Cycle (PTC-200), запрограммированный на 93oC в течение 3 минут с последующим трехстадийным профилем: 93oC в течение 30 секунд, 65oC в течение 45 секунд, и 72oC в течение 45 секунд на общие 30 или 35 циклов. 5 унл 6X нагрузочного буфера (0.25% бромфенолового голубого, 0.25% цианол ксилола FF и 15% Ficoll (Type 400; Pharmacia) в воде) добавляли в каждую пробирку к концу PCR опыта и загружали в 1% агарозу, 1X TBE гель, содержащий этидиний бромид (0.6 мкг/мл). Образцы были подвергнуты электрофорезу при 100 В в течение 1-1.5 часов и затем сфотографированы под УФ светом.
Реакция полимеразной цепи (PCR). Только 5 мкл приготовленной ДНК может быть использовано в единичной реакции полимеразной цепи. Для сыворотки PCR проводили в 20-мкл объеме, содержащем 2 mM MgCl2, 50 mM KCl, 0.1% Тритон X-100, 200 мкМ каждого из деоксирибонуклеазидных трифосфатов (dNTP), 10 mM Трис-HCl (pH 9.0), 5 мкМ каждого из праймеров, и 1.7 единиц Taq ДНК полимеразы (Promega). Реакции проводили при 94oC в течение 30 секунд, 58oC в течение 30 секунд и 72oC в течение 60 секунд для 35 циклов, за которым следовал 10-минутный промежуток при 72oC. PCR праймеры были выбраны из последовательности Ad5CMV-p53 со смысловым праймером, расположенным при 3' конце p53 кДНК (5'-GCCTGTCCTGGGAGAGACCG-3', SEQ ID NO:7), и антисмысловой праймер был выбран из E1B области аденовируса типа 5 (5'-CCCTTAAGCCACGCCCACAC-3', SEQ ID NO: 8). PCR продукт (фрагмент 838 пар оснований) был выделен в 1% агарозном геле. Тот же PCR продукт был субклонирован в pCR-Script векторе (Stratagene), секвенирован и инсерт, очищенный в геле, был использован в качестве зонда для обнаружения PCR продукта. Для мочи PCR проводили в 20-мкл объеме, содержащем 2 mM MgSO4, 10 mM (NH4)2SO4, 10 mM KCl, 0.1% Тритон X-100, 20 мМ Трис-HCl (pH 8.8), 0.1 мг/мл бычьего сывороточного альбумина, 200 мкМ каждого из деоксирибонуклеазидных трифосфатов (dNTP), 5 мкМ каждого из праймеров и 2.5 единиц Taq Plus long ДНК полимеразы (Stratagen). Реакции проводили при 93oC в течение 60 секунд и затем при 93oC в течение 30 секунд, 65oC в течение 45 секунд и 72oC в течение 45 секунд для 35 циклов с последующим 10-минутным промежутком при 72oC. PCR праймеры были выбраны из последовательности Ad5CMV-p53 со смысловым праймером, расположенным при 3' конце p53 кДНК (5'-CACTGCCCAACAACACCA-3', SEQ ID NO:9), и антисмысловой праймер был выбран из E1B области аденовируса типа 5 (5'-GCCACGCCCACACATTT-3', SEQ ID NO:10). PCR продукт (фрагмент 724 пар оснований) был выделен в 1% агарозном геле.
Саузерн блоттинг-анализ. В анализе Саузерн блоттинг, используемом для проверки специфичности продуктов PCR, ДНК в геле денатурировали и нейтрализовали до блоттинга на нейлоновой мембране (Hybond-N+, Amersham) посредством капиллярной абсорбции. Мембрану предварительно гибридизовали в течение 15 мин при 65oC в Rapid-hyb буфере (Amersham) и гибридизовали в том же самом буфере, содержащем 32P-меченый зонд, в течение 1-2 часов. Мембрану промывали в 0.1 x SSC и 0.1% SDS при комнатной температуре дважды, и снова дважды при 65oC (15 мин на промывание). Промытую мембрану выдерживали на рентгеновской пленке при -70oC в течение 1-16 часов с усиливающим экраном.
Контроль и оценка испытываемых образцов. Следующие виды контроля включаются с каждой серией образцов. На стадии выделения ДНК применялись два "негативных" контроля сыворотки (фондированная и приведенная к аликвотной пробе сыворотка от владельцев Introgen) и два позитивных контроля, содержащие негативную сыворотку, доведенную до пика 10 pfu или 100 pfu вируса AdCMV-p53, который был применен. Это делалось для получения окна чувствительности там, где 10 (и 100) pfu контроли являлись позитивными, но негативные контроли являлись негативными. Если негативные контроли были позитивными, PCR повторялась лишь 30 циклов. Если 10 pfu был негативным, ДНК далее чистили дополнительным осаждением этанола, и повторяли PCR. Вышеуказанные две стадии всегда помещали экспериментальные параметры в соответствующее окно чувствительности.
На стадии PCR были применены позитивный контроль 1ng AdCMV-p53 ДНК (выделенной из клинической партии) и негативный (H2O) контроль. PCR серии повторяли, если какой-либо из контролей был ошибочным. Неудач с негативными контролями не было. Для подтверждения предполагаемых позитивов ДНК повторно выделяли из сыворотки (наряду с несколькими временно смежными образцами) и повторяли PCR этой ДНК. Образцы оценивали как позитивные, только если результаты можно было воспроизвести. Те образцы, которые были позитивными в одном из двух анализов образцов, считались негативными для целей отчетности. Предполагаемые позитивы, которые невозможно было повторить (из-за отсутствия дополнительного необработанного образца), не включались в базу данных.
Измерение эффективности генного переноса. Хирургически удаленные образцы ткани помещали в криостаты, немедленно мгновенно замораживали и затем хранили в хранилище с жидким азотом до использования. Замороженные образцы декантировали в отверстие пульверизатора для ткани Bessman из нержавеющей стали (Spectrum, Houston, TX), который предварительно охлаждали погружением в жидкий азот. Ткань измельчали до тонкого порошка, ударяя пестиком Bessman со стальной головкой пять-десять раз. Пульверизованную ткань переносили в стеклянный тканевый гомогенизатор (Fisher Scientific, Pittsburg, PA), содержащий 1 мл TRI реагента (Molecular Research, Cincinnati, OH) на 50 мг ткани, и гомогенизировали путем пяти-десяти ударов сверху и снизу тефлоновым пестиком.
Вслед за гомогенизацией РНК выделяли согласно инструкциям, приложенным к TRI реагенту. Гомогенаты переносили в полипропиленовые центрифужные пробирки (Molecular Research) и выдерживали в течение 5 мин при комнатной температуре до добавления хлороформа (0.2 мл на 1 мл TRI реагента). Образцы затем энергично перемешивали, инкубировали дополнительно 15 мин при комнатной температуре и центрифугировали при 12000 • g в течение 15 мин при 4oC для отделения РНК, содержащей водный слой, от фенол-хлороформовой фазы. Добавляли изопропанол к жидкой фазе и РНК осаждали инкубированием при комнатной температуре в течение 15 мин. Гранулы РНК выделяли центрифугированием при 12000 • g в течение 15 мин при 4oC, промывали один раз 75% этанолом, высушивали воздухом, растворяли в диэтил пирокарбонате (DEPC)-обработанной воде и количественно определяли путем измерения поглощения при 260 nm. Загрязняющую ДНК удаляли инкубированием вплоть до 50 мкг РНК с 60 U DNase I (Pharmacia, Piscataway, NJ) в течение 25 мин при 37oC в общем реакционном объеме 260 мкл. РНК затем экстрагировали смесью фенолахлороформа, осаждали этанолом, промывали один раз 75% этанолом, гранулировали центрифугированием при максимальной скорости в микроцентрифуге в течение 15 мин при 4oC, сушили воздухом, ресуспендировали в DEPC-воде и хранили при -80oC. Качество РНК оценивали испытанием образцов на обычном неденатурированном 0.8% геле агарозы и проявлением 28S и 18S рибосомных полос окрашиванием этидиум бромидом. Для исключения перекрестного заражения между образцами и для сведения к минимуму RNase активности все пригодные для повторного использования инструменты, которые использовались для выделения РНК, обмакивались в 2% Liqui-Nox (Fisher Scientific) детергентный раствор в течение минимум 5 минут, очищались от дебриса, переносились в 10% отбеливающий раствор в течение 3 мин, интенсивно промывались деионизированной водой, опрыскивались 100% этанолом, сушились, погружались в хлороформ и высушивались снова перед использованием.
Была осуществлена обратная транскрипция с использованием 1.5 мкг полной клеточной РНК в 23.5 мкл реакционной смеси, содержащей 111 нг статистических гексамеров (Gibco BRL, Grand Island, NY). 40 единиц RNase ингибитора (Boehringer Mannheim, Indianapolis, IN), 0.4 мМ каждого dNTP (Perkin Elmer, Foster City, CA), и 300 единиц Superscript II Rnase H- обратной транскриптазы (Gibro BRL) в 1 x RT буфере (50 мМ Tris, pH 8.3, 75 мМ хлористого калия, 3 мМ хлористого магния и 20 мМ дитиотрейтола). РНК и статистические гексамеры нагревали до 70oC в течение 10 мин и охлаждали во льду до добавления остатка реакционной смеси. Реакцию инкубировали при 25oC в течение 5 мин 200 единицами обратной транскриптазы и затем дополнительные 10 мин при 25oC вслед за добавлением следующих 100 единиц обратной транскриптазы для облегчения отжига праймера перед инкубированием при 42oC в течение 50 мин. RT-реакции обрывали посредством тепловой инактивации обратной транскриптазы в течение 15 мин при 70oC. РНК комплементарную кДНК удаляли путем гидролиза одной единицей RNase H (Boehringer Mannheim) в течение 20 мин при 37oC. РНК из линии TU167 сквамозной клеточной карциномы (HNSCC) головы и шеи, зараженная рекомбинантным аденовирусным Ad5CMV-p53 (множественность инфекции 100:1), использовалась в качестве позитивного контроля для обнаружения вирусно транскрибированного p53 и TU167 клеток, инфецированных вариантным аденовирусным векторным d1312 (1), который не содержит p53 транскрипциональной единицы, которая была использована в качестве негативного контроля.
Для обнаружения Ad5CMV-p53 транскрипта PCR осуществляли в реакционном объеме 30 мкл, содержащем 0.2 мМ каждого dNTP, 1.5 мМ хлористого магния, 1 единицу полимеразы taq (Promega, Madison, WI) и 0.5 мМ каждого праймера CMV2 (5'-GGTGGATTGGAACGCGGATT, SEQ ID NO:11) и P53EX3 (5'-GGGGACAGAACGTTGTTTTC, SEQ ID NO: 12) в 1 x PCR буфере (50 мМ хлористого калия, 10 мМ Tris pH 9.0, 0.1% Triton X-100). Праймеры CMV2 и P53EX3 амплифицируют 295-оснований фрагмент, специфичный к аденовирусу, создавшему p53 транскрипт. PCR условия для обнаружения Ad5CMV-p53 транскриптов были следующие: 94oC в течение 1 мин с последующими 94oC в течение 30 сек, 58oC в течение 40 сек, 70oC в течение 1 мин для 35 циклов и 70oC в течение 10-минутного промежутка времени.
Для обеспечения того, чтобы продукт, амплифицированный в процессе PCR, обнаруживал мРНК и не загрязнял ДНК в препарате РНК, PCR проводили также с использованием RT продуктов из параллельных реакций, в которых не добавляли обратную транскриптазу.
RT-PCR, специфичная для глицеральдегид-3-фосфат дегидрогеназы (GAPDH), проводили для того, чтобы контролировать целостность RT реакций. 3 мкл объема RT реакции разбавляли в 30 мкл PCR смеси, содержащей 0.2 мМ каждого DNTR, 2 мМ хлористого магния, 1 taq единицу полимеразы и 0.5 мМ каждого праймера GAPDH1 (5'-ACGGATTTGGTCGTATTGGG, SEQ ID NO:13) и GAPDH2 (5'-TGATTTTGGAGGGATCTCGC, SEQ ID NO:14) в 1 x PCR буфере. GAPDH праймеры охватывают 3 эксона в GAPDH гене человека и амплифицируют продукт 231-основания специфичный для мРНК. Условия PCR для обнаружения GAPDH были следующие: 94oC в течение 1 мин с последующими 94oC в течение 30 сек, 60oC в течение 12 сек, 72oC в течение 1 мин для 35 циклов и 72oC в течение 7-минутного промежутка времени.
PCR осуществляли с использованием Perkin Elmer Gene Amp 9600 термоциклер, и все праймеры были коммерчески синтезированными (Genosys, Woodlands, TX).
Иммуногистохимическое определение внутриопухолевого гена. Иммунопироксидазные исследования были осуществлены на формалин-фиксированных, включенных в парафин тканевых срезах, используя способ авидин-биотин-пироксидазного комплекса (ABC) (1). Образцы нарезали толщиной 3-4 мкм, депарафинировали в ксилоле и повторно гидратировали в снижающихся концентрациях (100-70%) этанола. Активность эндогенной пироксидазы блокировалась 3% пироксидом водорода в метаноле. После нескольких промываний в дистиллированной воде и фосфатном забуференном солевом растворе (PBC) срезы инкубировали с 1:10 разбавлением нормальной лошадиной сывороткой, чтобы свести к минимуму фоновое окрашивание. За этим следовала инкубация в течение ночи при 4oC моноклональными антителами с p53 (DO-1, Oncogene Science, Inc., Uniondale, NY; 1:80 разбавлением) и p21 (Oncogene Science, Inc., 1:100). Процедура пироксидазного окрашивания проводилась с использованием наборов ABC Elite (Vector Laboratories, Burlingame, CA). Реакция иммуноокрашивания визуализовалась с использованием 0: 05% 3.3'-диаминобензидина в Tris-HCl буфере, содержащем 0.01% пироксида водорода, pH 7.6. Срезы противоокрашивали 0.01% толуидинового голубого и помещали в пермаунт. Оценку осуществляли с помощью подсчета позитивного окрашивания ядер в 200 клетках 10 последовательных высокомощных полей двумя независимыми наблюдателями.
TUNEL анализ для фрагментации ДНК. TUNEL анализ осуществлялся с использованием ApoptagTM PLUS набора (Oncor, Gaithersburg, M.D.) согласно инструкциям, обеспеченным производителем. Слайды противоокрашивали 4% метиленовым зеленым. Соответственно окрашенные гематоксилином и эозином слайды оценивались на присутствие инфильтратов воспалительных клеток и оценивались по шкале 1-4.
Процедура анализа на цитопатический эффект. Образцы мочи пациента также рассматривались на присутствие Ad5p53 с помощью анализа, в котором любому вирусу в образце позволялось заражать клеточный монослой реципиента. Эти клетки рассматриваются на появление цитопатического эффекта (CPE): клетки округляются сверху и отрываются от поверхности. Образцы мочи пациента, которые подвергались анализу на CPE, были из образцов первой утренней мочи, собранных в течение второй недели первого цикла обработки и на день 0 или 1 (предобработки). Образцы, приблизительно 15 мл каждый, хранили в стерильных 15-мл конических пробирках при -80oC до использования. IT293 клетки, которые образуют клеточный монослой реципиента в этом анализе, содержатся в DMEM плюс 10% FBS в инкубаторе при увлажнении 10% CO2, при температуре 37oC. За 2 дня до тестирования образца пациента клетки высевались при 2 • 105 на лунку в 12-луночные планшеты.
Ко времени анализа образцы мочи оттаивали в ледяной бане, и аликвотную пробу смешивали 1:1 с DMEM и стерильно фильтровали, используя 0.22 мкм шприцевой фильтр. 350 мкл аликвотной пробы этой 1:1 смеси медленно добавляли в каждую лунку после удаления среды роста. Планшет осторожно встряхивали спустя 15 минут. После 30 минут добавляли 2.0 мл DMEM плюс 10% FBS в каждую лунку для разбавления образца. На 3-й день (72 часа спустя) и 6-й день анализа добавляли 0.5 мл аликвотных проб свежей среды в каждую лунку для поддержания клеточного монослоя в течение максимум 6-7 дней.
Образцы пациентов подвергались анализам трижды. Разбавленный предварительно обработанный образец анализировали как есть, а также с убывающей концентрацией от 105 pfu Ad5p53 на лунку для регистрации любого компонента мочи, который мог бы препятствовать обнаружению вируса с помощью этой процедуры. Контрольные лунки инкубировали с одним лишь DMEM с убывающей концентрацией 105, 104, 103, 102, или 101 pfu на лунку, каждая дважды. Пик 105 pfu вызывает CPE при этих условиях на 2-й день анализа; в каждый последующий день следующий убывающий контроль будет показывать CPE. Следовательно, время, в которое CPE обнаруживается в каждом образце пациента, показывает уровень Ad5p53 в этом образце.
Рекомбинантный компетентный аденовирус. Аденовирусный p53, использованный в клиническом исследовании, испытывали на присутствие RCA, используя A549 клетки, с помощью Biotechnology Services Division of Microbiological Associates, Inc., (Rockville, Maryland).
Статистический анализ. Был применен односерийный исследовательский проект. Для предотвращения вовлечения большего числа пациентов, чем необходимо в исследовании избыточной токсичности, которая была обнаружена, применялось правило ранней остановки Bayesian. Обозначенный как WILCOXON рядовой тест и назначенный тест применялись для сравнения между до и после обработки процентным содержанием клеток, показывающих TUNEL и иммуногистохимическое окрашивание соответственно. Статистический анализ осуществлялся с использованием блока Survpac SPSS-Statistical.
Ответ и токсичность. Выживаемость и ответ оценивались в анализе в интервале между ними (interim), но не рассматривались цели этого анализа. Целью этого анализа являлось определение потенциала трансдукции этой стратегии генного переноса. Пациенты являлись ценными для ответа и токсичности вслед за 30-дневным наблюдением после одного цикла генного переноса. Эффекты токсичности терапии оценивали согласно критерию общей токсичности Национального института рака. (Xref.) Ответ на терапию оценивался с помощью CT скана или ультразвука шеи перед каждым курсом обработки. Пациенты оценивались на ответ, если они получали по крайней мере один курс терапии, за которым следовала соответствующая документация ответа. Пациенты, подвергавшиеся хирургической резекции возратных опухолей, не могли оцениваться на ответ, так как хирургия проводилась до 30-го дня периода наблюдения. Все CT сканы оценивались одним радиологом, а ультразвуки - другим. Частичный ответ определяли как 50-процентное или большее уменьшение результата произведения диаметров измеримых опухолей; незначительный ответ оценивали как от 25% до менее чем на 50% уменьшение результата произведения диаметров измеримого патологического изменения. Развитие заболевания оценивается как 25-процентное или большее увеличение результата произведений диаметров.
Продолжительность выживаемости измеряли от времени входа протокола до экспирации. Каждый ответ пациента повторно рассматривался с использованием данных комитета управления, включающего онколога хирургии головы и шеи, радиолога и онколога по лекарственным препаратам.
Результаты
Обнаружение вектора. Аденовирусную векторную ДНК обнаруживали с помощью PCR в сыворотке, а также в пробах мочи пациентов вплоть до 48 часов после переноса гена. Предел обнаружения для вектора составляет 1-5 вирусных частиц. Вирусную ДНК выделяли в моче среди пациентов, которые подвергались переносу гена при каждой дозе вируса, но не обнаруживалась после 48 часов генного переноса. Обнаружение вирусной ДНК сыворотки увеличивалось с увеличением вирусной дозы сверх 107 PFU, но также была необнаруживаемой спустя 48 часов после переноса гена.
Образцы мочи сначала анализировали на инфекционный вирус, измеренным CPE на клеточных монослоях, до PCR нуклеотидного анализа. Присутствие вируса в моче относили к 293 клеткам, как описано выше, для того, чтобы наблюдать CPE, которое наблюдается в то время, как клетки округляются сверху и появляются из чашки петри. CPE редко идентифицировали в образцах. CPE находили позже в вышерасположенных культурах (более чем через шесть дней) и эти результаты рассматривались как поддающиеся выражению. Не случайным был CPE, подтвержденный с помощью PCR и Саузерн-блоттингом на той же самой пробе мочи для регистрации аденовирусных нуклеотидов.
Анализ вирусной ДНК других систем органов. PCR анализ демонстрировал вирусную ДНК более чем два месяца после того, как 109 Ad5CMVp53 ген был перенесен в кожу, кардиальную мышцу, легкое и тестикулярные ткани в быстрозамороженных аутопсических образцах, тщательно проанализированных на предотвращение перекрестного заражения между тканями. Почечная паренхима, надпочечные железы и панкреатические ткани не показали вирусных ДНК последовательностей, специфичных для этого вектора. Иммуногистохимический анализ для этих аутопсичных образцов не выявил подтверждения сверхэкспрессии белкового продукта p53 дикого типа.
Оценка переноса гена. Все анализы проводились спустя по крайней мере 1 час после выдерживания тканей с вектором. мРНК продукт обнаруживался на 4 час и 48 час через RT-PCR. Напротив, образцы биопсии, замороженные спустя 1 час после подвергания воздействию или меньше, не показали p53 мРНК. Кроме того, пробы негативного контроля, полученные с операционного участка, которые не подвергались переносу гена, были также негативными на PCR продукт. Нетрансдуцированные и трансдуцированные образцы биопсии от четырех пациентов анализировали посредством RT-PCR на присутствие Ad5CMVp53 транскрибированной мРНК. Трансдуцированные образцы от двух пациентов были позитивными на EtBr окрашенных гелях, в то время как никакой Ad5CMVp53 продукт не был обнаружен ни в каком из нетрансдуцированных образцов, кроме факта, что GAPDH может быть амплифицированным из всех образцов. Специфичность 295 br PCR продукта от двух позитивных пациентов подтверждалась Саузерн-блоттингом. Так как PCR продукт не наблюдался, когда обратную транскриптазу исключили из RT реакций, 295 br продукт, обнаруженный на рисунке, должен был скорее генерироваться из мРНК, чем из загрязняющей ДНК.
Иммуногистохимический анализ. Все образцы пациентов с пре- и постгенным переносом одновременно анализировались позитивными и негативными контролями в каждом эксперименте. Множество срезов каждого образца анализировались и сравнивались с гематоксилиновыми и эозин-окрашенными, а также TDT концевомечеными образцами. Образцы биопсии, подвергнутые поствекторному инъецированию подтверждали генный перенос у трех пациентов, чьи образцы с предварительно перенесенной биопсией не показали эндогенно сверхэкспрессированного p53 белка. У 5 пациентов из 21 пациента (27%) p21 (CIP/WAFI) не был значительно эндогенно экспрессированным; это доказывало информативность генного переноса в образцах биопсии у пациентов с постгенным переносом. Сверхэкспрессию p53 ядерного белка также наблюдали в связанных с опухолью лимфоцитах так же, как в клетках опухоли стромы, тем самым указывая также на трансдукцию неопухолевых клеток.
Серологический отклик антитела. Аденовирусные серотипные 5 антитела индуцировали у всех пациентов вслед за повторной инъекцией вектора. Однако эффективность трансдукции, как было найдено, не значительно не изменялась по сравнению с начальным циклом генного переноса и последующими циклами. Слабая опухолевая эритема в месте инъекции обнаруживалась, начиная с 3•109 PFU, однако эти локальные реакции не ограничивали векторную толерантность. Отсутствие доказательства системной гиперчувствительности обнаруживали у каждого пациента, несмотря на повторное вирусное дозирование в продолжение такого длительного периода, как шесть последующих месяцев, у пациента, обработанного 109 PFU.
Патологические наблюдения. Следы иглы были идентифицированы в большинстве образцов биопсии, и экспрессия генного продукта демонстрировалась в более глубоких тканях за пределами участков инъекции. Аналогично нахождение клеток с концевым мечением в трансдуцированных зонах предполагало индукцию апоптоза в опухолевых клетках, но отсутствие апоптоза в стромальных или воспалительных клетках. Воспалительные клетки отмечались у пациентов и становились выдающимися гистологическими находками среди пациентов, которые получали дозы 109 PFU или больше. Интересно, что пациент под номером 7, из которого был взят образец экспрессированного эндогенного p53 дикого типа, демонстрировал некроз кровотечения с отсутствием доказательства жизнеспособной опухоли в сериально нарезанных хирургических образцах в массе его рецидивирующего 2-см образца шеи после одного цикла генного переноса. Патологические находки некроза так же, как индукции апоптоза, часто наблюдались в образцах.
Клинические наблюдения. Пациенты переносили прямые опухолевые инъекции вектора с наиболее частым побочным эффектом, связанным с беспокойством в участке инъекции. Признак локальной эритемы в участке инъекции отмечается при 109 PFU вектора и выше, хотя более значительный системный признак сверхчувствительности не замечен, несмотря на очевидность высоких значений титра системного антитела. Пациенты под номерами 5, 10, и 16 оставались без признака заболевания в среднем в последующие семь месяцев. Пациенты 7 и 13 проявляли устойчивое заболевание в течение 3 и 5 месяцев соответственно в показательной зоне повреждения. Пациент под номером 20 оказывал частичный ответ, когда он проверялся с помощью CAT Scan и ультразвука, но у него развивался спинальный и плевральный метастаз, требующий прекращения исследования из-за усиления заболевания.
В заключение, пациенты с рецидивирующими чешуйчатыми карциномами головы и шеи, способствующие с помощью аденовируса генному переносу p53 дикого типа, являются безопасными и могут эффективно трансдуцировать раковые и нормальные клетки посредством прямых опухолевых инъекций или интраоперационного введения вектора. Никто из пациентов не проявил токсичности, которая могла бы ограничить введение вектора увеличение дозы. Экспрессия трансгенного продукта не изменялась, несмотря на улучшение ответа системного антитела. Ответы местных воспалений, обнаруженные патологически внутри образцов иссеченных опухолей, могут, в действительности, являться ценными.


















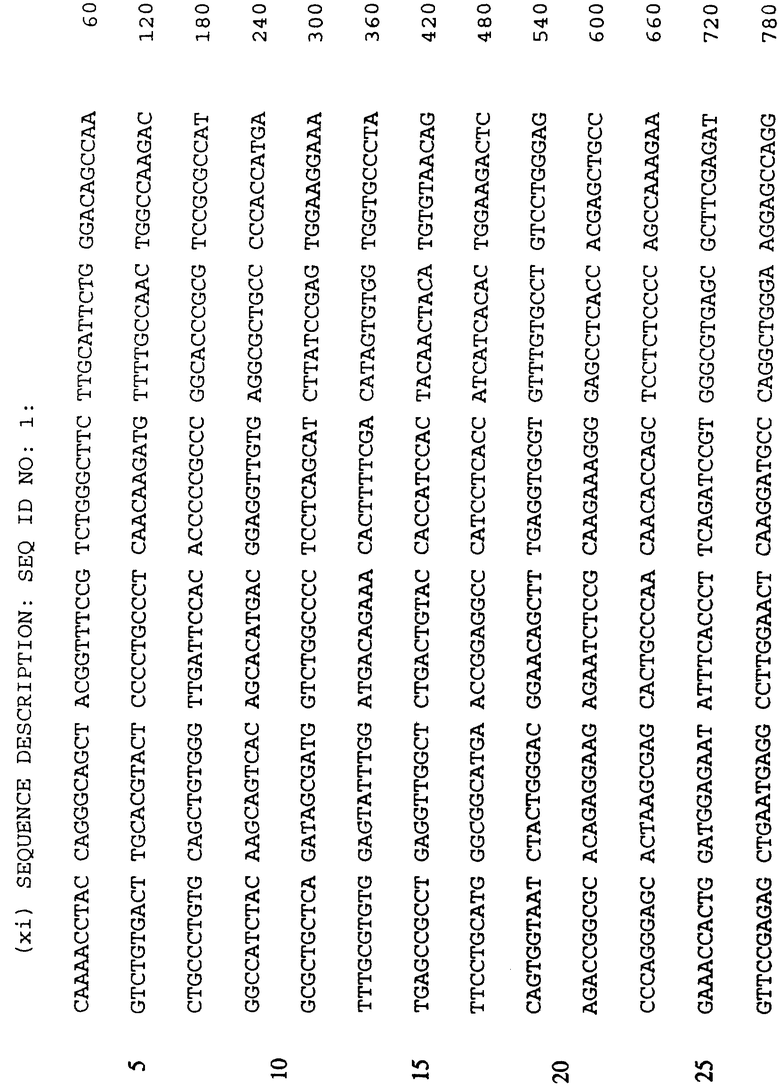














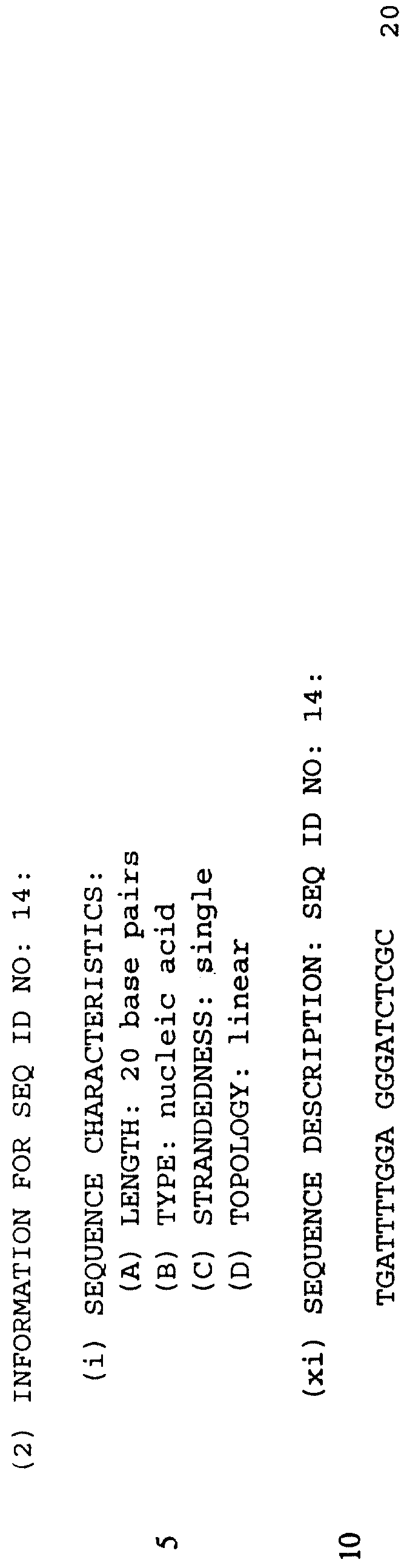
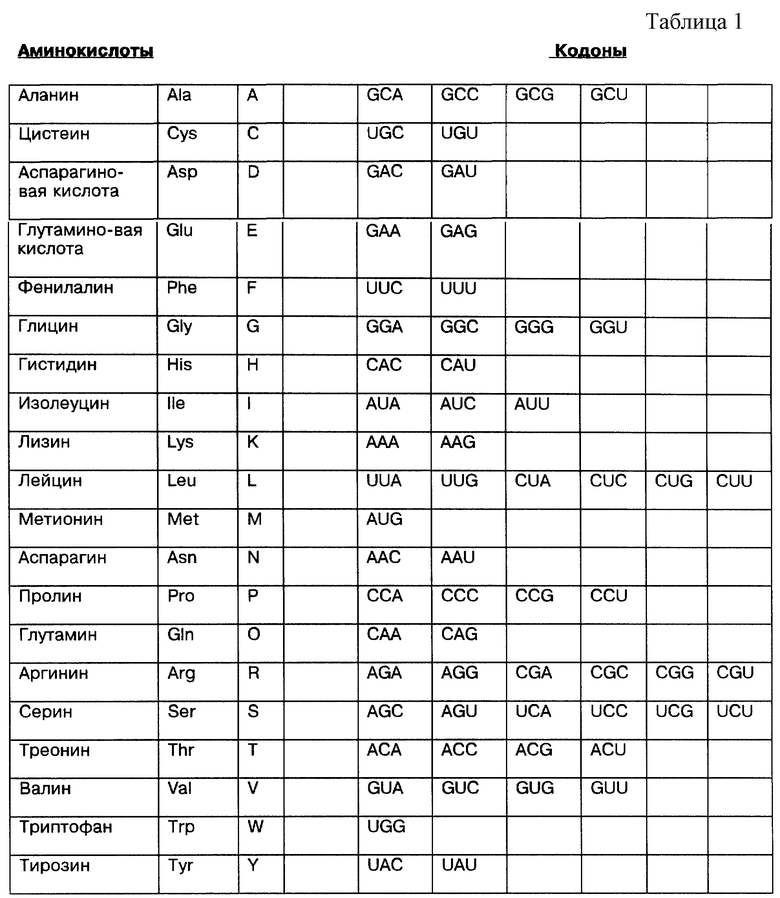



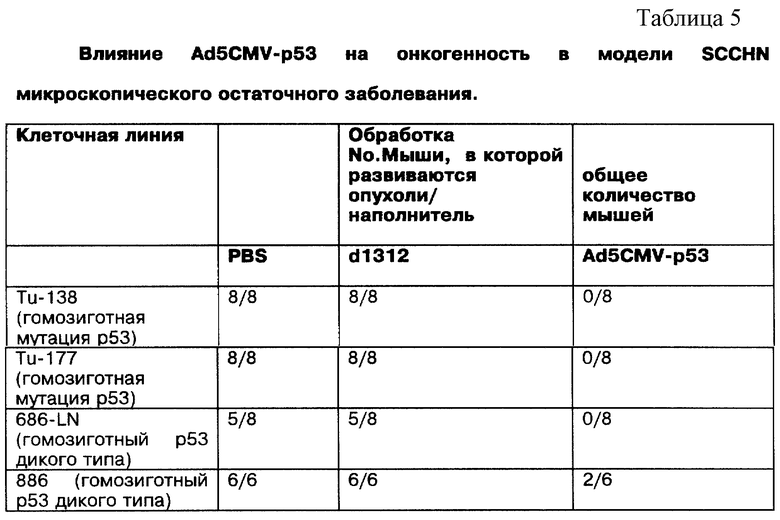
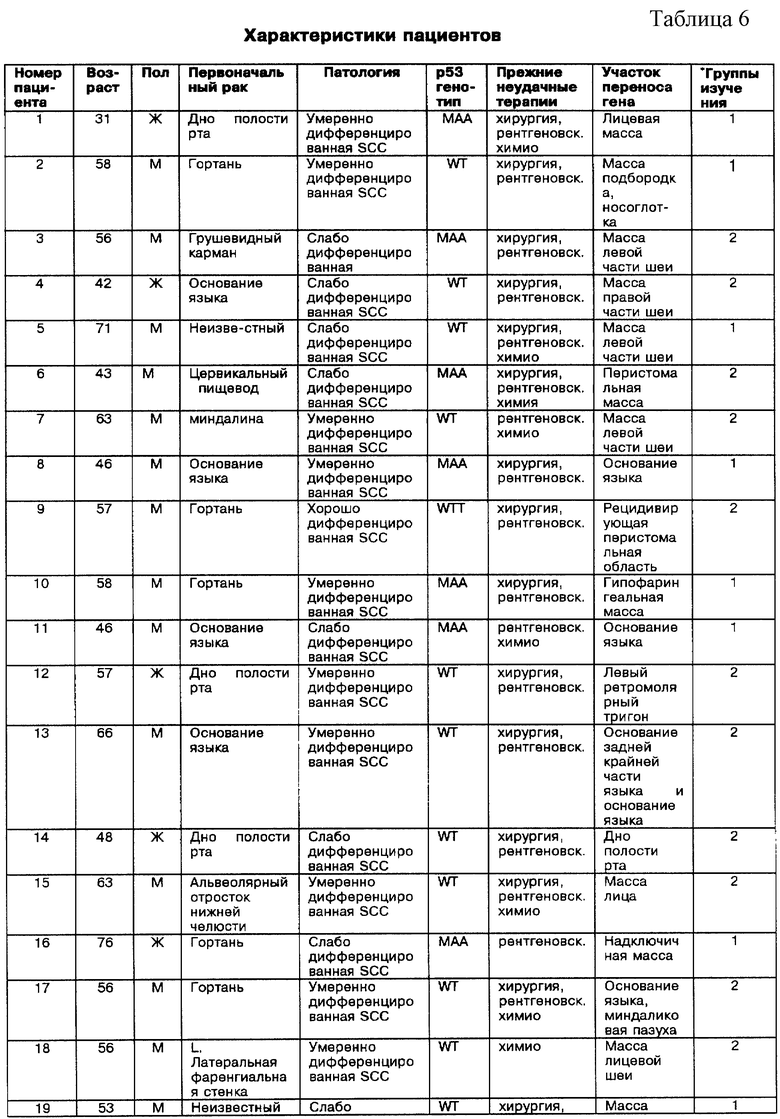
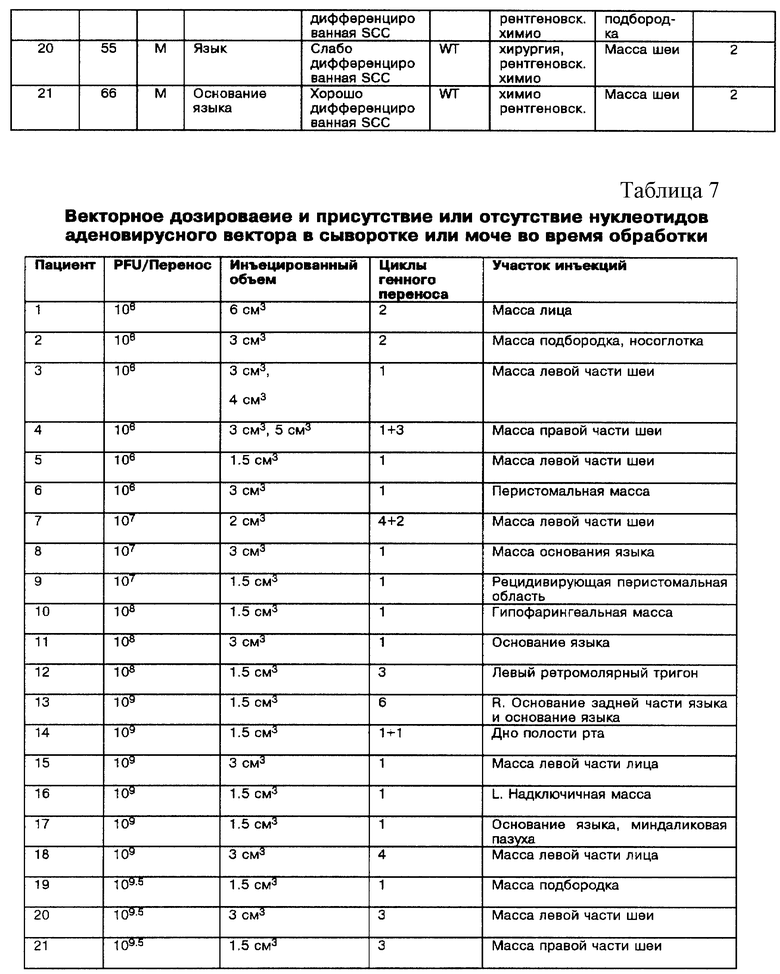
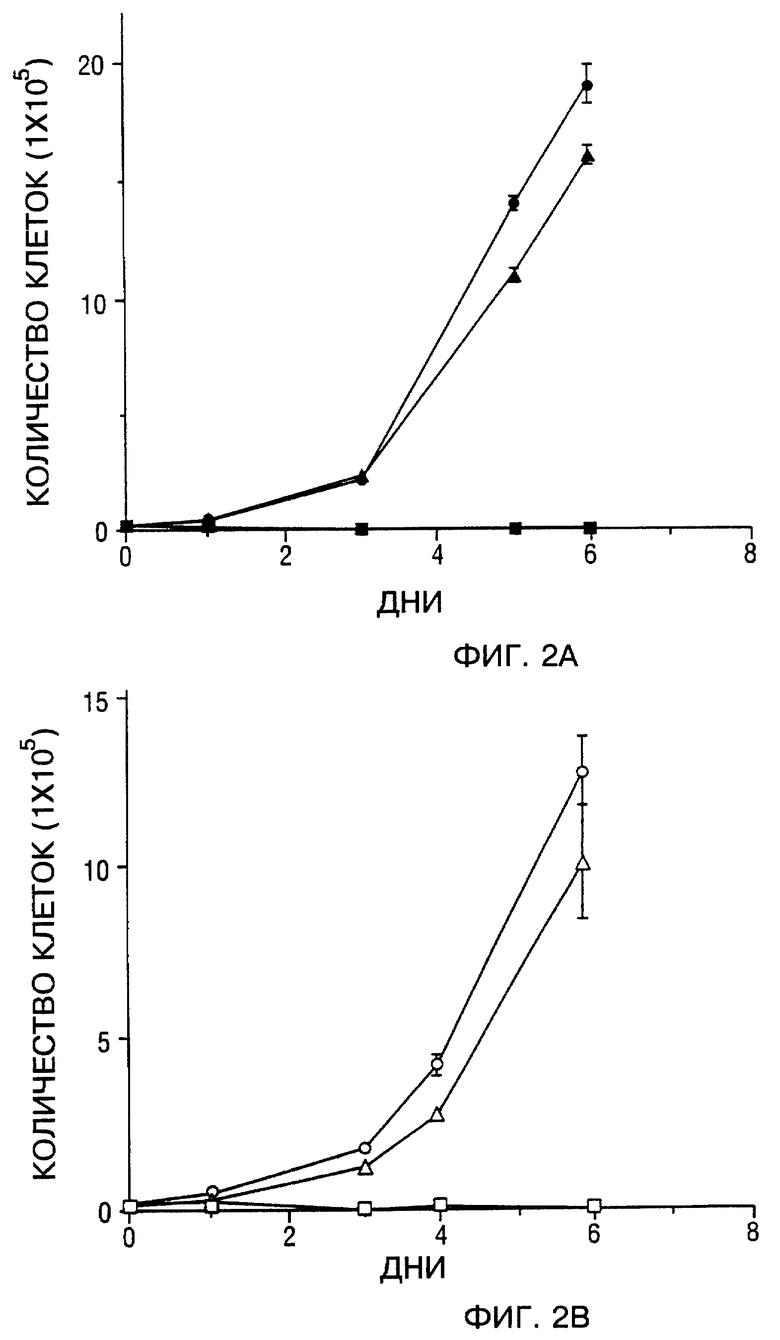
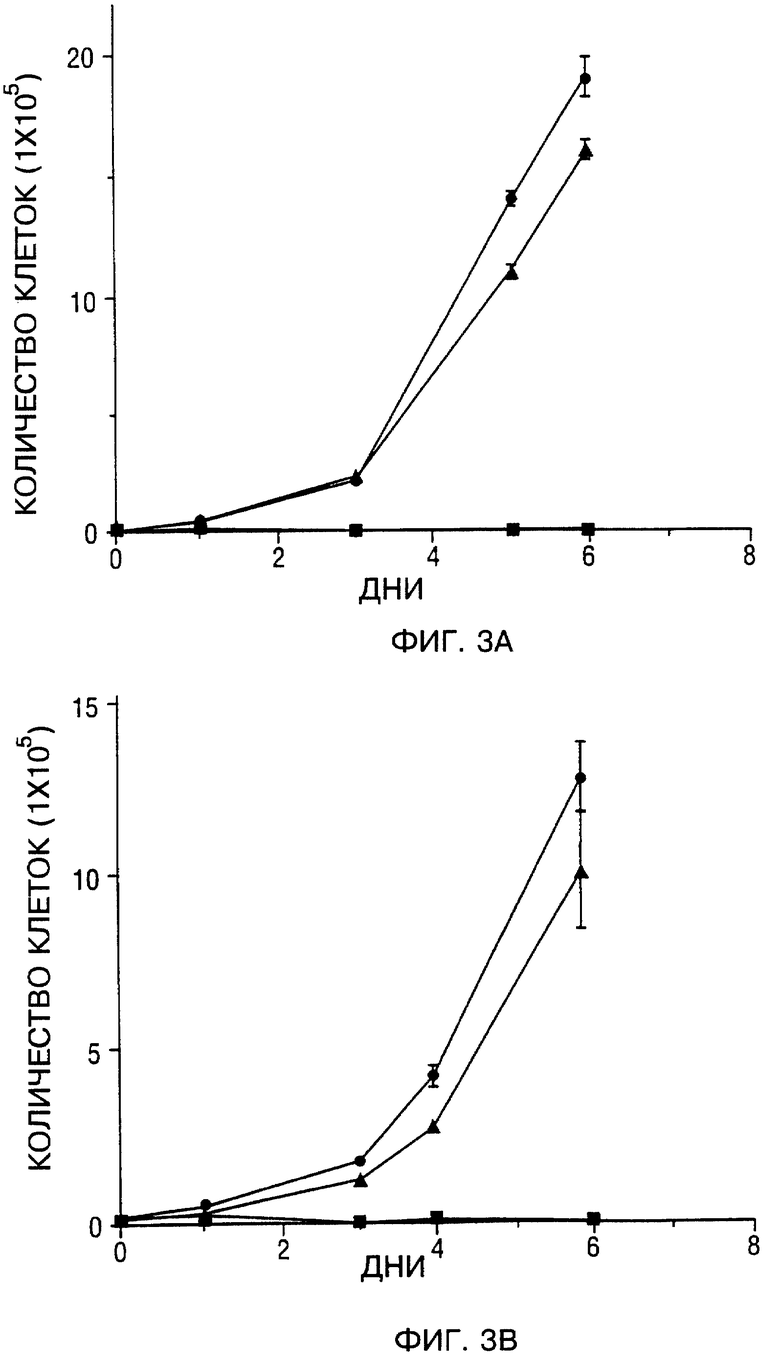


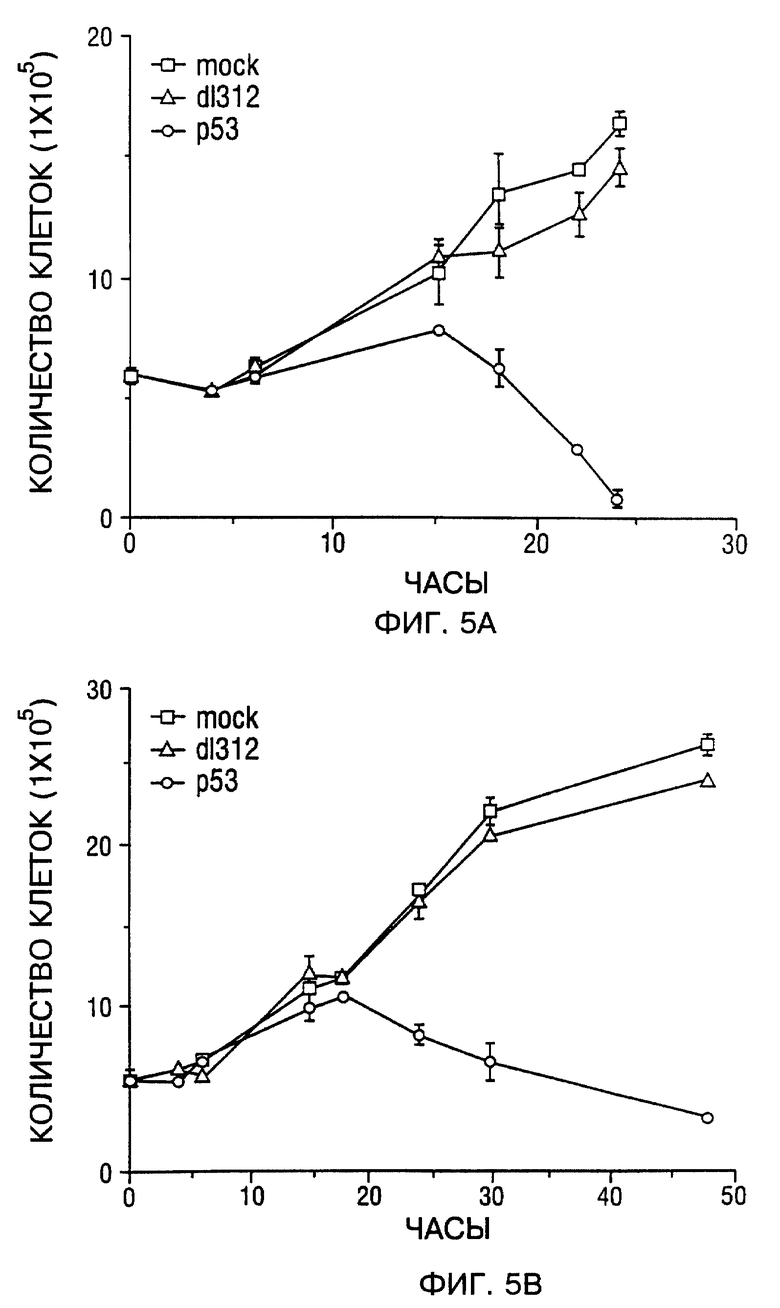
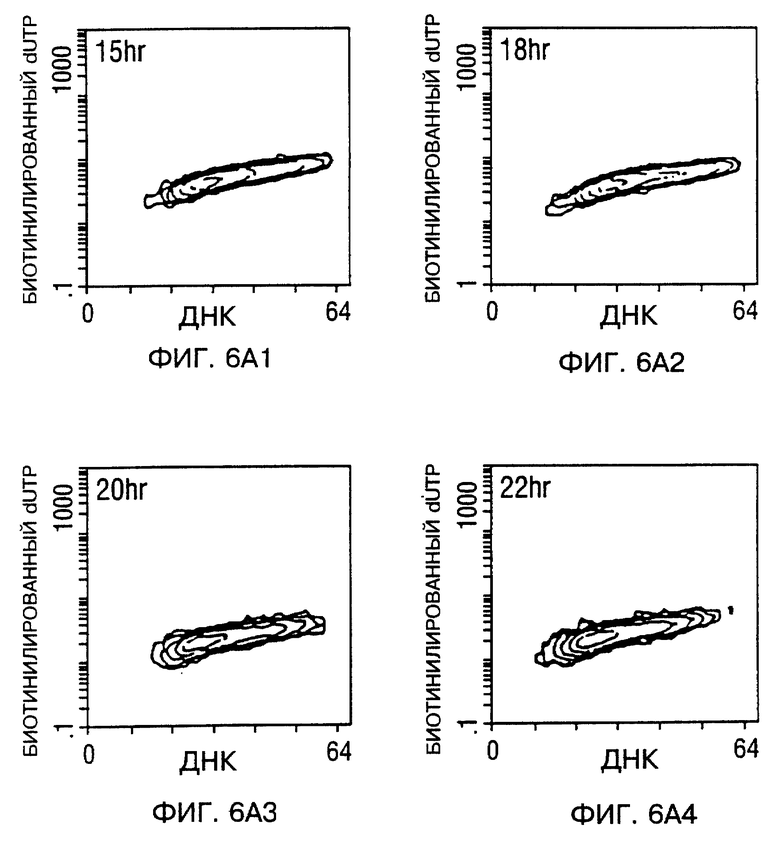
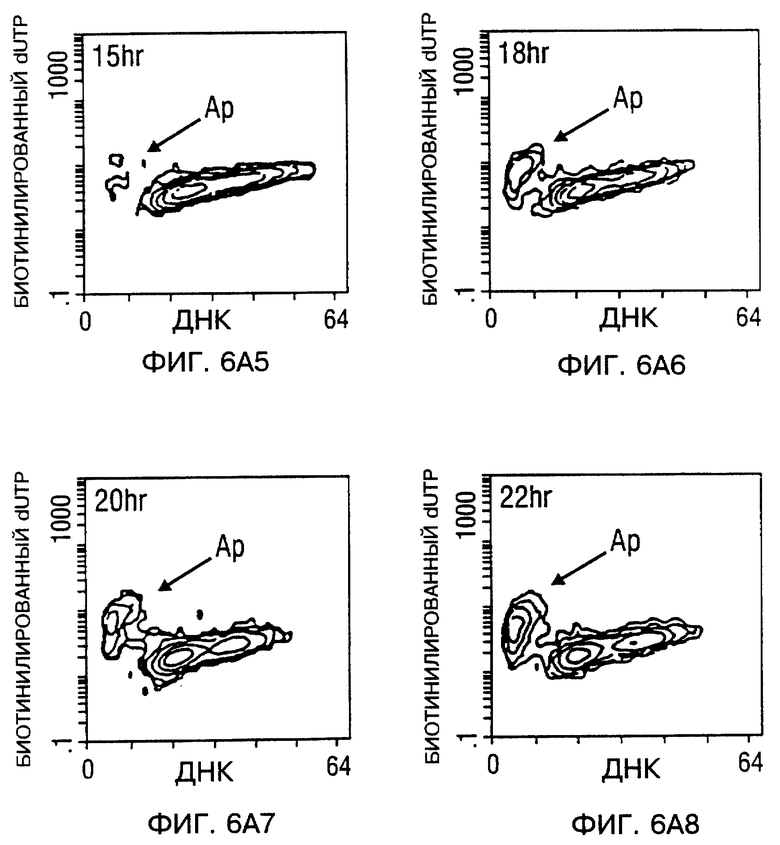
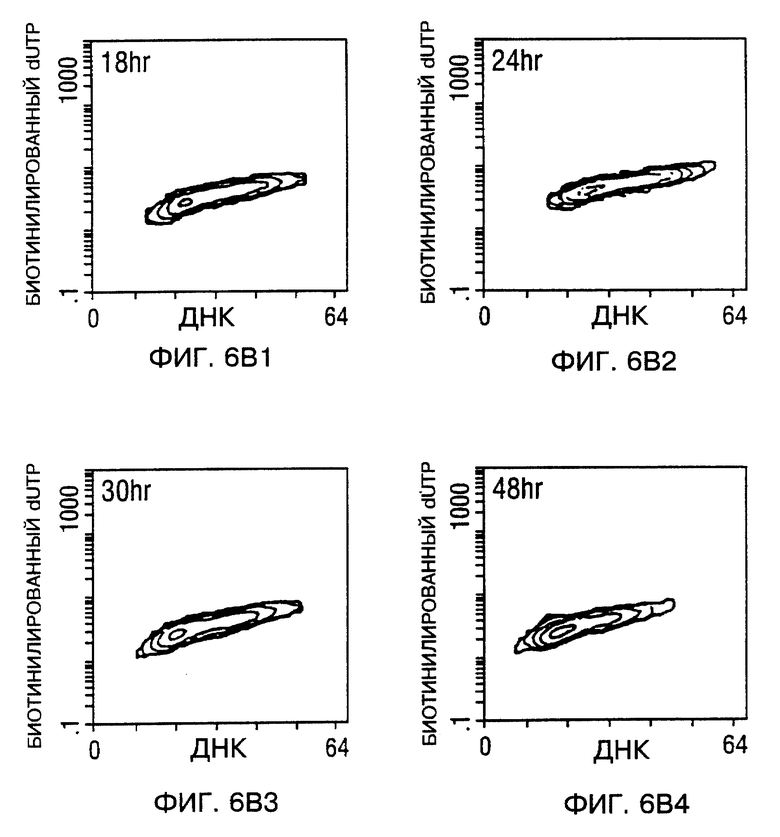
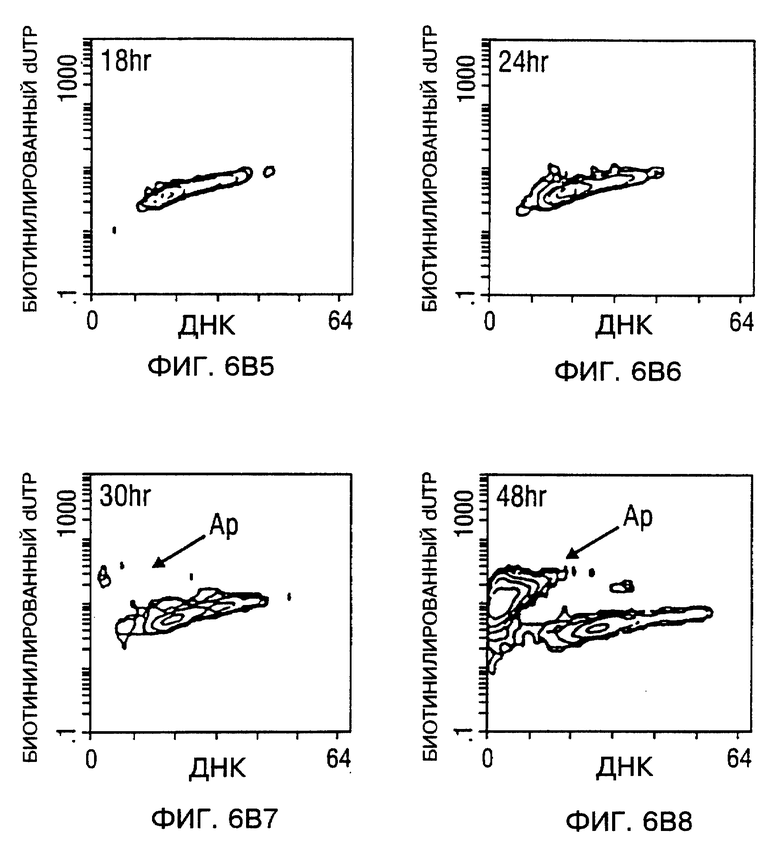
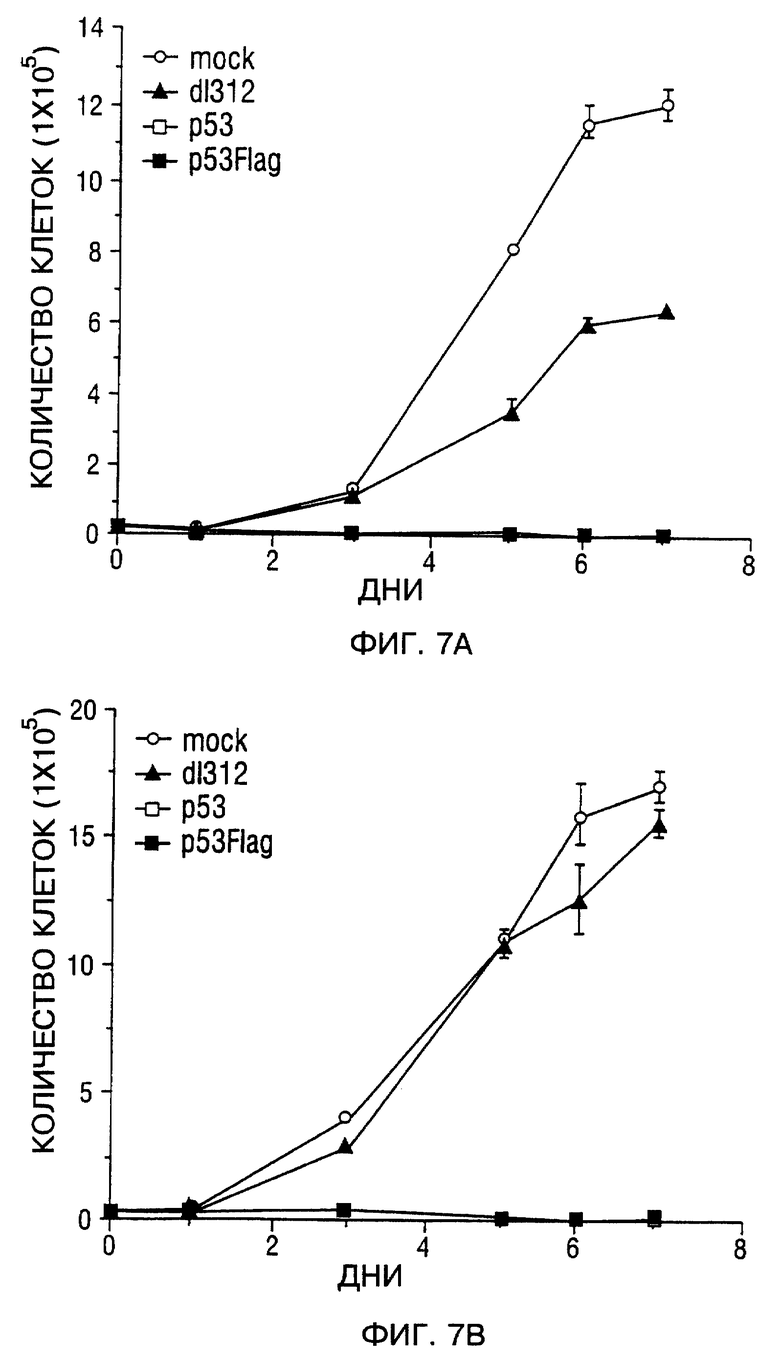
Изобретение относится к области медицины, в частности к биологии опухоли, а именно к способам и композициям лечения сквамозной (чешуйчатой) клеточной карциномы. Обеспечивается также животная модель для изучения микроскопических остаточных опухолей и опухолевого обсеменения в полостях тела, а также способы их лечения. Сущность изобретения: разработана экспрессионная конструкция, содержащая промотор, функциональный в эукариотических клетках, и полинуклеотид, кодирующий р53, причем полинуклеотид расположен в смысловой ориентации к промотору. Техническим результатом изобретения является расширение арсенала средств против раковых опухолей. 2 с. и 41 з.п. ф-лы, 7 табл., 28 ил.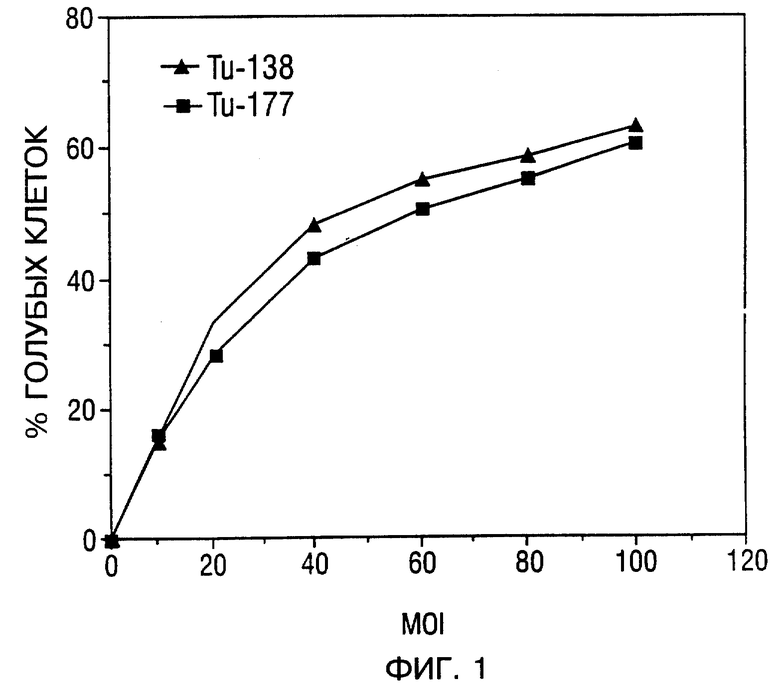
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Petty R | |||
| et al | |||
| Веникодробильный станок | 1921 |
|
SU53A1 |
| Biochemical and Biophys | |||
| Res, 199(1), 1994, 264-270. | |||
