Настоящее изобретение относится к классу комплексных соединений металлов группы 4 и к полученным из них катализаторам полимеризации олефинов, особенно пригодным для применения в процессах полимеризации для получения полимеров путем полимеризации α-олефинов и смесей α-олефинов.
Металлокомплексы ограниченной геометрии и способы их получения раскрыты в EP-A-416815, EP-A-468651, EP-A-514828, EP-A-520732 и WO 93/19104, а также US-A-5055438, US-A-5057475, US-A-5096867, US-A-5064802, US-A-5132380 и WO 95/00526.
В соответствии с настоящим изобретением предлагаются металлокомплексы, соответствующие формуле:  ,
,
где:
М представляет титан, цирконий или гафний в формальной степени окисления +2, +3 или +4;
A' представляет замещенную инденильную группу, замещенную по крайней мере в положении 2 или 3 группой, выбранной из гидрокарбила, фторзамещенного гидрокарбила, гидроксикарбилоксизамещенного гидрокарбила, диалкиламинозамещенного гидрокарбила, силила, гермила и их смесей, причем указанная группа содержит до 40 неводородных атомов, а указанная группа A' дополнительно ковалентно связана с
М посредством двухвалентной группы Z;
Z представляет двухвалентный фрагмент, связанный и с A' и с М посредством σ-связей, причем Z включает бор или элемент группы 14 Периодической таблицы элементов, а также азот, фосфор, серу или кислород;
X представляет анионную или дианионную лигандную группу, имеющую до 60 атомов, исключая класс лигандов, представляющих собой циклические делокализованные π-связанные лигандные группы;
X' в каждом случае появления представляет связывающее нейтральное основание Льюиса, имеющее до 20 атомов;
P представляет 0, 1 или 2, причем значение его на два меньше, чем значение формальной степени окисления металла М, при условии, что, когда X представляет дианионную лигандную группу, то p = 1; и
q представляет 0, 1 или 2.
Вышеописанные комплексы могут существовать в виде отдельных кристаллов, необязательно в чистой форме, или в виде смеси с другими комплексами, в форме сольватированного аддукта, необязательно в растворителе, особенно органической жидкости, а также в форме димера или его хелатного производного, в котором хелатообразующим агентом является органический материал, предпочтительно нейтральное основание Льюиса, в частности тригидрокарбиламин, тригидрокарбилфосфин или его галогенированное производное.
В соответствии с настоящим изобретением предлагается также способ получения полимеров из олефиновых мономеров, включающий контактирование одного или нескольких таких мономеров с катализатором, содержащим:
1) металлокомплекс, соответствующий формуле:  , где:
, где:
М представляет титан, цирконий или гафний в формальной степени окисления +2, +3 или +4;
A' представляет замещенную инденильную группу, замещенную по крайней мере в положении 2 или 3 группой, выбранной из гидрокарбила, фторзамещенного гидрокарбила, гидроксикарбилоксизамещенного гидрокарбила, диалкиламинозамещенного гидрокарбила, силила, гермила и их смесей, причем указанная группа содержит до 40 неводородных атомов, а указанная группа A' дополнительно ковалентно связана с М посредством двухвалентной группы Z;
Z представляет двухвалентный фрагмент, связанный и с A', и с М посредством σ-связей, причем Z содержит бор или элемент группы 14 Периодической таблицы элементов, а также азот, фосфор, серу или кислород;
X представляет анионную или дианионную лигандную группу, имеющую до 60 атомов, исключая класс лигандов, представляющих собой циклические делокализованные π- связанные лигандные группы;
X' в каждом случае появления представляет связывающее нейтральное основание Льюиса соединение, имеющее до 20 атомов;
p представляет 0, 1 или 2, причем значение его на два меньше, чем значение формальной степени окисления металла М, при условии, что, когда X представляет дианионную лигандную группу, то p = 1; и
q представляет 0, 1 или 2; и
2) активирующий сокатализатор при молярном отношении 1) к 2); составляющим от 1:10000 до 100:1, или продукт реакции, образованный путем превращения 1) в активный катализатор с использованием технологии активации.
Предлагаемые катализаторы и способ обеспечивают высокоэффективное производство высокомолекулярных олефиновых полимеров в широких пределах условий полимеризации, в частности при повышенных температурах. Они особенно пригодны для полимеризации в растворе этилена и пропилена (ЭП полимеры) и этилена, пропилена и диена (ЭПДМ полимеры), где диен представляет собой этилиденнорборнен, 1,4-гексадиен или подобный неконъюгированный (с несопряженными двойными связями) диен. Использование повышенных температур очень сильно повышает производительность такого процесса вследствие того, что повышенная растворимость полимера при повышенных температурах позволяет использовать повышенные степени конверсии (более высокая концентрация полимерного продукта) в рамках ограничений, налагаемых на оборудование для полимеризации в отношении вязкости раствора, и снижает затраты на энергию, необходимую на обезгаживание продукта реакции.
Все ссылки в данном описании на Периодическую таблицу элементов относятся к Периодической таблице элементов, опубликованной с предупреждением об авторском праве издательством CRC Press, Inc., 1989. Кроме того, всякая ссылка на группу или группы подразумевает группу или группы в представленном в этой таблице виде с использованием системы ИЮПАК (IUPAC) для нумерации групп.
Олефины, о которых идет речь в данном описании, представляют собой C2-20 алифатические или ароматические соединения с винильной ненасыщенностью, а также циклические соединения, такие, как циклобутен, циклопентен и норборнен, включая норборнен, замещенный в положениях 5 и 6 C1-20 гидрокарбильными группами. Включены также смеси таких олефинов, а также смеси таких олефинов с C4-40 диолефинами. Примеры последних соединений включают этилиденнорборнен, 1,4-гексадиен, норборнадиен и тому подобное. Предлагаемые в данном описании катализаторы и способ особенно пригодны для получения сополимеров этилена и 1-бутена, этилена и 1-гексена, этилена и стирола и этилена и 1-октена, а также тройных сополимеров этилена, пропилена и неконъюгированного диена, т. е. терполимеров ЭПДМ.
Предпочтительными группами X' являются моноксид углерода; фосфины, в частности триметилфосфин, триэтилфосфин, трифенилфосфин и бис (1,2-диметилфосфино)этан; P(OR)3, где R - такой, как определенный выше; простые эфиры, в частности тетрагидрофуран; амины, в частности пиридин, бипиридин, тетраметилэтилендиамин (ТМЭДА, TMEDA) и триэтиламин; олефины; и конъюгированные диены, имеющие от 4 до 40 углеродных атомов. Комплексы, содержащие последние группы X', включают те, в которых металл находится в формальной степени окисления +2.
Предпочтительными координационными комплексами по настоящему изобретению являются комплексы, соответствующие формуле:
где:
R1 и R2 независимо представляют группы, выбранные из водорода, гидрокарбила, перфторзамещенного гидрокарбила, силила, гермила и их смесей, причем указанные группы содержат до 20 неводородных атомов, при условии, что по крайней мере один из R1 или R2 не является водородом;
R3, R4, R5 и R6 независимо представляют группы, выбранные из водорода, гидрокарбила, перфторзамещенного гидрокарбила, силила, гермила и их смесей, причем указанные группы содержат до 20 неводородных атомов;
М представляет титан, цирконий или гафний;
Z представляет двухвалентный фрагмент, содержащий бор или элемент группы 14 Периодической таблицы элементов, а также содержащий азот, фосфор, серу или кислород, причем указанный фрагмент имеет до 60 неводородных атомов;
p представляет 0, 1 или 2;
q представляет ноль или единицу; при условии, что: когда р = 2, q = 0, М находится в формальной степени окисления +4 и X представляет анионный лиганд, выбранный из группы, состоящей из галогенида, гидрокарбила, гидрокарбилокси, ди (гидрокарбил)амидо, ди(гидрокарбил)фосфидо, гидрокарбилсульфидо и силила, а также их галоген-, ди-(гидрокарбил)амино-, гидрокарбилокси- и ди(гидрокарбил)фосфинозамещенных производных; причем указанная группа X имеет до 20 неводородных атомов,
когда р = 1, q = 0, М находится в формальной степени окисления +3 и X представляет стабилизирующий анионный лиганд, выбранный из группы, состоящей из аллила, 2-(N, N-диметиламинометил)фенила и 2-(N,N-диметил)аминобензила, или М находится в формальной степени окисления +4 и X представляет двухвалентное производное конъюгированного диена, причем М и X вместе образуют металлоциклопентановую группу, и
когда р = 0, q = 1, М находится в формальной степени окисления +2 и X' представляет нейтральный конъюгированный или неконъюгированный диен, необязательно замещенный одной или несколькими гидрокарбильными группами, причем указанная группа X' имеет до 40 углеродных атомов и образует α-комплекс с М.
Более предпочтительными комплексами по настоящему изобретению являются комплексы, соответствующие формуле:
где:
R1 и R2 представляют водород или C1-6 алкил, при условии, что по крайней мере один из R1, или R2 не является водородом;
R3, R4, R5 и R6 независимо представляют водород или C1-6 алкил;
М представляет титан;
Y представляет -О-, -S-, -NR* -, - PR*-;
Z* представляет SiR* 2, CR* 2, SiR* 2SiR* 2, CR* 2=CR* 2, CR*=CR*, CR* 2SiR* 2 или GeR* 2;
R* в каждом случае независимо представляет водород или элемент, выбранный из гидрокарбила, гидрокарбилокси, силила, галогенированного алкила, галогенированного арила и их сочетаний, причем указанная группа R* имеет до 20 неводородных атома и, хотя это и не обязательно, две группы R* из Z (когда R* не является водородом) или группа R* из Z и группа R* из Y образуют кольцевую систему;
p представляет 0, 1 или 2;
q представляет ноль или единицу; при условии, что:
когда р = 2, q = 0, М находится в формальной степени окисления +4 и X в каждом случае независимо представляет метил или бензил,
когда р = 1, q = 0, М находится в формальной степени окисления +3 и X представляет 2-(N,N-диметил)аминобензил или М находится в формальной степени окисления +4 и X представляет 1,4-бутадиенил, и
когда p = 0, q = 1, М находится в формальной степени окисления +2 и X' представляет 1,4-дифенил-1,3-бутадиен или 1,3-пентадиен. Последний диен является иллюстрацией несимметрической диеновой группы, приводящей к получению металлокомплексов, фактически являющихся смесями соответственных геометрических изомеров.
Наиболее предпочтительными металлокомплексами являются;
2-метилинденильные комплексы:
(трет-бутиламидо)диметил (η5 -2-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)диметил( η5 -2-метилинденил)силантитан(II) 1,3-пентадиен,
(трет-бутиламидо)диметил( η5 -2-метилинденил)силантитан(III) 2-(N,N-диметиламино)бензил,
(трет-бутиламидо)диметил( η5 -2-метилинденил)силантитан(IV) диметил,
(трет-бутиламидо)диметил( η5 -2-метилинденил)силантитан(IV) дибензил,
(н-бутиламидо)диметил( η5 -2-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диметил( η5 -2-метилинденил)силантитан(II)1,3-пентадиен,
(н-бутиламидо)диметил( η5 -2-метилинденил)силантитан(III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)диметил( η5 -2-метилинденил)силантитан(IV) диметил,
(н-бутиламидо)диметил( η5 -2-метилинденил)силантитан(IV) дибензил,
(циклододециламидо)диметил( η5 2-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диметил( η5 -2-метилинденил)силантитан(II) 1,3-пентадиен,
(циклододециламидо)диметил( η5 -2-метилинденил)силантитан(III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)диметил( η5 -2-метилинденил)силантитан(IV) диметил,
(циклододециламидо)диметил( η5 -2-метилинденил)силантитан(IV) дибензил,
2,4,6-триметиланилидо)диметил(η5 -2-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диметил(η5 -2-метилинденил) силантитан(II)1,3-пентадиен,
(2,4,6-триметиланилидо)диметил(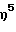 -2-метилинденил)силантитан(III) 2-(N, N-диметиламино)бензил,
-2-метилинденил)силантитан(III) 2-(N, N-диметиламино)бензил,
(2,4,6-триметиланилидо)диметил(η5 -2-метилинденил) силантитан(IV)диметил,
(2,4,6-триметиланилидо)диметил(η5 -2-метилинденил)силантитан(IV) дибензил,
(1-адамантиламидо)диметил( η5 -2-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диметил( η5 -2-метилинденил)силантитан(II) 1,3-пентадиен,
(1-адамантиламидо)диметил( η5 -2-метилинденил)силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диметил( η5 -2-метилинденил)силантитан(IV) диметил,
(1-адамантиламидо)диметил( η5 -2-метилинденил)силантитан(IV) дибензил,
(н-бутиламидо)диизопропокси( η5 -2-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо) диизопропокси( η5 -2-метилинденил) силантитан(II)1,3-пентадиен,
(н-бутиламидо)диизопропокси( η5 -2-метилинденил) силантитан(III) 2-(N, N-диметиламино)бензил,
(н-бутиламидо)диизопропокси( η5 -2-метилинденил) силантитан(IV)диметил,
(н-бутиламидо)диизопропокси( η5 -2-метилинденил) силантитан(IV)дибензил,
(циклододециламидо)диизопропокси( η5 -2-метилинденил)- силантитан(II) 1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диизопропокси( η5 -2-метилинденил)-силантитан(II) 1,3-пентадиен,
(циклододециламидо)диизопропокси( η5 -2-метилинденил)- силантитан(III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)диизопропокси( η5 -2-метилинденил)-силантитан(IV)диметил,
(циклододециламидо)диизопропокси( η5 -2-метилинденил)- силантитан(IV)дибензил,
(2,4,6-триметиланилидо)диизопропокси( η5 -2-метилинденил)- силантитан(II) 1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диизопропокси(η5 -2-метилинденил)-силантитан(II) 1,3-пентадиен,
(2,4,6-триметиланилидо)диизопропокси( η5 -2-метилинденил)- силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диизопропокси( η5-метилинденил)- силантитан(IV)диметил,
(2,4,6-триметиланилидо)диизопропокси(η5 -2-метилинденил)-силантитан(IV) дибензил,
(1-адамантиламидо)диизопропокси( η5 -2-метилинденил)- силантитан(II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диизопропокси( η5 -2-метилинденил)-силантитан(II) 1,3-пентадиен,
(1-адамантиламидо)диизопропокси( η5 -2-метилинденил)- силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диизопропокси( η5-2-метилинденил)-силантитан(IV) диметил,
(1-адамантиламидо)диизопропокси( η5 -2-метилинденил)-силантитан(IV) дибензил,
(н-бутиламидо)диметокси( η5 -2-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диметокси( η5 -2-метилинденил)силантитан(II) 1,3-пентадиен,
(н-бутиламидо)диметокси( η5 -2-метилинденил)силантитан(III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)диметокси( η5 -2-метилинденил) силантитан(IV) диметил,
(н-бутиламидо)диметокси( η5 -2-метилинденил)силантитан(IV) дибензил,
(циклододециламидо)диметокси( η5 -2-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диметокси( η5 -2-метилинденил) силантитан(II) 1,3-пентадиен,
(циклододециламидо)диметокси( η5 -2-метилинденил) силантитан(III) 2-(N, N-диметиламино)бензил,
(циклододециламидо)диметокси( η5 -2-метилинденил) силантитан(IV) диметил,
(циклододециламидо)диметокси( 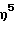 -2-метилинденил) силантитан(IV) дибензил,
-2-метилинденил) силантитан(IV) дибензил,
(2,4,6-триметиланилидо)диметокси(η5 -2-метилинденил)- силантитан(II) 1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диметокси(η5 -2-метилинденил)-силантитан(II) 1,3-пентадиен,
(2,4,6-триметиланилидо)диметокси(η5 -2-метилинденил) -силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диметокси(η5 -2-метилинденил)- силантитан(IV) диметил,
(2,4,6-триметиланилидо)диметокси(η5 -2-метилинденил)- силантитан(IV) дибензил,
(1-адамантиламидо)диметокси( η5 -2-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диметокси( η5 -2-метилинденил)- силантитан(II) 1,3-пентадиен,
(1-адамантиламидо)диметокси( η5 -2-метилинденил)силантитан(III) 2-(N, N-диметиламино) бензил,
(1-адамантиламидо)диметокси( η5 -2-метилинденил)силантитан(IV) диметил,
(1-адамантиламидо)диметокси( η5 -2-метилинденил)силантитан(IV) дибензил,
(н-бутиламидо)этоксиметил( η5 -2-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)этоксиметил( η5 -2-метилинденил)силантитан(II) 1,3-пентадиен,
(н-бутиламидо)этоксиметил( η5 -2-метилинденил)силантитан(III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)этоксиметил( η5 -2-метилинденил)силантитан(IV) диметил,
(н-бутиламидо)этоксиметил( η5 -2-метилинденил)силантитан(IV) дибензил,
(циклододециламидо)этоксиметил( η5 -2-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(циклододециламидо)этоксиметил( η5 -2-метилинденил)силантитан(II) 1,3-пентадиен,
(циклододециламидо)этоксиметил( η5 -2-метилинденил)силантитан(III) 2-(N, N-диметиламино)бензил,
(циклододециламидо)этоксиметил( η5 -2-метилинденил)силантитан (IV)диметил,
(циклододециламидо)этоксиметил( η5 -2-метилинденил)силантитан(IV) дибензил,
(2,4,6-триметиланилидо)этоксиметил( η5 -2-метилинденил)-силантитан(II) 1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)этоксиметил( η5 -2-метилинденил)-силантитан(II) 1,3-пентадиен,
(2,4,6-триметиланилидо)этоксиметил( η5 -2-метилинденил)-силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)этоксиметил( η5 -2-метилинденил)-силантитан(IV) диметил,
(2,4,6-триметиланилидо)этоксиметил( η5 -2-метилинденил)-силантитан(IV) дибензил,
(1-адамантиламидо)этоксиметил( η5 -2-метилинденил)-силантитан(II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)этоксиметил( η5 -2-метилинденил)-силантитан(II) 1,3-пентадиен,
(1-адамантиламидо)этоксиметил( η5 -2-метилинденил)-силантитан(III) 2-(N, N-диметиламино)бензил,
(1-адамантиламидо)этоксиметил( η5 -2-метилинденил)-силантитан(IV) диметил,
(1-адамантиламидо)этоксиметил( η5 -2-метилинденил)- силантитан(IV)дибензил,
2,3-диметилинденильные комплексы:
(трет-бутиламидо)диметил( η5 -2,3-диметилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)диметил(( η5 -2,3-диметилинденил)силантитан(II) 1,3-пентадиен,
(трет-бутиламидо)диметил( η5 -2,3-диметилинденил)силантитан (III) 2-(N, N-диметиламино)бензил,
(трет-бутиламидо)диметил( η5 -2,3-диметилинденил)силантитан(IV) диметил,
(трет-бутиламидо)диметил( η5 -2,3-диметилинденил)силантитан(IV) дибензил,
(н-бутиламидо)диметил( η5 -2,3-диметилинденил)силантитан (II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диметил( η5 -2,3-диметилинденил)силантитан(II) 1,3-пентадиен,
(н-бутиламидо)диметил( η5 -2,3-диметилинденил)силантитан(III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)диметил( η5 -2,3-диметилинденил)силантитан(IV) диметил,
(н-бутиламидо)диметил( η5 -2,3-диметилинденил)силантитан(IV) дибензил,
(циклододециламидо)диметил( η5 -2,3-диметилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диметил( η5 -2,3-диметилинденил)силантитан (II) 1,3-пентадиен,
(циклододециламидо)диметил( η5 -2,3-диметилинденил)силантитан(III) 2-(N, N-диметиламино)бензил,
(циклододециламидо)диметил( η5 -2,3-диметилинденил)силантитан(IV) диметил,
(циклододециламидо)диметил( η5 -2,3-диметилинденил)силантитан(IV) дибензил,
(2,4,6-триметиланилидо)диметил( η5 -2,3-диметилинденил)-силантитан(II) 1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диметил( η5 -2,3-диметилинденил)-силантитан(II) 1,3-пентадиен,
(2,4,6-триметиланилидо)диметил( η5 -2,3-диметилинденил)-силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диметил( 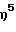 -2,3-диметилинденил)-силантитан(IV) диметил,
-2,3-диметилинденил)-силантитан(IV) диметил,
(2,4,6-триметиланилидо)диметил( η5 -2,3-диметилинденил)-силантитан(IV) дибензил,
(1-адамантиламидо)диметил( η5 -2,3-диметилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диметил( η5 -2,3-диметилинденил)силантитан(II) 1,3-пентадиен,
(1-адамантиламидо)диметил( η5 -2,3-диметилинденил)силантитан(III) 2-(N, N-диметиламино)бензил,
(1-адамантиламидо)диметил( η5 -2,3-диметилинденил)силантитан(IV) диметил,
(1-адамантиламидо)диметил( η5 -2,3-диметилинденил)силантитан(IV) дибензил,
(н-бутиламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан(II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан(II) 1,3-пентадиен,
(н-бутиламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан(III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан(IV) диметил,
(н-бутиламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан (IV) дибензил,
(циклододециламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан(II) 1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан (II) 1,3-пентадиен,
(циклододециламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан (III) 2-(N,N-диметиламино) бензил,
(циклододециламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан (IV)диметил,
(циклододециламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан(IV) дибензил,
(2,4,6-триметиланилидо)диизопропокси(η5 -2,3-диметилинденил)-силантитан(II) 1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диизопропокси(η5 -2,3-диметилинденил)силантитан (II) 1,3-пентадиен,
(2,4,6-триметиланилидо)диизопропокси(η5 -2,3-диметилинденил)силантитан (III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диизопропокси(η5 -2,3-диметилинденил)силантитан (IV) диметил,
(2,4,6-триметиланилидо)диизопропокси(η5 -2,3-диметилинденил)силантитан (IV) дибензил,
(1-адамантиламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан (II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан(II) 1,3-пентадиен,
(1-адамантиламидо)диизопропокси( η5 -2,3-диметилинденил)силантитан (III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан(IV) диметил,
(1-адамантиламидо)диизопропокси( η5 -2,3-диметилинденил)-силантитан(IV) дибензил,
(н-бутиламидо)диметокси( η5 -2,3-диметилинденил)-силантитан (II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диметокси( η5 -2,3-диметилинденил)-силантитан(II) 1,3-пентадиен,
(н-бутиламидо)диметокси( η5 -2,3-диметилинденил)-силантитан (III) 2-(N, N-диметиламино)бензил,
(н-бутиламидо)диметокси( η5 -2,3-диметилинденил)- силантитан(IV) диметил,
(н-бутиламидо)диметокси( η5 -2,3-диметилинденил)-силантитан(IV) дибензил,
(циклододециламидо)диметокси( η5 -2,3-диметилинденил)-силантитан(II) 1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диметокси( η5 -2,3-диметилинденил)-силантитан(II) 1,3-пентадиен,
(циклододециламидо)диметокси( η5 -2,3-диметилинденил)-силантитан(III) 2-(N,N-диметиламино) бензил,
(циклододециламидо)диметокси( η5 -2,3-диметилинденил)-силантитан(IV) диметил,
(циклододециламидо)диметокси( η5 -2,3-диметилинденил)-силантитан(IV) дибензил,
(2,4,6-триметиланилидо)диметокси( η5 -2,3-диметилинденил)силантитан (II) 1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диметокси( η5 -2,3-диметилинденил)- силантитан(II) 1,3-пентадиен,
(2,4,6-триметиланилидо)диметокси( η5 -2,3-диметилинденил)-силантитан (III) 2-(N,N-диметиламино) бензил,
(2,4,6-триметиланилидо)диметокси( η5 -2,3-диметилинденил)- силантитан (IV) диметил,
(2,4,6-триметиланилидо)диметокси( η5 -2,3-диметилинденил)- силантитан(IV) дибензил,
(1-адамантиламидо)диметокси( η5 -2,3-диметилинденил) силантитан(II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диметокси( η5 -2,3-диметилинденил)-силантитан(II) 1,3-пентадиен,
(1-адамантиламидо)диметокси( η5 -2,3-диметилинденил)-силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диметокси( η5 -2,3-диметилинденил)-силантитан (IV)диметил,
(1-адамантиламидо)диметокси( η5 -2,3-диметилинденил)-силантитан(IV) дибензил,
(н-бутиламидо)этоксиметил( η5 -2,3-диметилинденил)-силантитан(II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)этоксиметил( η5 -2,3-диметилинденил)-силантитан(II) 1,3-пентадиен,
(н-бутиламидо)этоксиметил( η5 -2,3-диметилинденил)-силантитан(III) 2-(N, N-диметиламино)бензил,
(н-бутиламидо)этоксиметил( η5 -2,3-диметилинденил)-силантитан(IV) диметил,
(н-бутиламидо)этоксиметил( η5 -2,3-диметилинденил)-силантитан(IV)дибензил,
(циклододециламидо)этоксиметил( η5 -2,3-диметилинденил)- силантитан(II) 1,4-дифенил-1,3-бутадиен,
(циклододециламидо)этоксиметил( η5 -2,3-диметилинденил) силантитан(II) 1,3-пентадиен,
(циклододециламидо)этоксиметил( η5 -2,3-диметилинденил)силантитан (III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)этоксиметил( η5 -2,3-диметилинденил)-силантитан(IV) диметил,
(циклододециламидо)этоксиметил( η5 -2,3-диметилинденил)силантитан(IV) дибензил,
(2,4,6-триметиланилидо)этоксиметил( η5 -2,3-диметилинденил)силантитан (II) 1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)этоксиметил( η5 -2,3-диметилинденил) силантитан (II) 1,3-пентадиен,
(2,4,6-триметиланилидо)этоксиметил( η5 -2,3-диметилинденил) силантитан (III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)этоксиметил( η5 -2,3-диметилинденил) силантитан(IV)диметил,
(2,4,6-триметиланилидо)этоксиметил( η5 -2,3-диметилинденил) силантитан(IV)дибензил,
(1-адамантиламидо)этоксиметил( η5 -2,3-диметилинденил)- силантитан (II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)этоксиметил( η5 -2,3-диметилинденил)-силантитан(II) 1,3-пентадиен,
(1-адамантиламидо)этоксиметил( η5 -2,3-диметилинденил)- силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)этоксиметил( η5 -2,3-диметилинденил)- силантитан(IV)диметил,
(1-адамантиламидо)этоксиметил( η5 -2,3-диметилинденил)-силантитан(IV) дибензил,
3-метилинденильные комплексы:
(трет-бутиламидо)диметил( η5 -3-метилинденил)силантитан (II) 1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)диметил( η5 -3-метилинденил)силантитан(II) 1,3-пентадиен,
(трет-бутиламидо)диметил( η5 -3-метилинденил)силантитан(III) 2-(N,N-диметиламино)бензил,
(трет-бутиламидо)диметил( η5 -3-метилинденил)силантитан(IV)диметил,
(трет-бутиламидо)диметил( η5 -3-метилинденил)силантитан(IV)дибензил,
(н-бутиламидо)диметил( η5 -3-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диметил( η5 -3-метилинденил)силантитан(II) 1,3-пентадиен,
(н-бутиламидо)диметил( η5 -3-метилинденил)силантитан(III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)диметил( 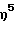 -3-метилинденил)силантитан(IV)диметил,
-3-метилинденил)силантитан(IV)диметил,
(н-бутиламидо)диметил( η5 -3-метилинденил)силантитан(IV)дибензил,
(циклододециламидо)диметил( η5 -3-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диметил( η5 -3-метилинденил)силантитан(II) 1,3-пентадиен,
(циклододециламидо)диметил( η5 -3-метилинденил)силантитан(III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)диметил( η5 -3-метилинденил)силантитан(IV) диметил,
(циклододециламидо)диметил( η5 -3-метилинденил)силантитан(IV) дибензил
(2,4,6-триметиланилидо)диметил(η5 -3-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диметил(η5 -3-метилинденил)-силантитан(II) 1,3-пентадиен,
(2,4,6-триметиланилидо)диметил(η5 -3-метилинденил)-силантитан (III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диметил(η5 -3-метилинденил)-силантитан(IV) диметил,
(2,4,6-триметиланилидо)диметил(η5 -3-метилинденил)- силантитан(IV)дибензил,
(1-адамантиламидо)диметил( η5 -3-метилинденил)силантитан (II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диметил( η5 -3-метилинденил)силантитан(II) 1,3-пентадиен,
(1-адамантиламидо)диметил( η5 -3-метилинденил)силантитан (III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диметил( η5 -3-метилинденил)силантитан(IV)диметил,
(1-адамантиламидо)диметил( η5 -3-метилинденил)силантитан(IV) дибензил,
(н-бутиламидо)диизопропокси( η5 -3-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диизопропокси( η5 -3-метилинденил)силантитан(II) 1,3-пентадиен,
(н-бутиламидо)диизопропокси( η5 -3-метилинденил)силантитан(III) 2-(N, N-диметиламино)бензил,
(н-бутиламидо)диизопропокси( η5 -3-метилинденил) силантитан(IV)диметил,
(н-бутиламидо)диизопропокси( η5 -3-метилинденил) силантитан(IV)дибензил,
(циклододециламидо)диизопропокси( η5 -3-метилинденил)- силантитан(II) 1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диизопропокси( η5 -3-метилинденил)-силантитан(II) 1,3-пентадиен,
(циклододециламидо)диизопропокси( η5 -3-метилинденил)- силантитан(III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)диизопропокси( η5 -3-метилинденил)-силантитан(IV) диметил,
(циклододециламидо)диизопропокси( η5 -3-метилинденил)-силантитан(IV) дибензил,
(2,4,6-триметиланилидо)диизопропокси(η5 -3-метилинденил)-силантитан (II) 1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диизопропокси(η5 -3-метилинденил)- силантитан(II)1,3-пентадиен,
(2,4,6-триметиланилидо)диизопропокси(η5 -3-метилинденил)- силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диизопропокси(η5 -3-метилинденил)-силантитан (IV)диметил,
(2,4,6-триметиланилидо)диизопропокси(η5 -3-метилинденил)-силантитан (IV)дибензил,
(1-адамантиламидо)диизопропокси( η5 -3-метилинденил)- силантитан(II)1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диизопропокси( η5 -3-метилинденил)-силантитан(II)1,3-пентадиен,
(1-адамантиламидо)диизопропокси( η5 -3-метилинденил) -силантитан (III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диизопропокси( η5 -3-метилинденил)-силантитан(IV)диметил,
(1-адамантиламидо)диизопропокси( η5 -3-метилинденил)-силантитан (IV)дибензил,
(н-бутиламидо)диметокси( η5 -3-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диметокси( η5 -3-метилинденил)силантитан(II) 1,3-пентадиен,
(н-бутиламидо)диметокси( η5 -3-метилинденил)силантитан (III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)диметокси( η5 -3-метилинденил)силантитан(IV)диметил,
(н-бутиламидо)диметокси( η5 -3-метилинденил)силантитан(IV)дибензил,
(циклододециламидо)диметокси( η5 -3-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диметокси( η5 -3-метилинденил) силантитан(II)1,3-пентадиен,
(циклододециламидо)диметокси( η5 -3-метилинденил)силантитан (III)2-(N, N-диметиламино)бензил,
(циклододециламидо)диметокси( η5 -3-метилинденил)силантитан (IV)диметил,
(циклододециламидо)диметокси( η5 -3-метилинденил)силантитан(IV)дибензил,
(2,4,6-триметиланилидо)диметокси( η5 -3-метилинденил)-силантитан (II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диметокси( η5 -3-метилинденил)-силантитан(II) 1,3-пентадиен,
(2,4,6-триметиланилидо)диметокси( η5 -3-метилинденил)-силантитан (III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диметокси( η5 -3-метилинденил)- силантитан(IV)диметил,
(2,4,6-триметиланилидо)диметокси( η5 -3-метилинденил)-силантитан (IV)дибензил,
(1-адамантиламидо)диметокси( η5 -3-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диметокси( η5 -3-метилинденил)силантитан(II) 1,3-пентадиен,
(1-адамантиламидо)диметокси( η5 -3-метилинденил)силантитан(III) 2-(N, N-диметиламино) бензил,
(1-адамантиламидо)диметокси( η5 -3-метилинденил)силантитан(IV) диметил,
(1-адамантиламидо)диметокси( η5 -3-метилинденил)силантитан(IV) дибензил,
(н-бутиламидо)этоксиметил( η5 -3-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)этоксиметил( η5 -3-метилинденил)силантитан(II) 1,3-пентадиен,
(н-бутиламидо)этоксиметил( η5 -3-метилинденил)силантитан(III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)этоксиметил( η5 -3-метилинденил)силантитан(IV) диметил,
(н-бутиламидо)этоксиметил( η5 -3-метилинденил)силантитан(IV)дибензил,
(циклододециламидо)этоксиметил( η5 -3-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(циклододециламидо)этоксиметил( η5 -3-метилинденил)силантитан(II) 1,3-пентадиен,
(циклододециламидо)этоксиметил( η5 -3-метилинденил)силантитан (III)2-(N, N-диметиламино)бензил,
(циклододециламидо)этоксиметил( η5 -3-метилинденил)силантитан(IV) диметил,
(циклододециламидо)этоксиметил( η5 -3-метилинденил)силантитан(IV) дибензил,
(2,4,6-триметиланилидо)этоксиметил( η5 -3-метилинденил)- силантитан(II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)этоксиметил( η5 -3-метилинденил)-силантитан(II) 1,3-пентадиен,
(2,4,6-триметиланилидо)этоксиметил( η5 -3-метилинденил)-силантитан (III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)этоксиметил( η5 -3-метилинденил)-силантитан(IV) диметил,
(2,4,6-триметиланилидо)этоксиметил( η5 -3-метилинденил)-силантитан(IV) дибензил,
(1-адамантиламидо)этоксиметил( η5 -3-метилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)этоксиметил( η5 -3-метилинденил)силантитан(II) 1,3-пентадиен,
(1-адамантиламидо)этоксиметил( η5 -3-метилинденил)силантитан (III) 2-(N, N-диметиламино)бензил,
(1-адамантиламидо)этоксиметил( 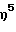 -3-метилинденил)силантитан(IV) диметил,
-3-метилинденил)силантитан(IV) диметил,
(1-адамантиламидо)этоксиметил( η5 -3-метилинденил)силантитан(IV) дибензил,
2-метил-3-этилинденильные комплексы:
(трет-бутиламидо)диметил( η5 -2-метил-3-этилинденил)-силантитан(II) 1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)диметил( η5 -2-метил-3-этилинденил)-силантитан(II) 1,3-пентадиен,
(трет-бутиламидо)диметил( η5 -2-метил-3-этилинденил)-силантитан(III) 2-(N,N-диметиламино)бензил,
(трет-бутиламидо)диметил( η5 -2-метил-3-этилинденил)-силантитан(IV) диметил,
(трет-бутиламидо)диметил( η5 -2-метил-3-этилинденил)-силантитан(IV) дибензил,
(н-бутиламидо)диметил( η5 -2-метил-3-этилинденил)силантитан(II) 1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диметил( η5 -2-метил-3-этилинденил)силантитан(II) 1,3-пентадиен,
(н-бутиламидо)диметил( η5 -2-метил-3-этилинденил)силантитан(III) 2-(N, N-диметиламино)бензил,
(н-бутиламидо)диметил( η5 -2-метил-3-этилинденил)силантитан (IV)диметил,
(н-бутиламидо)диметил( η5 -2-метил-3-этилинденил)силантитан(IV)дибензил,
(циклододециламидо)диметил( η5 -2-метил-3-этилинденил)-силантитан (II)1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диметил( η5 -2-метил-3-этилинденил)- силантитан(II)1,3-пентадиен,
(циклододециламидо)диметил( η5 -2-метил-3-этилинденил)- силантитан(III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)диметил( η5 -2-метил-3-этилинденил)- силантитан(IV)диметил,
(циклододециламидо)диметил( η5 -2-метил-3-этилинденил)-силантитан(IV) дибензил,
(2,4,6-триметиланилидо)диметил( η5 -2-метил-3-этилинденил)силантитан (II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диметил( η5 -2-метил-3-этилинденил)силантитан (II)1,3-пентадиен,
(2,4,6-триметиланилидо)диметил( η5 -2-метил-3-этилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диметил( η5 -2-метил-3-этилинденил)силантитан (IV)диметил,
(2,4,6-триметиланилидо)диметил( η5 -2-метил-3-этилинденил)силантитан (IV)дибензил,
(1-адамантиламидо)диметил( η5 -2-метил-3-этилинденил)- силантитан(II)1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диметил( η5 -2-метил-3-этилинденил) силантитан(II)1,3-пентадиен,
(1-адамантиламидо)диметил( η5 -2-метил-3-этилинденил)- силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диметил( η5 -2-метил-3-этилинденил)- силантитан(IV)диметил,
(1-адамантиламидо)диметил( η5 -2-метил-3-этилинденил)- силантитан(IV)дибензил,
(н-бутиламидо)диизопропокси( η5 -2-метил-3-этилинденил)- силантитан(II)1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диизопропокси( η5 -2-метил-3-этилинденил)- силантитан(II)1,3-пентадиен,
(н-бутиламидо)диизопропокси( η5 -2-метил-3-этилинденил)-силантитан(III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)диизопропокси( η5 -2-метил-3-этилинденил)- силантитан(IV)диметил,
(н-бутиламидо)диизопропокси( η5 -2-метил-3-этилинденил)- силантитан(IV)дибензил,
(циклододециламидо)диизопропокси( η5 -2-метил-3-этилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диизопропокси( η5 -2-метил-3-этилинденил) силантитан(II)1,3-пентадиен,
(циклододециламидо)диизопропокси( η5 -2-метил-3-этилинденил) силантитан(III)2-(N,N-диметиламино)бензил,
(циклододециламидо)диизопропокси( η5 -2-метил-3-этилинденил) силантитан(IV)диметил,
(циклододециламидо)диизопропокси( η5 -2-метил-3-этилинденил) силантитан(IV)дибензил,
(2,4,6-триметиланилидо)диизопропокси(η5 -2-метил-3- этилинденил)силантитан(II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диизопропокси(η5 -2-метил-3-этилинденил) силантитан(II)1,3-пентадиен,
(2,4,6-триметиланилидо)диизопропокси(η5 -метил-3-этилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диизопропокси(η5 -2-метил-3-этилинденил) силантитан(IV)диметил,
(2,4,6-триметиланилидо)диизопропокси(η5 -2-метил-3-этилинденил) силантитан(IV)дибензил,
(1-адамантиламидо)диизопропокси( η5-2-метил-3-этилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диизопропокси( η5 -2-метил-3-этилинденил) силантитан(II)1,3-пентадиен,
(1-адамантиламидо)диизопропокси( η5 -2-метил-3-этилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диизопропокси(η5 - 2-метил-3-этилинденил) силантитан(IV)диметил,
(1-адамантиламидо)диизопропокси( η5 -2- метил-3-этилинденил) силантитан(IV)дибензил,
(н-бутиламидо)диметокси( η5 -2-метил-3-этилинденил)-силантитан (II)1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диметокси( η5 -2-метил-3-этилинденил)-силантитан(II)1,3-пентадиен,
(н-бутиламидо)диметокси( η5 -2-метил-3-этилинденил)- силантитан(III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)диметокси( η5 -2-метил-3-этилинденил)-силантитан(IV) диметил,
(н-бутиламидо)диметокси( η5 -2-метил-3-этилинденил)-силантитан(IV) дибензил,
(циклододециламидо)диметокси( η5 -2-метил-3-этилинденил)- силантитан(II)1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диметокси( η5 -2-метил-3-этилинденил)-силантитан(II)1,3-пентадиен,
(циклододециламидо)диметокси( η5 -2-метил-3-этилинденил)-силантитан (III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)диметокси( η5 -2-метил-3-этилинденил)- силантитан(IV)диметил,
(циклододециламидо)диметокси( η5 -2-метил-3-этилинденил)-силантитан(IV) дибензил,
(2,4,6-триметиланилидо)диметокси( η5 - 2-метил-3-этилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диметокси( η5 -2-метил-3-этилинденил) силантитан(II)1,3-пентадиен,
(2,4,6-триметиланилидо)диметокси( η5 -2-метил-3-этилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диметокси( η5 -2-метил-3-этилинденил) силантитан(IV)диметил,
(2,4,6-триметиланилидо)диметокси( η5 -2-метил-3-этилинденил) силантитан(IV)дибензил,
(1-адамантиламидо)диметокси( η5 -2-метил-3-этилинденил)-силантитан (II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диметокси( η5 -2-метил-3-этилинденил)-силантитан(II)1,3-пентадиен,
(1-адамантиламидо)диметокси( η5 -2-метил-3-этилинденил)-силантитан (III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диметокси( η5 -2-метил-3-этилинденил)- силантитан(IV)диметил,
(1-адамантиламидо)диметокси( η5 -2-метил-3-этилинденил)-силантитан(IV) дибензил,
(н-бутиламидо)этоксиметил( η5 -2-метил-3-этилинденил)- силантитан(II)1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)этоксиметил( η5 -2-метил-3-этилинденил)-силантитан(II) 1,3-пентадиен,
(н-бутиламидо)этоксиметил( η5 -2-метил-3-этилинденил)-силантитан (III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)этоксиметил( η5 -2-метил-3-этилинденил)-силантитан(IV)диметил,
(н-бутиламидо)этоксиметил( η5 -2 -метил-3-этилинденил)-силантитан (IV)дибензил,
(циклододециламидо)этоксиметил( η5 -2-метил-3-этилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(циклододециламидо)этоксиметил( η5 -2-метил-3-этилинденил) силантитан(II)1,3-пентадиен,
(циклододециламидо)этоксиметил( η5 -2-метил-3-этилинденил) силантитан(III)2-(N,N-диметиламино)бензил,
(циклододециламидо)этоксиметил( 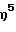 -2-метил-3-этилинденил) силантитан(IV)диметил,
-2-метил-3-этилинденил) силантитан(IV)диметил,
(циклододециламидо)этоксиметил( η5 -2-метил-3-этилинденил) силантитан(IV)дибензил,
(2,4,6-триметиланилидо)этоксиметил( η5 -2-метил-3-этилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)этоксиметил( η5 -2-метил-3-этилинденил) силантитан(II)1,3-пентадиен,
(2,4,6-триметиланилидо)этоксиметил( η5 -2-метил-3-этилинденил) силантитан(III)2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)этоксиметил( η5 -2-метил-3-этилинденил) силантитан(IV)диметил,
(2,4,6-триметиланилидо)этоксиметил( η5 -2-метил-3-этилинденил) силантитан(IV)дибензил,
(1-адамантиламидо)этоксиметил( η5 -2 -метил-3-этилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)этоксиметил( η5 -2-метил-3-этилинденил)- силантитан(II)1,3-пентадиен,
(1-адамантиламидо)этоксиметил( η5 -2-метил-3-этилинденил)-силантитан (III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)этоксиметил( η5 -2-метил-3-этилинденил)- силантитан(IV)диметил,
(1-адамантиламидо)этоксиметил( η5 -2-метил-3-этилинденил)-силантитан (IV)дибензил,
2,3,4,6-тетраметилинденильные комплексы:
(трет-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)- силантитан(II)1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)-силантитан(II) 1,3-пентадиен,
(трет-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)-силантитан(III) 2-(N,N-диметиламино)бензил,
(трет-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)-силантитан(IV) диметил,
(трет-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)-силантитан(IV) дибензил,
(н-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)- силантитан(II)1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)-силантитан(II) 1,3-пентадиен,
(н-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)-силантитан (III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)-силантитан(IV) диметил,
(н-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)-силантитан(IV) дибензил,
(циклододециламидо)диметил( η5 -2,3,4,6-тетраметилинденил)силантитан (II)1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диметил( η5 -2,3,4,6-тетраметилинденил)силантитан (II)1,3-пентадиен,
(циклододециламидо)диметил( η5 -2,3,4,6-тетраметилинденил)силантитан (III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)диметил( η5 -2,3,4,6-тетраметилинденил)силантитан (IV)диметил,
(циклододециламидо)диметил( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
(2,4,6-триметиланилидо)диметил( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диметил( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,3-пентадиен,
(2,4,6-триметиланилидо)диметил( η5 -2,3,4,6-тетраметилинденил) силантитан(III)2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диметил( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)диметил,
(2,4,6-триметиланилидо)диметил( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
(1-адамантиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)-силантитан (II) 1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)- силантитан(II)1,3-пентадиен,
(1-адамантиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)- силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)- силантитан(IV)диметил,
(1-адамантиламидо)диметил( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
(трет-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)- силантитан(II)1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)- силантитан(II)1,3-пентадиен,
(трет-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)- силантитан(III)2-(N,N-диметиламино)бензил,
(трет-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)- силантитан(IV)диметил,
(трет-бутиламидо)диметил( η5 -2,3,4,6-тетраметилинденил)- силантитан(IV)дибензил,
(циклододециламидо)диизопропокси( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диизопропокси( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,3-пентадиен,
(циклододециламидо)диизопропокси( η5 -2,3,4,6-тетраметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)диизопропокси( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)диметил,
(циклододециламидо)диизопропокси( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
(2,4,6-триметиланилидо)диизопропокси(η5 -2,3,4,6- тетраметилинденил)силантитан(II)1,4-дифенил-1,3-бутандиен,
(2,4,6-триметиланилидо)диизопропокси(η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,3-пентадиен,
(2,4,6-триметиланилидо)диизопропокси(η5 -2,3,4,6-тетраметилинденил)силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диизопропокси(η5 -2,3,4,6-тетраметилинденил) силантитан(IV)диметил,
(2,4,6-триметиланилидо)диизопропокси(η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
(1-адамантиламидо)диизопропокси( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диизопропокси( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,3-пентадиен,
(1-адамантиламидо)диизопропокси( η5 -2,3,4,6-тетраметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диизопропокси( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)диметил,
(1-адамантиламидо)диизопропокси( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
(н-бутиламидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диметокси( η5 -2,3,4,6-тетраметилинденил)-силантитан(II) 1,3-пентадиен,
(н-бутиламидо)диметокси( η5 -2,3,4,6-тетраметилинденил)силантитан (III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)диметокси( η5 -2,3,4,6-тетраметилинденил)силантитан (IV)диметил,
(н-бутиламидо)диметокси( η5 -2,3,4,6-тетраметилинденил)силантитан (IV)дибензил,
(циклододециламидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,3-пентадиен,
(циклододециламидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)диметил,
(циклододециламидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
(2,4,6-триметиланилидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,3-пентадиен,
(2,4,6-триметиланилидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)диметил,
(2,4,6-триметиланилидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
(1-адамантиламидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,3-пентадиен,
(1-адамантиламидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диметокси( 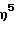 -2,3,4,6-тетраметилинденил) силантитан(IV)диметил,
-2,3,4,6-тетраметилинденил) силантитан(IV)диметил,
(1-адамантиламидо)диметокси( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
(н-бутиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,3-пентадиен,
(н-бутиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)диметил,
(н-бутиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
(циклододециламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(циклододециламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,3-пентадиен,
(циклододециламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)диметил,
(циклододециламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
(2,4,6-триметиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,3-пентадиен,
(2,4,6-триметиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(III)2-(N,N-диметиламино)бензил,
(2,4,6-триметиламидо)этоксиметил( η5-2,3,4,6-тетраметилинденил) силантитан(IV)диметил,
(2,4,6-триметиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
(1-адамантиламидо)этоксиметил( η5 2,3,4,6-тетраметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(II)1,3-пентадиен,
(1-адамантиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)диметил,
(1-адамантиламидо)этоксиметил( η5 -2,3,4,6-тетраметилинденил) силантитан(IV)дибензил,
2,3,4,6,7-пентаметилинденильные комплексы:
(трет-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил)силантитан (II)1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(трет-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III)2-(N,N-диметиламино)бензил,
(трет-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(трет-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил)силантитан (IV)дибензил,
(н-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил)- силантитан(II)1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил)-силантитан (II)1,3-пентадиен,
(н-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил)силантитан (III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил)силантитан (IV)диметил,
(н-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил)- силантитан(IV)дибензил,
(циклододециламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(циклододециламидо)диметил( η5 -2,3,4,6,7-пентаметлинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(циклододециламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(2,4,6-триметиланилидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(2,4,6-триметиланилидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(2,4,6-триметиланилидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(1-адамантиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(1-адамантиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III)2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(1-адамантиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(трет-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(трет-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан (III) 2-(N,N-диметиламино)бензил,
(трет-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(трет-бутиламидо)диметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(циклододециламидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(циклододециламидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(циклододециламидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(2,4,6-триметиланилидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(2,4,6-триметиланилидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(2,4,6-триметиланилидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(1-адамантиламидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диизопропокси( η5-2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(1-адамантиламидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(1-адамантиламидо)диизопропокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(н-бутиламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(н-бутиламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил
(н-бутиламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил)силантитан(IV)дибензил,
(циклододециламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(циклододециламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(циклододециламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)диметокси( 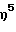 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
-2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(циклододециламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(2,4,6-триметиланилидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(2,4,6-триметиланилидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(2,4,6-триметиланилидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(1-адамантиламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(1-адамантиламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(1-адамантиламидо)диметокси( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(н-бутиламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(н-бутиламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(н-бутиламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(н-бутиламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(н-бутиламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(циклододециламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(циклододециламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(циклододециламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(циклододециламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(циклододециламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(2,4,6-триметиланилидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(2,4,6-триметиланилидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(2,4,6-триметиланилидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(2,4,6-триметиланилидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(2,4,6-триметиланилидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил,
(1-адамантиламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,4-дифенил-1,3-бутадиен,
(1-адамантиламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(II)1,3-пентадиен,
(1-адамантиламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(III) 2-(N,N-диметиламино)бензил,
(1-адамантиламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)диметил,
(1-адамантиламидо)этоксиметил( η5 -2,3,4,6,7-пентаметилинденил) силантитан(IV)дибензил.
Комплексы могут быть получены с использованием хорошо известных методов синтеза. Можно, но не обязательно, использовать восстановитель для получения комплексов с более низкой степенью окисления. Такой способ раскрыт в USSN 8/241523, поданной 13 мая 1994 г, опубликованной как WO 95-00526. Реакции проводят в подходящем растворителе, не оказывающем вредного влияния на реакции, при температуре от -100 до 300oC, предпочтительно от -78 до 100oC, а наиболее предпочтительно от 0 до 50oC. Под используемым в данном описании термином "восстановитель" подразумевается металл или соединение, который(ое) в условиях восстановления заставляет металл М восстанавливать от более высокой степени окисления до более низкой. Примерами подходящих металлических восстановителей являются щелочные металлы, щелочно-земельные металлы, алюминий и цинк, сплавы щелочных или щелочно-земельных металлов, такие как ртутно-натриевая амальгама и натрий-калиевый сплав. Примерами восстановителей-соединений являются нафталинид натрия, калийграфит, литийалкилы, литий- или калийалкадиенилы и реактивы Гриньяра. Наиболее предпочтительными восстановителями являются щелочные или щелочно-земельные металлы, в частности литий и магний.
Подходящая реакционная среда для образования комплексов включает алифатические и ароматические углеводороды, простые эфиры и циклические простые эфиры, особенно разветвленные углеводороды, такие как изобутан, бутан, пентан, гексан, гептан, октан и их смеси; циклические и алициклические углеводороды, такие как циклогексан, циклогептан, метилциклогексан, метилциклогептан и их смеси; ароматические и гидрокарбилзамещенные ароматические соединения, такие как бензол, толуол и ксилол, C1-4 диалкиловые эфиры, C1-4 диалкилэфирные производные (поли)алкиленгликолей и тетрагидрофуран. Пригодны также их смеси.
Комплексы делают каталитически активными путем сочетания с активирующим сокатализатором или путем применения активирующей технологии. Подходящие для этой цели активирующие сокатализаторы включают полимерные и олигомерные алюмоксаны, в частности метилалюмоксан, метилалюмоксан, модифицированный триизобутилалюминием, или изобутилалюмоксан; нейтральные кислоты Льюиса, такие как гидрокарбилзамещенные соединения группы 13, в частности соединения три(гидрокарбил)алюминия и три(гидрокарбил)бора и их галогенированные (включая пергалогенированные) производные, имеющие от 1 до 10 углеродных атомов в каждой гидрокарбильной или галогенированной гидрокарбильной группе, более предпочтительно перфорированные соединения три(арил)бора, а наиболее предпочтительно трис(пентафторфенил)боран; неполимерные совместимые некоординирующиеся ионообразующие соединения (включая применение таких соединений в окислительных условиях), в частности применение аммониевых, фосфониевых, оксониевых, карбониевых, силилиевых или сульфониевых солей совместимых некоординирующихся анионов или ферроцениевых солей совместимых некоординирующихся анионов; электролиз в массе (описанный более подробно далее) и сочетание указанных выше сокатализаторов и технологий. Указанные активирующие сокатализаторы и активирующие технологии уже были описаны по отношению к другим металлокомплексам в следующих ссылочных материалах EP-A-277003, US-A-5153157, US-A-5064802, EP-A-468651 (эквивалент U.S. серийный N 07/547718), EP-A-520732 (эквивалент U.S. сер. N 07/876268) и EP-A-520732 (эквивалент U. S. сер. N 07/884966, поданной 1 мая 1992 г.).
Особенно желательными активирующими сокатализаторами являются комбинации нейтральных кислот Льюиса, в частности комбинация триалкилалюминия, имеющего от 1 до 4 углеродных атомов в каждой алкильной группе, и галогенированного соединения три(гидрокарбил)бора, имеющего от 1 до 20 углеводородных атомов в каждой гидрокарбильной группе, в частности трис(пентафторфенил)борана, другие комбинации таких смесей нейтральных кислот Льюиса с полимерным или олигомерным алюмоксаном и комбинации отдельной нейтральной кислоты Льюиса, в частности трис(пентафторфенил)борана, с полимерным или олигомерным алюмоксаном. Преимущество настоящего изобретения заключается в открытии того, что наиболее эффективная активация катализатора с использованием такой комбинации трис(пентафторфенил)борана с алюмоксаном происходит при пониженных уровнях содержания алюмоксана. Предпочтительные молярные отношения металлический (группа 4) комплекс: трис(пентафторфенил)боран:алюмоксан находятся в пределах от 1:1:1 до 1:5:5, а более предпочтительно от 1:1:1,5 до 1:5:3. Неожиданно эффективное применение более низких уровней содержания алюмоксана с металлокомплексами по настоящему изобретению позволяет получить олефиновые полимеры при высокой каталитической эффективности, используя меньше дорогого алюмоксанового сокатализатора. Кроме того, это обеспечивает получение полимеров с более низкими остаточными содержаниями алюминия и, следовательно, с большей чистотой.
Подходящие ионообразующие соединения, полезные в качестве сокатализаторов, в одном из вариантов осуществления настоящего изобретения содержат катион, который является кислотой Бренстеда, способной отдавать протон и совместимый некоординирующийся анион А. Используемый в данном описании термин "некоординирующийся" означает анион или вещество, который(ое) либо не координируется с комплексом- предшественником, содержащим металл группы 4, и его каталитическим производным, либо лишь слабо координируется с такими комплексами, в результате чего остается достаточно лабильным, чтобы быть замещенным нейтральным основанием Льюиса. Некоординирующийся анион, в частности, относится к аниону, который, действуя как зарядоуравновешивающий анион в катионном металлокомплексе, не переносит анионный заместитель или его фрагмент к указанному катиону, в результате чего образуются нейтральные комплексы. "Совместимые анионы" являются анионами, которые не деградируют до нейтральности при распаде первоначально образованного комплекса и не создают помех при требуемой последующей полимеризации или других применениях комплекса.
Предпочтительными анионами являются те, которые содержат один координационный комплекс с зарядонесущим металлическим или металлоидным ядром, которое (анион) способно уравновешивать заряд активных видов катализаторов (катион металла), которые могут быть образованы при объединении двух компонентов. Кроме того, указанный анион должен быть достаточно лабильным, чтобы быть замещенным олефиновыми, диолефиновыми и ацетиленоненасыщенными соединениями или другими нейтральными основаниями Льюиса, такими как простые эфиры или нитрилы. Подходящие металлы включают (но не ограничиваются ими) алюминий, золото, платину. Подходящие металлоиды включают (но не ограничиваются ими) бор, фосфор и кремний. Соединения, содержащие анионы, которые имеют координационные комплексы с одним атомом металла или металлоида, разумеется, хорошо известны, и многие, особенно соединения, содержащие один единственный атом бора в анионной части, коммерчески доступны.
Предпочтительно такие сокатализаторы могут быть представлены следующей общей формулой:
(L* - H)d + (A)d-,
где: L* представляет нейтральное основание Льюиса;
(L* - H) представляет кислоту Брэнстеда;
Ad- представляет некоординирующийся, совместимый анион, имеющий заряд d-, и
d представляет целое число от 1 до 3.
Более предпочтительно Ad- соответствует формуле: [М'Q4]-,
где: М представляет бор или алюминий в формальной степени окисления +3; и
Q в каждом случае появления независимо выбран из гидрида, диалкиламидо, галогенида, гидрокарбила, гидрокарбилоксида, галогензамещенного гидрокарбила, галогензамещенного гидрокарбилокси и галогензамещенного силилгидрокарбила (включая пергалогенированный гидрокарбил, пергалогенированный гидрокарбилокси и пергалогенированный силилгидрокарбил), причем Q имеет до 20 углеродных атомов, при условии, что Q представляет галогенид в не более чем одном случае появления. Примеры подходящих гидрокарбилоксидных групп Q раскрыты в патенте США N 5296433.
В более предпочтительном случае d = 1, то есть противоион имеет единственный отрицательный заряд и является анионом A-. Содержащие бор активирующие сокатализаторы, которые особенно пригодны для получения катализаторов по настоящему изобретению, могут быть представлены следующей формулой:
(L* - H)+ (BQ4)-,
где L* - такой, как определено выше;
B представляет бор в формальной степени окисления +3; и
Q представляет гидрокарбильную, гидрокарбилокси-, фторированную гидрокарбильную или фторированную силилгидрокарбильную группу, имеющую до 20 неводородных атомов, при условии, что Q представляет гидрокарбил не более чем в одном случае.
Наиболее предпочтительно Q в каждом случае представляет фторированную арильную группу, в частности пентафторфенильную группу.
Иллюстративными, но не ограничивающими, примерами соединений бора, которые могут быть использованы в качестве активирующего сокатализатора при получении усовершенствованных катализаторов по настоящему изобретению, являются
соли трехзамещенного аммония, такие как:
триметиламмония тетракис(пентафторфенил)борат,
триэтиламмония тетракис(пентафторфенил)борат,
трипропиламмония тетракис(пентафторфенил)борат,
три(н-бутил)аммония тетракис(пентафторфенил)борат,
три(втор-бутил)аммония тетракис(пентафторфенил)борат,
N,N-диметиланилиния тетракис(пентафторфенил)борат,
N,N-диметиланилиния н-бутилтрис(пентафторфенил)борат,
N,N-диметиланилиния бензилтрис(пентафторфенил)борат,
N, N-диметиланилиния тетракис(4-(трет-бутилдиметилсилил)-2,3,5,6- тетрафторфенил)борат,
N,N-диметиланилиния тетракис(4-(триизопропилсилил)-2,3,5,6- тетрафторфенил)борат,
N,N-диметиланилиния пентафторфенокситрис(пентафторфенил)борат,
N,N-диметиланилиния тетракис(пентафторфенил)борат,
N,N-диметил-2,4,6-триметиланилиния тетракис(пентафторфенил)борат,
триметиламмония тетракис(2,3,4,6-тетрафторфенил)борат,
триэтиламмония тетракис(2,3,4,6-тетрафторфенил борат,
трипропиламмония тетракис(2,3,4,6-тетрафторфенил)борат,
три(н-бутил)аммония тетракис(2,3,4,6-тетрафторфенил)борат,
диметил(трет-бутил)аммония тетракис(2,3,4,6-тетрафторфенил)борат,
N,N-диметиланилиния тетракис(2,3,4,6-тетрафторфенил)борат,
N,N-диэтиланилиния тетракис(2,3,4,6-тетрафторфенил)борат, и
N, N-диметил 2,4,6-триметиланилиния тетракис(2,3,4,6-тетрафторфенил)борат;
соли диалкиламмония, такие как:
ди(изопропил)аммония тетракис(пентафторфенил)борат, и
дициклогексиламмония тетракис(пентафторфенил)борат;
соли трехзамещенного фосфония, такие как:
триметилфосфония тетракис(пентафторфенил)борат,
три(о-толил)фосфония тетракис(пентафторфенил)борат, и
три(2,6-диметилфенил)фосфония тетракис(пентафторфенил)борат;
соли двухзамещенного оксония, такие как:
дифенилоксония тетракис(пентафторфенил) борат,
ди(о-толил)оксония тетракис(пентафторфенил)борат, и
ди(2,6-диметилфенил)оксония тетракис(пентафторфенил)борат;
соли двухзамещенного сульфония, такие как:
дифенилсульфония тетракис(пентафторфенил)борат,
ди(о-толил)сульфония тетракис(пентафторфенил)борат, и
бис(2,6-диметилфенил)сульфония тетракис(пентафторфенил)борат.
Предпочтительными катионами (L*-H)+ являются N, N- диметиланилиний и трибутиламмоний.
Другой подходящий ионообразующий активирующий сокатализатор содержит соль катионного окислителя и некоординирующегося совместимого аниона, представленную формулой:
(Oxe+)d (Ad-)e,
где Oxe+ представляет катионный окислитель, имеющий заряд e+;
e представляет целое число от 1 до 3; и
Ad- и d - такие, как определенные ранее.
Примеры катионных окислителей включают ферроцений, гидрокарбилзамещенный ферроцений, Ag+ или Pb+2. Предпочтительными анионами Ad- являются анионы, определенные выше по отношению к активирующим сокатализаторам, содержащим кислоту Бренстеда, в частности тетракис(пентафторфенил)борат.
Еще один подходящий ионообразующий активирующий сокатализатор содержит соединение, являющееся солью иона карбения и некоординирующегося совместимого аниона, представленной формулой: 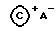
где:  представляет C1-20 карбениевый ион; и
представляет C1-20 карбениевый ион; и
A- - такой, как определенный ранее.
Предпочтительным карбениевым ионом является тритильный катион, а именно трифенилметилий.
И еще один подходящий ионообразующий активирующий сокатализатор содержит соединение, являющееся солью иона силилия и некоординирующегося совместного аниона, представленной формулой:
R3Si(X')q +A-,
где:
R представляет C1-10 гидрокарбил, а X', q и A- - такие, как определенные ранее.
Предпочтительными активирующими сокатализаторами, содержащими соль силилия, являются тетракиспентафторфенилборат триметилсилилия, тетракиспентафторфенилборат триэтилсилилия и их эфирозамещенные аддукты. Соли силилия уже были в общем описаны ранее в J. Chem. Soc. Chem. Comm., 1993, 383-384, а также Lambert, J.B., et al., Organometallics, 1994, 13, 2430-2443. Применение вышеуказанных солей силилия в качестве активирующих сокатализаторов в дополнение к катализаторам полимеризации заявлено в заявке на патент США под названием "Silylium Cationic Polimerization Activators For Metallocene Complexes", поданной от имени David Neithamer, David Devore, Robert LaPointe and Robert Mussel 12 сентября 1994 г.
Некоторые комплексы спиртов, меркаптанов, силанолов и оксимов с трис(пентафторфенил)бораном также являются эффективными активаторами катализаторов и могут быть использованы в соответствии с настоящим изобретением. Такие сокатализаторы раскрыты в патенте США USP 5296433.
Метод электролиза в массе включает электрохимическое окисление металлокомплекса при условиях электролиза в присутствии фонового электролита, содержащего некоординирующийся инертный анион. В этом методе используют такие растворители, фоновые электролиты и электролитические потенциалы, чтобы во время реакции, по существу, не образовывались побочные продукты электролиза, делающие металлокомплекс каталитически неактивным. В частности, подходящими растворителями являются материалы, которые представляют собой жидкости в условиях электролиза (обычно при температурах от 0 до 100oC), способные растворять фоновый электролит и инертные. "Инертные растворители" - это те, которые не подвержены восстановлению или окислению в условиях реакции, используемых для электролиза. Для осуществления требуемой реакции электролиза обычно можно выбрать растворитель и фоновый электролит, на которые не оказывает вредного влияния электрический потенциал, используемый для требуемого электролиза. Предпочтительные растворители включают дифторбензол (все изомеры), диметоксиэтан (DME, ДМЭ) и их смеси.
Электролиз может быть проведен в стандартной электролитической ячейке, содержащей анод и катод (называемые также рабочим электродом и противоэлектродом соответственно). Подходящими конструкционными материалами для ячейки являются стекло, пластмасса, керамика и металл, покрытый стеклом. Электроды изготавливают из инертных электропроводящих материалов, под которыми подразумевают электропроводящие материалы, не подверженные вредному влиянию реакционной смеси или условий реакции. Предпочтительными инертными электропроводящими материалами являются платина и палладий. Обычно ячейка разделена посредством ионопроницаемой мембраны, такой как фритта из тонкодисперсного стекла, на отдельные камеры - камеру для рабочего электрода и камеру для противоэлектрода. Рабочий электрод погружают в реакционную среду, содержащую активируемый металлокомплекс, растворитель, фоновый электролит и любые другие материалы, необходимые для обеспечения умеренных условий электролиза или стабилизации полученного комплекса. Противоэлектрод погружают в смесь растворителя и фонового электролита. Требуемое напряжение может быть определено теоретическими расчетами или экспериментально путем развертки ячейки с использованием электрода сравнения (такого как серебряный электрод), погруженного в электролит в ячейке. Определяют также базовый (фоновый) ток ячейки, т.е. ток, утекающий в отсутствие требуемого электролиза. Электролиз завершают при падении тока от требуемого уровня до базового уровня. Таким образом можно легко обнаружить полную конверсию исходного металлокомплекса.
Подходящими фоновыми электролитами являются соли, содержащие катион и совместимый некоординирующийся анион A-. Предпочтительными фоновыми электролитами являются соли, соответствующие формуле G+A-, где
G+ представляет катион, не реакционноспособный по отношению к исходному и полученному комплексам, и
A- - такой, как определенный ранее.
Примеры катионов (G+) включают тетрагидрокарбилзамещенные аммониевые или фосфониевые катионы, имеющие до 40 неводородных атомов. Предпочтительными катионами являются тетра(н-бутил)аммоний- и тетраэтиламмоний-катионы.
Во время активации комплексов по настоящему изобретению путем электролиза в массе катион фонового электролита проходит к противоэлектроду, а анион A- мигрирует к рабочему электроду и становится анионом полученного окисленного продукта. Растворитель или катион фонового электролита восстанавливается у противоэлектрода в молярном количестве, равном количеству окисленного металлокомплекса, образованного у рабочего электрода. Предпочтительными фоновыми электролитами являются такие соли, как тетракис(перфторарил)бораты тетрагидрокарбиламмония, имеющие от 1 до 10 углеродных атомов в каждой гидрокарбильной или перфторарильной группе, в частности тетра-(н-бутиламмоний) тетракис(пентафторфенил)борат.
Другим ранее раскрытым электрохимическим методом получения активирующих сокатализаторов является электролиз дисиланового соединения в присутствии источника некоординирующегося совместимого аниона. Этот метод более полно раскрыт и заявлен в ранее упомянутой заявке на патент США под названием "Silylium Cationic Polimerization Activators For Metallocene Complexes, поданной 12 сентября 1994 г.
Вышеописанную технологию электрохимического активирования и активирующие сокатализаторы можно также использовать в комбинации. Особенно предпочтительной комбинацией является смесь три(гидрокарбил)алюминия или три(гидрокарбил)борана, имеющего от 1 до 4 углеродных атомов в каждой гидрокарбильной группе, с олигомерным или полимерным алюмоксаном.
Используемое молярное отношение катализатор/ сокатализатор предпочтительно находится в пределах от 1:10000 до 100:1, более предпочтительно от 1: 5000 до 10:1, наиболее предпочтительно от 1:1000 до 1:1. Алюмоксан при использовании в качестве активирующего сокатализатора только его самого берут в большом количестве, обычно в молярном количестве, превышающем молярное количество металлокомплекса по крайней мере в 100 раз. Трис(пентафторфенил)боран при использовании в качестве активирующего сокатализатора берут в молярном отношении к металлокомплексу в пределах от 0,5:1 до 10:1, более предпочтительно от 1: 1 до 6:1, а наиболее предпочтительно от 1:1 до 5:1. Остальные активирующие сокатализаторы обычно используют в приблизительно эквимолярном с металлокомплексом количестве.
Процесс полимеризации может быть использован для полимеризации этиленненасыщенных мономеров, имеющих от 3 до 20 углеродных атомов, либо в отдельности, либо в сочетании. Предпочтительные мономеры включают моновинилиденовые ароматические мономеры, 4-винилциклогексен, винилциклогексан, норборнадиен и C3-10 алифатические α-олефины (в частности, этилен, пропилен, изобутилен, 1-бутен, 1-гексен, 3-метил-1-пентен, 4-метил-1-пентен и 1-октен), C4-40 диены и их смеси. Наиболее предпочтительными мономерами являются этилен и смеси этилена, пропилена и неконъюгированного диена, в частности этилиденнорборнена.
В общем, полимеризация может быть осуществлена при хорошо известных в данной области техники условиях для реакций полимеризации с помощью катализаторов Циглера-Натта или Каминского-Синна, а именно при температурах 0-250oC, предпочтительно 30-200oC, и давлениях от атмосферного до 10000 атмосфер. Можно использовать, по желанию, условия процесса полимеризации в суспензии, в растворе, в газовой фазе, в твердой порошкообразной фазе или другого процесса. Можно, а при использовании катализаторов в газофазном процессе полимеризации желательно применять носитель, в частности диоксид кремния, оксид алюминия или полимер (особенно поли(тетрафторэтилен) или полиолефин). Носитель предпочтительно используют в количестве, обеспечивающем массовое отношение катализатор (на основе металла): носитель в пределах от 1:100000 до 1:10, более предпочтительно от 1:50000 до 1:20, а наиболее предпочтительно от 1:10000 до 1:30.
В большинстве реакций полимеризации используемое молярное отношение катализатор: полимеризуемые соединения находится в пределах от 10-12:1 до 10-1:1, более предпочтительно от 10-9:1 до 10-5:1.
Подходящими растворителями для полимеризации являются инертные жидкости. Примеры включают углеводороды с неразветвленной и разветвленной цепью, такие как изобутан, бутан, пентан, гексан, гептан, октан и их смеси; циклические и алициклические углеводороды, такие как циклогексан, циклопентан, метилциклогексан, метилциклогептан и их смеси; перфорированные углеводороды, такие как перфорированные C4-10 алканы и тому подобное, и ароматические и алкилзамещенные ароматические соединения, такие как бензол, толуол, ксилол, этилбензол и тому подобное. Подходящими растворителями являются также жидкие олефины, которые могут действовать как мономеры или сомономеры и включают этилен, пропилен, бутадиен, циклопентен, 1-гексен, 1-гексан, 4-винилциклогексен, винилциклогексан, 3-метил-1-пентен, 4-метил-1-пентен, 1,4-гексадиен, 1-октен, 1-децен, стирол, дивинилбензол, аллилбензол, винилтолуол (включая все изомеры в отдельности или в смеси) и тому подобное. Пригодны также смеси указанных растворителей.
Катализаторы можно использовать в сочетании с по крайней мере одним дополнительным гомогенным или гетерогенным катализатором полимеризации в отдельных реакторах, соединенных последовательно или параллельно, для получения смесей полимеров, имеющих требуемые свойства. Пример такого процесса раскрыт в WO 94/00500, эквиваленте заявке U.S сер. N 07/904770, а также заявке U.S. сер. N 08/10958, поданной 29 января 1993 г.
Используя катализаторы по настоящему изобретению, можно легко получить сополимеры с высоким включением сомономеров и соответственно низкой плотностью, но все же имеющие низкий индекс расплава. То есть использование предлагаемых катализаторов, даже при повышенных температурах а реакторе, позволяет легко получать высокомолекулярные полимеры. Этот результат весьма желателен, потому что молекулярная масса α-олефиновых сополимеров может быть легко уменьшена путем использования водорода или подобного агента для передачи цепи, но увеличение молекулярной массы α-олефиновых сополимеров обычно может быть обеспечено только путем снижения температуры полимеризации в реакторе. А это не выгодно, потому что работа полимеризационного реактора при пониженных температурах значительно повышает расходы, так как для поддержания пониженной температуры реакции нужно отводить из реактора тепло, но в то же время необходимо добавлять тепло к вытекающему из реактора потоку, чтобы испарить растворитель. Кроме того, благодаря улучшенной растворимости полимера, пониженной вязкости раствора и более высокой концентрации полимера повышается производительность. Используя предлагаемые катализаторы, можно легко получить α-олефиновые гомополимеры и сополимеры с плотностями от 0,85 г/см3 до 0,96 г/см3 и скоростями течения расплава от 0,001 до 10,0 дг/мин при осуществлении высокотемпературного процесса.
Катализаторы по настоящему изобретению особенно выгодны для производства этиленовых гомополимеров и сополимеров этилена и α-олефина с высокими уровнями длинноцепной разветвленности. Применение катализаторов по настоящему изобретению в непрерывных процессах полимеризации, в частности в непрерывном процессе полимеризации в растворе, позволяет использовать повышенные температуры в реакторе, которые благоприятствуют образованию полимерных цепей с концевым винилом, которые могут быть включены в растущий полимер с образованием длинноцепного ответвления. Применение системы катализаторов по настоящему изобретению выгодно тем, что обеспечивает возможность экономичного производства сополимеров этилена и α-олефина с обрабатываемостью, подобной той, какую демонстрирует полиэтилен низкой плотности, полученный путем свободно-радикальной полимеризации при высоком давлении.
Предлагаемая система катализаторов может быть с выгодой использована для получения олефиновых полимеров с улучшенной обрабатываемостью путем полимеризации только этилена или смесей этилена и α-олефина с низкими уровнями содержания вызывающего "Н"-разветвление диена, такого как норборнадиен, 1,7-октадиен или 1,9-декадиен. Уникальное сочетание повышенных температур в реакторе, высокой молекулярной массы (или низких индексов расплава) при высоких температурах в реакторе и высокой реакционноспособности сомономеров выгодно тем, что обеспечивает возможность экономичного производства полимеров, обладающих превосходными физическими свойствами и способностью к переработке. Такие полимеры предпочтительно содержат этилен, C3-20 α-олефин и "Н"-разветвляющийся сомономер. В соответствии с предпочтительным вариантом такие полимеры получают путем полимеризации в растворе, а наиболее предпочтительно - путем осуществления непрерывного процесса полимеризации в растворе.
Как было указано ранее, предлагаемая каталитическая система особенно полезна в производстве сополимеров ЭП и ЭПДМ с высокими выходом и производительностью. Используемый процесс полимеризации может быть осуществлен либо в растворе, либо в суспензии, как уже известно в данной области техники. Kaminsky, J. Poly. Sci., том 23, страницы 2151-64 (1985) описал применение растворимой каталитической системы бис(циклопентадиенил)цирконийдиметил-
алюмоксан для полимеризации в растворе эластомеров ЭП и ЭПДМ. В патенте USP 5229478 раскрыт процесс полимеризации в суспензии с использованием подобных каталитических систем на основе бис(циклопентадиенил) циркония.
Вообще говоря, является желательным получать такие ЭП ЭПДМ эластомеры в условиях повышенной реакционноспособности диенового мономерного компонента. Причина этого была объяснена в указанном выше патенте USP 5229478 следующим образом (что все еще остается справедливым, несмотря на достигнутый в этом деле прогресс). Главным фактором, влияющим на производственные затраты и, следовательно, на полезность ЭПДМ, является стоимость диенового мономера. Диен более дорогой мономерный материал, чем этилен или пропилен. Кроме того, реакционноспособность диеновых мономеров при использовании уже известных металлоценовых катализаторов ниже, чем у этилена и пропилена. Следовательно, чтобы достигнуть требуемой степени включения диена для получения ЭПДМ с приемлемо высокой скоростью отверждения, необходимо использовать концентрацию диенового мономера (выраженную в процентах от общей концентрации присутствующих мономеров) в значительном избытке по сравнению с процентным содержанием диена, который должен быть включен в конечный ЭПДМ продукт. Поскольку значительные количества непрореагировавшего диенового мономера должны быть уловлены из вытекающего потока из полимеризационного реактора для возврата в цикл, то производственные затраты необязательно должны увеличиться.
Другим фактором увеличения стоимости производства ЭПДМ является то, что воздействие диена на катализатор полимеризации олефинов, особенно высоких концентраций диенового мономера, необходимых для обеспечения требуемого уровня включения диена в конечный ЭПДМ продукт, часто уменьшает скорость или активность, с которой катализатор заставляет протекать процесс полимеризации мономеров этилена и пропилена. Поэтому требовалось больше пропускать материала через реактор и больше времени на реакцию по сравнению с производством этилен-пропиленового эластомерного сополимера или другого α-олефинового эластомерного сополимера.
Предлагаемая каталитическая система выгодна тем, что позволяет повышать реакционную способность диена и благодаря этому получать ЭПДМ полимеры с высокими выходом и производительностью. Кроме того, каталитическая система по настоящему изобретению обеспечивает возможность экономичного производства ЭПДМ полимеров с содержанием диена до 20 массовых процентов или выше, которые обладают весьма желательными высокими скоростями отверждения.
Неконъюгированный диеновый мономер может быть неразветвленным, разветвленным или циклическим углеводородным диеном, имеющим от 6 до 15 углеродных атомов. Примерами подходящих неконъюгированных диенов являются неразветвленные ациклические диены, такие, как 1,4-гексадиен и 1,6-октадиен; разветвленные ациклические диены, такие как 5- метил-1,4-гексадиен, 3,7-диметил-1,6-октадиен, 3,7-диметил-1,7- октадиен и смешанные изомеры дигидромирицена и дигидрооцинена; одноядерные алициклические диены, такие как 1,3-циклопентадиен, 1,4-циклогексадиен, 1,5-циклооктадиен и 1,5-циклододекадиен; и многоядерные алициклические (конденсированные и с мостиковой связью) диены, такие как тетрагидроинден, метилтетрагидроинден, дициклопентадиен; бицикло-(2,2,1)-гепта-2,5-диен; алкенил-, алкилиден-, циклоалкенил- и циклоалкилиденнорборнены, такие, как 5-метилен-2-норборнен (MNB), 5-пропенил-2-норборнен, 5-изопропилиден-2-норборнен, 5-(4-циклопентил)-2-норборнен, 5-циклогексилиден-2-норборнен, 5-винил-2-норборнен и норборнадиен.
Из диенов, обычно используемых для получения ЭПДМ сополимеров, предпочтительными являются 1,4-гексадиен (HD), 5-этилиден-2-норборнен (ENB), 5-винилиден-2-норборнен (VNB), 5-метилен-2-норборнен (MNB) и дициклопентадиен (DCPD). Особенно предпочтительными диенами являются 5-этилиден-2-норборнен (ENB) и 1,4-гексадиен (HD).
Предпочтительные ЭПДМ эластомеры могут содержать 20-90, более предпочтительно 30-85, а наиболее предпочтительно 35-80 массовых процентов этилена.
Альфа-олефинами, пригодными для получения эластомеров с этиленом и диенами, предпочтительно являются C3-16 альфа-олефины. Иллюстративными, не ограничивающими примерами таких альфа-олефинов являются пропилен, 1-бутен, 1-пентен, 1-гексен, 1-октен, 1-децен и 1-додецен. Альфа-олефин обычно включают в ЭПДМ полимер в количестве 10-80, более предпочтительно 20-65 массовых процентов. Неконъюгированные диены обычно включают в ЭПДМ полимер в количестве 0,5-20, более предпочтительно 1-15, а наиболее предпочтительно 3-12 массовых процентов. При желании можно включить одновременно более одного диена, например HD и ENB, с общим содержанием диенов в указанных выше пределах.
Каталитическая система может быть приготовлена в виде гомогенного катализатора путем добавления необходимых компонентов к раствору, в котором будут осуществлять полимеризацию методом полимеризации в растворе. Каталитическая система может быть также приготовлена и использована в виде гетерогенного катализатора путем адсорбирования необходимых компонентов на носителе, таком как силикагель, оксид алюминия и другой подходящий неорганический носитель. При изготовлении каталитической системы в гетерогенной форме или на носителе в качестве носителя предпочтительно используют диоксид кремния. Гетерогенную форму каталитической системы используют в процессе полимеризации в суспензии. Практическим ограничением является то, что полимеризацию в суспензии осуществляют в жидких разбавителях, в которых полимерный продукт, по существу, нерастворим. Разбавителем для полимеризации в суспензии предпочтительно является один или несколько углеводородов с менее чем 5 углеродными атомами. При желании в качестве разбавителя можно использовать (полностью или частично) насыщенные углеводороды, такие как этан, пропан или бутан. Аналогичным образом, можно использовать (полностью или частично) в качестве разбавителя α-олефиновый мономер или смесь разных α-олефиновых мономеров. В соответствии с наиболее предпочтительным вариантом разбавитель содержит, по крайней мере в качестве основной части, α-олефиновый мономер или полимеризуемые мономеры.
Наоборот, при полимеризации в растворе используют растворитель для соответственных компонентов реакции, в частности ЭП или ЭПДМ полимера. Предпочтительные растворители включают минеральные масла и различные углеводороды, которые представляют собой жидкость при температурах реакции. Иллюстративные примеры подходящих растворителей включают алканы, такие как пентан, изопентан, гексан, гептан, октан и нонан, а также смеси алканов, включая керосин и Изопар Е, поставляемый ф. "Exxon Chemicals Inc."; циклоалканы, такие как циклопентан и циклогексан; и ароматические соединения, такие как бензол, толуол, ксилол, этилбензол и диэтилбензол.
Индивидуальные компоненты, а также регенерированные компоненты катализаторов всегда должны быть защищены от воздействия кислорода и влаги. Следовательно, компоненты катализаторов и катализаторы необходимо приготовлять и регенерировать в свободной от кислорода и влаги атмосфере. Поэтому реакции предпочтительно выполняют в присутствии сухого инертного газа, такого, как, например, азот.
Этилен подают в реактор в количестве, обеспечивающем избыточное давление этилена по отношению к суммарному давлению паров α-олефиновых и диеновых мономеров. Содержание этилена в полимере определяется отношением избыточного давления этилена к общему давлению в реакторе. Обычно процесс полимеризации осуществляют при избыточном давлении этилена в пределах от 10 до 1000 фунтов на квадратный дюйм (70-7000 кПа), а наиболее предпочтительно от 40 до 400 фунтов на квадратный дюйм (30-300 кПа). Полимеризацию обычно проводят при температуре от 25 до 200oC, предпочтительно от 75 до 170oC, а наиболее предпочтительно от более чем 95 до 140oC.
Процесс полимеризации может быть периодическим или непрерывным. Предпочтительным является непрерывный процесс, при котором катализатор, этилен, α-олефин и, необязательно, растворитель и диен непрерывно подают в реакционную зону и непрерывно отводят из нее полимерный продукт.
Один из способов осуществления такого процесса полимеризации, никоим образом не ограничивающий объем изобретения, состоит в следующем. В реактор с мешалкой непрерывно подают пропиленовый мономер вместе с растворителем, диеновым и этиленовым мономерами. Реактор содержит жидкую фазу, состоящую в основном из этиленового, пропиленового и диенового мономеров вместе с растворителем или дополнительным разбавителем. При необходимости может быть добавлено небольшое количество вызывающего "Н"-разветвление диена, такого как норборнадиен, 1,7-октадиен или 1,9-декадиен. В жидкую фазу, находящуюся в реакторе, непрерывно вводят катализатор и сокатализатор. Температуру и давление в реакторе можно регулировать путем регулирования отношения растворитель/мономеры, скорости подачи катализаторов, а также посредством охлаждающих или нагревающих змеевиков, рубашек или того и другого. Скорость полимеризации регулируют изменением скорости подачи катализаторов. Содержание этилена в полимерном продукте определяется отношением этилена к пропилену в реакторе, которое регулируют путем изменения соответственных скоростей подачи этих компонентов в реактор. Молекулярную массу полимерного продукта регулируют, необязательно, путем регулирования других переменных полимеризации, таких как температура, концентрация мономеров, или потоком водорода, вводимого в реактор, как хорошо известно в данной области техники. Вытекающий поток из реактора вводят в контакт с подавителем катализаторов, таким как вода. Полимерный раствор нагревают, хотя это и не обязательно, и извлекают полимерный продукт путем отгона газообразного этилена и пропилена, а также остаточного растворителя или разбавителя, при пониженном давлении и, если нужно, проведения другого удаления летучих веществ в оборудовании, таком как обезгаживающий экструдер. В непрерывном процессе среднее время пребывания катализатора и полимера в реакторе обычно составляет от 5 минут до 8 часов, предпочтительно от 10 минут до 6 часов.
При предпочтительном способе работы полимеризацию проводят в системе непрерывного действия для полимеризации в растворе, содержащем два реактора, соединенных последовательно или параллельно. В одном реакторе образуется продукт с относительно высокой молекулярной массой (Mw составляет от 300000 до 600000, более предпочтительно от 400000 до 500000), в то время как во втором реакторе образуется продукт с относительно низкой молекулярной массой (Mw 50000-300000). Конечный продукт представляет собой смесь из двух вытекающих из реактора потоков, которые объединяют до обезгаживания с получением однородной смеси двух полимерных продуктов. Такой процесс с двумя реакторами позволяет получать продукты с улучшенными свойствами. В предпочтительном варианте реакторы соединяют последовательно, то есть вытекающий поток из первого реактора направляют во второй реактор и в этот второй реактор добавляют свежий мономер, растворитель и водород. Условия в реакторах регулируют так, чтобы массовое отношение полимера, полученного в первом реакторе, к полимеру, полученному во втором реакторе, находилось в пределах от 20:80 до 80: 20. Кроме того, регулируют температуру во втором реакторе так, чтобы получить продукт с более низкой молекулярной массой. Эта система обеспечивает возможность выгодного производства ЭПДМ продуктов, имеющих широкий диапазон вязкостей по Муни, а также превосходные прочность и способность к переработке. Вязкость по Муни (ASTM D1646-94, ML 1+4 @ 125oC) полученного продукта предпочтительно регулируют так, чтобы она находилась в пределах от 1 до 200, предпочтительно от 5 до 150, а наиболее предпочтительно от 10 до 110.
ПРИМЕРЫ
Специалистам в данной области техники будет понятно, что раскрытое в данном описании изобретение может быть осуществлено в отсутствие какого-либо компонента, который не был конкретно раскрыт. Следующие далее примеры являются дополнительной иллюстрацией изобретения и не должны рассматриваться как ограничивающие. Если не указано иное, все части и проценты выражены по массе.
Спектры 1H и 13C ЯМР были записаны на спектрометре Varian XL (300 МГц). Химические сдвиги определялись относительно ТМС или через остаточный CHCL3 в CDCl3 или остаточный C6HD5 в C6D5 относительно ТМС. Использовали тетрагидрофуран (ТГФ), диэтиловый эфир, толуол и гексан, проходящие через сдвоенные колонки, загруженные активированным оксидом алюминия и смешанным металлооксидным катализатором, нанесенным на оксид алюминия (катализатор Q-5 производства ф. "Engelhard Corp."). Все соединения н-BuLi, КН, реактивы Гриньяра и 1,4-дифенил-1,3-бутадиен использовали в состоянии поставки фирмой "Aldrich Chemical Company". Все процессы синтеза осуществляли в атмосфере сухого азота с использованием комбинации бокса с перчатками и высоковакуумной технологии.
Пример 1. Получение (2-метилинденил)диметил(тpет-бутиламидо) силантитандихлорида
Получение 2-броминдена
В колбу емкостью 500 мл с магнитной мешалкой вводили (+/-)транс-2-бром- 1-инданол (50,0 г, 235 ммоль), моногидрат п-толуолсульфоновой кислоты (0,50 г, 2,6 ммоль) и толуол (300 мл). На колбу устанавливали ловушку Дина-Старка и обратный холодильник и реакционную смесь нагревали с обратным холодильником в течение 16 часов. Переносили реакционную смесь в делительную воронку, прибавляли хлороформ (200 мл) и полученную смесь промывали водным раствором бикарбоната натрия (3 х 200 мл). Затем органический слой промывали насыщенным водным раствором хлорида натрия (1 х 300 мл), сушили над безводным сульфатом магния и фильтровали. Отгоняли растворители с получением 40,6 г (88,7%) желтоватого кристаллического вещества, собранного при 72-105oC и 3 мм рт. столба.
1H ЯМР (300 МГц, CDCl3, ТМС): δ 7,4-7,1 (м, 4H) 6,93 (с, 1H), 3,60 (с, 2H).
13C ЯМР (75 МГц, CDCl3): δ 143,62, 142,22, 132,64, 126,38, 124,59, 124,49, 122,85, 119,88, 45,40.
ГХ-МС: Вычислено для C9H7 79/Br 193,97, найдено 194,00.
Вычислено для C9H7 81Br 195,90, найдено 195,90.
Получение 2-метилиндена через 2-броминден
2-Броминден (24,4 г, 0,125 моль) и Ni(dppp)Cl2 (0,925 г, 1,71 • 10-3 моль) (dppp = 1,3-бис(дифенилфосфино)пропан) перемешивали в диэтиловом эфире (200 мл) при -78oC под средой азота, прибавляя при этом метилMgBr (0,150 моль, 50,00 мл 3,0 М раствора в диэтиловом эфире). После этого сразу же убирали баню с сухим льдом и давали реакционной смеси согреться до комнатной температуры. Реакционная смесь сначала представляла собой гетерогенный раствор кирпично-красного цвета, а затем становилась гомогенным желто-золотистым раствором. После часового перемешивания в этом состоянии появился экзотермический эффект, приведший к некоторому кипению эфира в колбе. Раствор снова превращался в гетерогенную смесь кирпично-красного цвета. Общее время перемешивания смеси составляло 3 часа, после чего убирали ледяную баню, и проведенный после этого ГХ (газовая хроматография) анализ показал, что превращение 2-броминдена в 2-метилинден было, по существу, количественным. По истечении времени реакции смесь выливали на лед и затем экстрагировали 1 М HCl (1 х 100 мл) и 1 М NaHCO3 (1 х 100 мл), после чего сушили сульфатом магния и фильтровали. В результате был получен целевой продукт в виде светло-желтого масла (14,0 г, 86,2%).
1H ЯМР (300 МГц, CDCl3): δ 2,18 (с, 3H), 3,32 (с, 2H), 6,51 (с, 1H), 7,08-7,40 (м, 4H).
13C ЯМР (75 МГц, CDCl3): δ 17,02, 42,90, 119,71, 123,30, 123,49, 126,22, 127,16, 143,30, 145,90, 146,04.
ГХ-МС: Вычислено для C10H10 130,19, найдено 130,00.
Получение 2-метилиндена через 2-метилинданон
2-Метилинданон (20,0 г, 0,137 моль) и NaBH4 (5,175 г, 0,137 моль) смешивали вместе и перемешивали в ТГФ (200 мл). Затем медленно прибавляли безводный этанол (100 мл) и давали смеси перемешиваться в течение 16 часов при комнатной температуре. По истечении времени реакции смесь гасили медленным добавлением 1 М HCl и затем экстрагировали диэтиловым эфиром (3 х 100 мл). Удаление растворителя дало белое твердое вещество, которое вновь растворяли в диэтиловом эфире (300 мл) и перемешивали над катионообменными шариками (ионообменные шарики DowexTM DR-2030 производства The Dow Chemical Company) в течение 48 часов с мониторингом реакции путем газовой хроматографии. Затем смесь фильтровали и, удалив летучие вещества, получили целевой продукт в виде бледно-желтого масла (16,8 г, 94,3%).
Получение литий-2-метилинденида
2-Метилинден (15,5 г, 0,114 моль) перемешивали в диэтиловом эфире (250 мл), прибавляя при этом по каплям н-BuLi (0,120 моль, 60,0 мл 2,0 М раствора в циклогексане). Смеси давали перемешиваться в течение 3 часов при комнатной температуре. По истечении времени реакции удаляли летучие вещества и твердое вещество хорошо промывали гексаном и собирали фильтрованием под разрежением в виде светло-желтого порошка, который использовали без дальнейшей очистки или анализа (15,1 г, 97,0%).
Получение хлор (трет-бутиламино)диметилсилана
Перемешивали в пентане (2 л) Me2SiCl2 (151,50 г, 1,17 моль) с медленным добавлением при этом N(C2H5)3 (119,62 г, 1,18 моль). Затем прибавляли по каплям трет-бутиламин (85,85 г, 1,17 моль) в пентане (100 мл) и давали реакционной смеси перемешиваться при комнатной температуре в течение 16 часов. По истечении времени реакции смесь фильтровали и концентрировали до объема 600 мл, и в это время начинали выпадать в осадок дополнительные соли. Далее смесь вновь фильтровали и концентрировали до 250 мл, после чего переносили в круглодонную колбу емкостью 250 мл, снабженную микроперегонным аппаратом и термометром. Осуществляли перегонку до тех пор, пока температура перегонки не достигала уровня 50oC. Определение методом ЯМР показало, что оставшаяся прозрачная бесцветная жидкость была чистым продуктом и выход ее был, по существу, количественным.
1H ЯМР (300 МГц, CDCl3): δ 0,31 (с, 6H), 1,10 (с, 6H), 1,89 (с, 1H)
Получение (2-метилинденил)(трет-бутиламино)диметилсилана
Хлор(трет-бутиламино)диметилсилан (9,57 г, 0,058 моль) перемешивали в диэтиловом эфире (150 мл) при 0oC, добавляя при этом литий-2-метилинденид (7,68 г, 0,058 моль) в виде твердого вещества в течение 15 минут. Затем смеси давали перемешиваться в течение 16 часов при комнатной температуре. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление летучих веществ привело к выделению целевого продукта в виде бледно-желтого масла (9,99 г, 67,5%).
1H ЯМР (300 МГц, CDCl3, ТМС): δ 0,089 (с, 3H), 0,12 (с, 3H), 1,03 (с, 9H), 2,14 (с, 3H), 3,22 (с, 1H), 6,54 (с, 1H), 7,14- 7,55 (м, 4H).
Получение Li2[(2-метилинденил)(трет-бутиламидо)диметилсилан] •0,75 Et2O
(2-Метилинденил)(трет-бутиламино)диметилсилан (5,00 г, 0,0192 моль) перемешивали в диэтиловом эфире (60 мл), медленно прибавляя при этом н-BuLi (0,0405 моль, 16,2 мл 2,5 М раствора в гексане). Затем смеси давали перемешиваться в течение 16 часов. По истечении времени реакции удаляли летучие вещества и остаток промывали гексаном и затем собирали путем фильтрования в виде твердого вещества, которое использовали без дальнейшей очистки или анализа (5,70 г, 90,5%).
Получение (2-метилинденил)диметил(трет-бутиламидо)силантитандихлорида окислением PbCl2
Li2[(2-Метилинденил)(трет-бутиламидо)диметилсилан] • 3/4 Et2O (5,70 г, 0,0174 моль) медленно добавляли в виде твердого вещества к суспензии TiCl3 (ТГФ)3 (6,46 г, 0,0174 моль) в ТГФ (80 мл). Этой смеси давали перемешиваться в течение 45 минут. Затем добавляли к смеси PbCl2 (2,76 г, 0,00995 моль), и давали смеси перемешиваться еще 45 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием толуола. Затем удаляли толуол и остаток суспендировали в гексане и затем собирали путем фильтрования в виде красно-коричневого кристаллического твердого вещества. Второй сбор получали путем концентрирования и охлаждения фильтрата до -20oC в течение 16 часов с последующим вторым фильтрованием. Объединив сборы, определяли, что это был целевой продукт (4,88 г, 74,2%).
1H ЯМР (300 МГц, C6D6): δ 0,42 (с, 3H), 0,56 (с, 3H), 1,34 (с, 3H), 2,14 (с, 9H), 6,71 (с, 1H), 6,92 (т, 1H), 7,04 (т, 1H), 7,25 (д, 1H), 7,59 (д, 1H).
Получение (2-метилинденил)диметил(трет-бутиламидо)титан-хлорида путем окисления с использованием метиленхлорида
Li2[(2-Метилинденил)(трет-бутиламидо)диметилсилан] • Et2O (2,00 г, 0,00612 моль) и TiCl3 (ТГФ)3 (2,32 г, 0,00612 моль) объединяли в виде твердых веществ. К полученной смеси прибавляли ТГФ (100 мл), после чего смеси давали перемешиваться в течение 30 минут. Затем прибавляли CH2Cl2 (0,00306 моль) и давали смеси перемешиваться еще 2 часа. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием толуола через вспомогательное фильтровальное вещество марки Целит. Затем удаляли толуол и остаток промывали гексаном с получением целевого продукта (0,900 г, 40%). Результаты спектроскопического анализа были такими же, как описанные выше.
Получение (2-метилинденил)диметил(трет-бутиламидо) силантитандихлорида из TiCl4
Li2[(2-Метилинденил)(трет-бутиламидо)диметилсилан] • 0,75 Et2O (2,00 г, 0,00612 моль) растворяли в ТГФ (10 мл) и перемешивали, медленно прибавляя при этом TiCl4 (ТГФ)2 (2,043 г, 0,00612 моль) в ТГФ (15 мл). Этой смеси давали перемешиваться в течение 2 часов. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием толуола через слой Целита. Затем удаляли толуол и остаток промывали гексаном с получением целевого продукта (1,43 г, 62%). Результаты анализа были такими же, как описанные выше.
Пример 2. Получение (2-метилинденил)диметил(третбутиламидо) силантитандиметила
[(2-Метилинденил)диметил(трет-бутиламидо)силан] TiCl2 (0,800 г, 0,00211 моль) перемешивали в диэтиловом эфире (30 мл), прибавляя при этом по каплям MeMgI (0,00454 моль, 1,50 мл 3,00 М раствора в диэтиловом эфире). Этой смеси давали перемешиваться в течение 30 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление летучих дало целевой продукт в виде желтого твердого вещества (0,220 г, 30,8%).
1H ЯМР (300 МГц, C6D6): δ 0,11 (с, 3H), 0,46 (с, 3H), 0,56 (с, 3H), 0,85 (с, 3H), 1,47 (с, 9H), 2,00 (с, 3H), 6,75 (с, 1H), 6,88 (т, 1H), 7,06 (т, 1H), 7,42 (д, 1H), 7,50 (д, 1H).
Пример 3. Получение (2-метилинденил)диметил(трет-бутиламидо) силантитан (II) (1,4-дифенил-1,3-бутадиена)
[(2-Метилинденил)диметил(трет-бутиламидо)силан)TiCl2 (2,20 г, 0,00583 моль) перемешивали в гексане (150 мл) с 1,4- дифенил-1,3-бутадиеном (1,20 г, 0,00583 моль), медленно прибавляя при этом н-BuLi (0,0117 моль, 4,67 мл 2,5 М раствора в гексане). Затем смесь нагревали с обратным холодильником в течение 2 часов. По истечении времени реакции смесь охлаждали до комнатной температуры и затем фильтровали через Целит. Удаление летучих веществ дало целевой продукт в виде красно-коричневого порошка (1,81 г, 60,6%).
1H ЯМР (250 МГц, C6D6, ТМС): δ 0,61 (с, 3H), 0,72 (с, 3H), 1,25 (с, 9H), 1,78 (с, 3 Н), 3,35 (д, 1H), 3,70 (д, 1H), 3,85 (м, 1H), 5,08 (м, 1H), 5,42 (с, 1H), 7,40-6,15 (м, 14H).
Пример 4. Получение (3-метилинденил)диметил(трет-бутиламидо) силантитандихлорида
Получение 3-метилиндена
К раствору диметилсульфата (9,91 г, 0,0786 моль) в диэтиловом эфире (125 мл) прибавляли по каплям в течение 15 минут литийинденид (9,60 г, 0,0786 моль) в диэтиловом эфире (100 мл). По окончании прибавления к реакционной смеси прибавляли H2O (150 мл). Сушка MgSO4 с последующим фильтрованием и удалением растворителя дали целевой продукт в виде желтого масла (9,68 г, 94,7%).
1H ЯМР (300 МГц, CDCl3, ТМС): δ 1,20 (д, 3H), 3,90 (кв., 1H), 6,37 (дд, 1H), 6,68 (дд, 1 Н), 7,05-7,19 (м, 2H), 7,26 (д, 1H), 7,30 (д, 1H).
13C ЯМР (75 МГц, CDCl3): δ 15,94, 45,07, 120,96, 122,56, 124,72, 126,35, 130,23, 141,27, 143,88, 149,16.
Получение литий-1-метилинденида
3-Метилинден (9,68 г, 0,0745 моль) перемешивали в гексане (300 мл), прибавляя при этом по каплям н-BuLi (0,0745 моль, 29,78 мл 2,5 М раствора в гексане). Смеси давали перемешиваться при комнатной температуре 48 часов, в течение которых выпадало в осадок твердое вещество. По истечении времени реакции твердое вещество собирали фильтрованием под разрежением в виде светло-желтого порошка, который использовали без дальнейшей очистки или анализа (9,38 г, 92,5%).
Получение (3-метилинденил)(трет-бутиламино)диметилсилана
Хлор(трет-бутиламино)диметилсилан (5,47 г, 0,033 моль) перемешивали в ТГФ (200 мл), добавляли при этом литий-1- метилинденид (4,51 г, 0,033 моль) в ТГФ (50 мл) по каплям. Затем смеси давали перемешиваться в течение 16 часов при комнатной температуре. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление летучих веществ привело к выделению целевого продукта в виде желтого масла (7,24 г, 84,5%).
1H ЯМР (300 МГц, C6D6): δ -0,077 (с, 3H), -0,053 (с, 3H), 1,05 (с, 9H), 2,15 (с, 3H), 3,41 (с, 1H), 6,31 (с, 1H), 7,14-7,64 (м, 5H).
Получение Li2[(3-метилинденил)(трет-бутиламидо)диметилсилан] •0,75 Et2O
(3-Метилинденил)(трет-бутиламино)диметилсилан (7,24 г, 0,0279 моль) перемешивали в диэтиловом эфире (75 мл), медленно прибавляя при этом н-BuLi (0,0586 моль, 23,40 мл 2,5 М раствора в гексане). Затем смеси давали перемешиваться в течение 16 часов. По истечении времени реакции удаляли летучие вещества и остаток промывали гексаном и затем собирали путем фильтрования в виде твердого вещества, которое использовали без дальнейшей очистки или анализа (7,01 г, 76,9%).
Получение (3-метилинденил)диметил(трет-бутиламидо) силантитандихлорида
Li2[(1-Метилинденил)(трет-бутиламидо)диметилсилан] • 0,75 Et2O (7,01 г, 0,0214 моль) медленно добавляли в виде твердого вещества к суспензии TiCl3 (ТГФ)3 (7,94 г, 0,0214 моль) в ТГФ (75 мл). Этой смеси давали перемешиваться в течение 45 минут. Затем добавляли к смеси PbCl2 (2,98 г, 0,0107 моль) и давали смеси перемешиваться еще 45 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием толуола. Затем удаляли толуол и остаток суспендировали в гексане и затем собирали путем фильтрования в виде красно-коричневого кристаллического твердого вещества. Второй сбор получали путем концентрирования и охлаждения фильтрата с последующим вторым фильтрованием. Объединив сборы, определили, что это был целевой продукт (4,67 г, 57,9%).
1H ЯМР (300 МГц, C6D6): δ 0,36 (с, 3H), 0,55 (с, 3H) 1,32 (с, 9H), 2,37 (с, 3H), 6,08 (с, 1H), 6,97 (т, 1H), 7,11 (т, 1H) 7,27 (д, 1H), 7,55 (д, 1H).
Пример 5. Получение (3-метилинденил)диметил(тpет-буниламидо) силантитандиметила
[(3-Метилинденил)диметил(трет-бутиламидо)силан] TiCl2 (0,500 г, 0,00132 моль) перемешивали в диэтиловом эфире (35 мл), прибавляя при этом по каплям MeMgI (0,00292 моль, 1,00 мл 3,00 М раствора в диэтиловом эфире). Этой смеси давали перемешиваться в течение 35 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление летучих веществ с последующим повторным фильтрованием также с использованием гексана дало после удаления гексана целевой продукт в виде желтого масла (0,230 г, 51,3%).
1H ЯМР (300 МГц, C6D6): δ 0,16 (с, 3H), 0,38 (с, 3H), 0,57 (с, 3H), 0,70 (с, 3H), 1,46 (с, 9H), 2,34 (с, 3H), 5,83 (с, 1H), 6,91 (т, 1H), 7,11 (т, 1H),7,41 (д, 1H), 7,46 (д, 1H)
Пример 6. Получение (2,3-диметилинденил)диметил(трет-бутиламидо) силантитандихлорида
Получение 2,3-Диметилиндена
В перемешиваемый раствор 15,02 г (103 ммоль) (+/-)-2-метил- 1-инданона в 200 мл безводного диэтилового эфира при -78oC под средой аргона вводили 50 мл 3,0 М раствора метилMgI в эфире (150 ммоль MeMgI). Реакционной смеси давали медленно согреться до комнатной температуры в течение трех часов и затем нагревали ее при 35oC в течение 1 часа. Реакционную смесь вливали в 1 л воды и медленно прибавляли к ней концентрированную HCl, пока не был достигнут pH 1. Смесь переносили в делительную воронку и интенсивно встряхивали. Разделяли слои и водный слой экстрагировали эфиром. Объединенные органические слои промывали водой (1 х 500 мл), водным раствором NaHCO3 (1 x 500 мл) и насыщенным водным раствором NaCl (1 х 500 мл). Органический слой сушили над безводным MgSO4 и фильтровали. ГХ показала, что все еще присутствует некоторая часть спирта, и поэтому смесь перемешивали с 100 мл 10%-ного (по массе) водного раствора HCl в течение 1 часа. Смесь переносили в делительную воронку и разделяли слои. Органическую фазу промывали водой (1 х 200 мл), водным раствором NaHCO3 (1 х 300 мл) и насыщенным водным раствором NaCl (1 х 250 мл) Сушка над MgSO4 с последующим фильтрованием и удалением растворителя дали 14,7 г (99%) 1,2-диметилиндена.
1H ЯМР (300 МГц, CDCl3, ТМС): δ 7,4-7,0 (м, 4H, ароматическая), 3,23 (с, 2H, аллильная CH2), 2,04 (с, 3H, CH3), 2,01 (с, 3H, CH3),
13C ЯМР (75 МГц, CDCl3): δ 147,1, 141,9, 137,3, 132,1 125,7 123,3, 122,6, 117,6, 42,2, 13,6, 10,0
ГХ-МС: вычислено для C11H12 144,09, найдено: 144,10.
Получение литий-2,3-диметилинденида
2,3-Диметилинден (24,11 г, 0,1659 моль) перемешивали в гексане (400 мл), прибавляя при этом по каплям н-BuLi (0,20 моль, 80,0 мл 2,5 М раствора в гексане). Смеси давали перемешиваться при комнатной температуре 16 часов, в течение которых выпадало в осадок твердое вещество. По истечении времени реакции твердое вещество собирали фильтрованием под разрежением в виде белого порошка, который использовали без дальнейшей очистки или анализа (20,64 г, 82,3%).
Получение (2,3-диметилинденил)(трет-бутиламино)диметилсилана
Хлор(трет-бутиламино)диметилсилан (6,48 г, 0,039 моль) перемешивали в ТГФ (100 мл), добавляя при этом литий-2,3- диметилинденйд (5,651 г, 0,0373 моль) в ТГФ (25 мл) по каплям. Этой смеси давали перемешиваться в течение 16 часов при комнатной температуре. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление летучих веществ привело к выделению целевого продукта в виде желтого масла (9,64 г, 94,7%).
1H ЯМР (300 МГц, CDCl3, ТМС): δ -0,062 (с, 3H), 0,043 (с, 3H), 0,58 (с, 1H), 1,18 (с, 9H), 2,09 (с, 3H), 2,18 (с, 3H), 3,33 (с, 1H), 7,07-7,28 (м, 3H), 7,44 (д, 3Jнн= 7,4 Гц, 1H).
13C ЯМР (75 МГц, CDCl3): δ 0,0040, 0,90, 10,38, 15,39, 33,96, 49,66, 50,60, 117,73, 122,22, 122,88, 124,42, 130,45, 140,38, 144,14, 146,47.
Получение Li2[(2,3-диметилинденил)(трет-бутиламидо) диметилсилан] • 0,75 Et2O
(2,3-Диметилинденил)(трет-бутиламино)диметилсилан (7,28 г, 0,0266 моль) перемешивали в диэтиловом эфире (80 мл), медленно прибавляя при этом н-BuLi (0,0559 моль, 22,4 мл 2,5 М раствора в гексане). Затем смеси давали перемешиваться в течение 16 часов. По истечении времени реакции удаляли летучие вещества и остаток промывали гексаном и затем собирали путем фильтрования в виде твердого вещества, которое использовали без дальнейшей очистки или анализа (8,34 г, 92,0%).
Получение (2, 3-диметилинденил)диметил(трет-бутиламидо) силантитандихлорида
Li2[(2,3-диметилинденил)(трет-бутиламидо)диметилсилан] • 0,75 Et2O (8,34 г, 0,0245 моль) медленно добавляли в виде твердого вещества к суспензии TiCl3 (ТГФ)3 (9,07 г, 0,0245 моль) в ТГФ (75 мл). Этой смеси давали перемешиваться в течение 30 минут. Затем добавляли к смеси PbCl2 (3,40 г, 0,0123 моль) и давали смеси перемешиваться еще 30 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием толуола. Затем удаляли толуол и остаток суспендировали в гексане и затем собирали путем фильтрования в виде твердого вещества. Второй сбор получали путем концентрирования и охлаждения фильтрата с последующим вторым фильтрованием. Объединив сборы, определили, что это был целевой продукт (2,87 г, 30,0%).
1H ЯМР (300 МГц, C6D6): δ 0,48 (с, 3H), 0,60 (с,3H), 1,33 (с, 9H), 2,09 (с, 3H), 2,26 (с, 3H), 6,94-7,15 (м, 2H), 7,28 (д, 1H) 7,63 (д, 1H).
Пример 7. Получение (2,3-диметилинденил)диметил(трет-бутиламидо) силантитандиметила
[(2,3-Диметилинденил)диметил(трет-бутиламидо)силан] TiCl (0,750 г, 0,00191 моль) перемешивали в диэтиловом эфире (50 мл), прибавляя при этом по каплям метилMgI (0,00402 моль, 1,34 мл 3,0 М раствора в диэтиловом эфире). Этой смеси давали перемешиваться в течение 30 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление летучих веществ с последующим повторным фильтрованием также с использованием гексана дало после удаления гексана целевой продукт в виде желтого масла (0,620 г, 92,1%).
1H ЯМР (300 МГц, C6D6): δ -0,13 (с, 3H), 0,50 (с, 3H), 0,60 (с, 3H), 0,66 (с, 3H), 1,47 (с, 9H), 1,93 (с, 3H), 2,24 (с, 3H), 6,93 (т, 1H), 7,12 (т, 1H), 7,39 (д, 1H), 7,55 (д, 1H).
Пример 8. Получение (2,3-диметилинденил)диметил(циклододециламидо) силантитандихлорида
Получение Li2[(2,3-диметилинденил)(циклододециламидо) диметилсилан]•0,75 Et2O
(2,3-Диметилинденил)(циклододециламидо)диметилсилан (5,47 г, 0,0142 моль) перемешивали в диэтиловом эфире (25 мл), медленно прибавляя при этом н-BuLi (0,030 моль, 11,94 мл 2,5 М раствора в гексане). Затем смеси давали перемешиваться в течение 16 часов. По истечении времени реакции удаляли летучие вещества и остаток промывали гексаном и затем собирали путем фильтрования в виде твердого вещества, которое использовали без дальнейшей очистки или анализа (5,47 г, 85,2%).
Получение (2,3-диметилинденил)диметил(циклододециламидо) силантитандихлорида
Li2[(2,3-диметилинденил)(циклододециламидо) диметилсилан]•3/4 Et2O (5,47 г, 0,0121 моль) медленно добавляли в виде твердого вещества к суспензии TiCl3 (ТГФ)3 (4,48 г, 0,0121 моль) в ТГФ (75 мл). Этой смеси давали перемешиваться в течение 45 минут. Затем к смеси добавляли PbCl2 (1,68 г, 0,00604 моль) и давали смеси перемешиваться еще 45 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием толуола. Затем удаляли толуол и остаток суспендировали в гексане и затем собирали путем фильтрования в виде красно-коричневого кристаллического твердого вещества. Второй сбор получали путем концентрирования и охлаждения фильтрата с последующим вторым фильтрованием. Объединив сборы, определяли, что это был целевой продукт (0,457 г, 7,6%).
1H ЯМР (300 МГц, C6D6): δ 0,52 (с, 3H), 0,63 (с, 3H), 1,15-1,91 (м, 23H), 2,11 (с, 3H), 2,23 (с, 3H), 5,31 (м, 1H), 6,83-7,12 (м, 2H), 7,29 (д, 1H), 7,63 (д, 3H).
Пример 9. Получение (2,3-диметилинденил)диметил(циклододециламидо) силантитандиметила
(2, 3-диметилинденил)диметил(циклододециламидо)силан TiCl2 (0,200 г, 0,000400 моль) перемешивали в диэтиловом эфире (50 мл), прибавляя при этом по каплям метилMgI (0,00084 моль, 0,28 мл 3,0 М раствора в диэтиловом эфире). Этой смеси давали перемешиваться в течение 30 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление летучих веществ с последующим повторным фильтрованием также с использованием гексана дало после удаления гексана целевой продукт в виде оранжевого кристаллического твердого вещества (0,134 г, 73,2%).
1H ЯМР (300 МГц, C6D6): δ -0,11 (с, 3H), 0,53 (с, 3H), 0,61 (с, 3H), 0,65 (с, 3H), 1,10-1,90 (м, 23H), 1,98 (с, 3H), 2,26 (с, 3H), 5,12-5,25 (м, 1H) 6,91 (т, 1H) 7,09 (т, 1H), 7,45 (д, 1H), 7,58 (д, 1H).
Пример 10. Получение (2-этилиндeнил)диметил(тpет-бутиламидо) силантитандихлорида
Получение 2-этилиндена
2-Броминден (8,2135 г, 0,04211 моль) и Ni(dppp)Cl2 (0,1536 г, 2,834 х 10-4 моль) перемешивали в диэтиловом эфире (100 мл) при -78oC под средой азота, прибавляя при этом этилMgBr (0,045 моль, 15,00 мл 3,0 М раствора в диэтиловом эфире). После этого убирали баню с сухим льдом и давали реакционной смеси согреться до комнатной температуры. Реакционная смесь сначала представляла собой гетерогенный раствор кирпично-красного цвета, затем становилась гомогенным желто-золотистым раствором, а в ходе подогрева снова превращалась в гетерогенную смесь кирпично-красного цвета. Анализ путем газовой хроматографии через 2 часа перемешивания при комнатной температуре показал, что реакция была, по существу, количественной. По истечении времени реакции смесь выливали на лед и затем экстрагировали 1М HCl (1 х 100 мл) и 1 М NaHCO3 (1 х 100 мл), после чего сушили сульфатом магния и фильтровали. В результате был получен целевой продукт в виде светло-желтого масла (5,65 г, 93,1%).
1H ЯМР (300 МГц, CDCl3, ТМС): δ 1,31 (т, 3Jнн = 7,4 Гц, 3H), 2,59 (кв, 3Jнн = 7,4 Гц, 2H), 3,39 (с, 2H), 6,59 (с, 1H), 7,16-7,38 (м, 3H), 7,46 (д, 3Jнн = 7,4 Гц, 1H).
13C ЯМР (75 МГц, CDCl3): δ 13,65, 24,63, 41,23, 119,96, 123,47, 123,60, 125,25, 126,29, 143,12, 145,76, 152,47.
ГХ-МС: Вычислено для C11H12 144,22, найдено 144,10.
Получение литий-2-этилинденида
2-Этилинден (7,10 г, 0,049 моль) перемешивали в гексане (100 мл), прибавляя при этом по каплям н-BuLi (0,050 моль, 25,00 мл 2,0 М раствора в циклогексане). Смеси давали перемешиваться в течение 16 часов при комнатной температуре. По истечении времени реакции твердое вещество собирали фильтрованием под разрежением в виде светло-желтого порошка, который использовали без дальнейшей очистки или анализа (5,21 г, 70,5%).
Получение (2-этилинденил)(трет-бутиламино)диметилсилана
Хлор(трет-бутиламино)диметилсилан (6,0038 г, 0,03623 моль) перемешивали в ТГФ (100 мл), прибавляя при этом по каплям литий-2-этилинденид (4,96 г, 0,033 моль) в ТГФ (25 мл). Этой смеси давали перемешиваться в течение 16 часов при комнатной температуре. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление летучих веществ привело к выделению целевого продукта в виде желтого масла (8,64 г, 95,7%).
1H ЯМР (300 МГц, CDCl3): δ 0,067 (с, 3H), 0,085 (с, 3H), 1,18 (с, 9H), 1,25 (т, 3Jнн = 7,5, Гц, 3H), 2,46-2,54 (м, 1H), 2,54-2,82 (м, 1H), 3,47 (с, 1H), 6,57 (с, 1H), 7,04-7,45 (м, 4H).
Получение Li2[(2-этилинденил)(трет-бутиламидо)диметилсилан] •0,75 Et2O
(2-Этилинденил)(трет-бутиламино)диметилсилан (7,24 г, 0,026 моль) перемешивали в диэтиловом эфире (50 мл), медленно прибавляя при этом н-BuLi (0,0556 моль, 22,2 мл 2,5 М раствора в гексане). Этой смеси давали перемешиваться в течение 16 часов. По истечении времени реакции удаляли летучие вещества и остаток промывали гексаном и затем собирали путем фильтрования в виде твердого вещества, которое использовали без дальнейшей очистки или анализа (6,79 г, 75,2%).
Получение (2-этилинденил)диметил(трет-бутиламидо) силантитандихлорида
Li2[(2-Этилинденил)(трет-бутиламидо)диметилсилан] • 0,75 Et2O (6,79 г, 0,0199 моль) медленно добавляли в виде твердого вещества к суспензии TiCl3 (ТГФ)3 (7,37 г, 0,0199 моль) в ТГФ (75 мл). Этой смеси давали перемешиваться в течение 45 минут. Затем добавляли к смеси PbCl2 (2,76 г, 0,00995 моль) и давали смеси перемешиваться еще 45 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием толуола. Затем удаляли толуол и остаток суспендировали в гексане и затем собирали путем фильтрования в виде красно-коричневого кристаллического твердого вещества. Второй сбор получали путем концентрирования и охлаждения фильтрата с последующим вторым фильтрованием. Объединив сборы, определили, что это был целевой продукт (3,15 г, 40,6%).
1H ЯМР (300 МГц, C6D6): δ 0,45 (с, 3H), 0,57 (с, 3H), 1,19 (т, 3H), 1,34 (с, 9H), 2,43-2,70 (м, 2H), 6,81 (с, 1H), 6,90-7,09 (м, 2H), 7,28 (д, 1H). 7,62 (д, 1H).
Пример 11. Получение (2-этилинденил)диметил(трет-бутиламидо) силантитандиметила
(2-Этилинденил)диметил(трет-бутиламидо)силан TiCl2 (0,500 г, 0,00128 моль) перемешивали в диэтиловом эфире (50 мл), прибавляя при этом по каплям MeMgI (0,00269 моль, 0,900 мл 3,0 М раствора в диэтиловом эфире). Этой смеси давали перемешиваться в течение 30 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление летучих веществ с последующим повторным фильтрованием также с использованием гексана дало после удаления гексана целевой продукт в виде желтого масла (0,310 г, 62,2%).
1H ЯМР (300 МГц, C6D6): δ -0,11 (с, 3H), 0,49 (с, 3H), 0,57 (с, 3H), 0,83 (с, 3H), 1,14 (т, 3H), 1,47 (с, 9H), 2,20-2,34 (м, 1H), 2,36-2,51 (м, 1H), 6,83 (с, 1H), 6,85- 6,94 (м, 1H), 7,03-7,12 (м, 1H), 7,46 (д, 1H), 7,53 (д, 1H).
Пример 12. Получение (2-пpoпилинденил)диметил)трет-бутиламидо) силантитандихлорида
Получение 2-пропилиндена
В высушенную в печи круглодонную колбу емкостью 250 мл, снабженную магнитной мешалкой, обратным холодильником и вакуумным переходником, загружали 2-броминден (15,0 г, 76,9 нмоль) и Ni(dppp)Cl2 (0,42 г, 0,77 ммоль) (dppp= 1,3-бис(дифенилфосфино)пропан. Колбу закупоривали и создавали в ней разрежение. Прибавляли посредством канюли дезоксигенированный безводный диэтиловый эфир (150 мл) под аргоном при -78oC. Реакционную смесь перемешивали под аргоном без внешнего охлаждения, прибавляя при этом посредством шприца 42 мл 2,0 М пропилмагнийхлорида в эфирном растворе (84 ммоль пропилмагнийхлорида). При наступлении интенсивного кипения помещали реакционную смесь в баню с сухим льдом и ацетоном. Спустя 2 минуты баню убирали и реакционную смесь перемешивали при комнатной температуре под аргоном в течение 90 минут. Осторожно вливали реакционную смесь в воду и прибавляли 10%-ную (по массе) водную HCl, пока смесь не становилась кислой. Смесь экстрагировали эфиром (3 х 200 мл) и объединенные органические слои промывали водой (1 х 250 мл), водным раствором бикарбоната натрия (1 х 250 мл) и насыщенным водным раствором хлорида натрия (1 х 250 мл). Сушка над безводным сульфатом натрия с последующими фильтрованием и удалением растворителя дали 12,14 г (99,7%) целевого продукта.
1H ЯМР (300 МГц, CDCl3, ТМС): δ 7,4-7,0 (м, 4H), 6,48 (с, 1H), 3,26 (с, 2H), 2,43 (т, 3Jнн = 7,4 Гц, 2H), 1,61 (с, 3Jнн = 7,4 Гц, 2H), 0,96 (т, 3Jнн = 7,4 Гц, 3H).
13C ЯМР (75 МГц, CDCl3): δ 150,17, 145,46, 142,83, 126,03, 125,96, 123,30, 123,10, 119,64, 40,81, 33,23, 22,15, 13,95
ГХ-МС: Вычислено для C12H14 158,11, найдено 158,05
Получение литий-2-пропилинденида
2-Пропилинден (11,0 г, 0,068 моль) перемешивали в гексане (500 мл), прибавляя при этом по каплям н-BuLi (0,076 моль, 30,6 мл, 2,5 М раствора в гексане). Затем смеси давали перемешиваться при комнатной температуре 16 часов, в течение которых выпадало в осадок твердое вещество. По истечении времени реакции твердое вещество собирали фильтрованием под разрежением в виде светло-желтого порошка, который использовали без дальнейшей очистки для анализа (10,8 г, 94,3%).
Получение (2-пропилинденил)(трет-бутиламино)диметилсилана
Диметилсилил(трет-бутиламино) хлорид (3,03 г, 0,018 моль) перемешивали в ТГФ (100 мл), прибавляя при этом по каплям литий-2-пропилинденил (3,00 г, 0,018 моль) в ТГФ (20 мл). Этой смеси давали перемешиваться в течение 16 часов при комнатной температуре. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление гексана привело к выделению целевого продукта в виде желтого масла (4,67 г, 89,0%). Это соединение использовали без дальнейшей очистки или анализа.
Получение Li2[(2-пропилинденил)(трет-бутиламидо)диметилсилан] •3/4 Et2O
(2-Пропилинденил)(трет-бутиламино)диметилсилан (4,67 г, 0,0162 моль) перемешивали в диэтиловом эфире (75 мл), медленно прибавляя при этом н-BuLi (0,0341 моль, 13,70 мл 2,5 М раствора в гексане). Этой смеси давали перемешиваться в течение 16 часов. По истечении времени реакции удаляли летучие вещества и остаток промывали гексаном и затем собирали путем фильтрования в виде твердого вещества, которое использовали без дальнейшей очистки или анализа (4,92 г, 85,3%).
Получение (2-пропилинденил)диметил(трет-бутиламидо) силантитандихлорида
Li2[(2-Пропилинденил)(трет-бутиламидо)
диметилсилан] • 3/4 Et2O (4,92 г, 0,0138 моль) медленно добавляли в виде твердого вещества к суспензии TiCl3 (ТГФ)3 (5,12 г, 0,0138 моль) в ТГФ (75 мл). Этой смеси давали перемешиваться в течение 45 минут. Затем добавляли к смеси PbCl2 (1,92 г, 0,00691 моль) и давали смеси перемешиваться еще 45 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием толуола. Затем удаляли толуол и остаток суспендировали в гексане и затем собирали путем фильтрования в виде красно-коричневого кристаллического твердого вещества. Второй сбор получали путем концентрирования и охлаждения фильтрата с последующим вторым фильтрованием. Объединив сборы, определили, что это был целевой продукт (2,20 г, 39,4%).
1H ЯМР (300 МГц, C6D6): δ 0,49 (с, 3H), 0,58 (с, 3H), 0,80 (т, 3H), 1,35 (с, 9H), 1,47-1,64 (м, 2H), 2,51-2,73 (м, 2H), 6,83 (с, 1H), 6,93 (т, 1H), 7,05 (т, 1H), 7,29 (д, 1H), 7,63 (д, 1H).
Пример 13. Получение (2-пропилинденил)диметил(трет-бутиламидо)силантитандиметила
(2-Пропилинденил)диметил(трет-бутиламидо)силан TiCl2 (0,500 г, 0,00124 моль) перемешивали в диэтиловом эфире (50 мл), прибавляя при этом по каплям MeMgI (0,00260 моль, 0,870 мл 3,0 М раствора в диэтиловом эфире). Этой смеси давали перемешиваться в течение 30 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление летучих веществ с последующим повторным фильтрованием также с использованием гексана дало после удаления гексана целевой продукт в виде желтого масла (0,340 г, 75,6%).
1H ЯМР (300 МГц, C6D6): δ -0,11 (с, 3H), 0,52 (с, 3H), 0,57 (с, 3H), 0,85 (т, 3H), 1,48 (с, 9H), 1,56-1,70 (м, 2H), 2,20-2,32 (м, 1H), 2,40-2,52 (м, 1H), 6,84 (с, 1H), 6,90 (т, 1H), 7,08 (т, 1H), 7,46 (д, 1H), 7,53 (д, 1H).
Пример 14. Получение (2-метил-4-фенилинденил)диметил (трет-бутиламидо)силантитанхлорида
Получение (2-метил-4-фенилинденил)(трет-бутиламино) диметилсилана
2-Метил-4-фенилинден (синтезированный по существу в соответствии с методикой, описанной в патенте USP 5329033) (3,00 г, 0,014 моль) в ТГФ (10 мл) прибавляли по каплям к перемешиваемому раствору КН (0,601 г, 0,0150 моль) в ТГФ (50 мл). Этой смеси давали перемешиваться в течение 16 часов. Затем раствор фильтровали и добавляли по каплям к раствору диметилсилил(трет-бутиламидо) хлорида (2,41 г, 0,0145 моль) в ТГФ (75 мл). Этой смеси давали перемешиваться в течение 16 часов. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление летучих веществ привело к выделению целевого продукта в виде светло-желтого масла (4,00 г, 82,0%).
1H ЯМР (300 МГц, CDCl3): δ -0,0056 (с, 3H), 0,18 (с, 3H), 1,21 (с, 9H), 1,46 (с, 1H), 2,29 (с, 3H), 3,50 (с, 1H), 6,73 (с, 1H), 7,11-7,61 (м, 8H).
Получение (2-метил-4-фенилинденил)диметил(трет-бутиламидо) силантитандихлорида
(2-Метил-4-фенилинденил)(трет-бутиламино)диметилсилан (1,13 г, 0,00338 моль) перемешивали в диэтиловом эфире (50 мл), прибавляя при этом по каплям н-BuLi (0,00676 моль, 2,71 мл 2,5 М раствора в гексане). Этому раствору давали перемешиваться в течение 3 часов, после чего прибавляли его по каплям к суспензии TiCl3 (ТГФ)3 (1/25 г, 0,00338 моль) в ТГФ (75 мл). Этому раствору давали перемешиваться в течение 3 часов. Затем прибавляли метиленхлорид (0,50 мл) и давали раствору перемешиваться в течение еще 30 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление гексана дало темный остаток, который вновь растворяли в ТГФ (50 мл) и перемешивали с PbCl2 (1,0186 г, 0,003663 моль) в течение 30 минут. По истечении времени реакции удаляли летучие вещества и смесь экстрагировали и фильтровали с использованием гексана. Концентрирование этого раствора и последующее охлаждение до -78oC привели к выделению целевого продукта в виде кристаллического твердого вещества красно-коричневого цвета (0,8493 г, 55,5%).
1H ЯМР (300 МГц, C6D6): δ 0,43 (с, 3H), 0,60 (с, 3H), 1,36 (с, 9H), 2,09 (с, 3H), 6,98-7,29 (м, 7H), 7,61 (д, 1H), 7,67 (д, 1H).
Пример 15. Получение (2-метил-4-фенилинденил)диметил (трет-бутиламиндо)силантитандиметила
(2-Метил-4-фенилинденил)диметил(трет-бутиламидо)силан TiCl2 (0,254 г, 0,000563 моль) перемешивали в толуоле (50 мл) при 0oC, прибавляя при этом по каплям MeMgBr (0,00113 моль, 0,38 мл 3,0 М раствора в диэтиловом эфире). Этой смеси давали перемешиваться в течение 16 часов. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление гексана дало целевой продукт в виде аморфного твердого вещества (0,149 г, 64,3%).
1H ЯМР (300 МГц, C6D6): δ 0,029 (с, 3H), 0,48 (с, 3H), 0,61 (с, 3H), 0,86 (с, 3H), 1,49 (с, 9H), 1,96 (с, 3H), 6,90-7,35 (м, 7H), 7,53 (д, 1H), 7,67 (д, 1H).
Пример 16. Получение (η5 2,3,4,6,7-пентаметилинденил)-диметил (трет-бутиламидо)силантитандихлорида
Получение E-1-(2,3,5,6-тетраметилфенил)-2-метил-2-бутен-1-ола
Литиймезитилен (7,55 г, 0,0588 моль) суспендировали в диэтиловом эфире (50 мл) при 0oC, прибавляя при этом по каплям транс-2-метил-2-бутенол (5,04 г, 0,0588 моль). Этому раствору давали перемешиваться в течение 16 часов при комнатной температуре. По истечении времени реакции смесь вливали в ледяную воду, отделяли органический слой и промывали его водой и затем сушили над MgSO4. Фильтрование и удаление летучих веществ с последующей перекристаллизацией из гексана привели к выделению целевого продукта (5,88 г, 57,4%).
1H ЯМР (300 МГц, CDCl3, ТМС): δ 1,40-1,66 (м, 6H), 1,82 (ш, 1H), 2,25 (с, 3H), 2,31 (с, 6H), 5,41-5,51 (м, 1H), 6,81 (с, 1H).
ГХ-МС: Вычислено для C14H20O 204,32, найдено 204,15.
Получение 1,2,4,5,7-пентаметилиндена
E-1-(2,3,5,6-Тетраметилфенил)-2-метил-2-бутен-1-ол (1,50 г, 0,00734 моль) в гексане (20 мл) прибавляли по каплям к концентрированной H2SO4 (20 мл) при 0oC. Полученному красному раствору давали согреться до комнатной температуры, после чего гасили его добавлением к раствору Na2CO3 (300 мл 1,89 М раствора) при 0oC. Отделяли органический слой и водный слой экстрагировали пентаном (3 х 100 мл). Затем органические слои объединяли и сушили над MgSO4, после чего фильтровали и удаляли растворитель с получением целевого продукта (1,22 г, 89,7%).
1H ЯМР (300 МГц, CDCl3): δ 1,55 (с, 3H), 2,04 (с, 3H), 2,24 (с, 3H), 2,28 (с, 3H), 2,46 (с, 3H), 3,07 (с, 2H), 6,75 (с, 1H).
Получение литий-2,3,4,5,7-пентаметилинденида
1,2,4,5,7-Пентаметилинден (1,22 г, 0,00655 моль) перемешивали в пентане (250 мл), прибавляя при этом по каплям н-BuLi (0,00655 моль, 2,61 мл, 2,50 М раствора в гексане). Смеси давали перемешиваться при комнатной температуре 48 часов, в течение которых выпадало в осадок твердое вещество. По истечении времени реакции твердое вещество собирали фильтрованием под разрежением и использовали без дальнейшей очистки или анализа (1,07 г, 85,6%).
Получение (2,3,4,6,7-пентаметилинденил)(трет-бутиламино) диметилсилана
Хлор(трет-бутиламино)диметилсилан (0,922 г, 0,0556 моль) перемешивали в ТГФ (50 мл), прибавляя при этом по каплям литий-2, 3,4,6,7-пентаметилинденид (1,07 г, 0,055 моль) в ТГФ (20 мл). Затем эту смесь нагревали с обратным холодильником в течение 30 минут и давали ей перемешиваться при комнатной температуре. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление гексана привело к выделению целевого продукта (1,76 г, 99,9%).
ГХ-МС: Вычислено для C20H33NSi 315,58, найдено 315,25.
Получение Li2[(2,3,4,6,7-пентаметилинденил)(трет-бутиламидо)диметилсилан] • 0,75 Et2O
(2,3,4,6,7-Пентаметилинденил)(трет-бутиламино)диметилсилан (1,76 г, 0,00558 моль) перемешивали в диэтиловом эфире (35 мл), медленно прибавляя при этом н-BuLi (0,0112 моль, 4,46 мл 2,50 М раствора в гексане). Этой смеси давали перемешиваться в течение 16 часов. По истечении времени реакции удаляли летучие вещества и остаток промывали гексаном и затем собирали путем фильтрования в виде твердого вещества, которое использовали без дальнейшей очистки или анализа (1,32 г, 72,1%).
Получение (2,3,4,6,7-пентаметилинденил)диметил(трет-бутиламидо)силантитандихлорида
Li2[(2,3,4,6,7-Пентаметилинденил)(трет-бутиламидо)диметилсилан] • 0,75 Et2O (1,32 г, 0,0403 моль) медленно добавляли в виде твердого вещества к суспензии TiCl3 (ТГФ)3 (1,49 г, 0,0403 моль) в ТГФ (75 мл). Этой смеси давали перемешиваться в течение 45 минут. Затем добавляли к смеси РbСl2 (0,560 г, 0,00201 моль) и давали смеси перемешиваться еще 45 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием пентана. Затем пентановый экстракт концентрировали и охлаждали до -20oC с собиранием целевого продукта путем фильтрования в виде красно-коричневого микрокристаллического твердого вещества (0,33 г, 19%).
1H ЯМР (300 МГц, C6D6): δ 0,56 (с, 3H), 0,62 (с, 3H), 1,39 (с, 9H), 2,10 (с, 3H), 2,16 (с, 3H), 2,30 (с, 3H), 2,37 (с, 3H), 2,53 (с, 3H), 6,71 (с, 1H).
Пример 17. Получение (2,3,4,6,7-пентаметилинденил)диметил(трет-бутиламидо)силантитандиметила
(2,3,4,6,7-Пентаметилинденил)диметил
(трет-бутиламидо)- силан TiCl2 (0,243 г, 0,000562 моль) перемешивали в диэтиловом эфире (30 мл), прибавляя при этом по каплям MeMgI (0,00112 моль, 0,380 мл 3,0 М раствора в диэтиловом эфире). Этой смеси давали перемешиваться в течение 30 минут. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием пентана. Удаление пентана дало целевой продукт в виде желтого твердого вещества (0,181 г, 82,3%).
1H ЯМР (300 МГц, C6D6): δ -0,14 (с, 3H), 0,57 (с, 3H), 0,61 (с, 3H), 0,63 (с, 3H), 1,50 (с, 9H), 1,99 (с, 3H), 2,14 (с, 3H), 2,33 (с, 3H), 2,38 (с, 3H), 2,46 (с, 3H), 6,66 (с, 1H).
Пример 18. Получение (2,3-диметилинденил)диметил (трет-бутиламидо)силантитан (III) 2-(N,N-диметил)аминобензила
В сухом боксе перемешивали 0,543 г (1,5 ммоль) TiCl3 (ТГФ)3 в приблизительно 60 мл ТГФ. Добавляли при перемешивании дилитий (N-трет-бутиламидо)(диметил)(2,3-диметилинденил)силан (3/4 Et2O) (0,50 г, 1,5 ммоль) в виде твердого вещества. Перемешивание продолжали 15 минут, после чего добавляли 0,207 г (1,5 ммоль) литий (2-N,N-диметиламино)бензила и продолжали перемешивание еще 30 минут. Затем удаляли при пониженном давлении ТГФ. К остатку прибавляли гексан. Красно-коричневый осадок собирали путем фильтрования и промывали холодным гексаном. Твердый продукт высушивали при пониженном давлении с получением 0,593 г (89,2%) продукта.
Пример 19. Получение (2,3-диметилинденил)диметил(адамантиламидо) силантитандихлорида
Получение литий-1-адамантанамида
1-Адамантанамин (14,1 г, 0,0931 моль) перемешивали в гексане (300 мл), прибавляя при этом по каплям н-BuLi (0,0978 моль, 39,0 мл 2,50 М раствора в гексане). Смеси давали перемешиваться при комнатной температуре 16 часов, в течение которых выпадало в осадок твердое вещество. По истечении времени реакции твердое вещество собирали фильтрованием под разрежением в виде белого порошка, который использовали без дальнейшей очистки или анализа (13,44 г, 91,9%).
Получение (1-адамантиламино)хлордиметилсилана
В сухом боксе перемешивали 20,53 г дихлордиметилсилана (20,5 г, 0,159 моль) в ТГФ (150 мл), медленно добавляя при этом литий-1-адамантанамид (10,0 г, 0,064 моль) в ТГФ (100 мл) в виде суспензии. Этой смеси давали перемешиваться в течение 2,5 часов при комнатной температуре. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление гексана привело к выделению целевого продукта в виде белого твердого вещества (14,3 г, 92,1%).
1H ЯМР (300 МГц, CDCl3): δ 0,46 (с, 6H), 1,28 (ш, 1H), 1,62 (с, 6H), 1,74 (с, 6H), 2,04 (с, 3H).
1H ЯМР (15 МГц, CDCl3): δ 4,97, 30,12, 36,41, 46,74, 50,67.
Получение (2,3-диметилинденил)(1-адамантиламино)диметилсилана
(1-Аламантиламино)хлордиметилсилан (5,48 г, 0,0225 моль) перемешивали в ТГФ (100 мл), добавляя при этом по каплям 2,3- диметилинденид лития (3,40 г, 0,0225 моль) в ТГФ (25 мл). Этой смеси давали перемешиваться в течение 8 часов. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление гексана привело к выделению целевого продукта в виде твердого вещества (7,69 г, 97,0%).
1H ЯМР (300 МГц, CDCl3): δ -0,053 (с, 3H), 0,022 (с, 3H) 1,61 (с, 6H), 1,66 (с, 6H), 2,03 (с, 3H), 2,08 (с, 3H), 2,18 (с, 3H), 3,33 (с, 1H), 7,04-7,27 (м, 3H), 7,45 (д, 3Jнн = 7,4 Гц, 1H).
Протон для амина не может быть отделен от остальной части спектра.
Получение дилитий[(2,3-диметилинденил)(1-адамантиламидо)диметилсилана]
(2,3-Диметилинден)(1-адамантиламино) диметилсилан (7,69 г, 0,0218 моль) перемешивали в гексане (150 мл), медленно прибавляя при этом н-BuLi (0,0436 моль, 17,4 мл 2,50 М раствора в гексане). Затем этой смеси давали перемешиваться в течение 16 часов. По истечении времени реакции смесь фильтровали с получением целевого продукта в виде бледно-желтого порошка, который использовали без дальнейшей обработки или анализа (7,68 г, 96,6%).
Получение (2,3-диметилинденил)диметил(1-адамантиламидо) силантитандихлорида
Дилитий(2,3-диметилинденил)(1-адамантиламидо)диметилсилан (7,68 г, 0,0211 моль) в ТГФ (50 мл) добавляли по каплям к суспензии TiCl3 (ТГФ)3 (7,81 г, 0,0211 моль) в ТГФ (100 мл). Этой смеси давали перемешиваться в течение 3 часов. Затем добавляли к смеси PbCl2 (3,18 г, 0,0114 моль) и давали смеси перемешиваться еще час. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием толуола. Затем удаляли толуол и остаток суспендировали в гексане и затем охлаждали до -15oC. Фильтрование дало целевой продукт в виде красно-коричневого кристаллического твердого вещества (7,70 г, 77,9%).
1H ЯМР (300 МГц, C6D6): δ 0,55 (с, 3H), 0,67 (с, 3H), 1,49 (кв, 3Jнн= 10,6 Гц, 6H), 1,93 (с, 3H), 2,02 (с, 6H), 2,14 (с, 3H), 2,30 (с, 3H), 7,01 (т, 3Jнн = 7,2 Гц, 1H), 7,13 (т, 3Jнн = 8,3 Гц, 3H), 7,31 (д, 3Jнн = 8,5 Гц, 1H), 7,69 (д, 3Jнн = 8,6 Гц, 1H).
Пример 20. Получение (2,3-диметилинденил)диметил-(1-адамантиламидо) силантитандиметила
(2,3-Диметилинденил)диметил(1-адамантиламидо)силантитандихлорид (0,300 г, 0,000640 моль) перемешивали в ТГФ (60 мл), прибавляя при этом по каплям MeMgBr (0,00192 моль, 1,40 мл 1,40 М раствора в смеси толуол-ТГФ). Этой смеси давали перемешиваться в течение 1 часа. По истечении времени реакции удаляли летучие вещества и остаток экстрагировали и фильтровали с использованием гексана. Удаление гексана дало целевой продукт в виде желтого твердого вещества (0,228 г, 83,2%).
1H ЯМР (300 МГц, C6D6): δ -0,079 (с, 3H), 0,57 (с, 3H), 0,66 (с, 3H), 0,71 (с, 3H), 1,61 (ш.с, 6H), 1,98 (с, 3H), 2,03 (ш.с, 3H), 2,11 (с, 6H), 2,27 (с, 3H), 6,96 (т, 3Jнн = 7,4 Гц, 1H), 7,09-7,21 (м, 1H), 7,41 (д, 3Jнн = 8,2 Гц, 1H), 7,60 (д, 3Jнн = 8,3 Гц, 1H).
Полимеризация
В двухлитровый реактор Парра загружали 740 г смешанного алканового растворителя Изопар-Е (ф. "Exxon Chemicals Inc.") и 118 г 1-октенового сомономера. Из питательного резервуара емкостью 75 мл при 25 фунтах на кв. дюйм (2070 кПа) добавляли водород как средство регулирования молекулярной массы в результате расширения от перепада давлений. Реактор нагревали до температуры полимеризации 140oC и насыщали этиленом при избыточном (манометрическом) давлении 500 фунтов на кв. дюйм (3,4 МПа). Предварительно смешивали в сухом боксе по 2,0 мкмоль катализатора и сокатализатора в виде 0,005 М растворов в толуоле. Через необходимый для предварительного смешивания промежуток времени раствор переносили в резурвуар для ввода катализаторов и вводили в реактор. В течение 15 минут поддерживали условия полимеризации, подавая этилен по потребности. Полученный раствор отводили из реактора и добавляли к нему защищенный от внешнего воздействия фенольный антиокислитель (Ирганокс 1010 производства Ciba Geigi Corporation). Образовавшиеся полимеры высушивали в вакуумной печи при 120oC в течение 20 часов. Результаты представлены в таблице 1.
Эффективность в опытах 1-8 в среднем составляла приблизительно 80% от достигнутой в сравнительном опыте 9. Результаты опытов полимеризации показывают, что при использовании предлагаемых металлокомплексов, содержащих замещенный инденил, образуются полимеры с существенно более высокими молекулярными массами по сравнению с уже известными металлокомплексами ограниченной геометрии или на основе моноциклопентадиенила с амидосилановым мостиком при тех же самых условиях реакции. Такие результаты весьма желательны, особенно в реакции полимеризации в растворе, потому что оператор теперь может получать полимер с данной молекулярной массой при более высокой температуре, что повышает производительность и снижает затраты на переработку. Кроме того, используя такие каталитические системы, можно легко получить ранее недостижимые сополимеры этилена и α-олефина (в частности, ЭП и ЭПДМ сополимеры) с низким индексом расплава, высоким содержанием сомономера и высокой молекулярной массой.
Пример 21. Получение (2,3,4,6-тетраметилинденил)диметил-(тетра-бутиламидо)силантитандиметила
Получение 2,4,6-триметилинданона
м-Ксилол (34,1 г, 0,32 моль) и 2-бромизобутирилбромид (73,9 г, 0,32 моль) перемешивали в метиленхлориде (500 мл) при 0oC, медленно добавляли при этом AlCl3 (108,98 г, 0,82 моль) в виде твердого вещества под потоком азота в течение 20 минут. Реакционной смеси давали перемешиваться при 0oC в течение 1 часа и затем при 20oC в течение 16 часов. По истечении времени реакции смесь выливали на раздробленный лед и затем фильтровали через диатомовую землю (марки ЦелитTM). Далее смесь экстрагировали 1М HCl (2 х 100 мл), 1М NaHCO3 (1 х 100 мл) и H2O (1 х 100 мл) и органическую фазу сушили над MgSO4. Фильтрование с последующим удалением летучих веществ дало желтое масло. Вакуумная перегонка дала целевой продукт в виде бледно-желтого масла (50,4 г, выход 89,9%).
Получение 2,3,4,6-тетраметилиндена
2,4,6-Триметилинданон (30,0 г, 0,17 моль) перемешивали в диэтиловом эфире (300 мл) при 0oC, прибавляя при этом по каплям MeMgI (0,24 моль, 80,00 мл 3,0 М раствора в диэтиловом эфире). Эту смесь перемешивали еще 30 минут при 0oC и затем еще 3 часа при 20oC. По истечении времени реакции смесь выливали на раздробленный лед, подкисляли HCl и экстрагировали 1 М HCl (2 х 100 мл), 1 М NaHCO3 (1 х 100 мл) и затем H2O (1 х 100 мл). Сушка над MgSO4 с последующими фильтрованием и удалением летучих веществ дала, светло-коричневое масло. Вакуумная перегонка дала целевой продукт в виде бледно-желтого масла (28,0 г, выход 94,3%).
Получение литий-2,3,4,6-тетраметилинденида
2,3,4,6-Тетраметилинден (11,12 г, 64,52 ммоль) перемешивали в гексане (250 мл), медленно прибавляя при этом н-BuLi (70 ммоль, 28 мл, 2,5 М раствора в гексане). Смеси давали перемешиваться всю ночь. По истечении времени реакции выделяли путем фильтрования целевой продукт в виде не совсем белого твердого вещества, которое использовали без дальнейшей очистки или анализа (10,98 г, выход 95,56%).
Получение диметилсилил (2,3,4,6-тетраметилинденил)хлорида
Литий-2,3,4,6-тетраметилинденид (10,98 г, 61,6 моль) в ТГФ (50 мл) прибавляли по каплям к раствору Me2SiCl2 (25,4 г, 0,2 моль) в ТГФ (50 мл) при 0oC. Этой смеси давали перемешиваться при 20oC в течение 16 часов. По истечении времени реакции удаляли летучие вещества и остаток фильтровали с использованием гексана. Удаление гексана дало целевое соединение в виде бледно-желтого масла (16,1 г, выход 99,4%).
Получение диметилсилил(2,3,4,6-тетраметилинденил)(трет-бутиламина)
Диметилсилил(2,3,4,6-тетраметилинденил)Cl (16,1 г, 60,8 ммоль) перемешивали в гексане (200 мл), добавляя при этом NEt3 (6,51 г, 64,4 ммоль) и затем трет-бутиламин (5,61 г, 76,8 ммоль). Смеси давали перемешиваться в течение 24 часов. По истечении времени реакции смесь фильтровали и после удаления летучих веществ получили целевой продукт в виде бледно-желтого масла (18,24 г, выход 99,5%).
Получение дилитий(N-трет-бутиламидо)(диметил)(2,3,4,6-тетраметилинденил)силана
В сухом боксе растворили 7,4 г (25,4 ммоль) (N-трет-бутиламино) (диметил) (2,3,4,6-тетраметилинденил)силана в 300 мл гексана. К этому раствору прибавляли по каплям 24,5 мл (70,6 ммоль) н-BuLi (2,00 М). По окончании добавления н-BuLi раствор перемешивали 12 часов, после чего удаляли при пониженном давлении растворитель с получением 7,79 г (выход 100%) желто-оранжевого порошка.
Получение [(N-трет-бутиламидо)(диметил)(2,3,4,6-тетраметилинденил)силан] титандихлорида
В сухом боксе растворяли 9,21 г (24,8 ммоль) TiCl3 (ТГФ)3 в 75 мл ТГФ. К этому раствору добавляли при перемешивании 7,79 г (24,8 ммоль) дилитий(N-трет-бутиламидо) (диметил) (2,3,4,6-тетраметилинденил)силана в виде твердого вещества. Затем раствор перемешивали в течение 45 минут. Затем добавляли к смеси 3,45 г PbCl2 (12,4 ммоль) и перемешивали раствор еще 45 минут. Удаляли при пониженном давлении ТГФ. Остаток экстрагировали толуолом, фильтровали раствор и удаляли при пониженном давлении толуол. Затем остаток растирали с гексаном и уменьшали объем раствора, в результате чего образовывался красный осадок, который собирали путем фильтрования и промывали холодным гексаном. Твердый продукт высушивали под вакуумом с получением 5,63 г (выход 53%) продукта.
Пример 22. Получение [(N-трет-бутиламидо)(диметил)(2,3,4,6- тетраметилинденил)силан]титандиметила
В сухом боксе суспендировали в 50 мл Et2O 0,400 г [(N-трет- бутиламидо)(диметил)(2,3,4,6-тетраметилинденил)силан] титандихлорида (0,9 ммоль). К этой суспензии прибавляли по каплям 0,67 мл MeMgI (3,0 М) при перемешивании в течение 20 минут. По окончании прибавления MeMgI раствор перемешивали 40 минут. Затем удаляли при пониженном давлении Et2O, остаток экстрагировали гексаном, раствор фильтровали и, досуха выпарив при пониженном давлении фильтрат, получили 0,28 г (выход 77%) продукта.
Пример 23. Получение [(N-циклогексиламидо)(диметил) (2,3,4,6-тетраметилинденил)силан]титандиметила
Получение диметилсилил(2,3,4,6-тетраметилинденил)(циклогексиламина)
Диметилсилил(2,3,4,6-тетраметилинденил)C1 (9,95 г, 37,8 ммоль) перемешивали в гексане (200 мл), добавляя при этом Net3 (4,1 г, 40,6 ммоль) и затем циклогексиламин (4,05 г, 40,8 ммоль). Этой смеси давали перемешиваться в течение 24 часов при 20oC. По истечении времени реакции смесь фильтровали и после удаления летучих веществ получили целевой продукт в виде бледно-желтого масла (10,98 г, выход 89,3%).
Получение дилитий(N-циклогексиламидо)(диметил) (2,3,4,6- тетраметилинденил)силана
В сухом боксе растворяли 4,0 г (12,6 ммоль) (N-циклогексиламино) (диметил)(2,3,4,6-тетраметилинденил)силана в 300 мл гексана. К этому раствору прибавляли по каплям при 20oC 12,6 мл (25,2 ммоль) н-BuLi (2,00 М). После окончания добавления н-BuLi раствор перемешивали 12 часов, после чего удаляли при пониженном давлении растворитель с получением 4,12 (выход 96%) желто-оранжевого порошка.
Получение [(N-циклогексиламидо)(диметил)(2,3,4,6- тетраметилинденил)силан]титандихлорида
В сухом боксе растворяли 4,63 г (12,5 ммоль) TiCl3 (ТГФ)3 в 75 мл ТГФ. К этому раствору добавляли при перемешивании при 20oC 4,12 г (12,5 ммоль) дилитий(N-циклогексиламидо)(диметил)(2,3,4,6-тетраметилинденил)силана в виде твердого вещества. Затем раствор перемешивали в течение 45 минут. После этого добавляли к смеси 1,73 г PbCl2 (6,25 ммоль) и перемешивали раствор еще 45 минут. Удаляли при пониженном давлении ТГФ. Остаток экстрагировали толуолом, фильтровали раствор и удаляли при пониженном давлении толуол. Затем остаток растирали с гексаном и уменьшали объем раствора, в результате чего образовывался красный осадок, который собирали путем фильтрования и промывали холодным (0oC) гексаном. Твердый продукт высушивали под вакуумом с получением 1,70 г (выход 31%) продукта.
Пример 24. Получение [N-циклогексиламидо)(диметил)(2,3,4,6-тетраметилинденил)силан]титандиметила
В сухом боксе суспендировали 0,300 г [(N-циклогексиламидо)(диметил)(2,3,4,6-тетраметилинденил)силан] титандихлорида (0,675 ммоль) в 50 мл Et2O при 20oC. К этой суспензии прибавляли по каплям 0,45 мл MeMgI (3,0 М) при перемешивании в течение 20 минут. По окончании прибавления MeMgI раствор перемешивали 40 минут. Затем удаляли при пониженном давлении Et2O, остаток экстрагировали гексаном, раствор фильтровали и, досуха выпарив при пониженном давлении фильтрат, получили 0,27 г (выход 100%) продукта.
Пример 25. Получение [(N-трет-бутиламидо)(диметил)(2-пропилинденил)силан]титан(II)(1,4-дифенил-1,3-бутадиена)
В колбе емкостью 100 мл перемешивали 0,500 г (N-трет-бутиламидо)(диметил)(2-пропилинденил)силантитандихлорида (1,23 ммоль, из примера 12) с 0,225 г 1,4-дифенил-1,3-бутадиена (1,23 ммоль) в 70 мл гексана. К этому раствору прибавляли 1,0 мл 2,5 М н-BuLi (в гексане) и смесь нагревали с обратным холодильником в течение 1 часа. После охлаждения раствора до комнатной температуры его фильтровали. Остаток из фильтра промывали гексаном. Затем удаляли при пониженном давлении гексан с получением 0,460 г (выход 69%) продукта.
Пример 26. Получение [(N-циклогексиламидо)(диметил)(2,3- метилинденил)силан]титан(II) (1,4-дифенил-1,3-бутадиена)
В колбе емкостью 100 мл перемешивали 0,300 г (N-циклогексиламидо)(диметил)(2,3-метилинденил)силантитандихлорида (0,720 ммоль, из примера 23) с 0,149 г 1,4-дифенил-1,3-бутадиена (0,720 ммоль) в 70 мл гексана при 0oC. К этому раствору прибавляли 0,577 мл 2,5 М н-BuLi в гексане и смесь нагревали с обратным холодильником в течение 2 часов. После охлаждения раствора до 20oC его фильтровали. Остаток из фильтра промывали гексаном. Затем удаляли при пониженном давлении гексан с получением 0,109 г (выход 27%) продукта.
Опыты полимеризации
В двухлитровый реактор Парра загружали 740 г смешанного алканового растворителя (Изопар-Е) и 118 г 1-октенового сомономера. Из питательного резервуара емкостью примерно 75 мл при 25 фунтах на кв. дюйм (2070 кПа) добавляли водород как средство регулирования молекулярной массы в результате расширения от перепада давлений. Реактор нагревали до температуры полимеризации 140oC и насыщали этиленом при избыточном давлении 500 фунтов на кв. дюйм (3,4 МПа). Предварительно смешивали в сухом боксе по 2,0 ммоль катализатора и сокатализатора в виде 0,005 М растворов в толуоле. Через необходимый для предварительного смешивания промежуток времени раствор переносили в резервуар для ввода катализаторов и вводили в реактор. В течение 15 минут поддерживали условия полимеризации, подавая этилен по потребности. Полученный раствор отводили из реактора и добавляли к нему защищенный от внешнего воздействия фенольный антиокислитель (Ирганокс 1010 производства Ciba, Geigi Corporation). Образовавшиеся полимеры высушивали в вакуумной печи при 120oC в течение 20 часов. Результаты представлены в таблице 2.
Пример 27. Получение [(N-изопропиламидо)(диметил) (2,3,4,6-тетраметилинденил)силан]титандиметила
Получение диметилсилил(2,3,4,6-тетраметилинденил)(изопропиламина)
Диметилсилил(2,3,4,6-тетраметилинденил)C1 (22,29 г, 84,17 ммоль) перемешивали в ТГФ, добавляя при этом i-PrNH2 (28,68 мл, 336,7 ммоль). Смесь перемешивали в течение 16 часов. Удаляли при пониженном давлении летучие вещества. Остаток экстрагировали гексаном и фильтровали через диатомовую землю на 10-15 мм стеклянной фритте. Удаляли при пониженном давлении гексан с получением продукта в виде желтого масла (17,23 г, 71%).
Получение [(N-изопропиламидо)(диметил)(2,3,4,6- тетраметилинденил)силан] титандихлорида
В сухом боксе растворяли 17,23 г (59,93 ммоль) диметилсилил(2,3,4,6-тетраметилинденил)(изопропиламина) в 350 мл гексана в круглодонной 500 мл колбе. Прибавляли посредством шприца два эквивалента н-BuLi (47,94 мл, 2,5 М в гексане). Перемешивали реакционную смесь 12 часов. Удаляли при пониженном давлении растворитель с получением оранжевого порошка. Порошок растворяли в 250 мл ТГФ. Добавляли TiCl3 (ТГФ)3 (22,2 г, 59,93 ммоль) в виде твердого вещества. Через 15 минут добавляли CH2Cl2 (2,48 мл, 29,97 ммоль). Через 2 часа удаляли при пониженном давлении растворитель. Остаток экстрагировали толуолом и фильтровали через диатомовую землю на 10-15 мм стеклянной фритте. Удаляли при пониженном давлении толуол. Остаток суспендировали в гексане и фильтровали на 10-15 мм стеклянной фритте. Остаток высушивали при пониженном давлении с получением красного порошка. Выход: 12,3 г, 51%.
Получение [(N-изопропиламидо)(диметил)(2,3,4,6- тетраметилинденил)силан] титандиметила
В сухом боксе суспендировали [(N-изопропиламидо)(диметил) (2,3,4,6-тетраметилинденил)силан] титандихлорид (6,92 г, 17,21 ммоль) в 150 мл Et2O в 250 мл круглодонной колбе. Прибавляли два эквивалента 3,0 М раствора MeMgCl (11,41 мл, 34,23 ммоль) в ТГФ. Перемешивали смесь в течение одного часа. Удаляли при пониженном давлении летучие вещества. Остаток экстрагировали гексаном и фильтровали через диатомовую землю на 10-15 мм стеклянной фритте. Удаляли при пониженном давлении гексан с получением оранжевого порошка. Выход: 5,8 г, 93%.
Пример 28. Получение [(N-изопропиламидо)(диметил)(2,3,4,6- тетраметилинденил)силан]титан(1,4-дифенил-1,3-бутадиена)
В сухом боксе суспендировали 0,50 г (1,24 ммоль) [(N- изопропиламидо)(диметил)(2,3,4,6-тетраметилинденил)силан] титандихлорида в 60 мл циклогексана в 100 мл круглодонной колбе. Добавляли 1,4-дифенил-1,3-бутадиен (0,255 г, 1,24 ммоль) в виде твердого вещества. Прибавляли посредством шприца два эквивалента н-BuLi (0,989 мл, 2,5 М в гексанах). Колбу снабжали обратным холодильником и нагревали в течение одного часа. Реакционную смесь, охладив, фильтровали через диатомовую землю (ЦелитTM) на 10-15 мм стеклянной фритте. Соли и вспомогательное фильтровальное вещество промывали 50 мл пентана. Удалив при пониженном давлении растворитель, получили красно-коричневый порошок. Выход: 300 мг, 45%.
Опыты полимеризации
Полимеризацию проводили в реакторе с мешалкой емкостью 3,8 литра, в который загружали 1440 г Изопара Е (смешанные алканы: производство Exxon Chemicals Inc. ), 132 г 1-октена и 10 ммоль водорода. Реактор нагревали до 130 г и насыщали этиленом до избыточного давления 450 фунтов на квадратный дюйм (4,5 МПа). Катализатор приготавливали в сухом боксе путем набора в шприц вместе 5,0 ммоль (1,0 мл, 0,005 М) металлокомплекса, 15,0 ммоль (1,0 мл, 0,015 М) сокатализатора, триспентафторфенилборана (TPFPB) и 50,0 ммоль (1,0 мл, 0,05 М) модифицированного выносителя (метилалюмоксан производства Akzo-Nobel) с дополнительным Изопаром Е для получения общего объема 17 мл. Затем каталитический раствор переносили в шприце в контур ввода катализатора и вводили в реактор примерно за 4 минуты с использованием потока растворителя под высоким давлением. Полимеризацию продолжали 10 минут, подавая этилен по потребности, чтобы поддерживать избыточное давление 445 фунтов на квадратный дюйм (4,5 МПа). Затем полимерный раствор выливали из реактора в продуваемый азотом стеклянный бачок, содержащий примерно 15 мл изопропанола. Добавляли 20 мл аликвоту стабилизирующего раствора, приготовленного путем растворения 6,66 г ИргафосаTM 168 и 3,33 г ИрганоксаTM 1010 в 500 мл толуола. Выливали полимерный раствор в лоток, сушили на воздухе всю ночь, после чего тщательно высушивали в вакуумной печи в течение двух дней. Результаты опытов полимеризации с использованием металлокомплексов по настоящему изобретению и опыта для сравнения представлены в таблице 3.
Сравнивая приведенные выше результаты, можно видеть, что катализаторы по настоящему изобретению дают полимерный продукт, имеющий значительно более низкий индекс расплава при сравнимых условиях, что указывает на их значительно более высокую каталитическую активность при сравнимых условиях полимеризации.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПЛЕКСЫ МЕТАЛЛОВ, СОДЕРЖАЩИЕ ЛИГАНДЫ 3-АРИЛЗАМЕЩЕННОГО ИНДЕНИЛА, КАТАЛИЗАТОР ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ И СПОСОБ ПОЛИМЕРИЗАЦИИ | 1997 |
|
RU2186073C2 |
| КОМПЛЕКСЫ ТИТАНА (II) ИЛИ ЦИРКОНИЯ (II), КАТАЛИТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛИМЕРИЗАЦИИ ЭТИЛЕННЕНАСЫЩЕННЫХ ОЛЕФИНОВ | 1994 |
|
RU2135509C1 |
| АКТИВАТОР КАТАЛИЗАТОРОВ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ, КАТАЛИТИЧЕСКАЯ СИСТЕМА И СПОСОБ ПОЛИМЕРИЗАЦИИ | 1997 |
|
RU2178422C2 |
| ПОЛИОЛЕФИНОВАЯ СОПОЛИМЕРНАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛИМЕРИЗАЦИИ (ВАРИАНТЫ), МЕТАЛЛОЦЕНОВЫЙ КАТАЛИЗАТОР, ПЛЕНКА НА ОСНОВЕ КОМПОЗИЦИИ И СМЕСЬ ДВУХ ИЛИ БОЛЕЕ ПОЛИМЕРНЫХ КОМПОНЕНТОВ | 1997 |
|
RU2190632C2 |
| БИСЦИКЛОПЕНТАДИЕНИЛДИЕНОВЫЕ КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ, СПОСОБ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1995 |
|
RU2135508C1 |
| ТЕРМОРЕАКТИВНЫЕ ЭЛАСТОМЕРЫ | 1995 |
|
RU2159779C2 |
| МОНООЛЕФИН/ПОЛИЕНОВЫЕ СОПОЛИМЕРЫ, СПОСОБ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ МОНООЛЕФИН/ПОЛИЕНОВЫЕ СОПОЛИМЕРЫ, И ИЗДЕЛИЯ, ИЗГОТОВЛЕННЫЕ ИЗ НИХ | 1996 |
|
RU2167885C2 |
| ГАЗОФАЗНАЯ ПОЛИМЕРИЗАЦИЯ ОЛЕФИНОВ (ВАРИАНТЫ) | 1994 |
|
RU2139296C1 |
| ЗАМЕЩЕННЫЙ МОНОЦИКЛОПЕНТАДИЕНИЛМЕТАЛЛОКОМПЛЕКС, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ (СО)ПОЛИМЕРОВ ЭТИЛЕНА | 1990 |
|
RU2095363C1 |
| КОМПОЗИЦИЯ КАТАЛИЗАТОРА, СОДЕРЖАЩАЯ АГЕНТ ЧЕЛНОЧНОГО ПЕРЕНОСА ЦЕПИ ДЛЯ ОБРАЗОВАНИЯ СОПОЛИМЕРА ЭТИЛЕНА | 2005 |
|
RU2359979C2 |


Описываются новые катализаторы координационной полимеризации, содержащие металлокомплекс и активирующий сокатализатор. Отличие состоит в том, что металлокомплекс имеет общую формулу (1), где R1 и R2 независимо означают водород или C1-6 алкил, при условии, что R1 и R2 одновременно не являются водородом; R3, R4, R5 и R6 независимо означают водород, C1-6 алкил или фенильную группу; М означает титан; Y означает -NR*-; Z* означает SiR2; R* означает гидрокарбил; р=0, 1 или 2; q=0 или 1; при условии, что когда р=2, q= 0, М находится в формальном состоянии окисления +4, и Х означает галогенид или гидрокарбид; когда р=1, q=1, М находится в формальном состоянии окисления +2 и X' означает нейтральный сопряженный диен, замещенный двумя гидрокарбильными группами, причем указанный X' образует π-комплекс с М; активирующий сокатализатор содержит алюмоксан и триспентафторфенилборан. Описывается также способ полимеризации олефинов. Предлагаемые катализаторы и способ обеспечивают высокоэффективное производство высокомолекулярных олефиновых полимеров в широких пределах условий полимеризации, в частности при повышенных температурах. 2 с. и 5 з.п. ф-лы, 3 табл.
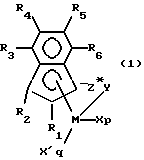
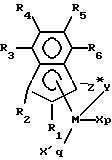
R1 и R2 независимо означают водород или С1-6 алкил при условии, что R1 и R2 одновременно не являются водородом;
R3, R4, R5 и R6 независимо означают водород, С1-6 алкил или фенильную группу;
М означает титан;
Y означает -NR*-;
Z* означает SiR2;
R* означает гидрокарбил;
p = 0, 1 или 2;
q = 0 или 1, при условии, что когда р = 2, q = 0, М находится в формальном состоянии окисления +4 и Х означает галогенид или гидрокарбил;
когда р = 1, q = 0, М находится в формальном состоянии окисления +3 и Х означает 2-(N,N-димeтил)aминoбeнзил; когда р = 0, q = 1, М находится в формальном состоянии окисления +2 и Х' означает нейтральный сопряженный диен, замещенный двумя гидрокарбильными группами, причем указанный Х' образует π-комплекс с М;
и активирующий сокатализатор содержит алюмоксан и триспентафторфенилборан.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Катализатор высокотемпературной полимеризации этилена и сополимеризации этилена с @ -олефинами | 1987 |
|
SU1641193A3 |
| US 5026798 A, 25.06.1991 | |||
| 1972 |
|
SU416815A1 | |
| US 5096867 A, 17.03.1992. | |||

