Изобретение относится к 6-диметиламинометил-1-фенилциклогексановым соединениям, способу их получения и к применению этих соединений в лекарственных средствах.
Лечение хронических и нехронических болезненных состояний играет в медицине важную роль. В настоящее время во всем мире существует большая потребность в дополнительном, и не только за счет применения опиоидных препаратов, но и высокоэффективном обезболивающем лечении. Настоятельная необходимость в осуществлении практических мер по целенаправленному, с учетом индивидуальных особенностей пациента лечению хронических и нехронических болезненных состояний, причем под этим следует понимать успешное и удовлетворительное для пациентов лечение их болезненных ощущений, находит свое отражение в появившихся в последнее время многочисленных научных публикациях, посвященных прикладной анальгетике, а также фундаментальным исследованиям по проблемам ноцицепции.
Опиоиды применяют в течение многих лет для обезболивающего лечения, хотя они и вызывают целый ряд побочных эффектов, таких как психическая зависимость, депрессия дыхания, ингибирующее воздействие на жедудочно-кишечный тракт и запоры. По этой причине их применение допустимо только лишь при соблюдении соответствующих мер предосторожности, требующих специальных предписаний, особенно в тех случаях, например, когда препараты назначают на длительный период времени или в высокой дозировке (см. Goodman, Gilman "The Pharmacological Basis of Therapeutics", изд-во Pergamon Press, Нью-Йорк, 1990).
Трамадолгидрохлорид, т. е. гидрохлорид (1RS, 2RS)-2-[(диметиламино)метил] -1-(3-метоксифенил)циклогексанола, занимает среди основных эффективных анальгетических средств особое место, поскольку это активное вещество обладает сильным обезболивающим действием, не вызывая, в отличие от опиоидов, известных побочных явлений (см. Journ. Pharmacol. Exp. Ther. 267, 331 (1993)). Трамадол представляет собой рацемат и состоит из равных количеств (+)-и (-)-энантиомеров. In vivo это действующее вещество образует метаболит O-десметилтрамадол, также представленный в виде смеси энантиомеров. Проведенные исследования показали, что как энантиомеры трамадола, так и энантиомеры метаболитов трамадола способствуют достижению анальгетического эффекта (см. Journ. Pharmacol. Exp. Ther. 260, 275 (1992)).
Из публикации Chem. Pharm. Bull. 32, 2279 (1984) известны соединения формулы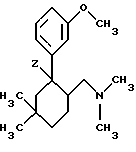
в которой Z означает H или OH. Эти субстанции обладают заметно более слабым анальгетическим действием по сравнению с трамадолом.
Положенная в основу изобретения задача состояла в разработке и создании обладающих анальгезирующим действием субстанций, предназначенных для лечения сильных болей и не вызывающих при этом побочных эффектов, типичных для опиоидов. Кроме того, создаваемые субстанции не должны были обладать свойствами, способствующими, как это наблюдается в ряде случаев при лечении трамадолом, проявлению побочных действий, таких, например, как тошнота и рвота.
Было найдено, что требованиям, выдвинутым при создании новых субстанций, отвечают определенные 6-диметиламинометил-1-фенилциклогексановые соединения. Эти соединения отличаются ярко выраженным анальгезирующим действием и по своей эффективности заметно превосходят трамадол, равно как и известные из публикаций Arzneim. -Forsch. /Drug. Res. 28 (IA) 107 (1978) соединения формулы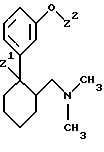
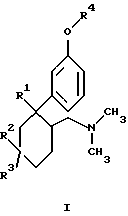
где Z1 означает H, OH или Cl, а Z2 означает CH3, или где Z1 означает OH, а Z2 означает H. Предметом изобретения являются в соответствии с этим 6-диметиламинометил-1-фенилциклогексановые соединения формулы I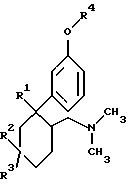
в которой
R1 представляет собой H, OH, Cl или F,
R2 и R3 являются идентичными или разными и означают H, C1-C4алкил, бензил, CF3, OH, OCH2-C6H5, O-C1-C4алкил, Cl или F при условии, что по крайней мере один из радикалов R2 или R3 означает H,
R4 означает H, CH3, CO(OC1-C4алкил)2, СO(OC1-C5алкил), CO-NH-C6H4-C1-C3алкил), CO-C6H4-R5, CO-C1-C5далкил, CO-CHR6-NHR7 или незамещенную, либо замещенную пиридиловую, тиениловую, тиазоиловую или фенильную группу,
R5 означает OC(O)C1-C3алкил в орто-положении или CH2-N(R8)2 в мета- либо пара-положении, причем R8 представляет собой C1-C4алкил или оба радикала R8 вместе с N представляют собой 4-морфолиновый радикал, и
R6 и R7 являются идентичными либо разными и означают H или C1-C6алкил при условии, что если оба радикала R2 и R3 означают H, то R4 не является CH3, если R1 означает H, OH или Cl, либо R4 не является H, если R1 означает OH,
в виде их оснований или солей физиологически приемлемых кислот.
Предпочтительные 6-диметиламинометил-1-фенилциклогексановые соединения соответствуют формуле I, где R1 означает H, OH или F. К особенно предпочтительным относятся 6-диметиламинометил-1-фенилциклогексановые соединения в виде их диастереомеров с конфигурацией согласно формуле Ia, в которой фенильное кольцо и диметиламинометиловая группа находятся в трансположении относительно друг друга: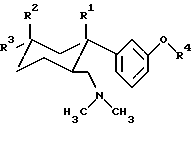
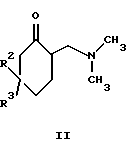
Другим предметом настоящего изобретения является способ получения 6-диметиламинометил-1-фенилциклогексановых соединений формулы I, в которой R1 означает OH, а R2 и R3 являются идентичными либо разными и означают H, C1-C4алкил, бензил, CF3, Cl или F при условии, что по крайней мере один из радикалов R2 или R3 является H, а R4 означает H, CH3 или незамещенную либо замещенную пиридиловую, тиениловую, тиазоиловую или фенильную группу, при условии, что R4 не представляет собой ни CH3, ни H, если оба радикала R2 и R3 означают H, причем способ отличается тем, что β-диметиламинокетон формулы II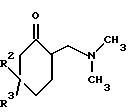
подвергают взаимодействию с металлорганическим соединением формулы III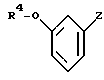
в которой Z означает MgCl, MgBr, MgI или Li, с получением соединения формулы I.
Реакция β-диметиламинокетона с соединением Гриньяра формулы III или с литийорганическим соединением формулы III может проводиться в простом алифатическом эфире, например в диэтиловом эфире, и/или тетрагидрофуране в диапазоне температур от -70oC до +60oC. Литийорганические соединения формулы III могут быть получены взаимодействием соединения формулы III, в которой Z означает Cl, Br или I, например, с раствором п-бутиллития/гексана за счет обменной реакции между галогеном и литием. При взаимодействии β-диметиламинокетона формулы II с металлорганическим соединением получают 6-диметиламинометил-1-фенилциклогексановые соединения с предпочтительной относительной конфигурацией согласно формуле Ia.
β-Диметиламинокетоны формулы II получают из кетонов формулы IV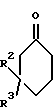
взаимодействием с диметиламиногидрохлоридом и формальдегидом в ледяном уксусе либо в C1-C4алкиловом спирте или взаимодействием с метиленхлоридом диметиламмония в ацетонитриле при ацетилхлоридном катализе (см. Synthesis 1973, 703; Tietze, Eicher в "Reaktionen und Synthesen im Organisch-Chemischen Praktikum", изд-во Thieme-Verlag, Штутгарт 1991, стр. 189). Образующиеся в ходе реакции аминометилирования диастереомерные β-диметиламинокетоны могут быть получены в виде чистых диастереомеров либо путем разделения с помощью хроматографии на колонке, либо путем фракционной кристаллизации их гидрохлоридов из органического растворителя, например, из 2-бутанона и/или ацетона.
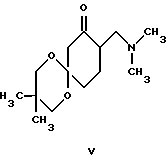
Еще одним предметом изобретения является способ получения 6-диметиламинометил-1-фенилциклогексановых соединений формулы I, в которой R1 является OH, один из радикалов R2 или R3 означает H, а другой означает OH, O-C1-C4алкил или OCH2-C6H5, и R4 означает H, CH3 или незамещенную, либо замещенную пиридиловую, тиениловую, тиазоиловую или фенильную группу, отличающийся тем, что β-диметиламинокетон, имеющий спироциклическую ацеталевую структуру формулы V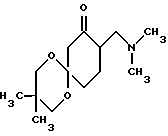
подвергают взаимодействию с металлорганическим соединением формулы III
в которой Z означает MgCl, MgBr, MgI или Li, с получением соединения формулы VI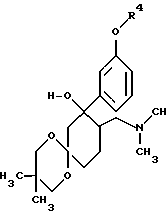
после чего полученное соединение формулы VI путем катализированной протонами реакции деацеталирования переводят в соответствующее производное кетона формулы VIII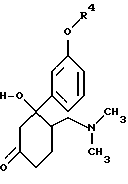
и затем полученное производное кетона с помощью комплексного гидрида щелочного металла восстанавливают до соединения формулы I, в которой один из радикалов R2 или R3 означает OH, и при необходимости полученное восстановлением соединение формулы I после перевода в соль щелочного металла переводят с помощью C1-C4алкил- либо бензилгалогенида в соединение формулы I, в которой один из радикалов R2 или R3 означает O-C1-C4алкил или OCH2-C6H5.
Реакцию восстановления соединения формулы VIII осуществляют предпочтительно с помощью борогидрида натрия либо алюмогидрида лития в органическом растворителе, как, например, тетрагидрофуран, простой диэтиловый эфир, и/или в C2-C4алкиловом спирте. Если по способу согласно изобретению требуется получить соединение, где R2 или R3 означает O-C1-C4алкил или OCH2Ph, то полученное восстановлением соединение с помощью гидрида щелочного металла, например гидрида натрия и/или гидрида калия, в растворителе, таком как диметилформамид, переводят в соответствующее соединение, представляющее собой соль щелочного металла, после чего его подвергают взаимодействию с C1-C4алкил-либо бензилгалогенидом.
β-Диметиламинокетоны со спироциклической ацеталевой структурой формулы V могут быть получены из 9-диметиламинометил-3,3-диметил-1,5-диоксаспиро[5.5] ундекан-8-она формулы VII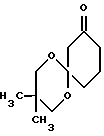
получаемого целевым моноацеталированием циклогексан-1,3-диона, взаимодействием с метиленхлоридом диметиламмония в ацетонитриле при ацетилхлоридном катализе (см. Synthesis 1973. 703; Tietze, Eicher в "Reaktionen und Synthesen im Organisch-Chemischen Praktikum", изд-во Thieme-Verlag, Штутгарт 1991, стр. 189).
Другим предметом изобретения является способ получения 6-диметиламинометил-1-фенилциклогексановых соединений формулы I, в которой R1 представляет собой H, а R2 и R3 являются идентичными либо разными и означают H, C1-C4алкил, бензил, CF3, OCH2-C6H5 или F при условии, что по крайней мере один из радикалов R2 или R3 является H и R4 означает H, CH3, незамещенную либо замещенную пиридиловую, тиениловую, тиазоиловую либо фенильную группу, отличающийся тем, что соединение формулы I, в которой R1 представляет собой Cl, подвергают взаимодействию с борогидридом цинка, цианборогидридом цинка или цианборогидридом олова в простом эфире или соединение формулы I, в которой R1 представляет собой OH, подвергают взаимодействию с никелем Ренея в C2-C4алкиловом спирте.
Реакцию соединения формулы I, в которой R1 представляет собой Cl, с борогидридом проводят предпочтительно в простом диэтиловом эфире и/или тетрагидрофуране в диапазоне температур от 0 до 30oC. Обменную реакцию между соединением формулы I, в которой R1 представляет собой OH, и никелем Ренея проводят предпочтительно в C2-C4алкиловом спирте в диапазоне температур от 70 до 100oC (см. Journ. Org. Chem. 59, 6895 (1994) и Angew. Chem. 95, 568 (1983)).
Циклогексановые соединения формулы I, в которой R1 является H, один из радикалов R2 или R3 означает H, а другой означает Cl и R4 представляет собой H, CH3 или незамещенную, либо замещенную пиридиловую, тиениловую, тиазоиловую или фенильную группу, могут быть получены по известной методике из соответствующих циклогексановых соединений формулы I, в которой один из радикалов R2 или R3 является H, а другой представляет собой OH и R1 и R4 имеют одно из указанных выше значений, взаимодействием с тионилхлоридом или со смесью соляная кислота/хлорид цинка (см. Journ. Chem. Soc. 1943. 636; Journ. Org. Chem. 17, 1116 (1952)).
Еще одним предметом настоящего изобретения является способ получения 6-диметиламинометил-1-фенилциклогексановых соединений формулы I, в которой R1 представляет собой H, R2 и R3 являются идентичными либо разными и означают H, C1-C4алкил, бензил, CF3 или F при условии, что по крайней мере один из радикалов R2 или R3 является H, а R4 означает CH3, отличающийся тем, что соединение формулы I, где R1 представляет собой Cl, гидрируют в присутствии палладиевого катализатора в C1-C4алкиловом спирте. Гидрирование осуществляют предпочтительно при давлении в пределах от 1 до 100 бар и в диапазоне температур от 20 до 80oC.
Соединения формулы I, в которой R1 представляет собой H, R2 и R3 являются идентичными либо разными и означают H, C1-C4алкил, бензил, CF3 или F, а R4 представляет собой H, могут быть получены также из соответствующих метоксифенильных соединений путем продолжительного, в течение нескольких часов, нагрева с концентрированной бромистоводородной кислотой (см. Chem. Rev. 54, 615 (1954); Journ. Am. Chem. Soc. 74, 1316 (1952)).
Циклогексановые соединения формулы I, в которой R1 является Cl и ни один из обоих радикалов R2 и R3 не означает OH, могут быть получены в виде свободного основания либо в виде гидрохлорида взаимодействием соединения формулы I, в которой R1 представляет собой OH, с тионилхлоридом при отсутствии растворителя в диапазоне температур от 0 до 20oC. В этом способе реакция хлоробмена протекает с образованием соответствующей конфигурации. Циклогексановые соединения формулы I, где R1 является Cl, а R2 или R3 представляют собой OH, могут быть получены по известной методике из соответствующих соединений, где R1 является Cl, а R2 или R3 означают OCH2-C6H5.
Предметом изобретения является далее способ получения 6-диметиламинометил-1-фенилциклогексановых соединений формулы I, в которой R1 представляет собой F, R2 и R3 являются идентичными либо разными и означают H, C1-C4алкил, бензил, CF3, OCH2-C6H5, Cl или F при условии, что по крайней мере один из радикалов R2 или R3 представляет собой H, а R4 означает CH3 или незамещенную, либо замещенную пиридиловую, тиениловую, тиазоиловую или фенильную группу, отличающийся тем, что соединение формулы I, в которой R1 представляет собой OH, подвергают взаимодействию с диметиламиносульфотрифторидом. Эту реакцию проводят предпочтительно в органическом растворителе, например дихлорметане, 1,1,2-трихлорэтане и/или толуоле, в диапазоне температур от -50oC до +30oC (см. Org. Reac. 35, 513 (1988)).
Соединения формулы I, в которой R1 представляет собой F, R2 и R3 являются идентичными либо разными и означают H, C1-C4алкил, бензил, CF3, OCH2-C6H5, Cl или F при условии, что по крайней мере один из радикалов R2 и R3 представляет собой H, a R4 означает H, могут быть получены взаимодействием соединений формулы I, в которой R1 означает OH, а R4 означает триалкилсилиловую группу, с диметиламиносульфотрифторидом и последующим расщеплением силилового эфира с помощью водных минеральных кислот. Предпочтительной триалкилсилиловой группой является диметил-трет-бутилсилиловая группа.
Другая возможность получения 6-диметиламинометил-1-фенилциклогексановых соединений формулы I, в которой R1 означает OH или H, R4 означает H и ни один из обоих радикалов R2 и R3 не означает Cl, F или CF3, состоит в селективном расщеплении эфира с помощью гидрида диизобутилалюминия 6-диметиламинометил-1-(3-метоксифенил)циклогексанового соединения, осуществляемого предпочтительно в ароматическом углеводороде, например толуоле, в диапазоне температур от 60 до 130oC (см. Synthesis 1975, 617).
Кроме того, 6-диметиламинометил-1-фенилциклогексановые соединения формулы I, в которой R1 означает OH, H или F, R4 означает H, а R2 и R3 являются идентичными либо разными и означают H, C1-C4алкил, бензил, CF3, F, Cl, OH или O-C1-C4алкил, могут быть получены из соответствующих 6-диметиламинометил-1-(3-бензилоксифенил)циклогексановых соединений путем восстановительного дибензилирования. Это дибензилирование осуществляют предпочтительно в присутствии платины либо палладия на носителе в присутствии водорода в растворителе, например, в уксусной кислоте и/или C1-C4алкиловом спирте, при давлении в пределах от 1 до 100 бар и в диапазоне температур от 20 до 100oC.
6-диметиламинометил-1-фенилциклогексановые соединения формулы I, в которой OR4 представляет собой фосфатную, карбонатную, карбаматную, карбоксилатную, арилокси- или гетероарилоксигруппу, могут быть получены взаимодействием соответствующих 6-диметиламинометил-1-(3-гидроксифенил)циклогексановых соединений в виде их солей щелочных металлов с солью щелочного металла диалкилхлорфосфата, с алкилхлорформиатом, с арил- либо гетероарилизоцианатом, с хлоридом карбоновой кислоты или с арил- , либо гетероарилгалогенидом. Эти реакции проводят обычно в растворителе, например толуоле, дихлорметане, простом диэтиловом эфире и/или тетрагидрофуране, в диапазоне температур от -15oC до +110oC (см. Drugs of the Future 16, 443 (1991); Journ. Med. Chem. 30, 2008 (1989) и 32, 2503 (1989); Journ. Org. Chem. 43, 4797 (1978); Tetrahedron Lett. 1977. 1571; Journ. Pharm. Sci. 51, 774 (1968)). Взаимодействие с арил- либо гетероарилгалогенидом проводят в присутствии медного порошка и/или галогенида меди (I) в качестве катализатора.
6-диметиламинометил-1-фенилциклогексановые соединения формулы I, в которой OR4 представляет собой α-аминокислотную группу, могут быть получены взаимодействием соответствующего 6-диметиламинометил-1-(3-гидроксифенил)циклогексанового соединения с соответствующей 2-трет-бутоксикарбониламинокарбоновой кислотой с использованием триэтиламина и реагентов сочетания, как, например, гексафторфосфат бензтриазол-1-илокситрипирролидинфосфония, в растворителе, например в дихлорметане.
Предлагаемые согласно изобретению соединения с помощью физиологически приемлемых кислот, таких, как соляная кислота, бромистоводородная кислота, серная кислота, метансульфоновая кислота, муравьиная кислота, уксусная кислота, щавелевая кислота, янтарная кислота, винная кислота, миндальная кислота, фумаровая кислота, молочная кислота, лимонная кислота, глутаминовая кислота и/или аспарагиновая кислота, могут переводиться по известной методике в их соли. Реакции солеобразования осуществляют предпочтительно в растворителе, например в диэтиловом эфире, диизопропиловом эфире, алкиловом эфире уксусной кислоты, ацетоне и/или 2-бутаноне. Для получения гидрохлоридов может использоваться, кроме того, триметилхлорсилан в присутствии воды в одном из вышеуказанных растворителей.
Предлагаемые согласно изобретению соединения обладают ярко выраженным анальгетическим эффектом и токсикологически совершенно безопасны. Их можно применять поэтому в качестве фармацевтических действующих веществ. Исходя из этого, предметом изобретения является также применение 6-диметиламинометил-1-фенилциклогексановых соединений формулы I в качестве действующих веществ в лекарственных средствах, предпочтительно в качестве таковых в анальгетиках.
Наряду по крайней мере с одним 6-диметиламинометил-1-фенилциклогексановым соединением формулы I лекарственные средства по изобретению содержат также носители, наполнители, растворители, разбавители, красители и/или связующие вещества. Выбор этих вспомогательных веществ, равно как и применяемые количества зависят от способов введения лекарственного средства, т. е. назначают ли его для орального, внутривенного, внутрибрюшинного, внутрикожного, внутримышечного, внутриназального или местного введения, например, при кожных инфекциях, поражениях слизистых оболочек и глаз. Для орального введения пригодны композиции в виде таблеток, драже, капсул, гранулятов, капель, микстур и сиропов; для парентерального, местного и ингаляционного введения могут применяться растворы, суспензии, легко реконструируемые сухие композиции, а также аэрозоли. В качестве примеров готовых препаративных форм, предназначенных для чрезкожного введения, можно назвать соединения по изобретению в депо в растворимой форме или в пластыре, при необходимости с добавками средств, способствующих кожной пенетрации. Из вводимых оральным путем или через кожу препаративных форм высвобождение соединений по изобретению может происходить постепенно.
Назначаемое пациентам количество действующих веществ варьируют в зависимости от веса пациента, от способа введения, от показания и степени тяжести заболевания. Обычно назначают от 10 до 500 мг/кг веса тела по крайней мере одного 6-диметиламинометил-1-фенилциклогексанового соединения формулы I.
Примеры
Если не указано иное, то использовали петролейный эфир с температурой кипения 50-70oC. Термин простой эфир означает диэтиловый эфир.
В качестве неподвижной фазы в хроматографии на колонках использовали силикагель 60 (0,040-0,063 мм), фирма E. Merck, Дармштадт.
Исследования с помощью тонкослойной хроматографии проводили с использованием пластинок для высокоэффективной тонкослойной хроматографии, силикагель 60 F 254, фирма E. Merck, Дармштадт.
Разделение рацематов осуществляли на колонке Chiracel OD.
Соотношения в смесях системы растворителей для всех хроматографических исследований указаны во всех случаях в отношении объема (объем/объем).
КТ означает комнатную температуру, tпл означает температуру плавления.
Пример 1
(-)-(1S, 2S)-3-(2-диметиламинометил-1-фторциклогексил)фенол, гидрохлорид (-1)
Стадия 1:
(-)-(1S, 2S)-1-(3-бензилоксофенил)-2-диметиламинометилциклогексанол, гидрохлорид (-2)
Из (-)-(1S, 2S)-3-(2-диметиламинометил-1-гидроксициклогексил)фенола, гидрохлорида с помощью водного раствора гидрокарбоната натрия/дихлорметана выделяли основание и после сушки раствора удаляли перегонкой дихлорметан. 135 г (545 ммолей) основания растворяли в 675 мл сухого диметилформамида и смешивали с 29,1 г 50%-ного гидрида натрия несколькими порциями. После добавки 69 мл (594 ммоля) бензоилхлорида нагревали в течение 3 ч до 70oC. Затем охлаждали до комнатной температуры и реакционную смесь сливали на ледяную воду. Далее трижды экстрагировали порциями по 150 мл этилацетата соответственно. После сушки органических фаз над сульфатом магния растворитель отгоняли. Остаток (204 г) растворяли в 1000 мл 2-бутанона и смешивали с 76 мл (600 ммолей) триметилхлорсилана и 10,9 мл воды. При комнатной температуре выпадали в виде кристаллов 190 г (93% от теории) гидрохлорида (-2) с температурой плавления 207-210oC.
[α]
Стадия 2:
(-)-(1S, 2S)-[2-(3-бензилоксифенил)-2-фторциклогексилметил] диметиламин (-3)
К раствору из 80,6 г (500 ммолей) диэтиламиносульфотрифторида в 450 мл сухого дихлорметана при -40oC по каплям добавляли 147,7 г (435 ммолей) соединения (-2), растворенных в 1500 мл сухого дихлорметана. После полного введения добавки перемешивали в течение 120 мин при указанной температуре и затем нагревали до комнатной температуры. После дальнейшего перемешивания еще в течение 1 ч при комнатной температуре охлаждали до 0-5oC и гидролизовали 500 мл воды. Водную фазу дважды экстрагировали 200 мл дихлорметана. После сушки органических фаз растворитель отгоняли перегонкой. Полученную сырую смесь (185 г) разделяли на четыре порции. Каждую из этих порций подавали на колонку 8 х 50 см, заполненную силикагелем, и элюировали этилацетатом/метанолом (соотношение 1: 1). В общей сложности получили 103 г (69% от теории) основания соединения (-3) в виде светло-желтого вязкого масла.
Стадия 3:
(-)-(1S, 2S)-3-(2-диметиламинометил-1-фторциклогексил)фенол, гидрохлорид (-1)
7,75 г (22,7 ммоля) соединения (-3) растворяли в 40 мл сухого метанола и смешивали в аппарате для гидрогенизации с 2,0 г палладия ца активированном угле (10% Pd). После перемешивания в течение 35 мин при комнатной температуре было использовано 430 мл водорода. Катализатор удаляли путем фильтрации, а метанол перегонкой. В результате получали 6,3 г основания, из которого затем с помощью триметилхлорсилана/воды в 2-бутанон/ацетоне (1: 1) было получено 4,9 г (75% от теории) гидрохлорида (-1).
tпл 188-190oC
[α]
Пример 2
(+)-(1R, 2R)-3-(2-диметиламинометил-1-фторциклогексил)фенол, гидрохлорид (+1)
Исходя из (+)-(1R, 2R)-3-(2-диметиламинометил-1-гидроксициклогексил)фенола, гидрохлорида, в условиях, описанных в примере 1, получали энантиомер (+1) с выходом 48% от теории.
tпл 188-190oC
[α]
Пример 3
(+)-(1R, 2R)-[2-хлор-2-(3-метоксифенил)циклогексилметил] диметиламин, гидрохлорид (+4)
10 г (33,4 ммоля) (+)-(1R, 2R)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола, гидрохлорида, при комнатной температуре смешивали с 10 мл тионилхлорида. Затем для удаления избыточного тионилхлорида в течение двух часов через реакционную смесь пропускали азот. После повторной добавки 10 мл тионилхлорида реакционную смесь оставляли на 12 ч и затем в течение 2,5 ч с помощью потока азота повторно удаляли избыточный тионилхлорид. После сушки остаток растворяли в 50 мл ледяного 2-бутанона и при перемешивании смешивали с 50 мл диизопропилового эфира, при этом гидрохлорид выпадал в виде кристаллов. Для полного завершения реакции суспензию перемешивали еще в течение двух часов при охлаждении ледяной баней. В результате получали 5,9 г (55% от теории) гидрохлорида (+4).
tпл 120-121oC (разложение)
[α]
Пример 4
(-)-(1S, 2S)-[2-хлор-2-(3-метоксифенил)циклогексилметил] диметиламин, гидрохлорид (-4)
Исходя из (-)-(1S, 2S)-2-диметиламинометил-1-(3-метоксифенил)циклогексанола, гидрохлорида, в условиях, описанных в примере 3, получали энантиомер соединения (-4) с выходом 55% от теории.
tпл 120-122oC
[α]
Пример 5
(+)-(1R, 2R)-3-(1-хлор-2-диметиламинометилциклогексил)фенол, гидрохлорид (+5)
3,6 г (12,6 ммолей) (+)-(1S, 2S)-3-(2-диметиламинометил-1-гидроксициклогексил)фенола, гидрохлорида, смешивали при комнатной температуре с 3 мл тионилхлорида, после чего в течение одного часа перемешивали при комнатной температуре. Для удаления избыточного тионилхлорида через реакционную смесь в течение двух часов пропускали азот. После повторной добавки 4 мл тионилхлорида перемешивали в течение двух часов при комнатной температуре и затем в течение двух часов с помощью потока азота удаляли избыточный тионилхлорид. Остаток растворяли в 70 мл 2-бутанона и при перемешивании смешивали с 50 мл диизопропилового эфира. Выпавший в виде кристаллов гидрохлорид трижды промывали при декантировании 25 мл 2-бутанона. После сушки получали 1,8 г (46% от теории) гидрохлорида (+5).
tпл 145-146oC (разложение)
[α]
Пример 6
(-)-(1S, 2S)-3-(1-хлор-2-диметиламинометилциклогексил)фенол, гидрохлорид (-5)
Исходя из (-)-(1S, 2S)-3-(2-диметиламинометил-1-гидроксициклогексил)фенола, гидрохлорида, в условиях, описанных в примере 5, получали энантиомер соединения (-5) с выходом 48% от теории.
tпл 146-147oC (разложение)
[α]
Пример 7
(+)-(1S, 2R)-[2-(3-метоксифенил)циклогексилметил] диметиламин, гидрохлорид (+6)
46 г высушенного хлорида цинка растворяли в 580 мл сухого простого эфира и затем добавляли по каплям к взвеси из 31 г борогидрида натрия в 1800 мл простого эфира. После перемешивания в течение 12 ч декантировали 500 мл полученной суспензии борогидрида натрия/хлорида натрия и по каплям добавляли к 10,2 г (32 ммоля) соединения (+4) в 200 мл сухого простого эфира. Реакционную смесь перемешивали в течение 48 ч при комнатной температуре и затем при охлаждении ледяной баней по каплям смешивали с 40 мл насыщенного раствора хлорида аммония. После разделения фаз эфирную фазу дважды промывали насыщенным раствором поваренной соли и после сушки над сульфатом натрия растворитель отгоняли под вакуумом. Таким путем получали 9,6 г комплекса амин-боран, который для выделения свободного основания растворяли в 100 мл сухого метанола. После добавки 7,5 г трифенилфосфина нагревали в течение 18 ч с обратным холодильником. После удаления перегонкой растворителя остаток смешивали со 100 мл 5%-ной соляной кислоты, после чего солянокислотную фазу еще дважды промывали 50 мл простого эфира. Затем солянокислотную фазу при охлаждении ледяной баней подщелачивали концентрированным едким натром и дважды экстрагировали путем встряхивания в 50 мл дихлорметана. После сушки объединенных органических фаз над сульфатом натрия растворитель отгоняли под вакуумом и образовавшийся остаток (7,8 г) растворяли в 2-бутаноне. После добавки триметилхлорсилана/воды выпадали в виде кристаллов 6,9 г (76% от теории) гидрохлорида (+6).
tпл 203-204oC (разложение)
[α]
Пример 8
(-)-(1R, 2S)-[2-(3-метоксифенил)циклогексилметил] диметиламин, гидрохлорид (-6)
Исходя из 10,2 г (32 ммоля) соединения (-4), в условиях, описанных в примере 7, получали энантиомер соединения (-6) с выходом 75% от теории.
tпл 201-203oC (разложение)
[α]
Пример 9
(+)-(1S, 2R)-3-(2-диметиламинометилциклогексил)фенол, гидрохлорид (+7)
4,3 г (15 ммолей) гидрохлорида (+6), полученного согласно примеру 7, смешивали со 100 мл концентрированной бромистоводородной кислоты. Затем в течение двух часов нагревали с обратным холодильником. После охлаждения до комнатной температуры реакционную смесь концентрировали в водоструйном вакууме. Остаток смешивали до момента начала щелочной реакции с концентрированным раствором гидрокарбоната натрия. После двухкратной экстракции порциями по 50 мл дихлорметана соответственно объединенные органические фазы сушили над сульфатом натрия. Затем дихлорметан отгоняли под вакуумом и остаток (4 г) растворяли в 2-бутаноне. После добавки триметилхлорсилана/воды выпадали в виде кристаллов 3,96 г (98% от теории) гидрохлорида (+7).
tпл 177-178oC (разложение)
[α]
Пример 10
(-)-(1R, 2S)-3-(2-диметиламинометилциклогексил)фенол, гидрохлорид (-7)
Исходя из соединения (-6), полученного согласно примеру 8, в условиях, описанных в примере 9, получали энантиомер соединения (-7) с 95%-ным выходом.
tпл 174-176oC (разложение)
[α]
Пример 11
(-)-(1R, 2S)-3-(2-диметиламинометилциклогексил)фениловый эфир 2,2-диметилпропионовой кислоты, гидрохлорид (-8)
Из энантиомера соединения (-7), полученного согласно примеру 10, с помощью дихлорметана/водного раствора гидрокарбоната натрия выделяли основание и после сушки раствора дихлорметан удаляли перегонкой. 1,7 г (7,3 ммолей) полученного основания растворяли в 10 мл сухого диметилформамида и по каплям добавляли к суспензии из 400 мг гидрида натрия (50%-ного) в 5 мл сухого диметилформамида. Затем перемешивали еще в течение 30 мин при 50oC. После охлаждения до комнатной температуры по каплям добавляли 1,03 мл (8,4 ммолей) 2,2-диметилпропионилхлорида, продолжали перемешивать еще в течение двух часов при комнатной температуре и затем реакционную смесь сливали на лед/воду. Водную фазу трижды экстрагировали 50 мл простого эфира. Объединенные органические фазы сушили над сульфатом натрия. После удаления растворителя перегонкой получали 2,3 г сырой смеси, которую подавали на колонку 4х30 см, заполненную силикагелем. Путем элюирования диизопропиловым эфиром/метанолом в соотношении 7: 1 получали 1,75 г основания, из которого с помощью триметилхлорсилана/воды в 2-бутаноне/диизопропиловом эфире получали 1,75 г (70% от теории) гидрохлорида (-8) с температурой плавления 218-219oC.
[α]
Пример 12
(-)-(1R, 2S)-{ 2-[3-(параизопропилфенилкарбамоил)оксифенил] циклогексилметил} диметиламин, гидрохлорид (-9)
Из энантиомера соединения (-7), полученного согласно примеру 10, с помощью дихлорметана/водного раствора гидрокарбоната натрия выделяли основание и после сушки раствора удаляли перегонкой дихлорметан. 2,1 г (9,0 ммолей) полученного основания растворяли в 20 мл сухого толуола и смешивали с 1,62 г (10 ммолей) 4-изопропилфенилизоцианата. После перемешивания в течение 20 ч при комнатной температуре толуол удаляли перегонкой. Остаток (2,5 г) подавали на колонку 5,5 х 15 см, заполненную силикагелем, и элюировали метанолом/этилацетатом в соотношении 1: 1. В результате получали 1,94 г основания, из которого с помощью триметилхлорсилана/воды в н-пропилацетате получали 1,8 г (46% от теории) гидрохлорида (-9).
tпл 156oC
[α]
Пример 13
(-)-(1R, 2S)-3-(2-диметиламинометилциклогексил)фениловый эфир 2-ацетоксибензойной кислоты, гидрохлорид (-10)
Из энантиомера соединения (-7), полученного согласно примеру 10, с помощью дихлорметана/водного раствора гидрокарбоната натрия выделяли основание и после сушки растворителя дихлорметан удаляли перегонкой. 0,7 г (3,0 ммоля) полученного основания растворяли в 7 мл сухого дихлорметана и смешивали при комнатной температуре с 0,6 г (3,24 ммоля) 2-ацетилбензоилхлорида, растворенного в 3 мл сухого дихлорметана. После перемешивания в течение 20 ч при комнатной температуре реакционную смесь смешивали с 20 мл раствора гидрокарбоната натрия и водную фазу дважды экстрагировали 10 мл дихлорметана. Органические фазы объединяли и сушили над сульфатом натрия. После удаления растворителя перегонкой получали 1,1 г сырой смеси, которую подавали на колонку 3х8 см, заполненную силикагелем. Путем элюирования простым эфиром получали 0,77 г основания, из которого с помощью триметилхлорсилана/воды в простом эфире получали 0,77г (54% от теории) гидрохлорида (-10).
tпл 171-174oC,
[α]
Пример 14
Изобутиловый эфир (-)-(1R, 2S)-[3-(2-диметиламинометилциклогексил)фенилового эфира угольной кислоты, гидрохлорид (-11)
Из энантиомера соединения (-7), полученного согласно примеру 10, с помощью дихлорметана/водного раствора гидрокарбоната натрия выделяли основание и после сушки раствора дихлорметан удаляли перегонкой. 2,34 г (10 ммолей) полученного основания растворяли в 11 мл сухого диметилформамида и по каплям добавляли к суспензии из 0,54 г гидрида натрия (50%-ного) в 5 мл сухого диметилформамида. Затем перемешивали в течение 30 мин при комнатной температуре. Далее по каплям добавляли 1,44 мл (11 ммолей) изобутилхлорформиата и продолжали перемешивание в течение двух часов при комнатной температуре, после чего реакционную смесь смешивали с 40 мл воды. Водную фазу трижды экстрагировали 50 мл простого эфира. Объединенные органические фазы сушили над сульфатом натрия. После удаления растворителя перегонкой получали 3,8 г сырой смеси, которую подавали на колонку 3 х 15 см, заполненную силикагелем. Путем элюирования простым эфиром получали 2,17 г основания, из которого с помощью триметилхлорсилана/воды в простом эфире получали 1,5 г (41% от теории) гидрохлорида (-11) в виде бесцветного сиропа.
[α]
Пример 15
(-)-(1R, 2S)-3-(2-диметиламинометилциклогексил)фениловый эфир 4-морфолин-4-илметилбензойной кислоты, дигидрохлорид (-12)
Из энантиомера соединения (-7), полученного согласно примеру 10, с помощью дихлорметана/водного раствора гидрокарбоната натрия выделяли основание и после сушки раствора дихлорметан удаляли перегонкой. 1,9 г (8,1 ммолей) полученного основания растворяли в 20 мл сухого дихлорметана и при комнатной температуре смешивали с 2,2 г (9,2 ммолей) 4-морфолин-4-илметилбензоилхлорида, гидрохлорида (получение согласно патенту США 4623486). После перемешивания в течение 20 ч при комнатной температуре реакционную смесь смешивали с 50 мл раствора гидрокарбоната натриия и водную фазу трижды экстрагировали 10 мл дихлорметана. Органические фазы объединяли и сушили над сульфатом натрия. После удаления растворителя перегонкой получали 2,9 г сырой смеси, которую подавали на колонку 4 х 20 см, заполненную силикагелем. Путем элюирования диизопропиловым эфиром/метанолом в соотношении 2: 1 получали 0,77 г основания, из которого с помощью триметилхлорсилана/воды в простом эфире получали 0,41 г (10% от теории) дигидрохлорида (-12).
tпл 234-236oC
[α]
Пример 16
(+)-(1R, 2S)-3-(2-диметиламинометилциклогексил)фениловый эфир (2S, 3S)-2-амино-3-метилпентановой кислоты, дигидрохлорид (+13)
Стадия 1:
(-)-(1R, 2S)-3-(2-диметиламинометилциклогексил)фениловый эфир (2S, 3S)-2-трет-бутоксикарбониламино-3-метилпентановой кислоты (-14)
Из энантиомера соединения (-7), полученного согласно примеру 10, с помощью дихлорметана/водного раствора гидрокарбоната натрия выделяли основание и после сушки раствора дихлорметан удаляли перегонкой. 2,1 г (9,8 ммолей) полученного основания растворяли в 140 мл сухого дихлорметана и при комнатной температуре перемешивали последовательно с 2,19 г (9,5 ммолей) (-)-(2S, 3S)-2-трет-бутоксикарбониламино-3-метилпентановой кислоты, моногидратом, 2,63 мл (19 ммолей) триэтиламина и 4,94 г (9,5 ммолей) гексафторфосфата бензтриазол-1-илокситрипирролидинфосфония. После перемешивания в течение двух часов при комнатной температуре растворитель отгоняли и остаток (10,1 г) подавали на колонку 7 х 40 см, заполненную силикагелем. Путем элюирования этилацетатом/метанолом в соотношении 1: 1 получали 2,66 г основания соединения (-14).
Стадия 2:
(+)-(1R, 2S)-3-(2-диметиламинометилциклогексил)фениловый эфир (2S, 3S)-2-амино-3-метилпентановой кислоты, дигидрохлорид (+13)
2,66 г (5,7 ммолей) соединения (-14) растворяли в 60 мл сухого дихлорметана и смешивали с 0,23 мл воды (13 ммолей) и 2,52 мл (19,5 ммолей) триметилхлорсилана. Затем перемешивали в течение 20 ч при комнатной температуре. После добавки 100 мл простого эфира выпадали в виде кристаллов 2,1 г (56% от теории) гидрохлорида (+13).
tпл 154oC (разложение)
[α]
Пример 17
(-)-(1R, 2S)-диметил-{ 2-[3-(6-метилпиридин-2-илокси)фенил] циклогексилметил)амин, дигидрохлорид (-15)
Из энантиомера соединения (-7), полученного согласно примеру 10, с помощью дихлорметана/водного раствора гидрокарбоната натрия выделяли основание и после сушки раствора дихлорметан удаляли перегонкой. 2,1 г (9,0 ммолей) полученного основания растворяли в 10 мл сухого диметилформамида и по каплям добавляли к суспензии из 475 мг гидрида натрия (50%-ного) в 5 мл сухого диметилформамида. Затем перемешивали в течение 10 мин при 60oC. При этой температуре по каплям добавляли 1,5 мл (13,7 ммолей) 2-хлор-6-метилпиридина. После добавки 30 мг медного порошка и 30 мг хлорида меди (1) перемешивали в течение 7 ч при 140oC и затем охлаждали до комнатной температуры. Реакционную смесь смешивали с 50 мл воды и водную фазу трижды экстрагировали 50 мл простого эфира. Органические фазы объединяли, промывали сначала 10 мл едкого натра, а затем 10 мл воды и сушили над сульфатом натрия. После удаления растворителя перегонкой получали 3,2 г сырой смеси, которую подавали на колонку 5 х 20 см, заполненную силикагелем. Путем элюирования простым эфиром/концентрированным раствором аммиака в соотношении 99,5: 0,5 получали 1,0 г основания, из которого с помощью триметилхлорсилана/воды в 2-бутаноне/этилацетате получали 1,89 г (53% от теории) дигидрохлорида (-15).
tпл 60oC (спекание)
[α]
Пример 18
(1RS, 3SR, 6RS)-6-диметиламинометил-1-(3-метоксифенил)циклогексан-1,3-диол, гидрохлорид (16)
и
(1RS, 3RS, 6RS)-6-диметиламинометил-1-(3-метоксифенил)циклогексан-1,3-диол, гидрохлорид (17)
Стадия 1:
9-диметиламинометил-3,3-диметил-1,5-диоксаспиро[5.5] ундекан-8-он, гидрохлорид (18)
125 г (630 ммолей) 3,3-диметил-1,5-диоксаспиро[5.5] ундекан-8-она, полученного азеотропным ацеталированием циклогексан-1,3-диона 2,2-диметилпропан-1,3-диолом в толуоле в качестве растворителя с использованием пара-толуолсульфоновой кислоты в качестве катализатора, и 59 г (630 ммолей) метиленхлорида диметиламмония перемешивали в 400 мл сухого ацетонитрила при комнатной температуре. После добавки 1 мл ацетилхлорида продолжали перемешивание еще в течение 3 ч при комнатной температуре, при которой образовывался бесцветный прозрачный раствор. Затем в реакционную смесь по каплям добавляли 800 мл сухого простого эфира и при этом гидрохлорид выпадал в виде кристаллов. Таким путем получали 158 г (98% от теории) указанного гидрохлорида (18).
Стадия 2:
(8RS, 9RS)-9-диметиламинометил-8-(3-метоксифенил)-3,3-диметил-1,5- диоксаспиро[5.5] ундекан-8-ол (19)
К 3,88г (160 ммолей) магниевой стружки в 10 мл сухого тетрагидрофурана по каплям добавляли 20 мл (158 ммолей) 1-бром-3-метоксибензола, растворенного в 100 мл сухого тетрагидрофурана, таким образом, чтобы происходило легкое кипение реакционной смеси. После полного завершения добавки 1-бром-3-метоксибензола нагревали в течение одного часа с обратным холодильником, после чего охлаждали до 5-10oC. Из гидрохлорида (18) из стадии 1 с помощью дихлорметана/едкого натра выделяли основание и после сушки раствора удаляли перегонкой дихлорметан. 32,7 г (150 ммолей) полученного основания растворяли в 50 мл сухого тетрагидрофурана и добавляли к раствору Гриньяра. Реакционную смесь оставляли на ночь и затем повторно охлаждали до 5-10oC. Добавкой 140 мл 20%-ного раствора хлорида аммония раствор Гриньяра разлагали. Реакционную смесь разбавляли 200 мл простого эфира/тетрагидрофурана (соотношение 1: 1), органическую фазу отделяли и дважды экстрагировали 100 мл простого эфира. Объединенные органические фазы сушили над сульфатом натрия. После удаления перегонкой растворителя остаток (43,6 г) подавали на колонку 8 х 50 см, заполненную силикагелем, и экстрагировали этилацетатом/метанолом (соотношение 1: 1). Полученное основание повторно подавали на колонку 5 х 13 см, заполненную силикагелем, и элюировали диизопропиловым эфиром/метанолом (соотношение 1: 1). В результате получали 22,1 г (42% от теории) основания в виде светло-желтого вязкого масла.
Стадия 3:
(3RS, 4RS)-4-диметиламинометил-3-гидрокси-3-(3-метоксифенил)циклогексанон (20)
61,8 г (176 ммолей) основания соединения (19) из стадии 2 растворяли в 800 мл тетрагидрофурана и охлаждали до 0-5oC. При этой температуре в течение 30 мин добавляли 800 мл водной соляной кислоты (концентрированная соляная кислота/вода 1: 5). Затем продолжали перемешивание в течение одного часа при комнатной температуре, после чего повторно охлаждали до 0-5oC. При этой температуре добавляли 200 мл концентрированного едкого натра. Затем реакционную смесь трижды экстрагировали 250 мл простого эфира. Объединенные органические фазы сушили над сульфатом натрия. После удаления перегонкой растворителя остаток (55 г) подавали на колонку 8 х 50 см, заполненную силикагелем, и элюировали сначала диизопропиловым эфиром/метанолом (соотношение 7: 1), а затем этилацетатом/метанолом (соотношение 4: 1). Полученное основание (24,7 г) растворяли в 1000 мл 2-бутанона и смешивали с триметилхлорсиланом/водой. При этом выпадали в виде кристаллов 16,5 г (24% от теории) гидрохлорида (20) с температурой плавления 161-163oC.
Стадия 4:
(1RS, 3SR, 6RS)-6-диметиламинометил-1-(3-метоксифенил)циклогексан-1,3-диол, гидрохлорид (16)
и
(1RS, 3RS, 6RS)-6-диметиламинометил-1-(3-метоксифенил)циклогексан-1,3-диол, гидрохлорид (17)
Из гидрохлорида (20), полученного на стадии 3, с помощью дихлорметана/едкого натра выделяли основание и после сушки раствора удаляли перегонкой дихлорметан. 27 г (97 ммолей) полученного основания растворяли в 300 мл изопропанола и при комнатной температуре порциями смешивали с 1,8 г (47,5 ммолей) борогидрида натрия. В течение одного часа перемешивали при комнатной температуре, после чего охлаждали до 0-5oC. При этой температуре добавляли 68 мл разбавленной соляной кислоты (концентрированная соляная кислота/вода в соотношении 1: 3). Непосредственно после добавки реакционную смесь подщелачивали концентрированным едким натром. После удаления перегонкой растворителей остаток (40 г) растворяли в 200 мл воды и трижды экстрагировали 50 мл дихлорметана. Объединенные органические фазы сушили над сульфатом натрия и перегонкой удаляли растворитель. Остаток (29,6 г) подавали на колонку 7 х 45 см, заполненную силикагелем, и элюировали сначала метанолом, а затем метанолом/концентрированным раствором аммиака в соотношении 99,5: 0,5. Таким путем получали 11,3 г основания соединения (16) и 13,5 г основания соединения (17). Полученные основания растворяли в 2-бутаноне и смешивали с триметилхлорсиланом/водой и при этом гидрохлориды выпадали в виде кристаллов.
(16): Выход: 9,9 г (32% от теории)
tпл 263-264oC
(17): Выход: 13,7 г (45% от теории)
tпл 197-198oC.
Пример 19
Энантиомеры соединения (17):
(+)-(1R, 3R, 6R)-6-диметиламинометил-1-(3-метоксифенил)циклогексан-1,3-диол, гидрохлорид (+17)
и
(-)-(1S, 3S, 6S)-6-диметиламинометил-1-(3-метоксифенил)циклогексан-1,3-диол, гидрохлорид (-17)
Из соединения (17) с помощью дихлорметана/едкого натра выделяли основание. После сушки раствора дихлорметан отгоняли под вакуумом. Затем рацемат разделяли на хиральной колонке ЖХВД. Из полученных энантиомеров взаимодействием с триметилхлорсиланом/водой в 2-бутаноне получали гидрохлориды с температурой плавления 232-233oC.
(+17): Выход: 42% от теории
[α]
(-17): Выход: 44% от теории
[α]
Пример 20
(1RS, 3RS, 6RS)-6-диметиламинометил-1-(3-гидроксифенил)циклогексан-1,3-диол, гидрохлорид (21)
Из полученного согласно примеру 18 соединения (17) с помощью дихлорметана/едкого натра выделяли основание и после сушки раствора перегонкой удаляли дихлорметан. 8,06 г (28,8 ммолей) полученного основания растворяли в 70 мл сухого толуола и медленно добавляли по каплям к 120 мл (144 ммоля) 1,2-молярного раствора диизобутилалюмогидрида в толуоле. После полного введения добавки нагревали в течение 8 ч с обратным холодильником и затем охлаждали до комнатной температуры. Далее реакционную смесь разбавляли 50 мл толуола. При охлаждении ледяной баней по каплям добавляли сначала 13 мл этанола, а затем 13 мл воды. После перемешивания в течение одного часа при охлаждении ледяной баней из реакционной смеси отфильтровывали алюминиевые соли и остаток трижды промывали порциями по 50 мл этилацетата соответственно. Затем объединенные органические фазы сушили и растворитель удаляли перегонкой. Из основания в ацетоне с помощью водного раствора соляной кислоты получали 7,3 г (84% от теории) указанного в заголовке соединения с температурой плавления 226-228oC.
Пример 21
Энантиомеры соединения (21):
(+)-(1R, 3R, 6R)-6-диметиламинометил-1-(3-гидроксифенил)циклогексан-1,3-диол, гидрохлорид (+21)
и
(-)-(1S, 3S, 6S)-6-диметиламинометил-1-(3-гидроксифенил)циклогексан-1,3-диол, гидрохлорид (-21)
Из соединения (21) с помощью дихлорметана/водного раствора гидрокарбоната натрия выделяли основание. После сушки растворителя дихлорметан отгоняли под вакуумом. Затем рацемат разделяли на хиральной колонке для ЖХВД. Из полученных энантиомеров в ацетоне с помощью водной соляной кислоты получали гидрохлориды с температурой плавления 217-219oC.
(+21): Выход: 40% от теории
[α]
(-21): Выход: 40% от теории
[α]
Пример 22
(1RS, 2RS, 5RS)-5-бензилокси-2-диметиламинометил-1-(3- метоксифенил)циклогексанол, гидрохлорид (22)
Из гидрохлорида (17), полученного согласно примеру 18, с помощью дихлорметана/едкого натра выделяли основание и после сушки раствора дихлорметан удаляли перегонкой. 4,0 г (14,3 ммолей) полученного основания растворяли в 30 мл сухого диметилформамида и по каплям добавляли к суспензии из 690 мг гидрида натрия (50%-ного) в 5 мл сухого диметилформамида. Затем перемешивали в течение двух часов при комнатной температуре. После нагревания до 50oC по каплям добавляли 1,81 г (14,3 ммолей) бензилхлорида и продолжали перемешивание еще в течение двух часов при 65oC и в течение 15 ч при комнатной температуре. Затем реакционную смесь сливали на лед/воду. Водную фазу трижды экстрагировали 50 мл простого эфира. Органические фазы объединяли и сушили над сульфатом натрия. После удаления перегонкой растворителя получали 4,6 г сырой смеси, которую подавали на колонку 4 х 30 см, заполненную силикагелем. Путем элюирования этилацетатом/метанолом в соотношении 4: 1 получали 1,5 г основания, из которого с помощью триметилхлорсилана/воды в 2-бутаноне/диизопропиловом эфире получали 1,38 г (24% от теории) гидрохлорида (22) с температурой плавления 138-139oC.
Пример 23
(1RS, 2RS, 5SR)-2-диметиламинометил-1-(3-метоксифенил)-5-метилциклогексанол, гидрохлорид (23)
95 мл (750 ммолей) 1-бром-3-метоксибензола растворяли в 425 мл сухого тетрагидрофурана и охлаждали до -75oC. После добавки 469 мл (750 ммолей) 1,6-молярного раствора н-бутиллития в гексане перемешивали в течение одного часа при -75oC. Затем по каплям добавляли 82 г (484 ммоля) (2RS, 5SR)-диметиламинометил-5-метилциклогексанона, полученного из 3-метилциклогексанона, гидрохлорида диметиламина и пара-формальдегида в ледяной уксусной кислоте, растворенных в 120 мл сухого тетрагидрофурана. В течение 2,5 ч реакционную смесь нагревали до комнатной температуры.
Для дальнейшей переработки по каплям добавляли при охлаждении ледяной баней 200 мл воды так, чтобы внутренняя температура не превышала 15oC. После разделения фаз водную фазу трижды экстрагировали 50 мл этилацетата. Объединенные органические фазы сушили над сульфатом натрия. После удаления перегонкой растворителя остаток (148,3 г) растворяли в 700 мл ацетона и смешивали с триметилхлорсиланом/водой. При 4-5oC выпадали в виде кристаллов 67 г (48% от теории) гидрохлорида (23) с температурой плавления 173-175oC.
Пример 24
Энантиомеры соединения (23):
(+)-(1R, 2R, 5S)-2-диметиламинометил-1-(3-метоксифенил)-5-метилциклогексанол, гидрохлорид (+23)
и
(-)-(1S, 2S, 5R)-2-диметиламинометил-1-(3-метоксифенил)-5-метилциклогексанол, гидрохлорид (-23)
Энантиомеры соединений (+23) и (-23) получали в условиях, описанных в примере 19.
(+23): Выход: 43% от теории
tпл 151-152oC
[α]
(-23): Выход: 44% от теории
tпл 151-153oC
[α]
Пример 25
(+)-(1R, 2R, 5S)-3-(2-диметиламинометил-1-гидрокси-5-метилциклогексил)фенол, гидрохлорид (+24).
В условиях, описанных в примере 20, из полученного согласно примеру 24 метоксильного соединения (+ 23) получали энантиомер соединения (+24).
Выход: 87% от теории
tпл 221-223oC
[α]
Пример 26
(-)-(1S, 2S, 5R)-3-(2-диметиламинометил-1-гидрокси-5-метилциклогексил)фенол, гидрохлорид (-24)
В условиях, описанных в примере 20, из полученного согласно примеру 24 метоксильного соединения (-23) получали энантиомер соединения (-24).
Выход: 87% от теории
tпл 220-222oC
[α]
Пример 27
(1RS, 2RS, 5SR)-3-(2-диметиламинометил-1-гидрокси-5- трифторметилциклогексил)фенол, гидрохлорид (25)
Стадия 1:
(1RS, 2RS, 5SR)-1-(3-бензилоксифенил)-2-диметиламинометил-5- трифторметилциклогексанол (26)
К 4,06 г (167 ммолей) магниевой стружки в 40 мл сухого тетрагидрофурана по каплям добавляли 43,9 г (167 ммолей) 3-бензилокси-1-бромбензола, растворенного в 200 мл сухого тетрагидрофурана, таким образом, чтобы реакционная смесь слегка кипела. После полного введения добавки 3-бензилокси-1-бромбензола нагревали в течение одного часа с обратным холодильником и затем охлаждали до 5-10oC. При этой температуре добавляли 30,8 г (139 ммолей) (2RS, 5SR)-2-диметиламинометил-5-трифторметилциклогексанона, полученного из 3-трифторметилциклогексанона и диметиламинометиленхлорида в ацетонитриле, растворенных в 80 мл сухого тетрагидрофурана. Реакционную смесь оставляли на ночь, после чего повторно охлаждали до 5-10oC. Добавкой 150 мл 20%-ного раствора хлорида аммония раствор Гриньяра разлагали. Реакционную смесь разбавляли 200 мл простого эфира, органическую фазу отделяли, а водную фазу дважды экстрагировали 100 мл простого эфира. Объединенные органические фазы сушили над сульфатом натрия. После удаления перегонкой растворителя остаток (60,6 г) подавали на колонку 8 х 50
см, заполненную силикагелем, и элюировали этилацетатом/метанолом. Таким путем получали 27,8 г (50% от теории) основания соединения (26).
Стадия 2:
(1RS, 2RS, 5SR)-3-(2-диметиламинометил-1-гидрокси-5-трифторметилциклогексил)фенол, гидрохлорид (25)
Из соединения (26) из стадии 1 получали соединение (25) в условиях, описанных в примере 1 (стадия 3), с 64%-ным выходом и температурой плавления 228-230oC.
Пример 28
(1S, 2RS, 5RS)-3-(2-диметиламинометил-1-гидрокси-5-трифторметилциклогексил)фенол, гидрохлорид (27)
Исходя из (2RS, 5RS)-2-диметиламинометил-5-трифторметилциклогексанона, полученного из 3-трифторметилциклогексанона и диметиламинометиленхлорида в ацетонитриле, в условиях, описанных в примере 27, получали 5-эпимер (27) по отношению к соединению (25) с выходом 27% от теории и температурой плавления 221-223oC.
Пример 29
(1RS, 2RS, 5SR)-3-(2-диметиламинометил-1-фтор-5-трифторметилциклогексил)фенол, гидрохлорид (28)
Исходя из полученного согласно примеру 27 (стадия 1) основания соединения (26), в условиях, описанных в примере 1 (стадии 2 и 3), получали гидрохлорид (28) с выходом 24% от теории и температурой плавления 204-205oC.
Пример 30 (IRS, 2RS, 5RS)-3-(2-диметиламинометил-1-фтор-5-трифторметилциклогексил)фенол, гидрохлорид (29)
Исходя из полученного согласно примеру 27 (стадия 1) основания (1RS, 2RS, 5RS)-1-(3-бензилоксифенил)-2-диметиламинометил-5- трифторметилциклогексанола, в условиях, описанных в примере 29, получали гидрохлорид (29) с выходом 22% от теории и температурой плавления 204oC.
Фармакологические исследования
Испытания анальгезии на мышах в тесте на вздрагивание хвоста
Соединения по изобретению испытывали на анальгетическую эффективность на мышах в тесте на вздрагивание хвоста с использованием теплового излучения по методу D'Amour и Smith (Journ. Pharm. Exp. Ther. 72. 74-79 (1941). Эксперименты проводили на мышах линии NMRI весом от 20 до 24 г. Подопытных животных помещали поодиночке в специальные клетки и на основание хвоста воздействовали сфокусированным тепловым лучом от электрической лампы (анальгезиметр Rhema 3010). Интенсивность излучения лампы устанавливали таким образом, что время с момента включения лампы до момента внезапного, судорожного вздрагивания хвоста (болевая латентность) у необработанных животных составляло 3-5 сек. До введения одного из соединений по изобретению животных в течение пяти минут дважды подвергали предварительному тестированию и по среднему значению этих измерений рассчитывали исходное среднее значение перед тестированием. Болевые ощущения фиксировали соответственно через 20, 40 и 60 минут после внутривенного введения соединений по изобретению. При повышении болевой латентности максимальное время облучения ограничивали 12 с и увеличение латентного периода до >150% от исходного среднего значения перед тестированием принимали за значение, при котором наступает анальгетическое действие. Для определения зависимости от дозировки соответствующее соединение по изобретению вводили 3-5 дозами в логарифмически возрастающем количестве, включавшем соответственно пороговую и максимально эффективную дозу и по числу анальгезированных животных по методу Litchfield и Wilcoxon (Journ. Pharm. Exp. Ther. 96, 99-113 (1949)) определяли значения ED50. Определение ED50 осуществляли в максимальном диапазоне действия по истечении 20 мин после внутривенного введения субстанций.
Все применявшиеся соединения по изобретению проявили ярко выраженную анальгетическую эффективность. Полученные результаты представлены в таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИМЕТИЛ(3-АРИЛБУТ-3-ЕНИЛ)АМИНОСОЕДИНЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2167146C2 |
| 1-ФЕНИЛ-2-ДИМЕТИЛАМИНОМЕТИЛЦИКЛОГЕКСАН-1-ОЛОВЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1996 |
|
RU2167148C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ БЕНЗОЦИКЛОАЛКЕНОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2197484C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОЛИДИН-2,4-ДИОНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ ДЕЙСТВУЮЩИХ ВЕЩЕСТВ | 1996 |
|
RU2163603C2 |
| ЗАМЕЩЕННЫЕ ЦИКЛОГЕПТЕНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1999 |
|
RU2233268C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЛИ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2002 |
|
RU2309942C2 |
| ЗАМЕЩЕННЫЕ АМИНОСОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2197474C2 |
| ПИРИДИЛТИОСОЕДИНЕНИЯ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBACTER | 1995 |
|
RU2149872C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ КАТЕХОЛА И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1997 |
|
RU2180898C2 |
| НОВЫЕ ПОЛИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ФТАЛАЗИНА И ИХ ПРИМЕНЕНИЕ | 1997 |
|
RU2181361C2 |

Изобретение относится к новым 6-диметиламинометил-1-фенилциклогексановым соединениям общей формулы 1 в виде оснований или их физиологически приемлемых солей, проявляющим анальгетическую активность и предназначенным для получения фармацевтической композиции, а также к способам их получения. В общей формуле I радикалы имеют следующие значения: R1 представляет собой Н, ОН, Сl или F;
R2 и R3 являются идентичными или разными и означают Н, С1-С4алкил, CF3, ОН, ОСН2-С6Н5, O-С1-С4алкил, при условии, что по крайней мере один из радикалов R2 или R3 означает Н; R4 означает Н, СН3, СО(ОС1-С5алкил), СО-NН-С6H4-С1-С3алкил, CO-C6H4-R5, СО-С1-С5алкил, CO-CHR6-NHR7 или незамещенную, либо замещенную С1-С4алкилом пиридиловую или фенильную группу; R5 означает ОС(O)С1-С3алкил в орто-положении или CH2-N(R8)2 в мета- либо пара-положении, причем оба радикала R8 вместе с N представляют собой 4-морфолиновый радикал; и R6, R7 являются идентичными или разными и означают Н или С1-С6алкил при условии, что если оба радикала R2 и R3 означают Н, то R4 не является CH3, если R1 означает Н, ОН или Сl, либо R4 не является Н, если R1 означает ОН. Описываются также различные способы получения соединений формулы I. Например, для получения соединений, где R1 означает ОН, диметиламинокетон формулы II подвергают взаимодействию с металлорганическим соединением формулы III, или диметиламинокетон со спироциклической ацетильной группой формулы V подвергают взаимодействию с металлорганическим соединением формулы III, с получением соединения формулы VI, которое деацетилированием переводят в соединение кетона VIII, с последующим восстановлением комплексным гидридом щелочного металла до соединения формулы I, которое после перевода в соль щелочного металла переводят в соответствующее O-алкил или O-бензилпроизводное. Описывается также способы получения соединений, где R1 означает Н, или соединений, где R1 означает F. 7 с. и 4 з. п. ф-лы, 1 табл.



в которой R1 представляет собой Н, ОН, Сl или F;
R2 и R3 являются идентичными или разными и означают Н, C1-С4алкил, CF3, ОН, ОСН2-С6Н5, O-С1-С4алкил, при условии, что по крайней мере один из радикалов R2 или R3 означает Н;
R4 означает Н, CH3, СО(ОС1-С5алкил), CO-NH-C6H4-С1-С3алкил, CO-C6H4-R5, СО-С1-С5алкил, CO-CHR6-NHR7 или незамещенную либо замещенную С1-С4алкилом пиридиловую или фенильную группу;
R5 означает ОС(O)С1-С3алкил в орто-положении или CH2-N(R8)2 в мета- либо пара-положении, причем оба радикала R8 вместе с N представляют собой 4-морфолиновый радикал; и
R6, R7 являются идентичными или разными и означают Н или C1-С6алкил при условии, что если оба радикала R2 и R3 означают Н, то R4 не является CH3, если R1 означает Н, ОН или Сl, либо R4 не является Н, если R1 означает ОН,
в виде их оснований или солей физиологически приемлемых кислот.
4. Способ получения 6-диметиламинометил-1-фенилциклогексанового соединения формулы I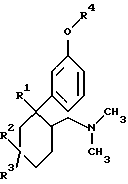
в которой R1 представляет собой ОН;
R2 и R3 являются идентичными или разными и означают Н, С1-С4алкил, CF3, при условии, что по крайней мере один из радикалов R2 либо R3 означает Н; и
R4 означает Н, CH3 или незамещенную либо С1-С4алкилзамещенную пиридиловую или фенильную группу при условии, что R4 не представляет собой ни CH3, ни Н, если оба радикала R2 и R3 означают Н,
отличающийся тем, что β-диметиламинокетон формулы II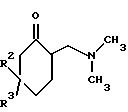
подвергают взаимодействию с металлорганическим соединением формулы III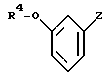
в которой Z означает MgCl, MgBr, MgI или Li,
с получением соединения формулы I, в которой R1 означает ОН.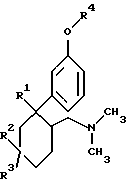
в которой R1 означает ОН;
один из радикалов R2 либо R3 означает Н, а другой означает ОН, O-С1-С4алкил или ОСН2-С6Н5;
R4 означает Н, CH3 или незамещенную либо С1-С4алкилзамещенную пиридиловую или фенильную группу,
отличающийся тем, что β-диметиламинокетон со спироциклической ацетальной структурой формулы V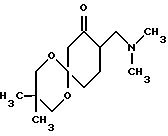
подвергают взаимодействию с металлорганическим соединением формулы III
в которой Z означает MgCl, MgBr, Mgl или L,
с получением соединения формулы VI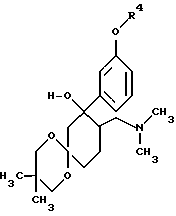
полученное соединение путем катализированной протонами реакции деацеталирования переводят в производное кетона формулы VIII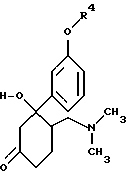
и затем полученное производное кетона с помощью комплексного гидрида щелочного металла восстанавливают до соединения формулы I, в которой один из радикалов R2 либо R3 означает ОН, и при необходимости полученное восстановлением соединение формулы I после перевода в соль щелочного металла превращают с помощью С1-С4алкил- или бензилгалогенида в соединение формулы I, в котором один из радикалов R2 либо R3 означает O-С1-С4алкил или ОСН2-С6Н5.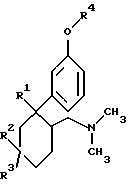
в которой R1 представляет собой Н;
R2 и R3 являются идентичными либо разными и означают Н, C1-С4алкил, CF3, ОСН2-С6Н5 при условии, что по крайней мере один из радикалов R2 либо R3 означает Н;
R4 означает Н, СН3 или незамещенную либо С1-С4алкилзамещенную пиридиловую или фенильную группу,
отличающийся тем, что соединение формулы I, в которой R1 означает Сl, подвергают взаимодействию с борогидридом цинка, цианборогидридом цинка или цианборогидридом олова в среде простого эфира.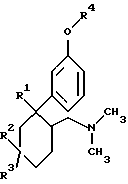
в которой R1 представляет собой Н;
R2 и R3 являются идентичными либо разными и означают Н, С1-С4алкил, CF3, ОСН2-С6Н5 при условии, что по крайней мере один из радикалов R2 либо R3 означает Н;
R4 означает Н, СН3 или незамещенную либо С1-С4алкилзамещенную пиридиловую или фенильную группу,
отличающийся тем, что соединение формулы I, в которой R1 означает ОН, подвергают взаимодействию с никелем Ренея в С2-С4алкиловом спирте.
в которой R1 представляет собой Н;
R2 и R3 являются идентичными либо разными и означают Н, C1-С4алкил, CF3 при условии, что по крайней мере один из радикалов R2 либо R3 представляет собой Н;
R4 означает CH3,
отличающийся тем, что соединение формулы I, где R1 означает Сl, гидрируют в присутствии палладиевого катализатора в С1-С4алкиловом спирте.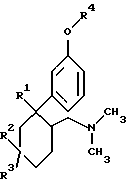
в которой R1 представляет собой F;
R2 и R3 являются идентичными либо разными и означают Н, C1-С4алкил, CF3, ОСН2-С6Н5, при условии, что по крайней мере один из радикалов R2 либо R3 представляет собой Н;
R4 означает CH3 или незамещенную либо С1-С4алкилзамещенную пиридиловую или фенильную группу,
отличающийся тем, что соединение формулы I, в которой R1 представляет собой ОН, подвергают взаимодействию с диметиламиносульфотрифторидом.
| US 3652569 A, 28.03.1972 | |||
| GB 997399 A, 07.07.1965 | |||
| ЛЕДОБУР | 0 |
|
SU376072A1 |
| Способ выделения аминоспиртов или их солей | 1974 |
|
SU546607A1 |
| Способ получения W-аминоалкоксиалканов или их солей | 1977 |
|
SU906367A3 |

