Изобретение относится к замещенным имидазолидин-2,4-дионовым соединениям, к способу их получения и к применению этих соединений в лекарственных средствах.
При патогенезе многих серьезных заболеваний чрезмерное образование цитокина, являющегося фактором некроза опухоли TNF-α (Tumor-Necrosis-Factor-α), играет главную роль. К таким заболеваниям относятся рассеянный склероз, синдром реакции "трансплантат против хозяина", отторжение трансплантата, афтозный стоматит, эритема нодозная лепроматозная, болезнь Бека, ревматоидный артрит и целый ряд других заболеваний, сопровождаемых воспалительными явлениями. Известный метод, используемый при терапии этих заболеваний, заключается в общем подавлении высвобождения TNF-α благодаря применению иммуномодуляторов супрессорного характера, таких, например, как дексаметазон.
Правда, при заболеваниях с доминантными в лейкоцитарном отношении васкулитами посткапиллярных венул, например афтозном стоматите, кожной красной волчанке, гангренозной пиодермии и орогенитальных язвах при болезни Бехчета, следует предпочесть целенаправленное вмешательство для предотвращения отрицательных последствий, обусловленных общей иммуносупрессией.
Патогенными факторами при этих болезнях считаются эндогенные медиаторы, оказывающие воздействие на эндотелий и циркулирующие лейкоциты. Локальное высвобождение TNF-α и других цитокинов приводит к фокальному повышению адгезивности эндотелия по отношению к лейкоцитам, что в решающей степени способствует образованию васкулитов [см. M.Clauss et al. в Tumor Necrosis Factors, издатель: B. Beutler, Raven, Нью-Йорк, 1992, стр. 49-64]. Субстанции, которые благодаря фокальному вмешательству могут подавить изменение эндотелия, не блокируя при этом одновременно специфическую клеточную иммунозащиту, превосходят по своей эффективности общие иммунодепрессанты, такие как дексаметазон, и могут тем самым открыть новые возможности терапии.
Класс гидантоиновых соединений, к которому относятся также соединения согласно изобретению, подвергался в прошлом интенсивным исследованиям. Так, был синтезирован целый ряд производных, находящих применение в косметических средствах, в качестве инсектицидов или гербицидов или же образующих основу для эпоксидных смол.
В фармацевтике гидантоиновые соединения известны прежде всего своим противосудорожным, противовоспалительным [см. Journ. Med. Chem. 8, 239 (1965); Arzneim. Forsch. /Drug. Res. 27(II), 1942 (1977); Pharmazie 38, 341 (1983); Journ. Med. Chem. 28, 601 (1985)] и противоопухолевым действием [см. Journ. Med. Chem. 18, 846 (1975); Arzneim. Forsch./Drug. Res. 34 (I), 663 (1984)].
Положенная в основу изобретения задача состояла в разработке новых, стабильных иммуномодуляторов, не вызывающих общей иммуносупрессии. Кроме того, создаваемые вещества должны были обладать противоваскулитным действием.
Было найдено, что требованиям, выдвинутым по отношению к создаваемым субстанциям, отвечают определенные замещенные имидазолидин-2,4-дионовые соединения. Эти относящиеся к классу гидантоинов соединения отличаются высокой иммуномодуляторной эффективностью. Они способны подавлять высвобождение TNF-α, не блокируя при этом одновременно в целом клеточную иммунозащиту. Соединения согласно изобретению проявляют, кроме того, противоваскулитное действие, которое не ограничивается только лишь ингибированием высвобождения TNF-α.
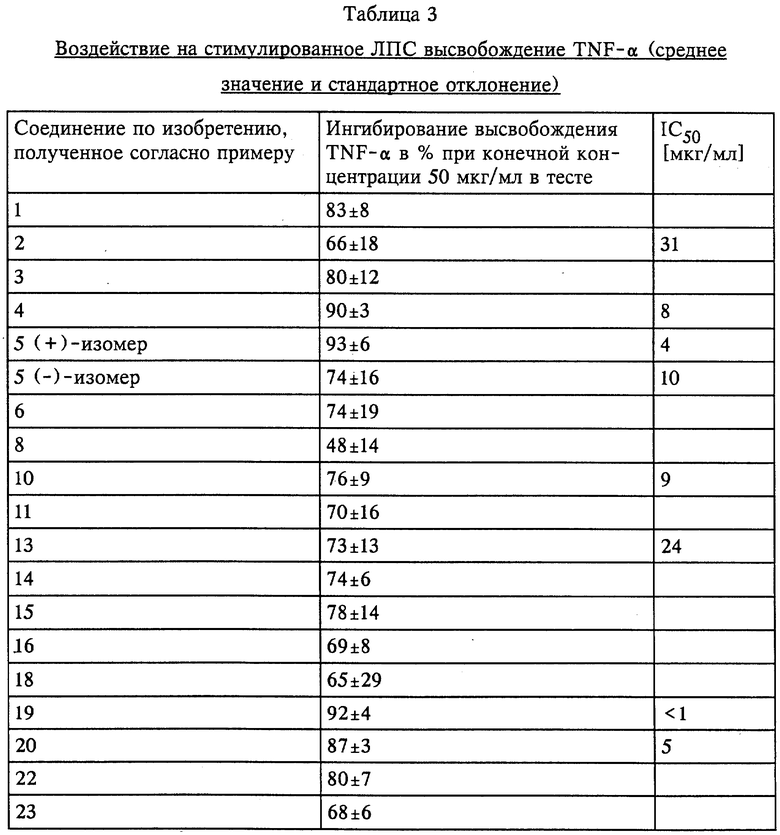
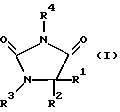
Предметом изобретения в соответствии с этим являются замещенные имидазолидин-2,4-дионовые соединения формулы I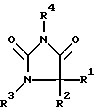
в которой R1 означает C1-C6алкил или С3-С6циклоалкил,
R2 означает С1-С6алкил, фенил, -(CH2)1-3-фенил или -(CH2)1-4-COOR5 или
R1 и R2 оба вместе означают -(CH2)4-6, -(CH2)2-О-(CH2)2- либо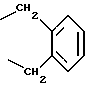
R3 означает H, C1-C5алкил или -(CH2)1-4-COOR5,
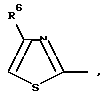
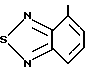
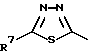
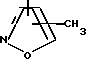
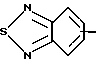
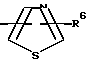
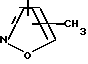
R4 представляет собой гетероароматический углеводород из группы, соответствующей формулам


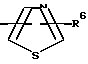

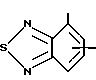
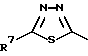
R5 представляет собой С1-C6алкил,
R6 означает H, С1-С4алкил, фенил или бензил и
R7 означает H, C1-С4алкил или трифторметил.
Предпочтительные замещенные имидазолидин-2,4-дионовые соединения соответствуют формуле I, в которой
R1 означает С1-С4алкил или С3- С4циклоалкил,
R2 означает С3-С6алкил, фенил, -(CH2)1-2-фенил или - (CH2)1-2-COOR5 или
R1 и R2 оба вместе означают -(CH2)5- или
R3 означает H, С1-С4алкил или -(CH2)1-2-COOR5,
R4 представляет собой гетероароматический углеводород из группы, соответствующей формулам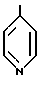





R5 представляет собой С1-С3алкил,
R6 означает H или фенил и
R означает H, метил, трет-бутил или трифторметил.
К особенно предпочтительным соединениям формулы I относятся те из них, в которых R1 представляет собой этил или циклобутил, R2 является фенилом или R1 и R2 оба вместе означают -(CH2)5-. Особенно предпочтительны соединения формулы I, в которых R1 и R2 оба вместе представляют собой -(CH2)5-.
Среди других особенно предпочтительных соединений формулы I следует назвать такие, в которых R3 представляет собой H, С1-С3алкил или -CH2- COOR5, a R5 означает этил. Из названных предпочтительны прежде всего соединения формулы I, в которых R3 является H.
К особенно предпочтительным соединениям формулы I относятся далее те из них, в которых R4 представляет собой гетероароматический углеводород из группы, включающей пиридин-4-ил, пиридин-3-ил, тиазол-2-ил, 3- метилизоксазол-5-ил или 5-метилизоксазол-3-ил. Среди указанных предпочтительны в первую очередь соединения формулы I, в которых R4, являющийся гетероароматическим углеводородом, представляет собой тиазол-2-ил.
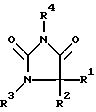
Еще одним предметом изобретения является способ получения замещенного имидазолидин-2,4-дионового соединения формулы I, в которой
R1 означает C1-С6алкил или С3-С6циклоалкил,
R2 означает C1-С6алкил, фенил, -(CH2)1-3-фенил или -(CH2)1-4-COOR5 или
R1 и R2 оба вместе означают -(CH2)4-6-, -(CH2)2-O-(CH2)2 - либо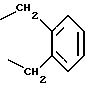
R3 означает H, С1-С5алкил или -(CH2)1-4-COOR5,
R4 представляет собой гетероароматический углеводород из группы, соответствующей формулам

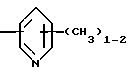


R5 представляет собой С1-С3алкил,
R6 означает H, С1-С4алкил, фенил или бензил и
R7 означает H, C1-С4алкил или трифторметил,
причем способ отличается тем, что к амину формулы II
R4-NH2
добавляют 1,1'-карбонилдиимидазол или дифениловый эфир угольной кислоты и затем взаимодействием с соединением формулы III
в которой R8 представляет собой H или С1-С3алкил, трансформируют в соединение формулы I, где R3 означает H, которое при необходимости депротонируют, после чего взаимодействием с соединением формулы IV
X-C1-С5алкил
либо с соединением формулы V
X-(CH2)1-4-COOR5,
в которых X означает хлор, бром или иод, трансформируют в соединение формулы I, в которой R3 означает С1-С5алкил или - (CH2)1-4-COOR5.
Взаимодействие амина формулы II с 1,1'-карбонилдиимидазолом или дифениловым эфиром угольной кислоты осуществляют по известной методике [см. Angew. Chem. 73. 66 (1961)]. Последующую реакцию с эфиром аминокислоты формулы III с конверсией в соединение формулы I, в которой R3 является H, проводят предпочтительно в апротонных растворителях, таких как простые эфиры, например в диэтиловом эфире или тетрагидрофуране, либо в ароматических углеводородах, например в толуоле, хлорбензоле или 1,2- дихлорбензоле, при температурах в интервале от 20oC до 180oC. В ходе этой реакции наряду с соединением формулы I, в которой R3 является H, может быть получено также соответствующее производное мочевины формулы VI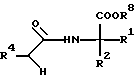
Соединение формулы VI, в которой R8 представляет собой H, взаимодействием с тионилхлоридом можно трансформировать в соединение формулы I, в которой R3 является Н. Соединение формулы VI, в которой R8 представляет собой С1-C3алкил, перед циклизацией до соединения формулы I, в которой R3 является H, подвергают омылению щелочью или путем нагревания с соляной кислотой переводят непосредственно в соединение формулы I, в которой R3 является Н.
Для получения соединения формулы I, в которой R3 означает С1-С5алкил или -(CH2)1-4-COOR5, депротонируют соединение формулы I, в которой R3 является H, предпочтительно с помощью гидрида натрия в диметилформамиде или тетрагидрофуране. Последующее взаимодействие с соединением формулы IV или V осуществляют при температурах в интервале от 20oC до 50oC.
Необходимый для получения соединения формулы I эфир аминокислоты формулы III можно получать этерификацией соответствующей аминокислоты, например, с помощью растворов хлористого водорода в соответствующем спирте или путем нагревания с соответствующим спиртом при кислотном катализе, например, с использованием серной или фосфорной кислоты.
Другая возможность получения соединения формулы III состоит в том, что эфир аминокислоты формулы VII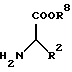
подвергают взаимодействию с бензальдегидом, получая таким путем соединение формулы VIII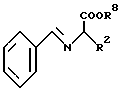
которое после депротонирования с помощью основания, предпочтительно диизопропиламида лития, в простых эфирах либо углеводородах, например в диэтиловом эфире, тетрагидрофуране или бензоле, алкилируют соединением формулы IX X-R1, в которой X означает хлор, бром или иод. Затем под воздействием кислот отщепляют бензилиденовую группу.
Предлагаемые согласно изобретению соединения токсикологически безопасны и пригодны поэтому для применения в качестве фармацевтических действующих веществ. В соответствии с этим предметом изобретения является также применение замещенного имидазолидин-2,4-дионового соединения формулы I в качестве действующего вещества в лекарственных средствах, предпочтительно в качестве иммуномодуляторов, или в лекарственных средствах с противоваскулитным действием.
Лекарственные средства согласно изобретению наряду с по крайней мере одним замещенным имидазолидин-2,4-дионовым соединением формулы I содержат также носители, наполнители, растворители, разбавители, красители и/или связующие вещества. Выбор этих вспомогательных веществ, равно как и применяемые количества зависят от того, назначено ли лекарственное средство для орального, внутривенного, интраперитонеального, интрадермального, внутримышечного, назального, щечного или локального введения. Для орального приема пригодны композиции в виде таблеток, жевательных таблеток, драже, капсул, гранулятов, капель, микстур или сиропов; для парентерального, местного и ингаляционного введения могут использоваться растворы, суспензии, легко реконструируемые сухие композиции, а также спреи. Соединения согласно изобретению в депо в растворенном виде, в пленке-носителе или в пластыре, при необходимости с добавками средств, способствующих кожной пенетрации, являются примерами форм, пригодных для чрескожного назначения. Высвобождение соединений согласно изобретению из композиций для орального или чрескожного применения может происходить постепенно.
Количество действующего вещества, назначаемого пациенту, варьирует в зависимости от веса пациента, метода введения, показания и степени серьезности заболевания. Обычно назначают от 1 до 150 мг/кг по крайней мере одного замещенного имидазолидин-2,4-дионового соединения формулы I.
Примеры
В качестве неподвижной фазы для хроматографии на колонке использовали силикагель 60 (0,040-0,0063 мм) фирмы E.Merck, Дармштадт.
Соотношение элюентов в смеси в хроматографических методах указаны всегда в отношении объем/объем.
Разделение рацематов осуществляли на колонке Chiracel OD фирмы Daicel Chemical Industries, LTD.
"tпл" означает температуру плавления и "КТ" означает комнатную температуру.
Получение соединений согласно изобретению
Пример 1A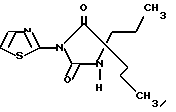
5,5-Дипропил-3-тиазол-2-илимидазолидин-2,4-дион
Стадия 1
Этиловый эфир 2-аминопентановой кислоты
Суспензию из 11,72 г DL-норвалина в 90 мл этанола смешивали с 3,6 мл концентрированной серной кислоты и смесь в течение восьми дней кипятили с обратным холодильником. В результате образовывался прозрачный раствор, из которого после охлаждения удаляли перегонкой этанол. Остаток растворяли в 200 мл дистиллированной воды и добавками карбоната калия устанавливали значение pH в пределах от 10 до 12. Затем трижды экстрагировали порциями по 50 мл этилового эфира уксусной кислоты соответственно, далее один раз промывали 50 мл насыщенного раствора хлорида натрия и сушили над сульфатом натрия. После удаления перегонкой растворителя получали 11,93 г этилового эфира 2-аминопентановой кислоты (82% от теории) в виде желтоватого масла.
Стадия 2
Этиловый эфир 2-(бензилиденамино)пентановой кислоты
Раствор из 11,90 г продукта из стадии 1 в 150 мл диэтилового эфира смешивали последовательно с 8,3 мл бензальдегида, 23 мл триэтиламина и 7,0 г безводного сульфата магния. Смесь перемешивали в течение 24 ч при комнатной температуре, затем фильтровали и промывали диэтиловым эфиром. После удаления перегонкой растворителя получали 18,40 г этилового эфира 2-(бензилиденамино) пентановой кислоты (96% от теории) в виде желтоватой вязкой массы.
Стадия 3
Этиловый эфир 2-амино-2-пропилпентановой кислоты
Раствор из 10,4 мл диизопропиламина в 200 мл тетрагидрофурана при 0oC при перемешивании и подаче сухого азота смешивали по каплям с 49 мл 1,6-молярного раствора н-бутиллития в н-гексане. После охлаждения до -78oC по каплям добавляли раствор из 18,31 г продукта из стадии 2 в 80 мл тетрагидрофурана. Всю реакционную смесь перемешивали в течение 30 мин, после чего по каплям добавляли раствор из 8,8 мл 1-йодистого пропана в 40 мл тетрагидрофурана. Реакционную смесь перемешивали в течение 16 ч, причем температура медленно повышалась до 20oC. Растворитель отгоняли. Полученный остаток оранжевого цвета растворяли в 500 мл 1 н. соляной кислоты. После перемешивания в течение одного часа при 20oC трижды экстрагировали порциями по 100 мл диэтилового эфира соответственно. Солянокислотную фазу с помощью гидроксида калия устанавливали на значение pH в пределах от 10 до 12, после чего трижды экстрагировали порциями по 100 мл диэтилового эфира соответственно. Экстракты объединяли, дважды промывали насыщенным раствором хлорида натрия и сушили над сульфатом натрия. Полученный после удаления перегонкой растворителя неочищенный продукт очищали на силикагельной колонке этиловым эфиром уксусной кислоты. Таким путем получали 9,84 г этилового эфира 2-амино-2-пропилпентановой кислоты (67% от теории) в виде слегка окрашенного масла.
Стадия 4
Этиловый эфир 2-пропил-2-(3-тиазол-2-илуреидо)пентановой кислоты
Раствор из 5,40 г 2-аминотиазола в 150 мл тетрагидрофурана смешивали при 20oC с 8,75 г 1,1'-карбонилдиимидазола и смесь перемешивали в течение 30 мин. Затем в течение 20 мин нагревали до температуры ванны 55-60oC и по каплям добавляли раствор из 9,80 г продукта из стадии 3 в 30 мл тетрагидрофурана. В результате образовывался прозрачный красно-коричневый раствор, который перемешивали в течение 60 ч при температуре в интервале от 55oC до 60oC. После удаления перегонкой растворителя остаток очищали на силикагельной колонке этиловым эфиром уксусной кислоты. Таким путем получали 10,20 г этилового эфира 2-пропил-2-(3-тиазол- 2- илуреидо)пентановой кислоты (62% от теории) в виде масла желтоватого цвета.
Стадия 5
2-Пропил-2- (3-тиазол-2- илуреидо)пентановая кислота
10,03 г продукта из стадии 4 при перемешивании растворяли в 200 мл полуконцентрированного едкого натра при температуре 20oC. Затем с помощью концентрированной соляной кислоты устанавливали значение pH 4 и трижды экстрагировали порциями по 50 мл дихлорметана соответственно. Экстракты один раз промывали насыщенным раствором хлорида натрия и сушили над сульфатом натрия. После удаления перегонкой растворителя получали 8,48 г 2-пропил-2-(3-тиазол-2- илуреидо) пентановой кислоты (93% от теории) в виде белых кристаллов (tпл 154-155oC).
Стадия 6
5,5-Дипропил-3-тиазол-2-илимидазолидин-2,4-дион
8,28 г продукта из стадии 5 смешивали с 20 мл тионилхлорида. Смесь перемешивали в течение 18 ч при 20oC. Затем для разложения добавляли лед, с помощью карбоната калия устанавливали щелочное значение pH и трижды экстрагировали порциями по 20 мл дихлорметана соответственно. После промывки экстрактов насыщенным раствором хлорида натрия и сушки над сульфатом натрия удаляли перегонкой растворитель. Полученный остаток очищали на силикагельной колонке этиловым эфиром уксусной кислоты. Таким путем получали 5,50 г 5,5-дипропил-3-тиазол-2-илимидазолидин-2,4-диона (71% от теории) в виде белых кристаллов (tпл 127-128oC).
Пример 1Б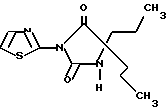
5,5-Дипропил-3-тиазол-2-илимидазолидин-2,4-дион
1,71 г 2-пропил-2-(3-тиазол-2-илуреидо)пентановой кислоты (продукт из примера 1А, стадия 4) смешивали с 30 мл 30%-ной соляной кислоты. Смесь кипятили в течение трех часов с обратным холодильником. После охлаждения устанавливали с помощью карбоната калия щелочное значение pH, трижды экстрагировали этиловым эфиром уксусной кислоты, дважды промывали насыщенным раствором поваренной соли и сушили над сульфатом натрия. Неочищенный продукт, полученный в результате последующего удаления перегонкой растворителя, очищали на силикагельной колонке этиловым эфиром уксусной кислоты. Таким путем получали 0,62 г 5,5-дипропил-3-тиазол-2-илимидазолидин-2,4- диона (42% от теории).
Пример 2
3-Тиазол-2-ил-1,3-диазаспиро[4.5]декан-2,4-дион
Стадия 1
Этиловый эфир 1-амино-1-циклогексанкарбоновой кислоты
В условиях, описанных в примере 1А, стадия 1, из 100 г 1-амино-1-циклогексанкарбоновой кислоты, гидрохлорида, 500 мл этанола и 20 мл концентрированной серной кислоты после очистки сырого продукта на силикагельной колонке этиловым эфиром уксусной кислоты/метанолом (соотношение 5:1) получали 75,5 г этилового эфира 1-амино-1- циклогексанкарбоновой кислоты (80% от теории) в виде желтоватого масла.
Стадия 2
3-Тиазол-2-ил-1,3-диазаспиро [4.5] декан-2,4-дион
44,4 г 2-аминотиазола, 71,9 г 1,1'-карбонилдиимидазола и 73,7 г продукта из стадии 1 с соблюдением условий, описанных в примере 1А, стадия 4, подвергали обменной реакции. Полученную смесь продуктов очищали на силикагельной колонке этиловым эфиром уксусной кислоты. Таким путем получали 71,8 г 3-тиазол-2-ил-1,3- диазаспиро [4.5] декан-2,4-диона (66% от теории) в виде белых кристаллов (tпл 213-215oC).
Пример 3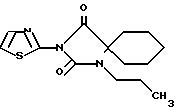
1-Пропил-3-тиазол-2-ил-1,3-диазаспиро [4.5]декан-2,4-дион
5,05 г продукта из примера 2, стадия 2, растворяли в 20 мл диметилформамида. Затем при перемешивании при температуре 20oC добавляли порциями 1,10 г гидрида натрия (50%-ная суспензия в минеральном масле). После перемешивания в течение одного часа добавляли 4 мл 1-йодистого пропана. Перемешивание продолжали еще в течение трех часов. Затем разбавляли 100 мл дистиллированной воды, трижды экстрагировали порциями по 30 мл этилового эфира уксусной кислоты соответственно, промывали насыщенным раствором хлорида натрия и сушили над сульфатом натрия. После удаления перегонкой растворителя остаток очищали на силикагельной колонке этиловым эфиром уксусной кислоты/н-гексаном (соотношение 8:5). Таким путем получали 3,95 г 1-пропил-3-тиазол-2-ил-1,3-диазаспиро [4.5] декан- 2,4-диона (67% от теории) в виде белых кристаллов (tпл 135-138oC).
Пример 4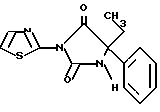
5-Этил-5-фенил-3-тиазол-2-илимидазолидин-2,4-дион
Стадия 1
Этиловый эфир 2-амино-2-фенилмасляной кислоты
10,0 г 2-амино-2- фенилмасляной кислоты перемешивали вместе со 100 мл этанольного раствора хлористого водорода (10% HCl) в течение 10 дней при температуре 30oC. Затем этанол отгоняли, остаток растворяли в 200 мл дистиллированной воды и с помощью карбоната калия устанавливали щелочное значение pH. После трехкратной экстракции этиловым эфиром уксусной кислоты, сушки экстрактов над сульфатом натрия и удаления перегонкой растворителя проводили очистку на силикагельной колонке этиловым эфиром уксусной кислоты. Таким путем получали 6,93 г этилового эфира 2-амино-2-фенилмасляной кислоты (62% от теории) в виде желтоватого масла.
Стадия 2
5-Этил-5-фенил-3-тиазол-2- илимидазолидин-2,4-дион
2,12 г 2-аминотиазола, 3,26 г 1,1'-карбонилдиимидазола и 4,16 г продукта из стадии 1 подвергали обменной реакции в условиях, описанных в примере 1А, стадия 4. После очистки сырой смеси на силикагельной колонке этиловым эфиром уксусной кислоты получали 3,90 г 5-этил-5-фенил-3-тиазол-2-илимидазолидин-2,4-диона (68% от теории) в виде белых кристаллов (tпл 150-152oC).
Пример 5
(+)-и (-)-5-Этил-5-фенил-3-тиазол-2-илимидазолидин-2,4-дион
Оба энантиомера получали в результате разделения рацемата из примера 4 на хиральной ЖХВД-колонке (элюенты: н-гексан/2-пропанол в соотношении 1:1; неподвижная фаза: целлюлоза-трис-3,5-диметилфенилкарбамат).
Пример 6
5-Этил-3-(5-метил[1.3.4]тиадиазол-2-ил)-5-фенилимидазолидин-2,4-дион
В атмосфере азота и при отсутствии влажности 2,30 г 2-амино-5-метил-1,3,4-тиадиазола растворяли при комнатной температуре в 40 мл сухого тетрагидрофурана. Затем добавляли 3,24 г 1,1'-карбонилдиимидазола и перемешивали в течение 30 мин при 50oC. К полученной суспензии добавляли по каплям раствор из 4,15 г этилового эфира 2-амино-2-фенилмасляной кислоты (продукт из примера 3, стадия 1) в 10 мл сухого тетрагидрофурана и перемешивали в течение 20 ч при 50oC. После удаления перегонкой растворителя остаток перекристаллизовывали из этанола. Таким путем получали 3,94 г 5-этил-3-(5-метил[1.3.4] тиадиазол-2-ил)-5-фенилимидазолидин-2,4-диона (58% от теории) в виде белых кристаллов (tпл 223-225oC).
Примеры 7-28
Представленные в таблице 1 соединения получали из соответствующих исходных соединений в условиях, описанных в примерах 1-6.
Разделением рацематов из примеров 17, 18 и 28 в условиях, описанных в примере 5, получали представленные в таблице 2 энантиомеры в виде вязких масел. При определении угла поворота плоскости поляризации [α]
Фармакологические исследования
Высвобождение TNF-α может исследоваться in vitro на мононуклеарных клетках периферической крови человека (Т-клетки, B-клетки и моноциты) после стимуляции липополисахаридом (ЛПС) (см. ниже раздел 1). ЛПС является составной частью бактериальной стенки клетки и стимулирует моноциты и макрофаги.
Наряду со стимуляцией с помощью ЛПС высвобождение TNF-α можно также провоцировать стимуляцией мононуклеарных клеток периферической крови человека специфическими по отношению к Т-клеткам моноклональными антителами против активирующих антигенов (antiCD2/antiCD28) или бактериальным суперантигеном токсином-1, вырабатываемым при токсическом шоке (Toxic Shock Syndrome Toxin-1 или TSST-1). Помимо высвобождения TNF-α эти стимуляторы приводят, в частности, также к образованию интерлейкина-2 (ИЛ-2). Соединения, обусловливающие общую иммуносупрессию, ингибируют высвобождение как ТNF-α, так и ИЛ-2. Соединения же, не обусловливающие блокирование клеточной иммунозащиты, должны, как полагают, хорошо ингибировать стимулированное ЛПС высвобождение TNF-α, тогда как при специфическом по отношению к Т-клеткам стимулированном высвобождении ИЛ-2 они индуцируют лишь незначительное ингибирование (см. ниже раздел 2).
1. Воздействие на высвобождение TNF-α (in vitro)
Ингибирующее действие соединений согласно изобретению по отношению к высвобождению TNF-α испытывали в in vitro-тесте с мононуклеарными клетками.
Мононуклеарные клетки получали из гепаринизированной крови по крайней мере трех добровольных доноров. С этой целью по 20 мл крови фракционировали по известной методике с помощью фиколл-пак- градиента. Затем клетки собирали и трижды промывали клеточно-культуральный средой. Используемая клеточно-культуральная среда состояла из RPMI 1640 с 2 мМ глутамином (фирмы Life Technologies, Эггенштейн), с добавками 10%-ной фетальной телячьей сыворотки (фирмы Life Technologies), 50 мкг/мл стрептомицина (фирмы Sigma, Дейзенхофен), 50 IU/мл пенициллина (фирмы Sigma) и 1 100 мкМ β-меркаптоэтанола (фирмы Merck, Дармштадт). Затем мононуклеарные клетки подвергали обработке в 15 мл клеточно-культуральной среды и распределяли порциями смеси по 1 мл в стерильных инкубационных планшетах с 24 ячейками (фирмы Sigma). К 1-миллилитровым порциям смеси, использовавшимися в качестве контроля, добавляли соответственно по 1 мл диметилсульфоксида (ДМСО, фирмы Merck). В тестируемую смесь добавляли по 1 мкл раствора соединения согласно изобретению (в ДМСО; конечные концентрации в тесте: 0,5; 5; 12,5 и 50 мкг/мл). Порции смеси инкубировали в течение одного часа в инкубаторном шкафу в атмосфере СО2 (5% СО2, 90% влажность воздуха). Затем добавляли, за исключением контроля, соответственно по 2,5 мкг ЛПС (Е. coli 0127:В8; фирмы Sigma, Дейзенхофен) в качестве стимулятора. Инкубацию смеси продолжали еще в течение 20 ч. Концентрацию TNF-α в надосадочной жидкости культуры клеток в тестируемой смеси определяли после окончания инкубации с помощью ферментного иммуносорбентного теста (ELISA; фирма Boehringer-Mannheim). На основе данных контроля и данных, полученных на порциях тестируемой смеси, инкубированных с помощью соединений согласно изобретению, рассчитывали степень ингибирования высвобождения TNF-α. C помощью прямой регрессии определяли концентрацию, обеспечивающую 50%-ное ингибирование высвобождения TNF-α (значения IC50).
Все применявшиеся соединения согласно изобретению проявляли ярко выраженное ингибирующее действие по отношению к стимулированному ЛПС высвобождению TNF-α. Полученные результаты приведены в таблице 3.
2. Воздействие на клеточную иммунозащиту (in vitro)
Для исследования воздействия соединений согласно изобретению на клеточную иммунозащиту в описанной ниже серии опытов in vitro использовали стимулированные различным образом мононуклеарные клетки.
Предлагаемые согласно изобретению соединения испытывали на их эффективность по ингибированию высвобождения TNF-α и ИЛ-2. Опыты проводили в соответствии с условиями, описанными в разделе 1. При этом в каждой серии экспериментов варьировали стимуляторы. В качестве таковых использовали либо моноклональные антитела antiCD2/antiCD28, суперантиген TSST-1, либо ЛПС.
В тестах применяли следующие конечные концентрации стимуляторов:
fntiCD2/antiCD28: 100 нг/мл AICD2.M1; 100 нг/мл AICD2.M2 (моноклональные антитела, оба направленные против CD2, источник: Немецкий центр по изучению рака, проф. д-р Мойер, Гейдельберг);
0,1% (объем/объем) antiCD28-асцит, жидкость (фирма CLB, Амстердам)
суперантиген: 0,1 мкг/мл TSST-1 (фирма Sigma, Дейзенхофен)
ЛПС: 2,5 мкг ЛПС (Е.coli 0127:B8; фирма Sigma, Дейзенхофен)
Соединения согласно изобретению применяли в концентрациях (см. таблицу 4, колонку 2), вызывавших 60-90%-ное ингибирование индуцированного ЛПС высвобождения TNF-α.
В тестируемых составах, стимулированных смесью антител antiCD2/antiCD28 или суперантигеном TSST-1, после завершения опытов определяли в надосадочной жидкости культуры клеток концентрацию ИЛ-2 с помощью ферментного иммуносорбентного анализа ELISA (фирма Boehringer-Mannheim).
Применявшиеся соединения согласно изобретению не проявляли общего иммуносупрессорного эффекта, поскольку в отличие от дексаметазона было индуцировано лишь относительно незначительное ингибирование высвобождения ИЛ-2.
Полученные результаты представлены в таблице 4.
3. Противоваскулитное действие на модели животного
Для выявления противоваскулитного действия соединений формулы I согласно изобретению in vivo использовали двухфазную модель, ведущую свое начало от локального генерализованного феномена Швартцмана [см. Exp. Toxic. Pathol., 47. 167 (1995)] . С помощью этой модели животного можно убедительно подтвердить ингибирование проницаемости эндотелия, обусловленной не только подавлением высвобождения TNF-α. Помимо количественного параметра проницаемости эндотелия можно установить также в качественном отношении значительное восстановление или отсутствие деструкции тканей, характерной для локального генерализованного феномена Швартцмана.
У мужских особей мышей линии NMRI при наркозе короткого действия удаляли с дорсальной поверхности волосяной покров. В расположенных симметрично по обе стороны местах чрескожным путем инъецировали 100 мкг липополисахарида (Salmonella typhosa; Sigma, Дейзенхофен) или, в качестве контроля, физиологический раствор поваренной соли. Через 24 ч через хвостовую вену вводили эванс голубой (фирма Merck, Дармштадт) в концентрации 1 мл/кг. Затем проводили подкожную инъекцию рекомбинантным, мышиным TNF-α (133 нг) под оба участка кожи, восприимчивых к ЛПС. Через четыре часа после провокации фактором TNF-α мышей безболезненно умерщвляли и затем вырезали участки кожи определенной площади. Содержание эванса голубого в пробах кожи определяли методом фотометрической экстинкции при 623 нм, которую осуществляли после 18-часовой экстракции в формамиде при 60oC.
Соединения согласно изобретению суспендировали в водном 1%-ном растворе карбоксиметилцеллюлозы и вводили внутрибрюшинным либо оральным путем. Введение соединений согласно изобретению при интраперитонеальном введении осуществляли соответственно за 10 мин до инъекции ЛПС или TNF-α, а при оральном введении - за 30 мин до этой стимуляции. Еще раз соединения согласно изобретению вводили во время препарационной стадии через 8 ч после инъекции ЛПС. Дозировка составляла 5-400 мг/кг. Для контроля предварительно обрабатывали также животных, которым вместо ЛПС вводили NaCl.
В таблице 5 представлены максимальные значения ингибирования в %, полученные на препарированных с использованием ЛПС животных, которых обрабатывали соединениями согласно изобретению, в сравнении с препарированными NaCl животными, которых также обрабатывали соединениями по изобретению. Процентные данные представляют собой средние значения по ≅10 животным каждой группы.
Предлагаемые согласно изобретению соединения обладают противоваскулитным действием, подтверждаемым количественными данными ингибирования проницаемости эндотелия. Полученные результаты приведены в таблице 5.
[Дополнительный пример 1]
Получение таблетки
К соединению примера 9 добавляют лактозу, кукурузный крахмал, кристаллическую целлюлозу, кармелозу кальция и стеарат магния в соотношениях, указанных ниже, и эти ингредиенты перемешивают в течение 15 мин с помощью Bore Container Mixer (продукция компании Kotobuki Giken Industries К.К.). Эту смесь формуют под давлением, используя установку для таблетирования, получая таблетки диаметром 8 мм и массой 250 мг. Полученная таблетка имеет удовлетворительную твердость, и, кроме того, было также установлено, что таблетка проявляет прекрасную способность к дезинтеграции сразу после начала пищеварения в желудке.
Состав:
Ингредиенты - Масса (%)
Соединение примера 9 - 40
Лактоза - 24
Кукурузный крахмал - 17
Кристаллическая целлюлоза - 12
Кармелоза кальция - 6
Стеарат магния - 1 - 100
[Дополнительный пример 2]
Получение раствора для инъекции
К 1 г соединения примера 9 добавляют 2 г глицерина (дополнительный растворитель), 0,3 г Полисольвата 80 (солюбилизатор) и дистиллированной воды для инъекции с получением однородного раствора. К этому раствору добавляют буфер и хлорид натрия (изотонический агент) с получением 300 мл изотонического раствора (pH 6,0).
Полученный изотонический раствор фильтруют через мембранный фильтр (0,22 мкм) и 30 мл полученного фильтрата помещают в стерилизованную виалу (коническую толстостенную пробирку с крышкой), получая раствор для инъекций.
[Дополнительный пример 3]
Получение мази
90 г гидрофильной пластичной основы добавляют к 1 г соединения примера 9 и диспергируют до состояния гомогенности с последующим добавлением 3 г легкой фракции безводного оксида кремния и 6 г октадодецилмиристата. Эту смесь перемешивают в течение 20 мин и 10 г этой смеси помещают в алюминиевую тубу с получением мази, содержащей соединение в концентрации 1%.
[Дополнительный пример 4]
Получение глазных капель
0,3 г соединения примера 9 растворяют в 100 мл стерилизованной очищенной воды и к полученному раствору добавляют буфер и хлорид натрия с получением изотонического раствора с pH 7,5. 5 мл этого раствора помещают в стерилизованный сосуд с получением глазных капель.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ ЗАМЕЩЕННЫХ ИМИДАЗОЛИДИН-2,4-ДИОНОВЫХ СОЕДИНЕНИЙ В КАЧЕСТВЕ ОБЕЗБОЛИВАЮЩИХ СРЕДСТВ | 1998 |
|
RU2195933C2 |
| ЗАМЕЩЕННЫЕ ПИПЕРИДИН-2,6-ДИОНЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2184733C2 |
| ЗАМЕЩЕННЫЕ ГЛУТАРИМИДЫ, СПОСОБЫ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2001 |
|
RU2278857C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОКСАЗИНДИОНА И ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1998 |
|
RU2184114C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ БЕНЗОЦИКЛОАЛКЕНОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2197484C2 |
| ДИМЕТИЛ(3-АРИЛБУТ-3-ЕНИЛ)АМИНОСОЕДИНЕНИЯ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2167146C2 |
| ЗАМЕЩЕННЫЕ АМИНОСОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2197474C2 |
| 6-ДИМЕТИЛАМИНОМЕТИЛ-1-ФЕНИЛЦИКЛОГЕКСАНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИ ДЕЙСТВУЮЩИХ ВЕЩЕСТВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1996 |
|
RU2178409C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗО-3-ИЛАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2000 |
|
RU2264402C2 |
| ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ АМИДЫ, СОДЕРЖАЩИЕ НАСЫЩЕННУЮ СВЯЗЫВАЮЩУЮ ГРУППУ, И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2006 |
|
RU2412181C2 |






Изобретение описывает замещенные имидазолидин-2,4-ционовые соединения общей формулы I, где R1, R2, R3, R4 имеют указанные в формуле изобретения значения, обладающие противоваскулитным действием и высокой иммуномодуляторной эффективностью по ингибированию высвобождения TNF-α и ИЛ-2, а также способ их получения. 4 с. и 8 з.п.ф-лы, 5 табл.
в которой R1 означает С1 - С6алкил или С3 - С6циклоалкил;
R2 означает С1 - С6алкил, фенил, -(СН2)1-3-фенил или -(СН2)1-4-СООR5
или R1 и R2 оба вместе означают -(СН2)4-6- либо
R3 означает Н, С1 - С5алкил или -(СН2)1-4-СООR5;
R4 представляет собой гетероароматический углеводород из группы, соответствующей формулам


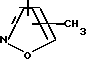
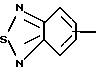

R5 представляет собой С1 - С3алкил;
R6 означает Н, фенил;
R7 означает Н, С1 - С4алкил или трифторметил.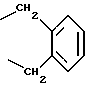
R3 означает Н, С1 - С4алкил или -(СН2)1-2-СООR5; R4 представляет собой гетероароматический углеводород из группы, соответствующей формулам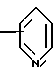

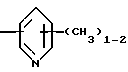


R5 представляет собой С1 - С3алкил;
R6 означает Н или фенил;
R7 означает Н, метил, трет-бутил или трифторметил.
в которой R1 означает С1 - С6алкил или С3 - С6циклоалкил;
R2 означает С1 - С6алкил, фенил, -(СН2)1-3-фенил или -(СН2)1-4-СООR5
или R1 и R2 оба вместе означают -(СН2)4-6- либо
R3 означает Н, С1 - С5алкил или -(СН2)1-4-СООR5;
R4 представляет собой гетероароматический углеводород из группы, соответствующей формулам

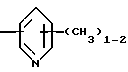
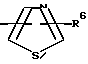

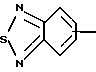

R5 представляет собой С1 - С3алкил;
R6 означает Н, фенил;
R7 означает Н, С1 - С4алкил или трифторметил,
отличающийся тем, что к амину формулы II R4 - NH2 добавляют 1,1'-карбонилдиимидазол или дифениловый эфир угольной кислоты и затем взаимодействием с соединением формулы III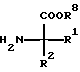
в которой R8 представляет собой Н или С1 - С3алкил, трансформируют в соединение формулы I, где R3 означает Н, которое при необходимости депротонируют, после чего взаимодействием с соединением формулы IV Х-С1 - С5алкил
либо с соединением формулы V
X - (CH2)1-4-COOR5,
в которых Х означает хлор, бром или иод,
трансформируют в соединение формулы I, в которой R3 означает С1 - С5алкил или -(СН2)1-4-СООR5.
| Способ получения производных пирролидина | 1983 |
|
SU1240359A3 |
| Способ получения гидантоиновых производных эфиров угольной или тиоугольной кислоты | 1970 |
|
SU333836A1 |
| Фунгицидное средство | 1976 |
|
SU622386A3 |
| 1970 |
|
SU436426A1 | |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |

