Настоящее изобретение относится к способу ацилирования гексакис(фенилметил)гексаазаизовюрцитана. Конкретнее, настоящее изобретение касается способа ацилирования гексакис(фенилметил) гексаазаизовюрцитана (в дальнейшем, часто относящегося просто к "WB6") восстановительным дефенилметилированием в присутствии ацилирующего реагента, включающего контактирование (a) WB6 и (b) гетерогенного катализатора восстановления в присутствии (с) ацилирующего агента и (d) восстановителя в (е) растворителе, пригодном для WB6, проводя таким образом реакцию восстановительного дефенилметилирования/ацилирования WB6 (а), причем в отсутствие одного из реагентов - ацилирующего реагента (с) и восстановителя (d) взаимодействие между WB6 (а) и гетерогенным катализатором восстановления (b) не происходит. В способе по настоящему изобретению, при получении производных тетраацилгексаазаизовюрцитана (которые пригодны в качестве предшественников гексанитрогексаазаизовюрцитана, используемого для улучшения эксплуатационных качеств обычных взрывчатых веществ) путем ацилирования WB6, можно эффективно подавлять разложение гексанитрогексаазаизовюрцитанового скелета, которое, вероятно, происходит на начальной стадии реакции ацилировния WB6 в качестве исходного вещества с получением желаемых производных тетраацилгексаазаизовюрцитана со стабильно высоким выходом. Следовательно, способ по настоящему изобретению является промышленно полезным. Коме того, способ по настоящему изобретению также является полезным тем, что по сравнению с общепринятыми способами позволяет эффективно подавлять снижение каталитической активности катализатора восстановления, имеющее место во время реакции.
В качестве общепринятого способа получения тетраацилбис(фенилметил)гексаазаизовюрцитана (в дальнейшем, часто относящегося просто к "WA4B2") известен способ, в котором гексакис(фенилметил)гексаазаизовюрцитан (т.е. WB6) подвергается восстановительному дефенилметилированию в присутствии ацилирующего реагента для получения таким образом WA4B2 (смотри "Tetrahedron", vol. 51, 16, 4711-4722 (1995), заявка на международный патент, публикации WО 96/23792 и WО 97/20785 и патент США 5693794).
Кроме того, известен способ, в котором WA4B2 подвергается восстановительному дефенилметилированию для получения таким образом тетраацилгексаазаизовюрцитана (в дальнейшем, часто относящегося просто к "WA4H2") (смотри вышеупомянутую WО 96/23792).
В каждом из этих патентных и непатентных документов получение WA4B2 проводится способом, в котором исходное вещество (т.е. WB6) и другие реагенты (включая ацилирующий агент, растворитель, катализатор и тому подобное) загружаются в реактор при относительно низкой температуре (т. е. от 5 до 25oС) для получения смеси, и затем в реактор вводится газообразный водород в качестве восстановителя, в то же время поддерживая температуру полученной смеси при вышеуказанной относительно низкой температуре с последующим перемешиванием, проводя таким образом реакцию (экзотермическую реакцию), причем температура реакционной системы повышается до желаемого уровня (т. е. от 40o до 70oС) за счет тепла, образующегося в реакции. В этом способе WB6 подвергается восстановительному дефенилметилированию в присутствии ацилирующего реагента для того, чтобы проводить ацилирование WB6 мягко, предотвращая таким образом разложение WB6. В этом способе, поскольку исходное вещество и другие реагенты загружаются в реактор при сравнительно низкой температуре, разложение WB6 под действием температуры можно подавить. Однако даже в этом способе невозможно с удовлетворительным результатом подавлять разложение WB6. Более того, этот способ сопровождается протеканием нежелательных побочных реакций таких, как побочная реакция восстановления, в которой ацильная группа, связанная с гексаазаизовюрцитановым скелетом, в результате ацилирования WB6 превращается в алкильную группу. Следовательно, в этом способе желаемый продукт невозможно получать с удовлетворительным выходом.
В вышеуказанном способе известного уровня техники реакция начинается при относительно низкой температуре (т.е. от 5 до 25oС). Однако возможно начинать реакцию таким образом, чтобы реакция начиналась уже после того, как температуру смеси исходного вещества и реагентов повышают до заранее определенного уровня (т. е. от 40 до 70oС). В качестве примера такого способа, в котором реакция начинается после повышения температуры смеси исходного соединения/других реагентов до заранее определенного уровня, можно упомянуть способ, в котором температуру смеси исходного вещества (т.е. WB6) и других реагентов (включая ацилирующий реагент, растворитель, катализатор и тому подобное) повышают до заранее определенного уровня без добавления к смеси газообразного водорода в качестве восстановителя, и затем уже к смеси добавляют газообразный водород для проведения реакции. Однако этот способ имеет ряд недостатков, поскольку происходит заметное разложение WB6, т.е. проблемы, сопутствующие вышеупомянутому способу известного уровня техники, нельзя решить.
Кроме того, недостатком вышеуказанного известного способа является также то, что каталитическая активность гетерогенного катализатора восстановления (состоящего, например, из редкого металла группы платины), используемого в реакции, вероятно, снижается. Обычно желательно, чтобы дезактивированный катализатор подвергался регенерации с целью его рециркуляции. Однако для регенерации дезактивированного катализатора необходимо проводить повторную активацию, которая является трудоемкой операцией и требует больших затрат. Следовательно, с промышленной точки зрения рециркуляция катализатора непрактична. Кроме того, вышеуказанная проблема снижения каталитической активности имеет другой недостаток, заключающийся в возникновении трудностей проведения промышленно-непрерывного способа. Следовательно, желательно предупредить снижение активности гетерогенного катализатора восстановления во время реакции.
Как упоминалось выше, известным способом, заключающемся в восстановительном дефенилметилировании WB6 в присутствии ацилирующего реагента, нельзя получать производные тетраацилгексаазаизовюрцитана со стабильно высоким выходом и предупреждать разложение гексаазаизовюрцитанового скелета. Кроме того, недостатком общепринятого способа также является снижение каталитической активности катализатора в ходе реакции. Следовательно, возникают трудности в проведении реакции промышленно-непрерывным способом.
Авторы настоящего изобретения провели экстенсивные и интенсивные исследования, направленные на решение вышеуказанных проблем, сопутствующих способам известного уровня техники. В результате неожиданно было установлено, что в способе получения реакционной смеси, содержащей, по меньшей мере, одно производное тетраацилгексаазаизовюрцитана, заключающемся в контактировании (а) гексакис(фенилметил)гексаазаизовюрцитана (WB6) и (b) гетерогенного катализатора восстановления в присутствии (с) ацилирующего реагента и (d) восстановителя в (е) растворителе, пригодном для WB6 (a), проводится реакция восстановительного дефенилметилирования/ацилирования WB6 (а), при которой исключается контакт между WB6 (а) и гетерогенным катализатором восстановления (b) в отсутствие одного из ацилирующих реагентов (с) и восстановителя (d), становится возможным предупреждать не только разложение гексаазаизовюрцитанового скелета (которое, вероятно, происходит на начальной стадии реакции ацилирования WB6), но также снижение активности катализатора восстановления в ходе реакции, получая желаемые производные тетраацилгексаазаизовюрцитана со стабильно высоким выходом. Настоящее изобретение основано на упомянутом выше открытии.
Основной задачей настоящего изобретения является создание способа ацилирования гексакис(фенилметил)гексаазаизовюрцитана (WB6), который можно использовать для стабильного получения производных тетраацилгексаазаизовюрцитана с высоким выходом, в то же время предупреждая разложение гексаазаизовюрцитанового скелета WB6 (которое, вероятно, происходит на начальной стадии реакции ацилирования WB6) и в то же время предупреждая снижение активности катализатора восстановления во время реакции.
Вышеупомянутые и другие задачи, признаки и преимущества настоящего изобретения станут очевидными из последующего детального описания вместе с сопровождающими рисунками и прилагаемой формулой изобретения.
Краткое описание рисунков
Фигура 1 является микрофотографией (х200), сделанной с помощью сканирующего электронного микроскопа (СЭМ) сырого гексабензилгексаазаизовюрцитана, полученного в сравнительном примере 3, в котором провели промывание;
Фигура 2 является микрофотографией, сделанной с помощью СЭМ (х200), кристаллов гексабензилгексаазаизовюрцитана, полученного в сравнительном примере 4, в котором сырой гексабензилгексаазаизовюрцитан, полученный в сравнительном примере 3, перекристаллизовывают;
Фигура 3 является микрофотографией, сделанной с помощью СЭМ (х200), кристаллов гексабензилгексаазаизовюрцитана, полученного в сравнительном примере 8, в котором сырой гексабензилгексаазаизовюрцитан, полученный в сравнительном примере 3, перекристаллизовывают и
Фигура 4 является микрофотографией, сделанной с помощью СЭМ (х200), кристаллов гексабензилгексаазаизовюрцитана, полученного в сравнительном примере 9, после промывания.
Детальное описание изобретения
В соответствии с настоящим изобретением создан способ ацилирования гексакис(фенилметил)гексаазаизовюрцитана восстановительным дефенилметилированием в присутствии ацилирующего реагента, гексакис(фенилметил)гексаазаизовюрцитана, представленного следующей формулой (1):
WB6,
где каждое В независимо представляет фенилметильную группу и W представляет остаток шестивалентного гексаазаизовюрцината, представленного следующей формулой (2):
который включает контактирование (а) гексакис(фенилметил)гексаазаизовюрцитана и (b) гетерогенного катализатора восстановления в присутствии (с) ацилирующего реагента и (d) восстановителя в (е) растворителе, пригодном для гексакис(фенилметил)гексаазаизовюрцитана (а), в результате которого происходит реакция восстановительного дефенилметилирования/ацилирования гексакис(фенилметил)гексаазаизовюрцитана (а) с получением реакционной смеси, содержащей, по меньшей мере, одно производное тетраацилгексаазаизовюрцитана, причем в отсутствие одного из ацилирующих реагентов (с) и восстановителя (d) взаимодействие между гексакис(фенилметил)гексаазаизовюрцитаном (а) и гетерогенным катализатором восстановления (b) отсутствует.
Для более легкого понимания настоящего изобретения основные признаки и различные предпочтительные воплощения настоящего изобретения перечисляются ниже:
1. Способ ацилирования гексакис(фенилметил)гексаазаизовюрцитана восстановительным дефенилметилированием в присутствии ацилирующего реагента, гексакис(фенилметил)гексаазаизовюрцитана, представленного следующей формулой (1):
WB6,
где каждое В независимо представляет фенилметильную группу и W представляет остаток шестивалентного гексаазаизовюрцитана, представленного следующей формулой (2):
который включает контактирование (а) гексакис(фенилметил)гексаазаизовюрцитана и (b) гетерогенного катализатора восстановления в присутствии (с) ацилирующего реагента и (d) восстановителя в (е) растворителе, пригодном для гексакис(фенилметил)гексаазаизовюрцитана (а), в результате чего происходит реакция восстановительного дефенилметилирования/ацилирования гексакис(фенилметил)гексаазаизовюрцитана (а) с получением реакционной смеси, содержащей, по меньшей мере, одно производное тетраацилгексаазаизовюрцитана, причем в отсутствие одного из ацилирующих реагентов (с) и восстановителя (d) взаимодействие между гексакис(фенилметил)гексаазаизовюрцитаном (а) и гетерогенным катализатором восстановления (b) отсутствует.
2. Способ по п.1 выше, в котором реакция восстановительного дефенилметилирования/ацилирования гексакис(фенилметил)гексаазаизовюрцитана (а) проводится при температуре от 40 до 160oС.
3. Способ по п.п.1 или 2 выше, в котором растворитель (е) является растворителем, содержащим амидную группу.
4. Способ по одному из п.п.1-3 выше, в котором гексакис(фенилметил)гексаазаизовюрцитан (а) и растворитель (е) представляют собой раствор (а) в (е) и гетерогенный катализатор восстановления (b) и восстановитель (d) представляют собой смесь (b) и (d), и где раствор (а) в (е) контактирует со смесью (b) и (d) в присутствии ацилирующего реагента (с).
5. Способ по одному из пп.1-3 выше, в котором гексакис(фенилметил)гексаазаизовюрцитан (а) и растворитель (е) представляют собой раствор (а) в (е) и гетерогенный катализатор восстановления (b), ацилирующий реагент (с) и восстановитель (d) представляют собой смеси (b), (с) и (d), и смесь (b), (с) и (d) готовят смешением гетерогенного катализатора восстановления (b) и восстановителя (d) с последующим добавлением ацилирующего реагента (с) к этому раствору, и где раствор (а) в (е) контактирует со смесью (b), (с) и (d).
6. Способ по одному из п.п.1-5 выше, в котором реакционная смесь содержит, по меньшей мере, одно производное тетраацилгексаазаизовюрцитана, представленного следующей формулой (3):
WA4BnH(2-n),
где n является целым числом от 0 до 2, каждое А независимо представляет C1-С10 ацильную группу, Н представляет атом водорода и каждый из В и W определены выше.
Характерным признаком способа по настоящему изобретению является то, что восстановительное дефенилметилирование WB6 проводится в присутствии ацилирующего реагента при контактировании WB6 и гетерогенного катализатора восстановления в условиях, при которых взаимодействие между WB6 и гетерогенным катализатором восстановления происходит только в присутствии как ацилирующего реагента, так и восстановителя. В результате разложение WB6, которое, вероятно, происходит во время реакции ацилирования WB6, может эффективно подавляться.
Известно, что, когда WB6 подвергается восстановительному дефенилметилированию при отсутствии ацилирующего реагента, образуются производные гексаазаизовюрцитана, содержащие вторичную аминогруппу (такие как WB5H, WB4H2 и WB3H3), которые имеют нестабильную структуру, и они легко претерпевают разложение их гексаазаизовюрцитанового скелета. С другой стороны, также известно, что, когда WB6 подвергается восстановительному дефенилметилированию в присутствии ацилирующего реагента, вышеуказанные нестабильные производные гексаазаизовюрцитана, содержащие вторичную аминогруппу, ацилируются сразу же после их образования с образованием стабильных ацилированных производных гексаазаизовюрцитана так, что разложение гексаазаизовюрцитанового скелета можно предотвратить (смотри заявку на международный патент, публикация WО 96/23792). Однако совершенно неожиданно, что если при проведении реакции восстановительного дефенилметилирования WB6 в присутствии ацилирующего реагента в то же время избегать взаимодействия между WB6 и гетерогенным катализатором восстановления в отсутствие одного из реагентов - ацилирующего реагента и восстановителя, то можно эффективно подавлять разложение WB6 по сравнению с общепринятыми способами.
Кроме того, способом по настоящему изобретению можно эффективно подавлять снижение каталитической активности в ходе реакции по сравнению с общепринятыми способами. В силу этого преимуществом способа по настоящему изобретению является то, что гетерогенный катализатор восстановления, используемый в реакции, можно рециркулировать без проведения трудоемкой обработки для регенерации катализатора. В настоящем изобретении, если желательно, катализатор, используемый в реакции, можно рециркулировать просто после промывания катализатора для удаления растворителя и других компонентов, прилипших к катализатору. (Обычно регенерацию дезактивированного гетерогенного катализатора для восстановления проводят с помощью промышленно непригодной операции, которая не только трудоемка, но также требует больших затрат. Например, в случае дезактивации катализатора на носителе его регенерацию проводят, например, громоздким и требующим больших затрат способом, в котором катализатор подвергается окислению, и затем обработанный катализатор растворяется в растворителе с последующей иммобилизацией катализатора на носителе). Кроме того, в силу вышеуказанного эффекта подавления снижения каталитической активности способ по настоящему изобретению можно применять в выгодном для промышленности непрерывном методе.
Ниже настоящее изобретение будет описано более детально.
Способ синтеза WB6, представленного формулой (1), не имеет ограничения. Однако предпочтительно, чтобы WB6, использованный в настоящем изобретении, синтезировали путем циклизации арилметиламина и глиоксаля с выделением воды в присутствии кислотного катализатора. Что касается чистоты WB6, то предпочтительно, чтобы WB6 был продуктом высокой чистоты, с чистотой порядка 95% или более. Использование продукта столь высокой чистоты выгодно для улучшения скорости реакции восстановительного деарилметилирования/ацилирования.
В способе по настоящему изобретению WB6 (а) формулы (1) обычно используется в количестве от 0,0001 до 0,4, предпочтительно от 0,001 до 0,3, более предпочтительно от 0,01 до 0,15, выраженном в весовом соотношении WB6 (а) к растворителю (е).
Предпочтительно, чтобы во время загрузки WB6 в реактор, WB6 находился в растворенном виде в растворителе (е). Однако WB6 может не полностью растворяться в растворителе и находиться в растворителе в виде суспензии, в которой только часть WB6 растворима в растворителе. Предпочтительно использовать раствор, в котором WB6 полностью растворяется в растворителе.
В отношении реакционного растворителя (е), используемого в способе по настоящему изобретению, не имеется особого ограничения при условии, что растворитель не влияет отрицательно на протекание реакции. В частности, органический растворитель, содержащий амидную группу, является предпочтительным для повышения скорости реакции и выхода производных тетраацилгексаазаизовюрцитана. Примеры органических растворителей, содержащих амидную группу, включают N,N-диметилацетамид, N,N-диметилформамид, 1,3-диметил-2-имидазолидон, N-метил-2-пирролидон и тому подобное. Среди этих растворителей N, N-диметилацетамид и N,N-диметилформамид являются предпочтительными. Вышеуказанные растворители можно использовать в отдельности или в комбинации.
В способе по настоящему изобретению в качестве восстановителя (d) обычно используется газообразный водород.
Восстановитель (d) обычно используется в количестве от 0,67 до 10000, предпочтительно от 0,67 до 1000, более предпочтительно от 2 до 50 в виде молярного соотношения восстановителя к арилметильным группам WB6. Когда в качестве восстановителя (d) используется газообразный водород, давление реакции обычно находится в пределах от 0,001 до 10 кгс/см2, предпочтительно от 0,01 до 30 кгс/см2, более предпочтительно от 0,01 до 10 кгс/см2, наиболее предпочтительно от 2 до 5 кгс/см2, выражен в виде парциального давления водорода. Когда в качестве восстановителя (d) используется газообразный водород, реакция может удовлетворительно протекать, даже если давление реакции (парциальное давление водорода) равняется 10 кгс/см2 или менее. Однако, когда используется реакционное оборудование, имеющее такое устройство, где скорость диффузии водорода в реакторе и скорость растворения водорода в реакционном растворе становятся низкими (например, при использовании автоклава), можно использовать высокое давление водорода (примерно до 50 кгс/см2) для того, чтобы поддерживать скорость диффузии водорода и скорость растворения водорода на высоком уровне. В дополнение к газообразному водороду в реакционной смеси могут присутствовать инертные газы такие, как азот, аргон и гелий.
В отношении гетерогенного катализатора восстановления (b), используемого в способе по настоящему изобретению, не имеется особого ограничения при условии, что он способен ускорять восстановительное дефенилметилирование WB6 и остается в гетерогенном состоянии в растворителе (е). В качестве гетерогенного катализатора восстановления (b) обычно используется катализатор, содержащий металл, принадлежащий к группе платины или содержащий ее производное. Предпочтительные примеры гетерогенных катализаторов восстановления включают соединения Pd (такие, как Pd(OAc)2, PdCl2, Pd(NO3)2, PdO, Pd(OH)2, Pd3Pb и Pd3Te), сплавы Pd и металлический Pd, и соединения Ru (такие, как RuCl3), сплавы Ru и металлический Ru. Из них соединения Pd (такие, как Pd(OAc)2 и PdCl2), сплавы Pd и металлический Pd являются более предпочтительными. Возможно, что среди вышеуказанных катализаторов некоторые катализаторы становятся гомогенными во время реакции в зависимости от типа используемого растворителя. В отношении такого катализатора предпочтительно, чтобы перед использованием катализатору придавали такой вид, при котором он не мог бы раствориться в использованном растворителе, т.е. катализатор подвергается восстановлению после нанесения его на носитель. Примеры носителей включают активированный уголь, двуокись кремния, окись алюминия, алюмосиликаты, цеолит, активированную глину, двуокись циркония и двуокись титана. Среди этих носителей активированный уголь является особенно полезным потому, что обладает низкой реакционной способностью в отношении карбоновой кислоты, производного ацилирующего агента в реакционной системе, и он также обладает относительно низкой реакционной способностью в отношении других химических веществ. Кроме того, для повышения каталитической активности предпочтительно, чтобы катализатор подвергался восстановлению перед использованием в реакции восстановительного дефенилметилирования. В качестве восстановителя для осуществления его восстановления предпочтительны газообразный водород, гидразин или формальдегид. Когда предполагается использовать катализатор на носителе, то поверхность носителя можно обработать таким образом, чтобы инактивированные кислотные участки, имеющиеся на поверхности носителя, обрабатывались силилированием, ацилированием и тому подобное, или таким образом, чтобы активировать кислотные участки, присутствующие на поверхности носителя, активированием (таким, как перемешивание в азотной кислоте), или таким образом, чтобы нейтрализовать кислотные участки на поверхности носителя адсорбцией вещества с основными свойствами (например, NaOH). Каждую из обработок для модификации кислотных участков на поверхности носителя можно осуществлять либо до, либо после нанесения катализатора на носитель.
Предпочтительно, чтобы гетерогенный катализатор восстановления (b) использовался в виде суспензии, полученной при диспергировании катализатора в жидкости такой, как дисперсионная среда. Дисперсионная среда для приготовления суспензии катализатора (b) не имеет особого ограничения, но предпочтительно использовать растворитель такой же, как растворитель (е).
Ацилирующий реагент (с), используемый в способе по настоящему изобретению, не имеет особого ограничения при условии, что он способен ацилировать вторичную аминогруппу с образованием связи N-ацил. В общем используется ацилирующий реагент такой, как ангидриды карбоновых кислот такие, как уксусный ангидрид, пропионовый ангидрид, муравьиный ангидрид, ангидрид молочной кислоты и ангидрид смеси уксусной кислоты и муравьиной кислоты. Эти ангидриды карбоновых кислот можно использовать в отдельности или в комбинации. В альтернативном случае вышеуказанные ангидриды карбоновых кислот можно использовать в комбинации с карбоновыми эфирами N-гидроксисукцинимида и/или ацилимидазолов. Примеры карбоновых эфиров N-гидроксисукцинимида включают N-ацетоксисукцинимид, N-пропионилоксисукцинимид и N-(2-фенилацетокси)сукцинимид; примеры ацилимидазолов включают ацетилимидазол и пропионилимидазол. Среди этих ацилирующих реагентов предпочтительно использовать только одни ангидриды карбоновой кислоты, и наиболее предпочтительным ангидридом карбоновой кислоты является уксусный ангидрид. Кроме того, когда используемый ацилирующий реагент является жидкостью (такой, как уксусный ангидрид, пропионовый ангидрид, ангидрид смеси уксусной кислоты/муравьиной кислоты), такой ацилирующий реагент можно также использовать в качестве растворителя.
Ацилирующий реагент (с) обычно используют в количестве от 0,67 до 50, предпочтительно от 0,67 до 5, более предпочтительно от 0,67 до 2, выражен в виде молярного соотношения ацилируюшего реагента к арилметильным группам WB6 (a).
Температура реакции при восстановительном дефенилметилировании/ацилировании WB6 в способе по настоящему изобретению обычно находится в пределах от 40 до 160oС, предпочтительно от 40 до 100oС, более предпочтительно от 40 до 80oС, еще более предпочтительно от 50 до 80oС и наиболее предпочтительно от 50 до 70oС. Когда эту реакцию проводят при относительно низкой температуре, а именно от 40oС до ниже, чем 80oС, скорость превращения WB6 становится относительно низкой, но разложение WB6 под действием температуры может быть подавлено. Следовательно, предпочтительно проводить восстановительное дефенилметилирование/ацилирование при сравнительно низкой температуре (от 40oС до ниже, чем 80oС). Когда реакцию проводят при относительно высокой температуре, а именно от 80 до 160oС, даже несмотря на то, что химическое разложение WB6 во время дефенилметилирования/ацилирования WB6 подавляется способом по настоящему изобретению, разложение WB6 под действием температуры протекает, и, следовательно, становится необходимым увеличить скорость превращения WB6. В этом случае является благоприятным увеличение растворимости WB6 при высокой температуре, способствуя таким образом протеканию реакции при сравнительно высокой концентрации WB6; однако с точки зрения подавления разложения WB6 под действием температуры предпочтительно избегать проведение реакции при высокой температуре от 80 до 160oС. Давление реакции восстановительного деарилметилирования/ацилирования в способе по настоящему изобретению обычно находится в пределах от 0,001 до 100 кгс/см2, предпочтительно от 0,01 до 30 кгс/см2, более предпочтительно от 0,01 до 10 кгс/см2, наиболее предпочтительно от 2 до 5 кгс/см2, выраженных в единицах парциального давления водорода. Не имеется особого ограничения в отношении времени реакции восстановительного дефенилметилирования/ацилирования в способе по настоящему изобретению при условии получения желаемых производных тетраацилгексаазаизовюрцитана. Однако в основном удовлетворительным является время реакции, равное 10 ч и менее.
Увеличение скорости превращения WB6 в реакции восстановительного дефенилметилирования/ацилирования можно достигнуть, повышая количество восстановителя и/или катализатора или повышая температуру реакции. В способе по настоящему изобретению наиболее предпочтительно, чтобы скорость реакции регулировалась увеличением или снижением количества катализатора. Количество катализатора в значительной степени варьируется в зависимости от его каталитической активности, но обычно составляет от 0,0001 до 1,0, предпочтительно от 0,01 до 0,8, более предпочтительно от 0,1 до 0,4, выражено в виде весового соотношения катализатора к используемому WB6.
Если желательно, то можно использовать промотор кислотного типа в качестве промотора реакции. Примеры промоторов кислотного типа включают органические кислоты такие, как карбоновые кислоты и фенолы, и бром(Вr)-содержащие промоторы кислотного типа. Среди этих промоторов органические кислоты имеют низкую способность вызывать разложение WB6, но обладают и низким промоторным действием в отношении реакции восстановительного дефенилметилирования/ацилирования. С другой стороны, бром(Вr)-содержащий промотор кислотного типа обладает высоким промоторным действием в отношении реакции восстановительного дефенилметилирования/ацилирования и сравнительно низкой способностью вызывать разложение WB6. Следовательно, бром(Вr)-содержащий промотор кислотного типа является более предпочтительным, чем органическая кислота. Примеры бром(Вr)-содержащих промоторов кислотного типа включают НВr и вещество, которое образует НВr, подвергаясь реакции гидрирования. Конкретнее, "веществом, которое образует НВr, подвергаясь реакции гидрирования" является вещество, имеющее такое свойство, что когда оно загружается в реактор, то оно находится в апротонной форме, и которое образует НВr, будучи гидрированным под действием гетерогенного катализатора для восстановления в атмосфере восстановителя (в атмосфере водорода). Примеры веществ, которые образуют НВr, подвергаясь реакции гидрирования, включают фенилбромид, бензилбромид, ацетилбромид и бром (Br2). Поскольку Вr-содержащий промотор кислотного типа имеет способность вызывать разложение WB6, то с точки зрения подавления разложения WB6, которое является предметом настоящего изобретения, предпочтительно не использовать бром(Вr)-содержащий промотор кислотного типа.
Для подавления разложения WB6 важно превратить WB6 (который является нестабильным при воздействии тепла и кислоты) в ацилированное соединение гексаазаизовюрцитана (которое является стабильным при воздействии тепла и кислоты) как можно быстрее. Когда два атома азота в структуре WB6 ацилированы, то полученное ацилированное производное гексаазаизовюрцитана обладает очень высокой стабильностью к воздействию тепла и кислоты по сравнению со стабильностью WB6. Для увеличения как скорости реакции восстановительного дефенилметилирования WB6, так и скорости ацилирования WB6 является предпочтительным, чтобы реакцию проводили при относительно высокой температуре. Для ускорения реакции ацилирующего реагента, использованного в способе по настоящему изобретению, предпочтительно выбирать температуру реакции, равную 40oС или выше. Однако поскольку разложение WB6 возрастает при температуре выше, чем 160oС, предпочтительно, чтобы температура реакции выбиралась в интервале от 40 до 160oС. В способе по настоящему изобретению предпочтительно регулировать условия реакции таким образом, чтобы температура реакции находилась в пределах от 40 до 160oС, когда WB6 контактирует с гетерогенным катализатором восстановления, или чтобы температура реакции в пределах от 40 до 160oС достигалась сразу же после того, как WB6 контактирует с гетерогенным катализатором восстановления.
Когда для превращения WB6 с высокой скоростью используется относительно высокая температура (от 40 до 160oС), предпочтительно использовать в качестве растворителя реакции растворитель, содержащий амидную группу. Органический растворитель, содержащий амидную группу, предпочтителен потому, что он является слабо основным и, следовательно, может нейтрализовать кислотный протон, образующийся при ацилировании в качестве побочного продукта. То есть, органический растворитель, содержащий амидную группу, может поддерживать реакционную систему около нейтральной точки, подавляя таким образом разложение WB6 под действием кислотного протона, даже в области высоких температур. Кроме того, основность растворителя, содержащего амидную группу, способствует ацилированию WB6. Причина этого заключается в следующем. Вторичный амин, образующийся в результате реакции восстановительного дефенилметилирования WB6, является нестабильным так, что вторичный амин, вероятно, претерпевает разложение гексаазаизовюрцитанового скелета, если только скелет сразу же не защищается ацилированием вторичного амина. Поскольку растворитель, содержащий амидную группу (который является слабощелочным), способствует ацилированию вторичного амина, разложение WB6 можно подавлять растворителем, содержащим амидную группу.
Скорость реакции восстановительного дефенилметилирования/ацилирования WB6 в способе по настоящему изобретению становится выше, когда гетерогенный катализатор восстановления уже находится в восстановленном состоянии перед началом реакции. Следовательно, наиболее предпочтительно использовать гетерогенный катализатор восстановления, который подвергся предварительной обработке с целью его восстановления. В качестве способа восстановления гетерогенного катализатора восстановителя используется способ, в котором гетерогенный катализатор восстановления контактирует с восстановителем. В отношении восстановителя, используемого в способе восстановления, не имеется особого ограничения при условии, что он обладает восстановительной активностью. Примеры восстановителей включают газообразный водород, муравьиную кислоту, гидразин, спирт, альдегид и тому подобное. Среди этих восстановителей предпочтительным является газообразный водород. Причина этого заключается не только в том, что газообразный водород обладает высокой восстановительной способностью, но также и в том, что после восстановления газообразным водородом нет нужды промывать восстановленный катализатор, и восстановленный катализатор, как таковой, можно использовать в способе по настоящему изобретению.
В качестве примеров способов для контактирования гексакис(фенилметил)гексаазаизовюрцитана (а) и гетерогенного катализатора восстановления (b) в присутствии ацилирующего реагента (с) и восстановителя (d) в растворителе (е), в то же время при выполнении требования, определенного в настоящем изобретении (т. е. требования об отсутствии контакта между WB6 (а) и гетерогенным катализатором восстановления (b) в отсутствие одного из ацилирующих реагентов (с) и восстановителя (d) ), можно упомянуть следующие способы от (А) до (Н):
(А) способ, в котором гексакис(фенилметил)гексаазаизовюрцитан (а) и растворитель (е) находятся в виде раствора (а) в (е) и гетерогенный катализатор восстановления (b) и восстановитель (d) находятся в виде смеси (b) и (d), где раствор (а) в (е) контактирует со смесью (b) и (d) в присутствии ацилирующего реагента (с);
(B) способ, в котором гексакис(фенилметил)гексаазаизовюрцитан (а) и растворитель (е) находятся в виде раствора (а) в (е) и гетерогенный катализатор восстановления (b), ацилирующий реагент (с) и восстановитель (d) находятся в виде смеси (b), (с) и (d), смесь (b), (с) и (d) готовят смешением гетерогенного катализатора восстановления (b) и восстановителя (d) с последующим добавлением ацилирующего реагента (с), причем раствор (а) в (е) контактирует со смесью (b), (с) и (d);
(C) способ, в котором гексакис(фенилметил)гексаазаизовюрцитан (а), гетерогенный катализатор восстановления (b), ацилирующий реагент (с), восстановитель (d) и растворитель (е) контактируют друг с другом одновременно;
(D) способ, в котором ацилирующий реагент (с), восстановитель (d) и растворитель (е) находятся в виде смеси (с), (d) и (е) и гексакис(фенилметил)гексаазаизовюрцитан (а) и гетерогенный катализатор восстановления (b) раздельно вводят в смесь (с), (d) и (е) так, чтобы (а) и (b) контактировали друг с другом в присутствии смеси (с), (d) и (е);
(Е) способ, в котором гетерогенный катализатор восстановления (b), ацилирующий реагент (с), восстановитель (d) и растворитель (е) находятся в виде смеси (b), (с), (d) и (e) и гексакис(фенилметил)гексаазаизовюрцитан (а) контактирует со смесью (b), (с), (d) и (е);
(F) способ, в котором гексакис(фенилметил)гексаазаизовюрцитан (а), ацилирующий реагент (с), восстановитель (d) и растворитель (е) находятся в виде смеси (а), (с), (d) и (е) и гетерогенный катализатор восстановления (b) контактирует со смесью (а), (с), (d) и (е);
(G) способ, в котором гетерогенный катализатор восстановления (b), восстановитель (d) и растворитель (е) находятся в виде смеси (b), (d) и (е) и гексакис(фенилметил)гексаазаизовюрцитан (а) и ацилирующий реагент (с) находятся в виде смеси (а) и (с), где смесь (b), (d) и (е) контактирует со смесью (а) и (с), и
(Н) способ, в котором гексакис(фенилметил)гексаазаизовюрцитан (а), восстановитель (d) и растворитель (е) находятся в виде смеси (а), (d) и (е) и гетерогенный катализатор восстановления (b) и ацилирующий реагент (с) находятся в виде смеси (b) и (с), где смесь (а), (d) и (е) контактирует со смесью (b) и (с).
Из этих способов (А), (В) и (G) являются предпочтительными.
В любом из вышеуказанных способов от (А) до (Н) вышеупомянутое взаимодействие обычно проводится в реакторе. Например, в способе (А) смесь (b) и (d) получают загрузкой восстановителя (d) (способом, описанным ниже) в реактор, содержащий гетерогенный катализатор восстановления (b), загруженный в него, и затем раствор, полученный при растворении гексакис(фенилметил)гексаазаизовюрцитана (а) и ацилирующего реагента (с) в растворителе (е) добавляют к вышеполученной смеси (b) и (d).
Предпочтительно для каждого из вышеуказанных способов от (А) до (Н), чтобы температура смеси (а), (b), (с), (d) и (е) находилась в пределах от 40 до 160oС и под давлением от 0,001 до 100 кгс/см2 (парциальное давление водорода), выполнялось по окончании приготовления смеси (а), (b), (с), (а) и (е) или выполнялось, как можно раньше (например, примерно в течение 10 мин) после приготовления вышеупомянутой смеси.
Реакционная смесь, полученная способом по настоящему изобретению, включает, по меньшей мере, одно производное тетраацилгексаазаизовюрцитана, гетерогенный катализатор восстановления (b), восстановитель (d) и растворитель (е). Реакционная смесь может содержать небольшое количество ацилирующего реагента (с).
По меньшей мере, одно производное тетраацилгексаазаизовюрцитана, содержащееся в реакционной смеси, полученное способом по настоящему изобретению, в общем представлено следующей формулой (3):
WA4BnH(2-n),
где n является целым числом от 0 до 2, каждое А независимо представляет C1-С10 ацильную группу, Н представляет атом водорода и каждый из В и W определены выше. Примеры производных тетраацилгексаазаизовюрцитана, представленных формулой (3), включают тетраацилбис(фенилметил)гексаазаизовюрцитан (WA4В2), тетраацилфенилметилгексаазаизовюрцитан (WA4BH) и тетраацилгексаазаизовюрцитан (WA4H2). WA4В2 превращается в WA4H2 через WA4BH при дальнейшем протекании реакции восстановительного дефенилметилирования.
Из этих трех производных тетраацилгексаазаизовюрцитана WA4H2 особенно пригоден в качестве предшественника гексанитрогексаазаизовюрцитана. В отношении способа нитрования WA4H2 можно обратиться, например, к WО 96/23792.
Также WA4B2 можно превратить в гексанитрогексаазаизовюрцитан нитрованием по общепринятому способу (смотри, например, WО 97/20785).
В отношении соотношений при образовании вышеупомянутых трех производных тетраацилгексаазаизовюрцитана желаемые соотношения образования можно легко достичь, соответствующим образом регулируя скорость реакции и время реакции. Скорость реакции можно регулировать изменением количества восстановителя, температуры реакции и активностью и количеством используемого катализатора. Наиболее эффективно регулировать скорость можно изменением активности и количества катализатора.
Скорость реакции имеет особо большое влияние на восстановительное дефенилметилирование WA4B2. Когда реакция проводится в условиях, при которых она является низкой, то соотношение образования WА4B2 становится высоким. В то же время, когда реакция проводится в условиях, где скорость реакции высокая, то скорость образования WA4H2 становится высокой. Кроме того, когда удлиняется время реакции, то соотношение образования WА4H2 увеличивается.
Например, когда реакция проводится в условиях, при которых через 1 ч после начала реакции общая масса WB6, WAB5, WA2B4 и WA3B3 в реакционной системе становится равной 10% или менее от общей массы всех производных гексаазаизовюрцитана в реакционной системе (такие условия можно достичь, используя в качестве гетерогенного катализатора восстановления (b) 10% Pd-C в количестве примерно 20 мас.%, основываясь на WB6), выходы WA4B2, WA4BH и WA4H2, полученные через 1 ч после начала реакции, составляют соответственно примерно 60%, примерно 15% и примерно 10%. Кроме того, когда реакция проводится в течение 4 ч в вышеуказанных условиях, выходы WA4B2, WA4BH и WA4H2 становятся равными соответственно примерно 10%, примерно 5% и примерно 70%.
Когда желательно получить только WA4H2 в качестве, по меньшей мере, одного производного тетраацилгексаазаизовюрцитана, представленного формулой (3), реакцию проводят с высокой скоростью реакции в течение длительного периода времени. В частности, например, когда реакцию проводят в течение от 5 до 6 ч в условиях, при которых общая масса WB6, WAB5, WA2B4 и WA3B3 становится равной 10% или менее от общей массы всех производных гексаазаизовюрцитана в реакционной системе в течение 1 ч после начала реакции (такие условия можно достичь, используя в качестве гетерогенного катализатора восстановления (b), 10% Pd-C в количестве примерно 20 мас.% или более от WB6), то можно получить только WA4H2 в качестве, по меньшей мере, одного тетраацилгексаазаизовюрцитана, представленного формулой (3).
С другой стороны, когда желательно получить только WA4В2 в качестве, по меньшей мере, одного производного тетраацилгексаазаизовюрцитана, представленного формулой (3), реакцию проводят с низкой скоростью реакции, например, используя катализатор, имеющий низкую каталитическую активность. В частности, например, когда реакцию проводят в условиях, при которых она протекает в течение 6 ч или более для получения общей массы WB6, WAB5, WA4B4 и WA3B3, равной 10% или менее от общей массы всех производных гексаазаизовюрцитана в реакционной системе (такие условия можно достичь, используя катализатор, имеющий очень низкую каталитическую активность), когда восстановительное дефенилметилирование полученного WA4B2 почти не происходит и, следовательно, WA4B2 можно получить в качестве, по меньшей мере, одного производного тетраацилгексаазаизовюрцитана, представленного формулой (3).
Образовавшийся WA4H2 можно выделить из реакционной смеси следующим образом. После завершения реакции к образовавшейся реакционной смеси добавляют воду (которая является хорошим растворителем для WA4H2) для растворения WA4H2 в воде, и затем гетерогенный катализатор восстановления (b) отделяют от реакционной смеси, получая жидкую смесь. Затем полученную жидкую смесь подвергают перегонке для удаления воды, в результате чего осаждаются кристаллы WA4H2 высокой чистоты (в последующем этот способ относится к "способу осаждения кристаллов при перегонке").
Следовательно, в способе по настоящему изобретению предпочтительно, чтобы гетерогенный катализатор восстановления (b) отделялся от реакционной смеси (содержащей WA4H2), полученной в реакции, для получения жидкой смеси, не содержащей катализатор, но содержащей растворитель (е), WА4H2 и воду, и затем полученную жидкую смесь подвергают перегонке для удаления воды, осаждая кристаллы (осаждение кристаллов при перегонке) тетраацилгексаазаизовюрцитана (WA4H2).
Наиболее характерным свойством способа осаждения кристаллов при перегонке является то, что кристаллы WA4H2 высокой чистоты можно осадить с высоким выходом просто при удалении из реакционной смеси компонентов (таких, как вода и арилметан) с помощью отгонки, имеющих точку кипения ниже, чем у растворителя (такого, как растворитель, содержащий амидную группу).
WA4H2, который осаждается в виде кристаллов, почти нерастворим в обычном органическом растворителе, но его можно легко растворить в протонном высокополярном растворителе таком, как вода и карбоновые кислоты, что отличает WA4H2 от других производных гексаазаизовюрцитана и продуктов разложения гексаазаизовюрцитанового скелета, где эти другие производные гексаазаизовюрцитана и продукты разложения скелета содержатся в растворителе в качестве примесей. Используя это свойство WA4H2, вода, используемая в качестве пригодного растворителя (в котором вещество растворяется) для желаемого WA4H2, удаляется отгонкой из смешанного растворителя, состоящего из воды и обычного органического растворителя, для осаждения кристаллов желаемого WA4H2, и выпавшие кристаллы выделяют из смешанного растворителя растворением вышеуказанных примесей в обычном органическом растворителе (например, растворителе, содержащем амидную группу). Таким образом, можно получить WA4H2 высокой чистоты.
В отношении жидкой смеси, используемой в вышеуказанном способе осаждения кристаллов перегонкой, не требуется полного растворения WA4H2 в жидкой смеси, и WA4H2 может присутствовать частично в нерастворенном виде (т.е. в виде суспензии). Однако предпочтительно, чтобы в вышеупомянутом способе осаждения кристаллов при перегонке использовалась жидкая смесь, не содержащая твердых частиц.
Что касается давления, при котором осуществляют отгонку пригодного растворителя (воды) для WA4H2 в вышеуказанном способе осаждения кристаллов перегонкой (пригодный растворитель соответствует "первому растворителю", описанному в примерах ниже), то можно использовать либо атмосферное давление, либо пониженное давление. В отношении температуры, при которой осуществляют отгонку, нет особого ограничения при условии, что пригодный растворитель можно отогнать при давлении, указанном для перегонки. Для проведения отгонки в течение короткого периода времени предпочтительно осуществлять перегонку при пониженном давлении, указанном для отгонки, и при температуре, которая равняется или выше, чем точка кипения пригодного растворителя, установленной при указанном пониженном давлении. Кроме того, предпочтительнее проводить отгонку при пониженном давлении, указанном для перегонки, и при температуре, которая равняется или выше точки кипения пригодного растворителя и которая равняется или ниже точки кипения неудовлетворительного растворителя (который соответствует "второму растворителю", описанному в примерах ниже), где каждая из точек пригодного и неудовлетворительного растворителя определяется при указанном пониженном давлении. Когда отгонку проводят при таком давлении и температуре, то становится возможным отделить пригодный растворитель от неудовлетворительного растворителя в этом способе очистки так, что каждый из растворителей можно легко рециркулировать. Отгонку проводят при давлении в пределах от 0,0000001 до 760 мм рт. ст. Чем меньше давление, тем требуется меньше времени для перегонки и требуется ниже температура для проведения перегонки, так что становится возможным с выигрышем снизить или подавить проявление разложения WA4H2 под действием температуры и гидролиз растворителя (е) (например, растворителя, содержащего амидную группу). Следовательно, предпочтительно проводить отгонку при пониженном давлении 200 мм рт. ст. или ниже.
В способе по настоящему изобретению, когда используется осаждение кристаллов при перегонке, некоторое количество неудовлетворительного растворителя может также отогнаться, когда пригодный растворитель удаляется отгонкой при условии, что примерно 10 мас.% или более неудовлетворительного растворителя, присутствующего в первоначальной жидкой смеси, остается неудаленным. Кроме того, полное удаление пригодного растворителя из жидкой смеси не является необходимым. Фактически, в зависимости от типа пригодного растворителя и типа неудовлетворительного растворителя очень трудно полностью отделить пригодный растворитель от неудовлетворительного растворителя при отгонке в промышленном масштабе. Следовательно, в способе осаждения кристаллов перегонкой отгонку можно проводить только в том случае, пока количество пригодного растворителя, остающееся в полученном остатке после перегонки, становится равным 0,2 или менее, выраженным в виде весового соотношения пригодного растворителя, оставшегося в неудовлетворительном растворителе, к неудовлетворительному растворителю. Для получения желаемого соединения с высоким выходом предпочтительно проводить отгонку, пока количество пригодного растворителя, остающееся в неудовлетворительном растворителе, становится равным 0,02 или менее, выраженным в виде весового соотношения пригодного растворителя к неудовлетворительному растворителю.
Способ выделения WA4H2 фильтрованием после осаждения кристаллов перегонкой кратко поясняется ниже.
Суспензия, содержащая WA4H2 в качестве основного твердого компонента и растворитель (е) (например, растворитель, содержащий амидную группу) в качестве основного жидкого компонента, который получается при проведении способа осаждения кристаллов при перегонке, подвергается фильтрованию с помощью вещества и устройства, пригодных для фильтрования суспензии.
Типичные примеры способов фильтрования включают способ, в котором фильтрование осуществляется с использованием фильтровальной бумаги, мембранного фильтра или металлокерамики. Среди этих способов фильтрования соответствующим образом выбирают способ, подходящий для диаметра частиц осажденных кристаллов WA4H2. Можно использовать фильтры, имеющие различный диаметр пор, в комбинации с многостадийной фильтрационной системой.
Выделение образовавшегося WA4В2 из реакционной смеси можно проводить, например, следующим образом. Когда в качестве растворителя реакции (е) используется растворитель, содержащий амидную группу, образовавшийся WA4В2 находится в растворенном состоянии в растворителе реакции (е). Следовательно, полученный WA4В2 можно выделить способом, в котором гетерогенный катализатор отфильтровывают из реакционной смеси, получая фильтрат, и затем полученный фильтрат подвергают перегонке, чтобы удалить растворитель (е), для получения таким образам WA4В2 в твердом виде. Кроме того, когда WA4B2 присутствует в реакционной смеси в такой высокой концентрации, что WA4В2 спонтанно выпадает в осадок, WA4В2 можно выделить способом, в котором хороший растворитель для WA4В2 добавляют к реакционной смеси, чтобы растворить осажденный там WA4В2, и гетерогенный катализатор восстановления отфильтровывают из реакционной смеси с последующим удалением растворителя из полученного фильтрата отгонкой, получая таким образом WA4В2 в твердом виде. Примеры хороших растворителей для WA4В2 включают растворители, содержащие амидную группу, такие как N,N-диметилацетамид, N,N-диметилформамид, 1,3-диметил-2-имидазолидон и N-метил-2-пирролидон; карбоновые кислоты такие, как муравьиная кислота, уксусная кислота и пропионовая кислота; амины такие, как триэтиламин и этилдиметиламин; галогенсодержащие растворители такие, как хлороформ, дихлорметан, четыреххлористый углерод и фенилбромид.
В способе по настоящему изобретению тетраацилгексаазаизовюрцитан (WA4H2), который является одним из производных тетраацилгексаазаизовюрцитана формулы (3) и который особенно полезен в качестве предшественника гексанитрогексаазаизовюрцитана, можно получить, проводя вышеуказанную реакцию восстановительного дефенилметилирования/ацилирования для получения, по меньшей мере, одного производного тетраацилгексаазаизовюрцитана формулы (3) в вышеуказанных условиях реакции (температура и давление), пока 100% производных гексаазаизовюрцитана, образовавшихся в реакционной системе, не превратилось в WA4H2.
Однако для увеличения скорости образования WA4H2 предпочтительно, чтобы температура реакции повышалась, когда общая масса WB6, WAB5, WA4В2 и WA3B3, которые образуются в реакционной системе, становится равной 10% или менее, предпочтительно 0%, основываясь на общей массе всех производных гексаазаизовюрцитана в реакционной системе, для проведения таким образом восстановительного дефенилметилирования производных тетраацилгексаазаизовюрцитана, образовавшихся в реакционной системе. То есть, в настоящем изобретении высокая скорость образования WA4Н2 становится возможной при способе, в котором реакция восстановительного дефенилметилирования WB6 формулы (1) проводится при относительно средней температуре (например, при от 40 до ниже, чем 80oС) для получения таким образом реакционной смеси, содержащей производные тетраацилгексаазаизовюрцитана формулы (3), и затем температуру реакции повышают (например, до 80-160oС) для проведения таким образом восстановительного дефенилметилирования производных тетраацилгексаазаизовюрцитана. В последующем, для удобства вышеуказанную реакцию восстановительного дефенилметилирования/ацилирования, проводимую при относительно средней температуре, часто относят к "реакции первой стадии" и последующую реакцию восстановительного дефенилметилирования, проводимую при более высокой температуре реакции, часто относят к "реакции второй стадии". В вышеуказанном способе по причине, описанной ниже, предпочтительно в реакционную систему добавлять воду в одной или более временных точек перед и во время реакции второй стадии (предпочтительно перед началом реакции второй стадии).
То есть, в способе по настоящему изобретению предпочтительно, чтобы реакционная смесь (содержащая, по меньшей мере, одно производное тетраацилгексаазаизовюрцитана), полученная в реакции первой стадии, являлась реакционной системой для восстановительного дефенилметилирования, и реакционная система для восстановительного дефенилметилирования нагревалась до и поддерживалась при температуре от 80 до 160oС, в то же время поддерживая количество восстановителя, находящегося в реакционной системе восстановительного дефенилметилирования, на уровне стехиометрического количества или выше, предпочтительно значительно выше, чем стехиометрическое количество, для восстановления, по меньшей мере, одного производного тетраацилгексаазаизовюрцитана, где к реакционной системе в одной или более временных точек добавляют воду перед и во время реакции второй стадии, получая таким образом реакционную смесь, содержащую тетраацилгексаазаизовюрцитан, представленный следующей формулой (4):
WA4H2,
где каждое из А, Н и W определены выше.
Примеры характерных способов для осуществления на практике вышеуказанного предпочтительного метода в способе по настоящему изобретению, который включает реакцию первой стадии и реакцию второй стадии (в последующем этот предпочтительный метод просто относится к "методу реакции первой стадии/второй стадии"), включают следующие способы от (1) до (5):
(1) способ, в котором после реакции первой стадии in situ проводят реакцию второй стадии, используя тот же реактор, который был задействован в реакции первой стадии;
(2) способ, в котором после реакции первой стадии реакционную смесь, полученную в реакции первой стадии, переносят в другой реактор, чем тот, который использовали в реакции первой стадии, и реакцию второй стадии проводят в этом другом реакторе (в способе (2), гетерогенный катализатор восстановления, содержащийся в реакционной смеси, полученной в реакции первой стадии, можно отделить и выделить из реакционной смеси фильтрованием и тому подобное, и к полученной смеси можно добавить свежий гетерогенный катализатор восстановления);
(3) способ, в котором реакцию первой стадии или реакцию второй стадии проводят непрерывно;
(4) способ, в котором каждую реакцию первой стадии и реакцию второй стадии проводят непрерывно, где реакционная смесь, полученная в реакции первой стадии, сохраняется перед реакцией второй стадии, и сохраняемая реакционная смесь подвергается реакции второй стадии и
(5) способ, в котором каждую реакцию первой стадии и реакцию второй стадии проводят непрерывно, где реакционная смесь, непрерывно удаляемая из реактора, используемого в реакции первой стадии, непрерывно переносится в другой реактор, чем тот, который использовали в реакции первой стадии.
Как детально описано ниже, способ (1) имеет то преимущество, что даже, когда часть продуктов WA4Н2, образовавшихся в реакции первой стадии, осаждается на гетерогенном катализаторе восстановления, продукты WA4Н2, осажденные на катализаторе, будут содержаться в реакционной смеси, полученной в реакции второй стадии, предотвращая таким образом потерю WA4Н2. В реакции второй стадии к реакционной системе добавляют воду, и, следовательно, состав реакционной системы реакции первой стадии отличается от такового для реакционной системы реакции второй стадии. Следовательно, в способе (1) для проведения реакции первой стадии при использовании реактора, который уже был задействован в реакции второй стадии, необходимо отмыть реактор внутри перед постановкой в нем реакции первой стадии. С другой стороны, в способе (2) реакция первой стадии и реакция второй стадии проводятся с использованием различных реакторов. Следовательно, способ (2) является выгодным в том, что при использовании двух различных реакторов, которые, соответственно, были задействованы только в реакции первой стадии и только в реакции второй стадии, операция по отмывке реактора отпадает. В случае способов (3)-(5) (в которых одна из или обе реакция первой стадии и реакция второй стадии проводится или проводится непрерывно) каждый из этих способов (непрерывные способы) выгодней в том, что даже, когда реакция первой стадии и/или реакция второй стадии проводится или проводятся с использованием реактора(ов), имеющих небольшую емкость, возможно достичь удовлетворительной скорости образования. Однако каждый из этих способов имеет недостаток, заключающийся в том, что выход желаемого продукта имеет тенденцию к снижению. Способы (1) и (2), каждый из которых является порционным способом (не непрерывный способ), не обладают вышеуказанным преимуществом, достигаемым при постановке непрерывных способов, однако эти порционные способы выгодны в том, что желаемый продукт можно получить с высоким выходом по сравнению с тем, как это имеет место в случае непрерывных способов.
Ниже приводится пояснение в отношении вышеуказанного предпочтительного метода (метода реакции первой стадии/второй стадии) в способе по настоящему изобретению, используя в качестве примеров способ (1) (в котором после реакции первой стадии in situ проводится реакция второй стадии с использованием того же реактора, который был задействован в реакции первой стадии) и способы, в которых, по меньшей мере, одна из реакции первой стадии и реакции второй стадии проводится непрерывно.
Один признак способа (1) реакции первой стадии/второй стадии (т.е. способа, в котором после реакции первой стадии in situ проводится реакция второй стадии, используя тот же реактор, который был задействован в реакции первой стадии) заключается в том, что гетерогенный катализатор восстановления, используемый в реакции первой стадии, так же используется в качестве гетерогенного катализатора восстановления в реакции второй стадии. Этот способ является очень выгодным по сравнению со способом, в котором катализатор, содержащийся в реакционной смеси первой стадии, отфильтровывают для получения фильтрата, и затем к полученному фильтрату добавляют свежий гетерогенный катализатор восстановления для проведения реакции второй стадии. То есть, в этом способе, когда растворитель, содержащий амидную группу, используется в качестве растворителя (е), вследствие низкой растворимости WA4Н2 в растворителе, содержащем амидную группу, часть продуктов WA4Н2, образовавшихся в реакционной системе в реакции первой стадии, осаждается на гетерогенном катализаторе восстановления в зависимости от используемых условий реакции. Даже в этом случае в способе (1), в котором гетерогенный катализатор восстановления, применяемый в реакции первой стадии, также используется в качестве гетерогенного катализатора восстановления в реакции второй стадии, продукты WA4Н2, осаждаемые на катализаторе, будут содержаться в реакционной смеси, полученной в реакции второй стадии, предотвращая таким образом потерю WA4Н2.
Кроме того, другой признак способа (1) метода реакции первой стадии/второй стадии заключается в том, что в течение периода времени между окончанием реакции первой стадии и началом реакции второй стадии и/или во время реакции второй стадии количество восстановителя в реакционной системе поддерживается на уровне, достаточном для восстановления производных тетраацилгексаазаизовюрцитана, содержащихся в реакционной смеси, полученной в реакции первой стадии (например, восстановитель может находиться в реакционной системе в количестве, которое составляет 80% или более от концентрации насыщения восстановителя в реакционной системе в используемых условиях реакции (температура и давление)). Метод реакции первой стадии/второй стадии можно выигрышно использовать для достижения высокой скорости образования желаемого WA4Н2. Однако в случае, когда в течение периода времени между окончанием реакции первой стадии и началом реакции второй стадии и/или во время реакции второй стадии количество восстановителя в реакционной системе становится недостаточным для восстановления производных тетраацилгексаазаизовюрцитана, содержащихся в реакционной смеси, полученной в реакции первой стадии, имеется опасность того, что будет иметь место снижение активности катализатора. Следовательно, для надежного достижения высокой скорости образования желаемого WA4Н2 важно поддерживать количество восстановителя в реакционной системе на уровне, который достаточен для восстановления производных тетраацилгексаазаизовюрцитана. Как детально описано ниже, в реакции второй стадии для легкого выделения образовавшегося WA4Н2 из реакционной системы к реакционной системе (реакционной смеси) добавляют воду. Однако добавление воды к реакционной системе приводит к снижению количества восстановителя (такого, как газообразный водород), растворенного в реакционной системе. Следовательно, в случае если имеется опасность того, что добавление воды будет приводить к снижению количества восстановителя, растворенного в реакционной системе, до уровня, который ниже, чем стехиометрическое количество для реакции второй стадии (реакция восстановительного дефенилметилирования производных тетраацилгексаазаизовюрцитана), необходимо увеличить добавление восстановителя в реакционную систему (например, при увеличении давления водорода, используемого в качестве восстановителя, до уровня, который выше, чем таковой в реакции первой стадии) для поддержания таким образом на удовлетворительном уровне количества восстановителя, растворенного в реакционном растворе.
Еще один признак способа (1) метода реакции первой стадии/второй стадии заключается в том, что после реакции первой стадии к реакционной системе добавляют воду в одной или более временных точек до или во время реакции второй стадии (предпочтительно перед началом реакции второй стадии). Причина добавления воды к реакционной системе заключается в следующем. WA4Н2 обладает таким свойством, что он почти нерастворим в растворителе, содержащем амидную группу, и других обычных органических растворителях; однако он растворим в воде. В настоящем изобретении, используя вышеуказанное свойство WA4Н2, воду используют для растворения в ней WA4Н2 для отделения таким образом WA4Н2 от катализатора. В отношении времени добавления воды к реакционной системе не имеется особого ограничения при условии, что добавление проводится после реакции первой стадии и перед отделением гетерогенного катализатора восстановления от реакционной смеси, содержащей WA4Н2, который описывается ниже. Однако для предотвращения сильной адсорбции WA4Н2 на гетерогенном катализаторе восстановления предпочтительно добавлять воду к реакционной системе перед или одновременно с началом реакции второй стадии.
Количество воды, добавляемое к реакционной смеси, обычно находится в пределах от 0,01 до 100, предпочтительно от 0,1 до 10, более предпочтительно от 0,2 до 5, выраженном в виде весового соотношения воды к растворителю (например, растворителю, содержащему амидную группу), использованному в реакции первой стадии.
Примеры восстановителей, используемых в реакции второй стадии, включают газообразный водород и гидразин. Из них газообразный водород является предпочтительным.
Когда в качестве восстановителя в реакции второй стадии используют газообразный водород, давление (давление реакции) обычно находится в пределах от 0,01 до 200 кгс/см2, предпочтительно от 0,1 до 100 кгс/см2, более предпочтительно от 1 до 50 кгс/см2, более предпочтительно от 8 до 12 кгс/см2 , выраженном в виде парциального давления газообразного водорода. В настоящем изобретении газообразный водород можно использовать в комбинации с инертным газом таким, как газообразный азот, газообразный аргон и газообразный гелий. Когда в реакции второй стадии используется иной восстановитель, чем газообразный водород (такой как гидразин), количество восстановителя (иного, чем газообразный водород) обычно находится в пределах от 1 до 10000, предпочтительно от 1 до 2000, при выражении в виде молярного соотношения восстановителя к фенилметильным группам WA4B2 и WA4BН, которые находятся в реакционной системе.
Температура реакции в реакции второй стадии обычно находится в пределах от 40 до 200oС, предпочтительно от 60 до 160oС, более предпочтительно от 80 до 160oС, более предпочтительно от 80 до 130oС.
После завершения реакции второй стадии WA4Н2 высокой чистоты можно получить следующим образом. Вначале гетерогенный катализатор восстановления (b) отделяется от реакционной смеси второй стадии для получения таким образом жидкой смеси. Затем полученная жидкая смесь подвергается вышеуказанному осаждению кристаллов при перегонке для осаждения таким образом WA4Н2 высокой чистоты.
Следовательно, в способе по настоящему изобретению предпочтительно, чтобы гетерогенный катализатор восстановления (b) отделялся от реакционной смеси (содержащей WA4Н2), полученной в реакции второй стадии в методе реакции первой стадии/второй стадии, для получения таким образом жидкой смеси, не содержащий катализатор (которая содержит растворитель (е), WA4Н2 и воду), и затем полученную жидкую смесь подвергают перегонке для удаления воды, осаждая таким образом кристаллы WA4Н2 (осаждение кристаллов при перегонке).
После осаждения кристаллов при перегонке выпавшие кристаллы WA4Н2 можно отделить фильтрованием таким же способом, как упомянутый выше.
В способе по настоящему изобретению наиболее предпочтительно рециркулировать растворитель реакции (е) и воду, используемые в реакции второй стадии.
После отделения гетерогенного катализатора восстановления (b) фильтрованием и удаления воды из реакционной смеси отгонкой растворитель реакции (е) можно отделить и выделить, подвергнув полученную жидкую смесь перегонке. Ниже, отделение и выделение растворителя реакции (е) поясняются, взяв в качестве примера случай, когда растворитель, содержащий амидную группу (который предпочтительно используется в настоящем изобретении), применяется в качестве растворителя (е).
Жидкая смесь, полученная после отделения гетерогенного катализатора восстановления (b) фильтрованием и удаления воды отгонкой, включает не только растворитель, содержащий амидную группу, но также карбоновую кислоту, образовавшуюся в качестве побочного продукта, производное ацилирующего реагента. Поскольку карбоновая кислота, образовавшаяся в качестве побочного продукта, вызывает разложение WB6, когда предполагается рециркулировать растворитель реакции (е), предпочтительно, как можно в большей степени удалить карбоновую кислоту, образовавшуюся в качестве побочного продукта. Количество карбоновой кислоты, образовавшейся в качестве побочного продукта, в рециркулируемом растворителе предпочтительно должно находиться в пределах от 0,000001 до 0,1, более предпочтительно от 0,000001 до 0,05, наиболее предпочтительно от 0,000001 до 0,02, выраженном в виде весового соотношения карбоновой кислоты к растворителю.
Некоторые растворители, содержащие амидную группу, образуют высококипящие азеотропные смеси с карбоновой кислотой, образовавшейся в качестве побочного продукта. В случае таких растворителей очень трудно удалить карбоновую кислоту из растворителей. Однако из таких растворителей карбоновую кислоту можно удалить способом, при котором другой растворитель, способный образовать низкокипящую азеотропную смесь с карбоновой кислотой, добавляется к растворителю, и образовавшуюся азеотропную смесь перегоняют. Имеются различные растворители, которые способны образовать низкокипящую азеотропную смесь с карбоновой кислотой, и любой из таких растворителей можно использовать в настоящем изобретении. Однако предпочтительно использовать арилметан такой, как толуол или ксилол, который является типичным примером растворителей, способных к образованию низкокипящей азеотропной смеси. Кроме того, особенно предпочтительно рециркулировать и использовать арилметан, образовавшийся в качестве побочного продукта, при восстановительном деарилметилировании в реакции первой стадии и/или реакции второй стадии. Время проведения азеотропной перегонки и способа проведения азеотропной перегонки не имеет особого ограничения. Например, предпочтительно, чтобы азеотропную перегонку проводили способом, где образовавшиеся в качестве побочных продуктов карбоновая кислота и арилметан удалялись азеотропной перегонкой из: х) реакционной смеси, полученной восстановительным деарилметилированием/ацилированием (реакция первой стадии); или у) реакционной смеси во время удаления воды отгонкой после реакции второй стадии и z) части реакционной смеси, часть которой остается после выделения из реакционной смеси выпавших кристаллов WA4Н2.
Кроме того, когда используется растворитель, содержащий амидную группу, который образует высококипящую смесь с карбоновой кислотой, удаление карбоновой кислоты, образовавшейся в качестве побочного продукта, можно также провести способом, в котором карбоновая кислота реагирует с соединением, имеющим основные свойства, для связывания таким образом карбоновой кислоты, образовавшейся в качестве побочного продукта, с основным соединением, и затем связанную карбоновую кислоту удаляют из растворителя.
Типичные примеры способа связывания карбоновой кислоты включают: 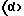 способ, в котором образуется соль карбоновой кислоты в реакции нейтрализации между основным соединением и карбоновой кислотой и (β) способ, в котором карбоновая кислота связывается путем адсорбции на адсорбенте таком, как анионообменник аминного типа, который способен адсорбировать на ней анионы карбоновых кислот.
способ, в котором образуется соль карбоновой кислоты в реакции нейтрализации между основным соединением и карбоновой кислотой и (β) способ, в котором карбоновая кислота связывается путем адсорбции на адсорбенте таком, как анионообменник аминного типа, который способен адсорбировать на ней анионы карбоновых кислот.
В отношении способа (α) не имеется особого ограничения в отношении основного соединения, используемого для образования соли карбоновой кислоты, в реакции нейтрализации; однако предпочтительно использовать основное соединение металла. В отношении основного соединения металла, используемого в способе (α), предпочтительно применять основное соединение металла, содержащее щелочной металл или щелочноземельный металл такой, как натрий, калий, литий, магний или кальций. Предпочтительные примеры основных соединений металла включают гидроокиси щелочного металла или щелочноземельного металла такие, как NaOH, КОН, LiOH, Mg(OH)2 и Са(ОН)2; и окиси щелочного металла или щелочноземельного металла такие, как МgО и СаО. Среди этих основных соединений металла особенно предпочтительны основные соединения магния (такие, как Мg(ОН)2 и МgО), поскольку эти соединения образуют соли магния карбоновой кислоты, которые обладают высоким сродством к растворителям, содержащим амидную группу. В отношении способа удаления растворителя, содержащего амидную группу, после образования соли карбоновой кислоты, предпочтительна отгонка. Когда растворитель, содержащий соль карбоновой кислоты, подвергается отгонке, то поскольку соль карбоновой кислоты, которая является основным компонентом остатка, оставшегося в перегонном аппарате, имеет высокое сродство к растворителю, содержащему амидную группу (магнивая соль карбоновой кислоты обладает особо высоким сродством к растворителю, содержащему амидную группу), остаток можно удалить в виде суспензии с низкой вязкостью. Подобные свойства соли карбоновой кислоты являются очень полезными потому, что не произойдет прилипания твердого вещества к внутренней стенке перегонного аппарата, которое изменяет коэффициент теплопередачи между источником тепла и растворителем, который отгоняется, приводя таким образом к созданию нестабильных условий при перегонке.
В последующем, дается пояснение в отношении условий реакции для связывания карбоновой кислоты в реакции нейтрализации с использованием основного соединения.
В отношении количества основного соединения основное соединение можно использовать в таком количестве, что нейтрализуется примерно 80% карбоновой кислоты, присутствующей в растворителе, или в количестве большем, чем вышеуказанное количество. Альтернативно добавление основного вещества можно проводить способом, при котором основное вещество прикапывают к растворителю, в то же время определяя значение рН полученной смеси, и добавление останавливают, когда значение рН смеси становится равным 6,5 или выше. В этом случае особенно предпочтительно остановить реакцию нейтрализации при значении рН 7 или выше (т. е. при щелочном рН) для того, чтобы увеличить соотношение нейтрализованной карбоновой кислоты.
В отношении температуры нейтрализации нейтрализацию можно проводить при от 20 до 160oС.
После удаления из жидкой смеси карбоновой кислоты, образовавшейся в качестве побочного продукта, способом (α) или (β), полученную смесь подвергают перегонке, получая таким образом растворитель, содержащий амидную группу, который можно рециркулировать. Полученный растворитель, содержащий амидную группу, включает не только образовавшуюся в качестве побочного продукта карбоновую кислоту, но также арилметан, образовавшийся в качестве побочного продукта во время деарилметилирования, и воду, используемую в реакции второй стадии. В отношении содержащего амидную группу растворителя, выделенного отгонкой, предпочтительно, чтобы содержание воды в растворителе находилось в пределах от 0,0001 до 0,1, более предпочтительно от 0,0001 до 0,05, наиболее предпочтительно от 0,0001 до 0,03, и чтобы содержание арилметана в растворителе находилось в пределах от 0,0000001 до 0,1, более предпочтительно от 0,0000001 до 0,05, наиболее предпочтительно от 0,0000001 до 0,01, выраженном в виде весового соотношения их к растворителю, содержащему амидную группу.
Вода, используемая в реакции второй стадии, удаляется из реакционной смеси при осаждении кристаллов при перегонке. Вода, удаленная из реакционной смеси при осаждении кристаллов при перегонке, имеет низкую чистоту, но подобную воду с низкой чистотой можно пускать в рециркуляцию, как таковую, в систему реакции второй стадии, не оказывая при этом отрицательного влияния на протекание реакции. Однако предпочтительно обработать такую воду с низкой чистотой способом, в котором вода с низкой чистотой (включающая отогнанную воду и образовавшийся в качестве побочного продукта арилметан) разделяется на две фазы, и затем воду (водную фазу) отделяют.
Способ (2) метода реакции первой стадии/второй стадии является способом, в котором после реакции первой стадии реакционную смесь, полученную в реакции первой стадии, переносят в другой реактор, чем тот, который использовали в реакции первой стадии, и реакцию второй стадии проводят в этом другом реакторе (в способе (2), гетерогенный катализатор восстановления, содержащийся в реакционной смеси, полученной в реакции первой стадии, можно отделить и выделить из реакционной смеси фильтрованием и тому подобное, и к полученной смеси можно добавить свежий гетерогенный катализатор восстановления.
В способе (2) предпочтительно предупреждать проявление такого феномена, что восстановитель (такой как водород) исчезает из реакционной смеси первой стадии после реакции первой стадии так, что производные тетраацилгексаазаизовюрцитана формулы (3) контактируют с гетерогенным катализатором восстановления в отсутствие восстановителя. Для этой цели предпочтительно непрерывно добавлять восстановитель в реакционную смесь, пока не начнется реакция второй стадии. Когда гетерогенный катализатор восстановления, содержащийся в реакционной смеси первой стадии, отделяют и выделяют из реакционной смеси и добавляют свежий гетерогенный катализатор восстановления к реакционной смеси, трудно продолжать добавление восстановителя к реакционной смеси. В этом случае предпочтительно после того, как начинается добавление восстановителя к реакционной смеси, добавить к реакционной смеси свежий катализатор для начала реакции.
Количество свежего катализатора, добавляемого к реакционной смеси, полученной в реакции первой стадии, варьирует в зависимости от каталитической активности используемого катализатора; однако количество свежего катализатора обычно находится в пределах от 0,0001 до 0,5, предпочтительно от 0,001 до 0,3, более предпочтительно от 0,01 до 0,2, выраженных в виде весового соотношения его к растворителю.
После реакции второй стадии можно провести осаждение кристаллов при перегонке таким же образом, как описано выше для способа (1), для получения таким образом кристаллов WA4Н2 высокой чистоты.
Далее, WA4Н2 можно получить фильтрованием после осаждения кристаллов при перегонке в основном таким образом, как описано выше для способа (1).
Кроме того, наиболее предпочтительно рециркулировать растворитель реакции (е) и воду, используемые в реакции второй стадии, в основном таким образом, как описано выше для способа (1).
Способ (3) метода реакции первой стадии/второй стадии (т.е. способ, в котором реакцию первой стадии или реакцию второй стадии проводят непрерывно) является способом, в котором реакция восстановительного деарилметилирования/ацилирования WB6 проводится непрерывно с использованием реактора с полным перемешиванием.
Вначале дается пояснение в отношении случая, когда реакция первой стадии проводится непрерывно.
Когда реакция первой стадии проводится непрерывно, предпочтительно, чтобы время нахождения реакционной смеси в реакторе находилось в пределах 10 ч.
Кроме того, предпочтительно, чтобы непрерывная реакция, при которой используется реактор с полным перемешиванием, осуществлялась с высокой скоростью так, что общая масса WB6, WAB5, WA4B2 и WA3B3, которые находятся в реакционной системе, становится равной 10% по массе или менее от общей массы производных гексаазаизовюрцитана, присутствующих в реакционной системе.
Скорость реакции можно регулировать, изменяя количество восстановителя, температуру реакции и активность, и количество используемого катализатора. Особенно эффективно регулировать скорость реакции изменением количества катализатора, который используется. На желаемое количество катализатора оказывает влияние активность катализатора. Однако обычно вышеуказанную высокую скорость реакции можно достичь при использовании катализатора в количестве от 0,01 до 0,4, предпочтительно от 0,02 до 0,2, выраженном в виде весового соотношения его к растворителю.
На скорость реакции большое влияние также оказывает весовое соотношение катализатора (b) к WB6 (a). Например, в случае, где катализатор (b) первым загружается в реактор, и затем реакцию проводят при непрерывной загрузке WB6 в реактор, вышеуказанную высокую скорость реакции можно достичь, загружая WB6 в количестве от 0,00001 до 0,5, предпочтительно от 0,00005 до 0,1, более предпочтительно от 0,0001 до 0,01, выраженном в виде соотношения скорости загрузки WB6 (a) (K1 (г/мин)) к количеству катализатора (Q (г)), находящегося в реакторе.
Когда реакция первой стадии осуществляется непрерывно, предпочтительно, чтобы количество карбоновой кислоты, образовавшейся в качестве побочного продукта (производное ацилирующего реагента), присутствующее в реакционной системе, равнялось 0,1 или менее, выраженном в виде весового соотношения ее к растворителю, находящемуся в реакционной системе. Для снижения количества карбоновой кислоты, образовавшейся в качестве побочного продукта, присутствующей в реакционной системе, до менее 0,1, выраженном в виде весового соотношения ее к растворителю, находящемуся в реакционной системе, WB6 применяется в таком количестве, что концентрация WB6 в реакционной системе становится равной от 0,0001 до 0,2, предпочтительно от 0,001 до 0,15, более предпочтительно от 0,01 до 0,1, выраженном в виде весового соотношения WB6 к растворителю, находящемуся в реакционной системе.
В способе (3), в котором реакция осуществляется при введении гетерогенного катализатора восстановления (b) в реактор, предпочтительно WA4BnH(2-n) (n равняется 1 или 2), растворенное в растворителе (е). На растворимость WA4BnH(2-n)(n равняется 1 или 2) в растворителе реакции оказывает влияние тип используемого растворителя в реакции и температура реакции; однако для получения реакционной смеси, где WA4BnH(2-n) (n равняется 1 или 2) находится в растворенном состоянии в растворителе реакции (е), предпочтительно реакцию проводить в таких условиях, чтобы весовое соотношение WA4BnH(2-n) (n равняется 1 или 2) в реакционной смеси к растворителю реакции (е) стало равным от 0,001 до 0,2, более предпочтительно от 0,01 до 0,1, наиболее предпочтительно от 0,02 до 0,07.
WA4BnH(2-n) (n равняется 1 или 2) является относительно стабильным, когда соединение находится в растворителе, содержащем амидную группу, и раствор является перенасыщенным. Следовательно, даже когда образуется большое количество WA4BnH(2-n) (n равняется 1 или 2) в растворителе, содержащем амидную группу, осаждение вещества происходит нелегко, и соединение остается в растворенном состоянии в растворителе в течение относительно длительного периода времени. Следовательно, при небольшом времени нахождения в реакторе возможно непрерывно удалять реакционную смесь, в которой WA4BnH(2-n) (n равняется 1 или 2) находится в состоянии перенасыщения, без осаждения вещества на катализатор, заключенный в реакторе.
Кроме того, в реакции первой стадии также образуется небольшое количество WA4Н2 в результате последующего восстановительного дефенилметилирования WA4BnH(2-n) (n равняется 1 или 2). Как в случае WA4BnH(2-n) (n равняется 1 или 2), WA4Н2 также относительно стабилен в растворителе, содержащем амидную группу, в состоянии перенасыщения. Следовательно, используя короткое время нахождения в реакторе, возможно удалять реакционную смесь, в которой WA4Н2 присутствует в состоянии перенасыщения, без осаждения соединения на гетерогенном катализаторе восстановления, находящемся в реакторе.
Альтернативно при проведении непрерывной реакции гетерогенный катализатор восстановления (b) может протекать через реактор вместо того, чтобы находиться в реакторе. В частности, например, непрерывную реакцию можно проводить способом, в котором раствор (полученный при растворении WB6 (a) в органическом растворителе, содержащем амидную группу (е)), ацилирующий реагент (с), гетерогенный катализатор восстановления (b) (который может быть в виде суспензии, полученной при диспергировании катализатора (b) в растворителе, содержащем амидную группу (е)) и восстановитель (а) загружают в реактор в условиях, определенных в настоящем изобретении, и реакционную смесь, содержащую гетерогенный катализатор восстановления, удаляют из реактора в виде суспензии. В этом способе даже когда WA4Н2, имеющий низкую растворимость в органическом растворителе, содержащем амидную группу (е), образуется в большом количестве в реакции первой стадии, реакционная смесь (содержащая как катализатор, так и WA4Н2), полученная в реакции первой стадии, подвергается реакции второй стадии, где в реакционную смесь добавляют воду в качестве хорошего растворителя для WA4Н2 с последующим отделением катализатора от реакционной смеси.
В отношении реактора для проведения реакции первой стадии в непрерывном режиме предпочтительно использовать многостадийный реактор, обычно применяемый в этой области для проведения непрерывной реакции. Не имеется особого ограничения в отношении числа стадий при условии, что число стадий равняется 2 или более. Однако чем больше число стадий, тем это более выгодно.
Кроме того, в настоящем изобретении реакцию первой стадии можно проводить периодически при повторении последовательности: реакция первой стадии в реакторе в течение заранее определенного периода времени; удаление из реактора заранее определенного количества реакционной смеси, полученной в реакции первой стадии; и введение в реактор жидкости с исходным веществом в количестве, соответствующем вышеуказанному заранее определенному количеству. Когда реакцию проводят таким образом, количество реакционной смеси, удаленное одновременно, предпочтительно находится в пределах от 1/1000 от общей реакционной смеси в реакторе.
При периодической загрузке в реактор WB6 количество катализатора (Q'1 (г)), находящегося в реакторе, количество WB6 (p1 (г)), загруженное в реактор одновременно, интервал (t1 (мин)) между загрузками WB6 в реактор выбираются так, что значение (p1/t1)/Q'1 обычно находится в пределах от 0,00001 до 0,5, предпочтительно от 0,00005 до 0,1, более предпочтительно от 0,0001 до 0,01.
В способе (3) реакцию первой стадии можно проводить непрерывно в основном таким же образом, как указано выше, в отношении реакции первой стадии способа (1), за исключением особых условий, упомянутых выше.
Затем дается пояснение в отношении случая, когда реакция второй стадии проводится непрерывно.
Когда реакция второй стадии проводится непрерывно, предпочтительно, чтобы WA4BnH(2-n) находился в растворенном состоянии в растворителе (е). На растворимость WA4BnH(2-n) в растворителе реакции оказывает влияние тип используемого в реакции растворителя и температура реакции; однако для получения реакционной смеси, где WA4BnH(2-n) (n равняется 1 или 2) находится в растворенном состоянии в растворителе реакции (е), предпочтительно реакцию проводить в таких условиях, что весовое соотношение WA4BnH(2-n) в реакционной смеси к растворителю реакции (е) стало равным от 0,001 до 0,1, предпочтительно от 0,005 до 0,07, более предпочтительно от 0,007 до 0,05.
WA4BnH(2-n) является относительно стабильным в растворителе при перенасыщении раствора. Следовательно, даже когда образуется большое количество WA4BnH(2-n) (n равняется 1 или 2) в растворителе, содержащем амидную группу, осаждение вещества происходит нелегко, и соединение остается в растворенном состоянии в растворителе в течение относительно длительного периода времени. Следовательно, при использовании небольшого промежутка времени нахождения в реакторе возможно непрерывно удалять реакционную смесь, в которой WA4BnH(2-n) (n равняется 1 или 2) находится в состоянии перенасыщения.
В отношении восстановителя и катализатора восстановления, которые можно использовать для проведения реакции второй стадии в непрерывном режиме, можно применять тот же восстановитель и тот же катализатор восстановления, что использовали в реакции первой стадии.
В реакции второй стадии катализатор, используемый в реакции первой стадии, можно использовать как таковой; однако если желательно, реакцию второй стадии можно проводить, добавляя свежий катализатор в реакционную систему второй стадии. Желаемое количество катализатора, находящегося в реакционной системе, варьирует в зависимости от каталитической активности катализатора. Однако свежий катализатор добавляют в реакционную систему в таком количестве, чтобы весовое соотношение катализатора в реакционной системе к растворителю стало равным от 0,0001 до 0,5, предпочтительно от 0,001 до 0,3, более предпочтительно от 0,01 до 0,2.
В отношении реактора для проведения реакции второй стадии в непрерывном режиме предпочтительно использовать многоступенчатый реактор, обычно применяемый в этой области для проведения непрерывной реакции. Не имеется особого ограничения в отношении числа стадий при условии, что число стадий равняется 2 или более. Однако чем больше число стадий, тем это более выгодно.
Кроме того, в настоящем изобретении реакцию второй стадии можно проводить в периодическом процессе при повторении последовательности: реакция второй стадии в течение заранее определенного периода времени; удаление из реактора заранее определенного количества реакционной смеси, полученной в реакции второй стадии; и введение в реактор реакционной смеси первой стадии в количестве, соответствующем вышеуказанному заранее определенному количеству. Когда реакцию проводят таким образом, количество реакционной смеси, удаленное одновременно, предпочтительно находится в пределах от 1/1000 от общего количества реакционной смеси в реакторе.
После реакции второй стадии можно провести осаждение кристаллов при перегонке в основном таким же образом, как описано выше для способа (1), для получения таким образом кристаллов WA4Н2 высокой чистоты.
Далее, WA4Н2 можно получить фильтрованием после осаждения кристаллов при перегонке в основном таким же образом, как описано выше для способа (1).
Кроме того, наиболее предпочтительно рециркулировать растворитель реакции (е) и воду, используемые в реакции второй стадии в основном таким же образом, как описано выше для способа (1).
Способ (4) метода реакции первой стадии/второй стадии (т.е. способ, в котором каждую реакцию первой стадии и реакцию второй стадии проводят непрерывно, где реакционная смесь, полученная в реакции первой стадии, сохраняется перед реакцией второй стадии, и сохраняемая реакционная смесь подвергается реакции второй стадии) является способом, в котором каждая реакция первой стадии и реакция второй стадии проводится непрерывно, как описано выше для способа (3), где реакционная смесь, полученная в реакции первой стадии, сохраняется перед реакцией второй стадии, и сохраняемая реакционная смесь подвергается реакции второй стадии. Этим способом можно эффективно получать желаемый WA4Н2.
После реакции второй стадии можно провести осаждение кристаллов при перегонке таким же образом, как описано выше для способа (1), для получения таким образом кристаллов WA4Н2 высокой чистоты.
Далее, WA4Н2 можно получить фильтрованием после осаждения кристаллов при перегонке в основном таким же образом, как описано выше для способа (1).
Кроме того, наиболее предпочтительно рециркулировать растворитель реакции (е) и воду, используемые в реакции второй стадии в основном таким же образом, как описано выше для способа (1).
Способ (5) метода реакции первой стадии/второй стадии (т.е. способ, в котором каждую реакцию первой стадии и реакцию второй стадии проводят непрерывно, в которой реакционная смесь, непрерывно удаляемая из реактора, используемого для реакции первой стадии, непрерывно переносится в другой реактор), является способом, в котором каждая реакция первой стадии и реакция второй стадии проводится непрерывно, как описано выше для способа (3), где реакционная смесь, непрерывно удаляемая из реактора, используемого для реакции первой стадии, непрерывно переносится в другой реактор. Этим способом можно очень эффективно получать желаемый WA4Н2 даже по сравнению со способом (4).
После реакции второй стадии можно провести осаждение кристаллов при перегонке аналогично описанному выше для способа (1) для получения таким образом кристаллов WA4H2 высокой чистоты.
Далее, WA4Н2 можно получить фильтрованием после осаждения кристаллов при перегонке аналогично способу (1).
Кроме того, наиболее предпочтительно рециркулировать и использовать повторно растворитель реакции (е) и воду, используемые в реакции второй стадии аналогично способу (1).
В способе по настоящему изобретению предпочтительно использовать WB6, имеющего чистоту 95% или более. Использование WB6 такой высокой чистоты является выгодным не только потому, что можно повысить скорость реакции восстановительного дефенилметилирования/ацилирования гексакис(арилметил)гексаазаизовюрцитана (т.е. WB6), но также потому, что желаемые производные тетраацилгексаазаизовюрцитана можно получить с высоким выходом.
WB6 можно синтезировать реакцией конденсации с глиоксаля и фенилметиламина с выделением воды. Реакцию проводят в растворителе в присутствии катализатора, и образовавшийся WB6 выпадает в виде кристаллов из полученной реакционной смеси. Чистота WB6 имеет большое влияние на скорость реакции восстановительного дефенилметилирования/ацилирования. Следовательно, способ стабильного получения WB6 высокой чистоты является очень важным методом для получения производных тетраацилгексаазаизовюрцитана в промышленном масштабе. Ниже дается пояснение в отношении способа получения WB6 высокой чистоты, в частности способа синтеза и кристаллизации WB6.
Кристаллы WB6 высокой чистоты можно получить, используя фенилметиламин и глиоксаль (т. е. исходные вещества) в таких количествах, что молярное соотношение фенилметиламина к глиоксалю становится равным 3 или более. Молярное соотношение фенилметиламина к глиоксалю предпочтительно находится в пределах от 3 до 100, более предпочтительно от 4 до 10.
Условия реакции для синтеза WB6 поясняются ниже.
В отношении растворителя, используемого для синтеза WB6, можно использовать растворитель, имеющий высокую полярность. Примеры высокополярных растворителей включают нитрилы, амиды, амины, спирты и воду. Эти растворители можно использовать в отдельности или в комбинации. Предпочтительно использовать, по меньшей мере, один растворитель, выбранный из группы, состоящей из спиртов, нитрилов и воды. В частности, предпочтительно использовать, по меньшей мере, один растворитель, выбранный из группы, состоящей из нитрилов таких, как ацетонитрил, пропионитрил и бутиронитрил; амидов таких, как N, N-диметилацетамид, N,N-диметилформамид и N-метил-2-пирролидон; аминов таких, как бензиламин; спиртов таких, как метанол, этанол, пропанол и бутанол; и воды. Более предпочтительным является смешанный растворитель из ацетонитрила и воды. Кроме того, использование растворителя, имеющего низкую способность растворения WB6, является выгодным в том, что выход осажденных кристаллов синтезированного WB6 становится высоким.
Примеры катализаторов, используемых для синтеза WB6, включают брендстедовы кислоты, соли аммония, соли алкиламинов, полимерные кислотные твердые вещества и их соли.
Примеры брендстедовых кислот включают карбоновые кислоты такие, как муравьиная кислота, уксусная кислота, пропионовая кислота и бензойная кислота; и неорганические кислоты такие, как серная кислота и азотная кислота. Среди них предпочтительными являются муравьиная кислота, уксусная кислота и пропионовая кислота. Примеры солей включают соли аммония такие, как формат аммония, ацетат аммония и пропионат аммония; соли алкиламина такие, как бензиламин гидрохлорид, бензиламин сульфат, этиламин гидрохлорид, пропиламин гидрохлорид, триэтиламин гидрохлорид, бензиламин пропионат, бензиламин ацетат, бензиламин формат, анилин ацетат, анилин формат, этиламин ацетат, пропиламин ацетат, бутиламин ацетат, диэтиламин ацетат и триэтиламин ацетат и соль ариламина.
В качестве полимерных кислотных твердых веществ и их солей можно использовать катионообменник кислотного или нейтрального солевого типа и анионообменник нейтрального солевого типа. Предпочтительно использовать слабокислый ионообменник кислотного типа и слабощелочной ионообменник нейтрального солевого типа.
Концентрация катализатора находится в пределах от 0,001 до 10, предпочтительно от 0,1 до 5, выраженных в виде молярного соотношения катализатора к глиоксалю. Когда в качестве катализатора используется ионообменник, то концентрация катализатора находится в пределах от 0,1 до 1000, предпочтительно от 0,1 до 20, выраженных в виде весового соотношения катализатора к глиоксалю.
Не имеется особого ограничения в отношении температуры в синтезе WB6 при условии, что температура находится в пределах от точки замерзания растворителя до точки кипения растворителя, но предпочтительно температура равняется от -10 до 60oС.
В отношении глиоксаля, используемого для синтеза WB6, можно использовать либо водный раствор глиоксаля, либо 100% глиоксаль. В случае водного раствора глиоксаля используют водный раствор глиоксаля, имеющий концентрацию 10 мас. % или более от общей массы водного раствора. Концентрация глиоксаля в растворе глиоксаля предпочтительно находится в пределах от 10 до 90 мас.%, более предпочтительно от 20 до 60 мас.%. Кроме того, когда глиоксаль используется в виде водного раствора, чистота глиоксаля в отношении его части, исключая воду, обычно находится в пределах от 80% или выше, предпочтительно от 90% и выше и более предпочтительно от 95% и выше.
Концентрация глиоксаля в реакционном растворе для синтеза WB6 находится в пределах от 0,001 до 0,5, предпочтительно от 0,005 до 0,4, более предпочтительно от 0,005 до 0,2, выраженных в виде весового соотношения глиоксаля к раствору.
Синтез WB6 включает следующие стадии от (I) до (III):
(I) добавление глиоксаля к раствору, полученному смешением фенилметиламина на, катализатора и растворителя, где молярное соотношение фенилметиламина к глиоксалю соответствующим образом выбирается таким, чтобы быть равным 3 или более, для получения таким образом WB6, который спонтанно кристаллизуется с получением кристаллов WB6;
(II) отделение кристаллов WB6, выпавших из реакционной смеси, и
(III) промывание кристаллов WB6 растворителем, содержащим органический растворитель.
Добавление глиоксаля в вышеуказанном способе (I) можно проводить в течение от 5 мин до 10 ч.
В отношении способа выделения кристаллов WB6 на вышеуказанной стадии (II) можно использовать общий способ выделения твердой фазы из жидкой фазы. Примеры таких способов включают фильтрование под вакуумом и фильтрование под давлением с использованием мембранного фильтра и центрифугирование.
В отношении растворителя для промывания кристаллов WB6 на вышеуказанной стадии (III) не имеется особого ограничения при условии, что растворимость WB6 в растворителе составляет 10 г/л или менее, и растворитель не взаимодействует с WB6. В частности, можно использовать тот же растворитель, что использовался для синтеза и кристаллизации WB6. Растворимость WB6 в таком растворителе в некоторых случаях выше 10 г/л при комнатной температуре. Однако в этом случае растворимость WB6 в подобном растворителе можно понизить до 10 г/л или менее проведением промывания при температуре примерно -20oС. Примеры растворителей, в которых растворимость WB6 в растворителе составляет 10 г/л или менее при комнатной температуре, включают нитрилы такие, как ацетонитрил, пропионитрил и бутиронитрил; спирты такие, как метанол, этанол, пропанол, бутанол, пентанол, гексанол, гептанол и октанол. Кроме того, более предпочтительно использовать смешанный растворитель из вышеуказанного нитрила и/или спирта и воды в качестве растворителя для промывания кристаллов WB6 вследствие превосходного промывающего эффекта такого смешанного растворителя и низкой растворимости WB6 в таком смешанном растворителе. Когда в качестве растворителя для промывания кристаллов WB6 используют смешанный растворитель, то содержание воды в смешанном растворителе обычно может составить 30 мас. % или менее, предпочтительно от 5 до 20 мас.% от общей массы смешанного растворителя. Наиболее предпочтительно использовать в качестве растворителя для промывания кристаллов WB6 смешанный растворитель, состоящий из ацетонитрила и воды, где содержание воды в смешанном растворителе составляет от 5 до 20 мас.%, от общей массы смешанного растворителя.
Вышеуказанный способ с использованием избыточного количества фенилметиламина (т.е. молярное соотношение фенилметиламина к глиоксалю составляет 3 или более) делает возможным получать WB6, имеющего чистоту выше 95% или более.
В случае, когда для получения WB6 используется иной способ, чистота конечного WB6 является низкой. Другими словами, когда для получения WB6 используют способ, в основном идентичный вышеупомянутому, за исключением того, что молярное соотношение фенилметиламина к глиоксалю составляет менее 3, получают WB6 с неудовлетворительно низкой чистотой. Однако примеси, содержащиеся в WB6 низкой чистоты, можно удалить соответствующей очисткой, используя органический растворитель, для получения WB6, имеющего высокую чистоту.
Один из общепринятых способов очистки WB6 описывается в Journal of Organic Chemistry, vol. 55, 1459-1466 (1990). Согласно этому способу сырой WB6 (гексабензилгексаазаизовюрцитан) суспендируют в холодном ацетонитриле и затем подвергают фильтрованию для сбора WB6 с последующим промыванием. Далее, промытый WB6 перекристаллизуют из ацетонитрила. Однако в вышеупомянутом документе нет описания, касающегося концентрации WB6 по отношению к растворителю и температуры кристаллизации. То есть, в вышеуказанном документе условия для удаления примесей, содержащихся в WB6 низкой чистоты, не указаны.
Авторы настоящего изобретения провели перекристаллизацию WB6 несколько раз согласно способу вышеуказанного документа. То есть, 16,68 г сырого WB6 подвергли перекристаллизации, используя 1 л ацетонитрила, для получения кристаллов WB6, и эту перекристаллизацию повторили несколько раз. Определили чистоту полученных кристаллов, и было подтверждено, что чистота WB6, полученного способом в соответствии с вышеуказанным документом, варьирует в пределах от 85 до 100%.
Кроме того, каждую партию кристаллов WB6, полученных вышеуказанным способом, по отдельности подвергли реакции дефенилметилирования в присутствии ацилирующего реагента (с использованием катализатора палладия) для получения производных гексаазаизовюрцитана, содержащих ацильную группу. В результате было установлено, что время реакции, необходимое для дефенилметилирования, в значительной мере варьируется в зависимости от партии кристаллов WB6, и при необходимости продолжительного времени для дефенилметилирования выходы желаемых производных гексаазаизовюрцитана, содержащих ацильную группу, очень занижены вследствие разложения гексаазаизовюрцитанового WB6.
В этой ситуации авторы настоящего изобретения провели экстенсивные и интенсивные исследования с целью разработки усовершенствованного способа перекристаллизации WB6, при котором можно было бы надежно получать WB6 высокой чистоты из сырого WB6. В результате было установлено, что поставленную задачу можно достичь следующим способом (в последующем, часто относящимся к "способу перекристаллизации для достижения высокой чистоты"). Способ перекристаллизации для достижения высокой чистоты является способом перекристаллизации WB6, который включает:
растворение сырого WB6 в органическом растворителе для получения раствора и
осаждение кристаллов WB6 высокой чистоты, где тип и количество органического растворителя выбирается таким образом, чтобы побочные продукты, содержащиеся в сыром WB6, полностью растворялись в органическом растворителе при температуре осаждения кристаллов WB6 высокой чистоты, причем побочные продукты определяются высокоэффективной жидкостной хроматографией.
Осаждение кристаллов WB6 можно проводить обычной перекристаллизацией. В качестве примера перекристаллизации можно упомянуть способ, основанный на различии в растворимости WB6 в зависимости от разницы температуры растворителя, используемого для перекристаллизации, который включает:
смешение сырого WB6 с соответствующим растворителем;
нагревание полученной смеси до температуры, равной или ниже точки кипения растворителя, до полного растворения WB6 в растворителе и
охлаждение полученного раствора до соответствующей температуры, при которой происходит осаждение кристаллов WB6 высокой чистоты.
В качестве примера другого способа перекристаллизации можно упомянуть способ, основанный на различии в растворимости WB6 в пригодном растворителе для WB6 и растворимости WB6 в неудовлетворительном растворителе для WB6, который включает:
растворение сырого WB6 в пригодном растворителе для WB6 при соответствующей температуре и
добавление порциями неудовлетворительного растворителя для WB6 к полученному раствору WB6 в пригодном растворителе для WB6 для осаждения таким образом кристаллов WB6 высокой чистоты.
Когда для растворения WB6 систему для перекристаллизации нагревают, то предпочтительно, чтобы избежать снижения выхода желаемого соединения вследствие разложения WB6 под действием температуры, проводить нагревание до 130oС или менее.
Осаждение кристаллов WB6 высокой чистоты из системы для перекристаллизации, содержащей WB6, обычно заканчивается в течение от одного часа до нескольких дней, причем систему для перекристаллизации, содержащую WB6, можно перемешивать или дать ей отстояться. Когда осаждение кристаллов проводится при выдерживании системы для перекристаллизации в спокойном состоянии, то имеется тенденция получения WB6 высокой чистоты в виде относительно крупных игольчатых кристаллов. С другой стороны, при осаждении кристаллов при перемешивании системы для перекристаллизации образуется WB6 высокой чистоты в виде относительно мелких кристаллов. Воспроизводимость выхода и чистоты кристаллов WB6 высокой чистоты можно улучшить поддержанием температуры системы для перекристаллизации, содержащей WB6, при соответствующей температуре во время выпадения кристаллов.
В вышеуказанном способе перекристаллизации для достижения высокой чистоты используется система растворителей, включающая, по меньшей мере, один тип органического растворителя, где растворитель имеет коэффициент растворения от 0,1 до 20, где коэффициент растворения Р представляется следующей формулой (5):
P=H/I,
где Н представляет растворимость WB6 (г/л) в органическом растворителе и I представляет растворимость примесей (г/л) (определенных ВЭЖХ) в органическом растворителе.
Ниже приводится краткое описание растворимости WB6 и растворимости примесей, детектируемых жидкостной хроматографией.
Растворимость WB6 высчитали на основании результатов анализа жидкостной хроматографии количества WB6, которое растворяется в растворителе. С другой стороны, растворимость примесей, упомянутых выше, высчитывали, основываясь на результатах жидкостной хроматографии количества примесей, растворенных в растворе, полагая, что количество примесей, детектируемых жидкостной хроматографией, представляет собой весь остаток, полученный при вычитании количества WB6 из количества сырого WB6.
Примеры органических растворителей, используемых в способе перекристаллизации для получения продукта высокой чистоты, включают ароматические углеводороды такие, как толуол и бензол, простые эфиры такие, как тетрагирофуран, диэтиловый эфир, дипропиловый эфир и дибутиловый эфир; нитрилы такие, как ацетонитрил, пропионитрил и бутиронитрил; амиды такие, как N,N-диметилацетамид, N, N-диметилформамид, 1,3-диметил-2-имидазолидон, N-метил-2-пирролидон и N,N-диэтилнипекотамид; спирты такие, как метанол, этанол, пропанол, бутанол, пентанол, гексанол, гептанол и октанол; сложные эфиры такие, как этилформат, этилацетат, метилацетат, пропилацетат, бутилацетат, метилпропионат, этилпропионат, пропилпропионат и бутилпропионат; галогенсодержащие углеводороды такие, как дихлорметан, хлороформ и 1,2-дихлорэтан. Эти растворители можно использовать в отдельности или в комбинации.
В способе перекристаллизации для достижения высокой чистоты предпочтительно использовать органический растворитель, имеющий небольшое значение коэффициента растворения Р, который представлен вышеуказанной формулой (5). Предпочтительные примеры растворителей включают спирты, нитрилы, сложные эфиры и амиды. Эти растворители можно использовать в отдельности или в комбинации. Более предпочтительны комбинация этанола и этилацетата, комбинация этанола и N,N-диметилацетамида и комбинация ацетонитрила и N,N-диметилацетамида. Наиболее предпочтительным является смешанный растворитель, содержащий этанол и этилацетат.
Предпочтительно, чтобы органический растворитель, используемый в способе перекристаллизации для достижения высокой чистоты, имел температуру в пределах от 5 до 60oС, при которой он растворяет примеси до 0,90 г/л или более, выраженных в виде растворимости примесей в сыром WB6. Когда используется такой органический растворитель, возможно снизить количество растворителя. Кроме того, для увеличения выхода при перекристаллизации для достижения высокой чистоты предпочтительно использовать растворитель, который обладает высокой растворяющей способностью в отношении примесей и низкой растворяющей способностью в отношении WB6. В частности, предпочтительно, чтобы растворитель имел температуру в пределах от 5 до 60oС, при которой значение вышеуказанного Р было менее 3,8. Следовательно, более предпочтительно, чтобы растворитель имел температуру в пределах от 5 до 60oС, при которой он растворяет примеси 0,90 г/л и более (в отношении примесей в сыром WB6), и растворение для WB6 менее 3,42 г/л.
В способе перекристаллизации для достижения высокой чистоты количество используемого растворителя особо не ограничивается при условии, что количество растворителя достаточно для растворения всех примесей в сыром WB6 при температуре перекристаллизации. Например, в случае перекристаллизации для достижения высокой чистоты по отношению к 100 г сырого WB6, который содержит 10% примесей по массе, когда используемый растворитель растворяет примеси 1 г/л при комнатной температуре, растворитель используют в количестве 10 л и более. Количество растворителя, используемого в способе перекристаллизации для достижения высокой чистоты, обычно находится в пределах от 10 до 10000, предпочтительно от 20 до 200, выраженном в виде весового соотношения растворителя к сырому WB6.
Ниже приводится пояснение в отношении структур вышеуказанных соединений WB6, WА4B2 и WA4H2.
Группа В имеет структуру, представленную следующей формулой (6):
-CH2Ar,
где Аr представляет фенил.
В отношении ацильной группы (А) в формулах (3) и (4) предпочтительно, чтобы ацильная группа имела от 1 до 10 атомов углерода. Примеры ацильных групп включают формильную группу, ацетильную группу, пропионильную группу, бутирильную группу, изобутирильную группу, валерильную группу, гексаноильную группу и 2-фенилацетильную группу. Из этих ацильных групп предпочтительными являются ацильные группы, имеющие от 1 до 3 атомов углерода, такие, как формильная группа, ацетильная группа и пропионильная группа. Более предпочтительной является ацетильная группа.
В отношении производного гексаазаизовюрцитана, представленного формулой WA4B2, можно допустить наличие множества стереоизомерных конфигураций, которые различаются по положению ацильных групп и арилметильных групп. Производное гексаазаизовюрцитана, представленное WА4B2, которое получают способом согласно настоящему изобретению, может представлять собой один из стереоизомеров. Это может быть изомер, имеющий стереоструктуру, представленную следующими формулами от (7-1) до (7-6), или их оптические изомеры.
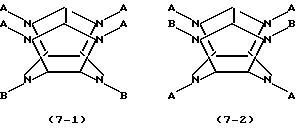
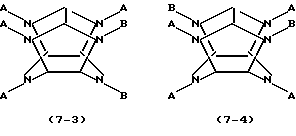
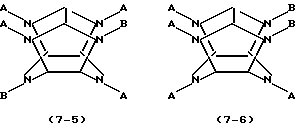
где А является вышеуказанной ацильной группой и В является вышеуказанной фенилметильной группой. Из этих соединений наиболее предпочтительным является соединение формулы (7-1).
Кроме того, WA4H2 может принимать множество стереоизомерных конфигураций, которые различаются по положению ацильных групп и атомов водорода. Производное гексаазаизовюрцитана, представленное WA4H2, которое получают способом по настоящему изобретению, может быть одним из стереоизомеров. В частности, эти стереоизомеры являются производными гексаазаизовюрцитана, представленные формулами от (7-1) до (7-6) выше, где каждая из арилметильных групп замещается атомом водорода. Из них наиболее предпочтительным является производное гексаазаизовюрцитана, представленное формулой (7-1) выше.
Реакция восстановительного дефенилметилирования/ацилирования в способе настоящего изобретения описывается ниже.
Эта реакция включает две стадии: 1) восстановительное дефенилметилирование WB6 в присутствии ацилирующего реагента для превращения N-арилметильной группы, содержащейся в нем, в NH-группу; и 2) последующее ацилирование полученного продукта для превращения NH-группы в N-ацильную группу. Кроме того, поскольку N-алкильная группа может образоваться при восстановлении (в качестве побочной реакции) N-ацильной группы, восстановление которой может происходить в зависимости от условий реакции, то в качестве побочного продукта образуется производное гексаазаизовюрцитана, содержащее N-алкильную группу. Схема реакции была предположительно составлена на основании продуктов реакции и указана ниже на схеме (8):
где А независимо представляет ацильную группу, имеющую от 1 до 10 атомов углерода, В представляет фенилметильную группу, R независимо представляет алкильную группу, имеющую от 1 до 10 атомов углерода, Н представляет атом водорода и W представляет шестивалентный гексаазаизовюрцитан, представленный формулой (2).
Следовательно, одно из соединений, представленных на схеме (8), может содержаться в реакционной смеси, полученной способом по настоящему изобретению.
Способ по настоящему изобретению является промышленно выгодным, поскольку производные тетраацетилгексаазаизовюрцитана можно стабильно получать с высоким выходом. Кроме того, способ по настоящему изобретению позволяет эффективно подавлять снижение каталитической активности катализатора восстановления в ходе реакции.
Предпочтительный способ реализации изобретения
Настоящее изобретение иллюстрируется более детально со ссылкой на примеры в соответствии с уровнем техники, примеры в соответствии с изобретением и сравнительные примеры, которые не следует истолковывать как ограничивающие объем настоящего изобретения.
(1) Сокращения, используемые в следующих примерах известного уровня техники, примерах и сравнительных примерах, представляют следующие соединения и аппаратуру для анализа.
WB6: гексабензилгексаазаизовюрцитан
WA4B2: тетраацетилдибензилгексаазаизовюрцитан
WA4BH: тетраацетилбензилгексаазаизовюрцитан
WA4H2: тетраацетилгексаазаизовюрцитан
WA3B3: триацетилтрибензилгексаазаизовюрцитан
WA2B4: диацетилтетрабензилгексаазаизовюрцитан
DMAc: N,N-диметилацетамид (в качестве органического растворителя)
Ас2О: уксусный ангидрид (в качестве ацилирующего реагента)
Pd-C: палладий на угле (в качестве гетерогенного катализатора восстановления)
ВЭЖХ: жидкостная хроматография высокого давления (высокоэффективная жидкостная хроматография)
ГХ: газовая хроматография
(2) Чистота WB6, использованного в каждом из следующих примеров и сравнительных примеров, составляет 100%, если не указано иначе.
(3) В примерах и сравнительных примерах количественные анализы различных производных гексаазаизовюрцитана проводили следующими методами.
(i) Количественный анализ WA4H2 проводили ВЭЖХ в следующих условиях, используя нижеописанную аппаратуру.
<Условия анализа ВЭЖХ>
Аппаратура для ВЭЖХ: система CLASS-LC-10 (произведено и поставлено Shimadzu Corporation, Япония)
Насос: LC10AD
Детектор: SPD-10A
Контроллер: CLASS-LC10
Колонка: TSK-GEL AMIDE-80 (произведено и поставлено TOSOH, Япония)
Размер: 4,6 мм•25 см
Температура колонки: 40oС
Подвижная фаза: тетрагидрофуран/Н2О (90/10) (объем/объем)
Скорость элюента: 1 мл/мин
Детектирование: УФ (220 нм)
Количество вводимой пробы: 5 мкл
<Подготовка пробы>
Пробу для ВЭЖХ готовили следующим образом, если не указано иначе.
К 0,5 мл реакционной смеси (содержащей гетерогенный катализатор восстановления), полученной деарилметилированием в присутствии или в отсутствие ацилирующего реагента, или фильтрат (не содержащий катализатор), полученный после фильтрования реакционной смеси, добавили 30,8 мл воды и затем полученную смесь подвергли обработке ультразвуком в течение 5 мин с помощью ультразвукового очистителя, и затем подвергли фильтрованию для получения таким образом фильтрата. Полученный фильтрат использовали в качестве пробы для анализа ВЭЖХ.
(ii) Количественные анализы WA2B4, WA3B3, WA4B2 и WA4BH проводили ГХ в следующих условиях, используя нижеописанную аппаратуру.
<Условия анализа ГХ>
Аппаратура для ГХ: газовый хроматограф типа GC-14B (произведено и поставлено Shimadzu Corporation, Япония)
Колонка: металлическая капиллярная колонка Ultra ALLOY (HT) (произведено и поставлено Frontier Lab, Япония)
Внутренний диаметр: 0,25 мм
Длина: 15 м
Толщина пленки, покрывающей внутреннюю стенку капиллярной колонки: 0,15 мкм
Детектирование: пламенно-ионизационный детектор (FID)
Температура:
Колонки: от 200 до 340oС (скорость повышения температуры: 15oС/мин) --> 340oС (10 мин)
Инжектор: 340oС
Детектор: 340oС
Газ-носитель: N2 (скорость газа: 100 мл/мин и внутреннее давление в колонке: 100 кПа)
Количество вводимой пробы: 5 мкл.
<Подготовка пробы>
К 0,5 мл реакционной смеси (содержащей гетерогенный катализатор для восстановления), полученной деарилметилированием в присутствии или в отсутствие ацилирующего агента, или фильтрат (не содержащий катализатор), полученный после фильтрования реакционной смеси, добавили 23,5 мл хлороформа и затем полученную смесь подвергли обработке ультразвуком в течение 5 мин с помощью ультразвукового очистителя, и затем отфильтровали с получением фильтрата. К 4 мл полученного фильтрата добавили 0,5 мл раствора трикосана в хлороформе (в качестве внутреннего стандарта) (который получили растворением 0,4 г трикосана в 100 мл хлороформа) с получением раствора. Полученный раствор использовали в качестве пробы для анализа ГХ.
(iii) Количественный анализ WB6 проводили ВЭЖХ в следующих условиях, используя аппаратуру, описанную ниже.
<Условия анализа ВЭЖХ>
Аппаратура для ВЭЖХ: система CLASS-LC-10 (произведено и поставлено Shimadzu Corporation, Япония)
Насос: LC10AD
Детектор: SPD-10A
Контроллер: CLASS-LC10
Колонка: TSK-GEL AMIDE-80 (произведено и поставлено TOSON, Япония) • 2
Размер: 4,6 мм•25 см
Температура колонки: 40oС
Подвижная фаза: тетрагидрофуран/Н2О (90/10) (объем/объем)
Скорость элюента: 1 мл/мин
Детектирование: УФ (254 нм)
Количество вводимой пробы: 5 мкл
<Подготовка пробы>
Пробу для ВЭЖХ готовили следующим образом, если не указано иначе.
Пробу, содержащую WB6, растворили в тетрагидрофуране с получением раствора, имеющего концентрацию WB6 примерно 0,01 мас.% Полученный раствор использовали в качестве пробы для анализа ВЭЖХ.
(4) Исследование с помощью сканирующего электронного микроскопа (СЭМ) проводили в следующих условиях.
Аппаратура: сканирующий электронный микроскоп для рентгеновского микроанализа типа Х-650 (произведено и поставлено Hitachi, Ltd., Япония).
Ускоряющее напряжение: 20 кВ
<Подготовка пробы для исследования с СЭМ>
Продукт WB6 поместили в графитную пасту и подвергли вакуумному напылению золотом, получая толщину слоя золота, равную примерно 300  , с помощью аппарата для напыления (аппарат для ионного покрытия; произведено и поставлено Eiko Engineering Co., Ltd., Япония), таким образом получают пробу для исследований с помощью СЭМ.
, с помощью аппарата для напыления (аппарат для ионного покрытия; произведено и поставлено Eiko Engineering Co., Ltd., Япония), таким образом получают пробу для исследований с помощью СЭМ.
Пример 1
(Способ, в котором гетерогенный катализатор восстановления и восстановитель загружают в реактор и предварительно нагревают до заранее определенной температуры реакции, далее в него загружают раствор, полученный при растворении WВ6 и ацилирующего реагента в растворителе).
В автоклав емкостью 100 мл загрузили 0,84 г 10% Pd-C (в качестве гетерогенного катализатора восстановления) и затем автоклав продули газообразным водородом, установив внутреннее давление в автоклаве, равное 1,1 кгс/см2. Затем содержимое автоклава перемешивали при скорости перемешивания 50 об/мин при 60oС в течение 2 ч. Раствор, полученный при растворении 2,1 г WB6 (гексабензилгексаазаизовюрцитана) и 1,82 г уксусного ангидрида (в качестве ацилирующего реагента) в 30 мл DMAc (N,N-диметилацетамид) (в качестве растворителя) и быстро загрузили в автоклав с помощью шприца. Затем скорость перемешивания увеличили до 700 об/мин, в то же время поддерживая температуру при 60oС и давление при 1,1 кгс/см2, и реакцию проводили в течение 1 ч, получая реакционную смесь. В дальнейшем эта реакция и полученная реакционная смесь относятся соответственно к "реакции первой стадии" и "реакционной смеси первой стадии".
Анализ полученной реакционной смеси первой стадии ГХ показал, что выходы WA4B2 (тетраацетилдибензилгексаазаизовюрцитана) и WA4BH (тетраацетилбензилгексаазаизовюрцитана) составили соответственно 39% и 21% (т. е. в целом 60%) от WB6, и анализ полученной реакционной смеси первой стадии ВЭЖХ показал, что выход WA4H2 (тетраацетилгексаазаизовюрцитана) составил 22% от WB6.
После завершения реакции первой стадии в автоклав, содержащий реакционную смесь первой стадии in situ, загрузили 30 мл воды, одновременно вводя (в автоклав) газообразный водород. Внутреннее давление водорода поднялось до 9 кгс/см2, и температуру реакции повысили до 90oС для проведения реакции в течение 1 ч, получая таким образом реакционную смесь. В дальнейшем эта реакция и полученная реакционная смесь относятся соответственно к "реакции второй стадии" и "реакционной смеси второй стадии".
Анализ полученной реакционной смеси второй стадии ВЭЖХ показал, что выход WA4H2 составил 82% от WB6.
Пример 2
(Способ, в котором гетерогенный катализатор восстановления и восстановитель загружают в реактор и предварительно нагревают до заранее определенной температуры реакции, и затем в него загружают ацилирующий реагент и раствор WB6 в растворителе).
8,4 г 10% Pd-C загрузили в автоклав емкостью 1 л, автоклав продули газообразным водородом, устанавливая внутреннее давление водорода в автоклаве, равное 1,1 кгс/см2. Затем содержимое автоклава нагревали при 60oС в течение 1 ч. В автоклав с помощью шприца загрузили 18,2 г уксусного ангидрида и раствор 21 г WB6 в 300 мл DMAc (предварительно нагретого до 55oС), перемешивая содержимое автоклава со скоростью 1500 об/мин, в то же время поддерживая температуру при 60oС и давление при 1,1 кгс/см2, реакцию первой стадии проводили в течение 1 ч, получая реакционную смесь первой стадии.
Анализ полученной реакционной смеси первой стадии ГХ показал, что выходы WA4B2 и WA4BH составили соответственно 35% и 26% (т.е. в целом 61%) в расчете на WB6, анализ реакционной смеси первой стадии ВЭЖХ показал, что выход WA4H2 составил 29% в расчете на WB6.
По завершении реакции первой стадии в автоклав, содержащий реакционную смесь первой стадии in situ, загрузили 300 мл воды, одновременно вводя в автоклав газообразный водород. Внутреннее давление водорода в автоклаве устанавливали до 10 кгс/см2 и температуру реакции повысили до 90oС для проведения реакции второй стадии в течение 1 ч, получая реакционную смесь второй стадии.
Анализ полученной реакционной смеси второй стадии ВЭЖХ показал, что выход WA4H2 составил 90% в расчете на WB6.
Пример 3
(Способ, в котором катализатор, содержащийся в реакционной смеси первой стадии, отфильтровывают с получением фильтрата, и реакцию второй стадии проводят, используя свежий катализатор и полученный фильтрат).
Реакцию первой стадии провели в основном аналогично примеру 1. Затем сразу же полученную реакционную смесь первой стадии удалили из автоклава, отфильтровали катализатор, содержащийся в реакционной смеси первой стадии, получая фильтрат. Используя полученный фильтрат, реакцию второй стадии провели следующим образом.
0,84 г 10% Pd-C загрузили в автоклав емкостью 100 мл, затем автоклав продули газообразным водородом, устанавливая внутреннее давление в автоклаве, равное 1,5 кгс/см2. Затем содержимое автоклава нагревали при 90oС в течение 1ч. В автоклав загрузили вышеуказанный фильтрат и 30 мл воды. Затем внутреннее давление водорода в автоклаве подняли до 10 кгс/см2, начали перемешивание содержимого автоклава со скоростью перемешивания 700 об/мин и температуру содержимого автоклава повысили до 90oС в течение 8 мин для проведения реакции второй стадии в течение 1 ч при 90oС, получая реакционную смесь второй стадии.
Анализ полученной реакционной смеси второй стадии ВЭЖХ показал, что выход WA4H2 составил 80%, основываясь на WB6.
Пример 4
(Способ, в котором катализатор, содержащийся в реакционной смеси первой стадии, отфильтровывают, получая фильтрат, и реакцию второй стадии проводят, используя свежий катализатор и полученный фильтрат).
Реакцию первой стадии провели в основном аналогично примеру 2. Затем сразу же из автоклава полученную реакционную смесь первой стадии удалили и отфильтровали катализатор, содержащийся в реакционной смеси реакции первой стадии с получением фильтрата. Используя полученный фильтрат, провели реакцию второй стадии следующим образом.
8,4 г 10% Pd-C загрузили в автоклав емкостью 1 л и затем автоклав продули газообразным водородом так, что внутреннее давление в автоклаве стало равным 1,5 кгс/см2. Затем содержимое автоклава нагревали при 90oС в течение 1 ч. В автоклав загрузили вышеуказанный фильтрат и 300 мл воды. Затем внутреннее давление водорода в автоклаве подняли до 10 кгс/см2, перемешивание содержимого в автоклаве начали со скоростью перемешивания 1500 об/мин и температуру содержимого автоклава подняли до 90oС в течение 8 мин для проведения реакции второй стадии в течение 1 ч при 90oС, получая реакционную смесь второй стадии.
Анализ полученной реакционной смеси второй стадии ВЭЖХ показал, что выход WA4H2 составил 90% в расчете на WB6.
Сравнительный пример 1
Этот сравнительный пример показывает, что при взаимодействии WB6 с гетерогенным катализатором восстановления в отсутствие, по крайней мере, одного из ацилирующего реагента и восстановителя, выход желаемого продукта заметно снижается.
2,1 г WB6, 3,15 г 10% Pd-C, 1,84 г уксусного ангидрида и 30 мл DMAc загрузили в автоклав емкостью 100 мл и автоклав продули газообразным азотом при комнатной температуре. Затем в автоклав ввели газообразный водород так, что внутреннее давление в автоклаве стало равным 1,1 кгс/см2. Реакцию первой стадии начали повышением температуры содержимого автоклава до 60oС в течение более 12 мин, одновременно перемешивая содержимое автоклава со скоростью 700 об/мин. Во время реакции в автоклав постоянно вводили газообразный водород, поддерживая внутреннее давление в автоклаве 1,1 кгс/см2, и реакцию первой стадии проводили в течение 3 ч, получая реакционную смесь первой стадии. Анализ полученной реакционной смеси первой стадии ГХ и ВЭЖХ показал, что конверсия WA3B3 полная и выходы WA4B2, WA4BH и WA4H2 составили соответственно 50%, 6% и 4% (т.е. в целом 60%) в расчете на WB6.
После окончания реакции первой стадии реакционную смесь первой стадии удалили из автоклава и отфильтровали катализатор, содержащийся в реакционной смеси, с получением фильтрата. Используя полученный фильтрат, провели реакцию второй стадии следующим образом. В автоклав емкостью 100 мл загрузили полученный фильтрат, 3,15 г 10% Pd-C и 30 мл воды. Автоклав продули газообразным азотом. Затем в автоклав ввели газообразный водород так, что внутреннее давление в автоклаве стало равным 3,3 кгс/см2. Реакцию второй стадии начали повышением температуры содержимого автоклава до 130oС в течение 20 мин при перемешивании содержимого автоклава со скоростью перемешивания 700 об/мин. Реакцию второй стадии проводили в течение 1 ч при 130oС (при непрерывном введении в автоклав газообразного водорода так, что внутреннее давление в автоклаве поддерживалось при 3,3 кгс/см2). Анализ реакционной смеси второй стадии ВЭЖХ показал, что выход WA4H2 составил 60% в расчете на WB6.
Сравнительный пример 2
Этот сравнительный пример показывает, что в случае взаимодействия WB6 и гетерогенного катализатора восстановления в отсутствие, по крайней мере, одного из ацилирующего реагента и восстановителя, выход желаемого продукта заметно снижается.
2,1 г WB6, 3,15 г 10% Pd-C, 1,84 г уксусного ангидрида и 30 мл DMAc загрузили в автоклав емкостью 100 мл и автоклав продули газообразным азотом при комнатной температуре. Затем в автоклав ввели газообразный водород так, что внутреннее давление в автоклаве стало равным 1,1 кгс/см2, перемешивая содержимое автоклава со скоростью 50 об/мин, при этом температура содержимого автоклава поднималась до 60oС в течение более 12 мин. Затем сразу же скорость перемешивания повысили до 700 об/мин, начав реакцию. Во время реакции в автоклав непрерывно вводили газообразный водород так, что внутреннее давление в автоклаве поддерживалось при 1,1 кгс/см2, и реакцию проводили в течение 3 ч, получая реакционную смесь. Анализ полученной реакционной смеси ГХ и ВЭЖХ показал, что соответственно выход WА3В3 составил 25% расчете на WB6 и выход WA4B2 составил 28% в расчете на WB6.
Пример 5
(Способ, в котором ацилирующий реагент и восстановитель загружают в реактор и затем в него загружают раствор WB6 и суспензию катализатора).
0,84 г 10% Pd-C подвергли восстановлению в 10 мл DMAc в течение 2 ч при давлении водорода 3 кгс/см2, температуре 60oС и скорости перемешивания 700 об/мин, получая суспензию катализатора.
1,82 г уксусного ангидрида загрузили в автоклав емкостью 100 мл и перемешивали в течение 30 мин со скоростью перемешивания 50 об/мин, поддерживая давление водорода при 1,1 кгс/см2 и температуру при 60oС. В автоклав последовательно загрузили с помощью шприца раствор WB6 в DMAc (полученный растворением 2,1 г WB6 в 20 мл DMAc) и вышеупомянутую суспензию катализатора. Затем сразу же скорость перемешивания повысили до 700 об/мин, поддерживая температуру при 60oС и давление при 1,1 кгс/см2, и реакцию проводили в течение 1 ч, в результате чего получали реакционную смесь. Анализ полученной реакционной смеси ГХ показал, что выходы WA4B2 и WA4BH составили соответственно 55% и 8% (т.е. в целом 63%) в расчете на WB6, и анализ полученной реакционной смеси ВЭЖХ показал, что выход WA4H2 составил 18% в расчете на WB6.
Пример 6
(Способ, в котором катализатор, используемый в реакции первой стадии, применяется в реакции второй стадии и последовательность реакций первой стадии и второй стадии проводится повторно при использовании того же катализатора без замены на свежий катализатор).
9,8 г 10% Pd-C (в качестве гетерогенного катализатора восстановления) диспергировали в 200 мл DMAc (N,N-диметилацетамид) (в качестве растворителя) для получения таким образом суспензии катализатора и полученную суспензию загрузили в автоклав емкостью 2 л. Автоклав продули газообразным азотом. Затем в автоклав ввели газообразный водород (в качестве восстановителя) так, что внутреннее давление в автоклаве стало равным 2 кгс/см2. Содержимое автоклава перемешивали со скоростью перемешивания 300 об/мин в течение 1 ч, поддерживая внутреннее давление при 2 кгс/см2 и температуру при 60oС, для проведения восстановления катализатора, содержащегося в суспензии катализатора.
49 г WB6 (гексабензилгексаазаизовюрцитана) растворили в 500 мл DMAc при 60o С для получения раствора. К полученному раствору добавили 42,4 г уксусного ангидрида (в качестве ацилирующего агента) и полученную жидкую смесь быстро загрузили в автоклав, содержащий суспензию катализатора. (Когда жидкую смесь загрузили в автоклав, температуру временно снизили, но примерно через 4 мин вновь подняли до 60oС). Затем сразу же скорость перемешивания увеличили до 1000 об/мин, в то же время поддерживая температуру при 60oС и давление при 2 кгс/см2, и проводили реакцию первой стадии в течение 1 ч, получая реакционную смесь первой стадии. Анализ реакционной смеси первой стадии ГХ показал, что выходы WA4B2 (тетраацетилдибензилгексаазаизовюрцитана) и WA4BH (тетраацетилбензилгексаазаизовюрцитана) составили соответственно 64% и 11% (т. е. в целом 75%) от WB6, и анализ реакционной смеси первой стадии ВЭЖХ показал, что выход WA4H2 (тетраацетилгексаазаизовюрцитана) составил 10% от WB6.
После реакции первой стадии давление водорода в автоклаве емкостью 2 л, в котором находилась реакционная смесь первой стадии, повысили до 9 кгс/см2 и температуру реакции повысили с 60oС до 90oС в течение 30 мин для начала реакции второй стадии. Введение воды в автоклав начали одновременно с началом подъема температуры, и в течение 1 ч с помощью насоса в автоклав ввели 700 мл воды. Реакцию второй стадии продолжали еще 40 мин после завершения введения воды для получения таким образом реакционной смеси второй стадии. Анализ полученной реакционной смеси второй стадии ВЭЖХ показал, что выход WA4H2 составил 84% от WB6.
Катализатор извлекали из реакционной смеси второй стадии следующим образом. Все операции проводились в атмосфере газообразного водорода, если не указано иначе. Реакционную смесь второй стадии перенесли в аппарат для фильтрования под давлением и катализатор, содержащийся в реакционной смеси второй стадии, отфильтровали под давлением, получая фильтрат. Катализатор, остающийся в аппарате для фильтрования под давлением, промыли 500 мл воды, воду отделили от катализатора фильтрованием под давлением. Затем катализатор промыли 500 мл DMAc в атмосфере газообразного азота, DMAc удалили фильтрованием под давлением.
Последовательность операций реакции первой стадии, операций реакции второй стадии и операций выделения катализатора повторили 10 раз вышеупомянутым образом. Каждую из реакционных смесей второй стадии, полученных при проведении последовательных операций, в отдельности анализировали ВЭЖХ для определения выхода WA4H2 от WB6. Результаты показали, что значения выходов WA4H2 с 1 по 10 стадий операций равнялись соответственно 84% (1-ая), 82% (2-ая), 85% (3-ья), 80% (4-ая), 79% (5-ая), 81% (6-ая), 84% (7-ая), 83% (8-ая), 84% (9-ая) и 80% (10-ая).
Как следует из вышеприведенного, выход WA4H2 не меняется, даже когда тот же катализатор используют повторно без замены на свежий катализатор.
Пример 7
(Способ, в котором реакцию проводят повторно, используя тот же катализатор).
Реакцию проводили аналогично реакции первой стадии примера 1, получая реакционную смесь. Катализатор, содержащийся в полученной реакционной смеси, выделили в основном аналогично примеру 6, за исключением того, что каждые количества воды и DMAc были по 50 мл.
Последовательность вышеуказанных операций реакции и операцию выделения катализатора повторили 5 раз вышеуказанным способом. Каждую из реакционных смесей, полученных при проведении последовательных стадий операций, в отдельности анализировали ГХ для определения выхода суммарного количества WA4B2 и WA4BH в расчете на WB6, и в отдельности анализировали ВЭЖХ для определения выхода WA4H2 в расчете на WB6. Результаты представлены в табл.1.
Как очевидно из вышеприведенного, выход производных тетраацетилгексаазаизовюрцитана не менялся, даже когда в реакции повторно использовали тот же катализатор.
Пример 8
(Способ, в котором реакцию непрерывно проводят с использованием того же катализатора (непрерывная реакция, в которой реакционную смесь фильтруют в реакторе, и полученный фильтрат, содержащий продукты реакции, периодически удаляется из реактора, в то же время в реактор периодически загружается раствор исходных веществ) ).
В качестве реактора использовали автоклав емкостью 200 мл, снабженный металлокерамическим фильтром (диаметр пор: 2 мкм) и трубкой для удаления жидкости, полученной из реакционной смеси фильтрованием через металлокерамический фильтр. Используя такой тип автоклава, становится возможным фильтровать реакционную смесь (которую получают в реакции, катализируемой гетерогенным катализатором восстановления, в таком автоклаве) и удалять полученный фильтрат (т. е. катализатор можно отфильтровать в автоклаве без удаления катализатора из автоклава так, чтобы реакцию можно было продолжать, используя тот же катализатор).
Используя вышеуказанный автоклав емкостью 200 мл, проводили непрерывную реакцию, где часть реакционной смеси, полученной в реакции, подвергается фильтрованию в реакторе, и полученный фильтрат, содержащий реакционные продукты периодически извлекается из реактора, в то же время периодически в реактор загружается раствор исходных веществ.
14,7 г WB6 растворили в 210 мл DMAc при примерно 60oС и полученному раствору затем дали охладиться до комнатной температуры, и добавили 12,74 г уксусного ангидрида, получая раствор исходных веществ.
Реакцию проводили аналогично реакции первой стадии в примере 1, за исключением того, что количества WB6, уксусного ангидрида, растворителя и катализатора были соответственно в четыре раза больше, чем количества, использованные в примере 1, и того, что давление водорода составило 4 кгс/см2 и скорость перемешивания была равной 1500 об/мин. Через 1 ч от начала реакции часть полученной реакционной смеси удалили из автоклава через металлокерамический фильтр, получая 40 г фильтрата, в то же время поддерживая давление водорода при 4 кгс/см2 и скорость реакции 1500 об/мин. Затем в автоклав загрузили 40 г раствора исходных веществ. Операцию по загрузке раствора исходных веществ в автоклав проводили, в то же время поддерживая давление водорода, скорость перемешивания и температуру реакционной смеси так, чтобы реакция могла продолжаться.
Через 40 мин после загрузки в автоклав раствора исходных веществ повторили последовательность реакции, фильтрование части реакционной смеси, удаление фильтрата из автоклава и загрузку раствора исходных веществ в автоклав, в то же время продолжая реакцию. Эту последовательность операций повторили 11 раз аналогично описанным выше, в то же время продолжая реакцию в автоклаве. Каждый из фильтратов, полученных при проведении последовательных стадий операций, в отдельности анализировали ГХ для определения выхода суммарного количества WA4В2 и WA4BH в расчете на WB6, и также анализировали ВЭЖХ для определения выхода WA4H2 в расчете на WB6. Результаты представлены в табл.2.
Как очевидно из вышепредставленного, выход производных тетраацетилгексаазаизовюрцитана не менялся, даже когда в реакции повторно использовали тот же катализатор.
Пример 9
(Способ, в котором реакцию непрерывно проводят, используя тот же катализатор (непрерывная реакция, где часть реакционной смеси, полученной в реакции, подвергается фильтрованию в реакторе, и полученный фильтрат, содержащий продукты реакции, непрерывно удаляется из реактора, в то же время в него непрерывно загружается раствор исходных веществ) ).
Используя тот же автоклав емкостью 200 мл, какой был использован в примере 8, проводили непрерывную реакцию, где часть реакционной смеси, полученной в реакции, фильтровали в реакторе, и полученный фильтрат, содержащий продукты реакции, непрерывно удаляется из реактора, в то же время в реактор непрерывно загружается раствор исходных веществ.
Реакцию проводили в основном аналогично реакции первой стадии примера 1, за исключением, что количества WB6, уксусного ангидрида, растворителя и катализатора были соответственно в четыре раза больше, чем в примере 1, и того, что давление водорода составило 4 кгс/см2 и скорость перемешивания была равной 1500 об/мин. Через 1 ч после начала реакции часть полученной реакционной смеси непрерывно удаляли из автоклава через металлокерамический фильтр так, что фильтрат непрерывно удалялся через трубку со скоростью 0,33 г/мин, в то же время поддерживая давление водорода при 4 кгс/см2, скорость перемешивания 1500 об/мин и температуру реакционной смеси и в то же время загружая раствор исходных веществ в автоклав со скоростью 0,33 г/мин. Эту непрерывную реакцию проводили в течение 20 ч, и часть фильтрата, удаленного из автоклава, отбирали для анализа ГХ и ВЭЖХ каждые 2 ч. Каждый из отобранных фильтратов в отдельности анализировали ГХ для определения выхода суммарного WA4B2 и WA4BH в расчете на WB6, и также анализировали ВЭЖХ для определения выхода WA4H2 в расчете на суммарное количество WA4B2 и WA4BH. Результаты представлены в табл.3.
Как очевидно из вышепредставленного, выход тетраацетилгексаазаизовюрцитанов не менялся, даже когда реакцию проводили непрерывно.
Пример 10
(Способ, в котором реакция второй стадии проводится повторно с использованием того же катализатора).
Все фильтраты (каждый из которых представлял собой реакционную смесь с отфильтрованным катализатором), полученные в примере 8, объединили и дали постоять при комнатной температуре для получения таким образом суспензии, содержащей выпавшие в осадок WA4B2, WA4BH и WA4H2 и тому подобное. Используя полученную суспензию, провели реакцию второй стадии следующим образом.
0,84 г 10% Pd-C загрузили в автоклав емкостью 100 мл, а затем в автоклав ввели газообразный водород таким образом, что внутреннее давление в автоклаве стало равным 1,1 кгс/см2. Содержимое автоклава перемешивали в течение 1 ч со скоростью перемешивания 50 об/мин, в то же время поддерживая давление при 1,1 кгс/см2 и температуру при 90oС. Смесь, состоящую из 30 г вышеуказанной суспензии и 10,5 г воды, быстро загрузили в автоклав с помощью шприца. Затем сразу же давление и скорость перемешивания подняли соответственно до 10 кгс/см2 и 700 об/мин для проведения реакции второй стадии в течение 1 ч при температуре 90oС под давлением 10 кгс/см2, получив таким образом реакционную смесь второй стадии. Затем сразу же реакционную смесь второй стадии фильтровали, получая фильтрат и катализатор.
Последовательность операций реакции второй стадии и операцию фильтрования повторили 5 раз вышеуказанным образом. Каждый из фильтров, полученных при проведении последовательностей операций, в отдельности анализировали ГХ для определения превращения суммарного количества WA4B2 и WA4BH и также анализировали ВЭЖХ для определения выхода WA4H2 в расчете на суммарное количество WA4B2 и WA4BH. Результаты представлены в табл.4.
Как очевидно из вышепредставленного, выход тетраацетилгексаазаизовюрцитанов не менялся, даже когда в реакции повторно использовали тот же катализатор.
Пример 11
(Способ, в котором реакция второй стадии проводится непрерывно с использованием того же катализатора).
Фильтрату, который получили в примере 9 (реакционная смесь), где катализатор отфильтровали, дали постоять при комнатной температуре для получения суспензии, содержащей выпавшие в осадок WA4B2, WA4BH, WA4H2 и тому подобное. 400 г полученной суспензии и 140 г воды перемешивали вместе, получая раствор исходных веществ. Анализ раствора исходных веществ ГХ показал, что суммарное количество WA4B2 и WA4BH, содержащихся в растворе исходных веществ, составило 13,9 г, и анализ раствора исходных веществ ВЭЖХ показал, что количество WA4H2, содержащегося в растворе исходной смеси, составило 1,7 г.
6,72 г 10% Pd-C загрузили в автоклав емкостью 200 мл, использованный в примере 8, и затем в автоклав ввели газообразный водород так, что внутреннее давление в автоклаве стало равным 1,1 кгс/см2. Содержимое автоклава перемешивали в течение 1 ч со скоростью 50 об/мин, в то же время поддерживая давление при 1,1 кгс/см2 и температуру при 90oС. 120 г раствора исходных веществ быстро загрузили в автоклав с помощью шприца. Затем сразу же скорость перемешивания и давление подняли соответственно до 1400 об/мин и 10 кгс/см2 для проведения реакции второй стадии в течение 30 мин. Через 30 мин от начала реакции часть реакционной смеси непрерывно удаляли из автоклава через металлокерамический фильтр так, что фильтрат непрерывно удалялся через трубку со скоростью 0,3 мл/мин, в то же время поддерживая давление водорода при 1,1 кгс/см2, скорость перемешивания 1400 об/мин и температуру реакционной смеси при 90oС и в то же время загружая раствор исходных веществ в автоклав со скоростью 0,3 мл/мин. Эту непрерывную реакцию проводили в течение 20 ч, и часть фильтрата, удаленного из автоклава, отбирали для анализов ГХ и ВЭЖХ каждые 2 ч. Каждый из отобранных фильтратов в отдельности анализировали ГХ для определения превращения суммарного количества WA4B2 и WA4BH и также анализировали ВЭЖХ для определения выхода WA4H2 в расчете на суммарное количество WA4B2 и WA4BH. Результаты представлены в табл.5.
Как очевидно из вышепредставленного, выход тетраацетилгексаазаизовюрцитанов не менялся, даже когда реакцию проводили непрерывно.
Сравнительный пример 3
Этот сравнительный пример 3 показывает, что в случае, когда реакционную смесь первой стадии подвергают реакции второй стадии без введения в реактор газообразного водорода, скорость реакции второй стадии становится заметно ниже.
Реакцию первой стадии проводили аналогично примеру 6, получая реакционную смесь первой стадии. Анализ полученной реакционной смеси первой стадии ГХ показал, что выходы WA4B2 и WA4BH составили соответственно 65% и 7% (т. е. в целом 72%) в расчете на WB6, и анализ полученной реакционной смеси первой стадии ВЭЖХ показал, что выход WA4H2 составил 10% в расчете на WB6.
После реакции первой стадии в автоклав ввели газообразный азот так, что внутреннее давление стало равным 9 кгс/см2. Температуру содержимого автоклава повысили с 60oС до 90oС в течение 30 мин. Введение воды в автоклав начали сразу же с началом повышения температуры и в течение 1 ч в автоклав ввели 700 мл воды с помощью насоса. Затем в автоклав ввели газообразный водород так, что внутреннее давление стало равным 9 кгс/см2, и реакцию второй стадии проводили в течение 2 ч для получения таким образом реакционной смеси второй стадии.
Анализ полученной реакционной смеси второй стадии ГХ показал, что выходы WA4B2 и WA4BH составили соответственно 42% и 3% (т.е. в целом 45%) в расчете на WB6, и анализ полученной реакционной смеси первой стадии ВЭЖХ показал, что выход WA4H2 составил 30% в расчете на WB6.
Как следует из вышесказанного, когда введение газообразного водорода в автоклав прерывается, то в то же время при изменении реакционных условий между окончанием реакции первой стадии и началом реакции второй стадии скорость реакции в реакторе второй стадии стала заметно ниже.
Сравнительный пример 4
Этот сравнительный пример показывает, что когда значение К (скорость потока при загрузке WB6)/Q (количество катализатора) равняется 0,5 или более, выход желаемого продукта становится заметно ниже.
Реакцию проводили аналогично примеру 9, за исключением того, что каждая скорость загрузки раствора исходных веществ и скорость удаления фильтрата (реакционная смесь с отфильтрованным катализатором) равнялась 30 мл/мин. Реакцию проводили непрерывно в течение 2 ч. Часть фильтрата, удаленного из автоклава, отобрали для анализов ГХ и ВЭЖХ через 30, 60 и 120 мин от начала реакции. Анализы фильтратов, удаленных из автоклава, показали, что как выход WA4B2, так и выход WA4BH составил 10% или менее в расчете на WB6 во всех фильтратах, полученных через 30 мин от начала реакции.
Пример 12
(Способ, в котором реакция проводится в присутствии карбоновой кислоты, где содержание карбоновой кислоты в растворителе, использованном для реакции, равняется примерно 10 мас.%).
Этот пример показывает, что даже когда реакционная система содержит карбоновую кислоту (производное ацилирующего реагента, использованного в реакции) в концентрации примерно 10 мас.% в расчете на массу растворителя, используемого в реакции, это не влияет отрицательно на способ по настоящему изобретению.
Реакцию проводили в основном аналогично реакции первой стадии примера 1, за исключением, что добавили 3,0 г уксусной кислоты к раствору, полученному при растворении WB6 и уксусного ангидрида в DMAc, сразу же перед тем, как раствор загрузили в автоклав. Анализ полученной реакционной смеси ГХ показал, что выходы WA4B2 и WA4BH составили соответственно 53% и 9% (т.е. в целом 62%) в расчете на WB6, и анализ полученной реакционной смеси ВЭЖХ показал, что выход WA4H2 составил 17% в расчете на WB6.
Сравнительный пример 5
Этот сравнительный пример показывает, что когда растворитель для реакции содержит уксусную кислоту в концентрации более чем 10 мас.% в расчете на массу растворителя, вход желаемого продукта заметно ниже.
Реакцию проводили в основном аналогично примеру 12, за исключением того, что использовали 6,0 г (вместо 3,0 г) уксусной кислоты. Анализ полученной реакционной смеси ГХ показал, что выход суммарного количества WA4B2 и WA4BH составил 40% или менее в расчете на WB6, и анализ полученной реакционной смеси первой стадии ВЭЖХ показал, что выход WA4H2 составил 5% или менее в расчете на WB6.
Пример 13
(Способ, в котором WB6 используют в высокой концентрации).
Этот пример показывает, что способ по настоящему изобретению можно проводить, даже когда реакционная система содержит WB6 (в качестве исходного вещества) в такой высокой концентрации, как 14 мас.% в расчете на растворитель.
Реакцию проводили в основном аналогично реакции первой стадии примера 1, за исключением того, что использовали 4,2 г (вместо 2,1 г) WB6, внутренняя температура в автоклаве перед загрузкой раствора WB6 и уксусного ангидрида DMAc в автоклав была равной 55oС (вместо 60oС), использовали 2,1 г 10% Pd-C (вместо 0,84 г), получая реакционную смесь. Анализ полученной реакционной смеси ГХ показал, что выходы WA4B2 и WA4BH составили соответственно 39% и 19% (т. е. в целом 58%) в расчете на WB6, и анализ полученной реакционной смеси первой стадии ВЭЖХ показал, что выход WA4H2 составил 20% в расчете на WB6.
Стандартный пример 1 (известный уровень техники)
(Растворимость производных гексаазаизовюрцитана различных типов, содержащих ацильную группу, в разных растворителях).
Исследовали растворимость производных гексаазаизовюрцитана различных типов, содержащих ацильную группу, в разных растворителях. Результаты представлены в табл.6.
Стандартный пример 2 (известный уровень техники)
(Осаждение кристаллов WA4H2 при перегонке)
1 г WA4H2 растворили в смешанном растворителе из первого и второго растворителей для получения раствора. Затем первый растворитель отогнали из раствора для осаждения кристаллов WA4H2. Соответствующие использованные количества (г) первого и второго растворителей и результаты осаждения кристаллов при перегонке представлены в таблице 7. В стандартном примере 2 перегонку для осаждения кристаллов проводили до такой степени, что количество первого растворителя стало равным 1 мас.% или менее в расчете на общую массу первого и второго растворителей.
Пример 14
(Способ, в котором реакционную смесь второй стадии (которую получили порционным способом при проведении реакции in situ с реакционной смесью первой стадии) подвергают способу осаждения кристаллов перегонкой).
Реакционную смесь второй стадии, полученную в примере 2, подвергли осаждению кристаллов перегонкой следующим образом.
300 мл реакционной смеси второй стадии, полученной в примере 2, фильтровали для удаления катализатора, находящегося в реакционной смеси, получая фильтрат. Полученный фильтрат загрузили в баклажановидную колбу емкостью 500 мл и подвергли перегонке (50oС, 10 мм рт. ст.), используя ротационный испаритель, снабженный баней с постоянной температурой, вакуумным насосом и вакуумным контроллером, для удаления 150 мл низкокипящих фракций (таких, как вода и толуол), имеющих точку кипения, которая ниже, чем у DMAc. После отгонки колбу отсоединили от роторного испарителя, и полученному после отгонки остатку в колбе дали охладиться до комнатной температуры и постоять при комнатной температуре в течение 12 ч для осаждения твердых частиц.
Выпавшие частицы собрали с помощью фильтрования под вакуумом и затем промыли примерно 10 мл DMAc, получая влажное твердое вещество. Влажное твердое вещество сушили с помощью вакуумной сушилки (70oС, 1 мм рт. ст. или менее), получая 4,4 г WA4H2 в виде белого твердого вещества (выход осаждения кристаллов: 98%; чистота: 98% (определено с помощью ВЭЖХ)).
Пример 15
(Способ, в котором реакционную смесь второй стадии (которую получают в непрерывным режиме путем непрерывного проведения реакции первой стадии) подвергают способу осаждения кристаллов перегонкой).
Фильтрат (реакционная смесь с отфильтрованным катализатором), полученный в примере 11, подвергли способу осаждения кристаллов перегонкой следующим образом.
Фильтрат (реакционная смесь с отфильтрованным катализатором) анализировали ВЭЖХ. В результате было установлено, что содержание WA4H2 в фильтрате составило 1,90 г/100 мл. 100 мл фильтрата загрузили в баклажановидную колбу емкостью 300 мл и перегоняли в основном аналогично примеру 14, используя ротационный испаритель, для удаления примерно 28 мл низкокипящих фракций (таких, как толуол и вода), имеющих точку кипения ниже, чем у DMAc. После перегонки колбу отсоединили от ротационного испарителя, и полученному после отгонки остатку в колбе дали охладиться до комнатной температуры и постоять в течение 12 ч при комнатной температуре для осаждения таким образом твердых частиц.
Выпавшие твердые частицы собрали фильтрованием под вакуумом и затем промыли примерно 5 мл DMAc, получая влажное твердое вещество. Влажное твердое вещество высушивают с помощью вакуумной сушилки (70oС, 1 мм рт. ст. или менее), получая 1,84 г WA4B2 в виде белого твердого вещества (выход осаждения кристаллов: 97%; чистота: 99% (определено с помощью ВЭЖХ) ).
Пример 16
(Способ, в котором растворитель для кристаллизации рециркулируется (способ нейтрализации NaOH) ).
В примере 14, когда осажденный WA4H2 собрали фильтрованием под вакуумом после проведения осаждения кристаллов WA4B2 при перегонке, получили также фильтрат, содержащий DMAc. Анализ фильтрата ГХ показал, что содержание уксусной кислоты в фильтрате составило 2,30 г по отношению к 100 мл фильтрата.
К 100 мл вышеуказанного фильтрата добавили 20 мл 2 н. водного раствора NaOH при перемешивании в течение 30 мин, нейтрализуя уксусную кислоту, содержащуюся в фильтрате. Затем полученную смесь перегоняли, получая 95 мл дистиллята, состоящего в основном из DMAc. Содержание уксусной кислоты в полученном дистилляте было менее 0,5 мас.%.
Вышеупомянутые количественные анализы на содержание уксусной кислоты в вышеуказанных фильтрате и дистилляте проводили ГХ в следующих условиях, используя описанную ниже аппаратуру.
<Условия анализа ГХ>
Аппарат для ГХ: газовый хроматограф типа GC-10 (произведено и поставлено Schimadzu Corporation, Япония)
Колонка: капиллярная колонка DB-1 (произведена и поставлена J & W Scientific Co., Ltd., США)
Внутренний диаметр: 0,25 мм
Длина: 30 м
Толщина пленки, покрывающей внутреннюю стенку капиллярной колонки: 0,25 мкм
Детектирование: пламенно-ионизационный детектор (FID)
Температура: 60oС (3 мин)
Колонка: поднимается до 300oС (скорость повышения температуры: 10oС/мин) --> 300oС (5 мин)
Инжектор: 300oС
Детектор: 300oС
Газ-носитель: N2 (скорость газа: 100 мл/мин и внутреннее давление в колонке: 100 кПа)
Количество вводимой пробы: 5 мкл (полученный фильтрат или дистиллят per se).
Пример 17
(Способ, в котором растворитель для кристаллизации рециркулируется (способ нейтрализации основным соединением Мg) ).
В примере 14, когда выпавшие кристаллы собрали фильтрованием в вакууме, после осаждения кристаллов WA4H2 при перегонке, получили также фильтрат, содержащий DMAc. Анализ фильтрата ГХ показал, что содержание уксусной кислоты в фильтрате равнялось 2,30 г по отношению к 100 мл фильтрата.
К 100 мл вышеуказанного фильтрата добавили Mg(OH)2 так, чтобы значение рН фильтрата стало равным 8,00, получая суспензию, содержащую Mg(OAc)2. (Примерно 2,4 г Mg(OH)2 необходимо для подведения значения рН фильтрата до 8,00.) Полученную суспензию перегоняют, используя перегонный аппарат при температуре, равной 100oС, и давлении 50 торр, получая 95 мл дистиллята, который в основном состоял из DMAc. Содержание уксусной кислоты в полученном дистилляте составило 0,5 мас.% или менее. Количественный анализ уксусной кислоты проводили ГХ в тех же условиях, что описаны в примере 16. В результате обследования внутренности перегонного аппарата после перегонки в нем обнаружили только бледно-коричневую жидкость и не обнаружили прилипшего к внутренней стенке перегонного аппарата вещества.
Стандартный пример 3 (известный уровень техники)
(Получение сырого WB6 общепринятым способом).
В съемную колбу емкостью 2 л, снабженную мешалкой, термометром, холодильником и капельной воронкой, загрузили 1,1 л ацетонитрила, 100 мл дистиллированной воды, 117,9 г (1,1 ммоль) бензиламина и 4,6 мл (0,11 моль) муравьиной кислоты, и полученную смесь охладили до 10oС в бане с постоянной температурой. Затем к смеси постепенно добавили 72,5 г 40% водного раствора глиоксаля (содержащего 0,5 моль глиоксаля) с помощью капельной воронки в течение 1 ч, в то же время перемешивая и поддерживая температуру смеси при 10oС. После добавления раствора глиоксаля полученную смесь нагрели до 25oС при перемешивании и затем дополнительно перемешивали в течение 18 ч для проведения таким образом реакции. В результате из полученной желтой реакционной смеси выпало белое твердое вещество. Реакционную смесь, содержащую белое твердое вещество, отфильтровали в вакууме, собирая твердое вещество, и затем собранное твердое вещество промыли смешанным растворителем (охлажденным до 0oС), содержащим 275 мл ацетонитрила и 25 мл дистиллированной воды. Промытое твердое вещество высушивали в течение ночи при комнатной температуре при пониженном давлении для получения 92,34 г сырого WB6 (гексабензилгексаазаизовюрцитана). Анализ полученного сырого WB6 ВЭЖХ показал, что чистота сырого WB6 была равной 90,2%.
Выше полученный сырой WB6 (в твердом виде) исследовали с помощью сканирующего электронного микроскопа (СЭМ) для получения микрофотографий (х200), и полученная микрофотография представлена на фиг.1.
Стандартный пример 4 (известный уровень техники)
(Очистка сырого WB6 общепринятой перекристаллизацией).
Ниже описывается общепринятый способ перекристаллизации для очистки WB6, в котором используется небольшое количество растворителя для кристаллизации.
16,58 г сырого WB6 (чистота: 90,2%), полученного в стандартном примере 3, и 1 л ацетонитрила загрузили в химический стакан емкостью 2 л. Полученную в химическом стакане смесь нагрели до 90oС на бане с постоянной температурой при перемешивании для полного растворения WB6 в ацетонитриле. После растворения WB6 в ацетонитриле температуру полученного раствора WB6 понизили до 20oС, и раствор перемешивали в течение 4 ч, в то же время поддерживая температуру раствора при 20o С для перекристаллизации WB6. Перекристаллизованный WB6 собрали фильтрованием под вакуумом и высушили для получения 14,52 г белых кристаллов WB6.
Анализ полученных кристаллов WB6 ВЭЖХ показал, что чистота кристаллов WB6 была равной 92,7%.
Кристаллы WB6, полученные выше, исследовали с помощью СЭМ для получения микрофотографий (х200), и полученная микрофотография представлена на фиг.2. Как четко видно из фиг.2, чистота кристаллов WB6, полученных общепринятым способом очистки, была удовлетворительно низкой.
Стандартный пример 5 (известный уровень техники)
(Растворимость WB6 в органических растворителях различных типов).
Используя сырой WB6, полученный в стандартном примере 3, определили растворимость WB6 (H) в различных органических растворителях и растворимость примесей (I) (содержащихся в сыром WB6) в различных органических растворителях определили следующим образом.
10 мл органического растворителя и сырой WB6, полученный в стандартном примере 3, загрузили в химический стакан, и полученную смесь перемешивали при заранее определенной температуре в течение 1 ч, получая суспензию. (Сырой WB6 использовали в таком количестве, что часть WB6 осталась диспергированной в органическом растворителе даже после перемешивания в течение 1 ч при заранее определенной температуре). Полученную суспензию отфильтровали, получая фильтрат. Часть фильтрата отобрали для анализа ВЭЖХ. Пробу для ВЭЖХ готовили разбавлением отобранного фильтрата смешанным растворителем, включающим тетрагидрофуран и воду (90/10 объем/объем) так, что содержание WB6 в полученном растворе стало равным 0,01 мас.% Пробу анализировали ВЭЖХ, получая хроматограммы. На хроматограмме был отмечен один пик, соответствующий WB6, и другой пик, соответствующий примесям. Время удерживания WB6 равнялось 2,4 мин, и время удерживания примесей равнялось 2,2 мин. Содержание WB6 в фильтрате определили по полученной хроматограмме. Определенное количество WB6 в фильтрате было принято в качестве показателя растворимости WB6 в органическом растворителе при заранее определенной температуре.
Растворимость примесей определяли следующим образом. К фильтрату, полученному выше, добавили заранее определенное количество сырого WB6 (содержащего примеси), полученного в стандартном примере 3. Затем полученную смесь отфильтровали вышеуказанным способом и полученный фильтрат анализировали ВЭЖХ, как указано выше. Добавление WB6 к фильтрату, фильтрование и анализ ВЭЖХ повторяли, пока соотношение интенсивности пика, соответствующего примесям, относительно пика, соответствующего WB6, на хроматограмме больше не увеличивалось, даже когда к фильтрату добавляли сырой WB6. Содержание примесей в фильтрате определили по хроматограмме, показывающей пик, соответствующий примесям, где соотношение интенсивности пика, соответствующего примесям, относительно пика, соответствующего WB6, было максимальным, и определенное количество примесей в фильтрате было принято в качестве растворимости примесей в органическом растворителе при заранее определенной температуре.
Используя растворимость WB6 (H) и растворимость примесей (I), определенные с помощью ВЭЖХ, вычисляли соотношение растворимостей (Р) по следующей формуле:
Р=H/I,
где Н представляет растворимость WB6 (г/л) в органическом растворителе и I представляет растворимость примесей (г/л) (детектируемых анализом ВЭЖХ) в органическом растворителе. Результаты представлены в табл.8.
Стандартный пример 6
(Очистка сырого WB6 усовершенствованным способом перекристаллизации).
Усовершенствованный способ перекристаллизации, используемый для очистки WB6, в котором используется большое количество растворителя для кристаллизации, описывается ниже.
3,9 г сырого WB6 (чистота: 90,2%), полученного в стандартном примере 3, и 1 л ацетонитрила загрузили в химический стакан емкостью 2 л. Полученную смесь нагревали в химическом стакане до 90oС на бане с постоянной температурой при перемешивании до полного растворения WB6 в ацетонитриле. После растворения WB6 в ацетонитриле температуру полученного раствора WB6 снизили до 20oС и раствор перемешивали в течение 4 ч, в то же время поддерживая температуру раствора при 20oС для перекристаллизации WB6. Перекристаллизированный WB6 собирали фильтрованием в вакууме и высушивали, получая 2,5 г белых кристаллов WB6.
Анализ полученных кристаллов WB6 ВЭЖХ показал, что чистота кристаллов WB6 составила 100%.
Стандартный пример 7
(Очистка сырого WB6 усовершенствованным способом перекристаллизации).
50 г сырого WB6 (чистота: 90,2%), полученного в стандартном примере 3, и 1 л этилацетата загрузили в химический стакан емкостью 2 л. Полученную в химическом стакане смесь нагревали до 60oС на бане с постоянной температурой при перемешивании до полного растворения WB6 в этилацетате. После растворения WB6 в этилацетате температуру полученного раствора WB6 снизили до 15oС и раствор перемешивали в течение 4 ч, в то же время поддерживая температуру раствора при 15oС для перекристаллизации WB6. Перекристаллизированный WB6 собирали фильтрованием в вакууме и высушивали, получая 22,6 г белых кристаллов WB6.
Анализ полученных кристаллов WB6 ВЭЖХ показал, что чистота кристаллов WB6 составила 100%.
Стандартный пример 8
(Очистка сырого WB6 усовершенствованным способом перекристаллизации).
25 г сырого WB6 (чистота: 90,2%), полученного в стандартном примере 3, и смешанный растворитель, состоящий из 500 мл этилацетата и 500 мл этанола, загрузили в химический стакан емкостью 2 л. Полученную в химическом стакане смесь нагревали до 60oС в бане с постоянной температурой при перемешивании до полного растворения WB6 в смешанном растворителе. После растворения WB6 в смешанном растворителе температуру полученного раствора WB6 снизили до 15oС и раствор перемешивали в течение 4 ч, в то же время поддерживая температуру раствора при 15oС для перекристаллизации WB6. Перекристаллизированный WB6 собирали фильтрованием в вакууме и высушивали, получая 18,0 г белых кристаллов WB6.
Анализ полученных кристаллов WB6 ВЭЖХ показал, что чистота кристаллов WB6 составила 100%.
Кристаллы WB6, полученные выше, исследовали с помощью СЭМ для получения микрофотографий (х200), и полученная микрофотография представлена на фиг.3. Как ясно видно из фиг.3, каждый из кристаллов WB6 имел четкую игольчатую форму, и чистота кристаллов WB6 была очень высокой.
Стандартный пример 9
(Получение высокоочищенного WB6 усовершенствованным способом).
В съемную колбу емкостью 1 л, снабженную мешалкой, термометром, холодильником и капельной воронкой, загрузили 514 мл ацетонитрила, 78 мл дистиллированной воды, 117,9 г (1,1 моль) бензиламина и 3,2 мл (0,055 мл) уксусной кислоты и полученную смесь в колбе охлаждали до 20oС в бане с постоянной температурой. Затем к смеси постепенно добавили 36,28 г 40% водного раствора глиоксаля (содержащего 0,25 моль глиоксаля) с помощью капельной воронки в течение 30 мин при перемешивании и поддержании температуры смеси при 20oС. После добавления раствора глиоксаля полученную смесь нагревали до комнатной температуры при перемешивании и затем продолжали перемешивание в течение 18 ч для проведения реакции. В результате из полученной желтой реакционной смеси выпало белое твердое вещество. Реакционную смесь, содержащую белое твердое вещество, фильтровали в вакууме, собирая белое твердое вещество, и затем собранное белое вещество промывали следующим образом. Собранное влажное белое твердое вещество диспергировали в смешанном растворителе, состоящем из 170 мл ацетонитрила и 30 мл дистиллированной воды, с последующим перемешиванием в течение 30 мин, получая суспензию. Полученную суспензию фильтровали в вакууме, собирая белые игольчатые кристаллы. Полученные кристаллы сушили в течение ночи при комнатной температуре при пониженном давлении, получая при этом 40,68 г белых игольчатых кристаллов WB6. Анализ полученных кристаллов WB6 ВЭЖХ показал, что чистота кристаллов WB6 составила 100%.
Белые игольчатые кристаллы WB6, полученные выше, исследовали с помощью СЭМ для получения микрофотографий (х200), и полученная микрофотография представлена на фиг.4. Как ясно видно из фиг.4, каждый из кристаллов WB6, полученных усовершенствованным способом, имел четкую игольчатую форму и чистота кристаллов WB6 была очень высокой.
Стандартный пример 10
(Получение высокоочищенного WB6 усовершенствованным способом).
Получение WB6 проводили в основном аналогичным стандартному примеру 9 способом, за исключением того, что использовали 235,8 г (2,2 моль) бензиламина (вместо 117,9 г (1,1 моль)) и температура содержимого в съемной колбе была равной 0oС (вместо 20oС), получая 32,5 г белых игольчатых кристаллов WB6. Анализ полученных кристаллов ВЭЖХ показал, что чистота кристаллов WB6 составила 100%.
Стандартный пример 11
(Получение высокоочищенного WB6 усовершенствованным способом).
Получение WB6 проводили в основном так же, как в стандартном примере 9, за исключением того, что использовали 85,7 г (0,8 моль) бензиламина (вместо 117,9 г (1,1 моль)) и температура содержимого в съемной колбе была 30oС (вместо 20oС), получая 38,2 г белых игольчатых кристаллов WB6. Анализ полученных кристаллов ВЭЖХ показал, что чистота кристаллов WB6 составила 98,2%.
Стандартный пример 12
(Получение высокоочищенного WB6 усовершенствованным способом).
Получение WB6 проводили в основном так же, как в стандартном примере 10, за исключением того, что использовали 4,8 мл (0,075 моль) уксусной кислоты (вместо 3,2 мл (0,055 моль)) и к смеси постепенно добавили 40% водный раствор глиоксаля с помощью капельной воронки в течение 6 ч (вместо 30 мин) при 35oС (вместо 20oС), получая 38,7 г белых игольчатых кристаллов WB6. Анализ полученных кристаллов ВЭЖХ показал, что чистота кристаллов WB6 составила 100%.
Стандартный пример 13
(Получение высокоочищенного WB6 усовершенствованным способом).
В съемную колбу емкостью 2 л, снабженную мешалкой, термометром, холодильником и капельной воронкой, загрузили 1100 мл ацетонитрила, 100 мл дистиллированной воды, 235,8 г (2,2 моль) бензиламина и 12,8 мл (0,22 моль) уксусной кислоты и полученную смесь в колбе охлаждали до 0oС на бане с постоянной температурой. Затем к смеси постепенно добавляли 72,6 г 40% водного раствора глиоксаля (содержащего 0,5 моль глиоксаля) с помощью капельной воронки в течение 2 ч при перемешивании и поддержании температуры смеси при 0oС. После добавления раствора глиоксаля полученную смесь нагревали до 25oС при перемешивании и затем продолжали перемешивание в течение 18 ч для проведения реакции. В результате из полученной желтой реакционной смеси выпадает в осадок белое твердое вещество. Реакционную смесь, содержащую белое твердое вещество, отфильтровывают в вакууме, собирая белое твердое вещество, и затем собранное белое твердое вещество диспергировали в смешанном растворителе, включающем 260 мл ацетонитрила и 40 мл дистиллированной воды, с последующим перемешиванием в течение 30 мин, получая суспензию. Полученную суспензию фильтровали в вакууме для сбора белых игольчатых кристаллов. Полученные белые игольчатые кристаллы сушили в течение ночи при комнатной температуре при пониженном давлении, получив таким образом 71,6 г белых игольчатых кристаллов WB6. Анализ полученных кристаллов WB6 ВЭЖХ показал, что чистота кристаллов WB6 составила 99,3%.
Пример 18
(Способ, в котором WB6 (чистота: 95% или более), полученный усовершенствованным способом перекристаллизации, использовали в качестве исходного вещества).
Этот пример 18 показывает, что желаемый продукт можно получить с высоким выходом при использовании WB6, имеющего чистоту 95% или более, который получают усовершенствованным способом перекристаллизации.
0,41 г 10% Pd-C (в качестве гетерогенного катализатора восстановления) загружали в автоклав емкостью 100 мл и автоклав продували газообразным азотом. Затем в автоклав вводили газообразный водород (в качестве восстановителя) так, что внутреннее давление в автоклаве стало равным 2 кгс/см2. Содержимое в автоклаве нагревали при 60oС в течение 1 ч. В автоклав загрузили жидкую смесь (поддерживаемую при 60oС), полученную при растворении 2,1 г WB6 (гексабензилгексаазаизовюрцитана) (чистота: 100%, полученный в стандартном примере 8) в 30 мл DMAc (в качестве растворителя) с последующим добавлением к полученному раствору 1,84 г уксусного ангидрида (в качестве ацилирующего агента). Затем сразу же начали перемешивать содержимое автоклава со скоростью перемешивания 700 об/мин, в то же время поддерживая температуру при 60oС и давление при 2 кгс/см2, и реакцию проводили в течение 1 ч, получая реакционную смесь.
Анализ полученной реакционной смеси ГХ показал, что выходы WA4B2 (тетраацетилдибензилгексаазаизовюрцитана) и WA4BH (тетраацетилбензилгексаазаизовюрцитана) составили соответственно 35% и 22% (т. е. в целом 57%) в расчете на WB6, и анализ реакционной смеси ВЭЖХ показал, что выход WA4H2 (тетраацетилгексаазаизовюрцитана) составил 24% в расчете на WB6.
Пример 19
(Способ, в котором WB6 (чистота: 95% или более), полученный усовершенствованным способом, используется в качестве исходного вещества).
Этот пример показывает, что желаемый продукт можно получить с очень высоким выходом при использовании WB6, имеющем чистоту 95% или более, который получают способом, примененным в настоящем изобретении.
Реакцию проводили аналогично примеру 18, за исключением того, что использовали WB6 (чистота: 100%), полученный в стандартном примере 9, получая реакционную смесь.
Анализ полученной реакционной смеси ГХ показал, что выходы WA4B2 и WA4BH составили соответственно 37% и 22% (т.е. в целом 59%) в расчете на WB6, и анализ реакционной смеси ВЭЖХ показал, что выход WA4H2 составил 24% в расчете на WB6.
Сравнительный пример 7
Этот сравнительный пример показывает, что выход желаемых продуктов заметно снижается, когда используется WB6, имеющий чистоту менее 95%, который получали и очищали известными способами.
Реакцию проводили в основном так же, как в примере 18, за исключением того, что использовали WB6 (чистота: 92,7%), полученный в стандартном примере 4, и реакцию проводили в течение 4 ч (вместо 1 ч), получая реакционную смесь.
Анализ полученной реакционной смеси ГХ и ВЭЖХ показал, что выход суммарного количества WA4B2, WA4BH и WA4H2 составил 10% или менее в расчете на WB6.
Пример 20
(Способ, в котором WA4B2 получают с высокой селективностью).
Этот пример показывает, что WA4B2 можно получить с высоким выходом, контролируя условия реакции (в частности, используя небольшое количество водосодержащего катализатора).
14,50 г 10% Pd-C катализатора, содержащего воду (содержание воды: 51,67%, содержание катализатора: 7,0 г) и 200 мл DMAc загрузили в автоклав емкостью 2 л и автоклав продували газообразным азотом. Затем в автоклав вводили газообразный водород так, что внутреннее давление в автоклаве стало равным 2 кгс/см2, и содержимое автоклава перемешивали со скоростью перемешивания 1000 об/мин при 60oС в течение 1 ч. Скорость перемешивания и внутреннее давление в автоклаве понизили соответственно до 300 об/мин и 1,1 кгс/см2, поддерживая температуру при 60oС. В автоклав с помощью шприца быстро загружали 60 г уксусного ангидрида и затем в автоклав загружали раствор (температура: 60oС), полученный при растворении 70 г WB6 (гексабензилгексаазаизовюрцитана) в 800 мл DMAc с помощью шприца. Затем сразу же скорость перемешивания и внутреннее давление подняли соответственно до 1000 об/мин и 2 кгс/см2, поддерживая температуру при 60oС, и реакцию проводили в течение 6 ч, получая реакционную смесь.
Анализ полученной реакционной смеси ГХ показал, что выход WA4B2 составил 83% в расчете на WB6.
Пример 21
(Способ, в котором WA4H2 получают с высокой селективностью).
0,42 г 10% Pd-C загружали в автоклав емкостью 100 мл, и автоклав продували газообразным азотом. Затем в автоклав вводили газообразный водород так, что внутреннее давление в автоклаве стало равным 1,1 кгс/см2 и содержимое автоклава перемешивали со скоростью перемешивания 300 об/мин при 60oС в течение 1 ч. Раствор, полученный при растворении 2,1 г WB6, и 1,82 г уксусного ангидрида в 30 мл DMAc быстро загружали в автоклав. Затем сразу же скорость перемешивания и внутреннее давление подняли соответственно до 2000 об/мин и 2 кгс/см2, поддерживая температуру при 60oС, и реакцию проводили в течение 5,5 ч, получая реакционную смесь.
Анализ полученной реакционной смеси ВЭЖХ показал, что выход WA4H2 составил 75% в расчете на WB6, и ни WA4B2, ни WA4BH не были обнаружены при анализе реакционной смеси ГХ.
В этом примере пробу для анализа ВЭЖХ готовили следующим образом. Реакционную смесь (включающую катализатор) выпаривали при пониженном давлении 1 мм рт. ст. или менее при 50oС, удаляя жидкие вещества, содержащиеся в реакционной смеси, получая остаток. К остатку добавили 60 мл воды таким образом, чтобы отсутствовало взаимодействие между твердыми частицами и воздухом (т.е. кислородом), получая таким образом смесь. Полученную смесь подвергли обработке ультразвуком в течение 10 мин с помощью ультразвукового очистителя и фильтровали, удаляя катализатор, с получением фильтрата. Полученный фильтрат использовали в качестве пробы для анализа ВЭЖХ.
Способом по настоящему изобретению при получении производных тетраацилгексаазаизовюрцитана (которые пригодны в качестве предшественников гексанитрогексаазаизовюрцитана, используемого для улучшения эксплуатационных качеств обычных взрывчатых веществ) из WB6 ацилированием можно очень эффективно подавлять разложение гексаазаизовюрцитанового скелета, которое, вероятно, происходит на начальной стадии реакции ацилирования WB6, в качестве исходного вещества, при этом желаемые производные тетраацилгексаазаизовюрцитана можно стабильно получать с высоким выходом. Следовательно, способ по настоящему изобретению промышленно применим. Кроме того, способ по настоящему изобретению также позволяет эффективно подавлять по сравнению с известными способами снижение каталитической активности гетерогенного катализатора восстановления, имеющее место в ходе реакции.

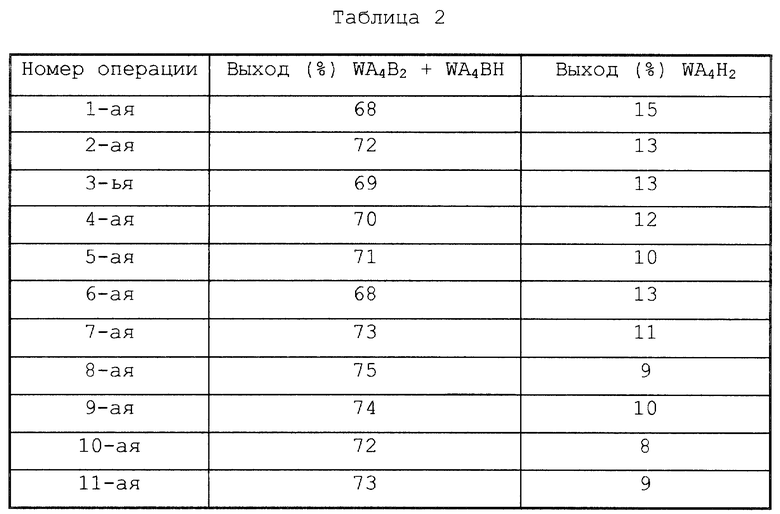
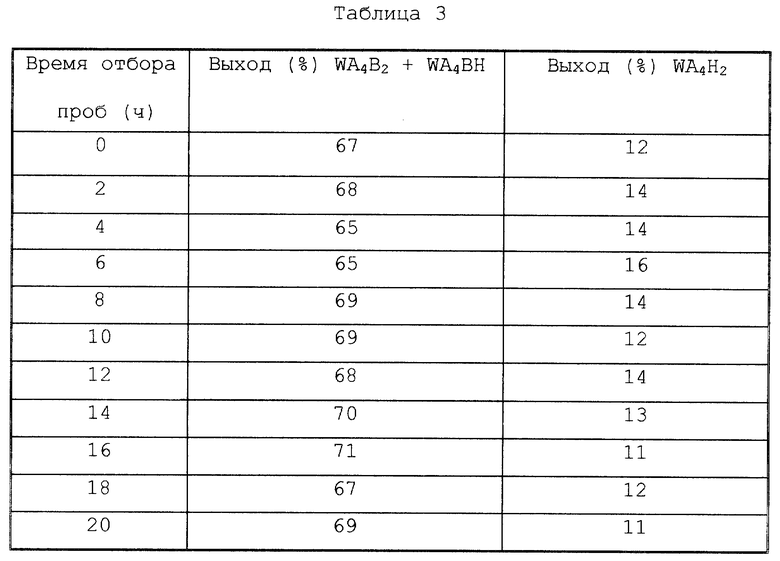

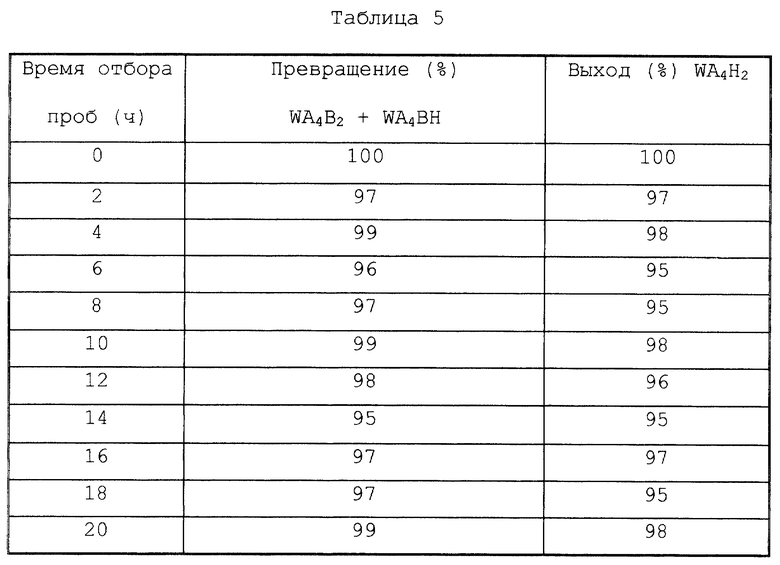



Описывается способ ацилирования гексакис(фенилметил)гексаазаизовюрцитана формулы (1): WB6(1), где В представляет фенилметил и W представляет остаток шестивалентного гексаазаизовюрцитана, представленного формулой (2)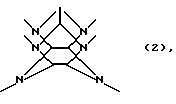
восстановительным дефенилметилированием, включающий контактирование гексакис(фенилметил)гексаазаизовюрцитана с гетерогенным катализатором восстановления в присутствии ацилирующего агента и восстановителя в растворителе для указанного гексакис(фенилметил)гексаазаизовюрцитана, отличающийся тем, что контактирование между WB6 и гетерогенным катализатором восстановления допускается только при обязательном присутствии обоих реагентов: ацилирующего агента и восстановителя с получением смеси, содержащей, по меньшей мере, одно производное тетраацилгексаазаизовюрцитана формулы (3) WA4BnH(2-n), где W, В как определено выше, Н представляет атом водорода, А представляет С1-С10 ацильную группу, n является 0-2. Технический результат - получение целевых продуктов со стабильно высоким выходом и эффективное подавление разложения гексаазаизовюрцитанового скелета. 4 з.п. ф-лы, 4 ил., 8 табл.
WB6,
где В представляет фенилметил и W представляет остаток шестивалентного гексаазаизовюрцитана, представленного следующей формулой II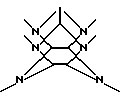
восстановительным дефенилметилированием, включающий контактирование гексакис(фенилметил)гексаазаизовюрцитана с гетерогенным катализатором восстановления в присутствии ацилирующего агента и восстановителя в растворителе для указанного гексакис(фенилметил)гексаазаизовюрцитана, отличающийся тем, что контактирование между WB6 и гетерогенным катализатором восстановления допускается только при обязательном присутствии обоих реагентов: ацилирующего агента и восстановителя с получением смеси, содержащей, по меньшей мере, одно производное тетраацилгексаазаизовюрцитана формулы (3)
WA4BnH(2-n),
где W, В как определено выше;
Н представляет атом водорода;
А представляет C1-С10 ацильную группу;
n является 0-2.
Приоритет по пунктам и признакам:
14.10.1997 по пп. 1-5;
26.12.1997 и 13.02.1998 - уточнение признаков.
| WO 9720785 A1, 12.06.1997 | |||
| WO 9623792 A1, 08.08.1996 | |||
| ANTHONY J | |||
| BELLAMY | |||
| Reductive Debenzylation of Hexabenzylhexaazaisowurtzitane, Tetrahedron | |||
| Топка с качающимися колосниковыми элементами | 1921 |
|
SU1995A1 |
| Способ запрессовки не выдержавших гидравлической пробы отливок | 1923 |
|
SU51A1 |

