Изобретение относится к новому классу стероидных соединений, а в частности к стероидным соединениям формулы (I)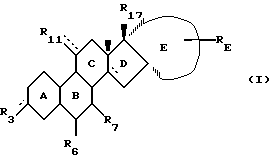
где 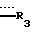 представляет =0, -ОН; =NOR;-OR или -OOCR, где R представляет алкильную группу, имеющую от 1 до 6 атомов углерода;
представляет =0, -ОН; =NOR;-OR или -OOCR, где R представляет алкильную группу, имеющую от 1 до 6 атомов углерода;
R6 представляет Н; =CH2 или -(СН2)mН, где m=1 или 2; причем указанное стероидное соединение может, но необязательно, иметь одну или несколько двойных связей, выбранных из группы, включающей: Δ9(10), Δ5(10), Δ4(5); Δ11(12); Δ14(15); либо любое из колец А или В может быть ароматическим; при этом присутствие или отсутствие атомов водорода не указано, поскольку это зависит от того, является ли данное кольцо насыщенным, ненасыщенным или ароматическим, и может быть легко определено самим специалистом;
R7 представляет Н; С1-4-алкил; С2-5-алкенил или С2-5-алкинил, где алкильная, алкенильная или алкинильная группа могут быть замещены 1-3 атомами галогена, независимо выбранными из атомов фтора и хлора;
R11 представляет Н; С1-4-алкил; С2-4-алкенил; С2-4-алкинил или С1-4-алкилиден, где алкильная, алкенильная, алкинильная или алкилиденовая группа могут быть замещены 1-3 атомами галогена, независимо выбранными из атомов фтора и хлора;
Е представляет, включая атомы углерода 16 и 17 кольца D, 4-7-членное кольцо, которое находится в α-положении по отношению к D-кольцу, замещено группой RE и необязательно содержит одну или две эндоциклические двойные связи; причем α-положение кольца Е по отношению к кольцу D является важным фактором, поскольку соответствующие стероиды, имеющие кольцо Е в β-положении, не обладают нужной биологической активностью. При этом следует отметить, что исходя из номенклатуры, некоторые соединения настоящего изобретения имеют название, в котором есть указание на 16β- и/или 17β-заместители. Однако независимо от этого во всех соединениях настоящего изобретения Е-кольцо находится в целом в положении α.
RE представляет Н; C1-6-алкил; С2-6-алкенил; С2-6-алкинил; С1-6-алкилиден, С2-6-спироаннелированный циклоалкил; -OR; -SR; -OOCR; -NHR; -NRR; -NHCOR, где R (а в случае, если RE является -NRR, то каждый R независимо друг от друга) представляет алкил с 1-6 атомами углерода, -NCO; -(СН2)n-N3 или -(CH2)n-CN, где n=0-5, и где алкильная, алкенильная, алкинильная, алкилиденовая или циклоалкильная группа могут быть замещены 1-3 заместителями, независимо выбранными из группы, включающей -OR; -SR; -OOCR; -NHR; -NRR; и -NHCOR, где R определен выше; атомы фтора и атомы хлора;
R17 представляет -ОН; -OCH2OR; -OR или -OOCR, где R представляет алкил с 1-6 атомами углерода. Любая алкильная, алкенильная, алкинильная и алкилиденовая группа в стероидном соединении формулы (I) может быть разветвленной или неразветвленной. Если R3, R6 или R11 связаны со стероидным каркасом посредством простой связи, то замещенный атом углерода стероидного каркаса либо имеет атом водорода, либо участвует в образовании двойной углерод-углеродной связи. Если RE связан с Е-кольцом посредством простой связи, то замещенный атом углерода Е-кольца также имеет атом водорода.
Неожиданно было обнаружено, что стероидные соединения настоящего изобретения обладают хорошими и представляющими интерес эстрогенными и/или прогестагенными свойствами. Благодаря этим специфическим свойствам стероидные соединения настоящего изобретения могут быть использованы для предупреждения или лечения расстройств, связанных с пери-менопаузой или постменопаузой, включая климактерические симптомы, такие, как приливы жара, нарушение душевного равновесия, расстройства мочеполового тракта, такие, как недержание мочи, атрофия кожи (и эпителия влагалища), а также другие симптомы, ассоциируемые с отсутствием или дефицитом эстрогена, такие, как остеопороз, атеросклероз, и болезнь Альцгеймера. Стероидные соединения настоящего изобретения могут быть использованы для предупреждения или лечения остеопороза, обусловленного дефицитом эстрогена.
Кроме того, стероидные соединения настоящего изобретения могут быть использованы в качестве контрацептивных средств.
Стероидные соединения, имеющие замещения в положении 16, 17 кольца, описаны в литературе. В Chemical Abstracts 89: 215660р (Kamernitskii A.V. et al. ) описано стероидное соединение, содержащее 16,17-аннелированное 5- или 6-членное кольцо и ацетильную группу в положении 17. Однако соединения, описанные в этой публикации, отличаются от соединений настоящего изобретения тем, что в них атом углерода в положении 11 несет атом водорода.
В "Chemical Abstracts 123: 285604t (Wang, J. et al.)" описаны стероидные соединения, имеющие 10-членное Е-кольцо с двумя тройными связями, гидроксильную группу в положении 17, и атом углерода в положении 11.
В ЕР 411733 (Schering AG) описано стероидное соединение, имеющее 6-членное Е-кольцо, и атом углерода в положении 17, участвующий в образовании СО-связи. Однако соединения, описанные в ЕР 411733, отличаются от стероидных соединений настоящего изобретения тем, что в них атом углерода в положении 11 имеет (замещенную) арильную группу. При этом указывается, что эти соединения являются конкурентными антагонистами по отношению к прогестерону.
Таким образом, ни в одной из предшествующих работ не описаны стероидные соединения настоящего изобретения. Стероидные соединения настоящего изобретения отличаются от известных соединений тем, что они имеют заместители в положениях 11, 16 и 17. Более конкретно стероидные соединения настоящего изобретения содержат кольцо Е, имеющее с 5-членным кольцом D общие атомы углерода в положении 16 и 17, и находящееся в положении α по отношению к указанному кольцу D. Кроме того, атом углерода в положении 17 замещен кислородсодержащей группой посредством связи СО. Атом углерода в положении 11 не имеет арильной группы.
Кроме того, ни в одной из вышеуказанных публикаций не было высказано предположения относительно представляющих интерес фармацевтических свойств, которыми обладают стероидные соединения настоящего изобретения. Поэтому соединения настоящего изобретения образуют новый класс стероидных соединений, отличающийся своей in vitro- и in vivo-активностью.
В частности, для получения избирательной эстрогенной активности предпочтительно, чтобы в стероидных соединениях настоящего изобретения Е-кольцо являлось 5-членным кольцом. Однако с точки зрения преимущественных эстроген/прогестогенных профилей соединений, которые имеют сильнодействующие селективные эстрогены и сильнодействующие смешанные эстрогенные/прогестогенные соединения, предпочтительно, чтобы Е-кольцо было 6-членным кольцом. В предпочтительном варианте изобретения А-кольцо является ароматическим, а остальные кольца являются насыщенными, при этом наиболее предпочтительно, чтобы R7 представлял собой α-пропил. Наиболее предпочтительное соединение, обозначенное Оrg 38515, характеризуется тем, что в нем R3 и R17 представляют ОН, a R6, R11 и RE представляют Н.
Настоящее изобретение также относится к фармацевтической композиции, содержащей стероидное соединение настоящего изобретения в смеси с фармацевтически приемлемыми добавками, такими, как, например, добавки, описанные в справочнике Gennaro et al., Remmington's Pharmaceutical Sciences (18th ed., Mack publishing Company, 1990, см., в частности, Part 8: Pharmaceutical Preparations and Their Manufacture). Смесь стероидных соединений настоящего изобретения и фармацевтически приемлемых добавок может быть спрессована с получением твердых унифицированных лекарственных форм, таких, как драже и таблетки, либо с получением капсул или суппозиториев. С использованием фармацевтически приемлемых жидкостей из соединений настоящего изобретения могут быть также получены препараты для инъекций в виде раствора, суспензии, эмульсии или аэрозоля, например для ингаляции через нос. Для изготовления унифицированных лекарственных форм, например таблеток, могут быть использованы стандартные добавки, такие, как наполнители, красители, полимерные носители, и т. п. Вообще говоря, для изготовления указанных лекарственных препаратов могут быть использованы любые фармацевтически приемлемые добавки при условии, что они не оказывают неблагоприятного воздействия на функцию активного соединения. Стероидные соединения настоящего изобретения могут быть также введены в имплантат, вагинальное кольцо, пластырь, гель и в любые другие препараты пролонгированного действия.
Подходящими носителями, которые могут быть введены в композиции настоящего изобретения, являются лактоза, крахмал, производные целлюлозы и т.п., либо их смеси, взятые в соответствующих количествах.
Кроме того, настоящее изобретение относится к использованию стероидных соединений настоящего изобретения для изготовления лекарственных препаратов, обладающих активностью, направленной на устранение или снижение расстройств, связанных с пери- и/или постменопаузой, а особенно антиостеопорозной активностью. Таким образом, препараты настоящего изобретения имеют показания при пери- и/или постменопаузе (при климактерическом синдроме) и остеопорозе, т. е. они могут быть использованы для лечения методом заместительной гормональной терапии (HRT), предусматривающей введение пациенту (женщине) вышеуказанного соединения (в подходящей фармацевтической форме).
Кроме того, настоящее изобретение относится к применению стероидного соединения настоящего изобретения для получения препарата, обладающего контрацептивным (противозачаточным) действием. Таким образом, настоящее изобретение может быть использовано для контрацепции, т.е. в способе предохранения от беременности, предусматривающем введение женщине или самке животного вышеописанного соединения (в подходящей фармацевтической форме).
И наконец, настоящее изобретение относится к использованию стероидного соединения для получения лекарственного препарата, обладающего избирательной эстрогенной активностью, например препарата, который может быть использован в HRT (заместительной гормональной терапии).
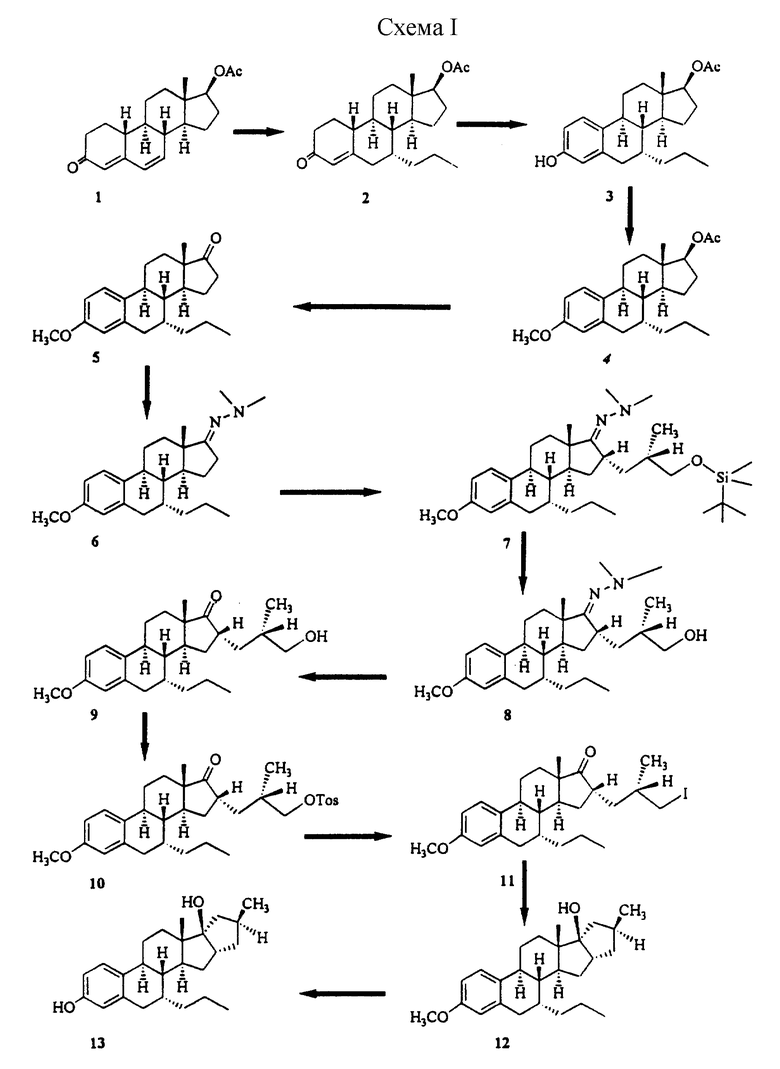
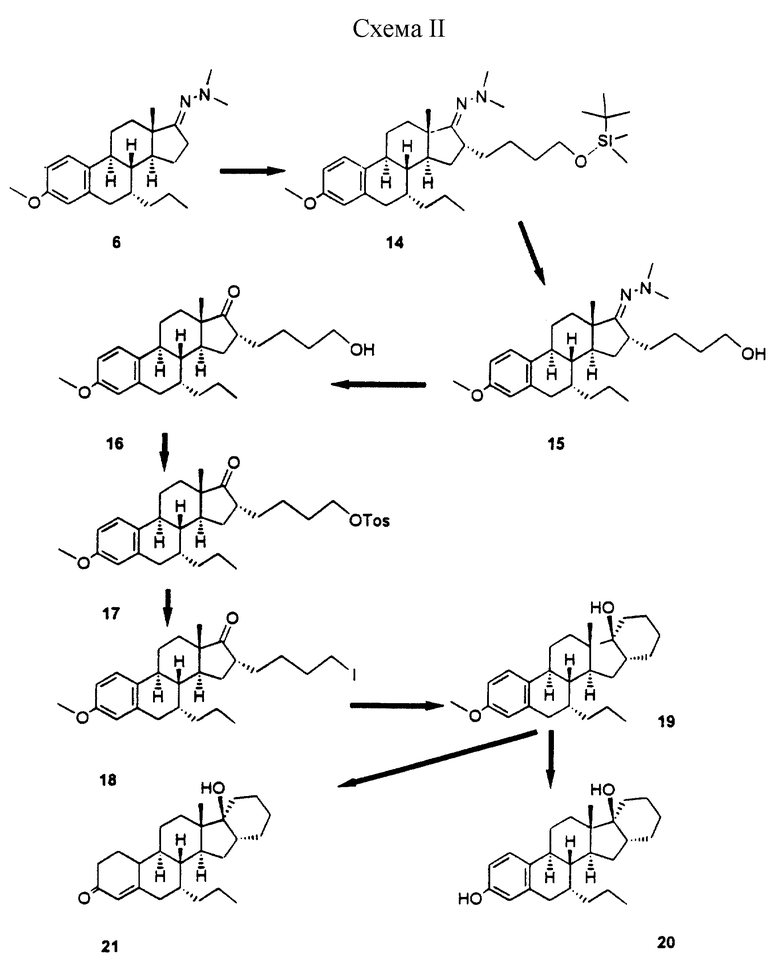
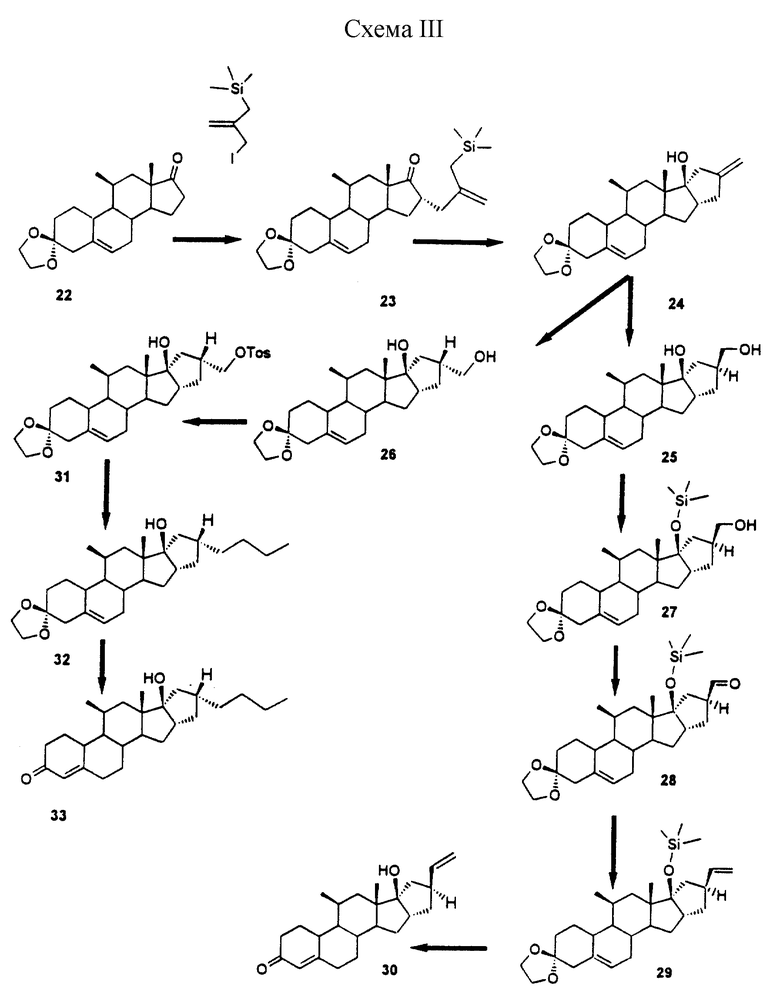
Синтез 16α,17α-аннелированных стероидов осуществляют сначала путем присоединения подходящим образом функционализированного С3- или С4-фрагмента к C16 α-положению стероида (для образования 5-членного или 6-членного колец соответственно). Для облегчения этого процесса функциональную 17-кетогруппу сначала превращают в диметилгидразон, который снова отщепляют после образования нужных функциональных групп боковой цепи. Замыкание кольца может быть осуществлено в основном методами, используемыми для образования металлоорганических соединений, такими, как обработка ω-иодалкиловых производных металлами переходного ряда такими, как самарий (в случае 5-членных колец, описанных в примере 1), либо путем образования литийорганических производных с использованием таких реактивов, как трет-бутиллитий (образование 6-членных колец описано в примере II). В альтернативном случае образование вышеупомянутых колец может быть осуществлено посредством продуцирования анионов путем отщепления кремниевых групп в ω-силиловых боковых цепях в присутствии фторида, как описано в примере III.
ω-Ацетилены могут с успехом служить в качестве субстратов при реакциях замыкания кольца в анионопосредованных реакциях, осуществляемых с использованием таких элементов, как натрий или литий, как описано в примере IV.
Совершенно иной способ предусматривает образование аннелированных колец путем олефинового обмена с использованием катализаторов на основе переходных металлов, таких, как рутений, молибден или вольфрам. Для этого в качестве субстратов служат 16α,17α- диалкенилированные стероиды. Они могут быть легко получены путем алкилирования стероидальных кетонов в положение С-16, с последующим введением алкенового фрагмента с помощью металлоорганических анионных производных (литиатов и т.п.). Такая реакция образования 5- и 6-членных колец была проиллюстрирована в примере V.
Таким образом, помимо получения вышеуказанных соединений и различных применений этих соединений, настоящее изобретение также относится к способам получения 16,17-аннелированных стероидов путем продуцирования кольца, присоединенного к стероидному каркасу, где указанное кольцо включает атомы углерода 16 и 17 указанного каркаса. Эти способы, которые не применяются в стероидной химии, позволяют получить широкий спектр 16,17-аннелированных стероидов. Так, например, в патенте DE 19709870 (ранее не опубликованном) описан способ, который ограничен в отношении синтеза конкретных соединений. Этот способ предусматривает осуществление реакции [4+2] циклоприсоединения бутадиена или диметилбутадиена с сильно активированной двойной связью у C16-17. Это означает, что у C17 всегда должен присутствовать сильный электроноакцепторный заместитель, например, такой заместитель, как -CN или -ацил, что в значительной степени ограничивает возможности этого способа. Кроме того, этот способ позволяет получать лишь 6-членные кольца, и ограниченное число вариантов этих соединений, а также требует симметричной бутадиеновой структуры, поскольку этот способ не является региоселективным. Способы настоящего изобретения не имеют указанных ограничений, и позволяют осуществлять стереоселективный и региоселективный синтез широкого ряда 5- и 6-кольцевых 16,17-аннелированных стероидов, описанных выше. Таким образом, эти способы вносят значительный вклад в область стероидной химии.
Настоящее изобретение проиллюстрировано схемами и примерами, которые не должны рассматриваться как ограничение данного изобретения рассмотренными конкретными вариантами.
Схемы:
Схема 1: схематическая иллюстрация (2-13) способа синтеза двух стероидных соединений (12 и 13) настоящего изобретения, описанного в примере 1.
Схема 2: схематическая иллюстрация (14-21) способа синтеза трех стероидных соединений (19, 20 и 21) настоящего изобретения, описанного в примере II.
Схема 3: схематическая иллюстрация (22-33) способа синтеза двух стероидных соединений (30 и 33) настоящего изобретения, описанного в примере III.
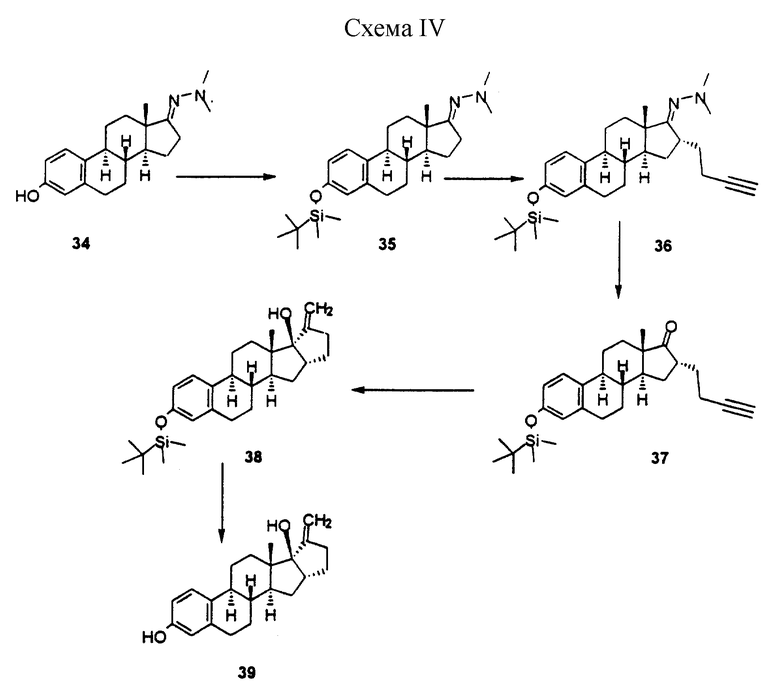
Схема 4: схематическая иллюстрация (34-39) способа синтеза стероидного соединения (39) настоящего изобретения, описанного в примере IV.
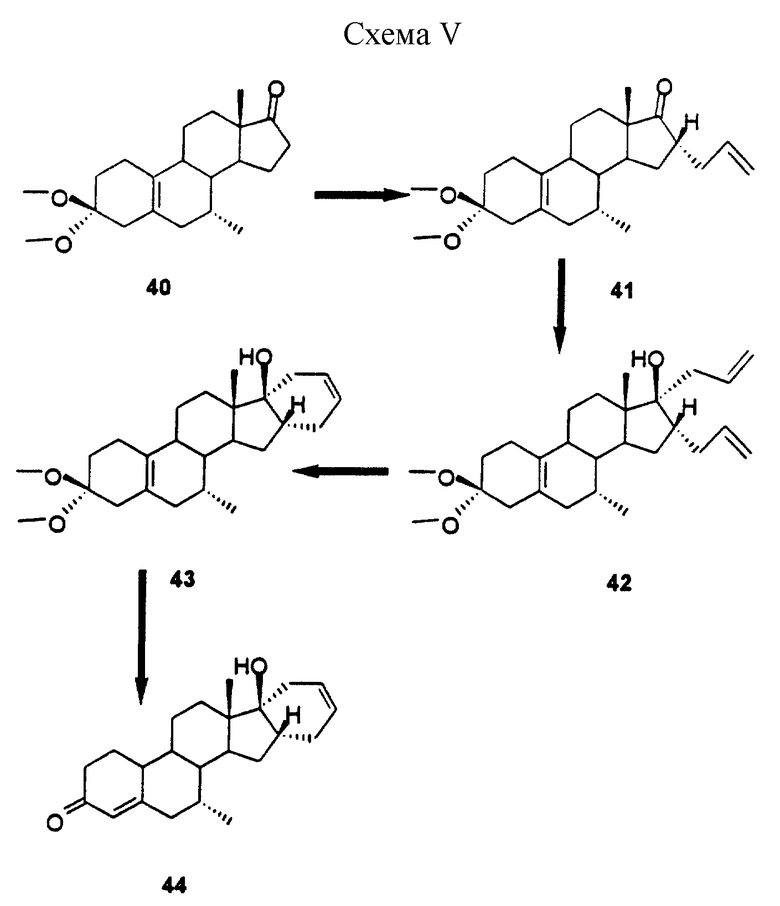
Схема 5: схематическая иллюстрация (40-44) способа синтеза стероидного соединения (44) настоящего изобретения, описанного в примере V.
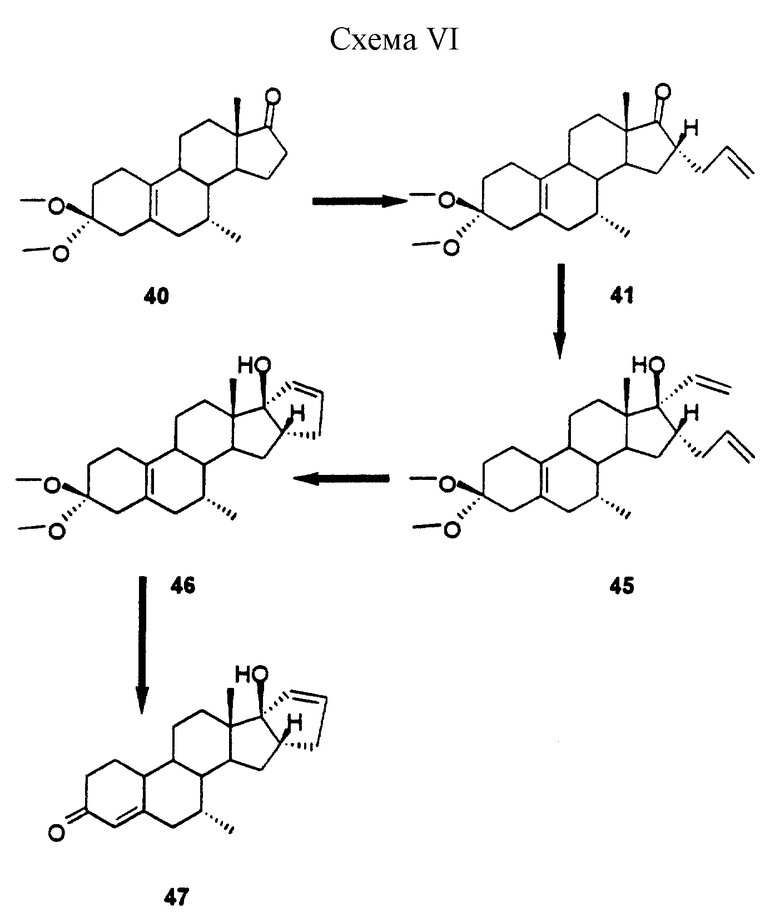
Схема 6: схематическая иллюстрация (40-47) способа синтеза стероидного соединения (47) настоящего изобретения, описанного в примере VI.
Число, указанное в скобках, относится к соответствующей структурной формуле соединений, представленных в схеме.
Пример 1
Хотя требуемый субстрат 1 может быть легко синтезирован путем дегидрирования стероидов у С6С7 методами, описанными в литературе (например, с использованием хлоранила или DDQ), однако, был разработан новый метод, позволяющий использовать различные 17-альфа-этинил,17-бета-гидроксистероиды, а также субстраты, в целях получения соответствующих 17-кетостероидов. Эти соединения могут быть деэтинилированы путем обработки карбонатом меди, осажденным на целите. Хотя аналогичное превращение с использованием карбоната серебра было описано в литературе, однако, метод, рассматриваемый в настоящем изобретении, имеет то преимущество, что в нем используется значительно более дешевый реактив. Партию СuСО3 на целите получали следующим образом. 100 г целита очищали путем перемешивания в смеси 500 мл метанола и 100 мл 6 н. НСl в течение 15 мин. Полученную смесь фильтровали и несколько раз промывали водой до тех пор, пока она не становилась нейтральной. Полученный таким образом продукт суспендировали в растворе 60 г Сu(NО3)2•3Н20 в 400 мл воды. К этому раствору по каплям при интенсивном перемешивании добавляли раствор 30 г Na2CO3•H2O в 200 мл воды. После перемешивания в течение еще 15 мин вещество фильтровали и промывали водой (для удаления основного количества воды перед сушкой полученное вещество суспендировали в ацетоне и фильтровали, а затем промывали пентаном). Конечную сушку проводили в вакууме при 80oС в течение ночи и получали 160 г реагента.
4 г (17β)-17-Гидроксипрегна-4,6-диен-20-ин-3-она и 20 г СuСО3-целита суспендировали в 100 мл толуола. Полученную смесь нагревали с обратным холодильником приблизительно в течение 6 ч с использованием ловушки Дина-Старка для удаления остаточного количества воды. За ходом реакции следили с помощью тонкослойной хроматографии (ТСХ). После завершения реакции реакционную смесь фильтровали через целит. Затем фильтрат концентрировали, а остаток обрабатывали изопропиловым эфиром/гексаном и получали 2,4 г прегна-4,6-диен-20-ин-3,17-диона, т. пл. 182-184oС. Полученное соединение подвергали реакции восстановления с боргидридом натрия и получали нужный 17-β-спирт, который после реакции ацетилирования с уксусным ангидридом давал нужный субстрат 1.
(7-альфа,17-бета)-17-(ацетилокси)-7-пропилэстр-4-ен-3-он (2)
Раствор пропиллития (полученный из 1,4 г лития и 9 мл пропилбромида в 60 мл эфира при -20oC) добавляли при температуре -40oС к 7,6 г CuI в 60 мл безводного ТГФ. После перемешивания в течение 0,5 ч к раствору по каплям при температуре -40oС добавляли раствор 5,2 г (17-бета)-17-(ацетилокси)эстра-4,6-диен-3-она (1) в 20 мл ТГФ. После еще 15-минутного перемешивания реакция была завершена и полученную смесь выливали в 300 мл насыщенного раствора NH4C1, а затем экстрагировали этилацетатом. Органическое вещество, выделенное после промывки, сушки и выпаривания растворителя, растворяли в 30 мл ТГФ, и перемешивали в присутствии 3 мл 6 н. H2SO4 для изомеризации определенного Δ5,6-изомера в Δ4,5-изомер. Через один час смесь нейтрализовали насыщенным раствором NаНСО3, а затем экстрагировали этилацетатом. После хроматографии неочищенного продукта на силикагеле (гептан/этилацетат = 8/2) получали 2,1 г соединения 2, т. пл. 97-100oC.
(7-альфа,17-бета)-7-пропилэстра-1,3,5(10)-триен-3,17-диол-17-ацетат (3)
К раствору 15 г соединения 2 в 300 мл ацетонитрила добавляли 12 г СuВr2. Полученную смесь перемешивали в течение 20 ч и ход реакции прослеживали с помощью ТСХ (ТСХ-пластины закупали у Merck A.G. Germany). Затем реакционную смесь выливали в воду и экстрагировали этилацетатом. Неочищенный продукт хроматографировали на короткой колонке с силикагелем (элюент: гептан/этилацетат = 4/1) и получали 13,5 г соединения 3 в виде белого аморфного вещества. Rf 0,57 (гептан/этилацетат = 7/3).
(7-альфа, 17-бета)-3-метокси-7-пропилэстра-1,3,5(10)-триен-17-олацетат (4)
К раствору 13,5 г соединения 3 в 60 мл ДМФ порциями добавляли 2,4 г NaH (60% дисперсия в минеральном масле). После перемешивания в течение 1 ч выделение водорода уменьшалось. Затем добавляли по каплям 3 мл метилиодида. После перемешивания в течение одного часа при комнатной температуре реакционную смесь выливали в 300 мл воды и полученный продукт экстрагировали этилацетатом. Остаток, образовавшийся в результате испарения летучих веществ, растворяли в 20 мл ТГФ, а затем вводили раствор 4 г NaOH в 80 мл СН3ОН. После перемешивания в течение одного часа реакция омыления была завершена. Реакционную смесь нейтрализовали путем добавления 1н. серной кислоты, и продукт экстрагировали этилацетатом с получением 11,5 г соединения 4, Rf 0,34 (гептан/этилацетат = 7/3).
(7-альфа)-3-метокси-7-пропилэстра-1,3,5(10)-триен-17-он (5)
К раствору 10,4 г 3-О-метил-7-α-пропилэстрадиола 4 в 50 мл метиленхлорида последовательно добавляли 15 г измельченного в порошок ацетата натрия, 30 г силикагеля, и 32 г хлорхромата пиридиния. После перемешивания в течение одного часа реакция окисления была завершена. Избыток реагента разлагали путем добавления 1 мл изопропанола и через 10 мин 150 мл гексана. Все осадки фильтровали через целит и фильтрат концентрировали досуха. Таким образом было получено 9,6 г в основном чистого кетона 5; Rf 0,54 (гептан/этилацетат = 7/3)
(7-альфа)-3-метокси-7-пропилэстра-1,3,5(10)-триен-17-она диметилгидразон (6)
К раствору 11,2 г 7α-пропил-3-0-метилэстрона 5 в 60 мл толуола добавляли 6 мл диметилгидразина и 0,5 мл трифторуксусной кислоты. Полученную смесь нагревали с обратным холодильником в течение 1,5 ч. После охлаждения до комнатной температуры реакционную смесь нейтрализовали 5%-ным NaHCO3, а органический слой несколько раз промывали водой и осушали сульфатом натрия. После концентрирования остаток хроматографировали и получали 11,4 г гидразона 6 в виде маслянистого продукта; Rf 0,30 (гептан/этилацетат = 7/3).
[7-альфа, 16-альфа(S)]3-16-[3-[[диметил(1,1-диметилэтил)-силил]окси]-2-метилпропил] -3-метокси-7-пропилэстр-1,3,5(10)-триен-17-она диметилгидразон (7)
К раствору 2,6 г соединения 6 в 30 мл сухого ТГФ добавляли при температуре -40oС 5,6 мл BuLi (1,5 н. раствор в гексане). После перемешивания в течение получаса при этой температуре добавляли 2,7 г (2R)-2-метил-3-иодпропанол-О-трет-бутилдиметилсилилового эфира (TBDMS) в 5 мл ТГФ. После перемешивания в течение еще одного часа при -20oС реакционную смесь выливали в воду и экстрагировали. В результате хроматографии было получено 4,6 г соединения 7; Rf 0,50 (гептан/этилацетат =7/3).
[7-альфа, 16-альфа(S)] -16-(3-гидрокси-2-метилпропил)-3-метокси-7-пропилэстра-1,3,5(10)-триен-17-она диметилгидразон (8)
Раствор 4,6 г соединения 7 в 5 мл ТГФ обрабатывали 15 мл 1М TBAF в ТГФ в течение одного часа при 50oС. Полученную смесь разбавляли 100 мл воды и экстрагировали этилацетатом. После пропускания продукта через короткую колонку с силикагелем получали 3,1 г соединения 8 в виде маслянистого продукта, Rf 0,18 (гептан/этилацетат = 7/3).
[7-альфа,16-альфа(S)]-16-[2-метил-3-[[(4-метилфенил)сульфонил] окси]пропил] -7-пропилэстра-1,3,5(10)-триен-17-он(10)Раствор 2,8 г соединения 9 в 1 мл пиридина при 0oС обрабатывали тозилхлоридом (2,6 г). После перемешивания в течение 2 ч, избыток реагента разлагали при перемешивании со льдом в течение получаса. Полученный продукт экстрагировали этилацетатом и очищали посредством хроматографии, в результате чего получали 3,2 г соединения 10 в виде бесцветного маслянистого вещества; Rf 0,35 (гептан/этилацетат = 7/3).
[7-альфа, 16-альфа(S)] -16-(3-гидрокси-2-метилпропил)-3-метокси-7-пропилэстра-1,3,5(10)-триен-17-он (9)
Смесь 3,1 г соединения 8 в 30 мл ацетона и 3 мл воды обрабатывали 3 г кислотной смолы Amberlist 15 (Fluka A.G.) в течение 2 ч при 55oС. Затем реакционную смесь фильтровали, концентрировали и получали 2,8 г соединения 9 в виде маслянистого вещества; Rf 0,75 (гептан/ацетон = 1/1).
[7-альфа,16-альфа(S)]-16-(3-иод-2-метилпропил)-7-пропилэстра-1,3,5(10)-триен-17-он (11)
Смесь 3,2 г соединения 10 и 10 г иодида натрия в 30 мл ацетона нагревали в течение 1 ч при 65oС. Затем реакционную смесь выливали в воду, экстрагировали этилацетатом и получали 2,9 г иодида 11; Rf 0,55 (гептан/этилацетат = 7/3).
(4'S, 7-альфа, 16-бета, 17-бета)-3',4',5',16-тетрагидро-3-метокси-4'-метил-7-пропил-17Н-циклопента[16,17]эстра-1,3,5(10)-триен-17-ол (12)
Раствор SmI2 получали из 3 г металлического самария и 4,7 г 1,2-дииодэтана в 70 мл безводного ТГФ. К этому раствору добавляли при 0oС 20 мг трис(дибензоил-метанато)железа, а затем раствор 2,8 г соединения 11 в 10 мл ТГФ. После перемешивания еще в течение 1 ч смесь выливали в воду, подкисляли 2 н. Н2S04 и экстрагировали эфиром.
Полученный таким образом неочищенный продукт хроматографировали для удаления 16,17-бета-изомера и получали 1,6 г соединения 12; Rf 0,32 (гептан/этилацетат = 7/3).
Родственный бета-изомер имеет Rf=0,37.
(4'S, 7-альфа, 16-бета, 17-бета)-3', 4',5',16-тетрагидро-4'-метил-7-пропил-17Н-циклопента[16,17]эстра-1,3,5 (10)-триен-3,17-диол (13)
К раствору 700 мг соединения 12 в 5 мл толуола добавляли 15 мл DIBAL (1M в толуоле). Смесь нагревали с обратным холодильником в течение 3 ч для отщепления эфира. Избыточный реагент разлагали путем добавления воды с последующим разбавлением 40 мл 2 н. НСl. Полученный продукт экстрагировали этилацетатом. После сушки и концентрирования остаток растирали с диизопропиловым эфиром и получали 460 мг кристаллического соединения 13; т. пл. 166-168oС; Rf 0,36 (гептан/этилацетат = 7/3).
Пример 11
[7-альфа, 16-альфа)-16-[4-[[диметил(1/1-диметилэтил)силил]окси] бутил]-3-метокси-7-пропилэстра-1,3,5(10)-триен-17-она диметилгидразон (14)
К раствору 3,9 г гидразона 6 в 45 мл безводного ТГФ добавляли при -60oС 8,5 мл 1,5 н. раствора бутиллития в гексане. После перемешивания в течение 0,5 ч по каплям добавляли раствор 4,2 г 4-иодбутанол-ТВDМS-эфира в 5 мл ТГФ. Полученную смесь перемешивали в течение 1 ч при -20oС, а затем выливали в воду (200 мл) и экстрагировали этилацетатом.
В результате очистки с помощью хроматографии на силикагеле получали 6,2 г соединения 14 в виде маслянистого вещества; Rf 0,52 (гептан/этилацетат = 7/3).
(7-альфа, 16-альфа)-16-(4-гидроксибутил)-3-метокси-7-пропилэстра-1,3,5(10)-триен-17-она диметилгидразон (15)
Раствор 6 г соединения 14 в 5 мл ТГФ обрабатывали 20 мл 1М фторида тетрабутиламмония в ТГФ в течение 2 ч. Реакционную смесь выливали в воду и экстрагировали этилацетатом. После хроматографии было получено 4,1 г соединения 15 в виде маслянистого продукта; Rf 0,17 (гептан/этилацетат = 7/3).
(7-альфа, 16-альфа)-16-(4-гидроксибутил)-3-метокси-7-пропилэстра-1,3,5(10)-триен-17-он (16)
Смесь, содержащую 4 г соединения 15, 40 мл ацетона, 4 мл воды и 4 г кислотной смолы Amberlist 15, перемешивали в течение 2 ч при температуре 50oС. Полученную смесь фильтровали, концентрировали, растворяли в 40 мл толуола, сушили и снова концентрировали, в результате чего получали 3,7 г в основном чистого соединения 16; Rf 0,61 (гептан/ацетон = 1/1); для исходного продукта Rf=0,65.
(7-альфа, 16-альфа)-16-[4-[[4-метилфенил)сульфонил] окси]бутил]-7-пропилэстра-1,3,5(10)-триен-17-он (17)
Смесь 3,7 г продукта 16 и 3,2 г тозилхлорида в 10 мл безводного пиридина перемешивали при 0-5oС в течение 3 ч. После разбавления водой продукт экстрагировали этилацетатом. После хроматографической очистки получали 4,6 г тозилата 17; Rf 0,45 (гептан/этилацетат = 7/3).
(7-альфа, 17-альфа)-16-(4-иодбутил)-3-метокси-7-пропил-эстра-1,3,5(10)-триен-17-он (18)
Смесь 4,6 г соединения 17 и 20 г иодида натрия в 50 мл ацетона нагревали при 60oС в течение 1,5 ч. Реакционную смесь концентрировали, разбавляли водой и экстрагировали толуолом. После сушки и концентрирования получали 4,4 г в основном чистого иодида 18; Rf 0,50 (гептан/этилацетат = 7/3).
(7-альфа, 16-альфа, 17-альфа)-3-метокси-7-пропил-16,24-цикло-19,21-динорхола-1,3,5(10)-триен-17-ол (19)
Раствор 3,8 г иодида 18 в 20 мл сухого ТГФ обрабатывали при -60oС 9 мл 1,7 М раствора трет-бутиллития в гептане. После перемешивания в течение 15 мин при -60oС, смесь выливали в воду и экстрагировали этилацетатом. Неочищенный продукт, полученный после удаления летучих веществ, растирали с гептаном, и получали 1,9 г в основном чистого продукта 19, т. пл. 161-162oС, Rf 0,40 (гептан/этилацетат = 7/3).
(7-альфа, 16-альфа, 17-альфа)-17-гидрокси-7-пропил-16,24-цикло-19,21-динорхол-4-ен-3-он (21)
К раствору 1 г лития в 90 мл жидкого аммиака добавляли при -33oС раствор 1,3 г продукта 19 в 30 мл сухого ТГФ. После перемешивания в кипящем аммиаке в течение 4 ч реакционную смесь обрабатывали 20 мл этанола, а затем аммиак выпаривали в постоянном потоке азота. Остаток разбавляли 50 мл воды и экстрагировали этилацетатом. После концентрирования органической фазы остаток растирали с гептаном и получали 1,1 г чистого промежуточного диенолового эфира, т. пл. 190-192oС.
Полученный продукт растворяли в 25 мл ТГФ и обрабатывали 5 мл 6 н. серной кислоты. После перемешивания в течение 6 ч, смесь нейтрализовали путем добавления Nа2СО3, и продукт экстрагировали этилацетатом. Неочищенный продукт подвергали хроматографической очистке и получали 610 мг соединения 21 в виде белой пены, Rf 0,25 (гептан/этилацетат = 7/3).
(7-альфа, 16-альфа, 17-альфа)-7-пропил-16,24-цикло-19,21-динорхола-1,3,5(10)-триен-3,17-диол (20)
К раствору 600 мг соединения 19 в 5 мл сухого толуола добавляли 12 мл 1М DIBAH (гидрид диизобутилалюминия) в толуоле. После нагревания с обратным холодильником в течение 2 ч реакция деметилирования была завершена; избыток реагента разлагали путем осторожного добавления воды, а затем смесь выливали в 50 мл 4 н. хлористоводородной кислоты и продукт экстрагировали этилацетатом. Органический слой сушили и концентрировали, после чего остаток обрабатывали диизопропиловым эфиром, и получали 310 мг продукта 20; т. пл. 240oС, Rf 0,20 (гептан/этилацетат = 7/3).
Пример III
(11-бета, 16-альфа)-11-метил-16-[2-[(триметилсилил)метил]проп-2-енил]-эстр-5-ен-3,17-диона 3-цикло(1,2-этандиил) ацеталь (23)
К раствору 12,7 мл гексаметилдисилазана в 50 мл ТГФ добавляли при -50oС 40 мл 1,5 М раствора BuLi в гептане. После перемешивания в течение 20 мин при -50oС медленно добавляли раствор 16,5 г соединения 22 в 100 мл ТГФ. После перемешивания в течение еще 0,5 ч вводили раствор 25 г 3-иод-2-триметилсилилметилпропена в 25 мл ТГФ. Реакционную смесь перемешивали в течение 3 ч при температуре -20oС, а затем выливали в 400 мл воды. Продукт экстрагировали этилацетатом и хроматографировали на силикагеле. После растирания с гептаном получали 12,5 г продукта 23;. т. пл. 184-185oС; Rf 0,55 (гептан/этилацетат = 7/3).
(11-бета, 16-бета,17-бета)-4',5',16,17-тетрагидро-17-гидрокси-11-метил-4'-метилен-3'Н-циклопента[16,17] эстра-5,16-диен-3-она 3-цикло(1,2-этандиилацеталь) (24)
Раствор 8,8 г продукта 23 в 200 мл сухого ТГФ обрабатывали 4 мл 1М фторида тетрабутиламмония (TBAF) в ТГФ. Полученную смесь нагревали с обратным холодильником в течение 15 мин до полного завершения реакции замыкания кольца. После добавления еще 15 мл 1М раствора TBAF нагревание с обратным холодильником продолжали еще 1 ч для отщепления 17-О-силилового эфира, образовавшегося в результате реакции. Затем полученную смесь концентрировали до небольшого объема и разбавляли водой, после чего экстрагировали этилацетатом. После хроматографической очистки получали 4,0 г продукта 24.: т. пл. 141-142oС, Rf 0,28 (гептан/этилацетат = 7/3).
(4'S, 11-бета, 16-бета, 17-бета)-4'5', 16,17-тетрагидро-17-гидрокси-4'-(гидроксиметил)-11-метил-3'Н-циклопента [16,17] -эстра-5,16-диен-3-она 3-цикло(1,2-этандиил-ацеталь)(25) и его 4'R-аналог (26)
Раствор борабициклононана (9-BBN) получали из 3 мл 10 М борандиметилсульфидного комплекса и 4 мл 1,5-циклооктадиена в 30 мл сухого ТГФ. К этому раствору добавляли 3,8 г соединения 24 в 10 мл ТГФ.
Полученную смесь перемешивали в течение 2 ч, а затем избыток реагента разлагали путем осторожного добавления 1 мл этанола с последующим добавлением 20 мл 2 н. раствора NaOH и 10 мл 30%-ного Н2О2. Эту смесь перемешивали еще 3 ч, а затем разбавляли водой и экстрагировали этилацетатом.
Неочищенный продукт хроматографировали на силикагеле (элюент: толуол/ацетон) и получали 2,1 г продукта 25 (т. пл. 178oС, Rf 0,47 (толуол/ацетон = 1/1) и 1,2 г продукта 26 (Rf 0,55, толуол/ацетон = 1/1)).
(4R', 11-бета, 16-бета,17-бета)-4'5',16,17-тетрагидро-17-гидрокси-11-метил-4-[[[(4-метилфенил)сульфонил] окси] метил] -3'Н-циклопента[16,17]эстра-5,16-диен-3-он-3-цикло(1,2-этандиилацеталь) (31)
Раствор 1,2 г продукта 26 и 0,8 г тозилхлорида в 5 мл пиридина перемешивали в течение 2 ч при 0-5oС. Затем смесь разбавляли водой со льдом, перемешивали в течение 15 мин и экстрагировали этилацетатом. Органическую фазу сушили и концентрировали с получением 1,6 г в основном чистого соединения 31; Rf 0,52 (толуол/этилацетат = 7/3).
(4'R, 11-бета, 16-бета, 17-бета)-4'-бутил-4'5'16,17-тетра-гидро-17-гидрокси-11-метил-3'Н-циклопента-[16,17]эстра-5,16-диен-3-он (32)
Купратный реактив получали путем добавления 12 мл 2М раствора бромида пропилмагния/эфира к 2,3 г CuI в 20 мл ТГФ при температуре -20oС. После перемешивания в течение 15 мин добавляли раствор 600 мг соединения 31 в 3 мл ТГФ. Перемешивание продолжали еще 2 ч при -20oС. К реакционной смеси добавляли 60 мл насыщенного NH4Cl и 10 мл 10%-ного аммиака, а затем смесь экстрагировали этилацетатом. Неочищенный продукт хроматографировали и получали 420 мг продукта 32; т. пл. 97-98oС. Rf 0,45 (гексан/этилацетат = 7/3).
(4'R, 11-бета, 16-бета,17-бета)-4'-бутил-4'5'16,17-тетрагидро-17-гидрокси-11-метил-3'Н-циклопента[16,17] эстра-4,16-диен-3-он (33)
Раствор 400 мг продукта 32 в 5 мл ацетона обрабатывали 2 мл 4 н. Н2S04. После выдерживания в течение 2 ч при комнатной температуре смесь разбавляли водой и экстрагировали этилацетатом. После хроматографической очистки получали 360 мг в основном чистого соединения 33 в виде аморфного продукта; Rf 0,27 (гептан/этилацетат = 7/3).
(4'S,11-бета,16-бета,17-бета)-4'5',16,17-тетрагидро-4'- (гидроксиметил)-11-метил-17- [(триметилсилил)окси] -3'Н-циклолента[16,17]-эстра-5,16-диен-3-она 3-цикло(1,2-этандиилацеталь) (27)
Реакцию защиты функциональной группы 17-ОН проводили в несколько стадий. Сначала первичный спирт ацетилировали. Для этого к раствору 750 мг соединения 25 в 2 мл пиридина добавляли 5 мг 4-диметиламинопиридина (DMAP), а затем 0,5 мл уксусного ангидрида. После перемешивания в течение 1 ч добавляли 10 г воды со льдом и полученный продукт экстрагировали этилацетатом. Органическое вещество концентрировали и остаток обрабатывали гептаном/диизопропиловым эфиром с получением 730 мг моноацетата, т. пл. 112oС.
Полученное вещество растворяли в 3 мл ДМФ, содержащего 200 мг имидазола. После добавления 240 мкл триметилсилилхлорида смесь перемешивали в течение 0,5 ч при комнатной температуре. После добавления 15 мл воды продукт экстрагировали эфиром. В результате сушки и концентрирования получали 900 мг в основном чистого производного силилового эфира; Rf 0,54 (гептан/этилацетат = 7/3). Этот продукт растворяли в 3 мл сухого ТГФ и добавляли 70 мг LiAlH4. После перемешивания в течение 10 мин смесь последовательно обрабатывали 0,3 мл воды и 0,1 мл 2 н. NaOH и 1 г NaS04. Остаток фильтровали через целит и концентрировали с получением 700 мг соединения 27 в виде аморфного вещества; Rf 0,29 (гептан/этилацетат = 7/3).
(4'S, 11-бета,16-бета,17-бета)-3,3-[1,2-этандиилбис(окси)]-4',5',16,17-тетрагидро-11-метил-17-[триметилсилил)окси] -3'Н-циклопента[16,17] -эстра-5,16-диен-4'-карбоксальдегид (28)
К раствору 600 мг продукта 27 в 15 мл метиленхлорида добавляли 1,5 г безводного ацетата натрия, 2,5 г силикагеля и 2 г хлорхромата пиридиния. Полученную смесь перемешивали в течение одного часа при комнатной температуре. После добавления 50 мл эфира и дополнительного перемешивания в течение 15 мин, реакционную смесь фильтровали через целит, а затем летучие вещества выпаривали и получали 420 мг в основном чистого карбоксальдегида 28; это вещество медленно отверждается при отстаивании; Rf 0,48 (гептан/этилацетат = 7/3).
(4'S, 11-бета, 16-бета, 17-бета)-4'-этенил-4',5',16,17-тетрагидро-11-метил-17-[(триметилсилил)окси] -3'Н-циклопента[16,17]эстра-5,16-диен-3-она 3-цикло(1,2-этан-диилацеталь) (29)
К 1,3 г хлорида метилтрифенилфосфония в 25 мл ТГФ добавляли 1,7 мл 1,5 М раствора BuLi в гексане при -40oС. После 30-минутного перемешивания добавляли 400 мг продукта 28 в 2 мл ТГФ. Полученную смесь нагревали до комнатной температуры в течение около получаса, а затем гасили путем выливания в 100 мл воды. Продукт экстрагировали диэтиловым эфиром и хроматографировали с получением 280 мг продукта 29 в виде маслянистого вещества; Rf 0,53 (гептан/этилацетат = 7/3); для исходного продукта Rf=0,23.
(4'S, 11-бета, 16-бета, 17-бета)-4'-этенил-4'5', 16,17-тетрагидро-11-метил-17-гидрокси-3'Н-циклопента[16,17]эстра-4,16-диен-3-он (30)
Раствор 260 мг продукта 29 в смеси 3 мл ТГФ и 3 мл 4 н. серной кислоты перемешивали в течение 2 ч при 45oС. Затем реакционную смесь нейтрализовали 5%-ным раствором NaHCO3 и продукт экстрагировали этилацетатом.
Остаток хроматографировали на короткой колонке с силикагелем и получали 150 мг соединения 3Q; Rf 0,25 (гептан/этилацетат = 7/3).
Пример IV
3-[[1,1-диметилэтил)диметилсилил] окси] эстра-1,3,5(10)-триен-17-она диметилгидразон (35)
К раствору 15,5 г диметилгидразона 3-гидроксиэстра-1,3,5 (10)-триен-17-она (34) в 200 мл ДМФ добавляли 13 г имидазола, а затем по каплям добавляли 15 г TBDMSC1 в 20 мл эфира. После перемешивания в течение еще 16 ч реакционную смесь выливали в 2 л воды и полученную смесь перемешивали еще 10 мин. Осадок фильтровали и сушили в вакууме с получением 20 г продукта 35, т. пл. 100-103oС.
(16-альфа)-3-[[1,1-диметилэтил)диметилсилил] окси] -16-(4-бутинил) эстра-1,3,5(10)-триен-17-она диметилгидразон (36)
Реакцию алкилирования стероида проводили с использованием аниона, образованного от 4-бром-1-бутина. Данный метод заключается в следующем. Раствор 11,9 г соединения 35 в 100 мл ТГФ обрабатывали при -20oС 20 мл 1,5 М раствора BuLi в гексане. После перемешивания в течение 1 ч при температуре -20oС реакционную смесь охлаждали до -70oС. К этой смеси по каплям добавляли холодный раствор аниона 4-бром-1-бутина (полученного путем добавления 36 мл BuLi к 7,7 г 4-бром-1-бутина в 50 мл ТГФ при -78oС), и реакционную смесь нагревали до комнатной температуры. Затем реакционную смесь перемешивали еще 1 ч и выливали в 300 мл 10%-ного водного раствора NH4Cl. Полученный продукт экстрагировали этилацетатом. После хроматографии получали 9,5 г соединения 36 в виде маслянистого вещества. Rf 0,85 (толуол/этилацетат = 6/4).
(16-альфа)-3-[[1,1-диметилэтил)диметилсилил] окси]-16-(4-бутинил)эстра-1,3,5(10)-триен-17-он (37)
К раствору 9 г соединения 36 в 100 мл ТГФ и 70 мл 1М ацетатного буфера (рН 4,5) добавляли 15 г периодной кислоты в 40 мл этанола. Полученную смесь перемешивали 24 ч. После добавления 500 мл воды продукт экстрагировали этилацетатом. Неочищенный продукт хроматографировали и получали 4,2 г соединения 37.
(16-альфа,17-альфа)-3-[[(1,1-диметилэтил)диметилсилил]окси]-16,23-цикла-19,24-динорхола-1,3,5(10),20-тетраен-17-ол (38)
Раствор нафталенида лития получали из 3,4 г нафталина и 150 мг литиевой стружки в 30 мл безводного ТГФ. Этот раствор по каплям добавляли к раствору 560 мг соединения 37 в 5 мл ТГФ до тех пор, пока не исчезал темно-зеленый цвет реакционной смеси. После перемешивания в течение еще 10 мин реакционную смесь
выливали в 30 мл NH4Cl и продукт экстрагировали этилацетатом.
После хроматографической очистки получали 150 мг кристаллического продукта 38.
(16-альфа, 17-альфа)-16,23-цикло-19,24-динорхола-1,3,5(10), 20-тетраен-3,17-диол (39)
Раствор 130 мг продукта 35 в 5 мл 5%-ной хлористо-водородной кислоты в метаноле перемешивали в течение 2 ч при комнатной температуре. Затем реакционную смесь обрабатывали 3 мл пиридина, концентрировали и разбавляли 10 мл воды. Продукт экстрагировали этилацетатом и очищали с помощью хроматографии, в результате чего получали 65 мг соединения 39, т. пл. 203-205oС.
Пример V
(7-альфа, 16-альфа)-7-метил-16-(проп-2-енил)-эстр-5(10)-ен-3,17-диона 3,3-диметилацеталь (41)
Раствор диизопропиламида лития получали из 16,6 мл 1,5 М бутиллития в гексане и 3,85 мл диизопропиламина в 35 мл ТГФ при -20oС. После перемешивания в течение 20 мин добавляли раствор 8,3 г стероида 40 в 30 мл ТГФ, и полученную смесь перемешивали в течение 20 мин при -20oС. После охлаждения до -40oС добавляли 2,2 мл аллилбромида, а затем перемешивание продолжали еще 4 ч при -20oС, после чего ТСХ указывала на завершение реакции. Реакционную смесь гасили путем добавления 200 мл 5%-ного раствора NaHCO3, а затем экстрагировали этилацетатом. В результате хроматографии на силикагеле (элюент: гексан/5% этилацетат) получали 1,2 г продукта 41 в виде белого твердого вещества, т. пл. 85-86oС.
(7-альфа, 16-альфа,17-бета)-7-метил-16,17-бис(проп-2-енил)-17-гидрокси-эстр-5(10)ен-3-она 3,3-диметилацеталь (42)
К раствору 15 мл 1 М бромида аллилмагния в 30 мл ТГФ добавляли при -40oС раствор 4,5 г соединения 41 в 30 мл ТГФ. После перемешивания в течение 30 мин при этой температуре смесь выливали в 250 мл 10%-ным раствора NH4Cl и экстрагировали этилацетатом. Полученный таким образом продукт хроматографировали и получали 3,2 г 16-альфа,17-альфа-диаллилового производного 42 в виде белого аморфного вещества.
(7-альфа, 16-альфа, 17-альфа)-7-метил-17-гидрокси-16,24-цикло-19,21-динорхола-5(10),22-диен-3-она 3,3-диметилацеталь (43)
К раствору 1,3 г соединения 42 в 30 мл метилендихлорида добавляли 200 мг дихлорида бис(трициклогексилфосфин)бензилиденрутения. Смесь перемешивали до полного завершения реакции. Растворитель частично удаляли путем концентрирования, и остаток продукта хроматографировали на колонке с силикагелем, в результате чего получали 1,1 г соединения 43 в виде белого аморфного вещества Rf 0,38 (гептан/этилацетат = 7/3, об/об).
(7-альфа, 16-альфа, 17-альфа)-7-метил-17-гидрокси-16,24-цикло-19,21-динорхола-4, 22-диен-3-он (44)
Раствор 1 г продукта 43 в 30 мл ацетона обрабатывали 5 мл 2 н. соляной кислоты. После перемешивания в течение 2 ч при комнатной температуре реакция была завершена. Смесь нейтрализовали 5%-ным раствором NаНСО3, экстрагировали этилацетатом и полученный продукт пропускали через короткую колонку с силикагелем. Полученный таким образом продукт обрабатывали диизопропиловым эфиром и получали 0,65 г соединения 44, т. пл. 130-131oС, Rf 0,14 (гептан/этилацетат = 7/3).
Пример VI
(7-альфа, 16-альфа, 17-альфа)-7-метил-16-(проп-2-енил)-17-гидрокси-прегна-5(10),20-диен-3-она 3,3-диметилацеталь (45)
Раствор виниллития получали путем добавления 0,8 мл 1,6М раствора бутиллития в гексане к 0,32 мл винилтрибутилолова в 3 мл ТГФ при -50oС. После перемешивания в течение 20 мин по каплям добавляли раствор 300 мг соединения 41 в 2 мл ТГФ. После еще 15-минного перемешивания, реакцию гасили путем добавления 20 мл 10%-ного раствора NH4Cl и продукт экстрагировали этилацетатом. После очистки посредством хроматографии получали 120 мг продукта 45 в виде аморфного вещества, Rf 0,56 (гептан/этилацетат = 7/3 об/об).
(7альфа, 16бета, 17бета)-16,17-дигидро-17-гидрокси-5'Н-циклопента[16,17] эстра-5(10),16-диен-3-она 3,3-диметил-ацеталь (46)
К раствору 120 мг соединения 45 в 4 мл метилендихлорида добавляли 30 мг дихлорида бис(три-циклогексилфосфин)бензилиденрутения. После перемешивания в течение 2 ч смесь концентрировали и фильтровали через колонку с силикагелем, в результате чего получали 80 мг соединения 46, Rf 0,40 (гептан/этилацетат = 7/3 об/об).
(7-альфа, 16-бета, 17-бета)-7-метил-16,17-дигидро-17-гидрокси-5'Н-циклопента[16,17]эстра-4,16-диен-3-он (47)
Раствор 80 мг продукта 46 в 2 мл ацетона обрабатывали 0,2 мл 2 н. НСl. После перемешивания в течение 2 ч при комнатной температуре реакционную смесь нейтрализовали путем добавления NаНСО3 и разбавляли водой. Продукт экстрагировали этилацетатом, пропускали через короткую колонку с двуокисью кремния и получали 45 мг соединения 47: т. пл. 175-176oС. Rf 0,49 (гептан/этилацетат = 1/1 об/об).
Пример VII
Тест на предупреждение индуцированного овариэктомией разрежения костей у крыс (тест на антиостеопорозную активность)
Введение
Удаление яичников (овариэктомия) у крыс приводит к разрежению кости (остеопорозу), обусловленному дефицитом эстрогена. Введение эстрогенных соединений позволяет предупреждать этот эффект. Этот тест проводили для оценки соединения на его антиостеопорозную активность у овариэктомизированных крыс. Влияние этого соединения на костную массу может быть определено путем измерения минеральной плотности губчатого костного вещества с помощью периферической количественной компьютерной томографии (pQCT).
Экспериментальные животные
Использовали предпочтительно зрелых неоплодотворенных самок крыс Wistar, весом 225-250 г. Штамм: Hsd/Cpd:Wu, SPF-скрещенных Harlan, СРВ, Zeist, The Netherlands.
Эксперимент
В день 1 эксперимента крыс взвешивали и распределяли по клеткам в соответствии с массой их тела. Крыса с самой низкой массой тела находилась в первой клетке, а крыса с самой высокой массой тела находилась в последней клетке. Обработку крыс каждого блока проводили в произвольном порядке. Блок (группа из 3 + n обработок) состоял из 1 крысы, у которой не были удалены яичники и которой вводили плацебо, 1 овариэктомизированной крысы, которой вводили плацебо; 1 овариэктомизированной крысы, которой вводили известное соединение; и 1 крысы, которую подвергали каждой из n обработок.
Ложную операцию (т.е. операцию, при которой не были удалены яичники) и операцию по овариэктомии проводили под анестезией. После восстановления животных от анестезии, через 24 ч животным в течение 4 недель один или два раза в день вводили соответственно наполнитель, известное соединение или испытуемое соединение.
Измерение минеральной плотности кости с помощью компьютерной томографии (pQCT)
Минеральная плотность (мг/см3) губчатого костного вещества в зоне метафиза бедренной кости измеряли с помощью pQCT (периферическое устройство для компьютерной томографии, ХСТ 960А, Stratec, Birkenfeld, Germany) на свежей ткани, взятой непосредственно после аутопсии. Были взяты два скана (360oС), которые имели благодаря рентгеновскому лучу стандартную толщину 1 мм. Эти сканы имели разрешение 0,148 х 0,148 мм. Один скан был взят при 5,5 мм от дистального конца бедренной кости, где была измерена минеральная плотность губчатого костного вещества в зоне метафиза. Другой скан был взят в зоне диафиза на расстоянии 13,5 мм от дистального конца, который не содержал губчатого костного вещества. При последнем сканировании определяли минеральную плотность кортикального слоя кости и геометрические параметры, такие, как толщина кортикального слоя, полная площадь поверхности кости, а также внешний и внутренний диаметр. Внутренние и внешние экспериментальные отклонения при измерении минеральной плотности губчатого костного вещества составляли около 2-3%. ХСТ-960А был прокалиброван с использованием стандартного гидроксиапатита в акриловом полимере.
Интерпретация результатов
Овариэтомия приводила к статистически значимому уменьшению минеральной плотности губчатого костного вещества (Р≤0,05, двухфакторный дисперсионный анализ ANOVA). Испытуемые соединения считались активными, если средние значения минеральной плотности губчатого костного вещества дистального отдела бедренной кости были статистически значимо более высокими, чем значения, полученные для овариэктомизированной контрольной группы.
Активная доза (ED50) означает дозу, при которой достигается среднепропорциональное расхождение в минеральной плотности губчатого костного вещества на 40-60% по сравнению с ложнооперированной и овариэктомизированной группами.
Библиография
- Wronski T.J. & Yen C.F. The ovariectomised rat as an animal model for postmenopausal bone loss. Cell and Materials, Supp., 1 (1991): 69-76.
- Yamasaki I. & Yamaguchi H. Characteristics of ovariectomised osteopenic rat model. J. Bone Min. Res. 4 (1989): 12-22.
Ederveen A.G.H., Spanjers C.P.M., Quaijtaal J.H.M. & Kloosterboer H.J.: Effect of treatment with tibolone (Org OD 14) or 17α-ethinyl estradiol on bone mass, bone turnover and biomechanical quality of cortical and trabecular bone in mature ovariectomised rats Osteoporosis Int. in press, 1998.
Пример VIII
Тест на связывание с рецептором in vitro
Относительные аффинности связывания соединений настоящего изобретения с рецептором прогестерона измеряли для цитоплазматических рецепторов прогестерона, присутствующих в клетках опухолей молочной железы человека (клетки MCF-7, время инкубирования 16 ч, температура 4oС) и сравнивали с аффинностью (16α)-16-этил-21-гидрокси-19-норпрегн-4-ен-3,20-диона (в соответствии с методикой, описанной Е. W. Bergink et al. , J. Steroid Biochem., Vol. 19, 1563-1570 (1983)). Относительную аффинность связывания с рецептором эстрадиола измеряли таким же способом, за исключением того, что в качестве стандартного соединения использовали 17β-эстрадиол.
Тест на эстрогенную активность in vivo
Эстрогенную активность in vivo определяли с помощью хорошо известного теста Allen Doisy, описанного в F. Allen, L. A. Doisy, J. Amer. Med. Assoc., 81, 819-821 (1923).
Тест на прогестагенную активность in vivo
Прогестагенную активность in vivo определяли с помощью хорошо известного теста McPhail, описанного в McPhail, M.K.: The assay of progestin. Journal of Physiology, 1934, 83: 145-156.
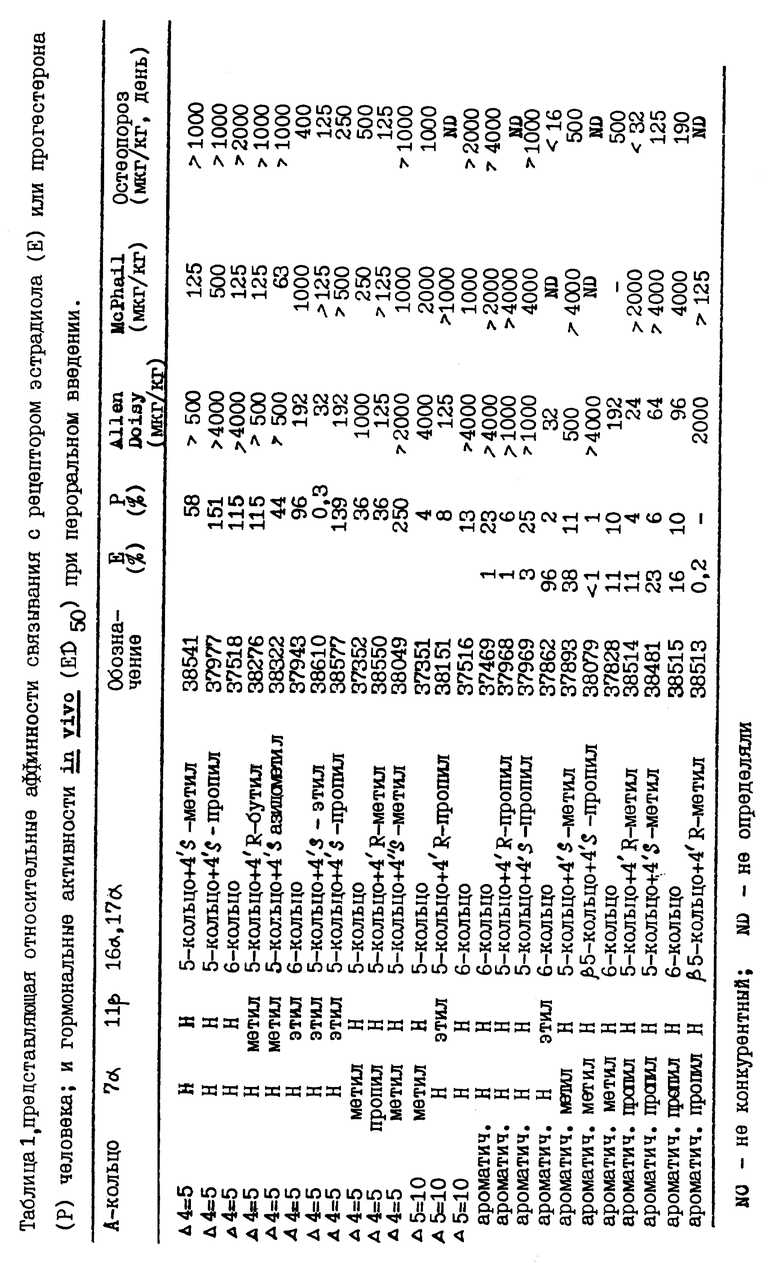
Несколько соединений примеров I-VI, а также другие соединения настоящего изобретения, синтезированные аналогичным способом, были подвергнуты испытаниям, описанным в примерах VII и VIII. Результаты этих испытаний представлены в таблице, где указаны тип А-кольца и замещения у атомов углерода 7, 11 и 17. В колонках, озаглавленных Е и Р, приводятся относительные аффинности связывания с рецепторами эстрогена и прогестерона; результаты, полученные для ED50 в тестах, проведенных методами Allen Doisy и McPhail, указаны в мкг/кг. В колонке, озаглавленной "Остеопороз", приводятся величины ED50, полученные в тесте на антиостеопорозную активность (доза в мкг/кг в день, как описано выше).
Следующие соединения были приготовлены и испытаны. При этом было найдено, что они проявляют эстрогенные и/или прогестагенные свойства:
Org 37351:
Т.пл. 145-146
Rf: (гепт/эт.сп.7/3) 0,25
ЯМР: δ 2,72 (м, СН2С4), 0,90 (с, 3, 18 метил), 0,81 (3, д, 7 СН3)
Org 37363:
Rf: (гепт/эт. спирт 1/1) 0,78
ЯМР: δ 3,80 (м, 1, СНОН), 0,89 (с, 3, 18СН3), 0,77 (д, 3, 7 СН3),
Org 38706: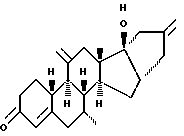
Rf:(тол./эт.сп. 7/3) 0,40
ЯМР: δ 5,88 (м, 1, Н4), 4,90 и 4,85 (м, 4Н'с, 2х метилен), 0,89 (с, 3, СН3)
Org 38707: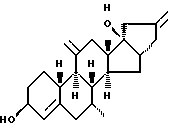
Т.пл.92-93
Rf: (СН2С12/ац 9/1) 0,49
ЯМР: δ 4,20 (м, 1, СНОН), 4,85 (м, 4Н'с, 2х метилен), 5,48 (м, 1, Н4), 0,86 (с, 3, СН3)
Org 38710: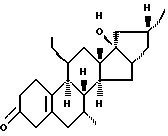
Т.пл. 145-147
Rf: (гепт/эт.сп. 7/3) 0,33
ЯМР: δ 2,78 (шс, 2, Н4), 1,02 (с, 3, СН3)
Org 38738: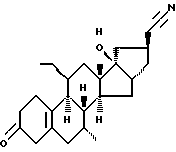
Т.пл.218-219
Rf: (гепт/эт. спирт 1/1) 0,53
ЯМР: δ 2,78 (шс, 2, Н4), 0,90 (т, 3, этил), 1,04 (с, 18-СН3)
Org 38739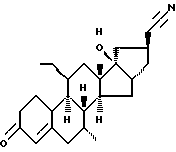
Rf: (гепт/эт. спирт 1/1) 0,40
ЯМР: δ 0,98 (т.3 этил), 1,03 (с, 3, 18-СН3), 5,86 (м, 1, Н4)
Org 38840: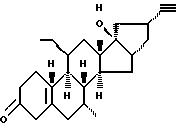
Т.пл. 130-132
Rf:(гепт/эт. спирт 6/4) 0,44
ЯМР: δ 0,98 (3, т, этил), 1,0 (с, 3, СН3), 4,17 (м, 1, CHOH), 5,85 (м, 1, Н4)
Org 38842: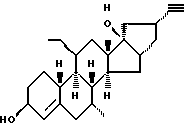
Т.пл.149-151
Rf: (гепт/эт. спирт 6/4) 0,39
ЯМР: δ 0,98 (c, 3, 18 СН3), 4,17 (м, 1, СНОН), 5,42 (м, 1, Н4)
Org 38843: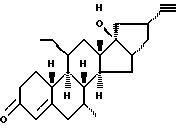
Rf: (гепт/эт. спирт 6/4) 0,48
ЯМР: δ 2,20 (д, 1, ацетилен), 1,07 (с, 3, 18 СН3), 5,85 (м, 1, Н4)
Org 38962: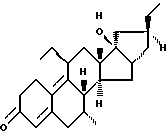
(D): -167 (c=1, СНС13)
ЯМР: δ 5,69 (м, 1, Н44), 1,12 (с, 3, СН3), 0,89 и 0,94 (2 х т, 6, 2 х С2Н5).
| название | год | авторы | номер документа |
|---|---|---|---|
| СТЕРОИДНЫЕ СОЕДИНЕНИЯ, СПОСОБ СЕЛЕКТИВНОЙ МОДИФИКАЦИИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2255090C2 |
| ПРОИЗВОДНОЕ 11-(ФЕНИЛЗАМЕЩЕННЫЙ)ЭСТРА-4,9-ДИЕНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2135514C1 |
| ПРОИЗВОДНЫЕ 16-ГИДРОКСИ-11-(ЗАМЕЩЕННЫЙ ФЕНИЛ)-ЭСТРА-4,9-ДИЕНА, СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2187510C2 |
| СТЕРОИД С 17-СПИРОМЕТИЛЕНЛАКТОНОВОЙ ГРУППОЙ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2168516C2 |
| НЕАРОМАТИЧЕСКИЙ ЭСТРОГЕННЫЙ СТЕРОИД, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2245886C2 |
| АКТИВНЫЙ КОМПОНЕНТ ЛЕКАРСТВЕННОГО ПРЕПАРАТА, СТЕРОИД, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2172740C2 |
| 3-МЕТИЛЕНСТЕРОИДНЫЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2248358C2 |
| ИНГИБИТОРЫ ПРОТЕАЗЫ СЕРИНА | 1997 |
|
RU2178419C2 |
| СОЕДИНЕНИЯ, НАБОР, АНДРОГЕННАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2242479C2 |
| ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ | 1997 |
|
RU2172321C2 |
Изобретение относится к стероидному соединению общей формулы I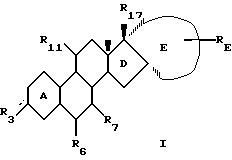
где  представляет = О, -ОН, ОR или -ООСR, где R представляет алкильную группу, имеющую от 1 до 6 атомов углерода; R6 представляет Н или -(СН2)mН, где m = 1 или 2; R7 представляет Н, С1-4-алкил, С2-4-алкенил или С2-4-алкинил; R11 представляет Н, С1-4-алкил, С2-4-алкенил, С2-4-алкинил; Е представляет, включая атомы углерода 16 и 17 кольца D, 4-7-членное углеводородное кольцо, где указанное кольцо находится в α-положении по отношению к D-кольцу, замещенному группой RE, и необязательно содержит одну эндоциклическую двойную связь; RE представляет Н, С1-5-алкил, С2-5-алкенил, С2-5-алкинил, С1-5-алкилиден, -(СН2)n-N3 или -(СН2)n-СN, где n = 1 или 2, и где алкильная группа может быть замещена -OR, -OOCR, где R является алкилом с 1-6 атомами углерода; R17 представляет -ОН, -ОR или -ООСR, где R представляет алкил с 1-6 атомами углерода, где указанное стероидное соединение может иметь, но необязательно, одну двойную связь, Δ5(10), Δ4(5), либо кольцо А может быть ароматическим. Изобретение также относится к фармацевтической композиции, содержащей указанное стероидное соединение. Стероидные соединения изобретения являются в высокой степени эффективными при использовании их в целях предупреждения или лечения нарушений, связанных с пери- и постменопаузой, а более предпочтительно в целях предупреждения или лечения остеопороза. Кроме того, стероидные соединения могут быть использованы в качестве контрацептивных средств. 4 с. и 6 з.п.ф-лы, 1 табл.
представляет = О, -ОН, ОR или -ООСR, где R представляет алкильную группу, имеющую от 1 до 6 атомов углерода; R6 представляет Н или -(СН2)mН, где m = 1 или 2; R7 представляет Н, С1-4-алкил, С2-4-алкенил или С2-4-алкинил; R11 представляет Н, С1-4-алкил, С2-4-алкенил, С2-4-алкинил; Е представляет, включая атомы углерода 16 и 17 кольца D, 4-7-членное углеводородное кольцо, где указанное кольцо находится в α-положении по отношению к D-кольцу, замещенному группой RE, и необязательно содержит одну эндоциклическую двойную связь; RE представляет Н, С1-5-алкил, С2-5-алкенил, С2-5-алкинил, С1-5-алкилиден, -(СН2)n-N3 или -(СН2)n-СN, где n = 1 или 2, и где алкильная группа может быть замещена -OR, -OOCR, где R является алкилом с 1-6 атомами углерода; R17 представляет -ОН, -ОR или -ООСR, где R представляет алкил с 1-6 атомами углерода, где указанное стероидное соединение может иметь, но необязательно, одну двойную связь, Δ5(10), Δ4(5), либо кольцо А может быть ароматическим. Изобретение также относится к фармацевтической композиции, содержащей указанное стероидное соединение. Стероидные соединения изобретения являются в высокой степени эффективными при использовании их в целях предупреждения или лечения нарушений, связанных с пери- и постменопаузой, а более предпочтительно в целях предупреждения или лечения остеопороза. Кроме того, стероидные соединения могут быть использованы в качестве контрацептивных средств. 4 с. и 6 з.п.ф-лы, 1 табл.
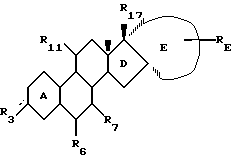
где  представляет = О, -ОН, ОR или -ООСR, где R представляет алкильную группу, имеющую от 1 до 6 атомов углерода;
представляет = О, -ОН, ОR или -ООСR, где R представляет алкильную группу, имеющую от 1 до 6 атомов углерода;
R6 представляет Н или -(СН2)mН, где m = 1 или 2;
R7 представляет Н, С1-4-алкил, С2-4-алкенил или С2-4-алкинил;
R11 представляет Н, С1-4-алкил, С2-4-алкенил, С2-4-алкинил;
Е представляет, включая атомы углерода 16 и 17 кольца D, 4-7-членное углеводородное кольцо, где указанное кольцо находится в α-положении по отношению к D-кольцу, замещенному группой RE, и необязательно содержит одну эндоциклическую двойную связь;
RE представляет Н, С1-5-алкил, С2-5-алкенил, С2-5-алкинил, С1-5-алкилиден, -(СН2)n-N3 или -(СН2)n-СN, где n = 1 или 2, и где алкильная группа может быть замещена -OR, -OOCR, где R является алкилом с 1-6 атомами углерода;
R17 представляет -ОН, -ОR или -ООСR, где R представляет алкил с 1-6 атомами углерода, где указанное стероидное соединение может иметь, но необязательно, одну двойную связь, Δ5(10), Δ4(5), либо кольцо А может быть ароматическим.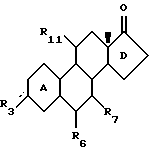
где замещающие группы определены в предыдущих пунктах, присоединяют у атома углерода в положении 16, т. е. смежного с 17-кеточастью, замещенную или незамещенную и соответствующим образом функционализированную алкильную цепь, так, что в результате этого получают ω-иодалкилную группу, и эту ω-иодалкильную группу подвергают реакции замыкания кольца путем обработки металлоорганическим реактивом.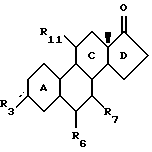
где замещающие группы определены в предыдущих пунктах, присоединяют у каждого из атомов углерода в положении 16 и 17 замещенную или незамещенную алкенильную цепь, и подвергают замыканию кольцо посредством олефиновой реакции обмена с использованием катализатора на основе переходного металла.
| ЕР 0411733, 1991 | |||
| ФИЗЕР Л., ФИЗЕР М | |||
| Стероиды.-М., 1964, с.462,555 | |||
| GENNARO ET AL "Remmington's Pharmacentical Sciences, MacK publishig Co., 1990, р.8. |

