Настоящее изобретение относится к применению стероидов для получения лекарственного препарата, предназначенного для лечения или предупреждения менопаузных заболеваний, особенно для лечения или предупреждения остеопороза.
Многие из стероидов настоящего изобретения, которые применяют для лечения менопаузных заболеваний, являются известными соединениями.
11β-Алкилстероиды известны, например, из патента США 3983144. Эти стероиды описаны как имеющие интересную противозачаточную активность. Другие 11β-алкилстероиды, обладающие анаболической, андрогенной и гестагенной активностью, описаны в патенте США 3325520. Родственные гестагенные соединения для регулирования менструации и овуляции известны также из родовой заявки Австралии AL 66114974, европейской родовой заявки 0145493 и патента США 3465010. Стероиды, имеющие ненасыщенные углеводородные группы в положении II стероидного скелета, известны из патента США 4292251, в котором указывается, что эти стероиды обладают гистеротропной активностью и ингибируют овуляцию, и из указанного выше европейского патента 0145493.
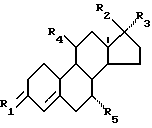
В данном патенте найдено, что соединения, имеющие общую формулу I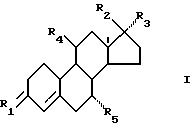
где R1 представляет собой O, (H, OH) или два атома водорода,
R2 представляет собой гидроксигруппу, возможно превращенную в группу простого или сложного эфира,
R3 представляет собой (2-6 C)-алкинил, возможно замещенный гидроксигруппой,
R4 представляет собой CN или одну из углеводородных групп, выбранных из (1-6 C)алкила, (1-6 C)-алкоксигруппы, (2-6 C)-алкенил, (2-6 C)-алкинила и (2-6 C)-алкилидена, причем каждая из этих углеводородных групп возможно замещена галогеном, гидрокси- или (1-6 C)-алкоксигруппой, и
R5 представляет собой водород или (1-6 C)-алкил,
можно применять для лечения или предупреждения послеменопаузных заболеваний, особенно остеопороза.
Одним из наиболее серьезных менопаузных нарушений является разрежение кости (остеопороз), которое характеристично поражает женщин. Целью настоящего изобретения является предоставление лекарственного средства, которое способно предотвращать разрежение кости и, возможно, повышать костную массу и еще лечить климактерические нарушения. Предпочтительно активные компоненты этих лекарственных препаратов обладают сильной эстрогенной и слабой (или совсем не обладают) андрогенной активностью. Предпочтительные лекарственные препараты обладают также благоприятными, способствующими кровотечению свойствами, не индуцируют пролиферацию эндометриоидной ткани и имеют благоприятное соотношение HDL/LDL (соотношение липидов высокой и низкой плотности).
В предпочтительном варианте осуществления изобретения соединения имеют общую формулу I, где R1 представляет собой O или два атома водорода, R2 представляет собой гидроксигруппу, R3 представляет собой этинил, R4 выбран из группы, состоящей из метила, (2-6 C)-алкинила, (2-6 C)-алкилидена и одного из (2-6 C)-алкила, (2-7 C)-алкоксиалкила, (1-6 C)-алкоксигруппы или (2-6 C)алкенила, каждый из которых может быть замещен галогеном, и R5 представляет собой водород или (1-6 C)-алкил.
Более предпочтительно применение стероидов, имеющих общую формулу I, где R1 представляет собой O, R2 представляет собой гидроксигруппу, R3 представляет собой этинил, R4 представляет собой этил, 2-фторэтил, этинил, (2-6 C)-алкенил, возможно замещенный фтором, или (2-6 C)-алкилиден, возможно замещенный фтором, и R5 представляет собой водород или метил. В предпочтительном варианте R5 представляет собой водород.
Еще более предпочтительно применение стероидов, имеющих формулу I, где R1 представляет собой O, R2 представляет собой гидроксигруппу, R3 представляет собой этинил, R4 представляет собой этил или этинил и R5 представляет собой водород.
Изобретение, кроме того, относится к новым стероидам, имеющим формулу I, где
R1 представляет собой O,
R2 представляет собой гидроксигруппу, возможно превращенную в группу простого или сложного эфира,
R3 представляет собой (2-6 C)-алкинил, возможно замещенный гидроксигруппой,
R4 представляет собой CN, (2-6 C)-алкил, возможно замещенный галогеном, или (2-6 C)-алкенил, замещенный галогеном, и
R5 представляет собой водород или (1-6 C)-алкил, или где
R1 представляет собой два атома водорода,
R2 представляет собой гидроксигруппу, возможно превращенную в группу простого или сложного эфира,
R3 представляет собой (2-6 C)-алкинил, возможно замещенный гидроксигруппой,
R4 представляет собой CN или одну из углеводородных групп, выбранных из (2-6 C)-алкила, (1-6 C)-алкоксигруппы, (2-6 C)-алкенила, (2-6 C)-алкинила и (2-6 C)-алкилидена, причем каждая из этих углеводородных групп возможно замещена галогеном, гидрокси- или (1-6 C)-алкоксигруппой, и
R5 представляет собой (1-6 C)-алкил.
Стероиды общей формулы I, где R1 представляет собой O, R2 представляет собой гидроксигруппу, R3 представляет собой этинил, R4 представляет собой 2-фторэтил или 2-фторэтенил и R5 представляет собой водород, являются предпочтительными стероидами.
Другими предпочтительными стероидами являются стероиды общей формулы I, где R1 представляет собой два атома водорода, R2 представляет собой гидроксигруппу, R3 представляет собой этинил, R4 представляет собой (2-6 C)-алкил, (2-6 C)-алкилиден или (2-6 C)-алкенил, каждый из которых может быть замещен фтором, и R5 представляет собой метил.
В определении для R2 гидроксигруппа может быть превращена в группу простого или сложного эфира. Термин превращение в группу простого эфира обозначает, что атом водорода гидроксигруппы замещен на низший алкил, предпочтительно имеющий 1-6 атомов углерода, например метил, пропил, втор-бутил и подобный алкил. Термин превращение в группу сложного эфира обозначает, что гидроксигруппа этерифицирована низшим алканоилом, предпочтительно имеющим 2-6 атомов углерода, например ацетилом, пропионилом и подобным алканоилом. В принципе пригоден любой сложный эфир, у которого эфирная группа отщепляется, когда соединение вводят.
(1-6 C)-Алкил в определении формулы I является алкилом нормального или разветвленного строения, имеющим 1-6 атомов углерода, например метилом, этилом, пропилом, бутилом, трет-бутилом, пентилом и гексилом. Предпочтительно алкил является метилом (особенно для R5) и этилом (особенно для R4). Термин (2-6 C)-алкил имеет такое же значение за исключением метила.
(2-6 C)-Алкенил является алкенилом нормального или разветвленного строения, имеющий 2-6 атомов углерода, например винилом, 2-пропенилом и 1,3-бутадиенилом.
(2-6 C)-Алкинил является алкинилом нормального или разветвленного строения, имеющим 2-6 атомов углерода, например этинилом, пропинилом, бутинилом и подобным алкинилом.
(2-6 C)-Алкилиден является алкилиденом нормального или разветвленного строения, имеющим 2-6 атомов углерода, например этилиденом, пропилиденом, 2-метилпропилиденом и подобным алкилиденом.
Термин галоген, применяемый в определении формулы I, обозначает фтор, хлор, бром или иод. Предпочтительным галогеном является фтор.
Термин (1-6 C)-алкоксигруппа обозначает группу, алкил которой является обозначенным выше (1-6 C)-алкилом.
Термин (2-7 C)-алкоксиалкил обозначает определенный выше (1-6C)-алкил, замещенный обозначенной выше (1-6 C)-алкоксигруппой, причем общее число атомов углерода в алкоксиалкиле между 2 и 7.
Новые соединения настоящего изобретения можно получить конденсацией 11-кетостероидов общей формулы II
где R1 представляет собой O,
R2 представляет собой гидроксигруппу, возможно превращенную в группу простого или сложного эфира,
R3 представляет собой (2-6 C)-алкинил, возможно замещенный гидроксигруппой, и
R5 представляет собой водород или (1-6 C)-алил,
или где
R1 представляет собой два атома водорода,
R2 представляет собой гидроксигруппу, возможно превращенную в группу простого или сложного эфира,
R3 представляет собой (2-6 C)-алкинил, возможно замещенный гидроксигруппой, и
R5 представляет собой (1-6 C)-алкил,
активные группы которых возможно защищены, с соединением (подобным соединению Виттига) формулы R4'R''4-CH-W, где R'4R''4C образуют группу R4, которая является возможно замещенным галогеном, гидрокси- или (1-6 C)-алкоксигруппой определенным выше (2-6 C)-алкилиденом, или соединением формулы R'''4Li, где R'''4 представляет собой (2-6 C)-алкил или (2-6 C)-алкенил, возможно замещенный галогеном, гидрокси- или (1-6 C)-алкоксигруппой, у которого реакционноспособные группы могут быть защищены защитными группами, которые известны в органической химии (смотри, например, T.W. Green. Protective Groups in Organic Synthesis, Wiley, NY, 1981), и W представляет собой остаток соединения, подобного соединению Виттига, Виттинга-Хорнера или Петерсона, с последующим возможным галогенированием и дегидратацией или гидратацией, после которой полученное соединение превращают в нитрил или конденсируют с соединением Виттига или подобным ему соединением формулы R6W, где W имеет указанные выше значения и R6 представляет собой независимо водород, галоген или (1-6 C)-алкил, с последующим гидроборированием и возможно затем алкилированием, галогенированием или галогенированием и дегидрогалогенированием, или (частичным) гидрированием, после чего возможно удаляют присутствующие защитные группы.
Пригодными реагентами являются трифенилфосфораны, например, формулы R'4R''4CH-P(Hal)Ph3 и подобные соединения, и пригодные реагенты Петерсона, например триметилсилановые реагенты формулы R'4R''4C(MgHal)Si(CH3)3, где Hal обозначает галоген, например хлор или бром.
Стероиды изобретения можно применять для предупреждения или лечения индуцированных дефицитом эстрогена нарушений, например менопаузных нарушений, как показано в анализе для определения индуцированного эстрогеном разрежения кости. В этом определении у молодых зрелых самок крыс Вистара удаляют яичники и вводят им испытываемое соединение в течение 1 месяца. Через 1 месяц отбирают кровь и в литий-гепариновой плазме определяют параметр костного функционального цикла (остеокальцин) по методу Verhaegne et al., J. Endrocrinol. 120, 143-151. При вскрытии трупа правую бедренную кость рассекают и костную плотность дистальной части метафиза измеряют рентгеновским денситометрическим методом. Костную плотность в мм алюминиевого эквивалента выражают в процентах относительно костной плотности интактной контрольной группы, которую определяли как 100%, и относительно костной плотности контрольной группы с удаленными яичниками, которую определили как 0%.
Величину остеокальцина определяют как 100% для группы с удаленными яичниками и 0% для интактной группы. Активные соединения ингибируют функциональный цикл кости и, следовательно, имеют величину остеокальцина менее 100%.
В таблице 1 приведены результаты этого анализа.
Соединения изобретения можно вводить энтерально или парентерально, предпочтительно для человека в виде суточной дозы 0,001-10 мг на кг массы тела. В смеси с фармацевтически приемлемыми вспомогательными компонентами, например описанными в стандартной ссылке Gennaro et al., Remington's Pharmaceutical Sciences (18th ed., Mack Publishing Compani, 1990, see especially Part 8: Pharmaceutical Preparations and Their Manufacture), соединения можно прессовать в твердые препараты в дозах на один прием, например пилюли, таблетки, или обрабатывать для превращения в капсулы или суппозитории. При помощи фармацевтически приемлемых жидкостей соединения можно также применять в качестве инъецируемого препарата в форме раствора, суспензии, эмульсии или разбрызгиваемого препарата, например, для назального введения. Для изготовления препаратов в дозах на один прием, например таблеток, предполагается применение обычных добавок, например наполнителей, красителей, полимерных связующих и подобных средств. В общем, можно применять любую фармацевтически приемлемую добавку, которая не препятствует функционированию активных соединений.
Приемлемые носители, с которыми можно вводить композицию, включают лактозу, крахмал, производные целлюлозы и подобные вещества или их смеси.
Изобретение далее иллюстрируется следующими примерами.
Пример 1.
В раствор 115 г (7 α, 11 α )-11-гидрокси-7-метилэстр-4-ен-3,17-диона в 5 мл ацетона добавляли по каплям при 0,5oC 110 мл 8 N хромовой кислоты. После перемешивания 1 час добавляли 50 мл пропанола-2 и через 15 мин смесь частично концентрировали и разбавляли 3 л воды. После перемешивания в течение нескольких часов осадок отделяли фильтрованием, переносили в небольшой объем дихлорметана и сушили над сульфатом натрия, получая после выпаривания органического растворителя 107 г трикетона- (7 α)-7-метилэстр-4-ен-3,11,17-триона. Rf = 0,54 (при элюировании смесью толуола и этилацетата, 4:6, объем/объем).
Смесь 115 г (7 α)-7-метилэстр-4-ен-3,11,17-триона, 8 г п-толуолсульфокислоты и 40 мл этандитиола в 1 л абсолютного этанола кипятили с обратным холодильником в течение 1 часа. После охлаждения смесь разбавляли 1 л воды и перемешивали на холоде несколько часов. Осадок отделяли фильтрованием и промывали 1N NaOH, водой и холодным метанолом. После высушивания получали 134 г (7 α)-3,3-этандитио-7-метилэстр-4-ен-11,17-диона. Rf = 0,62 (толуол-этилацетат, 8:2).
Раствор содержащий 36 г (7 α)-3,3-этандитио-7-метилэстр-4-ен-11,17-диона, 300 мл дихлорметана, 85 мл триэтилортоформиата, 70 мл этиленгликоля и 2 г п-толуолсульфокислоты перемешивали в течение 7 час. Затем смесь промывали 10%-ным раствором карбоната натрия и сушили, концентрировали и пропускали через колонку с силикагелем, получая 44 г продукта, (7 α)-3,3-этандитио-17,17-этилендиокси-7-метилэстр-4-ена. Rf = 0,65 (толуол-этилацетат, 8:2).
Смесь 9,2 г трет-бутоксида калия, 34,8 г бромистого метилтрифенилфосфония в 260 мл толуола кипятили с обратным холодильником в течение 1 часа. Затем добавляли 7,8 г (7 α)-3,3-этандитио-17,17-этилендиокси-7-метилэстр-4-ена и смесь кипятили еще 2 часа. Затем реакционную смесь охлаждали, промывали водой, сушили и концентрировали. Остаток хроматографировали, получая 6,1 г (7 α )-3,3-этандитио-17,17-этилендиокси-7-метил-11-метиленэстр-4-ена. Rf = 0,77 (толуол-этилацетат, 9:1).
Смесь 6,1 г (7 α)-3,3-этандитио-17,17-этилендиокси-7-метил-11-метиленэстр-4-ена, 90 мл ацетона, 50 мл тетрагидрофурана и 3 мл 6N соляной кислоты перемешивали при комнатной температуре. Через 1 час смесь разбавляли добавлением 700 мл 5%-ного раствора карбоната натрия и перемешивали 1/2 часа. Осадок отделяли фильтрованием и сушили, получая 5,4 г (7 α)-3,3-этандитио-7-метил-11-метиленэстр-4-ен-17-она. Rf = 0,74 (толуол-этиленацетат, 9:1).
Газообразный ацетилен пропускали через раствор 50 г трет-бутоксида калия в 500 мл сухого тетрагидрофурана. Через 2 часа ввели по каплям при 0oC раствор 30 г (7 α)-3,3-этандитио-7-метил-11-метиленэстр-4-ен-17-она в 400 мл тетрагидрофурана. После перемешивания дополнительно в течение 1 часа смесь выливали в 7 л воды и перемешивали еще 1/2 часа. Осадок отделяли фильтрованием и сушили, получая 31 г (7 α , 17 α) -3,3-этандитио-17-гидрокси-7-метил-11-метилен-19-норпрегн-4-ен-20-она. Rf = 0,64 (толуол-этилацетат, 9:1).
В раствор 8 г натрия в 300 мл жидкого аммиака при -50oC по каплям добавляли раствор 16 г (7 α , 17 α)-3,3-этандитио-7-метил-11-метилен-19-норпрегн-4-ен-20-ин-17- ола в 75 мл тетрагидрофурана. После перемешивания реакционной смеси в течение 1 часа избыток натрия ликвидировали добавлением 10 мл этанола. После выпаривания аммиака остаток распределили между дихлорметаном и водой. Органический слой отделяли, промывали и сушили. Остаток после выпаривания растворителя хроматографировали, получая 5,4 г (7 α , 17 α)-7-метил-11-метилен-19-норпрегн-4-ен-20-ин-17-ол. Rf = 0,59 (толуол-этилацетат, 9:1).
Пример 2.
Смесь 15 г (7 α , 17 α)-3,3-этандитио-7-метил-11-метилен-19-норпрегн-4-ен-20-ин-17-ола, 300 мл метанола, 20 мл воды, 6 г карбоната кальция и 51 мл иодистого метила кипятили с обратным холодильником в течение 6 часов. После фильтрования смеси через гифлоу фильтрат концентрировали, переносили в дихлорметан, промывали, сушили и выпаривали. Полученный остаток хроматографировали, получая 6,5 г (7 α , 17 α)-17-гидрокси-7-метил-11-метилен-19-норпрегн-4-ен-20-ин-3-она. Rf = 0,40 (толуол-этилацетат, 9:1).
Пример 3.
В раствор 134 г (7 α)-3,3-этандитио-7-метилэстр-4-ен-11,17-диона в 5 л сухого тетрагидрофурана добавляли по частям 110 г три-трет-бутоксиалюмогидрида лития при 0-5oC. После перемешивания в течение 3 час смесь выливали в 10 л ледяной воды и слегка подкисляли добавлением 1 л 2N соляной кислоты. Продукт экстрагировали этилацетатом. После сушки сульфатом натрия органический материал обрабатывали диэтиловым эфиром, получая 133 г по существу чистого (7 α , 17 β)-3,3-этандитио-17-гидрокси-7-метилэстр-4-ен-11-она. Rf = 0,45 (толуол-этилацетат, 6:4).
В раствор 132 г (7 α , 17 β)-3,3-этандитио-17-гидрокси-7-метилэстр-4-ен-11-она в 800 мл пиридина добавляли при 0oC 182 мл хлористого триметилсилила. После перемешивания в течение 1 часа смесь выливали в ледяную воду и продукт экстрагировали этилацетатом. После промывания, высушивания и выпаривания растворителя полученный остаток снова выпаривали с толуолом и затем обрабатывали гексаном, получая 137 г (7 α , 17 β)-3,3-этандитио-7-метил-17-триметилсилилокси-7-метилэстр-4-ен-11-она. Rf = 0,63 (толуол-этилацетат, 6: 4).
В суспензию 334 г хлористого метоксиметилтрифенилфосфония в 6 л сухого диэтилового эфира добавляли по каплям 600 мл 1,6 М бутиллития при 0,5oC. После перемешивания 1 час добавляли раствор 44,6 г (7 α , 17 β )-3,3-этандитио-17-триметилсилилокси-7-метилэстр-4-ен-11-она в 1,5 л диэтилового эфира и смесь перемешивали 24 час. Органический раствор затем промывали водой и сушили. Остаток, который получали после концентрирования органической фазы, распределяли между системой гексан-метанол-вода (1: 0,7:0,3, соотношения объемные) и перемешивали в течение 15 мин. Гексановую фазу сушили и концентрировали, получая 47 г (7 α , 17 β)-3,3-этандитио-11-метоксиметилен-17-триметилсилилокси-7-метилэстр-4-ена. Rf = 0,50 (гексан-этилацетат, 3:1).
В раствор 265 г (7 α , 17 β)-3,3-этандитио-11-метоксиметилен-17-триметилсилилокси-7-метилэстр-4-ена в 800 мл ацетона добавляли 80 мл концентрированной соляной кислоты и смесь перемешивали при комнатной температуре. После перемешивания в течение 1 часа смесь выливали в воду и экстрагировали этилацетатом. После промывания, сушки и выпаривания органического растворителя полученный остаток пропускали через колонку с силикагелем и элюировали смесью дихлорметан-ацетон (9:1), получая 63 г (7 α, 11 β, 17 β)-3,3-этандитио-17-гидрокси-7-метилэстр-4-ен-11-карбоксальдегида. Rf = 0,38 (толуол-этилацетат, 7:3).
В раствор 55 г (7 α, 11 β, 17 β)-3,3-этандитио-17-гидрокси-7-метилэстр-4-ен-11-карбоксальдегида и 165 мл дигидропирана в 1100 мл сухого тетрагидрофурана добавляли 1,3 г п-толуолсульфокислоты. После перемешивания в течение 2 час смесь выливали в 5 л 5%-ного раствора бикарбоната натрия и продукт экстрагировали этилацетатом. После концентрирования органической фазы выделяли (7 α, 11 β, 17 β)-3,3-этандитио-17-тетрагидропиранилокси-7-метилэстр-4-ен-11-карбоксальдегид.
Смесь 1 г (7 α, 11 β, 17 β)-3,3-этандитио-17-тетрагидропиранилокси-7-метилэстр-4-ен-11-карбоксальдегида и 2 г гидрохлорида гидроксиламина в 12 мл пиридина перемешивали при 80oC в течение 1 часа. Затем реакционную смесь охлаждали, выливали в воду и экстрагировали этилацетатом. После промывания, сушки и концентрирования получали 0,9 г аморфного оксима (7 α, 11 β, 17 β)-3,3-этандитио-17-тетрагидропиранилокси-7-метилэстр-4-ен-11-карбоксальдегида, Rf = 0,60 (толуол-этилацетат, 8:2).
Реакцию дигидратации 0,8 г оксима (7 α, 11 β, 17 β)-3,3-этандитио-17-тетрагидропиринилокси-7-метилэстр-4-ен-11-карбоксальдегида проводили в 8 мл уксусного ангидрида в течение 45 мин. Наблюдалось сопутствующее замещение 17-тетрагидропиранилоксигруппы на ацетилоксигруппу. Реакционную смесь выливали в 50 мл ледяной воды и перемешивали 30 мин. После нейтрализации 2N раствором едкого натра и экстракции этилацетатом получали 0,75 г (7 α, 11 β, 17 β)-3,3-этандитио-17-ацетилокси-7-метилэстр-4-ен-11-карбонитрила. Rf = 0,58 (толуол-этилацетат, 9:1).
Ацетатную группу омыляли перемешиванием 0,75 г (7 α , 11 β , 17 β )-3,3-этандитио-17-ацетилокси-7-метилэстр-4-ен-11-карбонитрила в течение 30 мин в смеси 20 мл тетрагидрофурана и 10 мл воды, содержащей 1 г едкого натра. Смесь разбавляли и экстрагировали этилацетатом. После сушки и выпаривали получали 0,50 г (7 α , 11 β , 17 β )-3,3-этандитио-17-гидрокси-7-метилэстр-4-ен-11-карбонитрила. Rf = 0,34 (толуол-этилацетат, 9:1).
В растворе 20 г (7 α , 11 β , 17 β )-3,3-этандитио-17-гидрокси-7-метилэстр-4-ен-11-карбонитрила в 600 мл сухого дихлорметана добавляли 20 г ацетата натрия, затем 85 г хлорхромата пиридиния. После перемешивания в течение 3 час реакцию заканчивали. Избыток окислителя удаляли добавлением 40 мл пропанола-2. Смесь фильтровали через гифлоу, концентрировали и хроматографировали, получая 13 г (7 α , 11 β )-3,3-этандитио-17-кето-7-метилэстр-4-ен-11-карбонитрила. Rf = 0,75 (толуол-этилацетат, 8:2).
Через раствор 10,5 г трет-бутоксида калия в 60 мл сухого тетрагидрофурана в течение 1 часа при 0oC пропускали газообразный ацетилен. Затем по каплям добавляли раствор 9,3 г (7 α , 11 β )-3,3-этандитио-17-оксо-7-метилэстр-4-ен-11-карбонитрила в 100 мл тетрагидрофурана. После перемешивания 1 час при 0-5oC смесь выливали в 500 мл насыщенного раствора хлористого аммония и продукт экстрагировали этилацетатом. После промывания, высушивания и концентрирования получали 9,5 (7 α , 11 β , 17 β )-3,3-этандитио-17-гидрокси-7-метил-19-норпрегн-4-ен-20-ин-11-карбонитрила. Rf = 0,28 (толуол-этилацетат, 9:1).
Смесь 6,5 г (7 α , 11 β , 17 α )-3,3-этандитио-17-гидрокси-7-метил-19-норпрегн-4-ен-20-ин-11-карбонитрила, 200 мл метанола, 100 мл тетрагидрофурана, 2,4 г карбоната кальция, 8,5 мл воды и 35 мл метанола кипятили с обратным холодильником в течение нескольких часов, добавляли время от времени дополнительное количество метанола и после исчезновения исходного материала смесь охлаждали, фильтровали и концентрировали. Остаток пропускали через колонку с силикагелем, получая 2,5 г чистого ( 7 α , 11 β , 17 α )-17-гидрокси-7-метил-3-оксо-19-норпрегн-4-ен-20-ин-11-карбонитрила. Т. пл. 234oC. Rf= 0,33 (толуол-этилацетат, 7:3).
Пример 4.
Гидроборирование 11-метиленовой группы 3,3,17,17-бис(этилендиокси)-11-метиленэстр-5-ена проводили следующим образом.
В раствор 2,18 мл 10 М комплекса гидрид бора-диметилсульфоксид в 10 мл сухого тетрагидрофурана добавляли при 0oC 2,7 мл циклооктадиена. После дополнительного кипячения с обратным холодильником в течение 1 часа в смесь добавляли раствор 2,7 г 3,3,17,17-бис(этилендиокси)-11-метиленэстр-5-ена в 30 мл тетрагидрофурана. Смесь перемешивали 16 час и затем обрабатывали 10 мл 10%-ного раствора едкого натра и затем 10 мл 30%-ного пероксида водорода. После перемешивания еще в течение дополнительных 4 час смесь выливали в воду и продукт экстрагировали дихлорметаном. Конечную очистку проводили хроматографией, получая 2 г (11 β )-3,3,17,17-бис-(этилендиокси)-11-(гидроксиметил)эстр-5-ена. Rf=0,25 (толуол-этилацетат, 1:1).
В суспензию 20 г хлорхромата пиридиния в 200 мл дихлорметана добавляли 9,3 г (11 β )-3,3,17,17-бис(этилендиокси)-11-(гидроксиметил)эстр-5-ена в 100 мл дихлорметана. После перемешивания в течение 1 часа избыток окислителя разрушали добавлением 40 г бисульфита натрия в 200 мл воды и затем продукт экстрагировали этилацетатом. После сушки и концентрирования органической фазы остаток очищали хроматографией, получая 5,4 г (11 β )-3,3,17,17-бис(этилендиокси)-эстр-5-ен-11-карбоксальдегида. Rf=0,50 (гексан-этилацетат, 1:1).
Раствор 1,6 М бутиллития в гексане (44 мл) добавляли по каплям в суспензию 24,4 г хлористого хлорметилтрифенилфосфония в 500 мл диэтилового эфира. После перемешивания 15 мин по каплям добавляли раствор 5,4 г (11 β )-3,3,17,17-бис(этилендиокси)эстр-5-ен-11-карбоксальдегида в 30 мл тетрагидрофурана. После 12 час смесь выливали в 0,5 л воды и органическую фазу отделяли, промывали, сушили и концентрировали. Остаток хроматографировали, получая 3,7 г E/Z (11 β )-3,3,17,17-бис(этилендиокси)-11-(2-хлорэтенил)экстр-5-ена. Rf=0,4 (гексан-этилацетат, 7:3).
В суспензию амида лития, полученную из 920 мг лития в 130 мл аммиака (жидкий), добавляли при -45oC раствор 3,6 г (11 β )-3,3,17,17-бис(этилендиокси)-11-(2-хлорэтенил)экстр-5-ена в 30 мл тетрагидрофурана. После перемешивания 1 час избыток реагента разрушали добавлением 15 г хлористого аммония и последующим выпариванием аммиака. Остаток распределяли между дихлорметаном и водой. Органическую фазу сушили, концентрировали и хроматографировали, получая 1,8 г (11 β )-3,3,17,17-бис(этилендиокси)-11-этинилэстр-5-ена. Т.пл. 200oC. Rf=0,45 (гексан-этилацетат, 7:3).
Смесь 8 г (11 β )-3,3,17,17-бис(этилендиокси)-11-этинилэстр- 5-ена, 200 мл ацетона, 100 мл метанола и 100 мл тетрагидрофурана обрабатывали 5 мл 6 N соляной кислоты и перемешивали в течение ночи. После обработки бикарбонатом натрия и концентрирования остаток хроматографировали, получая 5,1 г (11 β )-11-этинилэстр-4-ен-3,17-диона. Rf=0,48 (гексан-этилацетат, 1:1).
Через смесь 2,7 г трет-бутоксида калия в 12 мл тетрагидрофурана и 5 мл трет-бутанола в атмосфере азота в течение 1,5 часа при 0oC пропускали пузырьки газообразного ацетилена. Затем вводили суспензию 1,85 г (11 β) -11-этинилэстр-4-ен-3,17-диона в 5 мл тетрагидрофурана и перемешивание продолжали еще в течение 1 часа. Смесь разбавляли водой (200 мл), нейтрализовали добавлением 2N соляной кислоты и экстрагировали этилацетатом, сушили и концентрировали. Остаток хроматографировали, получая 1,6 г (11 β , 17 α )-11-этинил-17-гидрокси-19-норпрегн-4-ен-20-ин-3-она. Т. пл. 168oC. Rf= 0,60 (гексан-этилацетат, 1:1).
Пример 5.
В раствор 4 мл боран-метилсульфида (10 М в тетрагидрофуране) в 20 мл тетрагидрофурана добавляли по каплям 5 мл 1,5-циклооктадиена. После перемешивания в течение 1 часа по каплям добавляли 5 г 11 β -винил-3,3,17,17-бис(этилендиокси)эстр-5-ена (получен селективным гидрированием по методике Линдлара 11 β -винил-3,3,17,17-бис(этилендиокси)-11-этинилэстр-5-ена) в 25 мл тетрагидрофурана. После перемешивания дополнительно в течение 1 часа добавляли 20 мл 15%-ного водного раствора едкого натра и 20 мл 30%-ного пероксида водорода. Смесь перемешивали в течение ночи, затем продукт экстрагировали этилацетатом и полученный таким образом органический слой очищали хроматографией, получая 3,4 г 11 β -(2-гидроксиэтил)-3,3,17,17-бис(этилендиокси)эстр-5-ена. Т.пл. 190oC.
В раствор 5 г 11 β -(2-гидроксиэтил)-3,3,17,17-бис(этилендиокси)эстр-5-ена в 7 мл тетрагидрофурана добавляли при -30oC 500 мг 2,6-ди-трет-бутилпиридина и затем 800 мг ангидрида трифторметансульфокислоты. После перемешивания в течение дополнительных 15 мин при -30oC добавляли 10 мл 1 М раствора фтористого тетрабутиламмония в тетрагидрофуране и смесь перемешивали 2 часа при комнатной температуре и затем выливали в 30 мл 10%-ного раствора бикарбоната натрия. Затем смесь экстрагировали этилацетатом и органический слой очищали хроматографией, получая 600 мг 11 β -(2-фторэтил)-3,3,17,17-бис(этилендиокси)эстр-5-ена. Т.пл. 196oC.
В раствор 580 мг 11 β -(2-фторэтил)-3,3,17,17-бис(этилендиокси)эстр-5-ена в смеси 3 мл ацетона и 3 мл тетрагидрофурана добавляли 6 мл 3 М соляной кислоты. После перемешивания в течение 2 час добавляли бикарбонат натрия и воду и продукт экстрагировали этилацетатом. Остаток, полученный после выпаривания растворителя, обрабатывали диизопропиловым эфиром, получая 400 мг 11 β -(2-фторэтил)эстр-5-ен-3,17-диона. Т.пл. 85oC.
В раствор 0,7 г трет-бутоксида калия в 5 мл тетрагидрофурана и 1 мл трет-бутанола пропускали газообразный ацетилен. Через 15 мин добавляли раствор 400 мг 11 β -(2-фторэтил)эстр-5-ен-3,17-диона в 5 мл тетрагидрофурана. После перемешивания дополнительно в течение 15 мин раствор выливали в воду и продукт экстрагировали этилацетатом. Полученную таким образом органическую часть очищали колоночной хроматографией и обрабатывали эфиром, получая 320 мг (11 β , 17 α )-11-(2-фторэтил)-17-гидрокси-19-норпрегн-4-ен-20-ин-3-она. Т.пл. 212oC.
Пример 6.
В раствор диизопропиламида лития (получен из 250 мг диизопропиламина и 1,6 мл 1,6 М раствора бутиллития в гексане) в 3 мл тетрагидрофурана добавляли раствор 600 мг оксида дифторметилдифенилфосфина в 2 мл тетрагидрофурана при -50oC. После перемешивания 15 мин добавляли 800 мг 11 β -3,3,17,17-бис(этилендиокси)эстр-5-ен-11-карбоксальдегида в 3 мл тетрагидрофурана и смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в воду и экстрагировали этилацетатом. Хроматографией органической части получали 465 мг 11 β -(2,2-дифторэтенил)-3,3,17,17-бис(этилендиокси)эстр-5-ена. Т.пл. 180-181oC.
Раствор 430 мг указанного выше продукта в смеси 3 мл ацетона и 2 мл тетрагидрофурана обрабатывали 2 мл 4 N соляной кислоты. После перемешивания 2 часа смесь нейтрализовали твердым бикарбонатом натрия и продукт экстрагировали этилацетатом. Полученный таким образом органический материал кристаллизовали из диизопропилового эфира, получая 270 мг 11 β -(2,2-дифторэтилен)эстр-5-ен-3,17-диона. Т.пл. 150oC.
В раствор 0,48 г трет-бутоксида калия в смеси 5 мл тетрагидрофурана и 0,5 мл трет-бутанола пропускали газообразный ацетилен. Через 15 мин добавляли раствор 250 мг 11 β -(2,2-дифторэтилен)эстр-5-ен-3,17-диона в 3 мл тетрагидрофурана и еще через 15 мин смесь обрабатывали водой, продукт экстрагировали этилацетатом.
Выделенный таким образом органический материал обрабатывали диизопропиловым эфиром, получая 160 мг (11 β , 17 α )-11-(2,2-дифторэтенил)-17-гидрокси-19-норпренг-4-ен-20-ин-3-она. Т.пл. 196oC.
Пример 7.
Смесь E/Z 11 β -3,3,17,17-бис(этилендиокси)-11-(2-хлорэтенил)эстр-4-ена разделяли на колонке с силикагелем, получая чистый E-изомер (т.пл. 143oC) и Z-изомер (т. пл. 182oC). Обработкой раствора 3 г (E)-11 β -3,3,17,17-бис(этилендиокси)-11-(2-хлорэтенил)эстр-4-ена в 20 мл ацетона 3 мл концентрированной соляной кислоты в течение 1 часа с последующей нейтрализацией реакционной смеси раствором бикарбонатом натрия и экстракцией этилацетатом получали целевой дикетон-(E)-11 β -11-(2-хлорэтенил)эстр-4-ен-3,17-дион (2,3 г).
Этот продукт растворяли в 10 мл тетрагидрофурана и по каплям добавляли в раствор ацетилида калия в смеси 14 мл тетрагидрофурана и 3 мл трет-бутанола (ацетилид калия получали пропусканием газообразного ацетилена в раствор 2,9 г трет-бутоксида калия в указанной выше смеси трет-бутанола и тетрагидрофурана). После перемешивания 1/2 часа смесь обрабатывали водой и продукт экстрагировали этилацетатом. Органический материал очищали хроматографией, получая 1,9 г (11 β , E, 17  )-11-(2-фторэтенил)-17-гидрокси-19-норпрегн-4-ен-20-ин-3-она. Т.пл. 180oC.
)-11-(2-фторэтенил)-17-гидрокси-19-норпрегн-4-ен-20-ин-3-она. Т.пл. 180oC.
Пример 8.
Аналогично получали соединения, приведенные в таблице 2.
Пример 9.
Получали таблетки, имеющие следующий состав:
(11 β, 17 α ) - 17-Гидрокси-11-этил-19-норпрегн-4-ен-20-ин-3-он - 2,5 мг
Крахмал - 10 мг
Аскорбилпальмитат - 0,2 мг
Стеарат магния - 0,5 мг
Лактоза - До общей массы 100 мг
Гранулы основы получали смешиванием лактозы с частью крахмала. Оставшуюся часть крахмала смешивали с водой для образования крахмальной суспензии и добавляли в смесь. Всю смесь гранулировали и сушили. Эти гранулы основы смешивали с аскорбилпальмитатом и активным компонентом, просеивали, тщательно смешивали со стеаратом магния и затем таблетировали.
Пример 10.
Таблетки, имеющие такой же состав, как в примере 9, получали с применением в качестве активного компонента (11 β , 17 α )-11-этинил-17-гидрокси -19-норпрегн-4-ен-20-ин-3-она.
Пример 11.
Таблетки, имеющие такой же состав, как в примере 9, получали с применением (7 α, 11 β, 17 α )-11-метилен-17-гидрокси-7-метил-19-норпрегн-4-ен-20-ина в качестве активного компонента сначала смешиванием активного компонента с 10% лактозы и аскорбилпальмитатом и последующим смешиванием этой смеси с лактозой, крахмалом и крахмальной суспензий. Смесь сушили, тщательно смешивали со стеаратом магния и таблетировали.
Пример 12.
Получали капсулы, имеющие следующий состав:
(11 β , 17 α)-11-Этил-17-гидрокси-19-норпрегн-4-ен-20-ин-3-он - 2,5 мг
Крахмал - 10 мг
Аскорбилпальмитат - 0,2 мг
Стеарат магния - 0,5 мг
Авицел - До общей массы 100 мг
Компоненты смешивали друг с другом таким же образом, как в примере 9, гранулировали и наполняли гранулами желатиновые капсулы.
В таблице 3 приведены дополнительные данные, характеризующие соединения изобретения, обладающие эстрогенной активностью, определенной методом связывания эстрогена и/или методом Allty Doisy in vivo, и необязательно прогестогенной активностью, определенной методом связывания рецептора прогестерона.
Приведены вещества формулы, приведенной в описании, где R2 означает OH, а R3 означает этинил.
Методики
Относительная аффинность связывания для рецептора прогестагена человека.
Оценку аффинности связывания тестируемого соединения для рецептора прогестагена из цитоплазмы, полученной из клеток опухоли молочной железы MCF-7 человека, проводили по сравнению с аффинностью стандартного соединения - 16 α -этил-21-гидрокси-19-норпрегн-4-ен-3,20-диона (Org 2058). Тестируемое соединение исследовали в трех концентрациях, в соотношении 1:2:4 относительно трех концентраций стандартного соединения Org 2058, также в соотношении 1:2: 4. Эффективность вытеснения меченого соединения Org 2058 из рецептора прогестагена тестируемым соединением и стандартным соединением измеряли после инкубации в течение 16 ч при 4oC. Для каждой концентрации стандартного или испытуемого соединения рассчитывали радиоактивность, связавшуюся с рецептором (с учетом неспецифического связывания), как процент от суммарной радиоактивности (также с учетом неспецифического связывания), связавшийся с рецептором. Затем проценты были заменены соответствующими логарифмическими значениями и оценены статистически, используя анализ трех точечных параллельных линий. Если не была обнаружена существенная кривизна и отсутствовало значительное отклонение параллелей на уровне 0,01, то рассчитывали относительную аффинность связывания тестируемого соединения по сравнению с Org 2058.
Аналогично для определения относительной аффинности тестируемого соединения к экстрогенным рецепторам человека, по сравнению с эстрадиолом (E2), использовали цитоплазматические рецепторы эстрогена, присутствующие в цитоплазме рекомбинантных клеток яичника китайского хомячка (CHO), стабильно трансфецированных эстрогенным рецептором человека. Клеточная линия CHO-ER-(2B1) была получена в Отделении биотехнологии и биохимии (BBC, N.V.Organon). Эталонными соединениями были этинилэстрадиол и эстриол.
Тест Allen Doisy для определения эстрогенной активности in vivo.
Этот метод основан на оценке ороговения вагинального эпителия на вагинальных мазках. У зрелых самок крыс удаляли яичники и на третьей неделе после операции животных подвергали воздействию одной дозы (1 мкг) эстрадиола (1-е сутки). Через 7 суток после введения вновь вводили испытуемое соединение 1 раз на 8-е сутки и 2 раза на 9-е сутки. Вагинальные мазки отбирали во второй половине 10-х суток, 2 раза на 11-е сутки и снова утром на 12-е сутки. Эти мазки окрашивали по Гимзе и определяли число положительно окрашенных мазков (Van dervies J., De Visser J. 1983. Эндокринологические исследования в дезогестрелом. Drug. Res., т. 33, с. 231-236). Активность соединения выражена в виде минимальной эффективной дозы.


Описываются новые стероиды общей формулы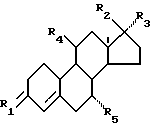
где R1 - O, (H,OH) или H2; R2 - OH, возможно этерифицированная или эстерифицированная; R3 - C2-C6-алкинил, возможно замещенный гидроксигруппой; R4 - CN, C1-C6-алкил, C1-C6-алкокси, C2-C6-алкенил, C2-C6-алкинил, C2-C6-алкилиден, возможно замещенные галогеном, гидрокси или C1-C6-алкокси; R5 - H или C1-C6-алкил. Стероиды являются активным компонентом лекарственного препарата или фармацевтической композиции, предназначенных для лечения или предупреждения менопаузных заболеваний, особенно остеопороза. Описан также способ получения этих стероидов. 4 с. и 10 з.п. ф-лы, 3 табл.
где R1 представляет собой О, (H, ОН) или два атома водорода;
R2 представляет собой гидроксигруппу, необязательно этерифицированную или эстерифицированную;
R3 представляет собой (С2-С6)-алкинил, необязательно замещенный гидроксигруппой;
R4 представляет собой CN или одну из углеводородных групп, выбранных из (С1-С6)-алкила, (С1-С6)-алкоксигруппы, (С2-С6)-алкенила, (C2-С6)-алкинила и (С2-С6)-алкилидена, причем каждая из этих углеводородных групп может быть замещена галогеном, гидрокси- или (С1-С6)-aлкoкcигpуппoй;
R5 представляет собой водород или (С1-С6)-алкил.
где R1 представляет собой О;
R2 представляет собой гидроксигруппу, необязательно этерифицированную или эстерифицированную;
R3 представляет собой (С2-С6)-алкинил, необязательно замещенный гидроксигруппой;
R4 представляет собой CN, (C2-C6)-алкил, замещенный галогеном или (С2-С6)-алкенил, замещенный галогеном;
R5 представляет собой водород или (С1-С6)-алкил.
в которой R1 представляет собой O;
R2 представляет собой гидроксигруппу, необязательно этерифицированную или эстерифицированную;
R3 представляет собой (C2-C6)-алкинил, необязательно замещенный гидроксигруппой;
R5 представляет собой водород или (С1-С6)-алкил,
или
где R1 представляет собой два атома водорода;
R2 представляет собой гидроксигруппу, необязательно этерифицированную или эстерифицированную;
R3 представляет собой (С2-С6)-алкинил, необязательно замещенный гидроксигруппой;
R5 представляет собой (С1-С6)-алкил, активные группы которого возможно защищены,
конденсируют с подобным соединению Виттига соединением формулы R'4R''4-CH-W, где R'4R''4C образует группу R4, которая является замещенным галогеном, гидрокси- или (С1-С6)-алкоксигруппой, определенным выше (C1-C6)-алкилиденом, или формулы R'''4Li, где R'''4'Li представляет собой (C2-С6)-алкил или (С2-С6)-алкенил, замещенный галогеном, гидрокси- или (С1-С6)-алкоксигруппой, у которого реакционноспособные группы могут быть защищены защитными группами, которые известны в органической химии; W - представляет собой остаток соединения, подобного соединению Виттига, Виттига-Хорнера или Петерсона, с последующим возможным галогенированием и дегидратацией или гидратацией, после которой полученное соединение превращают в нитрил или конденсируют с соединением Виттига или подобным ему соединением формулы R6W, где W имеет указанные выше значения и R6 представляет собой независимо водород, галоген или (С1-C6)-алкил, с последующим гидроборированием и возможно затем алкилированием, галогенированием и дегидрогалогенированием или (частичным) гидрированием, после чего возможно удаляют присутствующие защитные группы.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| КРЫМСКАЯ М.Л | |||
| Климактерический период | |||
| - М.: Медицина, 1989, с | |||
| Камневыбирательная машина | 1921 |
|
SU222A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US 3405010, 02.09.1969 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| DE 3702383 A, 04.08.1988 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Berqink E.W | |||
| et al | |||
| J | |||
| Steroid biochemistry, 1981, 14, № 2, p | |||
| Ручной прибор для загибания кромок листового металла | 1921 |
|
SU175A1 |

