Данное изобретение относится к области эстрогенных соединений со стероидной структурой, имеющей неароматическое А-кольцо и свободную или блокированную гидроксильную группу при атоме углерода 3. Широко известно, что эстрогенные соединения полезны в контрацепции, также при лечении нарушений, связанных с эстроген-дефицитом, таких как симптомы менопауз и остеопороз.
Известны из литературы многие эстрогенные соединения. Например, публикацией, дающей информацию по эстрогенным соединениям с неароматическим А-кольцом и свободной или блокированной гидроксильной группой при атоме углерода 3, является патент США 3413287. Другими документами, описывающими эстрогенное или гормональное воздействие неароматических стероидов с 3-гидроксил-замещенной группой и двойной связью в положении 4-5, являются патенты WO 94 18224, US 3465010, FR 2099385, US 3652606 и ЕР 145 493. Документом, в котором раскрыты неароматические стероиды с 3-кето-замещенной группой и двойной связью в положении 5-10, является патент США 3377366, Baran и др. Такие соединения описаны в целом как агенты, обладающие, кроме прочих свойств, эстрогенными или антиэстрогенными эффектами. В последнее время в области разработки лекарственных препаратов, имеющих сродство к рецепторам эстрогенов (ER), внимание сфокусировано на открытии двух особых типов рецепторов эстрогенов, обозначенных 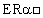 и ERβ (Mosselman и др., FEBS Letters 392 (1996) 49-53, а также ЕР-А-0 798 378). Так как указанные рецепторы по-разному распространены в тканях человека, то обнаружение соединений, которые обладают селективным сродством к каждому из двух, является важным вкладом в данную область, т.к. делает возможным проведение более селективного лечения эстроген-дефицитных состояний с менее тяжелыми побочными эффектами, связанными с эстрогенами.
и ERβ (Mosselman и др., FEBS Letters 392 (1996) 49-53, а также ЕР-А-0 798 378). Так как указанные рецепторы по-разному распространены в тканях человека, то обнаружение соединений, которые обладают селективным сродством к каждому из двух, является важным вкладом в данную область, т.к. делает возможным проведение более селективного лечения эстроген-дефицитных состояний с менее тяжелыми побочными эффектами, связанными с эстрогенами.
Данное изобретение касается эстрогенов, отвечающих общей формуле:

Формула I
в которой R1 обозначает Н, (C1-С3)алкил или (С2-С3)ацил;
R2 обозначает Н, α-(C1-C4)алкил, α-(C2-C4)алкенил или α-(C2-С4)алкинил;
R3 обозначает Н или (C1-C4)алкил, (С2-С4)алкенил или (C2-С4)алкинил, каждый может находиться в положении 15 или 16 стероидного скелета;
R4 обозначает Н или (C1-C5)алкил, (C2-C5)алкенил или (С2-C5)алкинил, каждый из которых необязательно замещен галогеном, предпочтителен этинил;
R5 обозначает Н, (С1-С3)алкил или (С2-С3)ацил;
R6 обозначает (C1-C5)алкил, (C2-C5)алкенил, (C2-C5)алкинил или (C1-C5)алкилиден, каждый из которых необязательно замещен галогеном или (C1-С3)алкилоксигруппой, предпочтителен аллил; предпочтительными галогенами в R6 являются фтор и хлор.
Связи, обозначенные точками, необязательно являются двойными связями. Если R6 обозначает алкилиден, то обозначенная точками линия к R6 является дополнительной связью, присутствующей в алкилиденовом звене, а если R6 обозначает алкил или алкенил, связь от атома 11 к R6 является простой связью.
Было обнаружено, что такие неароматические производные эстрадиола с заместителем в положении 11 стероидной структуры обладают селективным сродством к ERα-рецепторам.
Соединения настоящего изобретения представляют собой усовершенствованные эстрогены, в том смысле, что их можно использовать при эстроген-зависимых нарушениях, таких как симптомы менопауз и остеопороз. Они также находят применение в контрацепции, а, кроме того, могут быть полезны при лечении или профилактике болезни Альцгеймера, опухоли молочной железы, доброкачественной гипертрофии предстательной железы и сердечно-сосудистых нарушений. Соединения данного изобретения особо подходят для лечения и профилактики нарушений, вызванных эстроген-дефицитом, при ослаблении связанных с эстрогеном побочных эффектов.
Приведенные в данном описании термины имеют следующие значения:
(С1-С5)алкил представляет разветвленную, неразветвленную или циклическую алкильную группу, имеющую 1-5 атомов углерода, например метил, этил, изопропил, 2-метилциклопропил, бутил, втор-бутил, трет-бутил и др.
(C2-C5)алкенил представляет разветвленную, неразветвленную или циклическую алкенильную группу, имеющую от 2 до 5 атомов углерода, такую как этенил, 2-бутенил и др.
(С2-С5)алкинил представляет разветвленную или неразветвленную алкинильную группу, имеющую 2-5 атомов углерода, такую как этинил и пропинил.
(С2-С3)ацил представляет группу, имеющую 2-3 атома углерода и являющуюся производной из алкилкарбоновой кислоты, причем алкильная часть имеет значение, определенное выше.
(C1-C5)алкилиден представляет разветвленную, неразветвленную или циклическую алкилиденовую группу, имеющую 1-5 атомов углерода, такую как метилен и этилиден.
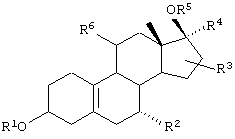
Из соединений приведенной выше общей формулы предпочтительны соединения, отвечающие общей формуле II:
Формула II
в которой R1 обозначает Н, (C1-С3)алкил, (С2-С3)ацил;
R2 обозначает Н, α-(C1-C4)алкил, α-(C2-C4)алкенил, α-(С2-С4)алкинил;
R3 обозначает Н или (С1-С4)алкил в положении 16 стероидного скелета;
R4 обозначает этинил;
R5 обозначает Н или (C1-С3)алкил, (С2-С3)ацил;
R6 обозначает (C1-C5)алкил, (С2-С5)алкенил, (C2-C5)алкинил, каждый из которых может быть замещен хлором или фтором. Если R6 обозначает (C1-C2)алкил, этенил или этинил, каждый из которых необязательно замещен хлором или фтором, то предпочтительно, чтобы R3 являлся метилом в положении 16 стероидного скелета.
Более предпочтительны стероиды данного изобретения указанной формулы II, в которой R1 обозначает Н;
R2 обозначает Н;
R3 обозначает Н или 16α-метил;
R4 обозначает этинил;
R5 обозначает Н;
R6 обозначает пропенил, аллил или бутенил.
Соединения данного изобретения можно получить различными способами, известными в органической химии вообще и, главным образом, в химии стероидов. Смотри, например: Fried J. and Edwards J.A. "Organic-Reactions in Steroid. Chemistry", Volumes I and II, Van Nostrand Reinhold Company, New York, 1972; и С.Djerassi "Steroid Reactions", Holden-Day, Inc., San Francisco, 1963.
Подходящим, например, является синтез стероидов с конкретными заместителями по положению С7 через сопряженное присоединение металлоорганических веществ к соответствующим 4,6-диен-3-оновым стероидам, обычно с получением 7α-производных стероидов (наряду с получением в меньшем количестве 7β-стероидов, которые можно легко удалить кристаллизацией или хроматографией). Многие примеры такого синтеза известны из литературы. Введение заместителей по положению С11 стероидного скелета можно выполнить несколькими способами. Таким способом является сопряженное присоединение металлоорганических веществ к защищенному соответствующим образом 5α,10α-эпокси, 9(11)-олефину, которое описано Teutsch et al., Steroids, 37, 361 (1981), но для получения заявленных соединений можно применять другие способы, используя 11-оксо-функциональную группу соответственно защищенного 19-норандрост-5-ена в качестве реакционно-способной группы для внутренней конверсии функциональной группы, например, до С11-альдегида, согласно хорошо известной химической методике (смотри Е. Ottow et al., Tetr. Lett., 5253 (1993) и др.). Конечно, также хорошо служит достижению цели комбинация обоих описанных методов. Введение двойной связи по положению 5(10) выполняют, применяя так называемое восстановление по Берчу А-кольца ароматических частей стероидов с подходящими функциональными группами, растворяя восстанавливающий металл Δ-4,5-9,11-диенонов, или кетализацией 3-кето-Δ-4,5-стероидов. Последняя методика дает либо непосредственно требуемые селективные Δ-5(10)-изомеры, либо смеси кеталей, которые можно разделить хроматографией или кристаллизацией на соответствующих стадиях синтеза. Осторожный гидролиз кеталя по С3 обычно дает требуемые 3-оксо-Δ-5(10)-изомеры, которые можно превратить в 3-ОН-соединения путем восстановления гидридами. Насыщенные стероиды (а именно, 5αН-производные) легко получить в восстановительных условиях, например при растворении щелочных металлов в аминах или аммиаке. Введение двойных связей по С14, С15 обычно проводят, вводя сначала двойную связь по положению С15, С16, с последующей изомеризацией данной связи в положение С14, С15 известными способами. В качестве альтернативы двойную связь, введенную по положению С15, С16, можно использовать для проведения сопряженного присоединения, например, с цианидом, что позволяет в дальнейшем ввести заместители по С15. Введение С16-заместителя легко выполнить путем алкилирования подходящими основаниями и электрофилами.
Настоящее изобретение касается также фармацевтической композиции, включающей стероидное соединение согласно изобретению в смеси с фармацевтически приемлемыми вспомогательными агентами, такими, которые описаны в работе Gennaro et al., Remington's Pharmaceutical Sciences (18th ed., Mack publishing Company, 1990, главным образом Part 8: Pharmaceutical Preparations and Their Manufacture), которую можно использовать в качестве ссылки. Смесь стероидных соединений согласно изобретению и фармацевтически приемлемого вспомогательного агента можно прессовать в виде твердых стандартных дозированных форм, таких как гранулы, таблетки, или получать капсулы или суппозитории. Используя фармацевтически приемлемые соответствующие жидкости, данные соединения можно также применять в препаратах для инъекций в виде растворов, суспензий, эмульсий или спреев, например спреев для носа. Для получения стандартных дозированных форм, например таблеток, можно использовать обычные добавки, такие как наполнители, красители, полимерные связующие и подобные. Вообще можно использовать любую фармацевтически приемлемую добавку, которая не мешает действию активных соединений. Стероидные соединения согласно изобретению можно также включать в имплантаты, вагинальные кольца, пластыри, гели и любые другие препараты продолжительного действия.
Подходящие носители, с которыми можно применять данные композиции, включают лактозу, крахмал, производные целлюлозы и подобные им вещества, или их смеси, используемые в подходящих количествах.
Данное изобретение касается также применения стероидного соединения согласно изобретению для получения лекарственного препарата для лечения нарушений, связанных с эстроген-дефицитом, таких как симптомы пери- и/или постменопаузы. Таким образом, данное изобретение может рекомендоваться к таким медицинским показаниям, как симптомы пери- и/или постменопаузы (климактерические) и остеопороз, и именно к способу лечения путем HRT (гормонозаместительной терапии), включая введение пациенту (женщине) описанного выше соединения (в виде подходящей фармацевтической дозированной формы).
Кроме того, данное изобретение касается применения стероидного соединения данного изобретения в качестве активного вещества в производстве лекарственного препарата с контрацептивной активностью. Таким образом, данное изобретение может применяться при контрацепции, а именно в способе контрацепции, включающем введение пациенту (женщине или самке животного) гестагена и эстрогена, как это принято в данной области, причем эстроген представляет собой описанное выше соединение (в виде подходящей фармацевтической дозированной формы).
В заключение, данное изобретение касается применения указанного стероидного соединения для получения лекарственного препарата, обладающего селективной эстрогенной активностью, который мог бы найти широкое применение в области гормонозаместительной терапии (HRT).
Дозированные количества настоящих стероидов находятся в диапазоне, обычном для эстрадиолов, примерно от 0,01 до 10 мг на прием.
Ниже данное изобретение иллюстрируется с помощью некоторых примеров, не имеющих ограничительного характера, и соответствующих схем формул.
ПРИМЕР 1
Синтез соединений (3α,11β,17β)-11-(2-пропенил)-19-норпрегн-5(10)-ен-20-ин-3,17-диола (соединение 11) и (3α,11β,16α,17β)-16-метил-11-(2-пропенил)-19-норпрегн-5(10)-ен-20-ин-3,17-диола (соединение 16) описан со ссылкой на схему I. Соединения обозначены номерами. Номера относятся к соответствующим структурным формулам на схемах I-VII.
Соединение 2
К раствору 17,3 г CuI и 3,84 г LiCl в 250 мл ТГФ добавляют при -70°С 90,6 мл 1М раствора аллилмагнийбромида в диэтиловом эфире. После перемешивания еще в течение 20 мин добавляют 11,4 мл триметилхлорсилана, а затем раствор 7,5 г стероида 1 в 100 мл ТГФ. Температуру реакционной смеси все время поддерживают ниже -60°С. После перемешивания в течение 1 час. реакционную смесь гасят, выливая в насыщенный водный раствор NH4C1. Продукт экстрагируют этилацетатом и затем очищают способом колоночной хроматографии, получая 6,25 г 2 в виде бесцветного масла. ЯМР: 5,20 (м, СН, аллил), 5,0 (СН2, аллил), 3,04 (м, Н11).
3
К раствору 9,6 г 2 в смеси 100 мл метанола и 30 мл метиленхлорида, содержащему 800 мг NaOH, добавляют 0,4 г боргидрида натрия при 0-5°С. После перемешивания в течение 1,5 ч реакция завершается и смесь обрабатывают 20 мл ацетона в течение 0,5 ч. Затем реакционную смесь выливают в воду и экстрагируют этилацетатом, получая 9,5 г 3. ЯМР: 3,59 (т, СНОН), 2,98 (м, Н11), 0,92 (с, СН3).
Схема I

4
К раствору 9,5 г 3 в 100 мл ацетона добавляют 8 мл 6N НСl. После перемешивания в течение 2 ч смесь нейтрализуют при помощи NаНСО3 и концентрируют до небольшого объема, который разбавляют водой и экстрагируют этилацетатом. Это дает 8,2 г 4 в виде бесцветного аморфного вещества. ЯМР: 5,68 (м, Н4), 3,10 (м, Н11), 3,65 (м, СНОН).
5
Раствор 8,2 г 4 в 100 мл сухого ТГФ добавляют к 500 мл жидкого NH3 при -70°С. Данную смесь обрабатывают некоторым количеством металлического лития (примерно 500 мг), пока синее окрашивание реакционной смеси не будет сохраняться в течение, по меньшей мере, 15 мин. Реакционную смесь гасят, добавляя порцию NH4Cl. Остаток, который остается после выпаривания NН3, разбавляют водой и экстрагируют этилацетатом. Хроматографическая очистка дает 4,0 г 5 в виде бесцветного масла. Rf 0,55 (гептан/этилацетат = 1/1 об./об.). ЯМР: 2,80 (ab, CH2 при С4), 0,93 (с, СН3).
6
К раствору 4,0 г 5 в 80 мл метанола добавляют 6 мл триметилортоформиата, а затем 0,8 г толуолсульфоновой кислоты. После перемешивания в течение 2 ч выполняют кетализацию. Данную смесь обрабатывают 6 мл пиридина и концентрируют до небольшого объема. Остаток разбавляют водой и экстрагируют этилацетатом. Полученный остаток 4,7 г состоит из почти чистого 6; ТСХ: Rf 0,78 (гептан/этилацетат = 1/1 об./об.). ЯМР: 3,22, 3,25 (2×с, ОСН3).
7
К раствору 33 г 6 в 50 мл ацетона добавляют 6 г молекулярных сит (4А), а затем 3,2 г N-метилморфолин-N-оксида и 150 мг перрутената тетрапропиламмония. Смесь перемешивают в течение 1 ч. К реакционной смеси добавляют 5 г силикагеля, а затем 50 мл гептана и перемешивают еще 5 мин. Данную смесь фильтруют посредством высокоскоростной фильтрации и после частичного концентрирования помещают в этилацетат, промывают водой и концентрируют. Остаток пропускают через короткую колонку с диоксидом кремния и получают 2,9 г 7; Rf 0,52 (гептан/этилацетат = 7/3). ЯМР: 1,02 (с, СН3).
8
Для этинилирования получают литийацетилид из дибромэтена и бутиллития.
К раствору 0,74 мл 1,2-дибромэтена в 20 мл ТГФ добавляют при -70°С 11 мл 1,6М раствора BuLi в гексане. После перемешивания в течение 15 мин добавляют раствор 800 мг 7 в 2 мл ТГФ. Смеси дают нагреться до комнатной температуры в течение 15 мин и еще через 15 мин при комнатной температуре реакционную смесь гасят водой и продукт экстрагируют этилацетатом. Концентрирование с последующим пропусканием через короткую колонку с силикагелем дает 810 мг 8 в виде белого аморфного вещества. Rf 0,48 (гептан/этилацетат = 7/3), Rf исходного вещества 0,52. ЯМР: 2,61 (с, ацетилен).
9
К суспензии 3,2 г 8 в 60 мл этанола добавляют 0,16 г щавелевой кислоты в 16 мл воды. Смесь перемешивают в течение 2,5 ч, и она постепенно становится гомогенной. Реакционную смесь обрабатывают NаНСО3 и концентрируют до небольшого объема. Затем добавляют воду и экстрагируют продукт этилацетатом. Выделенный таким образом сырой продукт пропускают через короткую колонку с силикагелем и кристаллизуют из диизопропилового эфира, получая 2,3 г 9, т. пл. 136°С. Rf 0,66 (гептан/этилацетат = 1/1). ЯМР: 2,78 (ab, 2, Н4), 2,61 (с, ацетилен).
10, 11
К раствору 1 г 9 в 12 мл ТГФ добавляют 1,6 г три-трет-бутоксиалюминийгидрида лития. После перемешивания в течение 1 ч при комнатной температуре смесь обрабатывают водой и нейтрализуют, добавляя 2N НСl. Продукт экстрагируют этилацетатом и хроматографируют на силикагеле (гептан/этилацетат = 8/2 в качестве элюента). Это дает 0,56 г 3β-спирта 10 (т.пл. 121-123°С) и 0,24 г 3α-спирта 11 (т.пл. 84-87°С). Rf 0,53 (10) и 0,45 (11), гептан/этилацетат = 1/1. ЯМР: (3αOН) 3,82 (м, СНОН); (3βОН): 4,08 (м, СНОН).
12
К раствору гексаметилдисилазида лития (получен из 1,9 мл 1,6М раствора BuLi в гексане и 0,71 мл гексаметилдисилазана в 4 мл сухого ТГФ) добавляют при -40°С 1 г 7 и 0,7 мл DMPU (N,N'-диметилпропиленмочевина) в 5 мл ТГФ. Смесь перемешивают в течение 0,5 ч при -40°С и затем добавляют шприцем 225 мкл СН3I. После перемешивания в течение еще 0,5 ч при -40°С реакция завершается. Смесь разбавляют водой и экстрагируют этилацетатом. Хроматография выделенного таким образом сырого продукта дает 1,3 г 12, Rf 0,43 (гептан/этилацетат = 8/2).
13
Согласно методике, описанной для получения 8, 1,3 г 12 превращают в требуемое соединение 13, получая 1,2 г, Rf 0,46 (гептан/этилацетат = 7/3); Rf (12) 0,55.
14
К раствору 800 мг 13 в 20 мл этанола добавляют раствор 80 мг щавелевой кислоты в 5 мл воды. Смесь перемешивают 1 ч и затем нейтрализуют, добавляя NaHCO3. После разбавления водой и экстракции этилацетатом остается 0,7 г 14 в виде кристаллического вещества. Rf 0,47 (гептан/этилацетат = 7/3).
15, 16
К раствору 725 мг 14 в 20 мл смеси 1/1 этанола и ТГФ добавляют 130 мг боргидрида натрия. После перемешивания в течение 1 ч добавляют 2 мл ацетона для разложения избытка реагента. Через 15 мин смесь выливают в воду и продукт экстрагируют этилацетатом. Полученное таким образом вещество очищают хроматографически на силикагеле, используя смесь метилен-хлорид/ацетон или смесь гексан/этилацетат в качестве элюента. Это дает 300 мг 16 (3α-ОН) и 75 мг 15 (3β-ОН). Rf (15) 0,47 (метиленхлорид/ацетон = 95/5) и Rf (16) 0,54 (метилен-хлорид/ацетон = 95/5).
ПРИМЕР 2
Синтез соединения (3β,5α,11β,17β)-11-дифторметил-19-норпрегн-20-ин-3,17-диола (соединение 21) описан со ссылкой на схему II.
Схема II

18
К раствору 4 г известного альдегида 17 в 200 мл метиленхлорида добавляют 10 мл диметилсератрифторида. Смесь перемешивают в течение 48 ч при температуре окружающей среды, а затем выливают в воду со льдом и экстрагируют метиленхлоридом, с последующим промыванием NаНСО3. После концентрирования и хроматографии (SiO2 - гептан/этилацетат = 2/1) получают 2,5 г 18, т.пл. 138-139°С. ЯМР (СDСl3): 6,05 м.д. двойной триплет CHF2, 5,9 (с, Н4), 0,95 (с, СН3).
19
Газообразный ацетилен пропускают в течение 45 мин через дегазированную смесь 1,3 г третбутилата калия и 1 г 18 в 7 мл ТГФ и 2 мл трет-бутанола при 0°С. Затем смесь выливают в воду и экстрагируют этилацетатом. После промывания и концентрирования оставшееся вещество обрабатывают диизопропиловым эфиром, получая 0,85 г 19 в виде белого твердого вещества. Т.пл. 178-180°С. Rf 0,43 (гептан/этилацетат = 1/1).
20
К раствору 20 мг Li в 10 мл жидкого NН3 добавляют при -60°С раствор 300 мг 19 в 6 мл ТГФ. После перемешивания в течение 1 мин смесь обрабатывают 0,5 г NH4Cl. NH3 выпаривают, а остаток обрабатывают водой и экстрагируют этилацетатом. Полученное таким образом вещество очищают хроматографически, получая 140 мг 20. Т.пл. 224-225°С. Rf 0,64 (гептан/этилацетат = 7/3).
21
К раствору 25 мг LiAlH4 в 4 мл ТГФ добавляют при -60°С 85 мг 20 в 1 мл ТГФ. После перемешивания в течение 5 мин смесь быстро нагревают до комнатной температуры и обрабатывают, добавляя 45 мкл воды, 45 мкл 3N раствора NaOH и 140 мкл воды. Осадки отфильтровывают, а фильтрат переносят в этилацетат и промывают 2N НСl и водой. Остаток, остающийся после сушки и концентрирования, обрабатывают диизопропиловым эфиром и получают 50 мг 21. Т.пл. 168-169°С. Rf 0,30 (толуол/этилацетат = 7/3). ЯМР: 3,6 м.д. СНОН, 2,65 (СН ацетилен), 6,0 (двойной триплет CHF2).
ПРИМЕР 3
Синтез соединений (3β,5α,11β,17β)-11-(2-фторэтил)-19-норпрегн-20-ин-3,17-диола (соединение 31) и (3α,5α,11β,17β)-11-(2-фторэтил)-19-норпрегн-20-ин-3,17-диола (соединение 32) описан со ссылкой на схему III.
23
К раствору 4,95 г 11β-гидроксиэтилэстрон-3-метилового эфира (22) в 115 мл ТГФ добавляют 66 г молекулярных сит 4А, а затем 45 мл 1М TBAF в ТГФ (сухом) и далее 5,3 г тозилфторида. Смесь перемешивают и кипятят с обратным холодильником в течение двух часов. Затем реакционную смесь охлаждают, выливают на 700 мл 10% водного раствора NaHCO3 и экстрагируют этилацетатом. После концентрирования раствора и хроматографии выделяют 3,4 г 23. Rf 0,39 (гептан/этилацетат = 7/3).
Схема III
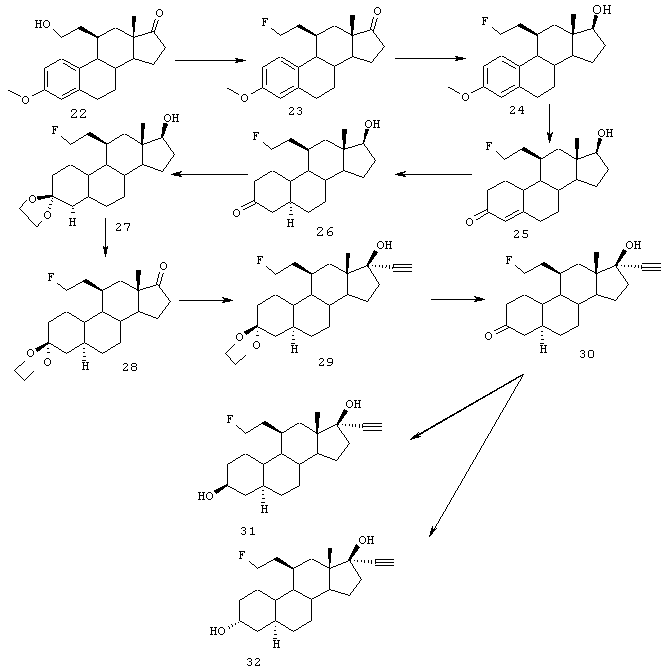
24
Раствор 4 г 23 в смеси 30 мл ТГФ и 30 мл метанола обрабатывают 0,1 мл 4N NaOH и затем 0,23 г NaBH4. После перемешивания в течение 1 ч реакционную смесь выливают в воду и экстрагируют этилацетатом. Сырое вещество фильтруют через короткую колонку с силикагелем, получая 3,8 г 24. Rf 0,26 (гептан/этилацетат = 7/3). Rf (23) 0,33.
25
К раствору 3,8 г 24 в смеси 40 мл ТГФ и 230 мл жидкого NН3 добавляют при -33°С 5 г Li. Смесь перемешивают в течение 5 ч. Затем избыток лития убирают посредством обработки 45 мл этанола. Аммиаку дают испариться, а остаток разбавляют водой и экстрагируют этилацетатом. После сушки и концентрирования получают 3,7 г сырого 1,2,5 (10)диенолэфира. Его растворяют в 30 мл ацетона и добавляют 3 мл 6N НСl. После перемешивания в течение 3 ч смесь нейтрализуют NаНСО3, затем разбавляют водой и экстрагируют этилацетатом, получая 2,8 г 25. Rf 0,10 (гептан/этилацетат = 4/6).
26
Раствор 0,84 г 25 в смеси 30 мл жидкого NН3 и 6 мл ТГФ обрабатывают при -60°С небольшими кусочками лития до тех пор, пока синее окрашивание реакционной смеси не будет сохраняться в течение, по меньшей мере, 5 мин. Затем избыток реагента убирают, добавляя небольшое количество NH4Cl, и выпаривают NН3. Остаток вещества разбавляют водой и экстрагируют этилацетатом. Концентрирование раствора дает 0,80 г практически чистого вещества; Rf 0,39 (гептан/этилацетат = 1/1), Rf (25) 0,24.
27
К раствору 0,8 г 26 в 8 мл дихлорметана добавляют 2,8 мл этиленгликоля, 2,5 мл триэтилортоформиата и 5 мг толуолсульфоновой кислоты. Смесь перемешивают в течение ночи, затем выливают в насыщенный раствор NаНСО3 и экстрагируют этилацетатом. Полученное таким образом сырое вещество очищают, пропуская через короткую колонку с силикагелем, получая 0,72 г 27. Rf 0,46 (гептан/этилацетат = 1/1), Rf (26) 0,38.
28
К раствору 0,72 г 27 в 15 мл ацетона добавляют 1 г молекулярных сит (4А), а затем 0,70 г N-метилморфолин-N-оксида и 30 мг перрутената тетрапропиламмония. Смесь перемешивают в течение 1 ч. К реакционной смеси добавляют 1 г силикагеля, затем 15 мл гептана и перемешивают еще 15 мин. Смесь фильтруют посредством высокоскоростной фильтрации и после частичного концентрирования помещают в этилацетат, промывают водой и концентрируют. Остаток пропускают через короткую колонку с диоксидом кремния и получают 0,59 г 28. Rf 0,62 (гептан/этилацетат = 1/1).
29
Для этинилирования получают литийацетилид из дибромэтена и бутиллития.
К раствору 0,74 мл 1,2-дибромэтена в 20 мл ТГФ добавляют при -70°С 11 мл 1,6М раствора BuLi в гексане. После перемешивания в течение 15 мин добавляют раствор 590 мг 28 в 2 мл ТГФ. Смеси дают нагреться до комнатной температуры в течение 15 мин и еще через 15 мин при комнатной температуре реакционную смесь гасят водой и экстрагируют продукт этилацетатом. Концентрирование с последующим пропусканием через короткую колонку с силикагелем дает 430 мг 29 в виде белого аморфного вещества. Rf 0,11 (гептан/ацетон = 9/1), Rf исходного вещества 0,21.
30
К раствору 0,43 г 29 в 15 мл ацетона добавляют 1 мл 2N НСl. Смесь перемешивают в течение 2 ч и затем обрабатывают насыщенным водным раствором NаНСО3 с последующей экстракцией этилацетатом, получая 0,40 г по существу чистого 30; Rf 0,18 (гептан/ацетон = 7/3), Rf (29) 0,23.
31/32
К раствору 0,40 г 30 в 4 мл ТГФ и 4 мл этанола добавляют 30 мг боргидрида натрия. После перемешивания в течение 0,5 ч добавляют несколько капель ацетона для разложения избытка реагента. Еще через 15 мин перемешивания реакционную смесь выливают в воду и экстрагируют этилацетатом, полученное таким образом сырое вещество подвергают колоночной хроматографии, получая 80 мг 3αOН изомера 32 и 160 мг 3βОН производного 31. Rf (31) 0,37, Rf (32) 0,42, Rf исходного вещества 0,48 (гептан/ацетон = 6/4).
ПРИМЕР 4
Синтез соединений (3α,11β,17β)-11-(3-бутенил)-19-норпрегн-5(10)-ен-20-ин-3,17-диола (соединение 42) и (3β,11β,17β)-11-(3-бутенил)-19-норпрегн-5(10)-ен-20-ин-3, 17-диола (соединение 43) описан со ссылкой на схему IV.
Схема IV

33
Раствор 55 г стероида 1 в смеси 300 мл ТГФ и 300 мл метанола обрабатывают раствором 2,7 г боргидрида натрия в 30 мл воды (содержащей 3 мг NaOH). После перемешивания в течение 1 ч восстановление завершается, и избыток гидрида удаляют, добавляя 75 мл ацетона. После перемешивания еще в течение 1 ч реакционную смесь выливают в воду и экстрагируют этилацетатом. Концентрирование органической фазы дает 54 г 33,
Rf 0,31 (гептан/этилацетат = 4/6), Rf исходного вещества 0,42.
34
К раствору 54 г 33 в 350 мл ДМФ добавляют 35 г имидазола, а затем 37 мл триметилсилилхлорида при -10°С. После перемешивания в течение 0,5 ч реакция завершается. Смесь выливают в 1,5 л воды и экстрагируют эфиром. Остаток, полученный после концентрирования органического вещества, растирают в 80% водном этаноле, получая 50 г чистого 34. Т.пл. 90-91°С, Rf 0,79 (гептан/этилацетат = 4/6), Rf исходного вещества 0,36.
35
К раствору бутенилмагнийбромида, полученному из 0,51 мл 4-бром-1-бутена и 119 мг Мg в 20 мл ТГФ, добавляют при -10°С 100 мг CuI. После перемешивания в течение 0,5 ч смесь охлаждают до -30°С и добавляют 1 г 34 в 5 мл ТГФ. Реакционной смеси дают нагреться до комнатной температуры примерно за 1 час. Затем добавляют 100 мл насыщенного водного раствора NH4Cl с последующей экстракцией этилацетатом. Хроматография дает 0,9 г 35. Rf 0,54 (гептан/этилацетат = 6/4), Rf исходного вещества 0,60.
36
К раствору 8,65 г 35 в 80 мл ацетона добавляют 2 мл 2N НСl. Смесь перемешивают в течение 2 ч, затем нейтрализуют насыщенным водным раствором NaHCO3, концентрируют до небольшого объема, разбавляют водой и далее экстрагируют этилацетатом. Полученное таким образом сырое вещество пропускают через короткую колонку с силикагелем, получая 5,3 г 36; Rf 0,18 (гептан/этилацетат = 7/3), Rf исходного вещества 0,51.
37
Небольшие кусочки металлического лития добавляют к раствору 5,3 г 36 в смеси 340 мл жидкого NН3, 110 мл ТГФ и 9 мл трет-бутанола при -70°С до тех пор, пока синее окрашивание реакционной смеси не будет сохраняться в течение 45 мин ± 350 мг). Избыток реагента убирают, добавляя NH4Cl. Аммиак выпаривают, а остаток разбавляют водой и экстрагируют этилацетатом. Полученное таким образом сырое вещество пропускают через короткую колонку с силикагелем, получая 3,8 г 37. Rf 0,46 (гептан/этилацетат = 6/4), Rf исходного вещества 0,22.
38
К раствору 3,3 г 37 в 60 мл метанола добавляют 5 мл триметилортоформиата и 0,3 г пара-толуолсульфоновой кислоты. После перемешивания при комнатной температуре в течение 1 ч смесь нейтрализуют, добавляя 1 мл пиридина. Данную смесь концентрируют до одной трети исходного объема, выливают в воду и экстрагируют этилацетатом, получая после хроматографической очистки 3,7 г 38. Rf 0,60 (гептан/этилацетат = 1/1), Rf исходного вещества 0,48.
39
К раствору 3,7 г 38 в 45 мл ацетона добавляют 5 г молекулярных сит (4А), а затем 3,6 г N-метилморфолин-N-оксида и 100 мг перрутената тетрапропиламмония. Смесь перемешивают в течение 1 ч. К реакционной смеси добавляют 5 г силикагеля, затем 100 мл гептана и перемешивают еще 5 мин. Смесь фильтруют посредством высокоскоростной фильтрации и после частичного концентрирования помещают в этилацетат, промывают водой и концентрируют. Остаток пропускают через короткую колонку с диоксидом кремния и получают 3,3 г 39. Rf 0,51 (гептан/этилацетат = 7/3), Rf исходного вещества 0,38.
40
Для этинилирования получают литийацетилид из дибромэтена и бутиллития.
К раствору 0,74 мл 1,2-дибромэтена в 20 мл ТГФ добавляют при -70°С 11 мл 1,6М раствора BuLi в гексане. После перемешивания в течение 15 мин добавляют раствор 0,8 г 39 в 2 мл ТГФ. Смеси дают нагреться до комнатной температуры в течение 15 мин и еще через 15 мин при комнатной температуре реакционную смесь гасят водой и экстрагируют продукт этилацетатом. Концентрирование с последующим пропусканием через короткую колонку с силикагелем дает 0,96 г 40 в виде белого аморфного вещества. Rf 0,15 (гептан/ацетон = 95/5), Rf исходного вещества 0,30.
41
К раствору 0,95 г 40 в 20 мл этанола добавляют 0,6 г щавелевой кислоты в 3 мл воды. Смесь перемешивают в течение 1,5 ч. Реакционную смесь обрабатывают NаНСО3 и концентрируют до небольшого объема. Затем добавляют воду и экстрагируют продукт этилацетатом. Выделенный таким образом сырой продукт пропускают через короткую колонку с силикагелем, получая 0,55 г 41. Rf 0,33 (гептан/этилацетат = 7/3), Rf исходного вещества 0,40.
42/43
К раствору 0,55 г 41 в 4 мл ТГФ и 4 мл этанола добавляют 30 мг боргидрида натрия. После перемешивания в течение 0,5 ч добавляют несколько капель ацетона для разложения избытка реагента. После перемешивания еще в течение 15 мин реакционную смесь выливают в воду и экстрагируют этилацетатом, полученное таким образом сырое вещество подвергают колоночной хроматографии, получая 90 мг 3αОН изомера 42, т.пл. 159°С и 150 мг 3βОН производного 43, т.пл. 90°С. Rf (42) 0,31, Rf (43) 0,23, Rf исходного вещества 0,41 (гептан/этилацетат = 6/4).
ПРИМЕР 5
Синтез соединений (3β,7α,11β,17β)-11-(2-фторэтил)-7-метил-19-норпрегн-5(10)-ен-20-ин-3,17-диола (соединение 54) и (3α,7α,11β,17β)-11-(2-фторэтил)-7-метил-19-норпрегн-5(10)-ен-20-ин-3,17-диола (соединение 55) описан со ссылкой на схему V.
45
К раствору 4 г 44 в 35 мл метанола добавляют 12 мл триметилортоформиата, затем 0,3 г толуолсульфоновой кислоты. После перемешивания в течение 1 ч исходное вещество расходуется, и реакционную смесь нейтрализуют, добавляя 1 г NaHCO3, и концентрируют. Остаток разбавляют водой и экстрагируют этилацетатом. Хроматография сырого вещества дает 3,4 г 45. Rf 0,61 (гептан/этилацетат = 7/3), Rf исходного вещества 0,35.
Схема V

46
К раствору 3,4 г 45 в 50 мл метанола добавляют 0,5 г NaOH. После перемешивания в течение 2 ч омыление завершается. Смесь концентрируют, разбавляют водой и экстрагируют этилацетатом, получая 3 г 46 в виде бесцветного масла. Rf 0,31 (гептан/этилацетат = 7/3), Rf исходного вещества 0,65.
47
К раствору 3,6 г 46 в 30 мл ДМФ добавляют 2,8 г имидазола, затем 2 г трет-бутилметилсилилхлорида. После перемешивания в течение 2 ч смесь выливают в воду и экстрагируют этилацетатом. Сырое вещество пропускают через короткую колонку с силикагелем, получая 3,8 г 47 в виде масла. Rf 0,70 (гептан/этилацетат = 7/3), Rf исходного вещества 0,61.
48
Раствор 9-BBN получают из 1,75 мл 1,5-циклооктадиена и 1,4 мл комплекса боран-диметилсульфоксид в 40 мл ТГФ. К нему добавляют 3,8 г 47 в 5 мл ТГФ. Смесь перемешивают 2 ч и затем гасят, осторожно добавляя воду (5 мл), а затем 10 мл 2N NaOH и 8 мл 30% H2O2. После энергичного перемешивания в течение 2 ч реакционную смесь еще разбавляют водой, экстрагируют этилацетатом и промывают 10% водным раствором Nа2SO3. После концентрирования и хроматографии выделяют 2,8 г 48. Rf 0,27 (гептан/этилацетат = 7/3), Rf исходного вещества 0,70.
49
Смесь 1 г 48 и 0,5 г тозилхлорида в 5 мл пиридина перемешивают в течение 5 ч при 0-5°С. Затем добавляют 1 мл воды и продолжают перемешивание в течение 15 мин. Реакционную смесь еще разбавляют водой, экстрагируют этилацетатом и сырой продукт очищают хроматографически, получая 1,1 г 49, т.пл. 120°С, Rf 0,57 (гептан/этилацетат = 7/3), Rf исходного вещества 0,30.
50
Раствор 400 мг 49 в 5 мл сухого 1М TBAF в ТГФ перемешивают в течение 5 ч, что приводит к образованию фторида и сопутствующему отщеплению силильной функции. Смесь выливают в воду и экстрагируют этилацетатом. Очистка способом хроматографии дает 185 мг 50, т.пл 161-162°С, Rf 0,35 (гептан/этилацетат = 7/3), Rf исходного вещества 0,53.
51
К раствору 180 мг 50 в 5 мл ацетона добавляют 0,5 г молекулярных сит (4А), а затем 200 мг N-метилморфолин-N-оксида и 10 мг перрутената тетрапропиламмония. Смесь перемешивают в течение 1 ч. Добавляют к реакционной смеси 0,5 г силикагеля, затем 10 мл гептана и перемешивают еще 5 мин. Смесь фильтруют посредством высокоскоростной фильтрации и после частичного концентрирования помещают в этилацетат, промывают водой и концентрируют, получая 150 мг 51, т.пл. 166°С, Rf 0,45 (гептан/этилацетат = 7/3), Rf исходного вещества 0,35.
52
Для этинилирования получают литийацетилид из дибромэтена и бутиллития.
К раствору 0,30 мл 1,2-дибромэтена в 6 мл ТГФ добавляют при -70°С 4,5 мл 1,6М раствора BuLi в гексане. После перемешивания в течение 15 мин добавляют раствор 150 мг 51 в 1 мл ТГФ. Смеси дают нагреться до комнатной температуры в течение 15 мин и еще через 15 мин при комнатной температуре реакционную смесь гасят водой и экстрагируют продукт этилацетатом. Концентрирование и обработка гептаном дают 140 мг 52 в виде белого твердого вещества, т.пл.168°С, Rf 0,38 (гептан/ацетон = 95/5), Rf исходного вещества 0,40.
53
К раствору 145 мг 52 в 3 мл этанола и 1,5 мл ТГФ добавляют 0,2 г щавелевой кислоты в 3 мл воды. Смесь перемешивают в течение 1,5 ч. Реакционную смесь обрабатывают NаНСО3 и концентрируют до небольшого объема. Затем добавляют воду и экстрагируют продукт этилацетатом. Выделенный таким образом сырой продукт пропускают через короткую колонку с силикагелем, получая 125 мг 53. Rf 0,23 (гептан/этилацетат = 7/3), Rf исходного вещества 0,38.
54/55
К раствору 125 мг 53 в 2 мл ТГФ и 1 мл этанола добавляют 20 мг боргидрида натрия. Восстановление завершают через 0,5 ч. Смесь разбавляют водой и экстрагируют этилацетатом. Выделенный таким образом сырой продукт очищают, пропуская через колонку с обращенной фазой С-18, используя в качестве элюента смесь ацетонитрил-вода и получая 40 мг 3αOН 55 и 20 мг 3βОН 54, оба аморфные вещества с одинаковыми значениями Rf на силикагеле; Rf 0,43 (CH2Cl2/ацетон = 9/1), Rf исходного вещества 0,70.
ПРИМЕР 6
Соединения данного примера получают по схеме VI.
57
Раствор 80 мг боргидрида натрия и 112 мг гидроксида натрия в 12 мл метанола добавляют по капле при 0°С к раствору 7α,11β-диметилэстр-4-ен-3,17-диона (56) в смеси 18 мл метанола и 4 мл метиленхлорида. После перемешивания в течение одного часа избыток боргидрида удаляют, добавляя 12 мл ацетона, и перемешивают еще 15 мин. Смесь выливают в воду и экстрагируют этилацетатом. Объединенные органические фазы промывают один раз насыщенным раствором NaCl и сушат, концентрируют и очищают на колонке с диоксидом кремния, получая 0,74 г 57, которому сопутствует некоторое количество 3,17-диола (последний удаляют на следующей стадии); Rf 0,40 (гептан/этилацетат = 6/4). ЯМР (CDCl3) δ: 3,60 (т, 1, 17αН), 0,77 (д, 3, 7αСН3), 1,08 (д, 3, 11βСН3).
58
К раствору 0,74 г 57 в 15 мл метанола и 1,1 мл триметилортоформиата добавляют 0,2 г пара-толуолсульфоновой кислоты. Смесь перемешивают в течение 2 ч. Затем добавляют 0,1 мл пиридина и выливают в воду. Продукт экстрагируют этилацетатом и очищают хроматографически на силикагеле, используя в качестве элюента смесь гептан/этилацетат и получая 0,59 г 3,3-диметилкеталя 58, Rf 0,47 (гептан/этилацетат = 6/4); ЯМР, δ: 3,20 (2 с, 6, ОСН3), 3,65 (т, 1, 17αН), 0,86 (с, 3, 18СН3), 0,77 (д, 3, 7αСН3), 0,90 (д, 3, 11βСН3).
Схема VI

59
К раствору 0,59 г стероида 58 в 10 мл ацетона последовательно добавляют 0,6 г N-метилморфолин-N-оксида и 40 мг перрутената тетрапропиламмония. После перемешивания в течение 1 ч добавляют 10 мл гептана и 1 г диоксида кремния. Смесь перемешивают еще 15 мин и затем фильтруют через целит, концентрируют и остаток пропускают через короткую колонку с диоксидом кремния, получая 0,56 г 59; Rf 0,55 (гептан/этилацетат = 6/4); ЯМР, δ: 0,98 (с, 3, 18 СН3), 0,82 (д, 3, 7αСН3), 0,92 (д, 3, 11βСН3), 3,20 (2×с, 6, ОСН3).
60
Гексаметилдисилазид лития получают, добавляя 1,24 мл 1,55М раствора BuLi в гексане при -40°С к раствору 0,44 мл бис-триметилсилиламина в 4 мл ТГФ. После перемешивания в течение 1/2 ч добавляют раствор 0,56 г 59 и 0,46 мл DMPU в 8 мл ТГФ. Перемешивание продолжают 45 мин при -40°С и затем добавляют 140 мкл метилйодида. После перемешивания в течение еще 30 мин алкилирование завершается. Смесь выливают в 40 мл насыщенного раствора NH4Cl и экстрагируют этилацетатом. Хроматографическая очистка дает 0,55 г 16α-метилированного продукта 60. Rf 0,73 (гептан/этилацетат = 1/1). Rf исходного вещества 0,69; ЯМР, δ: 1,11 (д, 3, 16αСН3), 1,02 (с, 3, 18СН3), 0,82 (д, 3, 7αСН3), 0,91 (д, 3, 11βСН3).
61
Получают литийацетилид, добавляя раствор 9,5 мл 1,55М BuLi в гексане к 0,60 мл 1,2-дибромэтана в 20 мл ТГФ при -60°С. После перемешивания в течение 1/2 ч добавляют раствор 0,55 г кетона 60 в 2 мл ТГФ и убирают охлаждение, чтобы смесь постепенно нагрелась до комнатной температуры. Затем добавляют воду и экстрагируют продукт этилацетатом. После хроматографической очистки получают 0,50 г 61; Rf 0,22 (гептан/этилацетат = 9/1), Rf исходного вещества 0,42; ЯМР, δ: 1,18 (д, 3, 16αСН3), 1,02 (с, 3, 18СН3), 0,78 (д, 3, 7αСН3), 0,94 (д, 3, 11βСН3), 2,66 (с, 1, ацетилен).
62
К раствору 0,49 г 61 в 20 мл этанола добавляют 50 мг щавелевой кислоты в 3 мл воды. Смесь перемешивают в течение 4 ч. Реакционную смесь обрабатывают 5% водным раствором бикарбоната натрия, а продукт экстрагируют этилацетатом и пропускают через колонку с диоксидом кремния, удаляя примеси и получая 250 мг 62. Rf 0,40 (гептан/этилацетат = 6/4), Rf исходного вещества 0,49; ЯМР, δ: 2,80 (м, 2, Н4), 1,19 (д, 3, 16αСН3), 1,04 (с, 3, 18СН3), 0,82 (д, 3, 7αСН3), 0,93 (д, 3, 11βСН3), 2,67 (с, 1, ацетилен).
63/64
К раствору 240 мг 62 в 5 мл ТГФ добавляют 360 мг литий(трет-бутокси)3АlН. Смесь перемешивают в течение 1 ч и выливают в воду, продукт экстрагируют этилацетатом. Полученные таким образом изомерные спирты разделяют путем препаративной ВЭЖХ (обращенная фаза С18), используя градиент ацетонитрила в воде. Полученные после концентрирования элюента продукты кристаллизуют из смеси вода/этанол. Это дает 68 мг 3α-гидрокси-производного 64 и 70 мг 3β-изомера 63, Rf (63/64) 0,36 (гептан/этилацетат = 6/4), Rf исходного вещества 0,54. Т.пл. (63) 165-167°С, т.пл. (64) 171-172°С. ЯМР (64), δ: 3,80 (м, 1, Н3), 2,67 (с, 1, ацетилен), 1,17 (д, 3, 16αСН3), 1,02 (с, 3, 18СН3), 0,77 (д, 3, 7αСН3), 0,90 (д, 3, 11βСН3).
ЯМР (63), δ: 4,05 (м, 1, Н3), 2,67 (с, 1, ацетилен), 1,18 (д, 3, 16αСН3), 1,02 (с, 3, 18СН3), 0,77 (д, 3, 7αСН3), 0,93 (д, 3, 11βСН3).
ПРИМЕР 7
Соединения данного примера получают по схеме VII
Схема VII

66
К раствору 10 г 11α-гидроксинордиендиона 65 в 45 мл ДМФ добавляют 10 г имидазола, затем при 0°С 8,6 мл триметилсилилхлорида. После перемешивания в течение 1 ч смесь выливают в ледяную воду и экстрагируют продукт этилацетатом. Сырое вещество хроматографируют и затем растирают с гептаном, получая 7,8 г 11-триметилсилилокси-производного 66. Т.пл. 89-90°С, ЯМР, δ: 4,08 (м, 1, 11αСН3), 5,80 (с, 1Н, Н4), 6,25 (м, 2, Н6,7), 0,15 (с, 9, триметилсилил).
67
К раствору 5,3 г 66 и 500 мг Cu(Oac)2 в 20 мл ТГФ добавляют по капле при -30°С 9,8 мл 1М метилмагнийхлорида в ТГФ. Смесь перемешивают в течение 1 ч и выливают в раствор 6 мл концентрированной серной кислоты в 300 мл воды и перемешивают в течение ночи. Продукт экстрагируют этилацетатом, сушат и концентрируют, получая 4,8 г твердого вещества, достаточно чистого для дальнейшего взаимодействия. ЯМР, δ: 5,85 (с, 1, Н4), 3,92 (м, 1, H11α), 0,83 (д, 3, 7αСН3), 0,95 (с, 3, 18СН3); Rf 0,22 (гептан/этилацетат=7/3), Rf исходного вещества 0,65.
68
К раствору 4,8 г 67 и 6,6 г NMO (N-метилморфолин-N-оксид) в 100 мл ацетона добавляют 130 мг перрутената тетрапропиламмония. После перемешивания в течение 2 ч добавляют 2 г силикагеля, затем 100 мл гептана. Смесь перемешивают еще 1/2 ч и фильтруют через целит. Фильтрат концентрируют, а остаток обрабатывают диизопропиловым эфиром, получая 4 г практически чистого триона 68. ЯМР, δ: 5,89 (с, 1, Н4), 0,89 (с, 3, 18СН3), 0,92 (д, 3, 7αСН3); Rf 0,46 (гептан/этилацетат = 7/3), Rf исходного вещества 0,24.
69
Смесь 3,5 г стероида 68, 1,3 мл этандитиола, 300 мг пара-толуолсульфоновой кислоты и 35 мл этанола кипятят с обратным холодильником в течение 1 ч. Реакционную смесь охлаждают до комнатной температуры и добавляют 80 мл холодного 0,5N NaOH. После перемешивания в течение 1 ч продукт фильтруют и промывают холодной водой. После сушки в вакууме при 50°С получают 4,6 г 69; ЯМР, δ: 5,66 (с, 1, Н4), 3,22 и 3,38 (м, 4, тиокеталь) 0,85 (с, 3, 18СН3), 0,88 (д, 3, 7αСН3); Rf 0,88 (толуол/ацетон = 7/3), Rf исходного вещества 0,59.
70
К раствору 4,0 г 69 в смеси 100 мл метанола и 50 мл метилен-хлорида добавляют при -20°С 450 мг NaBH4 небольшими порциями. После перемешивания в течение еще 1 ч восстановление завершается и реакционную смесь обрабатывают 5 мл ацетона, затем концентрируют до небольшого объема, разбавляют 100 мл воды и экстрагируют метиленхлоридом. После сушки и концентрирования получают 3,9 г по существу чистого 70; ЯМР, δ: 3,90 (м, 1, 17αН), 0,82 (с, 3, 7αСН3), 0,73 (с, 3, 18СН3); Rf 0,39 (гептан/этилацетат = 1/1), Rf исходного вещества 0,65.
71
Раствор 2,5 мл триметилсилилхлорида в 20 мл эфира добавляют по капле к смеси 4,7 г 70 и 2,6 г имидазола в 50 мл ДМФ при 0°С. После перемешивания в течение 1 ч реакционную смесь разбавляют ледяной водой и экстрагируют продукт этилацетатом. После сушки и концентрирования органической фазы остаток растирают с 80% водным этанолом на бане со льдом, фильтруют и сушат в вакууме при 50°С, получая 5,2 г 71; ЯМР, δ: 3,76 (т, 1, 17αН), 0,65 (с, 3, 18СН3), 0,80 (д, 3, 7αСН3), 5,60 (с, 1, Н4); Rf 0,86 (гептан/этилацетат = 1/1), Rf исходного вещества 0,37.
73
Суспензию 4,4 г трет-бутоксида калия и 16,2 г метилтрифенилфосфонийбромида в 130 мл толуола нагревают при 100°С в течение 1/2 ч в атмосфере азота. Желтую смесь охлаждают до 50°С и добавляют раствор 4,6 г стероида 71 в 20 мл толуола. Реакционную смесь перемешивают еще 1 ч при 100°С, охлаждают и выливают в 500 мл ледяной воды. Продукт экстрагируют толуолом, промывают, сушат и концентрируют. Остаток хроматографируют на силикагеле толуолом, удаляя большую часть загрязнений. Полученный таким образом сырой продукт 72 (6,3 г) растворяют в 20 мл ТГФ и добавляют раствор 18 мл 1М TBAF в ТГФ. После перемешивания в течение 1/2 ч смесь разбавляют водой и экстрагируют этилацетатом. Полученный таким образом продукт очищают способом хроматографии, получая 4,7 г 73; т.пл. 177-180°С; ЯМР, δ: 4,77 и 4,86 (АB, 2, метилен H'c), 5,60 (с, 1 Н4), 3,72 (м, 1, 17αН), 0,70 (с, 3, 18СН3), 0,77 (д, 3, 7α-СН3); Rf 0,10 (гептан/этилацетат = 9/1), Rf исходного вещества 0,73.
74
Раствор 2,45 г перйодной кислоты в смеси 12 мл воды и 12 мл метанола добавляют к раствору 4,7 г 73 в 40 мл метиленхлорида. Смесь перемешивают в течение 45 мин и затем разбавляют водой. Продукт экстрагируют метиленхлоридом. Органический слой промывают несколько раз 5% водным раствором тиосульфата натрия и водой, затем сушат и концентрируют. Остаток очищают способом колоночной хроматографии на силикагеле, получая 2,6 г 74; ЯМР, δ: 0,73 (с, 3, 18СН3), 0,79 (д, 3, 7αСН3), 5,88 (с, 1, Н4), 4,83, 4,94 (АВ, 2, метилен H'c), 3,78 (м, 1, 17αН); Rf 0,25 (гептан/этилацетат = 6/3), Rf исходного вещества 0,48.
75
Смесь 2,6 г 74, 7 мл триметилортоформиата, 480 мг паратолуолсульфоновой кислоты и 50 мл метанола перемешивают при комнатной температуре, контролируя реакцию способом ТСХ. Через 2,5 ч реакционную смесь выливают в насыщенный водный NaHCO3 и экстрагируют этилацетатом. После сушки и концентрирования в вакууме получают 3,1 г смеси 1/1 Δ5,6 и Δ5 (10)-кеталей. Rf 0,46 (гептан/этилацетат = 6/4), Rf исходного вещества 0,25; ЯМР (смесь кеталей 1/1), δ: 3,15, 3,22, 3,24 (с, 6, сигналы для ОСН3), 0,77, 0,87 (2×д, 3, 7αСН3), 0,65, 0,71 (2×с, 3, 18СН3).
76
Сырой продукт 75 растворяют в 60 мл ацетона. Добавляют 3,7 г N-метилморфолин-N-оксида и 75 мг перрутената тетрапропил-аммония. Смесь перемешивают в течение 2 ч, затем добавляют 1 г силикагеля и 60 мл гептана. После перемешивания в течение 15 мин реакционную смесь фильтруют через целит и фильтрат концентрируют досуха. Остаток пропускают через короткую колонку с диоксидом кремния и получают 1,9 г кетона 76 в виде смеси Δ5(10) и Δ5,6-кеталей; типичные сигналы ЯМР при δ: 0,82, 0,93 (2×д, 3, 7αСН3), 0,79, 0,85 (2×с, 3, 18СН3), 3,15, 3,22, 3,25 (с, 6, сигналы для ОСН3), 4,70, 4,85 и 4,87 и 4,93 (2×AВ, 2, метилен H'c); Rf 0,58 (гептан/этилацетат = 6/4), Rf исходного вещества 0,45.
77
Раствор 500 мг 76 в 10 мл ТГФ и 0,4 мл DMPU добавляют по капле при -40°С к 1,6 мл раствора 1М Li-гексаметилдисилазида в 10 мл ТГФ. После перемешивания в течение еще 45 мин добавляют 120 мкл метилйодида. Перемешивание продолжают еще 1 ч при -20°С и затем реакционную смесь выливают в воду и экстрагируют продукт этилацетатом. Полученное после промывания, сушки и концентрирования органической фазы вещество пропускают через колонку с диоксидом кремния и получают 510 мг 16α-метил-производного 77 в виде смеси изомеров по двойной связи; Rf 0,56 (гептан/этилацетат = 7/3), Rf исходного вещества 0,48. ЯМР, δ: 1,15, 1,17 (2×д, 16αСН3).
78
Получают Li-ацетилид, добавляя по капле 7,1 мл 1,6М BuLi в гексане к 0,46 мл 1,2-дибромэтена в 10 мл ТГФ при -60°С. После перемешивания в течение 1/2 ч добавляют раствор 500 мг стероида 77 в 10 мл ТГФ и реакционную смесь перемешивают в течение 1/2 ч, за это время температура поднимается до комнатной. Затем добавляют воду и экстрагируют продукт этилацетатом. Полученное таким образом вещество пропускают через короткую колонку с диоксидом кремния и получают 540 мг 78 в виде смеси изомеров по двойной связи. ЯМР, δ: 2,74 и 2,76 (2×с, 1, ацетилен), 1,17, 1,19 (2×д, 3, 16αСН3), 0,78, 0,88 (2×д, 3, 7αСН3); Rf 0,50 (гептан/этилацетат = 7/3), Rf исходного вещества 0,60.
79
К раствору 440 мг 78 в 20 мл ацетона добавляют 4 мл 4N НСl. После перемешивания в течение 1 ч реакция завершается, и смесь выливают в насыщенный водный NаНСО3 и экстрагируют этилацетатом. После промывания, сушки и концентрирования получают 380 мг по существу чистого 79, который непосредственно используют на следующей стадии; ЯМР, δ: 5,88 (с, 1, Н-4), 5,90 (АВ, 2, метилен), 2,76 (с, 1, ацетилен), 1,18 (д, 3, 16αСН3), 0,88 (с, 3, 18-СН3), 0,79 (д, 3, 7αСН3).
80
К раствору 280 мг 79 в смеси 30 мл жидкого NH3 и 10 мл ТГФ при -40°С добавляют небольшие кусочки Li-фольги, до тех пор пока синее окрашивание реакционной смеси не будет сохраняться в течение 15 мин. Затем небольшой избыток Li быстро гасят, добавляя твердый NH4Cl, и аммиаку дают испариться. К оставшемуся веществу добавляют 100 мл воды и смесь экстрагируют этилацетатом. Выделенное после промывания, сушки и концентрирования органическое вещество содержит почти чистый 80. Rf 0,57 (гептан/этилацетат = 6/4); ЯМР, δ: 4,78, 4,88 (АВ, 2, метилен), 2,66 (с, 1, ацетилен), 1,19 (д, 3, 16αСН3), 0,86 (с, 3, 18СН3), 0,90 (д, 3, 7αСН3).
81
К раствору 160 мг 80 в 10 мл ТГФ добавляют при 0°С небольшими порциями LiAlH4, пока не завершится реакция. После этого добавляют 0,1 мл насыщенного водного Na2SO4, а затем некоторое количество твердого Na2SO4. Смесь перемешивают 15 мин и затем фильтруют через целит. Фильтрат концентрируют, а остаток очищают, пропуская через препаративную ВЭЖХ-колонку с обращенной фазой С-18 диоксидом кремния, используя в качестве элюента смесь ацетонитрил-вода и получая 55 мг 81, т.пл. 198-199°С; Rf 0,33 (гептан/этилацетат = 6/4), Rf исходного вещества 0,57. ЯМР, δ: 3,67 (м, 1, 3αН), 4,70, 4,82 (АВ, 2, метилен), 2,65 (с, 1, ацетилен), 1,18 (д, 3, 16αСН3), 0,83 (с, 3, 18СН3), 0,89 (д, 3, 7αСН3).
ПРИМЕР 8
Соединения исследуют на их активность к рецепторам эстрогенов в тестах на связывание и в тестах на трансактивацию.
Определение конкурентного связывания с цитоплазматическим рецептором эстрогена человека из рекомбинантных клеток СНО используют для оценки относительного сродства (соотношение силы) исследуемого соединения к рецепторам эстрогена, присутствующим в цитозоле рекомбинантных клеток яичников китайского хомяка (СНО), постоянно трансфицируемых с рецептором эстрогена человека, по сравнению с эстрадиолом (E2). Цитозоль получают из рекомбинантных клеток СНО, постоянно трансфицируемых с рецептором эстрогена человека. Клеточная линия получена в Department of Biotechnology and Biochemistry (ВВС) (N.V.Organon) и известна под названием СНО-ER (2B1). Сравнительными соединениями являются этинилэстрадиол и эстриол.
Антиэстрогенную активность соединений определяют в биологических исследованиях in vitro с рекомбинантными клетками яичников китайского хомяка (СНО), постоянно сотрансфицируемыми с α (hERα) или β (hЕRβ) рецептором эстрогена человека, промотором окситоксина крыс (RO) и люциферазным репортерным геном (LUC). Антиэстрогенную активность (соотношение силы) исследуемого соединения по ингибированию трансактивации фермента люциферазы посредством рецептора эстрогена эстрогеном Org 2317 (эстрадиол, 1,3,5(10)-эстратриен-3,17β-диол) сравнивают со стандартным Org 34790 (ICI 164.384; (7α,17β)-N-бутил-3,17-дигидрокси-N-метилэстра-1,3,5(10)-триен-7-ундекан-амид).
Исследуемая среда: целые рекомбинантные клетки СНО, постоянно сотрансфицируемые с рецептором эстрогена человека, промотором окситоксина крыс и люциферазным репортерным геном. Клеточная линия продуцирована в Department of Biotechnology and Biochemistry (ВВС) (N.V.Organon) и известна под названием CHO-ERRO 2B1-1E9.
Результаты представлены ниже в таблице 1. Данные выражены в процентах от эффекта сравнительного соединения в данном исследовании.
Изобретение относится к новому типу селективных эстрогенов, имеющих стероидную структуру общей формулы I с неароматическим А-кольцом и свободной или блокированной гидроксильной группой при атоме углерода 3, в которой R1 - Н, (С1-С3)алкил или (С2-С3)ацил; R2 - Н, α-(C1-C4)алкил, α-(С2-С4)алкенил или α-(С2-С4)алкинил; R3 - Н или (C1-C4)алкил в положении 16 стероидного скелета; R4 - этинил, R5 - Н, (C1-С3)алкил или (С2-С3)ацил; R6 - (C1-C5) алкил, (С2-С5)алкенил, (С2-С5)алкинил, каждый из которых необязательно замещен хлором или фтором, пунктирная линия означает необязательную двойную связь. Соединения I обладают селективным сродством к ERα-рецепторам. 2 н. и 2 з.п. ф-лы, 1 табл. 

в которой R1 обозначает Н, (C1-С3)алкил, (С2-С3)ацил;
R2 обозначает Н, α-(C1-C4)алкил, α-(С2-С4) алкенил, α-(С2-С4)алкинил;
R3 обозначает Н или (C1-C4) алкил в положении 16 стероидного скелета;
R4 обозначает этинил;
R5 обозначает Н или (C1-С3)алкил, (С2-С3)ацил;
R6 обозначает (C1-C5)алкил, (C2-C5)алкенил, (C2-C5)алкинил, каждый из которых может быть замещен хлором или фтором,
пунктирная линия означает необязательную двойную связь.
| СПОСОБ ПОЛУЧЕНИЯ 17а-ЭТИНИЛЬНЫХ ПРОИЗВОДНЫХЭСТРАНА | 0 |
|
SU192202A1 |
| WO 9418224, A1, 18.08.1994 | |||
| US 3465010, A, 02.09.1969 | |||
| US 3652606, A, 28.03.1972. | |||

