Изобретение относится к способу получения тонкослойных молекулярных сит на различных подложках. Подложки, покрытые такими тонкими слоями, находят широкое применение в области мембранного разделения, технологии изготовления датчиков, катализа, электрохимии, электронике, а также в качестве армирующих наполнителей для полимеров.
Молекулярные сита характеризуются тем, что они представляют собой микропористые материалы с порами четко определенного размера в интервале 2-20 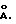 Большинство молекул продуктов, находятся ли они в газовой или жидкой фазе, как неорганических, так и минеральных, характеризуются размерами, которые при комнатной температуре находятся в этом интервале. Таким образом, наличие селективного молекулярного сита с соответствующим размером пор позволяет посредством селективной адсорбции выделять из смеси некоторые молекулы, поэтому его называют "молекулярным ситом". Помимо возможности селективной адсорбции и селективного выделения незаряженных материалов, система четко определенных пор молекулярного сита позволяет осуществлять селективный ионообмен заряженных материалов и селективный катализ. В этих последних двух случаях имеющими существенное значение свойствами, отличными от наличия микропористой структуры, являются, например, ионообменная способность, удельная площадь поверхности и кислотность. Молекулярные сита можно разделить на различные категории, например, в соответствии с их химическим составом и их структурными свойствами. Одной из категорий молекулярных сит, представляющих технический интерес, является группа, которую составляют цеолиты. Их определяют как кристаллические алюмосиликаты. Другую категорию молекулярных сит составляют силикаты металлов, которые в структурном отношении аналогичны цеолитам, за исключением того факта, что они не содержат алюминия (или содержат лишь очень малые его количества). Подробный обзор молекулярных сит приведен в работе "Molecular Sieves - Principies of Synthesis and Identification" (R. Szostak, Van Reinhold, Нью-Йорк, 1989).
Большинство молекул продуктов, находятся ли они в газовой или жидкой фазе, как неорганических, так и минеральных, характеризуются размерами, которые при комнатной температуре находятся в этом интервале. Таким образом, наличие селективного молекулярного сита с соответствующим размером пор позволяет посредством селективной адсорбции выделять из смеси некоторые молекулы, поэтому его называют "молекулярным ситом". Помимо возможности селективной адсорбции и селективного выделения незаряженных материалов, система четко определенных пор молекулярного сита позволяет осуществлять селективный ионообмен заряженных материалов и селективный катализ. В этих последних двух случаях имеющими существенное значение свойствами, отличными от наличия микропористой структуры, являются, например, ионообменная способность, удельная площадь поверхности и кислотность. Молекулярные сита можно разделить на различные категории, например, в соответствии с их химическим составом и их структурными свойствами. Одной из категорий молекулярных сит, представляющих технический интерес, является группа, которую составляют цеолиты. Их определяют как кристаллические алюмосиликаты. Другую категорию молекулярных сит составляют силикаты металлов, которые в структурном отношении аналогичны цеолитам, за исключением того факта, что они не содержат алюминия (или содержат лишь очень малые его количества). Подробный обзор молекулярных сит приведен в работе "Molecular Sieves - Principies of Synthesis and Identification" (R. Szostak, Van Reinhold, Нью-Йорк, 1989).
Процессы мембранного селективного разделения вызывают к себе значительный интерес, отчасти благодаря тому факту, что они потенциально более эффективны и экономически более выгодны в сравнении с применяемыми в настоящее время методами разделения и отчасти благодаря тому факту, что их применение позволяет открыть новые возможности разделения, которые с применением современной доступной техники невозможны. Существенный интерес представляет также создание каталитических мембранных реакторов и химических реакторов улучшенной селективности. Ограничения, с которыми связано применение мембран в различных областях техники, обусловлены прежде всего самими мембранами. Эксплуатационные характеристики современных доступных мембранных материалов обычно хуже оптимальных, если рассматривать производительность, селективность, тепловые и механические свойства, а также стойкость к биологическому разложению. Известно, что с использованием мембран на цеолитной основе могут быть достигнуты заметные усовершенствования.
Мембраны, которые состоят только из материала молекулярного сита, известны и описаны в различных патентных и иных публикациях. У Suzuki [(ЕР-А 180200 (1986)] описан способ приготовления цеолитной мембраны нанесением на подложку гелевого покрытия с последующей гидротермической обработкой гелевого покрытия с получением цеолитного тонкого слоя. В другом способе [US 4699892 (1987)] подложку вначале пропитывают синтезным гелем, который в дальнейшем в условиях гидротермического синтеза превращают в цеолит. В US 4800187 (1989) описан способ, в котором эти цеолитные тонкие слои получают взаимодействием поверхности подложки с активным диоксидом кремния.
В заявке WO 94/25151 предлагается наносить на подложку неорганический слой, включающий необязательно смежные частицы кристаллического молекулярного сита, причем средний размер таких частиц находится в интервале от 20 нм до 1 мкм. В предпочтительном варианте подложка является пористой. Когда поры подложки закрыты в такой степени, в которой они являются эффективно закрытыми, а подложка является непрерывной, образуется мембрана молекулярного сита; преимущество таких мембран состоит в том, что они при определенных условиях в состоянии обеспечить катализ и одновременное разделение. Хотя предлагаемые в этой заявке продукты эффективны во многих процессах разделения, кристаллы слоя характеризуются неупорядоченностью, в результате чего грани зерен могут препятствовать диффузии материалов через мембрану, а полости между кристаллами влияют на селективность.
В РСТ/ЕР93/01209 и PCT/US95/08514, опубликованной под номером WO 96/01687, описано применение слоев зародышеобразования на подложках для изготовления слоев молекулярных сит. Эти зародышеобразующие слои являются относительно толстыми и неупорядоченными, поэтому толщина слоев молекулярных сит относительно велика.
Согласно PCT/US95/08511, опубликованной под номером WO 96/01685, слои молекулярных сит синтезируют без применения зародышеобразующего слоя. Толщина образующегося слоя молекулярного сита относительно велика, составляя 2-100 мкм. В этом способе получают слои молекулярных сит, которые характеризуются относительно низкой плотностью молекулярного сита на границе с подложкой.
В заявке ЕР-А 87308451.1, опубликованной под номером ЕР 0270212, описан способ получения покрытия в виде закрытой упаковки коллоидных частиц. В виде начальной стадии в этом способе на границе жидкость/воздух предусмотрена необходимость формирования монослоя. Далее перед нанесением на подложку этот монослой подвергают сжатию.
Согласно заявке PCT/NL92/00029, опубликованной под номером WO 92/13631, неорганическую композитную мембрану изготовляют из предварительно сформированных кристаллов молекулярного сита, в результате чего между ними имеется газонепроницаемая матрица.
Более того, значительный интерес был проявлен к созданию новых и усовершенствованных композитных материалов, которые состоят главным образом из волокнистых неорганических материалов в сочетании с полимерами и пластиками различных типов. Для достижения предпочтительных механических свойств этих композитов были предприняты усилия с целью обеспечить совместимость, а там, где это целесообразно, создать между волокнами и полимерами наилучшие химические связи. Для неорганических волокон это может быть достигнуто с использованием пригодных для этой цели связующих веществ, т.е. соединений, характерная особенность которых состоит в том, что они могут содержать функциональные группы с сильным сродством как к волокну, так и к полимеру. Для достижения этой цели было разработано несколько способов. Известно также, что неорганические молекулярные сита некоторых типов проявляют высокое сродство к органическим соединениям, таким, как мономеры, используемые при получении полимеров, и известно, что волокна, покрытые молекулярными ситами, когда их используют в качестве наполнителей в полимерных матрицах, могут придать им армирующие свойства.
С использованием способов, известных в данной области техники, можно получать непрерывные тонкие слои молекулярных сит. Однако размеры этих тонких слоев таковы, что предел меньшей толщины находится в интервале, превышающем 1-10 мкм, причем конкретный предел зависит от типа используемого молекулярного сита. Попытки получить с использованием современной доступной технологии более тонкие слои ведут к образованию несплошных тонких слоев, которые находят очень ограниченное применение в современных областях техники. Так, например, известные до настоящего времени структуры молекулярных сит, применяемые в процессах разделения, могут ограничивать расход потока материала, проходящего через мембрану, из-за характеристик структуры, ограничивающих истечение, и/или мембране могут быть свойственны дефекты, которыми обусловлено наличие путей неселективного прохождения через мембрану. Тем не менее предпринимаются попытки получения еще более тонких слоев, поскольку благодаря техническим преимуществам таких тонких слоев они нашли бы несколько потенциальных областей применения. Однако на пути к получению очень тонких мембран возникают сочетания таких проблем, а в результате осуществления некоторых известных способов, которые упомянуты выше, получают мембранные структуры низкой плотности на границе с подложкой, что может быть устранено только применением более толстых мембран или серьезной компенсационной обработкой мембраны. При применении тонких слоев молекулярных сит в качестве мембран в процессах разделения на расход потока через мембрану влияет главным образом толщина такого тонкого слоя. Чем тоньше мембрана, тем больше расход потока. При применении в качестве химического датчика наибольшее значение имеет время отклика. Для конкретного молекулярного сита уменьшение толщины тонкого слоя сокращает время отклика. В области каталитических процессов усилия направлены на устранение низких скоростей реакций из-за ограниченного диффузионного переноса материалов в катализаторе. В реакторах с каталитическими мембранами, в которых активной фазой является молекулярное сито, уменьшением толщины тонкого слоя уменьшают сопротивление диффузии через поры.
Часто желательно получить тонкие слои неорганических материалов, которые не содержат трещин, а в большинстве случаев это является необходимым предварительным условием. При получении тонких слоев неорганических материалов в упомянутых областях применения часто целесообразно, а во многих случаях необходимо иметь тонкие слои, которые свободны от трещин и больших пор. Располагая имеющимися в настоящее время способами трудно получить цеолитные тонкие слои, которые свободны от трещин как непосредственно после получения, так и после воздействия высоких температур.
Другой проблемой, с которой связано получение мембран путем формирования тонких слоев молекулярных сит на пористых подложках в соответствии с известной технологией, является блокирование пористой системы подложки вследствие осаждения материала молекулярного сита на этой пористой системе, из-за чего действенно утолщается образующий слой.
Еще одна проблема, с которой связано получение тонких слоев молекулярных сит на подложках в соответствии с известной технологией, состоит в том, что число возможных сочетаний подложка/молекулярное сито ограничено фактором условий, необходимых для синтеза самых разнообразных сит, которые настолько жестки, что подложка растворяется или подвергается травлению.
Проблема использования волокон в композитных материалах или в качестве наполнителей в полимерах и пластиках заключается в низкой степени совместимости между полимером и наполнителем, вследствие чего в конечном счете получают материал с неудовлетворительными механическими свойствами. Однако с использованием известной технологии эту проблему несовместимости можно полностью или частично разрешить, но только за счет заметного повышения производственных затрат.
Согласно настоящему изобретению, позволяющему решить проблемы, с которыми связано осуществление известных способов получения тонких слоев молекулярных сит на подложках, предлагается новый способ получения тонких слоев молекулярных сит на подложках, прежде всего очень тонких слоев.
Задачей настоящего изобретения является устранение недостатков, которые свойственны известным способам, применяемым при получении тонких слоев молекулярных сит, и разработать новый способ, осуществление которого позволяет получать, в частности, очень тонкие непрерывные слои этого типа. Другой задачей настоящего изобретения является разработка способа осаждения очень тонких слоев молекулярных сит на поверхности пористой подложки без одновременного блокирования пор подложки вследствие осаждения на эти поры материала молекулярного сита. Еще одной задачей настоящего изобретения является разработка способа, осуществление которого дает возможность получать очень тонкие слои молекулярных сит на подложках, которые в условиях, обычно создаваемых для синтеза молекулярного сита, растворяются или подвергаются травлению. Кроме того, задачей настоящего изобретения является разработка способа получения очень тонких слоев молекулярных сит, которые свободны от трещин как непосредственно после получения, так и после термической обработки.
В основу настоящего изобретения также была положена задача обеспечить возможность получения очень тонких слоев молекулярных сит как на плоских, так и волокнистых подложках.
Таким образом, в изобретении предлагается способ получения тонких слоев молекулярных сит, прежде всего очень тонких слоев, при осуществлении которого отдельные микрокристаллы молекулярных сит связывают с поверхностью подложки, формируя монослой, включающий микрокристаллы молекулярного сита, который в дальнейшем заставляют расти с формированием сплошного слоя молекулярного сита.
Наиболее важным аспектом способа по настоящему изобретению является получение промежуточного продукта, в котором в качестве ключевого компонента имеется монослой, включающий микрокристаллы молекулярного сита на приемлемой подложке.
В контексте данного описания термин "монослой" обозначает слой, включающий микрокристаллы, которые наносят на подложку в практически одной и той же плоскости. Классический монослой могут образовывать микрокристаллы и другие материалы, если они имеются, которые плотно упакованы. В другом варианте микрокристаллы и другие материалы, если они имеются, упакованы неплотно, и, следовательно, они образуют субмонослой. Конкретно необходимая плотность упаковки в определенной мере зависит от природы микрокристаллов молекулярного сита и целевого тонкого слоя молекулярного сита, который необходимо вырастить из этих микрокристаллов. В любом случае плотность упаковки микрокристаллов в монослое должна быть такой, при которой обеспечивается возможность выращивания из этого слоя тонкого слоя молекулярного сита. Если микрокристаллы находятся в монослое в условиях неадекватной плотности, выращивание кристаллов тонкого слоя молекулярного сита в таком монослое, обеспечивающее получение плотного слоя молекулярного сита, осуществлялось бы в таких условиях, в которых образуется толстый слой молекулярного сита.
Согласно первому объекту настоящего изобретения предлагается способ получения монослойной структуры, содержащей микрокристаллы молекулярного сита, который включает:
а) приготовление дисперсии, содержащей дискретные микрокристаллы молекулярного сита, которые обладают поверхностным зарядом,
б) выбор или изготовление подложки, поверхностный заряд которой противоположен заряду дискретных микрокристаллов в дисперсии,
в) введение подложки в контакт с дисперсией, содержащей дискретные микрокристаллы молекулярного сита, вследствие чего эти дискретные микрокристаллы молекулярного сита притягиваются к подложке и сцепляются с ней в виде монослоя.
Согласно второму объекту настоящего изобретения предлагается способ получения структуры, содержащей тонкий слой молекулярного сита, который включает:
а) осаждение на подложке монослоя, содержащего микрокристаллы молекулярного сита, которые способны служить центрами кристаллизации, вызывающими рост тонкого слоя молекулярного сита,
б) приготовление синтезного раствора для молекулярного сита,
в) введение компонентов а) и б) в контакт и гидротермический рост молекулярного сита с получением на подложке тонкого слоя молекулярного сита.
Согласно третьему объекту настоящего изобретения предлагается структура, которая включает подложку и тонкий слой, представляющий собой кристаллическое молекулярное сито, и в которой этот тонкий слой молекулярного сита содержит введенный в его структуру монослой, содержащий микрокристаллы молекулярного сита.
Настоящее изобретение особенно эффективно при получении очень тонких слоев молекулярных сит. Термином "очень тонкие слои" обозначают тонкие слои, толщина которых составляет менее 2 мкм, в идеальном варианте менее 1 мкм, предпочтительно менее 0,25 мкм и наиболее предпочтительно менее 0,1 мкм.
Используемый в описании настоящего изобретения термин "микрокристалл" служит для обозначения кристаллов молекулярного сита с размерами менее 500 нм, предпочтительно менее 200 нм, кристаллическую структуру которых можно идентифицировать по дифракции рентгеновских лучей.
В заявке PCT/SE 93/00715, опубликованной под номером WO 94/05597, описание которой включено в настоящее описание в качестве ссылки, представлен способ, осуществление которого позволяет синтезировать коллоидные суспензии дискретных микрокристаллов молекулярного сита, пригодных для использования при получении монослойных структур по настоящему изобретению. Молекулярные сита, такие, как цеолиты и кристаллические микропористые силикаты металлов, обычно синтезируют гидротермической обработкой силикатного раствора четко определенного состава. Этот состав, а также параметры синтеза, такие, как температура, время и давление, определяют тип получаемого продукта и кристаллической формы.
Пригодные для использования по настоящему изобретению микрокристаллы молекулярного сита включают нанокристаллические цеолиты, которые представляют собой кристаллиты с размерами от примерно 10  до 1 мкм. Нанокристаллические цеолиты могут быть приготовлены, например, в соответствии со способами, приведенными в заявке РСТ/ЕР92/02386, опубликованной под номером WO 93/08125, описание которой включено в настоящее описание в качестве ссылки, или по другим способам, известным специалистам в данной области техники. Коллоидные частицы, размеры которых находятся в пределах 50-10000
до 1 мкм. Нанокристаллические цеолиты могут быть приготовлены, например, в соответствии со способами, приведенными в заявке РСТ/ЕР92/02386, опубликованной под номером WO 93/08125, описание которой включено в настоящее описание в качестве ссылки, или по другим способам, известным специалистам в данной области техники. Коллоидные частицы, размеры которых находятся в пределах 50-10000 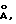 образуют стабильную дисперсию или раствор дискретных частиц. Предпочтительный размер коллоидных частиц составляет 250-5000
образуют стабильную дисперсию или раствор дискретных частиц. Предпочтительный размер коллоидных частиц составляет 250-5000  наиболее предпочтительно менее 1000
наиболее предпочтительно менее 1000 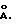 Коллоидные частицы с размерами <5000
Коллоидные частицы с размерами <5000  получить достаточно легко. После кальцинирования цеолит обычно представляет собой нанокристаллический цеолит или цеолит, размер частиц которого позволяет образовывать коллоид. Типичные примеры молекулярных сит (цеолитов), которые могут быть использованы, включают (хотя ими их список не ограничен) молекулярные сита структурных типов AFI, AEL, ВЕА, СНА. EUO, FAU, FER, KFI, LTA, LTL, MAZ, MOR, MEL, MTN, MTT, MTW, OFF, TON (включая цеолит Х и цеолит Y), бета-цеолит и прежде всего цеолиты MFI. В предпочтительном варианте обычно используют цеолит MFI со значением соотношения между кремнием и алюминием свыше 30, включая композиции без алюминия. В настоящем описании цеолиты MFI со значениями соотношения Si/Al свыше 300 названы силикалитами. Хотя некоторые из вышеупомянутых материалов истинными цеолитами не являются, в литературе их часто называют цеолитами, поэтому в настоящем описании понятие "цеолит" использовано в широком смысле, охватывающем такие материалы.
получить достаточно легко. После кальцинирования цеолит обычно представляет собой нанокристаллический цеолит или цеолит, размер частиц которого позволяет образовывать коллоид. Типичные примеры молекулярных сит (цеолитов), которые могут быть использованы, включают (хотя ими их список не ограничен) молекулярные сита структурных типов AFI, AEL, ВЕА, СНА. EUO, FAU, FER, KFI, LTA, LTL, MAZ, MOR, MEL, MTN, MTT, MTW, OFF, TON (включая цеолит Х и цеолит Y), бета-цеолит и прежде всего цеолиты MFI. В предпочтительном варианте обычно используют цеолит MFI со значением соотношения между кремнием и алюминием свыше 30, включая композиции без алюминия. В настоящем описании цеолиты MFI со значениями соотношения Si/Al свыше 300 названы силикалитами. Хотя некоторые из вышеупомянутых материалов истинными цеолитами не являются, в литературе их часто называют цеолитами, поэтому в настоящем описании понятие "цеолит" использовано в широком смысле, охватывающем такие материалы.
Синтезную смесь, предназначенную для получения микрокристаллов молекулярных сит, которую эффективно наносят на подложку, целесообразно готовить по способу, описанному в WO 93/08125. В таком способе синтезную смесь готовят кипячением водного раствора источника диоксида кремния и органического структуронаправляющего средства в пропорции, достаточной для того, чтобы вызвать практически полное растворение источника диоксида кремния. Органическое структуронаправляющее средство, если его используют, целесообразно вводить в синтезную смесь в форме основания, в частности в форме гидроксида, или в форме соли, например, галогенида, в частности бромида. Если целесообразно или необходимо регулировать рН смеси, можно применять смеси основания с солью.
Другие приемлемые микрокристаллы молекулярных сит могут быть получены по способам, представленным в заявках РСТ/ЕР96/03096, РСТ/ЕР96/03097 и РСТ/ЕР96/03098, описания которых включены в настоящее описание в качестве ссылок.
Структуронаправляющим средством может служить, например, гидроксид или соль тетраметиламмония (ТМА), тетраэтиламмония (ТЭА), триэтилметиламмония (ТЭМА), тетрапропиламмония (ТПА), тетрабутиламмония (ТБА), тетрабутилфосфония (ТБФ), триметилбензиламмония (ТМБА), триметилцетиламмония (ТМЦА), триметилнеопентиламмония (ТМНА), трифенилбензилфосфония (ТФБФ), биспирролидиния (БП), этилпиридиния (ЭП), диэтилпиперидиния (ДЭПП) или замещенного азонийбициклооктана, например, метил- или этилзамещенный хинуклидин или 1,4-диазобицикло(2,2,2)октан. Можно также использовать 1,6-диаминогексан, 1,8-диаминооктан или кронэфир.
Предпочтительными структурообразующими средствами являются гидроксиды и галогениды ТМА, ТЭА, ТПА и ТБА.
Монослой по настоящему изобретению может далее включать дополнительные материалы, такие, как диоксид кремния и/или оксид металла; металлические частицы; металлические частицы с оксидами металлов и/или диоксидом кремния. Монослой может быть получен из раствора, содержащего нанокристаллический или коллоидный цеолит или смесь оксида металла с нанокристаллическим или коллоидным цеолитом, или смесь нанокристаллического или коллоидного цеолита с коллоидным металлом. В предпочтительном варианте для получения монослоя обычно используют нанокристаллический или коллоидный цеолит или смесь оксида нанокристаллического или коллоидного цеолита с оксидом металла. Оксиды металлов, из которых получают монослой, представляют собой оксиды металлов или полимерные оксиды металлов, полученные по золь-гелевому методу. В соответствии с этой особенностью настоящего изобретения оксиды металлов, которые используют при его осуществлении, выбирают из группы, включающей коллоидный оксид алюминия, коллоидный диоксид кремния, коллоидный диоксид циркония, коллоидный диоксид титана, полимерные оксиды металлов, полученные по золь-гелевому методу, и их смеси. В предпочтительном варианте обычно используют коллоидный оксид алюминия. К коллоидным металлам, которые могут быть использованы, относятся медь, платина и серебро. В случае применения эти дополнительные материалы также должны обладать поверхностным зарядом, необходимым для подложки.
Регулирование соотношения между коллоидным цеолитом и оксидом металла позволяет регулировать плотность участков зародышеобразования в монослое. Такая плотность регулирует морфологию роста тонкого слоя цеолита по всему этому слою на последующей стадии гидротермического синтеза. Чем выше плотность зародышеобразования, тем меньшей шириной характеризуются кристаллы молекулярного сита на границе тонкий слой/подложка. Плотность зародышеобразования можно регулировать относительным содержанием микрокристаллов и оксидов металлов (с увеличением количества используемых оксидов металлов плотность увеличивается), а также размерами микрокристаллов в монослое. Таким образом, в этом слое используют микрокристаллы с размерами в интервале от 50 до 10000 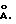 Чем больше используемые в этом слое микрокристаллы, тем обычно шире цеолитные кристаллы в верхнем слое. В предпочтительном варианте монослой по существу состоит из микрокристаллов молекулярного сита, а в наиболее предпочтительном варианте эти микрокристаллы молекулярного сита представляют собой цеолитные кристаллы, которые по природе являются коллоидными, с предпочтительными размерам менее 100 нм.
Чем больше используемые в этом слое микрокристаллы, тем обычно шире цеолитные кристаллы в верхнем слое. В предпочтительном варианте монослой по существу состоит из микрокристаллов молекулярного сита, а в наиболее предпочтительном варианте эти микрокристаллы молекулярного сита представляют собой цеолитные кристаллы, которые по природе являются коллоидными, с предпочтительными размерам менее 100 нм.
Осуществление этой стадии способа получения тонких слоев молекулярных сит в соответствии с настоящим изобретением обеспечивает сцепление с поверхностью подложки одного или нескольких почти непрерывных монослоев дискретных монокристаллов, которые в конечном итоге образуются и становятся составной частью тонкого слоя молекулярного сита. Для достижения наилучших результатов во время осаждения микрокристаллы должны образовывать дискретные частицы, а не соединяться в совокупности нескольких частиц.
В предпочтительном варианте размеры кристаллов среди микрокристаллов молекулярного сита в монослое составляют не более 500 нм, предпочтительно не более 300 нм, в идеальном случае находятся в интервале от 10 до 300 нм, предпочтительно от 20 до 200 нм и наиболее предпочтительно от 20 до 120 нм.
Предпочтительный размер дополнительных частиц, если они присутствуют, является таким же или аналогичным размерам микрокристаллов молекулярного сита.
Ключевым аспектом этой стадии предлагаемого способа является выбор и подготовка подложки, на которой необходимо осаждать монослой.
Выбор соответствующей подложки предполагает, что поверхности подложки следует или можно сообщить заряд, который достаточно силен и по знаку противоположен заряду адсорбируемых материалов. Большинство молекулярных сит, представляющих интерес для осуществления настоящего изобретения, представляют собой силикаты металлов, которые в нейтральных или щелочных водных суспензиях могут характеризоваться отрицательным зарядом. Поверхностный заряд обычно достигает максимальных значений в интервале рН 8-12, и, следовательно, этот интервал рН приемлем для адсорбции микрокристаллов на поверхностях подложки.
Молекулярные сита определенных типов готовят в присутствии стехиометрического избытка тетраалкиламмониевых ионов. В таких случаях адсорбции на поверхностях некоторых типов способствуют, заменяя избыток тетраалкиламмониевых ионов, например, аммониевыми ионами. Этого можно достичь пропусканием суспензии микрокристаллов через колонну, содержащую в качестве насадки органическую ионообменную смолу в аммониевой форме, или добавлением ионообменной смолы в такой форме в суспензию, содержащую микрокристаллы, и после завершения ионообмена выделением ионообменной смолы из суспензии, например, фильтрованием или центрифугированием.
Подложка, используемая по настоящему изобретению, может быть либо непористой, либо пористой. В качестве примеров непористых подложек можно назвать стекло, плавленый кварц, диоксид кремния, кремний, плотную керамику, например, глину, и металлы. В качестве примеров пористых подложек можно упомянуть пористое стекло, спеченные пористые металлы, например, сталь, никель и благородные металлы, и прежде всего неорганические окислы, например, альфа-алюминийоксид, диоксид титана, смесь оксид алюминия/диоксид циркония, кордиерит или цеолит, как он определен в настоящем описании.
Примеры подложек, которые представляют основной интерес для покрытий тонкими слоями молекулярных сит с целью применения в области технологии изготовления датчиков, включают кремниевые пластины, кварц, оксид алюминия, силикат алюминия и благородные металлы. Примерами материалов, которые могут быть использованы при изготовлении мембран для процессов разделения и в определенной мере каталитических мембран, являются подложки из пористого оксида алюминия, диоксида кремния, алюмосиликатов, спеченного металла и полимеров. В области электрохимии и электронике первостепенный интерес представляют подложки из металлов и сплавов. К примерам волокнистых материалов, на которые можно наносить покрытия в соответствии со способами, представленными в описании настоящего изобретения, относятся стекловолокно, керамические волокна, углеродное волокно, графитовое волокно, целлюлозные волокна и различные полимерные волокна. Все вышеупомянутые подложки могут быть изготовлены в соответствии с известными методами, а некоторые подложки технически доступны.
Подложкой может служить любой материал, совместимый с техникой осаждения монослоя и синтеза тонкого слоя, как это изложено, например, ниже, в частности пористый альфа-алюминийоксид, с размерами поверхностных пор в интервале от 0,004 до 100 мкм, преимущественно от 0,05 до 10 мкм, предпочтительно от 0,08 до 1 мкм, наиболее предпочтительно от 0,08 до 0,16 мкм, и предпочтительно с узким диапазоном распределения пор по размерам. Подложка может быть многослойной. Так, например, для улучшения характеристик массопереноса подложки малым диаметром пор может характеризоваться только та зона поверхности подложки, которая контактирует с монослоем, в то время как основная масса пор подложки в направлении к поверхности, удаленной от этого слоя, может обладать большим диаметром. Примером такой многослойной подложки является диск из альфа- алюминийоксида, поры диаметром приблизительно 1 мкм которого покрыты слоем альфа-алюминийоксида с размерами пор примерно 0,08 мкм.
Для получения слоев на пористых подложках с целью избежать образования тонкого слоя на пористой структуре подложки и блокирования пористой системы подложки часто целесообразно выбирать размер микрокристаллов или пористость подложки таким образом, чтобы размер микрокристаллов несколько превышал размеры пор подложки.
Многими материалами подложек, представляющих интерес для получения тонких слоев молекулярных сит в соответствии с настоящим изобретением, служат оксиды, или поверхность этих материалов покрыта оксидным слоем, а это означает, что в водной суспензии при рН в целевом интервале подложка также имеет отрицательный заряд. В таких случаях с той целью, чтобы сделать поверхность подложки приемлемой для адсорбции отрицательно заряженных микрокристаллов, ее можно зарядить противоположным зарядом, придав ей положительный заряд. Такое изменение знака заряда на противоположный может быть достигнуто обработкой подложки раствором, содержащим 0,1-4 мас.% катионоактивного полимера. Значение рН при изменении знака заряда на противоположный выбирают после оценки химической природы как подложки, так и полимера. Однако катионоактивные полимеры можно использовать в широком диапазоне рН. Повторяющимися звеньями в таких полимерах могут быть четвертичные амины с гидроксильными группами в главной цепи. Примером такого полимера является продукт Berocell 6100, водорастворимый полимер с повторяющимися звеньями [СН2СH(ОH)СН2N(СН3)2] n + и молекулярной массой 50000 г/моль, поставляемый на рынок фирмой Akzo Nobel АВ, Швеция. Другие приемлемые полимеры хорошо известны в данной области техники.
Альтернативным, но менее предпочтительным методом, является изменение знака заряда на противоположный не у поверхности подложки, а у микрокристаллов молекулярного сита. Этого можно достичь в соответствии с известными методами, аналогичными тем, которые применяют для изменения знака заряда подложки.
Для придания некоторым подложкам удовлетворительных поверхностных свойств может оказаться целесообразным предусмотреть для них одну или несколько стадий предварительной обработки, целью которой является очистка или химическая модификация их поверхности. В этих случаях подложку целесообразно обрабатывать осуществлением одной или нескольких таких стадий, как обработка щелочью, кислотой и окислительная очистка, или сочетания таких стадий. Эффективность адсорбции молекулярных сит на кварце и оксиде алюминия часто можно повысить обработкой подложек, включающей осаждение тонкого кремнийдиоксидного слоя, что обеспечивает получение поверхности с высокой плотностью гидроксильных групп и повышенной плотностью поверхностного заряда в условиях, в которых происходит адсорбция. Другой путь улучшения осаждения микрокристаллов состоит в осуществлении двух или большего числа стадий адсорбции, что может происходить в промежутке перед изменением знака заряда. В случае подложек некоторых типов, нанесение на которые очень тонких цеолитных слоев покрытий в соответствии с настоящим изобретением может представлять интерес, описанный выше метод осаждения более или менее завершенного микрокристаллического монослоя оказывается неудовлетворительным, поскольку при его осуществлении на поверхности подложки невозможно обеспечить создание достаточно большого поверхностного заряда. Примерами таких поверхностей являются поверхности благородных металлов и большинства органических полимеров. В таких случаях в соответствии с известной технологией могут быть использованы связующие вещества, например, силанового типа. Эти связующие вещества характеризуются тем, что они включают две функциональные группы, причем одна из них обладает сродством к поверхности подложки, а вторая присоединяется к поверхности микрокристалла. Для связи микрокристаллов цеолита или силиката металла с поверхностями благородных металлов часто приемлем силан, содержащий тиоловую группу. Связующие вещества выпускают, например, на фирмах Dow Corning и Union Carbide и их обычно используют для введения в органические полимеры неорганических наполнителей и армирующих добавок. Связующее вещество можно наносить на подложку и затем гидролизовать для придания требуемого поверхностного заряда, или оно может обладать характерными для него функциональными группами, которые придают требуемый заряд. Пригодные для этой цели связующие представляют собой химические агенты, которые хорошо известны в данной области техники, такие, как выпускаемые на фирме OSi specialties под названием "Silquest Silanes" и перечисленные в посвященной этим продуктам брошюре за 1994 г. Связующее вещество может быть использовано в сочетании с катионоактивными полимерами, которые упомянуты выше, для придания им требуемого поверхностного заряда. Таким образом, изменение знака или регулирование заряда может быть обеспечено созданием соответствующего рН раствора, в который погружают подложку и который содержит микрокристаллы, с целью сообщить кристаллам и поверхности подложки противоположные по знаку заряды; нанесением катионоактивного полимера, который придает соответствующий заряд, противоположный по знаку заряду микрокристаллов; или использованием связующего вещества с гидролизом или без него и/или с приемлемым катионоактивным полимером.
С целью обеспечить полноту формирования настоящего монослоя или добиться целевой плотности покрытия поверхности подложки под монослоем процесс осаждения монослоя можно повторять несколько раз.
В соответствии с одним из вариантов подложку с осажденным на ней монослоем помещают в синтезную смесь без какой-либо дальнейшей обработки этого монослоя. Даже при погружении в синтезную смесь микрокристаллы монослоя остаются сцепленными с подложкой, что упрощает рост тонкого слоя молекулярного сита. Однако в таких условиях, которые, например, создаются при перемешивании синтезной смеси в процессе гидротермического синтеза, прочность сцепления между частицами и подложкой может оказаться недостаточной, и потребуются шаги, необходимые для стабилизации монослоя и закрепления его положения.
Таким образом, в предпочтительном варианте перед помещением монослоя в синтезную смесь его стабилизируют или фиксируют. Согласно одному из вариантов эту стабилизацию можно обеспечить тепловой обработкой монослоя, например, при температуре 30-1000oС, предпочтительно свыше 50oС, более предпочтительно в пределах 200-1000oС и наиболее предпочтительно свыше 300oС. Предпочтительный диапазон составляет, по-видимому, 400-600oС. Такую тепловую обработку предпочтительно проводить в течение по меньшей мере двух часов с использованием водяного пара или без него.
По другому методу стабилизации монослой можно обрабатывать раствором, который модифицирует поверхностные характеристики микрокристаллов монослоя. Так, например, этот слой можно промывать раствором, который вызывал бы флокуляцию микрокристаллических частиц монослоя. Не основываясь на какой-либо теории, полагают, что процессы, аналогичные флокуляции в коллоидных растворах, также могут более прочно связывать микрокристаллы в монослоях. Приемлемые растворы включают те, которые содержат материалы, обычно участвующие в ионообмене с монослоем. К ним относятся растворы с ионами двухвалентных металлов, такие, как, например, растворы, содержащие соли щелочноземельных металлов. Среди таких процессов можно упомянуть, в частности, промывку разбавленным раствором соли Ca, например, раствором СаСl2. Этот вариант может предусматривать дополнительную стадию выдержки обработанного слоя при температуре до 300oС, предпочтительно до 200oС. Специалистам в данной области техники обычно известно, что для стабилизации монослоя могут быть использованы многие другие растворы и можно проводить многие другие процессы обработки.
При получении в соответствии с настоящим изобретением тонкого слоя молекулярного сита применяют монослойную структуру по настоящему изобретению. При осуществлении предлагаемого способа монослойную структуру вводят в контакт с синтезным раствором, предназначенным для целевого молекулярного сита.
При получении тонких слоев молекулярных сит в соответствии с настоящим изобретением осажденным микрокристаллам дают расти на поверхности подложки. Этот рост первоначально дискретных кристаллов ведет к их взаимному сцеплению, и более или менее завершенный монослой дискретных кристаллов на поверхности подложки преобразуется в сплошной и плотный тонкий слой молекулярного сита. Толщина самого тонкого слоя, необходимая для того, чтобы тонкий слой был сплошным и плотным, определяется как размерами осажденных кристаллов, так и плотностью упаковки таких кристаллов на поверхности подложки, а также их ориентацией на поверхности подложки. В случае максимальной плотности упаковки для получения сплошного и свободного от трещин тонкого слоя в большинстве случаев этого достаточно для роста кристаллов до толщины слоя, которая соответствует полуторакратной толщине монослоя, которая, когда кристаллы являются приблизительно сферическими, соответствует полуторакратному диаметру первоначально осажденных кристаллов. Когда геометрическая форма кристаллов отлична от сферической, их можно осаждать на подложке с получением монослоя, который состоит из кристаллов, которые сориентрированы и могут быть, кроме того, плотно упакованными. В этом случае верхние поверхности кристаллов, которые находятся в плоскости, отличной от плоскости поверхности монослоя, могут на самом деле представлять собой верхние поверхности кристаллов, из которых при образовании тонкого слоя рост нового молекулярного сита оказывается самым значительным. При такой схеме расположения кристаллы в монослое растут со стороны этих поверхностей в направлениях друг к другу и образуют плотный тонкий слой с незначительным ростом в направлении от плоскости поверхности монослоя или без такого роста. Таким образом, в этом случае может быть получен достаточно плотный тонкий слой, толщина которого по существу соответствует толщине первоначального монослоя.
Тонкий слой молекулярного сита получают помещением подложки с адсорбированными микрокристаллами молекулярного сита в раствор, состав которого пригоден для синтеза целевого молекулярного сита, с последующей его обработкой в условиях, приемлемых для синтеза целевого молекулярного сита. Поскольку синтез большинства молекулярных сит протекает в гелях, а не в растворах, необходимо подчеркнуть, что способ в соответствии с настоящим изобретением применим в случаях, в которых кристаллизацию предпочтительно осуществлять в прозрачном растворе. В таком случае применение раствора ведет к кристаллизации соответствующего молекулярного сита даже в отсутствии подложки.
Состав синтезной смеси варьируют в соответствии со способом; смесь часто содержит источник кремния и обычно включает структуронаправляющее средство, например, одно из упомянутых выше, и источник любого другого компонента, необходимого для состава конечного цеолита. В некоторых процессах в соответствии с изобретением требуется источник калия. Предпочтительным источником кремния служит коллоидный диоксид кремния, прежде всего коллоидный диоксид кремния, стабилизированный аммиаком, например, поставляемый на рынок фирмой DuPont под товарным знаком Ludox AS-40.
Источник кремния может также быть источником калия в форме силиката калия. Такой силикат удобен в форме водного раствора, в частности такого, как поставляемый на рынок фирмой Aremco Products, Inc. под товарным знаком CERAMA-BIND, который доступен в виде раствора с рН 11,3, с удельным весом 1,26 и вязкостью 40 мПa•с. К другим источникам кремния относится, например, кремниевая кислота.
В качестве примеров других источников калия, если они содержатся, можно упомянуть гидроксид. Независимо от того, содержит или не содержит синтезная смесь источник калия, для достижения целевой щелочности она может также содержать гидроксид натрия.
Структуронаправляющим средством, если оно содержится, может служить любое из вышеперечисленных для синтезной смеси, предназначенной для формирования кристаллов промежуточного слоя.
Пригодные для синтеза молекулярного сита растворы хорошо известны в данной области техники и описаны, например, в WO 94/25151, РСТ/ЕР93/01209. заявке PCT/US95/08514, опубликованной под номером WO 96/01687, и заявке PCT/US95/08511, опубликованной под номером WO 96/01685, сущность которых включена в настоящее описание в качестве ссылок.
В качестве материалов молекулярных сит для тонкого слоя молекулярного сита можно упомянуть силикат, алюмосиликат, алюмофосфат, силикоалюмофосфат, металлалюмофосфат или металлалюмофосфосиликат.
Предпочтительные параметры молекулярного сита обычно зависят от выбранной цели применения, такой, например, как разделение, катализ и совмещение реакции с разделением, и от размеров молекул обрабатываемых веществ. Существует множество известных путей придать молекулярным ситам целевые свойства, например, выбором типа структуры, химического состава, ионообменом и методами активации.
Типичными примерами являются молекулярные сита/цеолиты структурных типов AFI, AEL, ВЕА, СНА, EUO, FAU, FER, KFI, LTA. LTL, MAZ, MOR, MEL, MTT, MTW, OFF, TON и в особенности MFI. Хотя некоторые из этих материалов истинными цеолитами не являются, в литературе их часто называют цеолитами, поэтому в приведенной ниже части настоящего описания этот термин обычно использован в широком смысле. Примеры материалов молекулярных сит, которые представляют основной интерес для осуществления настоящего изобретения, включают силикалит, гидроксисодалит, TS-1, а также цеолиты А, бета-цеолит, L, X, Y, ZSM-2, ZSM-11, ZSM-22, ZSM-5 и SAPO-34.
Для получения цеолита типа MFI, прежде всего ZSM-5 или силикалита, например, силикалита-1, предпочтительная синтезная смесь имеет следующий молярный состав в пересчете на содержание оксидов:
M2O:SiO2 0-0,7:1, предпочтительно 0,016-0,350:1,
SiO:Al2O3 от 12 до бесконечности:1,
(ТПА)2O:SiO2 0-0,2:1, предпочтительно 0-0,075:1,
H2O:SiO2 7-1000:1, предпочтительно 9-300:1,
где ТПА обозначает тетрапропиламмоний, а М обозначает щелочной металл, предпочтительно натрий, калий, а также Li, Cs, или аммоний. В этих же пропорциях могут быть использованы другие шаблонные средства. Для получения слоя MFI предпочтительно используют гидроксид или галогенид тетрапропиламмония.
В настоящем описании соотношения с бесконечностью в качестве пропорции указывают на то, что один из материалов как компонент соотношения в смеси не содержится.
Когда в вышеописанном способе используют особые количества натрия, обычно образуется слой цеолита MFI, в котором КПО (расшифровка этой аббревиатуры указана ниже) оказывается такой, что кристаллографическая c-ось проходит перпендикулярно плоскости слоя. В структуре MFI система каналов, включающая прямые каналы, которые проходят параллельно b-оси, и синусоидальные каналы, которые проходят параллельно a-оси, лежит параллельно плоскости слоя.
Введение подложки с покрытием в контакт целесообразно осуществлять путем погружения или частичного погружения, причем подложку в синтезной смеси ориентируют и размещают таким образом, чтобы свести к минимальному влияние оседания кристаллов, образующихся в самой реакционной смеси, а не на покрытой поверхности. Так, например, с целью предупредить препятствующее влияние оседающих кристаллов и местное истощение смеси вследствие высокой концентрации растущих кристаллов, поверхность, на которую наносят покрытие, целесообразно размещать на расстоянии по меньшей мере 5 мм, предпочтительно по крайней мере 8 мм от стенки или прежде всего днища сосуда. Более того, покрытую поверхность целесообразно ориентировать под углом в интервале 90-270o, предпочтительно 180o, причем угол в 180o соответствует горизонтальной ориентации поверхности покрытия, которая при этом обращена вниз. В особом случае, когда покрытая поверхность структуры является трехмерной, например, сотовой, с целью ингибировать оседание можно применять другие средства, например, взбалтывание, перемешивание или перекачивание.
В том, что относится к представляемым в настоящем описании способам, то введение в контакт следует понимать как охватывающее погружение или частичное погружение подложки в смесь для синтеза соответствующего цеолита.
Гидротермическую обработку с получением тонкого слоя молекулярного сита целесообразно осуществлять контактированием подложки, несущей монослой, с синтезной смесью и выдержкой при повышенной температуре и в течение промежутка времени, которые необходимы для эффекта кристаллизации, целесообразно в автоклаве под самопроизвольно создающимся давлением. Длительность нагревания может составлять, например, от 1 ч до 14 дней, преимущественно от 1 ч до 6 дней. В случае применения нагрева токами сверхвысокой частоты его продолжительность можно сократить до уровня минут. Температура при этом составляет менее 200oС, предпочтительно менее 150oС, находясь в интервале 80-150oС, предпочтительно в пределах 80-125oС, и наиболее предпочтительно составляет менее 100oС.
При необходимости образование цеолитных кристаллов в самой синтезной смеси можно ингибировать поддержанием рН синтезной смеси в интервале 6-13. В таких слабощелочных синтезных смесях повышается эффективность действия кристаллов молекулярного сита в монослое в качестве затравочных кристаллов, что тем самым способствует росту тонкого слоя молекулярного сита. С другой стороны, при необходимости образование кристаллов молекулярного сита в самой синтезной смеси можно подавлять добавлением в эту синтезную смесь очень небольших количеств затравочных кристаллов коллоидного размера, благодаря чему уменьшается рост тонкого слоя молекулярного сита. Полагают, что введение регулируемых количеств коллоидных молекулярных сит в синтезную смесь позволяет регулировать толщину тонкого слоя молекулярного сита без изменения рН синтезной смеси, продолжительности кристаллизации и температуры кристаллизации.
Помимо образования при формировании тонкого слоя молекулярного сита на поверхности подложки, кристаллы молекулярного сита того же самого типа образуются в растворной фазе. Условия, создаваемые на этой стадии предлагаемого способа, в обычных случаях могут оказаться разрушительными для поверхностей подложек, например, вызывая их травление и растворение в случае определенных сочетаний подложки/молекулярного сита, когда используют сильнощелочные растворы. Некоторую защиту от такого агрессивного воздействия обеспечивают адсорбированные слои катионоактивного полимера и микрокристаллы молекулярного сита.
В случае молекулярных сит определенных типов необходима заключительная стадия кальцинирования для выжигания в пористой структуре органических молекул, обеспечивая таким образом доступность внутренней пористой структуры для адсорбции, катализа или ионообмена. Кальцинирование тонких слоев, полученных в соответствии с настоящим изобретением, чаще всего включающее обработку на воздухе при температуре, превышающей 400oС, не приводит к образованию трещин, которое можно наблюдать с помощью сканирующего электронного микроскопа.
Если подложка обладает пористостью, то перед нанесением микрокристаллов молекулярного сита из водной реакционной смеси или введением монослойной структуры в контакт с синтезным раствором эту подложку целесообразно обрабатывать нанесением барьерного слоя.
Задача барьерного слоя состоит в том, чтобы предотвратить преимущественное проникновение образующей покрытие смеси или ее компонентов в поры подложки, например, в такой степени, при которой цеолитные кристаллы образуют на подложке толстый гелевый слой.
Этот барьерный слой может быть временным или постоянным. В качестве материала временного слоя можно применять пропиточную жидкость, которая в процессе нанесения реакционной смеси способна удерживаться в порах и легко удаляться после такого нанесения и любой последующей обработки.
С целью улучшить пенетрацию барьерную жидкость можно применять под пониженным давлением или при повышенной температуре. Преждевременное испарение жидкости из самых внешних пор в процессе обработки можно уменьшить созданием атмосферы, насыщенной парами этой жидкости.
В качестве материала временного барьерного слоя, приемлемого, например, для альфа-алюминийоксидной подложки, можно упомянуть главным образом воду или гликоли. В качестве постоянного барьера для альфа-алюминийоксидной подложки можно упомянуть титандиоксидное, гамма-алюминийоксидное и альфа-алюминийоксидное покрытие на порах меньшего размера.
Более крупногабаритные подложки, например, сотовые реакторные секции, можно обрабатывать путем герметизации подложки в ее реакторном корпусе либо перед, либо после нанесения промежуточного слоя, после чего в корпус наливают или прокачивают через него синтезную смесь, причем кристаллизацию, промывку и кальцинирование проводят тогда, когда подложка уже находится в своем корпусе.
Если это целесообразно или необходимо, те участки подложки, будь она пористой или непористой, на которых нежелательно или нет потребности в получении монослоя и/или тонкого слоя молекулярного сита, перед их нанесением можно закрыть маскировочным покрытием, используя, например, воск, или же нежелательный цеолит можно удалять с таких участков после нанесения.
Было установлено, что несмотря на относительно малую толщину (менее 2 мкм) структуры тонкого слоя молекулярного сита по настоящему изобретению, она является сплошной, плотной и состоит из сросшихся кристаллов молекулярного сита. Кроме того, в отличие от некоторых ранее известных способов, для получения этой структуры хотя и используют зародышеобразующий затравочный слой, в готовом продукте он становится невидимым как четко определенный слой, поскольку микрокристаллы монослоя входят в состав тонкого слоя молекулярного сита. Конечный продукт обладает признаками ранее известных тонких слоев молекулярных сит, полученных с использованием нескольких зародышеобразующих слоев, но свободен от свойственных таким слоям недостатков, которыми являются, например, уменьшенный расход потока готовой мембраны и склонность к образованию трещин.
Необходимо отметить, что структура может характеризоваться любой формой; она, в частности может быть плоской, цилиндрической, чаще всего цилиндрической с круглым поперечным сечением, или может представлять собой сотовую структуру. Однако для ясности в приведенном ниже описании структура проиллюстрирована так, как если бы она была плоской, поэтому ссылки, как правило, сделаны в отношении плоскости слоя.
Этот тонкий слой молекулярного сита в такой структуре в определенной степени проявляет, как также было установлено, КПО (расшифровка этой аббревиатуры указана ниже) и ПОФ (расшифровка этой аббревиатуры указана ниже). Несмотря на то, что упомянутый слой является относительно тонким, по природе он, как было установлено, оказывается столбчатым.
Под термином "столбчатый" в этом контексте следует понимать указание на то, что тонкий слой молекулярного сита представляет собой кристаллическое молекулярное сито, в котором по меньшей мере 75%, как можно видеть с помощью сканирующего электронного микроскопа (СЭМ), предпочтительно не менее 85%, кристаллитов в самой верхней поверхности проходят в направлении границы между этим тонким слоем и подложкой.
По данным, полученным с помощью сканирующего электронного микроскопа (СЭМ), в предпочтительном варианте не менее 75% межзеренных границ верхнего слоя по меньшей мере в зоне верхней стороны сориентировано в пределах угла в 30o относительно перпендикуляра к плоскости слоя, предпочтительно по меньшей мере 90% сориентированы в пределах этого угла, более предпочтительно по меньшей мере 90% сориентированы в пределах угла в 25o и наиболее предпочтительно в пределах угла в 15o относительно перпендикуляра.
Направления межзеренных границ кристаллов в верхнем слое указывают на степень, с которой кристаллы характеризуются преимущественной ориентацией по форме (ПОФ).
Материалы, включающие несферические гранулы, могут проявлять преимущественную ориентацию по размерам или преимущественную
ориентацию по форме. ПОФ можно определить, например, как распределение по упорядоченной ориентации по самому большому размеру зерен или кристаллов. Такую ПОФ можно определить, например, по поперечным сечениям на фотографиях, выполненных с помощью электронного микроскопа; принимают во внимание только контуры зерен или кристаллов, причем с целью определить распределение по ориентации устанавливают и используют ориентацию по самому большому размеру каждого.
Поскольку форма зерна или кристалла необязательно связана с его кристаллографической ориентацией, ПОФ в принципе оказывается независящей от КПО, хотя во многих случаях ПОФ и КПО связаны между собой.
Помимо прочего продукты по изобретению можно охарактеризовать рентгенографией (РГФ). С этой целью можно применять обычный метод порошковой дифракции, в котором слоистую структуру в форме диска закрепляют в модифицированном держателе для порошкообразных образцов и выполняют обычное сканирование θ/2θ. Получаемые таким образом результаты измерений интенсивности цеолитных отражений сопоставляют с интенсивностью отражений неупорядоченно ориентированных частиц порошка цеолита тех же самых структуры и состава. Если одна или несколько групп отражений, связанных с одной или несколькими специфическими ориентациями кристалла, оказывается (или оказываются) значительно сильнее оставшихся отражений при сравнении с дифрактограммой неупорядоченно ориентированных частиц порошка, и это указывает на то, что распределение по ориентации в образце отклоняется от неупорядоченного. Это называют кристаллографически преимущественной ориентацией или КПО. Простым примером КПО служит случай, когда отражения 001 (например, 002, 004, 006 и т.д. для MFI) оказываются сильными, в то время как все другие отражения являются слабыми или отсутствуют. В этом случае кристаллографическая c-ось большей части кристаллов близка к перпендикуляру к плоскости слоя; этот случай часто называют а-осевой КПО. Другим примером является дифрактограмма, на которой отражения h00 (200, 400, 600, 800 и т.д. для MFI) являются доминирующими; этот случай называют a-осевой КПО. Могут также возникать более сложные ситуации, например, в случае дифрактограммы, на которой доминирующими являются отражения как 0k0, так и 001; этот случай называют смешанной b- и c-осевой КПО.
В случае КПО уникальная идентификация типа кристаллической структуры на основе РГФ дифрактограммы одного слоя может оказаться невозможной только из-за ограниченного числа отражений, которые могут быть определены. Строго говоря, материал этого слоя необходимо отделить от подложки, измельчить до порошкообразного состояния и для подтверждения типа структуры получить дифрактограмму неупорядоченно ориентированных частиц этого порошка. На практике это часто связано с серьезными затруднениями. Таким образом, если в результате синтеза получают какой-либо порошкообразный продукт или отложения на стенках или днище автоклава, этот материал используют для идентификации типа структуры. Если все отражения на дифрактограмме слоя обусловлены конкретными группами отражений на индексированной дифрактограмме порошка (например, отражения 001), это служит указанием на то, что слой характеризуется структурой того же типа, что и порошок.
Количественное определение степени КПО может быть основано на сравнении наблюдаемой РГФ дифрактограммы с РГФ дифрактограммой порошка из неупорядоченно ориентированных частиц. С целью определить число, которое может быть использовано в качестве параметра, характеризующего степень КПО, для каждого типа кристаллической структуры и КПО может быть выбрана специфическая группа отражений. Так, например, в случае системы, в которой самый верхний слой характеризуется структурой типа структуры цеолита MFI, а кристаллы характеризуются c-осевой КПО, КПО-параметр C001 можно определить с помощью значений интенсивности I отражения 002 и объединенных отражений 200 и 020 следующим образом: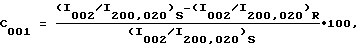
где I200,020 и I002 обозначают откорректированные с учетом фона соответственно значения высоты объединенных отражений MFI-I200,020 и отражения MFI-I002 для неупорядоченно ориентированных частиц порошка R и для исследуемого образца S перед кальцинированием.
Нулевое значение этого параметра C001 соответствует неупорядоченной ориентации, в то время как значение 100 соответствует фактическому отсутствию плоскостей 100 и 010, параллельных плоскости слоя. Отсутствие всех отражений MFI, исключая отражения 001, указывает на практически абсолютное выравнивание плоскостей 001 параллельно слою.
Подобным же образом в случае a-осевой КПО параметр Ch00 можно определить с использованием интенсивности отражения 10 0 0 относительно интенсивности значений, в частности суммы отражений 002 и 0 10 0 или отражения 133 (перед кальцинированием) с помощью следующей формулы: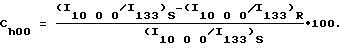
Для КПО других типов можно определить другие параметры. Можно также использовать другие пути определения КПО, например, гониометрией строения.
Для a-осевой КПО предпочтительный параметр С001 структуры в соответствии с изобретением составляет по меньшей мере 50, более предпочтительно не менее 95. Однако предпочтительный тонкий слой молекулярного сита должен проявлять сильные КПО и ПОФ.
В предпочтительном варианте кристаллы тонкого слоя молекулярного сита являются смежными, т.е. практически каждый кристалл находится в контакте с соседними с ним, хотя и необязательно в контакте с соседними с ним по всей его длине (кристалл находится в контакте с соседним с ним, если расстояние между ними в ширину составляет менее 2 нм). Предпочтительный тонкий слой молекулярного сита практически свободен от дефектов с поперечным сечением более 4 нм, проходящих по его толщине.
Способ по настоящему изобретению позволяет получать тонкие слои (менее 2 мкм) молекулярных сит на подложке с приемлемыми производительностью и свойствами без видимого наличия промежуточного затравочного слоя. Однако этот способ в равной мере применим к получению более толстых слоев молекулярного сита на подложке. Так, например, с применением такой технологии можно получать слои толщиной до 150 мкм. Полагают, что такие тонкие слои обладают также улучшенными свойствами и производительностью. Предпочтительно толщина тонкого слоя молекулярного сита и размеры кристаллитов молекулярного сита таковы, что эта толщина слоя приблизительно равна размеру самых длинных граней кристаллов, благодаря чему образуется по существу монослой со столбчатой структурой.
По изобретению предлагается также структура, в которой подложка, прежде всего пористая подложка, с каждой стороны снабжена тонкими слоями молекулярных сит, соответствующих изобретению, причем слои на обеих сторонах являются одинаковыми или различными; настоящее изобретение также предусматривает возможность получения на одной из сторон подложки тонкого слоя, который не соответствует изобретению, или введения в подложку других материалов, если она пористая.
Тонкий слой молекулярного сита может, а для многих областей применения это целесообразно, состоять по существу из материала молекулярного сита или может представлять собой композит из материала молекулярного сита и интеркалирующего материала, который может быть органическим или неорганическим. Интеркалирующим материалом может служить такой же материал, что и материал подложки. Этот материал можно наносить одновременно с осаждением молекулярного сита или после его осаждения, его можно наносить, например, по золь-гелевому методу с последующей термической обработкой. К приемлемым материалам относятся, например, неорганические окислы, в частности диоксид кремния, оксид алюминия и диоксид титана. Интеркалирующий материал целесообразно использовать в материале тонкого слоя при относительном содержании от общего количества материала, достаточно низком для того, чтобы кристаллы молекулярного сита оставались смежными.
Хотя полагают, что во многих случаях тонкий слой молекулярного сита обычно свободен от трещин и дефектов, в процессе получения тонкого слоя или во время последующей обработки, при обращении с ним или в применении в этом тонком слое могут образоваться локальные трещины или другие дефекты. В этих обстоятельствах остальная часть тонкого слоя может оказаться неповрежденной, высококачественной и работоспособной, хотя в целом из-за таких локальных дефектов тонкий слой считается несовершенным. В этих случаях тонкий слой необходимо подвергнуть компенсационной обработке. Методы такой компенсационной обработки неорганических мембран в данной области техники известны.
Каталитическое действие структуре тонкого слоя молекулярного сита по настоящему изобретению можно сообщить либо путем связывания катализатора с подложкой или свободной поверхностью тонкого слоя молекулярного сита, либо его размещением внутри трубки или соты, в виде которой выполнена структура, путем его введения в подложку, например, путем получения подложки из смеси образующего подложку и образующего каталитические участки материалов, или в монослой либо сам тонкий слой молекулярного сита. Если подложка является пористой, катализатор можно вводить в поры, причем катализатор необязательно представляет собой цеолит. В некоторых областях применения для структуры по изобретению достаточно, чтобы она находилась в непосредственной близости или в контакте с катализатором, например, в порошкообразной форме на поверхности структуры.
Каталитически активные участки могут быть введены в тонкий слой молекулярного сита этой структуры, например, посредством выбора в качестве цеолита материала с ограниченным соотношением SiO2/Аl2О3, предпочтительно менее 300. Посредством ионообмена можно также придать требуемую прочность этих участков. В синтезную смесь можно вводить металлсодержащие или металлоксидные предшественники для микрокристаллов монослоя, тонкого слоя молекулярного сита или для них обоих, или же металлы, металлоксиды, соли или органические комплексы можно вводить путем пропитки предварительно сформированного тонкого слоя молекулярного сита или посредством ионообмена с ним. Для регулирования свойств эту структуру можно также обрабатывать водяным паром или обрабатывать по другим известны методам.
Слои можно формировать в виде мембраны (причем этот термин использован в настоящем описании для обозначения барьера, обладающего разделительными свойствами), предназначенной для разделения смесей текучих сред (газообразных, жидких или смешанных), например, с целью выделения сырья для реакции из исходной смеси, или при применении с каталитическими целями, когда они способны, если это необходимо, сочетать каталитическое превращение реагента или реагентов с выделением реакционных продуктов.
Процессы разделения, которые можно проводить с использованием мембраны, содержащей структуру в соответствии с изобретением, включают, например, выделение нормальных алканов из кипящих в одном с ними температурном диапазоне углеводородов, в частности нормальных алканов из изоалканов в С4-С6cмесях и н-С10-С16алканов из керосина; отделение нормальных алканов и алкенов от соответствующего разветвленного алкана и алкановых изомеров; отделение ароматических соединений друг от друга, в особенности отделение ароматических С8изомеров друг от друга, более конкретно выделение параксилола из смеси ксилолов и необязательно этилбензола (например, выделение п-ксилола из богатой п-ксилолом смеси, полученной в процессе изомеризации ксилола), и разделение ароматических веществ с различным числом углеродных атомов, например, смесей бензола, толуола и смешанных ароматических С8соединений; отделение ароматических соединений от алифатических соединений, в особенности ароматических молекул с 6-8 углеродными атомами от алифатических С5-С10соединений (нафтенового ряда); отделение ароматических соединений от алифатических соединений и водорода в реакторе для реформинга; отделение олефиновых соединений от насыщенных соединений, прежде всего легких алкенов от алкан/алкеновых смесей, более конкретно этилена от этана и пропилена от пропана; выделение водорода из водородсодержащих потоков, прежде всего из газовых потоков легкокипящих фракций нефтепереработки и нефтехимических процессов, более конкретно от С2- и легкокипящих компонентов; удаление водорода из продуктов нефтепереработки и химических процессов, таких, как дегидрогенизация алканов с получением алкенов, дегидроциклизация легких алканов и алкенов с получением ароматических соединений и дегидрогенизация этилбензола с получением стирола; удаление спиртов из водных потоков и удаление спиртов из углеводородов, прежде всего алканов и алкенов, которые могут входить в состав смесей, образующихся во время получения спиртов.
Процессы превращения, которые можно проводить, включают изомеризацию, например, алканов и алкенов, превращение метанола или лигроина в алкены, гидрогенизацию, дегидрогенизацию, например, алканов, в частности пропана в пропилен, окисление, каталитический реформинг или крекинг, а также термический крекинг.
Разделению, например, посредством диффузии молекул, путем введения в контакт со структурой в соответствии с изобретением, целесообразно с такой, которая выполнена как мембрана, можно подвергать исходные материалы, полученные из углеводородов, например, в форме нефтяного или природного газа, или исходные материалы, полученные при переработке угля, битума или керогена, или с использованием воздуха, а также исходные материалы, содержащие продукты по меньшей мере с двумя различными разновидностями молекул, когда исходные продукты по меньшей мере одной разновидности молекул отделяют от продуктов по меньшей мере одной из других разновидностей молекул.
Примеры таких процессов разделения сведены в табл.1.
В табл. 2 представлены примеры химических реакций, которые можно проводить с использованием структуры по изобретению, целесообразно структуры, полученной в виде мембраны, в сочетании с катализатором (например, катализатором, который находится в модуле с этой структурой) или обработанной для придания этой структуре каталитического действия.
Структуру по изобретению можно использовать в качестве мембраны в таких процессах разделения без проблемы, связанной с повреждением при вхождении в контакт с материалами, которые необходимо разделить. Более того, многие из таких процессов разделения проводят при повышенных температурах, достигающих, например, 500oС, причем преимущество структуры по настоящему изобретению состоит в том, что ее можно применять при таких высоких температурах.
Таким образом, по настоящему изобретению предлагается также способ разделения смеси текучих сред, который включает введение этой смеси в контакт с одной стороной структуры в соответствии с изобретением в форме мембраны в таких условиях, в которых по меньшей мере один компонент этой смеси в стационарном режиме характеризуется способностью проникать через эту структуру, отличной от способности проникать другого компонента, и выделение компонента или смеси компонентов с другой стороны структуры.
Следовательно, по настоящему изобретению предлагается также способ разделения смеси текучих сред, который включает введение этой смеси в контакт со структурой в соответствии с изобретением в одном варианте в форме мембраны в таких условиях, в которых по меньшей мере один компонент этой смеси удаляют из смеси адсорбцией. Адсорбированный компонент необязательно выделяют и используют в химической реакции или его можно вводить в реакцию в виде материала, адсорбированного на такой структуре в соответствии с изобретением.
Более того, по изобретению предлагаются такие способы катализа химической реакции, в которых эта структура находится в непосредственной близости от катализатора или в контакте с ним.
По изобретению предлагается далее способ катализа химической реакции, который включает введение исходного материала в контакт со структурой в соответствии с изобретением, которая в условиях каталитического превращения находится в активной каталитической форме, и выделения композиции, включающей по меньшей мере один продукт превращения.
По изобретению предлагается, кроме того, способ катализа химической реакции, который включает введение в условиях каталитического превращения исходного материала в контакт с одной стороной структуры в соответствии с изобретением, которая находится в форме мембраны и в каталитически активной форме, и выделение с противоположной стороны этой структуры по меньшей мере одного продукта превращения, целесообразно в концентрации, отличной от его равновесной в реакционной смеси.
По изобретению предлагается также способ катализа химической реакции, который включает введение исходного материала в контакт с одной стороной структуры в соответствии с изобретением, которая находится в форме мембраны в таких условиях, в которых по меньшей мере один компонент этого исходного материала удаляют из сырья после прохождения через структуру в контакте с катализатором с противоположной стороны этой структуры в условиях каталитической конверсии.
Далее по изобретению предлагается способ катализа химической реакции, который включает введение одного реагента бимолекулярной реакции в контакт с одной стороной структуры в соответствии с изобретением, которая в условиях каталитического превращения находится в форме мембраны и в каталитически активной форме, и регулирование введения второго реагента за счет диффузии с противоположной стороны этой структуры с целью более точного регулирования реакционных условий. Примеры применения этого способа включают присоединение к бензолу этилена, пропилена и водорода при получении соответственно этилбензола, кумола и циклогексана.
Описание чертежей
На фиг. 1 представлена микрофотография монослоя из силикалита-1, связанного с поверхностью подложки из монокристаллического Si за счет электростатической адсорбции, полученная с помощью сканирующего электронного микроскопа.
На фиг. 2 представлена микрофотография тонкого слоя из ТПА-силикалита-1 на подложке из монокристаллического Si, полученная с помощью сканирующего электронного микроскопа.
На фиг. 3 представлена микрофотография, полученная с помощью сканирующего электронного микроскопа, полых волокон из силикалита-1 (с левой стороны изображения) и цеолита Y (с правой стороны изображения), полученных формированием тонких слоев молекулярных сит на поверхности углеродных волокон с последующим удалением углеродного волокна кальцинированием.
Определение характеристик
Способ по настоящему изобретению оценивали проверкой материалов с помощью сканирующего электронного микроскопа (СЭМ), по порошковой дифракции рентгеновских лучей (ДРЛ), спектроскопией, а также определением удельной площади поверхности по поглощению N2 и Кr.
Исследования с помощью сканирующего электронного микроскопа проводили с использованием образцов, покрытых углеродом или золотом (посредством соответственно техники осаждения из паровой фазы и металлизации напылением). В этих исследованиях применяли сканирующий электронный микроскоп типа Philips XL 30, снабженный источником излучения LaB6.
Исследования с дифракцией рентгеновских излучений проводили на образцах необработанных тонких слоев с помощью дифрактометра для порошковой дифракции Philips PW 1710-00.
Измерения адсорбции Кr для определения удельной площади поверхности проводили с помощью прибора ASAP 2010 фирмы Micromeritics Instruments Inc. Перед этими измерениями тонкие слои обезгаживали при 250oС в течение 3 ч.
Спектроскопические исследования некоторых образцов, полученных в соответствии с настоящим изобретением, проводили в ИК-спектрометре Perkin Elmer 2000 FT (для инфракрасной спектроскопии), а также в спектрометре Perkin Elmer Lambda 2S UV-VIS (для спектроскопии при длинах волн видимого и ультрафиолетового диапазонов излучения).
Анализ коллоидных суспензий молекулярных сит, использованных при получении тонких слоев молекулярных сит, представленных в описании настоящего изобретения, для определения размеров частиц и распределения частиц по размерам проводили по динамическому светорассеянию (ZetaPlus, Brookhaven Instruments).
Сущность изобретения проиллюстрирована ниже на нескольких примерах. Однако эти примеры на следует рассматривать как ограничивающие объем изобретения.
Пример 1
Этот пример иллюстрирует получение тонкого слоя из силикалита-1 толщиной приблизительно 100 нм на кристаллической кремниевой подложке.
Коллоидную суспензию дискретных частиц силикалита-1 готовили гидротермическим синтезом при 55oС синтезного раствора следующего состава: 9 ТПА-ОН/25 SiO2/480 H2O/100 EtOH, где ТПА-ОН обозначает гидроксид тетрапропиламмония, а EtOH обозначает этанол. Кремниевую кислоту добавляли в виде тетраэтоксисилана, гидролизованного в водном растворе ТПА-ОН. После синтеза образовавшийся золь очищали выделением из маточного раствора центрифугированием, после которого частицы золя вновь диспергировали в дистиллированной воде. Добавлением 0,10 M раствора аммиака рН доводили до 10,5. Размер полученных коллоидных частиц, как устанавливали по динамическому светорассеянию, составлял 51 нм.
В тефлоновом держателе вертикально устанавливали кристаллическую кремниевую подложку в форме тонкого диска (40х9х0,4 мм) и очищали ацетоном в ультразвуковой ванне в течение пяти минут. Далее кремниевую подложку кипятили в растворе следующего состава (в пересчете на объем): (5 Н2О/1 Н2О2 (30 мас. %)/1 NН3 (25 мас.%) в течение пяти минут и затем в растворе следующего состава: 6 Н2О/1 Н2О2 (30 мас.%)/1 НС1 (37 мас.%) в течение пяти минут. Между этими стадиями очистки подложку промывали дистиллированной водой. После операций очистки подложку в течение одного часа обрабатывали раствором, рН которого доводили до 8,0, содержавшим 0,4 мас.% катионоактивного полимера (продукт Berocell 6100 фирмы Akzo Nobel АВ, Швеция), с целью изменить знак поверхностного заряда подложки на обратный, т.е. первоначально отрицательный заменить положительным. Избыток катионоактивного полимера удаляли промывкой 0,1 М раствором аммиака. Подложку с модифицированной поверхностью переносили в вышеописанный золь (с содержанием сухого вещества 2,5 мас.%), включавший коллоидные кристаллы силикалита-1 с размерами 51 нм, и этим кристаллам давали адсорбироваться на поверхности подложки в течение одного часа. Избыток коллоидных кристаллов, если он был, удаляли промывкой 0,1 М раствором аммиака. Такой обработкой на поверхности подложки получали монослой из силикалитных кристаллов. На фиг. 1 представлена электронная микрофотография адсорбированных силикалитных кристаллов на поверхности подложки.
Далее подложку с адсорбированным монослоем из кристаллов силикалита-1 вначале обрабатывали при 550oС в атмосфере из 100%-ного водяного пара в течение одного часа. После охлаждения подложку подвергали дальнейшей обработке синтезным раствором следующего состава: 3 ТПА-ОН/25 SiO2/1500 Н2О/100 EtOH при 100oС в течение 13 ч. Результатом обработки являлись непрерывный рост и взаимное сцепление адсорбированных кристаллов с образованием на поверхности подложки плотного и сплошного силикалитного тонкого слоя. На фиг. 2 представлена выполненная с помощью СЭМ микрофотография (вид сбоку) этого тонкого слоя. Толщина тонкого слоя, которую вычисляли по этой выполненной с помощью СЭМ микрофотографии, составляла 100 нм.
Образец продукта анализировали по рентгеновской дифрактометрии и ИК-спектрометрии с преобразованием Фурье. По результатам анализов с применением обоих методов тонкий слой идентифицировали как состоявший из силикалита-1. Образец продукта кальцинировали при 600oС на воздухе в предварительно нагретой муфельной печи для удаления органического материала (ТПА +) и для получения силикалитной пористой структуры, доступной для адсорбции газов. Затем посредством адсорбции Кr определяли удельную площадь поверхности образца, и устанавливали, что она была равной 72 м2/(м2 поверхности подложки), значение, которое хорошо согласовывалось с расчетными данными для 100-нанометрового силикалитного слоя на кремниевой подложке этого типа.
Пример 2
Этот пример иллюстрирует получение тонких слоев толщиной около 100 нм на алюминийоксидных и кварцевых подложках.
Монокристаллические структуры в форме сапфирных (α- алюминийоксидных) и кварцевых дисков (10х10х1 мм) вертикально устанавливали в тефлоновом держателе и очищали ацетоном в ультразвуковой ванне в течение пяти минут. Далее подложки кипятили в растворе следующего состава (в пересчете на объем): 5 H2O/1 Н2О2 (30 мас. %)/1 NH3 (25 мас.%) в течение пяти минут и затем в растворе следующего состава: 6 Н2О/1 Н2О2 (30 мас.%)/1 НС1 (37 мас.%) в течение пяти минут. Между этими стадиями очистки подложки промывали дистиллированной водой. После операций очистки подложки в течение одного часа обрабатывали раствором, рН которого доводили до 8,0, содержавшим 0,4 мас.% катионоактивного полимера (продукт Berocell 6100 фирмы Akzo Nobel AB, Швеция), с целью изменить знак поверхностного заряда подложки на обратный, т.е. первоначально отрицательный заменить положительным. Избыток катионоактивного полимера удаляли промывкой 0,1 М раствором аммиака. Подложки с модифицированной поверхностью переносили в вышеописанный золь (с содержанием сухого вещества 2,5 мас.%), включавший коллоидные кристаллы силикалита-1 (см. пример 1) с размерами 51 нм, и этим кристаллам давали адсорбироваться на поверхности подложки в течение одного часа. Избыток коллоидных кристаллов, если он был, удаляли промывкой 0,1 М раствором аммиака. Такой обработкой на поверхности подложек получали монослой из силикалитных кристаллов.
Далее подложки с адсорбированным монослоем из кристаллов силикалита-1 вначале обрабатывали при 550oС в атмосфере из 100%-ного водяного пара в течение одного часа. После охлаждения подложки подвергали дальнейшей обработке синтезным раствором следующего состава: 3 ТПА-ОН/25 SiO2/1500 H2O/100 EtOH при 100oС в течение 13 ч. Результатом обработки являлись непрерывный рост и взаимное сцепление адсорбированных кристаллов с образованием на поверхностях подложек плотного и сплошного силикалитного тонкого слоя, что подтверждалось данными анализов посредством сканирующего электронного микроскопа, по рентгеновской дифрактометрии и измерениями адсорбции Кr (после кальцинирования при 600oС на воздухе). В ходе всех этих анализов получали результаты, аналогичные тем, которые были получены при использовании в качестве материала подложки кристаллического кремния (см. пример 1 практического осуществления); результаты показывают, что осуществление способа не зависит от химических свойств подложки.
Пример 3
Этот пример иллюстрирует получение тонкого слоя из силикалита-1 на углеродном волокне в качестве подложки.
Коллоидную суспензию дискретных частиц силикалита-1 готовили гидротермической обработкой (100oС) синтезного раствора следующего состава: 9 ТПА-ОН/25 SiO2/480 H2O/100 EtOH, где ТПА-ОН обозначает гидроксид тетрапропиламмония, а EtOH обозначает этанол. Кремниевую кислоту добавляли в виде тетраэтоксисилана, гидролизованного в воде. После синтеза образовавшийся золь очищали выделением из маточного раствора центрифугированием, после которого частицы золя вновь диспергировали в дистиллированной воде. Добавлением 0,10 М раствора аммиака рН очищенного золя доводили до 9,5. Размер полученных коллоидных частиц, как устанавливали по динамическому светорассеянию, составлял 98 нм.
Непрерывные углеродные волокна диаметром 7-15 мкм вначале очищали ацетоном в ультразвуковой ванне, а затем раствором следующего состава (в пересчете на объем): 5 H2O/1 Н2О2 (30 мас.%)/1 НС1 (37 мас.%). После обеих этих стадий очистки волокна отделяли вакуумным фильтрованием и промывали дистиллированной водой на фильтровальной бумаге. По завершении процесса очистки волокна в течение одного часа обрабатывали раствором, содержавшим 1,0 мас.% катионоактивного полимера (продукт Berocell 6100 фирмы Akzo Nobel АВ, Швеция), рН которого доводили до 8,0, с целью изменить знак поверхностного заряда подложки на обратный, т.е. первоначально отрицательный заменить положительным. Волокна с измененным знаком заряда на обратный переносили в коллоидный золь (с содержанием сухого вещества 4,6 мас.%), включавший кристаллы силикалита-1 с размерами 98 нм, которые адсорбировались на поверхности волокон. По завершении одного часа контактирования между волокнами и золем волокна отделяли фильтрованием и промывали 0,1 М раствором аммиака. Далее волокна переносили в синтезный раствор следующего состава: 9 ТПА-ОН/25 SiO2/480 H2O/100 EtOH и подвергали гидротермической обработке этим раствором в течение 24 ч при 100oС. Проверка волокон посредством сканирующего электронного микроскопа показывала, что в результате такой обработки на поверхности углеродного волокна образовывался плотный и сплошной тонкий силикалитный слой, образец продукта кальцинировали при 600oС на воздухе в течение трех часов. В результате такой обработки удаляли как органический материал из силикалитной пористой структуры, так и часть углеродного волокна, вокруг которого оборазовывался этот тонкий слой. Как следствие после такой обработки оставался волокнистый продукт, который состоял из тонкого силикалитного слоя, образовавшегося на поверхности волокна. На фиг. 3 представлена полученная на сканирующем электронном микроскопе микрофотография конечного материала. На этой микрофотографии можно ясно видеть, что тонкий слой действительно сплошной и плотный. Этот тонкий слой далее анализировали по рентгеновской дифрактометрии, ИК-спектрометрии с преобразованием Фурье и измерениями удельной площади поверхности с применением метода адсорбции азота. Результаты анализов показали, что тонкий слой состоял только из силикалита-1.
Пример 4
Этот пример иллюстрирует получение тонкого слоя цеолита Y на углеродном волокне в качестве подложки.
Коллоидную суспензию цеолита Y готовили гидротермической обработкой синтезной смеси следующего состава: 2,46 (ТМА)2О/0,04 Na2O/1,0 Аl2О3/3,4 SiO2/370 Н2О, где ТМА обозначает тетраметиламмониевый ион, который вводили в виде пентагидрата тетраметиламмония. Кремниевую кислоту добавляли в виде стабилизированного натрия кремнийдиоксидного золя, а алюминат - в виде стабилизированного щелочью алюминатного раствора. Стабилизированный щелочью алюминатный раствор готовили следующим образом: Al2(SO4)3 o18 Н2О при умеренном подогреве растворяли в дистиллированной воде. По завершении растворения при интенсивном перемешивании с целью вызвать осаждение Al(ОН)3 добавляли аммиачный раствор, содержавший 25 мас.% аммиака в воде. Образовавшийся гель выделяли вакуумным фильтрованием и фильтровальный пирог диспергировали в дистиллированной воде для растворения оставшегося сульфата. Затем этот гель вновь выделяли фильтрованием. Эти операции повторяли до тех пор, пока добавлением ВаСl2 невозможно было бы определить присутствие сульфатных ионов. Не содержавший сульфата гидроксид алюминия растворяли в водном растворе ТМА-ОН. Приготовленный таким образом стабилизированный щелочью алюминатный раствор при интенсивном перемешивании вводили в кремнийдиоксидный золь и при 100oС обрабатывали до тех пор, пока не образовывались (как это определяли по динамическому светорассеянию) кристаллы цеолита Y размерами 100 нм. По завершении синтеза золь очищали выделением из маточного раствора путем центрифугирования с последующим повторным диспергированием в дистиллированной воде. Добавлением 0,1 М раствора аммиака рН очищенного золя доводили до 9,5.
Углеродные волокна с измененным знаком заряда на обратный, полученные по методу, который описан в примере 3, переносили в коллоидный золь (с содержанием сухого вещества 2,4 мас.%), включавший 100-нанометровые кристаллы цеолита Y, которые были адсорбированы в виде монослоя на поверхности волокон. Эти волокна контактировали с золем в течение одного часа. После обработки золем волокна отделяли фильтрованием и избыток золя удаляли промывкой 0,1 М раствором аммиака. Волокна переносили в синтезный раствор следующего состава: 2,46 (TMA)2O/0,04 Na2O/1,0 Аl2О3/3,4 SiO2/370 Н2О, приготовленный по вышеописанной методике. С использованием этого раствора волокна подвергали гидротермической обработке в течение 96 ч при 100oС. Анализ продукта посредством сканирующего электронного микроскопа показал, что в результате на поверхности углеродного волокна образовался плотный и сплошной тонкий слой цеолита Y (см. фиг. 3). Наличие кристаллической структуры в материале тонкого слоя подтверждало дальнейшее определение характеристик дифрактометрией рентгеновских лучей и поглощением Кr.
Пример 5
Данный пример иллюстрирует нанесение тонкого слоя цеолита А на углеродное волокно в качестве подложки.
Коллоидную суспензию цеолита А готовили гидротермической обработкой синтезной смеси следующего состава: 1,2 (ТМА)2О/0,4 Na2O/1,0 Al2O3/3,4 SiO2/246 Н2О, где ТМА обозначает тетраметиламмониевый катион. Синтезный раствор готовили по методу, который описан в примере 4. По завершении синтеза конечный золь очищали в соответствии с методом, описанным в примере 1, и добавлением 0,1 М раствора аммиака рН очищенного золя доводили до 9,5. Углеродные волокна с измененным знаком заряда на обратный, полученные по методу, который описан в примере 3, переносили в коллоидный золь, включавший 120-нанометровые кристаллы цеолита А (содержания сухого вещества составляло 2,8 мас. %). В результате на поверхности углеродных волокон происходила адсорбция монослоя из кристаллов цеолита А. После такой обработки волокна отделяли фильтрованием и избыток золя удаляли промывкой 0,1 М раствором аммиака. Волокна переносили в синтезный раствор следующего состава: 1,2 (ТМА)2О/0,4 Na2O/1,0 Аl2О3/3,6 SiO2/246 Н2О, приготовленный по методу, изложенному в примере 4. Волокна обрабатывали этим раствором в течение 60 ч при 100oС. В результате такой обработки на поверхности углеродного волокна образовался плотный и сплошной тонкий слой цеолита А, что подтверждалось анализами с помощью сканирующего электронного микроскопа и дифрактометрией рентгеновских лучей и поглощением Кr.
Пример 6
Данный пример иллюстрирует нанесение тонкого слоя Ti-силикалита-1 на углеродное волокно в качестве подложки.
Коллоидную суспензию Ti-силикалита-1 готовили гидротермической обработкой синтезного раствора следующего состава: 9 ТПА-ОН/1,46 TiO2/25 SiO2/404 H2O/100 EtOH, где ТПА обозначает тетрапропиламмониевый катион, введенный в раствор гидроксида тетрапропиламмония в воде. TiO2 вводили в виде тетраэтилортотитаната (34,1 мас.% TiO2).
Синтезный раствор готовили следующим образом. При перемешивании тетраэтилортотитанат вводили в тетраэтоксисилан. В этот раствор при интенсивном перемешивании по каплям добавляли раствор ТПА-ОН. Образовывавшийся синтезный раствор подвергали гидротермической обработке при 100oС в течение 20 ч. В результате такой обработки происходила кристаллизация коллоидной суспензии дискретных кристаллов Ti-силикалита-1. По завершении синтеза центрифугированием из маточного раствора выделяли коллоидные частицы, после чего эти частицы вновь диспергировали в дистиллированной воде и добавлением 0,1 М раствора аммиака рН очищенного золя доводили до 9,5. Средний размер коллоидных кристаллов Ti-силикалита-1 составлял, как это устанавливали по динамическому светорассеянию, 90 нм.
Углеродные волокна с измененным знаком заряда на обратный, полученные по методу, который описан в примере 3, погружали в коллоидный золь (содержание сухого вещества: 1,9 мас.%) микрокристаллов Ti-силикалита-1, которым давали адсорбироваться на поверхности волокон. Продолжительность контактирования между золем и волокнами была равной одному часу. После завершения такой обработки волокна промывали в соответствии с методом, описанным в примере 3.
Далее волокна переносили в синтезный раствор следующего состава: 9 ТПА-ОН/1,46 ТiO2/25 SiO2/404 Н2О/100 EtOH. Этот синтезный раствор готовили по методу, изложенному выше. Волокна подвергали гидротермической обработке этим раствором при 100oС в течение 24 ч. Результаты анализа с помощью сканирующего электронного микроскопа этого продукта показали, что при такой обработке на поверхности углеродного волокна обеспечивалось образование плотного и сплошного тонкого слоя Ti-силикалита-1. Дальнейший анализ готового композитного материала проводили с использованием дифрактометрии рентгеновских лучей, инфракрасной спектроскопии по коэффициенту диффузного отражения (ИКСДО) и спектроскопии при длинах волн видимого и ультрафиолетового диапазонов излучения. Результаты этих анализов подтверждали, что полученный тонкий слой состоял из Ti-силикалита-1.
Пример 7
Этот пример демонстрирует применение повторных гидротермических обработок в адекватных синтезных растворах для увеличения толщины готового тонкого слоя до целевого значения.
Подложку из кристаллического кремния в форме пластины (40х9х0,4 мм) промывали по методу, который описан в примере 1. По завершении процесса промывки подложку обрабатывали в течение пяти минут раствором, рН которого доводили до 8,0, содержавшим 0,4 мас.% катионоактивного полимера (продукт Berocell 6100 фирмы Akzo Nobel, Швеция), с последующей промывкой 0,10 М раствором аммиака. Подложку с модифицированной поверхностью переносили в золь (с содержанием сухого вещества 2,5 мас.%), включавший микрокристаллы силикалита-1 с размерами приблизительно 30 нм, и выдерживали для контактирования с этим золем в течение пяти минут для адсорбции монослоя микрокристаллов на поверхности подложки. После промывки 0,10 М раствором аммиака и сушки на воздухе подложку с адсорбированными микрокристаллами кальцинировали при 250oС в течение 10 мин на воздухе и затем давали ей остывать до комнатной температуры.
Образец погружали в синтезный раствор следующего состава: 3 ТПА-ОН/25 SiO2/1500 H2O/100 EtOH и обрабатывали в этом растворе при 100oС в течение 62 ч. Результатом этой обработки являлись непрерывный рост и взаимное срастание адсорбированных кристаллов с образованием на поверхности подложки плотного и сплошного силикалитного тонкого слоя. Толщина тонкого слоя после этой обработки, как ее определяли с помощью СЭМ, составляла 530 нм. Далее этот образец подвергали гидротермической обработке по аналогичному методу (в течение 80 ч) с использованием свежеприготовленного синтезного раствора описанного выше состава по методике, описанной выше. После этой второй гидротермической обработки толщина тонкого слоя составляла, как ее определяли с помощью СЭМ, примерно 1200 нм.
Пример 8
Этот пример демонстрирует получение плотного и сплошного тонкого слоя силикалита-1 на поверхности пористой γ/α-алюминийоксидной мембране.
Обычную γ/α-алюминийоксидную мембрану с номинальным размером пор 5 нм вначале подвергали О2-плазменной обработке, а затем промывали в соответствии с методом, описанным в примере 1. После процесса такой промывки образец промывали 0,10 М раствором аммиака, профильтрованным через 0,1-микрометрическую поливинилиденфторидную мембрану. Затем алюминийоксидную мембрану в течение 10 мин обрабатывали раствором, рН которого доводили до 8,0, содержавшим 0,4 мас.% катионоактивного полимера (продукт Berocell 6100 фирмы Akzo Nobel, Швеция), с последующей промывкой профильтрованным 0,10 М раствором аммиака. Мембрану с модифицированной поверхностью переносили в золь (с содержанием сухого вещества 2,5 мас.%), включавший микрокристаллы силикалита-1 с размерами приблизительно 30 нм, и выдерживали для контактирования с этим золем в течение 10 мин для адсорбции монослоя микрокристаллов на поверхности подложки. После промывки профильтрованным 0,10 М раствором аммиака и сушки на воздухе мембрану с адсорбированными микрокристаллами кальцинировали, перенося в холодную печь, нагревая печь до 425oС в течение 20 мин и выдерживая при этой температуре в течение 10 мин. Затем печь выключали и давали ей остывать до комнатной температуры, после чего мембрану извлекали. Далее мембрану устанавливали в тефлоновом держателе, конструкция которого предназначалась для защиты обратной стороны мембраны от синтезного раствора. Установленную мембрану подвергали гидротермической обработке синтезным раствором, который характеризовался следующим составом: 3 ТПА-ОН/25 SiO2/1500 Н2О/100 EtOH при 100oС в течение 77 ч. Перед использованием этот синтезный раствор фильтровали через 0,1-микрометрическую поливинилиденфторидную мембрану. После гидротермической обработки мембрану извлекали и промывали 0,10 М раствором аммиака. Далее процесс синтеза повторяли с созданием тех же самых условий и с помощью такого же самого, что и описанный выше, (свежеприготовленного) синтезного раствора. Мембрану подвергали конечной промывке 0,10 М раствором аммиака и сушили на воздухе.
Некальцинированную мембрану испытывали проведением эксперимента на проницаемость при 70oС и с ΔP, равной 1,0 бар. Расход потока по данным измерений составлял <1,7•10-5 л/(м2•ч•бар), указывая на то, что тонкий слой оказывался по существу газонепроницаемым.
Пример 9
Этот пример иллюстрирует получение тонкого слоя гидроксисодалита на кварцевой подложке.
Коллоидную суспензию дискретных кристаллов гидроксисодалита готовили гидротермической обработкой раствора следующего молярного состава: 14 (ТМА)2O/0,85 Na2O/1,0 Al2O3/40 SiO2/805 Н2О. Золь очищали в соответствии с методом, описанным в примере 1, и добавлением 0,1 М NaOH pH золя доводили до 10,5. Размеры коллоидных микрокристаллов гидроксисодалита, определенные по динамическому светорассеянию, составляли 26 нм.
В тефлоновом держателе вертикально устанавливали монокристаллическую подложку в форме кварцевой пластины (10х10х1 мм) и промывали в соответствии с методом, описанным в примере 1. После процесса промывки подложку в течение 10 мин обрабатывали раствором, pH которого доводили до 8,0, содержавшим 0,4 мас. % катионоактивного полимера (продукт Berocell 6100 фирмы Akzo Nobel, Швеция), с последующей промывкой 0,10 М раствором аммиака. Подложку с модифицированной поверхностью переносили в золь (с содержанием сухого вещества 2,5 мас.%), включавший гидроксисодалитовые микрокристаллы, и выдерживали для контактирования с этим золем в течение 10 мин. С целью улучшить адсорбцию микрокристаллов на поверхности подложки обработку катионоактивным полимером и последующую адсорбцию микрокристаллов повторяли еще раз. После промывки 0,10 М раствором аммиака и сушки на воздухе подложку с адсорбированными микрокристаллами кальцинировали при 425oС на воздухе в течение 30 мин и затем ей давали остыть до комнатной температуры. Образец погружали в синтезный раствор следующего состава: 14 (ТМА)2O/1,9 Na2O/1,0 Аl2O3/40 SiO2/815 H2O и обрабатывали этим раствором при 100oС в течение 16 ч. В результате такой обработки происходили непрерывный рост и взаимное срастание адсорбированных кристаллов с образованием на поверхности подложки тонкого слоя гидроксисодалита.
Пример 10
Этот пример иллюстрирует получение тонкого сплошного слоя бета-цеолита на танталовой подложке.
В качестве подложек для выращивания бета-цеолитных тонких слоев применяли Та-пластинки (Plansee, Австрия, 99,9%) размерами 10x30 мм. Перед применением поверхность Та-пластинок очищали при комнатной температуре в течение 15 мин ацетоном (р.а.) с помощью ультразвука, а затем в течение 10 мин раствором, который характеризовался следующим молярным составом: 9 Н2О2/10 НСl/350 Н2О. После этих операций очистки Та-пластины несколько раз промывали дистиллированной водой. Знак поверхностного заряда Та-пластинок на обратный изменяли обработкой катионоактивным полимером (продукт Berocell 6100 фирмы Akzo Nobel, Швеция) с повторяющимися звеньями [СН2СH(ОH)СH2H(СH3)2]n + и средней молекулярной массой 50000 г/моль. Та-пластинки погружали на 1 ч в 30 мл раствора, содержавшего 0,5 мас.% такого катионоактивного полимера, после чего избыток полимера удаляли промывкой 0,1 М раствором аммиака.
Коллоидную суспензию бета-цеолита (со средним размером кристаллов 90 нм) готовили гидротермической обработкой раствора предшественника следующего молярного состава: 0,35 Na2O/9 ТЭА-ОН/0,25 Аl2O3/25 SiO2/295 Н2О в течение 6 дней при 100oС. Полученные таким образом кристаллы выделяли из маточного раствора центрифугированием и вновь диспергировали в дистиллированной воде, после чего суспензии вновь центрифугировали. Эти операции повторяли до тех пор, пока значение рН цеолитной суспензии не оказывалось в интервале 9,5-10,0. Та-подложку с модифицированной поверхностью вводили в контакт с очищенной бета-цеолитной суспензией (3,5 мас.%, добавлением 0,1 М аммиачного раствора рН доводили до 10) на 1 ч, после чего избыток цеолита удаляли промывкой 0,1 М раствора NН3. Анализ композита с помощью СЭМ показывал наличие бета-цеолитных кристаллов монослоя на Та-подложке.
Та-подложку с адсорбированными бета-цеолитными кристаллами кальцинировали на воздухе при 300oС в течение 1 ч, после чего композит погружали в раствор бета-цеолитного предшественника, молярный состав которого представлен выше, и подвергали гидротермической обработке с обратным холодильником в течение 6 дней при 100oС. Микрофотографии цеолитного тонкого слоя, выполненные с помощью сканирующего электронного микроскопа (СЭМ) после гидротермической обработки, демонстрируют поликристаллическую морфологию, а изображения сбоку показывают наличие сплошного тонкого слоя средней толщины 200 нм. Спектроскопические исследования бета-цеолитного тонкого слоя по отражению-поглощению инфракрасного излучения (ОПИK) в спектрометре Perkin Elmer 2000 (для ИК-спектрометрии с преобразованием Фурье). Угол падения составлял 83o, для каждого спектра получали по 500 интерферограмм при разрешающей способности 4 см-1. На спектре образца бета-цеолитного тонкого слоя были видны полосы поглощения при 450, 520, 570, 1150, 1175 и 1230 см-1, характерные для молекулярной структуры типа бета-диоксида кремния. Результаты РГФ-анализа подложки и тонкого слоя показали, что осажденный материал представлял собой бета-цеолит.
Пример 11
С использованием тетраэтоксисилана (ТЭОС, >98% сорта GС, <3 част./млн А1, фирма Aldrich-Chemie), гидроксида тетрапропиламмония (ТПА-ОН. 1,0 М в воде, 143 част./млн Na, 4200 част./млн K, <10 част./млн А1, фирма Sigma) и дистиллированной воды синтезировали коллоидные суспензии ТПА-силикалита-1.
Получение тонких слоев силикалита-1
Поверхность золотых подложек [выполненных нанесением золота на предварительно обработанные тонкие кремниевые диски (100) с TiN-покрытием с применением устройства для нанесения покрытий BAL-TRC MED 020, работавшей при базовом давлении приблизительно 2•10-2 мбар] модифицировали посредством контактирования подложки в течение 3 ч при комнатной температуре с 10 мМ раствором МПС [гамма-меркаптопропилтриметоксисилана (продукт MPS фирмы Osi Specialities)] в метаноле. Прилипавший к поверхности силан гидролизовали при комнатной температуре в кислом растворе (0,10 М НСl) в течение 15 ч. Избыток силана удаляли промывкой этанолом.
Суспензию дискретных коллоидных кристаллов среднего размера 90 нм готовили по методу, аналогичному описанным в предыдущих примерах. Золь затравочного предшественника обладал следующим молярным составом: 9 ТПА-ОН/25 SiO2/480 H2O/100 этанола. Добавлением сильной катионообменной смолы [DOWEX HCR-S(H+), фирма Dow Chemical] pH очищенного золя уменьшали до 3,4. Монослой силикалита-1 адсорбировали введением покрытой силаном подложки в контакт с 40 мл суспензии концентрацией 4,2 мас.% силикалита-1 при pH 3,4 [дзета-потенциал (z) силикалита-1: от +50 до +60 мВ] на 1 ч. Для удаления избыточного кристаллического материала пластинки несколько раз промывали дистиллированной водой. Эти тонкие диски кальцинировали на воздухе при 300oС в течение 1 ч. Композитные тонкие диски вводили в контакт с раствором ТПА-силикалита-1 в качестве предшественника следующего молярного состава: 3 ТПА-ОН/25 SiO2/1500 Н2О/100 этанола и подвергали гидротермической обработке при 100oС в течение 20 ч. После синтеза тонкие диски промывали дистиллированной водой и ацетоном и сушили при 100oС в течение 30 мин. На золотой подложке осаждались нанесенные тонкие слои силикалита-1.
Пример 12
Тонкие слои силикалита-1 на полированных, покрытых золотом QCM (фирма Cold Springs R& D, Марселлус, шт. Нью-Йорк; с резонансной частотой 10 МГц; смещение на 1 Гц соответствует 4,3 нг/см2 поверхности) получали по трехстадийному методу. Вначале QCM очищали в течение 30 мин в ультразвуковой ванне, содержавшей ацетон, а затем несколько раз промывали дистиллированной водой и дважды по 10 мин обрабатывали ультразвуком в дистиллированной воде. Монослой γ-меркаптопропилтриметоксисилана (МПС) на QCM получали контактированием подложки в течение 3 ч с 10 нМ раствором МПС в метаноле. В дальнейшем этот слой при комнатной температуре гидролизовали в 0,1 М НСl в течение 15 ч. Модифицированные силаном QCM при комнатной температуре в течение одного часа обрабатывали водным раствором концентрацией 0,4 мас.% катионоактивного полимера (продукт Berocell 6100 с молекулярной массой приблизительно 50000 фирмы Akzo Nobel).
Коллоидные кристаллы силикалита-1 (среднего размера 60 нм) адсорбировали на модифицированной поверхности QCM контактированием подложек в течение 1 ч с очищенной коллоидной суспензией (при значении рН 10) силикалита-1 в воде концентрацией 3 мас.%. После промывки несколько раз 0,1 М NН3 подложки кальцинировали на воздухе в течение 1 ч при 300oС. Далее QCM погружали в синтезный раствор силикалита-1. Синтезную смесь следующего молярного состава: 3 ТПА-ОН/25 SiO2/1500 Н2О/100 EtOH выдерживали в течение 20 ч на полиэтиленгликолевой бане при 100oС в полипропиленовом реакторе, снабженном обратным холодильником. Перед сорбционным анализом для удаления воды и органического шаблона снабженные покрытием QCM кальцинировали при 350oС в течение 12 ч в токе кислорода.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОЛЕКУЛЯРНЫЕ СИТА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2185228C2 |
| МОЛЕКУЛЯРНЫЕ СИТА, СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБ РАЗДЕЛЕНИЯ ТЕКУЧЕЙ СМЕСИ, СПОСОБ КАТАЛИЗА ХИМИЧЕСКОЙ РЕАКЦИИ (ВАРИАНТЫ) С ИХ ИСПОЛЬЗОВАНИЕМ | 1995 |
|
RU2169039C2 |
| МОЛЕКУЛЯРНЫЕ СИТА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2174044C2 |
| СПОСОБ КОНВЕРСИИ УГЛЕВОДОРОДОВ С ИСПОЛЬЗОВАНИЕМ СВЯЗАННОГО ЦЕОЛИТОМ ЦЕОЛИТНОГО КАТАЛИЗАТОРА | 1995 |
|
RU2177468C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПАРАКСИЛОЛА | 1997 |
|
RU2184106C2 |
| СОСТАВ ЖИДКОГО ТОПЛИВА И КОНЦЕНТРАТ ПРИСАДКИ | 1993 |
|
RU2114156C1 |
| ИОНОГЕННАЯ КАТАЛИТИЧЕСКАЯ КОМПОЗИЦИЯ НА НОСИТЕЛЕ | 1995 |
|
RU2183644C2 |
| ПРОИЗВОДНЫЕ АМИДОВ ИЛИ СОЛЕЙ АМИНОВ ФТАЛЕВОЙ КИСЛОТЫ, ДИСТИЛЛЯТНОЕ ЛЕГКОЕ ТОПЛИВО И КОНЦЕНТРАТ | 1991 |
|
RU2101276C1 |
| КОМПОЗИЦИЯ ЖИДКОГО ТОПЛИВА | 1996 |
|
RU2158750C2 |
| ВОЛОКНА И ТЕКСТИЛЬНЫЕ МАТЕРИАЛЫ ИЗ ПОЛИЭТИЛЕНА ВЫСОКОЙ ПЛОТНОСТИ И СПОСОБ ИХ ИЗГОТОВЛЕНИЯ | 1995 |
|
RU2164969C2 |
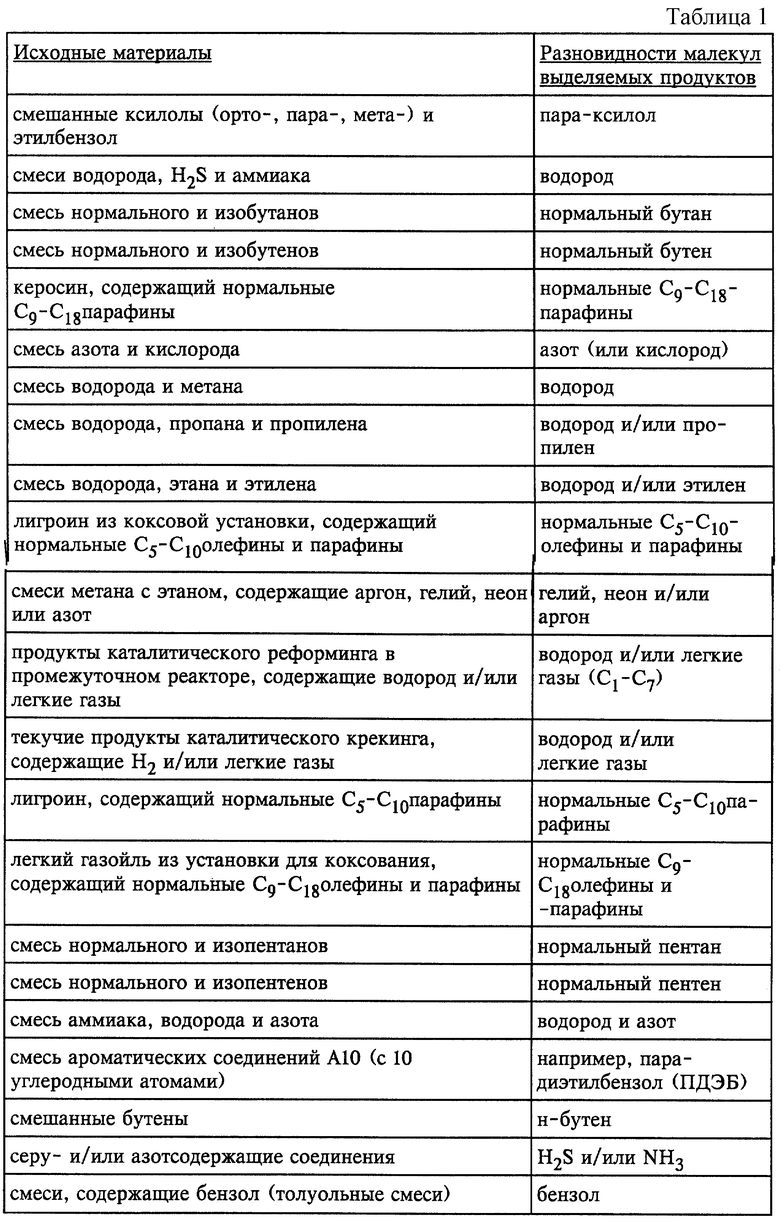


Изобретение относится к структурам молекулярных сит на подложках. Описан способ нанесения тонких слоев молекулярных сит на поверхность различных подложек. Способ характеризуется тем, что микрокристаллы предлагаемых молекулярных сит на первой стадии связывают с поверхностью подложки в виде монослоя, а затем на второй стадии дают ему расти с образованием тонкого сплошного и плотного слоя с толщиной 100 нм и меньше. Такие тонкие слои молекулярных сит эффективны для процессов изготовления мембран, катализаторов, датчиков и армирующих наполнителей для полимеров. 3 с. и 17 з.п. ф-лы, 3 ил. , 2 табл.
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Сборно-разъемное соединение тонкостенных труб | 1960 |
|
SU135069A1 |
| УСТАНОВКА ДЛЯ РЕЗКИ ЛИСТОВОГО СТЕКЛА | 0 |
|
SU270212A1 |
| Дытнерский Ю.И | |||
| Барометрические процессы | |||
| - М., 1986, с.33-34 | |||
| RU 94014484 А1, 20.02.1996. | |||

