Область техники, к которой относится изобретение.
Изобретение относится к каталитическим композициям, которые могут быть использованы в реакциях присоединения олефиновоненасыщенных мономеров, например, при полимеризации. Изобретение может быть использовано, в частности, в процессах координационной полимеризации, в которых применяют соединения на носителях, такие, как при суспензионной или газофазной полимеризации олефиновоненасыщенных мономеров. Изобретение относится также к самим способам такой полимеризации. Каталитические композиции включают металлоценовые катионоактивные катализаторы, анионоактивные активаторы и металлоксидные носители.
Предпосылки создания изобретения
Координационная полимеризация олефиновоненасыщенных мономеров хорошо известна, она привела к большому распространению в современном обществе эластомерных и пластических композиций, таких, как полиэтилен, полипропилен и этилен-пропиленовый каучук. Первоначально использовались соединения переходных металлов с такими активаторами, как алюминийалкилы, а следующие разработки расширили рамки этой работы до содержащих объемные лиганды (например, η5-циклопентадиенильные) соединений переходных металлов ("металлоценов") с активаторами, такими, как алкилалюмоксаны. Самые последние разработки показали эффективность ионогенных катализаторов, включающих металлоценовые катионы, активированные некоординационными анионами (см., например, европейскую заявку А-277004 и патент США 5198401). В этих ссылках описано протонирование металлоценовых соединений анионоактивными предшественниками с получением стойких ионогенных катализаторов.
Ранее известные ионогенные катализаторы в различной мере проявляют заметную чувствительность к каталитическим ядам, содержащимся в потоках мономерного сырья или в потоках рециркулирующей текучей среды в процессе полимеризации и создают проблемы при использовании совместно с инертными оксидными носителями, которые, как правило, характеризуются наличием либо удерживаемой влаги или полярных гидроксильных групп, либо и того, и другого. Соответственно были разработаны способы применения связывающих яды соединений, например, алюминийалкилов и алкилалюмоксанов, для проведения полимеризации в растворе и удаления или нейтрализации полярных групп, удерживаемых внутри или на металлоксидных носителях (см., например, патент США 5153157, в котором описаны очищающие соединения на основе металлов группы IIIА, и международные заявки WO 91/09882, 94/00500 и 94/03506, где описана методика нанесения на носители с использованием аналогичных соединений. В патенте США 5206197 описана улучшенная полимеризация стирола, где ионогенные каталитические системы включают гидрокарбил металла и могут быть нанесены на носитель. Все эти документы относятся к описанию металлоценовых соединений, ионогенных активаторов и пригодных для использования очищающих соединений.
Также хорошо известным классом металлоценовых катализаторов на носителях, которые, как было установлено, могут быть использованы, в частности, для получения линейного полиэтилена в промышленных процессах в растворе, суспензии или газовой фазе, являются катализаторы на носителях, приготовленные взаимодействием хромоценов с оксидами металлов (М), предложенные для получения хроматной или бихроматной структуры: -Сr(-О-М-)2 или (-М-O-Сr-)(O)(-Сr-O-М-) (см. , например, "Supported Chromium Catalysts for Ethylene Polymerization", McDaniel, Advances in Catalysis 1985, 33, 47-97. Диоксид кремния является менее предпочтительным носителем вследствие более низкой реакционной способности, чем, например, у фосфата алюминия, для которого, как правило, никакого другого активатора не требуется, а служащий центром металл металлоцена ковалентно связан через оксо-мостик (-O-) с металлом/металлоидом носителя.
Кроме того, в патенте США 5427991 и его аналоге - международной заявке 93/11172 описано химическое связывание некоординационных анионоактивных активаторов с носителями таким образом, чтобы получить полианионные активаторы, которые, если их используют с металлоценовыми соединениями, устраняют проблемы десорбции катализаторов, с которой приходится сталкиваться при использовании ионогенных катализаторов, физически адсорбированных на металлоксидных носителях, в процессе полимеризации в растворе или суспензии. Такие носители представляют собой основные компоненты инертных мономерных, олигомерных, полимерных или металлоксидных носителей, которые получают таким образом, чтобы ввести химически связанные некоординационные анионы. Описание приготовления полианионных активаторов из оксидов металлов (фиг. 8) включает взаимодействие гидроксилированной поверхности диоксида кремния с силановыми агентами сочетания, по меньшей мере некоторые из которых содержат галоидарильные остатки, которые можно литиировать с получением химически связанных ариллитиевых групп. В дальнейшем их обрабатывают объемным металлоидным трисперфторфенилборным [B(pfp)3] предшественником и вводят в ионообменную реакцию с диметиланилинийгидрохлоридом ([ДМAГ]+ [Cl]- в таких условиях, чтобы получить кремнийдиоксидную поверхность, на которой имеются ковалентно связанные активаторные группы [ДМАГ]+ [(pfp)3B]-. В примерах проиллюстрирована полимеризация в массе для получения полипропилена с использованием в автоклавных реакторах периодического действия полиионных каталитических систем на основе гидрокарбильного носителя.
В настоящее время существует необходимость как в приготовлении ионогенных каталитических систем на носителях, так и в поиске катализаторов на носителях, которым не свойственны проблемы, возникающие в процессе полимеризации и ведущие к нестабильности работы реактора в результате перепроизводства, засорения поверхностей полимеризационного оборудования и нежелательного получения полимерных частиц с плохой морфологией, неприемлемых для эффективного получения полимеров в промышленном масштабе.
Краткое описание изобретения
Целью настоящего изобретения является решение вышеуказанных проблем, а также проблем, которые описаны ниже, и создание каталитической композиции с переходным металлом, характеризующейся тем, что она включает металлоксидный носитель, содержащий ковалентно связанный с его поверхностью непосредственно через кислородный атом этого оксида металла противостоящий анион, который также ионно связан с каталитически активным соединением с катионом переходного металла. Кроме того, настоящее изобретение включает способ полимеризации, характеризующийся тем, что в нем предусмотрено введение одного или нескольких мономеров, способных полимеризоваться координационной или карбокатионной полимеризацией в традиционно приемлемых полимеризационных условиях, в контакт с каталитической композицией по изобретению. Настоящее изобретение включает способ приготовления каталитической композиции по изобретению, в котором как правило, предусмотрено взаимодействие кислоты Льюиса в форме объемного источника каталитического аниона с гидроксильными группами носителя, предпочтительно в присутствии основания Льюиса, с получением связанного с носителем анионоактивного активатора, который, когда его объединяют с приемлемым соединением переходного металла, протонирует его таким образом, что образуется ионогенная каталитическая система, включающая катион переходного металла и связанный с носителем анион.
Подробное описание изобретения и примеры
Каталитическая композиция по изобретению, описанная выше, может быть представлена общей химической формулой
[LnL'mM'R']+ [LA-O-M"-]-, (1)
где [LnL'mM'R']+ обозначает катион на основе каталитически активного переходного металла, а [LA-O-M"-]- обозначает связанный с металлоксидным носителем противостоящий анион. В частности, в этой формуле Ln обозначает один или несколько лигандов (n обозначает d0-1, где d0 обозначает высшее состояние окисления М'), ковалентно связанных с М', L'm обозначает нейтральный, неокисляющий лиганд, имеющий дативную связь с М' (как правило, m обозначает число 0-3), М' обозначает переходный металл группы 4, 5, 6, 9 или 10, R' обозначает лиганд, имеющий σ-связь с М', в который для координационной полимеризации можно ввести способный полимеризоваться мономер или макромономер. LA обозначает кислоту Льюиса, которая способна образовывать анионоактивный активатор, О обозначает кислород, а М" обозначает металл/металлоид металлоксидного носителя.
Металлоксидные носители по изобретению включают любые оксиды металлов/металлоидов, имеющих поверхностные гидроксильные группы, характеризующиеся величиной pKa, равной или меньше той, которой характеризуется аморфный диоксид кремния, т.е. рKa меньше или равна приблизительно 11. При получении по изобретению ковалентно связанного анионоактивного активатора по предпочтительному варианту LA выбирают таким образом, чтобы она была способна образовывать дативный комплекс с силанольной группой (которая действует как основание Льюиса), получая таким образом формально диполярную (цвиттерионную) структуру кислоты Бренстеда, связанную с металлом/металлоидом металлоксидного носителя. Следовательно, в соответствии с изобретением приемлем любой из общеизвестных кремнийдиоксидных материалов носителей, который удерживает гидроксильные группы после осуществления дегидратационной обработки. Благодаря доступности предпочтительны носители на основе как диоксида кремния, так и оксида металла, содержащего диоксид кремния, например, диоксид кремния/оксид алюминия. Наиболее типичными являются частицы диоксида кремния, гели и стеклянные шарики.
Эти металлоксидные композиции дополнительно могут включать оксиды других металлов, такие, как оксиды Аl, K, Mg, Na, Si, Ti и Zr, и предпочтительно должны быть обработаны термическими и/или химическими средствами с целью удалить воду и свободный кислород. Как правило, такую обработку проводят в вакууме внутри горячей сушильной печи, в горячем псевдоожиженном слое или с помощью обезвоживающих агентов, таких, как органосиланы, силоксаны, алюминийалкильные соединения и т.д. Степень обработки должна быть такой, чтобы при этом удалялось максимально возможное количество удерживаемый влаги и кислорода, но чтобы сохранялось химически значимое количество гидроксильных функциональных групп. Так, например, допустимо кальцинирование при температуре, доходящей до 800oС, или более высокой, доходящей до точки, близкой к точке разложения материала носителя, в течение нескольких часов, а если необходимо более высокое содержание нанесенного анионоактивного активатора, обычно приемлемо кальцинирование при более низких температурах и в течение более коротких периодов времени. В случае, когда оксидом металла является диоксид кремния, как правило, приемлема нагрузка, обеспечивающая достижение от менее 0.1 до 3,0 ммолей активатора/г SiO2, которую можно получить, например, варьированием температуры кальцинирования от 200 до 800oС и более. См., например, Zhuralev и др., Langmuir 1987, том 3, 316, где описана корреляция между температурой кальцинирования и ее продолжительностью и содержанием гидроксильных групп в диоксидах кремния с различной удельной площадью поверхности.
Согласно настоящему изобретению предварительная обработка перед добавлением LA менее чем стехиометрическим количеством химических обезвоживающих агентов позволяет также адаптировать гидроксильные группы, сделав их доступными в качестве участков присоединения. В предпочтительном варианте их обычно используют в недостаточном количестве и, как правило, применяют те, которые имеют единственный лиганд, способный вступать во взаимодействие с силанольными группами [например, (CH3)4SiCl] или гидролизоваться как-либо иначе, что позволяет минимизировать влияние фактора, препятствующего взаимодействию каталитических соединений переходных металлов со связанным активатором. В случае, когда кальцинирование проводят при температурах ниже 400oС, с целью создания превосходства над связанными водородом парами силанольных групп, которые присутствуют в этих менее жестких условиях кальцинирования, можно применять бифункциональные агенты сочетания (например, (CH3)3SiCl2). Касательно описания влияния силановых агентов сочетания на кремнийдиоксидные полимерные наполнители, которые обычно эффективны также при модификации силанольных групп на каталитических носителях по настоящему изобретению, см. , например, "Investigation of Quantitative SiOH Determination by the Silane Treatment of Disperse Silica", Gorski и др., Journ. of Colloid and Interface Science, том 126, 2, декабрь 1988 г. Аналогичным образом использование LA в избытке по сравнению со стехиометрическим количеством, необходимым для реакции с соединениями переходного металла, обычно обеспечивает нейтрализацию избытка силанольных групп без оказания существенного вредного влияния на приготовление катализатора или последующую полимеризацию.
Активаторные анионоактивные нейтральные предшественники, которые служат в качестве кислоты Льюиса (LA) по изобретению, включают любой из некоординационных анионоактивных предшественников достаточной кислотности для акцептирования доступной электронной пары кислородного атома гидроксильной группы и упрощения протонирования соединения переходного металла или вторичного протонного акцептора (см. ниже) протоном силанольной группы. Предпочтительные активаторные анионоактивные нейтральные предшественники, которые служат в качестве кислоты Льюиса (LA) по изобретению, представляют собой сильные кислоты Льюиса с негидролизуемыми лигандами, по меньшей мере один из которых оттягивает электроны, такие, как кислоты Льюиса, которые известны как способные отделять анионоактивный фрагмент от диметилцирконоцена (бициклопентадиенилцирко-нийдиметила), например, трисперфторфенилбор. Следовательно, эти предшественники не должны иметь никаких реакционноспособных лигандов, которые могут быть протонированы гидроксильными группами оксида металла (протоном силанольной группы). Так, например, любые кислоты Льюиса на основе элемента группы 13, содержащие только алкильные, галоидные, алкокси- и/или амидные лиганды, которые легко гидролизуются в водной среде, оказываются непригодны. По меньшей мере один лиганд LA должен обладать достаточной способностью оттягивать электроны для достижения требуемой кислотности, например, трисперфторфенилбор. Типичные металл/металлоидные центры LA включают бор, алюминий, сурьму, мышьяк, фосфор и галлий. Наиболее предпочтительной LA является нейтральное соединение, включающее в качестве центра металлоид группы 13 с дополнением из лигандов, которые совместно обладают достаточной способностью оттягивать электроны, благодаря чему его кислотность по Льюису превосходит или равна кислотности AlCl3. В качестве примеров можно назвать трисперфторфенилбор, трис[3,5-ди(трифторметил)фенил] бор, трис(ди-трет-бутилметилсилил)перфторфенилбор и другие высокофторированные трисарилборные соединения.
Кроме того, вероятно, что вследствие пространственно ограничивающего присутствия поверхности носителя в дополнение к пространственно ограничивающим стабилизирующим лигандам (которые не отделены или замещены протоном или алкильной группой в процессе протонирования или алкилирования) каталитически активных соединений переходных металлов противостоящие анионы по изобретению включают анионы, способные координировать с переходным металлом в обычных условиях ионной реакции в растворе, одновременно оставаясь лабильными. Термин "лабильный" является термином, известным в данной области техники, и означает, что в полимеризационных условиях этот анион лишь слабо координирован на участке каталитической активности, благодаря чему допустимо замещение полимеризуемым мономером в точке присоединения мономера. Примеры кислот Льюиса в качестве предшественников типичных координационных анионов по настоящему изобретению включают объемные алюминийсилоксиды, такие, как Al[OSi(C6H5)3] 3 или Al[OSi(O-трeт-бутил)3] 3, которые можно представить в общем виде как Al(OsiR'"3)3 или Al[OSi(O-R'")3]3, где каждый R'", которые одинаковые или различные, обозначает объемный заместитель С4 или с еще большим числом углеродных атомов, такой, как трет-бутил, или с более высокой Mw разветвленный алифатический, ароматический, ароматический, замещенный алифатической группой радикал, и т.д.
В соответствии с настоящим изобретением приемлемо любое каталитически активное соединение переходного металла, включая известные соединения переходных металлов, используемые при традиционной координационной полимеризации по Циглеру-Натта, а также металлоценовые соединения, которые также известны как пригодные для использования при координационной полимеризации, когда такие соединения способны к каталитической активации связанным с носителем анионоактивным активатором. Как правило, они включают соединения переходных металлов, металл которых находится в состоянии окисления d0, т.е. где металл имеет свою высшую степень окисления и где по меньшей мере один металлический лиганд может быть протонирован связанным с носителем анионоактивным активатором, в частности те лиганды. которые включают гидрид, алкил и силил. Лиганды, способные к протонированию, и содержащие их соединения переходных металлов включают таковые, описанные в известной литературе (см., например, европейские заявки А-277003 и А-277004 и патент США 5198401). Синтез таких соединений хорошо известен из опубликованной литературы. Кроме того, когда металлические лиганды включают галоидсодержащие остатки (например, бициклопентадиенилцирконийдихлорид), которые не способны к протонированию в стандартных условиях, их можно трансформировать с помощью известных реакций алкилирования с металлоорганическими соединениями, такими, как литий- или алюми-нийгидриды либо - алкилы, алкилалюмоксаны, реактивы Гриньяра и т.д. Касательно взаимодействия алюминийорганических соединений с дигалоидзамещенными металлоценовыми соединениями перед присоединением активирующих анионоактивных соединений, см., например, европейскую заявку А1-0570982.
Дополнительное описание металлоценовых соединений, которые включают или могут быть алкилированы с той целью, чтобы они включали по меньшей мере один лиганд, способный к отщеплению посредством протонирования с образованием катиона переходного металла, содержится в патентной литературе, например, в европейской заявке А-0129368, патентах США 4871705, 4937299 и 5324800, европейских заявках А-0418044 и А-0591736 и международных заявках 92/00333 и 94/01471. В описании настоящего изобретения такие металлоценовые соединения могут быть представлены как моно-, ди- и трициклопентадиенилзамещенные соединения переходных металлов группы 4, 5, 6, 9 или 10, у которых сами циклопентадиенильные заместители могут быть замещены одной или несколькими группами и могут быть связаны между собой мостиками или могут быть связаны мостиками через гетероатом с переходным металлом. Размеры и структура циклопентадиенильных заместителей и связывающих мостиковых элементов для приготовления ионогенных каталитических систем по изобретению решающего значения не имеет, но для повышения полимеризационной активности и придания полимеру предусмотренных характеристик их следует подбирать таким образом, как это описано в литературе. В предпочтительном варианте циклопентадиенильные (или замещенные циклопентадиенильные, такие, как инденильные или замещенные инденильные) кольца, когда они связаны мостиками между собой, обычно замещены низшими алкилами (С1-С6) в положении 2 и обычно дополнительно включают алкильные, циклоалкильные, арильные, алкиларильные и/или арилалкильные заместители, причем эти последние в виде либо сконденсированных, либо боковых подвешенных кольцевых структур включают полициклические структуры, например, такие, как представленные в патентах США 5278264 и 5304614. Каждый из таких заместителей должен обладать по существу гидрокарбильными характеристиками и, как правило, содержит до 30 углеродных атомов, но может содержать гетероатомы не более чем с 1-3 неводородными/углеродными атомами, например, N, S, О, Р и Si.
Металлоценовые соединения, пригодные для получения линейного полиэтилена или содержащих этиленовые звенья сополимеров (где термин "сополимер" означает содержание по меньшей мере двух различных мономерных звеньев), представляют собой по существу любые из соединений, которые известны в данной области техники (в частности, их конкретный перечень можно найти в указанных выше европейской заявке А-277004, международной заявке 92/00333 и патентах США 5198401, 5001205, 5324800, 5308816 и 5304614). Принципы выбора металлоценовых соединений для использования с целью получить изотактические или синдиотактические полипропилены и их синтез в данной области техники хорошо известны и в данном случае можно, в частности, сослаться как на патентную, так и научную литературу [см., например. Journal of Organmetallic Chemistry 369, 359-370 (1989)] . Такие катализаторы, как правило, представляют собой пространственно жесткие асимметричные хиральные или связанные мостиками хиральные металлоцены [см., например, патенты США 4892851, 5017714, 5296434 и 5278264, международные заявки PCT/US92/10066, 93/19103, европейские заявки А2-0577581 и А1-0578838 и научную литературу: "The Influence of Aromatic Substituents on the Polymerization Behavior of Bridged Zirconocene Catalysts", Spaleck, W. и др., Organometallics 1994, 13, 954-963, и "ansa-Zirconocene Polymerization Catalysts with Annelated Ring Ligands-Effects on Catalytic Activity and Polymer Chain Lengths, Brinzinger H. и др., Organometallics 1994, 13, 964-970, и упомянутые в них документы]. Хотя приведенное выше описание по большей части относится к каталитическим системам с алюмоксановыми активаторами, аналогичные металлоценовые соединения обычно могут быть использованы в сочетании с нанесенными на носитель противостоящими анионами по настоящему изобретению для приготовления активных координационных каталитических систем, когда по меньшей мере один из галоидсодержащих лигандов металлов (если это имеет место) замещен лигандами, способными к протонированию, например, в результате реакции алкилирования, как описано выше, а другой представляет собой группу, в которую можно вводить этиленовую группу -С= С-, например, гидридную, алкильную или даже менее эффективную силильную.
Неограничивающие примеры металлоценовых соединений включают моноциклопентадиенильные соединения, такие, как пентаметил циклопентадиенил титанизопропоксид, пентаметил циклопентадиенил трибензилтитан, диметил силилтетраметил циклопентадиенил -трет- бутиламидотитан дихлорид, пентаметил циклопентадиенил титантриметил, диметил силилтетраметилцикло пентадиенил- трет- бутиламидоцирконий диметил, диметил силилтетраметил циклопентадиенил додециламидогафний дигидрид, диметил силилтетраметил циклопентадиенил додециламидогафний диметил, не связанные мостиками бициклопентадиенильные соединения, такие, как бис(1,3-бутилметилциклопентадиенил) цирконийдиметил, пентаметил циклопентадиенил циклопентадиенил цирконийдиметил, связанные мостиками бициклопентадиенильные соединения, такие, как диметилсилилбис(тетрагидроинденил)цирконийдихлорид, связанные мостиками бисинденильные соединения, такие, как диметилсилилбисинденилцирконийдих-лорид, диметилсилилбисинденилгафнийдиметил, диметилсилилбис(2-метил-бензинденил) цирконийдихлорид, диметилсилилбис (2-метилбензинденил) цирконийдиметил, а также моно-, ди- и трициклопентадиенильные соединения, такие, как описанные в патенте США 5324800 и европейской заявке А-0591756.
Примеры традиционных соединений переходных металлов Циглера-Натта включают тетрабензилцирконий, тетрабис(триметилсилилметил) цирконий, оксотрис (триметилсилилметил) ванадий, тетрабензилгафний, тетрабензилтитан, бис (гексаметилдисилазидо) диметилтитан, трис (триметилсилилметил) ниобий -дихлорид, трис(триметилсилилметил)танталдихлорид. Важными особенностями таких композиций для координационной полимеризации являются лиганд, способный к отщеплению при протонировании, и тот лиганд, в который можно ввести этиленовую (олефиновую) группу. Такие особенности придают способность к протонированию соединения переходного металла и к сопутствующему образованию ионогенной каталитической композиции по изобретению.
Ионогенная каталитическая композиция на носителе по изобретению может быть приготовлена, например, выбором в качестве кислоты Льюиса такой кислоты, которая образует комплекс кислоты-основания Льюиса, т.е. комплекс, который при протонировании соединения переходного металла протоном содержащего гидроксильную группу комплекса кислоты-основания Льюиса, способен выполнять функции приемлемого противостоящего аниона. Такую реакционную последовательность можно представить с помощью нижеследующих уравнений химических реакций.
(2) LA + Н-О-М"- --> (LA)(H-O-M"-)
(3) (LA)(H-O-M"-) + LnL'mM'R'R -->
[LnL'mM'R']+ [LA-O-M"-]- + RH
где значения Ln, L'm, M', R', LA, О и М" указаны выше для уравнения (1), Н обозначает водород и R обозначает лиганд, способный к отщеплению при протонировании. Реакцию, как правило, проводят в растворе в углеводороде (гептане, толуоле и т.д.) при комнатной температуре и нормальном давлении, а нанесенный на носитель катализатор можно отделять, например, путем фильтрации.
Предпочтительный вариант способа получения состоит в присоединении дополнительного вторичного протонного акцептора (LB) с целью содействовать протеканию реакций по уравнениям (2) и (3). Такую реакцию можно представить с помощью нижеследующих уравнений химической реакции
(3а) (LA)(H-O-M"-) + LB --> [LA-O-M"-]-[LB-H]+
(4) [LA-O-M"-]-[LB-H]+ + LnL'mM'R'R-->
[LA-O-M"-]- [LnL'mM'R']+ + RH + LB
где все символы имеют значения, указанные выше. Эту реакцию можно проводить практически так же, как описано выше. LB представляет собой основание Бренстеда, такое, как диэтиланилин, который согласно уравнению (3а) с нанесенным на носитель анионоактивным активатором [LA-O-M"-]- образует аммониевую соль. Другие примеры LB включают диметиланилин, дифенилметиланилин, трибутиламин, дифенилметиламин, трифенилфосфин и триметилфосфит.
Очевидно, что продукт реакции (3а) связан ковалентно и, таким образом, в композиции явно отличается от физически нанесенных (т.е. адсорбированных) ионогенных материалов, таких, как описанные в международной заявке 91/09882. Это однозначно подтверждают результаты наблюдений того, что активные катализаторы получают даже после нескольких промывок растворителями, которые способны удалять индивидуальные компоненты LA и LB, и четко подтверждают результаты ЯМР-анализа в твердом состоянии. В частности, наличие предпочтительно связанной структуры у продуктов реакций (3а) и (4) и структурного различия между продуктом реакции (4) и физически адсорбированными материалами (см. выше) показала 11В-ЯМР-спектроскопия в твердом состоянии Magic Angie Spinning (см. нижеприведенный сравнительный пример 22).
11В-спектрограмма катализатора по изобретению состоит из единственного пика при -6,6 част./млн, тогда как 11B-спектрограмма катализатора, приготовленного в соответствии с примером 14 международной заявки 91/09882, характеризовалась единственным пиком при -17 част./млн. Это различие в химическом 11В-сдвиге согласуется с различием между ионогенными материалами, у которых имеется ковалентная связь с носителем, и ионогенными материалами, физически нанесенными на носитель в соответствии с принципами, известными специалистам в данной области техники и как это описано у Kidd R.G. в "NMR of Newly Accessible Nuclei", под ред. Laszlo P.; Academic Press: New York, 1983; том 2, стр. 49-77. Таким образом, каталитические материалы по изобретению, приготовленные в соответствии с данным описанием, отличны от материалов катализаторов, заявленных в международной заявке 91/09882.
Если результатом осуществления изобретения является активный катализатор, в отношении LB следует принять некоторые ограничительные условия: (а) LB должен представлять собой основание Бренстеда, достаточно сильное для отщепления протона, как показано в уравнении (3а), (б) LB не может быть настолько сильным основанием Бренстеда, чтобы сопряженная с ним кислота была бы не в состоянии протонировать соединения переходных металлов по изобретению, вследствие чего его величина рКbН+ должна быть меньше, чем у RH, (в) комплекс [LA-O-M"-] -[LB-H] + должен иметь кинетически доступный протон и (г) LB должно быть достаточно слабым основанием Льюиса (и/или быть достаточно пространственно объемным), чтобы не конкурировать эффективно с полимеризуемым мономером за свободный координационный участок при металле как катионоактивном центре. Необходимо отметить, что условие (а) в некоторой степени зависит от кислотности LA по Льюису, поскольку это определяет кислотность комплекса (LA) (H-O-M"-) по Бренстеду. Примерами потенциальных LB, которые не обеспечивают образования активных катализаторов, являются: перфтор(трибутил)амин (недостаточно сильное основание Бренстеда), "протонная губка" 1,9-N,N,N,N-тeтpa-метил-1,8-нафталиндиамин (протонированная форма не взаимодействует с LnL'mM'R'R) и хинолин (хорошее основание Льюиса, которое координирует с металлом М' как центром).
В результате осуществления таких способов получают ряд реакционных продуктов. Взаимодействие LA с LB позволяет с высоким выходом (>95%) получать промежуточный реакционный продукт, который представляет собой иммобилизованную активаторную предшествующую композицию, по существу все активаторные анионоактивные предшествующие материалы которой ковалентно связаны с металлоксидным носителем. В результате последующего взаимодействия с любым из описанных металлоценовых соединений получают аналогичное процентное количество иммобилизованной каталитической композиции на основе переходного металла. Для достижения такой чистоты может оказаться приемлемой, что очевидно для специалистов в данной области техники, достаточно тщательная промывка (промывки) после каждой реакции с целью удалить LA, LB или композиции переходного металла, которые не прореагировали с образованием предпочтительно связанной структуры.
С целью улучшить или упростить протекание любой реакции R можно выбрать таким образом, чтобы RH представлял собой газ, который можно легко отделить от реакционного растворителя. Таким образом, если R обозначает -Н или -CH3, при осуществлении обоих способов приготовления протонирование приводит к получению соответственно водорода или газообразного метана.
Этот катализатор по изобретению может быть использован для координационной полимеризации ненасыщенных мономеров, которые, как общеизвестно, способны полимеризоваться в условиях координационной полимеризации. Такие условия тоже хорошо известны; они включают полимеризацию в растворе, суспензионную полимеризацию и полимеризацию в газовой фазе под низким давлением. Таким образом, катализаторы на носителе по изобретению успешно могут быть использованы в известных режимах проведения процессов, в которых применяют неподвижный слой, подвижный слой, псевдоожиженный слой или суспензию, в одном и ряде последовательно или параллельно размещенных реакторов.
Линейные полиэтилены, включая высоко- и сверхвысокомолекулярные полиэтилены, включая также гомо- и сополимеры с другими альфа-олефиновыми мономерами, альфа-олефиновыми и/или несопряженными диолефинами, например, с С3-С20 олефинами/диолефинами, получают вводом этилена и необязательно других мономеров в реакционный сосуд под низким давлением (как правило, <50 бар) и при типичной температуре 20-250oС совместно с катализатором по изобретению, который предварительно суспендируют в растворителе, таком, как гептан или толуол. Теплоту полимеризации, как правило, отводят охлаждением. Полимер образуется в форме гранул, которые обычно нерастворимы в реакционном разбавителе. Полимеризацию обычно завершают посредством бета-гидридного элиминирования, результатом чего являются концевые олефиновые ненасыщенные группы, или добавлением регулятора степени полимеризации, такого, как водород или алюминийалкил, результатом чего является насыщенная концевая группа цепи. Полимер можно выделять фильтрованием или центрифугированием, промывать и сушить в случае суспензионных процессов. При необходимости сохранить ненасыщенные концевые группы цепи применение регулятора степени полимеризации, как указано выше, следует избегать. Полимеризацию в газовой фазе можно проводить, например, в газофазном реакторе непрерывного действия с псевдоожиженным слоем, работающим при 2000-3000 кПа и 60-160oС, используя водород в качестве реакционного модификатора (100-200 част./млн), поток C4-C8-сомономерного сырья (0,5-1,2 мол. %) и поток С2-сырья (25-35 мол.%) (см. патенты США 4543399, 4588790, 5028670 и совместно рассматриваемую заявку США 08/053067, поданную 26.04.93).
Как правило, полипропилен можно получать в основном тем же путем, как это описано выше для линейного полиэтилена. Реакционный разбавитель часто состоит из жидкого пропиленового мономера, в котором суспендирован ионогенный катализатор на носителе. Когда необходимо получить либо полиэтиленовые, либо полипропиленовые сополимеры, в реакционный разбавитель или растворитель можно вводить другие мономеры, как правило, низшие альфа-олефины (например, С2-С10) и/или несопряженные диолефины. Все реакции полимеризации для получения линейных полиэтиленовых, полипропиленовых и полиолефиновых полимеров можно проводить в любом пригодном реакторе, например, в установленных параллельно или последовательно реакторах периодического действия или в проточных реакторах непрерывного действия.
Введением газообразного этилена в суспензию с помощью α-олефина или его смеси с другими мономерами, способными или неспособными полимеризоваться, в качестве полимеризационного разбавителя, в котором суспендируют катализатор по изобретению, с использованием катализаторов по этому изобретению могут быть получены этилен-α-олефиновые (-диолефиновые) эластомеры высокой молекулярной массы и низкой кристалличности. Как правило, избыточное давление этилена составляет от 10 до 1000 фунтов/кв.дюйм (69-6895 кПа), а температура полимеризационного разбавителя обычно составляет от -10 до 100oС. Процесс можно проводить в реакторе с мешалкой или в нескольких реакторах, установленных последовательно или параллельно. Обеззоливание можно проводить в соответствии с практикой, известной в данной области техники, или, если его исключают, можно проводить процесс с использованием одного или нескольких колонных реакторов с неподвижным слоем или с насадкой (см. ниже). Общие условия процесса и принципы выбора предпочтительных соединений переходных металлов, которые, если у них имеются галоидные лиганды при переходном металле, предпочтительно следует алкилировать, как это описано выше, с целью обеспечить использование совместно с ионогенными каталитическими композициями по изобретению, можно найти в подробном описании к патенту США 5001205.
Для дополнительного регулирования морфологии полимерных частиц в типичных суспензионных и газофазных реакционных процессах в соответствии с обычной практикой можно также использовать предварительную полимеризацию катализатора на носителе по изобретению. Так, например, это можно осуществлять форполимеризацией С2-С6-альфа-олефина в течение ограниченного периода времени, например, введением этилена в контакт с катализатором на носителе при температуре от -15 до 30oС и под избыточным давлением этилена, достигающем приблизительно 250 фунтов/кв. дюйм (1724 кПа), в течение 75 мин с получением полимерного покрытия на носителе из полиэтилена молекулярной массы 30000-150000. После этого форполимеризованный катализатор готов для использования в процессах полимеризации, о которых речь шла выше. Аналогичным образом во время таких полимеризационных процессов может быть использован активированный катализатор на носителе, покрытом предварительно полимеризованным термопластичным полимером.
Кроме того, предпочтительно избегать влияния полимеризационных ядов, которые могут быть введены с потоками сырья, растворителями или разбавителями, удаляя или нейтрализуя эти яды. Так, например, потоки мономерного сырья или реакционный разбавитель может быть предварительно обработан или его можно обрабатывать в ходе реакции полимеризации приемлемым очищающим агентом. Как правило, им является металлоорганическое соединение, используемое в таких процессах, в которых применяют металлоорганические соединения с металлом группы 13, как описано в патенте США 5133157, в международных заявках 91/09882 и 94/03506, упомянутых выше, и в международной заявке 93/14132. Когда при проведении реакций полимеризации необходимо сохранить концевые ненасыщенные группы, предпочтительно количество очищающего агента должно быть минимальным или его следует избегать вовсе, поскольку он проявляет тенденцию действовать как регулятор степени полимеризации, результатом чего являются насыщенные концевые группы.
С использованием катализатора в соответствии с изобретением либо координационной, либо карбокатионной полимеризацией, помимо тех, которые уже описаны выше, могут быть полимеризованы другие олефиновоненасыщенные мономеры, например, стирол, алкилзамещенный стирол, этилиденнорборнен, норборнадиен, дициклопентадиен, циклопен и другие альфа-олефиновые ненасыщенные и циклические олефины деформированной геометрии, изобутилен, изопрен, бутадиен, простые виниловые эфиры, винилкарбазолы и т.д. Кроме того, благодаря способности катализаторов по изобретению на основе каталитически активных соединений переходных металлов вводить высшие альфа-олефиновые мономеры, сополимеризацией могут быть также введены альфа-олефиновые макромономеры, содержащие до 100 или более мономерных звеньев. Образующиеся полимеры могут представлять собой гомополимеры или сополимеры более одного мономера, и в зависимости от выбора металлоценового катионоактивного предшественника и мономера в соответствии с обычной практикой в данной области техники они могут характеризоваться любой из известных форм регулярности молекулярной структуры (см. , например, патенты США 5066741 и 5206197, в которых описано получение синдиотактических винилароматических полимеров с использованием единственных η5-циклопентадиенильных металлоценовых соединений, активированных некоординационными совместимыми анионами; патенты США 5278265 и 5304523, в которых описано получение изотактических и синдиотактических полипропиленов в условиях низкой температуры с использованием пространственно жестких металлоценов с некоординационными анионами, и патент США 5324801, в котором описано получение циклических олефинсодержащих сополимеров с использованием в качестве катализаторов особых металлоценовых соединений, каждое из которых может быть активировано в соответствии с настоящим изобретением.
Описанные особые структуры могут обеспечить достижение значительных промышленных положительных эффектов от использования изобретения. Благодаря тому, что гидроксильные группы равномерно закреплены на поверхности оксида металла, что обусловлено в свою очередь интервалами между смежными металлоксидными молекулами, катализатор обычно равномерно распределен на этой поверхности. Следовательно, полимеризация протекает относительно равномерно по всем поверхностям (вне и внутри пор), что позволяет избежать потенциальных затруднений, связанных с "горячими точками", где избыточная полимеризация ведет к местному перегреву. Это дает возможность избежать преждевременной каталитической фрагментации, которая является результатом "горячих точек", и свести к минимуму образование мелочи, с которой традиционно приходится сталкиваться в случае высокоактивных катализаторов и неравномерной адсорбции катализаторов на носителях. Кроме того, практически полностью устраняется десорбция вследствие потока текучей среды растворителя, разбавителя или мономера, благодаря чему полимеризация поддерживается на носителе, а не переносится на стенки реакторов, патрубков и т.д., из-за чего существует потенциальная возможность засорения и простоев на время очистки. Эта система фиксирования на носителе позволяет также упростить получение растворимых в углеводородах полимеров, практически не содержащих остаточного катализатора или золы, с использованием оборудования для неподвижного слоя оксида металла, так как мономер можно вводить, а полимер удалять в растворителе без попадания в него заметного количества катализатора вследствие удаления катализатора из такого неподвижного слоя.
Полученные с помощью каталитической системы по изобретению полимеры на альфа-олефиновой основе могут быть использованы в соответствии с их молекулярной массой, содержанием звеньев сомономеров, если их вводят, полидисперсностью ("ММР") и т.д. в обычных и известных для них областях применения. Так, например, типичными областями применения являются получение пленок, волокон и формуемых термопластов с помощью любого из известных средств формования из расплава, последующего экструдирования и/или формования листовых материалов. При этом, как известно, могут быть использованы добавки, такие, как вещества для улучшения технологических свойств, стабилизаторы, пигменты, наполнители. Конкретными примерами являются высокоплотные полиэтиленовые и изотактические полипропиленовые пленки, включая ориентированные в одном или обоих направлениях и модифицированные другими компонентами, такими, как повышающие клейкость углеводородные смолы.
Более того, можно вводить другие компоненты термопластических композиций как в больших, так и малых количествах, которые известны как обычно применяемые в различных полимерных смесях и композициях. Так, например, приемлемо использование эластомерных полиолефинов по изобретению для улучшения ударопрочных характеристик полярных конструкционных смол или в виде совулканизуемых эластомерных компаундов (обычно при наличии звеньев диолефинового сомономера и/или после дополнительной дериватизации, например, свободнорадикальной прививкой на полярные мономеры). Предпочтительный способ дериватизации описан в международной заявке 93/12148 и в ее аналоге - патенте США 5424367.
При получении с помощью катализаторов на носителе по изобретению низкомолекулярных альфа-олефиновых сополимеров, содержащих винилиденовые концевые ненасыщенные группы, можно успешно готовить композиции присадок к смазочным маслам. Так, например, в условиях суспензионной координационной полимеризации при типичной температуре 150-180oС и под давлением 10-20 бар можно получать низкомолекулярные этилен-пропиленовые или этилен-бутеновые сополимеры. Как правило, газообразный этилен вводят в поток жидкости, состоящей из или содержащей пропилен и/или 1-бутен (например, в пропан или бутан). Этот поток перед или после добавления этилена нагревают до температуры, близкой к точке кипения, и вводят в колонный реактор с неподвижным слоем или насадкой, содержащий катализатор на носителе. Катализатор представляет собой такой катализатор, который выбирают для наличия низкого соотношения между полимеризацией и обменной гидридизацией реакционной способности переходного металла. Так, например, катализатор на носителе можно приготовить в соответствии с приведенным ниже примером 5, но с использованием при этом бис(4, 5, 6, 7-тетрагидроинденил)цирконийдиметила с 1,1-диметилсилильным мостиком в качестве соединения переходного металла. Далее отходящий поток, содержащий этилен-бутеновый или этилен-пропиленовый сополимер, имеющий низкую молекулярную массу [среднечисленная молекулярная масса (Мn) менее 10000] и содержащий винилиденовые концевые ненасыщенные группы, можно отделять от растворителя отгонкой легких фракций с продувкой азотом.
После этого полученный сополимер можно дериватизировать обработкой в расплаве с использованием, например, измельченного малеинового ангидрида в баке с мешалкой, нагретом до температуры 220oС. Процесс можно завершить охлаждением до 60oС. Полученный жидкий продукт отделяют от непрореагировавшего малеинового ангидрида отгонкой легких фракций. Имидизацию как последующую реакцию реакционного продукта можно проводить в растворе вышеупомянутого реакционного продукта в минеральном масле совместно с полиамином, таким, как тетраэтиленпентамин. Полученный азотсодержащий низкомолекулярный маслорастворимый реакционный продукт пригоден для использования в композициях смазочных масел в качестве диспергатора. Касательно дополнительной информации о низкомолекулярных aльфа-олефиновых полимерах и соответствующих катализаторах, см. , например, совместно рассматриваемые заявки на патенты США 07/992192 и 07/992690, поданные 17.12.92, и ссылки, указанные в этих заявках.
Аналогичным путем, но с использованием катализатора, пригодного для получения более высокомолекулярных (10000 < Мn < 300000) альфа-олефин-диолефиновых сополимеров с кристалличностью, которая достаточна низка для обеспечения растворения в масле (например, с кристалличностью <40%), можно получать многофункциональные модифицирующие вязкость присадки к смазочным маслам (см. описания модификаторов смазочных масел и композиций смазочных масел в патентах США 4749505 и 4772406 и международной заявке 93/12148). Пригодные катализаторы с переходными металлами проиллюстрированы на примере одного или нескольких продуктов группы, включающей бис(циклопентадиенил)гафнийдиметил, бис(тетрагидроинденил)гафнийдиметил, этиленбис(тетрагидроинденил)гафнийдиметил и диметилсиланилен-бис(тетрагидроинденил)гафнийдиметил, бис(тетрагидроинденил) цирконий-диметил, этиленбис(тетрагидроинденил)цирконийдиметил и диметилсиланилен-бис(тетрагидроинденил)цирконийдиметил, нанесенных на носитель в соответствии с приведенным ниже примером 1 (см. вышеупомянутые патенты США 5001205 и 5198401).
Ионогенные каталитические композиции на носителях по изобретению могут быть использованы согласно описанному выше индивидуально для координационной или карбокатионной полимеризации или же могут быть смешаны для получения полимерных смесей. Выбор мономеров позволяет в полимеризационных условиях, аналогичных условиям, в которых используют индивидуальные каталитические композиции, получать смеси координационных полимеров и смеси карбокатионных полимеров или совместно те и другие. Таким образом, при получении полимеров с помощью смешанных каталитических систем можно получить полимеры, характеризующиеся повышенным ММР для улучшения технологичности и достижения других традиционных положительных эффектов.
Для улучшения каталитических свойств других известных дискретных каталитических катионов перед добавлением соединений переходных металлов можно дополнительно использовать нанесенный на диоксид кремния анионоактивный активатор, образующий промежуточные продукты по изобретению. В качестве примеров можно назвать катализаторы гидрогенизации на основе металлов группы 9, таких, как родий, например, [Rh(диен)(РРh3)2]+ и [Rh(дифос)]+; катализаторы димеризации олефинов, такие, как катализаторы на основе никеля, например, [Ni(L)4H] +; катализаторы димеризации метакрилата, такие, как основанные на родиевых металлоценах, например, [CpRh(L)(алкил)]+, и катализаторы с переходными металлами для полимеризации олефинов, такие, как кобальтовые металлоцены, например, [CpCo(L)(алкил)]+. Среди вышеприведенных стандартных химических символов использованы некоторые сокращенные обозначения заместителей, где Ph обозначает фенил, L обозначает ковалентный лиганд и Ср обозначает циклопентадиен. Предпочтительный способ применения заключается во взаимодействии кислоты Льюиса (LA) с неподвижным слоем или столбом ионообменного вещества, содержащим неподвижный диоксид кремния, с последующим введением соли отдельного каталитического катиона в раствор для ионообменного взаимодействия с иммобилизованным реакционным продуктом. После этого неподвижный слой или столб ионооменного вещества готов для ввода мономерных реагентов в реакционную среду, пригодную для проведения реакции [см. , например, "Comparison of Migratory Aptitudes of Hydrides and Alkyl Groups in бета-Migration Insertion Reactions of Cp* [P(OMe)3 Rh(C2H4)R]+ (R = -H, -CH2CH3)", M. Brookhart и D.M. Lincoln. J. Am. Chem. Soc.. 110, 8719-8720 (1988)].
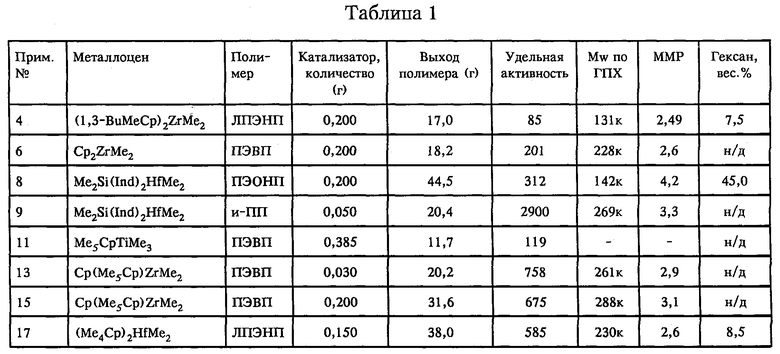
Вышеприведенное описание проиллюстрировано на следующих примерах. Результаты экспериментов в примерах 4, 6, 8, 13, 15 и 16 (полимеризация) являются усредненными для трех самостоятельных, но эквивалентных экспериментов. Во всех случаях, если не указано иное, количества выражены в весовых частях, долях и процентах. Хотя такие примеры могут относиться к некоторым конкретным вариантам выполнения изобретения, их не следует рассматривать как ограничивающие в каком-либо конкретном отношении объем изобретения. С целью упростить описание в этих примерах использованы некоторые аббревиатуры, где Me обозначает метил, Et обозначает этил, Вu обозначает бутил, Ph обозначает фенил, Ср обозначает циклопентадиенил, Ср* обозначает пентаметилциклопентадиенил, Ind обозначает инденил, Ti обозначает титан, Hf обозначает гафний, Zr обозначает цирконий и Si обозначает кремний. "Davison 948, кальцинированный при 800oС" представляет собой кремнийдиоксидный носитель и является коммерческим продуктом фирмы Grace Davison. Inc., который предварительно кальцинировали при 800oС в токе сухого N2 в течение 8-24 ч с целью обеспечить таким образом содержание гидроксильных групп 0,2-0,5 ммоля/г диоксида кремния.
Пример 1: Получение катализатора А
2,002 г диоксида кремния (Davison 948, кальцинированный при 800oС) при 25oС в токе азота суспендировали в 150 мл сухого толуола в 500-миллилитровой круглодонной колбе при одновременном перемешивании магнитной мешалкой с последующим добавлением 0,513 г трисперфторфенилбора (LA), который добавляли мелкими порциями в виде твердого вещества. Реакционную смесь перемешивали в течение 30 мин, а затем нанесенный на носитель активатор выделяли вакуумным фильтрованием и промывали тремя порциями по 50 мл сухого толуола с последующей кратковременной сушкой в вакууме. Далее нанесенный на носитель активатор повторно суспендировали в 150 мл толуола и добавляли 0,323 г циклопентадиенил (пентаметилциклопентадиенил) цирконийдиметила в виде твердого материала, получая при этом бесцветную взвесь. Перемешивание продолжали в течение 30 мин, а затем нанесенный на носитель катализатор выделяли фильтрованием и промывали тремя порциями по 50 мл толуола. После этого катализатор сушили в течение ночи в вакууме и получали 1,89 г готового катализатора, содержавшего 0,53 ммоля переходного металла на грамм готового катализатора.
Пример 2: Полимеризация этилена в суспензионной фазе
Полимеризацию проводили в суспензионной фазе в 1-литровом автоклаве, снабженном механической мешалкой, внешней водяной рубашкой для регулирования температуры, мембранным входным устройством и устройством для регулируемой подачи сухого азота и этилена. Реактор тщательно сушили и дегазировали при 115oС. В качестве разбавителя добавляли 400 мл гексана и, используя газонепроницаемый шприц, в качестве очищающего от примесей агента добавляли 0,2 мл раствора триэтилалюминия в гептане концентрацией 25 вес.%. Под избыточным давлением 75 фунтов/кв.дюйм (5,17 бар) в реактор при 60oС вводили этилен. В 10-миллилитровый сосуд высокого давления из нержавеющей стали загружали 0,2 г катализатора А и сосуд присоединяли к реактору с помощью хомута. Далее в реактор вводили катализатор. Полимеризацию продолжали в течение 30 мин, поддерживая в реакционном сосуде температуру 60oС и избыточное давление 75 фунтов/кв. дюйм (5,17 бар) постоянным током этилена. Реакцию прекращали резким охлаждением и вентиляцией. После этого выделяли полимер. Удельную полимеризационную активность рассчитывали аналогично тому, как это описано в примере 4.
Пример 3: Получение катализатора Б
50,0 г диоксида кремния (Davison 948, кальцинированный при 800oС) при 25oС в токе азота суспендировали в 350 мл сухого толуола в 1-литровой круглодонной колбе при одновременном перемешивании магнитной мешалкой с последующим добавлением с помощью шприца 0,90 мл диэтиланилина (LB). Перемешивание продолжали в течение 5 мин, причем за этот период времени мелкими порциями в виде твердого вещества добавляли трисперфторфенилбор (LA). Реакционную смесь перемешивали в течение 30 мин, а затем нанесенный на носитель активатор выделяли вакуумным фильтрованием и промыванием тремя порциями по 100 мл сухого толуола. Далее нанесенный на носитель активатор повторно суспендировали в 350 мл толуола и добавляли 2.38 г циклопентадиенил (1,3 -бутилметилциклопентадиенил) цирконийдиметила в виде толуольного раствора (приблизительно в 10 мл толуола). В течение 30 с добавления наблюдалось выделение газа, а реакционная смесь становилась светло-желтой. Перемешивание продолжали в течение 45 мин, а затем нанесенный на носитель катализатор выделяли фильтрованием и промывали тремя порциями по 100 мл толуола. После этого катализатор сушили в течение ночи в вакууме и получали 54,3 г готового катализатора, содержавшего 0.11 ммоля переходного металла на грамм готового катализатора.
Пример 4: Этилен-гексеновая полимеризация в суспензионной фазе
Полимеризацию проводили в суспензионной фазе в 1-литровом автоклаве, снабженном механической мешалкой, внешней водяной рубашкой для регулирования температуры, мембранным входным устройством и устройством для регулируемой подачи сухого азота и этилена. Реактор тщательно сушили и дегазировали при 115oС. В качестве разбавителя добавляли 400 мл гексана и, используя газонепроницаемый шприц, в качестве очищающего от примесей агента добавляли 0.2 мл раствора триэтилалюминия в гептане концентрацией 25 вес.% и с помощью полой иглы добавляли 50 мл гексена. Под избыточным давлением 75 фунтов/кв.дюйм (5,17 бара) в реактор при 60oС вводили этилен. В 10-миллилитровый сосуд высокого давления из нержавеющей стали загружали 0,2 г катализатора Б и этот сосуд присоединяли к автоклаву с помощью хомута. Далее в автоклав вводили катализатор. Полимеризацию продолжали в течение 30 мин, поддерживая в реакционном сосуде температуру 60oС и избыточное давление 75 фунтов/кв.дюйм (5,17 бара) постоянным током этилена. Реакцию прекращали резким охлаждением и вентиляцией. Выделяли 17,0 г этилен-гексенового сополимера. Средневесовая молекулярная масса такого полиэтилена составляла 131500, среднечисленная молекулярная масса составляла 53000, величина молекулярно-массового распределения была равной 2,5, а содержание гексеновых звеньев составляло 7,5 вес. %. Удельную полимеризационную активность рассчитывали как отношение выхода полимера к общему весу переходного металла, содержащегося в катализаторе, времени, выраженному в часах, и абсолютному давлению мономера, выраженному в атмосферах. Для примера 4 удельную активность рассчитывали следующим образом:
На этом примере проиллюстрировано введение 1-гексеновых звеньев при получении линейного полиэтилена низкой плотности (ЛПЭНП).
Пример 5: Получение катализатора В
0,640 г трисперфторфенилбора растворяли в 8,0 мл толуола и при перемешивании добавляли 8,0 мл толуола и 0,20 мл диэтиланилина, получая розовый раствор. Этот раствор добавляли по каплям при 25oС в токе азота в суспензию 1,00 г диоксида кремния (Davison 948, кальцинированный при 180oС в вакууме в течение 16 ч) в 30 мл сухого толуола в 100-миллилитровой круглодонной колбе с одновременным перемешиванием магнитной мешалкой до тех пор, пока явно сохранялась розовая окраска (6,0 мл). Раствор фильтровали с получением розового фильтрата и белого твердого материала (активатора на носителе). Далее нанесенный на носитель активатор повторно суспендировали в 10 мл толуола и добавляли 0,236 г диметилцирконоцена в виде твердого материала. При этом наблюдалось выделение газа, а реакционная смесь становилась желтой. После перемешивания в течение 15 мин нанесенный на носитель катализатор выделяли фильтрованием и промывали тремя порциями по 5 мл толуола. После этого катализатор сушили в течение 1 ч в вакууме и получали 1,25 г готового катализатора, содержавшего 0,76 ммоля переходного металла на грамм готового катализатора.
Этот пример подтверждает, что для приготовления активных полимеризационных катализаторов могут быть использованы диоксиды кремния с высокой концентрацией гидроксильных групп, а также иллюстрирует другой способ приготовления, в котором для оптимизации содержания гидроксильных групп их "титровали" окрашенным в розовый цвет комплексом кислоты-основания Льюиса PhEt2N:B(C6F5)3.
Пример 6: Полимеризация этилена в суспензионной фазе с использованием катализатора В
Процессы полимеризации проводили аналогично тому, как это описано в примере 2. Данные этой полимеризации представлены в таблице 1. На этом примере проиллюстрировано получение полиэтилена высокой плотности (ПЭВП) с использованием катализатора по изобретению.
Пример 7: Получение катализатора Г
Этот катализатор получали в соответствии с методикой из примера 3, за исключением того, что (1,3-BuMeCp)2ZrMe2 заменяли на Me2Si(Ind)2HfMe2 и каждый реагент использовали в нижеследующих количествах в сочетании с соответствующими количествами растворителя: 5,00 г диоксида кремния (Davison 948, кальцинированный при 800oС); 0,40 мл диэтиланилина; 1,28 г трисперфторфенилбора и 1,09 г Me2Si(Ind)2HfMe2, получая при этом содержание переходного металла в 0,28 ммоля на грамм готового катализатора. В этом примере проиллюстрировано протонирование менее реакционноспособной (в сравнении с Zr-) Hf-Me-связи с получением активного катализатора по изобретению.
Пример 8: Этилен-гексеновая полимеризация в суспензионной фазе с использованием катализатора Г
Процессы полимеризации проводили аналогично тому, как это описано в примере 4, за исключением того, что использовали 45 мл гексена. Данные полимеризаций представлены в таблице 1. В этом примере проиллюстрировано использование связанного мостиком металлоцена при получении сополимера очень низкой плотности (ПЭОНП) с высоким содержанием введенных гексеновых звеньев.
Пример 9: Полимеризация пропилена в массе с использованием катализатора Г
Полимеризацию проводили в суспензионной фазе в 1-литровом автоклаве, снабженном механической мешалкой, внешней водяной рубашкой для регулирования температуры, мембранным входным устройством и устройством для регулируемой подачи сухого азота и пропилена. Реактор тщательно сушили и дегазировали при 115oС. С помощью газонепроницаемого шприца вводили 400 мл пропилена совместно с 0,2 мл раствора триэтилалюминия в гептане концентрацией 25 вес.% в качестве очищающего от примесей агента. Далее в реактор в виде суспензии в толуоле (10 мг/мл) вводили катализатор. При этом вначале вводили 3 мл, а по истечении 15 мин еще 2 мл. Полимеризацию в реакционном сосуде проводили в течение 45 мин, поддерживая температуру 60oС. Реакцию прекращали резким охлаждением и вентиляцией. Полимеризационные данные представлены в таблице 1. Удельную полимеризационную активность рассчитывали как отношение выхода полимера к выраженному в миллимолях общему количеству переходного металла, содержащегося в катализаторе, и времени, выраженному в часах. В этом примере проиллюстрировано использование хирального связанного мостиком металлоцена при получении изотактического полипропилена (и-ПП).
Пример 10: Получение катализатора Д
Данный катализатор получали в соответствии с методикой из примера 3, за исключением того, что (1,3-BuMeCp)2ZrMe2 заменяли на Me5CpTiMe3 и каждый реагент использовали в нижеследующих количествах в сочетании с соответствующими количествами растворителя: 1,00 г диоксида кремния (Davison 948, кальцинированный при 800oС); 0,018 мл диэтиланилина; 0,58 г трисперфторфенилбора и 0,026 г Me5CpTiMe3, добиваясь при этом содержания переходного металла 0,10 ммоля на грамм готового катализатора. В этом примере проиллюстрировано получение моноциклопентадиенилтитанового соединения.
Пример 11: Полимеризация этилена в суспензионной фазе с использованием катализатора Д
Процессы полимеризации проводили аналогично тому, как это описано в примере 2. Полимеризационные данные представлены в таблице 1.
Пример 12: Получение катализатора Е
Данный катализатор получали в соответствии с методикой из примера 3, за исключением того, что (1,3-BuMeCp)2ZrMe2 заменяли на CpCp*ZrMe2 и каждый реагент использовали в нижеследующих количествах в сочетании с соответствующими количествами растворителя: 1,00 г диоксида кремния (Davison 948, кальцинированный при 800oС); 0,080 мл диэтиланилина; 0,256 г трисперфторфенилбора и 0,161 г CpCp*ZrMe2, получая при этом содержание переходного металла в 0,34 ммоля на грамм готового катализатора.
Пример 13: Полимеризация этилена в суспензионной фазе с использованием катализатора Е
Процессы полимеризации проводили аналогично тому, как это описано в примере 2. Полимеризационные данные представлены в таблице 1.
Пример 14: Получение катализатора Ж
Данный катализатор получали в соответствии с методикой из примера 12, за исключением того, что каждый реагент использовали в нижеследующих количествах в сочетании с соответствующими количествами растворителя: 1,00 г диоксида кремния (Davison 948, кальцинированный при 800oС); 0,016 мл диэтиланилина; 0,051 г трисперфторфенилбора и 0,032 г СрСр*ZrMe2, получая при этом содержание переходного металла в 0,09 ммоля на грамм готового катализатора.
Пример 15: Полимеризация этилена в суспензионной фазе с использованием катализатора Ж
Процессы полимеризации проводили аналогично тому, как это описано в примере 2. Полимеризационные данные представлены в таблице 1. Примеры 12-15 свидетельствуют о том, что максимальное содержание нанесенного на носитель активатора (и, следовательно, ионогенного катализатора) определяется скорее не количествами LA и LB, а содержанием гидроксильных групп на металлоксидном носителе. Как и ожидалось согласно изобретению, добавление избытка (относительно общей концентрации гидроксильных групп) кислоты Льюиса, вторичного акцептора протонов и соединения переходного металла в катализатор Е (из примера 12) не приводило к заметному повышению полимеризационной активности катализатора Е (из примера 13) в сравнении с катализатором Ж (из примера 15), который характеризовался стехиометрическим содержанием LA и LB относительно расчетных концентраций гидроксильных групп для диоксида кремния, использованного для приготовления катализаторов как Ж, так и Е.
Пример 16: Получение катализатора З
Данный катализатор получали в соответствии с методикой из примера 3, за исключением того, что (1,3-BuMeCp)2ZrMe2 заменяли на (Me4Cp)2HfMe2 и каждый реагент использовали в нижеследующих количествах в сочетании с соответствующими количествами растворителя: 5,00 г диоксида кремния (Davison 948, кальцинированный при 800oС); 0,393 г трибутиламина (LB); 1,075 г трисперфторфенилбора и 0,542 г (Me4Cp)2HfMe2, получая при этом содержание переходного металла в 0,2 ммоля на грамм готового катализатора.
Пример 17: Этилен-гексеновая полимеризация в суспензионной фазе с использованием катализатора З
Процессы полимеризации проводили аналогично тому, как это описано в примере 4, за исключением того. что использовали 45 мл гексена.
Полимеризационные данные представлены в таблице 1, и они свидетельствуют о том, что использование алкиламинов приводит к получению катализаторов, которые по меньшей мере так же активны, как и катализаторы, которые приготовлены с использованием замещенных анилинов, подтверждая тем самым, что при применении менее кислой (в сравнении с диэтиланилином) трибутиламмониевой соли химически связанного противостоящего аниона все еще происходит протонирование с получением активного катализатора по изобретению.
Пример 18: Получение катализатора И
25,01 г диоксида кремния (Davison 948, кальцинированный при 800oС) при 25oС в токе азота суспендировали в 400 мл сухого толуола в 1000-миллилитровой круглодонной колбе с одновременным перемешиванием магнитной мешалкой с последующим добавлением 1,56 г диэтиланилина и 5,38 г трисперфторфенилбора. Реакционную смесь перемешивали в течение 30 мин, а затем нанесенный на носитель активатор выделяли вакуумным фильтрованием, промывали тремя порциями по 100 мл сухого толуола и кратковременно сушили в вакууме. Далее нанесенный на носитель активатор повторно суспендировали в 400 мл толуола и добавляли 2,71 г бис(тетраметилциклопентадиенил)гафнийдиметила в виде твердого материала, получая при этом желто-оранжевую суспензию, окраска которой в течение одного часа темнела до оранжево-красной. После этого перемешивание прекращали и нанесенный на носитель катализатор выделяли фильтрованием и промывали четырьмя порциями по 100 мл толуола, а затем одной порцией пентана в 100 мл. Далее катализатор сушили в течение 12 ч в вакууме и получали 31,16 г готового катализатора.
Пример 19: Получение катализатора К
25,009 г диоксида кремния (Davison 948, кальцинированный при 800oС) при 25oС в токе азота суспендировали в 400 мл сухого толуола в 1000-миллилитровой круглодонной колбе при одновременном перемешивании магнитной мешалкой с последующим добавлением 1,58 г диэтиланилина и 5,38 г трисперфторфенилбора. Реакционную смесь перемешивали в течение 30 мин, а затем нанесенный на носитель активатор выделяли вакуумным фильтрованием, промывали тремя порциями по 100 мл сухого толуола и кратковременно сушили в вакууме. Далее нанесенный на носитель активатор повторно суспендировали в 400 мл толуола и добавляли 3,59 г бис(тетраметилциклопентадиенил)гаф-нийдиметила в виде твердого материала, получая желто-оранжевую суспензию, окраска которой в течение 70 мин темнела до оранжево-красной. После этого перемешивание прекращали и нанесенный на носитель катализатор выделяли фильтрованием и промывали четырьмя порциями по 100 мл толуола, а затем одной порцией пентана в 100 мл. Далее катализатор сушили в течение 2,5 ч в вакууме и суспендировали в 350 мл пентана. Затем в эту суспензию добавляли 4,55 г полипараметилстирола (Mw и Мn которого составляли 2000) ("ПМС") в 250 мл пентана, после чего пентан удаляли в вакууме с помощью роторного испарителя (в противоположность перемешиванию), в результате чего получали 35,02 г готового катализатора, причем этот готовый катализатор был покрыт полипараметилстиролом.
Пример 20: Получение катализатора Л
Данный катализатор получали аналогично получению катализатора К, за исключением того, что использовали нижеследующие количества материалов: 25,03 г диоксида кремния; 1,58 г диэтиланилина, 5,37 г трисперфторфенилбора; 2,71 г бис(тетраметилциклопентадиенил)гафнийдиметила; 4,5 г поли-пара-метилстирола. При этом получали 35,0 г готового катализатора с покрытием.
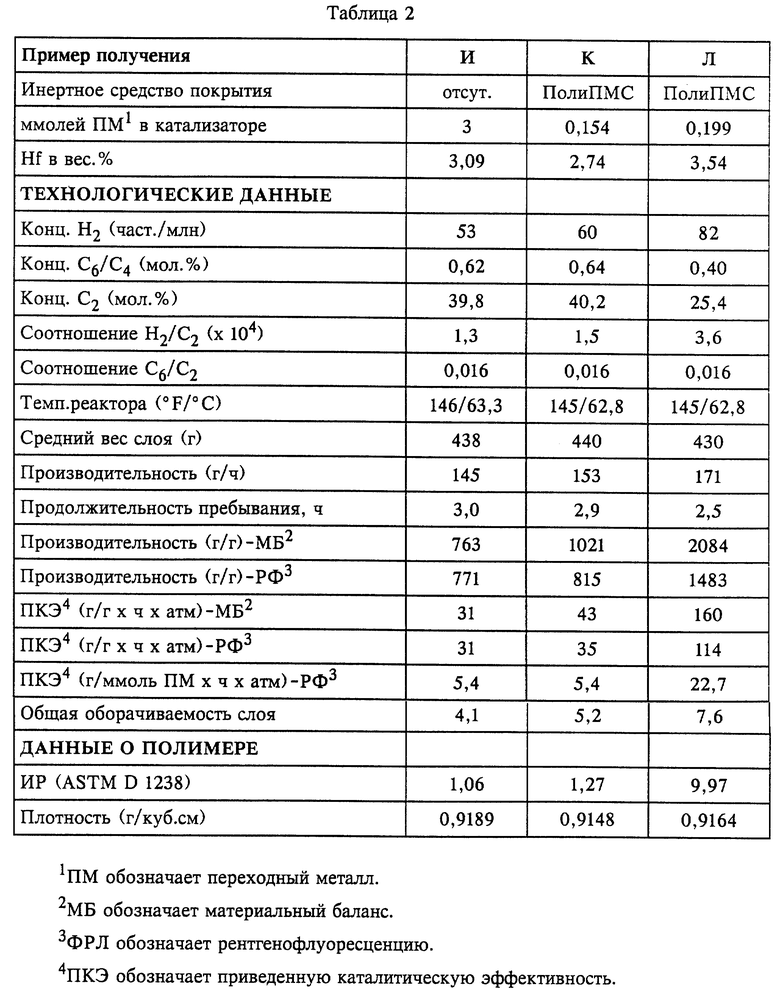
Примеры полимеризации в газовой фазе
В соответствии с приведенным ниже описанием для изучения сополимеризации этилена и 1-гексена использовали образцы каждого из вышеописанных катализаторов И, К и Л, нанесенных на носители. Для определения каталитической эффективности, способности вводить звенья и производительности в отношении молекулярной массы по звеньям сомономера, такого как 1-гексен. использовали газофазный реактор непрерывного действия с псевдоожиженным слоем, работавший под общим избыточным давлением 300 фунтов/кв.дюйм (20,7 бара), при температуре в реакторе 145o F (62,8oС) и скорости циркулировавшего газа 0,7 фут/с (21 см/с). В качестве очищающего от примесей агента в реактор вводили раствор триэтилалюминия в изопентане (концентрацией 1 вес.%) с расходом 1 см3/ч. Суммарные данные процесса представлены в таблице 2. После по меньшей мере трех проходов через слой собирали и анализировали образцы полимера.
Пример 21: Альтернативный способ получения
В том виде, в каком они представлены, приведенные выше уравнения химических реакций (2), (3), (3а) и (4) по существу являются количественными (т. е. реагенты превращаются в указанные реакционные продукты), как это определяли 11В-ЯМР-спектроскопией в твердом состоянии Magic Angle Spinning (см. выше). Для сравнения осуществляли нижеследующий альтернативный способ получения.
Сначала 0,153 г трисперфторфенилбора, 0,175 гдиэтиланилина и 0,322 г СрСр•ZrMe2 совместно растворяли в толуоле и затем при 25oС в токе азота добавляли в суспензию 3,01 г диоксида кремния (Davison 948, кальцинированный при 800oС) в 200 мл сухого толуола в 500-миллилитровой круглодонной колбе с одновременным перемешиванием магнитной мешалкой. Реакционную смесь перемешивали в течение 30 мин, а затем нанесенный на носитель активатор выделяли вакуумным фильтрованием и промывали тремя порциями по 50 мл сухого толуола. После этого катализатор сушили в течение ночи в вакууме и получали 3,47 г готового катализатора, содержащего 0,29 ммоля переходного металла на грамм готового катализатора. Этот продукт анализировали ЯМР-спектроскопией в соответствии с описанным выше, в результате чего определили, что он включал смесь борсодержащих материалов.
Процессы полимеризации проводили аналогично тому, как это описано в примере 4. Удельная активность согласно оценке составляла 130 г ПЭ/ммоль Zr-ч-атм. Средневесовая молекулярная масса этого полимера составляла 69600, среднечисленная молекулярная масса составляла 7300, величина молекулярно-массового распределения (ММР) была равной 9,5; полимер содержал 8,0 вес.% гексеновых звеньев.
Низкая Mw и широкое ММР позволили предположить, что отмеченная полимеризационная активность обусловлена не только единственным каталитически активным материалом, соответствующим изобретению. Вероятно, такая полимеризационная активность объясняется отчасти влиянием других каталитически активных материалов, что является ожидаемым следствием проведения такого процесса получения. Образование активного ионогенного катализатора fCpCp•ZrMe]+ [Me(pfp)3B] - вследствие взаимодействия СрСр•ZrMe2 с трисперфторфенилбором хорошо известно. Более того, полагают, что этот материал обычно нелегко удалить из диоксида кремния даже путем промывки большими объемами ароматического растворителя из-за взаимодействий с полярными функциональными группами носителя. Образование химически связанного материала с переходным металлом вследствие взаимодействия СрСр•ZrMe2 с гидроксильными группами на диоксиде кремния с выделением метана также известно.
Пример 22: Сравнительный пример и ЯМР-спектроскопия
Сравнительный катализатор готовили в соответствии с методикой. описанной в примере 14 международной заявки 91/09882, с использованием 5,0 г обработанного тиэтилалюминием диоксида кремния, 0,160 г N,N-диметилани-лийтетракис(пентафторфенил)бора и 0,080 г Cp2HfMe2, а также растворителя, количества которого увеличивали в той же процентной степени. Катализатор по изобретению готовили по описанной в вышеприведенном примере 1 методике, за исключением того, что использовали 5,00 г диоксида кремния, 0,179 г диэтиланилина, 0,614 г трисперфторфенилбора и 0,406 г Cp2HfMe2, а также растворителя, количества которого увеличивали в той же процентной степени. Высокоскоростную 11В-ЯМР-спектрометрию Magic Angle Spinning проводили в спектрометре Bruker MSL-400, настроенном на частоту 128,39 МГц. Образцы в токе азота вводили в воздухонепроницаемые цилиндрические роторы из диоксида циркония с внешним диаметром 5 или 7 мм, которые затем приводили во вращение со скоростями 5-8 кГц. Спектр получали с использованием одноимпульсного возбуждения. Все 11В-химические сдвиги определяли в сопоставлении с наивысшим экранирующим резонансом буры Na2B4O7•10H2O, для которой химический сдвиг составляет 2,0 част./млн относительно Et2O•BF3; эти данные приведены в данном случае относительно Et2O•BF3. Для увеличения соотношения S/N во всех спектрах применяли коэффициент уширения спектральных линий 25 Гц.
В нижеприведенной формуле изобретения, которая представляет изобретение в конкретных вариантах выполнения, каждый зависимый вариант для каждого из приведенных ниже независимых вариантов может быть практически осуществлен с одним или несколькими ограничениями других зависимых вариантов, что позволяет, таким образом, в рамках заявленного изобретения продемонстрировать другие рабочие варианты.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИТИЧЕСКИЕ СИСТЕМЫ ПОЛИМЕРИЗАЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 1995 |
|
RU2167883C2 |
| СПОСОБ НЕПРЕРЫВНОЙ ГАЗОФАЗНОЙ ПОЛИМЕРИЗАЦИИ АЛЬФА-ОЛЕФИНА(ОВ) | 1994 |
|
RU2125063C1 |
| СПОСОБ ПОЛИМЕРИЗАЦИИ | 1995 |
|
RU2165434C2 |
| ВОЛОКНА И ТЕКСТИЛЬНЫЕ МАТЕРИАЛЫ ИЗ ПОЛИЭТИЛЕНА ВЫСОКОЙ ПЛОТНОСТИ И СПОСОБ ИХ ИЗГОТОВЛЕНИЯ | 1995 |
|
RU2164969C2 |
| СВЯЗАННЫЕ МОСТИКАМИ МЕТАЛЛОЦЕНЫ, СПОСОБ ПОЛИМЕРИЗАЦИИ | 1999 |
|
RU2232766C2 |
| МЕТАЛЛООРГАНИЧЕСКОЕ СОЕДИНЕНИЕ | 1988 |
|
RU2139291C1 |
| СПОСОБ ПОЛИМЕРИЗАЦИИ МОНОМЕРОВ В ПСЕВДООЖИЖЕННОМ СЛОЕ | 1995 |
|
RU2140425C1 |
| КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ, ЕЕ ПОЛУЧЕНИЕ И СПОСОБ ПОЛИМЕРИЗАЦИИ ПРОПИЛЕНА | 1995 |
|
RU2153507C2 |
| МЕТАЛЛОЦЕНОВЫЕ КАТАЛИТИЧЕСКИЕ СИСТЕМЫ НА ПОДЛОЖКЕ ДЛЯ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ГОМО- ИЛИ СОПОЛИМЕРЫ ПРОПИЛЕНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2126017C1 |
| СПОСОБ СОПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ С МОСТИКОВЫМИ ГАФНОЦЕНАМИ | 1999 |
|
RU2228937C2 |
Изобретение относится к каталитическим композициям и может быть использовано в реакциях присоединения олефиновоненасыщенных мономеров, например, при полимеризации. Каталитическая композиция включает металлоксидный носитель, анион, полученный из кислоты Льюиса, связанный с каталитически активным соединением металла. Металлоксидный носитель имеет противостоящий анион, который получен из кислоты Льюиса, не имеющей легко гидролизуемых лигандов, и ковалентно связан с поверхностью носителя непосредственно через кислородный атом металла, где анион также ионно связан с каталитически активным соединением переходного металла. Способ полимеризации включает контактирование одного или нескольких мономеров с каталитической композицией. Используют мономеры, способные полимеризоваться при координационной полимеризации. Каталитическая активаторная композиция включает металлоксидный носитель и по меньшей мере один активаторный анионоактивный предшествующий материал, ковалентно связанный через атом кислорода с носителем. Изобретение позволяет получать качественный продукт полимеризации. 3 с. и 11 з.п. ф-лы, 2 табл.
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| ЕР 0529094 А1, 03.03.1993. | |||

