Область техники
Настоящее изобретение относится к сочетанию ингибитора повторного усвоения (поглощения) 5-НТ и селективного 5-HT1A антагониста, более конкретно, специфических (R)-5-карбамоил-8-фтор-3-(N, N-дизамещенный-амино)-3,4-дигидро-2Н-1-бензопи-ранов в форме свободного основания или фармацевтически приемлемых солей, к способу получения сочетания, фармацевтической композиции, содержащей указанное сочетание и к применению указанного сочетания путем либо совместного, либо индивидуального введения для улучшения лечения аффективных (эмоциональных) расстройств или заболеваний, таких как депрессия, страх, навязчивое компульсивное заболевание (НКЗ) и т.д.
Предпосылки изобретения
На сегодняшний день обычно считается, что для антидепрессантов, включая селективные ингибиторы повторного усвоения 5-НТ (SSRI), требуется 2-4 недели для достижения полного клинического эффекта. С другой стороны, побочные эффекты проявляются немедленно и уменьшаются в течение этого периода. Таким образом, медленное начало воздействия антидепрессантов приводит к появлению периода уязвимости для пациентов, при котором они подвергаются воздействию побочных эффектов при отсутствии лечебного действия лекарственных средств. Лечащему врачу часто трудно убедить пациента продолжить лечение в течение этого периода. Кроме того, так как начало действия лекарств наступает постепенно, пациенты, склонные к самоубийству, остаются инициативными без проявления обратных симптомов, что сохраняет риск суицида и приводит к необходимости частой госпитализации. Антидепрессанты с быстрым проявлением эффекта были бы не только предпочтительны из-за быстрой реализации действия, но также были бы более приемлемы для пациентов и врачей и снижали бы необходимость и длительность госпитализации. Такой же длительный период достижения полного клинического эффекта наблюдался и при лечении других аффективных заболеваний, таких как, например, тревожное состояние и НКЗ.
Описание прототипов
В работе Arch. Gen. Phsychiatry, vol.51. Mar. 1994, описано, что бета-блокирующее средство, такое как пиндолол, также обладает высоким сродством к 5-НТ рецепторам и антагонизирует 5-НТ1А-опосредуемые реакции, вызывает быстрое улучшение состояния пациентов, находящихся в депрессивном состоянии, которых лечат с помощью ингибиторов повторного усвоения серотонина.
Сущность изобретения
Антагонизм уменьшения, вызываемого ингибитором острого повторного усвоения 5-НТ, в обороте или круговороте 5-НТ, с помощью селективных 5-HT1A антагонистов.
Селективные ингибиторы повторного усвоения 5-НТ (SSRI) уменьшают распространение импульса в 5-НТ нервных клетках через негативную реакцию обратного питания, вероятно опосредуемую второстепенными 5-НТ аксонами, высвобождающими 5-НТ в ядра соединительных клеток. С помощью ингибирования соматодендритных 5-HT1A ауторецепторов селективные антагонисты противодействуют снижению оборота 5-НТ, индуцируемому ингибиторами повторного усвоения 5-НТ. Это указывает на то, что селективная блокада соматодендритного ауторецептора, то есть антагониста 5-HT1A, может иметь клинический потенциал для повышения эффективности ингибиторов повторного усвоения 5-НТ и обеспечивать новую возможность для быстрого проявления аффективных действий, например, антидепрессантных действий.
Сочетание
Таким образом, при комбинировании первого компонента (а), который представляет собой ингибитор повторного усвоения 5-НТ, со вторым компонентом (б), который является селективным 5-HT1A антагонистом формулы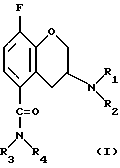
где заместитель R1 представляет собой н-пропил или циклобутил; заместитель R2 представляет собой изопропил, трет-бутил, циклобутил, циклопентил или циклогексил;
заместитель R3 представляет собой атом водорода;
заместитель R4 представляет собой атом водорода или метил;
причем указанный 5-HT1A антагонист находится в форме (R)-энантиомера и указанные компоненты (а) и (б) находятся в виде свободного основания или в виде их фармацевтически приемлемых солей, будет наблюдаться более быстрое проявление воздействия и, следовательно, более эффективное лечение пациентов.
В сочетание настоящего изобретения могут быть включены следующие 5-HT1A антагонисты в качестве компонента (б):
(R)-3-(N-циклопентил-N-н-пропиламино)-8-фтор-5-метилкарбамоил-3,4-дигидро-2Н-1-бензопиран,
(R)-8-Фтор-3-(N-изопропил-N-н-пропиламино)-5-карбамоил-3,4-дигидро-2Н-1-бензопиран,
(R)-5-Карбамоил-3-(N-трет. -бутил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран,
(R)-5-Карбамоил-3-(N, N-дициклогексиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран,
(R)-5-Карбамоил-3-(N-циклобутил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран,
(R)-5-Карбамоил-3-(N-циклобутил-N-изопропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран,
(R)-5-Карбамоил-3-(N-циклопентил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран,
(R)-5-Карбамоил-3-(N-циклогексил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран,
(R)-5-Карбамоил-3-(N-циклопентил-N-циклобутиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран.
Раскрытые здесь (R)-5-Карбамоил-8-фтор-3-(N,N-дизамещенный-амино)-3,4-дигидро-2Н-1-бензопираны, описаны в публикации WO 95/11891 (PCT/SE94/01010).
(R)-5-Карбамоил-8-фтор-3-(N, N-дизамещенный-амино)-3,4-дигидро-2Н-1-бензопираны находятся в виде свободного основания или в виде их фармацевтически приемлемых солей. Для образования нетоксичных фармацевтически приемлемых кислотно-аддитивных солей соединений настоящего изобретения могут быть использованы как органические, так и неорганические кислоты. В качестве примеров можно привести такие кислоты, как серная, азотная, фосфорная, щавелевая, соляная, муравьиная, бромистоводородная, лимонная, уксусная, молочная, винная, дибензоилвинная, памовая, этандисульфокислота, сульфаминовая, янтарная, пропионовая, гликолевая, яблочная, глюконовая, пировиноградная, фенилуксусная, 4-аминобензойная, антраниловая, салициловая, 4-аминосалициловая, 4-гидроксибензойная, 3,4-дигидроксибензойная, 3,5-дигидроксибензойная, 3-гидрокси-2-нафтойная, никотиновая, метансульфоновая, этансульфоновая, гидроксиэтансульфоновая, бензолсульфокислота, пара-толуолсульфокислота, сульфаниловая, нафталинсульфокислота, аскорбиновая, циклогексилсульфаминовая, фумаровая, малеиновая и бензойная. Эти соли легко получают известными методами.
Эти (R)-5-Карбамоил-8-фтор-3-(N, N-дизамещенный-амино)-3,4-дигидро-2Н-1-бензопираны проявляют высокое сродство к специфической подгруппе 5-HT1A рецепторов в центральной нервной системе (ЦНС) и действуют на этот 5-HT1A рецептор как антагонисты, а также обладают достаточной биодоступностью после перорального введения.
Приемлемыми известными ингибиторами повторного усвоения 5-НТ (SSRI), которые могут быть использованы в качестве компонента (а) в настоящем изобретении, являются норзимелдин, флуоксетин, пароксетин, циталопрам, кломипрамин, сертралин, флувоксамин, алапроклат, но не ограничиваются только этими SSRI.
Сочетание в соответствии с настоящим изобретением может быть приготовлено в одной фармацевтической рецептуре, которая содержит как активный первый компонент (а), так и активный второй компонент (б), или в виде двух фармацевтических рецептур, одна - для активного первого компонента (а) и одна - для активного второго компонента (б). Фармацевтическая рецептура может иметь форму таблеток или капсул, порошков, смесей, растворов или других приемлемых фармацевтических рецептурных форм.
Сочетание настоящего изобретения может быть приготовлено так, что ингибитор повторного усвоения 5-НТ вводится в тот же препарат, что и описанный выше селективный 5-HT1A антагонист, например, путем смешения обычным образом.
Настоящее изобретение также включает способ улучшения начала терапевтического действия путем совместного введения сочетания первого компонента (а), который представляет собой ингибитор повторного усвоения 5-НТ, и второго компонента (б), который представляет собой описанный здесь селективный антагонист 5-HT1A.
Еще один вариант осуществления настоящего изобретения представляет собой комплект, включающий сочетание первого компонента (а), который представляет собой ингибитор повторного усвоения 5-НТ, и второго компонента (б), который представляет собой селективный антагонист 5-HT1A, необязательно с инструкцией по применению.
Подробное описание изобретения
Способы получения (R)-5-Карбамоил-8-фтор-3-(N, N-дизамещенный-амино)-3,4-дигидро-2Н-1-бензопиранов
Получение (R)-3-амино-5-метокси-3,4-дигидро-2Н-1-бензопиран (соединение II) описано в публикации WO 93/07135, содержание которой включено в настоящее описание в качестве справочного материала. Предпочтительным методом введения атома фтора является бромирование ароматического кольца, которое протекает селективно в положение 8. Бромирование может быть осуществлено бромом с использованием катализатора или без него. В качестве других бромирующих агентов могут быть использованы НОВr и N-бромамиды (особенно N-бромсукцинимид). Приемлемыми растворителями для бромирования являются уксусная кислота, диоксан и хлорированные растворители, например, метиленхлорид.
Перед стадией фторирования первичная амино-группа должна быть полностью алкилирована заместителями R1 и R2, как указано выше, или защищена приемлемыми группами, которые позднее можно удалить, например, двумя бензильными группами. Введение алкильных групп по атому азота может быть осуществлено восстановительным аминированием из соответствующего альдегида или кетона с использованием подходящего восстановителя, например, NaCNBH3, или каталитически с использованием Н2 и соответствующего катализатора, содержащего палладий, платину или никель, в приемлемом растворителе, например, в тетрагидрофуране, диоксане, метаноле или этаноле. Введение алкильных групп также может быть осуществлено путем алкилирования соответствующим алкилгалогенидом, например, хлоридом, бромидом или иодидом, или активированным спиртом, например, алкил-мезилатом или-тозилатом, в подходящем растворителе, например, в диметилформамиде (ДМФА), ацетоне или ацетонитриле, в присутствии приемлемого основания, например, К2СО3.
Фторирование может быть проведено путем литирования бромпроизводного с помощью алкиллития, например, н-бутиллития, и последующей реакцией с подходящим фторирующим агентом, предпочтительно N-фтор-N-алкил/арилсульфонамидом, например, N-фторбензолсульфимидом. В качестве растворителя для этой реакции могут быть использованы безводные алифатические простые эфиры, например, тетрагидрофуран (ТГФ) или диэтиловый эфир, или апротонные растворители, например, гексан или бензол. Температура реакции может варьироваться от -100oС до комнатной, но предпочтительно от -78oС до -20oС. Соединения настоящего изобретения могут быть получены из (R)-3-амино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиранов (как это описано выше) в соответствии с известными методами, такими как восстановительное аминирование, N-алкилирование, деметилирование и превращение до уходящей группы Y с получением соединения IV.
Соединения формулы I могут быть получены в соответствии со следующими способами:
Метод i. Непосредственное превращение соединений формулы IV.

где заместитель Y представляет собой уходящую группу, такую как трифторметилсульфонатную (OSO2CF3), атом галогена, например, Сl, Вr или I при катализе переходным металлом (М) с нулевой валентностью, таким как Pd или Ni, который может генерироваться in situ, и подвергаться окислительному присоединению по связи арил-Y. Обработка оксидом углерода с последующим аминированием соответствующим амином (аммиаком или метиламином) дает соединения формулы I, которые затем при необходимости могут быть превращены в соли.
Метод ii. Соединения формулы IV могут быть превращены в соединения формулы V:
где заместитель Z представляет собой Сl, Вr, ОН или ORp, где заместитель Rp представляет собой C1-C6-алкил, при катализе переходным металлом с нулевой валентностью, способным подвергаться окислительному присоединению по связи арил-Y, например, по связи арил-OSO2CF3. При обработке монооксидом углерода (СО) образуется комплекс арил-СО-металл-Y.
Дополнительными реагентами являются спирт, такой как метанол, этанол, третичный амин, такой как триалкиламин, например, триэтиламин, в инертном органическом растворителе, предпочтительно в полярном апротонном растворителе, таком как диметилформамид (ДМФА), диметилсульфоксид (ДМСО), диоксан, тетрагидрофуран (ТГФ), ацетон, ацетонитрил и др. Реакцию обычно проводят при температуре в интервале от +40 до +120oС и давлении 100-500 кПа (Метод ii). Затем необязательно следует гидролиз и обработка тионилгалогенидом, например, тионилхлоридом, с получением соответствующего ацилгалогенидного производного.
Соединения формулы V аминируют (iib) подходящим амином (аммиаком или метиламином) в растворителе, например, толуоле, метиленхлориде, бензоле, воде, при кипячении с обратным холодильником в интервале температур 0-100oС с образованием соединений формулы I.
Фармацевтические рецептуры
В соответствии с настоящим изобретением соединения в сочетании будут обычно применяться перорально, ректально или путем инъекций, в форме фармацевтических рецептур, содержащих активный ингредиент в форме или свободного основания или нетоксичной фармацевтически приемлемой кислотно-аддитивной соли, например, в форме гидрохлорида, гидробромида, лактата, ацетата, фосфата, сульфата, сульфамида, цитрата, тартарата, оксалата и др., в фармацевтически приемлемой лекарственной форме. Лекарственная форма может быть твердой, полутвердой или жидкой рецептурой. Обычно активные вещества составляют от 0,1 до 99% вес. из расчета на вес рецептуры, более предпочтительно от 0,5 до 20% вес. для рецептур, предназначенных для инъекций, и от 0,2 до 50% вес. для рецептур, приемлемых для перорального применения.
Для получения фармацевтических рецептур сочетания настоящего изобретения в виде стандартных лекарственных форм для перорального применения выбранные соединения могут быть смешаны с твердым наполнителем, например, с лактозой, сахарозой, сорбитом, маннитом, крахмалами, такими как картофельный крахмал, кукурузный крахмал или амилопектин, производными целлюлозы, связующими веществами, такими как желатин или поливинилпирролидон, диспергирующими добавками, такими как крахмал-гликолат натрия, структурированный ПВП, кросс-кармелоза натрия, и смазывающими веществами, такими как стеарат магния, стеарат кальция, полиэтиленгликоль, воски, парафин и другие подобные вещества, а затем спрессованы в таблетки. Если необходимо приготовить таблетки с покрытием, то ядра, полученные по описанной выше методике, могут быть покрыты концентрированным раствором сахара, который может содержать аравийскую камедь, желатин, тальк, диоксид титана и др. С другой стороны, таблетки могут быть покрыты полимерами, которые известны специалистам в данной области, растворенными в легколетучем органическом растворителе или в смеси органических растворителей. К таким покрытиям могут быть добавлены красящие вещества для того, чтобы можно было легко различать таблетки, содержащие различные активные вещества или разные количества активных соединений.
Для рецептуры мягких желатиновых капсул активные вещества могут быть смешаны, например, с растительным маслом или полиэтиленгликолем. Твердые желатиновые капсулы могут содержать гранулы активного вещества, в которых используются любые описанные выше для таблеток наполнители, например, лактозу, сахарозу, сорбит, маннит, крахмалы (например, картофельный крахмал, кукурузный крахмал или амилопектин), производные целлюлозы или желатин. Также в твердые желатиновые капсулы может быть введено жидкое или полутвердое лекарство.
Стандартные лекарственные формы для ректального применения могут представлять собой растворы или суспензии или могут быть изготовлены в виде свечей, содержащих активные вещества в смеси с нейтральной жирной основой, или желатиновых ректальных капсул, содержащих активные вещества в смеси с растительным или парафиновым маслом. Жидкие препараты для перорального применения могут иметь форму сиропов или суспензий, например, растворов, содержащих приблизительно от 0,2 до 20% вес. описанных здесь активных веществ, причем в качестве балансовых ингредиентов используются сахар и смеси этанола, воды, глицерина и пропиленгликоля. Необязательно такие жидкие препараты могут содержать красящие агенты, корригирующие вкус и запах агенты, сахарин и карбоксиметилцеллюлозу в качестве загустителя или другие наполнители, хорошо известные квалифицированным в данной области специалистам.
Растворы для парентерального применения путем инъекций могут быть приготовлены в виде водного раствора водорастворимых фармацевтически приемлемых кислотно-аддитивных солей активных ингредиентов, предпочтительно с концентрацией приблизительно от 0,5 до 10% вес. Эти растворы могут также включать стабилизирующие агенты и/или буферные агенты и могут быть для удобства изготовлены в различных стандартных лекарственных ампулах.
Приемлемые дневные дозы активных соединений в сочетании настоящего изобретения при лечении человека составляют приблизительно 0,01-100 мг/кг веса тела при пероральном введении 0,001-100 мг/кг веса тела при парентеральном введении. Дневные дозы активного 5-HT1A антагониста могут сильно отличаться от дневных доз ингибитора повторного поглощения 5-НТ, но эти дозы могут быть одинаковыми для обоих активных соединений.
Получение исходных веществ для 5-НТ1A-антагонистов
Синтез 1
(R)-3-Амино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
а)(R)-3-Амино-8-бром-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-Амино-5-метокси-3,4-дигидро-2Н-1-бензопиран (25 г, 0,14 моля), полученный по методике публикации WO93/07135, и безводный ацетат натрия (34 г, 0,42 моля) растворяют в уксусной кислоте (500 мл). При комнатной температуре по каплям добавляют раствор брома (23,4 г, 0,15 моля) в уксусной кислоте (500 мл). Добавление брома осуществляют в течение приблизительно 7 дней. Растворитель упаривают в вакууме, а остаток растворяют в смеси 25%-ного раствора гидроксида натрия и диэтилового эфира (реакция экзотермична, смесь охлаждают на ледяной бане). Слои разделяют и водно-щелочную фазу дважды экстрагируют эфиром. Объединенные эфирные слои сушат (Na2SO4), растворитель упаривают, получают 35,5 г маслянистого остатка. Масло растворяют в диэтиловом эфире и раствор охлаждают на ледяной бане (0oС). По каплям добавляют раствор НСl в диэтиловом эфире до кислой реакции образующейся суспензии (контроль по рН-индикаторной бумаге). Кристаллы отфильтровывают и перекристаллизовывают из метанола, получают гидрохлорид названного соединения с выходом 70% (28,5 г) Т.пл. 281-282oС. Хлористоводородную соль распределяют между диэтиловым эфиром и 2 М водным NH3, свободное основание выделяют экстракцией водно-щелочной фазы диэтиловым эфиром.
[α]
б) (R)-8-бром-3-(N, N-дибензиламино)-5-метокси-3,4-дигидро-2Н-1-бензопиран
К раствору (R)-3-амино-8-бром-5-метокси-3,4-дигидро-2Н-1-бензопирана (11,5 г, 44 ммоля) в 400 мл сухого ацетонитрила добавляют бензилбромид (13 мл, 110 ммолей), безводный карбонат калия (измельченный) (16 г, 116 ммолей) и каталитическое количество KI и нагревают при 85oС в течение 48 часов.
Растворитель упаривают в вакууме, остаток обрабатывают 2М раствором NН3 и дважды экстрагируют эфиром. Объединенные эфирные слои промывают рассолом, сушат (МgSO4), фильтруют, растворитель упаривают в вакууме, получают сырой остаток. Хроматографирование на двуокиси кремния (элюент метиленхлорид) дает 15 г (выход 78%) названного соединения в виде прозрачного масла.
[α]
в) (R)-3-(N, N-Дибензиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-8-Бром-3-(N, N-дибензиламино)-5-метокси-3,4-дигидро-2Н-1-бензопиран (4,35 г, 9,9 ммоля) растворяют в 45 мл сухого ТГФ и охлаждают до -78oС. К раствору по каплям добавляют 1,6 М раствор н-бутиллития (6,8 мл, 10,9 ммоля) и перемешивают при -78oС в течение 1 часа. В течение 45 минут добавляют по каплям раствор фторбензолсульфонимида (3,8 г, 11,9 ммоля) в 30 мл сухого ТГФ и перемешивают при -78oС в течение 1 часа. Реакцию останавливают добавлением 3 мл насыщенного раствора NH4Cl, затем 9 мл раствора, содержащего 2 г NH2OH•HCl и 8 г Na2СО3 в 100 мл Н2O, после чего дают реакционной массе нагреться до комнатной температуры. К смеси добавляют 2М раствор NН3 и дважды экстрагируют эфиром, промывают рассолом, сушат (Na2SO4), фильтруют, упаривают в вакууме, получают сырой продукт. Очистку 8-фтор- (требуемый продукт от 8-бром-производного осуществляют хроматографированием (элюент 25%-ный метиленхлорид в гексане), получают 1,50 г (выход 40%) названного соединения в виде прозрачного масла.
[α]
г) (R)-3-Амино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N, N-Дибензиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран (13,0 г, 34,4 ммоля) растворяют в 265 мл метанола и 115 мл ТГФ. К раствору добавляют 10% Pd/C (4 г) и формиат аммония (51,5 г, 0,817 моля). Реакционную смесь нагревают при 50oС в течение 2,5 часов, после чего фильтруют, упаривают растворитель в вакууме, остаток забирают в 2 М раствор NaOH и дважды экстрагируют эфиром. Объединенные эфирные слои промывают рассолом, сушат (Nа2SО4), фильтруют, растворитель упаривают в вакууме, получают 6,2 г (выход 91%) названного соединения в виде прозрачного масла.
[α]
Синтез 2
(R)-8-Фтор-3-(N-изопропил-N-пропиламино)-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран
а) (R)-8-Бром-3-(N-изопропиламино)-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N-Изопропиламино)-5-метокси-3,4-дигидро-2Н-1-бензопиран (4,02 г, 18,2 ммоля, получен по методике публикации WO93/07135) и безводный ацетат натрия растворяют в уксусной кислоте (80 мл). К полученному раствору в течение 1,5 часа при перемешивании по каплям добавляют раствор брома (0,93 мл, 18,2 ммоля) в уксусной кислоте (40 мл).
Растворитель упаривают в вакууме, остаток забирают в 2 М раствор NaOH и дважды экстрагируют диэтиловым эфиром. Объединенные эфирные слои промывают рассолом, сушат (Na2SO4), фильтруют, растворитель упаривают, получают сырой остаток. Хлористоводородную соль получают растворением чистого основания в диэтиловом эфире и добавлением избытка раствора НСl в эфире, получают белое твердое вещество. Соль дважды перекристаллизовывают из смеси этанол-диэтиловый эфир, получают 3,8 г (выход 62%). Т.пл. 257-8oС.
[α]
Свободное основание, представляющее собой масло, получают из хлористоводородной соли. Масс-спектр (газовая хроматография) (70 эВ) М=301 (100%).
б) (R)-8-Бром-3-(N-изопропил-N-пропиламино)-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-8-Бром-3-(N-изопропиламино)-5-метокси-3,4-дигидро-2Н-1-бензопиран (3,8 г, 11,3 ммоля) растворяют в сухом метаноле (80 мл) и добавляют пропаналь (8,1 мл, 0,113 моля). Смесь охлаждают на ледяной бане и добавляют цианборгидрид натрия (1,3 г, 20,3 ммоля), доводят рН до 5 и перемешивают при комнатной температуре в течение ночи.
Растворитель упаривают в вакууме, остаток помещают в 1 М раствор Nа2СО3 и дважды экстрагируют эфиром. Объединенные эфирные слои промывают рассолом, сушат (Nа2SO4), фильтруют, растворитель упаривают, получают сырой остаток. Хроматографированием на двуокиси кремния (элюент - 7%-ный этилацетат в гексане) выделяют 3,75 г (выход 97%) названного соединения в виде прозрачного масла.
[α]
Хлористоводородную соль получают растворением основания в диэтиловом эфире и добавлением по каплям избытка раствора НС1 в эфире. Соль перекристаллизовывают из смеси этанол-диэтиловый эфир с получением белого твердого вещества. Т.пл. 177-9oС.
в) (R)-8-Фтор-3-(N-изопропил-N-пропиламино)-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-8-Бром-3-(N-изопропил-N-пропиламино)-5-метокси-3,4-дигидро-2Н-1-бензопиран (2,3 г, 6,72 ммоля) растворяют в 25 мл сухого ТГФ и охлаждают до -78oС. К полученному раствору по каплям добавляют 1,6 М раствор н-бутиллития (4,83 мл, 7,73 ммоля) и перемешивают при -78oС в течение 1 часа. В течение 20-30 мин добавляют по каплям раствор фторбензолсульфонимида (2,55 г, 8,06 ммоля) в 15 мл сухого ТГФ и перемешивают при -78oС в течение 4 часов. Реакцию останавливают добавлением 1 мл насыщенного водного раствора NH4C1, затем 3 мл раствора, содержащего 2 г NH2OH•HCl и 8 г Nа2СО3 в 100 мл H2O, после чего дают реакционной массе нагреться до комнатной температуры. К смеси добавляют 2 М раствор NH3 и дважды экстрагируют диэтиловым эфиром, промывают рассолом, сушат (Na2SO4), фильтруют, упаривают в вакууме, получают сырой продукт. Хроматографированием на двуокиси кремния (элюент - хлороформ) выделяют 1,0 г (выход 53%) названного соединения в виде прозрачного масла.
[α]
Хлористоводородную соль получают растворением чистого основания в диэтиловом эфире и добавлением по каплям избытка раствора НС1 в эфире, при этом образуется белое твердое вещество (спекается при 80oС).
г) (R)-8-фтор-3-(N-изопропил-N-пропиламино)-5-гидрокси-3,4-дигидро-2Н-1-бензопиран
Гидрохлорид (R)-8-Фтор-3-(N-изопропил-N-пропиламино)-5-метокси-3,4-дигидро-2Н-1-бензопирана (1,03 г, 3,24 ммоля) растворяют в сухом CH2Cl2 (30 мл) и охлаждают до -40oС. К раствору по каплям добавляют раствор ВВr3 (0,77 мл, 8,1 ммоля) в 5 мл СН2Сl2. Охлаждающую баню убирают и после 3 часов перемешивания при комнатной температуре реакция завершается.
Реакционную смесь выливают на лед с 2 М раствором NН3 и дважды экстрагируют эфиром. Объединенные эфирные экстракты промывают рассолом, сушат (Na2SO4), фильтруют, растворитель упаривают в вакууме, получают сырой продукт. Хроматографированием на двуокиси кремния (элюент - 20%-ный этилацетат в гексане) выделяют 0,84 г (выход 97%) названного соединения в виде прозрачного масла.
[α]
Хлористоводородную соль получают растворением чистого основания в диэтиловом эфире и добавлением по каплям избытка раствора НСl в эфире. Соль перекристаллизовывают из смеси СНСl3/диэтиловый эфир/этилацетат, получают белое твердое вещество. Т.пл. 220-2oС.
д) (R)-8-фтор-3-(N-изопропил-N-пропиламино)-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран
(R)-8-Фтор-3-(N-изопропил-N-пропиламино)-5-гидрокси-3,4-дигидро-2Н-1-бензопиран (0,71 г, 2,66 ммоля) и коллидин (0,49 мл, 3,72 ммоля) растворяют в 25 мл сухого СН2Сl2 и охлаждают до -40oС. К раствору по каплям добавляют ангидрид трифторметансульфокислоты (0,54 мл, 3,2 ммоля), охлаждение снимают, по достижении реакционной массы температуры 0oС реакция завершается. Реакционную смесь разбавляют CH2Cl2, промывают насыщенным водным раствором NаНСО3, сушат (MgSO4), фильтруют, упаривают в вакууме, получают сырой продукт. Хроматографированием на двуокиси кремния (элюент CH2Cl2) выделяют 0,82 г (выход 77%) названного соединения в виде прозрачного масла.
[α]
Пример 1.
(R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-N-метилкарбамоил-3,4-дигидро-2Н-1-бензопиран
а) (R)-3-N-Циклопентиламино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-Амино-8-Фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран (1,5 г, 7,6 ммоля), уксусную кислоту (0,45 г, 7,6 ммоля) и циклопентанон (2,5 г, 3 ммоля) растворяют в 30 мл метанола. При перемешивании порциями в течение нескольких минут добавляют цианборгидрид натрия (0,8 г, 13 ммолей) и перемешивают в течение 2 часов. Газовая хроматография показывает 100%-ное образование нового продукта. Растворитель упаривают и добавляют воду, 2 М раствор NН3 и этилацетат. Органический слой отделяют, промывают водой, сушат (Na2SO4), упаривают, получают 1,3 г (выход 64%) бесцветного масла. Масс-спектрометрия в сочетании с газовой хроматографией дает молекулярный пик 265, что подтверждает образование названного соединения.
б) (R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-N-Циклопентиламино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран (1,3 г, 5 ммолей, уксусную кислоту (0,3 г, 5 ммолей) и пропионовый альдегид (1,5 г, 25 ммолей) растворяют в 30 мл метанола. При перемешивании порциями в течение нескольких минут добавляют цианборгидрид натрия (0,8 г, 13 ммолей) и перемешивают в течение 3 часов, после чего газовая хроматография показывает 100%-ное образование нового продукта. Растворитель упаривают и добавляют воду, 2 М раствор NH3 и этилацетат. Органический слой отделяют, промывают водой до нейтральной реакции, сушат (Nа2SO4), упаривают, получают 1 г (выход 65%) бесцветного масла. Масс-спектрометрия в сочетании с газовой хроматографией дает молекулярный пик 307, что подтверждает образование названного соединения.
в) (R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран (1 г, 5 ммолей) растворяют в 25 мл CH2Cl2 и добавляют избыток раствора НСl в эфире с образованием гидрохлорида. К полученной смеси при перемешивании и охлаждении на ледяной бане по каплям добавляют раствор ВВr3 (4 г, 15 ммолей) в 10 мл СН2Сl2. Реакционной массе при перемешивании дают нагреться до комнатной температуры в течение 6 часов, после чего ее выливают в воду со льдом и подщелачивают водным аммиаком. Органический слой отделяют, сушат (Na2SO4), упаривают, получают темно-коричневое масло. Хроматографированием на двуокиси кремния (элюент - диизопропиловый эфир-гексан, 1:1) выделяют 1,1 г бесцветного масла. Хлористоводородную соль получают из основания и эфирного раствора НС1 и перекристаллизовывают из ацетонитрила, выделяют 0,85 г (выход 52%). Т.пл. 220-221oС
г) (R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран (0,7 г, 3 ммоля) растворяют в 25 мл СН2Сl2 и добавляют триэтиламин (0,3 г, 3 ммоля). К полученному раствору при -20oС в течение 10 минут по каплям добавляют раствор ангидрида трифторметансульфокислоты (1 г, 4 ммоля) в 5 мл СН2Сl2 и перемешивают в течение 1 часа. Реакционную смесь выливают в воду со льдом, добавлением водного аммиака доводят рН до 8 и экстрагируют эфиром. Органический слой отделяют, сушат (Na2SO4), упаривают масло коричневого цвета. Хроматографированием на двуокиси кремния (элюент СН2Сl2-гексан, 1:3) выделяют 0,5 г (выход 44%) бесцветного масла. Масс-спектрометрия в сочетании с газовой хроматографией дает молекулярный пик 425, что подтверждает образование названного соединения.
д) (R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-N-метилкарбамоил-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран (0,5 г, 1 ммоль) растворяют в 15 мл 1,4-диоксана. К раствору добавляют ацетат палладия (II) (10 мг), 1,3-бис(дифенилфосфино)пропан (20 мг) и метиламин (0,15 г, 5 ммолей) и смесь перемешивают в атмосфере монооксида углерода в течение ночи при 70oС. Упаривание и хроматографирование на двуокиси кремния (элюент - диэтиловый эфир-гексан, 1: 3) приводит к конечному соединению в виде бесцветного масла, из которого получают хлористоводородную соль в виде белых кристаллов. Т.пл. 108oС.
Пример 2.
(R)-8-Фтор-3-(N-изопропил-N-н-пропиламино)-5-карбамоил-3,4-дигидро-2Н-1-бензопиран
а) Метиловый эфир (R)-8-фтор-3-(N-изопропил-N-н-пропиламино)-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты
(R)-8-Фтор-3-(N-изопропил-N-пропиламино)-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран (2,4 г, 6,0 ммоля), триэтиламин (1,3 г, 12,9 ммоля), 1,3-бис(дифенилфосфино)-пропан (95 мг, каталитическое количество), ацетат палладия (II) (48 мг, каталитическое количество и 30 мл смеси ДМФА-метанол (3:1) смешивают в трехгорлой круглодонной колбе на 50 мл. Колбу вакуумируют и заполняют СО (операцию повторяют дважды). Реакционную смесь перемешивают при 70oС в течение 7,5 часов в атмосфере СО. Растворитель упаривают в вакууме, остаток обрабатывают смесью диэтиловый эфир-насыщенный раствор NаНСО3. Слои разделяют, водную фазу экстрагируют один раз эфиром. Объединенные эфирные экстракты сушат (МgSО4), растворитель упаривают в вакууме. Полученный сырой продукт очищают хроматографированием на двуокиси кремния (элюент - гексан-этилацетат, 9:1), получают 1,3 г названного соединения (выход 71%).
б) (R)-8-фтор-3-(N-изопропил-N-н-пропиламино)-5-карбамоил-3,4-дигидро-2Н-1-бензопиран
Метиловый эфир (R)-8-фтор-3-(N-изопропил-N-н-пропиламино)-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты (1,3 г, 4,2 ммоля) и КОН (0,52 г, 8,4 ммоля) смешивают в метаноле (6 мл) и кипятят с обратным холодильником в течение 2,5 часов. Растворитель упаривают в вакууме. Остаток растворяют в воде и подкисляют добавлением 2 М НСl. Растворитель упаривают в вакууме, остаток растворяют в SOCl2 (30 мл) и кипятят в течение 2,5 часов. Растворитель упаривают в вакууме, остаток растворяют в CH2Cl2 и растворитель упаривают в вакууме (операцию повторяют три раза для удаления избытка SOСl2). Остаток растворяют в диэтиловом эфире (50 мл), охлаждают до -30oС и пропускают газообразный NН3. К смеси добавляют воду, слои разделяют, водную фазу экстрагируют эфиром. Объединенные экстракты сушат (К2СО3), растворитель упаривают в вакууме. Полученный сырой продукт очищают быстрым хроматографированием на двуокиси кремния (элюент - этилацетат-гексан, 1:1), получают 1 г названного соединения (выход 80%). Перекристаллизация из смеси этилацетат-гексан дает кристаллы. Т.пл. 139-140oС.
Пример 3.
(R)-3-(N-трет-Бутил-N-н-пропиламино)-5-карбамоил-8-фтор-3,4-дигидро-2Н-1-бензопиран
а) (R)-8-фтор-5-метокси-3-[N-(4-метоксибензилиден)-амино]-3,4-дигидро-2Н-1-бензопиран
Смесь (R)-3-амино-8-Фтор-5-метокси-3,4-дигидро-2Н-1-бензопирана (7,85 г, 39,8 ммоля), 4-метоксибензальдегида (5,42 г, 39,8 ммоля), безводного карбоната калия (10,1 г) и абсолютного EtOH (200 мл) перемешивают при кипячении в течение ночи. Растворитель упаривают в вакууме и добавляют эфир (500 мл). После перемешивания в течение 15 минут соль отфильтровывают, фильтрат упаривают в вакууме, получают почти белое твердое вещество (12,4 г). Перекристаллизация из смеси i-Рr2О-гексан приводит к 10,8 г (выход 86%) названного соединения в виде бесцветных игольчатых кристаллов. Т.пл. 96,8-97,3oС.
[α]
б) (R)-8-Фтор-3-гидроксиламино-5-метокси-3,4-дигидро-2Н-1-бензопиран
К раствору (R)-8-фтор-5-метокси-3-[N-(4-метоксибензилиден)-амино]-3,4-дигидро-2Н-1-бензопирана (10,8 г, 34 ммоля) в метиленхлориде (65 мл) при перемешивании и охлаждении (+4oС) порциями добавляют 3-хлорнадбензойную кислоту (85%;
7,6 г, 37,6 ммоля) и смесь перемешивают в течение ночи при комнатной температуре. Выделившийся осадок 3-хлорбензойной кислоты отфильтровывают, а прозрачный фильтрат желтого цвета упаривают в вакууме. Маслянистый остаток забирают в раствор хлоргидрата гидроксиламина (2,83 г, 40,8 ммоля) в сухом метаноле (60 мл) и полученный раствор перемешивают при комнатной температуре в течение 2 часов. Растворитель упаривают в вакууме, получают густое масло оранжевого цвета. К маслу добавляют воду, добавлением водного раствора Na2СО3 доводят рН до 8-9 и смесь экстрагируют эфиром (3х150 мл). Объединенные экстракты промывают рассолом, сушат (Na2SO4), фильтруют и растворитель упаривают в вакууме. Сырой продукт подвергают быстрому хроматографированию на двуокиси кремния (элюент - от 15 до 50% этилацетата в гексане). Полученный загрязненный продукт подвергают повторному быстрому хроматографированию на двуокиси кремния (элюент EtOH-CHCl3, 1:99), получают 6,45 г (выход 89%) названного соединения в виде бесцветного кристаллического вещества. Т. пл. 111-113oС.
[α]
в) (R)-3-трет-Бутиламино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-8-Фтор-3-гидроксиламино-5-метокси-3,4-дигидро-2Н-1-бензопиран (6,30 г, 29,6 ммоля), безводный сульфат натрия (20 г) и ацетон (500 мл) кипятят с обратным холодильником в атмосфере азота в течение 4 дней до окончания реакции в соответствии с данными ТСХ. Отфильтровывают соль, к фильтрату добавляют эфир (300 мл) и раствор, еще содержащий суспендированные мелкие частицы соли, фильтруют через фильтр Шотта (номер 4). Прозрачный фильтрат упаривают в вакууме. Добавляют сухой бензол (30 мл, высушен ситами 3А) и полученный раствор упаривают в вакууме (окончательно на насосе). Стеклоподобный остаток растворяют в сухом бензоле (150 мл, высушен ситами 3А) в атмосфере азота и раствор охлаждают на ледяной бане (+4oС). К описанному выше раствору при перемешивании добавляют МеМgВr в Et2O (3,0 М; 32,0 мл, 96 ммолей) с такой скоростью, чтобы внутренняя температура была ниже +5oС (реакция экзотермична). По окончании добавления (30 минут) раствор перемешивают при +4oС в течение 0,5 часа. Охлаждающую баню убирают и через 15 минут раствор выливают на насыщенный раствор NаНСО3 и лед (суммарно 300 мл). Смесь промывают несколько раз эфиром (3х150 мл). Органические фазы объединяют, промывают рассолом, сушат (Na2SO4), фильтруют и упаривают в вакууме. Быстрая хроматография на двуокиси кремния (элюент - 2 и 10% EtOAc в СНСl3) дает 2,9 г трет-бутил-гидроксиламинопроизводного. Последнее растворяют в CS2 (100 мл) в атмосфере азота и раствор перемешивают при комнатной температуре в течение 4,5 ч. Растворитель упаривают в вакууме и получают оранжевое масло. К маслу добавляют ацетон (приблизительно 50 мл) и раствор недолго (15 мин) перемешивают при комнатной температуре (для осаждения элементарной серы), затем фильтруют и упаривают, получают масло. Быстрая хроматография на двуокиси кремния (элюент - 10-25% EtOAc в гексане) дает 2,34 г (общий выход 31%) названного соединения в виде желтого масла.
[α]
г) (R)-3-(N-трет-Бутил-N-н-пропиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-трет-Бутиламино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран (2,20 г, 8,7 ммоля), аллилбромид (7,5 мл, 87 ммолей), тонкоизмельченный безводный карбонат калия (6,0 г, 43 ммоля) и сухой ДМФА (6,0 мл) перемешивают в атмосфере азота при 65oС. Через 70 часов ГХ-анализ показывает частичную конверсию исходного материала (67% продукта и 30% исходного материала). На этом этапе реакцию останавливают. Соль отсасывают, промывают небольшим количеством ДМФА и прозрачный фильтрат упаривают. Полученное таким образом масло распределяют между насыщенным водным раствором Nа2СО3 и диэтиловым эфиром (4х70 мл). Органические фазы объединяют, промывают рассолом, сушат (МgSO4), фильтруют и упаривают в вакууме. Быстрая хроматография на двуокиси кремния (элюент - 2 и 15% ацетон в гексане) дает 0,80 г исходного материала и 1,47 г (выход 87% из расчета на выделенный исходный материал) аллилированного продукта в виде бесцветного масла.
[α]
Аллилированный продукт (1,30 г) смешивают с ДМФА (50 мл) и 5%-ным Rh на окиси алюминия (0,090 г) и гидрируют при температуре и давлении окружающей среды (21oС). Через 5 часов по данным ГХ и ТСХ реакция завершается. Катализатор отфильтровывают на целитах, слой промывают небольшими порциями ДМФА и полученный фильтрат упаривают в вакууме. Быстрая хроматография на двуокиси кремния сырого продукта (элюент - 0 и 3% EtOAc в CH2Cl2) дает 1,27 г (выход 97%) насыщенного соединения.
Масс-спектр (газовая хроматография) (70 эВ) М = 295 (28%).
[α]
Полученное основание растворяют в сухом диэтиловом эфире, раствор охлаждают на ледяной бане и к раствору при перемешивании добавляют эфирный раствор НСl. Полученную соль отфильтровывают, промывают сухим диэтиловым эфиром и сушат в вакууме при 50oС, получают 1,39 г (выход 98%) названного соединения в виде белых кристаллов. Т.пл. 175-176oС.
д) (R)-3-(N-трет-Бурил-N-н-пропиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран
Гидрохлорид (R)-3-(N-трет-Бутил-N-н-пропиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопирана (1,3 г, 3,9 ммоля) в сухом метиленхлориде (40 мл) охлаждают в атмосфере азота до -50oС на бане сухой лед/EtOH. К полученному раствору при перемешивании по каплям (в 1 минуту) добавляют трибромид бора (0,75 мл, 7,8 ммоля). Через 5 минут добавление трибромида бора завершают, баню с сухим льдом заменяют на ледяную баню (+4oС). После перемешивания в течение 4 часов при той же температуре раствор выливают на лед (100 г) и добавляют твердый NаНСО3 до получения рН 8-9. После того, как лед растает, смесь экстрагируют эфиром (4х75 мл). Эфирные экстракты объединяют, промывают рассолом, сушат (Na2SO4), фильтруют и упаривают в вакууме. Получают 1,1 г (выход 96%) названного соединения в виде светло-желтого масла.
[α]
е) (R)-3-(N-трет-Бутил-N-н-пропиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N-трет-Бутил-N-н-пропиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран (1,0 г, 3,6 ммоля) и 2,4,6-коллидин (0,52 мл, 3,9 ммоля) растворяют в безводном метиленхлориде (40 мл) и охлаждают до -30oС. Добавляют по каплям в течение 20 минут ангидрид трифторметансульфокислоты (0,66 мл, 3,9 ммоля), растворенный в безводном метиленхлориде (10 мл). Полученному раствору дают нагреться до комнатной температуры и после достижения 0oС реакция завершается. Реакционную смесь разбавляют метиленхлоридом и промывают насыщенным водным раствором NаНСО3 (50 мл). Водную фазу повторно экстрагируют эфиром (2х40 мл). Объединенные органические фазы сушат (МgSO4), фильтруют и упаривают в вакууме. Получают сырой продукт. Быстрая хроматография на двуокиси кремния (элюент - этилацетат:гексан, 3:97) дает 1,40 г (выход 95%) названного соединения в виде бесцветного масла.
[α]
ж) Метиловый эфир (R)-3-(N-трет-Бутил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты
(R)-3-(N-трет-Бутил-N-н-пропиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран (1,4 мл, 3,3 ммоля) и триэтиламин (1,0 мл, 7,4 ммоля) растворяют в смеси ДМФА/МеОН (6:2; 30 мл) и затем дегазируют и пропускают монооксид углерода (4 раза). При небольшом избыточном давлении монооксида углерода добавляют ацетат палладия (II) (0,030 г) и 1,3-бис(дифенилфосфино)пропан (0,060 г) и реакционную смесь дегазируют и снова подвергают воздействию монооксида углерода. Далее реакционную смесь нагревают до 70oС (температура масляной бани) в атмосфере монооксида углерода при интенсивном перемешивании в течение 12 часов. Данные ГХ указывают на неполное протекание реакции (68% продукта относительно 21% исходного материала), раствор охлаждают и затем фильтруют через целиты. Добавляют еще ацетат палладия (II) (0,030 г) и 1,3-бис(дифенилфосфино)пропан (0,060 г) и реакцию возобновляют в соответствии с описанной методикой. Через 3 часа данные ГХ показывают небольшое улучшение в соотношении продукта и исходного материала (55% относительно 12%) и реакционную массу снова охлаждают. На следующий день растворитель упаривают в вакууме. Оставшееся красно-коричневое масло забирают в насыщенный водный раствор NaHCO3 и экстрагируют этилацетатом (3х50 мл). Объединенные органические фазы промывают рассолом, сушат (MgSO4), фильтруют и упаривают в вакууме. Получают сырой эфир. Быстрая хроматография на двуокиси кремния (элюент - 15% и 30% EtOAc в гексане) дает 0,178 г исходного материала и 0,842 г (выход 89% из расчета на выделенный исходный материал) названного соединения в виде прозрачного масла.
[α]
з) (R)-3-(N-трет-Бурил-N-н-пропиламино)-5-карбамоил-8-фтор-3,4-дигидро-2Н-1-бензопиран
Метиловый эфир (R)-3-(N-трет-Бутил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты (0,84 г, 2,6 ммоля), метанол (10 мл) и 1,7 М водный раствор NaOH (3,0 мл, 5,2 ммоля) кипятят с обратным холодильником в течение 3 часов. Полученный прозрачный раствор охлаждают, метанол отпаривают, водный остаток промывают два раза смесью эфир:гексан (1:1), затем подкисляют с помощью 2 М НСl (рН<2). Воду упаривают в вакууме и остающуюся соль сушат в вакууме при 50oС в течение 2 часов. Добавляют сухой метиленхлорид (20 мл) и тионилхлорид (3,0 мл, 41 ммоль), полученную смесь кипятят с обратным холодильником в атмосфере азота в течение 11 часов. Летучие компоненты упаривают, добавляют еще сухой метиленхлорид и упаривают. Процедуру повторяют еще раз. Сырой хлорангидрид кислоты растворяют (суспендируют) в сухом метиленхлориде (50 мл) и добавляют по каплям при перемешивании к концентрированному водному раствору аммиака (40 мл), охлажденного на ледяной бане. Смеси дают нагреться до температуры окружающей среды, органическую фазу отделяют, а водную фазу промывают метиленхлоридом (100 мл) и эфиром (50 мл). Органические порции объединяют, сушат (MgSO4), фильтруют и упаривают. Получают сырой амид. Быстрая хроматография на двуокиси кремния (элюент - EtOAc:гексан, 4:5) дает 0,73 г (выход 91%)
(R)-3-(N-трет-Бутил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоксамида в виде твердого продукта. Т.пл. 70-75oС.
[α]
Основание растворяют в сухом эфире, полученный раствор охлаждают баней с сухим льдом (-20oС) и при перемешивании добавляют избыток эфирного раствора НСl. Соль отфильтровывают, промывают сухим эфиром и сушат в вакууме до 50oС, получают 0,78 г (выход 96%) хлористоводородной соли в виде белых кристаллов. Т.пл. 120oС (спекание).
Пример 4.
(R)-5-Карбамоил-3-(N, N-дициклобутиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран
а) (R)-3-(N-Циклобутиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран и
(R)-3-(N, N-Дициклобутиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-Амино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран (1,67 г, 8,47 ммоля) растворяют в безводном метаноле (20 мл) и добавляют циклобутанон (5,0 г, 71,3 ммоля). Реакционную массу охлаждают (ледяная баня), а затем добавляют цианборгидрид натрия (0,96 г, 15,3 ммоля) и при перемешивании дают нагреться до комнатной температуры в течение ночи. Через 24 часа с помощью уксусной кислоты доводят рН до 4-5 и перемешивают в течение еще одного дня.
Растворитель упаривают в вакууме, остаток обрабатывают 2 М раствором NН3 и затем экстрагируют диэтиловым эфиром. Объединенные эфирные порции сушат (Na2SO4), фильтруют и растворитель упаривают в вакууме, получают сырой остаток. При хроматографировании на двуокиси кремния (элюент - 15% этилацетата в гексане для диалкилированного продукта, а затем этилацетат для моноалкилированного продукта) получают 1,01 г (выход 48%) моноалкилированного названного соединения в виде прозрачного масла. [Масс-спектроскопия (газовая хроматография) (70 эВ) М=251 (6%)] и 0,71 г (выход 27%) диалкилированного названного соединения в виде прозрачного масла.
[α]
б) (R)-8-фтор-3-(N, N-дициклобутиламино)-5-гидрокси-3,4-дигидро-2Н-1-бензопиран
Гидрохлорид (R)-8-фтор-3-(N, N-дициклобутиламино)-5-метокси-3,4-дигидро-2Н-1-бензопирана (0,77 г, 2,26 ммоля) растворяют в безводном метиленхлориде (20 мл) и охлаждают до -40oС. К раствору по каплям добавляют ВВr3 (0,54 мл, 5,7 ммоля), растворенного в безводном метиленхлориде (3 мл). Охлаждающую баню убирают и через 2 часа при комнатной температуре реакция завершается.
Реакционную массу выливают на смесь льда и 2 М раствора NН3 и полученную смесь экстрагируют дважды диэтиловым эфиром. Объединенные эфирные порции сушат (МgSO4), фильтруют и растворитель упаривают в вакууме, получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - 50% этилацетата в гексане) получают 0,58 г (выход 89%) названного соединения в виде белого твердого вещества. Т.пл. 170-2oС.
[α]
в) (R)-3-(N, N-Дициклобутиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N, N-Дициклобутиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран (0,59 г, 2,02 ммоля) и коллидин (0,37 мл, 2,8 ммоля) растворяют в безводном CH2Cl2 (40 мл) и охлаждают до -40oС. Добавляют по каплям ангидрид трифторметансульфокислоты (0,41 мл, 2,4 ммоля) и дают реакционной массе нагреться до температуры окружающей среды, после достижения температуры 0oС реакция заканчивается. Реакционную массу разбавляют метиленхлоридом и промывают насыщенным водным раствором NаНСО3, сушат (MgSO4), фильтруют и упаривают в вакууме, получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - метиленхлорид) получают 0,84 г (выход 99%) названного соединения в виде прозрачного масла.
[α]
г) Метиловый эфир (R)-3-(N,N-дициклобутиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты
(R)-3-(N, N-Дициклобутиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран (0,82 г, 1,94 ммоля) растворяют в смеси ДМФА/метанол (6: 2, 15 мл) и затем дегазируют и пропускают монооксид углерода (3 раза). При небольшом избыточном давлении монооксида углерода добавляют ацетат палладия (II) (14 мг), 1,3-бис(дифенилфосфино)пропан (25 мг) и триэтиламин (0,60 мл, 4,3 ммоля) и реакционную смесь дегазируют и снова подвергают воздействию монооксида углерода. Далее реакционную смесь нагревают при 70oС в атмосфере монооксида углерода при интенсивном перемешивании в течение 5,5 часа. Реакционной массе дают охладиться и в вакууме упаривают растворитель. Остаток забирают в 2 М раствор NН3 и затем дважды экстрагируют диэтиловым эфиром. Объединенные эфирные порции сушат (МgSO4), фильтруют и упаривают в вакууме. Получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - 12,5% этилацетата в гексане) получают 501 мг (выход 78%) названного соединения в виде прозрачного масла.
[α]
д) (R)-5-Карбамоил-3-(N, N-дициклобутиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран
Метиловый эфир (R)-3-(N,N-дициклобутиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты (490 мг, 1,47 ммоля) кипятят с обратным холодильником с 6 М раствором НСl (20 мл) в течение 3,5 часа. Раствор охлаждают, упаривают досуха в вакууме, добавляют безводный толуол и растворитель упаривают в вакууме (4 раза).
К полученному белому твердому продукту добавляют тионилхлорид (15 мл) и полученный раствор перемешивают при комнатной температуре в течение ночи. Избыток тионилхлорида упаривают в вакууме, добавляют безводный толуол и растворитель упаривают в вакууме.
Хлорангидрид растворяют в СН2Сl2 (20 мл) и по каплям добавляют к охлажденному (ледяная баня) раствору концентрированного NН3 (20 мл). Реакционную массу перемешивают при комнатной температуре в течение 30 минут. Метиленхлоридную фазу отделяют, а водную фазу экстрагируют СН2Сl2 (3 раза). Объединенные метиленхлоридные порции сушат (MgSO4), фильтруют и упаривают в вакууме, получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - этилацетат) получают 430 мг (выход 92%) белого твердого продукта. Т.пл. 141,2-142,2oС.
[α]
Хлористоводородную соль получают при растворении чистого основания в эфире и добавлении по каплям избытка эфирного раствора НСl. Соль промывают диэтиловым эфиром, получают белый твердый продукт. Т.пл. 120oС (спекание).
Пример 5.
(R)-5-Карбамоил-3-(N-циклобутил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран
а) (R)-3-(N-Циклобутил-N-н-пропиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N-Циклобутиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран (1,01 г, 4,02 ммоля) растворяют в безводном метаноле (20 мл) и к раствору добавляют н-пропиональдегид (3,0 мл, 40,2 ммоля). Через 1 час реакционную массу охлаждают (ледяная баня) и добавляют цианборгидрид натрия (0,46 г, 7,24 ммоля), с помощью уксусной кислоты доводят рН до 4-5 и перемешивают при комнатной температуре в течение двух дней. Растворитель упаривают в вакууме, остаток забирают в 2 М раствор NН3 и затем экстрагируют трижды диэтиловым эфиром. Объединенные эфирные порции сушат (МgSO4), фильтруют и растворитель упаривают в вакууме, получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - 11% этилацетата в гексане) получают 0,95 г (выход 80%) названного соединения в виде прозрачного масла.
[α]
б) (R)-3-(N-Циклобутил-N-н-пропиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран
Гидрохлорид (R)-3-(N-Циклобутил-N-н-пропиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопирана (1,0 г, 3,03 ммоля) растворяют в безводном СН2Сl2 (25 мл) и охлаждают до -40oС. К полученному раствору по каплям добавляют ВВr3 (0,72 мл, 7,6 ммоля), растворенного в безводном CH2Cl2 (4 мл). Охлаждающую баню убирают и через два часа выдерживания при комнатной температуре реакция завершается.
Реакционную массу выливают на смесь льда и 2 М раствора NH3, отделяют метиленхлоридную фазу, а водную фазу экстрагируют дважды СН2Сl2. Объединенные метиленхлоридные порции сушат (МgSO4), фильтруют и растворитель упаривают в вакууме, получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - 25% этилацетат в гексане, затем 50% этилацетата в гексане) получают 0,83 г (98%) названного соединения в виде смолы.
[α]
в) (R)-3-(N-Циклобутил-N-н-пропиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N-Циклобутил-N-н-пропиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран (0,80 г, 2,86 ммоля) и коллидин (0,53 мл, 4,0 ммоля) растворяют в безводном CH2Cl2 (30 мл) и охлаждают до -40oС. Добавляют по каплям ангидрид трифторметансульфокислоты (0,60 мл, 3,6 ммоля) и дают реакционной массе нагреться до температуры окружающей среды, по достижении температуры 0oС реакция заканчивается. Реакционную массу разбавляют метиленхлоридом и промывают насыщенным водным раствором NаНСО3, водную фазу дважды экстрагируют метиленхлоридом. Объединенные метиленхлоридные порции сушат (МgSO4), фильтруют и упаривают в вакууме, получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - метиленхлорид) получают 1,01 г (выход 86%) названного соединения в виде прозрачного масла.
[α]
г) Метиловый эфир (R)-3-(N-Циклобутил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты
(R)-3-(N-Циклобутил-N-н-пропиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран (1,00 г, 2,43 ммоля) растворяют в смеси ДМФА/метанол (6:2, 20 мл) и затем дегазируют и пропускают монооксид углерода (3 раза). При небольшом избыточном давлении монооксида углерода добавляют ацетат палладия (II) (18 мг), 1,3-бис(дифенилфосфино)пропан (25 мг) и триэтиламин (0,75 мл, 5,3 ммоля) и реакционную смесь дегазируют и снова подвергают воздействию монооксида углерода. Далее реакционную смесь нагревают до 70oС в атмосфере монооксида углерода при интенсивном перемешивании в течение 6 часов. Реакционной массе дают охладиться и в вакууме упаривают растворитель. Остаток забирают в 2 М раствор NН3 и затем дважды экстрагируют диэтиловым эфиром. Объединенные эфирные порции сушат (МgSO4), фильтруют и упаривают в вакууме. Получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - 15% этилацетата в гексане) получают 0,73 мг (выход 94%) названного соединения в виде прозрачного масла.
[α]
д) (R)-5-Карбамоил-3-(N-циклобутил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран
Метиловый эфир (R)-3-(N-Циклобутил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты (0,71 мг, 2,21 ммоля) кипятят с обратным холодильником с 6 М раствором НСl (30 мл) в течение 3,5 часа. Раствор охлаждают, упаривают досуха в вакууме, добавляют безводный толуол и растворитель упаривают в вакууме (4 раза).
К полученному белому твердому продукту добавляют тионилхлорид (20 мл) и раствор перемешивают при комнатной температуре в течение ночи. Избыток тионилхлорида упаривают в вакууме, добавляют безводный толуол и в вакууме упаривают растворитель.
Хлорангидрид растворяют в CH2Cl2 (30 мл) и по каплям добавляют к охлажденному (ледяная баня) раствору концентрированного NН3 (30 мл). Реакционную массу перемешивают при комнатной температуре в течение 40 минут. Метиленхлоридную фазу отделяют, а водную фазу экстрагируют СН2Сl2 (3 раза). Объединенные метиленхлоридные порции сушат (MgSO4), фильтруют и упаривают в вакууме, получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - этилацетат) получают 622 мг (выход 92%) белого полукристаллического продукта, часть которого перекристаллизовывают из смеси этилацетата и гексана, получают легкий белый твердый продукт. Т.пл. 107-9oС.
[α]
Пример 6.
(R)-5-Карбамоил-3-(N-циклобутил-N-изопропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран
а) (R)-8-фтор-3-(N-изопропиламино)-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-Амино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран (1,62 г, 8,21 ммоля) растворяют в безводном метаноле (20 мл) и к раствору добавляют ацетон (6,0 мл, 82,1 ммоля). Реакционную массу охлаждают (ледяная баня) и затем добавляют цианборгидрид натрия (0,92 г, 14,8 ммоля), с помощью уксусной кислоты доводят рН до 4-5 и перемешивают при комнатной температуре в течение ночи. Растворитель упаривают в вакууме, остаток забирают в 2 М раствор NН3 и затем экстрагируют трижды диэтиловым эфиром. Объединенные эфирные порции сушат (MgSO4), фильтруют и растворитель упаривают в вакууме, получают сырой остаток, который используют без дополнительной обработки в следующей реакции.
Масс-спектр (газовая хроматография) (70 эВ) М=239 (81%).
б) (R)-3-(N-Циклобутил-N-изопропиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
(R)-8-Фтор-3-(N-изопропиламино)-5-метокси-3,4-дигидро-2Н-1-бензопиран (1,96 г, 8,19 ммоля) растворяют в безводном метаноле (20 мл) и к раствору добавляют циклобутанон (6,1 мл, 81,9 ммоля). Реакционную массу охлаждают (ледяная баня) и добавляют цианборгидрид натрия (2,0 г, 16,4 ммоля), с помощью уксусной кислоты доводят рН до 4-5, добавляют молекулярные сита 3А и смесь перемешивают при комнатной температуре в течение ночи. Через 24 часа рН снова доводят до 4-5 и перемешивают реакционную массу в течение 3 дней. Реакционную массу фильтруют, растворитель упаривают в вакууме, остаток забирают в 2 М раствор NН3 и затем экстрагируют трижды диэтиловым эфиром. Объединенные эфирные порции сушат (МgSO4), фильтруют и растворитель упаривают в вакууме, получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - 10% этилацетата в гексане) получают 1,60 г (выход 77%) названного соединения в виде прозрачного масла.
[α]
в) (R)-3-(N-Циклобутил-N-изопропиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран
Гидрохлорид (R)-3-(N-циклобутил-N-изопропиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопирана (1,76 г, 5,34 ммоля) растворяют в безводном СН2Сl2 (45 мл) и охлаждают до -40oС. К полученному раствору по каплям добавляют ВВr3 (1,3 мл, 13,4 ммоля), растворенного в безводном CH2Cl2 (7 мл). Охлаждающую баню убирают и через два часа выдерживания при комнатной температуре реакция завершается.
Реакционную массу выливают на смесь льда и 2 М раствора NН3, отделяют метиленхлоридную фазу, а водную фазу экстрагируют дважды СН2Сl2. Объединенные метиленхлоридные порции сушат (МgSО4), фильтруют и растворитель упаривают в вакууме, получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - 30% этилацетата в гексане) получают 1,46 г (выход 98%) названного соединения в виде смолы.
[α]
Хлористоводородную соль получают при растворении чистого основания в эфире и добавления по каплям избытка эфирного раствора НСl. Соль промывают диэтиловым эфиром, получают белый твердый продукт. Т.пл. 120oС (спекание).
г) (R)-3-(N-Циклобутил-N-изопропиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N-Циклобутил-N-изопропиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран (1,36 г, 4,87 ммоля) и коллидин (0,90 мл, 6,8 ммоля) растворяют в безводном CH2Cl2 (50 мл) и охлаждают до -40oС. Добавляют по каплям ангидрид трифторметансульфокислоты (1,05 мл, 6,1 ммоля) и дают реакционной массе нагреться до температуры окружающей среды, по достижении температуры 0oС реакция заканчивается. Реакционную массу разбавляют метиленхлоридом и промывают насыщенным водным раствором NaHCO3, водную фазу дважды экстрагируют метиленхлоридом. Объединенные метиленхлоридные порции сушат (МgSO4), фильтруют и упаривают в вакууме, получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - 70% гексана в метиленхлориде) получают 1,67 г (выход 83%) названного соединения в виде светло-желтого масла.
[α]
д) Метиловый эфир (R)-3-(N-Циклобутил-N-изопропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты
(R)-3-(N-Циклобутил-N-изопропиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран (1,65 г, 4,01 ммоля) растворяют в смеси ДМФА/метанол (6:2, 30 мл) и затем дегазируют и пропускают монооксид углерода (3 раза). При небольшом избыточном давлении монооксида углерода добавляют ацетат палладия (II) (30 мг), 1,3-бис(дифенилфосфино)пропан (55 мг) и триэтиламин (1,25 мл, 8,8 ммоля) и реакционную смесь дегазируют и снова подвергают воздействию монооксида углерода. Далее реакционную смесь нагревают до 70oС в атмосфере монооксида углерода при интенсивном перемешивании в течение 6 часов. Реакционной массе дают охладиться и в вакууме упаривают растворитель. Остаток забирают в 2 М раствор NН3 и затем дважды экстрагируют диэтиловым эфиром. Объединенные эфирные порции сушат (MgSO4), фильтруют и упаривают в вакууме. Получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - 8% этилацетата в гексане) получают 1,18 мг (выход 94%) названного соединения в виде прозрачного масла.
[α]
e) (R)-5-Карбамоил-3-(N-циклобутил-N-изопропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран
Метиловый эфир (R)-3-(N-Циклобутил-N-изопропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты (1,16 мг, 3,61 ммоля) кипятят с обратным холодильником с 6 М раствором НС1 (30 мл) в течение 3,5 часа. Раствор охлаждают, упаривают досуха в вакууме, добавляют безводный толуол и растворитель упаривают в вакууме (4 раза).
К полученному белому твердому продукту добавляют тионилхлорид (35 мл) и раствор перемешивают при комнатной температуре в течение ночи. Избыток тионилхлорида упаривают в вакууме, добавляют безводный толуол и растворитель упаривают в вакууме.
Хлорангидрид растворяют в CH2Cl2 (50 мл) и по каплям добавляют к охлажденному (ледяная баня) раствору концентрированного NН3 (50 мл). Реакционную массу перемешивают при комнатной температуре в течение 40 минут. Метиленхлоридную фазу отделяют, а водную фазу экстрагируют CH2Cl2 (3 раза). Объединенные метиленхлоридные порции сушат (МgSO4), фильтруют и упаривают в вакууме, получают сырой остаток. После хроматографирования на двуокиси кремния (элюент - этилацетат) получают 1,06 г (выход 95%) белой пены. Пену перекристаллизовывают из смеси метиленхлорида и гексана, получают легкий белый твердый продукт. Т.пл. 127,8-128,4oС.
[α]
Хлористоводородную соль получают при растворении чистого основания в эфире и добавления по каплям избытка эфирного раствора НСl. Соль промывают диэтиловым эфиром, получают белый твердый продукт. Т.пл. 120oС (спекание).
Пример 7.
(R)-5-Карбамоил-3-(N-циклопентил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран
а) (R)-3-(N-Циклопентиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
К раствору (R)-3-Амино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопирана (2,5 г, 12 ммолей), циклопентанона (3,3 г, 36 ммолей) и НОАс (0,7 г, 12 ммолей) в метаноле (25 мл) при комнатной температуре добавляют порциями NaCNBH3 (2,5 г, 40 ммолей). Раствор перемешивают при комнатной температуре в течение ночи, получают количественный выход названного соединения. Масс-спектр (газовая хроматография) (70 эВ) М= 265 (30%).
б) (R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
К раствору (R)-3-(N-циклопентиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопирана в метаноле (25 мл) добавляют пропаналь (2 г, 36 ммолей) и NаCNВН3 (2 г, 40 ммолей). Раствор перемешивают в течение ночи и получают названное соединение с выходом 97% (данные ГХ). Растворитель упаривают в вакууме, получают сырой остаток, который экстрагируют и выделяют 3,7 г названного соединения в виде бесцветного масла. Масс-спектр (газовая хроматография) (70 эВ) М=307 (40%).
в) (R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран
Хлористоводородную соль (R)-3-(N-циклопентил-N-н-пропиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопирана получают при добавлении избытка эфирного раствора НСl к эфирному раствору основания. Растворитель упаривают в вакууме, остаток растворяют в 48%-ном водном НВr (50 мл). Полученный раствор перемешивают при 120oС в течение 1,5 часа, нейтрализуют путем осторожного добавления концентрированного аммиака, экстрагируют и выделяют коричневое масло, которое фильтруют через слой двуокиси кремния (элюент - этилацетат). Выделяют названное соединение (3,7 г) в виде светло-желтого масла.
Масс-спектр (газовая хроматография) (70 эВ) М=293 (40%).
г) (R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран растворяют в диэтиловом эфире (100 мл). Добавляют триэтиламин (3 г, 30 ммолей) и полученную смесь охлаждают до -20oС. Затем добавляют по каплям (5 минут) ангидрид трифторметансульфокислоты (4,2 г, 15 ммолей), растворенный в диэтиловом эфире (20 мл). После перемешивания в течение 30 минут темно-коричневую смесь выливают в воду. Органический слой отделяют. После быстрого хроматографирования (элюент - этилацетат) получают 3,7 г (выход 69%) названного соединения в виде желтого масла. Масс-спектр (газовая хроматография) (70 эВ) М=425 (10%).
д) Метиловый эфир (R)-3-(N-циклопентил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты
(R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран (3,7 г, 8,7 ммоля), ДМФА (50 мл), триэтиламин (2,5 г, 25 ммолей), метанол (4 г, 130 ммолей), ацетат палладия (II) (100 мг, 0,45 ммоля) и 1,3-бис(дифенилфосфино)пропан (200 мг, 0,48 ммоля) помещают в круглодонную колбу. Полученный раствор перемешивают при 75oС в атмосфере монооксида углерода в течение 4 часов. После упаривания в вакууме и быстрого хроматографирования выделяют 2,5 г (выход 86%) названного соединения в виде бесцветного масла. Масс-спектр (газовая хроматография) (70 эВ) М=335 (20%).
е) (R)-5-Карбамоил-3-(N-циклопентил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран
Метиловый эфир (R)-3-(N-Циклопентил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты (1,4 мг, 4 ммоля) гидролизуют (кипятят с обратным холодильником с 6 М раствором НСl в течение 2 часов), растворитель упаривают. Сырую кислоту обрабатывают SOCl2 (комнатная температура, 5 минут), получают хлорангидрид, который после удаления избытка SOCl2 в вакууме добавляют к концентрированному аммиаку, получают амид. Выделяют сырой продукт и подвергают быстрой хроматографии. Хлористоводородную соль получают путем добавления избытка эфирного раствора НСl к эфирному раствору чистого основания, выделяют названное соединение (0,5 г, выход 35%) в виде белого кристаллического продукта. Т.пл. 85oС (разл.).
[α]
Пример 8.
(R)-5-Карбамоил-3-(N-циклогексил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран
а) (R)-3-(N-Циклогексиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
К раствору (R)-3-Амино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопирана (0,45 г, 2,2 ммоля), циклогексанона (0,7 г, 7,2 ммоля) и НОАс (0,14 г, 2,3 ммоля) в метаноле (25 мл) при комнатной температуре добавляют порциями NaCNBH3 (0,5 г, 8 ммолей). Раствор перемешивают при комнатной температуре в течение ночи, получают количественный выход названного соединения. Масс-спектр (газовая хроматография) (70 эВ) М=279 (30%).
б) (R)-3-(N-Циклогексил-N-н-пропиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
К раствору (R)-3-(N-циклогексиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопирана в метаноле (25 мл) добавляют пропаналь (1,3 г, 23 ммоля) и NаСNВН3 (0,15 г, 2,3 ммоля). Раствор перемешивают в течение ночи и получают названное соединение с выходом 97% (данные ГХ). Растворитель упаривают в вакууме, получают сырой остаток, который экстрагируют и выделяют 0,7 г названного соединения в виде бесцветного масла. Масс-спектр (газовая хроматография) (70 эВ) М=321 (40%).
в) (R)-3-(N-Циклогексил-N-н-пропиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран
Хлористоводородную соль (R)-3-(N-циклогексил-N-н-пропиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопирана получают при добавлении эфирного раствора НСl к эфирному раствору основания. Растворитель упаривают в вакууме, остаток растворяют в 48%-ном водном НВr (20 мл). Полученный раствор перемешивают при 120oС в течение 1,5 часа, нейтрализуют путем осторожного добавления концентрированного аммиака и экстрагируют. Выделенное коричневое масло фильтруют через слой двуокиси кремния (элюент - этилацетат). Выделяют названное соединение (0,6 г) в виде светло-желтого масла. Масс-спектр (газовая хроматография) (70 эВ) М=307 (40%).
г) (R)-3-(N-Циклогексил-N-н-пропиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N-Циклогексил-N-н-пропиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран растворяют в диэтиловом эфире (30 мл). Добавляют триэтиламин (0,8 г, 8 ммолей) и полученную смесь охлаждают до -20oС. Затем добавляют по каплям (5 минут) ангидрид трифторметансульфокислоты (0,8 г, 28 ммоля), растворенный в диэтиловом эфире (10 мл). После перемешивания в течение 30 минут темно-коричневую смесь выливают в воду. Органический слой отделяют. После быстрого хроматографирования (элюент - этилацетат:гексан, 1:1) получают 0,8 г названного соединения в виде желтого масла. Масс-спектр (газовая хроматография) (70 эВ) М=439 (20%).
д) Метиловый эфир (R)-3-(N-циклогексил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты
(R)-3-(N-Циклогексил-N-н-пропиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран (0,8 г, 1,8 ммоля) (4), ДМФА (30 мл), триэтиламин (0,5 г, 5 ммолей), метанол (0,8 г, 13 ммолей), ацетат палладия (II) (30 мг, 0,14 ммоля) и 1,3-бис(дифенилфосфино)пропан (60 мг, 0,14 ммоля) помещают в круглодонную колбу. Полученный раствор перемешивают при 75oС в атмосфере монооксида углерода в течение 4 часов. После упаривания в вакууме и быстрого хроматографирования выделяют 0,6 г (выход 76%) названного соединения в виде бесцветного масла. Масс-спектр (газовая хроматография) (70 эВ) М=349 (30%).
е) (R)-5-Карбамоил-3-(N-циклогексил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран
Метиловый эфир (R)-3-(N-Циклогексил-N-н-пропиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты (5) (0,6 мг, 1,7 ммоля) подвергают щелочному гидролизу (кипятят с обратным холодильником с 2%-ным раствором КОН EtOH в течение 2 часов). Растворитель упаривают в вакууме и сырую кислоту обрабатывают SOCl2 (комнатная температура, 5 минут), получают хлорангидрид, который после удаления в вакууме избытка SOCl2 добавляют к концентрированному аммиаку, получают амид. Выделяют сырой продукт и очищают с помощью быстрой хроматографии. Хлористоводородную соль получают путем добавления избытка эфирного раствора НСl к эфирному раствору чистого основания, выделяют названное соединение (86 мг, выход 14%) в виде белого кристаллического продукта. Т.пл. 75oС (разл.).
[α]
Пример 9.
(R)-5-Карбамоил-3-(N-циклопентил-N-цилобутиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран
а) (R)-3-(N-Циклопентиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
К раствору (R)-3-Амино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопирана (0,7 г, 3,4 ммоля), циклопентанона (0,7 г, 8,3 ммоля) и НОАс (0,2 г, 3,5 ммоля) в метаноле (25 мл) при комнатной температуре добавляют порциями NаСNВН3 (0,7 г, 10 ммолей). Раствор перемешивают при комнатной температуре в течение ночи, метанол упаривают. Полученный остаток растворяют в этилацетате и промывают водой. Органический слой сушат Na2SO4, растворитель упаривают, получают 0,9 г (выход 100%) названного соединения в виде бесцветного масла. Данные ГХ указывают на чистоту 99,6%.
Масс-спектр (газовая хроматография) (70 эВ) М=265 (30%).
б) (R)-3-(N-Циклопентил-N-циклобутиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран
К раствору (R)-3-(N-циклопентиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопирана (0,9 г, 3,4 ммоля), НОАс (0,22 г, 3,6 ммоля) и циклобутанона (2 г, 30 ммолей) в метаноле (25 мл) при комнатной температуре порциями добавляют NаСNВН3(1 г, 16 ммолей). После перемешивания в течение нескольких дней данные ТСХ указывают на образование 37% продукта реакции. Доводят рН до 4-5 (НОАс) и добавляют дополнительное количество циклобутанона (1 г, 15 ммолей). После перемешивания в течение еще 6 дней наблюдается конверсия 64% (данные ТСХ). Растворитель упаривают в вакууме, получают сырой остаток, который экстрагируют и подвергают быстрой хроматографии ('люент - этилацетат: петр. эфир, 1: 1), выделяют 0,53 г (выход 53%) названного соединения в виде бесцветного масла. Масс-спектр (газовая хроматография) (70 эВ) М=319 (3%).
в) (R)-3-(N-Циклопентил-N-циклобутиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N-циклопентил-N-циклобутиламино)-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран (0,53 г, 1,6 ммоля) растворяют в 47%-ном растворе НВr (15 мл) и перемешивают при 120oС в течение 1,5 часа. Раствор охлаждают путем добавления льда и подщелачивают 14 М раствором аммиака. После экстрагирования получают 0,5 г названного соединения в виде светло-коричневого масла. В ИК-спектре присутствует типичная полоса поглощения ОН-группы при 3654 см-1.
г) (R)-3-(N-Циклопентил-N-циклобутиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран
(R)-3-(N-Циклопентил-N-циклобутиламино)-8-фтор-5-гидрокси-3,4-дигидро-2Н-1-бензопиран растворяют в смеси диэтилового эфира и метиленхлорида (90+10 мл, 20 мл) и добавляют триэтиламин (0,7 г, 7 ммолей). Полученную смесь охлаждают до -20oС. Затем добавляют по каплям (5 минут) ангидрид трифторметансульфокислоты (0,85 г, 3 ммоля), растворенный в диэтиловом эфире (10 мл). После перемешивания в течение 30 минут темно-коричневую смесь выливают в воду. Растворитель упаривают. Остаток растворяют в гексане и обрабатывают активированным углем. Фильтруют через целиты и упаривают растворитель. Получают 0,67 г названного соединения в виде бесцветного масла. Масс-спектр (газовая хроматография) (70 эВ) М=437 (1%).
д) Метиловый эфир (R)-3-(N-циклопентил-N-циклобутиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты
(R)-3-(N-Циклопентил-N-циклобутиламино)-8-фтор-5-трифторметилсульфонилокси-3,4-дигидро-2Н-1-бензопиран (0,67 г, 1,5 ммоля) (4), ДМФА (20 мл), триэтиламин (0,6 г, 6 ммолей), метанол (0,8 г, 12,7 ммоля), ацетат палладия (II) (22 мг, 0,1 ммоля) и 1,3-бис(дифенилфосфино)пропан (44 мг, 0,1 ммоля) помещают в круглодонную колбу. Полученный раствор перемешивают при 75oС в атмосфере монооксида углерода в течение 4 часов. После упаривания в вакууме растворителя, остаток растворяют в диэтиловом эфире и обрабатывают активированным углем. После упаривания растворителя получают 380 мг названного соединения в виде бесцветного масла. Масс-спектр (газовая хроматография) (70 эВ) М=347 (3%).
е) (R)-5-Карбамоил-3-(N-циклопентил-N-циклобутиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран
Метиловый эфир (R)-3-(N-Циклопентил-N-циклобутиламино)-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоновой кислоты (1,4 мг, 4 ммоля) подвергают гидролизу (кипятят с обратным холодильником с 6 М НСl в течение 2 часов). Растворитель упаривают в вакууме. После сушки на воздухе при комнатной температуре в течение ночи сырой гидрохлорид аминокислоты обрабатывают SOCl2 (комнатная температура, 5 минут), получают хлорангидрид, который после удаления избытка SOCl2 растворяют в CH2Cl2 и добавляют к концентрированному аммиаку, получают амид. Выделяют сырой продукт и очищают с помощью быстрой хроматографии, получают 220 мг бесцветного масла, которое со временем кристаллизуется.
Перекристаллизация из смеси диэтилового эфира и гексана дает названное соединение в виде белого кристаллического продукта. Выход 110 мг, т.пл. 138-140oС.
[α]
Фармакология
Методы испытаний
(I) Оценка связывания 5-HT1A рецепторов
Для оценки сродства к 5-HT1A рецептору можно использовать описанный ниже метод оценки на головном мозге крыс.
Получение тканей. Кору головного мозга и гиппокамп крыс Sprague-Dawley рассекают и гомогенизируют в течение 10 сек с помощью Ultra-Turrax (Janke and Kunkel, Staufer, ФРГ) в 15 мл охлажденного льдом 50 мМ буфера Tris-HCl, pH 7,5, содержащего 4,0 мМ CaCl2: и 5,7 мМ аскорбиновой кислоты ("Буфер А"). После центрифугирования в течение 12,5 мин при 17000 об/мин (38000 g) в центрифуге Бекмана с охлажденным ротором JA-17 (Beckman, Palo Alto, CA, США), полученные гранулы повторно гомогенизируют и центрифугируют. Каждую гранулу суспендируют в 5 мл охлажденного льдом 0,32 М раствора сахарозы и гомогенизируют в течение 5 сек. Гомогенизированные образцы замораживают при -70oС. При использовании их разбавляют буфером А из расчета 8 мг ткани на 1 мл и гомогенизируют в течение 10 сек. Гомогенаты тканей выдерживают в течение 10 мин при 37oС и затем дополняют 10 мкМ паргилина и снова выдерживают в течение 10 минут.
Методика оценки связывания соответствует методике, описанной Peroutka, J.Neurochem. 47, 529-540 (1986). По этой методике фактически измеряется способность конкретной конкурирующей молекулы ингибировать связывание 3H-8-OH-DPAT с рецепторами 5-HT1A. Инкубационная смесь (2 мл) содержит 3H-8-OH-DPAT (0,25-0,8 нМ), необходимую концентрацию испытуемого (конкурирующего) соединения и 5 мг/мл гомогената ткани в 50 мМ буфера Tris-HCl, pH 7,5, содержащего 4,0 мМ CaCl2 и 5,7 мМ аскорбиновой кислоты. Анализируют 6 различных концентраций 3H-8-OH-DPAT. Оценку связывания начинают путем добавления гомогената ткани и последующего инкубирования при 37oС в течение 10 мин. Инкубационные смеси фильтруют через стеклянные фильтры Whatman GF/B со сборщиком клеток Брандела (Gaithersburg, MD, США). Фильтры дважды промывают 5 мл охлажденного льдом буфера 50 мМ Tris-HCl, pH 7,5, и подсчитывают с 5 мл Ultima GoldTM (Packard) с помощью сцинтилляционного счетчика Beckman LS 3801. Неспецифическое связывание измеряют при добавлении к реакционной смеси 10 мкМ 5-НТ. Данные по связыванию обрабатывают с помощью компьютерного нелинейного метода наименьших квадратов (Munson and Rodbard, Anal.Biochem. 107, 220-239, 1980).
(ii) Антагонизм 8-OH-DPAT, индуцированный снижением синтеза 5-НТ через блокаду пресинаптических 5-НТ рецепторов.
Скорость синтеза 5-гидрокситриптамина (5-НТ: серотонин) и допамина/норадреналина (ДА/НА) измеряют по накоплению 5-гидрокситриптофана (5-ГТФ) и 3,4-дигидроксифенилаланина (ДОФА), соответственно в течение 30 минут после ингибирования декарбоксилазы ароматической L-аминокислоты с помощью м-гидроксибензилгидразина (х2НСl) (100 мг/кг а.и.), поставляемого фирмой Sigma. Испытуемое соединение вводят за 75 минут до, 8-OH-DPAT за 60 минут до и м-гидроксибензилгидразин (х2НСl) за 30 минут до умерщвления крыс. Область головного мозга, которую необходимо изучить, быстро рассекают, замораживают в сухом льду и хранят при -70oС до проведения анализа.
ДОФА, 5-ГТФ и их метаболиты экстрагируют из тканей головного мозга с помощью перхлорной кислоты, содержащей внутренний стандарт (Изопреналин). Надосадочную жидкость от гомогената головного мозга вводят в жидкостную хроматографирующую систему, содержащую предварительную колонку и аналитическую колонку. Путем калориметрического окисления определяют катехол- и индоламины.
(iii) Антагонизм индуцированного ингибитором острого повторного усвоения 5-НТ (SSRI), снижения оборота 5-НТ с помощью селективных антагонистов 5-HT1A.
Животные
Самцов крыс Sprague-Dawley (B&K International АВ, Sollentuna, Sweden), весом 180-220 г, размещают по 5 штук на клетку в условиях с контролируемой температурой (21oС) и влажностью с 12-ти часовым циклом свет/темнота (освещение на 6 ч после полудня). Вода и пища свободно доступны. При этих условиях животных выдерживают по меньшей мере 4 дня перед проведением опыта. Соединения вводя перорально (п/о) или подкожно (п/к).
Оборот 5-НТ
Группе из 5 крыс вводят 5-HT1A антагонист за 15 минут до введения ингибитора повторного усвоения 5-НТ (например, норзимелдинa=(Z)-3-(4-бромфенил)-N-метил-3-(3-пиридил)-аллиламин, моногидрат дигидрохлорида). м-Гидроксибензилгидразин х2НСl (NSD 1015) вводят через 30 минут и крыс умерщвляют путем гильотинирования через 30 минут после введения ингибитора повторного усвоения (NSD 1015). Головной мозг быстро удаляют и рассеченные участки немедленно замораживают сухим льдом. Образцы до проведения анализа хранят при -70oС.
На различных участках головного мозга определяют эндогенные соединения, 5-НТ и 5-HT1A, с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) с электрохимическим детектором в соответствии со способом, описанным Magnusson, J. Chromatogr. 1980; 221:237-247. В качестве подвижной фазы используют смесь 0,1 М фосфатный буфер (рН 2,5): метанол: ацетонитрил, 89:9:2 (об./об. ), содержащую 1 нМ октилсульфата. Замороженные образцы взвешивают и гомогенизируют в 0,1 М перхлорной кислоте, содержащей 2,5 нМ бисульфита натрия, 1 нМ этилендиаминтетрауксусной кислоты (ЭДТУК). Надосадочную жидкость вводят непосредственно на колонку Supelcosil C18 (3 мкМ), присоединенную к детектору (ESA Coulochem 5100F), установленному на 0,05/0,30 В. Путем калориметрического окисления определяют катехол- и индоламины.
Фиг. 1 показывает, что блокирование соматодендритных рецепторов 5-HT1A с помощью селективного антагониста, такого как (R)-3-(N-циклопентил-N-н-пропиламино)-8-фтор-5-N-метилкарбамоил-3,4-дигидро-2Н-1-бензопирана (пример 1, NDL 249), зависимо от дозы увеличивает антидепрессантную эффективность блокаторов повторного усвоения 5-НТ, за счет обеспечения возможности серптонинергических окончаний высвобождать больше 5-НТ и тем самым создания основы для быстрого наступления терапевтического действия.

Изобретение относится к области медицины и касается комбинации первого компонента (а), который представляет собой ингибитор повторного усвоения 5-НТ со вторым компонентом (б), который является селективным 5-НТ1А антагонистом формулы (I), где R1 представляет собой н-пропил или циклобутил; R2 представляет собой изопропил, трет-бутил, циклобутил, циклопентил или циклогексил; R3 представляет собой атом водорода; R4 представляет собой атом водорода или метил; в форме (R)-энантиомера в виде свободного основания или его фармацевтически приемлемых солей, к его получению, к фармацевтическим препаратам для лечения аффективных нарушений, содержащим указанное сочетание. Описанная комбинация более эффективна при лечении аффективных состояний. 7 c. и 12 з.п. ф-лы, 1 ил.
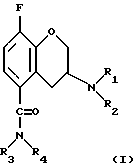

в которой
R1 представляет н-пропил или циклобутил;
R2 представляет изопропил, трет-бутил, циклобутил, циклопентил или циклогексил;
R3 представляет атом водорода;
R4 представляет атом водорода или метил;
причем указанный 5-НТ1А антагонист находится в форме (R)-энантиомера и указанные компоненты (а) и (б) находятся в форме свободных оснований или их фармацевтически приемлемых солей.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Samuel Perry et al | |||
| Pirdolol onduces a repid impovemnt of depressed patients treated with serotonin reuptake inhibitors | |||
| Arch | |||
| Gen | |||
| Psychiatry | |||
| Прибор для охлаждения жидкостей в зимнее время | 1921 |
|
SU1994A1 |
| Крылов Ю.Ф | |||
| Энциклопедия лекарств | |||
| - М., 2000, с.990. | |||
