Область изобретения
Данное изобретение относится к некоторым новым производным бензойной кислоты, способам получения таких соединений, их применению в лечении клинических состояний, ассоциированных с резистентностью к инсулину, способам их терапевтического применения и фармацевтическим композициям, которые их содержат.
Предшествующий уровень техники
Синдром резистентности к инсулину (СРИ), включающий в себя сахарный диабет типа 2, относится к кластеру проявлений, включающих в себя резистентность к инсулину с сопутствующей гиперинсулинемией, возможный сахарный диабет типа 2, артериальную гипертензию, центральное (висцеральное) ожирение, дислипидемию, наблюдаемую как ненормальные уровни липопротеинов, в типичных случаях характеризуемые повышенными ЛОНП (липопротеины очень низкой плотности), малыми плотными частицами ЛНП и сниженными концентрациями ЛВП (липопротеины высокой плотности) и сниженным фибринолизом.
Недавние эпидемиологические исследования документировали то, что индивиды с резистентностью к инсулину подвержены сильно увеличенному риску сердечно-сосудистой заболеваемости и смертности, особенно страдая от инфаркта миокарда и инсульта. Состояния, имеющие отношение к атеросклерозу, при сахарном диабете типа 2 причиняют до 80% всех смертей.
В клинической медицине имеет место осознание необходимости увеличения чувствительности к инсулину у пациентов, страдающих от СРИ, и, таким образом, коррекции дислипидемии, которую считают обуславливающей ускоренное прогрессирование атеросклероза. Однако в настоящее время она не является заболеванием с универсальным четким определением.
Соединения, которые являются модуляторами рецепторов, активируемых пролифераторами пероксисом (PPAR, обзор по PPAR см. в T.M.Willson et al, J Med Chem 2000, Vol 43, 527), эффективны в лечении состояний, ассоциированных с резистентностью к инсулину.
Неожиданно найдена серия соединений, которые являются модуляторами активности PPARα и/или PPARγ.
Описание изобретения
Данное изобретение предлагает соединение формулы I,
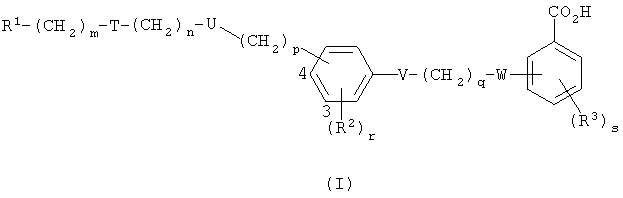
где R1 представляет собой арил, возможно замещенный гетероциклической группой, или гетероциклическую группу, возможно замещенную арилом, причем каждый арил или гетероциклическая группа возможно замещен(а) одной или более чем одной из нижеследующих групп:
С1-6алкильная группа;
С1-6ацильная группа;
арилС-1-балкил, причем алкильная, арильная или алкиларильная группа возможно замещена одним или более чем одним из Rb;
галоген,
-CN и NO2;
-NRcOORa;
-NRcCORa;
-NRcRa;
-NRcSO2Rd;
-NRcCONRkRc;
-NRcCSNRaRk;
-ORa;
-OSO2Rd;
-SO2Rd;
-SORd;
-SRc;
-SO2NRaRf;
-SO2ORa;
-CONRcRa;
-OCONRfRa;
где Ra представляет собой Н, С1-6алкильную группу, арильную или арилС1-6алкильную группу, причем алкильную, арильную или арилС1-6алкильную группу возможно замещает один или более чем один раз Rb, где Rb представляет собой С1-6алкил, арил, арилС1-6алкил, циано, -NRcRd, =O, галогено, -ОН, -SH, -ОС1-4алкил, -Оарил, -ОС1-4алкиларил, -CORc, -SRd, -SORd, или -SO2Rd, где Rc представляет собой Н, С1-4алкил, арил, арилС1-4алкил, и Rd представляет собой С1-4алкил, арил, арилС1-4алкил;
где Rf представляет собой водород, С1-4алкил, С1-4ацил, арил, арилС1-4алкил, и Ra является таким, как определено выше; и
Rk представляет собой водород, С1-4алкил, арил, арилС1-4алкил;
группа -(CH2)m-T-(CH2)n-U-(CH2)p- присоединена либо в 3-й, либо в 4-й позиции в фенильном кольце, как указано цифрами в формуле I, и представляет собой группу, выбранную из одной или более чем одной из нижеследующих: O(СН2)2, O(СН2)3, NC(O)NR4(CH2)2, CH2S(O2)NR5(CH2)2, CH2N(R6)C(O)CH2, (CH2)2N(R6)C(O)(CH2)2, C(O)NR7CH2, C(O)NR7(CH2)2 и CH2N(R6)C(O)CH2O;
V представляет собой О, S, NR8 или одинарную связь;
q представляет собой 1, 2 или 3;
W представляет собой О, S, N(R9)C(O), NR10 или одинарную связь;
R2 представляет собой галогено, С1-4алкильную группу, которая возможно замещена одним или более чем одним атомом фтора, С1-4алкоксильную группу, которая возможно замещена одним или более чем одним атомом фтора, С1-4ацильную группу, арил, арилС1-4алкильную группу, CN или NO2;
r представляет собой 0, 1, 2 иди 3;
R3 представляет собой галогено, С1-4алкильную группу, которая возможно замещена одним или более чем один атом фтора, С1-4алкоксильную группу, которая возможно замещена одним или более чем одним атомом фтора, С1-4ацильную группу, арил, арилС1-4алкильную группу или CN;
s представляет собой 0, 1, 2 или 3; и
R4, R5, R6, R7, R8, R9 и R10 независимо представляют собой Н, С1-10алкильную группу, арильную или арилС1-4алкильную группу или, если m представляет собой 0 и Т представляет собой группу N(R6)C(O) или группу (R5)NS(O2), то R1 и R6 или R1 и R5 вместе с атомом азота, к которому они присоединены, представляют собой гетероарильную группу;
и ее фармацевтически приемлемые соли;
при том условии, что
1) если R1 представляет собой, фенил, возможно замещенный одной или двумя группами, независимо выбранными из галогено, С1-4алкильной группы, которая возможно замещена одним или более чем одним атомом фтора, С1-4алкоксильную группу, которая возможно замещена одним или более чем одним атомом фтора;
m представляет собой 1;
Т представляет собой N(R6)C(O), причем R6 представляет собой С2-8алкильную группу, которую возможно прерывает кислород;
n представляет собой 1;
U отсутствует или представляет собой метилен;
p представляет собой 0;
r представляет собой 0;
V представляет собой О или S;
q представляет собой 1; и
W представляет собой одинарную связь, присоединенную в орто-положении относительно группы карбоновой кислоты;
то s не представляют собой 0; и
2) если R1 представляет собой фенил, возможно замещенный одной или двумя группами, независимо выбранными из галогено, С1-4алкильной группы, которая возможно замещена одним или более чем одним атомом фтора, С1-4алкоксильную группу, которая возможно замещена одним или более чем одним атомом фтора,
m представляет собой 1;
Т представляет собой N(R6)C(O), где R6 представляет собой неразветвленную С2-7алкильную группу;
n представляет собой 1;
U представляет собой О;
р представляет собой 0;
r представляет собой 0 или 1;
и, когда r представляет собой 1, R2 присоединен в 3-м положении и представляет собой ОСН3;
V представляет собой одинарную связь;
q представляет собой 2; и
W представляет собой атом О или S, присоединенный в орто-положении относительно группы карбоновой кислоты;
то s не представляют собой 0.
Примеры C1-6алкила включают в себя метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил и пентил с прямой и разветвленной цепью и гексил, а также циклопропил, циклобутил, циклопентил и циклогексил. Предпочтительными алкильными группами являются метил, этил, пропил, изопропил и трет-бутил.
Если не указано или не обозначено иначе, термин "галоген" должен означать фтор, хлор, бром или йод, предпочтительно фтор.
Если не указано или не обозначено иначе, термин "арил" означает замещенный или незамещенный фенил или сопряженную кольцевую систему, такую как нафтил.
Если не указано или не обозначено иначе, термин "гетероциклическая группа" представляет собой насыщенное, частично насыщенное или ненасыщенное моно- или бициклическое кольцо, содержащее 4-12 атомов, из которых по меньшей мере один атом выбран из азота, серы или кислорода, которые могут, если не указано иначе, быть связанными с углеродом или азотом, причем группа -СН2- может быть возможно замененной на -С(O)- и атом серы в кольце можно быть возможно окисленным с образованием S-оксида(ов). Примерами и подходящими значениями термина "гетероциклическая группа" являются морфолино, пиперидил, пиридил, пиранил, пирролил, имидазолил, тиазолил, индолил, хинолил, изохинолил, тиенил, 1,3-бенздиоксолил, 1,3-диоксоланил, тиадиазолил, пиперазинил, изотиазолидинил, 1,3,4-триазолил, тетразолил, пирролидинил, 2-оксазолидинонил, 5-изоксазолонил, бенз-3-азепинил, 1,4-бенздиоксанил, тиоморфолино, пирролинил, гомопиперазинил, 3,5-диоксапиперидинил, 3-пиразолин-5-онил, тетрагидропиранил, бензимидазолил, бензтиазолил, имидазо[1,2-а]пиридил, пиримидил, пиразинил, пиридазинил, изоксазолил, 4-пиридон, 1-изохинолон, 2-пирролидон, 4-тиазолидон, дигидроизохинол-2(1Н)-ил, 2,3-дигидро-1,5-бензтиазепин-4(5Н)-он. Предпочтительно "гетероциклическая группа" представляет собой пиридил, имидазолил, тиазолил, хинолил, тиенил, 1,3-бенздиоксолил, 1,3-диоксоланил, изотиазолидинил, 1,3,4-триазолил, тетразолил, 2-оксазолидинонил, 5-изоксазолонил, бенз-3-азепинил, гидантоинил, 1,4-бенздиоксанил, тиоморфолино, 3-пиразолин-5-онил, бензимидазолил, бензтиазолил, имидазол[1,2-а]пиридил, пиримидил, пиразинил и 2,3-дигидро-1,5-бензотиазепин-4(5Н)-он.
Далее следуют дополнительные значения R1, Т, U, V, W, R2, R3, m, n, p, q, r и s в соединениях Формулы I. Надо понимать, что такие значения можно использовать с любым из определений, пунктов формулы изобретения или воплощений, определенных здесь выше или ниже.
В первом аспекте R1 представляет собой фенил, который возможно замещен одним или более чем одним из нижеследующего: галогено, гидрокси, С1-4алкильная группа, которая возможно замещена одним или более чем одним атомом фтора, С1-4алкоксильная группа, которая возможно замещена одним или более чем одним атомом фтора, бензилокси, С1-4алкилсульфонилокси группа, фенильная или гетероарильная группа; или R1 представляет собой гетероциклическую группу, которая возможно замещена одним или более чем одним из нижеследующего: галогено, С1-4алкильная группа, которая возможно замещена одним или более чем одним атомом фтора, С1-4алкоксильная группа, которая возможно замещена одним или более чем одним атомом фтора, или фенил, возможно замещенный одним или более чем одним из нижеследующего: галогено, С1-4алкильная группа, которая возможно замещена одним или более чем одним из атомов фтора, С1-4алкоксильная группа, которая возможно замещена одним или более чем одним атомом фтора. Конкретный R1 представляет собой фенил, фурил, пиридил или тиазолил, каждый из которых возможно замещен одним или более чем одним из нижеследующего: галогено (в частности фтор), С1-4алкильная группа, трифторметил, С1-4алкоксильная группа, метансульфонилокси, гидрокси, бензилокси, имидазолил или фенил.
Во втором аспекте группа -(CH2)m-T-(CH2)n-U-(CH2)p присоединена в положении 4 фенильного кольца, как указано цифрами в формуле I, то есть в пара-положении относительно группы V.
В третьем аспекте группа -V-(CH2)q-W- представляет собой группу, выбранную из одного или более чем одного из нижеследующего: ОСН2, SCH2, NHCH2, CH2CH2S или СН2СН2О.
В четвертом аспекте группа -V-(CH2)q-W- представляет собой группу ОСН2.
В пятом аспекте группа -V-(CH2)q-W- присоединена в орто-положении относительно группы карбоновой кислоты.
В шестом аспекте R2 представляет собой галогено, С1-4алкильную группу или С1-4алкоксильную группу и г представляет собой 0 или 1.
В седьмом аспекте s представляет собой 0.
Специалистам должно быть понятно, что некоторые соединения формулы I могут содержать оптически-активный центр и поэтому могут существовать в виде энантиомеров, которые можно разделять как описано ниже. Ожидается, что большая часть активности, если не вся активность, соединения формулы I сосредоточена в одном энантиомере: либо S-, либо R-энантиомере или (+)- или (-)-энантиомере. Энантиомеры, которые являются более активными в анализах, которые описаны ниже, являются предпочтительными формами данного изобретения. Надо понимать, что настоящее изобретение включает в себя все смеси этого активного энантиомера с другими энантиомерами, например рацемическую смесь.
Активные энантиомеры можно изолировать разделением рацемата, например, фракционной кристаллизацией, повторным растворением или ВЭЖХ на хиральной колонке (например, колонки Chiralpak™ AD 250×50). Или же активные энантиомеры можно получать хиральным синтезом из хиральных исходных материалов в условиях, которые не вызовут рацемизацию или эпимеризацию, или дериватизацией хиральным реагентом.
Пролекарственные формы соединений формулы I также образуют часть данного изобретения. Термины "пролекарственная форма" или "пролекарство", использованные в этом описании эквивалентны и включают в себя производные группы карбоновой кислоты, которые в млекопитающем, в частности в человеке, превращаются в группу карбоновой кислоты или ее соль или конъюгат. Следует понимать, что, при отсутствии теоретических ограничений считают, что большая часть активности, ассоциированной с пролекарственными формами, происходит из активности соединения формулы I, в которое превращаются Пролекарственные формы. Пролекарственные формы можно получать рутинными способами вполне в пределах способностей специалиста. В данной области известны различные Пролекарственные формы карбокси. Примеры таких производных, представляющих собой Пролекарственные формы, см. в:
a) Design of Prodrugs, edited by H.Bundgaard, (Elsevier, 1985), и Methods in Enzymology. 42: 309-396, edited by K.Widder, et al. (Academic Press, 1985);
b) A Textbook of Prodrug Design and Development, edited by Krogsgaard-Larsen and H.Bundgaard, Chapter 5 "Design and Application of Prodrugs", by H.Bundgaard p.113-191 (1991);
c) H.Bundgaard, Advanced Drug Delivery Reviews, 8: 1-38 (1992);
d) H.Bundgaard, et al., Journal of Pharmaceutical Sciences, 77: 285 (1988); и
e) N.Kakeya, et al., Chem Pharm Bull, 32: 692 (1984).
Вышеуказанные документы инкорпорированы сюда ссылкой.
Расщепляемые in vivo сложные эфиры представляют собой только один тип пролекарственной формы исходной молекулы.
Соединения формулы I имеют активность как медикаменты. В частности, соединения формулы I являются селективными агонистами либо PPARα, либо PPARγ, в частности PPARα, или являются агонистами PPARα и PPARγ. Термин агонисты при использовании здесь включает в себя частичные агонисты.
Настоящее изобретение предлагает одно или более чем одно из соединений, выбранных из следующего:
3-{[(3-{[(1,1′-бифенил-4-илкарбонил)-амино]-метил)-фенил)-амино]-метил}-бензойная кислота;
2-{[4-(2-оксо-2-{[4-(трифторметил)-бензил]-амино}-этил)-фенокси]-метил}-бензойная кислота;
2-[(3-{2-[бензил(гексил)-амино]-2-оксоэтил}-фенокси)-метил]-бензойная кислота;
2-{[3-(2-оксо-2-{[4-(трифторметил)-бензил]-амино}-этил)-фенокси]-метил}-бензойная кислота;
2-[(4-{3-[[2-(3,4-диметоксифенил)-этил](метил)-амино]-3-оксопропил)-фенокси)-метил]-бензойная кислота;
2-[(4-{2-[({4-метил-2-[4-(трифторметил)-фенил]-1,3-тиазол-5-ил}-карбонил)-амино]-этил}-фенокси)-метил]-бензойная кислота;
2-({4-[2-({[(2,4-дифторфенил)-амино]-карбонил)-амино)-этил]-фенокси}-метил)-бензойная кислота;
2-[(4-{2-[(2-метил-5-фенил-3-фуроил)-амино]-этил}-фенокси)-метил]-бензойная кислота;
2-[(4-(2-[(бензилсульфонил)-амино]-этил}-фенокси)-метил]-бензойная кислота;
2-[(4-{2-[бензил(гексил)-амино]-2-оксоэтил}-2-фторфенокси)-метил]-бензойная кислота;
2-[(4-{2-[бензил(гексил)-амино]-2-оксоэтил}-2-метоксифенокси)-метил]-бензойная кислота;
2-({4-[3-(3,4-дигидроизохинолин-2(1H)-ил)-3-оксопропил]-фенокси}-метил)-бензойная кислота;
2-[(4-{2-[4-(1H-имидазол-1-ил)-фенокси]-этил}-феноксиметил]-бензойная кислота;
2-{[4-(2-{4-[(метилсульфонил)-окси]-фенокси}-этил)-фенокси]-метил)-бензойная кислота;
2-[(3-{2-[4-(бензилокси)-фенокси]-этил}-фенокси)-метил]-бензойная кислота;
2-{[3-(2-{4-[(метилсульфонил)-окси]-фенокси}-этил)-фенокси]-метил}-бензойная кислота;
2-({3-[2-(4-гидроксифенокси)-этил]-фенокси}-метил)-бензойная кислота;
2-[(4-{3-[4-(бензилокси)-фенокси]-пропил}-фенокси)-метил]-бензойная кислота;
2-{[4-(3-{4-[(метилсульфонил)-окси]-фенокси}-пропил)-фенокси]-метил}-бензойная кислота;
2-({4-[3-(4-гидроксифенокси)-пропил]-фенокси}-метил)-бензойная кислота;
2-{[4-(3-{[2-(2-этоксифенил)-этил]-амино}-3-оксопропил)-фенокси]-метил}-бензойная кислота;
2-[(4-{3-[этил(2-пиридин-2-илэтил)-амино]-3-оксопропил}-фенокси)-метил]-бензойная кислота;
2-{[2-(3-{2-[бензил(гексил)-амино]-2-оксоэтокси}-фенил)-этил]-тио}-бензойная кислота;
2-{[4-(2-{гептил[2-(2-метоксифенил)-этил]-амино}-2-оксоэтил)-фенокси]-метил}-бензойная кислота;
2-[(4-{2-[[2-(4-хлорфенил)-этил](гептил)-амино]-2-оксоэтил}-фенокси)-метил]-бензойная кислота;
2-[(4-{2-[гептил(2-фенилэтил)-амино]-2-оксоэтил}-фенокси)-метил]-бензойная кислота;
2-[(4-{2-[этил(2-фторбензил)-амино]-2-оксоэтокси}-фенокси)-метил]-бензойная кислота;
2-[(4-{2-[этил(2-фторбензил)-амино]-2-оксоэтил}-бензил)-окси]-бензойная кислота;
2-[(4-{2-[гептил(2-фенилэтил)-амино]-2-оксоэтил}-бензил)-окси]-бензойная кислота;
2-{2-[4-(2-{изобутил[4-(трифторметил)-бензил]-амино}-2-оксоэтокси)-фенил]-этокси}-бензойная кислота; и
2-[(4-{2-[[2-(4-хлорфенил)-этил](гептил)-амино]-2-оксоэтил}-бензил)-окси]-бензойная кислота
и его фармацевтически приемлемые соли.
Также надо понимать, что некоторые соединения по данному изобретению могут существовать в сольватированных, а также несольватированных формах. Надо понимать, что настоящее изобретение охватывает все такие сольватированные формы. Некоторые соединения по данному изобретению могут существовать как таутомеры. Надо понимать, что настоящее изобретение охватывает все такие таутомеры.
Способы получения
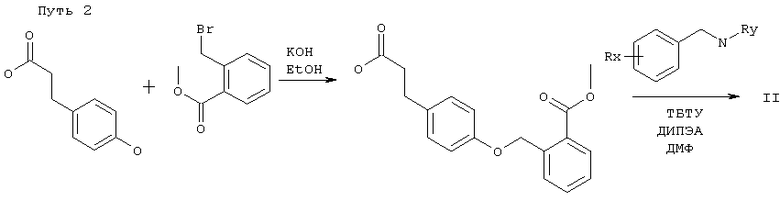
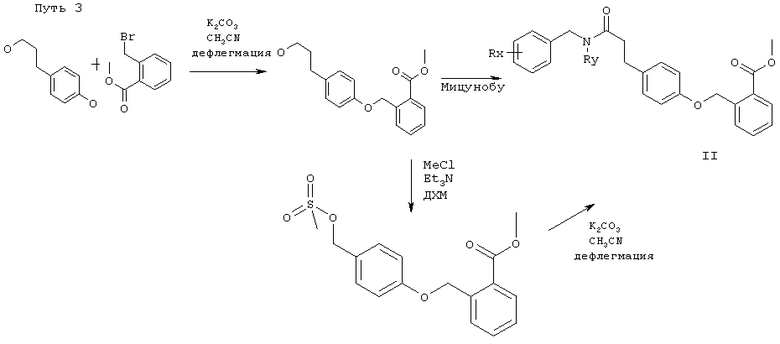
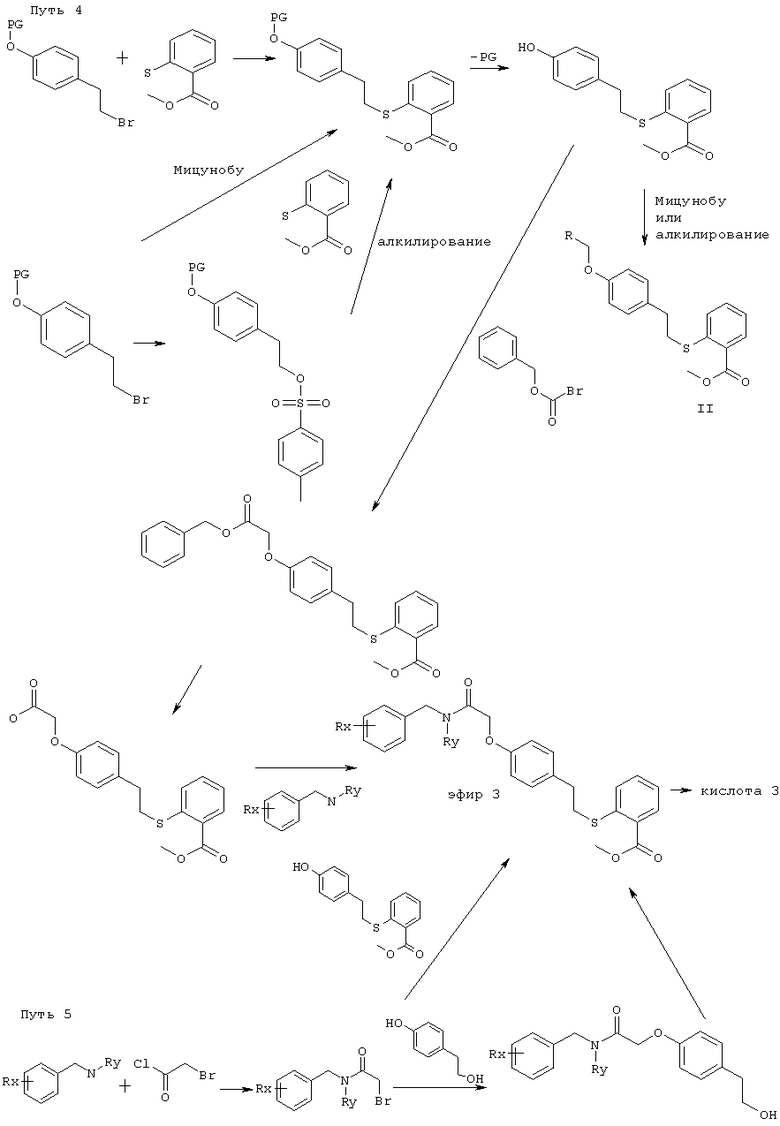
Соединения по этому изобретению могут получать как обрисовано ниже. Однако это изобретение не ограничено этими способами, эти соединения можно также получать как описано для структурно родственных соединений в уровне техники. Взаимодействие можно проводить в соответствии со стандартными процедурами или как описано в экспериментальной секции. Соединения формулы I можно получать взаимодействием соединения формулы II,
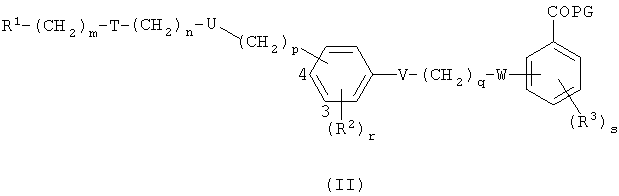
в которой R1, Т, U, V, W, R2, R3, m, n, p, q, r и s являются такими, как определено выше, и PG представляет собой защитную группу для гидроксила карбоксильной группы, как описано в стандартном тексте "Protective Groups in Organic Synthesis", 2nd Edition (1991) Greene и Wuts, с агентом снятия защиты. Защитной группой также может быть смола, такая как смола Вонга (Wang), или смола, представляющая собой хлорид 2-хлотритила. Защитные группы можно удалять в соответствии с методиками, которые хорошо известны специалистам. Одна такая защитная группа является такой, где PG представляет собой группу C1-6алкокси или группу арилалкокси, например бензилокси, такую, что COPG представляет собой сложный эфир. Такие сложный эфиры можно приводить во взаимодействие с гидролизующим агентом, например гидроксидом лития, в присутствии растворителя, например смеси тетрагидрофурана и воды, или гидроксидом калия в C1-3спирте, например метаноле, при температуре в диапазоне 0-200°С или микроволновой радиацией с получением соединения формулы I. Соединения формулы II можно получать в соответствии с одним из нижеследующих путей 1-5. Специалисты поймут, что способы, аналогичные данным в путях 1-5, можно использовать для получения соединений Формулы I, в которых R1 представляет собой гетероциклическую группу. Также пути, аналогичные путям 1-5, можно использовать для получения соединений Формулы I, в которых атом кислорода в связующей цепочке заменен на S или NR.
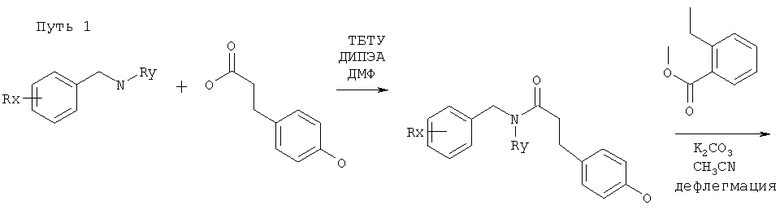
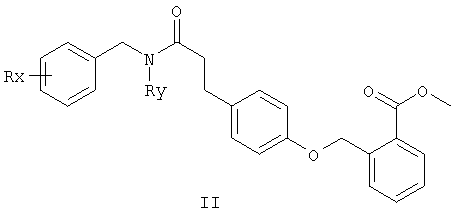
Исходные амины можно получать как описано в Ralph N Salvatore et. al., Tetrahedron, 57, 7758-7811, 2001 или способами, которые даны ниже.
1. Получение амида с последующим восстановлением.
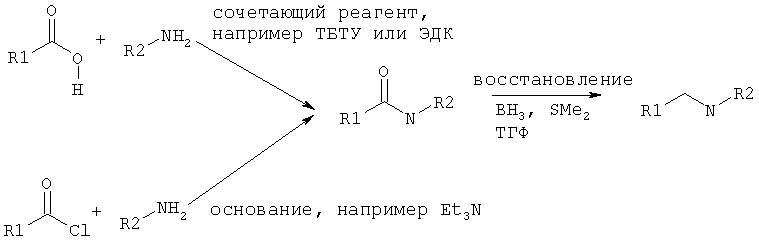
2. Восстановительное аминирование.
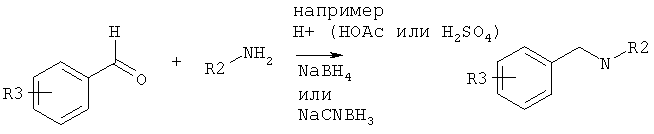
3. N-алкилирование.
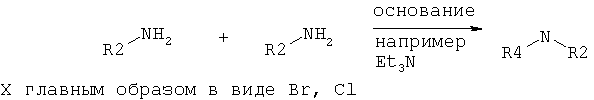
Соединения формулы II считаются новыми и заявлены здесь как полезные промежуточные соединения в получении соединения формулы I.
Соединения по этому изобретению можно изолировать из их реакционных смесей при использовании общепринятых методик.
Специалист должен понимать, что для получения соединений по этому изобретению альтернативным и в некоторых случаях более удобным образом индивидуальные стадии способов, упомянутые здесь выше, можно осуществлять в разном порядке, и/или индивидуальные взаимодействия можно осуществлять на разных стадиях всего пути (т.е. химические превращения можно осуществлять на иных промежуточных соединениях, нежели на тех, что ассоциированы здесь выше с конкретным взаимодействием).
В любом из предыдущих способов получения, если необходимо, можно защищать гидрокси, амино или другие взаимодействующие группы при использовании защитной группы Rр как описано в стандартном тексте "Protective groups in Organic Synthesis", 2nd Edition (1991) Greene и Wuts.
Защитной группой также может быть смола, такая как смола Вонга (Wang), или смола, представляющая собой хлорид 2-хлортритила. Защита функциональных групп и снятие защиты с них могут иметь место до или после любой из стадий взаимодействия, описанных здесь выше. Защитные группы можно удалять в соответствии с методиками, которые хорошо известны специалистам.
Выражение "инертный растворитель" относится к растворителю, который не взаимодействует с исходными материалами, реагентами, промежуточными соединениями или продуктами таким образом, который нежелательно влияет на выход желаемого продукта.
Фармацевтические препараты
Соединения по этому изобретению в норме надо вводить пероральным, парентеральным, внутривенным, внутримышечным, подкожным или другими инъекционными путями, защечным, ректальным, вагинальным, чрескожным и/или носовым путем, и/или ингаляцией в форме фармацевтического препарата, содержащего активный ингредиент либо как свободную кислоту, либо как фармацевтически приемлемую соль, в фармацевтически приемлемой лекарственной форме. В зависимости от нарушения и пациента, подлежащего лечению, и пути введения, эти композиции можно вводить в варьирующих дозах.
Подходящие суточные дозы соединения по этому изобретению в терапевтическом лечении людей составляют около 0,0001-100 мг/кг массы тела, предпочтительно 0,001-10 мг/кг массы тела.
Предпочтительными являются пероральные препараты, в частности таблетки или капсулы, которые можно составлять способами, известными специалистам, для предоставления доз активного соединение в диапазоне от 0,5 мг до 500 мг, например 1 мг, 3 мг, 5 мг, 10 мг, 25 мг, 50 мг, 100 мг и 250 мг.
Таким образом, в соответствии с дополнительным аспектом этого изобретения предложен фармацевтический препарат, включающий в себя любое из соединений по этому изобретению или его фармацевтически приемлемое производное в смеси с фармацевтически приемлемыми адъювантами, разбавителями и/или носителями.
Фармакологические свойства
Данные соединения формулы (I) полезны для профилактики и/или лечения клинических состояний, ассоциированных с врожденной или индуцированной сниженной чувствительностью к инсулину (резистентностью к инсулину), и ассоциированных метаболических нарушений (также известных как метаболический синдром). Эти клинические состояния включают в себя следующие без ограничения ими: общее ожирение, абдоминальное ожирение, артериальная гипертензия, гиперинсулинемия, гипергликемия, диабет типа 2 и дислипидемия, которая характерным образом появляется при резистентности к инсулину. Эта дислипидемия, также известная как атерогенный липопротеиновый профиль, характеризуется умеренно повышенными уровнями неэстерифицированных жирных кислот, повышенными уровнями богатых триглицеридами частиц, представляющих собой липопротеины очень низкой плотности (ЛОНП), и аполипопротеинов Аро В, низкими уровнями липопротеинов высокой плотности (ЛВП), ассоциированными с низкими уровнями частиц, представляющих собой аро AI, и с высокими уровнями Аро В, в присутствии мелких, плотных частиц, представляющих собой липопротеины низкой плотности (ЛНП) фенотипа В.
Ожидается, что соединения по данному изобретению полезны в лечении пациентов с комбинированными или смешанными гиперлипидемиями или различными степенями гипертриглицеридемий и послепищевой дислипидемии при наличии или отсутствии других проявлений метаболического синдрома.
Ожидается, что лечение данными соединениям из-за их антидислипидемических, а также противовоспалительных свойств снизит сердечно-сосудистую заболеваемость и смертность, ассоциированную с атеросклерозом. Состояния, представляющие собой сердечно-сосудистые заболевания, включают в себя макроангиопатии различных внутренних органов, вызывающие инфаркт миокарда, застойную сердечную недостаточность, заболевание церебральных сосудов и недостаточность периферических артерий нижних конечностей. По причине их действия, состоящего в сенсибилизации к инсулину, ожидается, что соединения формулы I предотвращают и замедляют развитие диабета типа 2 из метаболического синдрома и диабета беременных. Поэтому ожидается, что будет замедлено развитие долгосрочных осложнений, ассоциированных с хронической гипергликемией, таких как микроангиопатии, вызывающие заболевания почек, повреждение сетчатки и заболевания периферических сосудов нижних конечностей, при сахарном диабете. Более того, эти соединения могут быть полезными в лечении различных состояний вне сердечно-сосудистой системы, независимо от их связи с резистентностью к инсулину, таких как синдром поликистоза яичников, ожирение, рак и состояния, представляющие собой воспалительные заболевания, в том числе нейродегенеративные нарушения, такие как легкие умственные расстройства, болезнь Альцгеймера, болезнь Паркинсона и рассеянный склероз.
Ожидается, что соединения по данному изобретению полезны в контролировании уровней глюкозы у пациентов, страдающих диабетом типа 2.
Настоящее изобретение предлагает способ лечения или предотвращения дислипидемий, синдрома резистентности к инсулину и/или метаболических нарушений (какие определены выше), при котором соединение формулы I вводят млекопитающему (в частности, человеку), которое в этом нуждается.
Настоящее изобретение предлагает способ лечения или предотвращения диабета типа 2, при котором соединение формулы I вводят млекопитающему (в частности, человеку), которое в этом нуждается.
В дополнительном аспекте настоящее изобретение предлагает применение соединения формулы I в качестве медикамента.
В дополнительном аспекте настоящее изобретение предлагает применение соединения формулы I в изготовлении медикамента для лечения резистентности к инсулину и/или метаболических нарушений.
Комбинационная терапия
Соединения по этому изобретению можно комбинировать с другим терапевтическим агентом, который является полезным в лечении нарушений, ассоциированных с развитием и прогрессированием атеросклероза, таких как гипертензия, гиперлипидемии, дислипидемии, диабет и ожирение. Соединения по этому изобретению можно комбинировать с другим терапевтическим агентом, который снижает соотношение ЛНП:ЛВП, или агентом, который вызывает уменьшение уровней ЛНП-холестерина в крови. У пациентов с сахарным диабетом соединения по этому изобретению можно также комбинировать с терапевтическими агентами, применяемыми для лечения осложнений, имеющих отношение к микроангиопатиям.
Соединения по этому изобретению можно использовать наряду с другими способами терапии метаболического синдрома или диабета типа 2 и ассоциированных с ним осложнений, что включает в себя лекарственные средства, представляющие собой бигуаниды, например метформин, фенформин и буформин, инсулин (синтетические аналоги инсулина, амилин) и пероральные антигипергликемические средства (из разделяют на регуляторы глюкозы при приеме пищи и ингибиторы альфа-глюкозидазы). Примером ингибитора альфа-глюкозидазы является акарбоза или воглибоза или миглитол. Примером регулятора глюкозы при приеме пищи является репаглинид или натеглинид.
В другом аспекте этого изобретения соединение формулы I или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарственную форму можно вводить в ассоциации с другим PPAR-модулирующим агентом. PPAR-модулирующие агенты включают в себя следующие без ограничения ими: агонист PPARα и/или PPARγ, и/или PPARδ, или его фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарственные формы. Подходящие агонисты PPARα и/или PPARγ, их фармацевтически приемлемый соли, сольваты, сольваты таких солей или пролекарственные формы хорошо известны в данной области. Они включают в себя соединения, раскрытые в WO 01/12187, WO 01/12612, WO 99/62870, WO 99/62872, WO 99/62871, WO 98/57941, WO 01/40170, J Med Chem, 1996, 39, 665, Expert. Opinion on Therapeutic Patents, 10 (5), 623-634 (в частности, соединения, описанные в заявках на патент, перечисленных на стр.634) и в J Med Chem, 2000, 43, 527, которые все инкорпорированы сюда ссылкой на них. В частности, агонист PPARα и/или PPARγ относится к BMS 298585, клофибрату, фенофибрату, безафибрату, гемфиброзилу и ципрофибрату; GW 9578, пиоглитазону, розиглитазону, ривоглитазону, балаглитазону, KRP-297, JTT-501, SB 213068, GW 1929, GW 7845, GW 0207, L-796449, L-165041 и GW 2433. В частности, агонист PPARα и/или PPARγ относится к (S)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил)-этокси)-фенил]-пропаноевой кислоте и ее фармацевтически приемлемым солям.
Кроме того, комбинацию по этому изобретению можно использовать в сочетании с сульфонилмочевиной, например глимепиридом, глибенкламидом (глибуридом), гиклазидом, глипизидом, гликвидоном, хлорпропамидом, толбутамидом, ацетогексамидом, гликопирамидом, карбутамидом, глибонуридом, глизоксепидом, глибатиазолом, глибузолом, глигексамидом, глимидином, глипинамидом, фенбутамидом, толцинамидом и толазамидом. Предпочтительно сульфонилмочевина представляет собой глимепирид или глибенкламид (глибурид). Более предпочтительно сульфонилмочевина представляет собой глимепирид. Поэтому данное изобретение включает в себя введение соединения по данному изобретению в сочетании с одним, двумя или более чем двумя из существующих терапевтических средств, описанных в этом параграфе. Дозы других существующих терапевтических средств лечения диабета типа 2 и ассоциированных с ним осложнений должны быть теми, которые известны в данной области и которые одобрили для применения регулятивные органы, например FDA, и их можно найти в Orange Book, публикуемой FDA. Или же можно использовать меньшие дозы в результате пользы от этой комбинации. Настоящее изобретение также включает в себя соединение по настоящему изобретению в комбинации с агентом понижения холестерина. Агенты понижения холестерина, подразумеваемые в этой заявке, включают в себя без ограничения ими ингибиторы редуктазы ГМГ-КоА (редуктаза 3-гидрокси-S-метилглутарил-коэнзима А). Подходящим образом ингибитор редуктазы ГМГ-КоА представляет собой статин, который выбирают из группы, состоящей из аторвастатина, бервастатина, церивастатина, далвастатина, флувастатина, итавастатина, ловастатина, мевастатина, никостатина, нивастатина, правастатина и симвастатина или его фармацевтически приемлемой соли, в частности натриевой или кальциевой, или сольвата или сольвата такой соли. Конкретным статином является аторвастатин или его фармацевтически приемлемая соль, сольват, сольват такой соли или пролекарственная форма. Более конкретным статином является кальциевая соль аторвастатина. Однако в особенности предпочтительным статином является соединение с химическим названием (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)-амино]-пиримидин-5-ил]-(3R,5S)-3,5-дигидроксигепт-6-еноевая кислота [также известное как (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[N-метил-N-(метилсульфонил)-амино]-пиримидин-5-ил]-(3R,5S)-3,5-дигидроксигепт-6-еноевая кислота] или ее фармацевтически приемлемая соль или сольват или сольват такой соли. Соединение (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил-(метилсульфонил)-амино]-пиримидин-5-ил]-(3R,5S)-3,5-дигидроксигепт-6-еноевая кислота и его кальциевая и натриевая соли раскрыты в Заявке на европейский патент, Публикация №ЕР-А-0521471, и в Bioorganic and Medicinal Chemistry, (1997), 5 (2), 437-444. Этот последний статин сейчас известен под названием розувастатин (название дженерика).
В данной заявке термин "агент понижения холестерина" также включает в себя химические модификации ингибиторов редуктазы ГМГ-КоА, такие как сложные эфиры, пролекарственные формы и метаболиты, как активные, так и неактивные.
Настоящее изобретение также включает в себя соединение по настоящему изобретению в комбинации с агентом связывания желчных кислот, например колестиполом или холестирамином или холестагелем.
Настоящее изобретение также включает в себя соединение по настоящему изобретению в комбинация с ингибитором системы транспорта желчных кислот в тонком кишечнике (IBAT-ингибитором).
Были раскрыты подходящие соединения, имеющие активность IBAT-ингибитора, см., к примеру, соединения, раскрытые в WO 93/16055, WO 94/18183, WO 94/18184, WO 94/24087, WO 96/05188, WO 96/08484, WO 96/16051, WO 97/33882, WO 98/07749, WO 98/38182, WO 98/40375, WO 98/56757, WO 99/32478, WO 99/35135, WO 99/64409, WO 99/64410, WO 00/01687, WO 00/20392, WO 00/20393, WO 00/20410, WO 00/20437, WO 01/34570, WO 00/35889, WO 00/47568, WO 00/61568, WO 01/68637, WO 01/68096, WO 02/08211, WO 00/38725, WO 00/38726, WO 00/38727, WO 00/38728, WO 00/38729, DB 19825804, JP 10072371, US 5070103, BP 251315, EP 417725, EP 489423, EP 549967, EP 573848, EP 624593, EP 624594, EP 624595, EP 869121, EP 864582 и ЕР 1070703, и содержание этих заявок на патенты, в частности соединения, раскрытые в п.1 формулы, и поименованные примеры, инкорпорированы сюда ссылкой на них.
Конкретные классы IBAT-ингибиторов, подходящие для применения в настоящем изобретении, представляют собой бензотиепины, и соединения, описанные в формуле изобретения, в частности п.1, в WO 00/01687, WO 96/08484 и WO 97/33882, инкорпорированы сюда ссылкой на них. Другие подходящие классы IBAT-ингибиторов представляют собой 1,2-бензотиазепины, 1,4-бензотиазепины и 1,5-бензотиазепины. Дополнительный подходящий класс IBAT-ингибиторов представляет собой 1,2,5-бензотиадиазепины.
Одним конкретным подходящим соединением, имеющим активность IBAT-ингибитора, является (3R,5R)-3-бутил-3-этил-1,1-диоксидо-5-фенил-2,3,4,5-тетрагидро-1,4-бензотиазепин-8-ил-β-D-глюкопиранозидуроновая кислота (ЕР 864582). Другие подходящие IBAT-ингибиторы включают в себя одно из следующего:
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-1′-фенил-1′-[N′-(карбоксиметил)-карбамоил]-метил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-(N′-(карбоксиметил)-карбамоил]-4-гидроксибензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-1′-фенил-1′-[N′-(2-сульфоэтил)-карбамоил]-метил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-1′-фенил-1′-[N′-(2-сульфоэтил)-карбамоил]-метил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N′-(2-сульфоэтил)-карбамоил]-4-гидроксибензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-(α-[N′-(2-сульфоэтил)-карбамоил]-4-гидроксибензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-α-[N′-(2-карбоксиэтил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N′-(2-карбоксиэтил)-карбамоил]-4-гидроксибензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензтиазепин;
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-α-[N′-(5-карбоксипентил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-(α-[N′-(2-карбоксиэтил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{α-[N′-(2-сульфоэтил)-карбамоил]-2-фторбензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-α-[N′-(R)-(2-гидрокси-1-карбоксиэтил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензтиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N′-(R)-(2-гидрокси-1-карбоксиэтил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-{N-[(R)-α-(N′-{(R)-1-[N"-(R)-(2-гидрокси-1-карбоксиэтил)-карбамоил-2-гидроксиэтил}-карбамоил)-бензил]-карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метиллтио-8-(N-{α-[N′-(карбоксиметил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{α-[N′-((этокси)(метил)фосфорил-метил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-{N-[(R)-α-(N′-{2-[(гидрокси)(метил)-фосфорил]-этил}-карбамоил)-бензил]-карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N′-(2-метилтио-1-карбоксиэтил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-{N-[(R)-α-(N′-{2-[(метил)(этил)-фосфорил]-этил)-карбамоил)-4-гидроксибензил]-карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-{N-[(R)-α-(N′-{2-[(метил)(гидрокси)-фосфорил]-этил}-карбамоил)-4-гидроксибензил]-карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[(R)-N′-(2-метилсульфинил-1-карбоксиэтил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метокси-8-[N-{(R)-α-[N′-(2-сульфоэтил)-карбамоил]-4-гидроксибензил}-карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((R)-1-карбокси-2-метилтио-этил)-карбамоил]-4-гидроксибензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбокси-2-(R)-гидроксипропил)-карбамоил]-4-гидроксибензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбокси-2-метилпропил)-карбамоил]-4-гидроксибензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбоксибутил)-карбамоил]-4-гидроксибензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбоксипропил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбоксиэтил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,6-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбокси-2-(R)-гидроксипропил)-карбамоил]-бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-(2-сульфоэтил)-карбамоил]-4-гидроксибензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбоксиэтил)-карбамоил]-4-гидроксибензил}-карбамоилметоксй)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((R)-1-карбокси-2-метилтиоэтил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-{(S)-1-[N-((S)-2-гидрокси-1-карбоксиэтил)-карбамоил]-пропил}-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-кэрбокси-2-метилпропил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбоксипропил)-карбамоил]-4-гидроксибензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-((R/S)-α-{N-[1-(R)-2-(S)-1-гидрокси-1-(3,4-дигидроксифенил)-проп-2-ил]-карбамоил}-4-гидроксибензил)-карбамоилметокси-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-(2-(S)-3-(R)-4-(R)-5-(R)-2,3,4,5,6-пентагидроксигексил)-карбамоил]-4-гидроксибензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин; и
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-(2-(S)-3-(R)-4-(R)-5-(R)-2,3,4,5,6-пентагидроксигексил)-карбамоил]-бензил}-карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепин;
или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарственную форму.
В соответствии с еще одним дополнительным аспектом данного изобретения предложено комбинационное лечение, при котором вводят эффективное количество соединения формулы I или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарственной формы, возможно вместе с фармацевтически приемлемым разбавителем или носителем, при одновременном, последовательном или раздельном введении одного или более чем одного из нижеследующих агентов, выбранных из следующего:
ингибитор СЕТР (белка переноса эфиров холестерина), например те, что упомянуты и раскрыты в WO 00/38725, стр.7, строка 22 - стр.10, строка 17, которые инкорпорированы сюда ссылкой на них;
антагонист всасывания холестерина, например азетидиноны, такие как SCH 58235 и те, что описаны в US 5767115, которые инкорпорированы сюда ссылкой на них;
ингибитор МТР (белка микросомального переноса), например те, что в описаны в Science, 282, 751-54, 1998, которые инкорпорированы сюда ссылкой на них;
производное никотиновой кислоты, что включает в себя продукты для медленного высвобождения и комбинационные, например никотиновая кислота (ниацин), аципимокс и ницеритрол;
соединение, представляющее собой фитостерол, например станолы;
пробукол;
соединение против ожирения, например орлистат (ЕР 129748) и сибутрамин (GB 2184122 и US 4929629);
ω3-жирная кислота, например Omacor™;
антигипертензивное соединение, например ингибитор ангиотензинконвертирующего энзима (АСЕ), антагонист рецептора ангиотензина II, адренергический блокатор, α-адренергический блокатор, β-адренергический блокатор, например метопролол, смешанный α/β-адренергический блокатор, адренергический стимулятор, блокатор каналов для кальция, блокатор АТ-1, салуретическое средство, диуретическое средство или сосудорасширяющее средство;
антагонист или обратный агонист СВ1, например, какой описан в WO 01/70700 и ЕР 65635;
аспирин;
антагонист меланин-концентрирующего гормона (МСН);
ингибитор PDK; или
модуляторы нуклеарных рецепторов, например LXR, FXR, RXR, и RORα;
или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарственной формы, возможно вместе с фармацевтически приемлемым разбавителем или носителем, теплокровному животному, такому как человек, нуждающемуся в таком терапевтическом лечении.
Конкретные ингибиторы АСЕ или их фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарственные формы, в том числе активные метаболиты, которые можно использовать в комбинации с соединением формулы I, включают в себя нижеследующие соединения без ограничения ими: алацеприл, алатриоприл, кальциевая. соль алтиоприла, анковенин, беназеприл, беназеприла гидрохлорид, беназеприлат, бензоилкаптоприл, каптоприл, каптоприл-цистеин, каптоприл-глутатион, церанаприл, цераноприл, церонаприл, цилазаприл, цилазаприлат, делаприл, делаприл-дикислота, эналаприл, эналаприлат, энаприл, эпикаптоприл, фороксимитин, фостеноприл, фосеноприл, натриевая соль фосеноприла, фосиноприл, натриевая соль фосиноприла, фосиноприлат, фосиноприловая кислота, гликоприл, геморфин-4, идраприл, имидаприл, индолаприл, индолаприлат, либензаприл, лизиноприл, лициумин А, лициумин В, миксанприл, моэксиприл, моэксиприлат, мовелтиприл, мурацеин А, мурацеин В, мурацеин С, пентоприл, периндоприл, периндоприлат, пивалоприл, пивоприл, квинаприл, квинаприла гидрохлорид, квинаприлат, рамиприл, рамиприлат, спираприл, спираприла гидрохлорид, спираприлат, спироприл, спироприла гидрохлорид, темокаприл, темокаприла гидрохлорид, тепротил, трандолаприл, трандолаприлат, утибаприл, забициприл, забициприлат, зофеноприл и зофеноприлат. Предпочтительными ингибиторами АСЕ для применения в настоящем изобретении являются рамиприл, рамиприлат, лизиноприл, эналаприл и эналаприлат. Более предпочтительными ингибиторами АСЕ для применения в настоящем изобретении являются рамиприл и рамиприлат.
Предпочтительные антагонисты ангиотензина II, их фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарственные формы для применения в комбинации с соединением формулы I включают в себя, но без ограничения ими соединения: кандесартан, кандесартана цилексетил, лозартан, валсартан, ирбесартан, тасосартан, телмисартан и эпросартан. Особенно предпочтительными антагонистами ангиотензина II или их фармацевтически приемлемыми производными для применения в настоящем изобретении являются кандесартан и кандесартана цилексетил.
Поэтому в дополнительном аспекте этого изобретения предложен способ лечения диабета типа 2 и ассоциированных с ним осложнений у теплокровного животного, такого как человек, нуждающегося в таком лечении, при котором вышеуказанному животному вводят эффективное количество соединения формулы I или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарственной формы одновременным, последовательным или раздельным введением с эффективным количеством одного из других соединений, описанных в этой секции о комбинациях, или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарственной формы.
Поэтому в дополнительном отличии этого изобретения предложен способ лечения гиперлипидемических состояний у теплокровного животного, такого как человек, нуждающегося в таком лечении, при котором вышеуказанному животному вводят эффективное количество соединения формулы I или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарственной формы одновременным, последовательным или раздельным введением с эффективным количеством одного из других соединений, описанных в этой секции о комбинациях, или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарственной формы.
В соответствии с дополнительным аспектом этого изобретения предложена фармацевтическая композиция, которая содержит соединение формулы I или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарственную форму и одно из других соединений, описанных в этой секции о комбинациях или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарственную форму в ассоциации с фармацевтически приемлемым разбавителем или носителем.
В соответствии с дополнительным аспектом данного изобретения предложен набор, содержащий соединение формулы I или его фармацевтически приемлемую соль, сольват, сольват такой соль или пролекарственную форму и одно из других соединений, описанных в этой секции о комбинациях, или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарственную форму.
В соответствии с дополнительным аспектом данного изобретения предложен набор, содержащий:
a) соединение формулы I его или фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарственную форму в первой единичной стандартной лекарственной форме;
b) одно из других соединений, описанных в этой секции о комбинациях, или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарственную форму во второй единичной стандартной лекарственной форме; и
c) контейнерное средство для вмещения вышеуказанных первой и второй лекарственных форм.
В соответствии с дополнительным аспектом данного изобретения предложен набор, содержащий:
a) соединение формулы I или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарственную форму вместе с фармацевтически приемлемым разбавителем или носителем в первой единичной стандартной лекарственной форме;
b) одно из других соединений, описанных в этой секции о комбинациях, или его фармацевтически приемлемую соль, сольват, сольват такой соли или пролекарственную форму во второй единичной стандартной лекарственной форме; и
c) контейнерное средство для вмещения вышеуказанных первой и второй лекарственных форм.
В соответствии с другим отличием этого изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарственной формы и одного из других соединений, описанных в этой секции о комбинациях, или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарственной формы в изготовлении медикамента для применения в лечении метаболического синдрома или диабета типа 2 и ассоциированных осложнений у теплокровного животного, такого как человек.
В соответствии с другим отличием этого изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарственной формы и одного из других соединений, описанных в этой секции о комбинациях или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарственной формы в изготовлении медикамента для применения в лечении гиперлипидемических состояний у теплокровного животного, такого как человек.
В соответствии с дополнительным аспектом данного изобретения предложено комбинационное лечение, при котором вводят эффективное количество соединения формулы I или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарственной формы, возможно вместе с фармацевтически приемлемым разбавителем или носителем, при одновременном, последовательном или раздельном введении эффективного количества одного из других соединений, описанных в этой секции о комбинациях, или его фармацевтически приемлемой соли, сольвата, сольвата такой соли или пролекарственной формы, возможно вместе с фармацевтически приемлемым разбавителем или носителем, теплокровному животному, такому как человек, нуждающемуся в таком терапевтическом лечении.
Рабочие примеры
Осуществляли измерения 1Н-ЯМР и 13С-ЯМР на спектрометрах Varian Mercury 300 или Varian UNITY plus 400, 500 или 600, работающих для 1H на частотах 300, 400, 500 и 600 мГц соответственно, и для 13С на частотах 75, 100, 125 и 150 мГц соответственно. Делали измерения по дельта-шкале (δ).
Если не указано иначе, химические сдвиги даны в м.д. с растворителем в качестве внутреннего стандарта.
Сокращения
ISOLUTE® FLASH Si представляет собой колонку с кремнеземом, подходящую для хроматографии.
Боргидрид на полимерной подложке представляет собой боргидрид на Амберлите IRA-400 от Aldrich.
Пример 1
a) трет-Бутил-3-{[(1,1'-бифенил-4-илкарбонил)-амино]-метил}-фенилкарбамат
Смешивали в диметилформамиде (10 мл) бифенил-4-карбоновую кислоту (981 мг, 4,949 ммоль) и 3-(аминометил)-1-N-boc-анилин (1,0 г, 4,499 ммоль). При перемешивании добавляли гексафторфосфат бензотриазол-1-илокситрипирролидинофосфония (2,343 г, 4,504 ммоль) и затем N,N-диизопропилэтиламин (1,164 г, 9,007 ммоль). Эту смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду и этилацетат. Органическую фазу промывали водой, гидрокарбонатом натрия (насыщенный) и водой (×2) и сушили сульфатом магния. Растворитель удаляли. В остаток добавляли диэтиловый эфир. Твердый продукт фильтровали, промывали небольшим количеством диэтилового эфира и сушили. Получали 1,44 г продукта. Фильтрат выпаривали досуха. К остатку добавляли ДХМ. Фильтрация давала еще 0,12 г твердого продукта. В сумме получали 1,56 г желаемого продукта, выход 86%.
1H-ЯМР (400 МГц, CDCl3): δ 3.61 (s, 9H), 4.66 (d, 2Н), 6.43 (s, br, 1H), 6.50 (s, 1H), 7.06-7.09 (m, 1H), 7.27-7.30 (m, 2H), 7.38-7.50 (m, 4H), 7.61-7.64 (m, 2H), 7.67 (d, 2H), 7.88 (2H).
b) N-(3-Аминобензил)-1,1′-бифенил-4-карбоксамид
трет-Бутил-3-{[(1,1′-бифенил-4-илкарбонил)-амино]-метил}-фенилкарбамат (250 мг, 0,6 ммоль) растворяли в ДХМ (10 мл). Добавляли трифторуксусную кислоту (0,2 мл). Эту смесь перемешивали в течение ночи. ВЭЖХ показала, что более чем 50% исходного материала не провзаимодействовали. Добавляли еще трифторуксусную кислоту (0,3 мл). Эту смесь опять перемешивали в течение ночи. В эту смесь добавляли воду. Фазы были непрозрачными. ДХМ выпаривали в вакууме. К остатку добавляли этилацетат. Полученную органическую фазу промывали водой (×3) и сушили сульфатом магния. Затем упаривали растворитель. Получали твердый продукт (185 мг), выход 99%.
1H-ЯМР (500 МГц, CD3OD): δ 4.64 (s, 2H), 7.27 (d, 1H), 7.37-7.40 (m, 2H), 7.45-7.53 (m, 4H), 7.66 (d, 2H), 7,74 (d, 2H), 7.95 (d, 2H).
с) 3-{[(3-{[(1,1′-Бифенил-4-илкарбонил)-амино]-метил}-фенил)-амино]-метил}-бензойная кислота
N-(3-аминобензил)-1,1′-бифенил-4-карбоксамид (20 мг, 0,07 ммоль) растворяли в уксусной кислоте (0,5 мл). Добавляли 3-карбоксибензальдегид (14 мг, 0,09 ммоль) и затем боргидрид натрия (11 мг, 0,28 ммоль). Эту смесь перемешивали при комнатной температуре 2 часа и упаривали досуха. Добавляли в остаток ДХМ. Эту смесь наносили на колонку (ISOLUTE® SI, 500 мг/3 мл). Ее элюировали дихлорметилом, смесью МеОН/ДХМ (0,5:99,5) и затем смесью МеОН/ДХМ (1:99). Объединяли фракции продукта и удаляли растворитель. Повторная хроматография остатка на колонке (ISOLUTE® SI, 1 г/6 мл) при использовании ДХМ, МеОН/ДХМ (0,5:99,5) и затем МеОН/ДХМ (1:99) в качестве элюанта давала 9 мг желаемого продукта, выход 31%.
1H-ЯМР (500 МГц, CD3OD): δ 4.37 (s, 2H), 4.46 (d, 2H), 6.53 (d, 1H), 6.59-6.61 (m, 2H), 7.04 (t, 1H), 7.30-7.38 (m, 2H), 7.46 (t, 2H), 7.55 (d, 1H), 7.65-7.70 (m, 4H), 7.83 (t, 3Н), 8.02 (s, 1H), 8.80 (br, 1H).
Пример 2
а) Метил-2-{[4-(2-оксо-2-{[4-(трифторметил)-бензил)-амино}-этил)-фенокси]-метил}-бензоат
Растворяли (4-{[2-(Метоксикарбонил)-бензил]-окси}-фенил)-уксусную кислоту (50 мг, 0,167 ммоль) в ДХМ (2 мл), добавляли 4-(трифторметил)-бензиламин (35 мг, 0,2 ммоль), затем добавляли ЭДК (38 мг, 0,2 ммоль) и затем добавляли ДМАП (24,4 мг, 0,2 ммоль). Эту смесь перемешивали при комнатной температуре в течение ночи. Добавляли в эту смесь 1%-ную соляную кислоту (1 мл) и воду (1 мл). Разделяли две фазы при использовании фильтровальной трубки Whatman. Полученный органический раствор выпаривали в вакууме и твердый продукт (72 мг) оставляли, выход 95%.
1H-ЯМР (500 мГц, CDCl3): δ 3.63 (s, 2H), 3.93 (s, 3Н), 4.50 (d, 2H), 5.53 (s, 2Н), 5.79 (br, 1H), 7.02 (d. 2H), 7.22 (d, 2H), 7.32 (d, 2H), 7.42 (t, 1H), 7.57-7.61 (m, 3Н), 7.77 (d, 1H), 8.07 (d, 1H).
b) 2-{[4-(2-Оксо-2-{[4-(трифторметил)-бензил]-амино}-этил)-фенокси]-метил)-бензойная кислота
Метил-2-{[4-(2-оксо-2-{[4-(трифторметил)-бензил]-амино}-этил)-фенокси]-метил}-бензоат (71 мг, 0,155 ммоль) в тетрагидрофуране (1,5 мл) охлаждали на ледяной бане. Каплями добавляли гидроксид лития (7,5 мг, 0,310 ммоль) в воде (1,5 мл). Охладительную баню затем удаляли и смесь перемешивали в течение ночи. ВЭЖХ показала, что взаимодействие было неполным. Добавляли еще гидроксид лития (0,2М, 0,5 мл). Эту реакционную смесь перемешивали еще 4 дня. Затем ее упаривали в вакууме для удаления тетрагидрофурана. Остаток подкисляли 1%-ой соляной кислотой, рН 3-4 и затем экстрагировали этилацетатом. Органическую фазу сушили сульфатом магния и упаривали. Хроматография остатка на колонке (ISOLUTE®-SI, 2 г/6 мл) при использовании ДХМ, МеОН/ДХМ (0,5:99,5, затем 1:99 и затем 2:98) в качестве элюанта давала 39 мг белого твердого продукта, выход 57%.
1H-ЯМР (500 мГц, CD3OD): δ 3.50 (s, 2H), 4.42(d, 2H), 5.47 (s, 2H), 6.94 (d, 2H), 7.22 (d, 2H), 7.37-7.40 (m, 3Н), 7.53-7.58 (m, 3Н), 7.69 (d, 1H), 8.01 (d, 1H).
Пример 3
а) (3-{[2-(Метоксикарбонил)-бензил]-окси}-фенил)-уксусная кислота
3-Гидроксифенилуксусную кислоту (760 мг, 5 ммоль) растворяли в этаноле (99,5%, 20 мл). Добавляли гидроксид калия (560 мг, 10 ммоль). Эту смесь перемешивали 30 минут. Затем каплями добавляли метиловый эфир 2-бромметилбензойной кислоты (1,144 г, 5 ммоль). Получившуюся смесь нагревали при дефлегмации 2 часа и затем упаривали в вакууме досуха. Добавляли в остаток воду и этилацетат и разделяли фазы. Водную фазу подкисляли 10%-ой соляной кислотой, рН˜5 и затем экстрагировали этилацетатом. Органическую фазу сушили (сульфат магния) и упаривали в вакууме досуха. Хроматография остатка на колонке (ISOLUTE® SI, 5 г/25 мл) при использовании ДХМ, МеОН/ДХМ (1:99) в качестве элюанта давала 213 мг желаемого продукта, выход 14%.
1H-ЯМР (500 мГц, CDCl3): δ 3.65 (s, 2H), 3.91 (s, 3H), 5.51 (s, 2H), 6.90-6.96 (m, 3H), 7.27 (t, 1H), 7.39 (t, 1H), 7.57 (t, 1H), 7.77 (d, 1H), 8.04 (d, 1H).
b) Метил-2-[(3-{2-бензил(гексил)-амино]-2-оксоэтил}-фенокси)-метил]-бензоат
Растворяли (3-{[2-(метоксикарбонил)-бензил]-окси)-фенил)-уксусную кислоту (60 мг, 0,2 ммоль) в ДХМ (2 мл), добавляли N-гексилбензиламин (46 мг, 0,24 ммоль), затем добавляли ЭДК (46 мг, 0,24 ммоль) и затем добавляли ДМАП (29,3 мг, 0,24 ммоль). Эту смесь перемешивали при комнатной температуре в течение ночи. Добавляли в эту смесь 1%-ую соляную кислоту (1 мл) и воду (1 мл). Разделяли две фазы при использовании фильтровальной трубки Whatman. Выпаривали полученную органическую часть в вакууме и оставляли 59 мг неочищенного продукта в виде масла. Его затем использовали прямо в следующей стадий.
c) 2-[(3-{2-[Бензил(гексил)-амино]-2-оксоэтил}-фенокси)-метил]-бензойная кислота
Метил-2-[(3-{2-[бензил(гексил)-амино]-2-оксоэтил}-фенокси)-метил]-бензоат (59 мг, 0,125 ммоль) в тетрагидрофуране (1 мл) охлаждали на ледяной бане. Каплями добавляли гидроксид лития (6 мг, 0,249 ммоль) в воде (1 мл). Охладительную баню затем удаляли и смесь перемешивали 13 дней и затем упаривали в вакууме для удаления тетрагидрофурана. Остаток подкисляли 1%-ой соляной кислотой, рН˜4 и экстрагировали этилацетатом. Органическую фазу сушили (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 1 г/6 мл) при использовании ДХМ и МеОН/ДХМ (0,5:99,5, затем 1:99) в качестве элюанта давала 7 мг желаемого продукта, выход 8% (две стадии).
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 0.85-0.90 (m, 3Н), 1.20-1.30 (m, 6H), 1.45-1.57 (m, 2Н), 3.20, 3.40 (t, t, 2H), 3.70, 3.80 (s, s, 2H), 4.51, 4.65 (s, s, 2H), 5.51, 5.52 (s, s, 2H), 6.83-7.00 (m, 3H), 7.14-7.43 (m, 7H), 7.59 (t, 1H), 7.78 (d, 1H), 8.13 (d, 1H).
Пример 4
a) Метил-2-{[3-(2-оксо-2-{[4-(трифторметил)-бензил]-амино}-этил)-фенокси]-метил}-бензоат
(3-{[2-(Метоксикарбонил)-бензил]-окси}-фенил)-уксусную кислоту (60 мг, 0,2 ммоль) растворяли в ДХМ (2 мл). Добавляли 4-(трифторметил)-бензиламин (42 мг, 0,24 ммоль), затем добавляли ЭДК (46 мг, (0,24 ммоль) и затем добавляли ДМАП (29,3 мг, 0,24 ммоль). Эту смесь перемешивали при комнатной температуре в течение ночи. Добавляли к этой смеси 1%-ную соляную кислоту (1 мл) и воду (1 мл). Разделяли две фазы при использовании фильтровальной трубки Whatman. Выпаривали полученную органическую часть в вакууме и оставляли 82 мг твердого продукта. Его затем использовали прямо в следующей стадии.
b) 2-{[3-(2-Оксо-2-{[4-(трифторметил)-бензил]-амино}-этил)-фенокси]-метил}-бензойная кислота
Метил-2-{[3-(2-оксо-2-{[4-(трифторметил)-бензил]-амино}-этил)-фенокси]-метил}-бензоат (82 мг, 0,18 ммоль) в тетрагидрофуране (2 мл) охлаждали на ледяной бане. Каплями добавляли гидроксид лития (8,6 мг, 0,36 ммоль) в воде (1 мл). Охладительную баню затем удаляли и эту смесь перемешивали 7 дней и затем упаривали в вакууме для удаления тетрагидрофурана. Остаток подкисляли 1%-ной соляной кислотой, рН˜3 и экстрагировали этилацетатом. Органическую фазу сушили (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ и МеОН/ДХМ (1:99, затем 2:98) в качестве элюанта давала 20 мг желаемого продукта, выход 22,5% (две стадии).
1H-ЯМР (500 мГц, CD3OD): δ 3.55 (s, 2H), 4.43 (s, 2H), 5.47 (s, 2H), 6.90 (t, 2Н), 6.98 (s, 1H), 7.23 (t, 1H), 7.38-7.42 (m, 3Н), 7.53-7.59 (m, 3H), 7.70 (d, 1H), 8.03 (d, 1H).
Пример 5
a) N-[2-(3,4-диметоксифенил)-этил]-3-(4-гидроксифенил)-N-метилпропанамид
3-(4-Гидроксифенил)-пропионовую кислоту (166,2 мг, 1 ммоль) растворяли в диметилформамиде (4 мл). Добавляли 2-(3,4-диметоксифенил)-N-метилэтиламин (211 мг, 1,05 ммоль). Эту смесь охлаждали на ледяной бане. Добавляли ТБТУ (337 мг, 1,05 ммоль) и далее ДИПЭА (0,37 мг, 2,1 ммоль). Перемешивали эту смесь в течение ночи и давали температуре дойти до комнатной температуры. Добавляли этилацетат и водный раствор гидрокарбоната натрия (насыщенный) и затем разделяли две фазы. Водную фазу экстрагировали этилацетатом. Органические фазы объединяли и сушили (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 5 г/15 мл) при использовании ДХМ и затем МеОН/ДХМ (1:99) в качестве элюанта давала 333 мг желаемого продукта, выход 97%.
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 2.35, 2.59 (t, t, 2H), 2.71-2.80, 2.90 (m, t, 4H), 2.84, 2.97 (s, s, 3H), 3.45, 3.58 (t, t, 2H), 3.84-3.86 (m, 6Н), 6.61-6.83 (m, 4Н), 6.95 (d, 1H), 7.05 (d, 1H), 7.50, 7.56 (s, s, 1H).
b) Метил-2-[(4-{3-[(2-(3,4-диметоксифенил)-этил1(метил)-амино]-оксопропил}-фенокси)-метил]-бензоат
Смешивали в ацетонитриле (15 мл) N-[2-(3,4-диметоксифенил)-этил]-3-(4-гидроксифенил)-N-метилпропанамид (198 мг, 0,577 ммоль), метиловый эфир 2-бромметилбензойной кислоты (139 мг, 0,605 ммоль) и карбонат калия безводный (120 мг, 0,864 ммоль) Эту смесь нагревали при дефлегмации в течение ночи и затем упаривали досуха. Добавляли воду и этилацетат и разделяли две фазы. Органическую фазу сушили (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании смеси гептан/ДХМ (50:50), затем ДХМ и затем смеси МеОН/ДХМ (0,5:99,5) в качестве элюанта давала 172 мг желаемого продукта, выход 61%.
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 2.34, 2.58 (t, t, 2H), 2.70-2.97 (m, 7Н), 3.44, 3.53 (t, t, 2H), 3.84-3.92 (m, 9Н), 5.49 (s, br, 2H), 6.61-6.82 (m, 3Н), 6.92 (t, 2H), 7.05 (d, 1Н), 7.15 (d, 1H), 7.38 (t, 1Н), 7.55 (t, 1H), 7.74-7.77 (m, 1H), 8.03 (d, 1H).
с) 2-[(4-{3-[{2-(3,4-Диметоксифенил)-этил](метил)-амино]-3-оксопропил}-фенокси)-метил]-бензойная кислота
К метил-2-[(4-{3-[[2-(3,4-диметоксифенил)-этил](метил)-амино]-3-оксопропил}-фенокси)-метил]-бензоату (120 мг, 0,244 ммоль), растворенному в тетрагидрофуране (2 мл), добавляли гидроксид лития (12 мг, 0,488 ммоль) в воде (1 мл). Эту смесь затем облучали в микроволновой печи (синтезатор Смита) при 150°С в течение 7 минут и затем упаривали для удаления тетрагидрофурана. Остаток подкисляли 1%-ной соляной кислотой, рН˜4 и экстрагировали этилацетатом (×2). Объединяли органические экстракты, промывали их рассолом, сушили сульфатом магния и затем упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ, затем МеОН/ДХМ (1:99) в качестве элюанта давала 102 мг желаемого продукта, выход 87,5%.
1Н-ЯМР (ротамеры, 500 мГц, CDCl3): δ 2.40, 2.64 (t, t, 2H), 2.73-3.01 (m, 7H), 3.47, 3.63 (t, t, 2H), 3.85-3.88 (m, 6H), 5.56, 5.57 (s, s, 2H), 6.63-6.83 (m, 3H), 6.93-6.97 (m, 2H), 7.07 (d, 1H), 7.17 (d, 1H), 7.41 (t, 1H), 7.57-7.61 (m, 1H), 7.81 (t, 1H), 8.18 (d, 1H).
Пример 6
а) Метил 2-[(4-{2-(трет-бутоксикарбонил]-амино]-этил}-фенокси)-метил]-бензоат
Смешивали в ацетонитриле (50 мл) трет-бутил-2-(4-гидроксифенил)-этилкарбамат (3,534 г, 14,9 ммоль), метиловый эфир 2-бромметилбензойной кислоты (3,582 г, 15,6 ммоль) и карбонат калия безводный (3,087 г, 22,3 ммоль). Эту смесь нагревали при дефлегмации в течение ночи и затем упаривали досуха. Добавляли воду и этилацетат и разделяли две фазы.
Органическую фазу was сушили (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 20 г/70 мл) при использовании ДХМ и затем МеОН/ДХМ (1:99) в качестве элюанта давала 5,427 г желаемого продукта, выход 94,5%.
1H-ЯМР (500 мГц, CDCl3): δ 1.44 (s, 9Н), 2.72 (t, 2H), 3.30-3.35 (m, 2H), 3.86 (s, 3Н), 4.87 (s, br, 1Н), 5.46 (s, 2H), 6.92 (d, 2H), 7.09 (d, 2H), 7.33 (t, 1H), 7.51 (t, 1H), 7,74 (d, 1H), 8.00 (d, 1H).
b) Метил-2-{[4-(2-аминоэтокси)-фенокси]-метил}-бензоата гидрохлорид
Метил-2-[(4-{2-[(трет-бутоксикарбонил)-амино]-этил}-фенокси)-метил]-бензоат (5,1 г, 13,2 ммоль) растворяли в этилацетате (50 мл) и охлаждали на ледяной бане. Добавляли соляную кислоту (4 М в диоксане, 30 мл, 120 ммоль). Охладительную баню удаляли через 30 минут. Смесь перемешивали еще 3 часа, и в течение этого времени выпадал белый преципитат. Эту реакционную смесь выпаривали досуха. Добавляли в остаток этилацетат (20 мл). Затем это фильтровали. Получали белый твердый продукт (3,785 г), выход 89%.
1H-ЯМР (500 мГц, CDCl3): δ 3.06 (t, 2H), 3.23 (t, 2H), 3.90 (s, 3Н), 5.45 (s, 2H), 6.93 (d, 2H), 7.18 (d, 2H), 7.37 (t, 1H), 7.55 (t, 1H), 7.73 (d, 1H), 8.02 (d, 1H), 8.35 (s, br, 2H).
c) Метил-2-[(4-{2-[({4-метил-2-[4-(трифторметил)-фенил]-1,3-тиазол-5-ил}-карбонил)-амино]-этил}-фенокси)-метил]-бензоат
Смешивали в диметилформамиде (4 мл) 4-метил-2-[4-(трифторметил)-фенил]-1,3-тиазол-5-карбоновую кислоту (50 мг, 0,174 ммоль) и метил 2-{[4-(2-аминоэтил)-фенокси]-метил}-бензоата гидрохлорид (59 мг, 0,183 ммоль) и затем охлаждали эту смесь на ледяной бане. Добавляли ТБТУ (59 мг, 0,183 ммоль) и далее ДИПЭА (47,2 мг, 0,366 ммоль). Эту смесь перемешивали при комнатной температуре в течение ночи. Добавляли этилацетат и водный раствор гидрокарбоната натрия (насыщенный). Разделяли две фазы. Органическую фазу промывали водой и сушили (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ и затем МеОН/ДХМ (0,5:99,5) в качестве элюанта давала 59 мг желаемого продукта белого твердого вещества, выход 61%.
1H-ЯМР (400 мГц, CDCl3): δ 2.62 (s, 3Н), 2.87 (t, 2Н), 3.67 (dt, 2H), 3.89 (s, 3Н), 5.49 (s, 2H), 5.86 (t, 1Н), 6.97 (d, 2H), 7.15 (d, 2H), 7.36 (t, 1Н), 7.54 (1, 1Н), 7.67 (d, 2H), 7.74 (d, 2H), 7.99-8.03 (m, 3Н).
d) 2-[(4-{2-[({4-Метил-2-[4-(трифторметил)-фенил]-1,3-тиазол-5-ил}-карбонил)-амино]-этил}-фенокси)-метил]-бензойная кислота
Гидроксид лития (4 мг, 0,166 ммоль) в воде (1 мл) добавляли в метил-2-[(4-{2-[({4-метил-2-[4-(трифторметил)-фенил]-1,3-тиазол-5-ил}-карбонил)-амино]-этил}-фенокси)-метил]-бензоат (46 мг, 0,083 ммоль), растворенный в тетрагидрофуране (2 мл). Эту смесь затем облучали в микроволновой печи (синтезатор Смита) при 150°С в течение 7 минут и затем упаривали для удаления ТГФ. Остаток подкисляли 1%-ной соляной кислотой, рН˜4, и экстрагировали этилацетатом (×2). Объединяли органические экстракты и сушили сульфатом магния и затем упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ, затем МеОН/ДХМ (1:99) в качестве элюанта давала 38 мг желаемого продукта, выход 85%.
1H-ЯМР (400 мГц, ТГФ-d8): δ 2.65 (s, 3Н), 2.85 (t, 2H), 3.54 (dt, 2H), 5.52 (s, 2H), 6.94 (d, 2H), 7.17 (d, 2H), 7.36 (t, 1Н), 7.41 (t, 1H), 7.53 (t, 1Н), 7.75-7.79 (m, 3Н), 8.06 (d, 1H), 8.14 (d, 2H).
Пример 7
а) Метил-2-({4-[2-({[2,4-дифторфенил)-амино]-карбонил}-амино)-этил]-фенокси}-метил)-бензоат
Смешивали в ДХМ (4 мл) 2,4-дифторфенилизоцианат (26,5 мг, 0,171 ммоль) и метил-2-{[4-(2-аминоэтил)-фенокси]-метил}-бензоата гидрохлорид (55 мг, 0,171 ммоль). Добавляли поддерживаемый полимером ДИЭА (PS-ДИЭА) (3,66 ммоль/г, 140 мг, 0,512 ммоль). Эту смесь встряхивали при комнатной температуре в течение ночи. Выпадал белый преципитат. Эту смесь выпаривали досуха. Остаток (с добавлением ДХМ, суспензия) наносили на колонку (ISOLUTE® SI, 2 г/6 мл) и элюировали дихлорметилом и затем смесью МеОН/ДХМ (1:99). Получали белый твердый продукт, 51 мг, выход 68%.
1H-ЯМР (400 МГц, AMCO-d6): δ 2.65 (t, 2H), 3.26-3.31 (m, 2H), 3.79 (s, 3H), 5.36 (s, 2H), 6.49 (t, 1H), 6.88-6.97 (m, 3H, 7.12-7.22 (m, 3H), 7.44 (t, 1H), 7.58-7.65 (m, 2H), 7.88 (d, 1H), 8.01-8.07 (m, 1H), 8.23 (s, br, 1H).
b) 2-({4-[2-({[(2,4-Дифторметил)-амино]-карбонил}-амино)-этил]-фенокси}-метил)-бензойная кислота
Гидроксид лития (3,7 мг, 0,154 ммоль) в воде (1 мл) добавляли в метил-2-({4-[2-({[(2,4-дифторфенил)-амино]-карбонил}-амино)-этил]-фенокси}-метил)-бензоат (34 мг, 0,077 ммоль), растворенный в тетрагидрофуране (2 мл). Эту смесь затем облучали в микроволновой печи (синтезатор Смита) при 150°С в течение 7 минут и затем упаривали для удаления ТГФ. Остаток подкисляли 1%-ной соляной кислотой, рН˜4 и экстрагировали этилацетатом (×2). Объединяли органические экстракты, сушили их сульфатом магния и затем упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 1 г/6 мл) при использовании МеОН/ДХМ (1:99,2:98, 4:96 и затем 10:90) в качестве элюанта давала 18 мг желаемого продукта, выход 55%.
1Н-ЯМР (400 МГц, ТГФ-d8): δ 2.76 (t, 2H), 3.39-3.44 (m, 2H), 5.51 (s, 2H), 6.40 (t, 1H), 6.79-6.94 (m, 4Н), 7.16 (d, 2H), 7.35 (t, 1H), 7.53 (t, 1H), 7.76 (d, 1H), 7.93 (s, br, 1H), 8.06 (d, 1H), 8.29-8.35 (m, 1H).
Пример 8
а) Метил-2-[(4-{2-[(2-метил-5-фенил-3-фуроил)-амино]-этил}-фенокси)-метил]-бензоат
Смешивали в ДХМ (4 мл) 2-метил-5-фенилфуран-3-карбонилхлорид (36,4 мг, 0,165 ммоль) и метил-2-{[4-(2-аминоэтил)-фенокси]-метил}-бензоата гидрохлорид (53 мг, 0,165 ммоль). Добавляли Р8-ДИЭА (3,66 ммоль/г, 135 мг, 0,494 ммоль). Эту смесь встряхивали при комнатной температуре в течение ночи. ЖХ-МС показала, что присутствовали только следовое количества желаемого продукта и большой пик 2-метил-5-фенил-3-фуроевой кислоты. Добавляли ТБТУ (55 мг, 0,17 ммоль). Эту смесь встряхивали 2 часа и фильтровали. Фильтрат выпаривали досуха. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ и затем МеОН/ДХМ (0,5:99,5) в качестве элюанта давала 34 мг желаемого продукта, выход 44%.
1H-ЯМР (500 мГц, CDCl3): δ 2.61 (s, 3H), 2.85 (t, 2H), 3.60-3.66 (m, 2H), 3.89 (s, 3H), 5.49 (s, 2H), 5.82 (t, 1Н), 6.55 (s, 1Н), 6.96 (d, 2H), 7.15 (d, 2H), 7.25 (t, 1Н), 7.36 (t, 3H), 7.52-7.63 (m, 3H), 7.75 (d, 1H), 8.02 (d, 1H).
b) 2-[(4-{2-[(2-Метил-5-фенил-3-фуроил)-амино]-этил}-фенокси)-метил]-бензойная кислота
Гидроксид лития (3,3 мг, 0,136 ммоль) в воде (1 мл) добавляли в метил-2-[(4-{2-[(2-метил-5-фенил-3-фуроил)-амино]-этил}-фенокси)-метил]-бензоат (32 мг, 0,068 ммоль), растворенный в тетрагидрофуране (2 мл). Эту смесь затем облучали в микроволновой печи (синтезатор Смита) при 150°С в течение 7 минут и затем упаривали для удаления тетрагидрофурана. Остаток подкисляли 1%-ной соляной кислотой, рН˜4 и экстрагировали этилацетатом (×2). Объединяли органический экстракты и промывали их рассолом, сушили сульфатом магния и затем упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ, затем МеОН/ДХМ (1:99 и 2:98) в качестве элюанта давала 22 мг желаемого продукта, выход 71%.
1H-ЯМР (400 мГц, CDCl3): δ 2.60 (s, 3H), 2.84 (t, 2H), 3.60-3.65 (m, 2H), 5.51 (s, 2H), 5.90 (t, 1H), 6.56 (s, 1H), 6.94 (d. 2H), 7.13 (d, 2H), 7.23 (t, 1H), 7.32-7.40 (m, 3H), 7.55-7.60 (m, 3H), 7.77 (d, 1H), 8.13 (d, 1H).
Пример 9
а) Метил-2-[(4-{2-[(бензилсульфонил)-амино]-этил)-фенокси)-метил]-бензоат
Смешивали в ДХМ (3 мл) α-толуолсульфонилхлорид (38 мг, 0,199 ммоль) и метил-2-{[4-(2-аминоэтил)-фенокси]-метил}-бензоата гидрохлорид (64 мг, 0,199 ммоль). Добавляли PS-ДИЭА (3,66 ммоль/г, 272 мг, 0,997 ммоль). Эту смесь встряхивали при комнатной температуре в течение уикэнда. Затем ее наносили на колонку (ISOLUTE® SI, 1 г/6 мл) и элюировали дихлорметилом. Фракции продукта объединяли и упаривали. Получали продукт в виде масла (17 мг), выход 19%.
1H-ЯМР (500 МГц, CDCl3): δ 2.74 (t, 2H), 3.19-3.23 (m, 2H), 3.92 (s, 3H), 4.12 (t, 1Н), 4.21 (s, 2H), 5.50 (s, 2H), 6.94 (d, 2H), 7.07 (d. 2H), 7.32-7.42 (m, 6H), 7.57 (t, 1Н), 7.75 (d, 1H), 8.05 (d, 1H).
b) 2-[(4-{2-[(Бензилсульфонил)-амино]-этил}-фенокси)-метил]-бензойная кислота
Гидроксид лития (2 мг, 0,077 ммоль) в воде (0,5 мл) добавляли в метил-2-[(4-{2-[(бензилсульфонил)-амино]-этил}-фенокси)-метил]-бензоат (17 мг, 0,038 ммоль), растворенный в ТГФ (1 мл). Эту смесь затем облучали в микроволновой печи (синтезатор Смита) при 150°С в течение 7 минут и затем упаривали для удаления тетрагидрофурана. Остаток подкисляли 1%-ой соляной кислотой, рН˜4, и экстрагировали этилацетатом (×2). Объединяли органические экстракты, сушили их сульфатом магния и затем упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 500 мг/3 мл) при использовании ДХМ, затем МеОН/ДХМ (0,5:99,5) в качестве элюанта давала 10 мг желаемого продукта, выход 61%.
1H-ЯМР (400 мГц, CDCl3): δ 2.70 (t, 2H), 3.16-3.21 (m, 2H), 4.18 (s, 2H), 4.29 (t, 1Н), 5.48 (s, 2H), 6.91 (d, 2H), 7.05 (d, 2H), 7.29-7.35 (m, 5H), 7.41 (t, 1H), 7.59 (t, 1H), 7.76 (d, 1H), 8.14 (d, 1H).
Пример 10
а) N-Бензил-2-(3-фтор-4-гидроксифенил)-N-гексилацетамид
3-Фтор-4-гидроксифенилуксусную кислоту (170 мг, 0,999 ммоль), растворенную в диметилформамиде (3 мл), охлаждали на ледяной бане. Добавляли N-гексилбензиламин (201 мг, 1,049 ммоль) и затем ТБТУ (337 мг, 1,049 ммоль) и далее ДИПЭА (407 мг, 3,147 ммоль). Эту смесь перемешивали при комнатной температуре в течение ночи и упаривали. Добавляли в остаток водный раствор гидрокарбоната натрия (насыщенный). Эту смесь затем экстрагировали этилацетатом (×2). Объединяли экстракты, промывали их водой и рассолом, сушили (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 5 г/15 мл) при использовании ДХМ и затем МеОН/ДХМ (1:99) в качестве элюанта давала 265 мг желаемого продукта, выход 77%.
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 0.83-0.89 (m, 3H), 1.22-1.29 (m, 6H), 1.48-1.58 (m, 2H), 3.21, 3.40 (t, t, 2Н), 3.58, 3.68 (s, s, 2H), 4.54, 4.63 (s, s, 2H), 6.72-6.97 (m, 3H), 7.15 (d, 1Н), 7.21-7.32 (m, 3H), 7.35-7.39 (m, 1 Н).
b) Метил-2-[{4-{2-[бензил(гексил)-амино]-2-оксоэтил}-2-фторфенокси)-метил]-бензоат
Смешивали в ацетонитриле (и мл) N-бензил-2-(3-фтор-4-гидроксифенил)-N-гексилацетамид (142 мг, 0,414 ммоль), метиловый эфир 2-бромметилбензойной кислоты (99,4 мг, 0,434 ммоль) и карбонат калия безводный (86 мг, 0,620 ммоль). Эту смесь нагревали при дефлегмации в течение ночи и затем упаривали досуха. Добавляли этилацетат и воду и разделяли две фазы. Органическую фазу промывали рассолом и сушили (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ, МеОН/ДХМ (0,5:59,5) в качестве элюанта давала 144 мг желаемого продукта, выход 71%.
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 0.83-0.89 (m, 3H), 1.20-1.29 (m, 6H), 1.45-1.58 (m, 2H), 3.18, 3.37 (t, t, 2H), 3.58, 3.69 (s, s, 2H), 3.90 (s, 3H), 4.50, 4.60 (s, s, 2H), 5.53, 5.55 (s, s, 2H), 6.82-7.39 (m, 9H), 7.56 (t, 1H), 7.78-7.82 (m, 1H), 8.02 (d, 1H).
c) 2-[(4-{2-[Бензил(гексил)-амино]-2-оксоэтил}-2-фторфенокси)-метил]-бензойная кислота
Метил-2-[(4-{2-[бензил(гексил)-амино]-2-оксоэтил}-2-фторфенокси)-метил]-бензоат (109 мг, 0,222 ммоль) растворяли в тетрагидрофуране (2 мл). Добавляли гидроксид лития (10,6 мг, 0,444 ммоль), растворенный в воде (1 мл). Эту смесь помещали в микроволновую печь (синтезатор Смита) при 150°С на 7 минут. Затем ее подкисляли 1%-ной соляной кислотой, рН˜3 и экстрагировали этилацетатом. Органический экстракт промывали рассолом и сушили (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ и затем МеОН/ДХМ (0,5:99,5, затем 1:99) в качестве элюанта давала 89 мг желаемого продукта, выход 84%.
1H-ЯМР (ротамеры, 300 мГц, CDCl3): δ 0.84-0.92 (m, 3H), 1.26 (s, br, 6H), 1.48-1.60 (m, 2H), 3.21, 3.41 (t, t, 2Н), 3.65, 3.75 (s, s, 2H), 4.55, 4.66 (s, s, 2H), 5.60 (s, 2H), 6.84-7.43 (m, 9Н), 7.62 (t, 1H), 7.85 (d, 1H), 8.16.
Пример 11
a) N-Бензил-N-гексил-2-(4-гидрокси-3-метоксифенил)-ацетамид
Гомованильную кислоту (182 мг, 0,999 ммоль) растворяли в диметилформамиде (3 мл), охлажденном на ледяной бане. Добавляли N-гексилбензиламин (201 мг, 1,049 ммоль) и затем ТБТУ (337 мг, 1,049 ммоль) и далее ДИПЭА (407 мг, 3,147 ммоль). Эту смесь перемешивали при комнатной температуре в течение ночи и упаривали. Добавляли в остаток водный раствор гидрокарбоната натрия (насыщенный). Эту смесь затем экстрагировали этилацетатом (×2). Объединяли экстракты, промывали их водой и рассолом, сушили (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 5 г/15 мл) при использовании ДХМ и затем МеОН/ДХМ (1:99) в качестве элюанта давала 264 мг желаемого продукта, выход 74%.
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 0.83-0.89 (m, 3H), 1.20-1.29 (m, 6H), 1.44-1.56 (m, 2H), 3.18, 3.37 (t, t, 2H), 3.60, 3.71 (s, s, 2H), 3.81 (s, br, 3H), 4.50, 4.61 (s, s, 2H), 5.98 (s. br, 1H), 6.62-6.85 (m, 3H), 7.11-7.36 (m, 5H).
b) Метил-2-[(4-{2-[бензил(гексил)-амино}-2-оксоэтил}-2-метоксифенокси)-метокси]-бензоат
Смешивали в ацетонитриле (5 мл) N-бензил-N-гексил-2-(4-гидрокси-3-метоксифенил)-ацетамид (84 мг, 0,236 ммоль), метиловый эфир 2-бромметилбензойной кислоты (57 мг, 0,248 ммоль) и карбонат калия безводный (49 мг, 0,355 ммоль). Эту смесь нагревали при дефлегмации в течение ночи и затем упаривали досуха. Добавляли этилацетат и воду и разделяли две фазы. Органическую фазу промывали рассолом и сушили (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ, МеОН/ДХМ (0,5:99,5) в качестве элюанта давала 102 мг желаемого продукта, выход 86%.
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 0.82-0.88 (m, 3H), 1,18-1.28 (m, 6H), 1.43-1.55 (m, 2H), 3.17, 3.35 (t, t, 2H), 3.61, 3.71 (s, s, 2H), 3.86 (s, 3H), 3.88 (s, 3Н), 4.48, 4.60 (s, s, 2H), 5.55, 5.56 (s, s, 2H), 6.61-7.35 (m, 9H), 7.52 (t, 1H), 7.78 (d, 1H), 8.01 (d, 1H).
с) 2-[(4-{2-[Бензил(гексил)-амино]-2-оксоэтил}-2-метоксифенокси)-метил]-бензойная кислота
Метил-2-[(4-{2-[бензил(гексил)-амино]-2-оксоэтил}-2-метоксифенокси)-метил]-бензоат (98 мг, 0,195 ммоль) растворяли в тетрагидрофуране (2 мл). Добавляли гидроксид лития (9,3 мг, 0,389 ммоль), растворенный в воде (1 мл). Эту смесь помещали в микроволновую печь (синтезатор Смита) при 150°С на 7 минут. Затем ее подкисляли 1%-ной соляной кислотой, рН˜3 и экстрагировали этилацетатом. Органический экстракт промывали рассолом и сушили (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ и затем МеОН/ДХМ (0,5: 99,5, затем 1:99) в качестве элюанта давала 43 мг желаемого продукта, выход 45%.
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 0.82-0.88 (m, 3Н), 1,18-1.28 (m, 6H), 1.44-1.57 (m, 2H), 3.18, 3.37 (t, t, 2H), 3.64, 3.74 (s, s, 2H), 3.86 (s, 3H), 4.50, 4.62 (s, s, 2H), 5.57, 5.58 (s, s, 2H), 6.63-7.39 (m, 9H), 7.56 (t, 1H), 7.80 (d, 1H), 8.12 (d, 1H).
Пример 12
а) 4-[3-(3,4-Дигидроизохинолин-2(1Н)-ил)-3-оксопропил]-фенол
3-(4-Гидроксифенил)-пропионовую кислоту (202 мг, 1,216 ммоль) в диметилформамиде (3 мл) охлаждали в ледяной бане. Добавляли 1,2,3,4-тетрагидроизохинолин (170 мг, 1,276 ммоль), затем ТБТУ (410 мг, 1,276 ммоль) и далее ДИПЭА (330 мг, 2,553 ммоль). Эту смесь перемешивали при комнатной температуре в течение ночи. Добавляли водный раствор гидрокарбоната натрия (насыщенный). Эту смесь экстрагировали этилацетатом (×2). Объединяли экстракты, сушили их (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ и затем МеОН/ДХМ (1:99) в качестве элюанта давала 303 мг желаемого продукта, выход 89%.
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 2.72-2.77 (m, 2H), 2.83-2.90 (m, 2H), 2.95-3.01 (m, 2H), 3.63, 3.88 (t, t, 2H), 4.57, 4.79 (s, s, 2H), 6.85-6.90 (m, 2H), 7.07-7.26 (m, 6H).
b) Метил-2-({[4-[3-(3,4-дигидроизохинолин-2(1H)-ил)-3-оксопропил]-фенокси}-метил)-бензоат
4-[3-(3,4-дигидроизохинолин-2(1Н)-ил)-3-оксопропил]-фенол (155 мг, 0,551 ммоль) растворяли в ацетонитриле (10 мл). Добавляли 2-бромметилбензойной кислоты метиловый эфир (126 мг, 0,551 ммоль) и далее карбонат калия безводный (114 мг, 0,826 ммоль). Эту смесь нагревали при дефлегмации в течение ночи и затем упаривали досуха. Добавляли воду и этилацетат и разделяли две фазы. Органическую фазу сушили (сульфат магния) и упаривали. Колоночная хроматография остатка на силикагеле при использовании смеси этилацетат/гептан (40:60) в качестве элюанта давала 135 мг желаемого продукта, выход 57%.
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 2.69-2.74 (m, 2H), 2.82-2.87 (m, 2H), 2.95-3.01 (m, 2H), 3.62, 3.85 (t, t, 2H), 3.93 (s, 3H), 4.52, 4.73 (s, s, 2H), 5.48, 5.50 (s, s, 2H), 6.91-6.95 (m, 2H), 7.03-7.24 (m, 6H), 7.39 (t, 1H), 7.57 (t, 1H), 7.77 (d, 1Н) и 8.05 (d, 1Н).
с) 2-({4-[3-(3,4-Дигидроизохинолин-2(1Н)-ил)-3-оксопропил]-фенокси}-метил)-бензойная кислота
Растворяли гидроксид лития (14,4 мг, 0,6 ммоль) в воде (1 мл) и добавляли в 70377 метил-2-({4-[3-(3,4-дигидроизохинолин-2(1Н)-ил)-3-оксопропил]-фенокси}-метил)-бензоат (129 мг, 0,3 ммоль) в ТГФ (2 мл). Эту смесь помещали в микроволновую печь (синтезатор Смита) и облучали при 150°С в течение 7 минут и затем упаривали для удаления ТГФ. Остаток подкисляли 1%-ной соляной кислотой, рН˜5, и экстрагировали этилацетатом (×2). Объединяли экстракты, сушили их (сульфат магния) и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ, МеОН/ДХМ (1:99) в качестве элюанта давала 111 мг желаемого продукта, выход 89%.
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 2.70-2.73 (m, 2H), 2.79-2.83 (m, 2H), 2.92-3.00 (m, 2H), 3.58, 3.84 (t, t, 2H), 4.50, 4.76 (s, s, 2H), 5.50, 5.53 (s, s, 2H), 6.87-6.93 (m, 2H), 6.99-7.22 (m, 6H), 7.39 (t, 1H), 7.57 (t, 1H), 7.78 (d, 1H), 8.16 (d, 1Н).
Пример 13
a) Растворяли 4-(2-Гидроксиэтил)-фенол (2 г, 14,48 ммоль) и метил-2-(бромметил)-бензоат (3,48 г, 15,20 ммоль) в ацетонитриле (20 мл). Добавляли карбонат калия безводный (4,0 г, 28,95 ммоль). После перемешивания при 60°С в течение трех часов добавляли PS-трисамин (0,2 экв) и перемешивали эту смесь в течение ночи. PS-трисамин отфильтровывали и ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органический слой тремя порциями воды (3×10 мл). Органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием с получением 3,732 г метил-2-{[4-(2-гидроксиэтил)-фенокси]-метил}-бензоата (выход 90%).
1H-ЯМР (300 мГц, CDCl3): δ 2.37 (bs, 1H). 2.8 (t, 2H), 3.8 (bm, 2H), 3.9 (s, 3Н), 5.5 (s, 2H), 6.95 (d, 2H), 7.15 (d, 2H), 7.25 (t, 1H), 7.55 (t, 1H), 7.75 (d, 1H), 8.05 (d, 1H).
b) Метил-2-{[4-(2-гидроксиэтил)-фенокси]-метил}-бензоат (1,2 г, 4,19 ммоль) растворяли в дихлорметане (20 мл) и этот раствор охлаждали до -20°С. Добавляли каплями триэтиламин (0,64 г, 6,29 ммоль) и затем добавляли каплями метилсульфонилхлорид (0,53 г, 4,61 ммоль). Водяную баню удаляли и смесь перемешивали при комнатной температуре один час. Добавляли диэтиловый эфир (5 мл) и отфильтровывали преципитат. Органическую фазу промывали двумя порциями рассола, сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 M); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Это давало 0,703 г метил-2-[(4-{2-[(метилсульфонил)-окси]-этил}-фенокси)-метил]-бензоата (выход 46%).
1H-ЯМР (300 МГц, CDCl3): δ 2.8 (s, 3H), 2.95 (t, 2H), 3.85 (s, 3H), 4.35 (t, 2Н), 5.45 (s, 2H), 6.9 (d, 2H), 7.15 (d, 2H), 7.35 (t, 1H), 7.5 (t, 1H), 7.7 (d, 1H), 7.98 (d, 1H).
c) Растворяли метил-2-[(4-{2-[(метилсульфонил)-окси]-этил}-фенокси)-метил]-бензоат (0,2 г, 0,55 ммоль) и 4-(1H-имидазол-1-ил)-фенол (0,11 г, 0,66 ммоль) в ацетонитриле (10 мл) и добавляли карбонат калия (0,09 г, 0,66 ммоль). Эту смесь перемешивали в течение ночи при 75°С. Ацетонитрил удаляли упариванием, разбавляли этилацетатом (10 мл) и органическую фазу промывали рассолом три раза, сушили (сульфат магния) и упаривали. Это давало 0,268 г метил-2-[(4-{2-[4-(1Н-имидазол-1-ил)фенокси]-этил}-фенокси)-метил]-бензоата (выход 90%).
1H-ЯМР (300 МГц, CDCl3): δ 3.05 (t, 2H), 3.9 (s, 3H), 4.05-4.2 (bm, 2H), 5.55 (s, 2H), 6.9-7.0 (bm, 4H), 7.1-7.4 (bm, 7H), 7.52 (t, 2H), 7.75 (m, 2H), 7.98 (d, 1H).
d) Растворяли метил-2-[(4-{2-[4-(1H-имидазол-1-ил)фенокси]-этил}-фенокси)-метил]-бензоат (0,12 г, 0,28 ммоль) в смеси тетрагидрофуран/вода (7/1, 5 мл) и добавляли LiOH (0,03 г, 1,13 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Проводили обработку добавлением соляной кислоты (1 мл, 1М), экстрагировали продукт добавлением двух порций EtOAc (5 мл). Объединенные в пул органические фазы сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%, буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 M); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Упаривание давало 6 мг 2-[(4-{2-[4-(1H-имидазол-1-ил)-фенокси]-этил}-феноксиметил]-бензойной кислоты (выход 4,7%).
1H-ЯМР (500 МГц, CDCl3): δ 3.05 (t, 2H), 4.18 (t, 2H), 5.55 (s, 2H), 6.95 (d, 2H), 7.05 (m, 3Н), 7.25 (d, 2H), 7.3-7.45 (bm, 4Н), 7.55 (t, 1H), 7.78 (d, 1H), 7.85 (s, 1H), 8.07 (d, 1H).
Пример 14
a) Растворяли 4-(2-гидроксиэтил)-фенол (2 г, 14,48 ммоль) и метил-2-(бромметил)-бензоат (3,48 г, 15,20 ммоль) в ацетонитриле (20 мл). Добавляли карбонат калия безводный (4,0 г, 28,95 ммоль). После перемешивания при 60°С в течение трех часов добавляли PS-трисамин (0,2 экв) и перемешивали в течение ночи. PS-трисамин отфильтровывали и ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органический слой тремя порциями воды (3×10 мл). Органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием с получением 3,732 г метил-2-{[4-(2-гидроксиэтил)-фенокси]-метил}-бензоата (выход 90%).
1H-ЯМР (300 МГц, CDCl3): δ 2.37 (bs, 1H), 2.8 (t, 2H), 3.8 (bm, 2H), 3.9 (s, 3Н), 5.5 (s, 2H), 6.95 (d, 2H), 7.15 (d, 2H), 7.25 (t, 1H), 7.55 (t, 1H), 7.75 (d, 1H), 8.05 (d, 1H).
b) Добавляли метил-2-{[4-(2-гидроксиэтил)-фенокси]-метил}-бензоат (1 г, 3,49 ммоль), 4-(бензилокси)-фенол (0,7 г, 3,49 ммоль) и трифенилфосфин (1,01 г, 3,84 ммоль) в сухую круглодонную колбу, снабженную перегородкой. Пропускали через колбу в течение 5 минут N2 и далее добавляли сухой толуол (30 мл) и диизопропилазодикарбоксилат (0,78 г, 3,84 ммоль). Эту реакционную смесь перемешивали при 55°С в течение ночи. Растворитель удаляли упариванием и неочищенный материал очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером, была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 M); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). После удаления растворителя упариванием изолировали 0,7 г метил-2-[(4-{2-[4-(бензилокси)-фенокси]-этил}-фенокси)-метил]-бензоата (выход 42,8%).
1H-ЯМР (300 МГц, CDCl3): δ 3.05 (t, 2H), 3.95 (s, 3H), 4.07 (t, 2H), 5.03 (s, 2H), 5.55 (s, 2H), 6.85 (d, 2Н), 6.95 (d, 2H), 6.98 (d, 2Н), 7.23 (d, 2H), 7.3-7.5 (bm, 6Н), 7.6 (t, 1Н), 7.8 (d, 1Н), 8.07 (d, 1Н).
c) Растворяли метил-2-[(4-{2-[4-(бензилокси)-фенокси]-этил}-фенокси)-метил]-бензоат (0,80 г 1,71 ммоль), боронтрифторидэтерат (2,42 г, 17,09 ммоль) и диметилсульфид (1,27 г, 20,51 ммоль) в дихлорметане (25 мл). Эту смесь перемешивали 6 часов при комнатной температуре. Добавляли EtOAc (20 мл) и промывали эту смесь тремя порциями воды, органический слой сушили (сульфат магния) и растворитель удаляли упариванием.
1H-ЯМР (300 мГц, CDCl3): δ 3.0 (t, 2H), 3.9 (s, 3H), 4.1 (t, 2), 5.5 (s, 2H), 5.55 (s, 2H), 6.8 (bm, 4Н), 7.0 (d, 2H), 7.2 (d, 2H), 7.4 (t, 1H), 7.55 (t, 1Н), 7.8 (d, 1H), 8.05 (d.
d) Метил-2-({4-[2-(4-гидроксифенокси)-этил]-фенокси}-метил)-бензоат (0,547 г, 1,45 ммоль) растворяли в дихлорметане (10 мл) и этот раствор охлаждали до -20°С. Добавляли каплями триэтиламин (0,22 г, 2,17 ммоль) и далее добавляли каплями метилсульфонилхлорид (0,18 г, 1,59 ммоль). Водяную баню удаляли и смесь перемешивали в течение ночи при комнатной температуре. Избыток триэтиламина удаляли добавлением диэтилового эфира (5 мл) и отфильтровыванием преципитата. Органическую фазу промывали тремя порциями рассола (10 мл) и сушили (сульфат магния). Растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%, буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М), колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин)). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 0,317 г метил-2-{[4-(2-{4-[(метилсульфонил)-окси]-фенокси)-этил)-фенокси]-метил}-бензоата (выход 48%).
1H-ЯМР (500 МГц, CDCl3): δ 3.07 (t, 2H), 3.15 (s, 3Н), 3.95 (s, 3H), 4.17 (t, 2H), 5.55 (s, 2H), 6.95 (d, 2H), 7.0 (d, 2H), 7.23 (m, 4H), 7.42 (t, 1Н), 7.6 (t, 1H), 7.8 (d, 1H), 8.08 (d, 1H).
е) Метил-2-{[4-(2-(4-[(метилсульфонил)-окси]-фенокси}-этил)-фенокси]-метил}-бензоат (0,18 г, 0,38 ммоль) растворяли в смеси тетрагидрофурана и воды 7:1 (5 мл). Взаимодействие осуществляли в одноузловой микроволновой печи (150°С за 7 минут). Смесь разбавляли соляной кислотой (2 мл, 1М) и органическую фазу изолировали. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%, буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М), колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 13 мг 2-{[4-(2-{4-[(метилсульфонил)-окси]-фенокси}-этил)-фенокси]-метил}-бензойной кислоты (выход 7,7%).
1H-ЯМР (300 мГц, CDCl3): δ 3.07 (t, 2H), 3.15 (s, 3Н), 4.17 (t, 2H) 5.6 (s, 2H), 6.95 (d, 2H), 7.0 (d, 2H), 7.23 (m, 4H), 7.42 (t, 1H), 7.65 (t, 1H), 7.82 (d, 1H), 8.2 (d, 1H).
Пример 15
а) Растворяли 3-(2-гидроксиэтил)-фенол (1,0 г, 7,24 ммоль) и метил-2-(бромметил)-бензоат (1,74 г, 7,6 ммоль) в ацетонитриле (10 мл). Добавляли карбонат калия безводный (2,0 г, 14,48 ммоль). После перемешивание при 60°С в течение трех часов добавляли PS-трисамин (0,3 экв) и перемешивали в течение ночи. PS-трисамин отфильтровывали и ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органический слой тремя порциями воды (3×10 мл). Органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием с получением 1,99 г метил-2-{[3-(2-гидроксиэтил)-фенокси]-метил}-бензоата (выход 90%).
1Н-ЯМР (300 мГц, CDCl3): δ 2.95 (t, 2Н), 3.45 (s, 1H), 3.9-4.0 (bm, 5H), 5.58 (s, 2Н), 6.95 (m, 2H), 7.05 (s, 1H), 7.3 (t, 1H), 7.45 (t, 1H), 7.6 (t, 1H), 7.85 (d, 1H), 8.05 (d, 1H).
b) Добавляли метил-2-{[3-(2-гидроксиэтил)-фенокси]-метил}-бензоат (0,5 г, 1,75 ммоль), 4-(бензилокси)-фенол (0,35 г, 1,75 ммоль) и трифенилфосфин (0,5 г, 1,92 ммоль) в сухую круглодонную колбу, снабженную перегородкой. Добавляли сухой толуол (10 мл) и пропускали через эту смесь N2 в течение 5 минут. Добавляли каплями диизопропил-(Е)-диазен-1,2-дикарбоксилат (0,39 г, 1,92 ммоль) и перемешивали раствор при комнатной температуре. После трех часов добавляли еще эквивалент реагентов и перемешивали в течение одного часа. После удаления растворителя упариванием неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до100%, буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М), колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 0,319 г метил-2-[(3-{2-[4-(бензилокси)-фенокси]-этил}-фенокси)-метил]-бензоата (выход 39%).
1H-ЯМР (300 мГц, CDCl3): δ 3.15 (t, 2H), 3.95 (s, 3Н), 4.2 (t, 2H), 5.07 (s, 2H), 5.6 (s, 2H), 6.9-7.1 (bm, 7H), 7.3-7.55 (bm, 7Н), 7.6 (t, 1H), 7.85 (d. 1H), 8.05 (d, 1H).
c) Растворяли метил-2-[(3-{2-[4-(бензилокси)-фенокси]-этил}-фенокси)-метилбензоат (20 мг, 0,043 ммоль) в смеси тетрагидрофуран/Н2О (7/1, 3 мл) и добавляли LiOH (4,1 мг, 0,17 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Смесь подкисляли (соляная кислота, 1 мл, 1 М) и водную фазу промывали двумя порциями EtOAc (3×5 мл). После удаления растворителя упариванием неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%, буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 19 мг 2-[(3-{2-[4-(бензилокси)-фенокси]-этил}-фенокси)-метил]-бензойной кислоты (выход 98%).
1H-ЯМР (300 МГц, CDCl3): δ 3.1 (t, 2H), 4.15 (t, 2Н), 5.03 (s, 2H), 5.57 (s, 2Н), 6.8-7.0 (bm, 7H), 7.2-7.5 (bm, 7H), 7.6 (t, 1Н), 7.85 (d. 1H), 8.15 (d, 1H).
Пример 16
a) Растворяли 3-(2-гидроксиэтил)-фенол (1,0 г, 7,24 ммоль) и метил 2-(бромметил)-бензоат (1,74 г, 7,6 ммоль) в ацетонитриле (10 мл). Добавляли карбонат калия безводный (2,0 г, 14,48 ммоль). После перемешивания при 60°С в течение трех часов добавляли PS-трисамин (0,3 экв) и перемешивали в течение ночи. PS-трисамин отфильтровывали и ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органический слой тремя порциями воды (3×10 мл). Органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием с получением 1,99 г метил-2-{[3-(2-гидроксиэтил)-фенокси]-метил}-бензоата (выход 90%).
1H-ЯМР (300 МГц, CDCl3): δ 2.95 (t. 2H), 3.45 (s, 1H), 3.9-4.0 (bm, 5H), 5.58 (s, 2H), 6.95 (m, 2H), 7.05 (s, 1H), 7.3 (t, 1H), 7.45 (t, 1H), 7.6 (t, 1H), 7.85 (d. 1H), 8.05 (d, 1H).
b) Добавляли метил-2-{[3-(2-гидроксиэтил)-фенокси]-метил}-бензоат (0,5 г, 1,75 ммоль), 4-(бензилокси)-фенол (0,35 г, 1,75 ммоль) и трифенилфосфин (0,5 г, 1,92 ммоль) в сухую круглодонную колбу, снабженную перегородкой. Добавляли сухой толуол (10 мл) и пропускали через эту смесь N2 в течение 5 минут. Добавляли каплями диизопропил-(Е)-диазен-1,2-дикарбоксилат (0,39 г, 1,92 ммоль) и перемешивали этот раствор при комнатной температуре. После трех часов добавляли еще эквивалент реагентов и перемешивали один час. После удаления растворителя упариванием неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм, скорость тока 40 мл/мин)). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 0,319 г метил-2-[(3-{2-[4-(бензилокси)-фенокси]-этил}-фенокси)-метил]-бензоата (выход 39%).
1H-ЯМР (300 мГц, CDCl3): δ 3.15 (t, 2H), 3.95 (s, 3Н), 4.2 (t, 2H), 5.07 (s, 2Н), 5.6 (s. 2H), 6.9-7.1 (bm, 7H), 7.3-7.55 (bm, 7H), 7.6 (t, 1H), 7.85 (d. 1H), 8.05 (d, 1Н).
с) Растворяли метил-2-[(3-{2-[4-(бензилокси)-фенокси]-этил}-фенокси)-метил]-бензоат (0,275 г, 0,59 ммоль) в дихлорметане (10 мл), добавляли диметилсульфид (0,44 г, 7,0 ммоль) и боронтрифторидэтерат (0,83 г, 5,87 ммоль) и перемешивали эту смесь при комнатной температуре в течение шести часов. Добавляли EtOAc (10 мл) и промывали органическую фазу водой (3×10 мл), сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). После удаления растворителя упариванием получали 0,096 граммов метил-2-({3-[2-(4-гидроксифенокси)-этил]-фенокси}-метил)-бензоата (выход 43,2%). Этот продукт использовали прямо в следующей стадии.
d) Метил-2-({3-[2-(4-гидроксифенокси)-этил]-фенокси}-метил)-бензоат (0,096 г, 0,25 ммоль) растворяли в дихлорметане (10 мл) и охлаждали до -20°С. Добавляли каплями триэтиламин (0,039 g, 0,38 ммоль) и дооавляли каплями метансульфонилхлорид (0,032 г, 0,28 ммоль). Водяную баню удаляли и смесь нагревали до комнатной температуры. Добавляли диэтиловый эфир (5 мл) и отфильтровывали преципитат. Органическую фазу промывали двумя порциями рассола (5 мл) и сушили (сульфат магния). Растворитель удаляли упариванием с получением 0,109 граммов метил-2-{[3-(2-{4-[(метилсульфонил)-окси]-фенокси}-этил)-фенокси]-метил}-бензоата (выход 94,1%).
1H-ЯМР (500 мГц, CDCl3): δ 3.15 (m, 5Н), 3.95 (s, 3H), 4.2 (t, 20), 5.55 (s, 2Н), 6.95 (s, 4Н), 7.0 (s, 1H), 7.25 (d, 2H), 7.3 (t, 1H), 7.42 (t, 1H), 7.6 (t, 1H), 7.8 (d, 1H), 8.1 (d, 1H).
e) Метил-2-{[3-(2-{4-[(метилсульфонил)-окси]-фенокси}-этил)-фенокси]-метил}-бензоат (0,109 г, 0,24 ммоль) растворяли в смеси тетрагидрофуран/вода (7/1, 2,5 мл). Добавляли гидроксид лития (23 мг, 0,96 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Смесь подкисляли (соляная кислота, 1 мл, 1 M) и водную фазу экстрагировали двумя порциями EtOAc (2×5 мл). Органические фазы объединяли, сушили (сульфат магния) и растворитель удаляли упариванием, что давало 17 мг 2-{[3-(2-{4-[(метилсульфонил)-окси]-фенокси}-этил)-фенокси]-метил}-бензойной кислоты (выход 16%).
1H-ЯМР (500 мГц, CDCl3): δ 3.15 (m, 5H), 4.2 (t, 2H), 5.55 (s, 2H), 6.95 (s, 4Н), 7.0 (s, 1H), 7.25 (d, 2H), 7.3 (t, 1H), 7.42 (t, 1H), 7.6 (t, 1H), 7.8 (d. 1H), 8.1 (d, 1H).
Пример 17
a) Растворяли 3-(2-гидроксиэтил)-фенол (1,0 г, 7,24 ммоль) и метил 2-(бромметил)-бензоат (1,74 г, 7,6 ммоль) в ацетонитриле (10 мл). Добавляли карбонат калия безводный (2,0 г, 14,48 ммоль). После перемешивание при 60°С в течение трех часов добавляли PS-трисамин (0,3 экв) и перемешивали в течение ночи. PS-трисамин отфильтровывали и ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органический слой тремя порциями воды (3×10 мл). Органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием с получением 1,99 г метил-2-{[3-(2-гидроксиэтил)-фенокси]-метил}-бензоата (выход 90%).
1H-ЯМР (300 мГц, CDCl3): δ 2.95 (t. 2H), 3.45 (s, 1H), 3.9-4.0 (bm, 5H), 5.58 (s, 2H), 6.95 (m, 2H), 7.05 (s, 1H), 7.3 (t, 1H), 7.45 (t, 1H), 7.6 (t, 1H), 7.85 (d, 1H), 8.05 (d, 1H).
b) Добавляли метил-2-{[3-(2-гидроксиэтил)-фенокси]-метил}-бензоат (0,5 г, 1,75 ммоль), 4-(бензилокси)-фенол (0,35 г, 1,75 ммоль) и трифенилфосфин (0,5 г, 1,92 ммоль) в сухую круглодонную колбу и снабжали ее перегородкой. Добавляли сухой толуол (10 мл) и пропускали через эту смесь N2 в течение 5 минут. Добавляли каплями диизопропил-(Е)-диазен-1,2-дикарбоксилат (0,39 г, 1,92 ммоль) и перемешивали этот раствор при комнатной температуре. После трех часов добавляли еще эквивалент реагентов и перемешивали один час. После удаления растворителя упариванием неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем увеличивали концентрацию ацетонитрила до 100%, буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 M); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 0,319 г метил-2-[(3-{2-[4-(бензилокси)-фенокси]-этил}-фенокси)-метил]-бензоата (выход 39%).
1H-ЯМР (300 мГц, CDCl3): δ 3.15 (t, 2H), 3.95 (s, 3Н), 4.2 (t, 2H), 5.07 (s, 2Н), 5.6 (s, 2Н), 6.9-7.1 (bm, 7H), 7.3-7.55 (bm, 7Н), 7.6 (t, 1H), 7.85 (d, 1H), 8.05 (d, 1H).
c) Растворяли метил-2-[(3-{2-[4-(бензилокси)-фенокси]-этил}-фенокси)-метил]-бензоат (0,275 г, 0,59 ммоль) в дихлорметане (10 мл), добавляли диметилсульфид (0,44 г, 7,0 ммоль) и боронтрифторидэтерат (0,83 г, 5,87 ммоль) и перемешивали эту смесь при комнатной температуре в течение шести часов. Добавляли EtOAc (10 мл) и промывали органическую фазу водой (3×10 мл) и сушили ее (сульфат магния), растворитель удаляли упариванием и неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). После удаления растворителя упариванием получали 0,096 граммов метил-2-({3-[2-(4-гидроксифенокси)-этил]-фенокси}-метил)-бензоата (выход 43,2%). Этот продукт использовали прямо в следующей стадии.
d) Метил-2-({3-[2-(4-гидроксифенокси)-этил]-фенокси)-метил)-бензоат (0,096 г, 0,25 ммоль) растворяли в дихлорметане (10 мл) и охлаждали до -20°С. Добавляли каплями триэтиламин (0,039 g, 0,38 ммоль) и добавляли каплями метансульфонилхлорид (0,032 г, 0,28 ммоль). Водяную баню удаляли и смесь нагревали до комнатной температуры. Добавляли диэтиловый эфир (5 мл) и отфильтровывали преципитат, промывали органическую фазу двумя порциями рассола (5 мл) и сушили (сульфат магния). Удаление растворителя упариванием давало 0,109 граммов метил-2-{[3-(2-{4-[(метилсульфонил)-окси]-фенокси }-этил)-фенокси]-метил}-бензоата (выход 94,1%).
1H-ЯМР (500 мГц, CDCl3): δ 3.15 (m, 5Н), 3.95 (s, 3Н), 4.2 (t, 2Н), 5.55 (s, 2Н), 6.95 (s, 4H), 7.0 (s, 1H), 7.25 (d, 2H), 7.3 (t, 1H), 7.42 (t, 1H), 7.6 (t, 1H), 7.8 (d, 1H), 8.1 (d, 1H).
f) Метил-2-([3-(2-{4-[(метилсульфонил)-окси]-фенокси)-этил)-фенокси]-метил}-бензоат (0,109 г, 0,24 ммоль) растворяли в смеси тетрагидрофуран/вода (7/1, 2,5 мл). Добавляли гидроксид лития (23 мг, 0,96 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Смесь подкисляли (соляная кислота, 1 мл, 1М) и водную фазу экстрагировали двумя порциями EtOAc. Органические фазы объединяли, сушили (сульфат магния) и растворитель удаляли упариванием, что давало 16 мг 2-({3-[2-(4-гидроксифенокси)-этил]-фенокси}-метил)-бензойной кислоты (выход 18%).
1H-ЯМР (400 мГц, CDCl3): δ 3.0 (t, 2Н), 4.1 (t, 2H), 5.55 (s, 2H), 6.7 (m, 4H), 6.8-6.95 (bm, 3Н), 7.4 (m, 1Н), 7.6 (m, 1Н), 7.8 (m, 1Н), 8.15 (m, 1H).
Пример 18
a) Растворяли 4-(3-гидроксипропил)-фенол (1,0 г, 6,57 ммоль) и метил-2-(бромметил)-бензоат (1,66 г, 7,23 ммоль) в ацетонитриле (10 мл). Добавляли карбонат калия (1,82 г, 13,14 ммоль) и перемешивали эту смесь при 60°С три часа. Добавляли поддерживаемый полимером трисамин (0,3 экв.) и перемешивали раствор при комнатной температуре в течение ночи. PS-трисамин отфильтровывали и ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органический слой тремя порциями воды (3×10 мл). Органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием с получением 1,66 граммов метил-2-{[4-(3-гидроксипропил)-фенокси]-метил}-бензоата (выход 84,2%).
1H-ЯМР (500 мГц, CDCl3): δ 1.9 (m, 2H), 2.65 (t, 2H), 3.25 (s, 1H), 3.65 (t, 2H), 3.85 (s, 3Н), 5.45 (s, 2H), 6.95 (d, 2H), 7.15 (d, 2H), 7.35 (t, 1H), 7.5 (t, 1H), 7.75 (d, 1H), 8.05 (d, 1H).
b) Добавляли метил-2-{[4-(3-гидроксипропил)-фенокси]-метил}-бензоат (0,50 г, 1,66 ммоль) и 4-(бензилокси)-фенол (0,33 г, 1,66 ммоль) в сухую круглодонную колбу, снабженную перегородкой. Добавляли сухой толуол (10 мл) и пропускали через эту смесь N2 в течение 5 минут. Добавляли каплями (трибутилфосфоранилиден)-ацетонитрил (0,80 г, 3,33 ммоль) и осуществляли взаимодействие в одноузловой микроволновой печи. После удаления растворителя упариванием неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм, скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 0,515 граммов метил-2-[(4-{3-[4-(бензилокси)-фенокси]-пропил}-фенокси)-метил]-бензоата (выход 64,1%).
1H-ЯМР (500 МГц, CDCl3): δ 2.15 (m, 2H), 2.85 (t, 2Н), 4.0 (m, 5H), 5.1 (s, 2Н), 5.6 (s, 2H), 6.9-7.1 (bm, 6Н), 7.22 (d, 2H), 7.35-7.55 (bm, 6H), 7.62 (t, 1H), 7.9 (d, 1H), 8.15 (d, 1H).
с) Растворяли метил-2-[(4-{3-[4-(бензилокси)-фенокси]-пропил)-фенокси)-метил]-бензоат (0,047 г, 0,097 ммоль) в смеси тетрагидрофуран/вода (7/1,2 мл) и добавляли гидроксид лития (9,3 мг, 0,39 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут; 150°С). Эту реакционную смесь подкисляли (соляная кислота, 1М, 1 мл) и водную фазу промывали двумя порциями EtOAc (2×5 мл). Органические фазы объединяли, сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%, буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 2 мг 2-[(4-(3-[4-(бензилокси)-фенокси]-пропил}-фенокси)-метил]-бензойной кислоты (выход 4,4%).
1H-ЯМР (500 мГц, CDCl3): δ 2.05 (m, 2H), 2.75 (t, 2H), 4.0 (t, 2H), 5.05 (s, 2H), 5.6 (s, 2H), 6.8-7.0 (bm, 6H), 7.15 (d, 2H), 7.35-7.55 (bm, 6H), 7.55 (t, 1H), 7.8 (d, 1H), 8.15 (d, 1H).
Пример 19
a) Растворяли 4-(3-гидроксипропил)-фенол (1,0 г, 6,57 ммоль) и метил-2-(бромметил)-бензоат (1,66 г, 7,23 ммоль) в ацетонитриле (10 мл). Добавляли карбонат калия (1,82 г, 13,14 ммоль) и перемешивали эту смесь при 60°С в течение трех часов. Добавляли поддерживаемый полимером трисамин (0,3 экв.) и перемешивали раствор при комнатной температуре в течение ночи. PS-трисамин отфильтровывали и ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органический слой тремя порциями воды (3×10 мл). Органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием с получением 1,66 граммов метил-2-{[4-(3-гидроксипропил)-фенокси]-метил}-бензоата (выход 84,2%).
1H-ЯМР (500 мГц, CDCl3): δ 1.9 (m, 2Н), 2.65 (t, 2H), 3.25 (s, 1H), 3.65 (t, 2H), 3.85 (s, 3Н), 5.45 (s, 2H), 6.95 (d, 2H), 7.15 (d, 2H), 7.35 (t, 1H), 7.5 (t, 1H), 7.75 (d, 1H), 8.05 (d, 1H).
b) Добавляли метил-2-{[4-(3-гидроксипропил)-фенокси]-метил}-бензоат (0,50 г, 1,66 ммоль) и 4-(бензилокси)-фенол (0,33 г, 1,66 ммоль) в сухую круглодонную колбу и снабжали ее перегородками. Добавляли сухой толуол (10 мл) и пропускали через эту смесь N2 в течение 5 минут. Добавляли каплями (трибутилфосфоранилиден)-ацетонитрил (0,80 г, 3,33 ммоль) и осуществляли взаимодействие в одноузловой микроволновой печи. После удаления растворителя упариванием неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%, буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 M); колонка KR-100-7-C8, 50 мм ×250 мм, скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 0,515 граммов метил-2-[(4-{3-[4-(бензилокси)-фенокси]-пропил}-фенокси)-метил]-бензоата (выход 64,1%).
1H-ЯМР (500 МГц, CDCl3): δ 2.15 (m, 2H), 2.85 (t.2H), 4.0 (m, 5H), 5.1 (s, 2Н), 5.6 (s, 2Н), 6.9-7.1 (bm, 6H), 7.22 (d, 2Н), 7.35-7.55 (bm, 6H), 7.62 (t, 1H), 7.9 (d, 1H), 8.15 (d, 1H).
c) Метил-2-[(4-{3-[4-(бензилокси)-фенокси]-пропил}-фенокси)-метил]-бензоат (0,70 г, 1,45 ммоль) растворяли в дихлорметане (10 мл). Добавляли диметилсульфид (1,08 г, 17,4 ммоль) и боронтрифторидэтерат (2,06 г, 14,5 ммоль) и перемешивали эту смесь при комнатной температуре в течение шести часов. Добавляли EtOAc (10 мл) и промывали органическую фазу водой (3×10 мл), сушили ее (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 M); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). После удаления растворителя упариванием получали 0,328 граммов метил-2-({4-[3-(4-гидроксифенокси)-пропил]-фенокси)-метил)-бензоата (выход 57,6%).
1H-ЯМР (500 мГц, CDCl3): δ 2.05 (m, 2Н), 2.75 (t, 2H), 3.9 (m, 5H), 5.5 (s, 2H), 6.65-6.8 (bm, 4Н), 6.95 (d, 2H), 7.15 (d, 2H), 7.4 (t, 1H), 7.55 (t, 1H), 7.8 (d, 1H), 8.05 (d, 1H).
d) Метил 2-({4-[3-(4-гидроксифенокси)-пропил]-фенокси}-метил)-бензоат (0,32 г, 0,81 ммоль) растворяли в дихлорметане (10 мл) и охлаждали до -20°С. Добавляли каплями триэтиламин (0,123 г, 1,22 ммоль) и добавляли каплями метансульфонилхлорид (0,10 г, 0,89 ммоль). Водяную баню удаляли и смесь нагревали до комнатной температуры. Добавляли диэтиловый эфир (5 мл) и отфильтровывали преципитат. Органическую фазу промывали двумя порциями рассола (5 мл) и сушили (сульфат магния). Удаление растворителя упариванием давало 0,37 граммов метил-2-{[4-(3-{4-[(метилсульфонил)-окси]-фенокси}-пропил)-фенокси]-метил}-бензоата (выход 97,3%).
1H-ЯМР (500 МГц, CDCl3): δ 2.1 (m, 2H), 2.75 (t. 2H), 3.15 (s, 3H), 3.9 (m, 5Н), 5.5 (s, 2H), 6.9-7.0 (bm, 4Н), 7.15 (d, 2H), 7.22 (d, 2H), 7.4 (t, 1Н), 7.55 (t, 1H), 7.8 (d, 1H), 8.05 (d, 1H).
e) Растворяли метил-2-{[4-(3-{4-[(метилсульфонил)-окси]-фенокси}-пропил)-фенокси]-метил}-бензоат (0,38 г, 0,81 ммоль) в смеси тетрагидрофуран/вода (7/1, 4 мл) и добавляли гидроксид лития (9,3 мг, 0,39 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Реакционную смесь подкисляли (соляная кислота, 1 M, 1 мл) и водную фазу промывали двумя порциями EtOAc (2×5 мл). Объединяли органические фазы, сушили их (сульфат магния) и удаляли растворитель упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 M); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 88 мг 2-{[4-(3-{4-[(метилсульфонил)-окси]-фенокси)-пропил)-фенокси]-метил)-бензойной кислоты (выход 23,7%).
1H-ЯМР (500 мГц, CDCl3): δ 2.1 (m, 2H), 2.75 (t, 2H), 3.15 (s, 3H), 3.95 (t, 2H), 5.58 (s, 2H), 6.9-7.05 (bm, 4H), 7.15-7.25 (bm, 4H), 7.45 (t, 1H), 7.65 (t, 1H), 7.85 (d, 1H), 8.2 (d, 1H).
Пример 20
а) Растворяли 4-(3-гидроксипропил)-фенол (1,0 г, 6,57 ммоль) и метил 2-(бромметил)-бензоат (1,66 г, 7,23 ммоль) в ацетонитриле (10 мл). Добавляли карбонат калия (1,82 г, 13,14 ммоль) и перемешивали эту смесь при 60°С в течение трех часов. Добавляли поддерживаемый полимером трисамин (0,3 экв.) и перемешивали раствор при комнатной температуре в течение ночи. PS-трисамин отфильтровывали и ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органический слой тремя порциями воды (3×10 мл). Органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием с получением 1,66 граммов метил-2-{[4-(3-гидроксипропил)-фенокси]-метил}-бензоата (выход 84,2%).
1H-ЯМР (500 мГц, CDCl3): δ 1.9 (m, 2Н), 2.65 (t, 2H), 3.25 (s, 1H), 3.65 (t, 2H), 3.85 (s, 3Н), 5.45 (s, 2H), 6.95 (d, 2H), 7.15 (d, 2H), 7.35 (t, 1H), 7.5 (t, 1H), 7.75 (d, 1H), 8.05 (d, 1H).
b) Добавляли метил-2-([4-(3-гидроксипропил)-фенокси]-метил}-бензоат (0,50 г, 1,66 ммоль) и 4-(бензилокси)-фенол (0,33 г, 1,66 ммоль) в сухую круглодонную колбу и снабжали ее перегородкой. Добавляли сухой толуол (10 мл) и пропускали через эту смесь N2 в течение 5 минут. Добавляли каплями (трибутилфосфоранилиден)-ацетонитрил (0,80 г, 3,33 ммоль) и осуществляли взаимодействие в одноузловой микроволновой печи. После удаления растворителя упариванием неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 M); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мм). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 0,515 граммов метил-2-[(4-{3-[4-(бензилокси)-фенокси]-пропил}-фенокси)-метил]-бензоата (выход 64,1%).
1H-ЯМР (500 мГц, CDCl3): δ 2.15 (m, 2H), 2.85 (t, 2H), 4.0 (m, 5H), 5.1 (s, 2H), 5.6 (s, 2H), 6.9-7.1 (bm, 6H), 7.22 (d, 2H), 7.35-7.55 (bm, 6H), 7.62 (t, 1H), 7.9 (d, 1H), 8.15 (d, 1H).
c) Метил-2-[(4-{3-[4-(бензилокси)-фенокси]-пропил)-фенокси)-метил]-бензоат (0,70 г, 1,45 ммоль) растворяли в дихлорметане (10 мл). Добавляли диметилсульфид (1,08 г, 17,4 ммоль) и боронтрифторидэтерат (2,06 г, 14,5 ммоль) и перемешивали эту смесь при комнатной температуре в течение шести часов. Добавляли EtOAc (10 мл) и промывали органическую фазу водой (3×10 мл), сушили ее (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). После удаления растворителя упариванием получали 0,328 граммов метил-2-({4-[3-(4-гидроксифенокси)-пропил]-фенокси}-метил)-бензоата (выход 57,6%).
1H-ЯМР (500 мГц, CDCl3): δ 2.05 (m, 2H), 2.75 (t, 2H), 3.9 (m, 5H), 5.5 (s, 2H), 6.65-6.8 (bm, 4Н), 6.95 (d, 2H), 7.15 (d, 2H), 7.4 (t, 1Н), 7.55 (t, 1H), 7.8 (d, 1H), 8.05 (d, 1H).
d) Метил-2-({4-[3-(4-гидроксифенокси)-пропил]-фенокси}-метил)-бензоат (0,32 г, 0,81 ммоль) растворяли в дихлорметане (10 мл) и охлаждали до -20°С. Добавляли каплями триэтиламин (0,123 г, 1,22 ммоль) и добавляли каплями метансульфонилхлорид (0,10 г, 0,89 ммоль). Водяную баню удаляли и смесь нагревали до комнатной температуры. Добавляли диэтиловый эфир (5 мл) и отфильтровывали преципитат. Органическую фазу промывали двумя порциями рассола (5 мл) и сушили (сульфат магния). Удаление растворителя упариванием давало 0,37 граммов метил-2-{[4-(3-{4-[(метилсульфонил)-окси]-фенокси)-пропил)-фенокси]-метил}-бензоата (выход 97,3%).
1H-ЯМР (500 мГц, CDCl3): δ 2.1 (m, 2H), 2.75 (t, 2H), 3.15 (s, 3Н), 3.9 (m, 5H), 5.5 (s, 2H), 6.9-7.0 (bm, 4H), 7.15 (d, 2H), 7.22 (d, 2H), 7.4 (t, 1H), 7.55 (t, 1H), 7.8 (d, 1H), 8.05 (d, 1H).
e) Растворяли метил-2-{[4-(3-{4-[(метилсульфонил)-окси]-фенокси}-пропил)-фенокси]-метил)-бензоат (0,38 г, 0,81 ммоль) в смеси тетрагидрофуран/вода (7/1, 4 мл) и добавляли гидроксид лития (9,3 мг, 0,39 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Реакционную смесь подкисляли (соляная кислота, 1М, 1 мл) и водную фазу промывали двумя порциями EtOAc (2×5 мл). Объединяли органические фазы, сушили их (сульфат магния) и удаляли растворитель упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь а цетонитр ил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 63 мг 2-({4-[3-(4-гидроксифенокси)-пропил]-фенокси}-метил)-бензойной кислоты (выход 20,5%).
1H-ЯМР (500 МГц, CDCl3): δ 2.05 (m, 2H), 2.75 (t, 2Н), 3.9 (t, 2H), 5.58 (s, 2Н), 6.65-6.8 (bm, 4H), 6.95 (d. 2H), 7.15 (d, 2H), 7.4 (t, 1H), 7.65 (t, 1H), 7.85 (d, 1H), 8.2 (d, 1H).
Пример 21
a) Растворяли 2-(2-этоксифенил)-этанамин (0,55 г, 3,33 ммоль) и 3-(4-гидроксифенил)-пропаноевую кислоту (0,50 г, 3,00 ммоль) в диметилформамиде (5 мл) и охлаждали до 0°С. Добавляли тетрафторборат N-[(1H-1,2,3-бензотриазол-1-илокси)(диметиламино)-метилен]-N-метилметан-аминия (1,18 г, 3,66 ммоль) и диизопропилэтиламин (0,90 г, 7,0 ммоль) и нагревали раствор до комнатной температуры и перемешивали в течение ночи. Добавляли EtOAc (15 мл) и промывали органическую фазу двумя порциями гидрокарбоната натрия (водный, 10 мл). Органическую фазу сушили (сульфат магния) и EtOAc удаляли упариванием с получением 0,98 граммов N-[2-(2-этоксифенил)-этил]-3-(4-гидроксифенил)-пропанамида (выход 93,9%).
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 1.42 (t, 3H), 2.42 (t, 2H), 2.8-2.92 (m, 4H), 3.55 (q, 2H), 4.05 (q, 2H), 6.08 (m, 1H), 6.82-6.93 (m, 4H), 6.96-7.1 (m, 3H), 7.22 (t, 1H).
b) Растворяли N-[2-(2-этоксифенил)-этил]-3-(4-гидроксифенил)-пропанамид (0,35 г, 1,12 ммоль) и метил-2-(бромметил)-бензоат (0,28 г, 1,23 ммоль) в ацетонитриле (5 мл) и добавляли карбонат калия (324 мг, 2,34 ммоль). Эту смесь перемешивали при 60°С в течение трех часов. Добавляли поддерживаемый полимером трисамин (0,3 экв.) и перемешивали в течение ночи. Полимер отфильтровывали, растворитель удаляли упариванием, добавляли EtOAc (10 мл) и промывали органическую фазу тремя порциями воды. После сушки неочищенного продукта (сульфат магния) и удаления растворителя упариванием неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 26 мг метил-2-{[4-(3-{[2-(2-этоксифенил)-этил]-амино}-3-оксопропил)-фенокси]-метил}-бензоата (выход 50,4%).
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 1.38 (t, 3Н), 2.35 (t, 2H), 2.76 (t, 2H), 2.85 (t, 2H), 3.45 (q, 2H), 3.86 (s, 3Н), 3.99 (q, 2H), 5.44 (s, 2H). 5.84 (m, 1H), 6.78-6.9 (m, 4H), 6.96-7.03 (m, 3Н), 7.15 (t, 1H), 7.32 (t, 1H), 7.5 (t, 1H), 7.72 (d, 1H), 8.0 (d, 1H).
с) Растворяли метил-2-{[4-(3-{[2-(2-этоксифенил)-этил]-амино}-3-оксопропил)-фенокси]-метил}-бензоат (0,26 г, 0,56 ммоль) в смеси ТГФ/вода (7/1, 5 мл) и добавляли гидроксид лития (54 мг, 2,25 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Реакционную смесь подкисляли (соляная кислота, 1М, 1 мл) и водную фазу промывали двумя порциями EtOAc (2×5 мл). Объединяли органические фазы, сушили их (сульфат магния) и удаляли растворитель упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 136 мг 2-{[4-(3-{[2-(2-этоксифенил)-этил]-амино}-3-оксопропил)-фенокси]-метил}-бензойной кислоты (выход 53,9%).
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 1.42 (t, 3H), 2.38 (t, 2H), 2.8 (t, 2H), 2.9 (t, 2H), 3.42 (q, 2H), 3.99 (q,2H), 5.55 (s, 2H), 6.81-6.95 (m, 4H), 7.05-7.17 (m, 4H), 7.38 (t, 1Н), 7.55 (m, 1Н), 7.81 (d, 1Н), 8.11 (d, 1Н).
Пример 22
a) Растворяли N-этил-N-(2-пиридин-2-илэтил)-амин (0,5 г, 3,32 ммоль) и 3-(4-гидроксифенил)-пропаноевую кислоту (0,50 г, 3,00 ммоль) в диметилформамиде (5 мл) и охлаждали до 0°С. Добавляли тетрафторборат N-[(1Н)-1,2,3-бензотриазол-1-илокси)(диметиламино)-метилен]-N-метилметанаминия (1,18 г, 3,66 ммоль) и диизопропилэтиламин (0,90 г, 7,0 ммоль) нагревали раствор до комнатной температуры и перемешивали в течение ночи. Добавляли EtOAc (15 мл) и промывали органическую фазу двумя порциями гидрокарбоната натрия (водный, 10 мл). Органическую фазу сушили (сульфат магния) и EtOAc удаляли упариванием с получением 0,913 граммов N-этил-3-(4-гидроксифенил)-N-(2-пиридин-2-илэтил)-пропанамида (выход 91,9%).
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 0.97, 1.05 (t, t, 3H), 2.41, 2.52 (t, t, 2H), 2.75-3.0 (m, 4H), 3.09, 3.33 (q, q, 2H), 3.54, 3.61 (t. t. 2H), 6.74-6.82 (m, 2H), 6.93-7.2 (m, 4H), 7.58 (m, 1H), 8.48 (m, 1H).
b) Растворяли N-этил-3-(4-гидроксифенил)-N-(2-пиридин-2-илэтил)-пропанамид (0,35 г, 1,17 ммоль) и метил-2-(бромметил)-бензоат (0,30 г, 1,29 ммоль) в ацетонитриле (5 мл) и добавляли карбонат калия (324 мг, 2,34 ммоль). Эту смесь перемешивали при 60°С в течение трех часов. Добавляли поддерживаемый полимером трисамин (0,3 экв.) и перемешивали в течение ночи. Отфильтровывали полимер, удаляли растворитель упариванием, добавляли EtOAc (10 мл) и промывали органическую фазу тремя порциями воды. После сушки органического слоя (сульфат магния) растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 135 мг метил-2-[(4-{3-[этил-(2-пиридин-2-илэтил)-амино]-3-оксопропил)-фенокси)-метил]-бензоата (выход 25,8%).
1H-ЯМР (ротамеры, 600 мГц, CDCl3): δ 0.97, 1.05 (t, t, 3H), 2.41, 2.52 (t, t, 2H), 2.75-3.0 (m, 4H), 3.09, 3.33 (q, q, 2H), 3.54, 3.61 (t, t, 2H), 3.83 (s, 3H), 5.43 (s, 2H), 6.75-6.85 (m, 2H), 6.93-7.2 (m, 4H), 7.3 (t, 1Н), 5.42 (m, 2H), 7.7 (d, 1H), 7.97 (d, 1H), 8.48 (m, 1H).
с) Растворяли метил-2-[(4-{3-[этил-(2-пиридин-2-илэтил)-амино]-3-оксопропил}-фенокси)-метил]-бензоат (0,135 г, 0,30 ммоль) в смеси тетрагидрофуран/вода (7/1, 5 мл) и добавляли гидроксид лития (29 мг, 1,2 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Реакционную смесь подкисляли (соляная кислота, 1М, 1 мл) и водную фазу промывали двумя порциями EtOAc (2×5 мл). Объединяли органические фазы, сушили их (сульфат магния) и удаляли растворитель упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали со смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Удаление растворителя упариванием давало 27 мг 2-[(4-{3-[этил-(2-пиридин-2-илэтил)-амино]-3-оксопропил}-фенилокси)-метил]-бензойной кислоты (выход 20,6%).
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 1.02, 1,12 (t, t, 3H), 2.48, 2.59 (t, t, 2H), 2.85-3.43 (m, 6H), 3.58, 3.66 (t, t, 2H), 5.51, 5.53 (s, s, 2H), 6.86-6,96 (m, 2H), 7.06-7.33 (m, 4H), 7.4 (t, 1H), 5.56 (t, 1H), 7.64-7.75 (m, 2H), 8.14 (m, 1H), 8.64 (m, 1Н).
Пример 23
a) 1-(2-Бромэтил)-3-трет-бутоксибензол
3-(2-Бромэтил)-фенол (1,349 г, 6,709 ммоль) в ДХМ (7 мл) охлаждали под аргоном до -78°С. При перемешивании пробулькивали через эту смесь изобутен, пока не добавляли больше, чем 5 мл. Каплями добавляли трифторметансульфоновую кислоту (50 мкл). Эту смесь перемешивали под аргоном при -78°С в течение 4,5 часов. Добавляли триэтиламин (120 мкл). Эту реакционную смесь доводили до комнатной температуры и затем фильтровали. Фильтрат выпаривали досуха и в остаток добавляли петролейный эфир (25 мл). Затем это фильтровали и упаривали. Полученное масло растворяли в этилацетате, промывали водой, сушили (сульфат натрия) и упаривали. Остаток растворяли в CDCl3 и затем упаривали. Оставляли 1,223 г желаемого продукта, выход 71%.
1H-ЯМР (500 МГц, CDCl3): δ 1.42 (s, 9H), 3.17 (t, 2H), 3.59 (t, 2H), 6.92-6.98 (m, 3H), 7.25 (t, 1H).
b) Метил-2-{[2-(3-трет-бутоксифенил)-этил]-тио}-бензоат
1-(2-Бромэтил)-3-трет-бутоксибензол (320 мг, 1,244 ммоль) растворяли в ацетонитриле (15 мл). Добавляли метилтиосалицилат (209 мг, 1,244 ммоль) и затем добавляли карбонат калия безводный (258 мг, 1,866 ммоль). Эту смесь нагревали при дефлегмации в течение 3 часов и затем упаривали под вакуумом досуха. Хроматография остатка на колонке (ISOLUTE® SI, 5 г/25 мл) при использовании смеси этилацетат/гептан (5:95) в качестве элюанта давала 421 мг желаемого продукта, выход 98%.
1H-ЯМР (500 мГц, CDCl3): δ 1.39 (s, 9H), 3.00 (t, 2H), 3.21 (t, 2H), 3.96 (s, 3Н), 6.90-6.93 (m, 2H), 7.01 (d, 1H), 7.19-7.26 (m, 2H), 7.38 (d, 1H), 7.48 (t, 1H), 8.00 (d, 1H).
c) Метил-2-([2-(3-гидроксиФенил)-этил]-тио}бензоат
Метил-2-{[2-(3-трет-бутоксифенил)-этил]-тио}-бензоат (402 мг, 1,167 ммоль) растворяли в ДХМ (3 мл). Добавляли трифторуксусную кислоту (3 мл). Эту смесь перемешивали в течение ночи и затем упаривали досуха. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании смеси этилацетат/гептан (5:95, затем 10:90 и затем 25:75) в качестве элюанта давала 260 мг желаемого продукта, выход 77%.
1H-ЯМР (500 МГц. CDCl3): δ 2.94 (t, 2Н), 3.17 (t, 2H), 3.95 (s, 3Н), 6.18 (s, 1H), 6.77-6.83 (m, 3Н), 7.18 (t, 2H), 7.34 (d, 1H), 7.44 (t, 1Н), 7.99 (d, 1H).
d) N-бензил-2-бром-N-гексилацетамид
Смешивали N-гексилбензиламин (4,2 г, 21,953 ммоль) и триэтиламин (3,98 мл, 28,539 ммоль) в ДХМ (20 мл) и охлаждали на ледяной бане. Добавляли бромацетилхлорид (3,455 мг, 21,953 ммоль) в ДХМ (5 мл). Перемешивали эту смесь в течение уикэнда и давали температуре дойти до комнатной температуры. Эту смесь промывали водой, смешанной с 1%-ной соляной кислотой (водная фаза с рН˜4-5), и рассолом, сушили сульфатом магния и упаривали. Колоночная Хроматография остатка на силикагеле при использовании смеси этилацетат/гептан (10:90, затем 20:80) в качестве элюанта давала 4,0 г желаемого продукта, выход 58%.
1H-ЯМР (ротамеры, 300 мГц, CDCl3): δ 0.85-0.92 (m, 3Н), 1.28 (s, br, 6H), 1.53-1.62 (m, 2H), 3.25, 3.39 (t, t, 2H), 4.05, 4.16 (s, s, 2H), 4.61, 4.63 (s, s, 2H), 7.19-7.42 (m, 5H).
е) Метил 2-{[2-{3-{2-[бензил(гексил)-амино]-оксоэтил}-фенил]-тио}-бензоат
Смешивали метил 2-{[2-(3-гидроксифенил)-этил]-тио}-бензоат (129 мг, 0,447 ммоль), N-бензил-2-бром-N-гексилацетамид (154 мг, 0,492 ммоль) и карбонат калия безводный (93 мг, 0,671 ммоль) в ацетонитриле (10 мл). Эту смесь нагревали при дефлегмации в течение ночи и затем упаривали досуха. Добавляли в остаток воду и этилацетат. Разделяли две фазы. Органическую фазу промывали рассолом и сушили сульфатом магния и затем упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании смеси этилацетат/гептан (10:90, затем 25:75) в качестве элюанта давала 208 мг желаемого продукта, выход 89,5%.
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 0.86-0.91 (m, 3H), 1.24-1.32 (m, 6H), 1.53-1.64 (m, 2H), 2.94-3.03 (m, 2Н), 3.13-3.20 (m, 2H), 3.29, 3.41 (t, 4 2Н), 3.93 (s, 3H), 4.64, 4.65 (s, s, 2H), 4.71, 4.81 (s, s, 2H), 6.75-6.77 (m, 1H), 6.85-6.93 (m, 2H), 7.17-7.39 (m, 8H), 7.46 (t, 1Н), 7.99 (d, 1Н).
13C-ЯМР (ротамеры, 125 мГц, CDCl3): δ 13.85, 13.86, 22.39, 26.38, 27.03, 28.27, 31.31, 31.37, 33.28, 34.51, 34.57, 46.34, 48.13, 50.33, 51.94, 67.13, 67.38, 112.33, 112.39, 114.84, 114.96, 121.48, 121.56, 123.79, 125.49, 126.40, 127.24, 127.54, 127.64, 127.89, 128.42, 128.74, 129.48, 129.57, 131,17, 132.22, 136.51, 137.03, 141.30, 141.74, 141.87, 158.06, 158.15, 166.75, 167.74, 167.88.
f) 2-{[2-(3-{2-[Бензил(гексил)-амино]-2-оксоэтокси}-фенил)-этил]-тио}-бензойная кислота
Метил-2-{[2-(3-{2-[бензил(гексил)-амино]-2-оксоэтокси}-фенил)-этил]-тио}-бензоат (80 мг, 0,154 ммоль) в тетрагидрофуране (3 мл) охлаждали на ледяной бане. Добавляли гидроксид лития (7,4 мг, 0,308 ммоль) в воде (3 мл). Затем удаляли охладительную баню и перемешивали смесь 12 дней и затем упаривали в вакууме для удаления тетрагидрофурана. Остаток подкисляли 1%-ной соляной кислотой, рН 3 и экстрагировали этилацетатом. Органическую фазу сушили сульфатом магния и упаривали. Хроматография остатка на колонке (ISOLUTE® SI, 2 г/6 мл) при использовании ДХМ, МеОН/ДХМ (1:99, и затем 2:98) в качестве элюанта давала 27 мг смеси продуктов. Повторная хроматография этой смеси на колонки (ISOLUTE® SI, 1 г/6 мл) при использовании ДХМ и затем МеОН/АсОН/ДХМ (0,25:0,25:99,5) в качестве элюанта давала 17 мг желаемого продукта, выход 22%.
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 0.85-0.90 (m, 3H), 1.23-1.31 (m, 6H), 1.53-1.63 (m, 2H), 2.91-3.00 (m, 2H), 3.11-3.20 (m, 2H), 3.29, 3.41 (t, t, 2H), 4.65, 4.66 (s, s, 2H), 4.74, 4.83 (s, s, 2H), 6.72-6.93 (m, 3H), 7.20-7.32 (m, 6H), 7.37 (t, 2H), 7.47 (t, 1H), 8.09 (d, 1H).
13С-ЯМР (ротамеры, 125 мГц, CDCl3): δ 13.98, 22.51, 26.51, 27.11, 28.36, 31.42, 31.49, 34.16, 34.60, 34.66, 46.52, 48.35, 50.52, 67.20, 67.47, 112.60, 115.15, 115.29, 121.66, 121.75, 124.46, 126.54, 126.80, 127.42, 127.70, 128.04, 128.56, 128.89, 129.59.129.67, 132.13, 132.79, 136.46, 136.98, 141,16, 141.75, 141.89, 158.08, 158.17, 168.25, 168.37, 169.60.
Пример 24
AR-H072686
a) Растворяли гептан-1-амин (1 г, 8,679 ммоль) в диметилформамиде (10 мл), добавляли (2-метоксифенил)-уксусную кислоту (1,587 г, 9,547 ммоль) и охлаждали эту смесь до 0°С. Добавляли тетрафторборат N-[(1H-1,2,3-бензотриазол-1-илокси)(диметиламино)-метилен]-N-метилметанаминия (3,065, 9,547 ммоль]) и N-этил-N,N-диизопропиламин (2,356 г, 18,226 ммоль). Этот раствор перемешивали в течение ночи при комнатной температуре. Добавляли EtOAc (20 мл) и промывали органическую фазу двумя порциями NaHCO3 (2×20 мл, водный). Органический слой сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили ее (сульфат магния) и удаляли растворитель упариванием с получением 2,00 г N-гептил-2-(2-метоксифенил)-ацетамида (выход 87,5%).
1H-ЯМР (500 мГц, CDCl3): δ 0.75 (t, 3H), 1,10 (m, 8H), 1.28 (m, 2H), 3.02 (q, 2Н), 3.39 (s, 2H), 3.45 (s, 3H), 6.2 (bs, 1H), 6.75 (m, 2H), 7.08 (m, 2H).
b) N-Гептил-2-(2-метоксифенил)-ацетамид (2,00 г, 7,594 ммоль) растворяли в ТГФ (10 мл) и охлаждали до нуля градусов под атмосферой аргона. Добавляли комплекс (метилтио)метана с бораном (1:1) (1,442 г, 18,984 ммоль) и эту смесь держали при температуре дефлегмации в течение 5 часов. После охлаждения смеси до комнатной температуры осторожно добавляли 5 мл соляной кислоты (10%) и перемешивали эту смесь в течение ночи. Растворитель удаляли упариванием. Добавляли EtOAc (20 мл) и промывали органическую фазу карбонатом калия (2 М, 2×20 мл). Неочищенный продукт очищали флэш-хроматографией (начинали с изократической смеси гептан/EtOAc 50/50 и затем увеличивали концентрацию EtOAc до 100%; силикагель 60, 0,004-0,063 мм). Фракции, содержащие продукт, объединяли в пул, EtOAc удалили упариванием с получением 1,037 г N-[2-(2-метоксифенил)-этил]-гептан-1-амина (выход 54,8%).
1H-ЯМР (300 мГц, CDCl3): δ 0.83 (t, 3Н), 1.07 (s, 1H), 1,13 (m, 8H), 1.43 (m, 2H), 2.57 (m, 2Н), 2.80 (s, 4Н), 3.72 (s, 3Н), 6.8 (bm, 2H), 7.08 (m, 2H).
с) Растворяли N-[2-(2-метоксифенил)-этил]-гептан-1-амин (0,091 г, 0,366 ммоль) в диметилформамиде (5 мл), добавляли (4-{[2-(метоксикарбонил)-бензил]-окси}-фенил)-уксусную кислоту (0,100 г, 0,333 ммоль) и охлаждали эту смесь до 0°С. Добавляли N-[(1Н-1,2,3-бензотриазол-1-илокси)(диметиламино)-метилен]-N-метилметанаминия тетрафторборат (0,118 г, 0,366 ммоль) и N-этил-N,N-диизопропиламин (0,090 г, 0,699 ммоль). Этот раствор перемешивали в течение ночи при комнатной температуре. Добавляли EtOAc (20 мл) и промывали органическую фазу двумя порциями воды (2×20 мл). Органический слой сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-1QO-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния) и растворитель удаляли упариванием с получением 0,094 г метил-2-([4-(2-{гептил[2-(2-метоксифенил)-этил]амино}-2-оксоэтил)-фенокси]-метил)-бензоата (выход 53,1%).
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 0.85 (t, 3Н), 1,13 (m, 8H), 1.43 (m, 2H), 2.7-3.75 (m, 8H), 3.77-3.95 (bm, 6Н), 5.5 (m, 2H), 6.8-7.45 (bm, 9H), 7.55 (q, 1Н), 7.73 (q, 1H), 8.03 (3Н).
d) Растворяли метил-2-{[4-(2-{гептил[2-(2-метоксифенил)-этил]-амино}-2-оксоэтил)-фенокси]-метил)-бензоат (0,094 г, 0,177 ммоль) в EtOH (5 мл, 95%), добавляли гидроксид калия (0,015 г, 0,265 ммоль) Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Проводили обработку удалением растворителя, упариванием и добавлением соляной кислоты (2 мл, 1М). Водную фазу экстрагировали двумя порциями EtOAc (20 мл), органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/96 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Растворитель удаляли упариванием с получением 0,011 г 2-{[4-(2-{гептил[2-(2-метоксифенил)-этил]-амино)-2-оксоэтил)-фенокси]-метил}-бензойной кислоты (выход 12%).
1Н-ЯМР (ротамеры, 500 мГц, CDCl3): δ 0,85 (t, 3Н), 1,13 (m, 8H), 1,43 (m, 2H), 2,7-3,75 (m, 8H), 3,77-3,95 (bm, 3Н), 5,5 (m, 2H). 6,8-7,45 (bm, 9H), 7,55 (q, 1Н), 7,73 (q, 1H), 8,03 (3Н).
Пример 25
AR-H072687
а) Растворяли гептан-1-амин (1 г, 8,679 ммоль) в сухом тетрагидрофуране под N2 и добавляли поддерживаемый полимером N-бензил-N,N-диизопропиламин (4,955 г, 26,038 ммоль). Эту смесь перемешивали в течение 30 минут и охлаждали до 0°С и добавляли к ней (4-хлорфенил)-ацетилхлорид (1,969 г, 10,415 ммоль). Этот раствор перемешивали в течение ночи при комнатной температуре. Избыток (4-хлорфенил)-ацетилхлорида удаляли фильтрованием этой смеси через NH2-картридж. Растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния) и растворитель удаляли упариванием с получением 1,045 г 2-(4-хлорфенил)-N-гептилацетамида (выход 45,0%).
1Н-ЯМР (500 мГц, CDCl3): δ 0.82 (t, 3H), 1.2 (m, 8H), 1.39 (m, 2H). 3.12 (t, 2Н), 3.43 (s, 2H), 6.35 (bs, 1Н), 7.15 (d, 2H), 7.23 (d, 2H).
b) 2-(4-Хлорфенил)-N-гептилацетамид (0,8 86 г, 3,797 ммоль) растворяли в тетрагидрофуране (10 мл) и охлаждали до нуля градусов под атмосферой аргона. Добавляли комплекс (метилтио)метана с бораном (1:1) (0,741 г, 9,755 ммоль) и нагревали эту смесь при температуре дефлегмации 5 часов. После охлаждения этой смеси до комнатной температуры осторожно добавляли 5 мл соляной кислотой (10%) и перемешивали эту смесь в течение ночи. Растворитель удаляли упариванием. Добавляли EtOAc (20 мл) и промывали органическую фазу карбонатом калия (2М, 2×20 мл). Неочищенный продукт очищали флэш-хроматографией (начинали с изократической смеси гептан/EtOAc 50/50 и затем концентрацию EtOAc увеличивали до 100% (силикагель 60; 0,004-0,063 мм). Фракции, содержащие продукт, объединяли в пул, EtOAc удаляли упариванием с получением 0,609 г N-[2-(4-хлорфенил)-этил]-N-гептиламина (выход 61,5%).
1Н-ЯМР (300 мГц, CDCl3): δ 0.85 (t, 3H), 1.22 (m, 8H), 1.39 (m, 2H), 2.52 (t, 2H), 2.45-2.6 (t, 2H), 2.6-2.82 (m, 4H), 7.0-7.2 (bm, 2H).
c) N-[2-(4-Хлорфенил)-этил]-N-гептиламин (0,093 г, 0,366 ммоль) растворяли в диметилформамиде (5 мл). Добавляли (4-{[2-(метоксикарбонил)-бензил]-окси}-фенил)-уксусную кислоту (0,100 г, 0,333 ммоль) и охлаждали эту смесь до 0°С. Добавляли тетрафторборат N-[(1Н-1,2,3-бензотриазол-1-илокси)(диметиламино)-метилен]-N-метилметанаминия (0,118 г, 0,366 ммоль) и N-этил-N,N-диизопропиламин (0,090 г, 0,699 ммоль). Этот раствор перемешивали в течение ночи при комнатной температуре. Добавляли EtOAc (20 мл) и промывали органическую фазу двумя порциями воды (2×20 мл). Органический слой сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния) и растворитель удаляли упариванием с получением 0,112 г метил-2-[(4-{2-[[2-(4-хлорфенил)-этил](гептил)-амино]-2-оксоэтил}-фенокси)-метил]-бензоата (выход 62,7%).
1Н-ЯМР (ротамеры, 300 мГц, CDCl3): δ 0.85 (t, 3Н), 1.22 (m, 8H), 1.39 (m, 2H), 2.62-3.7 (bm, 8H), 3.9 (m, 3Н), 5.45-5.55 (m, 2H), 6.89-7.43 (bm, 9H), 7.52 (m, 1H), 7.75 (t, 1H), 8.02 (t, 1H).
d) Растворяли метил-2-[(4-{2-[[2-(4-хлорфенил)-этил]-(гептил)-амино]-2-оксоэтил}-фенокси-)метил]-бензоат (0,112 г, 0,209 ммоль) в EtOH (5 мл, 95%) и добавляли гидроксид калия (0,018 г, 0,313 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Проводили обработку удалением растворителя упариванием и добавлением соляной кислоты (2 мл, 1 М). Водную фазу экстрагировали двумя порциями EtOAc (20 мл) и органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием, Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Растворитель удаляли упариванием с получением 0,006 г 2-[(4-{2-[[2-(4-хлорфенил)-этил](гептил)-амино]-2-оксоэтил)-фенокси)-метил]-бензойной кислоты (выход 5,5%).
1H-ЯМР (ротамеры, 300 мГц, CDCl3): δ 0.85 (t, 3H), 1.22 (m, 8H), 1.39 (m, 2H), 2.62-3.7 (bm, 8H), 5.45-5.55 (m, 2Н), 6.89-7.43 (bm, 9H), 7.52 (m, 1Н), 7.78 (t, 1H), 8.13 (t, 1H).
Пример 26
AR-H072688
a) Растворяли гептан-1-амин (1 г, 8,679 ммоль) в сухом тетрагидрофуране под N2 и добавляли поддерживаемый полимером /V-бензил-N,N-диизопропиламин (4,955 г, 26,038 ммоль). Эту смесь перемешивали в течение 30 мм и охлаждали до 0°С и добавляли к ней фенилацетилхлорид (1,610 г, 10,415 ммоль). Этот раствор перемешивали в течение ночи при комнатной температуре. Избыток фенилацетилхлорида удаляли фильтрованием этой смеси через NH2-картридж. Растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 M); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния) и растворитель удаляли с получением 0,886 г N-гептил-2-фенилацетамида (выход 43,7%).
1H-ЯМР (500 мГц, CDCl3): δ 0.82 (t, 3H), 1.22 (m, 8H), 1.39 (m, 2H), 3.12 (t, 2H;, 3.45 (s, 2H), 6.45 (bs, 1H), 7.18-7.3 (bm, 5H).
b) N-Гептил-2-фенилацетамид (0,886 г, 3,797 ммоль) растворяли в тетрагидрофуране (10 мл) и охлаждали до нуля градусов под атмосферой аргона. Добавляли комплекс (метилтио) метан с бораном (1:1) (0,721 г, 9,492 ммоль) и нагревали эту смесь при температуре дефлегмации в течение 5 часов. После охлаждения этой смеси до комнатной температуры осторожно добавляли 5 мл соляной кислоты (10%) и перемешивали в течение ночи. Растворитель удаляли упариванием. Добавляли EtOAc (20 мл) и промывали органическую фазу карбонатом калия (2М, 2×20 мл). Неочищенный продукт очищали флэш-хроматографией (начинали с изократической смеси гептан/EtOAc 50/50 и затем концентрацию EtOAc увеличивали до 100%; силикагель 60, 0,004-0,063 мм). Фракции, содержащие продукт, объединяли в пул, EtOAc удаляли упариванием с получением 0,584 г N-(2-фенилэтил)гептан-1-амина (выход 70,1%).
1H-ЯМР (300 мГц, CDCl3): δ 0.85 (t, 3H), 1.25 (m, 8H), 1.43 (m, 2H), 2.6 (t, 2Н), 2.7-2.95 (bm, 4H), 7.1-7.35 (bm, 5H).
с) Растворяли N-(2-фенилэтил)-гептан-1-амин (0,080 г, 0,366 ммоль) в диметилформамиде (5 мл), добавляли (4-{[2-(метоксикарбонил)-бензил]-окси}-фенил)-уксусную кислоту (0,100 г, 0,333 ммоль) и охлаждали эту смесь до 0°С. Добавляли тетрафторборат N-[(1Н-1,2,3-бензотриазол-1-илокси)(диметиламино)-метилен]-N-метилметанаминия (0,118 г, 0,366 ммоль) и N-этил-N,N-диизопропиламин (0,090 г, 0,699 ммоль). Этот раствор перемешивали в течение ночи при комнатной температуре. Добавляли EtOAc (20 мл) и промывали органическую фазу двумя порциями воды (2×20 мл). Органический слой сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мм). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния) и растворитель удаляли упариванием с получением 0,146 г метил-2-[(4-{2-[гептил(2-фенилэтил)-амино]-2-оксоэтил}-фенокси)-метил]-бензоата (выход 87,4%).
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 0.85 (t, 3H), 1,1-1.35 (bm, 8H), 1.4-1.6 (bm, 2Н), 2.65-65 (m, 8H), 3.9 (m, 3H), 5.43-5.56 (m, 2H), 6.85-7.4 (bm, 10Н), 7.52 (m, 1H), 7.73 (t, 1H), 8.02 (t, 1H).
d) Растворяли метил-2-[(4-{2-[гептил-(2-фенилэтил)-амино]-2-оксоэтил}-фенокси)-метил]-бензоат (0,146 г, 0,291 ммоль) в EtOH (5 мл, 95%), добавляли гидроксид калия (0-025 г, 0,437 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Проводили обработку удалением растворителя упариванием и добавлением соляной кислоты (2 мл, 1 M). Водную фазу экстрагировали двумя порциями EtOAc (20 мл) и органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетон итрил/вода 10/90 с ацетатом аммония (0,1 M); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Растворитель удаляли упариванием с получением 0,043 г 2-[(4-{2-[гептил(2-фенилэтил)-амино]-2-оксоэтил}-фенокси)-метил]-бензойной кислоты (выход 30,3%).
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 0.85 (t, 3H), 1,1-1.35 (bm, 8H), 1.4-1.6 (bm, 2H), 2.65-65 (m, 8H), 5.43-5.56 (m, 2H), 6.9-7.45 (bm, ЮН), 7.57 (m, 1H), 7.78 (t, 1H), 8.17 (t, 1H).
Пример 27
AR-H075101
а) Растворяли N-(2-фторбензил)-этанамин (0,248 г, 0,993 ммоль) в диметилформамиде (10 мл), добавляли (4-гидроксифенокси)-уксусную кислоту (0,150 г, 0,903 ммоль) и охлаждали эту смесь до 0°С. Добавляли тетрафторборат N-[(1H-1,2,3-бензотриазол-1-илокси)(диметиламино)-метилен]-N-метилметанаминия (0,319 г, 0,993 ммоль) и N-этил-N-диизопропиламин (0,245 г, 1,896 ммоль). Этот раствор перемешивали в течение ночи при комнатной температуре. Добавляли EtOAc (20 мл) и промывали органическую фазу двумя порциями Na2СО3 (2×20 мл, водный). Органический слой сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния) и растворитель удаляли упариванием с получением 0,177 г N-этил-N-(2-фторбензил)-2-(4-гидроксифенокси)-ацетамида (выход 64,6%).
1H-ЯМР (ротамеры, 300 мГц, CDCl3): δ 1.02-1.25 (m, 3H), 3.4 (q, 2H), 4.68 (m, 4H), 6.65-6.85 (bm, 4Н), 6.95-7.4 (m, 4H).
b) Растворяли N-этил-N-(2-фторбензил)-2-(4-гидроксифенокси)-ацетамид (0,177 г, 0,586 ммоль) в ацетонитриле (10 мл), добавляли метил-2-(бромметил)-бензоат (0,147 г, 0,642 ммоль) и гидрокарбонат калия (0,161 г, 1,167 ммоль). Этот раствор перемешивали в течение 2 часов при 60°С. Добавляли EtOAc (20 мл) и промывали органическую фазу двумя порциями рассола (2×20 мл, водный). Органический слой сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%, буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния) и растворитель удаляли упариванием с получением 0,242 г метил-2-[(4-{2-[этил(2-фторбензил)-амино]-2-оксоэтокси}-фенокси)-метил]-бензоата (выход 91,9%).
1H-ЯМР (ротамеры, 300 мГц, CDCl3): δ 1.02-1.28 (m, 3H), 3.39 (m, 2H), 3.89 (s, 3H) 4.6-4.75 (m, 4H), 5.42 (d, 2H), 6.75-7.42 (bm, 9Н), 7.55 (t, 1H), 7.72 (t, 1H), 8.0 (d, 1H).
с) Растворяли метил-2-[(4-{2-[этил-(2-фторбензил)-амино]-2-оксоэтокси}-фенокси)-метил]-бензоат (0,242 г, 0,536 ммоль) в EtOH (5 мл, 95%), добавляли гидроксид калия (0,060 г, 1,072 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Проводили обработку удалением растворителя, упариванием и добавлением соляной кислоты (2 мл, 1 M). Водную фазу экстрагировали двумя порциями EtOAc (20 мл), органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали флэш-хроматографией (начинали с изократического ДХМ 100% и затем концентрацию МеОН увеличивали от 0,5% до 20% (силикагель 60; 0,004-0,063 мм). Фракции, содержащие продукт, объединяли в пул, EtOAc удаляли упариванием с получением 0,160 г 2-[(4-{2-[этил-(2-фторбензил)-амино]-2-оксоэтокси)-фенокси)-метил]-бензойной кислоты (выход 68,2%).
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 1.07-1.28 (m, 3H), 3.42 (m, 2H), 4:62-4.78 (m, 4H), 5.48 (d, 2H), 6.78-7.37 (bm, 8H), 7.40 (m, 1H), 7.60 (q, 1H), 7.80 (t, 1H), 8.18 (d, 1H).
Пример 28
AR-H075104
а) Растворяли N-(2-фторбензил)-этанамин (0,077 г, 0,500 ммоль) в диметилформамиде (10 мл,), добавляли (4-{[2-(метоксикарбонил)-фенокси]-метил}-фенил)-уксусную кислоту (0,150 г, 0,500 ммоль) и охлаждали эту смесь до 0°С. Добавляли тетрафторборат N-[(1Н-1,2,3-бензотриазол-1-илокси)(диметиламино)-метилен]-N-метилметанаминия (0,176 г, 0,549 ммоль) и N-этил-N,N-диизопропиламин (0,136 г, 1,049 ммоль). Этот раствор перемешивали в течение ночи при комнатной температуре. Добавляли EtOAc (20 мл) и промывали органическую фазу двумя порциями карбоната натрия (2×20 мл, водный). Органический слой сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетон итрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм, скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния) и растворитель удаляли упариванием с получением 0,146 г метил-2-[(4-{2-[этил-(2-фторбензил)-амино]-2-оксоэтил}-бензил)-окси]-бензоата (выход 67,1%).
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 1.02-1,15 (m, 3H), 3.25-3.5 (m, 2H), 3.65-3.8 (m, 2H), 3.88 (s, 3H), 4.5-4.7 (bm, 2H), 5.17 (m, 2H), 6.92-7.5 (bm, 11H), 7.8 (m, 2H).
b) Растворяли метил-2-[(4-(2-[этил-(2-фторбензил)-амино]-2-оксоэтил}-бензил)-окси]-бензоат (0,242 г, 0,555 ммоль) в EtOH (5 мл, 95%), добавляли гидроксид калия (0,062 г, 1,111 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Проводили обработку удалением растворителя упариванием и добавлением соляной кислоты (2 мл, 1 М). Водную фазу экстрагировали двумя порциями EtOAc (20 мл) и органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали флэш-хроматографией (начинали с изократического ДХМ 100% и затем концентрацию МеОН увеличивали от 0,5% до 20% (силикагель 60; 0,004-0,063 мм). Фракции, содержащие продукт, объединяли в пул, EtOAc удаляли упариванием с получением 0,013 г 2-[(4-{2-[этил-(2-фторбензил)-амино]-2-оксоэтил}-бензил)-окси]-бензойной кислоты (выход 5,6%).
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 1,1 (t, 3H), 3.3-3.5 (bm, 2H), 3.7-3.82 (m, 2Н), 4.55-4.7 (bm. 2H), 5.23 (d, 2H), 6.95-7.45 (bm, 11H), 7.55 (t, 3Н), 8.22 (d. 1H).
Пример 29
AR-H075106
a) Растворяли гептан-1-амин (1 г, 8,679 ммоль) в сухом тетрагидрофуране под N2 и добавляли поддерживаемый полимером N-бензил-N,N-диизопропиламин (4,955 г, 26,038 моль). Перемешивали эту смесь в течение 30 мин и охлаждали до 0°С и добавляли к ней фенилацетилхлорид (1,610 г, 10,415 ммоль). Этот раствор перемешивали в течение ночи при комнатной температуре. Избыток фенилацетилхлорида удаляли фильтрованием этой смеси через NH2-картридж. Растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 M); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл), промывали органическую фазу двумя порциями рассола и сушили (сульфат магния); растворитель удаляли упариванием с получением 0,886 г N-гептил-2-фенилацетамида (выход 43,7%).
1H-ЯМР (500 мГц, CDCl3): δ 0.82 (t, 3Н), 1.22 (m, 8Н), 1.39 (m, 2H), 3.12 (t. 2H), 3.45 (s, 2H), 6.45 (bs, 1H), 7.18-7.3 (bm, 5H).
b) N-Гептил-2-фенилацетамид (0,886 г, 3,797 ммоль) растворяли в тетрагидрофуране (10 мл) и охлаждали до 0°С под атмосферой аргона. Добавляли комплекс (метилтио)метана с бораном (1:1) (0,721 г, 9,492 ммоль) и нагревали эту смесь при температуре дефлегмации в течение 5 часов. После охлаждения этой смеси до комнатной температуры осторожно добавляли 5 мл соляной кислотой (10%) и перемешивали эту смесь в течение ночи. Растворитель удаляли упариванием. Добавляли EtOAc (20 мл) и промывали органическую фазу карбонатом кальция (2М, 2×20 мл). Неочищенный продукт очищали флэш-хроматографией (начинали с изократической смеси гептан/EtOAc 50/50 и затем концентрацию EtOAc увеличивали до 100% (силикагель 60; 0,004-0,063 мм). Фракции, содержащие продукт, объединяли в пул, EtOAc удаляли упариванием с получением 0,584 г N-(2-фенилэтил)-гептан-1-амина (выход 70,1%).
1Н-ЯМР (300 мГц, CDCl3): δ 0.85 (t, 3H), 1.25 (m, 8H), 1.43 (m, 2H), 2.6 (t, 2Н), 2.7-2.95 (bm, 4H), 7.1-7.35 (bm, 5H).
c) Растворяли N-(2-Фенилэтил)-гептан-1-амин (0,110 г, 0,500 ммоль) в диметилформамиде (10 мл), добавляли (4-{[2-(метоксикарбонил)-фенокси]-метил}-фенил)-уксусную кислоту (0,150 г, 0,500 ммоль) и охлаждали эту смесь до 0°С. Добавляли тетрафторборат N-[(1H-1,2,3-бензотриазол-1-илокси)(диметиламино)-метилен-N-метилметанаминия (0,176 г, 0,549 ммоль) и N-этил-N,N-диизопропиламин (0,136 г, 1,049 ммоль). Этот раствор перемешивали в течение ночи при комнатной температуре. Добавляли EtOAc (20 мл) и промывали органическую фазу двумя порциями Na2CO3 (2×20 мл, водный). Органический слой сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер. 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл), промывали органическую фазу двумя порциями рассола и сушили (сульфат магния); растворитель удаляли упариванием с получением 0,229 г метил-2-[(4-{2-[гептил-(2-фенилэтил)-амино]-2-оксоэтил}-бензил)-окси]-бензоата (выход 91,4%).
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 0.85 (m, 3H), 1.25 (m, 8H), 1.40-1.6 (m, 2H), 2.75-3.7 (bm, 8H), 3.9 (m, 3H), 5.17 (d, 2H), 6.96-7.5 (bm, 12H), 7.8 (t, 1H).
d) Растворяли метил-2-[(4-{2-[гептил(2-фенилэтил)-амино]-2-оксоэтил}-бензил)-окси]-бензоат (0,229 г, 0,457 ммоль) в EtOH (5 мл, 95%) и добавляли гидроксид калия (0,051 г, 0,913 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Проводили обработку удалением растворителя упариванием и добавлением соляной кислоты (2 мл, 1 М). Водную фазу экстрагировали двумя порциями EtOAc (20 мл) и органическую фазу сушили (сульфат магния); растворитель удаляли упариванием. Неочищенный продукт очищали флэш-хроматографией (начинали с изократического ДХМ 100% и затем концентрацию МеОН увеличивали от 0,5% до 20% (силикагель 60; 0,004-0,063 мм). Фракции, содержащие продукт, объединяли в пул, EtOAc удаляли упариванием. Продукт не был чистым и поэтому его чистили препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%, буфером была смесь ацетон итрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мм). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл), промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Растворитель удаляли упариванием с получением 0,025 г 2-[(4-{2-[гептил(2-фенилэтил)-амино]-2-оксоэтил}-бензил)-окси]-бензойной кислоты (выход 11,2%).
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 0.85 (m, 3Н), 1.25 (m, 8H), 1.40-1.62 (m, 2Н), 2.75-3.7 (bm, 8H), 5.23 (d, 2H), 7.05-7.6 (bm, 12H), 8.2 (t, 1H).
Пример 30
AR-H075107
а) Растворяли гептан-1-амин (1 г, 8,679 ммоль) в сухом тетрагидрофуране под N2 и добавляли поддерживаемый полимером N-бензил-N,N-диизопропиламин (4,955 г, 26,038 ммоль). Эту смесь перемешивали в течение 30 минут, охлаждали до 0°С и добавляли к ней (4-хлорфенил)-ацетилхлорид (1,969 г, 10,415 ммоль). Этот раствор перемешивали в течение ночи при комнатной температуре. Избыток (4-хлорфенил)-ацетилхлорида удаляли фильтрованием этой смеси через NH2-картридж. Растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь а цетонитр ил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм, скорость тока 40 мл/мм). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния); растворитель удаляли упариванием с получением 1,045 г 2-(4-хлорфенил)-N-гептилацетамида (выход 45,0%).
1H-ЯМР (500 мГц, CDCl3): δ 0.82 (t, 3H), 1.22 (m, 8H), 1.35 (m, 2H), 3.12 (q, 2Н), 3.40 (s, 2H), 6.35 (bs, 1H), 7.1-7.3 (bm, 4H).
b) 2-(4-Хлорфенил)-N-гептилацетамид (1,045 г, 3,902 ммоль) растворяли в тетрагидрофуране (10 мл) и охлаждали до 0°С под атмосферой аргона. Добавляли комплекс (метилтио)-метана с бораном (1:1) (0,741 г, 9,756 ммоль) и нагревали эту смесь при температуре дефлегмации в течение 5 часов. После охлаждения этой смеси до комнатной температуры осторожно добавляли 5 мл соляной кислоты (10%) и перемешивали в течение ночи. Растворитель удаляли упариванием. Добавляли EtOAc (20 мл) и промывали органическую фазу карбонатом калия (2М, 2×20 мл). Неочищенный продукт очищали флэш-хроматографией (начинали с изократической смеси гептан/EtOAc 50/50 и затем концентрацию EtOAc увеличивали до 100% (силикагель 60, 0,004-0,063 мм). Фракции, содержащие продукт, объединяли в пул, EtOAc удаляли упариванием с получением 0,609 г N-[2-(4-хлорфенил)-этил]-N-гептиламина (выход 61,5%).
1H-ЯМР (300 мГц, CDCl3): δ 0.82 (t, 3H), 1.22 (m, 8H), 1.38 (m, 2H), 2.52 (t, 2H), 2.6-2.85 (bm, 4H), 7.0-7.3 (bm, 4H).
c) Растворяли N-(4-хлорфенил)-этил]-N-гептиламин (0,127 г, 0,500 ммоль) в диметилформамиде (10 мл), добавляли (4-{[2-(метоксикарбонил)-фенокси]-метил)-фенил)-уксусную кислоту (0,150 г, 0,500 ммоль) и охлаждали эту смесь до 0°С. Добавляли N-[(1Н-1,2,3-бензотриазол-1-илокси)(диметиламино)-метилен]-N-метилметанаминия тетрафторборат (0,176 г, 0,549 ммоль) и N-этил-N,N-диизопропиламин (0,136 г, 1,049 ммоль) Этот раствор перемешивали в течение ночи при комнатной температуре. Добавляли EtOAc (20 мл) и промывали органическую фазу двумя порциями Na2СО3 (2×20 мл, водный). Органический слой сушили (сульфат магния) и растворитель удаляли упариванием. Неочищенный продукт очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм; скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния) и удаляли растворитель упариванием с получением 0,224 г метил-2-[(4-{2-[[2-(4-хлорфенил)-этил](гептил)-амино]-2-оксоэтил}-бензил)-окси]-бензоата (выход 83,7%).
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 0.85 (m, 3Н), 1.25 (m, 8H), 1.40-1.6 (m, 2Н), 2.7-3.75 (bm, 8H), 3.9 (m, 3Н), 5.17 (d, 2Н), 6.96-7.5 (bm, 11H), 7.8 (t, 1H).
d) Растворяли метил 2-[(4-{2-[[2-(4-хлорфенил)-этил](гептил)-амино]-2-оксоэтил}-бензил)-окси]-бензоат (0,224 г, 0,418 ммоль) в EtOH (5 мл, 95%) и добавляли гидроксид калия (0,047 г, 0,836 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (7 минут, 150°С). Проводили обработку удалением растворителя упариванием и добавлением соляной кислоты (2 мл, 1 М). Водную фазу экстрагировали двумя порциями EtOAc (20 мл) и органическую фазу сушили (сульфатом магния) и растворитель удаляли упариванием. Неочищенный продукт очищали флэш-хроматографией (начинали с изократического ДХМ 100% и затем концентрация МеОН увеличивали от 0,5% до 20% (силикагель 60: 0,004-0,063 мм). Фракции, содержащие продукт, объединяли в пул, EtOAc удаляли упариванием. Продукт не был чистым и поэтому его очищали препаративной ВЭЖХ (начинали с изократической смеси ацетонитрил/буфер 60/40 и затем концентрацию ацетонитрила увеличивали до 100%; буфером была смесь ацетонитрил/вода 10/90 с ацетатом аммония (0,1 М); колонка KR-100-7-C8, 50 мм ×250 мм, скорость тока 40 мл/мин). Фракции, содержащие продукт, объединяли в пул, ацетонитрил удаляли упариванием. Добавляли EtOAc (10 мл) и промывали органическую фазу двумя порциями рассола и сушили (сульфат магния). Растворитель удаляли упариванием с получением 0,154 г 2-[(4-{2-[[2-(4-хлорфенил)-этил]-гептил)-амино]-2-оксоэтил}-бензил)-окси]-бензойнои кислоты (выход 76,8%).
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 0.85 (m, 3Н), 1.25 (m, 11H), 1.40-1.62 (m, 2H), 2.7-3.7 (bm, 10Н), 4.5 (d, 2H), 7-7.4 (bm, 8H).
Пример 31
AR-H075135
a) Растворяли N-изобутил-N-[4-(трифторметил)-бензил]-амин (0,172 г, 0,746 ммоль) в сухом ацетонитриле под N2 и добавляли N-этил-N,N-диизопропиламин (0,371 г, 2,867 ммоль). Перемешивали эту смесь в течение 30 минут и добавляли метил 2-{2-[4-(2-хлор-2-оксоэтокси)-фенил]-этокси}-бензоат (0,200 г, 0,573 ммоль). Этот раствор перемешивали в течение ночи при комнатной температуре. Неочищенный продукт очищали флэш-хроматографией (начинали с изократической смеси гептан/EtOAc 50/50 и затем концентрацию EtOAc увеличивали до 100% (силикагель 60 0,004-0,063 мм). Фракции, содержащие продукт, объединяли в пул, EtOAc удаляли упариванием с получением 0,242 г метил-2-{2-[4-(2-{изобутил[4-(трифторметил)-бензил]-амино}-2-оксоэтокси)-фенил]этокси}-бензоата (выход 77,6%).
1H-ЯМР (ротамеры, 500 мГц, CDCl3): δ 0.82-0.97 (bm, 6H), 2.0 (m, 1H), 3.02-3.24 (m, 4H), 3.88 (s, 3Н), 4.18 (m, 2H), 4.62-4.8 (m, 4H), 6.78-7.0 (m, 4H), 7.1-7.32 (m. 4H) 7.35-7.62 (m, 3Н), 7.78 (d, 1H).
b) Растворяли метил-2-{2-[4-(2-{изобутил-[4-(трифторметил)-бензил]-амино}-2-оксоэтокси)-фенил]-этокси}-бензоат (0,242 г, 0,455 ммоль) в смеси тетрагидрофуран (свежеперегнанный)/вода (2/1, 3 мл), добавляли гидроксид лития (0,218 г, 0,909 ммоль). Взаимодействие осуществляли в одноузловой микроволновой печи (5 минут, 150°С). Тетрагидрофуран удаляли упариванием. Добавляли воду (10 мл) и промывали щелочную водную фазу диэтиловым эфиром (2×10 мл). Добавляли соляную кислоту (2 мл, 1 М, рН 1). Водную фазу экстрагировали двумя порциями ДХМ (20 мл), органическую фазу сушили (сульфат магния) и растворитель удаляли упариванием с получением 0,135 г 2-{2-[4-(2-{изобутил-[4-(трифторметил)-бензил]-амино}-2-оксоэтокси)-фенил]-этокси}-бензойной кислоты (выход 57,3%).
1H-ЯМР (ротамеры, 400 мГц, CDCl3): δ 0.82-0.97 (bm, 6H), 1.98 (m, 1H), 3.06-3.24 (m, 4Н), 4.4 (m, 2Н), 4.62-4.8 (m, 4Н), 6.8 (d, 1H), 6.92 (d, 1H), 7.01 (m, 1Н), 7.03-7.22 (m, 3Н), 7.27 (m, 2H), 7.5 (m, 2Н), 7.58 (d, 1Н), 8.1 (d, 1Н).
Биологическая активность
Препараты
Растворяли соединения в ДМСО с получением 16 мМ сток-растворов. Перед анализами дополнительно разбавляли сток-растворы диметилсульфоксидом и культуральной средой.
ОБЩИЕ ХИМИКАТЫ И РЕАГЕНТЫ
Реагент для определения люциферазы приобретали у Packard, USA. Рестрикционные энзимы были от Boehringer, a Vent-полимераза от New England Biolabs.
КЛЕТОЧНЫЕ ЛИНИИ И УСЛОВИЯ КУЛЬТУРЫ КЛЕТОК
U2-OS (клетки остеогенной саркомы человека) получали в АТСС, США. Размножали и снова замораживали клетки партиями с пересева номер шесть. Культивировали клетки в среде Игла при модификации по Дульбекко (DMEM) с 25 мМ глюкозой, 2 мМ глутамином или 4 мМ L-аланил-L-глутамином, 10%-ой фетальной телячьей сывороткой при 5% CO2. Использовали забуференный фосфатами физраствор (PBS) без добавления кальция или магния. Все реагенты для клеточных культур были от Gibco (USA), 96-луночные планшеты для клеточных культур были приобретены у Wallach.
ПЛАЗМИДНЫЕ КОНСТРУКТЫ ДЛЯ ГЕТЕРОЛОГИЧЕСКОЙ ЭКСПРЕССИИ
Применяли стандартные методики работы с рекомбинантной ДНК как описано в Ausubel (7). Люциферазный репортерный вектор pGL5UAS (клон состоит из пяти копий ДНК связывающей последовательности GAL4, 5′-CGACGGAGTACTGTCCTCCGAGCT-3′) клонировали в Sacl/Xhol-сайты pGL3-промотора (Promega). Sacl/Xhol-фрагмент, несущий UAS-сайты, конструировали при использовании отжига перекрывающихся олигонуклеотидов.
Использованные экспрессионные векторы основаны на pSG5 (Stratagene). Все векторы содержат EcoRI/Nhel-фрагмент, кодирующий ДНК-связывающий домен GAL4 (позиции кодирующих аминокислот 1-145 в базе данных под номером доступа Р04386), с последующим слиянием в рамке считывания с фрагментом, кодирующим последовательность локализации в ядре, из Т-антигена вируса полиомы. Последовательность локализации в ядре конструировали при использовании отожженных перекрывающихся олигонуклеотидов, что создает липкие Nhel/Kpnl-концы (5′-CTAGCGCTCCTAGAAGAAACGCAAGGTTGGTAC-3′). Лиганд-связывающие домены PPARα человека и мыши и PPARγ человека и мыши амплифицировали в ПЦР как Kpnl/Bamlll-фрагменты и клонировали в рамке в ДНК-связывающий домен GAL4 и последовательность локализации в ядре. Последовательность всех использованных плазмидных конструктов подтверждали секвенированием. Для кратковременной трансфекции использовали нижеследующие экспрессионные векторы:
КРАТКОВРЕМЕННЫЕ ТРАНСФИЦИРОВАНИЯ
Размораживали замороженные запасы клеток из пересева номер шесть и перед трансфицированием размножали до пересева номер восемь. Трипсинизировали конфлюэнтные клетки, промывали их и осаждали центифугированием при 270 g в течение 2 минут. Осадок клеток снова суспендировали в холодном PBS до концентрации клеток около 18×106 клеток/мл. После добавления ДНК инкубировали клеточные суспензии на льду приблизительно 5 минут перед электропорацией при 230 В, 960 микрофарад в Biorad′s Gen Pulser™ партиями по 0,5 мл. В сумме каждую партию по 0,5 мл клеток добавляли по 50 мкг ДНК, включающей в себя 2,5 мкг экспрессионного вектора, 25 мкг репортерного вектора и 22,5 мкг неспецифической ДНК (pBluescript, Stratagene).
После электропорации разбавляли клетки до концентрации 320000 клеток/мл в DMEM без фенола красного и высевали приблизительно 25000 клеток на лунку в 96-луночные планшеты. Для того, чтобы перед добавлением тестируемого соединения дать клеткам оправиться, инкубировали засеянные планшеты при 37°С в течение 3-4 часов. В анализах на PPARα среду клеток дополняли обедненной на смоле с активированным углем фетальной телячьей сывороткой (FCS) для того, чтобы избежать фоновой активации жирнокислотными компонентами FCS. Обедненную на смоле с активированным углем FCS получали следующим образом: к 500 мл инактивированной нагреванием FCS добавляли 10 г активированного угля и 25 г анионообменной смолы Bio-Rad для анализа с зернистостью 200-400 меш и этот раствор держали на магнитной мешалке при комнатной температуре в течение ночи. На следующий день FCS центрифугировали и процедуру обеднения повторяли в течение 4-6 часов. После второй обработки FCS центрифугировали и стилизовали филырацией для удаления остатков активированного угля и смолы.
ПРОЦЕДУРА АНАЛИЗА
Разбавляли в ДМСО сток-растворы соединений до надлежащих диапазонов концентраций в главных планшетах. Из главных планшетов разбавляли соединения в культуральной среде с целью получения растворов тестируемых соединений для конечных доз.
После доведения количества среды клеток до 75 мкл в каждой лунке добавляли по 50 мкл раствора тестируемого соединения. Кратковременно трансфицированные клетки экспонировали соединениям в течение около 24 часов перед определением люциферазы. Для определения люциферазы добавляли вручную 100 мкл реагента для анализа к каждой лунке и оставляли планшеты приблизительно на 20 минут для того, чтобы создать возможность для лизиса клеток. После лизиса измеряли активность люциферазы в счетчике 1420 Multiwell Victor от Wallach.
Референтные соединения
Пиоглитазон TZD использовали в качестве референтной субстанции для активации как человеческих, так и мышиных PPARγ. 5,8,11,14-Эйкозатетраеновую кислоту (ETYA) использовали как референтную субстанцию для человеческих PPARα.
Вычисления и анализ
Для вычисления величин EC50 строили кривую концентрация-эффект. Использованные величины производили из средних по двум или трем независимым измерениям (после вычитания средней величины фона) и выражали как проценты от максимальной активации, полученной с референтным соединением. Величины наносили против логарифма концентрации тестируемого соединения. Оценивали величины EC50 по линейной интеркаляции между точками, которые соответствовали данным, вычислением концентрации, требуемой для достижения 50% максимальной активации, полученной с референтным соединением.
Соединения формулы 1 имеют EC50 меньше, чем 50 мкмоль/л для PPARα и/или PPARγ. Предпочтительные соединения имеют EC50 меньше, чем 5 мкмоль/л для PPARα и/или PPARγ. Например, Пример 2 имеет EC50 2,96 мкмоль/л для человеческих PPARα и EC50 3,05 мкмоль/л для мышиных PPARγ.
Соединения формулы I являются низкотоксичными.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОРТОЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ БЕНЗОЙНОЙ КИСЛОТЫ, СПОСОБ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2003 |
|
RU2288912C2 |
| АГОНИСТ РЕЦЕПТОРА GLP-1, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО, И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 2021 |
|
RU2800290C1 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| 3,6-ДИАМИНОПИРИДАЗИН-3-ИЛЬНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ | 2020 |
|
RU2830186C2 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2760554C1 |
| 6,7-ДИГИДРО-5H-ПИРИДО[2,3-C]ПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛКОВ BCLXL И ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2020 |
|
RU2832191C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| ПРОИЗВОДНЫЕ ПРОПИОНОВЫХ КИСЛОТ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АКТИВАТОРОВ hPPARs. | 2003 |
|
RU2316539C2 |
| ИНГИБИТОРЫ TrkA КИНАЗЫ, ОСНОВАННЫЕ НА НИХ КОМПОЗИЦИИ И СПОСОБЫ | 2015 |
|
RU2672583C2 |
| СОЕДИНЕНИЯ | 2019 |
|
RU2800063C2 |
Изобретение относится к новым соединениям формулы I, где R1 представляет собой фенил, возможно замещенный фенилом или гетероциклической группой, или гетероциклическую группу, возможно замещенную фенилом, где указанная гетероциклическая группа представляет собой моно- или бициклическое кольцо, содержащее 4-12 атомов, из которых по меньшей мере один атом выбран из азота, серы или кислорода, причем каждый фенил или гетероциклическая группа возможно замещен(а) одной или более чем одной из нижеследующих групп: C1-6алкильная группа; фенилС1-6алкил, причем алкильная, фенильная или алкилфенильная группа возможно замещена одним или более чем одним из Rb; галоген; -ORa -OSO2Rd; -SO2Rd; -SORd; -SO2ORa; где Ra представляет собой Н, C1-6алкильную группу, фенильную или фенилС1-6алкильную группу; где Rb представляет собой галогено, -ОН, -ОС1-4алкил, -Офенил, -OC1-4алкилфенил, и Rd представляет собой С1-4алкил; группа -(CH2)m-T-(CH2)n-U-(CH2)p присоединена либо в 3-ем, либо 4-ом положении в фенильном кольце, как указано цифрами в формуле I, и представляет собой группу, выбранную из одной или более чем одной из нижеследующих: O(СН2)2, O(СН2)3, NC(O)NR4(CH2)2, CH2S(O2)NR5(CH2)2, CH2N(R6)C(O)CH2, (CH2)2N)(R6)С(O)(СН2)2, C(O)NR7CH2, C(O)NR7(CH2)2 и CH2N(R6)C(O)CH2O; V представляет собой О, NR8 или одинарную связь; q представляет собой 1, 2 или 3; W представляет собой О, S или одинарную связь; R2 представляет собой галогено или С1-4алкоксильную группу; r представляет собой 0, 1, 2 или 3; s представляет собой 0; и R6 независимо представляют собой Н или C1-10алкильную группу; R4, R5, R7 и R8 представляют собой атом водорода; и их фармацевтически приемлемым солям. Изобретение также относится к способам получения таких соединений, их применению в изготовлении лекарственного средства для лечения резистентности к инсулину и к фармацевтической композиции, обладающей активностью модулирования рецепторов, активируемых пролифераторами пероксисом, на их основе. 6 н. и 6 з.п. ф-лы.
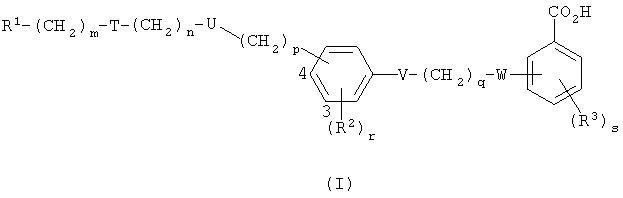
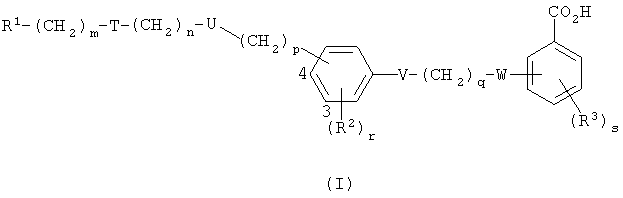
где R1 представляет собой фенил, возможно замещенный фенилом или гетероциклической группой, или гетероциклическую группу, возможно замещенную фенилом, где указанная гетероциклическая группа представляет собой моно- или бициклическое кольцо, содержащее 4-12 атомов, из которых по меньшей мере один атом выбран из азота, серы или кислорода, причем каждый фенил или гетероциклическая группа возможно замещен(а) одной или более чем одной из нижеследующих групп:
C1-6алкильная группа;
фенилС1-6алкил, причем алкильная, фенильная или алкилфенильная группа возможно замещена одним или более чем одним из Rb;
галоген;
-ORa;
-OSO2Rd;
-SO2Rd;
-SORd;
-SO2ORa;
где R3 представляет собой Н, C1-6алкильную группу, фенильную или фенилС1-6алкильную группу; где Rb представляет собой галогено, -ОН, -ОС1-4алкил, -Офенил, -ОС1-4алкилфенил, и Rd представляет собой С1-4алкил;
группа -(CH2)m-T-(CH2)n-U-(CH2)p- присоединена либо в 3-м, либо 4-м положении в фенильном кольце, как указано цифрами в формуле I, и представляет собой группу, выбранную из одной или более чем одной из нижеследующих: O(СН2)2, O(СН2)3, NC(O)NR4(CH2)2, CH2S(O2)NR5(CH2)2, CH2N(R)C(О)CH2, (CH2)2N(R6)C(O)(CH2), C(O)NR7CH2, C(O)NR7(CH2)2 и CH2N(R6)C(O)CH2O;
V представляет собой O, NR8 или одинарную связь;
q представляет собой 1, 2 или 3;
W представляет собой О, S или одинарную связь;
R2 представляет собой галогено или С1-4алкоксильную группу;
r представляет собой 0, 1, 2 или 3;
s представляет собой 0; и
R6 независимо представляют собой Н или C1-10алкильную группу;
R4, R5, R7 и R8 представляют собой атом водорода;
и его фармацевтически приемлемые соли;
при условии, что исключаются следующие соединения:
1) когда R1 представляет собой фенил, возможно замещенный одной или двумя группами, независимо выбранными из галогено, С1-4алкильной группы, которая возможно замещена одним или более чем одним атомом фтора, С1-4алкоксильной группы;
m представляет собой 1;
Т представляет собой N(R6)C(O), причем R6 представляет собой С2-8алкильную группу;
n представляет собой 1;
U отсутствует или представляет собой метилен;
р представляет собой 0;
r представляет собой 0;
V представляет собой О или S;
q представляет собой 1; и
W представляет собой одинарную связь, присоединенную в орто-положении относительно группы карбоновой кислоты;
2) когда R1 представляет собой фенил, возможно замещенный одной или двумя группами, независимо выбранными из галогено, С1-4алкильной группы, которая возможно замещена одним или более чем одним атомом фтора, С1-4алкоксильной группы;
m представляет собой 1;
Т представляет собой N(R6)C(O), где R6 представляет собой неразветвленную С2-7алкильную группу;
n представляет собой 1;
U представляет собой О;
р представляет собой 0;
r представляет собой 0 или 1;
и, когда г представляет собой 1, R2 присоединен в 3-м положении и представляет собой ОСН3;
V представляет собой одинарную связь;
q представляет собой 2; и
W представляет собой атом О или S, присоединенный в орто-положении относительно группы карбоновой кислоты.
3-{[(3-{[(1,1′-бифенил-4-илкарбонил)-амино]-метил}-фенил)-амино]-метил}-бензойная кислота;
2-{[4-(2-оксо-2-{[4-(трифторметил)-бензил]-амино}-этил)-фенокси]-метил}-бензойная кислота;
2-[(3-{2-[бензил(гексил)-амино]-2-оксоэтил}-фенокси)-метил]-бензойная кислота;
2-{[3-(2-оксо-2-{[4-(трифторметил)-бензил]-амино}-этил)-фенокси]-метил}-бензойная кислота;
2-[(4-{3-[[2-(3,4-диметоксифенил)-этил](метил)-амино]-3-оксопропил}-фенокси)-метил]-бензойная кислота;
2-[(4-{2-[({4-метил-2-[4-(трифторметил)-фенил]-1,3-тиазол-5-ил}-карбонил)-амино]-этил}-фенокси)-метил]-бензойная кислота;
2-({4-[2-({[(2,4-дифторфенил)-амино]-карбонил}-амино)-этил]-фенокси}-метил)-бензойная кислота;
2-[(4-{2-[(2-метил-5-фенил-3-фуроил)-амино]-этил}-фенокси)-метил]-бензойная кислота;
2-[(4-{2-[(бензилсульфонил)-амино]-этил}-фенокси)-метил]-бензойная кислота;
2-[(4-{2-[бензил(гексил)-амино]-2-оксоэтил}-2-фторфенокси)-метил]-бензойная кислота;
2-[(4-{2-[бензил(гексил)-амино]-2-оксоэтил}-2-метоксифенокси)-метил]-бензойная кислота;
2-({4-[3-(3,4-дигидроизохинолин-2(1H)-ил)-3-оксопропил]-фенокси}-метил)-бензойная кислота;
2-[(4-{2-[4-(1H-имидазол-1-ил)-фенокси]-этил}-феноксиметил]-бензойная кислота;
2-{[4-(2-{4-[(метилсульфонил)-окси]-фенокси}-этил)-фенокси]-метил}-бензойная кислота;
2-[(3-{2-[4-(бензилокси)-фенокси]-этил}-фенокси)-метил]-бензойная кислота;
2-{[3-(2-{4-[(метилсульфонил)-окси]-фенокси}-этил)-фенокси]-метил}-бензойная кислота;
2-({3-[2-(4-гидроксифенокси)-этил]-фенокси}-метил)-бензойная кислота;
2-[(4-{3-[4-(бензилокси)-фенокси]-пропил}-фенокси)-метил]-бензойная кислота;
2-{[4-(3-{4-[(метилсульфонил)-окси]-фенокси}-пропил)-фенокси]-метил}-бензойная кислота;
2-({4-[3-(4-гидроксифенокси)-пропил]-фенокси}-метил)-бензойная кислота;
2-{[4-(3-{[2-(2-этоксифенил)-этил]-амино}-3-оксопропил)-фенокси]-метил}-бензойная кислота;
2-[(4-{3-[этил(2-пиридин-2-илэтил)-амино]-3-оксопропил}-фенокси)-метил]-бензойная кислота;
2-{[2-(3-{2-[бензил(гексил)-амино]-2-оксоэтокси}-фенил)-этил]-тио}-бензойная кислота;
2-{[4-(2-{гептил[2-(2-метоксифенил)-этил]-амино}-2-оксоэтил)-фенокси]-метил}-бензойная кислота;
2-[(4-{2-[[2-(4-хлорфенил)-этил](гептил)-амино]-2-оксоэтил}-фенокси)-метил]-бензойная кислота;
2-[(4-{2-[гептил(2-фенилэтил)-амино]-2-оксоэтил}-фенокси)-метил]-бензойная кислота;
2-[(4-{2-[этил(2-фторбензил)-амино]-2-оксоэтокси}-фенокси)-метил]-бензойная кислота;
2-[(4-{2-[этил(2-фторбензил)-амино]-2-оксоэтил}-бензил)-окси]-бензойная кислота;
2-[(4-{2-[гептил(2-фенилэтил)-амино]-2-оксоэтил}-бензил)-окси]-бензойная кислота;
2-{2-[4-(2-{изобутил[4-(трифторметил)-бензил]-амино}-2-оксоэтокси)-фенил]-этокси}-бензойная кислота; и
2-[(4-{2-[[2-(4-хлорфенил)-этил](гептил)-амино]-2-оксоэтил}-бензил)-окси]-бензойная кислота
и их фармацевтически приемлемых солей.
где R1, Т, U, V, W, R2, m, n, p, q, r и s являются такими, как определено в п.1,
при котором проводят взаимодействие соединения формулы II,
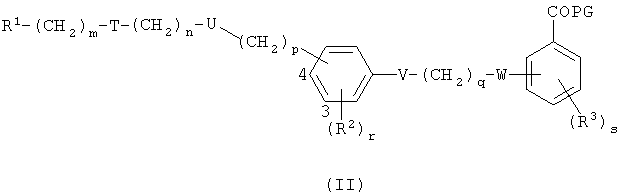
где R1, Т, U, V, W, R2, m, n, p, q, r и s являются такими, как определено в формуле (I), и PG представляет собой защитную группу для карбоксильной гидрокси-группы, с агентом снятия защиты.
где R1, Т, U, V, W, R2, m, n, p, q, r и s являются такими, как определено в формуле (I), и PG представляет собой защитную группу для карбоксильной гидрокси-группы.
| US 4113871 А, 12.09.1978 | |||
| Способ измерения амплитуды колебаний баланса часов | 1957 |
|
SU112187A1 |
| WO 9920275 A1, 29.04.1999 | |||
| Способ получения однохлористой меди | 1974 |
|
SU802186A1 |
| US 6306854 A, 23.10.2001 | |||
| 5-ФЕНОКСИАЛКИЛ-2,4-ТИАЗОЛИДИНДИОНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2169144C2 |

