Изобретение относится к замещенным в кольце производным К-252а для использования в способах, направленных на облегчение повреждающих воздействий различных заболеваний, расстройств и состояний.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
1. Индолкарбазол К-252а
К-252а представляет собой соединение, имеющее индолкарбазольный скелет, [Японские публикации непрошедших экспертизу патентных заявок 41489/85 (США 4555402)] со стереохимией, показанной в формуле I: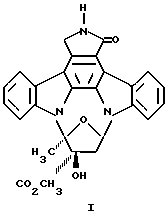
Ранее сообщалось, что К-252а является мощным ингибитором протеинкиназы С (РКС), играющей центральную роль в регуляции клеточных функций, и имеет различные виды активности, такие как ингибирующее действие на сокращение гладкой мускулатуры (Jpn. J. Pharmacol., 43 (suppl.): 284, 1987), ингибирующее действие на секрецию серотонина (Biochem. Biophys. Res. Commun., 144: 35, 1987), ингибирующее действие на удлинение аксонов (нейронов) (J. Neuroscience, 8: 715, 1988), ингибирующее действие на высвобождение гистамина (Allergy, 43: 100, 1988), ингибирующее действие на MLCK гладкой мускулатуры (J. Biol. Chem. , 263: 6215, 1988), противовоспалительное действие (Acta Physiol. Hung, 80: 423, 1992) и способность влиять на выживание клеток (J. Neurochemistry, 64: 1502, 1995). Также в Experimental Cell Research., 193: 175-182, 1991 было показано, что К-252а подавляет выработку интерлейкина 2.
Кроме того, сообщалось, что производные К-252а подавляют активность РКС, ингибируют секрецию гистамина [Японские публикации непрошедших экспертизу патентных заявок 295588/88] , имеют противоопухолевую активность [Японские публикации непрошедших экспертизу патентных заявок 168689/89 (США 4877776), WO 88/07045 (США 4923986), WО 94/04541, способны увеличивать количество тромбоцитов [WO 94/06799 (ЕР 630898 А)], имеют вазодепрессорную активность [Японские публикации непрошедших экспертизу патентных заявок 120388/87/89], способны ускорять холинергические функции нейронов [WО 94/02483 (США 5461146 и США 5621100)] и оказывать лечебный эффект при раке предстательной железы [WO 94/27982 (США 5516771)]. Отобранные аминсодержащие тринденовые соединения были получены перегруппировкой Бекмана из соответствующих стауроспориновых оксимов (WO 97/05140).
Индолкарбазолы в целом липофильны. Благодаря этому свойству индолкарбазолы способны относительно легко, по сравнению с белками, проникать через биологические мембраны. Также индолкарбазолы в целом имеют более длинный, чем у белков, период полувыведения in vivo.
Кроме того, сам К-252а, различные его производные были синтезированы и исследованы на биологическую активность. Показано, что среди производных К-252а биологическую активность имеет соединение, раскрытое Lewis et al. в патентах США 5461146 и 5621100 и РСТ публикации WO 94/02488 и именуемое в них как "соединение II-51". Показано, что соединение II-51 усиливает функцию холинергических нейронов, стриарных нейронов и сенсорных нейронов.
2. Нейродегенеративные заболевания и расстройства
Болезнь Паркинсона является нейродегенеративным расстройством, при котором происходит прогрессирующая и избирательная потеря допаминергических нейронов нигро-стриарного пути (Agid, Lancet: 337:1991). Введение 1-метил-4-фенил-1, 2, 3, 5-тетрагидропиридина (МРТР) мышам приводит к дегенерации допаминергических нейронов и служит в качестве экспериментальной модели потери допаминергических нейронов и нарушений поведения, наблюдаемых при болезни Паркинсона. Периферическое применение МРТР приводит к высокоизбирательной дегенерации системы допаминергических нигро-стриарных нейронов у людей, обезьян и мышей (Heikkila et al., Science 224: 1451-1453, 1984; Burns et al., Proc. Natl. Acad. Sci. USA 80: 4546-4550, 1983).
Дегенерация нейронов, вызванная МРТР на модели у мышей, достаточно полно описана. Системное введение МРТР вызывает избирательную селективную потерю содержания допамина (и метаболитов), активности тирозингидроксилазы и участков поглощения допамина в допаминергических нейронах полосатого тела мыши (Heikkila et al., Nature, 311: 467-469, 1984, a, b; Tipton et al., J. Neurochem., 61: 1191-1206, 1993). Этот эффект является дозозависимым. Максимальная потеря происходит между третьим и седьмым днем после поражения (Jackson-Lewis et al., Neurodegeneration, 4: 257-269, 1955). Органеллы допаминергических клеток черной субстанции менее чувствительны к токсическому действию МРТР, чем их соответствующие нервные окончания. При высоких дозах МРТР или множественных инъекциях МРТР существенная потеря ТН-иммуноположительных клеток в черной субстанции происходит в пределах 1 недели(Heikkila et al., Science, 224: 1451-1453, 1984; Jackson-Lewis et al., Neurodegeneration, 4: 257-269, 1995). При более низких дозах МРТР или при однократной инъекции потеря черной субстанции тирозингидроксилаза - положительных клеток происходит позднее (Tatton et al. , J. Neuroscience. Res., 30: 666-672, 1991). Таким образом, при более низких дозах МРТР и коротком периоде после повреждения стриарное поражение может наблюдаться в отсутствие потери тирозингидроксилаза - положительных клеток черной субстанции. Эта нейродегенеративная последовательность сходна с последовательностью, наблюдаемой при заболевании. Модель является признанной и широко используемой моделью для изучения болезни Паркинсона с использованием МРТР у мышей.
Нехолинергические нейроны, использующие в качестве нейротрансмиттера гамма аминомасляную кислоту (ГАМК) (так называемые ГАМК-ергические нейроны), распространены по всему объему мозга. Например, они обнаружены в крупноклеточном базальном узле у грызунов (аналогичная область в мозге человека называется базальное ядро Мейнерта), области базального отдела переднего мозга, играющей важную роль для функций внимания и памяти. Повреждение ГАМК-ергических нейронов в базальном отделе переднего мозга также может вызывать нарушение поведения при нейродегенеративных заболеваниях, таких как болезнь Альцгеймера (Dekker et ai., Neurosci and Biobeh. Rev., 15: 299-317, 1991; Gallagher et al., Seminars in Neuroscience, 6: 351-358, 1994, Torres et al., Neuroscience, 63: 95-122, 1994).
Нейроны в базальном отделе переднего мозга погибают при нескольких заболеваниях центральной нервной системы, в частности при болезни Альцгеймера. (Arendt et al., Acta Neuropathol. (Berl.), 61: 101-108, 1983; Iraizoz et al. , Neuroscience, 43: 33-40, 1991; Vogels et al., Neurobiol. Aging, 11: 3-13, 1990). Важным фактором, влияющим на гибель таких нейронов, является связанная с перевозбуждением токсичность глутамата, т.е. гиперстимуляция нейронов избытком глутамата (Choi, Neuron, 1: 623-634, 1988). Соответственно в ряде экспериментальных моделей болезни Альцгеймера глутамат или его аналоги используются для моделирования эксцитотоксической гибели клеток в базальных отделах переднего мозга, где и происходит гибель нейронов, т.е. в крупноклеточном базальном узле (Wenk, Ben. Brain Res., 72: 17-24, 1996).
Сначала патология нейронов при болезни Альцгеймера наблюдается в коре, и по мере прогрессирования заболевания потеря нейронов в этой области становится выраженной (Braak et al., Acta Neuropathol, 82: 239-259, 1991; Hyman et al., Ann Neurol. 20: 472-481, 1986). Нейроны во 2-ом слое энторинальной коры проецируются в зубчатую извилину гиппокампа, и этот нейронный путь играет важную роль в формировании памяти (Levisohnet et al., Brain Res., 564: 230-244, 1991; Olton et al., Brain. Res., 139: 295-308, 1978; Stewart et al. , Brain. Res. Bull. , 2: 41-48, 1977). Нейроны во 2-ом слое энторинальной коры, подобно множеству других нейронов коры головного мозга, используют глутамат в качестве нейротрансмиттера (Mattson et al., Neuron, 1: 865-876, 1988; White et al. , Nature, 270: 356-357, 1977). Таким образом, потеря глутаматергических нейронов в энторинальной коре влияет на нарушения поведения, наблюдаемые при болезни Альцгеймера и других неврологических расстройствах.
3. Периферические невропатии
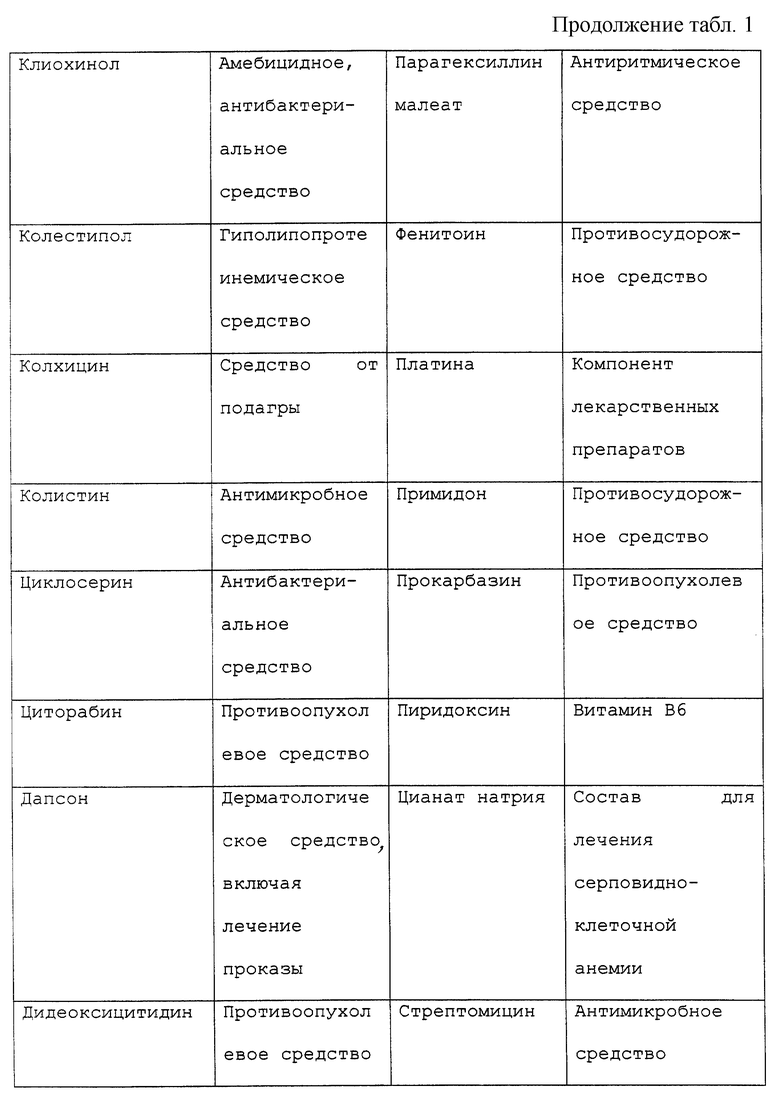
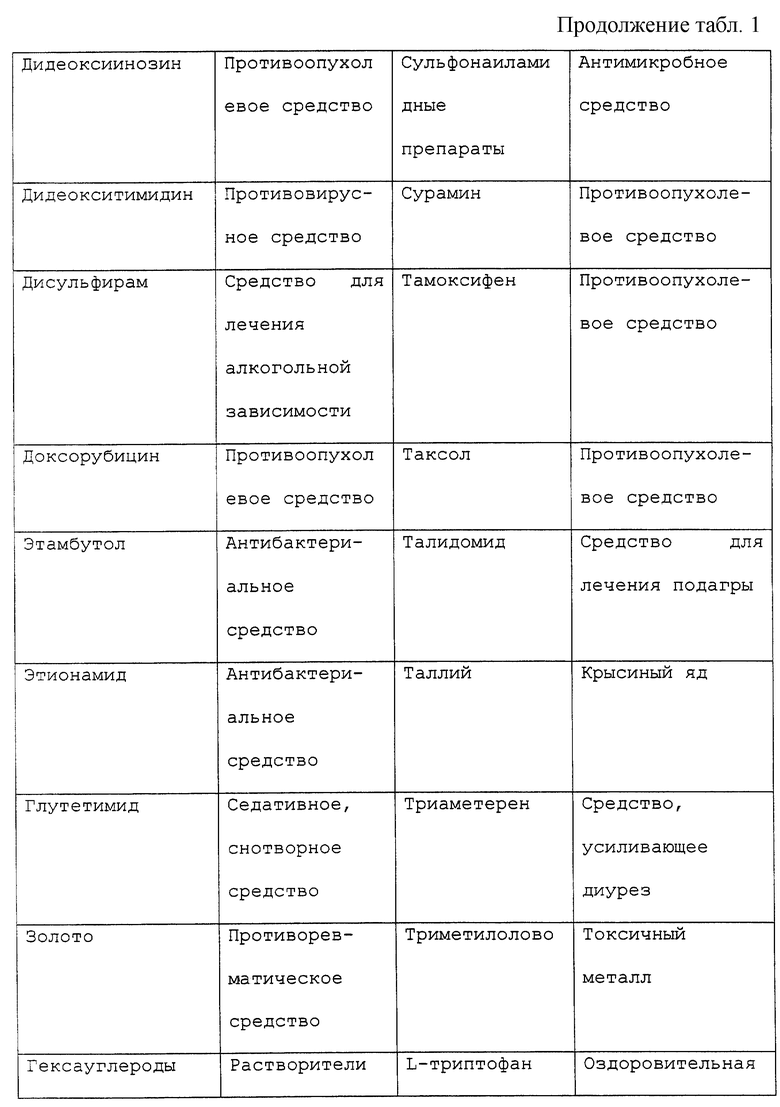
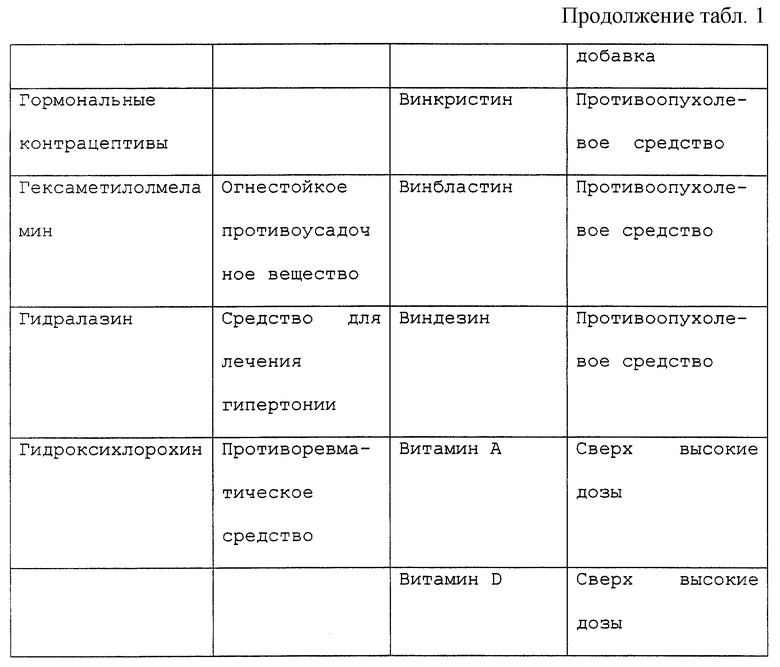
Периферической невропатией обычно именуют расстройство, поражающее периферические нервы, наиболее часто проявляющиеся как изолированное или комбинированное нарушение моторной, сенсорной, сенсорномоторной или автономной функций. Каждое из широкого разнообразия морфологии, проявляющееся при периферических невропатиях, может быть удивительно связано с большим множеством причин. Например, периферические невропатии могут быть приобретены генетически, могут возникать в результате системных заболеваний или могут быть вызваны токсическим агентом. Нейротоксические воздействия могут оказывать лекарственные препараты, противоопухолевые средства, примеси в пищевых продуктах или медикаментах, факторы загрязнения окружающей среды и промышленные загрязняющие факторы.
В частности, известны химиотерапевтические средства, являющиеся причиной сенсорных и/или моторных невропатий, включая винкристин, противоопухолевое средство, используемое для лечения злокачественных гематологических заболеваний и сарком. Нейротоксичность зависит от дозы и проявляется в виде сниженной моторики кишечника и периферической невропатии, в особенности дистальных мышц кистей рук и стоп как постуральной гипотонии и атонии мочевого пузыря. Сходные нарушения были отмечены при использовании таксола и цисплатина (Mollman, 1990, New Eng. Jour. Med., 322: 1266-1267), хотя связанная с цисплатином нейротоксичность может быть облегчена фактором роста нервов (NGF) (Apfel et al., 1992, Annals neurolology, 31: 76-80). Хотя нейротоксичность иногда является обратимой после отмены нейротоксичного агента, восстановление может быть очень медленным процессом (Legha, 1986, Medical Toxicology, 1: 421-427; Olsen et al., 1991, Drug Safety, 6: 302-314).
Существует ряд наследственных периферических невропатий, включая болезнь Рефсума, А-беталипопротеинемию, болезнь Танжера, болезнь Краббса, метахроматическую лейкодистрофию, болезнь Фабри, синдром Дежерена-Соттаса и другие. Среди всех наследственных невропатий, известных издавна, наиболее частым является болезнь Шарко-Мари-Туса (для дополнительной информации по периферическим невропатиям см. также патент США 5420112).
4. Цитокины
Как известно, фактор некроза опухолей альфа(TNF-α) и интерлейкин-1β (IL-1β) являются полипептидами, вовлеченными в ряд воспалительных и метаболических процессов in vivo. Информацию о роли TNF-α в воспалительных заболеваниях, включая септический шок, см. также в Ann. Rev. Imunnol. 7: 625 (1980) и Clinical Trials for Treament of Sepsis, Sibbald W.J. and Vincent J. -L. (Eds), Springer-Verlag, Berlin, Heidelberg, 1995. Принято считать, что избыточная или неадекватная выработка TNF-α ответственна за ряд патологических состояний, включая септический шок (Spooner et аl., Clinical Immunology and Immunopathology, 62: p. S11 (1992), и различные другие аллергические и воспалительные состояния или заболевания, включая, но не ограничиваясь ревматоидным артритом, остеоартритом, астмой, бронхитом, хроническим обструктивным заболеванием дыхательных путей, псориазом, аллергическим ринитом, дерматитом и воспалительным заболеванием кишечника и другими аутоиммунными заболеваниями (Immunol. Res. , 10: 122 (1991), Science, 229: 896 (1985) and Proc. Natl. Acad. Sci., 89: 7375 (1992).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В целом, изобретение представляет способы облегчения вредных эффектов различных заболеваний, расстройств и состояний с помощью лечения нуждающегося в нем субъекта терапевтически эффективным количеством соединения А.
Более конкретно, изобретение представляет способ лечения вредных эффектов заболеваний, расстройств и состояний, приводящих к смерти или являющихся ее причиной, либо приводящих к подавлению функций определенных нейронов или являющихся его причиной с помощью усиления функции или выживания допаминергических, ГАМК-ергических или глутаматергических нейронов у млекопитающих, включающий этап контактирования нейрона с соединением А. Обычно млекопитающим, у которого обнаруживается нейрон, является человек. Обычно функция допаминергических, ГАМК-ергических или глутаматергических нейронов, контактировавших с соединением А, нарушена или им угрожает риск гибели из-за нейродегенеративного заболевания. Обычно нейродегенеративным заболеванием является болезнь Паркинсона или болезнь Альцгеймера.
Более конкретно, изобретение также представляет способ уменьшения периферической невропатии, включающей введение млекопитающему количества соединения А, уменьшающего невропатию.
Хотя уже сообщалось, индолкарбазольное соединение К-252а снижает летальность в результате введения эндотоксина, эту способность связывали со способностью К-252а ингибировать протеинкиназы, в особенности протеинкиназу С (lhaba et al., Jpn. J. Surg., 23: 234 (1993). Неожиданно было обнаружено, что соединение А, которое обладает небольшим ингибирующим действием на РКС или вообще его не имеет, проявляет удивительно высокую активность в качестве ингибитора выработки TNF-α и цитокина IL-lβ. Поэтому еще одной особенностью изобретения является то, что оно представляет способ подавления выработки TNF-α и IL-lβ у млекопитающего и способ лечения или облегчения воспалительных состояний или заболеваний, включая, но не ограничиваясь септическим шоком, ревматоидным артритом, остеоартритом, астмой, бронхитом, хроническим обструктивным заболеванием дыхательных путей, псориазом, аллергическим ринитом, дерматитом, воспалительным заболеванием кишечника и другими аутоиммунными заболеваниями, причем способ включает введение указанному млекопитающему эффективного количества соединения А и его фармацевтически приемлемых солей в комбинации с фармацевтически приемлемым носителем.
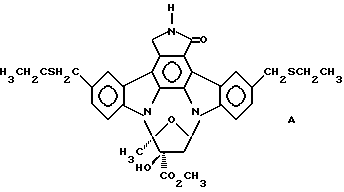
Использованный здесь термин "соединение А" обозначает соединение, химическая структура которого представлена ниже:
Соединение А также именуется соединением II-51 (Lewis et аl., патенты США 5461146 и 5621100; WO 94/02488).
Использованный здесь термин "облегчать" или "облегчающий" обозначают терапевтически улучшать и/или терапевтически уменьшить, и/или делать более терапевтически переносимым.
Использованный здесь термин "вредный" обозначает повреждающий и/или опасный, и/или отрицательный.
Использованное здесь слово "избыточная выработка", когда используется для обозначения изменений выработки TNF-α и/или IL-lβ, обозначает выработку TNF-α и/или IL-lβ, приводящую к патологическим состояниям, таким, например, как септический шок, аллергические состояния, воспалительные состояния и т. д.
Использованные здесь термины "ингибировать" или "ингибирующий" означают, что наличие соединения А оказывает сравнительно больший эффект на уменьшение и/или прекращение, и/или предотвращение выработки материала, контактирующего с соединением А, чем сравнительного материала, не контактировавшего с соединением А.
Использованные здесь термины "усиливать" или "усиливающий" при использовании для модификации терминов "функция" или "выживание" обозначает, что наличие соединения А имеет сравнительно больший эффект на функцию и/или выживание определенного нейрона, чем сравнительного нейрона, не контактировавшего с соединением А. Например (но не в виде ограничения), что касается выживания, например, допаминергического нейрона, соединение А должно свидетельствовать об усилении функции популяции допаминергических нейронов под угрозой гибели (вследствие повреждения, патологического состояния, дегенеративного состояния или естественного прогрессирования) при сравнении с популяцией допаминергических нейронов, не контактировавших с соединением А, если подвергшаяся его действию популяция имеет относительно больший период функциональности, чем не обработанная популяция.
Использованный здесь термин "допаминергический нейрон" обозначает нейрон, использующий в качестве нейротрансмиттера допамин.
Использованный здесь термин "ГАМК-ергический нейрон" обозначает нейрон, использующий в качестве нейротрансмиттера гамма-аминомасляную кислоту.
Использованный здесь термин "глутаматергический нейрон" обозначает нейрон, использующий в качестве нейротрансмиттера глутам.
Использованный здесь термин "nbm" обозначает крупноклеточное базальное ядро.
В случае отсутствия других определений все использованные здесь технические и научные термины имеют такое же значение, как и термины, обычно понятные рядовому специалисту в этой области, которому предназначено это изобретение. Хотя способы и материалы аналогичны или эквивалентны описанным здесь, далее могут использоваться на практике или при испытании настоящего изобретения, ниже описаны предпочтительные способы и материалы. Все публикации, заявки на патенты, патенты и другие ссылки, упомянутые здесь, включены в качестве ссылок. В случае конфликта настоящий документ, включая определения, будет служить контролем. При отсутствии других определений описанные здесь материалы, способы и примеры являются только иллюстративными и не предназначены для ограничения. Различные признаки и преимущества изобретения станут очевидны из следующего подробного описания и из формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ФИГУР
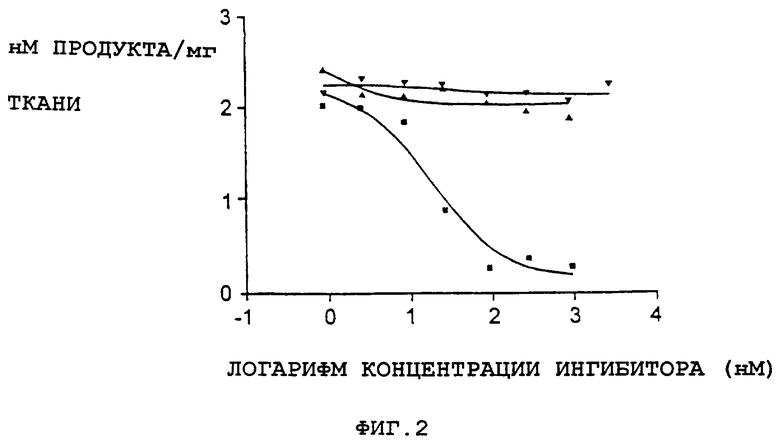
Фиг.1 представляет собой графическое представление данных, показывающих, что соединение А не является ингибитором моноаминоксидазы А. ИК50 для клоргилина составило 21 нмоль. Треугольники вверх вершиной - клоргилин; перевернутые треугольники - соединение А; квадраты - L-депренил.
Фиг.2 представляет собой графическое представление данных, показывающих, что соединение А не является ингибитором моноаминоксидазы В. ИK50 для клоргилина составило 21 нмоль. Треугольники вверх вершиной - клоргилин; перевернутые треугольники - соединение А; квадраты - L-депренил.
Фиг. 3 представляет собой столбцовую диаграмму, показывающую, что животные, получавшие соединение А (светлые столбики), имели значительно больше меченных фторзолотом нейронов в клювовидной части поврежденного nbm (более 400 мкм от средней точки повреждения), по сравнению с имеющими повреждение животными, получавшими только носитель (темные столбики). *=Р<0.05 по критерию Newman-Keuls.
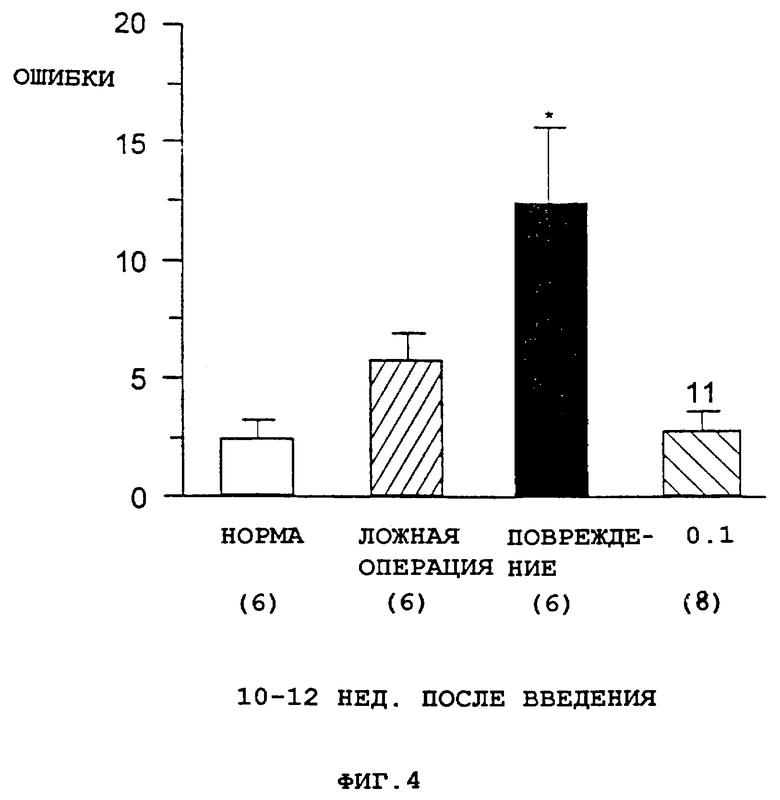
Фиг. 4 представляет собой столбцовую диаграмму, показывающую общее количество ошибок, совершенных при выполнении меняющегося задания в Т-образном лабиринте с вознаграждаемым чередованием, по сравнению с контрольной группой, ложно оперированными животными, животными с хирургическим повреждением, получавшими только носитель ("повр.") и животными с хирургическими повреждениями, получавшими соединение А в дозе 0.1 мг/кг 1 р/д ("0.1"). Число животных в каждой группе показано в скобках *=Р<0.05 по сравнению с ложно оперированными 11= Р<0.01 в сравнении с (повр.) с помощью критериев Newman-Keuls.
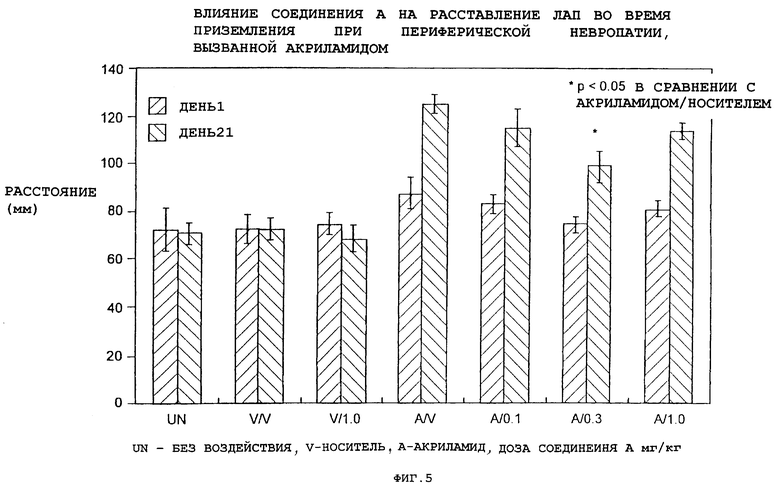
Фиг. 5 показывает влияние соединения А на вызванную акриламидом периферическую невропатию.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В целом, в изобретении описываются способы облегчения вредных воздействий различных заболеваний, расстройств и состояний с помощью лечения нуждающегося в нем субъекта терапевтически эффективным количеством соединения А.
Это изобретение представляет способ лечения вредных воздействий заболеваний, расстройств и состояний, которые отрицательно воздействуют на функцию и/или выживание нейронов, которым угрожает гибель вследствие таких заболеваний и расстройств путем усиления функции или выживания специфических типов нейронов центральной нервной системы млекопитающих. Более конкретно, изобретение представляет способ усиления функции или выживания допаминергических нейронов, ГАМК-ергических нейронов и глутаматергических нейронов у млекопитающего с помощью введения млекопитающему соединения А, замещенного в кольце производного К-252а со следующей химической структурой: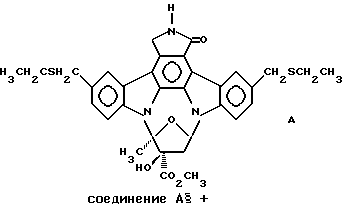
Допаминергические нейроны, ГАМК-ергические нейроны, глутаматергические нейроны широко распространены в центральной нервной системе млекопитающих. Каждый из этих трех типов нейронов претерпевает нарушение функций или даже гибель при одном или нескольких нейродегенеративных заболеваний центральной нервной системы. При болезни Паркинсона происходит прогрессирующая потеря допаминергических нейронов нигростриарного пути. При болезни Альцгеймера происходит гибель различных типов нейронов, включая ГАМК-ергические нейроны в базальном ядре Мейнерта базального отдела переднего мозга. При болезни Альцгеймера также происходит гибель глутаматергических нейронов в энторинальной области коры головного мозга.
Одним из методов лечения болезни Паркинсона или болезни Альцгеймера является введение соединения, которое усиливает функцию или выживание допаминергических нейронов, ГАМК-ергических нейронов или глутаматергических нейронов. Соединение А фармакологически активно в биологических количественных определениях и моделях in vivo усиленной функции или выживания допаминергических нейронов, ГАМК-ергических нейронов и глутаматергических нейронов. Таким образом, настоящее изобретение применимо для лечения болезни Паркинсона или болезни Альцгеймера. Изобретение, однако, не ограничено лечением этих болезней. Использование соединения А для усиления функций или выживания допаминергических нейронов, ГАМК-ергических нейронов или глутаматергических нейронов, чья нарушенная функция или риск гибели происходит в результате иных причин, нежели болезнь Паркинсона или болезнь Альцгеймера, также находится в объеме притязаний настоящего изобретения.
Изобретение также раскрывает способ уменьшения периферической невропатии. Способ включает введение млекопитающему уменьшающего невропатию количества соединения А. В различных предпочтительных вариантах реализации млекопитающее является человеком или сельскохозяйственным или домашним млекопитающим, у которого развивается невропатия, например, в результате лечения новообразования химиотерапевтическим средством. Соединение А может вводиться способом, считающимся эффективным, специалистом в данной области, предпочтительным способом введения является подкожная инъекция.
Используемым здесь термином "периферическая невропатия" именуется расстройство, поражающее сегмент периферической нервной системы. Изобретение включает использование соединения А для уменьшения нейротоксичности, включая, но не ограничиваясь, дистальную сенсорно-двигательную невропатию или вегетативные невропатии, такие как снижение сократительной способности желудочно-кишечного тракта или атонию мочевого пузыря.
Предпочтительные невропатии, которые могут эффективно излечиваться соединением А, включают невропатии, связанные с системным заболеванием, например постполиомиелитный синдром; генетически приобретенные невропатии, например болезнь Шарко-Мари-Туса; и невропатии, вызываемые токсическим агентом, например акриламидом, или химиотерапевтическим средством, например винкристином.
При использовании соединения А для лечения невропатии, вызванной токсическим агентом, оно может вводиться перед, одновременно или после воздействия токсического агента или перед, во время или после введения химиотерапевтического средства. Предпочтительно, соединение А и химиотерапевтическое средство вводятся через эффективные интервалы времени во время перекрывающего периода лечения. Соединение А может вводиться млекопитающему после воздействия нейротоксического агента или после химиотерапии для восстановления, по меньшей мере, части неврологической функции, нарушенной нейротоксическим или химиотерапевтическим агентом. Химиотерапевтическим может быть любой химиотерапевтический агент, оказывающий нейротоксическое действие, такой как винкристин, таксол, дидеоксиинозин или цисплатин.
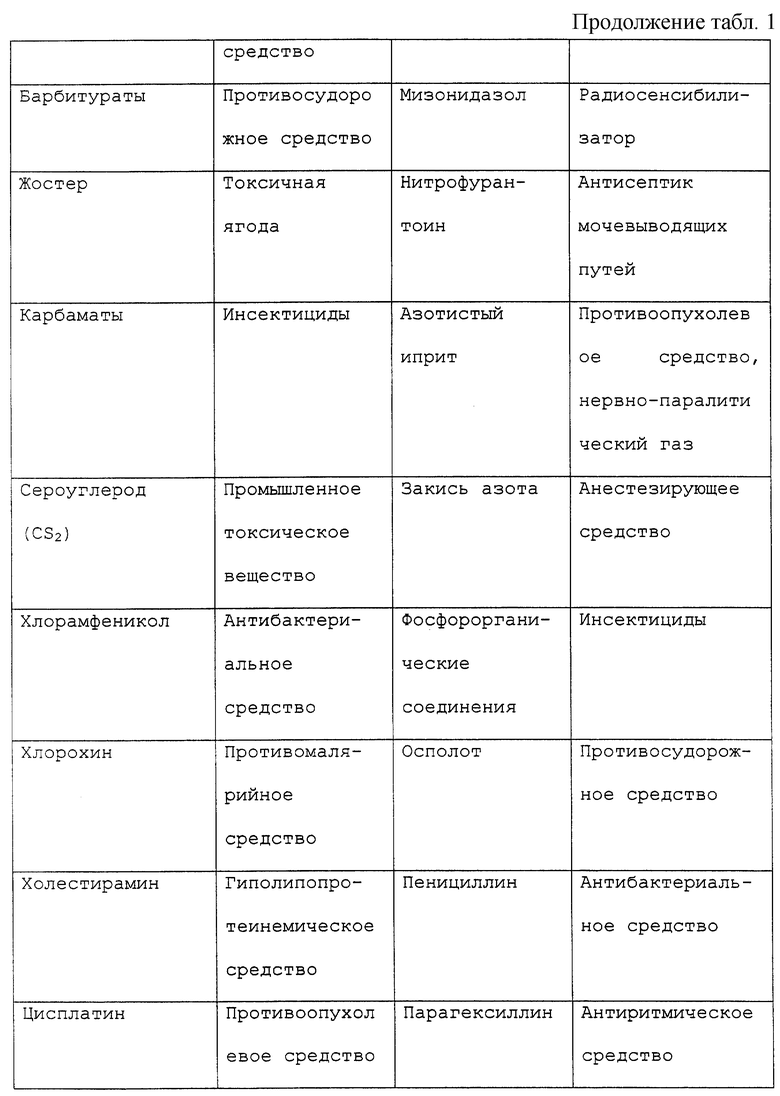
Под "токсическим агентом" или "нейротоксическим агентом" подразумевается вещество, которое посредством своего химического действия повреждает, нарушает или подавляет активность компонента нервной системы. Существует длинный перечень нейротоксических агентов, которые вызывают невропатии, включающий, но не ограничивающийся, противоопухолевые средства, такие как винкристин, винбластин, цисплатин, таксол или дидеокси соединения, например дидеоксиинозин; спирт; металлы; промышленные токсины, контакт с которыми имеется в промышленной или окружающей среде; примеси в пище или лекарственных средствах или передозировка витаминов или терапевтических препаратов, например антибиотиков, таких как пенициллин или хлорамфеникол, или мегадозы витаминов A, D или В6. Обширный, хотя и не полный перечень химических соединений с нейротоксическими побочными эффектами представлен в таблице 1. Хотя этот перечень представляет примеры нейротоксических соединений, он предназначен служить примером, а не ограничивать диапазон притязаний изобретения. Другие токсические агенты могут вызывать невропатии и могут характеризоваться с помощью способов, известных специалистам в этой области. Под "воздействием токсического агента" подразумевается, что токсический агент становится доступным или вступает в контакт с млекопитающим изобретения. Воздействие токсического агента может происходить путем прямого введения, например проглатыванием или введением с пищей, лекарственного или терапевтического средства, например химиотерапевтического средства, при случайном загрязнении или контакте с окружающей средой, например посредством контакта с воздухом или водой.
Несмотря на несопоставимую морфологию и причины, вызывающие периферические невропатии in vivo, заявители предположили, что соединение А может быть эффективным средством профилактики или лечения таких невропатий у млекопитающего.
Было также показано, что соединение А подавляет выработку цитокина TNF-α, несмотря на отсутствие или низкую подавляющую активность против РКС. Соединение А также подавляет выработку цитокина IL-1β. Выработка TNF-α связана с множеством заболеваний и расстройств, так что ее подавление с помощью соединения А может оказать благоприятное воздействие на субъекта, нуждающегося в подавлении выработки TNF-α.
Таким образом, соединение А может также использоваться для подавления выработки TNF-α у млекопитающего и/или в способе лечения или облегчения воспалительных состояний или заболевания, включая, но не ограничиваясь септическим шоком, ревматоидным артритом, остеоартритом, астмой, бронхитом, хроническим обструктивным заболеванием дыхательных путей, псориазом, аллергическим ринитом, дерматитом и воспалительным заболеванием кишечника и другими аутоиммунными заболеваниями, причем способ включает введение указанному млекопитающему терапевтически эффективного количества соединения А.
Для применения в настоящем изобретении соединение А может быть составлено в фармацевтическую композицию с помощью смешивания с фармацевтически приемлемыми не токсичными наполнителями и носителями. Такая композиция может быть получена для введения любым из различных путей. Пути введения и предпочтительные лекарственные формы включают следующие: парентеральный, предпочтительно в форме жидких растворов или суспензий; оральный, предпочтительно в форме таблеток или капсул; интраназальный, предпочтительно в форме порошков, капель в нос или аэрозолей; и дермальный, например, с помощью трансдермальных систем.
Соединение А может для удобства вводиться в виде стандартной дозы препарата с помощью любого способа, хорошо известного в фармации, например, как описано в Remington's Pharmaceutical Sciences (Mack Pub. Co., Easton, PA, 1980). Композиции для парентерального введения могут содержать в качестве обычных наполнителей стерильную воду или солевой раствор, полиалкиленгликоли, такие как полиэтиленгликоль, масла растительного происхождения, гидрогенизированные нафталины и им подобные. В частности, для контроля высвобождения активных соединений могут использоваться: биосовместимый, биодеградируемый лактидный полимер, лактид /гликолидный сополимер или полиоксиэтилен-полиоксипропиленовые сополимеры. Другие системы, которые потенциально могут использоваться для доставки соединения А, включают частицы этиленвинилацетатного сополимера, осмотические насосы, имплантируемые инфузионные системы и липосомы. Композиции для ингаляционного введения в качестве наполнителей содержат, например, лактозу или могут быть водными растворами, содержащими, например, лактозу, или могут быть водными растворами, содержащими, например, простой полиоксиэтилен-9-лауриловый эфир, гликохолат и дезоксихолат, или маслянистые растворы для введения в форме капель в нос, или в виде геля для интраназального введения. Композиции для парентерального введения могут также включать гликохолат для трансбуккального введения, салицилат для ректального введения или лимонную кислоту для вагинального введения. Композиции для трансдермальных систем представляют собой предпочтительно липофильные эмульсии.
Соединение А может применяться в качестве единственного активного ингредиента в фармацевтической композиции. Альтернативно, оно может применяться в комбинации с другими активными ингредиентами, как, например, факторами роста, способствующими выживанию нейронов или регенерации аксонов при заболеваниях или расстройствах.
Концентрация соединения А, применяемая в терапевтической композиции при осуществлении этого изобретения, может варьировать. Концентрация будет зависеть от таких факторов, как общая доза препарата, которую предполагается ввести, и путь введения. Обычно соединение А должно предоставляться в водном физиологическом буферном растворе для парентерального введения, содержащем приблизительно от 0.1 до 10 мас./об.%. Обычные диапазоны дозы составляют от приблизительно 1 мкг/кг до приблизительно 1 г/кг массы тела в сутки; предпочтительный диапазон дозы составляет от приблизительно 0.01 мг/кг до 100 мг/кг массы тела в сутки. Вероятно, предпочтительная доза соединения А, которую предполагается вводить, зависит от таких факторов, как тип и степень тяжести заболевания или расстройства, общего состояния здоровья конкретного пациента, относительной эффективности соединения А при подлежащем лечению конкретном заболевании или расстройстве, и пути введения.
Соединение А для применения в настоящем изобретении предпочтительно получают в соответствии со способами, описанными в патенте США 5461146, выданном Lewis et al.
Далее настоящее изобретение будет проиллюстрировано следующими примерами. Эти примеры не следует расценивать ни как ограничивающие диапазон притязаний изобретения, ни как ограничивающие диапазон притязаний прилагаемой к нему формулы изобретения.
Пример 1
Фармакологическая активность при действии на допаминергические нейроны
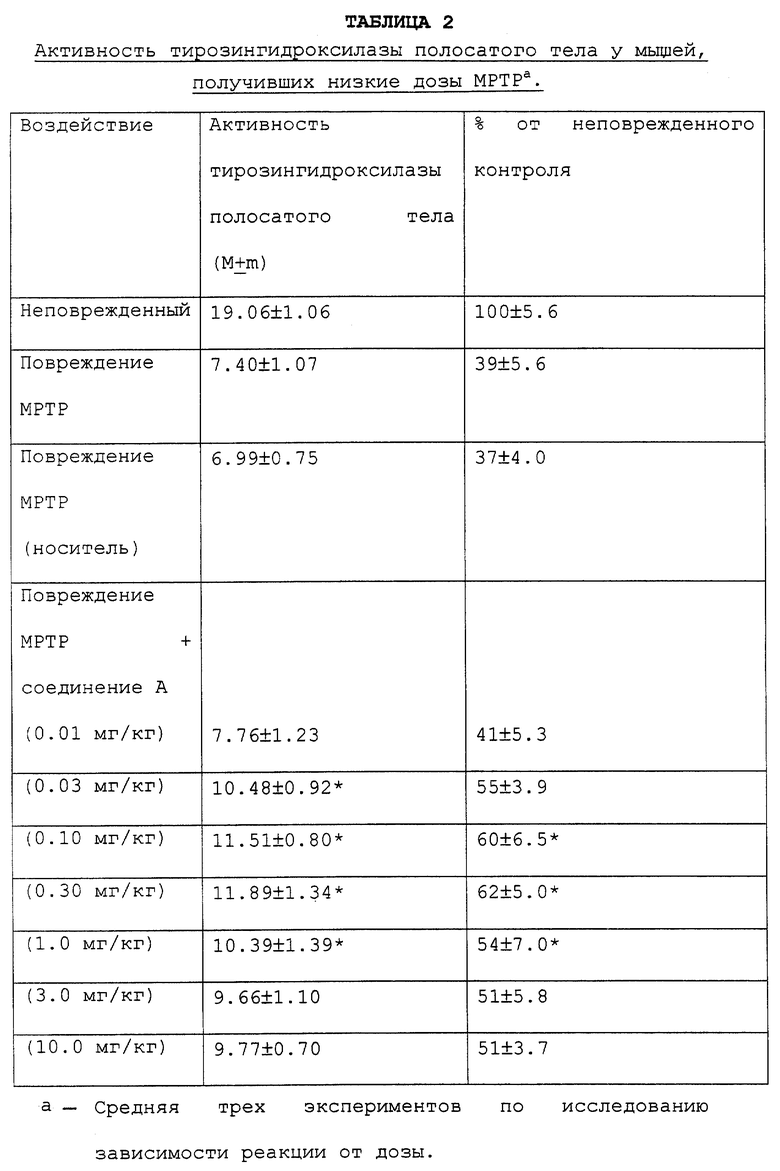
Эксперименты проводят с использованием соединения А на поврежденных МРТР допаминергических нейронах у мышей, модель с использованием МРТР у мышей. Однократная доза МРТР (20 мг/кг), введенная подкожно черным мышам линии С57, вызвала потерю активности приблизительно на 60% гидроксилазы полосатого тела. Введение соединения А в ежесуточной дозе в диапазоне от 0.01 до 10 мг/кг (подкожно) таким мышам, получавшим МРТР, снизило потерю активности тирозин гидроксилазы полосатого тела. Эти данные показаны в таблице 2.
Мышам вводят МРТР (20 мг/кг подкожно) через 4-6 часов после первой инъекции соединения А. Соединение А затем вводят ежедневно до конца эксперимента, продолжавшегося 7 дней. Проводят оценку активности фермента тирозингидроксилазы полосатого тела. Звездочка показывает статистически достоверное различие (р<0.05) от животных, получавших МРТР-носитель.
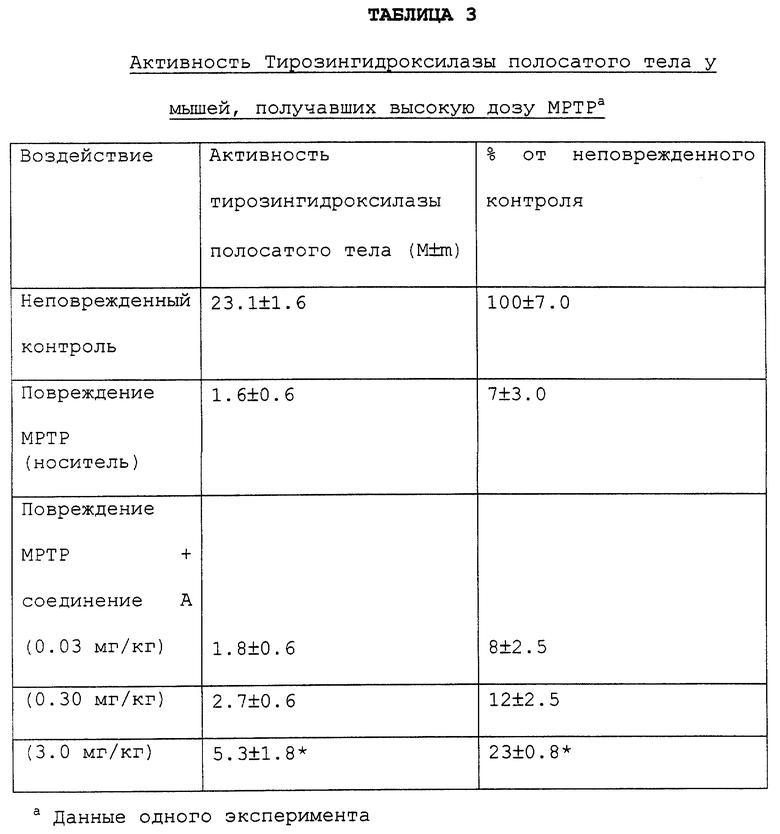
В отдельном эксперименте активность соединения А оценивают на модели с введением мышам высоких доз МРТР. Однократная инъекция 40 мг/кг МРТР вызывала через 7 дней после инъекций снижение активности фермента тирозингидроксилазы полосатого тела на 95%. Введение соединения А в дозе от 0.03 до 3.0 мг/кг в сутки дозозависимым образом уменьшало величину поражения. Эти данные представлены в таблице 3.
Мышам вводят МРТР (40 мг/кг подкожно) через 4-6 часов после первой инъекции соединения А. Соединение А затем вводят ежедневно до конца эксперимента, продолжавшегося 7 дней. Проводят оценку активности фермента тирозингидроксилазы полосатого тела. Звездочка показывает статистически достоверное различие (р<0.05) от животных, получавших МРТР-носитель.
Допаминергическая токсичность, опосредованная МРТР, зависит от опосредованного МАО-превращения МРТР в МРР+ (ион 1-метил-4-фенилпиридина) и захвата МРР+ в допаминергические нейроны. Соединения с обнаруженной активностью на модели у мышей, получавших МРТР, не должны определяться как ингибиторы МАО или захвата допамина. Как было установлено, соединение А не ингибирует моноаминоксидазу А и В in vitro (фиг.1 и 2) или не блокирует захват катехоламинов в нервные окончания, что указывает на то, что это соединение не предотвращает метаболическое превращение МРТР в МРР+ или не ингибирует активный захват МРР+ в допаминовые нейроны. Эти данные, подтверждающие эффективность соединения А на модели повреждения допаминергических нейронов МРТР у мышей, указывают на возможность использования соединения А в лечении нейродегенеративных расстройств, таких как болезнь Паркинсона.
Пример 2
Фармакологическая активность при действии на ГАМК-ергические нейроны
Соединение А исследуют на его способность предотвращать истощение или увеличивать выживание ГАМК-ергических нейронов в крупноклеточном базальном ядре, используя хорошо изученную модель поражения иботеновой кислотой. Иботеновая кислота (эксцитотоксин), как известно, уменьшает количество ГАМК-экспрессирующих нейронов в области nbm (Lindefors et al., Neurosci. Lett., 135: 262-264, 1992; Shaugnessy et al., Brain Res., 637: 12-26, 1994).
Взрослым самцам крыс линии Spraque-Dawley производят инъекцию иботеновой кислоты (5.0 г) в nbm, с одной стороны. Крысам затем путем подкожной инъекции вводят соединение А (0.03 мг/кг), начиная с 18 ч после поражения и продолжая введение 1 раз до 18 дней после поражения. Затем по всему рострально-каудальному протяжению nbm делают срезы и обрабатывают их для выявления декарбоксилазы глутаминовой кислоты, фермента, требуемого для биосинтеза ГАМК. Количество нейронов, экспрессирующих декарбоксилазу глутаминовой кислоты, затем подсчитывают по всему рострально-каудальному протяжению nbm в области, где нейроны nbm, экспрессирующие глутамат декарбоксилазу, легко отличимы от нейронов прилегающих структур, экспрессирующих глутамат декарбоксилазу (белое вещество медиальное и вентралльное от globus pallidus). Результат выражают в виде процента нейронов с пораженной стороны, экспрессирующих декарбоксилазу глутаминовой кислоты, от количества на противоположной, непораженной стороне. Результаты также рассчитывают отдельно для рострального отдела nbm, среднего отдела nbm и каудального отдела nbm.
В среднем по подсчетам в ростральном отделе nbm животных, получавших соединение А, обнаруживают 78±16% нейронов, содержащих декарбоксилазу глутаминовой кислоты, в сравнении с 34±19% у животных, получавших только носители, что, по критерию t Стьюдента, является статистически достоверным различием (р<0.05). В среднем или каудальном отделах nbm не наблюдают статистически достоверных различий между животными, получавшими соединение А, и животными, получавшими только носитель (контроль).
Эти результаты показывают, что соединение А может защищать от потери ГАМК-ергических нейронов в результате эксцитотоксического поражения и, следовательно, оказывает нейротрофическое воздействие на популяцию нейронов.
Пример 3
Фармакологическая активность при действии на нейроны nbm
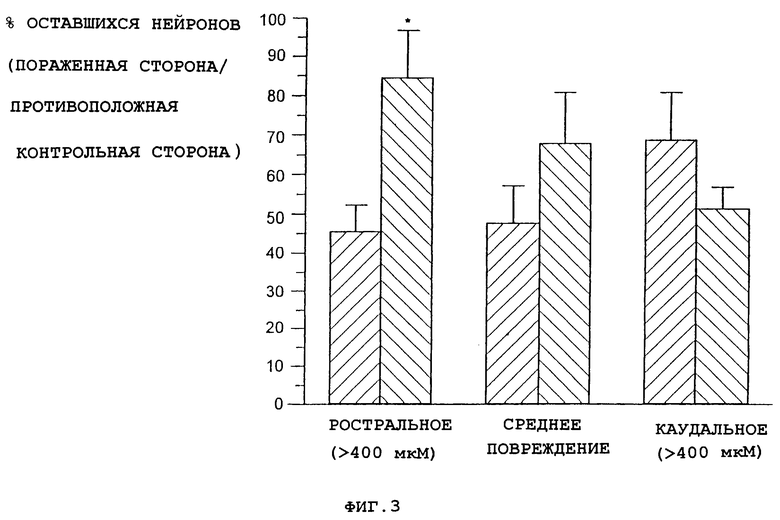
Для прямого исследования способности соединения А предотвратить эксцитотоксическую гибель нейронов нейроны nbm взрослых самцов крыс линии Spraque-Dawley сначала метят стойким маркером. Это осуществляют с помощью инъекции фторозолота (ФЗ), маркера нейронов, захватываемого нервными окончаниями и транспортируемого в обратном направлении к телу клеток (Book et al., J. Neuropath. Exp. Neurology, 53: 95-102, 1994), в области-мишени нейронов nbm в лобной и теменной коре. Спустя 7-10 дней у животных с помощью введения 5 мкг иботеновой кислоты вызывают одностороннее повреждение nbm. Через 18 ч после этого повреждения посредством подкожной инъекции животным начинают введение соединения А (0.03 мг/кг) и производят его 1 раз в 2 дня до 18 дней после повреждения. По всему рострально-каудальному протяжению nbm с обеих сторон мозга делают срезы ткани и подсчитывают количество нейронов nbm с меткой ФЗ. С помощью стандартных методик делают поправку расчетов на различия размера и результаты выражают в виде процентной доли меченных нейронов в nbm на стороне повреждения каждого животного относительно числа меченных нейронов в nbm на противоположной, не поврежденной стороне мозга.
В сравнении с животными, имеющими повреждения и получавшими только носитель, у животных, получавших соединение А, в ростральной части поврежденного nbm (более 400 мкм от средней точки повреждения) было значительно больше нейронов, меченных ФЗ (фиг.3). В повреждении средней и каудальной части эффект соединения А снижался.
Эти данные являются прямым доказательством того, что соединение А может предотвратить потерю предварительно меченных нейронов в nbm в результате эксцитотоксического поражения.
Пример 4
Длительное функциональное улучшение после кратковременного введения соединения А
Сначала взрослых самцов крыс линии Spraque-Dawley обучают чередующимся реакциям в стандартном задании в Т-образном лабиринте с вознаграждаемым чередованием (Helper et al., J. Neurosci., 5: 866-873, 1985). Затем для нарушения выполнения заданий в Т-образном лабиринте у животных вызывают двухсторонние повреждения крупноклеточного базального ядра (см., например, Salamone et al., Behav. Brain Res., 13: 63-70, 1984). Через 18 ч после этого повреждения посредством подкожной инъекции животным начинают введение соединения А (0.1 мг/кг) и производят его 1 раз в 2 дня до 12 дней после повреждения. Через 24 ч после последней инъекции всех животных один раз в сутки помещают в Т-образный лабиринт до тех пор, пока их чередующееся выполнение не достигнет предоперационного уровня, что занимает приблизительно от 3 до 10 дней. После достижения критерия животных 8-10 нед не подвергают дальнейшим исследованиям. В этот момент животных снова один раз в сутки помещают в Т-образный лабиринт до тех пор, пока они не достигнут предоперационного выполнения задания. Общее число ошибок, совершенных животными во время этого исследования, показано на фиг.4. Животные с повреждением, получавшие только носитель, совершили значительно больше ошибок, чем не оперированные или ложно оперированные животные, тогда как животные с повреждением, за 10-12 нед до этого получавшие соединение А, не отличались от нормальных животных.
Эти данные показывают, что соединение А вызывает стойкое улучшение поведения, отражающееся в улучшении внимания или памяти через несколько месяцев после прекращения введения.
Пример 5
Эффективность соединения А на модели повреждения энторинальной коры
Содержащие глутамат нейроны во 2 слое энторинальной коры проецируются в молекулярный слой зубчатой извилины, где они образуют синапсы на дендритах нервных клеток-зерен зубчатой извилины. Повреждение во 2 слое энторинальной коры может быть вызвано с помощью стереотаксической инъекции экситотоксина N-метил-D-аспартата (NMDA). В условиях анестезии пентобарбиталом натрия крысам производят инъекцию NMDA (15 нМ/участок) в каждый из 2 участков в энторинальной коре. Через 2 нед крыс забивают и перфузируют раствором, содержащим 50 мМ сульфида натрия. Мозг удаляют, производят срезы толщиной 40 мкм в горизонтальной плоскости и окрашивают крезилом фиолетовым или красителем Тимма. Выживание нейронов во 2 слое энторинальной коры оценивают в срезах, окрашенных крезилом фиолетовым, тогда как целостность окончаний их аксонов в молекулярном слое зубчатой извилины измеряют в чередующихся срезах, окрашенных красителем Тимма.
Инъекция NMDA разрушала 62±11% 2 слоя энторинальной коры и уменьшала площадь среднего молекулярного слоя зубчатой извилины на 19±6%. Ежесуточное введение соединения А (1 мг/кг подкожно) при первой инъекции, произведенной сразу после инъекции NMDA, уменьшало потерю нейронов 2 энторинального слоя до 22±10% (р<0.05 по t критерию Стьюдента) и предотвращало уменьшение площади среднего молекулярного слоя (р<0.05 по t критерию Стьюдента). Следовательно, соединение А защищало нейроны в энторинальной коре от эксцитотоксического повреждения и поддерживало целостность окончаний их аксонов в зубчатой извилине.
Эти данные показывают эффективность соединения А в защите глутаматергических нейронов энторинальной коры. На экспериментальных моделях было показано, что повреждения энторинальной коры вызывают нарушения памяти, а при болезни Альцгеймера происходят тяжелые дегенеративные изменения этих нейронов. Это подтверждает точку зрения, что соединение А может использоваться при лечении неврологических расстройств, связанных с потерей или повреждением глутаматергических нейронов и нейронов коры головного мозга, включая болезнь Альцгеймера, инсульт и черепно-мозговую травму, но не ограничиваясь ими.
Пример 6
Влияние соединения А на периферическую невропатию, вызванную акриламидом
Акриламид вызывает у людей и животных центрально-периферическую дистальную невропатию с "отмиранием в обратном направлении". Повреждение представляет собой смешанную сенсорно-двигательную невропатию, характеризующуюся у людей слабостью, тремором и атаксией. У животных акриламид вызывает изменения поведения (сенсорные, двигательные и проприоцептивные), паралогические изменения структуры и электрофизиологических характеристик ткани и потерю массы тела. На периферии акриламид преимущественно поражает волокна Аb большого диаметра с длинными аксонами. По данным измерения увеличения расставления лап при приземлении (РЛП), являющегося показателем проприоцепции, у крыс введение акриламида вызывает аксонопатию.
Невропатия, вызванная акриламидом, является моделью токсического воздействия химических соединений. Она отличается от других моделей действия химических соединений (таких как химиотерапевтические модели) тем, что ее продолжительность относительно короткая (3 нед) и животные относительно хорошо себя чувствуют. Акриламид не вызывает значительное системное токсическое действие.
В начале эксперимента используют самцов крыс линии Spraque-Dawley с массой тела 250 г. Акриламид (50 мг/кг внутрибрюшинно) вводят 3 раза в неделю в течение трех недель. Животных бросают с высоты 30 см и регистрируют расстояние между следами задних лап (Edwards P.M. and Parker V.H. (1977): "Простой, чувствительный и объективный способ ранней оценки акриламидной невропатии у крыс" (Toxicol Appl Pharmacol, 40: 589-591). Соединение А (0.1, 0.3 или 1.0 мг/кг подкожно в 5% солутоле) вводят один раз в сутки в течение всего эксперимента. У животных, получавших акриламид, расстояние РЛП было больше, чем у животных, получавших носитель. Соединение А (0.3 мг/кг/сутки) уменьшало степень увеличения РЛП, вызванного акриламидом (фиг.5).
Эти данные показывают, что соединение А может использоваться для лечения периферических невропатий.
Пример 7
Неспособность соединения А подавлять протеинкиназу in vitro
Количественное определение протеинкиназы С и подавление под воздействием протеинкиназы С К-252а раскрыто в патенте США 4923968 и в работе Kase H. et al. (eds.), Japan Scientific Societies Press, Tokyo, pp. 93-296 (1988). Посредством по существу аналогичного количественного определения было установлено, что К-252а подавляет протеинкиназу С с ПК50, составляющей 0.028 мМ, тогда как ПК50 соединения А составляет 16.0 мкМ, т.е. его активность ниже в 570 раз. В качестве ингибитора выработки TNF-α ПК50 соединения А составляет 139 нМ, а в качестве ингибитора выработки IL-lβ величина ПК50 составляет 261 нМ. Это концентрации, при которых соединение А не активно в качестве ингибитора протеинкиназы С.
Пример 8
Подавление in vitro выработки TNF-α и IL-lβ соединением А
Способность соединения А подавлять индукцию TNF-α была показана с помощью применения методики стандартного фармакологического исследования in vitro, как описано ниже. Маточные растворы, состоящие из 4 мМ соединения А в 100% диметилсульфоксиде (DMSO), хранят при 4oС. Среда RMPI 1640 для клеточной культуры (Media Tech, Herndon, Va) и фетальная телячья сыворотка (Hyclone, Logan, UT) составляют среду для исследования. Используют липополисахарид (LPS) (Sigma, St. Louis, МО) серотипа 0111:В4 Е. coli, экстрагированный трихлоруксусной кислотой. Маточные растворы LPS готовят и хранят при 4oС в солевом растворе с фосфатным буфером (PBS). Наборы иммуноферментного анализа для количественного определения фактора некроза опухоли альфа (TNF-β и IL-1α) закупают у компании Boehringer-Mannheim (Indianapolis, IN) и используют в соответствии с инструкциями производителя. Клетки ТНР-1, клеточную линию, происходящую из моноцитов человека, получают из American Type Culture Collection (ATCC TIB 202). Клетки выращивают в RMPI 1640, содержащей 10% фетальную телячью сыворотку (среду) в увлажненной атмосфере 5% СO2: 95% воздуха при 37oС. Эксперименты проводят в 24-ячеечных культуральных планшетах (Nunc) при концентрации 5•105 клеток ТНР-1 на 1 мл среды. Для индукции TNF--α клетки инкубируют с LPS при концентрации 2 мкг/мл. После 3-часового периода инкубации клетки и среду центрифугируют при 1000•g в течение 5 мин и либо сразу проводят количественное определение полученной в результате надосадочной жидкости на TNF-α, или хранят ее при -70oС и количественное определение проводят позднее. С помощью иммуноферментных количественных определений измеряют содержание TNF-α и IL-1β в образцах среды.
Для исследования соединения А на способность подавлять индукцию TNF-α клетки перед добавлением LPS инкубируют в течение 1 ч с меняющимися концентрациями соединения А. Соединения разводят так, что в среде для клеточной культуры никогда нет DMSO в концентрации выше 0.1%. Исследование и количественное определение соединения А проводят, как описано выше, на способность подавлять выработку TNF-α и IL-1β.
Пример 9
Подавление TNF-α и IL-1β, индуцированных LPS в сыворотке крови мышей
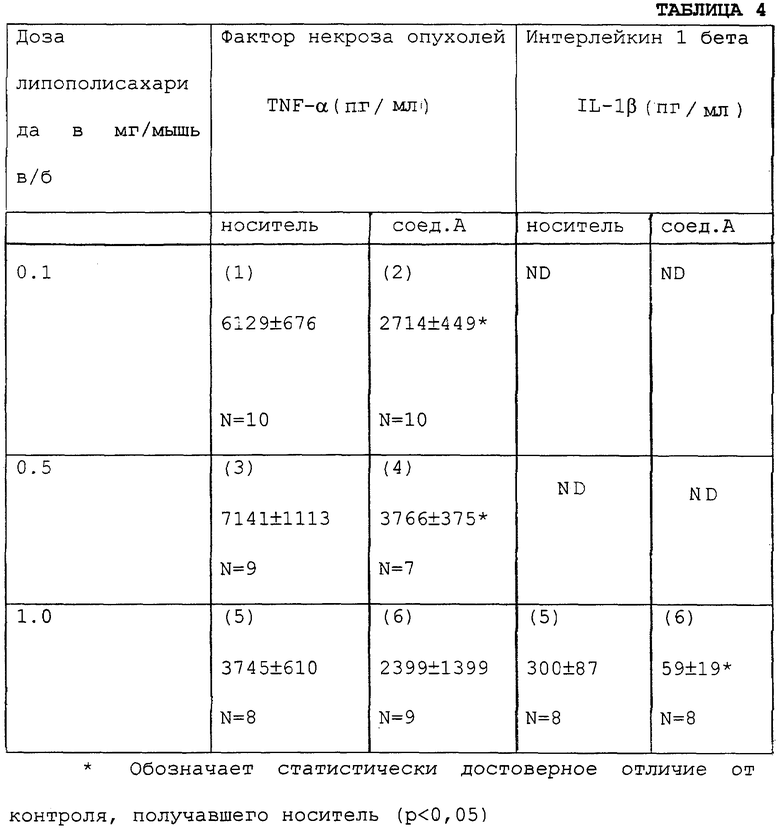
60 самок мышей линии C57BL (возраст 4-6 нед) делят на 6 групп, каждая из которых включает 10 животных. Трем группам (1, 3, 5) внутрибрюшинно (в/б) вводят только носитель (PBS, содержащую 0.1% TEA (тетраэтиламмоний гидрохлорид)) - по 200 мкл на животное. Другим трем группам (2, 4, 6) вводят соединение А (30 мг/кг в/б в 200 мкл) носителя (PBS, содержащей 10% сложного этилового эфира 12-гидроксистеариновой кислоты; солутол). Через 2 ч животным в/б вводят 0.1 мг/кг LPS (группы 1 и 2), 0.5 мг/кг LPS (группы 3 и 4) и 1.0 мг/кг LPS (группы 5 и 6). Через 2 ч после введения LPS всех животных забивают, получают образцы плазмы и проводят количественное определение содержания в них TNF-α и IL-lβ. Количественные определения проводят как описано в примере 8.
Данные сведены в таблице 4. Числа в скобках представляют собой номера описанных выше групп. N указывает количество животных, в плазме которых проводили определения. ND - определение не проводилось.
Как показано в таблице 4, введение LPS мышам вызвало выработку TNF-α у этих мышей, и количество выработанного TNF-α заметно уменьшалось при предварительном введении соединения А за 2 ч до исследования. Предварительное введение соединения А также значительно подавляло выработку IL-lβ в ответ на введение LPS в дозе 1.0 мг/кг.
Пример 10
Предотвращение гибели мышей, вызванной LPS
45 мышей делят на 3 группы, две из которых состоят из 10 мышей и одна группа из 20 мышей. Носители, LPS и соединение А вводят внутрибрюшинно. Группе 1 (n=10) исходно (время - 0) вводят носитель (солевой раствор с фосфатным буфером для LPS, 200 мкл на мышь), а через 2 ч - носитель для соединения А (200 мкл на мышь). Мышам в группе 2 (n=10) исходно (время - 0) вводят носитель для соединения А (200 мкл на мышь), а через 2 ч - LPS (2 мг в 200 мкл на мышь). Группе 3 (n=20) исходно (время - 0) вводят соединение А (30 мг/кг в 200 мкл носителя), а через 2 ч - LPS (2 мг в 200 мкл на мышь).
Введение LPS вызвало 90% смертность в пределах 20 ч и 100% смертность через 42 ч. В группе животных, за 2 ч до введения LPS получавших соединение А, через 20 ч после введения LPS смертность составила 25%, через 42 ч после введения LPS смертность была 75% и через 1 нед после введения LPS смертность составила 80%. Введение носителя хорошо переносилось и не вызывало гибели мышей.
Специалистам в этой области будет понятно, что в предпочтительные варианты реализации изобретения могут быть внесены многочисленные изменения и модификации и что такие изменения и модификации могут вноситься без отхода от сущности изобретения. Поэтому предполагается, что прилагаемая формула изобретения охватывает все эквивалентные варианты, укладывающиеся в истинную сущность и диапазон притязаний изобретения. Материалы, приводимые во всем описании этой патентной заявки, включены в него в виде ссылки.
Изобретение относится к медицине и касается способов подавления избыточной выработки фактора некроза опухолей альфа и интерлейкина-1 бета, а также лечения заболеваний, связанных с избыточным синтезом этих цитокинов, болезней Альцгеймера и Паркинсона c помощью производного индолкарбазола К-252a. Изобретение позволяет повысить эффективность лечения заболеваний. 6 с. и 2 з.п.ф-лы, 4 табл., 5 ил.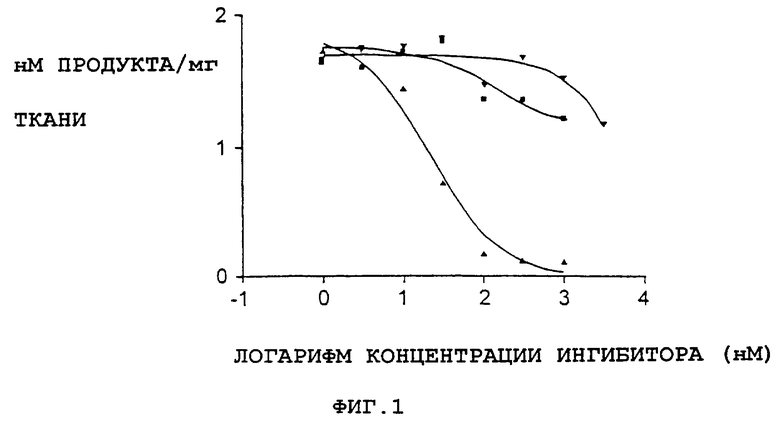
2. Способ облегчения вредных эффектов избыточной выработки фактора некроза опухолей альфа у млекопитающего, включающий стадию введения нуждающемуся в нем млекопитающему терапевтически- эффективного количества соединения А, представленного формулой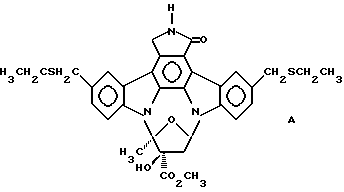
3. Способ подавления избыточной выработки интерлейкина-1 бета у млекопитающего, включающий стадию введения нуждающемуся в нем млекопитающему терапевтически-эффективного количества соединения А, представленного формулой
4. Способ облегчения вредных эффектов избыточной выработки интерлейкина-1 бета у млекопитающего, включающий стадию введения нуждающемуся в нем млекопитающему терапевтически-эффективного количества соединения А, представленного формулой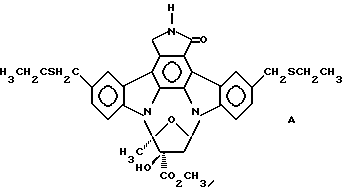
5. Способ по п. 2, где указанные вредные эффекты выбраны из группы, включающей септический шок, ревматоидный артрит, остеоартрит, астму, бронхит, хроническое обструктивное заболевание дыхательных путей, псориаз, аллергический ринит, дерматит и воспалительное заболевание кишечника.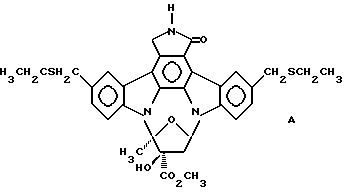
8. Способ уменьшения повреждающих воздействий болезни Паркинсона на допаминергические нейроны, включающий стадию введения нуждающемуся в нем млекопитающему терапевтически-эффективного количества соединения А, представленного формулой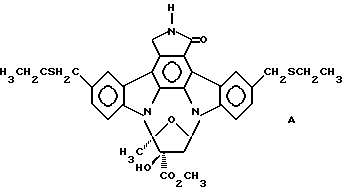
| РЕКОМБИНАТНАЯ ПЛАЗМИДНАЯ ДНК PPR - TGATG - HIL - 1 BETA - TSR, ОБЕСПЕЧИВАЮЩАЯ СИНТЕЗ РЕКОМБИНАНТНОГО ИНТЕРЛЕЙКИНА-1 И ШТАММ ESCHERICHIA COLI - ПРОДУЦЕНТ РЕКОМБИНАНТНОГО ИНТЕРЛЕЙКИНА-1 БЕТА ЧЕЛОВЕКА | 1992 |
|
RU2045575C1 |
| RU 94026088 Al, 20.05.1996 | |||
| US 4877776, 31.10.1989 | |||
| US 5461146, 24.10.1995 | |||
| WO 8807045, 08.05.1990. | |||
