Область изобретения
Настоящее изобретение относится к соединениям, композициям, способам лечения различных неврологических и психиатрических расстройств и применению соединений в комбинационной терапии. В частности, настоящее изобретение относится к таким соединениям, композициям и способам, в которых соединения представляют новые 4-фенилзамещенные тетрагидроизохинолиновые производные.
Предпосылки изобретения
Серотонин, допамин и норадреналин известны как важные химические медиаторы, участвующие в передаче нервных импульсов в мозге. Данные медиаторы высвобождаются в специфических (сайтах) участках на пресинаптических клетках и принимаются, для завершения передачи импульса, специфическими участками на постсинаптических клетках. Данный процесс далее завершается с помощью метаболизма или с помощью захвата пресинаптическими клетками. Лекарственные препараты, способные блокировать пресинаптический захват каждого из данных химических медиаторов в мозге, являются пригодными для облегчения заболеваний, связанных со сниженными уровнями данных химических медиаторов. Например, было обнаружено, что дулоксетин и флуоксетин, являющиеся известными ингибиторами обратного захвата серотонина, пригодны для лечения депрессии, ожирения и навязчиво-компульсивного состояния (Wong, et al. Патент США №5532244). Также Moldt, et al., Патент США № 5444070, описали применение ингибиторов обратного захвата допамина при лечении депрессии, болезни Паркинсона, пристрастия к наркотическим средствам и/или злоупотребления, кокаинового и/или амфетаминовой зависимости и/или злоупотребления. Freedman, et al., Патент США №6136803, также представляет ингибиторы синаптического захвата норадреналина или серотонина, подходящие для лечения депрессии у пациентов. Более того, Norden, Патент США №5789449, описывают применение ингибиторов обратного захвата серотонина при лечении психиатрических симптомов, включающих раздражительность, подавление чувствительности и недостаток психологической и физической энергии. Также Foster, et al., Патент США №4902710, представляет применение ингибиторов захвата серотонина и норадреналина для подавления тяги к курению и употреблению алкоголя человеком. Таким образом, продолжает сохраняться потребность в создании новых соединений, блокирующих обратный захват норадреналина, допамина или серотонина.
Соединения, ингибирующие обратный захват серотонина или норадреналина, также применяют в комбинированной терапии. Например, Glatt, et al., Патент США №6121261, представляют применение селективных ингибиторов обратного захвата серотонина или ингибиторов захвата норадреналина в комбинации с антагонистом рецептора нейрокинина-1 для лечения дефицита внимания у пациентов.
Также Hohenwarter, Патент США №4843071, описывает применение ингибитора обратного захвата норадреналина и предшественника норадреналина для лечения ожирения, лекарственного злоупотребления или нарколепсии. Кроме того, Wong, et al., Патент США №5532244, представляет применение ингибиторов обратного захвата серотонина в комбинации с антагонистом 1A-рецептора серотонина для увеличения количества серотонина, норадреналина и допамина в мозге.
Лечение ряда неврологических и психических расстройств характеризуется некоторым количеством побочных эффектов, предположительно являющихся следствием неспособности соединений селективно блокировать определенные нейромедиаторы, не действуя на другие. ADHD (синдром дефицита внимания/гиперактивности), например, является заболеванием, поражающим 3-6% детей школьного возраста и также зарегистрированным у некоторого процента взрослых. Помимо возникновения затруднений при нахождении в школе и на работе, ADHD является существенным фактором риска последующего развития тревожных состояний, депрессии, кондуктивного расстройства и злоупотребления наркотическими средствами. Поскольку имеющиеся в настоящий момент схемы лечения включают психостимуляторы и поскольку существенное количество пациентов (30%) невосприимчивы к стимуляторам или не толерантны к их побочным эффектам, существует необходимость в новом лекарственном препарате или классе лекарственных препаратов, пригодных для лечения ADHD и не имеющих проблем невосприимчивости или побочных эффектов. Кроме того, метилфенидат, являющийся в настоящее время препаратом выбора для лечения ADHD, обладает рядом побочных эффектов; они включают анорексию, бессонницу и ощущение тревоги, судороги, так же как и повышение кровяного давления и учащение сердечного ритма вследствие активации симпатической нервной системы. Однако метилфенидат также обладает более высокой селективностью по отношению к транспортному белку допамина, чем к транспортному белку норадреналина (соотношение Ki DAT/NET 0,1), которая может привести к склонности к зависимости и потребности в большом количестве доз в сутки для оптимальной эффективности. Таким образом, сохраняется необходимость создания новых соединений, блокирующих обратный захват норадреналина, допамина и серотонина с определенными соотношениями селективности.
Патент США №3947456 представляет тетрагидроизохинолины, которые имеют применение в качестве антидепрессантов. Патент США №3666763 представляет применение фенилтетрагидроизохинолиновых производных в качестве антидепрессантов и антигипотензивных препаратов. Заявка на выдачу патента Канады №2015114 представляет применение фенилтетрагидроизохинолиновых производных в качестве антидепрессантов; более того, описанные в этой заявке производные являются, по-видимому, неселективными по отношению к захвату норадреналина, допамина и серотонина. Заявка на выдачу патента Великобритании №2271566 представляет применение фенилтетрагидроизохинолиновых производных в качестве препаратов против ВИЧ. Международная РСТ-заявка №WO 98/40358 на выдачу патента представляет применение фенилтетрагидроизохинолиновых производных, применяемых для лечения расстройств, связанных с путями метаболизма глюкозы. WO 97/36876 представляет применение фенилтетрагидроизохинолиновых производных в качестве противоопухолевых препаратов. WO 97/23458 также описывает 4-фенилзамещенные тетрагидроизохинолины в качестве лигандов рецептора NMDA, применяемые при состояниях, связанных с разрушением нейронов. Фенилзамещенные тетрагидроизохинолины также описываются в Mondeshka et al Il Farmaco, 1994, 49, р.475-481.
Номофензин®, являющийся 4-фенилзамещенным тетрагидроизохинолиновым производным, известен как ингибитор нейронального захвата допамина и других катехоламинов и проявил клиническую эффективность в отношении ADHD. Однако длительный период приема Номофензина® приводит к летальной иммунной гемолитической анемии. Таким образом, сохраняется потребность в создании новых соединений, пригодных для лечения ADHD, но не имеющих серьезных побочных эффектов, связанных с Номофензином® или ранее описанными психостимуляторами.
Настоящее изобретение относится к новым арильным и гетероарильным замещенным производным тетрагидроизохинолина, блокирующим обратный захват норадреналина, допамина или серотонина и применяемым в качестве альтернативы метилфенидату и известным психостимуляторам для лечения ADHD и других неврологических и психических расстройств.
Авторы настоящего изобретения открыли, что представленные в формуле изобретения соединения, блокирующие обратный захват норадреналина, допамина и серотонина, обладают определенными селективными соотношениями, например обладают большей селективностью по отношению к транспортному белку норадреналина (NET), чем к транспортному белку допамина (DAT) или транспортному белку серотонина (SERT) (более низкая Ki для NET, чем для DAT и SERT). Предполагается, что соединения поэтому будут более эффективны для лечения ADHD в связи со сниженным уровнем склонности к привыканию. В частности, некоторые из соединений согласно данному изобретению являются неожиданно и особенно селективными по отношению к белку NET, чем к SERT, таким образом, также представляя соединения без побочных эффектов, известных для соединений класса селективных ингибиторов обратного захвата серотонина (SSRI).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к соединению, представленному формулой (IA-F):
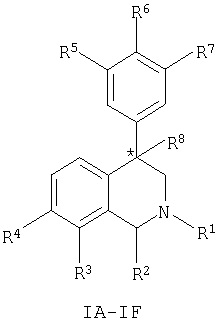
где:
атом углерода, обозначенный *, имеет R или S-конфигурацию;
R1 представляет собой C1-С6-алкил, С2-С6-алкенил, C2-C6-алкинил, С3-С6-циклоалкил или С4-С7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-С3-алкил, галоген, арил, -CN, -OR9 и -NR9R10;
R2 представляет собой Н, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил, С4-С7-циклоалкилалкил или C1-C6-галогеналкил;
R3 представляет собой Н, галоген, -OR11, -S(O)nR12, -S(O)nNR11R12, -CN, -C(O)R12, -С(О)NR11R12, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил, С4-С7-циклоалкилалкил, -О(фенил) или -О(бензил), где каждый из -О(фенил) и -О(бензил) необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, С1-С4-алкил, С1-С4-галогеналкил или С1-С4-алкокси, либо если R3 представляет собой C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или С4-С7-циклоалкилалкильную группу, указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10; при условии, что в соединениях, представленных формулой IA, R3 представляет собой C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или С4-С7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10;
при условии, что в соединениях, представленных формулой IB, R3 представляет собой -О(фенил), -О(бензил), -OC(O)R13 или -S(O)nR12, каждый из -О(фенил) и -О(бензил) необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, C1-C4-алкил, C1-C4-галогеналкил или C1-C4-алкокси;
R4 представляет собой Н, галоген, -OR11, -S(O)nR12, -S(O)NR11R12, -CN, -C(O)R12, -С(О)NR11R12, -NR11R12, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил, С4-С7-циклоалкилалкил, -О(фенил) или -О(бензил), где каждый из -О(фенил) и -О(бензил) необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, С1-С4-алкил, С1-С4-галогеналкил или С1-С4-алкокси, и когда R4 представляет собой C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или С4-С7-циклоалкилалкильную группу, то каждая указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10; при условии, что в соединениях, представленных формулой IC, R4 представляет собой C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или С4-С7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10; или R5 и R6 или R6 и R7 могут являться -O-C(R12)2-O-;
при условии, что в соединениях, представленных формулой ID, R4 представляет собой -О(фенил), -О(бензил), -OC(O)R13, -NR11R12 или -S(O)nR12, каждый из -О(фенил) и -О(бензил) необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, C1-С4-алкил, С1-С4-галогеналкил или С1-С4-алкокси;
каждый R5, R6 и R7 в соединениях, представленных каждой из формул IA, IB, IC, ID, IE и IF, независимо представляет собой Н, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -NR11R12, -C(O)NR11R12, -NR11C(O)R12, -NR11C(O)2R12, -NR11C(O)NR12R13, C1-C6-алкил, С2-С6-алкенил, С2-С6-алкинил, С3-С6-циклоалкил или С4-С7-циклоалкилалкил, и когда каждый из R5, R6 и R7 представляет собой C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или С4-С7-циклоалкилалкильную группу, указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10; или R5 и R6 или R6 и R7 могут являться -O-C(R12)2-O-;
при условии, что в соединениях, представленных формулой IE, по крайней мере, один из R5 или R7 представляет собой фтор, хлор или метил;
либо R5 и R6 каждый независимо представляют собой -O-C(R12)2-O- в соединениях, представленных формулой IE, но только в которых R7 представляет собой фтор, хлор или метил;
либо R7 и R6 также независимо могут представлять собой -O-C(R12)2-O- в соединениях, представленных формулой IE, но только в которых R5 представляет собой фтор, хлор или метил;
R8 представляет собой Н, галоген или -OR11, при условии, что в соединениях, представленных формулой IF, R8 представляет собой галоген;
R9 и R10 каждый независимо представляет собой Н, C1-C4-алкил, С1-С4-галогеналкил, С1-С4-алкоксиалкил, С3-С6-циклоалкил, С4-С7-циклоалкилалкил, -C(O)R13, фенил или бензил, при этом фенил или бензил необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, С1-С4-алкил, С1-С4-галогеналкил или С1-С4-алкокси;
либо R9 и R10 вместе с азотом, с которым они связаны, образуют пиперидин, пирролидин, пиперазин, N-метилпиперазин, морфолин или тиоморфолин;
R11 представляет собой Н, С1-С4-алкил, С1-С4-галогеналкил, С1-С4-алкоксиалкил, С3-С6-циклоалкил, С4-С7-циклоалкилалкил, -C(O)R13, фенил или бензил, и когда R11 представляет собой C1-С4-алкил, фенил или бензил, указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, С1-С4-алкил, C1-C4-галогеналкил или С1-С4-алкокси;
R12 представляет собой Н, амино, С1-С4-алкил, (C1-C4-алкил)амино, С1-С4-галогеналкил, С1-С4-алкоксиалкил, С3-С6-циклоалкил, С4-С7-циклоалкилалкил, фенил или бензил, где фенил или бензил является необязательно замещенным 1-3 заместителями, независимо выбранными из следующих: галоген, циано, С1-С4-алкил, С1-С4-галогеналкил и С1-С4-алкокси;
либо R11 и R12 вместе с азотом, с которым они связаны, образуют пиперидин, пирролидин, пиперазин, N-метилпиперазин, морфолин или тиоморфолин;
при условии, что только один из R9 и R10 или R11 и R12 вместе с азотом, с которым они связаны, образуют пиперидин, пирролидин, пиперазин, N-метилпиперазин, морфолин или тиоморфолин;
R13 представляет собой С1-С4-алкил, С1-С4-галогеналкил или фенил;
n равно 0, 1 или 2; и
арил представляет собой фенил, необязательно замещенный 1-3 заместителями из следующих: галоген, циано, С1-С4-алкил, С1-С4-галогеналкил и С1-С4-алкокси; или
его оксиду, фармацевтически приемлемой соли, сольвату или пролекарству.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Применяемые выше и в ходе всего описания изобретения следующие термины, если не указано иначе, будут иметь следующий смысл:
Термин «алкил» означает алифатическую углеводородную группу, которая может быть неразветвленной или разветвленной, содержащую от 1 до 6 атомов углерода в цепи. Разветвленная означает, что одна или несколько низших алкильных групп, таких как метил, этил или пропил, присоединены к линейной алкильной цепи. Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, н-пентил и 3-пентил.
Термин «алкенил» означает алифатическую углеводородную группу, содержащую двойную углерод-углеродную связь, и которая может быть неразветвленной или разветвленной, и содержащую от 2 до 6 атомов углерода в цепи. Предпочтительные алкенильные группы содержат от 2 до 4 атомов углерода в цепи. Разветвленная означает, что одна или несколько низших алкильных групп, таких как метил, этил или пропил, присоединены к линейной алкенильной цепи. Примеры алкенильных групп включают этенил, пропенил, н-бутенил и изобутенил.
Термин «алкинил» означает алифатическую углеводородную группу, содержащую тройную углерод-углеродную связь, и которая может быть неразветвленной или разветвленной, и содержащую от 2 до 6 атомов углерода в цепи. Предпочтительные алкинильные группы содержат от 2 до 4 атомов углерода в цепи. Разветвленная означает, что одна или несколько низших алкильных групп, таких как метил, этил или пропил, присоединены к линейной алкинильной цепи. Примеры алкинильных групп включают этинил, пропинил, н-бутинил, 2-бутинил, 3-метилбутинил и н-пентинил.
Термин «арил» означает ароматическую моноциклическую или полициклическую кольцевую систему из 6-14 атомов углерода, предпочтительно от 6 до 10 атомов углерода. Представители арильных групп включают фенил и нафтил.
Термин «гетероарил» означает ароматическую моноциклическую или полициклическую кольцевую систему из 5-14 атомов, предпочтительно от 5 до 10 атомов в кольце, в которой один или несколько атомов могут представлять собой иной элемент(ы), чем углерод, например азот, кислород или серу. Предпочтительные гетероарилы содержат от 5 до 6 атомов в кольце. Приставка аза-, окса- или тиа- перед гетероарилом означает, что, по крайней мере, атом азота, кислорода или серы соответственно входит в кольцо в качестве гетероатома. Атом азота гетероарила необязательно окислен до соответствующего N-оксида. Представители гетероарилов включают пиразинил; фуранил; тиенил; пиридил; пиримидинил; изоксазолил; изотиазолил; оксазолил; тиазолил; пиразолил; фуразанил; пирролил; пиразолил; триазолил; 1,2,4-тиадиазолил; пиразинил; пиридазинил; хиноксалинил; фталазинил; 1(2Н)-фталазинонил; имидазо[1,2-а]пиридин; имидазо[2,1-b]тиазолил; бензофуразанил; индолил; азаиндолил; бензимидазолил; бензотиенил; хинолинил; имидазолил; тиенопиридил; хиназолинил; тиенопиримидил; пирролопиридил; имидазопиридил; изохинолинил; бензоазаиндолил; азабензимидазолил; 1,2,4-триазинил; бензотиазолил и подобные.
Термин «алкокси» означает алкил-O-группу, в которой алкильная группа такая, как описано выше. Примеры алкоксигрупп включают метокси, этокси, н-пропокси, изопропокси, н-бутокси и гептоксигруппу.
Термин «соединения согласно изобретению» и эквивалентные выражения означают соединения, представленные общей формулой (IA-F), описанной здесь выше, включая пролекарства, фармацевтически приемлемые соли и сольваты, например, гидраты, где это допускается контекстом. То же самое относится и к промежуточным продуктам, представленным или не представленным в формуле изобретения, значение включает их соли и сольваты, где это допускается контекстом. С целью пояснения отдельные примеры, где это допускается контекстом, иногда приведены в тексте, но данные примеры являются исключительно иллюстративными и не означают исключение других примеров, где это допускается контекстом.
Термин «циклоалкил» означает неароматическую моно- или полициклическую кольцевую систему, состоящую из 3-7 атомов углерода, предпочтительно 5-7 атомов углерода. Примеры моноциклических циклоалкилов включают циклопентил, циклогексил, циклогептил и подобные.
Термин «циклоалкилалкил» означает циклоалкил-алкильную группу, в которой циклоалкил и алкил представляют собой описанные выше группы. Примеры циклоалкилалкильных групп включают циклопропилметил и циклопентилметил.
Термин «галоген» означает фтор, хлор, бром или иод.
Термин «галогеналкил» означает алкил как с разветвленной, так и неразветвленной цепью, замещенный одним или несколькими галогенами, при этом алкильная группа представляет собой такую, как описано выше.
Термин «галогеналкокси» означает C1-4-алкоксигруппу, замещенную, по крайней мере, одним атомом галогена, при этом алкоксигруппа представляет собой такую, как описано выше.
Термин «замещенный» атом или «замещение» атома означает, что один или несколько атомов водорода у обозначенного атома замещены группой, выбранной из указанных, учитывая нормальную валентность обозначенного атома. «Незамещенные» атомы несут все атомы водорода, предполагаемые согласно их валентности. В случае, когда заместитель представляет собой кето (т.е. =O), у атома замещаются 2 водорода. Комбинации заместителей и/или переменных являются допустимыми, только если подобные комбинации приводят к получению стабильных соединений; «стабильное соединение» или «стабильная структура» означают соединение, достаточно устойчивое к проведению стадий выделения из реакционной смеси до приемлемой степени очистки и преобразуемое в эффективное терапевтическое средство.
Термин «фармацевтические приемлемые соли» означает относительно нетоксичные соли, полученные с помощью неорганических и органических кислот, соли, полученные с помощью оснований, соединений согласно настоящему изобретению. Данные соли могут быть получены in situ в ходе окончательного выделения и очистки соединений. В частности, соли кислот могут быть получены с помощью отдельного взаимодействия очищенного соединения в форме свободного основания с подходящей органической или неорганической кислотой и выделения таким образом полученной соли. Примеры солей, полученных с помощью кислот, включают гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, оксалат, валерат, олеат, пальмитат, стеарат, лаурат, борат, бензоат, лактат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактиобионат, сульфаматы, малонаты, салицилаты, пропионаты, метилен-бис-b-гидроксинафтоаты, гентизаты, изотионаты, ди-пара-толуоилтартраты, метансульфонаты, этансульфонаты, бензолсульфонаты, пара-толуолсульфонаты, циклогексилсульфаматы и хинатлаурилсульфонатные соли и подобные (См., например, S.M.Berge, et al., «Pharmaceutical Salts,» J. Pharm. Sci., 66: с.1-19 (1977) и Remington's Pharmaceutical Sciences, 7th ed., Mack Publishing Company, Easton, PA, 1985, с.1418, которые включены здесь в ссылку). Соли основания также могут быть получены с помощью отдельного взаимодействия очищенного соединения в кислотной форме с подходящим органическим или неорганическим основанием и выделения таким образом полученной соли. Соли, полученные с помощью оснований, включают фармацевтически приемлемые соли металлов и аминов. Подходящие соли металлов включают соли натрия, калия, кальция, бария, цинка, магния и алюминия. Предпочтительными являются соли натрия и калия. Подходящие соли неорганических оснований получают из оснований металлов, включающих гидрид натрия, гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид алюминия, гидроксид лития, гидроксид магния, гидроксид цинка. Подходящие соли аминов получают из аминов, обладающих достаточной основностью для образования стабильной соли, и предпочтительно включают такие амины, которые часто применяются в медицинской химии вследствие их низкой токсичности и приемлемы для медицинских целей: аммоний, этилендиамин, N-метилглюкамин, лизин, аргинин, орнитин, холин, N,N'-дибензилэтилендиамин, хлоропрокаин, диэтаноламин, прокаин, N-бензилфенетиламин, диэтиламин, пиперазин, трис(гидроксиметил)аминометан, гидроксид тетраметиламмония, триэтиламин, дибензиламин, эфенамин, дегидроабиетиламин, N-этилпиперидин, бензиламин, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, этиламин, основные аминокислоты, например лизин и аргинин, и дициклогексиламин и подобные.
Применяемый здесь термин «фармацевтически приемлемые пролекарства» означает пролекарства соединений, применяемых соответственно настоящему изобретению, которые согласно медицинским правилам пригодны для использования в контакте с тканями человека и низших животных без неприемлемой токсичности, раздражимости, аллергических реакций и подобных эффектов, соответствуют приемлемому соотношению между выгодой и риском, и эффективны при их использовании по назначению, так же как и возможные цвиттерионные формы соединений согласно настоящему изобретению. Термин «пролекарство» означает соединения, быстро трансформирующиеся in vivo с получением исходного соединения, представленного вышеуказанной формулой, например, с помощью гидролиза в крови. Функциональные группы, которые могут быть легко преобразованы с помощью метаболического расщепления in vivo, образуют класс групп, вступающих в реакцию с карбоксильной группой соединений согласно настоящему изобретению. Они включают, но не ограничиваются такими группами, как алканоил (такие как ацетил, пропионил, бутирил и подобные), незамещенный или замещенный ароил (такой как бензоил и замещенный бензоил), алкоксикарбонил (такой как этоксикарбонил), триалкилсилил (такой как триметил- и триэтилсилил), сложные моноэфиры, образованные дикарбоновыми кислотами (такой как сукцинил) и подобные. Вследствие легкости, с которой метаболически расщепляемые группы соединений, применяемых согласно настоящему изобретению, расщепляются in vivo, соединения, несущие подобные группы, действуют как пролекарства. Соединения, несущие метаболически расщепляемые группы, имеют преимущество, выражающееся в том, что они могут проявлять улучшенную биодоступность, являющуюся результатом повышения растворимости и/или скорости абсорбции, присущих исходному соединению, благодаря присутствию метаболически расщепляемых групп. Исчерпывающее обсуждение пролекарств представлено в следующих источниках: Design of Prodrugs, H.Bundgaard, ed., Elsevier, 1985; Methods в Enzymology, К.Widder et al, Ed., Academic Press, 42, p.309-396, 1985; A Textbook of Drug Design и Development, Krogsgaard-Larsen и H.Bundgaard, ed., Chapter 5; «Design и Applications of Prodrugs» p.113-191, 1991; Advanced Drug Delivery Reviews, H.Bundgard, 8, p.1-38, 1992; Journal of Pharmaceutical Sciences, 77, p.285, 1988; Chem. Pharm. Bull., N.Nakeya et al, 32, p.692, 1984; Prodrugs as Novel Delivery Systems, T.Higuchi и V.Stella, Vol.14 of the A.C.S. Symposium Series, и Bioreversible Carriers в Drug Design, Edward B.Roche, ed., American Pharmaceutical Association и Pergamon Press, 1987, которые включены здесь в ссылки. Примеры пролекарств включают, но не ограничиваются ими, ацетатные, формиатные и бензоатные производные функциональной спиртовой группы и аминогруппы соединений согласно изобретению.
Термин «терапевтически эффективное количество» означает количество соединения согласно настоящему изобретению, эффективное для увеличения уровней серотонина, норадреналина или допамина в синапсе, и таким образом для достижения желаемого терапевтического эффекта. Подобные количества обычно изменяются в зависимости от ряда факторов, хорошо известных специалистам в данной области, которые представлены в описании для определения и принятия во внимание. Они включают, не ограничиваясь, конкретный объект и также его возраст, массу, рост, общее физическое состояние и медицинский анамнез; конкретное применяемое соединение, так же как и входящий с ним в состав носитель, и выбранный для него путь введения; и характер и тяжесть состояния, подвергаемого лечению.
Термин «фармацевтическая композиция» означает композицию, содержащую соединение, представленное формулой (IA-F), и, по крайней мере, один компонент, выбранный из группы, включающей фармацевтически приемлемые носители (carriers), разбавители, адъюванты, наполнители или носители (vehicles), такие как консерванты, наполнители, дезинтегрирующие вещества, смачивающие вещества, эмульгирующие вещества, суспендирующие вещества, подсластители, корригенты, отдушки, антибактериальные вещества, противогрибковые вещества, смазывающие вещества и диспергирующие вещества, в зависимости от пути введения и характера дозированных форм. Примеры суспендирующих веществ включают этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбита, микрокристаллическую целюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант или смеси данных веществ. Предупреждение действия микроорганизмов может быть обеспечено с помощью различных антибактериальных и противомикробных веществ, например парабены, хлорбутанол, фенол, сорбиновая кислота и подобные. Также может быть желательным включение изотонических веществ, например сахаров, хлорида натрия и подобных. Пролонгированная абсорбция инъекционной фармацевтической формы может обеспечиваться с помощью применения соединений, замедляющих абсорбцию, например, моностеарата алюминия и желатина. Примеры подходящих носителей (carriers), разбавителей, растворителей или носителей (vehicles) включают воду, этанол, полиолы, подходящие их смеси, растительные масла (такие как оливковое масло) и органические сложные эфиры, пригодные для инъекционного введения, такие как этилолеат. Примеры наполнителей включают лактозу, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция (dicalcium phosphate phosphate). Примеры дезинтегрирующих веществ включают крахмал, альгиновые кислоты и основной комплекс силикатов. Примеры смазывающих веществ включают стеарат магния, лаурилсульфат натрия, тальк, так же как и высокомолекулярные полиэтиленгликоли.
Термин «фармацевтически приемлемый» означает согласно медицинским правилам пригодность для использования в контакте с клетками человека и низших животных без неприемлемой токсичности, раздражимости, аллергических реакций и подобных эффектов и соответствие приемлемому соотношению между выгодой и риском.
Термин «фармацевтически приемлемые дозированные формы» означает дозированные формы соединений согласно настоящему изобретению и включает, например, таблетки, драже, порошки, эликсиры, сиропы, жидкие препараты, включающие суспензии, спреи, таблетки для ингаляций, лепешки, эмульсии, растворы, гранулы, капсулы и суппозитории, так же как и жидкие препараты для инъекций, включающие липосомные препараты. Способы и формы, в общем, могут быть найдены в Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA, latest edition.
ПРЕДПОЧТИТЕЛЬНЫЕ ВОПЛОЩЕНИЯ
Другим воплощением изобретения является соединение, представленное формулами IA-IF,
где:
атом углерода, обозначенный *, имеет R или S-конфигурацию.
Другим воплощением изобретения является соединение, представленное формулами IA, IB, IC, ID, IE и IF,
где:
R1 представляет собой C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или С4-С7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-С3-алкил, галоген, арил, -CN, -OR9 и -NR9R10.
Другим воплощением изобретения является соединение, представленное формулами IA, IB, IC, ID, IE и IF,
где:
R2 представляет собой Н, С1-С6-алкил, С2-С6-алкенил, С2-С6-алкинил, С3-С6-циклоалкил, С4-С7-циклоалкилалкил или C1-C6-галогеналкил.
Другим воплощением изобретения является соединение, представленное формулой IA,
где:
R3 представляет собой C1-C6-алкил, С2-С6-алкенил, С2-С6-алкинил, С3-С6-циклоалкил или С4-С7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-С3-алкил, галоген, арил, -CN, -OR9 и -NR9R10.
Другим воплощением изобретения является соединение, представленное формулой IB,
где:
R3 представляет собой -О(фенил), -О(бензил), -OC(O)R13 или -S(O)nR12, каждый из -О(фенил) и -О(бензил) необязательно замещены 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, С1-С4-алкил, C1-C4-галогеналкил или С1-С4-алкокси.
Другим воплощением изобретения является соединение, представленное формулами IC, ID, IE и IF,
где:
R3 представляет собой Н, галоген, -OR11, -S(O)nR12, -S(O)NR11R12, -CN, -C(O)R12, -С(O)NR11R12, -О(фенил), -О(бензил), -OC(O)R13 или -S(O)nR12, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил и С4-С7-циклоалкилалкил, где каждый из C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил и С4-С7-циклоалкилалкил необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10, и, когда R3 представляет собой -О(фенил) или -О(бензил)группу, указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, C1-С4-алкил, С1-С4-галогеналкил или С1-С4-алкокси.
Другим воплощением изобретения является соединение, представленное формулой IC, где:
R4 представляет собой C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или С4-С7-циклоалкилалкил, каждый из которых необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10.
Другим воплощением изобретения является соединение, представленное формулой ID, где:
R4 представляет собой -О(фенил), -О(бензил), -OC(O)R13, -NR11R12 или -S(O)nR12, и указанный -О(фенил) или -О(бензил) необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, C1-С4-алкил, С1-С4-галогеналкил или С1-С4-алкокси.
Другим воплощением изобретения является соединение, представленное формулами IA, IB, IE и IF, где:
R4 представляет собой Н, галоген, -OR11, -S(O)nR12, -S(O)NR11R12, -CN, -О(фенил), -О(бензил), -OC(O)R13, -C(O)R12, -C(O)NR11R12, -NR11R12, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил и С4-С7-циклоалкилалкил, и, когда R4 представляет собой C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или С4-С7-циклоалкилалкильную группу, указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10, и, когда R4 представляет собой -О(фенил) или -О(бензил)группу, указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, C1-С4-алкил, С1-С4-галогеналкил и С1-С4-алкокси.
Другим воплощением изобретения является соединение, представленное формулами IA, IB, IC, ID и IF, где:
R5, R6 и R7 каждый независимо представляет собой Н, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -NR11R12, -C(O)NR11R12, -NR11C(O)R12, -NR11C(O)2R12, -NR11C(O)NR12R13, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или С4-С7-циклоалкилалкил, и, когда каждый R5, R6 и R7 независимо представляет собой C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, С3-С6-циклоалкил или С4-С7-циклоалкилалкильную группу, указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-С3-алкил, галоген, арил, -CN, -OR9 и -NR9R10, или R5 и R6 или R6 и R7 могут являться -O-C(R12)2-O-.
Другим воплощением изобретения является соединение, представленное формулой IE, где:
R5 представляет собой фтор, хлор или метил; при этом R7 и R6 каждый независимо представляет собой Н, галоген, -OR11, -S(O)nR12, -CN, -C(O)R12, -NR11R12, -С(О)NR11R12, -NR11C(О)R12, -NR11C(O)2R12, -NR11C(O)NR12R13, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или С4-С7-циклоалкилалкил, где каждый из R7 и R6 представляет собой C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или С4-С7-циклоалкилалкильную группу, причем указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10, при условии, что R7 не представляет собой фтор, хлор или метил.
Другим воплощением изобретения является соединение, представленное формулой IE, где:
R7 представляет собой фтор, хлор или метил, при этом R5 и R6 вместе также могут являться -O-C(R12)2-O-.
Другим воплощением изобретения является соединение, представленное формулой IE, где:
R5 представляет собой фтор, хлор или метил, при этом R7 и R6 вместе также могут являться -O-C(R12)2-O-.
Другим воплощением изобретения является соединение, представленное формулами IA-IE, где:
R8 представляет собой Н, галоген или OR11.
Другим воплощением изобретения является соединение, представленное формулой IF, в которой R8 представляет собой галоген.
Другим воплощением изобретения является соединение, представленное формулами IA-F, где:
R9 и R10 каждый независимо представляет собой Н, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, С4-С7-циклоалкилалкил, -C(O)R13, фенил или бензил, при этом указанный фенил или бензил необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, С1-С4-алкил, С1-С4-галогеналкил или С1-С4-алкокси; или
R9 и R10 вместе с азотом, с которым они связаны, образуют кольца пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина.
Другим воплощением изобретения является соединение, представленное формулами IA-F, где
R11 представляет собой Н, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, С4-С7-циклоалкилалкил, -C(O)R13, фенил или бензил, при этом указанный фенил или бензил необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, C1-С4-алкил, С1-С4-галогеналкил или С1-С4-алкокси.
Другим воплощением изобретения является соединение, представленное формулами IA-F, где:
R12 представляет собой Н, C1-C4-алкил, C1-C4-галогеналкил, C1-C4-алкоксиалкил, C3-C6-циклоалкил, С4-С7-циклоалкилалкил, фенил или бензил, при этом указанный фенил или бензил необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, C1-C4-алкил, C1-C4-галогеналкил и C1-C4-алкокси; или
R11 и R12 вместе с азотом, с которым они связаны, образуют кольца пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина.
Другим воплощением изобретения является соединение, представленное формулами IA-F, где:
R13 представляет собой C1-C4-алкил, C1-C4-галогеналкил или фенил; и n равно 0, 1, или 2.
Другим воплощением изобретения является соединение, представленное формулами IA-F, где:
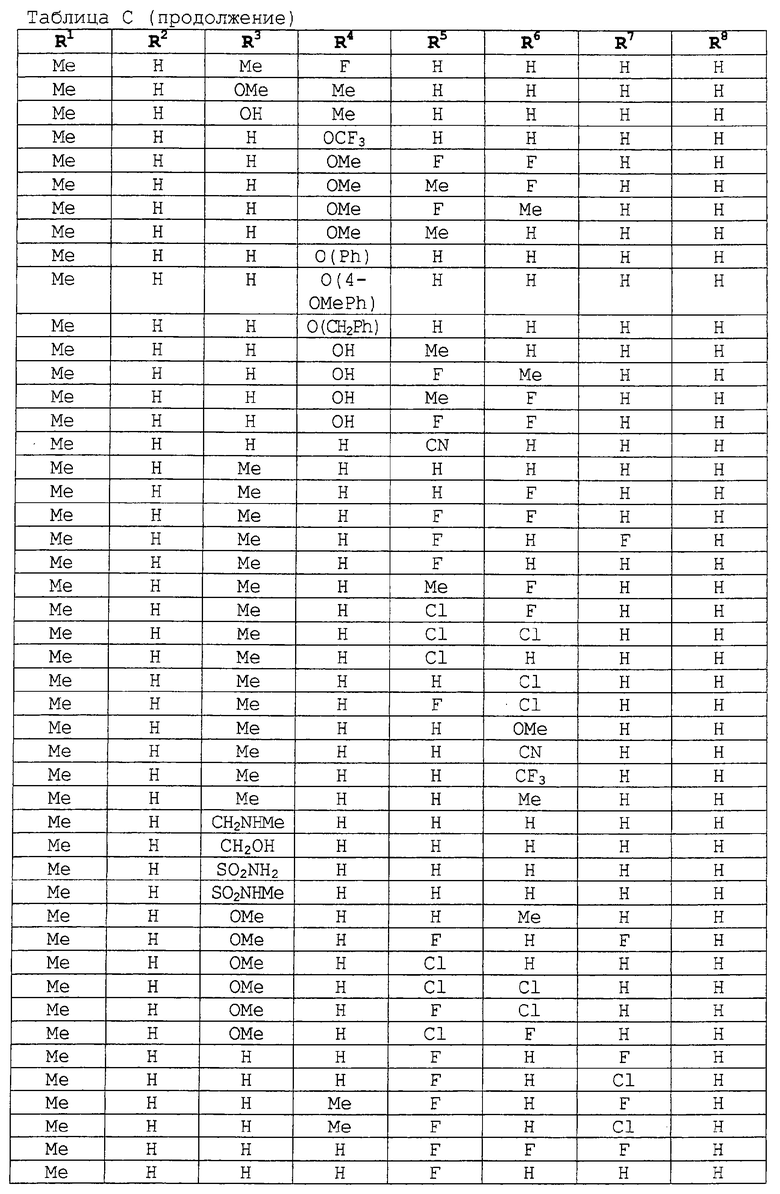
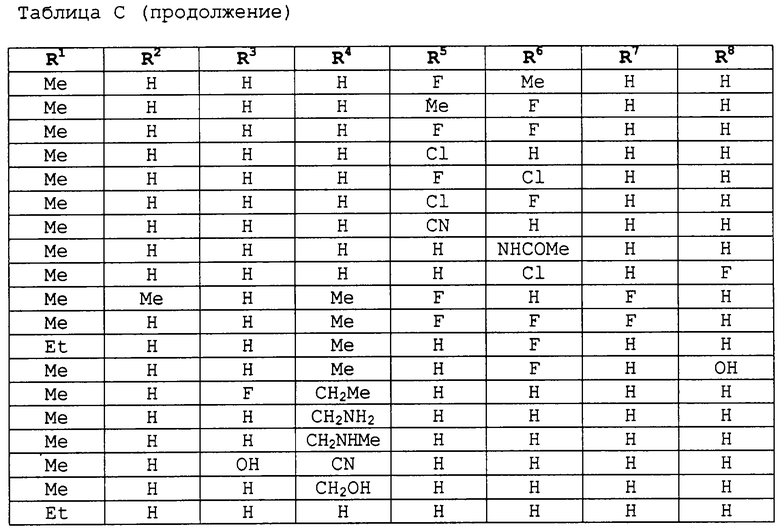
заместители R1-R8 представлены в следующей таблице:
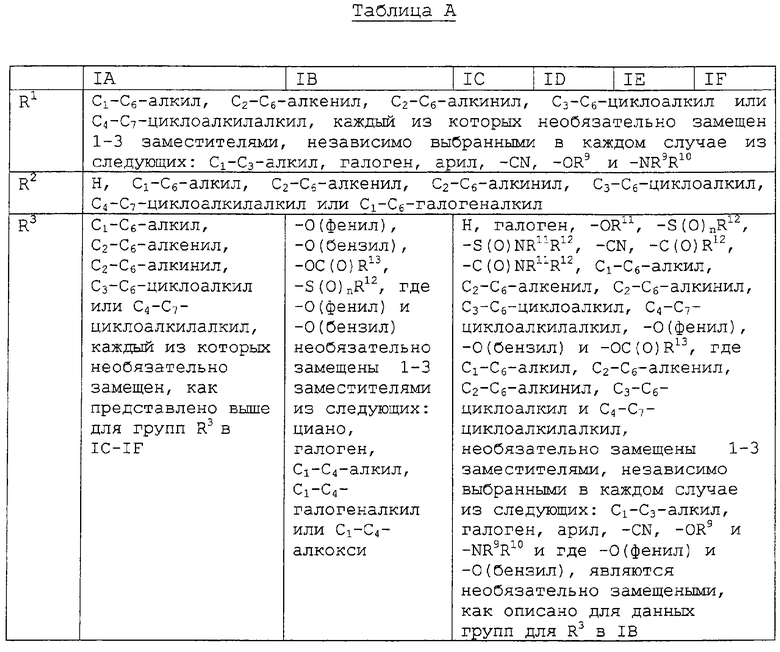
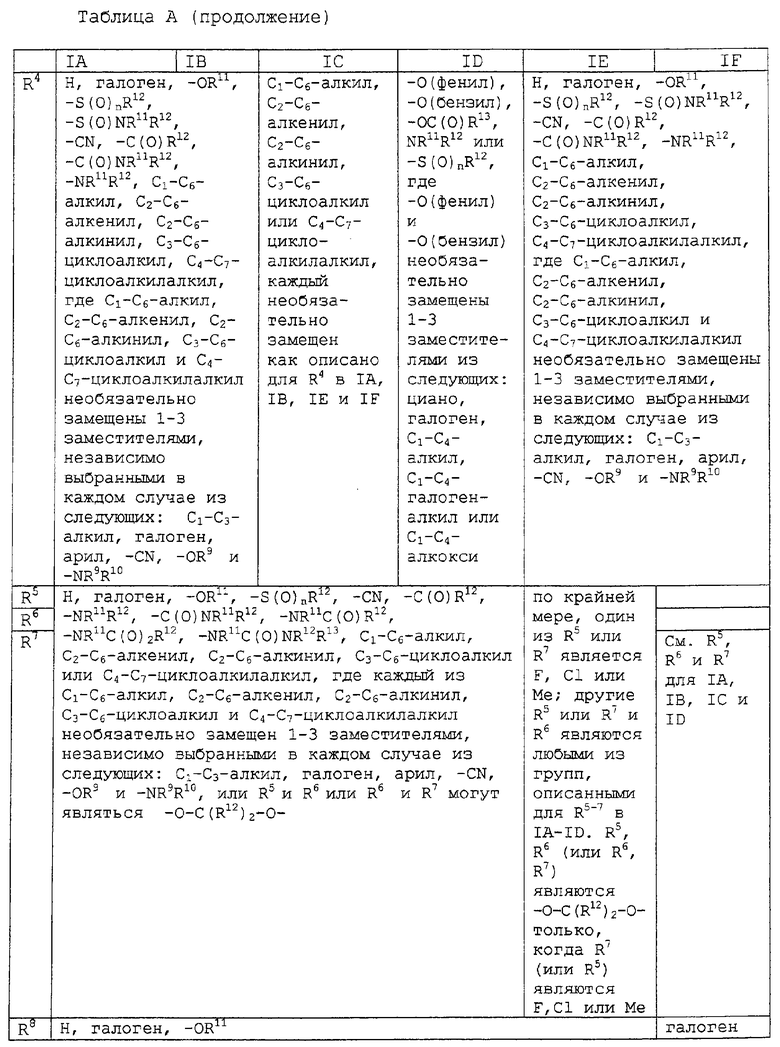
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IA-IF, где:
R1 представляет собой C1-C3-алкил;
R2 представляет собой Н, C1-C4-алкил или C1-C6-галогеналкил.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IA, IC, ID, IE и IF, где:
R3 представляет собой C1-C4-алкил, C3-C6-циклоалкил или С4-С7-циклоалкилалкил, каждая из данных групп необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулой IB, где:
R3 представляет собой -О(фенил) или -О(бензил), необязательно замещенный 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, C1-С4-алкил, С1-С4-галогеналкил или С1-С4-алкокси.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IC, ID, IE и IF, где:
R3 представляет собой -О(фенил) или -О(бензил), необязательно замещенный 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, C1-С4-алкил, С1-С4-галогеналкил или C1-C4-алкокси.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IC-IF, где:
R3 представляет собой Н.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IA, IB, IC, IE и IF, где:
R4 представляет собой C1-C4-алкил, C3-C6-циклоалкил или С4-С7-циклоалкилалкил, каждая из данных групп необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IA, IB, IE и IF, где:
R4 представляет собой Н.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IA, IB, IE и IF, где:
R4 представляет собой -NR11R12-, -О(фенил) или -О(бензил), каждая из данных арильных групп необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, C1-C4-алкил, C1-C4-галогеналкил или C1-C4-алкокси.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IE и IF, где:
R3 и R4 оба представляют собой галоген.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IA, IB, IC, ID и IF, где:
R5, R6 и R7 каждый представляют собой Н, галоген, -OR11, -NR11R12, C1-C6-алкил или C1-C6-алкил, необязательно замещенный 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IA, IB, IC, ID, IE и IF, где:
R5 представляет собой фтор, хлор или метил;
один из R6 или R7 представляет собой Н; и другой из R6 или R7, не являющийся Н, представляет собой галоген, -OR11, -NR11R12, C1-C6-алкил или C1-C6-алкил, каждый из которых необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-C3-алкил, галоген, арил, -CN, -OR9 и -NR9R10.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IA, IB, IC, ID и IE, где:
R8 представляет собой Н или галоген.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулой IF, где:
R8 представляет собой галоген.
Предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IA, IB, IC, ID, IE и IF, где:
заместители R1-R8 принимают значения, представленные в следующей таблице В:
алкил,
С3-С6-
цикло-
алкил или
С4-С7-
циклоалкилалкил, каждый
необязательно
замещенный
или
-О(бензил),
каждый
необязательно замещенный
циклоалкил или С4-С7-циклоалкилалкил,
каждый необязательно замещенный, или
-O(фенил), или
-О(бензил), каждый необязательно
замещенный
альтернативно
С1-С4-алкил,
С3-С6-циклоалкил или
С4-С7-
циклоалкилалкил,
каждый необязательно
замещенный, -NR11R12;
или -O(фенил), или
-О(бензил), каждый
необязательно
замещенный
алкил,
С3-С6-
цикло-
алкил или
С4-С7-
циклоал-
килалкил,
каждый
необязательно
замещенный
или
-О(бензил),
каждый
необязательно
замещенный
альтернативно
C1-С6-алкил,
С3-С6-циклоалкил или
С4-С7-
циклоалкилалкил,
каждый необязательно
замещенный, -NR11R12;
или -O(фенил), или
-О(бензил), каждый
необязательно
замещенный
C1-С6-алкил, необязательно замещенный
для
IA-ID
R7
собой Н и
другой
представляет
собой
галоген,
-OR11,
-NR11R12,
C1-С6-алкил
или C1-С6-
алкил,
необязательно
замещенный
для
IA-ID
Более предпочтительными воплощениями данного изобретения являются соединения, в которых:
R1 представляет собой C1-C3-алкил;
R2 представляет собой Н или C1-C3-алкил;
R3 представляет собой Н, C1-C4-алкил, -О(фенил) или необязательно замещенный -О(фенил), более предпочтительно галоген;
R4 представляет собой Н, C1-C4-алкил, -О(фенил) или необязательно замещенный -О(фенил), более предпочтительно галоген;
R5 представляет собой F, Cl или Me, более предпочтительно -OR11, где R11 представляет собой C1-C3-алкил;
R6 представляет собой Н или более предпочтительно Cl, F, C1-C3-алкил, галоген-замещенный C1-C3-алкил или -OR11, R11 представляет собой C1-C3-алкил или -NR11R12;
R7 представляет собой Н или более предпочтительно Cl, F, C1-C3-алкил или -OR11, где R11 представляет собой C1-C3-алкил.
Еще более предпочтительными воплощениями данного изобретения являются соединения, в которых:
R1 представляет собой СН3;
R2 представляет собой Н или СН3;
R3 представляет собой Н, СН3, или -О(фенил), или -О-СН2-(фенил), каждый из указанных -О(фенил) или -O-СН2-( фенил) необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, C1-С4-алкил, C1-C4-галогеналкил или C1-C4-алкокси;
R4 представляет собой Н, F, СН3, СН2СН3, СН2СН2СН3, СН2СН(СН3)СН3, -О (фенил) или -O-CH2- (фенил), где каждый из указанных -О(фенил) или -О-СН2-(фенил) необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, циано, C1-C4-алкил, C1-C4-галогеналкил или C1-C4-алкокси;
R5 представляет собой H, СН3, ОСН3, F или Cl;
R6 представляет собой H, СН3, -ОСН3, F, Cl или CF3;
R7 представляет собой H, F, Cl, СН3 или ОСН3; и
R8 представляет собой галоген.
Еще более предпочтительными воплощениями данного изобретения являются соединения, представленные формулами IA-IF, где:
R1-R8 являются следующими:
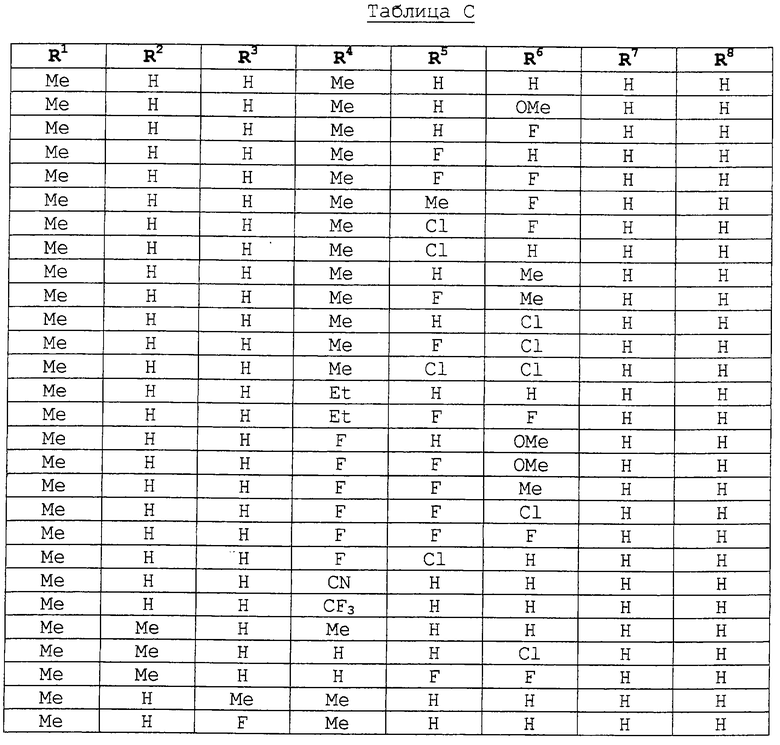
To есть, особенно предпочтительными соединениями являются:
2,7-диметил-4-фенил-1,2,3,4-тетрагидроизохинолин;
4-(4-метокси)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(4-фтор)фенил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(3-фтор)фенил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дифтор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(4-фтор-3-метил)фенил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор-4-фтор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(4-метил)фенил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(3-фтор-4-метил)фенил-1,2,3,4-тетрагидроизохинолин;
4-(4-хлор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(4-хлор-3-фтор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дихлор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
7-этил-2-метил-4-фенил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дифтор)фенил-7-этил-2-метил-1,2,3,4-тетрагидроизохинолин;
7-фтор-4-(4-метокси)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
7-фтор-4-(3-фтор-4-метокси)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
7-фтор-4-(3-фтор-4-метил)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
7-фтор-4-(4-хлор-3-фтор)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дифтор)фенил-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор)фенил-7-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
7-циано-2-метил-4-фенил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-фенил-7-трифторметил-1,2,3,4-тетрагидроизохинолин;
4-фенил-1,2,7-триметил-1,2,3,4-тетрагидроизохинолин;
4-(4-хлор)фенил-1,2-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дифтор)фенил-1,2-диметил-1,2,3,4-тетрагидроизохинолин;
4-фенил-2,1,8-трифторметил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-8-фтор-4-фенил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-7-фтор-4-фенил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-8-метокси-4-фенил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-8-гидрокси-4-фенил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-фенил-7-трифторметокси-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дифтор)фенил-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(4-фтор-3-метил)фенил-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3-фтор-4-метил)фенил-7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
7-метокси-4-(3-метил)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
2-метил-7-фенокси-4-фенил-1,2,3,4-тетрагидроизохинолин;
7-(4-метокси)фенокси-2-метил-4-фенил-1,2,3,4-тетрагидроизохинолин;
7-бензилокси-2-метил-4-фенил-1,2,3,4-тетрагидроизохинолин;
7-гидрокси-2-метил-4-(3-метил)фенил-1,2,3,4-тетрагидроизохинолин;
4-(3-фтор-4-метил)фенил-7-гидрокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(4-фтор-3-метил)фенил-7-гидрокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дифтор)фенил-7-гидрокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3-циано)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-4-фенил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-4-(4-фтор)фенил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дифтор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3,5-дифтор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-4-(3-фтор)фенил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-4-(4-фтор-3-метил)фенил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор-4-фтор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дихлор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(4-хлор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(4-хлор-3-фтор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-4-(4-метокси)фенил-1,2,3,4-тетрагидроизохинолин;
4-(4-циано)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-4-(4-трифторметил)фенил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-4-(4-метил)фенил-1,2,3,4-тетрагидроизохинолин;
2-метил-8-(N-метиламино)метил-4-фенил-1,2,3,4-тетрагидроизохинолин;
8-(гидрокси)метил-2-метил-4-фенил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-фенил-8-сульфонамид-1,2,3,4-тетрагидроизохинолин;
2-метил-8-(N-метил)сульфонамид-4-фенил-1,2,3,4-тетрагидроизохинолин;
8-метокси-2-метил-4-(4-метил)фенил-1,2,3,4-тетрагидроизохинолин;
4-(3,5-дифтор)фенил-8-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор)фенил-8-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дихлор)фенил-8-метокси-2-метил)-1,2,3,4-тетрагидроизохинолин;
4-(4-хлор-3-фтор)фенил-8-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор-4-фтор)фенил-8-метокси-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3,5-дифтор)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор-5-фтор)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3,5-дифтор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор-5-фтор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-(3,4,5-трифтор)фенил-1,2,3,4-тетрагидроизохинолин;
4-(3-фтор)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3-фтор-4-метил)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(4-фтор-3-метил)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дифтор)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(4-хлор-3-фтор)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор-4-фтор)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(3-циано)фенил-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(4-ацетанилид)-2-метил-1,2,3,4-тетрагидроизохинолин;
4-(4-хлор)фенил-4-фтор-2-метил-1,2,3,4-тетрагидроизохинолин;
(3,5-дифтор)-4-фенил-1,2,7-триметил-1,2,3,4-тетрагидроизохинолин;
(8-фтор-2-метил-4-фенил-1,2,3,4-тетрагидро-7-изохинолин)-N-метилметанамин;
(2-метил-4-фенил-7-изохинолинил)-N-метилметанамин;
N-метил(2-метил-4-фенил-7-изохинолинил)-N-метилметанамин;
8-гидрокси-2-метил-4-фенил-1,2,3,4-тетрагидро-7-изохинолинкарбонитрил;
(2-метил-4-фенил-1,2,3,4-тетрагидро-7-изохинолинил)-метанол; и
2-этил-4-фенил-1,2,3,4-тетрагидроизохинолин; или
его оксид, фармацевтически приемлемая соль, сольват или пролекарство.
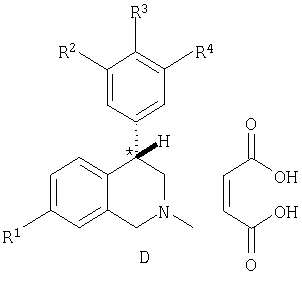
Еще более предпочтительные соединения согласно настоящему изобретению включают (+)-энантиомеры соединений, представленных формулами IA-IF, выбранные из таблицы D:
Другим предпочтительным аспектом изобретения является смесь соединений, представленных формулами (IA-F), в которой соединения, представленные формулами (IA-F), мечены радиоактивной меткой, т.е. в которых один или несколько описанных атомов замещены на радиоактивный изотоп данного атома (например, С замещен на 14С и Н замещен на 3H). Подобные соединения имеют ряд возможных применений, например в качестве стандартов или реагентов для определения способности потенциального лекарственного препарата связывать нейромедиаторные белки.
Другим аспектом изобретения является терапевтически эффективное количество соединения, представленного формулами (IA-F), и фармацевтически приемлемого носителя.
Другим аспектом изобретения является способ лечения расстройства, вызванного или зависящего от снижения уровня серотонина, норадреналина или допамина, который включает введение пациенту, нуждающемуся в подобном лечении, терапевтически эффективного количества соединения, представленного формулами (IA-F), или его фармацевтически приемлемой соли.
Другим аспектом изобретения является способ лечения расстройства, вызванного или зависящего от снижения уровня серотонина, норадреналина или допамина, который включает введение пациенту, нуждающемуся в подобном лечении, терапевтически эффективного количества соединения, представленного формулами (IA-F), или его фармацевтически приемлемой соли и терапевтически эффективного количества антагониста рецептора 1А серотонина или его фармацевтически приемлемой соли.
Другим аспектом изобретения является способ лечения расстройства, вызванного или зависящего от снижения уровня серотонина, норадреналина или допамина, который включает введение пациенту, нуждающемуся в подобном лечении, терапевтически эффективного количества соединения, представленного формулами (IA-F), или его фармацевтически приемлемой соли и фармацевтически эффективного количества соединения, выбранного из группы, включающей WAY 100135 и спиперон, или его фармацевтически приемлемой соли.
WAY 100135 (N-(трет-бутил)-3-[а-(2-метоксифенил)-пиперазин-1-ил]-2-фенилпропанамид) представлен в Abou-Gharbia et al., патент США №4988814, как обладающий аффинностью к рецептору 5-HT1A. Также Cliffe et al., J. Med. Chem. 36, 1509-10 (1993) показали, что соединение представляет собой антагонист 5-HT1A. Спиперон (8-[4-(4-фторфенил)-4-оксобутил]-1-фенил-1,3,8-триазоспиро[4,5]декан-4-он) представляет собой хорошо известное соединение и описан в патенте США №3155669 и 3155670. Активность спиперона как антагониста 5-HT1A показана в Middlemiss et al., Neurosci. и Biobehav. Rev.16, 75-82 (1992).
Другим аспектом изобретения является способ лечения расстройства, вызванного или зависящего от снижения уровня серотонина, норадреналина или допамина, который включает введение пациенту, нуждающемуся в подобном лечении, терапевтически эффективного количества соединения, представленного формулами (IA-F), или его фармацевтически приемлемой соли и терапевтически эффективного количества селективного антагониста рецептора нейрокинина-1 или его фармацевтически приемлемой соли.
Антагонисты рецептора нейрокинина-1, применяющиеся в сочетании с соединением, представленным формулами (IA-F), в настоящем изобретении, полностью описаны, например, в патенте США №5373003, 5387595, 5459270, 5494926, 5162339, 5232929, 5242930, 5496833, 5637699; международной публикации РСТ-патента №WO 90/05525, 90/05729, 94/02461, 94/02595, 94/03429, 94/03445, 94/04494, 94/04496, 94/05625, 94/07843, 94/08997, 94/10165, 94/10167, 94/10168, 94/10170, 94/11368, 94/13639, 94/13663, 94/14767, 94/15903, 94/19320, 94/19323, 94/20500, 91/09844, 91/18899, 92/01688, 92/06079, 92/12151, 92/15585, 92/17449, 92/20661, 92/20676, 92/21677, 92/22569, 93/00330, 93/00331, 93/01159, 93/01165, 93/01169, 93/01170, 93/06099, 93/09116, 93/10073, 93/14084, 93/14113, 93/18023, 93/19064, 93/21155, 93/21181, 93/23380, 93/24465, 94/00440, 94/01402, 94/26735, 94/26740, 94/29309, 95/02595, 95/04040, 95/04042, 95/06645, 95/07886, 95/07908, 95/08549, 95/11880, 95/14017, 95/15311, 95/16679, 95/17382, 95/18124, 95/18129, 95/19344, 95/20575, 95/21819, 95/22525, 95/23798, 95/26338, 95/28418, 95/30674, 95/30687, 95/33744, 96/05181, 96/05193, 96/05203, 96/06094, 96/07649, 96/10562, 96/16939, 96/18643, 96/20197, 96/21661, 96/29304, 96/29317, 96/29326, 96/29328, 96/31214, 96/32385, 96/37489, 97/01553, 97/01554, 97/03066, 97/08144, 97/14671, 97/17362, 97/18206, 97/19084, 97/19942, 97/21702, и 97/49710; и в заявках на выдачу патента Великобритании №2266529, 2268931, 2269170, 2269590, 2271774, 2292144, 2293168, 2293169, и 2302689; Европейская публикация патента №ЕР 0360390, 0517589, 0520555, 0522808, 0528495, 0532456, 0533280, 0536817, 0545478, 0558156, 0577394, 0585913, 0590152, 0599538, 0610793, 0634402, 0686629, 0693489, 0694535,0699655, 0394989, 0428434, 0429366, 0430771, 0436334, 0443132, 0482539, 0498069, 0499313, 0512901, 0512902, 0514273, 0514274, 0514275, 0514276, 0515681, 0699674, 0707006, 0708101, 0709375, 0709376, 0714891, 0723959, 0733632 и 0776893. Получение подобных соединений полностью описано в вышеуказанных патентах и публикациях.
Другим аспектом изобретения является способ лечения расстройства, вызванного или связанного со снижением уровней серотонина, норадреналина или допамина, который включает введение пациенту, нуждающемуся в подобном лечении, терапевтически эффективного количества соединения, представленного формулами (IA-F), или его фармацевтически приемлемой соли и терапевтически эффективного количества предшественника норадреналина или его фармацевтически приемлемой соли.
Другим аспектом изобретения является способ лечения расстройства, вызванного или зависящего от снижения уровня серотонина, норадреналина или допамина, который включает введение пациенту, нуждающемуся в подобном лечении, терапевтически эффективного количества соединения, представленного формулами (IA-F), или его фармацевтически приемлемой соли и терапевтически эффективного количества соединения, выбранного из L-тирозина и L-фенилаланина или его фармацевтически приемлемой соли.
Другим аспектом изобретения является способ лечения расстройства, относящийся к вышеупомянутым воплощениям, где расстройство выбирают из группы: дефицит внимания, гиперактивность, тревога, депрессия, посттравматический стресс, супрануклеарный паралич, расстройство приема пищи, навязчиво-компульсивное состояние, аналгезия, пристрастие к никотину, панические атаки. Паркинсонизм и фобия, ожирение, синдром поздней фазы желтого тела или нарколепсия, пристрастие к кокаину, пристрастие к амфетамину и психиатрические симптомы раздражительности, такие как подавление чувствительности и недостаток психологической и физической энергии.
Другим аспектом изобретения является способ ингибирования синаптического захвата норадреналина у пациента, нуждающегося в этом, включающий введение терапевтически эффективного в отношении ингибирования количества соединения, представленного формулами (IA-F).
Другим аспектом изобретения является способ ингибирования синаптического захвата серотонина у пациента, нуждающегося в этом, включающий введение терапевтически эффективного в отношении ингибирования количества соединения, представленного формулами (IA-F).
Другим аспектом изобретения является способ ингибирования синаптического захвата допамина у пациента, нуждающегося в этом, включающий введение терапевтически эффективного в отношении ингибирования количества соединения, представленного формулами (IA-F).
Другим аспектом изобретения является описанный здесь способ лечения, в котором применяется (+)-стереоизомер соединения, представленного формулами (IA-F).
Другим аспектом изобретения является описанный здесь способ лечения, в котором применяется (-)-стереоизомер соединения, представленного формулами (IA-F).
Другим аспектом изобретения является набор, включающий соединение, представленное формулами (IA-F), и, по крайней мере, одно соединение, выбранное из группы, включающей антагонист рецептора 1А серотонина, селективный антагонист рецептора нейрокинина-1 и предшественник норадреналина.
Другим аспектом изобретения является способ лечения депрессии у пациентов, нуждающихся в этом, заключающийся в ингибировании синаптического захвата серотонина и норадреналина с помощью введения терапевтически эффективного в отношении ингибирования количества соединения, представленного формулами (IA-F), которое действует как ингибитор захвата серотонина, так и норадреналина.
Другим аспектом изобретения является способ лечения депрессии у пациентов, нуждающихся в этом, заключающийся в ингибировании синаптического захвата серотонина и допамина с помощью введения терапевтически эффективного в отношении ингибирования количества соединения, представленного формулами (IA-F), которое действует как ингибитор захвата серотонина, так и допамина.
Другим аспектом изобретения является способ лечения депрессии у пациентов, нуждающихся в этом, заключающийся в ингибировании синаптического захвата допамина и норадреналина с помощью введения терапевтически эффективного в отношении ингибирования количества соединения, представленного формулами (IA-F), которое действует как ингибитор захвата допамина, так и норадреналина.
Другим аспектом изобретения является способ ингибирования захвата серотонина у млекопитающих, который включает введение млекопитающему, нуждающемуся в повышении нейропередачи серотонина, фармацевтически эффективного количества соединения, представленного формулами (IA-F).
Другим аспектом изобретения является способ ингибирования захвата допамина у пациентов, который включает введение млекопитающему, нуждающемуся в повышении нейропередачи допамина, фармацевтически эффективного количества соединения, представленного формулами (IA-F).
Другим аспектом изобретения является способ ингибирования синаптического захвата норадреналина у пациентов, который включает введение млекопитающему, нуждающемуся в повышении нейропередачи норадреналина, фармацевтически эффективного количества соединения, представленного формулами (IA-F).
Другим аспектом изобретения является способ подавления тяги человека к курению, включающий введение человеку, нуждающемуся в подобном подавлении, эффективной дозы соединения, представленного формулами (IA-F), для подавления тяги к курению.
Другим аспектом изобретения является способ подавления тяги человека к принятию алкоголя, включающий введение человеку, нуждающемуся в подобном подавлении, эффективной дозы соединения, представленного формулами (IA-F), для подавления тяги к принятию алкоголя.
Следует понимать, что основные аспекты изобретения, которые для ясности описаны в контексте отдельных воплощений, также могут быть представлены в виде комбинации в одном воплощении. И, напротив, различные аспекты изобретения, которые для краткости описаны в контексте одного воплощения, также могут быть представлены отдельно или в виде любой подходящей субкомбинации.
Получение соединений согласно настоящему изобретению
Соединения согласно изобретению, например исходные вещества, промежуточные продукты или конечные продукты, получали, как описано здесь или применяя или адаптируя известные способы, которые представляют собой методы, применявшиеся ранее или описанные в литературе.
Соединения, применяемые согласно изобретению, могут быть получены с помощью применения или адаптации известных способов, которые представляют собой методы, применявшиеся ранее или описанные в литературе, например описанные R.C.Larock в Comprehensive Organic Transformations, VCH publishers, 1989.
Соединение, представленное формулами (IA-F), включающее группу, содержащую один или несколько кольцевых атомов азота, может быть преобразовано в соответствующее соединение, в котором один или несколько кольцевых атомов азота данной группы окислен до N-оксида, предпочтительно взаимодействием с перкислотой, например с перуксусной кислотой в уксусной кислоте или мета-хлорпероксибензойной кислотой в инертном растворителе, таком как дихлорметан, при температуре от комнатной до температуры флегмы, предпочтительно при повышенной температуре.
В реакциях, описанных здесь и далее, может возникнуть необходимость в защите реактивных функциональных групп, например гидрокси, амино, имино, тио или карбоксигрупп, в случае их желательного присутствия в конечном продукте, для избежания их нежелательного участия в реакциях. Могут применяться обычные защитные группы в соответствии со стандартной практикой, например, см. Т.W.Green и Р.G.М.Wuts в «Protective Groups in Organic Chemistry» John Wiley и Sons, 1991; J.F.W.McOmie в «Protective Groups in Organic Chemistry» Plenum Press, 1973.
Представленные здесь соединения синтезировали, например, с помощью описанных ниже способов (см. схемы 1-4), вместе с известными способами в области химии органического синтеза, или их вариациями, хорошо понятными специалистам в данной области. Предпочтительные способы включают, но не ограничиваются ими, описанные выше способы.
Соединения, представленные формулами (IA-F), согласно данному изобретению, например, получали соответственно схеме 1. Обработка необязательно замещенного ацетофенона, представленного формулой (II), с помощью обычных бромирующих реагентов, таких как, но не ограничиваясь ими, бром, NBS или трибромид тетрабутиламмония, легко приводило к получению желаемых бромацетофенонов, представленных формулой (III, X=Br). Данные реакции оптимально проводили в уксусной кислоте или метиленхлориде, применяя метанол в качестве сорастворителя для трибромидного реагента, при температуре реакции, равной или ниже комнатной. Другое воплощение данной методики будет включать соединения, представленные формулой (III, Х=Cl).
Ацетофеноны, представленные формулой (II), являются доступными в коммерческих источниках или легко получены с помощью нескольких хорошо известных способов, включающих обработку соответствующих промежуточных продуктов бензойной кислоты с помощью двух стехеометрических эквивалентов метиллития, как полностью описано в обзоре Jorgenson, M.J. (Organic Reactions, 1970, 18, pg.1). Альтернативно можно обработать соответствующие бензальдегиды с помощью нуклеофилов, как алкильный реактив Гриньяра (например, MeMgBr) или алкиллития (например, MeLi) с последующим обычно проводимым окислением до кетона, как хорошо описано Larock, R.C (Comprehensive Organic Transformations, VCH Publishers, New York, 1989, p.604).
Обработка промежуточных продуктов, представленных формулой (III), с помощью продуктов, представленных формулой (R3,R4-Ph)-CH(R2)-NHR1, легко приводила к получению алкилированных продуктов, представленных формулой (V). Реакции алкилирования могут проходить в широком диапазоне условий, хорошо известных специалистам в области органического синтеза. Типичные растворители включают ацетонитрил, толуол, диэтиловый эфир, тетрагидрофуран, диметилсульфоксид, диметилформамид, метиленхлорид и низшие алкильные спирты, включающие этанол. Реакции могут успешно проходить при температуре от 0°С до точки кипения применяемого растворителя. Прохождение реакции обычно определяют с помощью стандартных хроматографических и спектроскопических методов. Реакцию алкилирования необязательно проводят с добавлением ненуклеофильного органического основания, такого как пиридин, триэтиламин и диизопропилэтиламин, но не ограничиваясь ими.
R1-замещенные N-бензиламины, представленные формулой (R3,R4-Ph)-CH(R2)-NHR1, могут быть получены из коммерческих источников или альтернативно получены с помощью простой реакции гидроаминирования. Таким образом, карбонил, включающий соединения, представленные формулой (IV, схема 1), может быть обработан с помощью H2N-R1 в присутствии низших алкильных спиртовых растворителей (предпочтительно метанол) при температуре, равной или ниже комнатной. Полученный имин может быть восстановлен, как правило, с помощью боргидридов щелочноземельных металлов (предпочтительно боргидрида натрия) с получением желаемого продукта амина.
Восстановление соединений, представленных формулой (V), до бензиловых спиртов, представленных формулой (VI), проходит с помощью многих восстанавливающих реагентов, включающих, например, боргидрид натрия, боргидрид лития, боран, диизобутилалюмогидрид и алюмогидрид лития. Реакции восстановления проводят в течение 1 часа - 3 дней при комнатной температуре или повышенной температуре до точки возгонки применяемого растворителя. В случае использования борана он может применяться в виде комплекса, например, но не ограничиваясь, боран-метилсульфидный комплекс, боран-пиперидиновый комплекс, боран-тетрагидрофурановый комплекс. Специалистам в данной области будут понятны оптимальные комбинации восстанавливающих реагентов и необходимые условия реакции, или они могут быть найдены в руководстве публикации Larock, R.С. (Comprehensive Organic Transformations, VCH Publishers, New York, 1989, p.527).
Соединения, представленные формулой (VI), могут быть циклизованы с получением целевых соединений согласно данному изобретению, представленных формулами IA-IF, с помощью непродолжительной обработки сильной кислотой. Подходящие кислоты включают, но не ограничиваясь такими, как концентрированная серная кислота, полифосфорная кислота, метансульфоновая кислота и трифторуксусная кислота. Реакции проводят при обязательном или необязательном присутствии сорастворителя, такого как, например, метиленхлорид и 1,2-дихлорэтан. Циклизации могут проводиться в температурном диапазоне от 0°С до температуры возгонки применяемого растворителя. Специалистам в области гетероциклической химии будут легко понятны данные условия или они могут быть взяты из руководства Mondeshka, et al. (Il Farmaco, 1994, 49, 475-480) или Venkov, et al. (Synthesis, 1990, 253-255). Циклизация также может проводиться взаимодействием соединений, представленных формулой (VI), с сильной кислотой Льюиса, такой как, например, хлорид алюминия, обычно в галогенированных растворителях, таких как метиленхлорид. Специалисту в данной области должны быть известны предыдущие работы Kaiser, et al. (J. Med. Chem., 1984, 27, 28-35) и Wyrick, et al. (J. Med. Chem., 1981, 24, 1013-1015).
Соединения, представленные формулами IA-IF, могут быть получены в виде чистых (R)- и (S)-энантиомеров с помощью кристаллизации с хиральными солями, что хорошо известно специалистам в данной области, или альтернативно могут быть выделены с помощью хиральной ВЭЖХ, применяя коммерчески доступные хиральные колонки.
Альтернативно соединения, представленные формулами (V) и (VI), могут быть получены, как представлено на схеме 2. Таким образом, галогенацетофеноны, представленные формулой, могут взаимодействовать с простыми аминами, представленными формулой H2N-R1, при условиях алкилирования, описанных выше (см. выше), с получением соединений, представленных формулой (VII). Далее может проводиться второе алкилирование с помощью реагентов, представленных формулой (VIII), где Х представляет собой уходящую группу, такую как, например, но не ограничиваясь ею, галоген, мезилат или тозилат с получением общего промежуточного продукта, представленного формулой (V). Реагенты, представленные формулой (VIII), соответственно получают из подходящего замещенного карбонильного соединения, представленного формулой (IV), посредством восстановления (см. выше) и активации.
Активация уходящей группы Х проводится взаимодействием спирта с метансульфонилхлоридом или пара-толуолсульфонилхлоридом в присутствии ненуклеофильного основания, такого как, но не ограничиваясь им, 1,5-диазабицикло[4,3,0]нон-5-ен (DBN), пиридин или триэтиламин. Реакцию обычно проводят в присутствии галогенированного органического растворителя, например метиленхлорида, и при температуре от -78°С до температуры кипения применяемого растворителя. Бензильная активация уходящей (удаляемой) группы Х также может выполняться с помощью обработки галогенирующими реагентами, такими как, но не ограничиваясь ими, SO2Cl2, Cl2, PCl5, Br2, CuBr2, NBS и CBr4. Ряд условий, необходимых для проведения данного преобразования, будет легко понятен специалистам в области органической химии и дополнительная ссылка относительно бензильной активации может быть найдена в Larock, R.C. (Comprehensive Organic Transformations, VCH Publishers, New York, 1989, p.313).
Гибкость синтеза, кроме того, проявляется наличием альтернативного ряда реакций, в ходе которых (VII) может быть восстановлено (см. выше) и или i) алкилировано, как описано выше с помощью (VIII) с получением (VI), или ii) конденсировано с (IV) с последующим восстановлением имина in situ также с получением (VI). R5=R6=R7=H и производное (метиламинометил)бензилового спирта могут быть получены из коммерческих источников.
Соединения, представленные формулами IA-IF, согласно данному изобретению также могут быть получены согласно схеме 3. Взаимодействие соответствующего замещенного 2-йодбензальдегида (или 2-бромбензальдегида) (X) с амином H2N-R1 в низших алкильных спиртовых растворителях с последующим восстановлением полученного имина, как описано выше на схеме 1 (см. выше), приводит к получению промежуточного продукта (2-1 или Br), R2, R3-PhCH2-NH-R1, которое при обработке необязательно замещенным бромацетофеноном (как описано для синтеза (V), схема 1) приводит к получению алкилированного продукта (XI).
Соединения, представленные формулой (XI), могут быть обработаны сильными основаниями, такими как, но не ограничиваясь ими, низшие алкил(С1-6) литиевые основания (предпочтительно трет-BuLi или н-BuLi), получая ожидаемую замену галоген-металл, с последующей внутримолекулярной циклизацией Barbier с получением соединений, представленных формулами (IA-IE, R8=OH). Инертные растворители, такие как диалкильные эфиры (предпочтительно диэтиловый эфир), циклические эфиры (предпочтительно тетрагидрофуран или 1,4-диоксан) и т.п. являются необходимыми для поддержания низкой температуры реакции (от -78°С до 25°С) для избежания побочных продуктов. Альтернативно замена галоген-металл также может проводиться в присутствии ноль валентного никеля, при этом в качестве идеальных растворителей подходят N,N-диалкилформамиды (предпочтительно диметилформамид). Специалистам в области органического синтеза будут понятны оптимальные сочетания условий, и дополнительные ссылки можно найти в Kihara, et al. (Tetrahedron, 1992, 48, 67-78) и Blomberg, et al. (Synthesis, 1977, p.18-30). Кроме того, соединения, представленные формулами (IA-E, R8=OH), могут быть легко алкилированы (см. выше) с получением соединений, представленных формулами (IA-E, R8=OR11). Наконец, дополнительная обработка соединений, представленных формулами (IA-E, R8=OH), галогенирующим реагентом или специфически фторирующим реагентом, таким как, но не ограничиваясь им, диэтиламиносерный трифторид (DAST), легко приводит к получению соединений, представленных формулами (IA-F, R8=F). Дополнительная ссылка может быть найдена в обзоре Hudlicky (Organic Reactions. 1985, 35, р.513-637).
Соединения, представленные формулами IA-F, согласно данному изобретению также могут быть получены соответственно схеме 4. 4-Бромизохинолины (XII) могут быть обработаны арилбороновой кислотой или сложным эфиром арилбороновой кислоты, где Y представляет собой В (ОН)2 или В (ORa) (ORb) (где Ra и Rb представляют собой низший алкил, т.е. C1-C6, или взятые вместе Ra и Rb представляют собой низший алкилен, т.е. C2-C12) в присутствии металлического катализатора вместе или в отсутствие основания в инертном растворителе с получением изохинолина, представленного формулой (XIII). Металлические катализаторы включают, но не ограничиваются ими, соли или комплексы фосфина с Cu, Pd или Ni (например, Cu(ОАс)2, PdCl2(PPh3)2, NiCl2(PPh3)2). Основания могут включать, но не ограничиваясь ими, карбонаты щелочноземельных металлов, бикарбонаты щелочноземельных металлов, гидроксиды щелочноземельных металлов, карбонаты щелочных металлов, бикарбонаты щелочных металлов, гидроксиды щелочных металлов, гидриды щелочных металлов (предпочтительно гидрид натрия), алкоксиды щелочных металлов (предпочтительно метоксид натрия или этоксид натрия), гидриды щелочноземельных металлов, диалкиламиды щелочных металлов (предпочтительно диизопропиламид лития), бис(триалкилсилил)амиды щелочных металлов (предпочтительно бис(триметилсилил)амид натрия, триалкиламины (предпочтительно диизопропилэтиламин или триэтиламин) или ароматические амины (предпочтительно пиридин). Инертные растворители могут включать, но не ограничиваясь, ацетонитрил, диалкильные эфиры (предпочтительно диэтиловый эфир), циклические эфиры (предпочтительно тетрагидрофуран или 1,4-диоксан), N,N-диалкилацетамиды (предпочтительно диметилацетамид), N,N-диалкилформамиды (предпочтительно диметилформамид), диалкилсульфоксиды (предпочтительно диметилсульфоксид), ароматические углеводороды (предпочтительно бензол или толуол) или галогеналканы (предпочтительно метиленхлорид). Предпочтительный температурный диапазон реакции изменяется от комнатной температуры до точки кипения применяемого растворителя. Реакции могут проводиться в обычной стеклянной посуде или в одной из множества коммерчески доступных параллельных ячеек для синтеза. Коммерчески недоступные бороновые кислоты или сложные эфиры бороновой кислоты могут быть получены из соответствующих необязательно замещенных арилгалогенидов, как описано в Gao, et al. (Tetrahedron, 1994, 50, 979-988).
Соединения, представленные формулой (XIII), преобразовывают в целевые тетрагидроизохинолины, представленные формулой, посредством двухстадийной процедуры, применяя первичную кватернизацию аминов с реагентом R1-LG, где LG представляет собой подходящую уходящую группу, такую как I, Br, O-трифлат, O-тозилат, O-метансульфонат и т.п. Реакции оптимально проводят в галогеналканах (предпочтительно метиленхлорид), диалкильных эфирах (предпочтительно диэтиловый эфир), циклических эфирах (предпочтительно тетрагидрофуран или 1,4-диоксан) или других инертных растворителях. Реакции оптимально проводят при комнатной температуре или ниже в течение от 10 минут до 24 часов. Вторая стадия последовательности включает восстановление тетрагидроизохинолинов, представленных формулами IA-F. Оптимально применяется слабый восстанавливающий реагент, такой как, например, цианоборгидрид натрия в присутствии кислотного катализатора для облегчения прохождения реакции. Дополнительное руководство для эффективного проведения данных химических реакций можно найти в работах Miller, et al. (Synthetic Communications, 1994, 24, 1187-1193) и Terashima, et al. (Heterocycles, 1987, 26, 1603-1610).
Схема 1
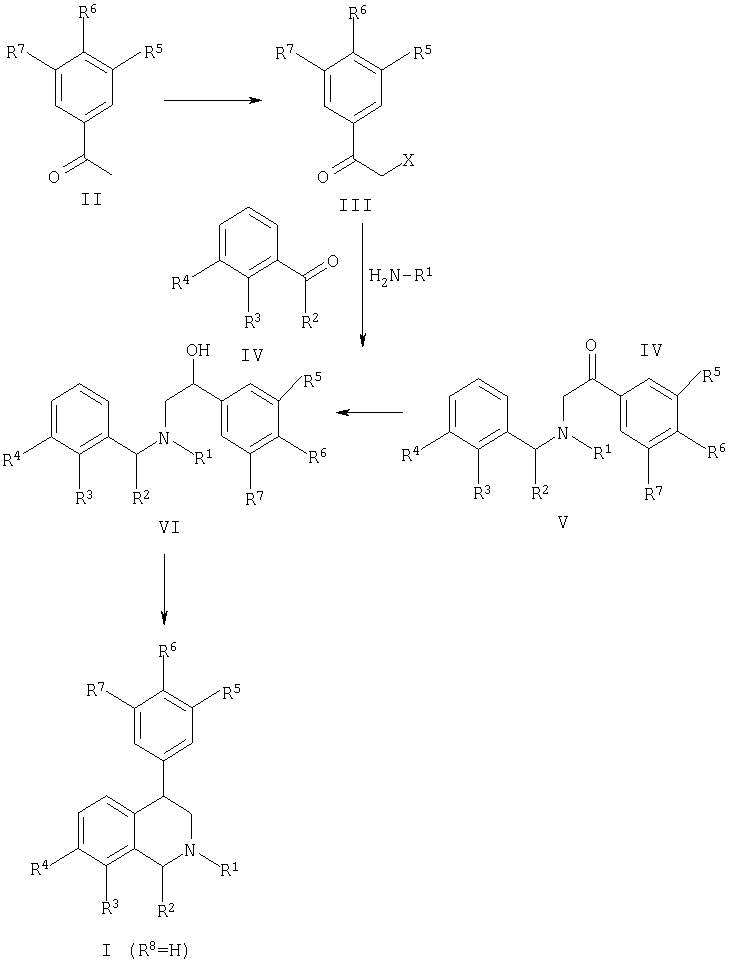
Схема 2
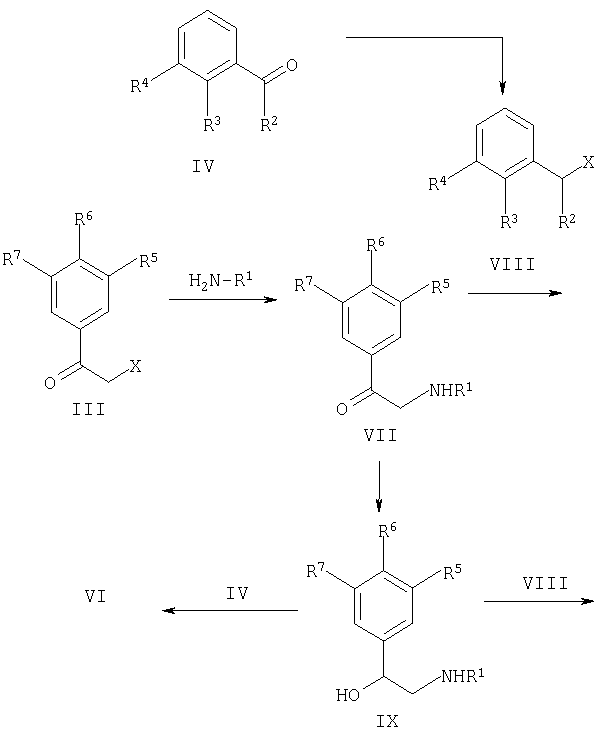
Схема 3
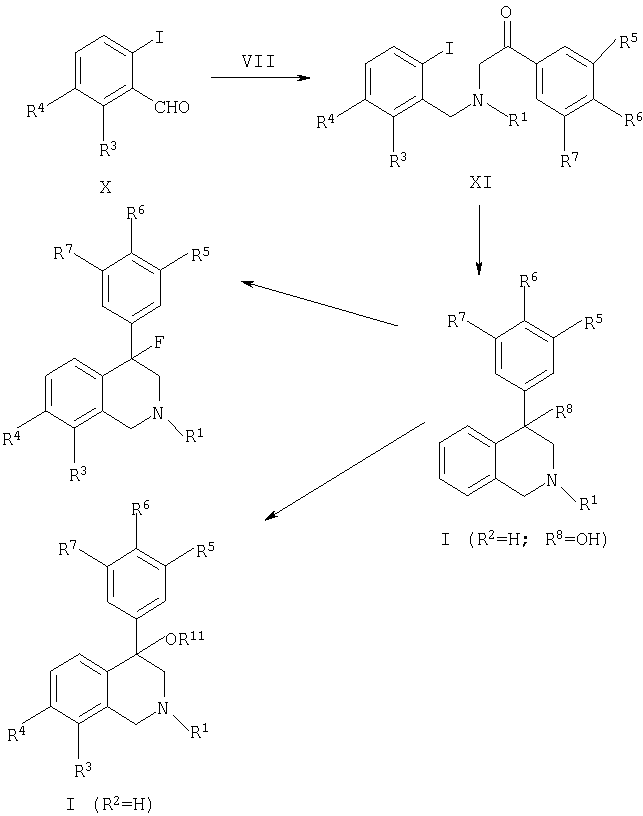
Схема 4
Следует понимать, что соединения, применяемые согласно настоящему изобретению, могут содержать центры асимметрии. Данные центры асимметрии могут независимо находиться в R- или S-конфигурации, и подобные соединения способны вращать плоскость поляризованного света в поляриметре. Если соединение вращает указанную плоскость поляризованного света в направлении против часовой стрелки, то соединение представляет собой (-)-стереоизомер. Если соединение вращает указанную плоскость поляризованного света в направлении по часовой стрелке, то соединение представляет собой (+)-стереоизомер. Для специалистов в данной области будет очевидно, что основные соединения, применяемые согласно изобретению, могут также обладать геометрической изомерией. Понятно, что настоящее изобретение включает индивидуальные геометрические изомеры и стереоизомеры и их смеси, включая рацемические смеси соединений, выше представленных здесь формулами (IA-F). Подобные изомеры могут быть выделены из смеси с помощью применения или адаптации известных способов, например хроматографии и перекристаллизации, или их получают отдельно из соответствующих изомеров промежуточных продуктов.
Радиоактивно меченные соединения согласно изобретению синтезировали с помощью ряда способов, хорошо известных специалистам в данной области, например, применяя исходные соединения, содержащие один или несколько радиоизотопов.
Данное соединение относится к композициям, содержащим описанные здесь соединения, включая, в частности, фармацевтические композиции, содержащие терапевтически эффективные количества соединений и фармацевтически приемлемых носителей.
Следующий объект согласно изобретению обеспечивает наборы, содержащие множество активных ингредиентов (вместе или без носителя), которые вместе могут эффективно применяться для проведения новой комбинированной терапии согласно изобретению.
Другим объектом изобретения является обеспечение новых фармацевтических композиций, которые эффективны вместе или сами по себе для применения при успешной комбинированной терапии, вследствие включения множества активных ингредиентов, которые могут применяться согласно изобретению.
Изобретение также обеспечивает наборы или отдельные упаковки, содержащие два или несколько активных ингредиентов, пригодных для лечения описанных здесь расстройств. Набор может обеспечивать (отдельно или в сочетании с фармацевтически приемлемым разбавителем или носителем) соединение, представленное формулами (IA-F), и дополнительный активный ингредиент (отдельно или в сочетании с разбавителем или носителем), выбранный из антагониста рецептора 1А серотонина, селективного антагониста рецептора нейрокинина-1 и предшественника норадреналина.
На практике соединения согласно настоящему изобретению могут, как правило, вводиться парентерально, внутривенно, чрескожно, внутримышечно, через ободочную кишку, назально, внутрибрюшинно, ректально или перорально.
Продукты согласно изобретению могут быть представлены в формах, позволяющих вводить их наиболее подходящим путем, и изобретение также относится к фармацевтическим композициям, содержащим, по крайней мере, один продукт согласно изобретению, подходящий для применения в ветеринарии и медицине. Данные композиции могут быть получены согласно обычным способам, применяя один или несколько фармацевтически приемлемых адъювантов или наполнителей. Адъюванты включают, между прочим, разбавители, стерильную водную среду и различные нетоксичные органические растворители. Композиции могут быть представлены в виде таблеток, пилюль, гранул, порошков, водных растворов или суспензии, растворов для инъекций, эликсиров или сиропов и могут содержать один или несколько агентов, выбранных из группы, включающей подсластители, корригенты, красители или стабилизаторы для получения фармацевтически приемлемых препаратов.
Выбор носителя и содержание активного вещества в носителе обычно определяют согласно растворимости и химическим свойствам продукта, особенностям пути введения и условиям, наблюдаемым в фармацевтической практике. Например, для получения таблеток могут применяться наполнители, такие как лактоза, цитрат натрия, карбонат кальция, дикальций фосфат и дезинтегрирующие вещества, такие как крахмал, альгиновые кислоты и основной комплекс силикатов в сочетании со смазывающими веществами, такими как стеарат магния, лаурилсульфат натрия и тальк. Для получения капсулы преимущественным является применение лактозы и высокомолекулярных полиэтиленгликолей. В случае применения водных суспензий они могут содержать эмульгирующие вещества или вещества, способствующие получению суспензии. Также могут применяться разбавители, такие как сахароза, этанол, полиэтиленгликоль, пропиленгликоль, глицерин и хлороформ или их смеси.
Для парентерального введения применяют эмульсии, суспензии или растворы продуктов согласно изобретению в растительном масле, например кунжутном масле, арахисовом масле или оливковом масле, или водно-органические растворы, такие как вода и пропиленгликоль, инъекционные органические сложные эфиры, такие как этилолеат, так же как и стерильные водные растворы фармацевтически приемлемых солей. Растворы солей продуктов согласно изобретению особенно пригодны для введения путем внутримышечных или чрескожных инъекций. Водные растворы, также включающие растворы солей в очищенной дистиллированной воде, могут применяться для внутривенного введения с условием, что их рН соответствующим образом подобрана, что они разумно забуферены и приведены в изотоническое состояние с помощью значительного количества глюкозы или хлорида натрия и они стерилизованы с помощью нагревания, облучения или микрофильтрации.
Подходящие композиции, содержащие соединения согласно изобретению, могут быть получены обычными способами. Например, соединения согласно изобретению могут быть растворены или суспендированы в подходящем носителе для применения в небулайзере или в виде аэрозольной суспензии или раствора или могут быть абсорбированы или адсорбированы на подходящий твердый носитель для применения в сухом порошковом ингаляторе.
Твердые композиции для ректального введения включают суппозитории, полученные согласно известным способам, и содержащие, по крайней мере, одно соединение, представленное формулами (IA-F).
Процентное содержание активного компонента в композициях согласно изобретению может варьировать, что является необходимым для установления такого соотношения, чтобы могла быть получена подходящая доза. Очевидно, что несколько отдельных дозированных форм могут вводиться одновременно. Применяющаяся доза будет определяться лечащим врачом и зависеть от желаемого терапевтического эффекта, пути введения и курса лечения и состояния пациента. Для взрослых дозы, в общем, составляют от 0,01 до 100, предпочтительно от 0,01 до 10 мг/кг массы тела в сутки при ингаляционном пути введения, от 0,01 до 100, предпочтительно от 0,1 до 70, более предпочтительно от 0,5 до 10 мг/кг массы тела в сутки при пероральном введении, и от 0,01 до 50, предпочтительно от 0,01 до 10 мг/кг массы тела в сутки при внутривенном введении. В каждом отдельном случае дозы будут определяться согласно с факторами, характерными для пациента, которому проводится лечение, такими как возраст, масса тела, общее состояние здоровья и другие характеристики, которые могут оказывать влияние на эффективность медицинского препарата.
Продукты согласно изобретению могут вводиться так часто, как это необходимо для получения желаемого терапевтического эффекта. Некоторые пациенты могут быстро отвечать на повышенную или пониженную дозу и могут признать адекватными более слабые поддерживающие дозы. Другие пациенты могут нуждаться в длительном курсе лечения с уровнем в 1-4 дозы в сутки согласно физиологическим потребностям каждого отдельного пациента. В общем, активный продукт может вводиться перорально 1-4 раза в сутки. Само собой разумеется, что для других пациентов может быть предписано не больше чем одна или две дозы в день.
Настоящее изобретение относится к соединениям, ингибирующим синаптический захват норадреналина, допамина и серотонина и поэтому предполагает пригодность для лечения расстройства, вызванного или зависящего от снижения уровня серотонина, норадреналина или допамина. Хотя соединения, представленные формулами (IA-F), ингибируют синаптический захват норадреналина, допамина и серотонина, для каждого отдельного соединения данные эффекты ингибирования могут проявляться при одинаковых или очень отличающихся концентрациях или дозах. В результате некоторые соединения, представленные формулами (IA-F), являются пригодными для лечения подобных расстройств в дозах, при которых может существенно ингибироваться синаптический захват норадреналина, но несущественно ингибироваться синаптический захват серотонина или допамина или наоборот. Также некоторые соединения, представленные формулами (IA-F), являются пригодными для лечения подобных расстройств в дозах, при которых может существенно ингибироваться синаптический захват допамина, но несущественно ингибироваться синаптический захват норадреналина или серотонина или наоборот. И, наоборот, некоторые соединения, представленные формулами (IA-F), являются пригодными для лечения подобных расстройств в дозах, при которых может существенно ингибироваться синаптический захват серотонина, но несущественно ингибироваться синаптический захват норадреналина или допамина или наоборот. Другие соединения, представленные формулами (IA-F), являются пригодными для лечения подобных расстройств в дозах, при которых существенно ингибируется синаптический захват норадреналина, допамина и серотонина.
Концентрации или дозы, при которых испытуемое соединение ингибирует синаптический захват норадреналина, допамина и серотонина, легко определяют с помощью стандартных тестов и способов, хорошо известных и понятных специалистам в данной области. Например, степень ингибирования для отдельной дозы у крыс можно определять способом, описанным Dudley, et al., J. Pharmacol. Exp. Ther. 217, 834-840 (1981), который включен в ссылку.
Терапевтически эффективная ингибирующая доза представляет собой дозу, эффективную для существенного ингибирования синаптического захвата норадреналина, синаптического захвата допамина или синаптического захвата серотонина или ингибирующую синаптический захват двух или нескольких из норадреналина, допамина и серотонина. Терапевтически эффективная ингибирующая доза может легко быть определена специалистами в данной области с помощью обычных способов определения и аналогичных результатов, полученных в тест-системах, описанных выше.
Соединения согласно данному изобретению обеспечивают определенный преимущественный терапевтический индекс (показатель) по сравнению с другими соединениями, имеющимися в наличии для лечения подобных расстройств. Не ограничиваясь теорией, предполагается, однако, что это обеспечивается благодаря, по крайней мере, частично наличию у некоторых соединений более высокого сродства к связыванию, например способности быть более селективным по отношению к транспортному белку норадреналина («NET»), чем к транспортным белкам других нейромедиаторов, например транспортному белку допамина («DAT») и транспортному белку серотонина («SERT»).
Аффинности связывания показаны с помощью ряда способов, хорошо известных специалистам в данной области, включая без ограничения способы, нижеописанные здесь в примерах. Кратко, например, содержащие белок экстракты клеток, например клеток НЕК293Е, экспрессирующих транспортные белки, инкубировали с радиомеченными лигандами белков. Связывание радиомеченных лигандов белков является обратимым в присутствии других белковых лигандов, например соединений согласно данному изобретению; указанная обратимость, как описано ниже, обеспечивает способ измерения аффинности связывания белков соединениями (Ki). Более высокое значение Ki для соединения означает, что соединение обладает меньшей аффинностью связывания белка, чем соединение с более низкой Ki; наоборот, более низкие значения Ki указывают на более высокую аффинность связывания.
Соответственно, различие в селективности соединений по отношению к белкам определяется с помощью более низкой Ki по отношению к белку, к которому соединение более селективно, и более высокой Ki по отношению к белку, к которому соединение менее селективно. Таким образом, более высокое соотношение Ki соединения по отношению к белку А по сравнению с белком В показывает более высокую селективность соединения по отношению к последнему, чем к первому (первый обладает более высокой Ki и последний более низкой Ki для данного соединения). Соединения, обеспеченные здесь, индуцируют меньше побочных эффектов в ходе терапевтического применения вследствие их селективности по отношению к транспортному белку норадреналина, как показывают их соотношения Ki, связывание NET преобладает над связыванием других транспортных белков, например DAT и SERT. В общем, некоторые соединения согласно данному изобретению обладают соотношением Ki для DAT/NET, равным, по меньшей мере, 2:1; также, в общем, соотношение SERT/NET составляет, по меньшей мере, 20:1.
Более того, оценка in vivo активности соединений по отношению к переносчикам NE и DA проводилась, например, с помощью определения их способности предотвращать седативный эффект тетрабеназина (TBZ) (см., например, G.Stille, Arzn. Forsch 14: 534-537, 1964, который включен здесь в ссылку). Рандомизированные и кодированные дозы испытуемых соединений вводили мышам после введения дозы тетрабеназина. Животных далее оценивали по проявлению антагонистического эффекта по отношению к вызванной тетрабеназином потере активности и птозу в конкретные интервалы времени после введения лекарственного препарата. Исследуемую активность оценивали, помещая животное в центр круга и далее оценивая период времени, необходимого животному, для пересечения периметра круга: как правило, чем больше времени было необходимо животному для пересечения, тем большей была потеря исследуемой активности. Кроме того, у животного отмечали наличие птоза, если веки были закрыты, по крайней мере, на 50%. Более чем 95% контрольных мышей (которым вводили носитель) ожидаемо проявляли исследуемую потерю активности и птоз; активность соединения далее вычисляли в виде процентной доли мышей с отсутствием ответа, вызываемого дозой тетрабеназина, с более терапевтически эффективными соединениями, ожидаемо лучше снижающими потерю исследуемой активности и птоз.
Соответственно, данное изобретение относится к способам лечения пациентов, страдающих различными неврологическими и психическими расстройствами, с помощью введения указанным пациентам дозы фармацевтического соединения, обеспеченного здесь. Указанные расстройства включают, не ограничиваясь ими, дефицит внимания/гиперактивность, тревогу, депрессию, посттравматический стресс, супрануклеарный паралич, расстройства приема пищи, навязчиво(обсессивно)-компульсивное состояние, аналгезию, тягу к курению, панические атаки, Паркинсонизм и фобию. Соединения, обеспеченные здесь, являются особенно пригодными для лечения данных и других расстройств вследствие, по крайней мере, частично, способности селективно связывать транспортные белки определенных нейромедиаторов с большей аффинностью, чем транспортные белки других нейромедиаторов.
Соединения согласно изобретению, способы их получения и их биологическая активность будут более понятны при изучении следующих примеров, представленных только в качестве иллюстрации, но не ограничивающих пределы изобретения.
ПРИМЕРЫ
Соединения, представленные в следующей таблице 1, получали с помощью описанных выше способов. Конкретные условия реакций и способов получения 2,7-диметил-4-фенил-1,2,3,4-тетрагидроизохинолина (пример 1), 2,7-диметил-4-(3-фторфенил)-1,2,3,4-тетрагидроизохинолина (пример 4), 2,7-диметил-4-(4-фтор-3-метилфенил)-1,2,3,4-тетрагидроизохинолина (пример 6), 2,7-диметил-8-фтор-4-фенил-1,2,3,4-тетрагидроизохинолина (пример 28), 4-(4-хлор-3-фторфенил)-2-метил-1,2,3,4-тетрагидроизохинолина (пример 70), 4-(3,4-дифторфенил)-2-метил-1,2,3,4-тетрагидроизохинолина (пример 78) и 4-(3,5-дифторфенил)-2-метил-1,2,3,4-тетрагидроизохинолина (пример 80) представлены в следующей таблице.
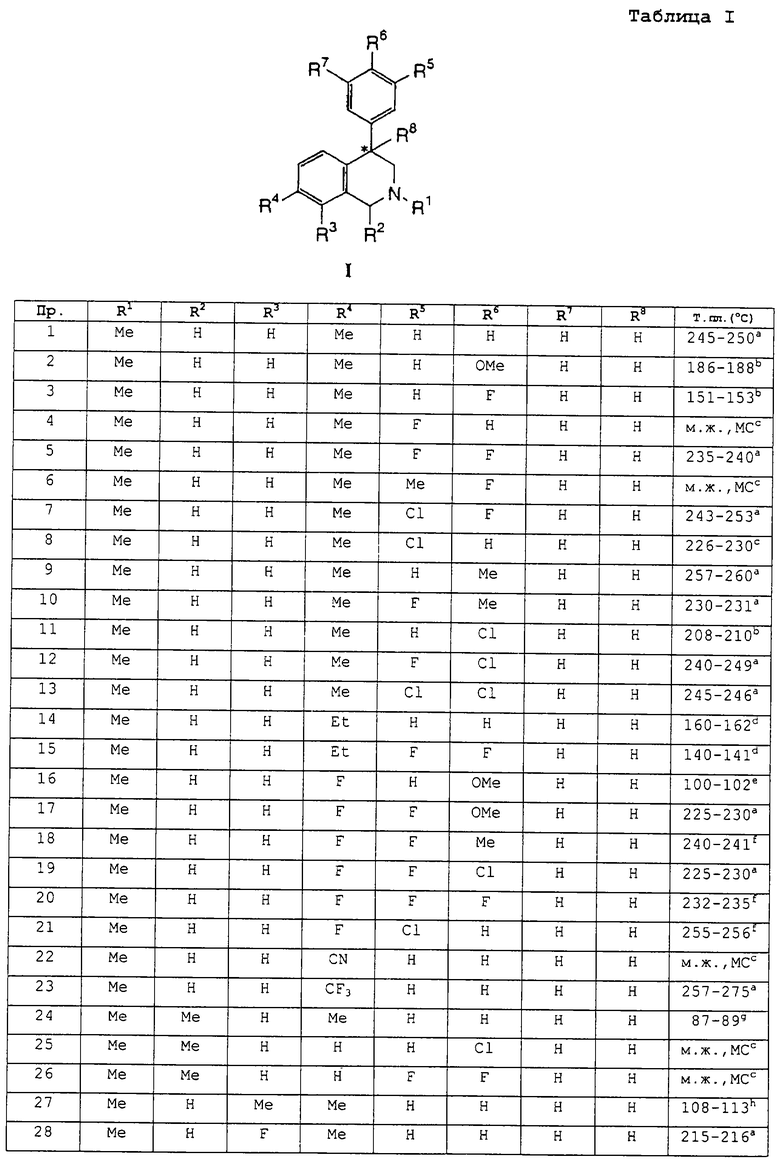
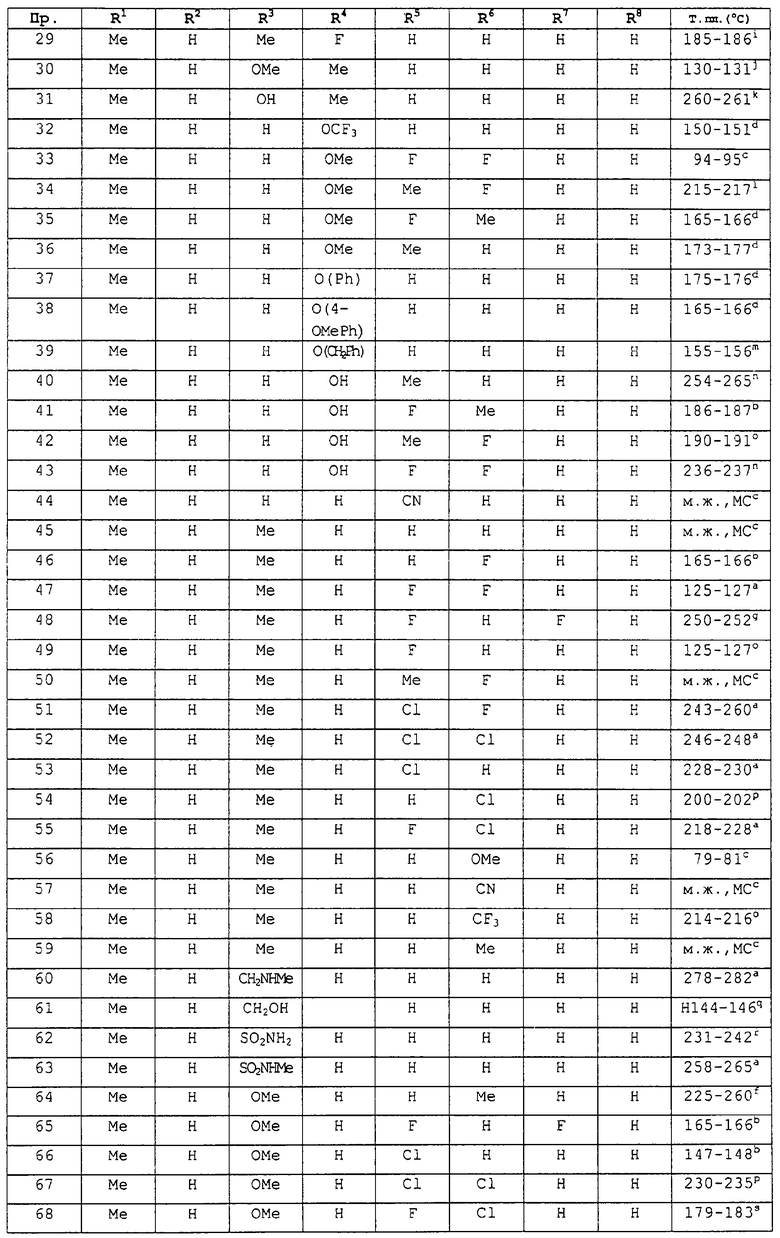
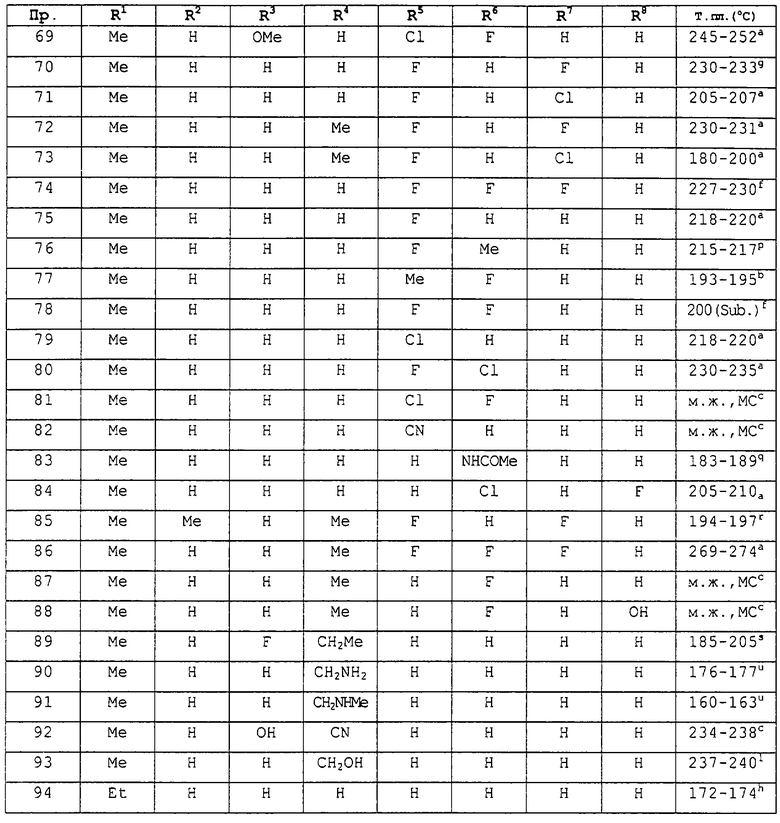
МС - масс-спектр
м.ж. - маслянистая жидкость
Сноски для таблицы 1 для примеров солевых форм:
a - моногидрохлорид
b - мономалеат
с - моногидрохлорид · 0,2 гидрата
d - монофумарат
е - свободное основание - молекулярный ион в показаниях масс-спектра
f - моногидрохлорид · 0,25 гидрата
g - моногидрохлорид · 0,10 гидрата
h - моногидрохлорид · 0,75 гидрата
l - 1,5 фумарат · 0,25 гидрата
j - монофумарат · 0,5 диэтилового эфира
k - моногидробромид · 0,25 гидрата
l-моногидрохлорид · 0,33 гидрата
m - монофумарат · 0,25 гидрата
n - моногидробромид
о - мономалеат · 0,25 гидрата
р - моногидрохлорид · 0,5 гидрата
q - 0,25 гидрат
r - мономалеат · 0,25 гидрата · 0,13 этанола
s - моносульфат
t - дигидрохлорид · 0,5 гидрата
u - бисмалеат
Пример 1
Получение 2,7-диметил-4-фенил-1,2,3,4-тетрагидроизохинолина
Стадия А: Раствор мета-толуальдегида (500 мг, 4,16 ммоль), D-(метиламинометил)бензилового спирта (630 мг, 4,16 ммоль) и уксусной кислоты (0,5 мл) перемешивали в метаноле (16 мл) при 0°С в атмосфере азота, добавляя небольшими порциями цианоборгидрид натрия (784 мг, 12,5 ммоль). Реакционную смесь перемешивали в течение 5 минут при 0°С и двух дней при комнатной температуре. рН реакционной смеси доводили до 12 с помощью 2 н. гидроксида натрия, разбавляли водой и экстрагировали с помощью диэтилового эфира (3Х). Объединенные органические экстракты промывали солевым раствором, сушили над безводным сульфатом магния и растворитель удаляли в вакууме с получением целевого промежуточного продукта (1,24 г): 1H-ЯМР (300 МГц, CDCl3) δ: 7,08-7,35 (м, 9Н), 4,73-4,77 (м, 1Н), 3,71 (д, J=13,0 Гц, 1Н), 3,50 (д, J=13,0 Гц, 1Н), 2,46-2,67 (м, 2Н), 2,36 (с, 3Н), 2,32 (с, 3Н); МС ХИ m/z=256 [С17Н21NO+Н]+.
Стадия В: Продукт, полученный на стадии А (1,24 г, 4,90 ммоль), перемешивали в метиленхлориде (208 мл) и обрабатывали концентрированной серной кислотой по капле (98%, 10 мл) в течение 3 минут. После перемешивания в течение 20 минут реакционную смесь разбавляли кусочками льда и подщелачивали с помощью 25% водного раствора гидроксида аммония. Реакционную смесь экстрагировали с помощью метиленхлорида (3×) и органические экстракты объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Проводили очистку с помощью колоночной хроматографии, применяя в качестве элюента гексан/этилацетат (5/1) с получением целевого тетрагидроизохинолина (0,23 г): 1H-ЯМР (300 МГц, CDCl3) δ: 7,17-7,31 (м, 5Н), 6,87-6,89 (м, 2Н), 6,75 (д, J=7,8 Гц, 1Н), 4,20-4,26 (м, 1Н), 3,72 (д, J=14,8 Гц, 1Н), 3,57 (д, J=14,8 Гц, 1Н), 2,96-3,10 (м, 1Н), 2,51-2,58 (м, 1Н), 2,42 (с, 3Н), 2,29 (С, 3Н).
Стадия С: Продукт, полученный на стадии В (0,23 г), обрабатывали с помощью эфирной HCl в метаноле (5 мл) с получением преципитата. Растворители и избыток HCl удаляли в вакууме и полученный твердый продукт перекристаллизовывали из этанол/диэтилового эфира с получением соли HCl целевого продукта (0,21 г) в виде белого твердого вещества: точка плавления 245-250°С; 1H-ЯМР (CD3OD) δ: 6,86-7,40 (м, 7Н), 6,74 (д, J=7,8 Гц, 1Н), 4,52-4,64 (м, 3Н), 3,72-3,88 (м, 1Н), 3,45-3,55 (м, 1Н), 3,08 (с, 3Н), 2,32 (с, 3Н); 13С-ЯМР (75 МГц, CD3OD) 130,6, 130,3, 129,1, 127,8, 59,3, 56,8, 44,5, 44,0, 21,1; ИК (KBr) 2937, 2474, 1454, 701 см-1; МС ХИ m/z=238 [C17H19N+H]+. Вычислено для C17H19N-HCl: С, 74,57; Н, 7,36; N 5,12. Найдено: С, 74,20; Н, 7,34; N 4,82.
Пример 4
Получение 2,7-диметил-4-(3-фторфенил)-1,2,3,4-тетрагидроизохинолина
Стадия А: мета-Толуальдегид (1,66 г, 14,0 ммоль) обрабатывали метиламином (40% водный раствор, 1,39 мл, 18,0 ммоль) в метаноле (20 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 20 минут и обрабатывали порциями боргидрида натрия (0,26 г, 7,0 ммоль). Реакционную смесь перемешивали в течение 1 часа и обрабатывали 3'-фтор-2-бромацетофеноном (3,0 г, 14,0 ммоль) с последующим перемешиванием в течение 45 минут при комнатной температуре. Реакционную смесь окончательно обрабатывали порциями боргидрида натрия (0,52 г, 14,0 ммоль) и продолжали перемешивание в течение ночи. Реакционную смесь разбавляли водой (100 мл) и экстрагировали с помощью метиленхлорида (3×100 мл). Объединенные органические экстракты промывали солевым раствором и сушили над безводным сульфатом натрия с последующей фильтрацией и концентрированием в вакууме. Очистку проводили с помощью колоночной хроматографии на силикагеле, применяя в качестве элюента гексан/этилацетат (3/1), с получением аминоспирта (4,3 г) в виде желтой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,08-7,30 (м, 7Н), 4,73 (т, J=6,0 Гц, 1Н), 3,60 (ABq, JAB=14,0 Гц, 2Н), 2,55 (д, J=8,0 Гц, 2Н), 2,36 (с, 3Н), 2,31 (с, 3Н); МС ХИ m/z=274 [C17H20NFO+H]+.
Стадия В: Продукт, полученный на стадии А (1,0 г, 4,0 ммоль), перемешивали в метиленхлориде (100 мл) и обрабатывали концентрированной серной кислотой по капле (98%, 7,0 мл) в течение 3 минут. После перемешивания в течение 1 часа реакционную смесь разбавляли кусочками льда и подщелачивали с помощью 25% водного раствора гидроксида аммония. Реакционную смесь экстрагировали с помощью метиленхлорида (3×100 мл) и органические экстракты объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Проводили очистку с помощью колоночной хроматографии, применяя в качестве элюента гексан/этилацетат (3/1) с получением целевого тетрагидроизохинолина в виде желтой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 6,89-7,00 (м, 5Н), 6,75 (д, J=8,0 Гц, 1Н), 4,21 (т, J=7,0 Гц, 1Н), 3,64 (ABq, JAB=15,0 Гц, 2Н), 3,02 (м, 1H), 2,56 (м, 1H), 2,41 (с, 3Н), 2,29 (с, 3Н); МС ХИ m/z=256 [C17H18NF+H]+.
Стадия С: Продукт, полученный на стадии В, подвергали разделению с помощью хиральной ВЭЖХ, применяя колонку Chiral Technologies Chiracel® AD (5 см×50 см), проводя элюцию с помощью гексан/изопропанола (9/1) с получением энантиомеров (R), [α]25 D-16,3 (с=0,498, МеОН) и (S), [α]25 D+16,3 (с=0,476, МеОН) в порядке проведения элюции. (S)-(+)-энантиомер обрабатывали малеиновой кислотой (1,0 эквивалент) и полученную соль малеиновой кислоты фильтровали и сушили до постоянной массы. (S)-(+)-2,7-диметил-4-(3-фторфенил)-1,2,3,4-тетрагидроизохинолин, малеат: точка плавления 172-173,5°С.
Пример 6
Получение 2,7-диметил-4-(4-фтор-3-метилфенил)-1,2,3,4-тетрагидроизохинолина
Стадия А: мета-Толуальдегид (4,0 г, 33,0 ммоль) обрабатывали метиламином (40% водный раствор, 3,36 мл, 43,0 ммоль) в метаноле (40 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 20 минут и обрабатывали порциями боргидрида натрия (0,64 г, 33,0 ммоль). Реакционную смесь перемешивали в течение 1 часа и обрабатывали 4'-фтор-3'-метил-2-бромацетофеноном (7,69 г, 33,0 ммоль) с последующим перемешиванием в течение 45 минут при комнатной температуре. Реакционную смесь окончательно обрабатывали порциями боргидрида натрия (1,0 г, 33 ммоль) и продолжали перемешивание в течение ночи. Реакционную смесь разбавляли водой (100 мл) и экстрагировали с помощью метиленхлорида (3×100 мл). Объединенные органические экстракты промывали солевым раствором и сушили над безводным сульфатом натрия с последующей фильтрацией и концентрированием в вакууме. Очистку проводили с помощью колоночной хроматографии на силикагеле, применяя в качестве элюента гексан/этилацетат (2/1), с получением аминоспирта (65,3 г) в виде желтой маслянистой жидкости; МС ХИ m/z=286 [C18H22NFO+H]+.
Стадия В: Продукт, полученный на стадии А (0,52 г, 2,0 ммоль), растворяли в метиленхлориде (20 мл) и обрабатывали концентрированной серной кислотой по капле (98%, 3 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем разбавляли кусочками льда и подщелачивали с помощью 25% водного раствора гидроксида аммония. Реакционную смесь экстрагировали с помощью метиленхлорида (3×50 мл) и органические экстракты объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Проводили очистку с помощью колоночной хроматографии, применяя в качестве элюента гексан/этилацетат (3/1) с получением целевого тетрагидроизохинолина (0,08 г): 1H-ЯМР (300 МГц, CDCl3) δ: 6,87-7,00 (м, 5Н), 6,74 (д, J=8,0 Гц, 1Н), 4,17 (т, J=7,0 Гц, 1Н), 3,64 (ABq, JAB=15,0 Гц, 2Н), 3,01 (м, 1Н), 2,53 (м, 1Н), 2,40 (с, 3Н), 2,29 (с, 3Н), 2,23 (с, 3Н); МС ХИ m/z=270 [C18H20NF+H]+.
Пример 28
Получение 2,7-диметил-8-фтор-4-фенил-1,2,3,4-тетрагидроизохинолина
Стадия А: Раствор D-(метиламинометил)бензилового спирта (745 мг, 4,9 ммоль) и триэтиламина (0,79 мл, 5,66 ммоль) в ацетонитриле (45 мл) при 0°С в атмосфере азота обрабатывали с помощью 2-фтор-3-метилбензил бромида (1,0 г, 4,9 ммоль) по капле в виде раствора в ацетонитриле (25 мл). Реакционную смесь перемешивали при 0°С в течение 1 часа и при комнатной температуре в течение 1,5 часов с последующим разбавлением водой и экстракцией метиленхлоридом (3×). Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме с получением алкилированного продукта (1,35 г): 1H-ЯМР (CDCl3) δ: 7,23 (м, 5Н), 7,08-7,17 (м, 2Н), 6,97-7,06 (м, 1Н), 4,71-4,82 (м, 1Н), 3,79 (д, J=13,1 Гц, 1Н), 3,62 (д, J=13,2 Гц, 1Н), 2,33 (с, 3Н), 2,29 (с, 3Н).
Стадия В: Продукт, полученный на стадии А (0,5 г, 1,8 ммоль), обрабатывали серной кислотой (3,7 мл) и очищали с помощью колоночной хроматографии, как описано в примере 1, стадия В с получением целевого продукта (0,33 г) в виде маслянистой жидкости: 1H-ЯМР (CDCl3) δ: 7,06-7,37 (м, 5Н), 6,88 (т, J=7,8 Гц, 1Н), 6,54 (д, J=7,8 Гц, 1Н), 4,18-4,27 (м, 1Н), 3,86 (д, J=15,6 Гц, 1Н), 2,94-3,04 (м, 1Н), 2,49-2,59 (м, 1Н), 2,45 (с, 3Н), 2,22 (с, 3Н).
Стадия С: Продукт, полученный на стадии В (0,33 г, 1,3 ммоль), обрабатывали эфирной HCl, как описано в примере 1, стадия С с получением целевого гидрохлорида (0,30 г): точка плавления 215-216°С; 1H-ЯМР (300 МГц, CD3OD) δ: 7,31-7,44 (м, 2Н), 7,21-7,28 (м, 2Н), 7,15 (т, J=7,9 Гц, 1Н), 6,61 (д, J=8,0 Гц, 1Н), 4,67-4,78 (м, 1Н), 4,42-4,62 (м, 2Н), 3,77-3,88 (м, 1Н), 3,55 (т, J=12,0 Гц, 1Н), 3,11 (с, 3Н), 2,26 (с, 3Н); ИК (KBr) 3432, 2954, 2376, 1497, 1457, 1216, 1043, 704 см-1; МС ХИ m/z=256 [C17H18NF+H]+. Вычислено для C17H18NF-HCl: С, 69,98; Н, 6,56; N, 4,80. Найдено: С, 69,64; Н, 6,49; N, 4,65.
Пример 70
Получение 4-(4-хлор-3-фторфенил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия А: Метилбромид магния добавляли по капле в течение 5 минут к перемешиваемому раствору 4-хлор-3-фторбензальдегида (10,86 г, 68,5 ммоль) в безводном тетрагидрофуране (100 мл) при -78°С в атмосфере азота. После перемешивания в течение 15 минут охлаждающую баню удаляли и раствор оставляли нагреваться до комнатной температуры. После перемешивания в течение 3 часов раствор медленно вливали при помешивании в насыщенный хлорид аммония (100 мл), затем разбавляли водой (50 мл) и экстрагировали диэтиловым эфиром. Органические экстракты промывали водой и насыщенным хлоридом натрия, сушили над безводным сульфатом натрия, фильтровали и растворитель удаляли в вакууме с получением бензилового спирта (11,89 г) в виде прозрачной желтой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,35 (т, J=7,8 Гц, 1Н), 7,18 (дд, J=2,0, 10,0 Гц, 1Н), 7,07 (дд, J=1,7, 8,1 Гц, 1Н), 4,83-4,92 (м, 1H), 2,01 (д, J=3,6 Гц, 1Н), 1,47 (д, J=6,3 Гц, 3Н), МС ХИ m/z=175 [C8H8ClFO+H]+.
Стадия В: Продукт, полученный на стадии А (9,0 г, 52,0 ммоль), в безводном метиленхлориде (60 мл) в атмосфере азота добавляли с помощью канюли к перемешиваемой суспензии хлорхромата пиридиния (16,7 г, 77,0 ммоль) и диатомовой земли (15 г) в безводном метиленхлориде (150 мл) при 0°С в атмосфере азота. После перемешивания в течение 26 часов гетерогенную смесь разбавляли диэтиловым эфиром (300 мл), перемешивали в течение 1 часа и фильтровали. Фильтрат концентрировали в вакууме и летучий продукт очищали с помощью колоночной хроматографии на силикагеле (60 г), применяя в качестве элюента гексан/этилацетат (9/1) с получением целевого ацетофенона в количественном выходе неочищенного продукта: 1H-ЯМР (300 МГц, CDCl3) δ: 7,65-7,75 (м, 2Н), 7,51 (т, J=7,6 Гц, 1H), 2,60 (с, 3Н), МС ХИ m/z=173 [C8H6ClFO+H]+.
Стадия С: Продукт, полученный на стадии В (52 ммоль), обрабатывали трибромидом тетрабутиламмония (25,5 г, 52,9 ммоль) в метаноле/метиленхлориде (1/3, 240 мл) в атмосфере азота. После перемешивания в течение 3 дней при комнатной температуре растворители удаляли в вакууме и остаток растворяли в диэтиловом эфире (200 мл), промывали водой (4×50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Проводили очистку с помощью колоночной хроматографии на силикагеле (120 г), применяя в качестве элюента гексан/этилацетат (30/1) с получением целевого α-бромацетофенона (6,23 г) в виде кристаллического твердого вещества: 1H-ЯМР (300 МГц, CDCl3) δ: 7,70-7,81 (м, 2Н), 7,55 (т, J=7,7 Гц, 1Н), 4,39 (с, 2Н); МС ХИ m/z=251 [C8H5BrClFO+H]+.
Стадия D: Метиламин (40% водный раствор, 18,0 ммоль) добавляли к перемешиваемому раствору бензальдегида (1,8 г, 17 ммоль) в метаноле (20 мл) в атмосфере азота. После перемешивания в течение 10 минут при комнатной температуре раствор охлаждали до 0°С и обрабатывали порциями боргидрида натрия (0,32 г, 8,5 ммоль). Реакционную смесь перемешивали в течение 15 минут, нагревали до комнатной температуры и дополнительно перемешивали в течение 1 часа, после чего добавляли продукт, полученный на стадии С (4,3 г, 17 ммоль). Реакционную смесь перемешивали в течение 1 часа, охлаждали до 0°С и снова обрабатывали боргидридом натрия (0,32 г, 8,5 ммоль) и оставляли перемешиваться в течение ночи при нагревании до комнатной температуры. Раствор разбавляли водой (100 мл) и экстрагировали метиленхлоридом (3×50 мл). Органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением целевого продукта в виде прозрачной желтой маслянистой жидкости (1,77 г): 1H-ЯМР (300 МГц, CDCl3) δ: 7,25-7,39 (м, 6Н), 7,17 (дд, J=1,8, 10,0 Гц, 1Н), 7,04 (д, J=8,3 Гц, 1Н), 4,69 (дд, J=5,8, 8,2 Гц, 1H), 3,74 (д, J=13,0 Гц, 1Н), 3,52 (д, J=13,0 Гц, 1Н), 2,45-2,57 (м, 2Н), 2,32 (с, 3Н), МС ХИ m/z=294 [C16H17ClFNO+H]+.
Стадия Е: Продукт, полученный на стадии D (1,77 г, 6,0 ммоль), перемешивали в концентрированной серной кислоте (4,0 мл) и метиленхлориде (40 мл) в течение 15 минут при комнатной температуре. Реакционную смесь помещали на лед, переводили в щелочное состояние с помощью концентрированного гидроксида аммония и экстрагировали диэтиловым эфиром. Объединенные эфирные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением неочищенного продукта в виде непрозрачной желтой маслянистой жидкости (1,7 г): 1H-ЯМР (300 МГц, CDCl3) δ: 7,30 (т, J=7,9 Гц, 1Н), 7,06-7,22 (м, 3Н), 6,92-7,03 (м, 2Н), 6,85 (д, J=7,4 Гц, 1Н), 4,28 (т, J=6,7 Гц, 1Н), 3,77 (д, J=15,1 Гц, 1Н), 3,70 (д, J=15,1 Гц, 1H), 3,05 (дд, J=5,6, 11,9 Гц, 1Н), 2,62 (дд, J=8,0, 11,5 Гц, 1Н), 2,46 (с, 3Н).
Стадия F: Продукт, полученный на стадии Е (1,7 г, 6,0 ммоль), обрабатывали эфирной HCl (1,0 М, 12,0 мл, 12,0 ммоль) в метаноле (20 мл) с получением преципитата. Растворители и избыток HCl удаляли в вакууме и полученное твердое вещество перекристаллизовывали из смеси метанол/диэтиловый эфир с получением соли HCl целевого продукта (1,1 г) в виде белого твердого вещества: точка плавления 230-235°С; 1H-ЯМР (CD3OD) δ: 7,51 (т, J=8,0 Гц, 1Н), 7,26-7,39 (м, 3Н), 7,18 (дд, J=2,0, 10,2 Гц, 1Н), 7,11 (дд, J=1,8, 8,3 Гц, 1H), 6,92 (д, J=7,9 Гц, 1Н), 4,68 (дд, J=6,3, 11,3 Гц, 1Н), 4,59 (ш.с, 2Н), 3,87 (дд, J=6,2, 12,4 Гц, 1Н), 3,56 (т, J=11,8 Гц, 1Н), 3,08 (с, 3Н), ИК (KBr) 3448, 2928, 2365, 1491, 1060, 747 см-1; МС ХИ m/z=276 [C16H15NClF+H]+. Вычислено для C16H15NClF-HCl: С, 61,55; Н, 5,17; N, 4,49. Найдено: С, 61,20; Н, 5,07; N, 4,32.
Стадия G: Продукт, полученный на стадии Е, подвергали разделению с помощью хиральной ВЭЖХ, применяя колонку Chiral Technologies Chiracel® OD (2 см×20 см), проводя элюцию с помощью гексана/изопропанола (9/1) с получением энантиомеров (S) и (R) в порядке проведения элюции. Каждый энантиомер обрабатывали малеиновой кислотой (1,0 эквивалент) и полученные соли малеиновой кислоты фильтровали и сушили до постоянной массы. (S)-(+)-4-(4-хлор-3-фторфенил)-2-метил-1,2,3,4-тетрагидроизохинолин, малеат: точка плавления 171-172°С; [α]25 D+16,0 (с=0,200, МеОН). (R)-(-)-4-(4-хлор-3-фторфенил)-2-метил-1,2,3,4-тетрагидроизохинолин, малеат: точка плавления 171-172°С; [α]25 D-15,5 (с=0,200, МеОН).
Пример 78
Получение 4-(3,4-дифторфенил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия А: 3,4-Дифторацетофенон (25,0 г, 160,0 ммоль) обрабатывали уксусной кислотой (250 мл) и бромином (8,23 мл, 160,0 ммоль, раствор в 13 мл уксусной кислоты) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали в вакууме для удаления уксусной кислоты. Остаток суспендировали в насыщенном карбонате натрия и экстрагировали метиленхлоридом несколько раз. Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением целевого производного бромацетофенона (37,0 г) в виде желтого кристаллического твердого вещества: 1H-ЯМР (300 МГц, CDCl3) δ: 7,81 (м, 2Н), 7,32 (м, 1Н), 4,39 (с, 2Н).
Стадия В: Продукт, полученный на стадии А (37,0 г, 158,0 ммоль), растворяли в метиленхлориде (290 мл) и добавляли по капле к раствору N-бензил-N-метиламина (20,3 мл, 158,0 ммоль) и триэтиламина (22,0 мл, 158,0 ммоль) в метиленхлориде (312 мл). Добавление проводили в течение 45 минут при 0°С, нагревали до комнатной температуры и оставляли перемешиваться дополнительно в течение 4 часов. Реакционную смесь разбавляли водой (300 мл) и экстрагировали метиленхлоридом. Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Продукт очищали с помощью колоночной хроматографии на силикагеле (600 г), проводя элюцию гексаном/этилацетатом (7/3) с получением целевого алкилированного продукта в виде прозрачной светло-коричневой маслянистой жидкости (30,2 г): 1H-ЯМР (300 МГц, CDCl3) δ: 7,87-7,73 (м, 2Н), 7,35-7,15 (м, 6Н), 3,68 (с, 2Н), 3,64 (с, 2Н), 2,34 (с, 3Н).
Стадия С: Продукт, полученный на стадии В (15,0 г, 54,0 ммоль), растворяли в метаноле (65 мл), охлаждали на ледяной бане и обрабатывали боргидридом натрия (1,38 г, 36,0 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 часа и при комнатной температуре в течение 1 часа, с последующим охлаждением водой и экстракцией метиленхлоридом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с непосредственным получением очищенного бензилового спирта (14,4 г) в виде желтой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,38-7,00 (м, 8Н), 4,67 (т, J=7,0 Гц, 1Н), 3,74 и 3,35 (ABq, JAB=13,2 Гц, 2Н), 2,50 (д, J=7,0 Гц, 2Н), 2,31 (с, 3Н). Вычислено для C16H17N1O1F2: С, 69,30; Н, 6,19; N, 5,05. Найдено: С, 68,94: Н 6,21; N, 4,94.
Стадия D: Продукт, полученный на стадии С (14,4 г, 52,0 ммоль), перемешивали в концентрированной серной кислоте (27,0 мл) и метиленхлориде (333 мл) в течение 15 минут при комнатной температуре. Реакционную смесь помещали на лед, приводили в щелочное состояние с помощью концентрированного гидроксида аммония и экстрагировали диэтиловым эфиром. Объединенные эфирные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Продукт очищали с помощью колоночной хроматографии на силикагеле, проводя элюцию гексаном/этилацетатом (1/1) с получением очищенного тетрагидроизохинолина (11,4 г): 1H-ЯМР (300 МГц, CDCl3) δ: 7,29-7,36 (м, 1Н), 6,83-7,20 (м, 6Н), 4,20 (т, J=6,3 Гц, 1Н), 3,66 (с, 2Н), 2,95 (дд, J=5,4, 11,5 Гц, 1Н), 2,58 (дд, J=7,4, 11,3 Гц, 1Н), 2,41 (с, 3Н).
Стадия Е: Продукт, полученный на стадии D (0,8 г, 3,0 ммоль), обрабатывали эфирной HCl, как описано в примере 1, стадия F, с получением ожидаемого гидрохлорида (0,6 г): точка плавления 200°С (чистый); 1H-ЯМР (300 МГц, CD3OD) δ: 7,24-7,39 (м, 4Н), 7,14-7,23 (м, 1Н), 7,06-7,13 (м, 1Н), 6,92 (д, J=7,8 Гц, 1Н), 4,65 (дд, J=6,1, 11,4 Гц), 4,58 (с, 2Н), 3,85 (дд, J=6,2, 12,4 Гц, 1Н), 3,54 (т, J=11,8 Гц, 1Н), 3,07 (с, 3Н). ИК (KBr) 3448, 2932, 2549, 1512, 1465, 1276, 742 см-1; МС ХИ m/z=260 [C16H15NF2+H]+. Вычислено для C16H15NF2-HCl-0,25H2O: С, 64,00, Н, 5,54; N, 4,66. Найдено: С, 64,11; Н, 5,30; N, 4,62.
Стадия F: Продукт, полученный на стадии D, подвергали разделению с помощью хиральной ВЭЖХ, применяя колонку Chiral Technologies Chiracel® OD (2 см×20 см), проводя элюцию гексаном/изопропанолом (9/1) с получением энантиомеров (S) и (R) в порядке проведения элюции. Каждый энантиомер обрабатывали малеиновой кислотой (1,0 эквивалент) и полученные соли малеиновой кислоты фильтровали и сушили до постоянной массы. (S)-(-)-4-(3,4-дифторфенил)-2-метил-1,2,3,4-тетрагидроизохинолин, малеат: точка плавления 138-139°С; [α]25 D-2,6 (с=0,366, МеОН). (R)-(+)-4-(3,4-дифторфенил)-2-метил-1,2,3,4-тетрагидроизохинолин, малеат: точка плавления 138-139°С; [α]25 D+2,5 (с=0,386, МеОН).
Пример 80
Получение 4-(3,5-дифторфенил)-2-метил-1,2,3,4-тетрагидроизохинолина
Стадия А: Трибромид тетрабутиламмония (18,6 г, 38,6 ммоль) добавляли к перемешиваемому раствору 3,5-дифторацетофенона (6,0 г, 38,6 ммоль) в метаноле/метиленхлориде (1/3, 180 мл) в атмосфере азота. После перемешивания при комнатной температуре в течение 72 часов растворители удаляли в вакууме. Остаток растворяли в диэтиловом эфире (200 мл), промывали водой (4×50 мл), сушили над безводным сульфатом натрия, фильтровали и растворитель удаляли в вакууме с получением смеси α-бромацетофенона и соответствующего диметилового кеталя (9,0 г): 1H-ЯМР (300 МГц, CDCl3) δ: 7,50 (дд, J=2,0, 4,0 Гц, 2Н), 7,08 (м, 1Н), 4,39 (с, 2Н).
Стадия В: К смеси продуктов, полученных на стадии А (3,5 г, 14,7 ммоль), и N-метил-N-бензиламина (1,8 г, 14,7 ммоль) в метиленхлориде (15 мл) добавляли диизопропилэтиламин (3,0 мл, 17 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5,5 часов, затем промывали водой и сушили над безводным сульфатом натрия. После фильтрации и концентрации в вакууме вещество очищали с помощью колоночной хроматографии на силикагеле (140 г), применяя в качестве элюента гексан/этилацетат/триэтиламин (9/1/0,1) с получением целевого алкилированного продукта (1,2 г) в виде оранжевой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,48 (дд, J=2,0, 4,0 Гц, 2Н), 7,33 (м, 5Н), 7,00 (м, 1Н), 3,69 (с, 2Н), 3,66 (с, 2Н), 2,36 (с, 3Н).
Стадия С: Продукт, полученный на стадии В (1,1 г, 4,0 ммоль), растворяли в метаноле, охлаждали на водяной бане и обрабатывали боргидридом натрия (0,1 г, 2,7 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 часа и при комнатной температуре в течение 1 часа с последующим гашением водой и экстракцией метиленхлоридом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением бензилового спирта (0,8 г) в виде оранжевой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,40-7,30 (м, 5Н), 6,90-6,82 (м, 1Н), 6,70-6,60 (м, 1H), 4,70 (м, 1Н), 3,73 (д, J=14,0 Гц, 1H), 3,52 (д, J=14,0 Гц, 1H), 2,55-2,40 (м, 2Н), 2,29 (с, 3Н).
Стадия D: Продукт, полученный на стадии С (0,4 г, 1,4 ммоль), перемешивали в концентрированной серной кислоте (1,5 мл) и метиленхлориде (10 мл) в течение 15 минут при комнатной температуре. Реакционную смесь помещали на лед, приводили к щелочному состоянию с помощью концентрированного гидроксида аммония и экстрагировали диэтиловым эфиром. Объединенные эфирные экстракты сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистку проводили с помощью колоночной хроматографии на силикагеле (15 г), применяя в качестве элюента гексан/этилацетат/триэтиламин (9/1/0,1) с получением целевого продукта (70 мг): 1H-ЯМР (300 МГц, CDCl3) δ: 7,40-7,07 (м, 4Н), 6,87 (д, J=7,0 Гц, 1H), 6,77-6,62 (м, 2Н), 4,21 (т, J=6,0 Гц, 1H), 3,66 (д, J=2,0 Гц, 2Н), 2,95 (дд, J=5,0, 6,0 Гц, 1H), 2,61 (дд, J=6,0 Гц, 7,0 Гц, 1H), 2,41 (с, 3Н).
Стадия Е: Продукт, полученный на стадии D (70 мг, 0,27 ммоль), обрабатывали эфирной HCl (1,0 М, 0,6 мл, 0,6 ммоль) в метаноле (1,4 мл) с получением преципитата. Растворители и избыток HCl удаляли в вакууме и полученное твердое вещество перекристаллизовывали из смеси метанол/диэтиловый эфир с получением соли HCl целевого продукта (53 мг) в виде белого твердого вещества: точка плавления 230-233°С; 1H-ЯМР (300 МГц, CD3OD) δ: 7,36-7,28 (м, ЗН), 6,99-6,90 (м, 4Н), 4,67 (дд, J=6,0, 6,0 Гц, 1Н), 4,58 (ш.с, 1Н), 3,87 (дд, J=6,0, 6,0 Гц, 1Н), 3,57 (м, 1Н), 3,08 (с, 3Н). ИК (KBr) 2931, 2473, 1625, 1598, 1462, 1119 см-1; МС ХИ m/z=260 [C16H15F2N+H]+; Вычислено для C16H15F2N-HCl-0,1H2O: С, 64,58; Н, 5,49; N, 4,71. Найдено: С, 64,45; Н, 5,43; N, 4,49.
Пример 85
Получение (3,5-дифтор)-4-фенил-1,2,7-триметил-1,2,3,4-тетрагидроизохинолина
Стадия А: Нитрометан (1,6 мл, 30 ммоль) добавляли по капле к охлажденному льдом раствору фторида тетрабутиламмония (7,5 ммоль) в безводном ТГФ (20 мл). Раствор 3,5-дифторбензальдегида (2,85 г, 20,1 ммоль) в безводном ТГФ (5 мл) добавляли по капле. Затем по капле добавляли триэтиламин (2,8 мл, 20 ммоль). Раствор трет-бутилдиметил-силилхлорида (4,54 г, 30,1 ммоль) в безводном ТГФ (15 мл) добавляли по капле, приводя к образованию белого преципитата. Реакционную смесь перемешивали при 0°С в течение 30 минут и затем фильтровали. Твердое вещество промывали смесью эфир/гексан. Фильтрат промывали (2х) водой. Органическую фазу сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением желтой маслянистой жидкости. Желтую маслянистую жидкость очищали с помощью колоночной хроматографии на силикагеле (300 г), применяя в качестве элюента 30% EtOAc/гексан с получением продукта (2,65 г, 65%) в виде бесцветной маслянистой жидкости: 1H-RMP (300 МГц, CDCl3) δ: 6,98-6,95 (м, 2Н), 6,80 (тт, J=8,8, 2,3 Гц, 1H), 5,49-5,44 (м, 1Н), 4,56-4,53 (м, 2Н), 3,00 (д, J=2,9 Гц, 1H).
Стадия В: Суспензию продукта, полученного на стадии А (2,35 г, 11,6 ммоль), и оксида платины (0,20 г) в абсолютном этаноле (20 мл) гидрировали при 40 фунт/кв. дюйм (изб.) в течение 4 часов. Реакционную смесь фильтровали через целитную подушку, которую дополнительно промывали абсолютным этанолом. Растворитель удаляли в вакууме с получением амина (1,97 г, 98%) в виде белого твердого вещества: точка плавления 54-58°С; 1H-ЯМР (300 МГц, CD3OD) δ: 7,01-6,98 (м, 2Н), 6,87-6,81 (м, 1H), 4,70 (дд, J=8,2, 3,8 Гц, 1H), 2,90 (дд, J=13,0, 3,8 Гц, 1H), 2,76 (дд, J=13,0, 8,2 Гц, 1H).
Стадия С: Раствор 3-метилацетофенона (1,36 г, 10,1 ммоль) и продукта, полученного на стадии В (1,75 г, 10,1 ммоль), в толуоле (20 мл) нагревали в колбе с обратным холодильником с азеотропным удалением воды в течение 4 часов в атмосфере азота. Толуол удаляли в вакууме, получая оранжевую маслянистую жидкость. К охлажденному льдом раствору оранжевой маслянистой жидкости в метаноле (10 мл) добавляли NaBH4 (0,44 г, 12 ммоль). Реакционную смесь перемешивали в течение 1 часа при 0°С и затем оставляли медленно нагреваться до комнатной температуры в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении. Остаток вносили в воду и экстрагировали эфиром (3×). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением продукта в виде смеси диастереомеров (3,00 г, >100%) в виде желтой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,22-7,18 (м, 2Н), 7,08-7,06 (м, 2Н), 6,91-6,81 (м, 2Н), 6,70-6,64 (м, 1Н), 4,69-4,45 (м, 1Н), 3,81-3,67 (м, 1Н), 2,83-2,75 (м, 1Н), 2,58-2,40 (м, 1Н), 2,34 (с, 3Н), 1,39-1,36 (м, 3Н).
Стадия D: Концентрированную H2SO4 (12,0 мл) добавляли к перемешиваемому, охлажденному льдом раствору неочищенного продукта, полученного на стадии С (3,00 г, 10,3 ммоль), в CH2Cl2 (105 мл). После перемешивания в течение 15 минут смесь помещали на лед, приводили к сильнощелочному состоянию с помощью избытка конц. NH4OH и экстрагировали (2х) с помощью Et2O. Объединенные органические экстракты сушили над Na2SO4, фильтровали и растворитель удаляли в вакууме. Остаток (1,75 г) очищали с помощью колоночной хроматографии на силикагеле (145 г), применяя в качестве элюента 10% EtOAc/гексан, содержащий 1% Et3N и далее 20% EtOAc/гексан, содержащий 1% Et3N, с получением продукта в виде смеси диастереомеров (426 мг, 15%) в виде желтой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,04-6,61 (м, 6Н), 4,22-3,99 (м, 2Н), 3,49-3,29 (м, 1Н), 3,19-2,92 (м, 1Н), 2,34-2,32 (м, 3Н), 1,52-1,47 (м, 3Н).
Стадия Е: Формальдегид (37% по массе, 0,70 мл, 9,4 ммоль) добавляли к раствору продукта, полученного на стадии D (426 мг, 1,56 ммоль), в метаноле (16 мл). После 1,5 часов добавляли никель Ренея (0,51 г) и реакционную смесь гидрировали при 35 фунт/кв. дюйм (изб.) в течение 21 часа. Реакционную смесь фильтровали через целитную подушку, которую промывали метанолом. Фильтрат упаривали в вакууме, получая молокообразную жидкость, которую экстрагировали эфиром. Эфирный экстракт сушили над Na2SO4, фильтровали и растворитель удаляли в вакууме. Остаток (392 мг) очищали с помощью колоночной хроматографии на силикагеле (150 г), применяя в качестве элюента 10% EtOAc/гексан, содержащий 1% Et3N, с получением целевого соединения (82 мг, 18%) в виде бесцветной маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 6,97 (с, 1Н), 6,92 (д, J=7,7 Гц, 1Н), 6,78-6,61 (м, 4Н), 4,11 (т, J=6,4 Гц, 1Н), 3,65 (кв, J=6,6 Гц, 1Н), 3,04-2,86 (м, 2Н), 2,45 (с, 3Н), 2,32 (с, 3Н), 1,45 (д, J=6,6 Гц, 3Н).
Стадия F: 1M раствор HCl в эфире (1,0 мл, 1,0 ммоль) добавляли по капле к перемешиваемому раствору продукта, полученного на стадии Е (82 мг, 0,28 ммоль), в метаноле (3 мл). После 30 минут растворители и избыток HCl удаляли в вакууме и остаток осаждали из эфира и обрабатывали ультразвуком в течение 30 минут. Выделяли с помощью фильтрации не совсем белое твердое вещество и затем высушивали при комнатной температуре в вакууме в течение 24 часов с получением продукта (78 мг, 83%) в виде не совсем белого твердого вещества: точка плавления 194-197°С (с разложением); 1H-ЯМР (300 МГц, CD3OD) δ: 7,14-7,12 (м, 2Н), 7,00-6,81 (м, 4Н), 4,65-4,59 (м, 2Н), 3,66-3,64 (м, 2Н), 3,03 (с, 3Н), 2,35 (с, 3Н), 1,75 (д, J=6,5 Гц, 3Н). ИК (KBr) 2928, 2480, 1624, 1599, 1464, 1119, 975, 859 см-1; МС ХИ m/z=288 [C18H19F2N+H]+; ВЭЖХ>99%, tr=16,96 минут. Вычислено для C18H19F2N-HCl-0,25H2O: С, 65,85; Н, 6,29; N, 4,2. Найдено: С, 65,98; Н, 6,12; N, 4,16.
Пример 89
Получение (8-фтор-2-метил-4-фенил-1,2,3,4-тетрагидро-7-изохинолинил)-N-метилметанамина
Стадия А: Метиламин (15,3 мл, 40% водный раствор, 177 ммоль) добавляли к перемешиваемому раствору 3-фторбензальдегида (20,0 г, 161 ммоль) в МеОН (150 мл) при комнатной температуре. После перемешивания в течение 6 часов реакционную смесь охлаждали до 0°С и затем порциями добавляли NaBH4 (6,10 г, 161 ммоль). Ледяную баню удаляли и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16,5 часов. Реакционную смесь гасили с помощью H2O и осторожно подкисляли с помощью 2 н. HCl и затем экстрагировали (3×) с помощью CH2Cl2. Водную фазу затем подщелачивали с помощью 6 н. NaOH и далее экстрагировали (4×) CH2Cl2. Последние органические экстракты объединяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением продукта (21,51 г, 96%) в виде прозрачной маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,32 (тд, J=7,5, 1,7 Гц, 1Н), 7,28-7,19 (м, 1Н), 7,14-6,98 (м, 2Н), 3,80 (с, 2Н), 2,45 (с, 3Н), 1,47 (ш.с, 1Н).
Стадия В: Триэтиламин (8,40 мл, 60,0 ммоль) добавляли к перемешиваемому раствору продукта, полученного на стадии А (8,35 г, 60,0 ммоль), и фенацилбромида (11,94 г, 60,0 ммоль) в СН2Cl2 (200 мл) при комнатной температуре в атмосфере N2. После перемешивания в течение 18 часов реакционную смесь гасили с помощью смеси Н2O/6н. NaOH (33 мл) 10:1 и органическую фазу сушили над Na2SO4, фильтровали и растворитель упаривали в вакууме, получая неочищенный продукт (17,08 г, теоретически = 15,44 г) в виде желтой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 8,00-7,94 (м, 2Н), 7,59-7,52 (м, 1Н), 7,48-7,37 (м, ЗН), 7,30-7,21 (м, 1Н), 7,15-7,10 (м, 2Н), 3,85 (с, 2Н), 3,79 (с, 2Н), 2,39 (с, 3Н). ИК (раствора СН2Cl2) 3055, 2925, 2850, 1682, 1598, 1490, 1450, 1266, 1225, 738, 703 см-1; МС ХИ m/z=258 [C16H16FNO+H]+. Данное вещество применяли без дальнейших манипуляций.
Стадия С: Боргидрид натрия (4,54 г, 120 ммоль) добавляли порциями к перемешиваемому раствору продукта, полученного на стадии В (17,1 г, ˜60,0 ммоль), в МеОН (150 мл), охлаждали до 0°С в атмосфере N2. После перемешивания в течение 4,5 часов при комнатной температуре реакционную смесь разбавляли Н2O (300 мл) и экстрагировали (4×) с помощью СН2Cl2. Органические экстракты объединяли, промывали насыщ. NaCl, сушили над Na2SO4, фильтровали и растворитель упаривали в вакууме. Проводили хроматографию полученной желтой маслянистой жидкости (15,81 г), применяя силикагель (200 г), и элюцию 50% EtOAc/гексаном с получением продукта (14,81 г, 95% в ходе 2 стадий) в виде желтой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,39-7,22 (м, 7Н), 7,15-7,01 (м, 2Н), 4,75 (дд, J=8,3, 5,6 Гц, 1Н), 3,79 (д, J=13,3 Гц, 1Н), 3,64 (д, J=13,3 Гц, 1Н), 2,65-2,53 (м, 2Н), 2,33 (с, 3Н). ИК (раствора СН2Cl2) 3062, 2849, 1587, 1491, 1455, 1333, 1266, 1228, 1094, 1062, 1023, 897, 877, 758, 738, 701 см-1; МС ХИ m/z=260 [C16H18FNO+H]+.
Стадия D: Конц. серную кислоту (24 мл) добавляли по капле к перемешиваемому раствору продукта, полученного на стадии С (14,8 г, 57,1 ммоль) в CH2Cl2 (280 мл), охлаждали до 0°С с помощью ледяной-водяной бани. Охлаждающую баню удаляли после завершения добавления и реакционную смесь энергично перемешивали при комнатной температуре в течение 20 минут. Реакционную смесь далее вносили в смесь лед/вода (400 мл) и полученную смесь подщелачивали с помощью конц. раствора NH4OH до рН ˜10. Водную фазу экстрагировали (3×) с помощью СН2Cl2. Органические экстракты объединяли, промывали с помощью смеси 2:1 насыщ. NaCl/1 н. NaOH, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Проводили хроматографию остатка (13,91 г) на силикагеле (450 г) и элюцию 33% EtOAc/гексан с получением продукта (12,66 г, 92%) в виде желтой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,33-7,15 (м, 5Н), 7,08-6,98 (м, 1Н), 6,90-6,82 (м, 1 Н), 6,66 (д, J=7,7 Гц, 1Н), 4,30-4,22 (м, 1Н), 3,86 (д, J=15,6 Гц, 1Н), 3,53 (д, J=15,6 Гц, 1Н), 3,02 (дд, J=11,4, 5,6, 1,1 Гц, 1Н), 2,57 (дд, J=11,6, 8,7 Гц, 1Н), 2,47 (с, 3Н). ИК (раствора CH2Cl2) 2941, 2782, 1583, 1494, 1468, 1457, 1378, 1248, 1139, 1040, 887, 792, 764, 736, 701 см-1; МС ХИ m/z=242 [C16H16FN+H]+.
Стадия Е: трет-Бутиллитий (30 мл, 1,7 М в пентане, 50,5 ммоль) добавляли по капле к перемешиваемому раствору продукта, полученного на стадии D (5,50 г, 22,8 ммоль), и TMEDA (7,6 мл, 50,2 ммоль) в Et2O (120 мл), охлажденному до -60°С в атмосфере N2. После перемешивания в течение 45 минут добавляли ДМФ (7,0 мл, 91,2 ммоль) и реакционную смесь перемешивали при -60°С в течение 1,5 часа. Реакцию гасили с помощью МеОН (10 мл), нагревали до комнатной температуры и затем разбавляли Н2О (200 мл) и водную фазу экстрагировали (4×) с помощью CH2Cl2. Объединенный СН2Cl2-экстракт сушили над Na2SO4, фильтровали и концентрировали в вакууме. Проводили хроматографию остатка (9,05 г) на силикагеле (350 г) и элюцию 33% EtOAc/гексан с получением продукта (1,21 г, 20%) в виде коричневой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 10,32 (с, 1Н), 7,56 (т, J=7,6 Гц, 1Н), 7,34-7,21 (м, 3Н), 7,19-7,10 (м, 2Н), 6,79 (д, J=8,2 Гц, 1Н), 4,31-4,23 (м, 1Н), 3,90 (д, J=15,8 Гц, 1Н), 3,58 (д, J=15,8 Гц, 1Н), 3,04 (дд, J=11,9, 5,6, 1,0 Гц, 1Н), 2,61 (дд, J=11,7, 8,3 Гц, 1Н), 2,49 (с, ЗН); МС ХИ m/z=270 [C17H16FNO+H]+.
Стадия F: Метиламин (0,05 мл, 40% водный раствор, 0,62 ммоль) добавляли к перемешиваемому раствору содержащего примеси альдегида 147 (0,15 г, ˜0,57 ммоль) в МеОН (3 мл) при комнатной температуре. После перемешивания в течение 6 часов реакционную смесь охлаждали до 0°С и далее добавляли NaBH4 (0,022 г, 0,57 ммоль). Охлаждающую баню удаляли и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 18 часов. Реакцию гасили с помощью Н2О, экстрагировали (4×) раствором СН2Cl2. Органические экстракты объединяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Проводили хроматографию остатка (0,18 г) на силикагеле (10 г) и элюцию с помощью CHCl3:МеОН:конц. NH4OH 88:12:1 с получением метиламина 147 (0,10 г) в виде коричневой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,32-7,12 (м, 5Н), 7,02 (т, J=7,8 Гц, 1Н), 6,63 (д, J=7,9 Гц, 1Н), 4,28-4,20 (м, 1Н), 3,86 (д, J=15,6 Гц, 1Н), 3,75 (с, 2Н), 3,52 (д, J=15,6 Гц, 1Н), 3,00 (дд, J=11,3, 5,6, 0,9 Гц, 1Н), 2,55 (дд, J=11,5, 8,7 Гц, 1Н), 2,46 (с, 3Н), 2,43 (с, 3Н); МС ХИ m/z=285 [C18H21FN2+H]+.
Стадия G: Добавляли эфирный раствор HCl (1,80 мл, 1 н., 1,80 ммоль) к раствору продукта, полученного на стадии F (0,10 г, 0,35 ммоль), в МеОН (0,5 мл) и Et2O (5 мл) при комнатной температуре с образованием не совсем белого твердого вещества. Твердое вещество выделяли и далее перекристаллизовывали из MeOH/Et2O (3×) и твердое вещество сушили в вакууме (54°С) с получением соли (0,083 г, 66%) в виде светло-зеленого твердого вещества: точка плавления 185-205°С; 1H-ЯМР (300 МГц, CD3OD) δ: 7,50-7,24 (м, 6Н), 6,86-6,78 (м, 1H), 4,80-4,50 (м, 3Н), 4,29 (с, 2Н), 3,92-3,83 (м, 1Н), 3,70-3,55 (м, 1Н), 3,15 (с, 3Н), 2,76 (с, 3Н); ИК (KBr) 3422, 2956, 2698, 1635, 1497, 1456, 1218, 1032, 895, 770, 703, 560 см-1; МС ХИ m/z=285 [C18H21FN2+H]+; ВЭЖХ 95,5%, tr=10,96 мин. Вычислено для C18H21FN2-2HCl·0,5H2O: С 59,02; Н, 6,60; N, 7,65. Найдено: С, 59,13; Н, 6,73; N, 7,42.
Пример 90
Получение (2-метил-4-фенил-7-изохинолинил)-N-метилметанамина
Стадия А: Метиламин (40% по массе водный раствор, 2,6 мл, 30 ммоль) добавляли к перемешиваемому раствору 3-бромбензальдегида (5,44 г, 29,4 ммоль) в МеОН (30 мл) в атмосфере N2. После перемешивания в течение 1 часа бесцветный раствор охлаждали до 0°С и затем порциями добавляли NaBH4 (0,60 г, 16 ммоль). После перемешивания в течение 1 часа охлаждающую баню удаляли. После перемешивания в течение 90 минут реакционную смесь охлаждали до 0°С и затем порциями добавляли фенацилбромид (5,90 г, 29,6 ммоль) в течение 30 минут. Реакционную смесь оставляли нагреваться до комнатной температуры. После перемешивания в течение 2 часов при комнатной температуре раствор охлаждали до 0°С и затем добавляли порциями NaBH4 (1,20 г, 31,7 ммоль) в течение 10 минут. Раствор перемешивали в течение 24 часов, в ходе которых температура повысилась от 0 до 25°С. Раствор разбавляли H2O (400 мл), экстрагировали (4×) с помощью эфира. Эфирные экстракты сушили над Na2SO4, фильтровали и растворитель удаляли в вакууме с получением продукта (9,21 г, 98%) в виде желтой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,47-7,21 (м, 9Н), 4,77 (дд, J=10,0, 4,0 Гц, 1Н), 3,71 (д, J=13,3 Гц, 1Н), 3,51 (д, J=13,3 Гц, 1Н), 2,61-2,49 (м, 2Н), 2,32 (с, 3Н).
Стадия В: Конц. H2SO4 (40,0 мл) добавляли по капле в течение 15 мин к перемешиваемому раствору продукта, полученного на стадии А (9,18 г, 28,7 ммоль), в CH2Cl2 (300 мл). После перемешивания в течение 45 мин смесь помещали на лед, сильно подщелачивали с помощью избытка конц. NH4OH, экстрагировали (3×) с помощью Et2O. Эфирные экстракты сушили над Na2SO4, фильтровали, растворитель удаляли в вакууме и остаток (7,29 г) очищали с помощью колоночной хроматографии на силикагеле (300 г), применяя в качестве элюента 10% EtOAc/гексан, содержащий 1% Et3N, с получением продукта (2,05 г, 24%) в виде оранжевой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,32-7,27 (м, 4Н), 7,25-7,14 (м, 3Н), 6,74 (д, J=8,3 Гц, 1Н), 4,22-4,17 (м, 1Н), 3,71 (д, J=15,1 Гц, 1Н), 3,57 (д, J=15,1 Гц, 1Н), 3,05-2,99 (м, 1Н), 2,54 (дд, J=11,5, 8,7 Гц, 1Н), 2,42 (с, 3Н).
Стадия С: Суспензию бромида продукта, полученного на стадии В (1,15 г, 3,81 ммоль), цианид цинка (271 мг, 2,31 ммоль) и тетракис(трифенилфосфин)палладия(0) (266 мг, 0,230 ммоль) в безводном ДМФ (5 мл) нагревали до 83°С в течение 24 часов. После оставляли реакционную смесь охлаждаться до комнатной температуры, реакционную смесь разбавляли толуолом и промывали 2 н. NaOH. Толуоловый экстракт сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток (1,20 г) очищали с помощью колоночной хроматографии на силикагеле (95 г), применяя в качестве элюента 20% EtOAc/гексан, содержащий 1% Et3N, с получением продукта (673 мг, 71%) в виде желтого твердого вещества: точка плавления 103-104°С; 1H-ЯМР (500 МГц, CDCl3) δ: 7,38 (с, 1Н), 7,34-7,23 (м, 4Н), 7,16-7,14 (м, 2Н), 6,98 (д, J=8,0 Гц, 1Н), 4,27 (т, J=7,0 Гц, 1Н), 3,75 (д, J=15,2 Гц, 1Н), 3,61 (д, J=15,2 Гц, 1Н), 3,07-3,03 (м, 1Н), 2,59 (дд, J=11,7, 8,4 Гц, 1Н), 2,44 (с, 3Н); МС ХИ m/z=249 [C17H16N2+H]+.
Стадия D: Раствор продукта, полученный на стадии С (201 мг, 0,809 ммоль) в безводном ТГФ (4 мл) добавляли по капле к охлажденной льдом суспензии алюмогидрида лития (61 мг, 1,6 ммоль) в безводном ТГФ (2 мл). Реакционную смесь перемешивали в течение 90 минут при охлаждении и затем оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали в течение 5 часов и затем гасили реакцию с помощью EtOAc и далее насыщенным раствором Na2SO4. Реакционную смесь разбавляли эфиром, сушили над твердым Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (26 г), применяя в качестве элюента 12% метанол/хлороформ, содержащий 1% конц. NH4OH, с получением продукта (134 мг, 66%) в виде бесцветной маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,31-7,18 (м, 5Н), 7,04 (с, 1Н), 7,00 (д, J=8,0 Гц, 1Н), 6,83 (д, J=8,0 Гц, 1H), 4,25 (т, J=7,0 Гц, 1Н), 3,81 (с, 2Н), 3,75 (д, J=14,9 Гц, 1Н), 3,60 (д, J=14,9 Гц, 1Н), 3,06-3,00 (м, 1Н), 2,56 (дд, J=11,4, 8,7 Гц, 1Н), 2,43 (с, 3Н).
Стадия Е: Суспензию продукта, полученного на стадии D (53 мг, 0,21 ммоль) и малеиновой кислоты (25 мг, 0,22 ммоль) в абсолютном EtOH (10 мл) нагревали на водяной бане при 40°С до полного растворения твердого вещества. После 1 часа реакционную смесь концентрировали в вакууме. Остаток перекристаллизовывали из смеси этанол/эфир с получением бисмалеата (43 мг, 42%) в виде зеленого твердого вещества: точка плавления 176-177°С (с разложением); 1H-ЯМР (300 МГц, CD3OD) δ: 7,40-7,30 (м, 5Н), 7,22 (дд, J=8,0, 1,3 Гц, 2Н), 6,97 (д, J=8,0 Гц, 1Н), 6,24 (с, 4Н), 4,58 (дд, J=11,3, 6,1 Гц, 1Н), 4,52 (с, 2Н), 4,12 (с, 2Н), 3,78 (дд, J=12,3, 6,2 Гц, 1Н), 3,45 (т, J=11,8 Гц, 1Н), 3,02 (с, 3Н); ВЭЖХ 95,8%, tr=10,81 минут. Вычислено для C17H20N2-2(C4H4O4): С, 61,98; Н, 5,82; N, 5,78. Найдено: С, 61,86; Н, 5,82; N, 5,60.
Пример 91
Получение N-метил-(2-метил-4-фенил-7-изохинолинил)-N-метилметанамин
Стадия А: 1 М раствор HCl в эфире (3,0 мл, 3,0 ммоль) добавляли по капле к раствору продукта, полученного на стадии С примера 90 (82 мг, 0,32 ммоль), в метаноле (6 мл). Растворители и избыток HCl удаляли в вакууме, получая зеленое твердое вещество. Суспензию из данного зеленого твердого вещества, карбоната калия (199 мг, 1,44 ммоль) и этилхлорформиата (0,20 мл, 2,1 ммоль) в метаноле (1 мл) и ацетоне (6 мл) нагревали при 50°С в течение 20 часов. После оставляли реакционную смесь охлаждаться до комнатной температуры, разбавляли реакционную смесь солевым раствором и экстрагировали (4×) с помощью EtOAc. Объединенные органические экстракты сушили над твердым Na2SO4, фильтровали и концентрировали в вакууме с получением карбамата (99 мг, 88%) в виде оранжевой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,31-7,14 (м, 5Н), 6,98-6,93 (м, 2Н), 6,83-6,76 (м, 1Н), 4,30-4,10 (м, 5Н), 3,77-3,58 (м, 2Н), 3,07-3,01 (м, 1Н), 2,61-2,54 (м, 1Н), 2,43 (с, 3Н), 1,24 (т, J=7,1 Гц, 3Н); МС ХИ m/z=325 [C20H24N2O2+H]+.
Стадия В: Алюмогидрид лития (60 мг, 1,6 ммоль) добавляли порциями к раствору продукта, полученного на стадии А (99 мг, 0,30 ммоль), в безводном ТГФ (5 мл). Реакционную смесь нагревали с обратным холодильником в течение 6 часов и далее оставляли охлаждаться до комнатной температуры. Реакцию гасили с помощью EtOAc и затем насыщенного раствора Na2SO4. Реакционную смесь разбавляли эфиром, сушили над твердым Na2SO4, фильтровали и концентрировали в вакууме. Остаток (81 мг) очищали с помощью колоночной хроматографии на силикагеле (8 г), применяя в качестве элюента 12% метанол/хлороформ, содержащий 1% конц. NH4OH, с получением продукта (49 мг, 61%) в виде бесцветной маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,32-7,17 (м, 5Н), 7,04 (с, 1Н), 7,00 (д, J=8,0 Гц, 1Н), 6,82 (д, J=8,0 Гц, 1Н), 4,26 (т, J=7,1 Гц, 1Н), 3,83-3,57 (м, 4Н), 3,07-3,01 (м, 1Н), 2,54 (дд, J=11,4, 8,9 Гц, 1Н), 2,45 (с, 3Н), 2,43 (с, 3Н): МС ХИ=267 [C18H22N2+H]+.
Стадия С: Суспензию продукта, полученного на стадии В (20 мг, 0,075 ммоль), и малеиновой кислоты (9 мг, 0,08 ммоль) в абсолютном EtOH (5 мл) нагревали в водяной бане при 40°С до полного растворения твердого вещества. После 2 часов реакционную смесь концентрировали в вакууме. Остаток перекристаллизовывали из смеси этанол/эфир с получением бисмалеата (13 мг, 35%) в виде желто-коричневого твердого вещества: точка плавления 160-163°С (с разложением); 1H-ЯМР (300 МГц, CD3OD) δ: 7,41-7,31 (м, 5Н), 7,24-7,21 (м, 2Н), 6,99 (д, J=8,0 Гц, 1Н), 6,24 (с, 4Н), 4,57 (дд, J=10,9, 5,7 Гц, 1Н), 4,50 (с, 2Н), 4,18 (с, 2Н), 3,76 (дд, J=12,3, 6,2 Гц, 1Н), 3,50-3,38 (м, 1Н), 3,00 (с, 3Н), 2,72 (с, 3Н); ВЭЖХ 95,8%, tr=11,09 минут.
Пример 92
Получение 8-гидрокси-2-метил-4-фенил-1,2,3,4-тетрагидро-7-изохинолинкарбонитрил
Стадия А: Раствор N-метил-2-метоксиамина (8,00 г, 52,9 ммоль) и триэтиламина (5,40 г, 53,0 ммоль) в дихлорметане (100 мл) охлаждали на ледяной бане. Добавляли 2-бромацетофенон (10,5 г, 53,0 ммоль) и реакционную смесь оставляли нагреваться до комнатной температуры. Реакционную смесь разбавляли водой (200 мл) и МТВЕ (200 мл). Фазы разделяли и органическую фазу промывали с помощью Н2О и солевого раствора. Органическую фазу сушили над MgSO4, фильтровали и концентрировали с получением красной маслянистой жидкости, которую подвергали хроматографии (SiO2, 20% EtOAc/гексан) с получением целевого аминокетона в виде желтой маслянистой жидкости (12,6 г, 89%): 1H-ЯМР (300 МГц, CDCl3) δ: 7,97 (д, J=7,4 Гц, 2Н), 7,53-7,50 (м, 1Н), 7,41 (т, J=7,5 Гц, 2Н), 7,32 (д, J=7,4 Гц, 1Н), 7,28-7,21 (м, 1Н), 6,92 (т, J=7,5 Гц, 1Н), 6,85 (д, J=8,1 Гц, 1Н), 3,81 (с, 2Н), 3,77 (с, 3Н), 3,73 (с, 2Н), 2,39 (с, 3Н).
Стадия В: Продукт, полученный на стадии А (12,6 г, 46,8 ммоль), вносили в метанол (120 мл) и охлаждали на ледяной бане. Добавляли порциями боргидрид натрия (1,76 г, 46,8 ммоль). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь концентрировали до половины исходного объема. Добавляли воду (100 мл) и смесь экстрагировали (3×) дихлорметаном. Объединенные органические фазы сушили над MgSO4, фильтровали и концентрировали с получением целевого аминоспирта в виде бледно-желтой маслянистой жидкости (10,0 г, 79%): 1H-ЯМР (300 МГц, CDCl3) δ: 7,39-7,21 (м, 6Н), 6,94-6,85 (м, 3Н), 4,78 (дд, J=4,3, 9,6 Гц, 1Н), 3,85 (с, 3Н), 3,82 (д, J=12,8 Гц, 1Н), 3,47 (д, J=12,8 Гц, 1Н), 2,62-2,57 (м, 2Н), 2,28 (с, 3Н).
Стадия С: Метансульфоновую кислоту (47,7 мл, 735 ммоль) добавляли при комнатной температуре к раствору продукта, полученного на стадии В (4,20 г, 13,7 ммоль), в дихлорметане (250 мл). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 24 часов. После завершения реакции реакционную смесь подщелачивали (рН˜11) с помощью 2 н. NaOH, и экстрагировали метиленхлоридом (3×). Объединенные органические фазы промывали солевым раствором, сушили над MgSO4 и концентрировали в вакууме. Остаток очищали с помощью хроматографии (SiO2, EtOAc/гексан, 2/3) с получением целевого продукта в виде желтой маслянистой жидкости (5,67 г, 61%): 1H-ЯМР (300 МГц, CDCl3) δ: 7,30-7,15 (м, 5Н), 7,02 (т, J=8,0 Гц, 1Н), 6,65 (д, J=8,1 Гц, 1Н), 6,47 (д, J=7,6 Гц, 1Н), 4,25 (т, J=6,8 Гц, 1Н), 3,82 (с, 3Н), 3,81 (д, J=16,2 Гц, 1Н), 3,36 (д, J=16,2 Гц, 1Н), 2,96 (дд, J=4,1, 15,3 Гц, 1Н), 2,58 (дд, J=8,5, 11,4 Гц, 1Н), 2,43 (с, 3Н).
Стадия D: Раствор продукта, полученного на стадии С (5,60 г, 22,1 ммоль), в 48% бромистоводородной кислоте (60 мл) нагревали с обратным холодильником при 100°С в течение 3 часов. Реакционную смесь концентрировали в вакууме и перекристаллизовывали из этанола с получением целевого продукта (4,74 г, 67): 1H-ЯМР (300 МГц, ДМСО-d6) δ: 9,92 (с, 1Н), 7,48-7,25 (м, 3Н), 7,21 (д, J=7,8 Гц, 1Н), 6,98 (т, J=7,7 Гц, 1Н), 6,67 (д, J=7,8 Гц, 1Н), 6,24 (д, J=7,7 Гц, 1Н), 4,26 (т, J=6,0 Гц, 1Н), 3,80 (д, J=15,8 Гц, 1Н), 3,32 (д, J=15,8 Гц, 1Н), 2,99 (дд, J=5,2, 11,3 Гц, 1Н), 2,66 (дд, J=7,1, 11,4 Гц, 1Н), 2,39 (с, 3Н).
Стадия Е: Смесь продукта, полученного на стадии D (4,70 г, 14,7 ммоль), и гексаметилентетрамина (2,06 г, 14,7 ммоль) в трифторуксусной кислоте (50 мл) нагревали до 80°С в течение 7 часов. Реакционную смесь концентрировали в вакууме, затем разбавляли водой (100 мл). Раствор подщелачивали с помощью твердого Na2CO3. Полученный раствор экстрагировали этиловым эфиром (3×) и объединенные органические фазы концентрировали в вакууме. Остаток очищали с помощью хроматографии (SiO2, EtOAc/гексан, 4/1) с получением целевого продукта в виде не совсем белого твердого вещества (2,47 мг, 49%): 1H-ЯМР (500 МГц, CDCl3) δ: 11,42 (ш.с, 1Н), 9,82 (с, 1Н), 7,28 (д, J=8,1 Гц, 1Н), 7,12-6,90 (м, 3Н), 6,54 (д, J=8,1 Гц, 1Н), 4,19 (т, J=6,1 Гц, 1Н), 3,72 (д, J=16,1 Гц, 1Н), 3,62 (д, J=16,2 Гц, 1Н), 2,93 (дд, J=11,9, 6,28 Гц, 1Н), 2,60 (дд, J=11,4, 7,0 Гц, 1Н), 2,47 (с, 3Н).
Стадия F: Продукт, полученный на стадии Е (1,00 г, 2,87 ммоль), растворяли в воде (20 мл) перед обработкой сульфатом натрия (100 мг) и гидроксиламинсульфонатом (0,32 мг 2,87 ммоль). Реакционную смесь перемешивали в течение 2 часов. Реакционную смесь охлаждали на ледяной бане и обрабатывали CH2Cl2 (20 мл). Добавляли бикарбонат натрия (600 мг) и реакционную смесь оставляли нагреваться до комнатной температуры. Отфильтровывали твердые вещества и объединяли с органической фазой. Смесь концентрировали и подвергали хроматографии (SiO2, EtOAc/гексан, 1/1). Два соединения элюировали одновременно. Смесь обрабатывали этанолом (5 мл) и фильтровали. Фильтрат концентрировали с получением целевого нитрила в виде не совсем белого порошка (130 мг, 17%): точка плавления 234-238°С (с разложением); 1H-ЯМР (300 МГц, CD3OD) δ: 7,31-7,14 (м, 6Н), 6,40 (д, J=8,1 Гц, 1Н), 4,21 (т, J=6,1 Гц, 1Н), 4,12 (ш.с, 1Н), 3,61-3,50 (м, 2Н), 2,72 (дд, J=5,4, 11,7 Гц, 1Н), 2,58 (дд, J=7,1, 11,5 Гц, 1Н), 2,38 (с, 3Н). ИК (KBr) 3427, 3026, 2940, 2207, 1590, 1454 см-1; ESI MS m/z 265 [C17H16N2O+H]+; ВЭЖХ 96,3%, tr=13,54 минут.
Пример 93 Получение (2-метил-4-фенил-1,2,3,4-тетрагидро-7-изохинолинил)метанола
Стадия А: Раствор, полученный на стадии С, примера 90 (127 мг, 0,511 ммоль) в безводном толуоле (13 мл) охлаждали до -16°С и затем добавляли по капле 1М DIBAL-H в толуоле (1,7 мл, 1,7 ммоль). Реакционную смесь перемешивали в течение 45 минут при охлаждении и затем добавляли EtOAc (1,1 мл). Реакционную смесь оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали в течение 45 минут и затем добавляли 1 н. H2SO4 (12 мл). Реакционную смесь нагревали с обратным холодильником в течение 30 минут. После оставляли реакционную смесь охлаждаться до комнатной температуры, разбавляли реакционную смесь водой, подщелачивали с помощью 2 н. NaOH и экстрагировали (2×) с помощью CH2Cl2. СН2Cl2-экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением целевого продукта (112 мг, 87%) в виде желтой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 9,95 (с, 1H), 7,62 (с, 1H), 7,59-7,56 (м, 1Н), 7,34-7,16 (м, 5Н), 7,05 (д, J=8,0 Гц, 1H), 4,32 (т, J=7,1 Гц, 1Н), 3,84 (д, J=15,1 Гц, 1H), 3,67 (д, J=15,1 Гц, 1Н), 3,10-3,04 (м, 1Н), 2,60 (дд, J=11,6, 8,6 Гц, 1Н), 2,46 (с, 3Н).
Стадия В: К охлажденному льдом раствору продукта, полученного на стадии А (110 мг, 0,438 ммоль), в метаноле (20 мл) добавляли NaBH4 (36 мг, 0,95 ммоль). Реакционную смесь оставляли медленно нагреваться до комнатной температуры в течение ночи. Реакцию гасили водой и солевым раствором и затем экстрагировали (3×) с помощью CH2Cl2. Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток (106 мг) очищали с помощью колоночной хроматографии на силикагеле (31 г), применяя в качестве элюента EtOAc, с получением целевого спирта (44 мг, 40%) в виде желтой маслянистой жидкости: 1H-ЯМР (300 МГц, CDCl3) δ: 7,32-7,22 (м, 3Н), 7,17 (дд, J=6,6, 1,6 Гц, 2Н), 7,03 (д, J=7,6 Гц, 1Н), 7,02 (с, 1Н), 6,83 (д, J=7,6 Гц, 1Н), 4,61 (с, 2Н), 4,26 (дд, J=8,6, 6,0 Гц, 1Н), 3,69 (д, J=14,9 Гц, 1Н), 3,55 (д, J=14,9 Гц, 1Н), 3,07-3,01 (м, 1Н), 2,53 (дд, J=11,5, 9,1 Гц, 1Н), 2,42 (с, 3Н).
Стадия С: 1 М раствор HCl в эфире (1,0 мл, 1,0 ммоль) добавляли по капле к перемешиваемому раствору продукта, полученного на стадии В (44 мг, 0,17 ммоль) в МеОН (2 мл). Растворители и избыток HCl удаляли в вакууме и остаток перекристаллизовывали из MeOH-Et2O с получением соли (32 мг, 62%) в виде зеленого твердого вещества: точка плавления 237-240°С (с разложением); 1H-ЯМР (300 МГц, CD3OD) δ: 7,42-7,31 (м, 3Н), 7,27-7,23 (м, 4Н), 6,88 (д, J=7,2 Гц, 1Н), 4,60 (ш.с, 5Н), 3,84 (дд, J=12,4, 6,0 Гц, 1H), 3,65-3,45 (м, 1Н), 3,08 (с, 3Н). ИК (KBr) 3356, 2934, 2596, 1495, 1456, 1428, 1049, 758, 703 см-1; ESI MS m/z=254 [C17H19NO+H]+; ВЭЖХ 94,9%, tr=12,83 минуты. Вычислено для C17H19NO-HCl-0,33H2O: С, 69,03; Н, 7,04; N, 4,74. Найдено: С, 68,89; Н, 6,87; N, 4,61.
Пример 94
Получение 2-этил-4-фенил-1,2,3,4-тетрагидроизохинолина
Стадия А: Этиленгликольдиметиловый эфир (20 мл) и 2 н. Na2CO3 (12,2 мл) обрабатывали N2 и вносили в круглодонную колбу, содержащую 4-бромизохинолин (2 г, 9,6 ммоль), фенилбороновую кислоту (1,76 г, 14,4 ммоль) и Pd(PPh3)4 (1,11 г, 0,96 ммоль). Однородный раствор обрабатывали с помощью N2. Полученную реакционную смесь нагревали с обратным холодильником в атмосфере N2 в течение ночи. Раствор охлаждали, реакцию гасили с помощью NaHCO3 (230 мл) и экстрагировали пять раз этиловым эфиром. Объединенные органические экстракты сушили над Na2SO4, фильтровали и растворитель удаляли в вакууме с получением оранжевой маслянистой жидкости. Проводили колоночную хроматографию (этилацетат/гексан 1/1) с получением чистого изохинолина в виде желтой маслянистой жидкости, которую кристаллизовали с охлаждением (2,21 г). 1H-ЯМР (300 МГц, CDCl3) δ: 9,29 (с, 1H), 8,52 (с, 1H), 8,04 (д, 1H, J=8,4 Гц), 7,91 (д, 1H, J=8,1 Гц), 7,66 (м, 2Н), 7,46 (м, 5Н).
Стадия В: Этилтрифлат (383 мг, 2,15 ммоль) добавляли по капле к раствору продукта, полученного на стадии А (400 мг, 1,95 ммоль) в CH2Cl2 (24 мл) при 0°С в атмосфере N2. Раствор перемешивали в течение 15 минут при комнатной температуре. Растворитель удаляли в вакууме с получением желтой соли трифлата изохинолина в виде твердого белого вещества (420 мг, 56% выход). Трифлат (420 мг, 1,09 ммоль) растворяли в МеОН (16 мл) и добавляли к раствору NaCNBH3 (159 мг, 2,53 ммоль). Полученную реакционную смесь перемешивали в течение 5 минут и добавляли несколько капель бромкрезол-зеленого в МеОН. К раствору добавляли HCl в метаноле до получения желтой окраски. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, при этом добавление HCl в метаноле требовалось для сохранения желтой окраски. Реакцию гасили с помощью Н2О (100 мл) и подщелачивали с помощью 5% NaOH до получения голубой окраски. Полученный раствор экстрагировали четыре раза этиловым эфиром. Объединенные органические экстракты промывали солевым раствором, сушили над MgSO4, фильтровали и растворитель удаляли в вакууме с получением тетрагидроизохинолина в виде прозрачной маслянистой жидкости (140 мг, 30% выход).
Стадия С: Малеат получали добавлением малеиновой кислоты (68 мг, 0,59 ммоль) и EtOH (2 мл) к продукту, полученному на стадии В. После охлаждения и удаления EtOH получали белое твердое вещество (130 мг), точка плавления 172-174°С. Свободное основание: 1H-ЯМР CDCl3 δ: 7,17 (м, 8Н), 6,85 (д, 1Н, J=7,7 Гц), 4,28 (т, 1Н, J=7,5 Гц), 3,89 (д, 1Н, J=14,65 Гц), 3,62 (д, 1Н, J=14,65 Гц), 3,15 (дд, 1Н, J=5,7, 11,7 Гц), 2,57 (м, 2Н), 1,16 (т, 3Н, J=7,2 Гц).
Анализ связывания
Предварительный анализ связывания
Для оценки относительного сродства различных соединений к NE, DA и 5НТ создали клеточные линии НЕК293Е, экспрессирующие каждый из трех переносчиков человека. Амплифицировали кДНК, содержащие полные кодирующие области каждого переносчика, с помощью ПЦР, используя библиотеки мозга человека. Секвенировали кДНК, содержащие векторы pCRII, для проверки их идентичности и далее субклонировали в вирус Эпштейна-Барра, несущий экспрессирующую плазмиду (Е.Shen, GM Cooke, RA Horlick, Gene 156: 235-239, 1995). Данную плазмиду, содержащую кодирующую последовательность одного из переносчиков человека, трансфицировали в клетки НЕК293Е. Успешность трансфекции контролировали с помощью способности известных блокаторов обратного захвата ингибировать захват меченных тритием NE, DA или 5НТ.
Для связывания клетки гомогенизировали, центрифугировали и затем ресуспендировали в инкубирующем буфере (50 мМ Трис, 120 мМ NaCl, 5 мМ KCl, рН 7,4). Затем добавляли подходящую радиоактивную метку. Для связывания NET добавляли [3H]Nisoxetine (86,0 Ки/ммоль, NEN/DuPont) до конечной концентрации приблизительно 5 нМ. Для связывания DAT добавляли [3H]WIN 35428 (84,5 Ки/ммоль) до 15 нМ. Для связывания 5НТ добавляли [3H]Citolapram (85,0 Ки/ммоль) до 1 нМ. Далее добавляли различные концентрации (от 10^-5 до 10^-11 М) исследуемого соединения для замещения радиометки. Инкубацию проводили при комнатной температуре в течение 1 часа в 96-луночном планшете. После инкубации планшеты помещали в harvester и быстро промывали 4 раза (50 мМ Трис, 0,9% NaCl, pH 7,4), где клеточные мембраны, содержащие связанную радиоактивную метку, улавливались фильтрами Whatman GF/B. Добавляли сцинтилляционную смесь к фильтрам, которые затем помещали в Packard TopCount. Аффинность связывания исследуемых соединений определяли с помощью нелинейной кривой регрессии, применяя программу GraphPad Prism 2,01. Неспецифическое связывание определяли с помощью промывки (замещения) 10 микромолярным мазиндолом.
TBZ-тест
Для оценки in vivo активности соединений по отношению к медиаторам NE и DA определяли их способность предотвращать седативный эффект тетрабеназина (TBZ) (G.Stille, Arzn. Forsch 14: 534-537, 1964). Самцов мышей CFI (Charles River Breeding Laboratories), массой 18-25 г во время тестирования, выращивали минимум 60 дней при тщательно контролируемых условиях (22,2+1,1°С; 50% уровень влажности 12 часовой световой цикл/24 часа). Мышей не кормили в течение ночи (16-22 часов) перед тестированием. Мышей помещали в чистые поликарбонатные «обувные» ящики (17 см × 28,5 см × 12 см). Рандомизированные и кодированные дозы испытуемых соединений вводили п/о. За 30 минут до начала отсчета вводили в/б тетрабеназин в дозе 45 мг/кг. Все соединения вводили в объеме 0,1 мл/10 г массы тела. Животных оценивали по проявлению антагонистического эффекта в отношении тетрабеназина, вызывающего потерю активности и птоз, в конкретные интервалы времени после введения лекарственного препарата. В обозначенный интервал времени мышей оценивали по состоянию исследуемой активности и птоза. Исследуемую активность оценивали с помощью помещения животного в центр 5 дюймового круга. Животному предоставляли пятнадцать секунд для передвижения и пересечения периметра. Это рассматривали как антагонистический эффект по отношению к тетрабеназину и оценивали в 0 баллов. Неспособность покинуть круг расценивали как потерю исследования и оценивали в 4 балла. Наличие птоза у животного отмечали, если веки были закрыты, по крайней мере, на 50%, и оценивали в 4 балла в случае полного смыкания век. Незакрытые веки оценивали в 0 баллов. Ожидалось, что 95% контрольных мышей (которым вводили носитель) проявляли исследуемую потерю активности и птоз. Активность лекарственного препарата вычисляли в виде процентной доли мышей с отсутствием ответа, вызываемого дозой тетрабеназина.
Статистическая оценка
Средние эффективные дозы (ED50) и 95% доверительные интервалы определяли численно с помощью методов Томпсона (1947) и Личфилда и Вилкоксона (1949). Для 4-(3,5-дифтор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолина и 2,7-диметил-4-(4-фтор)фенил-1,2,3,4-тетрагидроизохинолина ED50 для птоза составляет 4,0 и 2,5 мг/кг соответственно.
Пример фармацевтической композиции
Таблетки, покрытые оболочкой.
А. Получение ядра таблетки.
Смесь 100 г активного компонента (соединение формулы IA-IF), 570 г лактозы и 200 г крахмала тонко измельчают и тщательно гомогенизируют, после чего увлажняют раствором 5 г додецилсульфата натрия и 10 г поливинилпирролидона в 200 мл воды. Влажную массу пропускают через сито, высушивают и просеивают вновь. Затем добавляют 100 г микрокристаллической целлюлозы и 15 г гидрированного растительного масла. Полученную смесь таблетитуют в обычной машине с получением 10000 таблеток, каждая из которых содержит 10 мг активного компонента.
Б. Нанесение покрытия.
К раствору 10 г метилцеллюлозы в 75 мл спирта добавляют раствор 5 г этилцеллюлозы в 150 мл дихлорметана. Раствор разбавляют 75 мл дихлорметана и добавляют в него 2,5 мл глицерина.
Расплавляют 10 г полиэтиленгликоля и растворяют их в 75 мл дихлорметана. Полученный раствор добавляют к первому раствору, послу чего в объединенный раствор вносят 2,5 г октадеканата магния, 5 г поливинилпирролидона и 30 мл концентрированной суспензии красителя, обычно используемой в таких целях. Полученную массу тщательно гомогенизируют и покрывают ей таблетки в обычном аппарате для нанесения покрытий.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ФОСФАТНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2617682C2 |
| АРИЛ- И ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ТЕТРАГИДРОИЗОХИНОЛИНЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ БЛОКИРОВАНИЯ ОБРАТНОГО ЗАХВАТА НОРЭПИНЕФРИНА, ДОПАМИНА И СЕРОТОНИНА | 2005 |
|
RU2388751C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ С ИХ ИСПОЛЬЗОВАНИЕМ | 2001 |
|
RU2265598C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ БЕНЗОПИПЕРАЗИН, В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНОВ ВЕТ | 2014 |
|
RU2720237C2 |
| НОВЫЕ АНТИГЕЛЬМИНТНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2828007C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ | 1993 |
|
RU2130453C1 |
| ЭНАНТИОМЕРНО ЧИСТЫЕ ОСНОВНЫЕ ЭФИРЫ АРИЛ-ЦИКЛОАЛКИЛГИДРОКСИКАРБОНОВЫХ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ | 1997 |
|
RU2238936C2 |
| ПРОИЗВОДНЫЕ ДИГИДРОИЗОХИНОЛИНОНА И ИЗОИНДОЛИНОНА И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2833422C1 |
| ЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ 11БЕТА-ГИДРОКСИСТЕРОИД-ДЕГИДРОГЕНАЗЫ 1 | 2009 |
|
RU2531272C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2586974C2 |
Изобретение относится к новым тетрагидроизохинолинам, которые пригодны для лечения различных неврологических и психических расстройств, например ADHD. Описывается производное тетрагидроизохинолина, представленное формулой IA-IF, со следующей структурой:
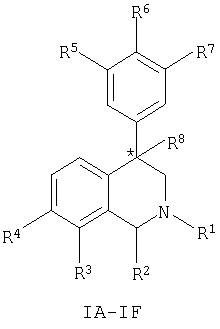
где атом углерода, обозначенный *, имеет R- или S-конфигурацию; R1 представляет собой C1-С6-алкил; R2 представляет собой Н, C1-С6-алкил; R3 представляет собой Н, -S(O)nNR11R12, C1-С6-алкил, либо, если R3 представляет собой C1-С6-алкил, указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-С6-алкил, -OR9 и -NR9R10; при условии, что в соединениях, представленных формулой IA, R3 представляет собой C1-С6-алкил, который необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-С6-алкил, -OR9 и -NR9R10; R4 представляет собой Н, C1-С6-алкил, и когда R4 представляет собой C1-С6-алкил, то указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-С3-алкил, галоген, -OR9 и -NR9R10; при условии, что в соединениях, представленных формулой ID, R4 представляет собой C1-С6-алкил, который необязательно замещен; и где R3 и R4 оба не являются водородом; каждый R5, R6 и R7 в соединениях, представленных каждой из формул IA, IB, IC, ID, IE и IF, независимо представляет собой Н, галоген, -OR11, -NR11C(O)R12, и C1-С6-алкил, и когда каждый из R5, R6 и R7 представляет собой C1-С6-алкил, то указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-С3-алкил, галоген; при условии, что в соединениях, представленных формулой IE, по крайней мере, один из R5 или R7 представляет собой фтор, хлор, или метил; R8 представляет собой Н или галоген, при условии, что в соединениях, представленных формулой IF, R8 представляет собой галоген; R9 и R10 каждый независимо представляет собой Н, С1-С4-алкил; R11 представляет собой Н, С1-С4-алкил, и когда R11 представляет собой С1-С4-алкил, указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, C1-C4-алкил; R12 представляет собой Н, С1-С4-алкил; n равно 2; его фармацевтически приемлемая соль или сольват. Также описываются фармацевтическая композиция, способ лечения расстройства, вызванного или связанного с недостаточностью серотонина, норадреналина или допамина, способ ингибирования синаптического захвата норадреналина, способ ингибирования синаптического захвата серотонина, способ ингибирования синаптического захвата допамина и набор, включающий, в том числе, соединение по п.1. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 7 н. и 37 з.п. ф-лы, 5 табл.
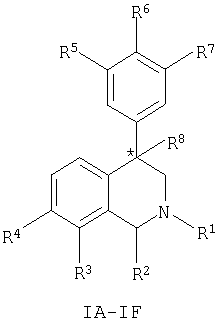
где атом углерода, обозначенный *, имеет R- или S-конфигурацию;
R1 представляет собой C1-С6-алкил;
R2 представляет собой Н, C1-С6-алкил;
R3 представляет собой Н, -S(O)nNR11R12, C1-С6-алкил, либо если R3 представляет собой C1-С6-алкил, указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-С6-алкил, -OR9 и -NR9R10;
при условии, что в соединениях, представленных формулой IA, R3 представляет собой C1-C6-алкил, который необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-С6-алкил, -OR9 и -NR9R10;
R4 представляет собой Н, C1-С6-алкил, и когда R4 представляет собой C1-С6-алкил, то указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-С3-алкил, галоген, -OR9 и -NR9R10;
при условии, что в соединениях, представленных формулой ID, R4 представляет собой C1-С6-алкил, который необязательно замещен;
и где R3 и R4 оба не являются водородом;
каждый R5, R6 и R7 в соединениях, представленных каждой из формул IA, IB, IC, ID, IE и IF, независимо представляет собой Н, галоген, -OR11, -NR11С(O)R12, и C1-С6-алкил, и когда каждый из R5, R6 и R7 представляет собой C1-С6-алкил, то указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: C1-С3-алкил, галоген;
при условии, что в соединениях, представленных формулой IE, по крайней мере, один из R5 или R7 представляет собой фтор, хлор, или метил;
R8 представляет собой Н или галоген, при условии, что в соединениях, представленных формулой IF, R8 представляет собой галоген;
R9 и R10 каждый независимо представляет собой Н, С1-С4-алкил;
R11 представляет собой Н, С1-С4-алкил, и когда R11 представляет собой C1-C4-алкил, указанная группа необязательно замещена 1-3 заместителями, независимо выбранными в каждом случае из следующих: галоген, С1-С4-алкил;
R12 представляет собой Н, С1-С4-алкил;
n равно 2;
его фармацевтически приемлемая соль или сольват.
2,7-диметил-4-фенил-1,2,3,4-тетрагидроизохинолин;
4-(4-метокси)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(4-фтор)фенил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(3-фтор)фенил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дифтор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(4-фтор-3-метил)фенил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор-4-фтор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(4-метил)фенил-1,2,3,4-тетрагидроизохинолин;
2,7-диметил-4-(3-фтор-4-метил)фенил-1,2,3,4-тетрагидроизохинолин;
4-(4-хлор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(4-хлор-3-фтор)фенил-2,7-диметил-1,2,3,4тетрагидроизохинолин;
4-(3,4-дихлор)фенил-2,7-диметил-1,2,3,4тетрагидроизохинолин;
7-этил-2-метил-4-фенил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дифтор)фенил-7-этил-2-метил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-фенил-7-трифторметил-1,2,3,4-тетрагидроизохинолин;
4-фенил-1,2,7-триметил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-4-(4-фтор)фенил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дифтор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3,5-дифтор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-4-(3-фтор)фенил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-4-(4-фтор-3-метил)фенил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор-4-фтор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3,4-дихлор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3 -хлор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(4-хлор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
4-(4-хлор-3-фтор)фенил-2,8-диметил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-4-(4-метокси)фенил-1,2,3,4-тетрагидроизохинолин;
2,8-диметил-4-(4-трифторметил)фенил-1,2,3,4-тетрагидроизохинолин;
2-метил-8-(N-метиламино)метил-4-фенил-1,2,3,4-тетрагидроизохинолин;
8-(гидрокси)метил-2-метил-4-фенил-1,2,3,4-тетрагидроизохинолин;
2-метил-4-фенил-8-сульфонамид-4-фенил-1,2,3,4-тетрагидроизохинолин;
2-метил-8-(N-метил)сульфонамид-4-фенил-1,2,3,4-тетрагидроизохинолин;
4-(3,5-дифтор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3-хлор-5-фтор)фенил-2,7-диметил-1,2,3,4-тетрагидроизохинолин;
4-(3,5-дифтор)фенил-1,2,7-триметил-1,2,3,4-тетрагидроизохинолин;
(2-метил-4-фенил-1,2,3,4-тетрагидро-7-изохинолинил)-N-метилметанамин;
N-метил(2-метил-4-фенил-7-изохинолинил)-N-метилметанамин;
(2-метил-4-фенил-1,2,3,4-тетрагидро-7-изохинолинил)-метанол; и
его фармацевтически приемлемая соль или сольват.
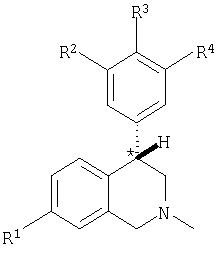
| MARYANOFF В.Е | |||
| ет al | |||
| J | |||
| Med | |||
| Chem | |||
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| УСТРОЙСТВО для БИОПСИИ | 0 |
|
SU400319A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| MASARU KINARA et al | |||
| Chem | |||
| Pharm | |||
| Bull | |||
| Топка с качающимися колосниковыми элементами | 1921 |
|
SU1995A1 |
| US 5654296 A, 05.08.1997 | |||
| ТЕТРАГИДРОИЗОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ СВЯЗЫВАНИЯ ДОПАМИНОВЫХ РЕЦЕПТОРОВ | 1992 |
|
RU2122999C1 |

