Изобретение относится к гетероциклическим соединениям. Главным образом настоящее изобретение относится к новым амидам и сложным эфирам, способам их получения, их применению и фармацевтическим композициям содержащим такие вещества. Новые соединения изобретения находят применение в качестве антагонистов специфических 5-гидрокситриптаминовых (5-НТ) рецепторов.
В ряде описаний известных патентов раскрываются 5-НТ3 антагонисты различных структур, например, в ЕР-А-0200444, [1] СВ-А-2153821 [2] СВ-А-2125398 [3] и ЕР-А-323077 [4]
Новые соединения настоящего изобретения отвечают общей формуле
R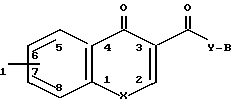 к ним также относятся фармацевтически применимые соли присоединения кислот. В указанной формуле R1 представляет собой атом водорода или один или более заместителей выбранных из низшего алкила, низшей алкокси, атома галогена, метилендиокси или гало(низшего)алкила, Х представляет собой -О- или -NR2, где R2 представляет собой низший алкил, низший алкенил, цикло(низший)алкил, цикло(низший)алкил низший алкил, фенил, необязательно замещенный атомом галогена, фенил (низший)алкил, группу формулы -(CH2)r-Y-R8, где r целое число имеющее значение в интервале 1-4, Y' представляет собой 0, или NR5, где R5 представляет собой атом водорода или низший алкил, а R8 представляет собой атом водорода, низший алкил или цикло(низший)алкил, или группу формулы Z -, которая связана с положением 8 ароматического кольца в результате чего образуется гетероциклическое кольцо из 5-7 элементов, в котором элементы кольца представленные индексом Z представляют собой одну или более метиленовых групп, необязательно замещенных одной или более низшими алкильными группами, Y представляет собой 0 или NR3, где R3 представляет собой атом водорода или низший алкил, а В представляет собой насыщенное азабициклическое кольцо или его N-оксид, причем насыщенное азабициклическое кольцо имеет формулу
к ним также относятся фармацевтически применимые соли присоединения кислот. В указанной формуле R1 представляет собой атом водорода или один или более заместителей выбранных из низшего алкила, низшей алкокси, атома галогена, метилендиокси или гало(низшего)алкила, Х представляет собой -О- или -NR2, где R2 представляет собой низший алкил, низший алкенил, цикло(низший)алкил, цикло(низший)алкил низший алкил, фенил, необязательно замещенный атомом галогена, фенил (низший)алкил, группу формулы -(CH2)r-Y-R8, где r целое число имеющее значение в интервале 1-4, Y' представляет собой 0, или NR5, где R5 представляет собой атом водорода или низший алкил, а R8 представляет собой атом водорода, низший алкил или цикло(низший)алкил, или группу формулы Z -, которая связана с положением 8 ароматического кольца в результате чего образуется гетероциклическое кольцо из 5-7 элементов, в котором элементы кольца представленные индексом Z представляют собой одну или более метиленовых групп, необязательно замещенных одной или более низшими алкильными группами, Y представляет собой 0 или NR3, где R3 представляет собой атом водорода или низший алкил, а В представляет собой насыщенное азабициклическое кольцо или его N-оксид, причем насыщенное азабициклическое кольцо имеет формулу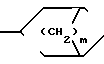 N-R4 в которой m равно 2, 3 или 4, a R4 представляет собой водород или (низший)алкил, или
N-R4 в которой m равно 2, 3 или 4, a R4 представляет собой водород или (низший)алкил, или или их фармацевтически приемлемые аддитивные соли кислоты, или N-оксид гетероциклического соединения или его аддитивная соль кислоты.
или их фармацевтически приемлемые аддитивные соли кислоты, или N-оксид гетероциклического соединения или его аддитивная соль кислоты.
Термин "низший", используемый в тексте относится к радикалу, содержащему до 6 углеродных атомов. Предпочтительно такой радикал содержит до 4 углеродных атомов. Так например, низшая алкильная группа может иметь нормальное или разветвленное строение и может представлять собой метил, этил, пропил или бутил. Предпочтительным примером низшего алкенила является аллил. Низшая алкокси группа может представлять собой, например, метокси, этокси, пропокси или бутокси. Цикло(низший) алкильная группа может представлять собой, например, циклопропил, циклобутил, циклопентил или циклогексил. Арильная группа, предпочтительно, представляет собой фенильную группу, которая может быть необязательно, замещенной одним или более заместителями, имеющими значения указанные выше для R1. В том случае когда R1 представляет собой один или более галогеновых заместителей, такие заместители предпочтительно представляют собой хлор или фтор. Гало(низший)алкильный заместитель, предпочтительно, представляет собой трифторметил. Предпочтительным примером цикло(низший)алкил низшего алкила является циклопропилметил. Арил(низший) алкильная группа, предпочтительно, представляет собой бензил или замещенный бензил, заместители в котором могут иметь значения, указанные для R1.
В том случае когда Х представляет собой NR2 где R2 обозначает -Z-, соединения отвечают формуле
R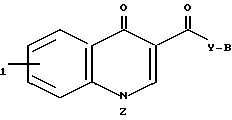
В этой формуле предпочтительные примеры Z включают -(CH2)n где n равно 2 или 3, алкил замещенную ди- или три-метиленовую цепь, например, -СН2СН (низший алки)-, -СН2 С(низший алки)2 или цепь содержащую алкиленовую группе (необязательно замещенную низшим алкилом).
В m предпочтительно равно 2, а R4 представляет собой низший алкил, предпочтительно, метил. Радикал, в котором m равно 2, а R4 метил, известен под названием тропан-3-ил, иначе -8-метил-8-азабицикло(3,2,1)октан-3-ил.
Радикал формулы III известен под названием хинуклидинил, или 1-азабицикло[2,2,2]октан-3-ил.
Соединения изобретения могут содержать один или более асимметричных углеродных атомов в связи с чем они могут существовать в различных стереоизомерных формах. Так например, такие соединения могут существовать в виде рацематов или в оптически активных формах. Оптически активные формы могут быть получены расщеплением рацематов или путем использования оптически активной формы исходного вещества в описанных ниже способах. Кроме этого радикалы формул II и IV могут иметь различные конфигурации соответствующие эндо-конфигурации, как в случае тропина, и экзо-конфигурации, как в случае псевдотропина. Эндо-конфигурация является предпочтительной.
Соединения изобретения могут быть получены известными методами из известных исходных веществ или из исходных материалов, которые могут быть получены традиционными способами. Согласно одному из способов получения амида формулы 1, в которой Y представляет собой NR3, амин формулы VI
NHR3B в которой R3 и В имеют указанные значения, ацилируют кислотой формулы
R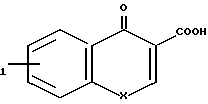 (где R1 и Х имеют указанные значения) или ее ацилирующим производным. Примерами ацилирующих производных могут служить галоидангидриды (например, хлорангидриды), азиды, ангидриды, имидазолиды (например, полученные из карбонилдиимидазола), активированные сложные эфиры или 0-ацил мочевины полученные из такого карбодиимида, как диалкилкарбодиимид, особенно дициклогексилкарбодиимид. Предпочтительно амин ацилируют кислотой в присутствии такого агента сочетания, как дициклогексилкарбодиимид, 1,1'-карбонилдиимидазол, изо-бутилхлорформиат или дифенилфосфинил хлорид.
(где R1 и Х имеют указанные значения) или ее ацилирующим производным. Примерами ацилирующих производных могут служить галоидангидриды (например, хлорангидриды), азиды, ангидриды, имидазолиды (например, полученные из карбонилдиимидазола), активированные сложные эфиры или 0-ацил мочевины полученные из такого карбодиимида, как диалкилкарбодиимид, особенно дициклогексилкарбодиимид. Предпочтительно амин ацилируют кислотой в присутствии такого агента сочетания, как дициклогексилкарбодиимид, 1,1'-карбонилдиимидазол, изо-бутилхлорформиат или дифенилфосфинил хлорид.
Сложный эфир по изобретению в котором Y представляет собой -О- может быть получен этерификацией кислоты формулы VII cпиртом формулы VIII
B-OH (в которой В имеет указанные значения). Этерификацию можно проводить обычными методами, известными в данной области. Так, например, спирт может реагировать с галоидангидридом, например, в присутствии акцептора кислоты.
Кислоты формулы VII являются известными соединениями или они могут быть получены известными методами. Так например, кислота, в которой Х представляет собой -NR2- может быть получена согласно следующей реакционной схеме:
R R
R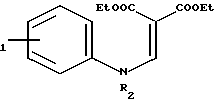
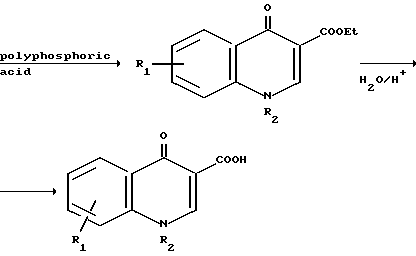
Другой способ получения амидов настоящего изобретения (Y= -NR3-) заключается в циклизации соединения формулы IX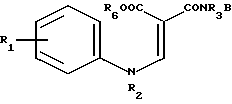 в которой R1, R2, R3 и В имеют указанные значения, R6 представляет собой (низший) алкил, например, этил. Циклизацию можно проводить в присутствии такого циклодегидратирующего агента, как полифосфорная кислота. Исходное вещество формулы IX может быть получено по реакции амина формулы
в которой R1, R2, R3 и В имеют указанные значения, R6 представляет собой (низший) алкил, например, этил. Циклизацию можно проводить в присутствии такого циклодегидратирующего агента, как полифосфорная кислота. Исходное вещество формулы IX может быть получено по реакции амина формулы
R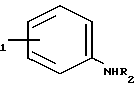 где R1 и R2 имеют указанные значения, с ненасыщенным соединением формулы
где R1 и R2 имеют указанные значения, с ненасыщенным соединением формулы
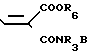 в которой R3, R6 и B имеют указанные значения, а R7 представляет собой (низший)алкил, предпочтительно, этил. Реакцию можно проводить нагреванием реагентов как таковых, или в присутствии подходящего растворителя.
в которой R3, R6 и B имеют указанные значения, а R7 представляет собой (низший)алкил, предпочтительно, этил. Реакцию можно проводить нагреванием реагентов как таковых, или в присутствии подходящего растворителя.
Другой способ получения соединений изобретения, в которых Х представляет собой -NR2-, где R2 представляет собой низший алкил, цикло(низший)алкил, цикло(низший)алкил-низший алкил, арил, арил(низший)алкил или -(СН2)r-Y-R8 заключается в циклизации соединения формулы
R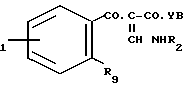 где R1, R2, Y и B имеют указанные значения; R9 представляет собой уходящую группу, например галоген (например, фтор или хлор), либо алкил- или арил-сульфонилокси группу. Такую циклизацию можно осуществлять обработкой таким сильным основанием, как гидрид натрия. Исходное вещество формулы (ХII) может быть получено методами известными для аналогичных соединений.
где R1, R2, Y и B имеют указанные значения; R9 представляет собой уходящую группу, например галоген (например, фтор или хлор), либо алкил- или арил-сульфонилокси группу. Такую циклизацию можно осуществлять обработкой таким сильным основанием, как гидрид натрия. Исходное вещество формулы (ХII) может быть получено методами известными для аналогичных соединений.
Соединения изобретения, в которых Х представляет собой -NR2-, где R2 представляет собой низший алкил, низший алкенил, низший алкинил, цикло(низший)алкил, цикло(низший)алкил-низший алкил, арил, арил(низший)алкил или -(CH2)r-Y-R8 могут быть получены алкилированием соответствующего соединения, в котором Х представляет собой -NH-. Такое алкилирование можно, если необходимо, проводить например по реакции с (низшим) алкилом, низшим алкенилом, низшим алкинилом, цикло(низшим) алкилом, цикло(низшим)алкил низшим алкилом, арилом, арил (низшим) алкилом или -(CH2)r-Y-R8 галогенидом в присутствии основания. Исходное соединение в котором Х представляет собой -NH- может быть получено методом аналогичным описанному выше из соответствующим образом замещенного соединения (VII). Соединения изобретения в которых В представляет собой N-оксид радикалов (II)-(V) могут быть получены окислением соединения, в котором В представляет собой радикалы (II)-(V), например, пероксидом водорода или перкислотой.
Если в любом из указанных способов реагент содержит группы, подтвержденные различным воздействиям в условиях используемых для проведения такой реакции, то такая группа может быть защищена, после чего защитная группа может быть снята.
Если в описанных способах соединение изобретения получают в виде соли присоединения кислоты, то свободное основание может быть получено воздействием на раствор соли присоединения кислоты основанием. Напротив, если продукт реакции представляет собой свободное основание, то соль присоединения кислоты, особенно фармацевтически применимая соль присоединения кислоты, может быть получена растворением свободного основания в подходящем органическом растворителе и обработкой раствора кислотой в соответствии с традиционными методиками получения солей присоединения кислот из оснований.
Примерами солей прикосновения кислот могут служить вещества, полученные из таких неорганических и органических кислот, как серная, хлористоводородная, бромистоводородная, фосфорная, винная, фумаровая, малеиновая, лимонная, уксусная, муравьиная, метансульфокислота, п-толуолсульфокислота, оксолиновая и янтарная кислота.
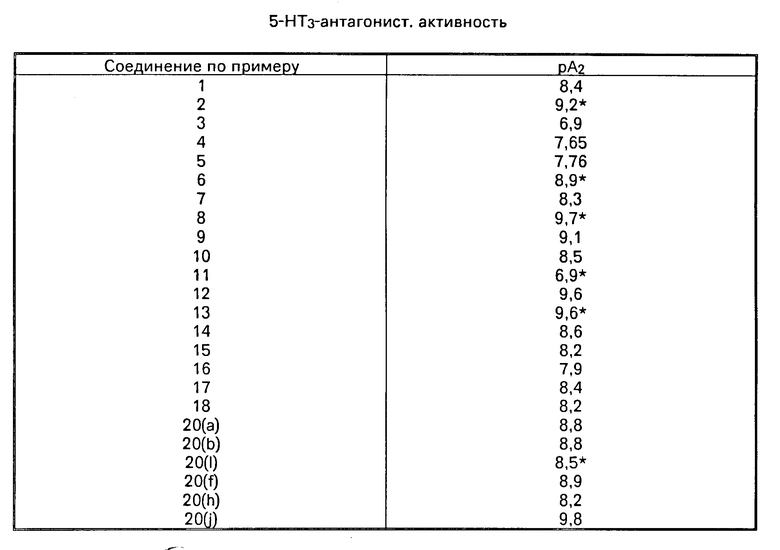
Соединения по изобретению обладают фармакологической активностью. Как правило, они являются антагонистами специфических 5-гидрокситриптаминовых рецепторов у теплокровных животных. Такие соединения обладают 5-НТ3 антагонистической активностью и, следовательно, являются ценными веществами в тех случаях когда желателен антагонизм 5-НТ3 рецепторов. 5-НТ3-антагонисты обозначаются также, как "антагонисты" "нейронных" 5-гидрокситриптаминовых рецепторов" и "серотонин (5-гидрокси-триптамин) М-рецепторные антагонисты".
Соединения изобретения испытывали на 5-НТ3 антагонистическую активность в блуждающем нерве крыс в соответствии со следующей методикой.
Такой метод аналогичен описанному Айрелэндом и Тирсом в Br.J.Pharmac, 1987, 90, 229-238 и зависит от способности 5-НТ деполяризовать блуждающий нерв in vitro.
Сегменты блуждающего нерва крыс разновидности Sprague-Dawley, помещали в перспексовую камеру и заливали раствором Кребса. Электроды помещенные на каждом конце сегмента нерва использовали для записи разности потенциалов, которая возникла в ходе добавления различных концентраций 5-НТ к одному из концов сегмента нерва. Этим методом получали зависимости концентрация реакция на действие 5-НТ до и после уравновешивания сегмента нерва раствором Кребса, содержащим испытуемое вещество. На базе этих результатов осуществляли анализ Шилда с целью получения меры мощности антагониста, выраженной величиной рА2. Результаты испытаний приведены в таблице.
Далее изобретение предусматривает использование соединения формулы I, или его фармацевтически применимой соли присоединения кислоты в качестве антагониста 5-НТ3 рецепторов у млекопитающих.
5-НТ3 антагонисты могут использовать в лечении нейропсихиатрических расстройств таких как беспокойство, психические расстройства (например, шизофрения), зависимость от лекарств или других веществ при злоупотреблении ими, нарушения сознания; в течении таких желудочно-кишечных нарушений, как рвота и тошнота и при лечении мигрени. Изобретение предусматривает использование соединения изобретения в одном или более указанных лечениях. Изобретение предусматривает также способ одного или более из указанных лечений, который состоит в применении на теплокровном животном эффективного количества соединения изобретения.
В случае некоторых из указанных состояний совершенно ясно, что соединения изобретения могут использоваться профилактически, а также для облегчения острых симптомов. Следует иметь в виду, что используемый в тексте термин "лечение" или аналогичный термин включают как профилактику, так и лечение острых состояний.
Противорвотные свойства таких соединений особенно полезны при лечении тошноты и рвоты связанных с противораковыми земотерапевтическими агентами и радиационной терапией. В связи с этим соединения изобретения используются при лечении рака хемотерапевтическими агентами (такие цитотоксические или цитостатические агенты, как цисплатин, доксорубицин и циклофосфамид), а также при облучении. В соответствии с этим, настоящее изобретение также предусматривает продукт, содержащий противораковый хемотерапевтический агент и соединение изобретения в виде объединенного препарата для одновременного, раздельного или последовательного использования в раковой терапии.
Согласно еще одному аспекту изобретение предусматривает фармацевтическую композицию, включающую соединение изобретения совместно с фармацевтически применимым носителем. Для получения такой фармацевтической композиции может использоваться любой подходящий носитель, известный в данной области. В такой композиции носитель обычно представляет собой твердое или жидкое вещество или смесь твердого вещества и жидкости.
Твердые формы таких композиций включают порошки, гранулы, таблетки, капсулы (например, твердые и мягкие желатиновые капсулы), свечи и маточные кольца. Твердый носитель может представлять собой одно или более веществ, действующих также в качестве отдушек, смазочных агентов, солюбилизаторов, суспендирующих агентов, наполнителей, агентов способствующих скольжению, компрессионных средств, связующих или разрушающих таблетки агентов; он может также представлять собой материал капсулы. В порошках носитель представляет собой тонкоизмельченное твердое вещество, находящееся в смеси с тонкоизмельченным активным ингредиентом. В таблетках активный ингредиент смешивают с носителем, обладающим необходимыми компрессионными свойствами и надлежащей пропорции, и формируют продукт желаемой формы и размера. Такие порошки и таблетки, предпочтительно, содержат до 99% например, 0,03-99% предпочтительно, 1-80% активного ингредиента. Подходящие твердые носители включают, например, фосфат кальция, стеарат магния, тальк, сахара, лактозу, декстрин, крахмал желатину, целлюлозу, метил целлюлозу, натрий карбоксиметил целлюлозу поливинилпирролидон, низко-плавкие воски и ионообменные смолы.
Под термином "композиция" подразумевается рецептура активного ингредиента в капсулу, в которой активный ингредиент (в присутствии или отсутствии носителей) окружен носителем, который таким образом связан с ним. Аналогичным образом готовят саше.
Жидкие формы композиций включают, например, растворы, суспензии, эмульсии, сиропы, эликсиры и композиции под давлением. Активные ингредиенты могут быть, например, растворены или суспендированы в таком фармацевтически применимом жидком носителе, как вода, органический растворитель, их смеси или фармацевтически применимые масла или жиры. Жидкий носитель может содержать другие фармацевтические присадки, такие как солюбилизаторы, эмульгаторы, буффера, предохраняющие агенты, подслащивающие агенты, отдушки, суспендирующие агенты, загустители, окрашивающие агенты, регуляторы вязкости, стабилизаторы или осморегуляторы. Подходящими примерами жидких носителей для орального и парентерального применения могут служить вода (особенно содержащая указанные добавки, например, производные целлюлозы, предпочтительно, раствор натрий карбоксиметилцеллюлозы) спирты (включая одноатомные спирты и многоатомные спирты, например глицерин и гликоли), а также их производные, и масла (например, фракционированное кокосовое и арахисовое масло). В случае парентерального применения носитель может также представлять собой маслянистый сложный эфир, например, этилолеат и изопропил миристат. Стерильные жидкие носители используются в стерильных жидких композициях для парентерального применения.
Жидкие фармацевтические композиции, которые являются стерильными растворами или суспензиями могут применяться, например, внутримышечно, внутрибрюшинно или подкожно. Стерильные растворы могут также применяться внутривенно. В случае когда соединение орально активно оно может применяться орально в жидкой или твердой композиционной форме.
Соединения по изобретению могут также применяться путем закапывания в нос. При формировании для носового применения такие композиции могут включать соединение изобретения в жидком носителе; такие композиции могут применяться, например, в виде спрэя или капель. В качестве жидкого носителя может использоваться вода (которая может содержать дополнительные компоненты обеспечивающие желательную изотоничность и вязкость композиции). Композиция может также содержать такие дополнительные эксипиенты, как предохраняющие агенты, поверхностно-активные агенты, и т.п. Такие композиции могут содержаться в устройстве для носового применения, которое позволяет применять такую композицию в виде капель или спрэя. При применении из аэрозольного резервуара композиция должна также содержать пропеллант.
Фармацевтические композиции для лечения и/или предотвращения тошноты или рвоты могут содержать помимо соединения изобретения цикло-оксигеназный ингибитор. Примерами цикло-оксигеназных ингибиторов могут служить системные NSAID, например, индометацин, пироксикам.
Предпочтительно, чтобы фармацевтическая композиция была в виде единичной дозировки, например в виде таблеток или капсул. В такой форме композицию делят на единичные дозы, включающие соответствующие количества активного ингредиента. Единичные дозировочные формы могут представлять собой упакованную композицию, например порошки в пакетах, ампулы, предварительно заполненные шприцы или саше, содержащие жидкости. Единичная дозировочная форма может представлять собой, например, капсулу или таблетку как таковые, или она может представлять собой соответствующее число любых таких композиций в упакованной форме.
Количество активного ингредиента в единичной дозе композиции может изменяться от 0,5 мг до менее 750 мг или более, в соответствии с конкретной необходимостью и активностью активного ингредиента.
Изобретение также охватывает соединения в отсутствии носителя когда сами такие соединения представляют собой единичную дозировочную форму.
П р и м е р 1. (Эндо)-N'-(8-метил-8-азабицикло[3.2.1]октан-3-ил)-1,4-дигидро-1-ме- тил-4- оксохинолин-3-карбоксамид.
Суспензию 1,4-дигидро-1-метил-4-оксохинолин-3-карбоновой кислоты (1,02 г, 5 ммоля) и карбонилдиимидазола (0,85 г, 5 ммоля) в диметилформамиде (15 мл) перемешивали и нагревали в течение 0,5 ч при 80оС с получением прозрачного раствора. (Эндо)-3-аминотропан дигидрохлорид (1,06 г, 5 ммоля) добавляли в систему после чего добавляли триэтиламин (1,3 г) и реакционную смесь перемешивали при 80оС еще в течение 2 ч. Затем смесь охлаждали, разбавляли водой (25 мл) и осажденный продукт (1,2 г) собирали и перекристаллизовывали из воды (150 мл) с получением 0,7 г целевого основания. Это основание растворяли в этаноле (мл) и подкисляли эфирным раствором хлористоводородной кислоты с осаждением целевого соединения в виде гидрохлорида (0,55е г), т. пл. 300оС.
П р и м е р 2. (Эндо)-11-(8-метил-азабицикло[3,2,1]октан-3-ил)-1,4-дигидро-1-бу- тил- 4- оксохинолин-3-карбоксамид.
Смесь 1-бутил-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (0,98 г, 4 ммоля), карбонилдимидазола (0,7 г, 4,4 ммоля) и диметилформамида (12 мл) перемешивали в течение 1,5 ч при 80оС. Затем добавляли (Эндо)-3-аминотропан (0,56 г, 4 ммоля) и перемешивание продолжали еще в течение 1,5 ч при той же температуре. Растворитель удаляли и остаток разбавляли водой со льдом (15 г). Осажденное твердое вещество собирали, промывали охлажденной льдом водой и сушили на воздухе. Основание растворяли в горячей смеси воды (15 мл) и этанола (3 мл), затем охлаждали льдом и подщелачивали до рН 11 путем добавления концентрированного водного раствора аммиака с целью осаждения кристаллического продукта, который собирали и промывали холодным разбавленным раствором аммиака. Затем очищенное основание (0,82 г) растворяли в этаноле (8 мл), подкисляли этанольным раствором HCl и разбавляли эфиром (3 мл).
В результате охлаждения получали продукт в виде гидрохлорида (0,49 г) т. пл. 267-268оС.
П р и м е р 3. (Эндо)-N-(8-метил-8-азабицикло[3.2.1]-октан-3-ил)-1-бензил-1,4-диги- дро-4- оксохинолин-3-карбоксамид.
1-Бензил-1,4-дигидро-4-оксохинолин-3- карбоновую кислоту (1,96 г, 7,03 ммоля) в сухом ДМФ (20 мл) обрабатывали карбонилдиимидазолом (1,14 г, 7,04 ммоля) при комнатной температуре и смесь в течение 3 часов нагревали при 80оС. Добавляли (эндо)-3-аминотропан (0,99 г, 7,0 ммоля) и нагревание продолжали в течение ночи (19 ч) с получением суспензии. Полученную смесь разбавляли водой (40 мл) и рН устанавливали равным 9-10 путем добавления концентрированного водного ртаствора карбоната калия. Твердое вещество собирали, промывали водой, сушили и перекристаллизовывали из этанола (20 мл) и воды (20 мл) с получением свободного основания (1,72 г). Это вещество растворяли в кипящем этаноле (15 мл) и раствор подкисляли этанольным раствором хлористого водорода. Полученный в результате осадок собирали, промывали этанолом и сушили при 80оС в вакууме с получением целевого соединения в виде гидрохлорида, гидрата, 1,3 этанолата (1,91 г) т.пл. 296-297оС.
П р и м е р 4. (Эндо)-N-(8-аза-8-метилбицикло[3.2.1]октан-3-ил)хромон-3-карбокса- мид.
а) Смесь хромон-3-карбоновой кислоты (1,25 г) и тионил хлорида (6 мл) нагревали с обратным холодильником в течение 5 мин. Затем реакционную смесь разбавляли циклогексаном (15 мл) и охлаждали льдом. Кристаллический остаток собирали фильтрацией, промывали циклогексаном и сушили в вакууме с образованием хромон-3-карбонил хлорида (1,2 г).
(б) Раствор хроном-3-карбонил хлорида (1,04 г, 5 ммоля) в СН2Сl2(20 мл) прикапывали в течение 5 мин с охлажденной льдом перемешиваемой смеси (эндо)-3-аминотропана (0,7 г, 5 ммоля), безводного К2СО3 (3 г) и СН2Cl2 (20 мл). После завершения добавления перемешивание продолжали в течение 0,5 ч и смесь разбавляли водой (50 мл). Органическую фазу отделяли, сушили (Na2SO4) и выпаривали с образованием твердого вещества (1,7 г). Основание растворяли в этаноле (15 мл) и подкисляли этанольным раствором HCl с целью осаждения сырого гидрохлорида (0,9 г). В результате трехкратной перекристаллизации из этанола получили целевое соединение в виде чистого гидрохлорида (0,3 г), т. пл. 300оС.
П р и м е р 5. (Эндо)-N-(9-метил-азабицикло[3,3,1]нона-3-ил)-1,4-дигидро-1-метил-4- оксохинолин-3-карбоксамид.
Суспензию 1,4-дигидро-1-метил-оксохинолин-3-карбоновой кислоты (1,02 г, 5 ммоля) и карбонилдиимидазола (0,85 г, 5 ммолей) в ДМФ (15 мл) перемешивали и нагревали в течение 3 ч при 85оС с получением прозрачного раствора. Добавляли (эндо)-3-аминогомотропан дигидрохлорид (1,13 г, 5 ммолей) и диизопропилэтиламин (1,29 г, 10 ммолей) и нагревание продолжали в течение ночи (19 ч). Раствор охлаждали до комнатной температуры и разбавляли водой (25 мл). рН устанавливали равным 10-11 путем добавления небольшого количества водного раствора гидроксида калия. Осажденное твердое вещество собирали, промывали водой и сушили с получением целевого основания (1,25 г), которое трижды перекристаллизовывали из смесей вода/этанол. Основание растворяли в горячем этаноле (12 мл) и подкисляли этанольным раствором хлористого водорода с получением целевого соединения в виде гидроьхлорида, 1,25 гидрата (0,92 г), т. пл. 278-81oС (разл.).
П р и м е р 6. (Эндо-N-(8-метил-8-азабицикло[3.2.1]октан-3-ил)-1,8-этанол-1,4-ди- гидро-4-оксохинолин-3-карбоксамид.
Суспензию 1,8-этано-1,4-1,4-дигидро-4-оксохинолин-3-карбоновой кислоты (1,08 г, 5 ммолей) и карбонилдиимидазола (0,89 г, 5,5 ммолей) в диметилформамиде (15 мл) перемешивали и нагревали в течение 1,25 ч при 80оС с получением прозрачного раствора. (Эндо)-3-аминотропан (0,7 г, 5 ммолей) добавляли в систему одной порцией и реакционную смесь перемешивали в течение 2 ч при 80оС. Реакционную смесь охлаждали льдом и разбавляли водой (25 мл) и осажденный продукт собирали и дважды перекристаллизовывали из смеси: вода (2: 1) с получением 0,7 г целевого основания. Основание растворяли в горячем этаноле (15 мл) и подкисляли этанольным раствором хлористого водорода с образованием целевого соединения в виде гидрохлорида (0,55 г), т.пл. 300оС.
П р и м е р 7. (Эндо)-N-(8-аза-8-метилбицикло[3,2,1]октан-3-ил)-1,4-дигидро-1,8- пропанхинолин- 4-он-3-карбоксамид.
Целевое соединение получали по методике примера 6, заменяя 1,8-этано-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту на 1,8-пропано-1,4-дигидро-4-оксохинолин-3-карбоновую кислоту. Продукт реакции получали в виде гидрохлорида, т.пл. 300оС.
П р и м е р 8. (Эндо)-N-(8-метил-8-азабицикло[3.2.1]октан-3-ил)-1,4-дигидро-4-оксо- 1-н- пропилхинолин-3-карбоксамид.
1,4-Дигидро-4-оксо-1-н-пропилхинолин -3-карбоновую кислоту 1,58 г, 6,82 ммоля) и триэтиламин (0,7 г, 7 ммоля) растворяли в дихлорметане (20 мл) в атмосфере аргона. Сразу же, при перемешивании добавляли хлорид дифенилфосфина (1,6 г, 6,76 ммоля). Полученный раствор оставляли на 6 ч и затем добавляли (эндо)-3-аминотропан (1,0 г, 7,14 ммоля) и триэтиламин (0,7 г, 7 ммолей). Полученный раствор оставляли на 3 дня и затем выпаривали. Остаток растворяли в воде и подкисляли концентрированной хлористоводородной кислотой. Остаток отфильтровывали, промывали водой и выбрасывали. Фильтрат подщелачивали карбонатом натрия и выпаривали. Остаток дважды обрабатывали этилацетатом, этилацетат выпаривали и остаток обрабатывали водой (6 мл) и концентрированным аммиаком (1 мл) с получением белого твердого вещества (1,34 г). Этот материал (3,68 ммоля) растворяли в горячем этаноле (15 мл) и добавляли горячий дигидрат оксалиновой кислотой (0,47 г, 3,73 ммоля). Полученный в результате раствор хранили в течение ночи в холодильнике. Осадок собирали, промывали этанолом и сушили с образованием целевого соединения в виде полугидрата оксалата (1,24 г), т.пл. 227-231оС.
П р и м е р ы 9-14. Следуя методике примера 1, но заменяя 1,4-дигидро-1-метил-4-оксохинолин-3-карбоновую кислоту на следующие реагенты, получали следующие ниже продукты.
П р и м е р 9. Реагент: 1,4-дигидро-1-этил-4-оксохинолин-3-карбоновая кислота.
Продукт: (эндо)-N-(8-метил-8-азабицикло[3,2,1]октан-3-ил)-1,4-дигидро-1-этил-4- оксохинолин-3-карбоксамид, гидрохлорид, полугидрат, т.пл. 298-302оС.
П р и м е р 10. Реагент: 1,4-дигидро-1-(2-метоксиэтил)-4-оксохинолин-3-карбоно- вая кислота.
Продукт: (эндо)-N-(8-метил-8-азабицикло[3,2,1]октан-3-ил)-1,4-дигидро-1-2-меток- сиэтил)-4-оксохинолин-3-карбоксамид, 1: 1 фумарат, т.пл. 243-245оС.
П р и м е р 11. Реагент: 9-фтор-6,7-дигидро-5-метил-1-оксо-1Н, 5Н-бензо(ij)хинолизин-2-карбоновая кислота.
Продукт: (эндо)-N-(8-метил-8-азабицикло[3,2,1]октан-3-ил)-9-фтор-6,7-дигид- ро- 5-метил-1-оксо-1Н, 5Н-бензо(ij)хинолизин-2-карбоксамид, гидрохлорид, 1/4 гидрат, т.пл. 320оС.
П р и м е р 12. Реагент: 1-циклогексил-1,4-дигидро-4-оксохинолин-3-карбоновая кислота.
Продукт: (эндо)-N-8-метил-8-азабицикло[3,2,1] октан-3-ил)-1-циклогексил-1,4- дигидро-4-оксохинолин-3-карбоксамид, 1:1 оксалат, 1,5 гидрат, т.пл. 217оС.
П р и м е р 13. Реагент: 1-(циклопропилметил)-1,4-дигидро-4-оксохинолин-3-карбо- новая кислота.
Продукт: (эндо)-N-(8-метил-8-азабицикло[3,2,1] октан-3-ил)-1- циклопропилметил-1,4-дигидро-4-оксохинолин-3-карбоксамид, гидрохлорид, 0,75 гидрат, т.пл. 164-166оС (разл.).
14. Реагент: 1-(4-фторфенил)-1,4-дигидро-4-оксахинолин-3-карбоновая кислота.
Продукт: (эндо)N-(8-метил-8-азабицикло[3,2,1]октан-3-ил)-1-(4-фторфенил)-1,4- дигидро-4-оксохинолин-3-карбоксамид, гидрохлорид, т.пл. 240оС (разл. ).
П р и м е р 15. Следуя методике примера 1, но заменяя (эндо)-3-аминотропан на 1-азабицикло[2.2.2] октан-3-амин (3-аминохинукледин) получали N-(1-азабицикло[2,2,2] октан-3-ил)-1,4-дигидро-1-метил-4-оксохинолин-3-карбок самигидрохлорид, гидрат, т.пл. 179-181оС.
П р и м е р 16. (Эндо)-N-(9-метил-9-азабицикло[3,3,1]нонан-3-ил)-1,4-дигидро-1- этил-4- оксохинолин-3-карбоксамид.
Суспензию 1,4-дигидро-1-этил-4-оксохинолин-3-карбоновой кислоты (1,52 г, 7 ммолей) и триэтиламина (0,7 г, 7 ммолей) в дихлорметане (20 мл) перемешивали при комнатной температуре в атмосфере аргона в течение 1 ч. Добавляли изобутил хлорформиат (0,96 г, 7,03 ммоля и смесь перемешивали в течение часа. Добавляли триэтиламин (1,4 г, 14 ммолей) и (эндо)-3-амино-9-метил-9-азабицикло[3.3.1] нонан дигидрохлорид (1,58 г, 6,96 ммоля). Через 3 дня реакционную смесь охлаждали метанолом и растворители выпаривали. Остаток обрабатывали водой (10 мл) и концентрированным аммиаком (2 мл), твердое вещество собирали, промывали концентрированным раствором аммиака и сушили. Это вещество превращали в его соль фумаровой кислоты в соотношении 1:1 в системе IРА: метанол (2:1, 15 мл) с получением целевого соединения 1:1 фумарата, полугидрата (73,1%), т.пл. 184-185оС.
П р и м е р 17. (Эндо)-N-(9-метил-9-азабицикло[3,3,1]нонан-3-ил)-1-бутил-1,4-ди- гидро- 4-оксо-хинолин-3-карбоксамид.
1,4-Дигидро-1-бутил-4-оксохинолин-3-карбоновая кислота реагировала с (эндо)-3-амино-9-метил-9-азабицикло[3.3.1] нонаном согласно методике примера 16 и целевое соединение получили в виде малеата 1:1, т.пл. 203-205оС.
П р и м е р 18. (Эндо)-N-(8-метил-8-азабицикло[3,2,1]октан-3- ил-1-этил-6-фтор-1,4-дигидро-4-оксохинолин-3-карбоксамид.
Проводили реакцию между 1,4-дигидро-1-этил-6-фтор-4-оксохинолин-3-карбоновой кислотой с (эндо)3-аминотропаном согласно методике примера 16 и целевое соединение получали в виде гидрохлорида, 0,75 гидрата, т.пл. 315-317оС.
П р и м е р 19. Следуя методикам приведенным выше и с использованием соответствующих реагенетов получали: (эндо)-N-(8-метил-8-азабицикло[3,2,1] октан 3-ил)-1,4-дигидро-1-циклопропил-4-оксохи- нолин-3-карбоксамид и соответствующие 1-циклобутил, 1-циклопентил, 1-трет-бутил и 1-(бут-3-енил)аналоги; (эндо)-N-(8-метил-8-азабицикло[3,2,1]октан-3-ил)-1,4-дигидро-1- этил-6,7-метилендиокси-4-оксихинолин-3-карбоксамид; (эндо)-N-(8- метил-8-азабицикло[3,2,1] октан-3-ил)-1,4-дигидро-1-этил- 7-фтор-4-оксохинолин-3+карбоксаид и аналоги, в которых 7-фтор заместитель заменен на 7-трифторметил, 8-фтор; 6,7-дифтор и 6-хлор-8-метил.
П р и м е р 20. Используя способ примера 16, получают следующие соединения:
а) (эндо)-N-(8-метил-8-азабицикло[3,2,1]октан-3-ил)-1,4-дигидро-1-этил- 8-фтор-4-оксохинолин-3-карбоксамид, гидрохлорид, четвертьгидрат, т. пл. 296-300оС (разл.).
(b) (эндо)-N-(8-метил-8-азабицикло[3,2,1]октан-3-ил)-1-(4-бутенил)-1,4- дигидро-4-оксохинолин-3-карбоксамид, бумарат, 0,75е гидрат, т.пл. 213-216оС.
с) (R)-(-)-N-) 1-азабицикло[2,2,2]октан-3-ил)-1-циклогексил-1,4-дигидро-4-оксохино- лин-3-карбоксамид, 1:1 оксалат, 0,75 гидрат, т.пл. 173-177оС.
(d) (S)-(+)-N-(1-азабицикло[2,2,2]октан-3-ил)-1-циклогексил-1,4-дигидро 4-оксохинолин-3-карбоксамид, 1:1 оксалат, 1,25 гидрат, т.пл. 179-183оС.
(е) (эндо)-N-(8-метил-8-азабицикло[3,2,1] октан-3-ил)циклопропил- 1,4-дигидро-4-оксохинолин-3-карбоксамид, 1: 1 фумарат, 0,75 гидрат, т.пл. 201-3оС.
(f) (эндо)-N-(8-метил-8-азабицикло[3,2,1] октан-3-ил)-1-циклобутил-1,4- дигидро-4-оксохинолин-3-карбоксамид, фумарат, 1,25 гидрат, т. пл. 232оС (разл.).
(g) (эндо)-N-(8-метил-8-азабицикло[3,2,1] октан-3-ил)-1,4-дигидро- 1-этил-4-оксо-7-трифторметилхинолин-3-карбокса- мид, фумарат, 0,25 гидрат, т. пл. 225-227оС.
(h) (эндо)-N-(8-метил-8-азабицикло[3,2,1] октан-3-ил(1-)1,1-диметилэтил)-1,4- дигидро-4-оксохинолин-3-карбоксамид, 1:1 малеат, т.пл. 226-229оС (разл.).
(i) (эндо)-N-(8-метил-8-азабицикло[3,2,1] октан-3-ил)-1,4-дигидро- 1-этил-6,7-метилендиокси-4-оксохинолин-4-карбокса- мид, 1:1 малеат, т.пл. 240оС (разл.).
(j) (эндо)-N-(8-метил-8-азабицикло[3,2,1]октан-3-ил-1-циклопентил-1,4-ди- гидро- 4-оксохинолин-3-карбоксамид, 1:1 малеат, т.пл. 181-184оС.
П р и м е р 21. Эндо-N-(8-этил-8-азабицикло[3,2,1]октан-3-ил)-1-циклогексил-1,4-дигидро- 4-оксохинолин-3-карбоксамид.
N-Метилморфолин (0,4 мл, 3,64 ммоль) добавляли к раствору 1-циклогексил-1,4-дигидро-4-оксахинолин-3-карбоновой кислоты (0,80 г, 2,95 ммоль) в безводном ТГФ (25 мл в атмосфере аргона. Раствор охлаждался до -15оС в течение 0,25 ч, прежде чем был добавлен холороформат изобутила (0,4 млл, 3,08 ммоль). К полученному раствору добавляли раствор дигидрохлорида эндо-N-(8-этил-8-азабицикло[3,2,1] октан-3-ил-)амина (0,71 г, 2,69 ммоль), N-метил морфолин (0,4 мл, 3,64 ммоль), безводный ДМФ (5 мл) и безводный ТГФ (10 мл) при -10оС. После перемешивания в течение 20 ч добавляли раствор триэтиламина (1,0 млл, 7,2 ммоль) в хлороформе (10 мл) и смесь перемешивалась при комнатной температуре в течение 25 ч. В эту реактивную смесь добавлялся хлороформ и разбавленный раствор NaOH. Органический слой удаляли, и водный слой промывали хлороформом. Объединенные органические экстракты промывали рассолом, высушивали (безводный Na2So4) и выпаривали в вакууме до получения прозрачной жидкости, из которой выделили соединение, указанное в заголовке, в виде основания (0,74 г). Основание растворяли в этанольном растворе хлористого водорода до выпадения в осадок целевого соединения в виде гидрохлорида, 11/4 гидрата, т.п. > 250оС (разл. свыше 202оС).
П р и м е р 22. (Эндо)-N-(8-азабицикло[3,2,1.октан-3-ил-1-циклогексан- 1,4-дигидро-4-оксохинолин-3-карбоксамид.
(Эндо)N-8-метил-8-азабицикло[3,2,1]ок- тан-3-ил)-1-циклогексил-1,4-дигидро-4- оксохинолин-3-карбоксамид (1,0 г, 2,54 ммоль) высушивали и суспендировали в 1,2-дихлорэтане (30 мл), в атмосфере аргона, и охлаждали льдом. Добавляли хлороформат 1-хлорэтила (0,28 мл, 0,37 г, 2,59 ммоль) и оставляли для согревания в течение 1 ч до комнатной температуры. Затем раствор кипятили с обратным холодильником в течение 1 ч и растворитель выпаривали. Остаток растворяли в метаноле и кипятили с обратным холодильником сутки (24 ч). Реакционную смесь выпаривали, остаток хроматографировали на основной окисиаммония (активность II-III), элюировали смесью хлороформ: метанол (10:0,1 ->> 0,4). Общий выделенный продукт (1,54 г, 4,06 ммоль) растворяли в горячем этаноле (10 мл) и добавляли фумаровую кислоту (0,45 г, 3,88 ммоль). Раствор фильтровали, охлаждали и рекристаллизировали из этанола (20 мл) и небольшого количества воды для получения (эндо)-N-(8-азабицикло[3,2,1]октан-3-ил)-1-циклогексил-1,4-дигидро-4-оксохин олин1:1 фумарат, полугидрат в виде белого твердого вещества (0,77 г), т.п. 237-239оС.
П р и м е р 23. (Эндо, антил)-и (эндо, син)-N-(8-метил-8-азабицикло[3.2.1] октан-3-ил-1-циклогексил-1,4- дигидро-4-оксохинолин-3-карбоксамид N-оксид.
(Эндо)-N-(8-метил-8-азабицикло[3,2,1] октан-3-ил)-1-циклогексил-1,4- дигидро-4-оксохинолин-3-карбоксамид (7,93 г, 10 ммоль) в метаноле (10 мл) обрабатывали 27,5 мас/мас. водной перекиси водорода (3,2 г). Раствор разбавляли водой (30 мл) и метанол выпаривали. Добавили шлам платина (углеродного катализатора с водой и смесь фильтровали. Фильтрат выпаривали досуха, остаток выпаривали со смесью толуол: этанол 4:1, получая твердый остаток (2,48 г), содержащий смесь анти и син-N-оксидов. Твердый остаток растворяли в горячем этилацетате (25 мл) и метаноле (1,5 мл) и кипятили до выпадения осадка, затем охлаждали. Твердое вещество собирали, промывали этилацетатом и высушивали. Получали (эндо, анти)-N-(8-метил-8-азабицикло[3,2,1] октан-3-ил)-1-циклогексил-1,4-дигидро- 4-оксохинолин-3-карбоксамид N-оксид, дигидрат в виде белого твердого вещества (0,91 г), т.п. 174-177оС. Порцию (250 мг) вторично собранного вещества, содержащую смесь син- и анти N-оксидов, разделяли посредством центрифуговой хроматографии на силикагеле (толщ. 2 мм) и элюировали смесью хлороформ: метанол: 0,888 г аммиака (10:1: 0,1). Син-N-оксид получался в виде стекломассы индуцировали для кристаллизации с помощью расстирания до порошка с холодным эфиром, а затем горячим этилацетатом. Твердое вещество собирали и высушивали в вакууме при 75оС в течение суток. Получали (эндо, син)-N-(8-метил-8-азабицикло[3,2,1]октан-3-ил)-1-цик- логексил-1,4- дигидро-4-оксохинолин-3-карбоксамид N-оксид, 0,9 гидрохлорид, в виде белого твердого вещества (50 мг) т.пл. 223-232оС
Использование: в медицине в качестве антагонистов специфических 5-гидрокситриптаминовых (5-НТ)рецепторов для лечения нейропсихиатрических устройств. Сущность изобретения: продукт-гетероциклические соединения ф-лы I. R1 X, Y, и B имеют соответствующие значения, или их фармацевтические приемлемые аддитивные соли кислоты, или n-оксид гетероциклического соединения, или аддитивная соль кислоты. Эти соединения используют в фармацевтических композициях в количестве от 0,5 750 мг на дозу. Структура соединения ф-лы I: 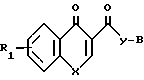
Гетероциклические соединения общей формулы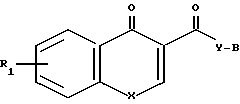
где R1 водород или один или два заместителя, выбранных из низшего алкила, атома галогена, низшей алкокси-, метилендиоксигруппы или гало (низшего) алкила,
X -O- или NR2, где R2 низший алкил, низший алкенил, цикло (низший)алкил, цикло(низший)алкил-низший алкил, фенил, необязательно замещенный атомом галогена, фенил(низший)алкил, группа общей формулы -(CH2)2 Y'-R8, где r=1-4 целое число, Y' О или R5, где R5 водород или низший алкил, R8 водород, низший алкил или цикло(низший)алкил, или X группа формулы -Z- которая связана с положением 8 ароматического кольца с образованием гетероциклического кольца из 5-7 кольцевых элементов, в котором кольцевые элементы, обозначенные индексом Z, представляют собой одну или более метиленовых групп, необязательно замещенные одной или более низшими алкильными группами, Y -О- или NR3, где R3 водород или низший алкил;
B насыщенное азабициклическое кольцо общей формулы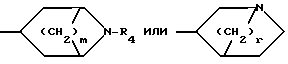
где m- 2,3 или 4,
R4 водород или низший алкил,
или их фармацевтически приемлемые аддитивные соли кислоты, или N-оксид гетероциклического соединения, или его аддитивная соль кислоты.
| Бесконтактный электропривод постоянного тока | 1969 |
|
SU323077A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Механизм для сообщения поршню рабочего цилиндра возвратно-поступательного движения | 1918 |
|
SU1989A1 |
