Изобретение относится к новым соединениям, способу их получения, их использованию и фармацевтическим композициям, содержащим новые соединения. Новые соединения можно использовать в терапии, особенно для лечения боли.
Предпосылки создания изобретения и известный уровень техники
δ-Рецептор был идентифицирован как играющий роль во многих функциях организма, таких как сердечно-сосудистая и болевая системы. Лиганды для δ-рецептора могут, следовательно, найти потенциальное использование в качестве анальгезирующих и/или антигипертензивных средств. Было также обнаружено, что лиганды для δ-рецептора обладают иммуномодулирующей активностью.
В настоящее время идентифицированы, по меньшей мере, три разные популяции опиоидных рецепторов (μ, δ и χ), и все три обнаруживаются как в центральной, так и периферической нервных системах многих видов животных, включая человека. Анальгезию наблюдали на различных моделях животных, когда были активированы один или несколько из этих рецепторов.
За редким исключением, доступные в настоящее время селективные опиоидные δ-лиганды являются по природе пептидными соединениями и не пригодны для введения системными способами. Некоторые непептидные δ-антагонисты стали доступны в течение некоторого последнего времени (см. Takemori and Portoghese, 1992, Ann. Rev. Pharmacol. Tox. , 32:239-269.). Эти соединения, например налтриндол, характеризуются весьма слабой (т.е. слабее в 10 раз) селективностью связывания δ-рецептора по сравнению с μ-рецептором и не проявляют анальгезирующую активность, факт, который подразумевает необходимость разработки высокоселективных непептидных δ-лигандов.
Таким образом, проблемой, лежащей в основе данного изобретения, было получение новых анальгетиков, имеющих повышенное анальгезирующее действие, а также улучшенный профиль побочного действия по сравнению с современными μ-агонистами и значительную пероральную эффективность.
Анальгетики, которые были идентифицированы и описаны в известном уровне техники, имеют много недостатков, проявляющихся в том, что они имеют плохую фармакокинетику и не проявляют анальгезирующее действие при введении системными способами. Кроме того, документально подтверждено, что предпочтительные соединения, описанные в известном уровне техники, проявляют также значительное судорожное действие при системном введении.
Проблема, указанная выше, решена путем разработки новых соединений, которые имеют кольцо пиперидина с экзоциклической двойной связью, как будет описано ниже.
Описание изобретения
Новые соединения по настоящему изобретению имеют общую формулу (I)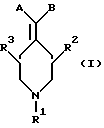
где R1 выбирают из водорода, разветвленного или неразветвленного C1-С6-алкила, C1-С6-алкенила, C3-С8-циклоалкила, C4-С8-(алкилциклоалкила), где алкил представляет C1-С2-алкил и циклоалкил представляет C3-С6-циклоалкил;
C6-С10-арила или гетероарила, имеющего от 5 до 10 атомов, выбранных из С, S, N и О, где арил или гетероарил может быть необязательно и независимо замещен 1 или 2 заместителями, независимо выбранными из водорода, СН3, -(СН2)pСF3, галогена, -CONR5R4, -COOR5, -COR5, -(CH2)pNR5R4,
-(CH2)рСН3(CH2)pSOR5R4, -(CH2)pSO2R5 и -(CH2)pSO2NR5, где R4 и R5, каждый независимо, такие, как определено выше для R1, и р равно 0, 1 или 2;
(C1-С2-алкил)-(C6-С10-арила) или (C1-С2-алкил)гетероарила, причем гетероарильные части имеют от 5 до 10 атомов, выбранных из С, S, N и О, и где арил или гетероарил может быть необязательно и независимо замещен 1 или 2 заместителями, независимо выбранными из водорода, СН3, -(CH2)qCF3, галогена, -CONR5R4, -COOR5, -COR5, -(CH2)qNR5 R4, -CH2)qCH3 (CH2)qSOR5R4,
-(CH2)qSO2R5, -(CH2)qSOgNR5 и -(CH2)pOR5, где R4 и R5, каждый независимо, такой, как определено выше для R1, и q равно 0, 1 или 2, и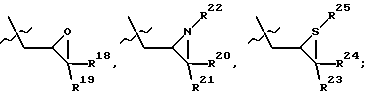
где R18, R19, R20, R21, R22, R23, R24 и R25, каждый независимо, представляют водород, C1-С6-алкил или C1-С6-алкенил;
R2 и R3, каждый независимо, представляют водород или C1-c6-алкил;
А выбирают из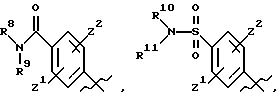
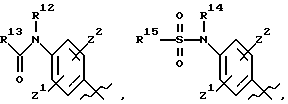
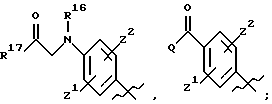
где R8, R9, R10, R11, R12, R13, R14, R15 , R16 и R17, каждый независимо, такие, как определено выше для R1, и где фенильное кольцо каждого заместителя А может быть необязательно и независимо замещено по любому положению фенильного кольца 1 или 2 заместителями Z1 и Z2, которые, каждый независимо, выбирают из водорода, СН3, -(СН2)qСF3, галогена, -CONR6R7, -COOR6, -COR6, -(CH2)rNR6R7, -(CH2)rСН3(CH2)rSOR6, -(CH2)rSO2R6 и -(СН2)rSO2NR6R7, где R6 и R7, каждый независимо, такие, как определено выше для R1, и r равно 0, 1 или 2;
Q представляет C5-C6-гидроарильный или гетерогидроароматический радикал, имеющий 5 или 6 атомов, выбранных из С, S, N и О; C5-C6-циклоалкил или гетероциклоалкил, имеющий 5 или 6 атомов, выбранных из С, S, N и О; и где каждый Q необязательно может быть замещен Z1 и Z2, как определено выше;
В представляет замещенную или незамещенную ароматическую, гетероароматическую, гидроароматическую или гетерогидро- ароматическую часть, имеющую от 5 до 10 атомов, выбранных из С, S, N и О, и необязательно и независимо замещенную 1 или 2 заместителями, независимо выбранными из водорода, СН3, -(CH2)tCF3, галогена, -(CH2)tCONR5R4, -(CH2)tNR5R4, -(CH2)tCOR5,
-(CH2) tCOOR5, -OR5, -(CH2) t SOR5, -(CH2)tSO2R5 и -(CH2)tSO2NR5R4, где R4 и R5, каждый независимо, такие, как определено выше для R1, и t равно 0, 1, 2 или 3; и R4 и R5, каждый независимо, такие, как определено выше для R1.
В объем данного изобретения включены также фармацевтически приемлемые соли соединений формулы (I), а также их изомеры, гидраты, изомерные формы и пролекарства.
Предпочтительные соединения изобретения представляют собой соединения формулы (I), где
А выбирают из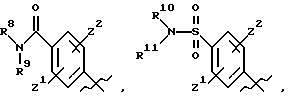
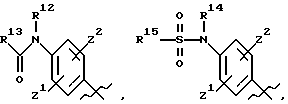
где R8, R9, R10 R11, R12, R13, R14, R15, R16 и R17, каждый независимо, такие как определено выше для R1, и где фенильное кольцо каждого заместителя А может быть необязательно и независимо замещено по любому положению фенильного кольца 1 или 2 заместителями Z1 и Z2, которые, каждый независимо, выбирают из водорода, СН3, -(СН2)qСF3, галогена, -CONR6R7, -COOR6, -COR6, -(CH2)rNR6R7, -(CH2)rСН3 (CH2)rSOR6, -(CH2)rSO2R6 и -(СН2)rSО2NR6R7, где R6 и R7, каждый независимо, такие, как определено выше для R1, и r равно 0, 1 или 2;
Q выбирают из морфолина, пиперидина и пирролидина; R1, R4 и R5, каждый независимо, выбирают из водорода, разветвленного или неразветвленного C1-C4-алкила, C3-C5-циклоалкила, C4-C8-алкилциклоалкила), где алкил представляет C1-C2-алкил, и циклоалкил представляет C3-C6-циклоалкил; C6-C10-арила или гетероарила, имеющего от 5 до 6 атомов, выбранных из С, S, N и О, и где арил или гетероарил может быть необязательно и независимо замещен 1 или 2 заместителями, независимо выбранными из водорода, СН3, -(СН2)pСF3, галогена, -CONR5R4, -COOR5, -COR5, -(СН2)рNR5R4, -(CH2)рСН3 (CH2)pSOR5R4, -(CH2)pSO2R5 и - (CH2)p SO2NR5, где R4 и R5, каждый независимо, такие, как определено выше для R1, и р равно 0, 1 или 2;
В выбирают из фенила, нафтила, индолила, бензофуранила, дигидробензофуранила, бензотиофенила, пиррила, фуранила, хинолинила, изохинолинила, циклогексила, циклогексенила, циклопентила, циклопентенила, инданила, инденила, тетрагидронафтила, тетрагидрохинила, тетрагидроизохинолинила, тетрагидрофуранила, пирролидинила и индазолинила, каждый из которых необязательно и независимо замещен 1 или 2 заместителями, независимо выбранными из водорода, СН3, СF3, галогена, -(CH2)qCONR5R4, -(CH2)qNR5R4, -(CH2)qCOR5,
-C(CH2)qCO2R5 и -OR5, где q равно 0 или 1 и где R4 и R5 такие, как определено выше;
R2 и R3, каждый независимо, представляют водород или метил.
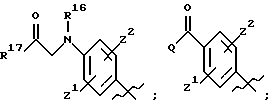
Особенно предпочтительные соединения изобретения представляют собой соединения формулы (I), где
А представляет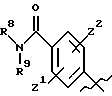
где R8 и R9 оба представляют этил и где фенильное кольцо необязательно и независимо может быть замещено по любому положению фенильного кольца двумя заместителями Z1 и Z2, которые, каждый независимо, выбирают из водорода, СН3, -(CH2)qCF3, галогена, -CONR6R7, -COOR6, -COR6, -(CH2)rNR6R7, -(CH2)rСН3(CH2)rSOR6, -(CH2)r SO2R6 и -(CH2)rSO2 NR6R7, где R6 и R7, каждый независимо, такие, как определено выше для R1, и r равно 0, 1 или 2;
R1 выбирают из водорода, метила, этила, -CH2CH=CH2, -CH2-циклопропил, -СН2-арил или CH2-гетероарил, причем гетероарильные части имеют от 5 до 6 атомов, выбранных из С, S, N и О;
В выбирают из фенила, нафтила, индолила, бензофуранила, дигидробензофуранила, бензотиофенила, фуранила, хинолинила, изохинолинила, циклогексила, циклогексенила, циклопентила, циклопентенила, инданила, инденила, тетрагидронафтила, тетрагидрохинила, тетрагидроизохинолинила, тетрагидрофуранила и индазолинила, каждый из которых необязательно и независимо замещен 1 или 2 заместителями, независимо выбранными из водорода, СН3, СF3, галогена, -(CH2)qCONR5R4, -(CH2)qNR5R4, -(CH2)qCOR5, -(CH2)qCO2R5 и -OR5, где q равно 0 или 1, и где R4 и R5 такие, как определено выше;
R2 и R3, каждый независимо, представляет водород или метил.
Заместители А и В соответственно могут быть необязательно замещены в любом положении кольца.
Термин "галоген" обозначает хлор, фтор, бром и иод.
Термин "арил" обозначает ароматическое кольцо, имеющее от 6 до 10 атомов углерода, такое как фенил и нафтил.
Термин "гетероарил" обозначает ароматическое кольцо, в котором один или несколько из 5-10 атомов в кольце представляют собой атомы, отличные от углерода, такие как N, S и О.
Термин "гидроароматический" обозначает частично или полностью насыщенное ароматическое кольцо, имеющее 5-10 атомов в кольце.
Термин "гетерогидроароматический" обозначает частично или полностью насыщенное ароматическое кольцо, в котором один или несколько из 5-10 атомов в кольце являются атомами, отличными от углерода, такими как N, S и О.
Термин "изомеры" обозначает соединения формулы (I), которые отличаются положением их функциональной группы и/или ориентацией. Термин "ориентация" обозначает стереоизомеры, диастереоизомеры, региоизомеры и энантиомеры.
Термин "изоформы" обозначает соединения формулы (I), которые отличаются кристаллической решеткой, такие как кристаллическое соединение и аморфные соединения.
Термин "пролекарство" обозначает фармакологически приемлемые производные, например сложные эфиры и амиды, такие что образующийся продукт биопревращения такого производного является активным лекарственным средством. Публикация Goodman and Gilmans, The Pharmacological basis of Therapeutics, 8th ed., McGraw-Hill, Jnt. Ed. 1992, "Biotransformation of Drugs, p. 13-15, описывающая пролекарства в общем, включена сюда в качестве ссылки.
Новые соединения данного изобретения полезны в терапии, особенно для лечения различных болезненных состояний, таких как хроническая боль, острая боль, боль при раке, боль, вызванная ревматоидным артритом, мигрень, висцеральная боль и т.д. Этот перечень, однако, не должен истолковываться как исчерпывающий.
Соединения изобретения полезны в качестве иммуномодуляторов, особенно при аутоиммунных болезнях, таких как артрит, при трансплантации кожи, органов-трансплантатов и для аналогичных хирургических потребностей, для коллагеновых болезней, различных аллергий, для использования в качестве противоопухолевых средств и антивирусных агентов.
Соединения изобретения полезны при болезненных состояниях, где имеет место дегенерация или дисфункция опиоидных рецепторов, или опиоидные рецепторы вовлечены в эту парадигму. Это применение может включать использование меченных изотопами вариантов соединений изобретения в диагностических методиках и в применениях для получения изображений, таких как позитронная эмиссионная томография (ПЭТ).
Соединения изобретения можно использовать для лечения диареи, депрессии, недержания мочи, различных психических болезней, кашля, отека легких, различных желудочно-кишечных нарушений, спинального повреждения и привыкания к чрезмерному употреблению лекарственных средств, включая лечение алкогольной, никотиновой, опиоидной и других зависимостей, и нарушений симпатической нервной системы, например гипертензии.
Соединения изобретения полезны в качестве анальгезирующего агента для применения во время общей анестезии и для оказания помощи контролируемой анестезией. Комбинации агентов с разными свойствами часто используют для достижения баланса действий, требуемых для поддержания анестезированного состояния (например, амнезии, анальгезии, мышечной релаксации и садативного эффекта). В эту комбинацию входят вводимые ингаляцией анестетики, снотворные средства, анксиолитики, нейромышечные блокаторы и опиоиды.
Соединения данного изобретения в меченной изотопом форме можно использовать в качестве диагностического агента.
В объем данного изобретения включено также использование любого из соединений приведенной выше формулы (I) для изготовления лекарственного средства для лечения любого из состояний, указанных выше.
Следующим аспектом изобретения является способ лечения субъекта, страдающего любым из указанных выше состояний, включающий введение эффективного количества соединения указанной выше формулы (I) пациенту, нуждающемуся в таком лечении.
Способы получения
Соединения данного изобретения можно получить, как описано в схемах I-IV.
Как показано выше на схемах I и II, соединения вышеприведенной формулы (I) можно получить дегидратацией гидроксисоединений (g) или (h), где R1, R2, R3, А и В такие, как определено выше в формуле (I). Последующую дегидратацию гидроксильных соединений (g) или (h), где R1, R2, R3, А и В такие, как определено выше в формуле (I), можно проводить без растворителей или в растворителе, таком как вода, спирты, сложные эфиры, НМРА, дихлорметан, толуол, простые эфиры, кетоны, карбоновые кислоты, или в смеси растворителей в присутствии кислот Бронстедта или Льюиса, таких как серная кислота, хлористоводородная кислота, трифторуксусная кислота, трихлорид алюминия, ZnCl2 или подобные, или в присутствии оксидов металлов, таких как Al2O3, Cr2O3, TiO2, WO3, P2O5 или подобные, или в присутствии других дегидратирующих агентов, таких как I2, диметилсульфоксид, KHSO4, CuSO4, фталевый ангидрид или подобные.
Заместители R1, R2 и R3 и заместители на А и В соединения (I), как определено выше, можно модифицировать способами, известными в данной области и проиллюстрированными в литературе, см., например, Protecting groups by Green, or Modern Synthesis Reactions by House, которые хорошо известны специалисту в данной области, после или в процессе получения (I) из (g) и (h).
Как показано в способе по схеме I, соединения формулы (g), как описано выше, можно получить реакцией между кетоном формулы (с), где R1, R2 и R3 такие, как определено в формуле (I), и соединением формулы (е), где А и В такие, как определено в формуле (I), и X представляет подходящую группу, такую как Н, Cl, Br, I, OSO2R или подобные.
Реакцию можно проводить без растворителей или в органическом растворителе, таком как ТГФ, толуол, простой эфир, диметилсульфоксид, или смеси растворителей обработкой подходящим металлом, таким как магний, литий, медь, церий или подобные, или обработкой галогенидом металла, таким как SmI2, CrCl2 или подобные, или обработкой металлорганическими агентами, такими как алкилмагнийгалогениды, алкиллитий или подобные.
R1, R2 и R3 и заместители на А и В соединения формулы (g), как определено выше, можно модифицировать способами, известными в данной области, после или в процессе реакции металлорганических соединений (March, 3., Advanced Organic Chemistry, 4th Ed, John Willey and Sons, 1992).
Соединения формул (с) или (е) могут быть коммерчески доступны или их можно получить способами, известными в данной области (March, J. Advanced Organic Chemistry, 4th Ed, John Willey and Sons, 1992).
Как показано в способе b на схеме (II), соединения формулы (h), как описано выше, можно получить реакцией между кетоном формулы (i), где R1, R2 и R3 и В такие, как определено в формуле (I), и металлорганическим реагентом формулы (j), где А такой, как определено в формуле (I), и М представляет группу подходящего металла, такого как магний, литий, цинк, медь, церий или подобные. Реакцию можно проводить без растворителей или в органическом растворителе, таком как ТГФ, толуол, простые эфиры, диметилсульфоксид, или в смесях растворителей.
Как показано в способе с на схеме II, соединения формулы (h) можно также получить реакциями между карбонильным соединением формулы (I), где R1, R2 и R3 такие, как определено в формуле (I), и Х представляет подходящую уходящую группу, такую как Сl, Вr, ОН, OR, SR, NR2, N(OR')R или подобные, и металлорганическими реагентами формул (j) и (k), где А и В такие, как определено в формуле (I), и М представляет группу подходящего металла, такого как магний, литий, цинк, медь, церий или подобные. Реакции можно проводить без растворителей или в растворителях, таких как ТГФ, толуол, простые эфиры, диметилформамид, диоксан, диметилсульфоксид, или в смесях растворителей.
R1, R2 и R3 и заместители на А и В соединений (h), как определено выше, можно модифицировать способами, известными в данной области и проиллюстрированными в литературе, см., например. Protecting groups by Green, or Modern Synthesis Reactions by House, которые хорошо известны специалисту в данной области, после или в процессе реакций металлорганических соединений.
Соединения формул (i), (j), (k) и (1) могут быть коммерчески доступны или их можно получить способами, известными в данной области (March, 3., Advanced Organic Chemistry, 4th Ed, John Willey and Sons, 1992).
Как показано выше на схеме III, соединения вышеприведенной формулы (I) можно получить сочетанием по Сузуки винилгалогенида (о) (X=Вr, I) с бороновой кислотой, боронатным эфиром (р) в присутствии основания, такого как Nа2СО3, К2СО3, К3РO4, триэтиламин, CsF, NaOH, или алкоксидов и палладиевого катализатора, такого как (РРh3)4Рd, бис(дибензилиденацетон)Рd(0), Pd на угле с РРh3; в качестве катализатора можно также использовать соединения Pd(II), в том числе (РРh3)2PdCl2, 1,4-бис(дифенилфосфинобутан)палладий(II)хлорид, ацетат палладия, бис(ацетонитрил)палладий(II)хлорид, дихлор[1,1'-бис(дифенилфосфино)ферроцен] палладий(II) и ацетат палладия-три(о-толил)фосфин, где R1, R2, R3, А и В такие, как определено выше в формуле (I). Сочетание по Сузуки можно проводить в толуоле, ксилоле, анизоле, ДМФ, ТГФ, спиртах, простых эфирах, воде или в смеси растворителей.
Соединения формулы (р), где В такой, как определено в формуле (I), и Z представляет B(OH)2, могут быть коммерчески доступны или их можно получить гидролизом боронатного эфира. Соединения формулы (р), где В такой, как определено в формуле (I), и Z представляет B(OR)2 (R = Me, Et), можно получить реакцией соединения формулы В-М и В(ОR)3, где R = Me или Et и М представляет группу подходящего металла, такого как литий или магний или подобное. Соединения формулы (р), где В такой, как определено в формуле (I), и Z представляет 9-борабицикло[3.3.1]нонан (9-ББН), можно получить реакцией алк-1-ина с борабицикло[3.3.1]нонаном.
Заместители R1, R2 и R3 и заместители на А и В соединения формулы (I), как определено выше, можно модифицировать способами, известными в данной области и проиллюстрированными в литературе, см., например. Protecting groups by Green, or Modern Synthesis Reactions by House, которые хорошо известны специалисту в данной области, после или в процессе получения (I) из (о) и (р).
Как показано на схеме III, соединения формулы (о), где Х представляет Вr или I, можно получить галогенированием и отщеплением алкена формулы (n), R1, R2, R3 и А такие, как определено в формуле (I). Галогенирование можно проводить в растворителе, таком как дихлорметан, хлороформ, тетрахлорид углерода, дихлорэтан или уксусная кислота, с использованием молекулярного брома или йода в качестве галогенирующего агента. Последующую стадию отщепления проводят в растворителе, таком как вода, спирты, ДМФ или простые эфиры, с использованием основания, такого как гидроксид натрия, гидроксид калия, алкоксиды металлов или триэтиламин.
Как показано на схеме III, соединения формулы (n), как описано выше, можно получить реакцией Виттига кетона формулы (с), где R1, R2 и R3 такие, как определено в формуле (I), и реагента формулы (m), где А такой, как определено в формуле (I), и Y представляет подходящую фосфонатную или фосфониевую соль. Реакцию Виттига можно проводить в различных условиях, известных в данной области и проиллюстрированных в литературе (March, J., Advanced Organic Chemistry, 4th Ed, John Willey and Sons, 1992).
Реагенты формул (с) и (m) могут быть коммерчески доступны или их можно получить способами, известными в данной области (March, J. Advanced Organic Chemistry, 4th Ed, John Willey and Sons, 1992).
Как показано на вышеприведенной схеме IV, соединения формулы (u) можно получить дегидратацией гидроксисоединения (t), где R1, R2, R3, R12, R13 и В такие, как определено выше. Дегидратацию можно проводить без растворителя или в растворителе, таком как вода, спирты, сложные эфиры, НМРА, дихлорметан, толуол, простые эфиры, кетоны, карбоновые кислоты, или в смеси растворителей в присутствии кислот Бронстедта или Льюиса, таких как серная кислота, хлористоводородная кислота, трифторуксусная кислота, трихлорид алюминия, ZnCl2 или подобные, или в присутствии оксидов металлов, таких как Аl2Оз, Сr2О3, TiO2, WO3, Р2O5 или подобные, или в присутствии других дегидратирующих агентов, таких как I2, диметилсульфоксид, KHSO4, CuSO4, фталевый ангидрид или подобные.
Заместители R1, R2 и R3 и заместитель В соединения (u), как определено выше, можно модифицировать способами, известными в данной области и проиллюстрированными в литературе, см. например. Protecting groups by Green, or Modern Synthesis Reactions by House, которые хорошо известны специалисту в данной области, после или в процессе получения (u) из (t).
Как показано на вышеприведенной схеме IV, соединения формулы (t) можно получить из соединения (s), где R1, R2, R3, R13 и В такие, как определено выше, с использованием реакции алкилирования алкилгалогенидом, таким как МеI, в присутствии основания, такого как гидроксид натрия, и агента переноса фаз, такого как Bu4NHSO4. Соединения формулы (s) можно получить реакцией между кетоном формулы (r), где R1, R2, R3, R13 такие, как определено выше, и металлорганическим реагентом формулы (k), где В такой, как определено в формуле (I), и M представляет группу подходящего металла, такого как магния, литий, цинк, медь, церий или тому подобное. Реакцию можно проводить без растворителя или в растворителях, таких как ТГФ, простые эфиры, диметилформамид, диоксан, диметил-сульфоксид, или в смеси растворителей.
Заместители R1, R2, R3, R13 соединения (s), как определено выше, можно модифицировать способами, известными в данной области и проиллюстрированными в литературе, см. например, Protecting groups by Green, or Modern Synthesis Reactions by House, которые хорошо известны специалисту в данной области, после или в процессе получения (s) из (r) и (k).
Как показано на схеме IV, соединение формулы (r) можно получить реакциями между карбонильным соединением формулы (I), где R1, R2 и R3 такие, как определено в формуле (I), и Х представляет подходящую уходящую группу, такую как С1, Вr, ОН, OR, SR, NR2, N(OR1)R или подобные, и металлорганическим реагентом, полученным сначала обработкой основанием, таким как NaH, соединения (q), где R13 такой, как определено выше, с последующим переметаллизированием с использованием алкил-лития, такого как BuLi. Реакцию можно проводить в растворителях, таких как ТГФ, толуол, простые эфиры, диметилформамид, диоксан, или в смеси растворителей. Заместители R1, R2, R3, R13 соединения (r), как определено выше, можно модифицировать способами, известными в данной области и проиллюстрированными в литературе, см., например. Protecting groups by Green, or Modern Synthesis Reactions by House, которые хорошо известны специалисту в данной области, после или в процессе получения (r) из (q) и (1).
Как показано на схеме IV, соединения формулы (q) можно получит ацилированием 4-иоданилина с использованием либо ацилангидрида, либо ацилхлорида в органическом растворителе, таком как дихлорметан. Заместитель R13 соединения (q) такой, как определено ранее.
Изобретение далее описано более подробно с помощью следующих примеров, которые не должны никоим образом рассматриваться как ограничивающие изобретение.
А) Схема синтеза соединений примеров 1-7
Соединения примеров 1-7 получали путем следующей методики, показанной на приведенной схеме 1.
Получение N-трет-бутоксилкарбонил-N'-метил-N'-метоксилизонипекотамида (соединение 2)
Смесь этилизонипекотата (соединение I) (4,71 г 30,0 ммоль), ди-трет-бутилдикарбоната (6,55 г, 30,0 ммоль) и Nа2СО3 (4,77 г, 45 ммоль) в Н2O-ТТФ (90/10 мл) кипятили с обратным холодильником в течение 2 часов. Реакционную смесь экстрагировали этилацетатом (150 мл). Органический слой промывали солевым раствором, сушили над MgSO4. Удаление растворителей давало N-трет-бутоксилкарбонилэтилизонипекотат (7,67 г).
δH (400 МГц, СDСl3): 1,25 (т, J=7,2 Гц, 3Н), 1,45 (с, 9Н), 1,62 (м, 2Н), 1,87 (м, 2Н), 2,43 (м, 1Н), 2,84 (м, 2Н), 4,02 (м, 2Н), 4,13 (кв, J=7,2 Гц, 2Н); δC-13 (100 МГц, CDCl3) δ : 14,0, 27,8, 28,2, 40,9, 42,9, 60,2, 79,2, 154,4, 174,2.
Вышеуказанный N-трет-бутоксилкарбонилэтилизонипекотат растворяли в ТГФ (60 мл) и смешивали с NHMe(OMe)HCl (4,39 г, 45,0 ммоль). Смесь обрабатывали изо-PrMgCl (2,0 М в ТГФ, 45 мл, 90 ммоль) при -20oС и полученный раствор перемешивали в течение 1 час при -5oС и затем гасили водным раствором NH4C1 и экстрагировали этилацетатом (2•100 мл). Объединенные органические слои промывали солевым раствором, сушили над MgSO4. Удаление растворителей давало N-трет-бутоксилкарбонил- N'-метил-N'-метоксилизонипекотамид (соединение 2) (8,0 г, 98%).
δH (400 МГц, CDCl3): 1,30 (с, 9Н), 1,54 (м, 4Н), 2,65 (м, 3Н), 3,02 (с, 3Н), 3,56 (с , 3Н), 3,99 (шир. с, 2Н); δC-13 (100 МГц, СDСl3) δ : 27,7, 28,1, 32,0, 37,8, 43,1, 61,3, 79,1, 154,4, 176,0.
(ii) Получение 4-(4'-N', N'-диэтиламинокарбонилбензоил)-N-трет-бутоксилкарбонилпиперидина (соединение 3)
К раствору 4-иод-N,N-диэтилбензамида (9,09 г, 30,0 ммоль) и ТМЭДА (6,96 г, 60,0 ммоль) в сухом ТГФ (60 мл) добавляли трет-бутиллитий (35,0 мл, 1,7 М, 60,0 ммоль) при -78oС. Через 30 мин по каплям добавляли N-трет-бутоксилкарбонил-N'-метил-N'-метоксилизонипекотамид (соединение 2) (8,0 г, 29,4 ммоль) в ТГФ (10 мл). Реакционную смесь нагревали до комнатной температуры и затем гасили водным раствором NH4C1, нейтрализовали хлористоводородной кислотой (концентрированной, 20 мл) при 0oС и экстрагировали этилацетатом (2•100 мл). Объединенные органические слои промывали солевым раствором, сушили над MgSO4. Удаление растворителей давало сырой продукт, который очищали на колонке с силикагелем с элюированием MeOH-CH2Cl2 (2:98), получая 4-(4' -N', N' -диэтиламинокарбонилбензоил)-N-трет-бутоксилкарбонилпиперидин (соединение 3) (3,15 г, 28%).
δH (400 МГц, СDСl3): 1,08 (шир.с, 3Н), 1,23 (шир.с, 3Н), 1,43 (с, 9Н), 1,61 (м, 2Н), 1,80 (м, 2Н), 2,89 (м, 2Н), 3,20 (шир.с, 2Н), 3,40 (м, 1Н), 3,53 (шир. с, 2Н), 4,11 (шир.с, 2Н), 7,44 (д, J=8,0 Гц, 2Н), 7,94 (д, J=8,0 Гц, 2Н).
(iii) Получение 4-(α-гидроксил-α-(4-N-трет-бутоксилкарбонилпиперидинил)-α-(1-нафтил)метил)-N,N-диэтилбензамида (соединение 4)
К раствору 1-бромнафталина (0,52 г, 2,5 ммоль) в сухом ТГФ (10 мл) добавляли н-бутиллитий (1,1 мл, 2,5 М, 2,75 ммоль) при -78oС. Через 30 мин по каплям добавляли 4-(4'-N', N'-диэтиламинокарбонилбензоил)-N-трет-бутоксилкарбонилпиперидин (соединение 3) (776 мг, 2,0 ммоль) в ТГФ (2 мл). Реакционную смесь нагревали до комнатной температуры и затем гасили водным раствором NH2Cl и экстрагировали этилацетатом (2•50 мл). Объединенные органические слои промывали солевым раствором, сушили над MgSO4. Удаление растворителей давало сырой продукт, который очищали на колонке с силикагелем с элюированием MeOH-CH2Cl2(0,5: 99,5-->5: 95), получая 4- (α-гидроксил-α-(4-N-трет-бутоксилкарбонилпиперидинил)-α-(1-нафтил)метил)-N,N-диэтилбензамид (соединение 4) (760 мг, 74%).
Т. пл. 121-124oС (CH2Cl2).
γmax(KBr), см-1: 3402, 2960, 1685, 1626, 1425, 1283, 1160.
Элементный анализ. Вычислено для C32H40N2O4• 0,50 H2O: С 73,11%; Н 7,86%; N 5,33%. Найдено С 72,86%; Н 7,64%; N 5,26%.
δH (400 МГц, CDCl3): 1,03 (шир.с, 3Н), 1,16 (шир.с, 3Н), 1,18-1,35 (м, 3Н), 1,95 (м, 1Н), 2,60 (м, 2Н), 2,75 (шир.с, 2Н), 3,15 (шир.с, 2Н), 3,42 (шир.с, 2Н), 4,10 (шир.с, 2Н), 7,10-7,50 (м, 7Н), 7,75 (м, 3Н), 8,27 (шир.с, 1Н); δC-13 (100 МГц, СDСl3) δ : 12,8, 14,1, 27,1, 27,2, 28,4, 39,2, 43,3, 45,4, 79,3, 80,4, 124,1, 124,9, 125,2, 125,3, 126,0, 127,3, 128,8, 129,2, 131,4, 135,0, 135,2, 139,4, 146,5, 154,6, 171,0.
(iv) Получение 4-(α-гидроксил-α-(4-N-трет-бутоксилкарбонилпиперидинил)-2,6-диметилбензил)-N,N-диэтилбензамида (соединение 5)
Получали по способу, описанному для соединения 4, за исключением использования 2-бром-м-ксилола (749 мг, 76%).
Т. пл. 92-96oС (CH2Cl2).
νmax(KBr), см-1: 3451, 2970, 1690, 1631, 1425, 1165.
Элементный анализ. Вычислено для C30H42N2O4• 0,50 H2O: С 71,54%, Н 8,61%; N 5,56%. Найдено: С 71,70%; Н 8,34%; N 5,62%.
δH (400 МГц, СDСl3): 1,10 (шир.с, 3Н), 1,21 (шир.с, 3Н), 1,32 (м, 2Н), 1,43 (с, 9Н), 1,69 (м, 1Н), 1,77 (м, 1Н), 2,32 (с, 6Н), 2,47 (с, 1Н), 2,75 (м, 3Н), 3,25 (шир.с, 2Н), 3,51 (шир.с, 2Н), 4,13 (шир.с, 2Н), 6,91 (м, 2Н), 7,00 (м, 1Н), 7,26 (д, J=8,4 Гц, 2Н), 7,39 (д, J=8,4 Гц, 2Н); δC-13 (100 МГц, СDСl3) δ : 12,6, 14,0, 25,0, 27,7, 28,2, 39,1, 42,9, 43,1, 44,4, 53,3, 79,1, 83,0, 125,8, 126,3, 127,2, 131,2, 135,3, 136,7, 142,9, 147,8, 154,5, 170,7.
Пример 1
Получение N,N-диэтил-4-(фенилпиперидин-4-илиденметил)бензамида (соединение 6)
К раствору 4-(α-гидроксил-α-(4-N-трет-бутоксилкарбонилпиперидинил)бензил)-N, N-диэтилбензамида (932 мг, 2,0 ммоль) в сухом дихлорметане (10 мл) при комнатной температуре добавляли трифторуксусную кислоту (10,0 мл). Реакционную смесь перемешивали в течение 16 час при комнатной температуре и затем концентрировали. Остаток растворяли в AcOEt (100 мл). Полученный раствор промывали 1 н. раствором NaOH, водным раствором NH4Cl и солевым раствором, сушили над MgSO4. Удаление растворителей давало сырой продукт, который очищали на колонке с силикагелем с элюированием MeOH-CH2Cl2 (20:80), получая (α-фенил-α-(4-N',N' -диэтиламинокарбонилфенил))-4-метиленпиперидин (соединение 6) (632 мг, 91%).
δH (400 МГц, СDСl3): 1,08 (шир.с, 3Н), 1,17 (шир.с, 3Н), 2,29 (м, 4Н), 2,86 (м, 4Н), 2,94 (шир.с, 1Н), 3,24 (шир.с, 2Н), 3,47 (шир.с, 2Н), 7,09 (м, 4Н), 7,15 (м, 1Н), 7,24 (м, 4Н); δC-13 (100 МГц, СDСl3) δ: 12,6, 14,1, 32,7, 32,8, 39,1, 43,2, 47,9, 126,0, 126,4, 127,9, 129,6, 134,9, 135,4, 135,9, 141,7, 143,2, 171,1.
Соль НСl: т. пл. 110-120oС (AcOEt-простой эфир-CH2Cl2).
νmax(KBr), см-1: 3440, 2970, 1617, 1438, 1289.
Элементный анализ. Вычислено для C23H28N2O•1,0 НСl•0,50 CH2Cl2•0,25 H2O: С 65,35%; Н 7,12%; N 6,49%. Найдено: С 65,14%; Н 7,08%; N 6,55%.
Пример 2
Получение N, N-диэтил-4-(1-нафтилпиперидин-4-илиденметил)бензамида (соединение 7)
Получали по способу, описанному в примере 1, с использованием соединения 4 (226 мг, 71%).
Т. пл. 80-85oС (MeOH-CH2Cl2).
νmax(KBr),см-1: 3052, 2970, 1628, 1431, 1286.
Элементный анализ. Вычислено для С27H30N2O•0,20 CH2Cl2 :С 78,62%; Н 7,37%; N 6,74%. Найдено: С 78,63%; Н 7,07%; N 6,54%.
δH (400 МГц, СDСl3): 1,06 (шир.с, 3Н), 1,16 (шир.с, 3Н), 2,00 (м, 2Н), 2,53 (м, 2Н), 2,64 (шир.с, NH), 2,77 (м, 2Н), 2,97 (м, 2Н), 3,20 (шир.с, 2Н), 3,47 (шир. с, 2Н), 7,26 (м, 5Н), 7,43 (м, 3Н), 7,74 (м, 2Н), 8,0 (м, 1Н); δC-13 (100 МГц, СDСl3) δ: 12,8, 14,1, 32,6, 33,5, 39,1, 43,2, 47,9, 48,2, 125,5, 125,7, 125,8, 126,1, 127,1, 127,2, 129,1, 131,9, 132,5, 133,8, 135,1, 138,3, 139,8, 142,6, 171,1.
Пример 3
Получение N,N-диэтил-4-(2,6-диметилфенилпиперидин-4-илиденметил)бензамида (соединение 8)
Получали по способу, описанному в примере 1, с использованием соединения 5 (242 мг, 80%).
Соль НСl: разлож. ≥115oС (AcOEt-простой эфир-СН2Cl2).
νmax(KBr), см-1: 2970, 2725, 1590, 1464, 1290, 1101.
Элементный анализ. Вычислено для C25H32N2O•1,0 НСl•0,50 CH2Cl2•0,25 H2O: С 65,94%; Н 7,60%; N 6,03%. Найдено: С 65,98%; Н 7,37%; N 5,81%.
Пример 4
Получение N,N-диэтил-4-(1-нафтил-N-аллилпиперидин-4-илиденметил)бензамида (соединение 9)
Смесь (α-(1-нафтил)-α-(4-N',N'-диэтиламинокарбонилфенил))-4-метиленпиперидина (соединение 7) (125 мг), аллилбромида (90 мг) и K2CO3 (138 мг) в MeCN (10 мл) перемешивали в течение 14 час при комнатной температуре и затем гасили 1 н. раствором NH4OH, экстрагировали AcOEt (100 мл). Органическую фазу промывали водным раствором NH4Cl и солевым раствором, сушили над MgSO4. Удаление растворителей давало сырой продукт, который очищали на колонке с силикагелем с элюированием MeOH-CH2Cl2 (2:98), получая (α-(1-нафтил)-α-(4-N', N'-диэтиламинокарбонилфенил))-4-метилен-N-аллилпиперидин (50 мг, 36%).
δH (400 МГц, СDСl3): 1,08 (шир.с, 3Н), 1,19 (шир.с, 3Н), 2,08 (м, 2Н), 2,39 (м, 2Н), 2,61 (м, 4Н), 3,01 (м, 2Н), 3,24 (шир.с, 2Н), 3,52 (шир. с, 2Н), 5,13 (м, 2Н), 5,90 (м, 1Н), 7,27 (м, 5Н), 7,45 (м, 3Н), 7,80 (м, 2Н), 8,04 (м, 1Н); δC-13 (100 МГц СDСl3) δ: 12,8, 14,1, 30,9, 32,0, 39,1, 43,2, 54,7, 54,9, 61,5, 117,8, 125,4, 125,6, 125,8, 126,0, 127,1, 128,2, 129,1, 131,8, 132,4, 133,7, 135,0, 138,0, 139,8, 142,6, 171,1.
Соль НСl: т. пл. 110-120oС (AcOEt-простой эфир-СН2Cl2).
νmax(KBr), см-1: 3416, 2961, 1620, 1430, 1288.
Элементный анализ. Вычислено для C30H34N2O•1,0 НСl•0,50 CH2Cl2•0,25 Н2О: С 70,17%; Н 7,05%; N 5,37%. Найдено: С 70,15%; Н 6,92%; N 5,24%.
Пример 5
Получение N,N-диэтил-4-(фенил-N-бензилпиперидин-4-илиденметил)бензамида (соединение 10)
Получали по способу, описанному в примере 4, с использованием соединения 6 и бензилбромида (215 мг, 98%):
δH (400 МГц, СDСl3): 1,09 (шир.с, 3H), 1,19 (шир.с, 3Н), 2,37 (м, 4Н), 2,47 (м, 4Н), 3,25 (шир. с, 2Н), 3,50 (шир.с, 4Н), 7,0-7,30 (м, 14Н); δC-13(100 МГц CDCl3) δ: 12,7,
14,0, 31,6, 39,1, 43,1, 54,9, 55,0, 62,8, 125,9, 126,2, 126,8, 127,8, 128,0, 128,9, 129,6, 129,7, 134,9, 135,0, 136,3, 138,2, 141,9, 143,3, 171,0.
Соль НСl: т. пл. 230-245oС (AcOEt-простой эфир-СН2Cl2).
νmax(KBr), см-1: 3423, 2976, 1624, 1434, 1288.
Элементный анализ. Вычислено для С30Н34N2О•1,0 НСl•0,25 CH2Cl2•0,25 H2O: С 72,55%; Н 7,25%; N 5,59%. Найдено: С 72,38%; Н 7,16%; N 5,50%.
Пример 6
Получение N, N-диэтил-4-(N-2,3-эпоксипропилфенилпиперидин-4-илиденметил)бензамида (соединение 11)
Получали по способу, описанному в примере 4, с использованием соединения 6 и эпибромгидрина (102 мг, 84%).
δH (400 МГц, СDСl3): 1,10 (шир.с, 3Н), 1,20 (шир.с, 3Н), 2,28 (м, 1Н), 2,39 (м, 4Н), 2,45 (м, 1Н), 2,54 (м, 2Н), 2,61 (м, 2Н), 2,74 (м, 2Н), 3,09 (м, 1Н), 3,26 (шир. с, 2Н), 3,50 (шир.с, 2Н), 7,10 (м, 4Н), 7,15 (м, 1Н), 7,25 (м, 4Н); δC-13(100 МГц, СDСl3) δ: 12,8, 14,1, 31,4, 39,1, 43,2, 44,9, 50,1, 55,5, 60,8, 126,0, 126,4, 127,9, 129,6, 129,7, 135,0, 135,3, 135,7, 141,8, 143,2, 171,1.
Пример 7
Получение N, N-диэтuл-4-(1-циклопропилметилфенилпиперидин-4-илиденметил)бензамида (соединение 12)
Получали по способу, описанному в примере 4, с использованием соединения 6 и циклопропилметилхлорида (104 мг, 86%).
δH (400 МГц, СDСl3): 0,20 (м, 2Н), 0,59 (м, 2Н), 1,04 (м, 1Н), 1,14 (шир. с, 3Н), 1,24 (шир.с, 3Н), 2,48 (д, J=6,4 Гц, 2Н), 2,56 (шир.с, 4Н), 2,80 (шир.с, 4Н), 3,29 (шир.с, 2Н), 3,53 (шир.с, 2Н), 7,14 (м, 4Н), 7,22 (м, 1Н), 7,27 (м, 4Н); δC-13 (100 МГц, СDСl3) δ: 4,18, 7,3, 12,8, 14,1, 30,3, 39,2, 43,2, 54,3, 62,7, 126,2, 126,6, 128,0, 129,5, 129,6, 134,1, 135,3, 136,3, 141,5, 142,9, 171,0.
Соль НСl: разлож.≥100oС (AcOEt-простой эфир-CH2Cl2).
νmax(KBr), см-1: 3027, 2359, 1620, 1439, 958.
Элементный анализ. Вычислено для C27H34N2O•1,0 НСl•0,50 CH2Cl2•0,75 H2O: С 66,73%; Н 7,64%; N 5,66%. Найдено: С 66,60%; Н 7,45%; N 5,78%.
В) Схема синтеза соединения примера 8
Соединение примера 8 получали, используя следующую методику, показанную на схеме 2.
(i) Получение 4-(2-бензофуроид)-N-трет-бутоксилкарбонилпиперидина (соединение 13)
К раствору 2,3-бензофурана (295 мг, 2,5 ммоль) в сухом ТГФ (10 мл) при -78oС добавляли трет-бутиллитий (1,5 мл, 1,7 М, 2,5 ммоль). Через 30 мин по каплям добавляли N-трет-бутоксилкарбонил-N-метил-N-метоксилизонипекотамид (544 мг, 2,0 ммоль) в ТГФ (2 мл), реакционную смесь нагревали до комнатной температуры и затем гасили водным раствором NH4Cl и экстрагировали этилацетатом (2•50 мл). Объединенные органические слои промывали солевым раствором, сушили над MgSO4. Удаление растворителей давало сырой продукт, который очищали на колонке с силикагелем с элюированием MeOH-CH2Cl2 (5:95), получая 4-(2-бензофуроил)-N-трет-бутоксилкарбонилпиперидин (13) (456 мг, 69%).
δH (400 МГц, СDСl3): 1,46 (с, 9Н), 1,75 (м, 2Н), 1,91 (м, 2Н), 2,91 (м, 2Н), 3,37 (м, 1Н), 4,20 (шир.с, 2Н), 7,29 (м, 1Н), 7,46 (м, 1Н), 7,53 (с, 1Н), 7,56 (м, 1Н), 7,69 (м, 1Н); δC-13(100 МГц CDCl3) δ: 27,8, 28,3, 43,1, 44,4, 79,5, 112,3, 112,9, 123,1, 123,8, 126,9, 128,2, 151,8, 154,5, 155,5, 192,8.
(ii) Получение 4-α-гидрокси-α-(4-N-трет-бутоксилкарбонилпиперидинил)-2-бензофурил)-N,N -диэтилбензамида (соединение 14)
Получали по способу, описанному в примере 4, с использованием 4-иод-N, N-диэтилбензамида (425 мг, 61%).
Т. пл. 102-106oС (CH2Cl2).
νmax(KBr), см-1: 3362, 2970, 1690, 1617, 1425, 1288, 1160.
δH (400 МГц, СDСlз): 1,06 (шир.с, 3Н), 1,20 (шир.с, 3Н), 1,24 (м, 2Н), 1,46 (м, 11Н), 2,42 (м, 1Н), 2,58 (шир.с, 2Н), 3,20 (шир.с, 2Н), 3,50 (шир. с, 2Н), 4,05 (шир.с, 2Н), 4,37 (с, 1Н), 6,70 (с, 1Н), 7,16 (м, 2Н), 7,23 (д, J= 8,0 Гц, 2Н), 7,41 (д, J=7,6 Гц, 1Н), 7,47 (д, J=7,6 Гц, 1Н), 7,58 (д, J= 8,0 Гц, 2Н); δC-13 (100 МГц, СDСl3) δ: 12,6, 13,9, 25,5, 26,3, 28,2, 39,0, 43,1, 44,9, 77,3, 79,0, 103,3, 110,9, 120,6, 122,5, 123,5, 125,6, 125,8, 127,9, 135,3, 144,0, 154,4, 154,5, 160,5, 170,9.
Пример 8
Получение N, N-диэтил-4-(2-бензофурилпиперидин-4-илиденметил)бензамида (соединение 15)
Получали по способу, описанному в примере 1, с использованием соединения 14 (135 мг, 88%).
δH (400 МГц, CDCl3): 1,20 (шир.с, 3Н), 1,24 (шир.с, 3Н), 2,36 (шир.с, 2Н), 3,00 (шир.с, 4Н), 3,15 (шир.с, 2Н), 3,33 (шир.с, 2Н), 3,56 (шир.с, 2Н), 4,45 (шир. с, 1Н), 6,25 (с,1Н), 7,24 (м, 4Н), 7,41 (м, 4Н); δC-13 (100 МГц, СDСl3) δ: 12,9, 14,2, 29,6, 32,0, 32,4, 39,3, 43,4, 47,2, 107,4, 111,0, 120,7, 122,7, 124,2, 126,0, 126,5, 128,2, 129,9, 136,1, 139,5, 140,5, 154,4, 156,2, 171,0.
Соль НСl: разлож.≥120oC (AcOEt-простой эфир-CH2Cl2).
νmax(KBr),см-1: 2977, 2801, 1586, 1449, 1257.
С) Схема синтеза соединений примеров 9-10
Соединения примеров 9 и 10 получали, используя следующую методику, приведенную на схеме 3.
(i) Получение 4-(4-фторбензоил)-N-трет-бутоксилкарбонилпиперидина (соединение 18)
Смесь гидрохлорида 4-(4-фторбензоил)пиперидина (соединение 16) (2,44 г, 10,0 ммоль), ди-трет-бутилдикарбоната (2,18 г, 10,0 ммоль) и Na2CO3 (1,59 г, 15 ммоль) в Н2О-ТГФ (50/5 мл) кипятили с обратным холодильником в течение 1 часа. Реакционную смесь экстрагировали этилацетатом (2•100 мл). Объединенные органические слои промывали солевым раствором, сушили над MgSO4. Удаление растворителей давало 4-(4-фторбензоил)-N-трет-бутоксилкарбонилпиперидин (0В 701-31, 2,28 г, 74%).
Т. пл. 80-83oС (CH2Cl2).
νmax(KBr), см-1: 2980, 2842, 1680, 1587, 1416, 1160.
δH (400 МГц, СDСl3) 1,44 (с, 9Н), 1,69 (м, 2Н), 1,79 (м, 2Н), 2,87 (м, 2Н), 3,34 (м, 1Н), 4,13 (шир.с, 2Н), 7,12 (м, 2Н), 7,95 (м, 2Н); δC-13(100МГц, СDСl3) δ: 27,4, 28,4, 43,2, 43,4, 79,6, 115,8, 115,9, 130,8, 130,9, 132,2, 154,6, 164,4, 166,9, 200,4.
(ii) Получение 4-(4-хлорбензоил)-N-трет-бутоксилкарбонилпиперидина (соединение 19)
Получали по способу, описанному для соединения 18, с использованием соединения 17 (1,23 г, 85%).
Т. пл. 122-125oС (CH2Cl2).
νmax(KBr), см-1: 2970, 2842, 1680, 1582, 1420, 1200.
δH (400 МГц, СDСl3): 1,47 (с, 9Н), 1,69 (м, 2Н), 1,81 (м, 2Н), 2,90 (м, 2Н), 3,36 (м, 1Н), 4,18 (шир.с, 2Н), 7,44 (м, 2Н), 7,88 (м, 2Н); δC-13(100 МГц, CDCl3) δ: 28,3; 28,4; 43,2; 43,4; 79,6; 129,0; 129,6; 134,1; 139,4; 154.6; 200,7.
(iii)Получение4-(α-гидрокси-α-(4-N-трет-бутоксилкарбонилпиперидинил)-4-фторбензил)-N,N-диэтилбензамида (соединение 20)
Получали по способу, описанному для соединения 4, с использованием соединения 18 и 4-иод-N,N-диэтилбензамида (454 мг, 47%).
Т. пл. 84-86oС (CH2Cl2).
νmax(KBr), см-1: 3421, 2970, 1685, 1612, 1430, 1288, 1165.
δH (400 МГц, СDСl3): 1,13 (шир.с, 3Н), 1,23 (шир.с, 3Н), 1,32 (м, 4Н), 1,44 (с, 9Н), 2,48 (м, 1Н), 2,68 (шир.с, 2Н), 3,26 (шир.с, 2Н), 3,54 (шир.с, 2Н), 3,57 (с, 1Н), 4,11 (шир. с, 2Н), 6,96 (м, 2Н), 7,27 (д, J=8,0 Гц, 2Н), 7,44 (м, 2Н), 7,47 (д, J=8,0 Гц, 2Н); δC-13(100 МГц, CDCl3) δ: 12,9; 14,0; 26,2; 28,2; 39,1; 43,2; 43,6; 44,3; 78,9; 79,1; 114,5; 114,7; 125,7; 126,1; 127,5; 127,6; 135,0; 141,2; 146,9; 154,5; 160,0; 162,5; 170,9.
(iv) Получение 4-(α-гидрокси-α-(4-N-трет-бутоксилкарбонилпиперидинил) - 4-хлорбензил) -N,N-диэтилбензамида (соединение 21)
Получали по способу, описанному для соединения 4, с использованием соединения 19 и 4-иод-N,N-диэтилбензамида (626 мг, 63%).
Т. пл. 100-105oС (CH2Cl2).
νmax(KBr),см-1: 3411, 2970, 1685, 1617, 1425, 1288, 1165, 1092.
δH(400 МГц, СDСl3): 1,08 (шир.с, 3Н), 1,20 (шир.с, 3Н), 1,33 (м, 4Н), 1,41 (с, 9Н), 2,44 (м, 1Н), 2,63 (шир.с, 2Н), 3,22 (шир.с, 2Н), 3,49 (шир.с, 2Н), 3,99 (с, 1Н), 4,05 (м, 2Н), 7,20 (м, 4Н), 7,39 (д, J=8,0 Гц, 2Н), 7,44 (д, J= 8,0 Гц, 2Н); δC-13 (100 МГц, CDCl3) δ: 12,5; 13,9; 25,9; 28,1; 39,0; 43,0; 44,1; 78,7; 79,0; 125,6; 126,0; 127,2; 127,8; 131,9; 134,8; 144,1; 146,6; 154,3; 170,7.
Пример 9
Получение N, N-диэтил-4-(4-фторфенилпиперидин-4-илиденметил)бензамида (соединение 22)
Получали по способу, описанному в примере 1 (соединение 6), с использованием соединения 20.
1H ЯМР (400 МГц, CDCl3)  : 1,12 (3Н, шир.м, СН3CH2-), 1,24 (3Н, шир.м, СН3СН2-), 2,32 (4Н, м, пиперидин-СН-), 2,54 (1Н, шир. м, NH), 2,91 (4Н, м, пиперидин-СН-), 3,27 (2Н, шир.м, CH2N-), 3,52 (2Н, шир.м, CH2N-), 7,00 (2Н, м, АrН), 7,09 (2Н, м, АrН), 7,11 (2Н, д, 3=8,0 Гц, АrН), 7,29 (2Н, д, J=8,0 Гц, АrН).
: 1,12 (3Н, шир.м, СН3CH2-), 1,24 (3Н, шир.м, СН3СН2-), 2,32 (4Н, м, пиперидин-СН-), 2,54 (1Н, шир. м, NH), 2,91 (4Н, м, пиперидин-СН-), 3,27 (2Н, шир.м, CH2N-), 3,52 (2Н, шир.м, CH2N-), 7,00 (2Н, м, АrН), 7,09 (2Н, м, АrН), 7,11 (2Н, д, 3=8,0 Гц, АrН), 7,29 (2Н, д, J=8,0 Гц, АrН).
Пример 10
Получение N, N-диэтил-4-(4-хлорфенилпиперидин-4-илиденметил)бензамида (соединение 23)
Получали по способу, описанному в примере 1 (соединение 6), с использованием соединения 21.
1H ЯМР (400 МГц, СDСl3) δ: 1,13 (3Н, шир.м, СН3CH2-), 1,22 (3Н, шир.м, СН3СН2-), 2,02 (1Н, шир. м, NH), 2,30 (4Н, м, пиперидин-СН-), 2,90 (4Н, м, пиперидин-СН-), 3,28 (2Н, шир.м, CH2N-), 3,53 (2Н, шир.м, CH2N-), 7,04 (2Н, д, J=8,0 Гц, АrН), 7,11 (2Н, д, J=8,0 Гц, АrН), 7,25 (2Н, д, J=8,0 Гц, АrН), 7,30 (2Н, д, J=8,0 Гц, АrН).
Соль НСl: т. пл. 115-120oС (H2O-CH2Cl2).
ИК (KBr): 3337, 2973, 1618, 1431, 1290, 1092 см-1.
Элементный анализ. Вычислено для С23Н27ClN2O•1,0 НСl•1,20 H2O: С 62,64%; Н 6,95%; N 6,35%. Найдено: С 62,53%; Н 6,91%; N 6,30%.
D) Схема синтеза соединения примера 11 (см. схему 4).
Пример 11
Получение N,N-диэтил-4- (фенил-N-аллилпиперидин-4-илиденметил)бензамида (соединение 25)
4-(α-Гидрокси-α-(4-N-аллилпиперидинил)бензил) -N, N-диэтилбензамид (соединение 24) (81 мг) растворяли в CH2Cl2 (10 мл) и обрабатывали тионилхлоридом (2 мл) при комнатной температуре. Реакционную смесь кипятили с обратным холодильником в течение 2 час и затем концентрировали. Остаток растворяли в этилацетате (50 мл) и полученный раствор промывали NH4OH (1 н.), водным раствором NH4Cl и солевым раствором, сушили над MgSO4. Удаление растворителей давало сырой продукт, который очищали на колонке с силикагелем с элюированием MeOH-CH2Cl2 (1:99-->5:95), получая α-фенил-α- (4-N',N' -диэтиламинокарбонилфенил))-4-метилен-N-аллилпиперидин (соединение 25, пример 11) (32 мг, 40%).
δH (400 МГц, СDСl3): 1,12 (шир.с, 3Н), 1,21 (шир.с, 3Н), 2,43 (м, 4Н), 2,55 (м, 4Н), 3,08 (д, J=6,8 Гц, 2Н), 3,25 (шир.с, 2Н), 3,53 (шир.с, 2Н), 5,18 (м, 2Н), 5,86 (м, 1Н), 7,12 (м, 4Н), 7,20 (м, 1Н), 7,27 (м, 4Н).
Соль НСl: т. пл. 85-95oС (AcOEt-CH2Cl2).
νmax(KBr), см-1: 3491, 2971, 1624, 1428, 1289, 1096.
Элементный анализ. Вычислено для C26H32N2O•HC1•0,25 Н2О•0,25 CH2Cl2:С 69,95%; Н 7,60%; N 6,21%. Найдено: С 70,00%; Н 7,73%; N 6,07%.
Пример 12
Получение N, N-диэтил-4-(4-хлорфенил-N-бензилпиперидин-4-илиденметил)бензамида (соединение 26)
N, N-Диэтил-4-(4-хлорфенил-N-бензилпиперидин-4-илиденметил)бензамид (110 мг, 93%) получали по способу, описанному в примере 4, с использованием соединения 23 (96 мг) и бензилбромида (43 мг).
1H ЯМР (400 МГц, СDСl3): δ 1,13 (3Н, шир.м, СН3CH2-), 1,23 (3Н, шир.м, СН3СН2-), 2,37 (4Н, м, пиперидин-СН-), 2,49 (4Н, м, пиперидин-СН-), 3,28 (2Н, шир.м, СН3СН2N-), 3,53 (4Н, шир.м, PhCH2N и СН3СН2N-), 7,04 (2Н, д, J= 8,0 Гц, АrН), 7,11 (2Н, д, J=8,0 Гц, АrН), 7,25 (2Н, д, J=8,0 Гц, АrН), 7,29 (7Н, м, АrН).
Соль (CHOHCO2H)2: т. пл. 100 - 110oС (МеОН).
ИК (КВr): 3368, 2977, 1728, 1603, 1433, 1290, 1087 см-1.
Элементный анализ. Вычислено для C34H39ClN2O7•1,50 H2O: С 62,81%; Н 6,51%; N 4,31%. Найдено: С 62,85%; Н 6,17%; N 4,21%.
Пример 13
Получение N, N-диэтил-4-[ (N-3-метил-2-бутенил)фенилпиперидин-4-илиденметил]бензамида (соединение 27)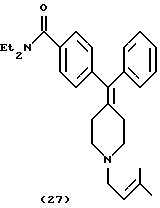
Получали по способу, описанному в примере 4, с использованием 1-бром-3-метил-2-бутена в качестве алкилирующего реагента.
ИК (NaCl, пленка): соль НСl, ν: 3432, 2976, 1623, 1434 см-1.
1Н ЯМР (основание) (СDСl3, ТМС) δ: 1,10-1,30 (6Н, шир. ОСNСН2СН3), 1,64 (3Н, с, = ССН3), 1,73 (3Н, с, =ССН3), 2,40 (4Н, м, NCH2CH2), 2,52 (4Н, м, = ССН2), 3,0 (2Н, д, J=7,6 Гц, NCH2CH=C), 3,20-3,60 (4Н, шир. OCNCH2СН3), 5,28 (1Н, м, NСН2СН=С), 7,16-7,45 (9Н, м, Аr) м.д.
Элементный анализ. Вычислено для C28H36N2O•1,8 НСl: С 69,74%; Н 7,90%; N 5,81%. Найдено: С 69,71%; Н 7,48%; N 5,58%.
Пример 14
Получение N,N-диэтил-4-[(1-циклогексилпиперидии-4-илиден)фенилметил]бензамида (соединение 28)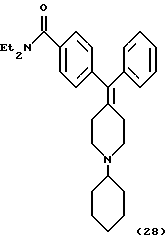
Смесь соединения 6 (100 мг, 0,29 ммоль), циклогексанона (36 мкл, 0,35 ммоль) и Ti(OPr-i)4 (0,17 мл, 0,58 ммоль) обрабатывали ультразвуком в течение 1 часа и затем перемешивали при комнатной температуре в течение ночи в атмосфере азота. Смесь разбавляли этанолом (5 мл) и затем добавляли NaBH4 (33 мг, 87 ммоль). Полученную смесь перемешивали в течение 12 час при комнатной температуре. Для гашения реакционной смеси добавляли 2 н. NН3•H2О и смесь фильтровали через целит. Фильтрат экстрагировали несколько раз этилацетатом и объединенные органические фазы промывали водой и солевым раствором и сушили над Na2SO4. Концентрирование в вакууме и очистка MPLC (силикагель 60, элюирование смесью EtOAc:гептан, от 0:100 до 100:0) давали указанное в заголовке соединение (24 мг, 20%).
Т. пл. (соль НС1): 105 - 109oС.
ИК (соль НС1, пленка), ν: 3394 (NH), 1620 (CONEt2) см-1.
1Н ЯМР (свободный амин, 400 МГц, СОСl3) δ: 1,00-1,25 (17Н, м, NCHCH2CH2CH2CH2CH2, 2 х СН3 и СН(СН)С=С), 1,60 (1Н, м, СН(СН)С=С), 1,75 (1Н, м, СН(СН)С=С), 1,80 (1Н, м, СН(СН)С=С), 2,30 (3Н, м, NCH2 и NCH), 2,60 (2Н, м, NCH2), 3,20 (2Н, шир.с, NСН2СН3), 3,50 (2Н, шир.с, NCH2СН3), 7,00-7,30 (9Н, м, Аr).
13 С ЯМР (свободный амин, 100 МГц, СОСl3)  : 12,7; 14,1; 25,9; 28,7; 32,0; 39,1; 43,2; 50,7; 50,8; 63,6; 126,0; 126,3; 127,9; 129,7; 129,8; 134,7; 134,9; 136,9; 142,0; 143,4; 171,2.
: 12,7; 14,1; 25,9; 28,7; 32,0; 39,1; 43,2; 50,7; 50,8; 63,6; 126,0; 126,3; 127,9; 129,7; 129,8; 134,7; 134,9; 136,9; 142,0; 143,4; 171,2.
Элементный анализ. Вычислено для С29Н40N2OCl2: С 69,17%; Н 8,01%; N 5,56%. Найдено: С 69,17%; Н 7,82%; N 5,18%.
Пример 15
Получение N,N-диэтил-4-[(N-бутил)фенилпиперидин-4-илиденметил]бензамида (соединение 29)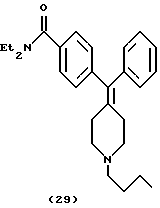
Получали по способу, описанному в примере 4, с использованием 1-иодбутана в качестве алкилирующего реагента.
ИК (NaCI, пленка) (соль НС1), ν: 3430, 2967, 2499, 1622, 1433 см-1.
1H ЯМР (СDСl3, ТМС) δ: 0,92 (3Н, т, J=7,2 Гц, CH2СН3), 1,10-1,26 (6Н, шир. OCNCH2CH3), 1,32 (2Н, м , CH2СН3), 1,53 (2Н, м, CH2CH2CH2), 2,42 (6Н, м, NCH2), 2,55 (4Н, м, =ССН2), 3,20-3,60 (4Н, шир. ОСNСН3СН3), 7,10-7,31 (9Н, м, Аr) м. д.
Элементный анализ. Вычислено для C27Н36N2O•HCl•0,4 CH2Cl2•0,4 H2O: С 68,24%; Н 8,07%; N 5,81%. Найдено: С 68,24%; Н 8,12%; N 5,89%.
Пример 16
Получение N,N-диэтил-4-[(N-4-метоксибензил)фенилпиперидин-4-илиденметид] беизамида (соединение 30)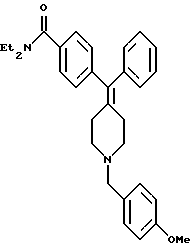
Соединение заголовка (160 мг, 68%) получали по способу, описанному в примере 4, с использованием соединения 6 (174 мг) и 4-метоксибензилхлорида (78 мг).
1Н ЯМР (400 МГц, CDCl3) δ: 1,11 (3Н, шир. СН3СН2N-), 1,20 (3Н, шир. СН3СН2N-), 2,38 (4Н, м, CCH2C), 2,46 (4Н, м, NCH2-), 3,26 (2Н, м, NCH2-), 3,47 (2Н, с, CH2N-), 3,49 (2Н, шир. СH3СН2N-), 3,77 (3Н, с, ОСН3), 6,83 (2Н, д, J=8,0 Гц, АrН), 7,05-7,30 (11Н, м, АrН).
Соль НСl: т. пл. 100-110oС (CH2Cl2).
ИК (КВr): 3425, 2974, 1618, 1515, 1434, 1255 см-1.
Элементный анализ. Вычислено для С31Н36N2O2•1,0 НСl•0,35 CH2Cl2: С 70,41%; Н 7,11%; N 5,24%. Найдено: С 70,46%; Н 7,10%; N 5,21%.
Пример 17
Получение N, N-диэтил-4-[(N-2,4-дихлорбензил)фенилпиперидин-4-илиденметил]бензамида (соединение 31)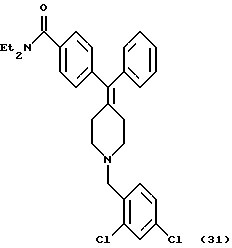
Соединение заголовка (206 мг, 81%) получали по способу, описанному в примере 4, с использованием соединения 6 (174 мг) и α,2,4-трихлортолуола (98 мг).
1H ЯМР (400 МГц, CDCl3)  : 1,12 (3Н, шир. CH3CH2N-), 1,21 (3Н, шир. СН3СН2N-), 2,39 (4Н, м, CCH2C), 2,52 (4Н, м, NCH2-), 3,28 (2Н, м, NCH2-), 3,53 (2Н, шир., CH3CH2N-), 3,57 (2Н, м, NCH2-), 7,05-7,48 (12Н, м, АrН).
: 1,12 (3Н, шир. CH3CH2N-), 1,21 (3Н, шир. СН3СН2N-), 2,39 (4Н, м, CCH2C), 2,52 (4Н, м, NCH2-), 3,28 (2Н, м, NCH2-), 3,53 (2Н, шир., CH3CH2N-), 3,57 (2Н, м, NCH2-), 7,05-7,48 (12Н, м, АrН).
Соль НСl: т. пл. 95-110oС (CH2Cl2).
ИК (КВr): 3408, 2976, 1620, 1472, 1436, 1288, 1101 см-1.
Элементный анализ. Вычислено для С30Н32N2OCl2•1,0 НСl•0,30 CH2Cl2: С 63,91%; Н 5,95%; N 4,92%. Найдено: С 63,81%; Н 6,03%; N 4,84%.
Пример 18
Получение N,N-диэтиk-4-[(1-метилпиперидин-4-илиден)фенилметил]бензамида (соединение 32)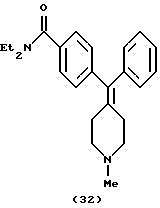
N, N-Диэтил-4-[(пиперидин-4-илиден)фенилметил] бензамид (0,34 г, 1,0 ммоль) растворяли в ацетонитриле (5 мл). При 25oС с перемешиванием добавляли карбонат калия (0,14 г, 1,0 ммоль) и метилиодид (63 мкл, 1,0 ммоль). Через 30 мин реакционную смесь выпаривали и вводили в колонку с силикагелем для очистки хроматографией с использованием от 0 до 10% МеОН (10% NH4OH) в CH2Cl2, получая 48 мг конечного продукта (28% превращение исходного материала), который превращали в гидрохлоридную соль обработкой НС1 в простом эфире.
Т. пл. 110oС (разлож.).
ИК (КВr), см-1: 2361, 1695, 1487, 1289.
МС (свободный амин): 362, 318, 219, 189, 165, 144.
lH ЯМР (амин, СDСl3): δ 1,1 (м, 6Н, амид-Ме), 2,40 (с, 3Н, МеN), 2,49, 2,60 (2 м, 8Н, пиперазин-Н), 3,40 (м, 4Н, амид-СН2), 7,08-7,34 (м, 9Н, ArH).
Элементный анализ. Вычислено для С24Н30N2O•0,1 H2O•3,1 НСl: С 60,39%; Н 7,03%; N 5,87%. Найдено: С 60,43%; Н 6,84%; N 5,45%.
Пример 19
Получение N, N-диэтил-4-[(N-трет-бутоксикарбонилпиперидин-4-ил)-8-хинолинилгидроксиметил]бензамида (соединение 33)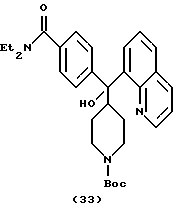
К раствору 4-иод-N,N-диэтилбензамида (1,52 г, 5,0 ммоль) и 8-бромхинолина (1,0 г) в сухом ТГФ (30 мл) добавляли при -78oС н-бутиллитий (7,0 мл, 2,5 М, 17,5 ммоль). Через 10 мин по каплям добавляли N-трет-бутоксилкарбонилэтилизонипекотат (2) (0,77 г, 3,0 ммоль) в ТГФ (5 мл). Реакционную смесь нагревали до 0oС и затем гасили водным раствором NH4C1 и экстрагировали ацетатом (2•100 мл). Объединенные органические слои промывали солевым раствором, сушили над MgSO4. Удаление растворителей давало сырой продукт, который очищали на колонке с силикагелем с элюированием MeOH-CH2Cl2 (2:98), получая MTL 0599 (145 мг, 9%).
Т. пл. 100 - 105oС.
ИК (NaCl): 2971, 1686, 1625, 1426, 1167 см-1.
Элементный анализ. Вычислено для С31Н39N3O4 0,20 H2O: C 71,43%; Н 7,62%. Найдено: С 71,50%; Н 7,75%.
1H ЯМР (400 МГц, СDСl3) δ: 1,07 (3Н, шир. CH3CH2N-), 1,19 (3Н, шир. СН3СН2М-), 1,24 (1Н, м, пиперидин-СН-), 1,43 (9Н, с, СН3С), 1,65 (1Н, м, пиперидин-СН-), 1,89 (2Н, м, пиперидин-СН-), 2,52 (1Н, м, пиперидин-СН-), 2,64 (1Н, шир. пиперидин-СН-), 2,78 (1Н, шир. пиперидин-СН-), 3,22 (2Н, шир. СН3СН2N-), 3,49 (2Н, шир. СН3СН2N-), 4,16 (2Н, шир. пиперидин-СН-), 7,24 (2Н, д, J=8,0 Гц, АrН), 7,35 (1Н, дд, J=8,0, 4,4 Гц, АrН), 7,55 (2Н, д, J= 8,0 Гц, АrН), 7,58 (1Н, д, J=8,0 Гц, АrН), 7,71 (1Н, д, J=8,0 Гц, АrН), 7,80 (1Н, д, J=8,0 Гц, АrН), 8,14 (1Н, д, J=8,0 Гц, АrН), 8,69 (1Н, м, АrН), 9,80 (1Н, с, ОН).
Пример 20
Получение N, N-диэтил-4-(8-хинолинилпиперидин-4-илиденметил)бензамида (соединение 34)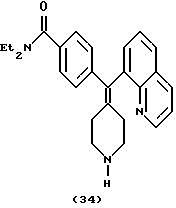
Смесь соединения примера 19 (45 мг), трифторуксусной кислоты (1,0 мл) и трифторметансульфоновой кислоты (1 мл) кипятили с обратным холодильником в течение 8 час и затем концентрировали. Остаток растворяли в AcOEt (50 мл). Полученный раствор промывали 1 н. раствором NaOH, водным раствором NH4Cl и солевым раствором, сушили над MgS04. Удаление растворителей давало сырой продукт, который очищали на колонке с силикагелем с элюированием NH4OH (1н.) - MeOH - CH2Cl2 (2,5:17,5:80), получая N,N-диэтил-4-(8-хинолинилпиперидин-4-илиденметил)бензамид (29 мг, 84%).
1H ЯМР (400 МГц, СDСl3), δ: 1,07 (3Н, шир.м, СН3СН2-), 1,20 (3Н, шир.м, СН3СН2-), 2,00 (2Н, м, пиперидин-СН-), 2,46 (1Н, с, NH), 2,52 (2Н, м, пиперидин-СН-), 2,75 (1Н, м, пиперидин-СН-), 2,92 (2Н, м, пиперидин-СН-), 3,05 (1Н, м, пиперидин-СН-), 3,22 (2Н, м, СН2N-), 3,49 (2Н, м, CH2N-), 7,23 (2Н, м, АrН), 7,32 (2Н, м, АrН), 7,36 (1Н, м, АrН), 7,49 (2Н, м, АrН), 7,72 (1Н, дд, J=6,4, 3,2 Гц, АrН), 8,11 (1Н, дд, J=8,4, 1,6 Гц, АrН), 8,91 (1Н, дд, J= 4,0, 1,6 Гц, ArH).
Соль НСl: т. пл. >170oС (разлож.)
ИК (КВr): 3410, 2973, 1614, 1551, 1436, 1284 см-1.
Элементный анализ. Вычислено для C26H29N3O• 2,0 HCl• 0,50 CH2Cl2•0,75 H2O: С 60,23%; Н 6,39%. Найдено: С 60,27%; Н 6,42%.
Пример 21
Получение N, N-диэтил-4-[(N-тpeт-бутоксикарбонилпиперидин-4-ил)-3-метоксифенилгидроксиметил]бензамида (соединение 35)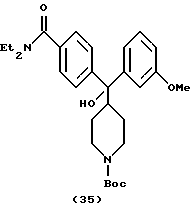
Получали по способу примера 19 с использованием 3-броманизола, что давало указанное в заголовке соединение (226 мг, 23%).
Т. пл. 95 - 103oС.
ИК (NaCl): 3422, 2973, 1684, 1614, 1429, 1289 см-1.
Элементный анализ. Вычислено для C29H40N2O5•0,60 Н2О: С 68,64%; Н 8,18%; N5,52%. Найдено: С 68,66%; Н 7,98%; N 5,64%.
1H ЯМР (400 МГц, СDСl3) δ: 1,07 (3Н, шир. СH3СH2N-), 1,19 (3Н, шир. СН3СН2N-), 1,31 (4Н, м, пиперидин-СН-), 1,41 (9Н, с, СН3С), 2,46 (1Н, м, пиперидин-СН-), 2,64 (2Н, шир. пиперидин-СН-), 3,22 (2Н, шир. СН3СН2N-), 3,49 (2Н, шир. СH3СН2N-), 3,65 (1Н, С, ОН), 3,72 (3Н, с, ОСН3), 4,06 (2Н, шир. пиперидин-СН-), 6,69 (1Н, м, АrН), 7,01 (1Н, д, J=7,6 Гц, АrН), 7,08 (1Н, с, АrН), 7,17 (1Н, Д, J=8,0 Гц, АrН), 7,21 (2Н, д, J=8,0 ГЦ, АrН), 7,48 (2Н, Д, J=8,0 Гц, АrН)
Пример 22
Получение N,N-диэтил-4-(3-метоксифенилпиперидин-4-илиденметил)бензамида (соединение 36)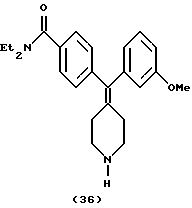
Получали по способу, описанному в примере 1, с использованием соединения 21 (100 мг), что давало N,N-диэтил-4-(3-метоксифенилпиперидин-4-илиденметил)бензамид (75 мг, 98%).
1H ЯМР (400 МГц, CDCl3) δ: 1,12 (3Н, шир. СН3СН2N-), 1,23 (3Н, шир. СН3СН2N-), 2,34 (4Н, м, пиперидин-СН-), 2,91 (4Н, шир. пиперидин-СН-), 3,17 (1Н, с, NH), 3,27 (2Н, шир. СН3СН2N-), 3,52 (2Н, шир. СН3СН2N-), 3,76 (3Н, с, ОСН3), 6,64 (1Н, с, ArH), 6,70 (1Н, д, J=8,0 Гц, АrН), 6,76 (1Н, д, J=7,6 Гц, ArH), 7,15 (2Н, д, J=8,0 Гц, ArH), 7,22 (1Н, м, ArH), 7,29 (2Н, д, J=8,0 Гц, ArH).
Соль НСl: т. пл. >90oС (разлож.).
ИК (NaCl): 2970, 1621, 1430, 1287 см-1.
Элементный анализ. Вычислено для С24Н30N2O2•НСl• 1,70 H2O: С 64,69%; Н 7,78%; N 6,29%. Найдено: С 64,82%; Н 7,60%; N 6,08%.
Пример 23
Получение N, N-диэтил-4-[(N-бензил)-3-метоксифенилпиперидин-4-илиденметил)бензамида (соединение 37)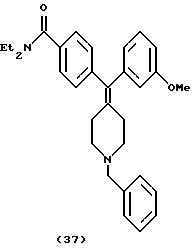
Использовали способ, описанный в примере 4, с использованием соединения примера 22 (38 мг), получая N,N-диэтил-4-[(N-бензил)-3-метоксифенилпиперидин-4-илиденметил)бензамид (46 мг, 98%).
1H ЯМР (400 МГц, СDС13) δ: 1,12 (3Н, шир. CH3CH2N-), 1,25 (3Н, шир. СH3СН2N-), 2,38 (4Н, м, пиперидин-СН-), 2,48 (4Н, шир. пиперидин-СН-), 3,27 (2Н, шир. CH3CH2N-), 3,52 (2Н, с, PhCH2N-), 3,53 (2Н, шир. CH3CH2N-), 3,75 (3Н, с, ОСН3), 6,65 (1Н, с, АrН), 6,69 (1Н, д, J = 8,0 Гц, АrН), 6,74 (1Н, д, J = 7,6 Гц, АrН), 7,13 (2Н, д, J = 8,0 Гц, АrН), 7,13-7,32 (8Н, м, АrН).
Соль НС1: т. пл. 100 - 110oС (CH2Cl2).
ИК (NaCl): 3421, 2972, 1619, 1430, 1287 см-1.
Элементный анализ. Вычислено для С31H36N2O2•НС1•0,40 CH2Cl2: С 69,96%; Н 7,07%; N 5,20%. Найдено: С 69,94%; Н 7,06%; N 5,15%.
Пример 24
Получение N, N-диэтил-4-[(N-трет-бутоксикарбонилпиперидин-4-ил)-3-флюрфенилгидроксиметил]бензамида (соединение 38)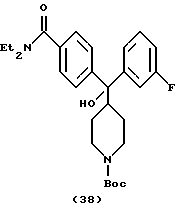
По способу примера 19 с использованием 3-бромфторбензола получали указанное в заголовке соединение (257 мг, 27%).
1H ЯМР (400 МГц, СDС13) δ: 1,03 (3Н, шир. СН3СН2N-), 1,15 (3Н, шир. СН3СН2Н-), 1,19-1,29 (4Н, м, пиперидин-СН-), 1,35 (9Н, с, СН3С), 2,39 (1Н, м, пиперидин-СН-), 2,59 (2Н, шир. пиперидин-СН-), 3,17 (2Н, шир. CH3CH2N-), 3,28 (1H, c, ОН), 3,45 (2Н, шир, СН3СН2N-), 4,02 (2Н, шир. пиперидин-СН-), 6,80 (1H, м, АrН), 7,15 (3Н, м, АrН), 7,18 (2Н, д, J = 8,0 Гц, АrН), 7,39 (2Н, д, J = 8,0 Гц, АrН).
Пример 25
Получение N, N-диэтил-4-(3-фторфенилпиперидин-4-илиденметил)бензамида (соединение 39)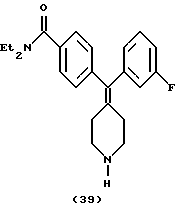
По способу примера 20 с использованием соединения примера 24 (165 мг) получали N,N-диэтил-4-(3-фторфенилпиперидин-4-илиденметил)бензамид (108 мг, 87%).
1H ЯМР (400 МГц, СDС13) δ: 1,08 (3Н, шир. CH3CH2N-), 1,19 (3Н, шир. СH3СН2N-), 2,09 (1H, с, NH), 2,25 (4Н, шир, пиперидин-СН-), 2,84 (4Н, шир. пиперидин-СН-), 3,23 (2Н, шир. СН3СН2N-), 3,47 (2Н, шир. CH3CH2N-), 6,74 (1H, м, АrН), 6,86 (2Н, м, АrН), 7,06 (2Н, д, J = 8,0 Гц, АrН), 7,18 (1H, м, АrН), 7,24 (2Н, д, J = 8,0 Гц, АrН).
Соль НС1: т. пл. > 70oС (разлож.).
ИК (NaCl) 2978, 1605, 1478, 1432, 1290 см-1.
Элементный анализ. Вычислено для С23H27N2OF•НС1•0,25 CH2Cl2•1,50 H2O: С 61,89%; Н 7,04%; N 6,21%. Найдено: С 61,97%; Н 6,95%; N 6,22%.
Е) Схема синтеза соединения примера 26
Соединение примера 26 получали по методике, как показано на схеме 5.
(i) Получение 4'-иодацетанилида (соединение 40)
К раствору 4-иоданилина (15 г, 69 ммоль) в сухом CH2Cl2 (100 мл) добавляли уксусный ангидрид (14,09 г, 138 ммоль) при комнатной температуре, реакционную смесь затем перемешивали в течение 2 час. Серый осадок, образовавшийся во время реакции, фильтровали, промывали простым эфиром и собирали, маточный раствор концентрировали досуха и добавляли AcOEt, полученный осадок фильтровали, промывали простым эфиром и объединяли с предыдущим твердым веществом, являющимся целевым продуктом (15,95 г, 88,7%).
1Н ЯМР (СDС13) δ: 2,19 (3Н, с, СОСН3), 7,2 (1Н, с, шир. -NH), 7,23 (2Н, м, Аr), 7,61 (2Н, м, Аr).
(ii) Получение 4-(4-ацетамидобензоил)-N-трет-бутоксилкарбонилпиперидина (соединение 41)
К раствору 4'-иодацетанилида (11,7 г, 45 ммоль) в сухом ТГФ (200 мл) при 0oС добавляли NaH (1,62 г, 67,5 ммоль), реакционную смесь перемешивали в течение 30 мин в то время, как температура повышалась до комнатной температуры, затем медленно при -78oС добавляли н-BuLi (1,6 М в гептане, 54 ммоль). Смесь перемешивали в течение 15 мин, затем по каплям через шприц добавляли N-трет-бутоксилкарбонил-N'-метил-N'-метоксилизонипекотамид (6,15 г, 30 ммоль) в ТГФ (10 мл). Реакционную смесь нагревали до комнатной температуры и затем гасили водным раствором NH4Cl и экстрагировали этилацетатом (2•100 мл). Органический слой промывали насыщенным (водным) NH4Cl, солевым раствором, сушили над MgSO4 и концентрировали, получая сырой продукт, который дополнительно очищали колоночной хроматографией на силикагеле с использованием MeOH-CH2Cl2 (0:100 ~ 5:95), получая целевой продукт (9,02 г, 87%).
1H ЯМР (CDCl3) δ: 1,47 (9Н, с (СН3)3). 1,6-1,8 (4Н, м, пиперидин), 2,21 (3Н, с, СОСН3), 2,9 (2Н, м, пиперидин), 3,37 (1Н, м, СОСН-), 4,15 (2Н, м, пиперидин), 7,64 (2Н, м, Аr), 7,86 (1Н, с, шир., -CONH), 7,91 (2Н, м, Аr).
(iii) Получение 4-(α-гидрокси-α-(4-'N-трет-бутоксилкарбонилпиперидинил)-3-фторбензил)ацетанилида (соединение 42)
По способу, описанному для получения соединения 4, но при замене 1-бромнафталина на 3-фтор-1-иодбензол, получали указанное в заголовке соединение (93%).
1H ЯМР (ДMCO-D6) δ: 1,2-1,3 (4Н, м, пиперидин), 1,37 (9Н, с, (СН3)3), 2,0 (3Н, с, СОСН3), 2,65 (3Н, шир., пиперидин), 3,95 (2Н, м, пиперидин), 6,98 (1Н, м, Аr), 7,21-7,50 (7Н, м, Аr), 9.85 (1Н, с, OC-NH).
(iv) Получение N-метил-4-(α-гидрокси-α-(4-N-трет-бутоксилкарбонилпиперидинил)-3-фторбензил)ацетанилида (соединение 43)
К 2 М (водному) раствору NaOH (10 мл) добавляли гидросульфат тетрабутиламмония (1,35 г, 3,97 ммоль), затем добавляли 4-α-гидрокси-α-(4-N-трет-бутоксилкарбонилпиперидинил)-3-фторбензил)ацетанилид (825 мг, 1,86 ммоль) и метилиодид (769 мг, 5,4 ммоль) в 10 мл дихлорметана. Реакционную смесь затем кипятили с обратным холодильником в течение 1 часа, охлаждали до комнатной температуры. Слой дихлорметана собирали и выпаривали до объема около 1 мл. Добавляли этилацетат и осадок отделяли фильтрованием. Органическую фазу промывали солевым раствором и сушили над MgSO4, концентрировали, получая твердый материал, который дополнительно очищали MPLC с использованием MeOH-CH2Cl2 (5: 95), чтобы получить чистое, указанное в заголовке соединение (770 мг, 93%).
1H ЯМР (СDС13) δ: 1,2-1,5 (4Н, м, пиперидин), 1,42 (9Н, с, (СН3)3), 1,83 (3Н, с, СОСН3), 2,52 (1Н, м, -СН-С-ОН), 2,70 (2Н, м, пиперидин), 2,86 (1Н, с, шир., -ОН), 3,21 (3Н, с, NCH3), 4,15 (2Н, с, шир., пиперидин), 6,90 (1Н, м, Аr), 7,12-7,60 (7Н, м, Аr).
Пример 26
Получение N-метил-4-(3-фторфенилпиперидин-4-илиденметил)ацетанилида (соединение 44)
КрастворуN-метил-4-α-гидрокси-α-(4-N-трет-бутоксилкарбонилпиперидинил)-3-фторбензил)ацетанилида (300 мг, 0,657 ммоль) в сухом дихлорметане (5 мл) добавляли трифторуксусную кислоту (50 мл) при комнатной температуре. Реакционную смесь кипятили с обратным холодильником в течение 4 часов и затем концентрировали. Остаток растворяли в AcOEt (50 мл). Полученный раствор промывали 2 н. (водным) NaOH, (водным) NH4C1 и солевым раствором, сушили над MgSO4. Удаление растворителей давало сырой продукт, который очищали MPLC с элюированием MeOH-CH2Cl2-NH4ОH (5: 95: 1), получая чистый продукт (176 мг, 79%).
Т. пл. 235-237oС (разлож.).
ИК (NaCl, пленка) (соль НС1),νmax:2961, 2722, 2480, 1658, 1608, 1580, 1507, 1429, 1381 см-1.
1H ЯМР (CDCl3) δ: 1,89 (3Н, с, СОСН3), 1,95 (1Н, с, -NH), 2,32 (4Н, м, пиперазин), 2,92 (4Н, м, пиперазин), 3,26 (3Н, с, N-СН3), 6,81-7,28 (8Н, м, Аr).
13C ЯМР (СDС13) δ: 22,4, 33,2, 33,3, 37,0, 48,3, 113,3 (м, C-F), 116,5 (м, C-F), 125,4, 126,6, 129,5, 129,6, 130,9, 133,7, 137,7, 141,2, 142,8, 144,2, 161,3, 163,8, 170,4.
Элементный анализ. Вычислено для C21H23N2FO•HC1: С 67,28%; Н 6,45%; N 7,47%. Найдено: С 66,88%; Н 6,44%; N 7,16%.
(F) Схема синтеза соединения примера 27
Соединение примера 27 получали по методике, как показано на схеме 6
(i) Получение N-трет-бутоксилкарбонил-4-пиперидона (соединение 46)
Смесь соединения 45 (50 г, 0,325 моль) и ди-трет-бутилдикарбоната (71 г, 0,325 моль) в 300 мл дихлорметана перемешивали при 0oС, по каплям добавляя триэтиламин (133 г, 1,32 моль). Смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 12 часов. Растворитель выпаривали и сырой продукт распределяли между водой (400 мл) и диэтиловым простым эфиром (400 мл). Водную фазу промывали дополнительной порцией диэтилового простого эфира (400 мл). Объединенный эфир промывали водой (400 мл) и солевым раствором (400 мл), сушили над MgSO4. Удаление растворителя давало соединение 46 в виде бледно-желтого твердого материала (55,3 г, 85%).
δH (400 МГц, СDС13): 1,50 (с, 9Н), 2,45 (т, 4Н, J=6,1 Гц), 3,72 (т, 4Н, J=6,1 Гц).
(ii) Получение трет-бутилового эфира 4-(4-метоксикарбонилбензилиден)пиперидин-1-карбоновой кислоты (соединение 49)
Метил-4-(бромметил)бензоат (соединение 47) (11,2 г, 49 ммоль) растворяли в 25 мл триметилфосфита и кипятили с обратным холодильником в атмосфере N2 в течение 5 часов. Избыток триметилфосфита удаляли совместной перегонкой с толуолом, получая сырой метиловый эфир 4-(диметоксифосфорилметил)бензойной кислоты (соединение 48).
δH (400 МГц, СDС13): 3,20 (д, 2Н, J=22 Гц), 3,68 (д, 3Н, 10,8 Гц), 3,78 (д, 3Н, 11,2 Гц), 3,91 (с, 3Н), 7,38 (м, 2Н), 8,00 (д, 2Н, J=8 Гц).
Сырой продукт (соединение 48) растворяли в сухом ТГФ (200 мл) в атмосфере N2 и охлаждали до -78oС. По каплям добавляли диизопропиламид лития (32,7 мл, 1,5 М в гексанах, 49 ммоль). Раствор оставляли для нагревания до комнатной температуры. К реакционной смеси по каплям добавляли раствор соединения 46 (9,76 г, 49 ммоль в 100 мл сухого ТГФ) и смесь перемешивали в атмосфере N2 в течение 12 час. К реакционной смеси добавляли воду (300 мл) и этилацетат (300 мл) и экстрагировали. Водную фазу промывали этилацетатом (2 • 300 мл). Объединенные слои этилацетата сушили над МgSО4 и выпаривали, получая сырой продукт, который очищали хроматографией на силикагеле (0-33% этилацетат в гексанах), получая соединение 49 в виде белого твердого материала (5,64 г, 35%).
δH (400 МГц, СDС13): 1,44 (с, 1Н), 2,31 (т, J=5,5 Гц, 2Н), 2,42 (т, J= 5,5 Гц, 2Н), 3,37 (т, J=5,5 Гц, 2Н), 3,48 (т, J=5,5 Гц, 2Н), 3,87 (с, 3Н), 6,33 (с, 1Н), 7,20 (д, J=6,7 Гц, 2Н), 7,94 (д, J=6,7 Гц, 2Н); δc-13 (СDС13): 28,3, 29,2, 36,19, 51,9, 123,7, 127,8, 128,7, 129,4, 140,5, 142,1, 154,6, 166,8 м.д.
νmax (NaCl), см-1: 3424, 2974, 2855, 1718, 1688, 1606, 1427, 1362, 1276.
Элементный анализ. Вычислено для С9Н25NО4: 68,86%; Н 7,60%; H 4,23%. Найдено: С 69,1%; Н 7,69%; N 4,25%.
(iii) Получение трет-бутилового эфира 4-бром-4-[бром-(4-метоксикарбонилфенил)метил]пиперидин-1-карбоновой кислоты (соединение 50)
К раствору соединения 49 (5,2 г, 16 ммоль) в сухом дихлорметане (200 мл) добавляли К2СО3 (1,0 г). Затем по каплям при 0oС добавляли раствор брома (2,9 г, 18 ммоль в 30 мл DCM) и смесь перемешивали в течение 1,5 часов при комнатной температуре. К2СО3 удаляли фильтрованием и раствор выпаривали досуха. Сырой продукт растворяли в этилацетате (200 мл) и промывали водой (200 мл), 0,5 М НС1 (200 мл) и солевым раствором (200 мл), сушили над MgSO4. Растворитель выпаривали, получая сырой продукт, который перекристаллизовывали из метанола, получая соединение 50 в виде белого твердого материала (6,07 г, 78%).
δH (400 МГц, СDС13): 1,28 (с, 9Н), 1,75 (м, 2Н), 1,90 (м, 2Н), 2,1 (м, 4Н), 3,08 (шир. 4Н), 3,90 (с, 3Н), 4,08 (шир., 4Н), 5,14 (с, 1Н), 7,57 (д, J= 8,4 Гц, 2Н), 7,98 (д, J=8,4 Гц, 2Н); δc-13 (400 МГц, СDС13): 28,3, 36,6, 38,3, 40,3, 52,1, 63,2, 72,9, 129,0, 130,3, 130,4, 141,9, 154,4, 166,3 м.д.
νmax (NaCl), см- 1: 3425, 2969, 1725, 1669, 1426, 1365, 1279, 1243.
Элементный анализ. Вычислено для С19Н25Br2NO4: С 46,6%; Н 5,13%; N 2,85%. Найдено: С 46,64%; Н 5,16%; N 2,89%.
(iv) Получение трет-бутилового эфира 4-[бром-(4-карбоксифенил)метилен] пиперидин-1-карбоновой кислоты (соединение 51)
К раствору соединения 50 (5,4 г, 11 ммоль) в метаноле (300 мл) при 40oС добавляли 2,0 М NaOH (100 мл). Реакционную смесь перемешивали в течение 3 часов при 40oС. Сырую соль выделяли фильтрованием. Твердый материал сушили в течение ночи в вакууме. Сухую соль растворяли в 40% смеси ацетонитрил/вода и рН раствора доводили до 2 с использованием концентрированной HCl. Целевой продукт (7) (3,8 г, 87%) выделяли в виде белого порошка фильтрованием.
δH (400 МГц, СDС13): 1,45 (с, 9Н), 2,22 (дд, J=5,5 Гц, 6,1 Гц, 2Н), 2,64 (дд, J=5,5 Гц, 6,1 Гц, 2Н), 3,34 (дд, J=5,5 Гц, 6,1 Гц, 2Н), 3,54 (дд, J=5,5 Гц, 6,1 Гц, 2Н), 7,35 (д, J=6,7 Гц, 2Н), 8,08 (д, J=6,7 Гц, 2Н); δc-13 (400 МГц, СDС13): 28,3, 31,5, 34,2, 44,0, 115,3, 128,7, 129,4, 130,2, 137,7, 145,2, 154,6, 170,3.
Элементный анализ. Вычислено для C18H22BrNO4: С 54,56%; Н 5,60%; N 3,53%. Найдено: С 54,66%; Н 5,68%; N 3,59%.
(v) Получение трет-бутилового эфира 4-[бром-(4-диэтилкарбамоилфенил)метилен]пиперидин-1-карбоновой кислоты (соединение 52)
К раствору соединения 51 (1,0 г, 2,5 ммоль) в сухом дихлорметане (10 мл) при -20oС добавляли изобутилхлорформиат (450 мг, 3,3 ммоль). Через 20 мин при -20oС добавляли диэтиламин (4 мл) и реакционную смесь оставляли для нагревания до комнатной температуры. Через 1,5 часа растворитель выпаривали и реакционную смесь распределяли между этилацетатом и водой. Слой этилацетата промывали водой и солевым раствором и сушили над MgSO4 и этилацетат удаляли выпариванием. Сырой продукт очищали хроматографией на силикагеле (0-60% этилацетат в гептанах), получая продукт (соединение 52) в виде белых игл (800 мг, 73%).
δH (400 МГц, СDС13): 1,13 (шир., 3Н), 1,22 (шир., 3Н), 1,44 (с, 9Н), 2,22 (т, J=5,5 Гц, 2Н), 2,62 (т, J=5,5 Гц, 2Н), 3,31 (т, J=5,5 Гц, 2Н), 3,52 (т, J= 5,5 Гц, 2Н), 7,27 (д, J=7,9 Гц, 2Н), 7,33 (д, J=7,9 Гц, 2Н); δc-13 (400 МГц, СDС13): 12,71, 14,13, 28,3, 31,5, 34,2, 39,1, 43,2, 79,7, 115,9, 126,3, 129,3, 136,8, 137,1, 140,6, 154,6, 170,5.
Элементный анализ. Вычислено для С22H31BrN2O3: С 58,3%; Н 6,92%; N 6,21%. Найдено: С 58,62%; Н 6,89%; N 6,21%.
Пример 27
Получение N, N-диэтил-4-[пиперидин-4-илиден-(3-трифторметилфенил)метил] бензамида (соединение 54 г Аr = 3-трифторметилфенил) (общая методика)
Сочетание по Сузуки соединения 52 с разными бороновыми кислотами с последующим удалением защитной группы у заместителя проводили параллельно в небольшом масштабе. Реакции и жидкостно-жидкостную экстракцию проводили в пробирках для культур размером 25 • 150 мм. Методика типичной реакции описана ниже.
К раствору соединения 52 (25 мг, 57 мкмоль) и тетракис(трифенилфосфин)палладия(0) (5 мг, 4,3 мкмоль) в ксилолах (дегазированы, 0,5 мл) добавляли 3-трифторфенилбороновую кислоту (28,5 мг, 150 мкмоль) в этаноле (дегазирован, 0,5 мл), затем 150 мкл 2 М Na2CO3 (водный) (300 мкмоль). Реакционную смесь оставляли для прохождения реакции при 80oС в течение 1,5 часов в атмосфере Ar. Реакционную смесь разбавляли водой (1 мл) и диэтиловым простым эфиром (1 мл) и перемешивали с завихрением. Органическую фазу отделяли и выпаривали, получая сырой продукт (соединение 9, Аr=3-трифторметилфенил).
Вос-группу удаляли обработкой сырого продукта 1 мл ТФУ. Через 30 минут ТФУ выпаривали при комнатной температуре, получая сырую соль ТФУ. Соль нейтрализовали 1 М NH4OH (1,0 М) и экстрагировали диэтиловым простыми эфиром (2 • 1 мл). Эфирную фазу подкисляли 4,0 М НС1 в диоксане (200 мкл) и соль НС1 экстрагировали водой (2 • 1 мл). Водный раствор соли промывали диэтиловым простым эфиром (2 • 1 мл) и лиофилизировали, получая продукт (соединение 54, Аr=3-трифторметилфенил) в виде белого порошка (10 мг, 39%).
1Н ЯМР (СDС13) (основание) δ: 1,11 (шир., 3Н), 1,20 (шир., 3Н), 2,26 (т, J= 5,6 Гц, 2Н), 2,31 (т, J=5,6 Гц, 2Н), 2,88-2,91 (м, 4Н), 3,27 (шир. 2Н), 3,52 (шир. 2Н), 7,10-7,47 (м, 8Н).
Элементный анализ. Вычислено для C24H28N2OF3Cl • 1,80 H2O: С 59,39%; Н 6,56%; N 5,77%. Найдено: С 59,39%; Н 5,90%; N 5,77%.
Примеры 28-52
По методике, описанной для соединения 54 примера 27, но с заменой 3-трифторметилфенилбороновой кислоты на соответствующие бороновые кислоты были также получены следующие соединения.
Пример 28
N, N-Диэтил-4-(3-нитрофенилпиперидин-4-илиденметил)бензамид (соединение 55)
Использовали 3-нитрофенилбороновую кислоту.
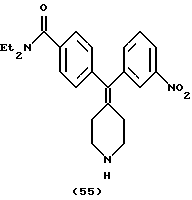
1Н ЯМР (СDС13) (основание) δ: 1,11 (шир. 3Н), 1,21 (шир. 3Н), 2,27-2,34 (м, 4Н), 2,92 (т, J=6,0 Гц, 4Н), 3,26 (шир. 2Н), 3,52 (шир. 2Н), 7,10 (д, J= 8,4 Гц, 2Н), 7,31 (д, J=8,4 Гц, 2Н), 7,40-7,50 (м, 2Н), 7,95-8,08 (м, 2Н).
Пример 29
N,N-Диэтил-4-(4-толуилпиперидин-4-илиденметил)бензамид (соединение 56)
Использовали п-толуилбороновую кислоту.
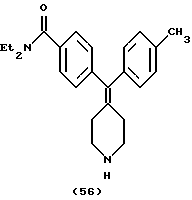
1Н ЯМР (СDС13) (основание) δ: 1,10 (шир. 3Н), 1,19 (шир. 3Н), 2,29 (с, 3Н), 2,26-2,31 (м, 4Н), 2,86-2,88 (м, 4Н), 3,25 (шир, 2Н), 3,49 (шир. 2Н), 6,95-7,28 (м, 8Н).
Пример 30
N, N-Диэтил-4-(4-формилфенилпиперидин-4-илиденметил)бензамид (соединение 57)
Использовали 4-формилфенилбороновую кислоту.

1Н ЯМР (СDС13) (основание) δ: 1,10 (шир. 3Н), 1,20 (шир. 3Н), 2,28-2,33 (м, 4Н), 2,89-2,92 (м, 4Н), 3,25 (шир. 2Н), 3,50 (шир. 2Н), 7,08-7,79 (м, 8Н), 9,95 (с, 1Н).
Пример 31
N, N-Диэтил-4-(3-хлор-4-фторфенилпиперидин-4-илиденметил)бензамид (соединение 58)
Использовали 3-хлор-4-фторфенилбороновую кислоту.
1Н ЯМР (СDС13) (основание) δ: 1,10 (шир. 3Н), 1,20 (шир. 3Н), 2,26-2,30 (м, 4Н), 2,86-2,91 (м, 4Н), 3,25 (шир. 2Н), 3,50 (шир. 2Н), 6,93-7,30 (м, 7Н).
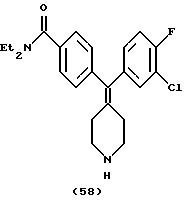
Пример 32
N,N-Диэтил-4-(фторфенилпиперидин-4-илиденметил)бензамид (соединение 59)
Использовали 4-фторфенилбороновую кислоту.
1H ЯМР (СDС13) (основание) δ: 1,11 (шир. 3Н), 1,16 (шир. 3Н), 2,25 (с, 4Н), 2,84 (с, 4Н), 3,20 (шир. 2Н), 3,47 (шир. 2Н), 6,92 (м, 2Н), 7,01 (м, 4Н), 7,23 (д, J=8,8 Гц, 2Н).
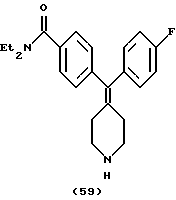
Пример 33
N, N-Диэтил-4-(2-фторфенилпиперидин-4-илиденметил)бензамид (соединение 60)
Использовали 2-фторфенилбороновую кислоту.
1H ЯМР (СDС13) (основание) δ: 1,11 (шир. 3Н), 1,15 (шир. 3Н), 2,10 (т, J= 5,2 Гц, 2Н), 2,27 (т, J=5,2 Гц, 2Н), 2,83 (м, 4Н), 3,20 (шир. 2Н), 3,45 (шир. 2Н), 6,94-7,03 (м, 3Н), 7,10-7,23 (м, 5Н).
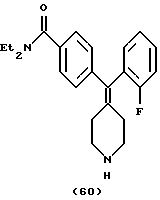
Пример 34
N,N-Диэтил-4-(2,4-дихлорфенилпиперидин-4-илиденметил)бензамид (соединение 61)
Использовали 2,4-дихлорфенилбороновую кислоту.
1H ЯМР (ДМСО) (соль НС1) δ: 1,07 (шир. 6Н), 2,24 (т, 2Н), 2,50 (т, 2Н), 3,10 (т, 2Н), 3,30 (т, 2Н), 3,31 (шир. 2Н), 3,43 (шир. 2Н), 7,25 (д, J=8,4 Гц, 2Н), 7,32 (д, J=8,4 Гц, 2Н), 7,43 (д, J=8,0 Гц, 1Н), 7,47 (д, J=8,0 Гц, 1Н), 7,68 (с, 1Н), 9,20 (шир. 2Н).
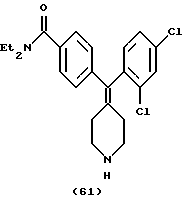
Пример 35
N,N-Диэтил-4-(3,5-дихлорфенилпиперидин-4-илиденметил)бензамид (соединение 62)
Использовали 3,5-дихлорфенилбороновую кислоту.

1H ЯМР (ДМСО) (соль НС1) δ: 1,03 (шир. 6Н), 2,36-2,38 (м, 4Н), 3,0-3,2 (м, 4Н), 3,2 (шир. 2Н), 3,38 (шир. 2Н), 7,19 (с, 1Н), 7,21 (д, J=8,0 Гц, 2Н), 7,29 (д, J=8,0 Гц, 2Н), 7,49 (с, 2Н), 9,10 (шир. 2Н).
Пример 36
N, N-Диэтил-4-(3-ацетилфенилпиперидин-4-илиденметил)бензамид (соединение 63)
Использовали 3-ацетилфенилбороновую кислоту.
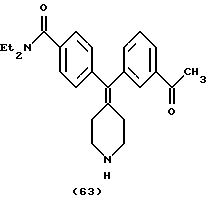
1Н ЯМР (CDC13) (основание) δ: 1,11 (шир. 3Н), 1,20 (шир. 3Н), 2,26 (т, J=5,6 Гц, 2Н), 2,32 (т, J=5,6 Гц, 2Н), 2,55 (с, 3Н), 2,92-2,88 (м, 4Н), 3,26 (шир. 2Н), 3,51 (шир. 2Н), 7,11 (д, J=8,0 Гц, 2Н), 7,29 (д, J=8,0 Гц, 2Н), 7,29 (д, J=7,2 Гц, 1Н), 7,37 (т, J=8,0 Гц, 1Н), 7,70 (с, 1Н), 7,79 (д, J=7,2 Гц, 1Н).
Пример 37
N, N-Диэтил-4-(3,5-трифторметилфенилпиперидин-4-илиденметид)бензамид (соединение 64)
Использовали 3,5-трифторметилфенилбороновую кислоту.
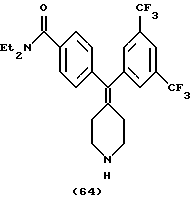
1H ЯМР (ДМСО) (соль НС1) δ: 1,06 (шир. 3Н), 1,08 (шир. 3Н), 2,33 (шир. 2Н), 2,41 (шир. 2Н), 3,12 (шир. 6Н), 3,38 (шир. 2Н), 7,24 (д, J=7,6 Гц, 2Н), 7,30 (д, J=7,6 Гц, 2Н), 7,84 (с, 2Н), 8,00 (с, 2Н), 8,9 (шир. 2Н).
Пример 38
N,N-Диэтил-4-(3-тиофенилпиперидин-4-илиденметил)бензамид (соединение 65)
Использовали 3-тиофенилбороновую кислоту.
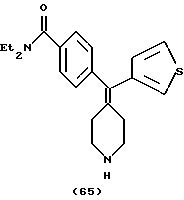
1H ЯМР (ДМСО) (соль НС1) δ: 1,10 (шир. 6Н), 2,44 (т, 2Н), 2,58 (т, 2Н), 3,10-3,15 (м, 4Н), 3,21 (шир. 2Н), 3,44 (шир. 2Н), 6,86 (д, J=4,8 Гц, 1Н), 7,20 (д, J=8,0 Гц, 2Н), 7,32 (д, J=8,0 Гц, 2Н), 7,33 (с, 1Н), 7,52 (д, J=4,8 Гц, 1Н).
Пример 39
N,N-Диэтил-4-(2-тиофенилпиперидин-4-илиденметил)бензамин (соединение 66)
Использовали 2-тиофенилбороновую кислоту.
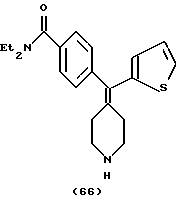
1H ЯМР (CDC13) (основание) δ: 1,12 (шир. 3Н), 1,20 (шир. 3Н), 2,24 (т, J= 5,2 Гц, 2Н), 2,50 (т, J=5,2 Гц, 2Н), 2,85 (т, J=5,6 Гц, 2Н), 2,92 (т, J= 5,6 Гц, 2Н), 3,27 (шир. 2Н), 3,51 (шир. 2Н), 6,75 (д, J=3,6 Гц, 1Н), 6,93 (т, J=3,6 Гц, 1Н), 7,16 (д, J=7,2 Гц, 2Н), 7,21 (д, J=3,6 Гц, 1Н), 7,30 (д, J=7,2 Гц, 2Н).
Пример 40
N,И-Диэтил-4-(4-метилтиофенилпиперидин-4-илиденметил)бензамид (соединение 67)
Использовали 4-метилтиофенилбороновую кислоту.
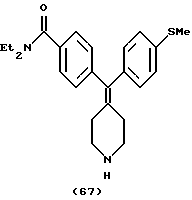
1H ЯМР (СDС13) (основание) δ: 1,11 (шир. 3H), 1,20 (шир. 3Н), 2,32-2,75 (м, 4Н), 2,45 (с, 3Н), 2,90-2,87 (м, 4Н), 3,26 (шир. 2Н), 3,51 (шир. 2Н), 7,01 (д, J=6,0 Гц, 2Н), 7,10 (д, J=6,0 Гц, 2Н), 7,15 (д, J=6,8 Гц, 2Н), 7,27 (д, J=6,8 Гц, 2Н).
Пример 41
N, N-Диэтил-4-(3-аминофенилпиперидин-4-илиденметил)бензамид (соединение 68)
Использовали 3-аминофенилбороновую кислоту.
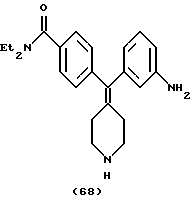
1Н ЯМР (СDС13) (основание) δ: 1,11 (шир. 3Н), 1,20 (шир. 3Н), 2,27-2,33 (м, 4Н), 2,86-2,90 (м, 4Н), 3,27 (шир. 2Н), 3,51 (шир. 2Н), 3,57 (шир. 2Н), 3,68 (с, 1Н), 6,39 (с, 1Н), 6,52 (дд, J=1,6 Гц, J=7,6 Гц, 2Н), 7,06 (т, J= 8,0 Гц, 1Н), 7,12 (д, J=6,4 Гц, 2Н), 7,26 (д, J=6,4 Гц, 2Н).
Пример 42
N, N-Диэтил-4-(4-трифторметилфенилпиперидин-4-илиденметил)бензамид (соединение 69)
Использовали 4-трифторметилфенилбороновую кислоту.
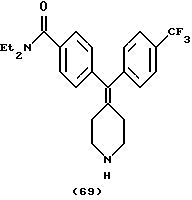
1Н ЯМР (ДМСО) (соль НСl) δ: 1,05 (шир. 6Н), 2,35 (т, 2Н), 2,40 (т, 2Н), 3,09 (м, 6Н), 3,35 (шир. 2Н), 7,17 (д, J=8,0 Гц, 2Н), 7,28 (д, J=8,0 Гц, 2Н), 7,35 (д, J=8,0 Гц, 2Н), 7,67 (д, J=8,0 Гц, 2Н), 8,71 (шир. 2Н).
Пример 43
N,N-Диэтил-4-(4-метоксифенилпиперидин-4-илиденметил)бензамид (соединение 70)
Использовали 4-метоксифенилбороновую кислоту.
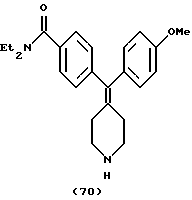
1Н ЯМР (СDСl3) (основание) δ: 1,12 (шир. 3Н), 1,19 (шир. 3Н), 2,29 (м, 4Н), 2,87 (м, 4Н), 3,27 (шир. 2Н), 3,51 (шир. 2Н), 3,77 (с, 3Н), 6,80 (м, 2Н), 7,00 (м, 2Н), 7,10 (д, J=8,4 Гц, 2Н), 7,26 (д, J=8,4 Гц).
Пример 44
N,N-Диэтил-4-(3,4-дихлорфенилпиперидин-4-илиденметил)бензамид (соединение 71)
Использовали 3,4-дихлорфенилбороновую кислоту.
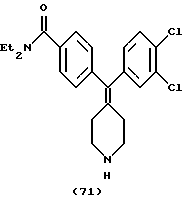
1Н ЯМР (СDCl3) (основание) δ: 1,12 (шир. 3Н), 1,20 (шир. 3Н), 2,28 (т, J= 5,6 Гц, 4Н), 2,89 (м, 4Н), 3,27 (шир. 2Н), 3,52 (шир. 2Н), 6,8-7,4 (м, 7Н).
Пример 45
N, N-Диэтил-4-(2-трифторметилфенилпиперидин-4-илидeнмeтил)бензамид (соединение 72)
Использовали 2-трифторметилфенилбороновую кислоту.
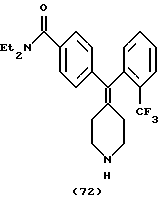
1Н ЯМР (СDС13) (основание) δ: 1,05 (шир. 3Н), 1,16 (шир. 3Н), 1,95 (м, 2Н), 2,35-2,41 (м, 2Н), 2,7-2,9 (м, 4Н), 3,20 (шир. 2Н), 3,48 (шир. 2Н), 7,2-7,6 (м, 8Н).
Пример 46
N,N-Диэтил-4-(3-толуилпиперидин-4-илиденметил)бензамид (соединение 73)
Использовали м-толилбороновую кислоту.
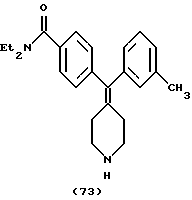
1H ЯМР (СDС13) (основание) δ: 1,11 (шир. 3Н), 1,19 (шир. 3H), 2,28 (с, 3Н), 2,29 (м, 4Н), 2,89 (м, 4Н), 3,27 (шир. 2Н), 3,51 (шир. 2Н), 6,8-7,3 (м, 8Н).
Пример 47
N,N-Диэтил-4-(2-метоксифенилпиперидин-4-илиденметил)бензамид (соединение 74)
Использовали 2-метоксифенилбороновую кислоту.
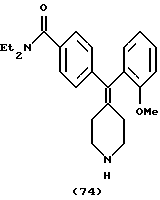
1Н ЯМР (СDС13) (основание) δ: 1,09 (шир. 3Н), 1,18 (шир. 3Н), 2,10 (кв, J= 4,8 Гц, 2Н), 2,31 (кв, J=4,8 Гц, 2Н), 2,8-2,9 (м, 4Н), 3,25 (шир. 2Н), 3,50 (шир. 2Н), 3,68 (с, 3Н), 6,83-6,90 (м, 2Н), 7,0 (д, 1Н), 7,15-7,25 (м, 5Н).
Пример 48
N, N-Диэтил-4-(3-формилфенилпиперидин-4-илиденметил)бензамид (соединение 75)
Использовали 3-формилфенилбороновую кислоту.
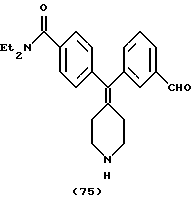
1Н ЯМР (СDС13) (основание) δ: 1,15 (шир. 3Н), 1,20 (шир. 3Н), 2,26-2,34 (м, 4Н), 2,90-2,92 (м, 4Н), 3,28 (шир. 2Н), 3,2 (шир. 2Н), 7,11-7,31 (м, 8Н), 9,96 (с, 1Н).
Пример 49
N,N-Диэтил-4-(2-нафтилпиперидин-4-илиденметил)бензамид (соединение 76)
Использовали 2-нафтилбороновую кислоту.

lН ЯМР (СDС13) (основание) δ: 1,11 (шир. 3Н), 1,20 (шир. 3Н), 2,35-2,39 ( м, 4Н), 2,91-2,96 (м, 4Н), 3,27 (шир. 2Н), 3,51 (шир. 2Н), 7,16-7,40 (м, 5Н), 7,42-7,44 (м, 2Н), 7,57 (с, 1Н), 7,72-7,79 (м, 2Н).
Пример 50
N, N-Диэтил-4-(2-формилфенилпиперидин-4-илиденметил)бензамид (соединение 77)
Использовали 2-формилфенилбороновую кислоту.
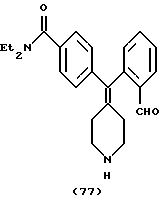
1Н ЯМР (СDСl3) (основание) δ: 1,09 (шир. 3Н), 1,18 (шир. 3Н), 1,70-2,10 (м, 2Н), 2,40-2,49 (м, 2Н), 2,76-2,84 (м, 2Н), 2,85-2,97 (м, 2Н), 3,23 (шир. 2Н), 3,48 (шир. 2Н), 7,13-7,40 (м, 6Н), 7,53-7,55 (м, 1Н), 7,90 (д, J=7,6 Гц, 1Н), 10,27 (с, 1Н).
Пример 51
N, N-Диэтил-4-(4-ацетилфенилпиперидин-4-илиденметил)бензамид (соединение 78)
Использовали 4-ацетилфенилбороновую кислоту.
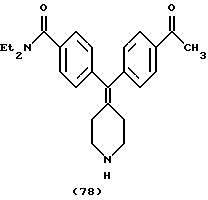
1Н ЯМР (СDС13) (основание) δ: 1,11 (шир. 3Н), 1,20 (шир. 3Н), 2,30-2,35 (м, 4Н), 2,56 (с, 3Н), 2,92 (м, 4Н), 3,27 (шир. 2Н), 3,52 (шир. 2Н), 7,10-7,30 (м, 6Н), 7,87 (д, J=7,2 Гц, 2Н).
Пример 52
N, N-Диэтил-4-(3-трифторметилфенилпиперидин-4-илиденметил)бензамид (соединение 79)
Использовали 3-трифторметилфенилбороновую кислоту.
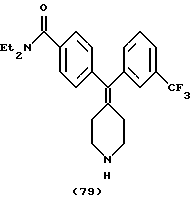
1Н ЯМР (СDС13) (основание) δ: 1,11 (шир. 3Н), 1,20 (шир. 3Н), 2,26 (т, J= 5,6 Гц, 2Н), 2,31 (т, J=5,6 Гц, 2Н), 2,88-2,91 (м, 4Н), 3,27 (шир. 2Н), 3,52 (шир. 2Н), 7,10-7,47 (м, 8Н).
Пример 53
Получение N, N-диэтил-4-([1-(2,6-диаминогексанол)пиперидин-4-илиден]фенилметил)бензамида (соединение 80)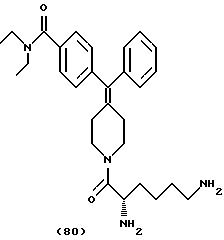
L-Boc-лизин (Cbz) (0,38 г, 1,0 ммоль) растворяли в сухом тетрагидрофуране (5 мл) в атмосфере азота при -15oС. Добавляли N-метилморфолин (0,11 мл, 1,0 ммоль), затем изобутилхлорформиат (0,13 мл, 1 ммоль). После перемешивания в течение 10 минут добавляли N,N-диэтил-4-(фенилпиперидин-4-илиденметил)бензамид (соединение 6) (0,35 г, 1,0 ммоль) в тетрагидрофуране (1 мл) и температуре давали повыситься до 25oС в течение 2 часов. Реакционную смесь выпаривали на силикагеле. MPLC на силикагеле (от 0 до 100% этилацетат в гептане) давала 0,4 г.
Продукт (0,40 г, 0,56 ммоль) растворяли в метиленхлориде (10 мл) и обрабатывали трифторуксусной кислотой (3 мл) в течение 30 мин, затем летучие компоненты выпаривали. Остаток растворяли в уксусной кислоте (25 мл) и подвергали гидрогенолизу в течение 1,5 часов водородом (1 атм) над палладием на угле (10%, 0,10 г). Растворитель выпаривали и остаток очищали хроматографией на короткой колонке с обращенной фазой (RP-18), элюируя 0-30% ацетонитрилом в воде. Свободный амин экстрагировали 5% раствором карбонат калия/метиленхлорид, получая 123 мг, и затем обрабатывали двумя эквивалентами хлористоводородной кислоты в смеси метанол/вода. Лиофилизация давала дигидрохлоридную соль.
1H ЯМР (свободный амин, СD3ОD): δ 1,0-1,7 (м, 16Н, амид-Ме, пиперидин-Н, лизин-Н), 2,3-2,7 и 3,0-4,5 (м, 11Н, амид-Н, пиперидин-Н, лизин-Н), 4,8 (с, 4Н, 2 NH2), 7,10-7,50 (м, 9Н, ArH).
Элементный анализ. Вычислено для C29H40N4O2 • 2,4 H2O • 2 HCl: С 58,76%; Н 7,96%; N 9,43%. Найдено: С 58,70%; Н 7,51%; N 9,33%.
Пример 54
Получение фосфонооксиметилового эфира 4-[(4-диэтилкарбамоилфенил)фенилметилен]пиперидин-1-карбоновой кислоты (соединение 81)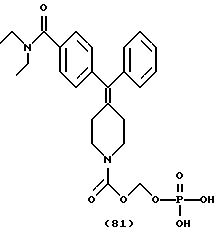
N,N-Диэтил-4-(фенилпиперидин-4-илиденметил)бензамид (соединение 6) (0,62 г, 1,8 ммоль) растворяли в метиленхлориде (10 мл) и добавляли 1,8-бисдиаминонафталин (0,42 г, 2,0 ммоль). Раствор охлаждали до 0oС и по каплям добавляли хлорметилхлорформиат (0,25 г, 2,0 ммоль) в метиленхлориде (1 мл). После выдерживания в течение 2 часов при 25oС добавляли дополнительную порцию сначала 1,8-бисдиаминонафталина (0,21 г, 1,0 ммоль), затем хлорметилхлорформиата (0,12 г, 1,0 ммоль). После общего времени выдерживания 4 часа раствор промывали 1 М НС1, солевым раствором, сушили (MgSO4) и выпаривали, получая 0,62 г. Остаток растворяли в толуоле (25 мл), добавляли дибензилфосфат серебра (0,81 г, 2,1 ммоль) и смесь нагревали 3 часа при 80oС. Раствор фильтровали, затем промывали 5% раствором карбоната калия, солевым раствором, сушили (К2СО3) и выпаривали. MPLC на силикагеле (от 0 до 100% этилацетат в гептане) давала 0,66 г (0,96 ммоль, 54%). Остаток растворяли в этилацетате (50 мл) и подвергали гидрогенолизу (1 атм водорода) с палладием на угле (10%, 0,3 г) в течение 2 час. После фильтрования и выпаривания растворителя продукт обрабатывали двумя эквивалентами гидроксида натрия в смеси метанол/вода. Лиофилизация давала двунатриевую соль продукта в виде белого твердого материала.
1H ЯМР (D2O): δ 1,03, 1,20 (2 м, 6Н, амид-Ме), 2,34 (м, 4Н, пиперидин-Н), 3,19-3,61 (м, 8Н, амин-СН2, пиперидин-Н), 5,44 (д, J=13 Гц, 2Н, ОСН2О), 7,18-7,36 (м, 9Н, ArH).
Соединения 80 и 81 соответственно представляют собой подходящие пролекарства соединений формулы (I).
(П) Схема, синтеза соединений примеров 55-57
Соединения примеров 55, 56 и 57 были получены по методике схемы 7.
(i) Получение трет-бутил-4-{бром[4-(морфолинокарбонил)фенил]метилен}-1-пиперидинкарбоксилата (соединение 82)
К раствору соединения 51, полученного по схеме 6 (0,25 г, 0,625 ммоль), и свежеперегнанного триэтиламина (0,5 мл) в дихлорметане (12 мл) добавляли по каплям при комнатной температуре оксалилхлорид (0,38 мл, 2,0 М, 0,75 ммоль). Раствор перемешивали в течение 10 минут при комнатной температуре и растворитель и избыточные реагенты удаляли в вакууме, получая хлорангидрид в виде сырого продукта, который использовали на следующей стадии без дополнительной очистки.
Морфолин (56 мг, 0,65 ммоль) добавляли к раствору хлорангидрида (0,65 ммоль) и триэтиламина (0,5 мл) в дихлорметане (5 мл). Реакционную смесь оставляли для прохождения реакции в течение одного часа при комнатной температуре. Растворитель затем удаляли в вакууме. Сырой продукт распределяли между этилацетатом (25 мл) и водой (25 мл). Водный слой промывали этилацетатом и объединенные слои этилацетата промывали 2 М NaOH (2 • 25 мл), 2 М НС1 (2 • 25 мл), солевым раствором (1 • 25 мл) и сушили над сульфатом магния. Растворитель удаляли в вакууме, получая продукт (соединение 82) (294 мг, выход 97%).
1H ЯМР (СDС13, 400 МГц): 1,44 (с, 9Н), 2,21 (т, J=5,6 Гц, 2Н), 2,62 (т, J= 5,6 Гц, 2Н), 3,31 (т, J=5,6 Гц, 2Н), 3,52 (т, J=5,6 Гц, 2Н), 3,69 (шир., 8Н), 7,31 (д, J=6,4 Гц, 2Н), 7,37 (д, J=6,4 Гц, 2Н).
(ii) Получение трет-бутил-4-{бром-[4-(пиперидинокарбонил)фенил]метилен} -1-пиперидинкарбоксилата (соединение 83)
Использовали ту же методику, как описана для получения соединения 82, но с применением пиперидина вместо морфолина.
1Н ЯМР (СDС13, 400 МГц): 1,44 (с, 9Н), 1,51 (шир., 2Н), 1,66 (шир., 4Н), 2,21 (т, J=5,6 Гц, 2Н), 2,62 (т, J=5,6 Гц, 2Н), 3,31 (т, J=5,6 Гц, 2Н), 3,33 (шир., 2Н), 3,52 (т, J=5,6 Гц, 2Н), 3,68 (шир., 2Н), 7,26 (д, J=8,4 Гц, 2Н), 7,35 (д, J=8,4 Гц, 2Н).
(iii) Получение трет-бутил-4-{ бром-[4-(тетрагидро-1H-1-пирролилкарбонил)фенил]метилен}-1-пиперидинкарбоксилата (соединение 84)
Использовали ту же методику, как описана для получения соединения 82, но с применением пирролидина вместо морфолина.
1H ЯМР (СDС13, 400 МГц): 1,44 (с, 9Н), 1,87 (к, J=6,8 Гц, 2Н), 1,95 (к, J= 6,8 Гц, 2Н), 2,20 (т, J=5,6 Гц, 2Н), 2,62 (т, J=5,6 Гц, 2Н), 3,31 (т, J= 5,6 Гц, 2Н), 3,43 (т, J=6,8 Гц, 2Н), 3,52 (т, 5,6 Гц, 2Н), 3,63 (т, J=6,8 Гц, 2Н), 7,27 (д, J=8,0 Гц, 2Н), 7,47 (д, J=8,0 Гц, 2Н).
Пример 55
Получение 4-[(3-фторфенил)пиперидин-4-илметил]фенилморфолин-4-илметанона (соединение 85)
К раствору соединения 82 (37 мг, 0,085 ммоль) и тетракис(трифенилфосфин)палладия(0) (5 мг, 0,0043 ммоль) в ксилолах (дегазированные, 0,5 мл) добавляли 3-фторфенилбороновую кислоту (25 мг, 0,18 ммоль) в этаноле (дегазирован, 0,5 мл), затем 150 мкл 2 М Na2SО4 (водный) (300 мкмоль). Реакционную смесь оставляли для прохождения реакции при 80oС в течение 2 час в атмосфере аргона. Реакционную смесь разбавляли водой (1 мл) и диэтиловым простым эфиром (1 мл) и перемешивали с завихрением. Органическую фазу отделяли и выпаривали, получая сырой продукт, который использовали без дополнительной очистки.
Вос-группу удаляли обработкой сырого продукта 1 мл ТФУ. Через 30 минут при комнатной температуре ТФУ выпаривали, получая сырую соль ТФУ. Соль нейтрализовали 1 М NH4OH (1,0 М) и экстрагировали диэтиловым простым эфиром (2•1 мл). Эфирную фазу подкисляли 4,0 М НС1 в диоксане (200 мкл) и соль НС1 экстрагировали водой (2•1 мл). Водный раствор соли промывали диэтиловым простым эфиром (2•1 мл) и лиофилизировали, получая продукт в виде белого порошка.
1Н ЯМР (СDС13, 400 МГц) δ: 2,67 (м, 4Н), 3,19 (м, 4Н), 3,45 (шир., 2Н), 3,68 (шир. , 6Н), 6,75 (д, J=9,6 Гц, 1Н), 6,85 (д, J=8,0 Гц, 1Н), 6,95 (м, 1Н), 7,11 (д, J=7,6 Гц, 2Н), 7,25 (с, 1Н), 7,35 (д, J=7,6 Гц, 2Н).
Пример 56
Получение 4-[(3-фторфенил)пиперидин-4-илметил] фенилпиперидин-4-илметанона (соединение 86)
Использовали методику, описанную для получения соединения 85, но с использованием соединения 83 в качестве исходного материала.
1H ЯМР (СDС13, 400 МГц): δ 1,51 (шир., 2Н), 1,65 (шир., 4Н), 2,60 (шир., 4Н), 3,14 (шир., 4Н), 3,33 (шир., 2Н), 3,68 (шир., 2Н), 6,67 (д, J=8,0 Гц, 1Н), 6,86 (д, J=8,0 Гц, 1Н), 6,93 (т, J=8,0 Гц, 1Н), 7,08 (д, J=8,4 Гц, 2Н), 7,25 (с, 1Н), 7,32 (д, J=8,4 Гц, 2Н).
Пример 57
Получение 4-[(3-фторфенил)пиперидин-4-илметил] фенилпирролидин-1-илметанона (соединение 87)
Использовали методику, описанную для получения соединения 85, но с использованием соединения 84 в качестве исходного материала.
1Н ЯМР (CDCl3, 400 МГц) δ: 1,84-1,89 (м, 2Н), 1,90-1,98 (м, 2Н), 2,60-2,63 (м, 4Н), 3,13-3,17 (м, 4Н), 3,41 (т, J=6,8 Гц, 2Н), 3,62 (т, J=6,8 Гц), 6,73 (д, J=8,8 Гц, 1Н), 6,86 (д, J=7,2 Гц, 1Н), 6,93 (м, 1Н), 7,10 (д, J=8,0 Гц, 2Н), 7,25 (с, 1Н), 7,45 (д, J=8,0 Гц, 2Н).
(Н) Схема синтеза соединений примеров 58-68
Соединения примеров 58-68 были получены по методике, приведенной на схемах 8(а) - (с).
(i) Получение трет-бутилового эфира 4-[бром-(4-этоксикарбониламинофенил)метил]пиперидин-1-карбоновой кислоты (соединение 88)
К смеси соединения 51, полученного по схеме 6 (0,25 г, 0,625 ммоль), в толуоле (5 мл) добавляли дифенилфосфорилазид (0,192 г, 0,70 ммоль) и триэтиламин (0,1 мл, 0,7 ммоль). После перемешивания смеси в атмосфере аргона при 95oС в течение двух часов добавляли избыток безводного этанола (2 мл) и триэтиламин (0,1 мл) и раствор перемешивали при 95oС в течение дополнительных 5 часов. После охлаждения до комнатной температуры реакционную смесь распределяли между водой и диэтиловым эфиром. Эфирный слой промывали водой, сушили над сульфатом магния и выпаривали в вакууме, получая продукт (соединение 88) в виде рыжевато-коричневой пены (300 мг, выход 99%).
1Н ЯМР (400 МГц, СDС13): 1,30 (т, J=7,2 Гц, 3Н), 1,44 (с, 9Н), 2,22 (т, J= 6,0 Гц, 2Н), 2,60 (т, J=6,0 Гц, 2Н), 3,31 (т, J=6,0 Гц, 2Н), 3,51 (т, J= 6,0 Гц, 2Н), 4,21 (кв, J=7,2 Гц, 2Н), 6,58 (с, 1Н), 7,19 (д J=8,4 Гц, 2Н), 7,33 (д, J=8,4 Гц, 2Н).
(ii) Получение трет-бутилового эфира 4-[(4-этоксикарбониламинофенил)-(3-фторфенил)метил]пиперидин-1-карбоновой кислоты (соединение 92)
Сочетание по Сузуки четырех винилбромидов (соединения 88-91) с 3-фторфенилбороновой кислотой проводили параллельно. Реакции и жидкостно-жидкостные экстракции проводили в пробирках для культур размером 25 мм•150 мм. Методика типичной реакции описана ниже.
К раствору соединения 88 (0,30 г, 0,625 ммоль) и тетракис(трифенилфосфин)палладия(0) (50 мг) в толуоле (дегазирован, 0,5 мл) добавляли 3-фторфенилбороновую кислоту (0,182 г, 1,3 ммоль) в этаноле (дегазирован, 0,5 мл), затем 0,75 мл 2 М Na2СО3 (водный) (1,5 ммоль). Реакционную смесь оставляли для прохождения реакции при 80oС в течение 3 часов в атмосфере аргона. Реакционную смесь разбавляли водой и диэтиловым простым эфиром и перемешивали с завихрением. Органическую фазу отделяли и выпаривали, получая сырой продукт. Сырой продукт очищали хроматографией на силикагеле (0-50% EtOAc в гексанах), получая продукт (соединение 92) в виде белого порошка (0,166 г, выход 58%).
1H ЯМР (400 МГц, СDС13) δ: 1,25 (т, J=7,2 Гц, 3Н), 1,44 (с, 9Н), 2,27-2,33 (м, 4Н), 3,41-3,44 (м, 4Н), 4,20 (кв, J=7,2 Гц, 2Н), 6,52 (с, 1Н), 6,76 (д, J= 10 Гц, 2Н), 6,85-6,89 (м, 2Н), 7,01 (д, J=8,8 Гц, 2Н), 7,19-7,23 (м, 1Н), 7,28 (д, J=8,8 Гц, 2Н).
Пример 58
Получение этилового эфира 4-[(3-фторфенил)пиперидин-4-илметил] фенилкарбаминовой кислоты (соединение 96)
Удаление Вос-защитной группы проводили в небольшом масштабе параллельно в пробирках для испытаний (13 мм•100 мм). Типичная методика описана ниже.
Вос-группу удаляли обработкой соединения 92 (50 мг, 0,11 ммоль) НС1 в диоксане (4,0 М, 2 мл). Смесь перемешивали при комнатной температуре в течение 30 минут. Растворитель и HCl удаляли в вакууме, получая соединение-продукт 96 в виде белого порошка после лиофилизации (40 мг, выход 99%).
1Н ЯМР (400 МГц, СDС13) δ: 1,28 (т, J=7,2 Гц, 3Н), 2,27-2,31 (м, 4Н), 2,85-2,91 (м, 4Н), 4,19 (кв, J=7,2 Гц, 2Н), 6,50 (с, 1Н), 6,76 (д, J=10 Гц, 1Н), 6,85-6,89 (м, 2Н), 7,01 (д, J=8,8 Гц, 2Н), 7,19-7,23 (м, 1Н), 7,28 (д, J=8,8 Гц, 2Н).
Пример 59
Получение этилового эфира 4-[(3-фторфенил)пиперидин-4-илметил]фенилметилкарбаминовой кислоты (соединение 100)
Алкилирование амидного азота проводили в небольшом масштабе параллельно в пробирках для испытания (13 мм • 100 мм). Типичная методика описана ниже.
К раствору соединения 92 (50 мг, 0,11 ммоль) в дихлорметане (1,5 мл) добавляли метилиодид (31 мг, 0,22 ммоль), водный гидроксид натрия (1,0 мл, 2 М) и сульфат тетрабутиламмония (44 мг, 0,13 ммоль). Раствор кипятили с обратным холодильником в течение одного часа. После охлаждения до комнатной температуры слой дихлорметана отделяли и выпаривали. К остатку добавляли простой эфир и белый иодид тетрабутиламмония удаляли фильтрованием. Эфир удаляли в вакууме, получая сырой продукт-соединение 100 в виде светлого масла. Вос-группу удаляли обработкой HC1 в диоксане, как описано выше, получая продукт в виде белого порошка после лиофилизации (17 мг, выход 42%).
1Н ЯМР (400 МГц, CDC13) δ: 1,23 (т, J=7,2 Гц, 3Н), 2,27-2,33 (м, 4Н), 2,85-2,91 (м, 4Н), 3,26 (с, 3Н), 4,15 (кв, J=7,2 Гц, 2Н), 6,78 (д, J=10 Гц, 1Н), 6,85-6,89 (м, 2Н), 7,05 (д, J=8,0 Гц, 2Н), 7,14 (д, J=8,0 Гц, 2Н), 7,19-7,23 (м, 1Н).
Пример 60
Получение этилового эфира 4-[(1-бензилпиперидин-4-ил)-(3-фторфенил)метил]фенилкарбаминовой кислоты (соединение 116)
Бензилирование соединения 100 проводили в небольшом масштабе параллельно в пробирках для испытания (13 мм • 100 мм). Типичная методика описана ниже.
Соединение 100 в форме свободного основания получали добавлением гидроксида аммония (1 М, 0,5 мл) к водному раствору соединения 100 (0,046 ммоль) и экстрагировали в простой эфир. Эфир удаляли в вакууме, получая масло, которое растворяли в дихлорметане и обрабатывали бензилбромидом (0,14 мл, 0,5 М в дихлорметане) и триэтиламином (0,05 мл). Раствор перемешивали при комнатной температуре в течение 5 часов. Растворитель удаляли в вакууме. Продукт растворяли в смеси вода/ацетонитрил/НСl (2:1:0,5 М) и лиофилизировали, получая продукт-соединение 108 в виде белого порошка.
1H ЯМР (400 МГц, СDС13) δ: 1,28 (т, J=7,2 Гц, 3Н), 2,33-2,36 (м, 4Н), 2,38-2,46 (м, 4Н), 3,51 (с, 2Н), 4,19 (кв, J=7,2 Гц, 2Н), 6,50 (с, 1Н), 6,78 (д, J= 10 Гц, 1Н), 6,85-6,89 (м, 2Н), 7,05 (д, J=8,0 Гц, 2Н), 7,19-7,30 (м, 7Н).
Примеры 61-68
Соединения также были получены синтетическими путями, описанными на схемах 8(а)-(с).
В предпочтительном варианте осуществления изобретения, известном в настоящее время, используют соединения 6, 7, 9, 10, 12, 26, 27, 34, 39, 44, 58, 59, 62, 69, 71, 104, 106 и 109.
Фармацевтические композиции
Новые соединения по настоящему изобретению можно вводить перорально, внутримышечно, подкожно, местно, интраназально, внутрибрюшинно, интраторакально, внутривенно, эпидурально, внутриоболочечно, интрацеребровентрикулярно и путем инъекции в суставы.
Предпочтительный способ введения пероральный, внутривенный или внутримышечный.
Доза будет зависеть от способа введения, тяжести болезни, возраста и веса пациента и других факторов, обычно рассматриваемых лечащим врачом, при определении индивидуального режима приема и дозы лекарственного средства, наиболее подходящих для конкретного пациента.
Для получения фармацевтических композиций из соединений данного изобретения инертные, фармацевтически приемлемые носители могут быть либо твердыми, либо жидкими. Препараты в твердой форме включают порошки, таблетки, диспергируемые гранулы, капсулы, саше и суппозитории.
Твердым носителем может быть одно или несколько веществ, которые могут действовать в качестве разбавителей, корригентов, солюбилизаторов, смазывающих веществ, суспендирующих агентов, связующих агентов или агентов, разрыхляющих таблетки; им может быть также капсулирующий материал.
В порошках носитель представляет собой тонкоизмельченный твердый материал, который находится в смеси с тонкоизмельченным активным компонентом. В таблетках активный компонент смешивают с носителем, обладающим необходимыми связующими свойствами, в подходящих пропорциях и прессуют для получения необходимой формы и размера.
Для получения композиций в форме суппозиториев низкоплавящийся воск, такой как смесь глицеридов жирных кислот и масла какао, сначала расплавляют и в ней диспергируют активный ингредиент, например, путем перемешивания. Расплавленную гомогенную смесь затем выливают в формы общепринятого размера и оставляют для охлаждения и затвердевания.
Подходящими носителями являются карбонат магния, стеарат магния, тальк, лактоза, сахар, пектин, декстрин, крахмал, трагакант, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, низкоплавкий воск, масло какао и подобные.
Фармацевтически приемлемые соли представляют собой ацетат, бензолсульфонат, бензоат, бикарбонат, битартрат, бромид, кальцийацетат, камсилат, карбонат, хлорид, цитрат, дигидрохлорид, этилендиаминтетраацетат, эдисилат, эстолат, эзилат, фумарат, глюкаптат, глюконат, глутаминат, гликолли-ларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, иодид, изотионат, лактат, лактобионат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, памоат (эмбонат), пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, субацетат (основная уксуснокислая соль), сукцинат, сульфат, таннат, тартрат, теоклат, триэтиодид, соль бензтина, хлорпрокаина, холина, диэтаноламина, этилендиамина, меглумина, прокаина, алюминия, кальция, лития, магния, калия, натрия и цинка.
Предпочтительными фармацевтически приемлемыми солями являются гидрохлориды и цитраты.
Термин композиция относится к готовой препаративной форме активного компонента с капсулирующим материалом в качестве носителя, обеспечивающего образование капсулы, в которой активный компонент (с другими носителями или без них) окружается носителем, который, таким образом, находится в ассоциации с ним. Аналогичным образом включаются саше.
В качестве твердых дозированных форм, подходящих для перорального введения, можно использовать таблетки, порошки и капсулы.
Композиции в жидкой форме включают растворы, суспензии и эмульсии. В качестве примеров жидких препаратов, подходящих для парентерального введения, можно упомянуть растворы активных соединений в стерильной воде или смеси вода/ пропиленгликоль. Жидкие композиции можно также изготовить в водном растворе полиэтиленгликоля.
Водные растворы для перорального введения можно получить растворением активного компонента в воде и добавлением подходящих красителей, корригентов, стабилизаторов и загустителей, если необходимо. Водные суспензии для перорального использования можно приготовить диспергированием тонкоизмельченного активного компонента в воде вместе с вязким материалом, таким как природные и синтетические камеди, смолы, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, и другими суспендирующими агентами, известными в области фармацевтических готовых препаративных форм.
Предпочтительными фармацевтическими композициями является единичная дозированная форма. В такой форме композиция разделена на единичные дозы, содержащие подходящее количество активного компонента. Единичная дозированная форма может быть упакованным препаратом, причем упаковка содержит дискретные количества препаратов, например упакованные таблетки, капсулы и порошки в пузырьках или ампулах. Единичная дозированная форма может сама также представлять капсулу, саше или таблетку или она может представлять подходящее число любых из этих упакованных форм.
Биологическая оценка
А) Модель in vitro
Клеточная культура
Клетки 293S человека, экспрессирующие клонированные рецепторы человека μ, δ и χ и устойчивые к неомицину, выращивали в суспензии при 37oС в атмосфере с 5% CO2 во встряхиваемых колбах, содержащих DMEM (модифицированную по способу Дульбекко среду Игла), не содержащую кальция, 10% ФТС, 5% BCS, 0,1% плюроник F-68 и 600 мкг/мл генетицина.
Препарат мембраны
Клетки собирали в виде осадка после центрифугирования и повторно суспендировали в буфере для лизиса (50 мМ Трис, рН 7,0, 2,5 мМ ЭДТУ с ФМСФ, добавленным непосредственно перед использованием до 0,1 мМ из 0,1 М исходного раствора в этаноле), инкубировали на льду в течение 15 мин, затем гомогенизировали с политроном в течение 30 сек. Суспензию центрифугировали при 1000 g (mах) в течение 10 мин при 4oС. Супернатант сохраняли на льду и осадок повторно суспендировали и центрифугировали, как указано ранее. Супернатанты двух центрифугирований объединяли и центрифугировали при 46000 g (mах) в течение 30 мин. Осадок повторно суспендировали в холодном буфере Трис (50 мМ Трис/Сl, рН 7,0) и центрифугировали снова. Конечный осадок повторно суспендировали в буфере для мембран (50 мМ Трис, 0,32 М сахароза, рН 7,0). Аликвоты (1 мл) в полиэтиленовых пробирках замораживали в системе сухой лед/этанол и сохраняли при -70oС до использования. Концентрации белка определяли модифицированным анализом Lowry с ДСН.
Анализы связывания
Мембраны оттаивали при 37oС, охлаждали на льду, пропускали 3 раза через иглу 25 G и разводили в буфере для связывания (50 мМ Трис, 3 мМ MgCl2, 1 мг/мл АБС (Sigma A-7888), рН 7,4, который сохраняли при 4oС после фильтрования через фильтр 0,22 м и к которому только добавляли 5 мкг/мл свежего апротинина, 10 мкМ бестатина, 10 мкМ дипротина А без ДТТ). В охлажденные на льду полипропиленовые пробирки размером 12•75 мм, содержащие 100 мкл подходящего радиоактивного лиганда (см. таблицу 1) и 10 мкл испытуемых пептидов при различных концентрациях, добавляли аликвоты 100 мкл (на 1 мкг белка, см. таблицу 1). Общее (ОС) и неспецифическое (НС) связывание определяли в отсутствие и в присутствии 10 мкМ налоксона соответственно. Пробирки встряхивали вортексом и инкубировали при 25oС в течение 60-75 мин, после этого времени содержимое быстро фильтровали в вакууме и промывали около 12 мл/пробирку охлажденного льдом буфера для промывки (50 мМ Трис, рН 7,0, 3 мМ MgCl2) через фильтры GF/B (Ватман), предварительно вымоченные в течение, по меньшей мере, 2 часов в 0,1% полиэтиленимине. Радиоактивность (число распадов в минуту), оставшуюся на фильтрах, измеряли бета-счетчиком после вымачивания фильтров в течение, по меньшей мере, 12 час в миниампулах, содержащих 6-7 мл сцинтилляционной жидкости. Если анализ проводили в планшетах с 96 глубокими лунками, фильтрование выполняли через 96-местный единый фильтр, предварительно вымоченный в ПЭИ, который промывали 3•1 буфером для промывания и сушили в сушильном шкафу при 55oС в течение 2 часов. Радиоактивность фильтра для планшета подсчитывали в TopCount (Packard) после добавления 50 мкл сцинтилляционной жидкости MS-20/лунку.
Анализ данных
Специфическое связывание (СС), вычисляемое как ОС-НС и СС в присутствии различных испытуемых пептидов, выражали как процент СС от контроля. Величины IC50 и коэффициент Хилла (nH) для лигандов и в вытесненном специфически связанном радиоактивном лиганде вычисляли по логарифмическому графику или программе для построения кривых, таких как Ligand, GraphPad Prism, SigmaPlot или RecepyorFit. Величины Кi вычисляли по уравнению Cheng-Prussoff. Величины средних значений ± среднеквадратичная ошибка для IC50, Кi и nH приводились для испытуемых лигандов, по меньшей мере, на основании трех кривых вытеснения.
Эксперименты по насыщению рецепторов
Величины Kδ радиоактивного лиганда определяли путем проведения анализов связывания на клеточных мембранах с подходящими радиоактивными лигандами при концентрациях в диапазоне от 0,2 до 5-кратных концентраций оцененного Kδ (вплоть до 10-кратного, если возможны такие количества требуемого радиоактивного лиганда). Специфическое связывание радиоактивного лиганда выражали как пмоль/мг мембранного белка. Величины Kδ и Вmах из отдельных экспериментов получали из нелинейной зависимости специфически связанного (В) лиганда от нМ свободного (F) радиоактивного лиганда из отдельного эксперимента согласно односайтовой модели.
В) биологическая модель (модель in vivo)
Механо-аллодиния у крысы, индуцированная полным адъювантом фрейнда (ПАФ) и манжеткой, помещенной на седалищный нерв
Животные
Использовали самцов крыс Sprague-Dawley (Charles River, St-Constant, Canada), весящих 175-200 г на момент хирургической операции. Их поселяли группами по три особи в боксах, в которых термостатически поддерживали 20oС и устанавливали суточный цикл освещение/темнота 12:12, причем крысам обеспечивали свободный доступ к корму и воде. После помещения в боксы животных оставляли для акклиматизации в течение, по меньшей мере, 2 дней до хирургической операции. Эксперименты были одобрены Medical Ethical Committee для исследования животных.
ЭКСПЕРИМЕНТАЛЬНАЯ МЕТОДИКА
ПОЛНЫЙ АДЪЮВАНТ ФРЕЙНДА
Крыс сначала анестезировали в камере галотаном, после чего в дорсальную область левой ступни, между вторым и третьим наружными пальцами, подкожно инъецировали 10 мкл ПАФ. Животных затем оставляли для восстановления от анестезии при наблюдении за ними в клетке.
МАНЖЕТКА ДЛЯ СЕДАЛИЩНОГО НЕРВА
Животных готовили по методу, описанному Mosconi and Kruger (1996). Крыс анестезировали внутрибрюшинно смесью кетамин/ксилазин (2 мл/кг) и помещали на их правый бок и делали разрез поперек и вдоль оси боковой стороны левого бедра. Мышцы верхних четырехглавых мышц были препарированы по отдельности при помощи иглы для высвобождения седалищного нерва, вокруг которого помещали пластиковую манжетку (трубка РЕ-60, длина 2 мм). Рану затем зашивали в двух слоях 3-0 викриловыми и шелковыми шовными материалами.
ОПРЕДЕЛЕНИЕ МЕХАНО-АЛЛОДИНИИ С ИСПОЛЬЗОВАНИЕМ ТЕСТА VON FREY
Испытание проводили между 08:00 и 16:00 часами, используя способ, описанный Chaplan et al. (1994). Крыс помещали в клетки из плексигласа на верх основания из проволочной сетки, которая обеспечивала доступ к лапке, и оставляли для привыкания на 10-15 мин. Испытуемой площадью была среднеплантарная часть левой задней лапки, что позволяло избежать использования менее чувствительных подушечек ступней. Лапку трогали волосками из группы с 8 von Frey с логарифмически увеличивающейся жесткостью (0,41, 0,69, 1,20, 2,04, 3,63, 5,50, 8,51 и 15,14 грамм; Stoelting, III, USA). Волосок von Frey применяли снизу сетчатого пола перпендикулярно плантарной поверхности с усилием, достаточным для вызывания слабого сгибания волоска относительно лапки, и это усилие поддерживали в течение приблизительно 6-8 секунд. Положительную ответную реакцию отмечали, если лапа была резко отдернута. Вздрагивание сразу после удаления волоска также рассматривали как положительную ответную реакцию. Перемещение крысы рассматривали как неясную реакцию и в таких случаях стимул повторяли.
ПРОТОКОЛ ИСПЫТАНИЯ
Животных испытывали в послеоперационный день 1 для ПАФ-обработанной группы и в послеоперационный день 7 для группы с манжеткой седалищного нерва. 50% порог одергивания определяли с использованием метода Диксона "вверх-вниз" (1980). Испытание начинали с волоска 2,04 г, среднего в серии. Стимулирование всегда осуществляли последовательно вне зависимости от того, были ли они повышающимися или понижающимися. При отсутствии ответной реакции отдергивания лапы на первоначально выбранный волосок давали более сильный стимул; в случае одергивания лапы выбирали следующий, более слабый стимул. Вычисление оптимального порога этим способом требует 6 ответных реакций в непосредственной близости от 50% порога, и подсчет этих 6 ответных реакций начинали, когда имело место первое изменение в ответной реакции, например, когда порог был впервые пересечен. В случаях, когда пороги находились вне пределов стимулов, были оценены величины 25,14 (нормальная чувствительность) или 0,41 (максимальная аллодиния) соответственно. Полученные результаты положительных и отрицательных ответных реакций были представлены в виде таблиц 1 и 2 с использованием стандартного метода, Х=без отдергивания, 0= отдергивание, и 50% порог отдергивания интерполировали с использованием формулы:
50% порог, г=10(хf+kd)/10000,
где xf - величина последнего использованного волоска von Frey (log-единицы); k - табличная величина (из Chaplan et al. (1994)) для результатов положительных/отрицательных ответных реакций и d - средняя разность между стимулами (log-единицы). Здесь d=0,224.
Пороги von Frey были превращены в процент максимально возможного эффекта (% МВЭ) по Chaplan et al., 1994. Для вычисления % МВЭ использовали следующее уравнение: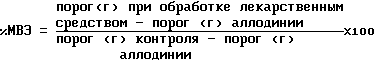
ВВЕДЕНИЕ ИСПЫТУЕМОГО ВЕЩЕСТВА
Крысам вводили инъекцией (подкожно, внутрибрюшинно или вводили перорально) испытуемое соединение до испытания по методу von Frey, время между введением испытуемого соединения и испытанием по способу von Frey варьировали в зависимости от природы испытуемого соединения.
Следующие аббревиатуры имеют указанные ниже значения:
Ac = ацетил
Аr = арил
t-BOC = третичный бутоксикарбонил
t-Bu =третичный бутил
Et = этил
iPr = изопропил
Me = метил
Ph = фенил
Рr = пропил
r.t = комнатная температура
ТФУ = трифторуксусная кислота
ТГФ = тетрагидрофуран
ТМЭДА = N,N,N'N'-тетраметилэтилендиаминн
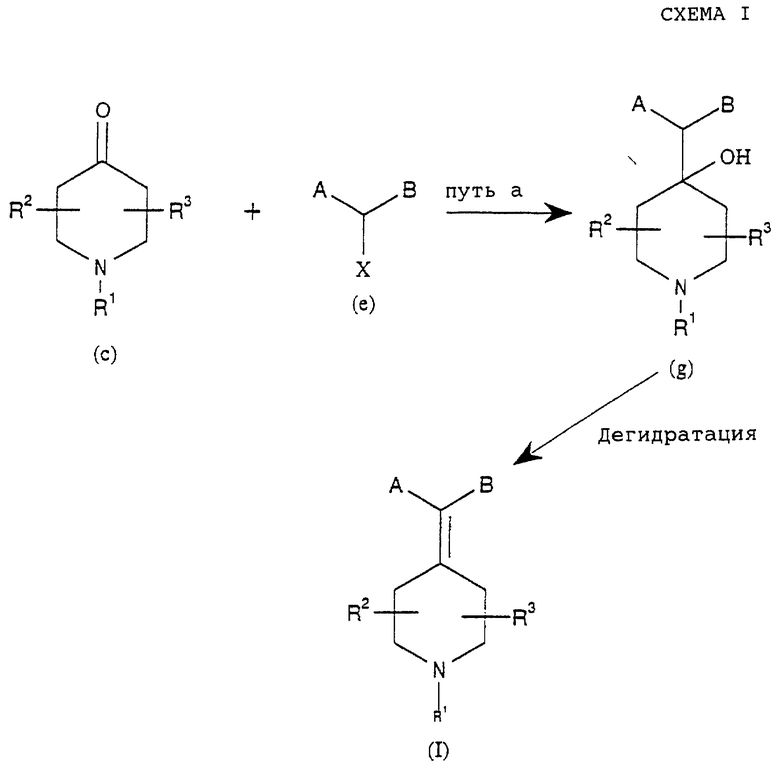
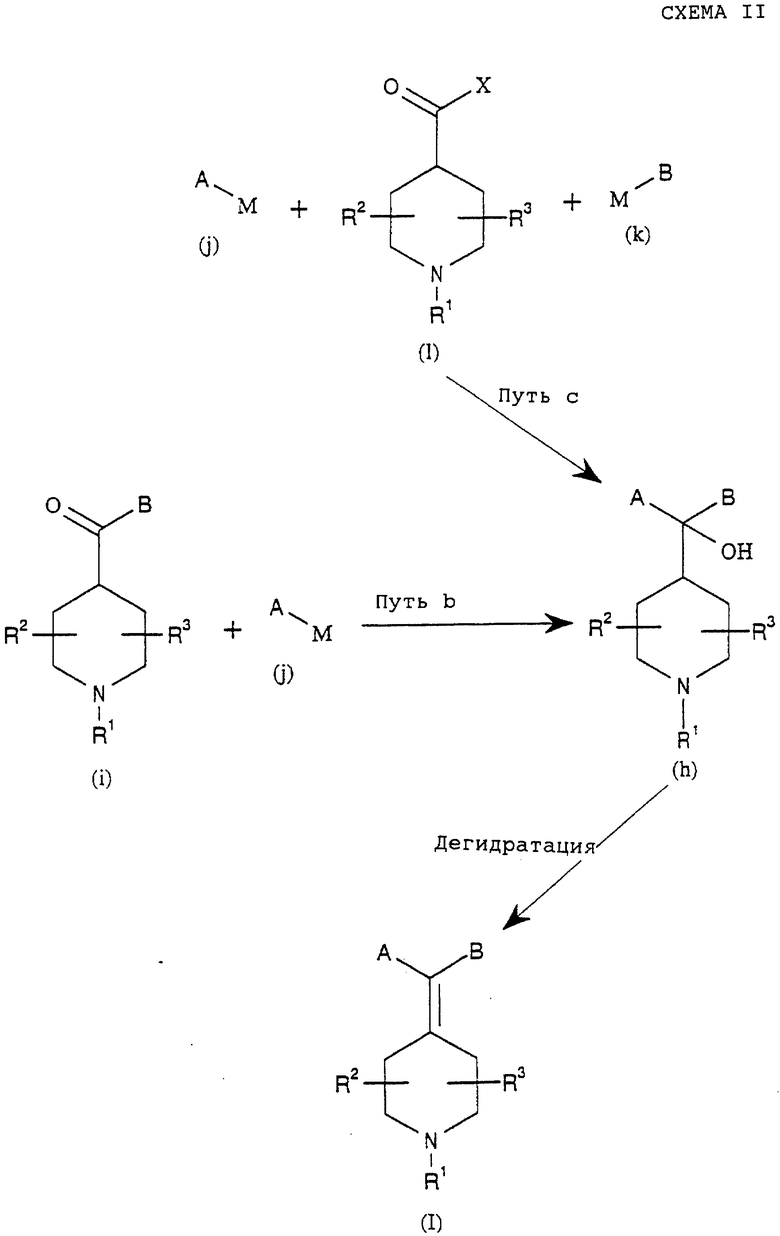
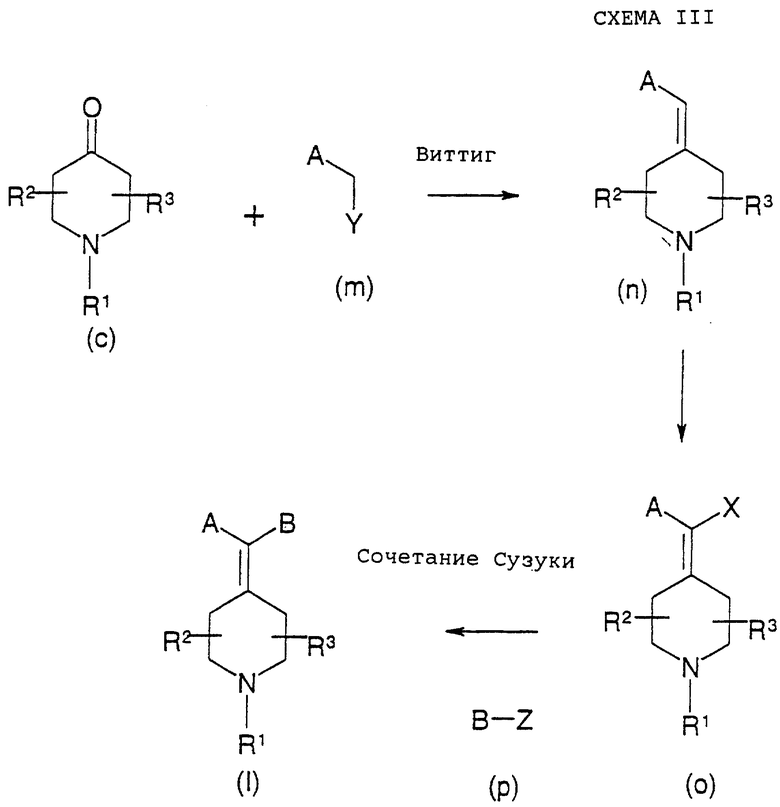
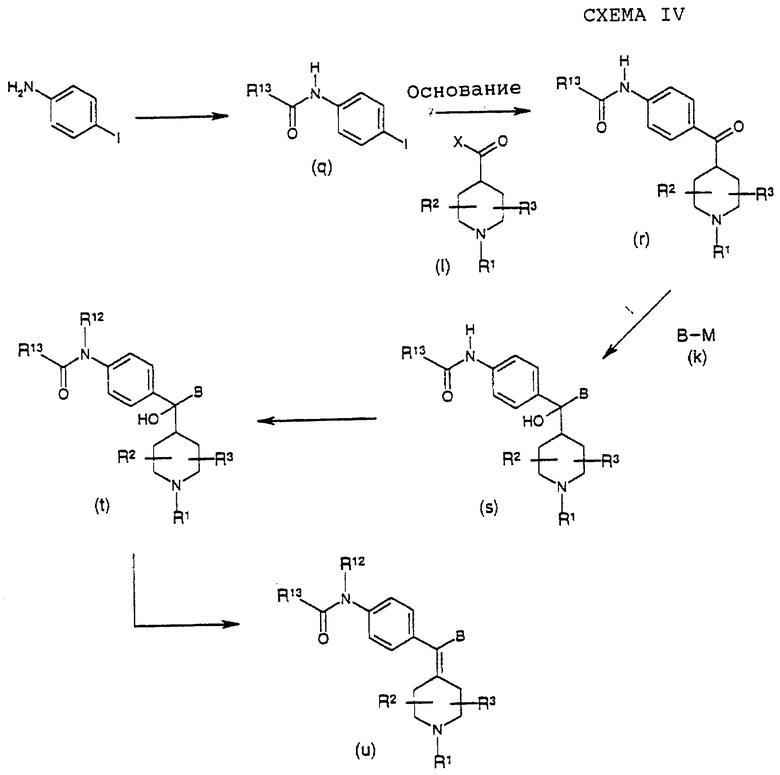
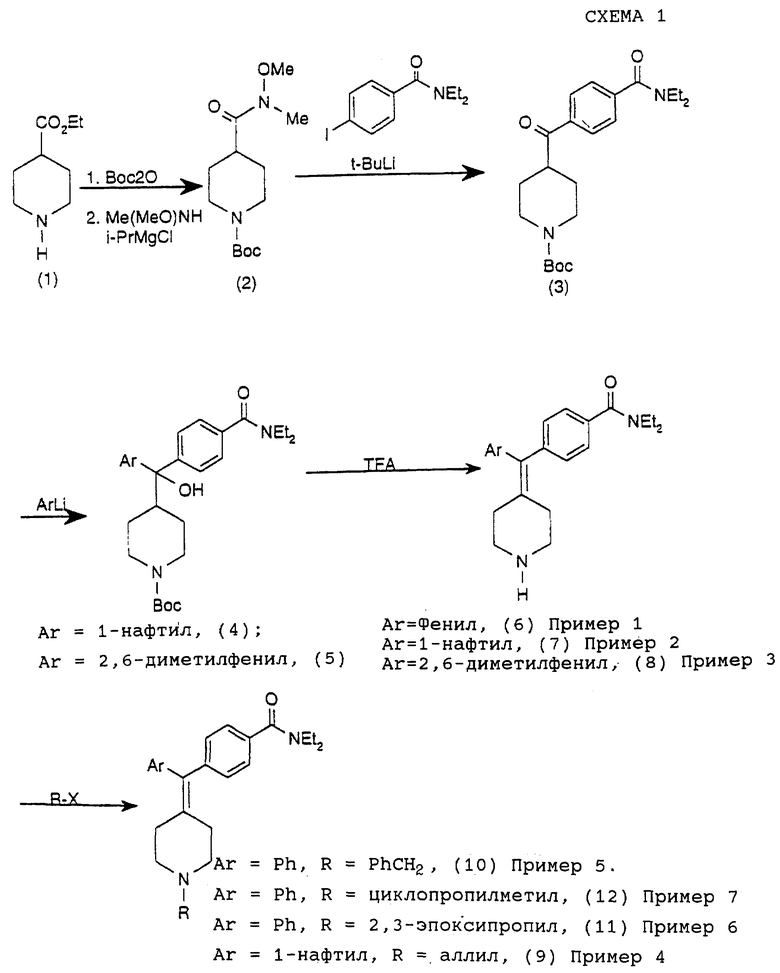
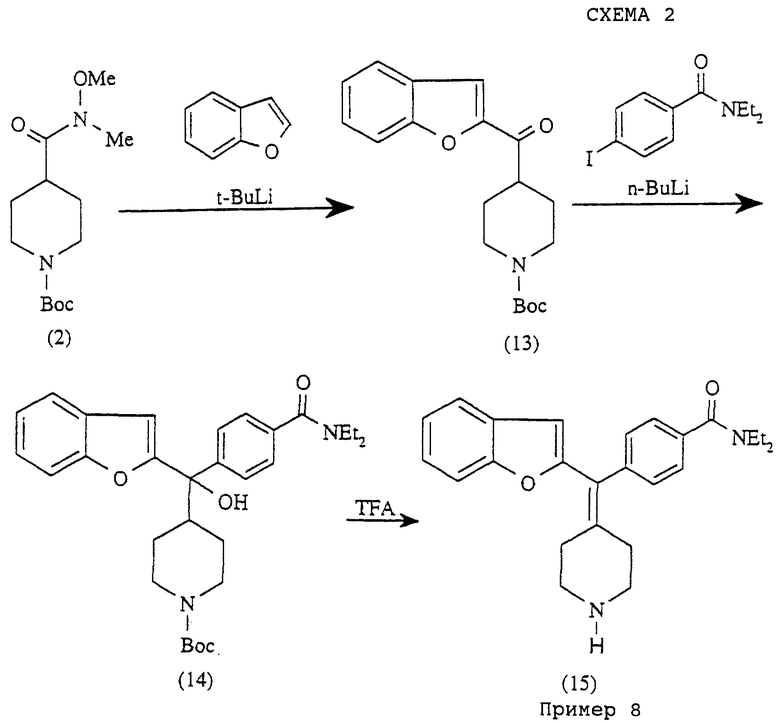
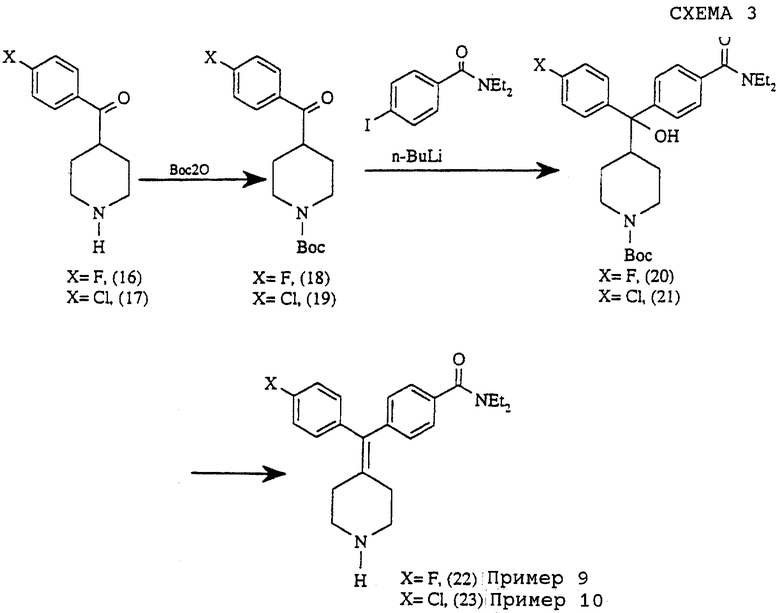
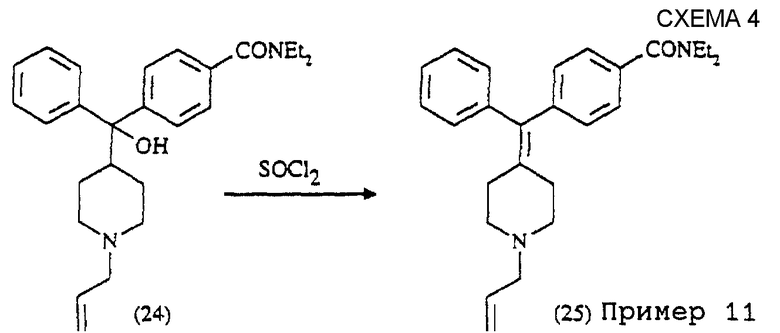
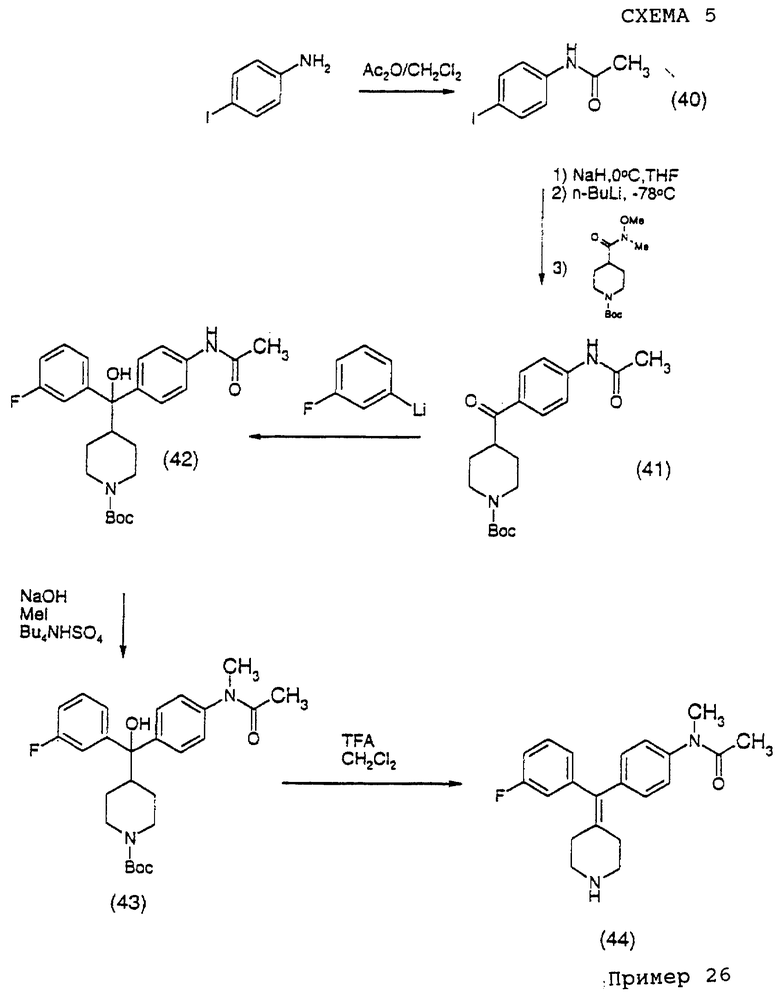
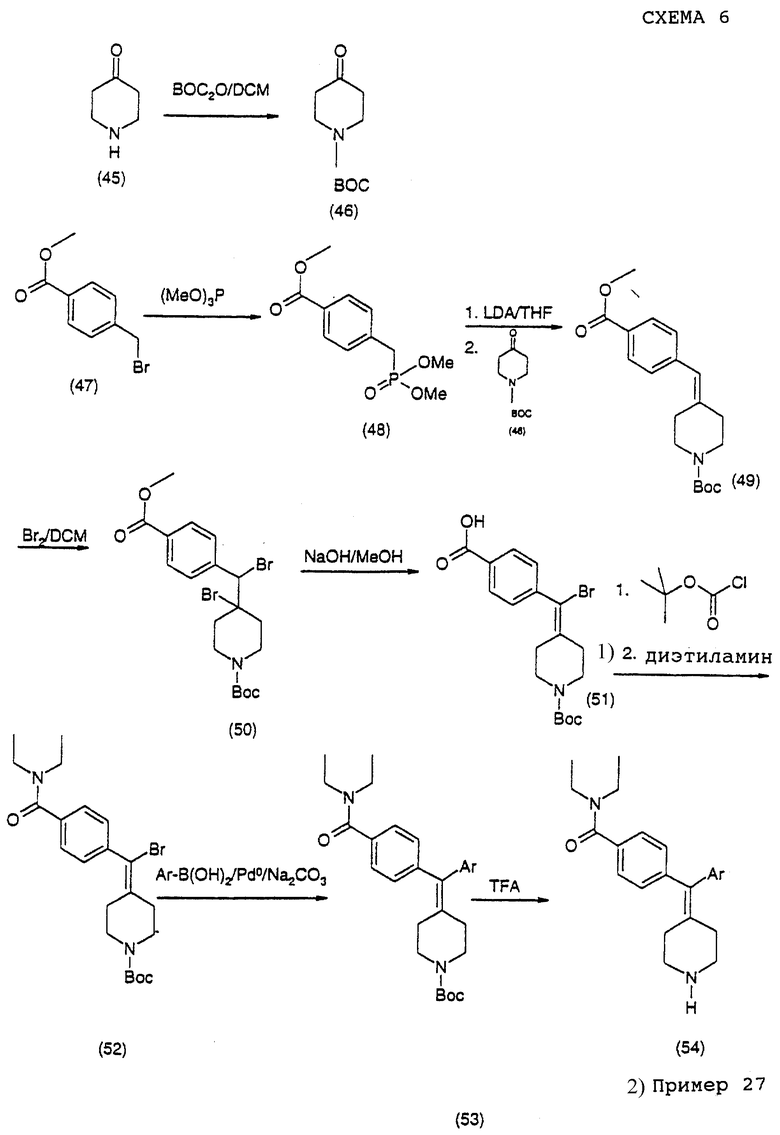
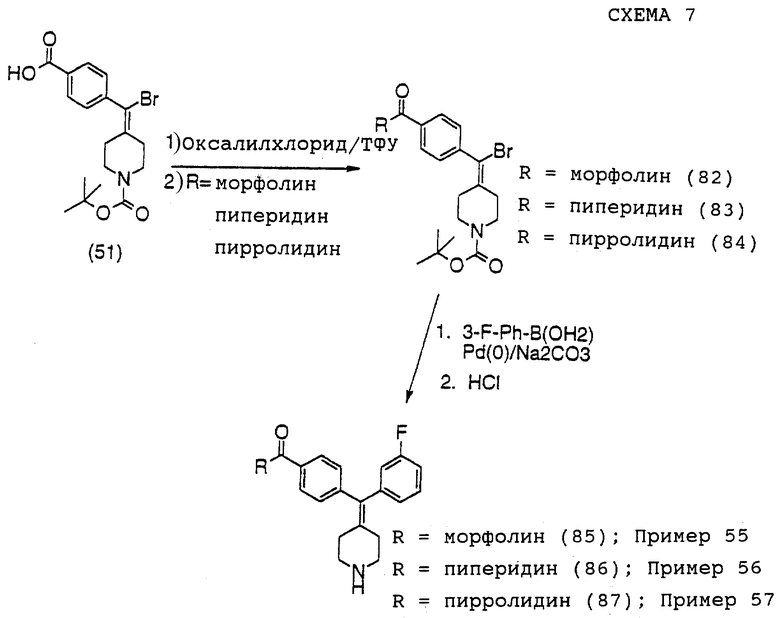
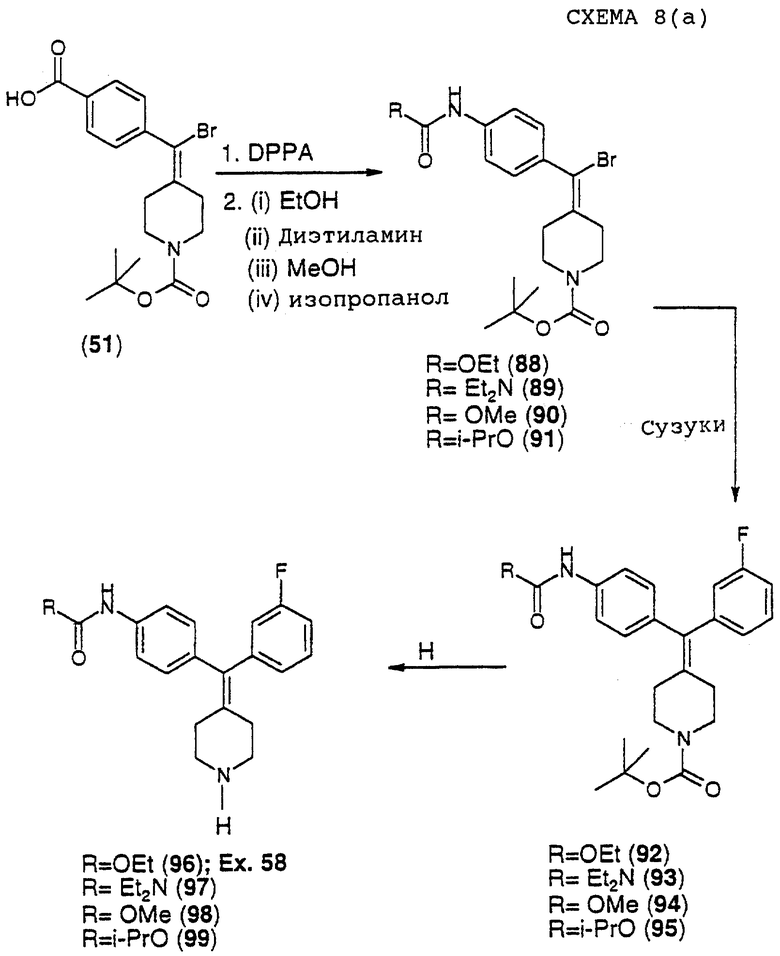
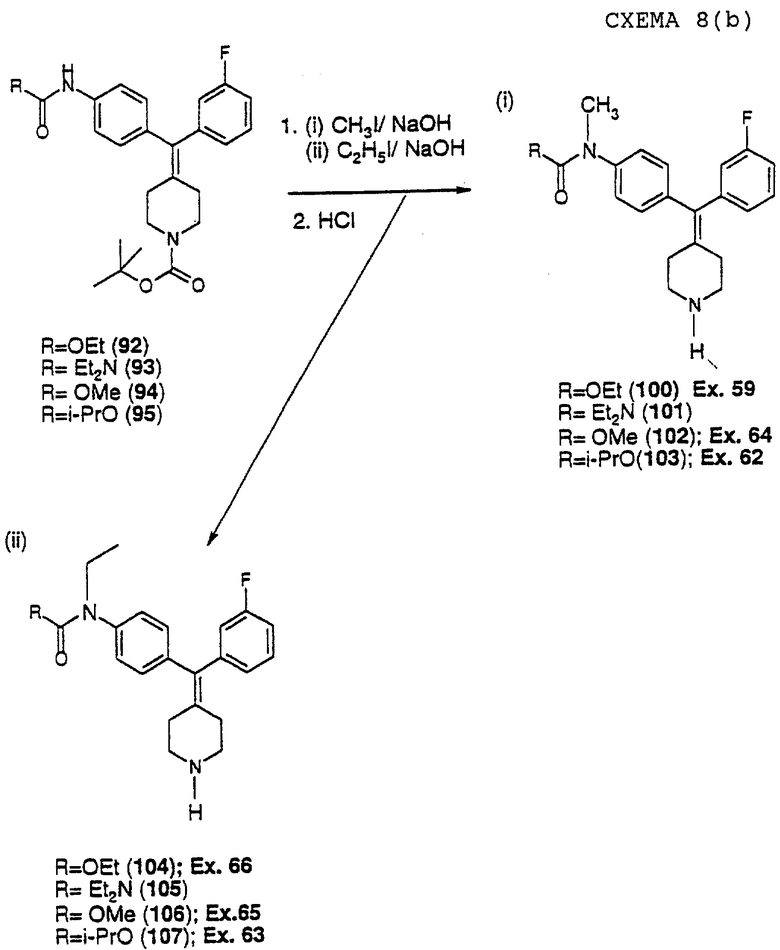
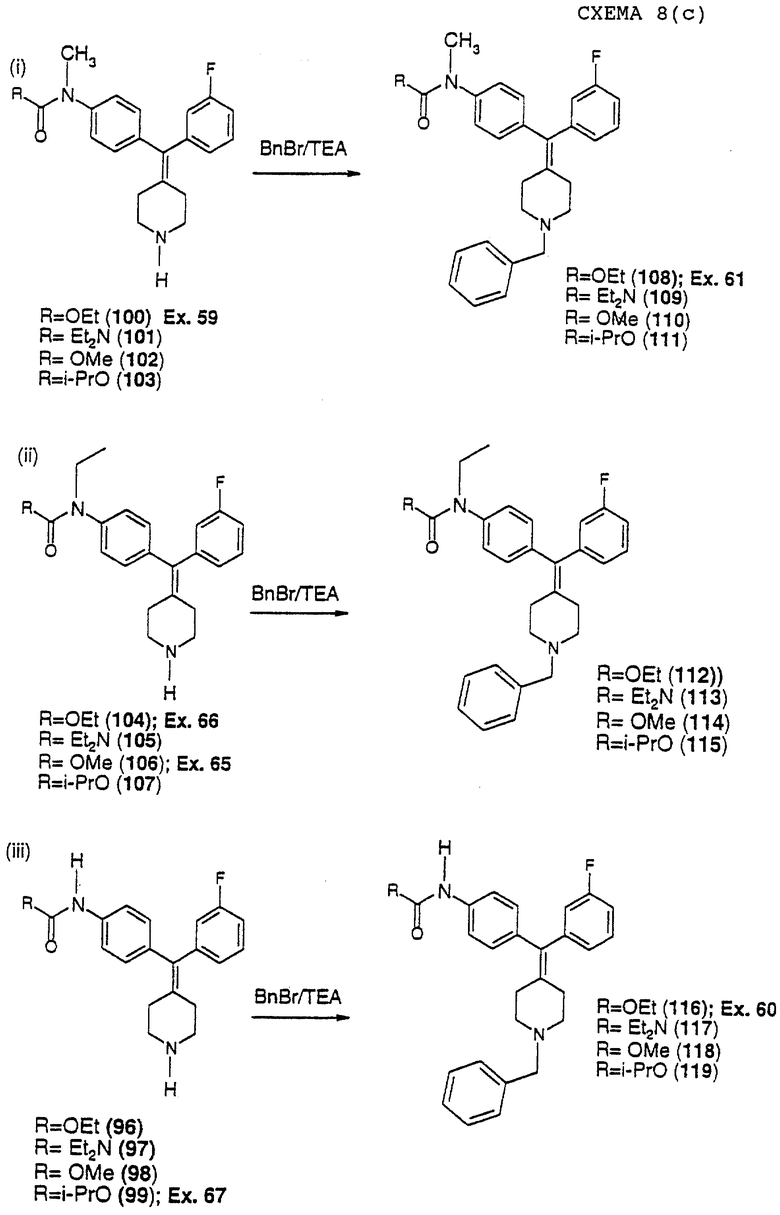
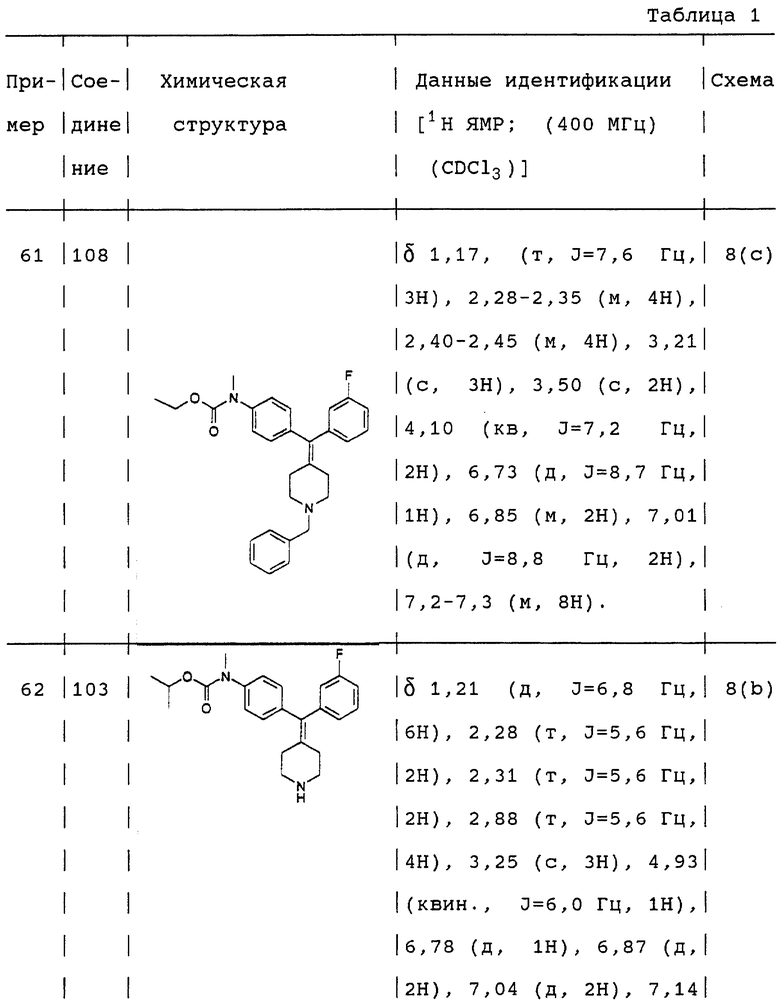
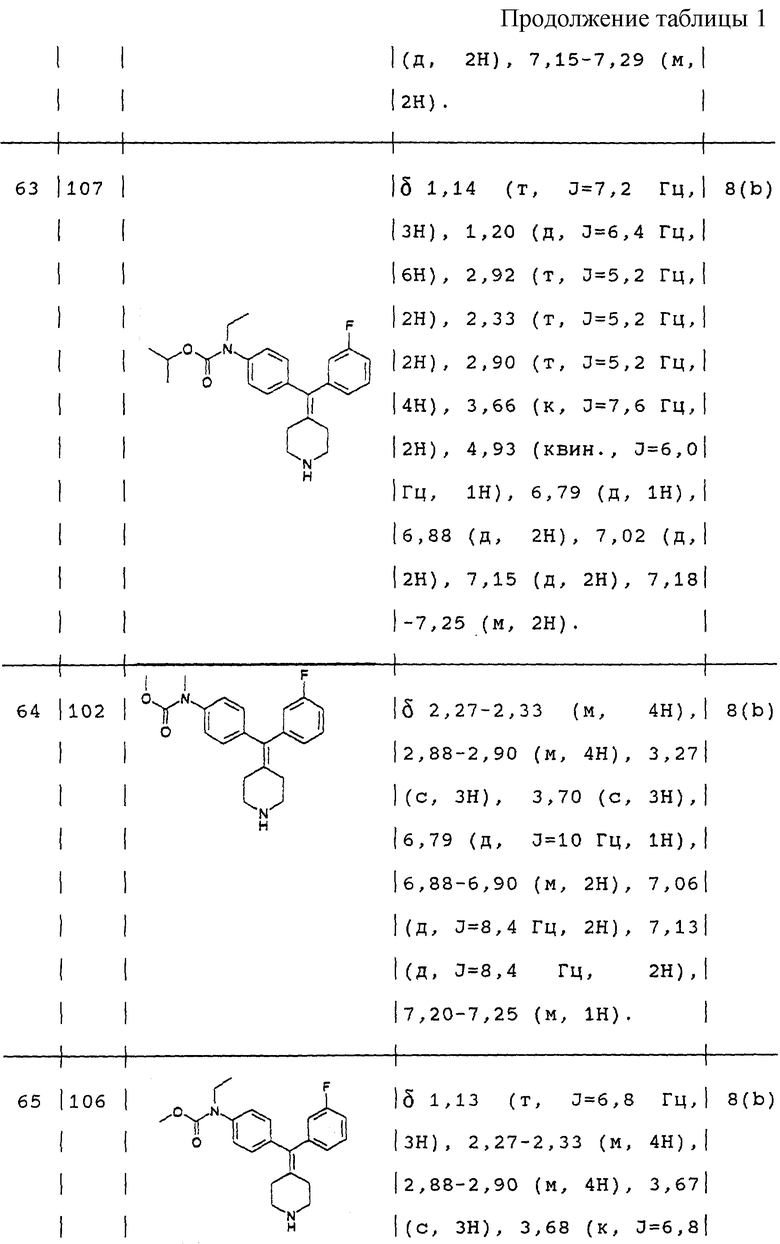
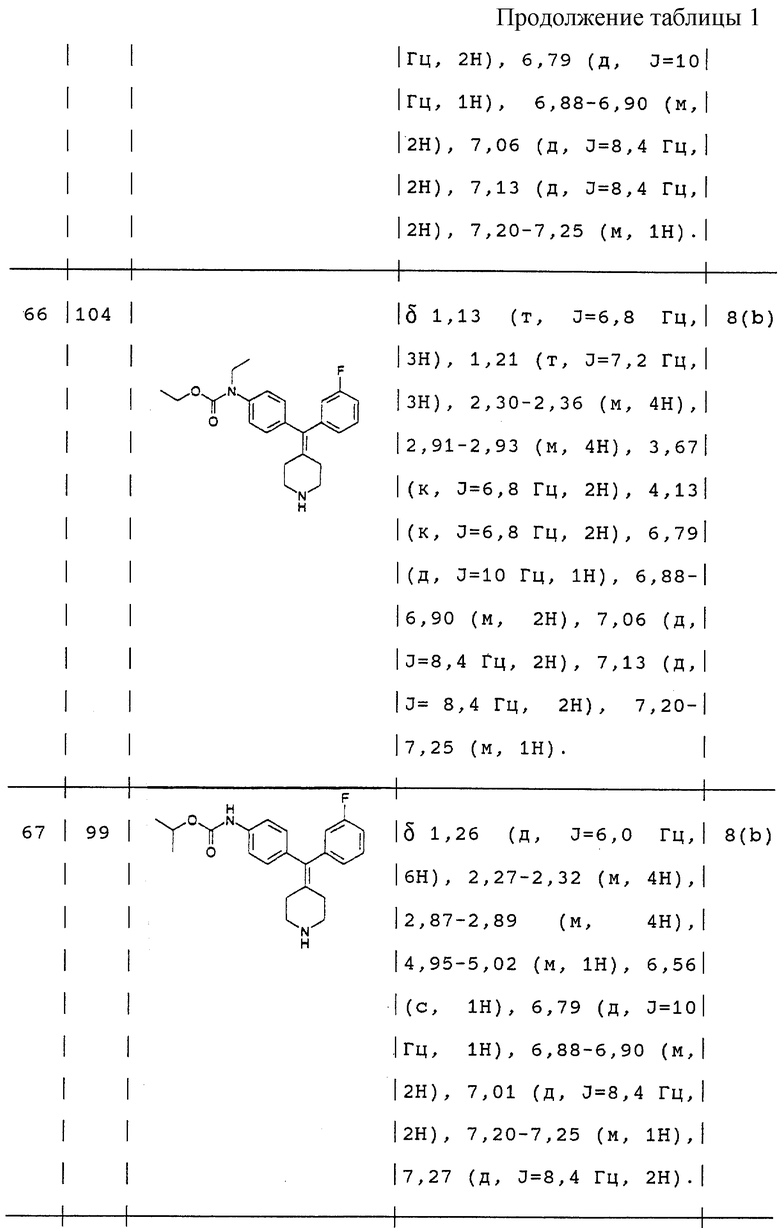
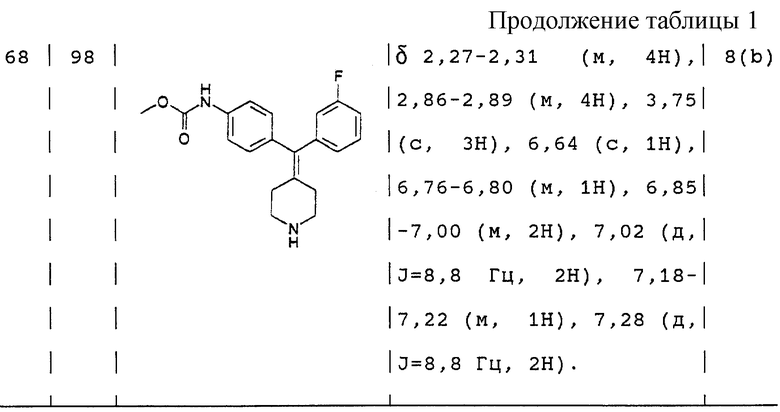
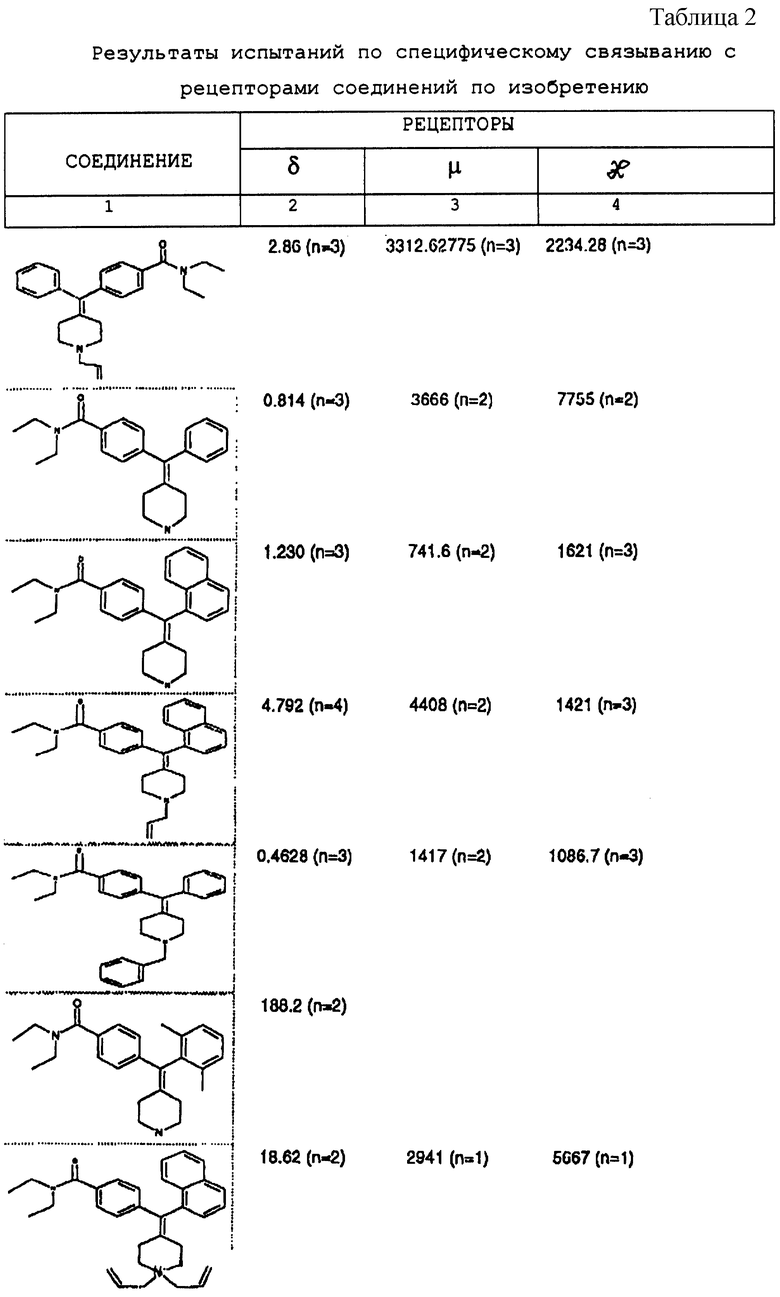
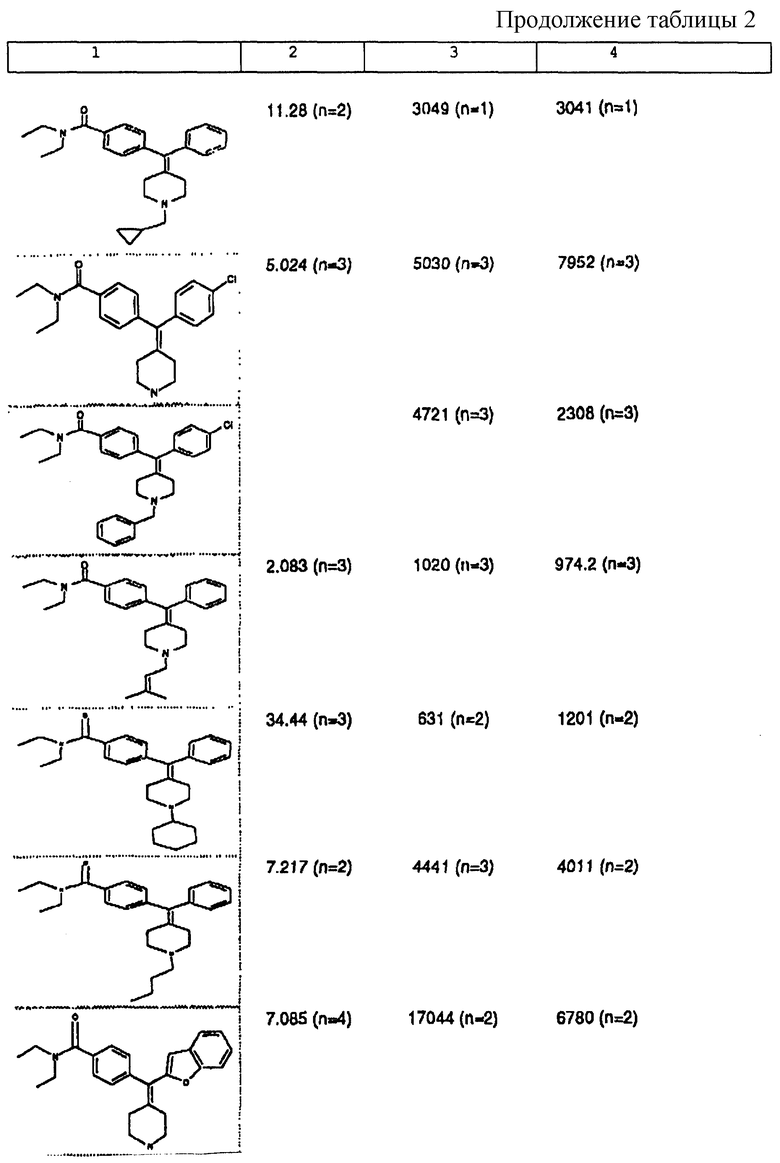
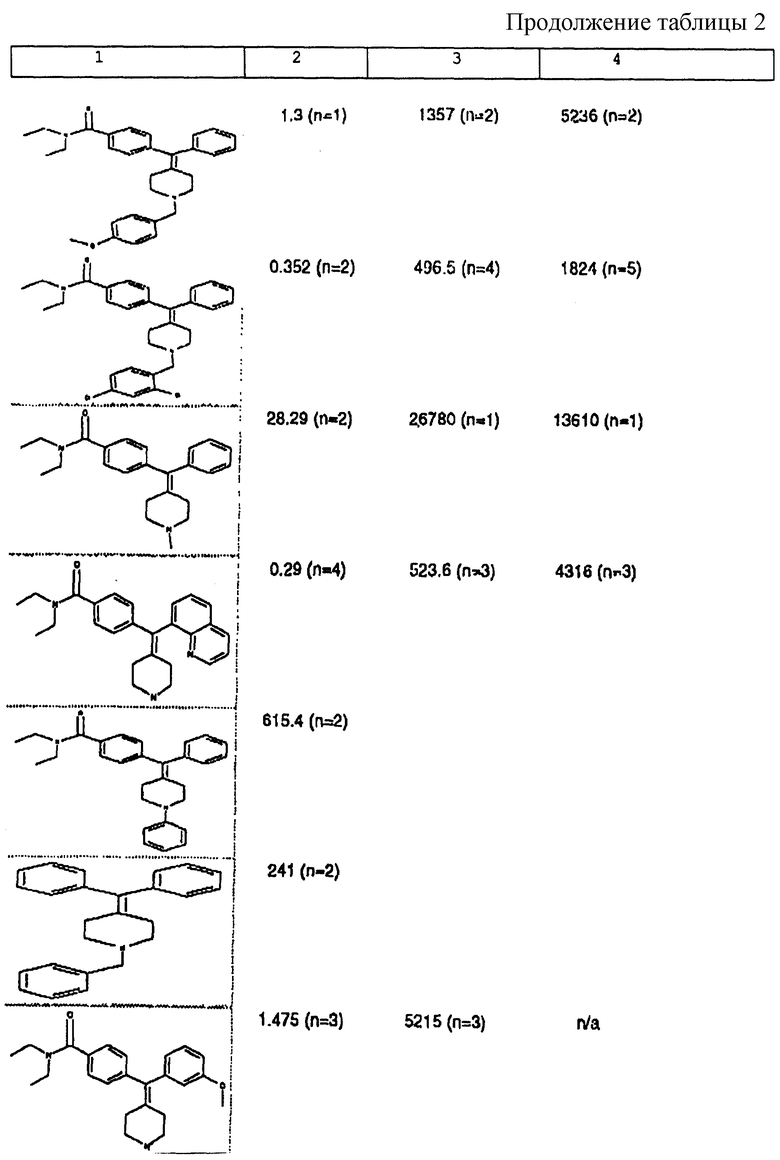
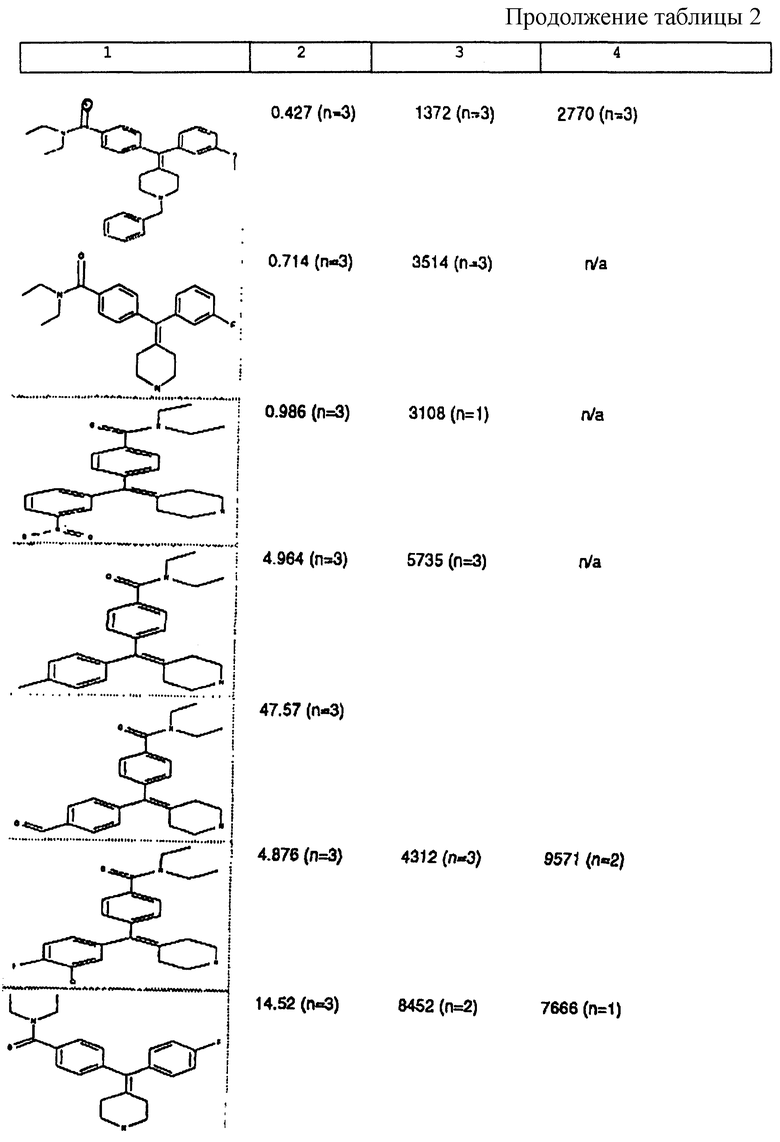
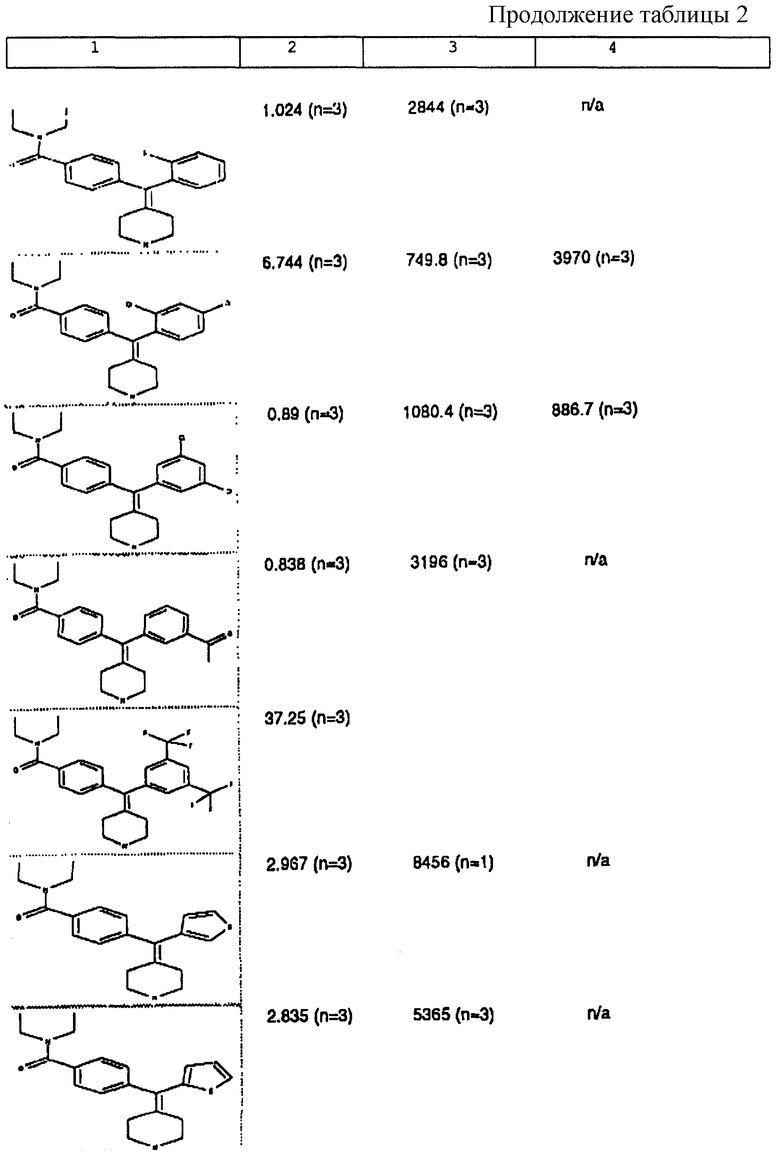
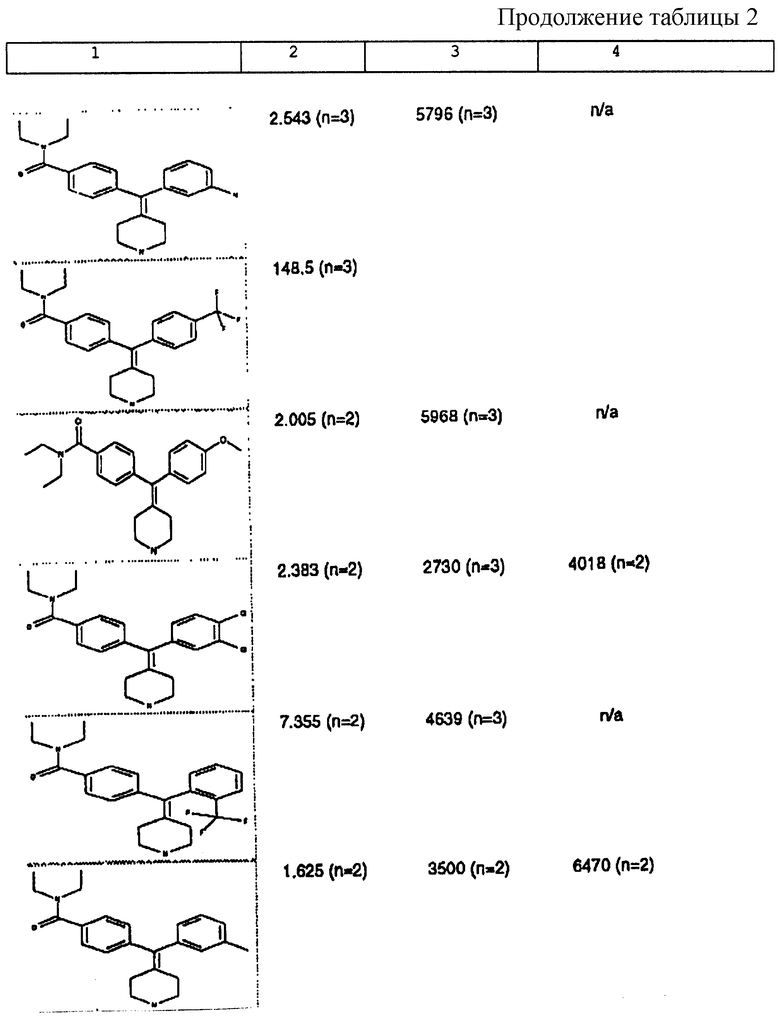
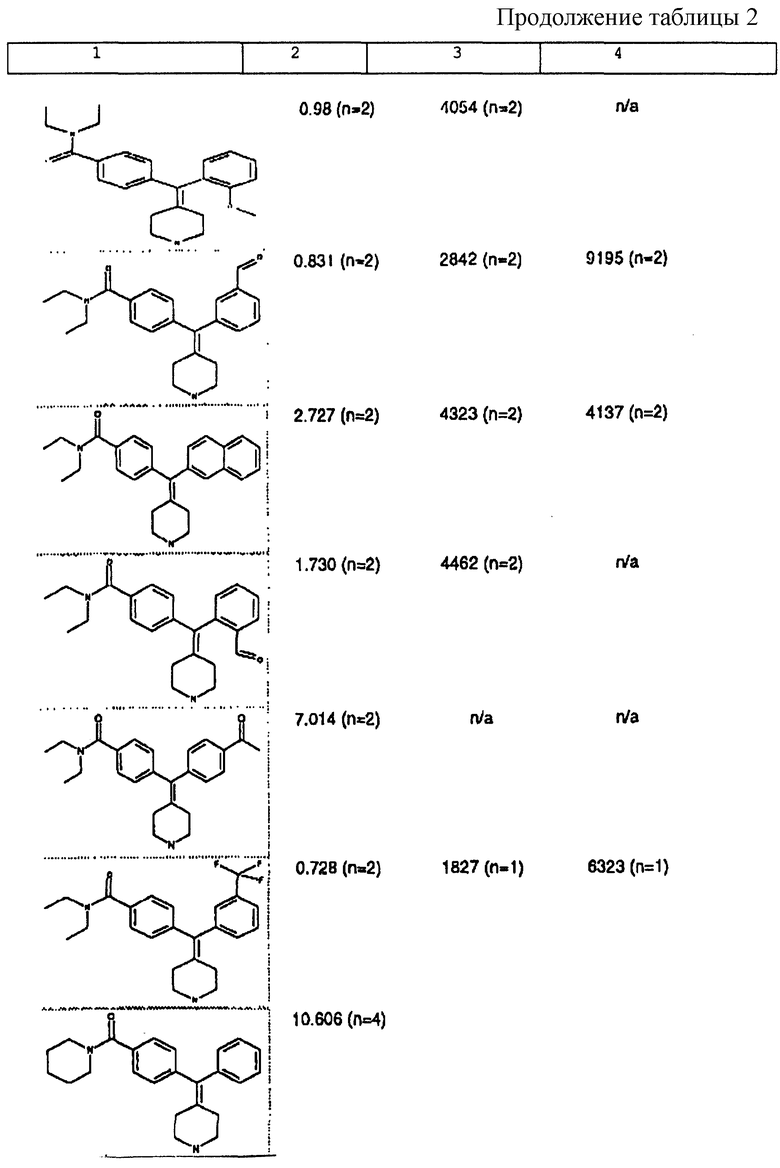
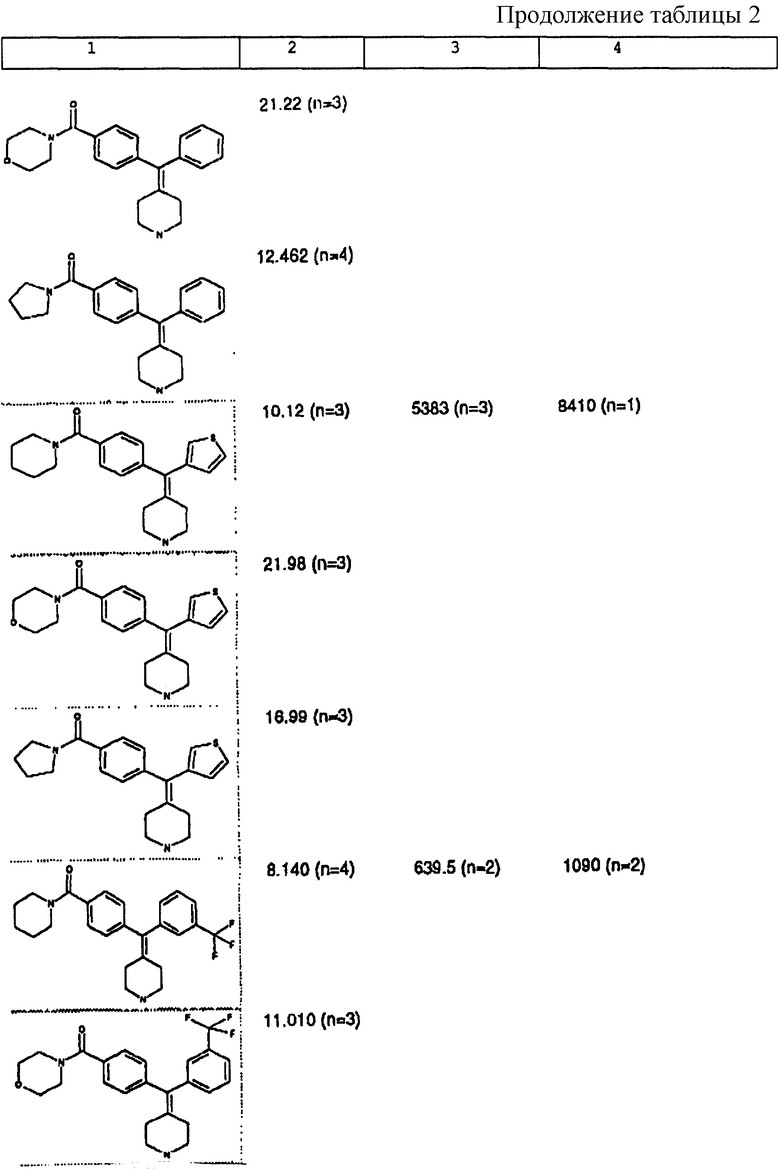
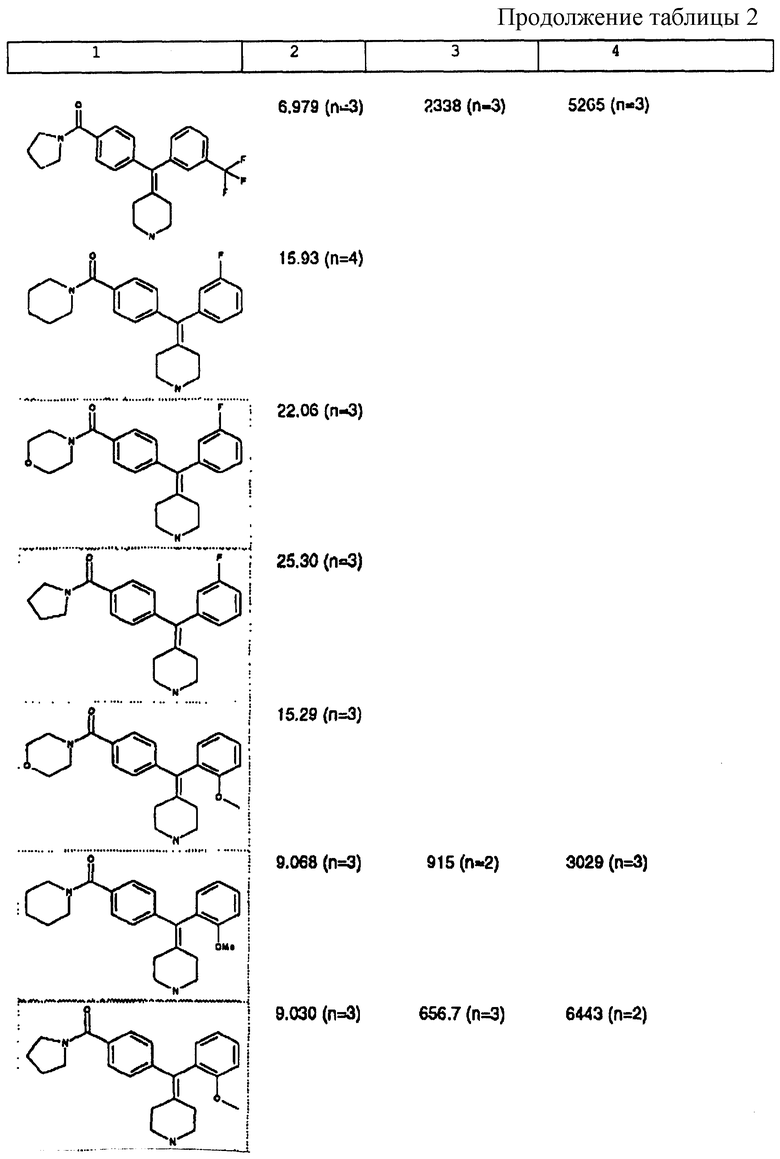
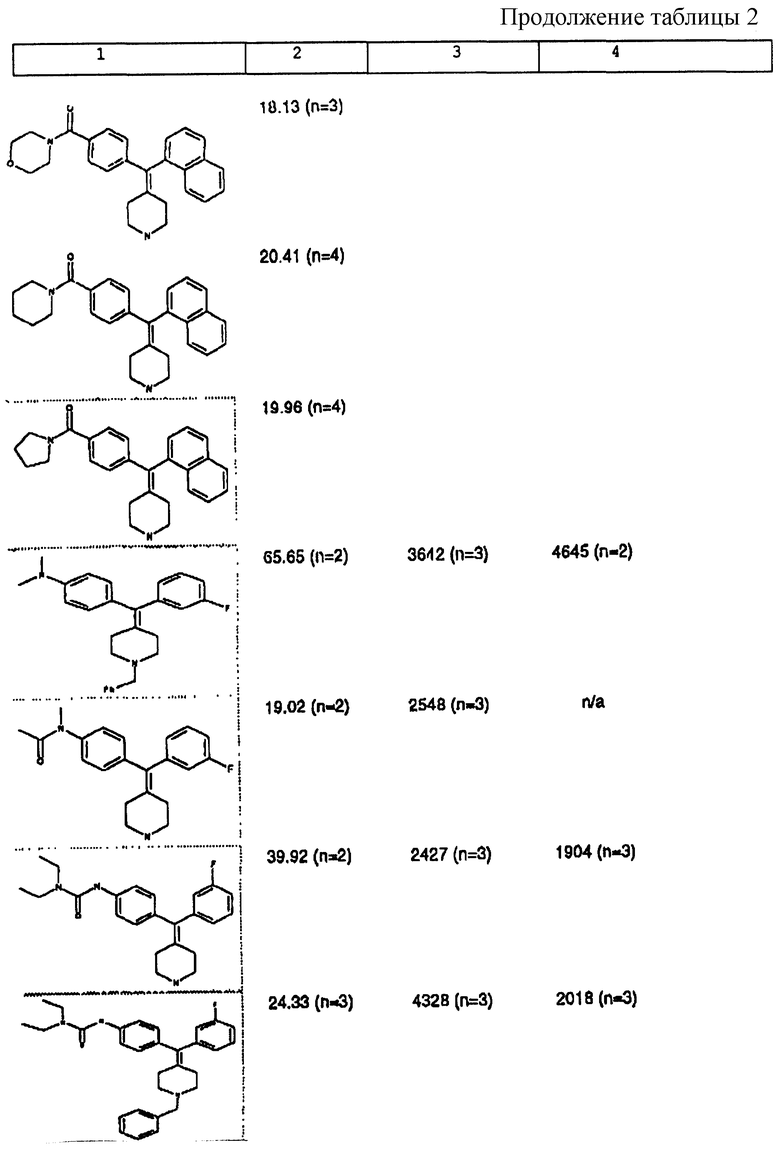
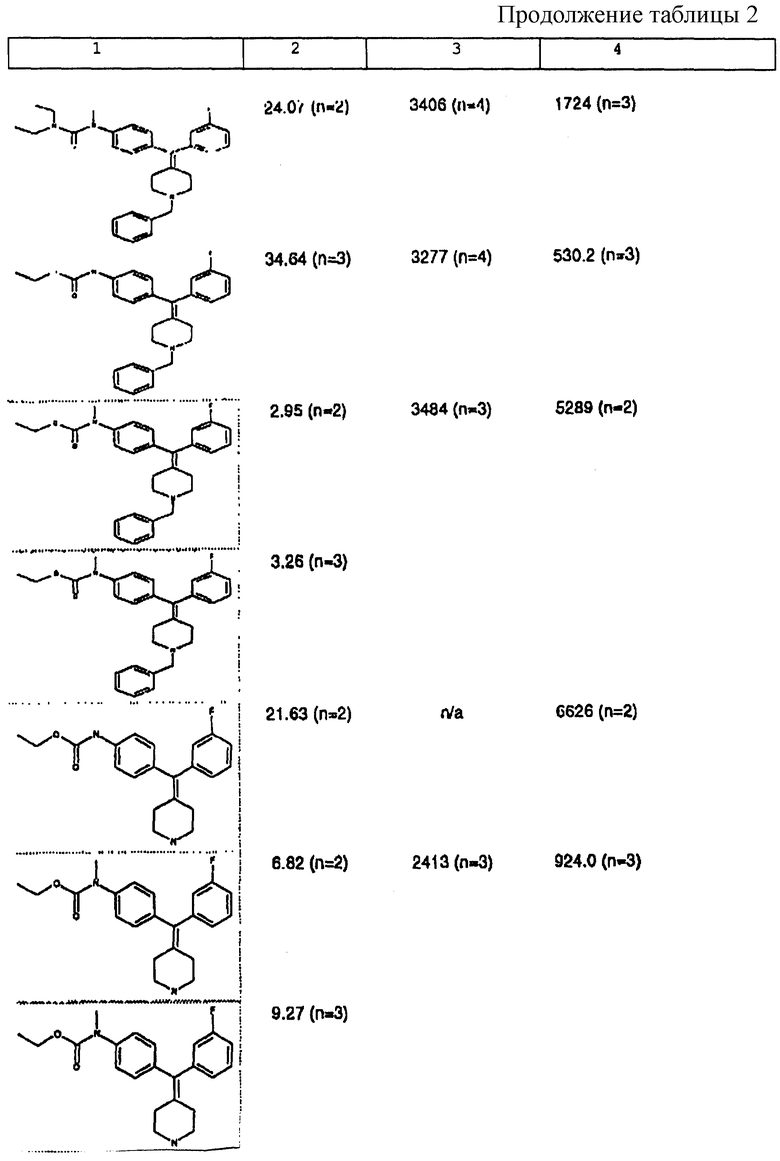
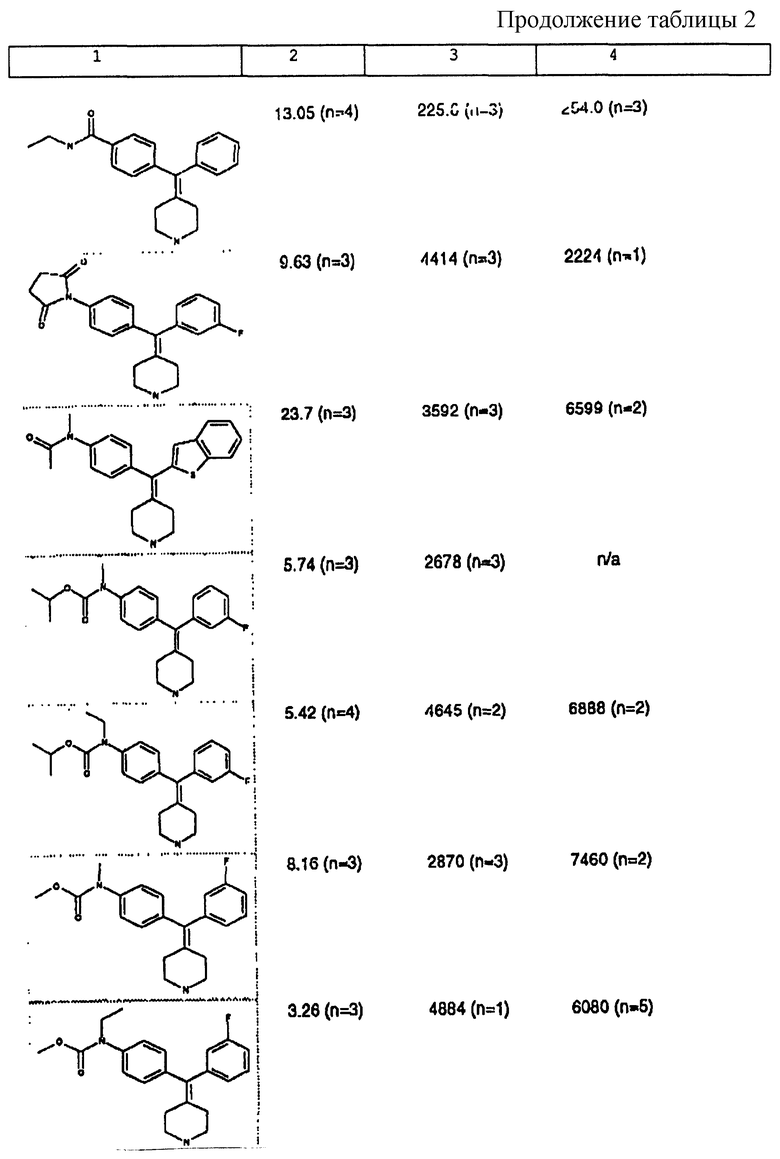

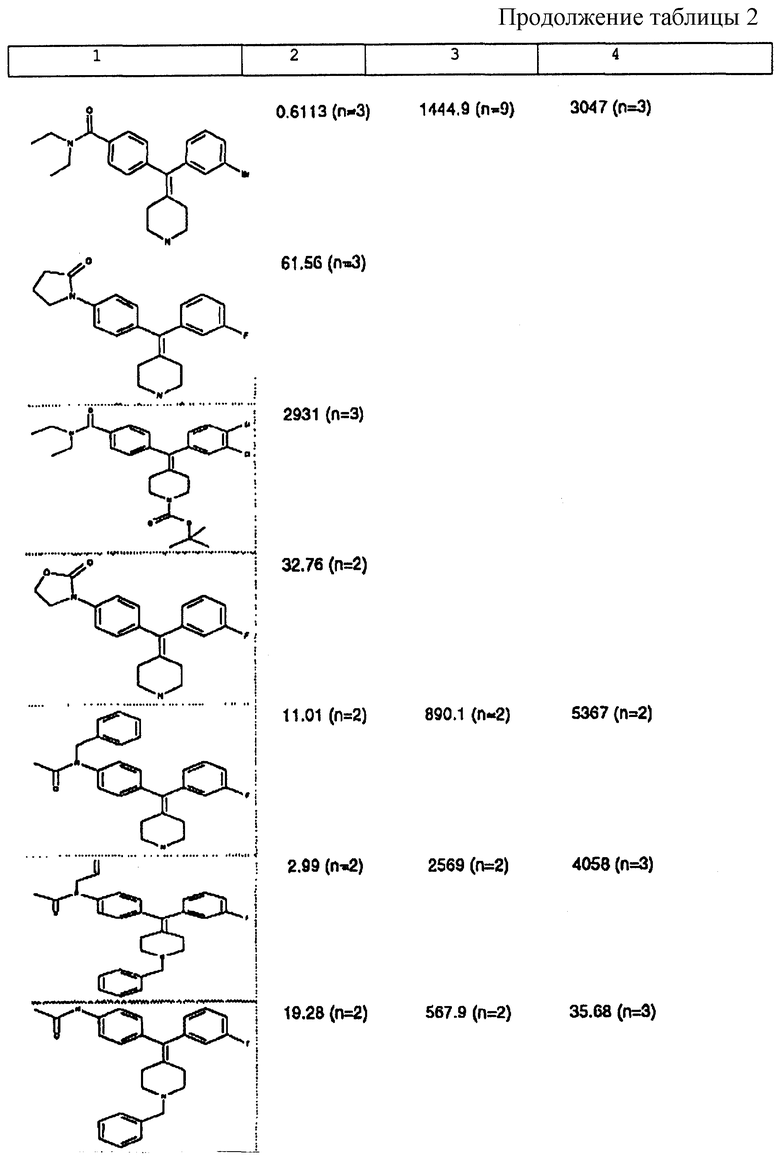

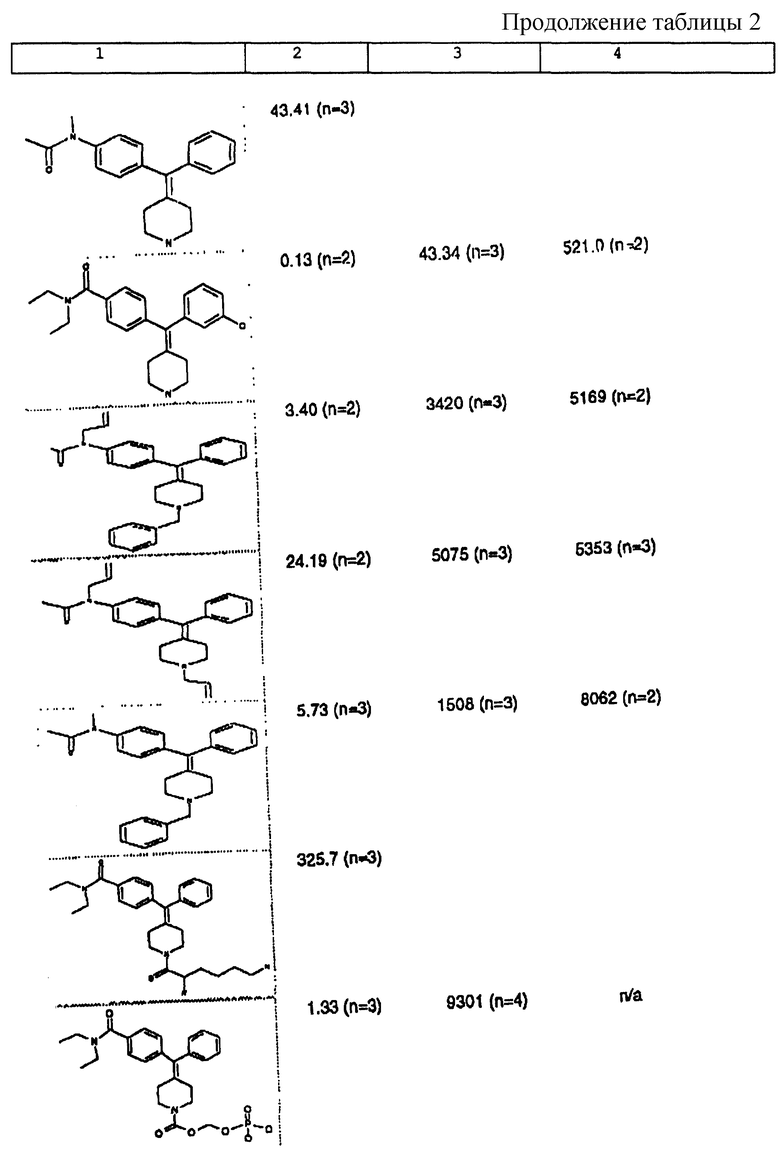
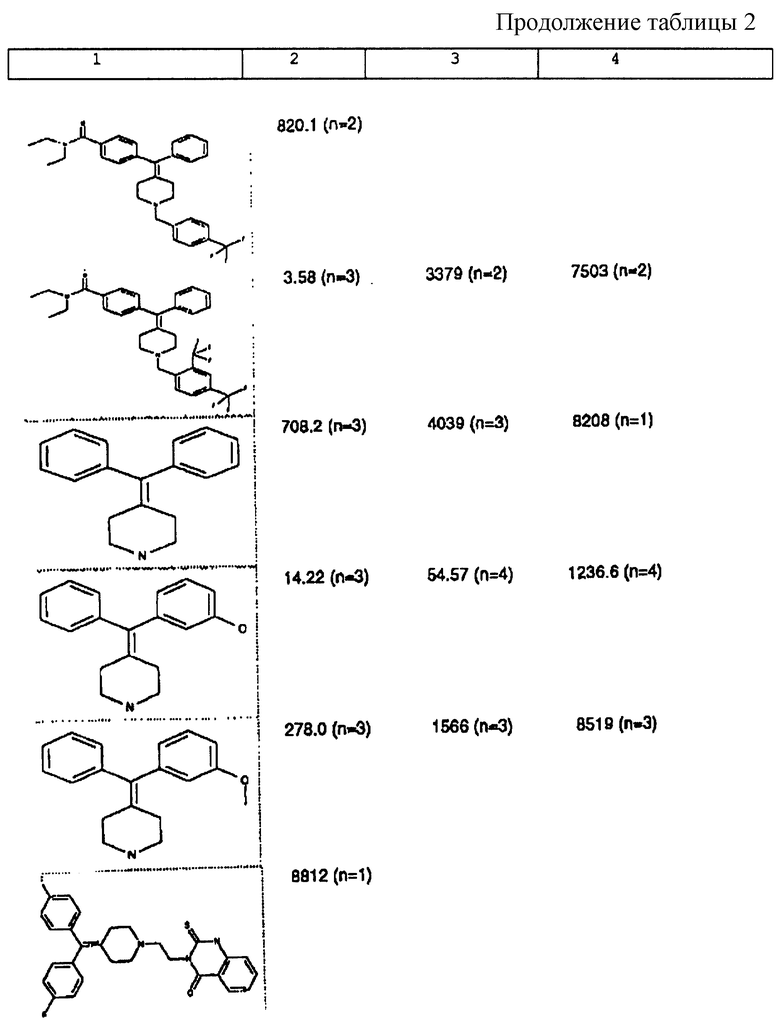
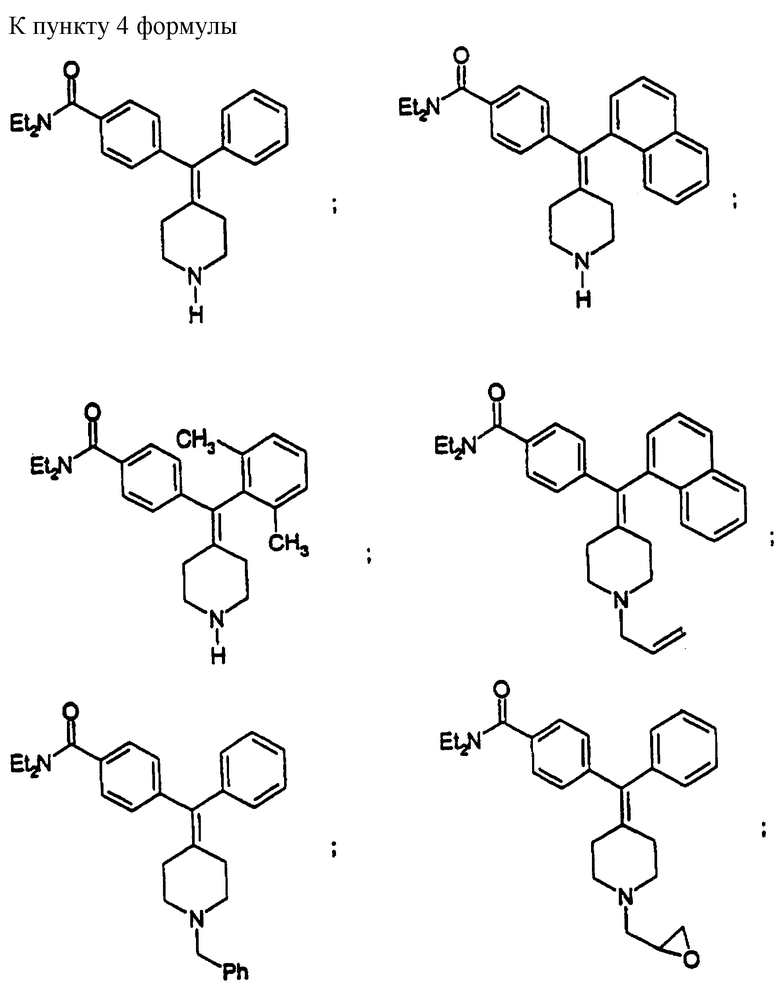
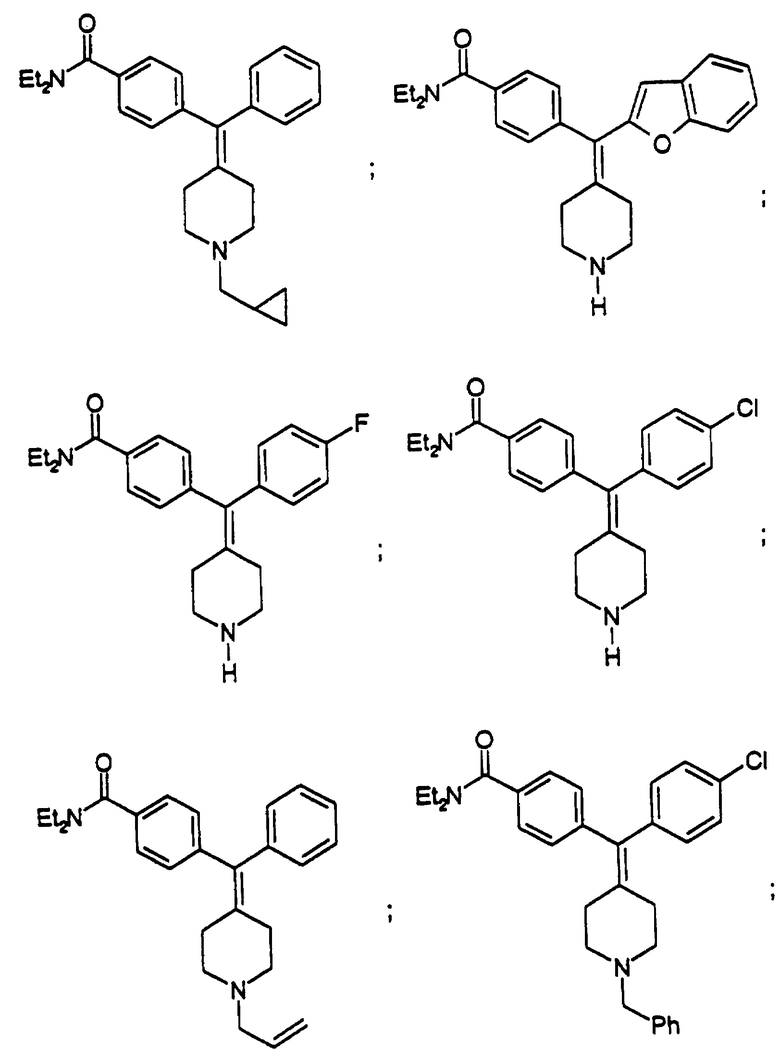
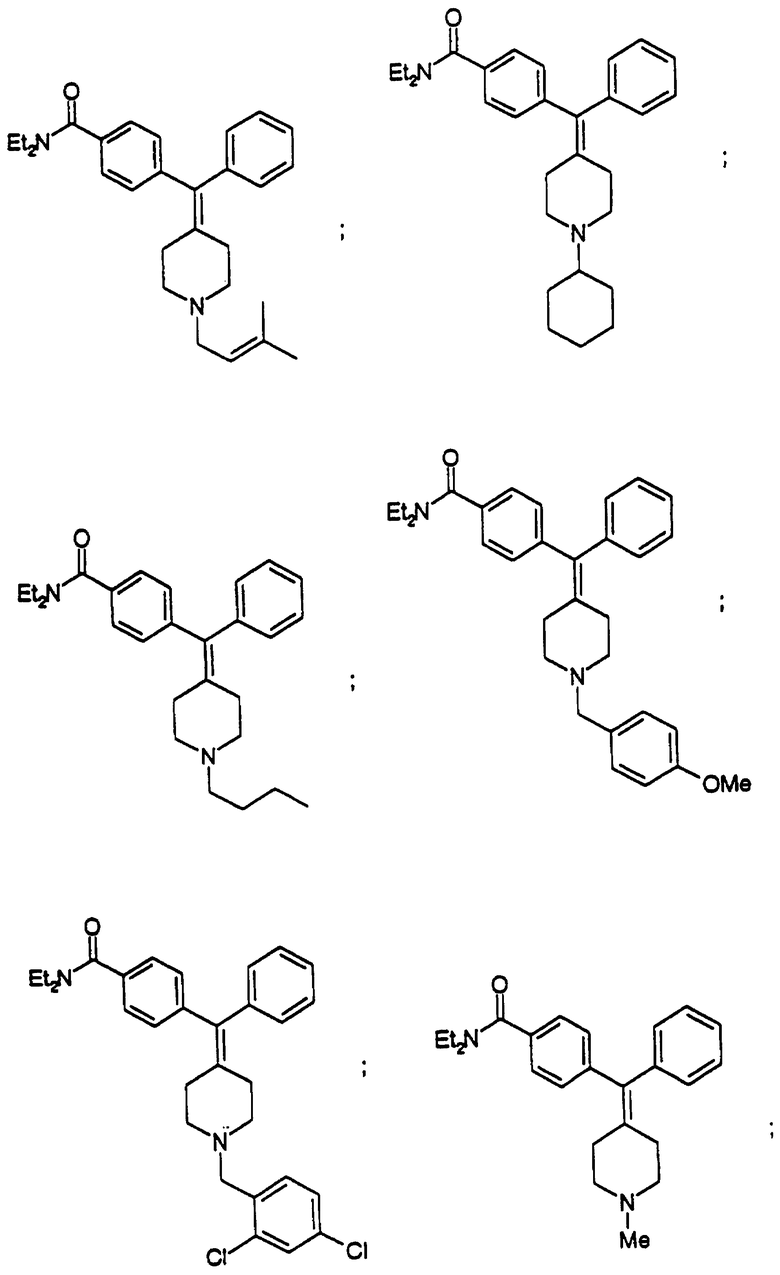
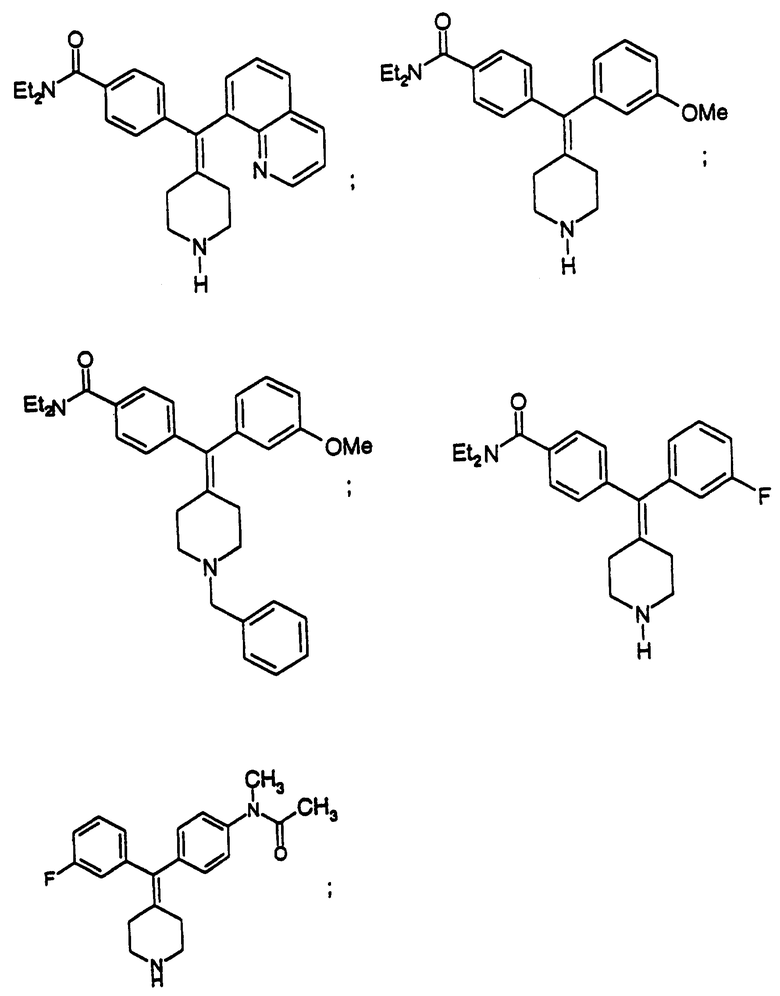
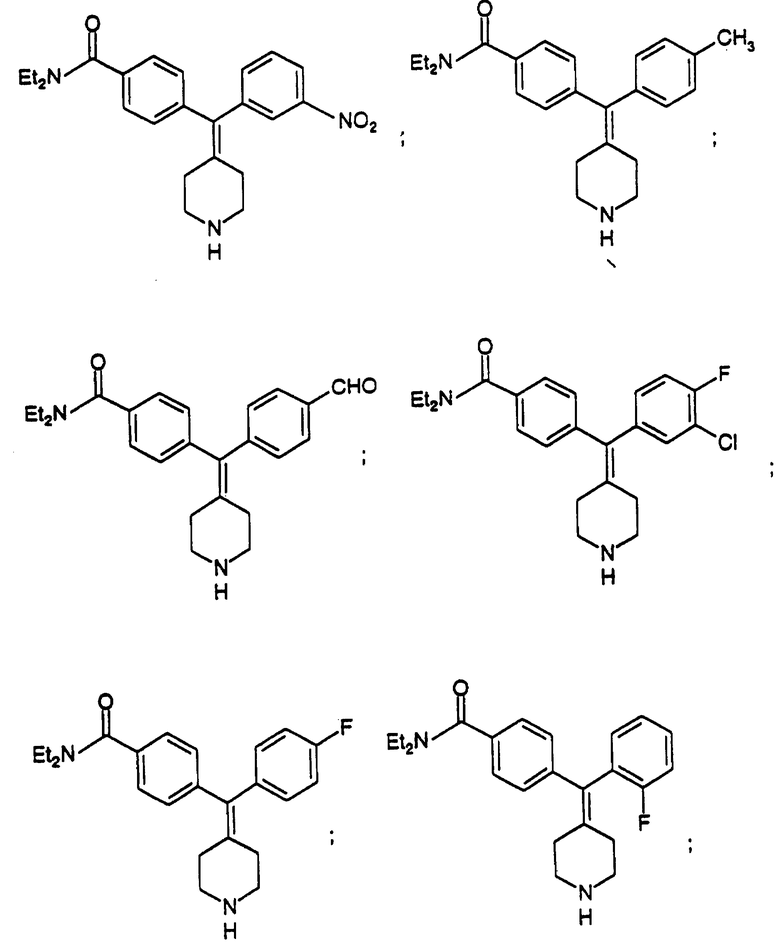
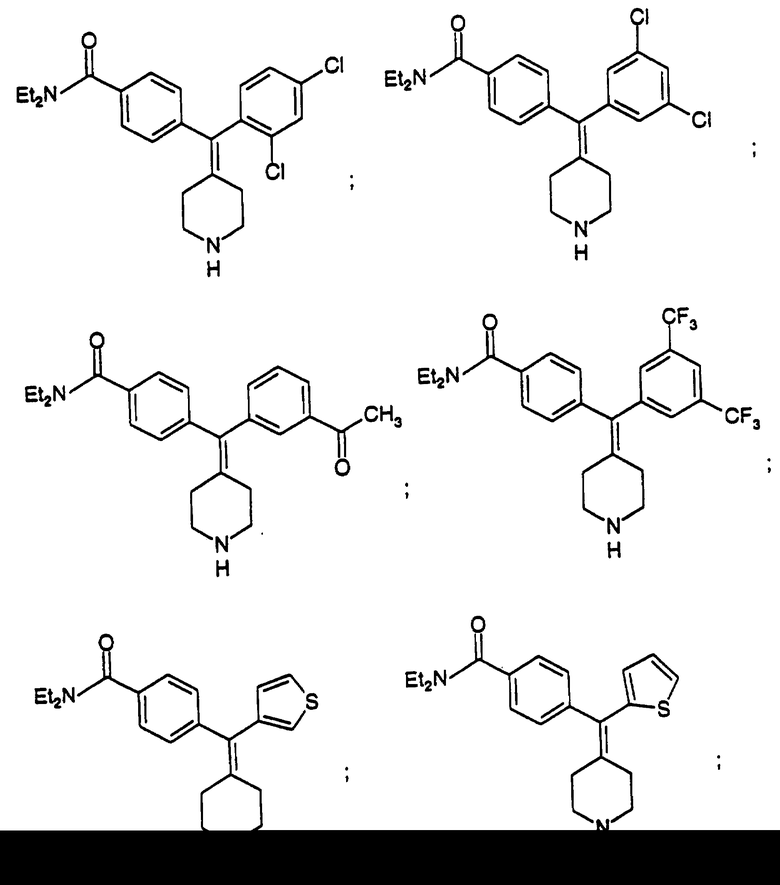
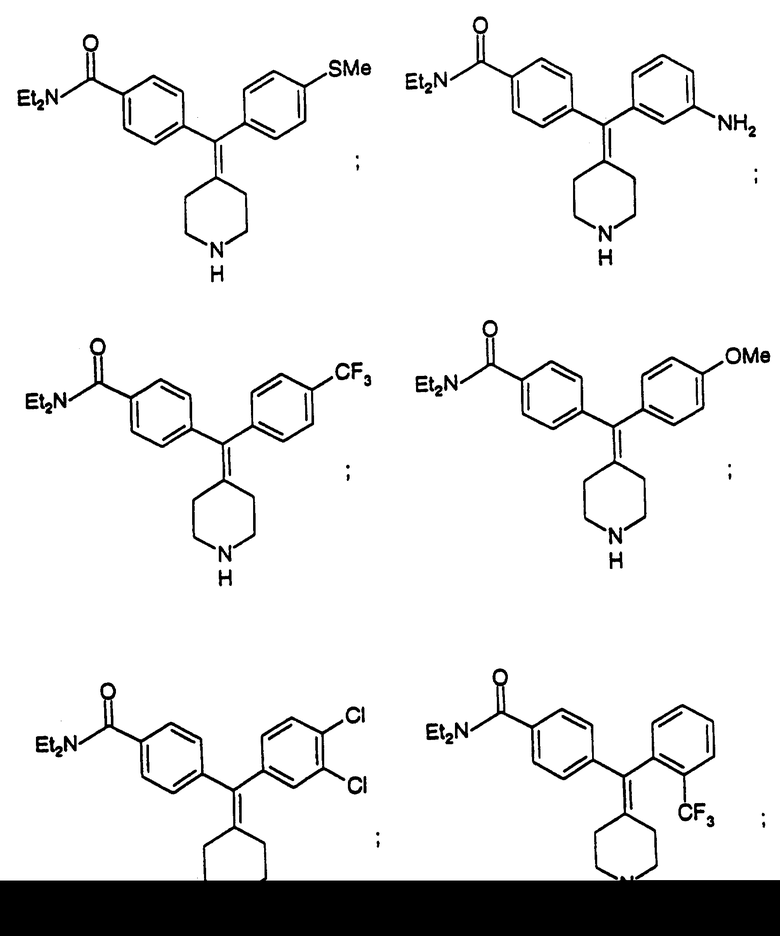
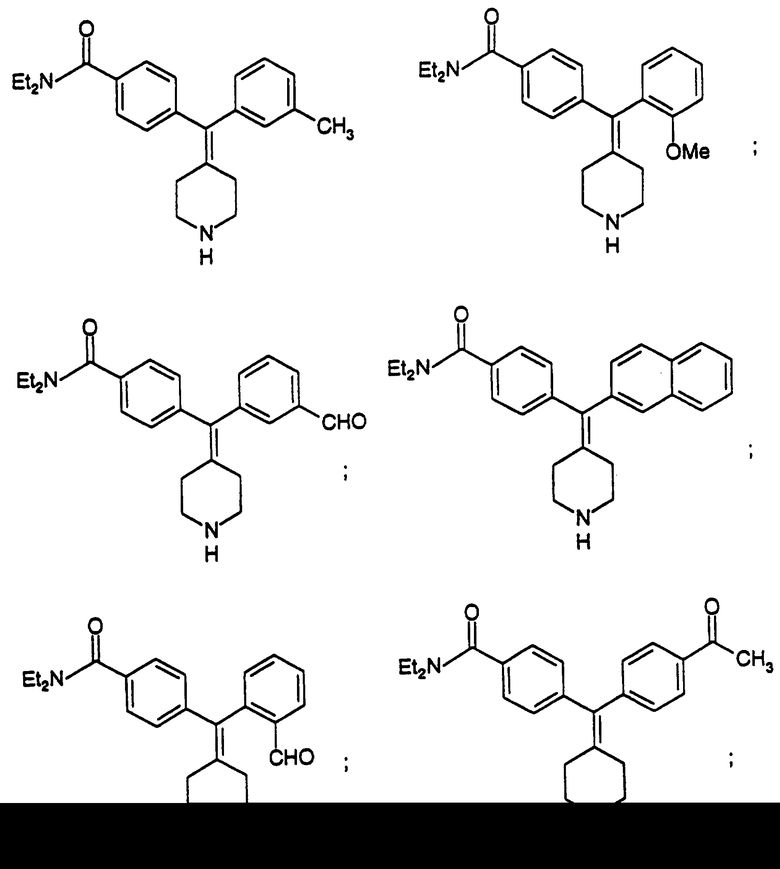
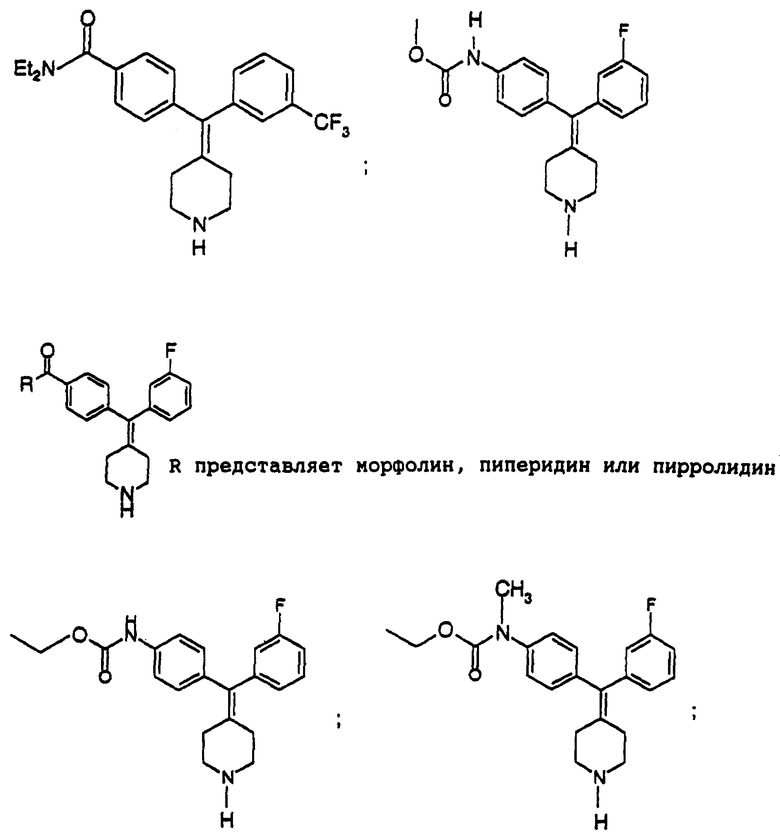
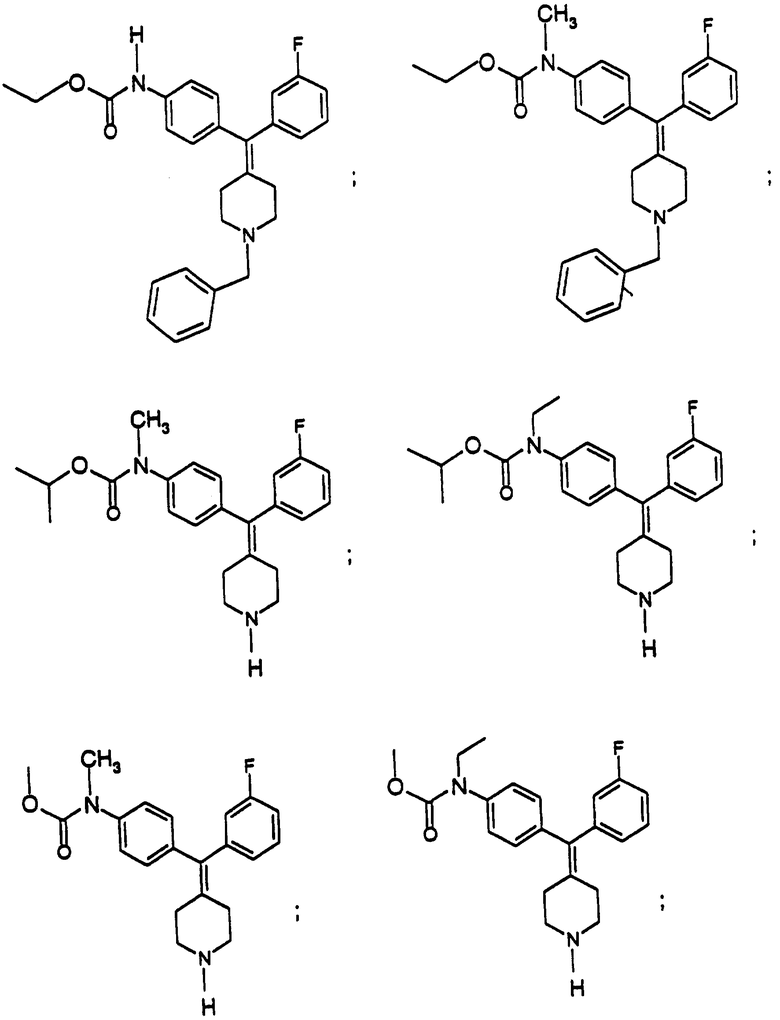
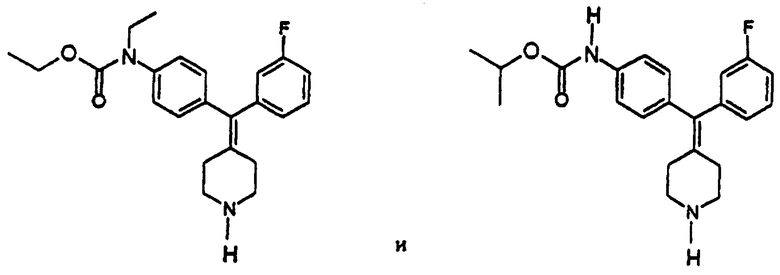
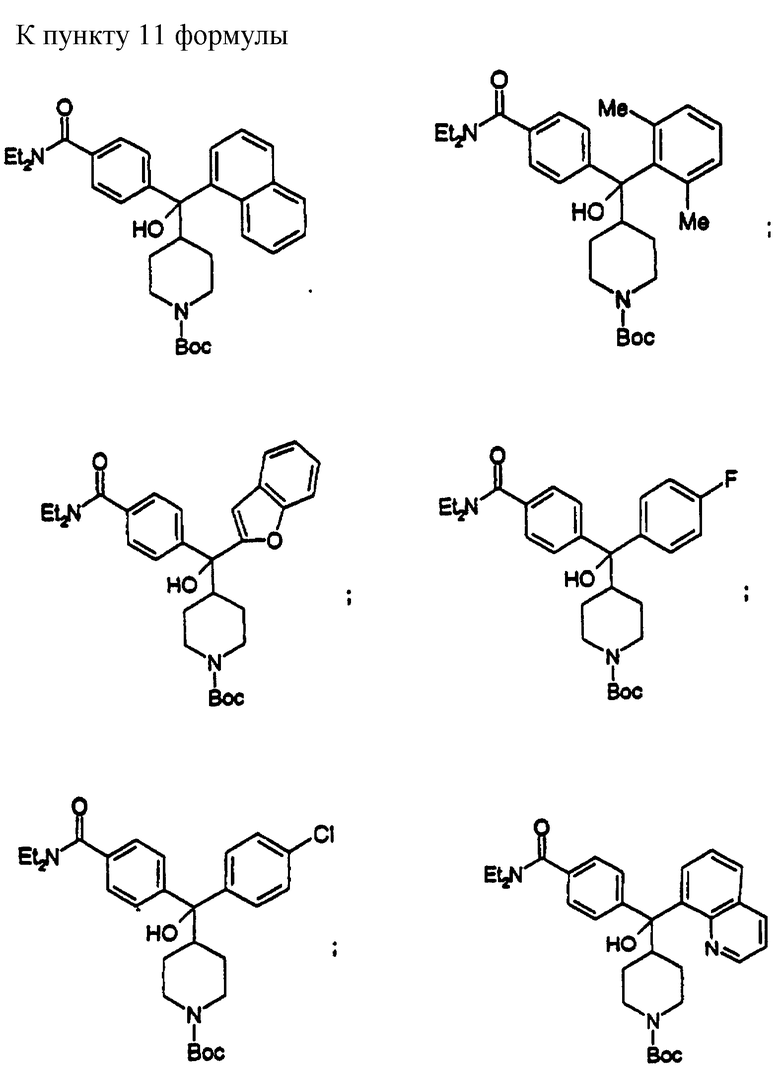
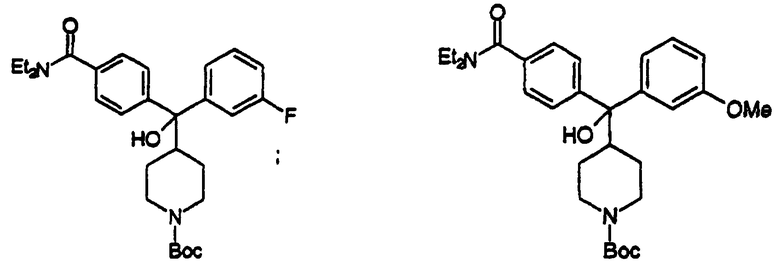
Изобретение относится к производным пиперидина общей формулы I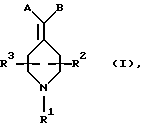
а также к их фармацевтически приемлемым солям, где R1 - водород, C1-С6-алкил, C2-С6-алкенил, C3-С8-циклоалкил, C6-С10-арил, который может быть замещен СН3, галогеном, OR5, где R5 - C1-С6-алкил, C1-С2-алкил-гетероарил, содержащий в качестве гетероатома S, N или О; А - фенил, замещенный карбонильной или аминогруппой; В - C6-С10-арил или C5-С10-гетероарил, содержащий в качестве гетероатома S, N или О. Изобретение относится также к способу получения соединений формулы I, а также к фармацевтическим композициям. Соединения формулы I обладают аналгезирующей активностью и могут найти применение в медицине. 4 с. и 7 з.п. ф-лы, 2 табл.
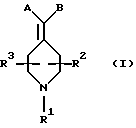
где R1 выбирают из водорода, разветвленного или неразветвленного C1-С6-алкила, C2-С6-алкенила, C3-С8-циклоалкила, C4-С8-(алкилциклоалкила), где алкил представляет C1-С2-алкил и циклоалкил представляет C3-С6-циклоалкил, C6-С10-арила, (C1-С2-алкил)-(C6-С10-арила), причем арил может быть замещен 1 или 2 заместителями, выбранными из СН3, OR5 или галогена, где R5 представляет C1-С6-алкил; (C1-С2-алкил) гетероарила, причем гетероарильная часть имеет от 5 до 10 атомов, выбранных из С, S, N и О; группы
где R18 и R19 представляют водород, C1-С6-алкил; группы
и
R2 и R3 представляют водород;
А выбирают из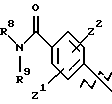
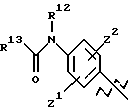

где R8, R9 и R12 представляют водород или C1-С6-алкил;
R13 представляет C1-С6-алкил или C1-С6-алкоксил;
Z1 и Z2 представляют водород;
Q представляет 5- или 6-тичленный гетероцикл, содержащий в качестве гетероатома О и/или N;
В представляет C6-С10-арил или C5-С10-гетероарил, содержащий в качестве гетероатома S, N или О, причем арил может быть замещен 1 или 2 заместителями, выбранными из СН3, СF3, NO2, галогена, ОН, COR4, NR4R5, ОСН3 или SСН3, где R4 и R5 представляют водород или C1-С6-алкил,
или их фармацевтически приемлемые соли и гидраты.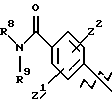

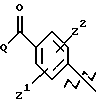
где R8, R9 и R12 представляют водород или C1-С6-алкил;
R13 представляет C1-С6-алкил или C1-С6-алкоксил;
Z1 и Z2 представляют водород;
Q выбирают из морфолина, пиперидина и пирролидина,
R1 выбирают из водорода, разветвленного или неразветвленного С1-С4-алкила, C3-С5-циклоалкила, C4-С8-(алкилциклоалкила), где алкил представляет C1-С2-алкил и циклоалкил представляет C3-С6-циклоалкил; C6-С10-арила, (C1-С2-алкил)-(C6-С10-арила), причем арил может быть замещен 1 или 2 заместителями, выбранными из СН3, OR5 или галогена, где R представляет C1-С6-алкил;
В выбирают из фенила, нафтила, бензофуранила, хинолинила, тиофенила, причем фенил или нафтил могут быть замещены 1 или 2 заместителями, выбранными из СН3, СF3, NО2, ОН, галогена, COR4, NR4R5, ОСН3 или SСН3, где R4 и R5 представляют водород или C1-С6-алкил.
где R8 и R9 представляют этил;
Z1 и Z2 представляют водород;
R1 выбирают из водорода, метила, этила, -СН2-СН-СН2, -СН2-циклопропила, -СН2-арила;
В выбирают из фенила, нафтила, бензофуранила, хинолинила, тиофенила, причем фенил или нафтил могут быть замещены 1 или 2 заместителями, выбранными из СН3, СF3, NO2, галогена, COR4, NR4R5, ОСН3 или SСН3, где R4 и R5 представляют водород или C1-С6-алкил.
и
6. Соединение по любому из предыдущих пунктов в форме его гидрохлоридной, сульфатной, тартратной или цитратной соли.
а) взаимодействие кетона формулы I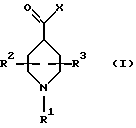
где R1, R2 и R3 имеют указанные в п. 1 значения и где R1 может быть также трет-бутоксикарбонилом;
Х представляет уходящую группу,
с металлоорганическими реагентами формул j и k
A-M(j)
и
В-М (k),
где А и В имеют указанные в п. 1 значения;
М представляет металлсодержащую группу,
в присутствии, в случае необходимости, растворителя с получением соединения формулы h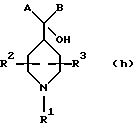
где А, В, R1, R2 и R3 имеют указанные в п. 1 значения и где R1 может быть также трет-бутоксикарбонилом;
b) дегидратацию соединения формулы h с получением соединения формулы I по п. 1.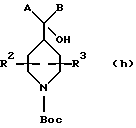
где А, В, R2 и R3 имеют указанные в п. 1 значения;
Воc обозначает трет-бутоксикарбонил.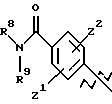
где R8 и R9 представляют этил;
и Z1 и Z2 представляют водород.
Приоритет по пунктам и признакам:
20.12.1996 по пп. 1-3, 6-10, когда R1, R2, R3, R4, R5, R8, R9, R12, R13, R18, R19, A, B, Z, 1 Z2 имеют указанные значения, за исключением случаев, когда R1 - группа:
и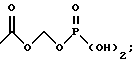
за исключением случаев, когда В-арил, который замещен 1 или 2 заместителями, выбранными из NО2 или SСН3; тиофенил; за исключением случаев, когда А - группа:
п. 4 - для первых 22 из указанных соединений, п. 11;
01.07.1997 по пп. 1-3, 6-10, когда В - тиофенил, п. 4 - для соединений 34 и 35 по ходу их изложения в этом пункте;
09.12.1997 (по дате подачи международной заявки) - по пп. 1-3, 6-10, когда R1 - группа:
и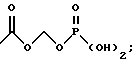
когда В - арил, который замещен 1 или 2 заместителями, выбранными из NO2 или SСН3; когда А - группа: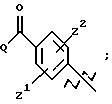
п. 4 - для соединений 23-33, 36-60 по ходу их изложения в этом пункте, п. 5.
| US 2898339 А, 04.08.1959 | |||
| WO 9315062 А1, 05.08.1993 | |||
| ПРОИЗВОДНОЕ N-ЗАМЕЩЕННОГО 1-ГЕКСИЛ-4-ФЕНИЛ-4-ПИПЕРИДИНКАРБОКСАМИДА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2039043C1 |

