Изобретение поддерживается частично денежными средствами Национального института здравоохранения (National Institutes of Health) и Фондом национальной науки (the National Science Fondation). В соответствии с этим, Правительство Соединенных Штатов Америки имеет определенные права на данное изобретение.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
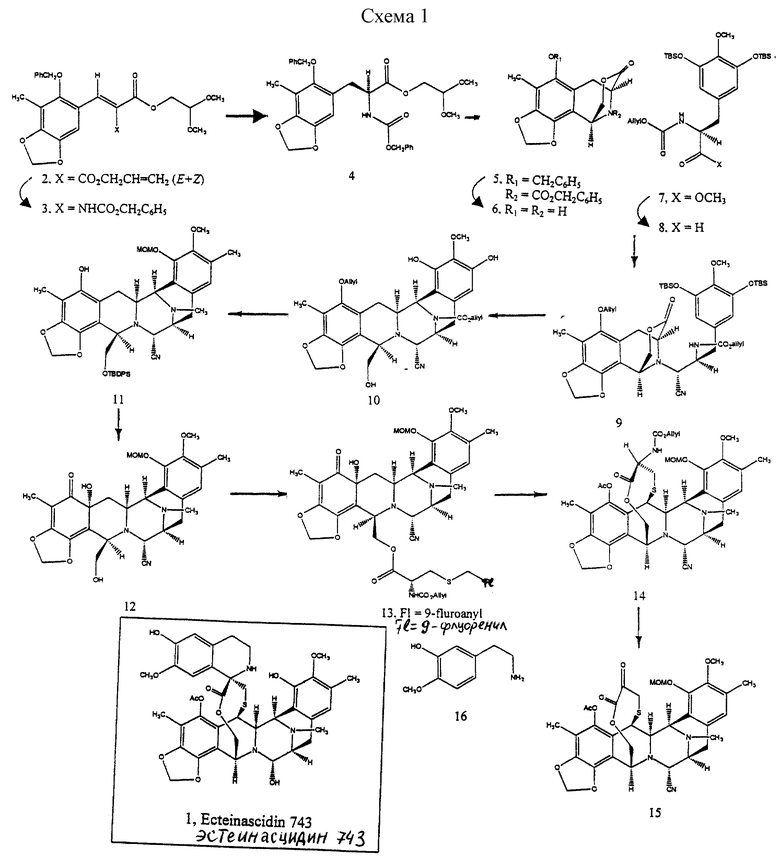
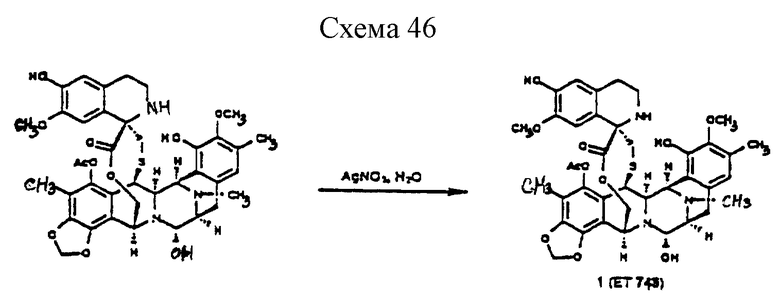
Данное изобретение относится к синтетическому способу получения соединений ряда эстеинасцидина и относящихся к ним структур, таких как сафрамицины. В одном особенно предпочтительном осуществлении данное изобретение относится к синтетическому пути получения эстеинасцидина 743(1)1.
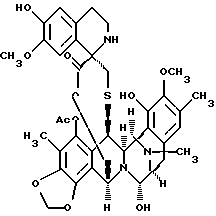
чрезвычайно сильнодействующего и редкого противоопухолевого средства морского происхождения, который предполагается для клинических исследований, когда адекватные количества его стали доступными2,3. Этот способ является энантио- и стереорегулируемым, конвергентным и коротким. Предпочтительное осуществление синтетического способа данного изобретения лучше всего представляется схемой 1 (см. в конце описания).
Как показано выше на схеме 1, предпочтительный способ синтетического получения эстеинасцидина 743 включает последовательные стадии:
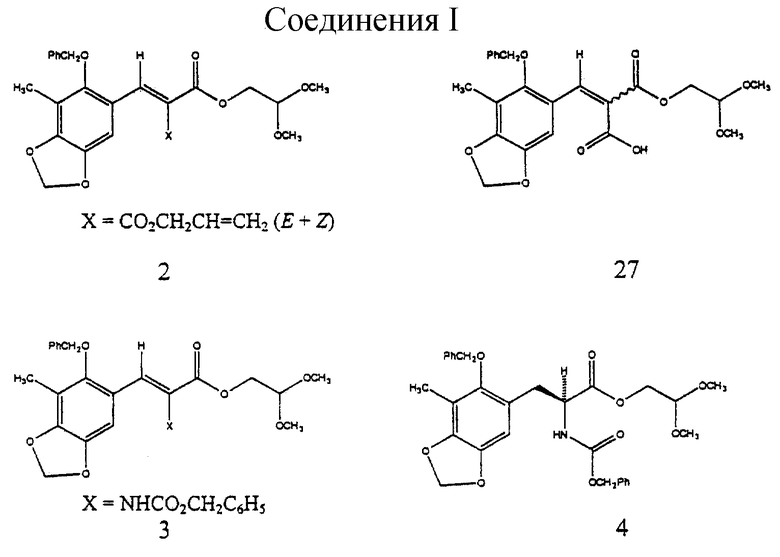
(a) образование α, β-ненасыщенного сложного эфира малоновой кислоты формулы 2 в виде смеси Е- и Z-изомеров из 2-бензилокси-3-метил-4,5-метилендиоксибензальдегида и аллил-2,2-диметокси-этилмалоката;
(b) стереоспецифическое преобразование соединения формулы 2 в соединение формулы 3 путем селективного отщепления аллилового сложного эфира, перегруппировки Курциуса и взаимодействия промежуточного изоцианата с бензиловым спиртом;
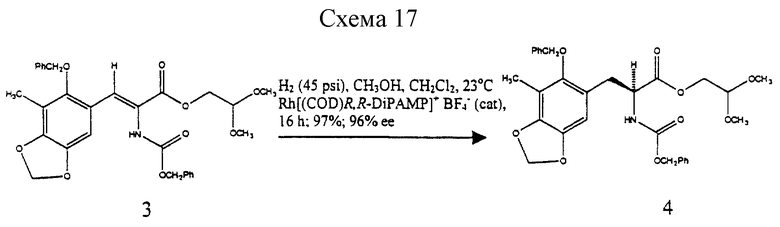
(c) преобразование соединения формулы 3 в соединение формулы 4 путем каталитического гидрирования над Rh [(COD) R1R-DIPAMP]+ВF4 -,
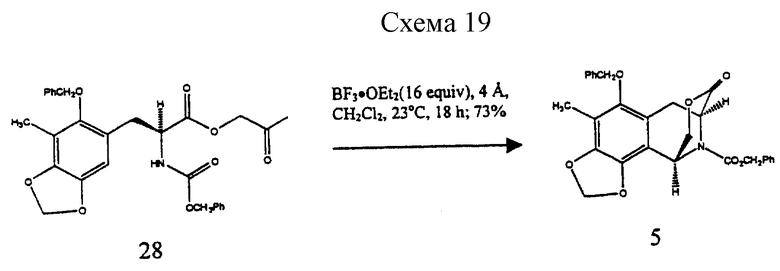
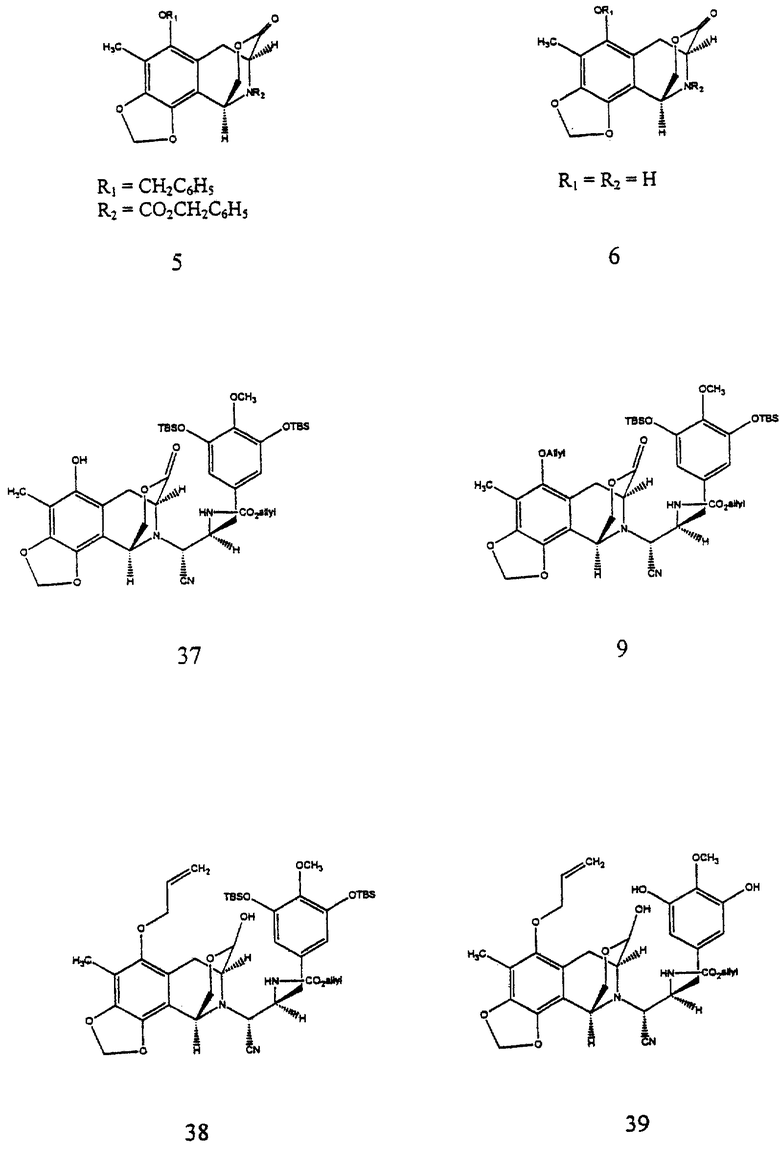
(d) преобразование соединения формулы 4 в соединение формулы 5 путем отщепления ацеталя, где выделение и воздействие на полученный альдегид ВF3•Еt2О и молекулярных сит  дает производное мостикового лактона формулы 5;
дает производное мостикового лактона формулы 5;
(e) преобразование производного мостикового лактона формулы 5 в производное свободного аминофенола формулы 6 гидрированием 10% Pd-C;
(f) образование производного защищенного сложного α-аминоэфира формулы 7 путем взаимодействия 3,5-бис-трет-бутилдиметилсилилокси-4-метоксибензальдегида с метилгидромалонатом;
(g) преобразование производного защищенного сложного α-аминоэфира формулы 7 в хиральный альдегид 8 восстановлением;
(h) связывание соединений формул 6 и 8 с получением ключевого мономостикового пентациклического промежуточного продукта формулы 10 следующим образом:
взаимодействием соединений формул 6 и 8 с получением связанного фенольного α-аминнитрила с последующим O-аллилированием и получением произвольного аллилового простого эфира формулы 9; селективным преобразованием лактоновой группы в соединении формулы 9 в лактол путем взаимодействия соединения формулы 9 c диизобутилалюминийгидридом; десилилированием производного лактола и циклизацией десилилированного производного с получением пентациклического соединения формулы 10 путем внутреннего бисаннулирования по Манниху;
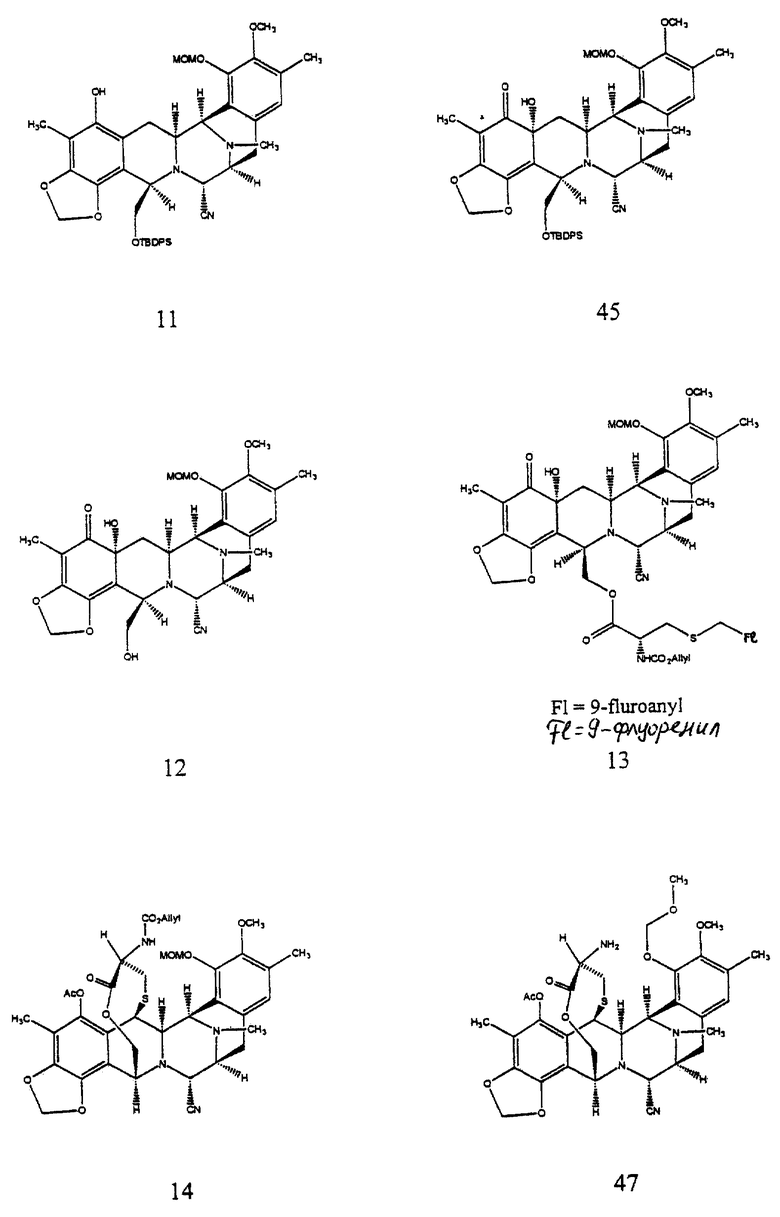
(i) преобразование пентациклического соединения формулы 10 в соединение формулы 11 путем селективного трифторметансульфирования по меньшей мере затрудненного фенольного гидроксила; с последующим
(1) селективным силилированием первичного гидроксила;
(2) защитой оставшейся фенольной группы в виде метоксиметилового эфира;
(3) двойным деаллилированием;
(4) восстановительным N-метилированием и
(5) замещением СF3SO3 на СН3;
(j) окисление производного фенола формулы 11, осуществляемого селективным по положению ангулярным гидроксилированием, с получением после десилилирования производного дигидроксидиенона формулы 12;
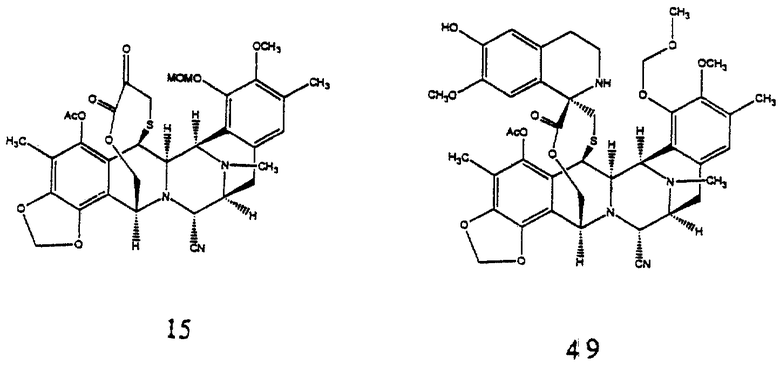
(k) образование соединения формулы 13 путем этерификации первичной гидроксильной группы соединения 12 с помощью (S)-N-аллилоксикарбонил-S-(9-флуоренилметил) цистеина;
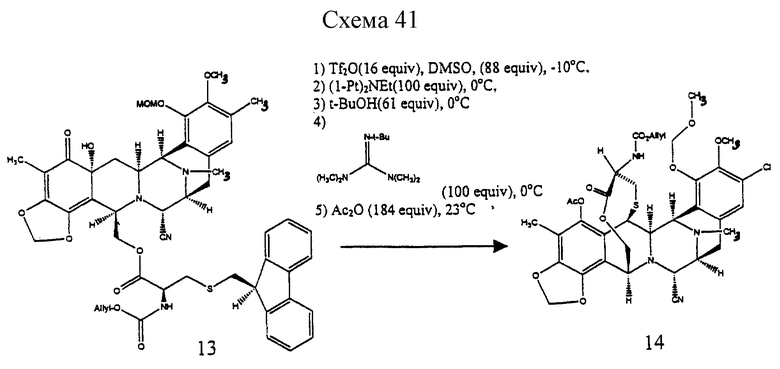
(l) преобразование соединения формулы 13 в производное мостикового лактона формулы 14:
(1) сначала взаимодействием соединения формулы 13 с образованным in situ реагентом Сверна (Swern); (2) затем образованием эксэндохинонметида; (3) разложением избытка реагента Сверна; (4) добавлением избытка N-трет-бутил-N',N''-тетраметилгуанидина для образования 10-членного лактонового мостика и (5) добавлением избытка Ac2O для ацетилирования полученной феноксидной группы;
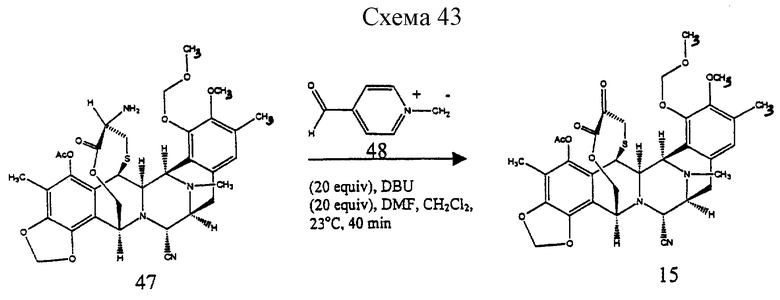
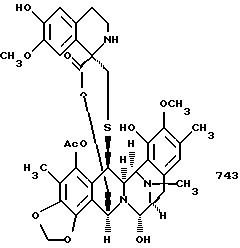
(m) отщепление N-аллилоксикарбонильной группы соединения формулы 14 и окисления полученного α-аминолактона в соответствующий α-кетолактон путем переаминирования с получением, таким образом соединения формулы 15;
(n) стереоспецифическое образование производного спиротетрагидроизохинолина путем взаимодействия соединения формулы 15 с 2-[3-гидрокси-4-метоксифенил] этиламином;
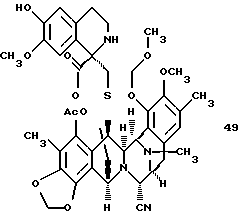
(о) последующее отщепление метоксиметила (образующеющего Et770) и далее замещение CN на НО с получением соединения формулы 1, эстеинасцидина 743.
Кроме предпочтительного способа по схеме 1 данное изобретение относится к новым промежуточным соединениям, используемым для синтеза известных соединений ряда эстеинасцидина, а также аналогов и производных указанных соединений. Эти новые промежуточные продукты включают соединения I (см. в конце описания)
Соединения I
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ОСУЩЕСТВЛЕНИЙ ИЗОБРЕТЕНИЯ
Предпочтительный способ по данному изобретению иллюстрируется на схеме 1. Как показано здесь и как обсуждается более подробно в примерах, которые следуют ниже, этот способ проводят следующим образом:
α, β-Ненасыщенный эфир малоновой кислоты 2 получали в виде смеси Е- и Z-изомеров из 2-бензилокси-3-метил-4,5-метилендиоксибензальдегида4a и аллил-2,2-диметоксиэтилмалоната4b (2 эквив. пиперидина и 4 эквив. уксусной кислоты в C6H6 или С7Н8 при 23oС в течение 18 ч; 99%), подвергали обработке для селективного отщепления аллилового сложного эфира (Еt3N-НСООН, катализатор Рd(РРh3)4, 23oС, 4 ч; выход 94%), перегруппировке Курциуса (1,2 эквив. (PhO)2Р(О)N3, 4 эквив. Еt3N, в С7Н8, содержанием молекулярные сита  при 70oС в течение 2ч) и промежуточный изоцианат подвергали взаимодействию с бензиловым спиртом при 23oС в течение 1 часа для стереоспецифического образования 3 (выход 93%)5.
при 70oС в течение 2ч) и промежуточный изоцианат подвергали взаимодействию с бензиловым спиртом при 23oС в течение 1 часа для стереоспецифического образования 3 (выход 93%)5.
Гидрирование 3 при 3 атм с использованием Rh[(COD)R,R-DIPAMP]+BF4 - в качестве катализатора при 23oС в течение 16 ч давало 4 с выходом 97% и энантиомерным избытком (эи) 96%6. Отщепление ацеталя от 4 (10 эквив. BF3•Et2O и 10 эквив. H2O в CH2Cl2 при 0oС в течение 10 мин), выделение и обработка полученного альдегида ВF3•Еt2O (17 эквив.) и молекулярными ситами  в СН2Сl2 при 23oС течение 18 ч давала мостиковый лактон 5 с выходом 73%7.
в СН2Сl2 при 23oС течение 18 ч давала мостиковый лактон 5 с выходом 73%7.
Гидрирование 5 (1 атм H2, 10% Pd-C, EtOAc, 23oC, 6 ч) давало свободный аминофенол 6 с выходом 100%. Защищенный сложный α-аминоэфир 7 синтезировали аналогичным путем, исходя из 3,5-бис-трет-бутилдиметилсилилокси-4-метоксибензальдегида и метилгидромалоната, и затем восстанавливали (2 эквив. диизобутилалюминийгидрида в CH2Cl2 при -78oС в течение 1 ч), получая хиральный альдегид 8 (выход > 90%).
Следующая стадия синтеза, которая включает связывание блоков построения 6 и 8 и последующее превращение для создания ключевого мономостикового пентациклического промежуточного продукта 10, начиналась с реакции 6 и 8 и НОАс, содержащей 25 экв. KCN, при 23oС в течение 18 ч для получения связанного фенольного α-аминонитрила (61%) и последующего O-аллилирования с получением аллилового простого эфира 9 с выходом 87% (2 эквив. Сs2СО3 и 5 эквив. аллилбромида в ДМФ при 23oС в течение 1 ч). Полагают, что исходя из промежуточного продукта 10, можно синтетически получить все известные соединения ряда эстеинасцидина, а также их аналоги и производные.
Обработка 9 1,2 эквив. диизобутилалюминийгидрида в толуоле при -78oС в течение 5 ч приводила к селективному преобразованию лактоновой функциональной группы в лактол, который десилилировали обработкой избытком KF•2H2O в СН3ОН при 23oС в течение 20 мин и циклизовали в пентацикл 10 внутренним бисаннелированием по Манниху 20 эквив. СН3SО3Н в CH2Cl2 в присутствии молекулярных сит  при 23oС в течение 5 ч (суммарный выход из 9 - 55%).
при 23oС в течение 5 ч (суммарный выход из 9 - 55%).
Селективное трифторметансульфирование по меньшей мере стерически затрудненного фенольного гидроксила (5 эквив. Tf2NPh, Et3N, 4,4-диметиламинопиридин (ДМАП)) в CH2Cl2 при 23oС в течение 6 ч, выход 72%) с последующим (1) селективным силилированием первичного гидроксила (избыток смеси трет-бутилдифенилсилилхлорид-ДМАП в CH2Cl2 при 23oС в течение 13 ч; 89%), (2) защитой оставшейся фенольной группы в виде метоксиметилового эфира (МеОСН2Вr и изо-Pr2NEt в CH2Cl2 при 23oС в течение 20 мин; 92%), (3) двойным деаллилированием (Вu3SnН, катализатор Cl2Pd (РРh3)2, избыток НОАс в CH2Cl2 при 23oС в течение 15 мин; 100%), (4) восстановительным N-метилированием (избыток формалина, NаВН3СN, НОАс в СН3СN при 23oС в течение 30 мин; 95%) и (5) заменой СF3SO3 на СН3 (избыток Me4Sn, Сl2Рd(Рh3H)2, LiCl, ДМФ, 80oС, 2 ч) давали 11 с выходом 83%.
Окислением фенола 11 1,1 эквив. (PhSeO)2O в CH2Cl2 при 23oС в течение 15 мин осуществляли селективное по положению ангулярное гидроксилирование, получая после десилилирования (2 эквив. Bu4NF в ТГФ при 23oС в течение 10 мин) дигидроксидиенон 12 (выход из 11 - 75%).
Последние три кольца эстеинасцидина 743, 10-членный лактоновый мостик и спиротетрагидроизохинолиновую подгруппу, затем присоединяли на последней стадии синтеза 1 следующей последовательностью реакций:
Первичную гидрокcильную функцию 12 этерифицировали (S)-N-аллилоксикарбонил-S-(9-флуоренилметил) цистеином с использованием 5 эквив. 1-(3-диметиламинопропил)-3-этилкарбодиимид•НСl и 5 эквив. ДМАП в CH2Cl2 при 23oС в течение 30 мин с получением 13 (91%), которое затем преобразовывали в одной колбе в мостиковый лактон с общим выходом 79% путем следующих операций: (1) взаимодействия 13 с реагентом Сверна, полученным in situ из избытка трифторметансульфонового ангидрида и ДМСО (диметилсульфоксида) при -40oС в течение 30 мин8a, (2) добавления изо-Pr2NEt и нагревания до 0oС в течение 30 мин для образования эксендохинонметида8b, (3) гашения смеси трет-бутиловым спиртом (для разрушения избытка реагента Сверна), (4) добавления избытка N-трет-бутил-N', N''-тетраметилгуанидина9 для превращения простого 9-флуоренилметилтиолэфира в тиолатион и промотирования нуклеофильного присоединения серы к хинонметиду для образования 10-членного лактонового мостика и (5) добавления избытка Ac2O для ацетилирования образуемой феноксидной группы. N-Аллилоксикарбонильную группу 14 отщепляли (избыток Вu3SnН, НОАс и катализатор Сl2Рd(РРh3)2 в CH2Cl2 при 23oС в течение 5 мин; 84%) и получаемый α-аминолактон окисляли в соответствующий α-кетолактон путем переаминирования метиодидом пиридин-4-карбоксальдегида, DBU и ДМФ в CH2Cl2 при 23oС в течение 40 мин, получая 15 (70%). Взаимодействие 15 с 2-[3-гидрокси-4-метоксифенил] этиламином (16) в EtOH в присутствии силикагеля при 23oС стереоспецифически давало спиротетрагидроизохинолин (82%), который затем подвергали отщеплению метоксиметила (смесь 4: 1:1 СF3СО2Н-Н2О-ТГФ при 23oС в течение 9 ч) и замене CN на ОН (АgNO3 в СН3СN-Н2О при 23oС в течение 11 ч), с получением с высоким выходом эстеинасцидина 741 (1), идентичного во всех отношениях с аутентичным образцом10.
Синтетический способ по данному изобретению делает доступным не только 1, но также множество других членов ряда эстеинасцидина и аналогов, а также относящихся к ним более простых структур, таких как сафрамицины11. Получение и исследование новых промежуточных продуктов, указанных выше, описывается подробно в примерах ниже.
Данное изобретение далее проиллюстрировано со ссылкой на следующие примеры, которые помогают понять данное изобретение, но которые не должны рассматриваться как его ограничения.
ПРИМЕРЫ
Общие методики. Все реакции проводили и высушенных огнем круглодонных или модифицированных колбах Шленка (форма Кьельдаля), снабженных каучуковыми перегородками, при положительном давлении аргона, если не оговорено особо. Чувствительные к воздуху и влаге жидкости и растворы переносили через шприц или канюлю из нержавеющей стали. Где необходимо (это указано), из растворов удаляли кислород циклами чередующихся операций откачка/промывание аргоном (более чем три повтора). Органические растворы концентрировали путем упаривания на роторе, при температуре ниже 30oС и приблизительно при 25 мм рт. ст. Колоночную флэш-хроматографию проводили, как описано Still et al., используя силикагель 230-400 меш12. Тонкослойную хроматографию (аналитическую и препаративную) проводили с использованием стеклянных пластин, предварительно покрытых на глубину 0,25 мм силикагелем 230-400 меш, пропитанных фпуоресцентным индикатором (254 нм).
Материалы. Коммерческие реагенты и растворители использовали такими, какими получали, со следующими исключениями. Тетрагидрофуран и этиловый простой эфир перегоняли из кетила бензофенона натрия. Дихлорметан, гексан, N, N-диизопропилэтиламин, диизoпропиламин, триэтиламин, пиридин, толуол, бензол, ТМЭДA (тетраметилэтилендиамин), пиперидин и ацетонитрил перегоняли с гидридом кальция при 760 мм рт.ст. Молярность растворов н-бутиллития определяли титрованием с использованием дифенилуксусной кислоты в качестве индикатора (среднее значение трех определений)13.
Измерительная аппаратура. Инфракрасные (ИК) спектры получали с использованием спектрофотометра Nicolet 5ZDX FT-IR, отнесенного к стандарту полистиролу. Данные представляются следующим образом: частота поглощения (см-1) и итенсивность поглощения (с.=сильное, ср.=среднее и cл.=слабое). Спектры протонного и углеродного-13 ядерного магнитного резонанса (1Н ЯМР или 13С ЯМР) регистрировали спектрометром Bruker AM500 (500 МГц), Bruker AM400 (400 МГц) или Bruker АМ300 (300 МГц); химические сдвиги выражаются в частях на миллион (шкала δ) по направлению от тетраметилсилана и относятся к остаточному прoтию в растворителе для ЯМР (СНСl3: δ 7,26, C6HD5: δ 7,20 CDHCl2: δ 5,38, СD3СОСD2Н: δ 2,04, CD2HOD: δ 3,30).
Данные представляются следующим образом: химический сдвиг, мультиплетность (с= синглет, д= дублет, т=триплет, м=мультиплет и/или мультиплетные резонансные сигналы), интегрирование, константа взаимодействия в герцах (Гц) и отнесение. Хиральную высокоэффективную жидкостную хроматографию (ВЭЖХ) проводили в приборе Isco 2350, снабженном определенной колонкой (см.ниже). Точки плавления регистрировали в аппаратуре Fisher-Johns для определения точки плавления, точки плавления некорректированы.
Схемы 2-7 иллюстрируют примеры, которые приводятся в конце описания.
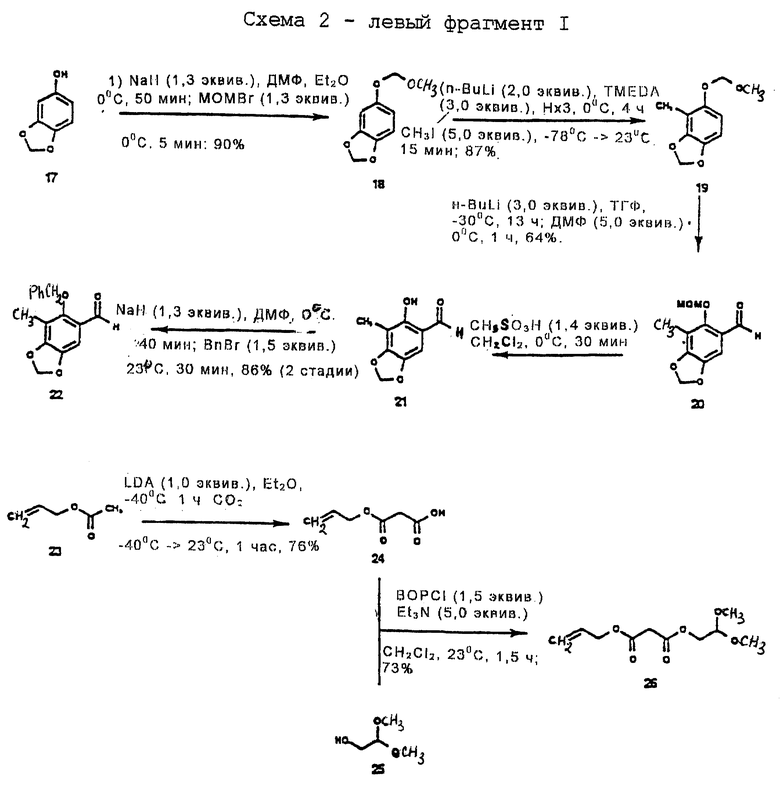
Левый фрагмент
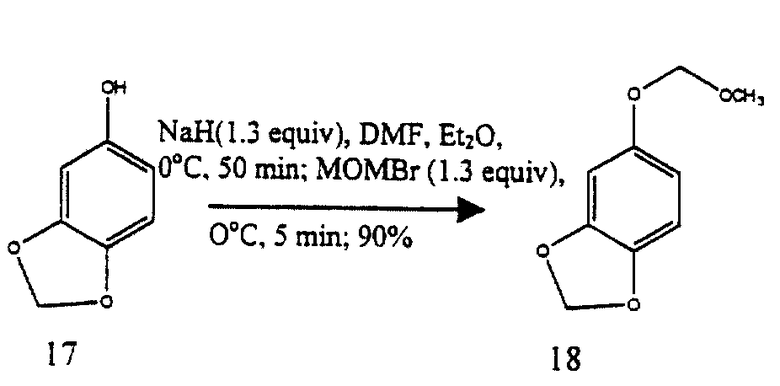
Пример 1 - Метоксиметиловый эфир 18 (см. схему 8 см. в конце описания).
К раствору 17 (10,2 г, 74,3 ммoль, 1 эквив.) в смеси этилового простого эфира и ДМФ (4: 1 (об./об.), 100 мл) при 0oС добавляли суспензию гидрида натрия в минеральном масле (57% (мас./мас.), 4,07 г, 96,6 ммсль, 1,3 эквив. ). Полученную суспензию перемешивали при 0oС в течение 35 мин и затем по каплям добавляли бромметилметиловый эфир (7,89 мл, 96,6 ммоль, 1,3 эквив.). Суспензию перемешивали при 0oС в течение 5 мин и затем при 23oС в течение 1 ч до того, как избыток гидрида натрия был нейтрализован медленным добавлением метилового спирта (5 мл) при 0oС. Раствор распределяли между этилацетатом (500 мл) и водой (300 мл) и органическую фазу, затем промывали насыщенным водным раствором хлорида натрия (200 мл), сушили (сульфат натрия) и концентрировали. Остаток очищали колоночной флэш-хроматографией (7% этилацетат в гексане), получая 18 (13,1 г, 90%) в виде бесцветного масла. Rf 0,32 (10% этилацетат в гексане); 1H ЯМР (500 МГц, CDCl3) δ 6,70 (д, 1Н, J=8/4 Гц, ArH), 6,62 (д, 1Н, J=2,4 Гц, ArH), 6,49 (дд, 1Н, J=8,4, 2,4 Гц, ArH), 5,91 (с, 2Н, ArOCH2OAr), 5,10 (с, 2H, MOM СН2), 3,50 (с, 3Н, ОСН3); 13С ЯМР (100 МГц, СDСl3) δ 152,5, 148,1, 142,5, 108,5, 108,0, 101,1, 99,7, 95,5, 60,3, 55,8, 14,1; ИК (пленка неразбавленного соединения) 2990 (ср.), 2847 (ср.), 2827 (ср. ), 1632 (ср.), 1611 (ср.), 1502 (с.), 1486 (с.), 1451 (ср.), 1245 (с. ), 1213 (с.), 1152 (с.), 1069 (с.), 1004 (с.), 922 (с.) см-1; HRMC (МС с высоким разрешением, МСВР) (EI+, ионизация элекронным ударом) m/z: вычислено для C9H10O4(M+) 182,0578, найдено 182,0582.
Пример 2 - Метоксиметиловый эфир 19 (см. схему 9 в конце описания).
К раствору 18 (6,76 г, 37,1 ммоль, 1 эквив.) и тетраметилэтилендиамина (16,8 мл, 111 ммoль, 3,0 эквив.) в гексанах (70 мл) при 0oС по каплям добавляли раствор н-бутиллития (1,55 М в гексанах, 72,0 мл, 74,2 ммоль, 2,0 эквив. ) и полученную желтую суспензию перемешивали при 0oС в течение 2,5 ч. По каплям добавляли раствор иодметана (11,5 мл, 186 ммоль, 5,0 эквив.) в диэтиловом простом эфире (12 мл) и полученную суспензию перемешивали при 23oС в течение 1 ч до того, как ее гасили медленным добавлением воды (10 мл). Реакционную смесь разбавляли диэтиловым простым эфиром (500 мл), раствор продукта промывали последовательно водой (50 мл) и насыщенным водным раствором хлорида натрия (50 мл) и затем сушили (сульфат натрия) и концентрировали. Остаток очищали колоночной флэш-хроматографией (градиентное элюирование 2%-->3% этилацетат в гексане), получая 19 (6,32 г, 87%) в виде бледно-желтого масла. Rf 0,31 (10% этилацетат в гексанах); 1H ЯМР (500 МГц, СDCl3) δ 6,57 (д, 1Н, J=8,5 Гц, АrН), 6,51 (д, 1Н, J=8,5 Гц, АrН), 5,91 (с, 2Н, ArOCH2OAr), 5,11 (с, 2Н, MOM CH2), 3,49 (с, 3Н, ОСН3), 2,14 (с, 3Н, АrСН3); 13С ЯМР (126 МГц, CDCl3) δ 151,0, 146,6, 141,9, 110,7, 106,7, 104,8, 100,9, 95,7, 56,0, 8,9; ИК (пленка неразбавленного соединения) 2928 (cл.), 1479 (с.), 1468 (с.), 1242 (с.), 1155 (ср.), 1103 (с.), 1068 (с.), 1020 (ср. ), 988 (ср. ), 793 (сл.) см-1; МСВР (EI+) m/z: вычислено для C10H12O4(M+) 196,0735, найдено 196,0729.
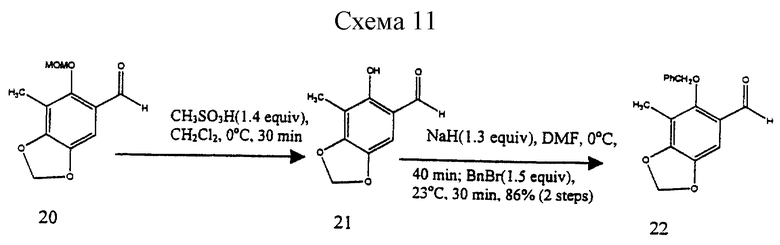
Пример 3 - Альдегид 20 (см. схему 10 в конце описания).
К раствору 19 (7,50 г, 38,3 ммоль, 1 эквив.) в смеси 1:1 (об./об.) диэтилового простого эфира и гексана (70 мл) при 0oС добавляли по куплям раствор н-бутиллития (1,50 М в гексане, 77,0 мл, 115 ммоль, 3,0 эквив.). Реакционную смесь оставляли для нагревания до 23oС и перемешивали при этой температуре в течение 5 ч. Желтую суспензию охлаждали до -10oС и затем добавляли N,N-диметилформамид (14,7 мл, 191 ммоль, 5,0 эквив.). Полученный раствор перемешивали при -10oС в течение 1 ч. Избыточное основание нейтрализовали медленным добавлением ледяной уксусной кислоты (10 мл) при -10oС и получаемую суспензию перемешивали в течение 5 мин. Реакционную смесь разбавляли этилацетатом (500 мл) и раствор продукта промывали последовательно насыщенным водным раствором бикарбоната натрия (400 мл), водой (400 мл) и насыщенным раствором хлорида натрия (300 мл). Органическую фазу сушили (сульфат натрия) и концентрировали и продукт 20 кристаллизовали из 10% этилацетата в гексанах (4,05 г). Маточный раствор очищали колоночной флэш-хроматографией (15% этилацетат в гексане), получая дополнительный 20 (1,35 г) (общий выход 64%) в виде бледно-желтого твердого продукта (т. пл. 91,5oС). Rf 0,22 (этилацетат в гексане). 1Н ЯМР (400 МГц, СDСl3) δ 10,15 (с, 1Н, СНО), 7,13 (с, 1Н, АrН), 6,03 (с, 2Н, ArOCH2OAr), 5,03 (с, 2Н, MOM CH2), 3,59 (с, 3Н, ОСН3), 2,19 (с, 3Н, АrCН3); 13С ЯМР (100 МГц СDСl3) δ 189,0, 157,0, 152,4, 144,2, 123,8, 113,7, 103,3, 102,1, 101,3, 58,0, 9,4, ИК (пленка неразбавленного соединения) 2925 (cл.), 1670 (с.), 1614 (cл.), 1473 (ср.), 1452 (ср.), 1399 (ср.), 1280 (ср.), 1155 (ср.), 1059 (ср.), 935 (с.), 927 (с.), 860 (ср.) см-1; МСВР (ЕI+) m/z: вычислено для С11Н12O5(М+) 224,0684, найдено 224,0684 (см. схему 11 в конце описания).
К раствору 20 (3,70 г, 16,5 ммоль, 1 эквив.) в дихлорметане (50 мл) и воде (1,0 мл) при 0oС добавляли метансульфоновую кислоту (1,50 мл, 22,5 ммоль, 1,4 эквив. ) Реакционную смесь затем нейтрализовали насыщенным водным раствором бикарбоната натрия (50 мл) при 0oС и получаемую смесь распределяли между насыщенным водным раствором бикарбоната натрия (400 мл) и дихлорметаном (3 х 200 мл). Объединенные органические слои сушили (сульфат натрия) и концентрировали, получая 21 в виде сырого промежуточного продукта. К раствору 21 в N,N-диметилформамиде (16,0 мл) при 0oС добавляли суспензию гидрида натрия в минеральном масле (57% (мас./мас.), 903 мг, 21,5 ммоль, 1,3 эквив.) и получаемую суспензию перемешивали при 0oС в течение 40 мин. К реакционной смеси при 0oС добавляли бензилбромид (2,94 мл, 24,8 ммоль, 1,5 эквив.) и получаемую суспензию перемешивали при 23oС в течение 30 мин. Избыточное основание нейтрализовали медленным добавлением метанола (2,0 мл) при 0oС и реакционную смесь разбавляли этилацетатом (250 мл). Раствор продукта промывали последовательно водой (200 мл) и насыщенным водным раствором хлорида натрия (200 мл), затем сушили (сульфат натрия) и концентрировали. Остаток очищали колоночной флэш-хроматографией (10% этилацетат в гексанах), получая 22 (3,85 г, 86%) в виде вязкого сиропа. Rf 0,18 (10% этилацетат в гексанах). 1H ЯМР (400 МГц, CDCl3) и 10,08 (с, 1Н, СНО), 7,40 (м, 5Н, Вn АrН), 7,12 (с, 1Н, АrН), 6,04 (с, 2Н, ArOCH2OAr), 4,93 (с, 2Н, Bn CH2), 1,60 (с, 3Н, АrСН3); 13С ЯМР (100 МГц, СDCl3) δ 188,5, 158,3, 152,6, 144,1, 135,7, 128,7, 128,3, 123,6, 113,8, 103,2, 102,1, 78,5, 11,8, 9,1, ИК (пленка неразбавленного соединения) 2923 (сл.), 1674 (с.), 1612 (сл.), 1470 (ср.), 1420 (ср. ), 1375 (ср. ), 1352 (ср.), 1278 (с.), 1170 (ср.), 1096 (с.), 1069 (ср.) см-1; МСВР (EI+) m/z: вычислено для C16H14O4 (M+) 270,0892, найдено 270,0892.
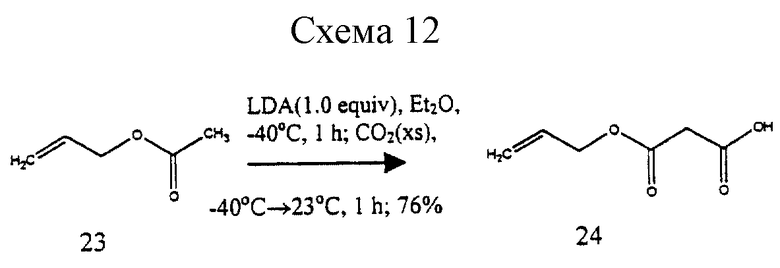
Пример 5 - Моноаллилмалонат 24 (см. схему 12 в конце описания).
Раствор н-бутиллития (1,56 М в гексанах, 19,2 мл, 30,0 ммоль, 1,0 эквив. ) добавляли к раствору диизопропиламина (5,47 мл, 39,0 ммоль, 1,3 эквив.) в этиловом простом эфире (30,0 мл) при -78oС. Реакционную колбу быстро переносили к ледяной бане (10 мин) и затем снова охлаждали до -78oС. К охлажденному раствору диизопропиламида лития добавляли аллилацетат 23 (3,23 мл, 30,0 ммоль, 1 эквив.) и получаемый раствор перемешивали при -40oС в течение 1 ч. Реакционную смесь охлаждали до -78oС и к реакционной смеси добавляли избыток твердого диоксида углерода до того, как ее оставляли для нагревания до 23oС в течение периода 1 ч. Мутный раствор разбавляли водoй (100 мл) и промывали этиловым простым эфиром (3х50 мл). Водный слой подкисляли при 0oС до рН 2 медленным добавлением концентрированной хлористоводородной кислоты и затем экстрагировали этилацетатом (3х50 мл). Объединенные органические слои сушили (сульфат натрия) и концентрировали, получая сырую кислоту 24 (3,35 г, 76%) в виде бледно-желтого масла, которое использовали без дальнейшей очистки. 1H ЯМР (400 МГц, СDСl3) δ 5,92 (м, 1Н, СН2=СН-), 5,36 (м, 1Н, СН2=СН-), 5,27 (м, 1Н, CH2= CH-), 4,68 (дт, 2Н, J=5,7, ~1 Гц, СН2=СНСН2-), 3,48 (с, 2Н, СН2), ИК (пленка неразбавленного соединения) 3300-2400 (ср.), 1744 (с.), 1322 (ср.), 1156 (ср.) см-1.
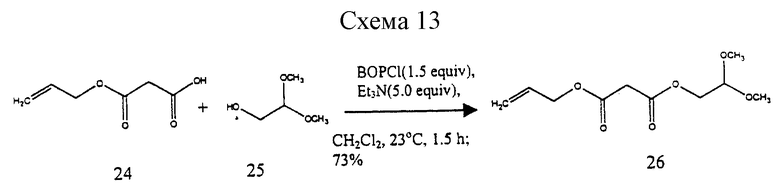
Пример 6 - Аллил-2,2-диметоксиэтилмаломанат 26 (см. схему 13 в конце описания).
К раствору кислоты 24 (7,50 г, 52,0 ммоль, 1 эквив.) 2,2-диметоксиэтанола (25) (5,50 г, 52,0 ммоль, 1,0 эквив.) и триэтиламина (36,0 г, 258 ммоль, 5,0 эквив.) в дихлорметане (100 мл) добавляли твердый BOPCI (20,0 мг, 78,7 ммоль, 1,5 эквив.) и получаемую суспензию перемешивали при 23oС в течение 1 ч. Реакционную смесь фильтровали, фильтрат разбавляли зтилацетатом (400 мл) и раствор продукта промывали последовательно водой (2 х 300 мл) и насыщенным водным раствором хлорида натрия (300 мл). Органический слой сушили (сульфат натрия) и концентрировали и остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 20-33% этилацетат в гексанах), получая 26 (8,81 г, 73%) в виде бесцветной жидкости. Rf 0,26 (25% этилацетат в гексанах); 1H ЯМР (300 МГц, СDСl3) δ 5,91 (м, 1Н, СН2=СН-), 5,34 (м, 1Н, СН2= СН-), 5,26 (м, 1Н, СН2=СН-), 4,64 (дт, 2Н, J=5,6, ~1 Гц, СН2=СНСН2), 4,58 (т, 1H, J= 5,3 Гц, СН(ОСН3)2), 4,17 (д, 2Н, J=5,3 Гц, СН2СН(ОСН3)2), 3,46 (с, 2Н, СН2), 3,39 (с, 6Н, ОСН3); 13С ЯМР (100 МГц, СDСl3) δ 166,0, 165,9, 131,5, 118,7, 101,0, 66,0, 63,8, 53,9, 41,2, FТИК (ИК с преобразователем Фурье, ИКПФ) (пленка неразбавленного соединения) 2955 (ср.), 1757 (с. ), 1738 (с.), 1447 (ср.), 1412 (ср.), 1367 (с.), 1340 (с.), 1323 (с.), 1276 (с. ), 1193 (с.), 1134 (с.), 1102 (с), 1078 (с.), 1046 (с.) см-1; МСВР (ЕI+) m/z: вычислено для C10H20NO6(M+NH4)+ 250,1291, найдено 250,1296.
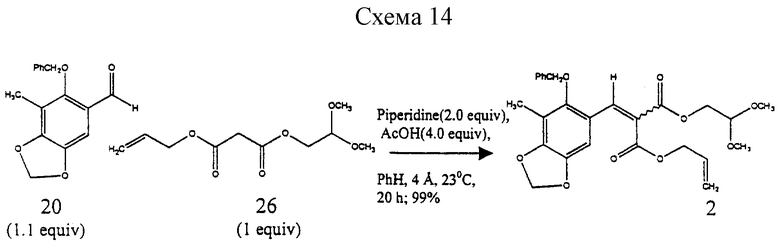
Пример 7 - α, β-Ненасыщенный сложный диэфир 2 (см. схему 14 в конце описания).
К смеси альдегида 20 (3,84 г, 14,2 ммоль, 1,1 эквив.), 26 (3,00 г, 12,9 ммоль, 1 эквив.), пиперидина (2,80 мл, 28,4 ммоль, 2,0 эквив.) и измельченных активированных молекулярных сит  (~6 г) в бензоле (40 мл) добавляли по каплям ледяную уксусную кислоту (3,25 мл, 56,8 ммоль, 4,0 эквив.) и получаемую суспензию перемешивали при 23oС в течение 18 ч. Реакционную cмесь фильтровали и фильтрат концентрировали. Остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 20-->33% этилацетат в гексанах),
(~6 г) в бензоле (40 мл) добавляли по каплям ледяную уксусную кислоту (3,25 мл, 56,8 ммоль, 4,0 эквив.) и получаемую суспензию перемешивали при 23oС в течение 18 ч. Реакционную cмесь фильтровали и фильтрат концентрировали. Остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 20-->33% этилацетат в гексанах),
получая 2 (6,20 г, 99%) и виде неразделяемой смеси E/Z-изомеров (1,3:1). Rf 0,62 (10% этиловый простой эфир в дихлорметане); 1H ЯМР (500 МГц, СDСl3) δ, изомер, присутствующий в большем количестве: 8,07 (с, 1Н, АrСН), 7,38 (м, 5Н, Ph-H), 6,83 (с. 1Н, АrН), 5,98 (с, 2Н, ArOCH2OAr), 5,75 (м, 1Н, СН2=СН), 5,34 (м, 1Н, СН2=СН), 5,24 (м, 1Н, СН2=СН), 4,77 (с, 2Н, Вn СН2), 4,72 (м, 2Н, CH2= CHCH2), 4,64 (т, 1Н, J=5,6 Гц, СН(ОСН3)2), 4,32 (д, 2Н, J=5,6 Гц, CH2CH (ОСН3)2, 3,41 (с, 6Н, ОСН3), 2,16 (с, 3Н, АrСН3), изомер, присутствующий в меньшом количестве: 8,06 (с, 1Н, АrСН), 7,38 (м, 5Н, Ph-H), 6,76 (с, 1Н, АrН), 5,98 (с, 2Н, ArOCH2OAr), 5,73 (м, 1Н, СН2=СН), 5,38 (м, 1Н, СН2= СН), 5,28 (м, 1Н, СН2= СН), 4,77 (с, 2Н, 3n СН2), 4,78 (м, 2Н, CH2= CHCH2), 4,59 (т, J=5,6 Гц, СН(ОСН3)2), 4,23 (д, 2Н, J=5,6 Гц, СН2СН(ОСН3)2), 3,40 (с, 6Н, ОСН3), 2,16 (с, 3Н, АrСН3); 13С ЯМР (100 МГц, СDСl3) δ 166,3, 166,2, 163,9, 163,8, 153,5, 149,5, 143,6, 139,1, 139,0, 136,3, 131,8, 131,4, 128,6, 128,4, 123,6, 119,4, 119,1, 118,2, 114,1, 104,7, 104,6, 101,7, 101,2, 101,0, 77,5, 77,4, 66,2, 65,8, 63,9, 63,8, 53,9, 53,8, 14,1, 9,3. ИК (пленка неразбавленного соединения) 2928 (cл.), 1732 (с.), 1609 (ср.), 1476 (ср.), 1423 (ср.), 1243 (с.), 1217 (с), 1186 (с.). 1096 (с.), 1079 (с.) см-1; MCBP (FAB+), бомбардировка ускоренными атомами, БУА) m/z: вычислено для С26Н28О9Nа (MNa+) 507,1631, найдено 507,1640.
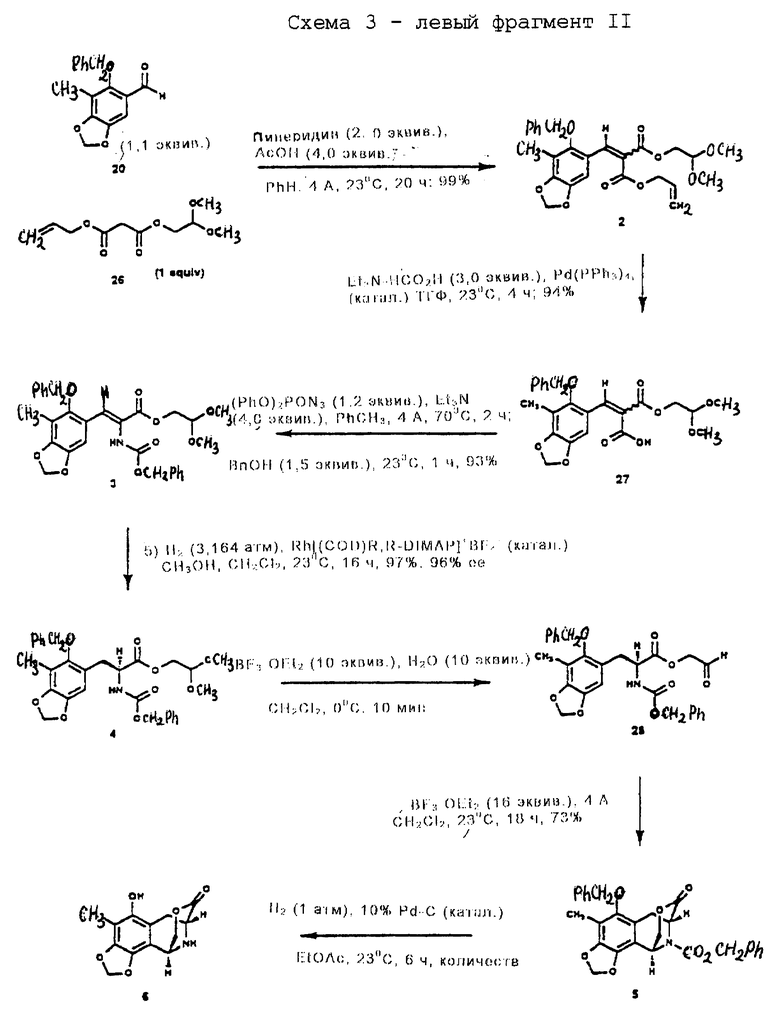
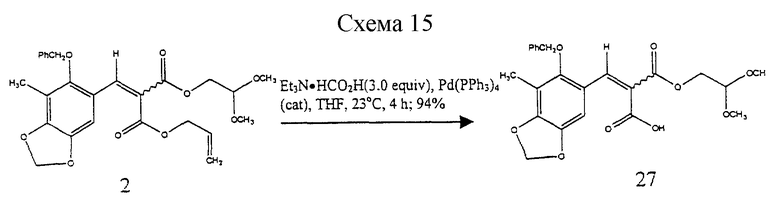
Пример 8 - α, β-Ненасыщенная кислота 27 (см. схему 15 в конце описания).
К раствору 2 (6,20 г, 12,8 ммоль, 1 эквив.) в тетрагидрофуране (30 мл) добавляли последовательно раствор формиата триэтиламмония (1M в тетрагидрофуране, 38,4 мл, 38,4 ммоль, 3,0 эквив.) и твердый тетракис(трифенилфосфин) палладий (120 мг) и получаемый раствор перемешивали при 23oС в течение 4 ч. Все летучие компоненты удаляли в вакууме и остаток очищали колоночной флэш-хроматографией (10% метиловый эфир в дихлорметане), получая желтое масло 27 (5,33 г, 94%) в виде смеси E/Z-изомеров (4:1). Rf 0,21 (10% метиловый спирт в дихлорметане); 1H ЯМР (500 МГц, CDCl3) δ, изомер, присутствующий в большем количестве: 8,19 (с, 1Н, АrСН), 7,40 (м, 5Н, Ph-H), 6,82 (с, 1Н, АrН), 6,00 (с, 2Н, ArOCH2OAr), 4,78 (с, 2Н, Вn СН2), 4,61 (т, 1Н, J= 5,8 Гц, СН(ОСН3)2), 4,29 (д, 2Н, J=5,8 Гц, СО2СН2), 3,40 (с, 6Н, ОСН3), 2,15 (с, 3Н, АrСН3), изомер, присутствующий в меньшем количестве: 8,21 (с, 1H, АrСН), 7,40 (м, 5Н, Ph-H), 7,13 (с, 1Н, АrН), 5,96 (с, 2Н, АrОСН2ОАr), 4,78 (с, 2Н, Вn СН2), 4,59 (т, 1Н, J=5,8 Гц, СН(ОСН3)2), 4,24 (д, 2Н, J=5,8 Гц, CO2CH2), 3,38 (с, 6Н, ОСН3), 2,15 (с, 3Н, АrСН3); 13С ЯМР (100 МГц, СОС13) δ 169,3, 168,9, 166,3, 164,8, 153,8, 149,9, 143,6, 143,5, 141,6, 141,4, 136,1, 135,9, 128,7, 128,5, 128,4, 128,3, 122,0, 121,5, 119,2, 119,1, 114,0, 113,8, 105,2, 104,7, 101,7, 101,0, 100,9, 77,6, 77,5, 63,9, 63,7, 53,9, 53,8, 53,3, 50,3, 9,2 ИК (пленка неразбавленного соединения) 3500-2500 (ср. ), 2958 (cp.), 1735 (с.), 1701 (с), 1608 (ср.), 1476 (с.), 1423 (с.), 1258 (с.), 1218 (ср.), 1188 (с.), 1135 (ср.), 1096 (с.) см-1, МС (ЕI+, ИЭУ) m/z: 444 (М+).
Пример 9 - Бензилкарбамат 3 (см. схему 16 в конце описания).
К смеси 27 (5,32 г, 11,2 ммоль, 1 эквив.), триэтиламина (6,24 мл, 44,8 ммоль, 4,0 эквив.) и измельченных активированных молекулярных сит  (~20 г) в толуоле (53 мл) добавляли дифенилфосфорилазид (3,10 мл, 14,4 ммоль, 1,2 эквив. ) и получаемую суспензию нагревали до 70oС в течение 2 ч. Реакционную смесь охлаждали до 23oС и затем добавляли бензиловый спирт (1,73 мл, 16,8 ммоль, 1,5 эквив. ). Суспензию перемешивали при 23oС и течение 1 ч, фильтровали и фильтрат концентрировали. Остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 20-->50% этилацетат в гексанах), получая 3 (5, 90 г, 93%) в виде бледно-желтого твердого продукта (т.пл. 102-103oС). Rf 0,25 (33% этилацетат в гексанах); 1H ЯМР (400 МГц, СDС13) δ 7,40 (м, 1Н, Ph-H и АrСН), 6,92 (с, 1Н, АrH), 6,70 (с (шир.), 1Н, NH), 5,99 (с, 2Н, ArOCH2OAr), 5,10 (с, 2Н, CbzCH2), 4,70 (м(шир.), 2Н, Вn СН2), 4,58 (т(шир.) 1Н, J= нерезон., CH(OCH3)2), 4,23 (д(шир.), 2Н, J=нерезон., СO2СH2СН), 3,39 (с, 6Н, ОСН3), 2,18 (г, 3Н, АrСН3), Z-конфигурация подтверждена путем 5,8% NOE Ar-H при облучении N-H; 13С ЯМР (100 МГц, CDC13) δ 165,0, 151,7, 148,1, 143,4, 136,3, 135,9, 128,6, 128,5, 128,4, 128,3, 128,1, 126,3, 123,6, 120,1, 113,9, 105,0, 101,5, 101,1, 67,3, 64,0, 53,9, 9,4, ИК (пленка неразбавленного соединения) 3350 (сл., шир.), 2940 (cл.), 1718 (с.), 1498 (ср.), 1473 (ср. ), 1423 (ср.), 1247 (с.), 1193 (с.), 1130 (ср.), 1094 (с.), 1069 (ср.) см-1; МСВР (FAB+) m/z: вычислено для С30Н31NО9Nа (MNa+) 572,1896, найдено 572,1909.
(~20 г) в толуоле (53 мл) добавляли дифенилфосфорилазид (3,10 мл, 14,4 ммоль, 1,2 эквив. ) и получаемую суспензию нагревали до 70oС в течение 2 ч. Реакционную смесь охлаждали до 23oС и затем добавляли бензиловый спирт (1,73 мл, 16,8 ммоль, 1,5 эквив. ). Суспензию перемешивали при 23oС и течение 1 ч, фильтровали и фильтрат концентрировали. Остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 20-->50% этилацетат в гексанах), получая 3 (5, 90 г, 93%) в виде бледно-желтого твердого продукта (т.пл. 102-103oС). Rf 0,25 (33% этилацетат в гексанах); 1H ЯМР (400 МГц, СDС13) δ 7,40 (м, 1Н, Ph-H и АrСН), 6,92 (с, 1Н, АrH), 6,70 (с (шир.), 1Н, NH), 5,99 (с, 2Н, ArOCH2OAr), 5,10 (с, 2Н, CbzCH2), 4,70 (м(шир.), 2Н, Вn СН2), 4,58 (т(шир.) 1Н, J= нерезон., CH(OCH3)2), 4,23 (д(шир.), 2Н, J=нерезон., СO2СH2СН), 3,39 (с, 6Н, ОСН3), 2,18 (г, 3Н, АrСН3), Z-конфигурация подтверждена путем 5,8% NOE Ar-H при облучении N-H; 13С ЯМР (100 МГц, CDC13) δ 165,0, 151,7, 148,1, 143,4, 136,3, 135,9, 128,6, 128,5, 128,4, 128,3, 128,1, 126,3, 123,6, 120,1, 113,9, 105,0, 101,5, 101,1, 67,3, 64,0, 53,9, 9,4, ИК (пленка неразбавленного соединения) 3350 (сл., шир.), 2940 (cл.), 1718 (с.), 1498 (ср.), 1473 (ср. ), 1423 (ср.), 1247 (с.), 1193 (с.), 1130 (ср.), 1094 (с.), 1069 (ср.) см-1; МСВР (FAB+) m/z: вычислено для С30Н31NО9Nа (MNa+) 572,1896, найдено 572,1909.
Пример 10 - Защищенная аминокислота 4 (см. схему 17 в конце описания).
Раствор 3 (800 мг, 1,46 ммоль, 1 эквив.) и Rh[(COD)) R,R- DiPAMP]+BF4 (20 мг) в смеси метилового спирта и дихлорметана (10:1 (об./об.), 13,0 мл) помещали в реактор Парра для высокого давления и продували газообразным водородом (5х3,515 атм (5х50 psi)). Реакционную смесь с водородом (3,515 атм) герметизировали и перемешивали при 23oС в течение 16 ч. Раствор концентрировали и остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 33-->50% этилацетат в гексанах), получая 4 (774 мг, 97%) в виде белого твердого продукта (т.пл. 93,5-94,0oС). Rf 0,25 (33% этилацетат в гекcанах); энатиомерный избыток (эи) 96% (ВЭЖХ, Chiracel OD, 10% изопропиловый спирт в гексанах); [α]
Пример 11 - Альдегид 28 (см. схему 18 в конце описания).
К раствору 4 (175 мг, 0,318 ммоль, 1 эквив.) и воды (57 мл, 3,18 ммоль, 10,0 эквив. ) в дихлорметане (10,0 мл) при 0oС добавляли эфират трифторида бора (392 мл, 3,18 ммоль, 10,0 эквив.) и получаемый раствор перемешивали при этой температуре в течение 10 мин. Кислоту Льюиса нейтрализовали медленным добавлением насыщенного водного раствора бикарбоната натрия (10,0 мл) и получаемую смесь затем распределяли между насыщенным водным раствором бикарбоната натрия (80 мл) и дихлорметаном (40 мл). Водную фазу экстрагировали далее этилацетатом (2х50 мл) и объединенные органические слои сушили (сульфат натрия) и концентрировали, получая сырой альдегид 28 достаточной чистоты. Rf 0,24 (50% этилацетат в гексанах): 1Н ЯМР (500 МГц, СDС13) δ 9,44 (с, 1Н, СНО), 7,32 (м, 10Н, Ph-H), 6,50 (с, 1Н, АrН), 5,95 (с, 2Н, ArOCH2OAr), 5,72 (д, 1Н, J=7,4 Гц, NH), 5,07 (д, 1Н, J=10,7 Гц, Cbz СН2), 5,02 (д, 1H, J=10,7 Гц, Cbz CH2), 4,78 (д, 1Н, J=10,2 Bn СН2), 4,74 (д, 1Н, J= 10,2 Bn СН2), 4,58 (м, 1Н, СНСО2), 4,53 (д, 1Н, J=16,8 Гц, СН2СНО), 4,48 (д, 1Н, J= 16,8 Гц, СН2СНО), 3,04 (м, 2Н, АrСН2), 2,20 (с, 3Н, АrСН3), ИК (пленка неразбавленного соединения) 3353 (сл., шир.), 2913 (сл.), 1724 (с.), 1476 (ср. ), 1254 (ср.), 1215 (ср.), 1184 (ср.), 1090 (с), 1063 (ср.), 1027 (ср.) см-1.
Пример 12 - Лактон 5 (см. схему 19 в конце описания).
Эфират трифторида бора (640 мл, 5,20 ммаль, 16,4 эквив.) добавляли к смеси сырого альдегида 28 (0,318 ммоль, 1 эквив.) и размолотых, активированных молекулярных сит  (2,8 г) в дихлорметане (32 мл) при 0oС и получаемую суспензию перемешивали при 23oС в течение 18 ч. Реакционную смесь гасили добавлением насыщенного водного раствора бикарбоната натрия (100 мл) и распределяли. Водный слой далее экстрагировали этилацетатом (3х50 мл) и объединенные органические слои сушили (сульфат натрия) и концентрировали. Остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 0-->5% этилацетат в дихлорметане), получая 5 (113 мг, 73%) в виде белого твердого продукта (т.пл. 53-55oС). Rf 0,19 (дихлорметан);
(2,8 г) в дихлорметане (32 мл) при 0oС и получаемую суспензию перемешивали при 23oС в течение 18 ч. Реакционную смесь гасили добавлением насыщенного водного раствора бикарбоната натрия (100 мл) и распределяли. Водный слой далее экстрагировали этилацетатом (3х50 мл) и объединенные органические слои сушили (сульфат натрия) и концентрировали. Остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 0-->5% этилацетат в дихлорметане), получая 5 (113 мг, 73%) в виде белого твердого продукта (т.пл. 53-55oС). Rf 0,19 (дихлорметан);
[α]
Пример 13 - Аминофенол 6 (см. схему 20 в конце описания).
Смесь лактона 5 (240 мг, 0,493 ммоль, 1 эквив.) 10% палладия на угле (20 мг) в этилацетате (10,0 мл) перемешивали при давлении 1 атм водорода при 23oС в течение 6 ч. Реакционную смесь фильтровали и фильтрат концентрировали, получая 6 (131 мг, колич.) в виде бесцветной пленки. Rf 0,20 (этилацетат); 1H ЯМР (400 МГц, СDС13) δ 5,94 (д, 1Н, J ~ 1 Гц, ОСН2О), 5,91 (д, 1Н, J~ 1 Гц, OCH2O), 4,76 (дд, 1Н, J=3,7, 10,6 Гц, СН2О2С), 4,43 (д, 1Н, J= 10,6 Гц, CH2O2C), 4,38 (д, 1Н, J=3,7 Гц, ArCH), 4,29 (д.(шир.), 1Н, J=6,2 Гц, СНСО2), 3,00 (дд, 1Н, J=1,1, 16,9 Гц, АrСН2), 2,91 (дд, 1Н, J=6,2, 16,9 Гц, АrСН2); ИКПФ (пленка неразбавленного соединения) 3360 (cл.,шир,), 2951 (cл. ), 1731 (с.), 1461 (с.), 1432 (с.), 1241 (ср.), 1117 (ср.), 1096 (с.), 1076 (ср. ), 1048 (с. ), 1025 (ср.) см-1; МСВР (El+) m/z: вычислено для C13H13NO5 (М+) 263,0794, найдено 263,0802.
Правый фрагмент
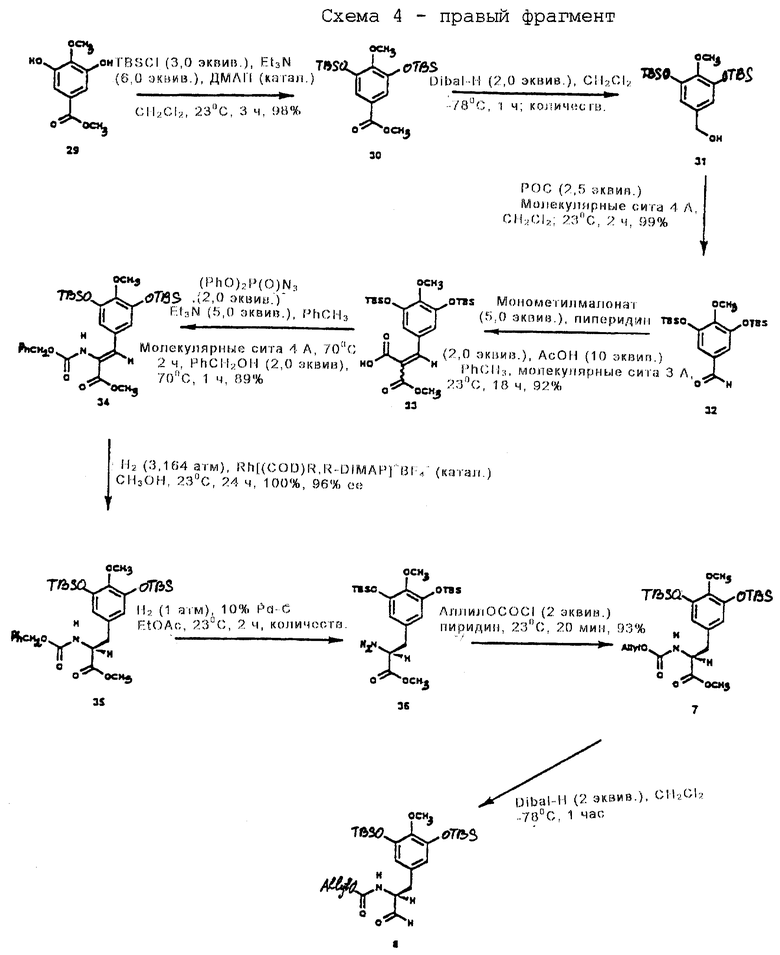
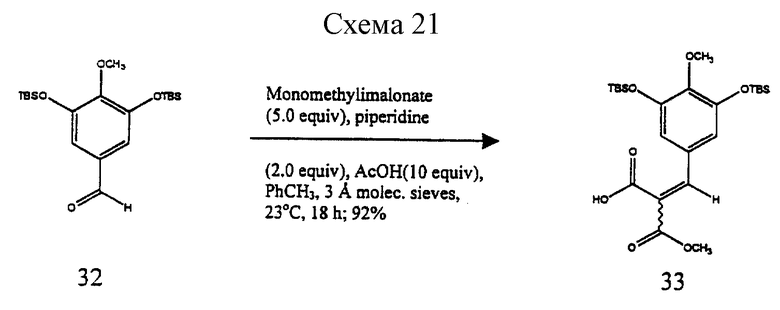
Пример 14 - Кислота 33 (см. схему 21 в конце описания).
Пиперидин (1,01 мл, 10,2 ммоль, 2,0 эквив.) добавляли к суспензии 32 (2,02 г, 5,10 ммоль, 1 эквив.) монометилмалоната (3,01 г, 25,5 ммоль, 5,0 эквив. ), уксусной кислоты (2,92 мл, 51,0 ммоль, 10,0 эквив.) и размолотых, активированных молекулярных сит  (~12 г) в толуоле (25,0 мл) и получаемую суспензию перемешивали при 23oС в течение 18 ч. Реакционную смесь фильтровали, хорошо промывали этилацетатом (100 мл). Фильтрат концентрировали и остаток очищали колоночной флэш-хроматографией (4% метиловый эфир в дихлорметане), получая кислоту 33 (2,32 г, 92%) в виде неразделяемой смеси E/Z-изомеров. Rf 0,42 (10% метиловый спирт в дихлорметане); 1H ЯМР (500 МГц, СDС13) δ (изомер, присутствующий в большем количестве): 7,71 (с, 1Н, АrСН), 6,83 (с, 2Н, АrН), 3,90 (с, 3Н, ОСН3), 3,75 (с, 3Н, ОСН3), 1,00 (с, 18Н, трет-бутил, 0,18 (с, 12Н, SiCH3), d (изомер, присутствующий в меньшем количестве): 7,71 (с, 1Н, АrСН), 6,65 (с, 2Н, ArH), 3,81 (с, 3Н, ОСН3), 3,77 (с, 3Н, ОСН3), 1,00 (с, 18Н, трет-бутил), 0,18 (с, 12Н, SiСН3); 13С ЯМР (126 МГц, СDС13) δ 169,9, 165,3, 150,0, 145,8, 144,5, 127,4, 122,5, 116,8, 60,0, 52,8, 25,6, 18,2, -4,7; ИК (пленка неразбавленного соединения) 3600-2600 (ср., шир.), 2955 (с.), 1741 (с.), 1713 (с.), 1569 (с.), 1493 (с.), 1253 (с.), 1219 (ср. ), 1096 (с. ), 864 (с.) см-1, МСВР (FAB+) m/z: вычислено для С24Н39O7Si2 (М-Н-) 495,2234, найдено 495,2253.
(~12 г) в толуоле (25,0 мл) и получаемую суспензию перемешивали при 23oС в течение 18 ч. Реакционную смесь фильтровали, хорошо промывали этилацетатом (100 мл). Фильтрат концентрировали и остаток очищали колоночной флэш-хроматографией (4% метиловый эфир в дихлорметане), получая кислоту 33 (2,32 г, 92%) в виде неразделяемой смеси E/Z-изомеров. Rf 0,42 (10% метиловый спирт в дихлорметане); 1H ЯМР (500 МГц, СDС13) δ (изомер, присутствующий в большем количестве): 7,71 (с, 1Н, АrСН), 6,83 (с, 2Н, АrН), 3,90 (с, 3Н, ОСН3), 3,75 (с, 3Н, ОСН3), 1,00 (с, 18Н, трет-бутил, 0,18 (с, 12Н, SiCH3), d (изомер, присутствующий в меньшем количестве): 7,71 (с, 1Н, АrСН), 6,65 (с, 2Н, ArH), 3,81 (с, 3Н, ОСН3), 3,77 (с, 3Н, ОСН3), 1,00 (с, 18Н, трет-бутил), 0,18 (с, 12Н, SiСН3); 13С ЯМР (126 МГц, СDС13) δ 169,9, 165,3, 150,0, 145,8, 144,5, 127,4, 122,5, 116,8, 60,0, 52,8, 25,6, 18,2, -4,7; ИК (пленка неразбавленного соединения) 3600-2600 (ср., шир.), 2955 (с.), 1741 (с.), 1713 (с.), 1569 (с.), 1493 (с.), 1253 (с.), 1219 (ср. ), 1096 (с. ), 864 (с.) см-1, МСВР (FAB+) m/z: вычислено для С24Н39O7Si2 (М-Н-) 495,2234, найдено 495,2253.
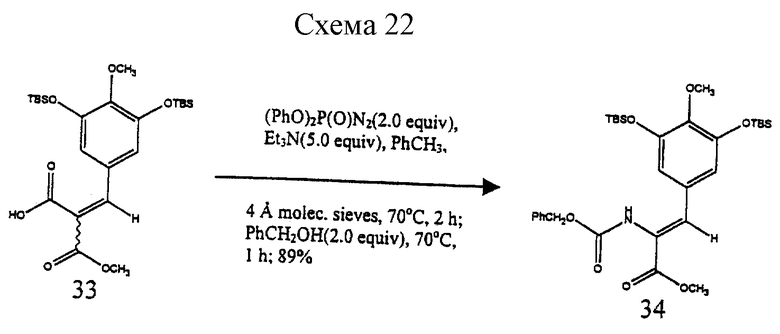
Пример 15 - бензилкарбамат 34 (см. схему 22 в конце описания).
К суспензии 33 (3,35 г, 6,75 ммоль, 1 эквив.), триэтиламина (4,71 мл, 33,8 ммоль, 5,0 эквив.) и размолотых, активированных молекулярных сит  (~ 15 г) в толуоле (50 мл) добавляли дифенилфосфорилазид (2,90 мл, 13,5 ммоль, 2,0 эквив.) и получаемую суспензию нагревали при 70oС в течение 2 ч. К реакционной смеси затем добавляли бензиловый спирт (1,40 мл, 13,5 ммоль, 2,0 эквив. ), и суспензию перемешивали при 70oС в течение 1 ч. Реакционную смесь фильтровали, тщательно промывали этилацетатом (100 мл) и фильтрат концентрировали. Остаток очищали колоночной флэш-хроматографией (25% этилацетат в гексане), получая 34 в виде бледно-желтого масла (3,62 г, 89%). Rf 0,53 (25% этилацетат в гексане); 1Н ЯМР (500 МГц, СDС13) δ 7,34 (м, 5Н, CbzArH), 7,18 (с, 1Н, АrСН), 6,77 (с, 2Н, АrH), 6,14 (с(шир.) 1Н, NH), 5,13 (с, 2Н, Cbz CH2), 3,81 (с(шир.), 3Н, ОСН3), 3,75 (с, 3Н, ОСН3), 1,00 (с, 18Н, трет-бутил), 0,16 (с, 12Н, SiСН3), Z-конфигурация подвержена путем 11,6% NOE АrН при облучении NH, 13С ЯМР (100 МГц, СDС13) δ 165,8, 149,8, 144,4, 135,8, 132,5, 130,0, 128,5, 128,4, 128,2, 126,1, 123,4, 120,2, 116,4, 67,6, 60,0, 52,5, 25,7, 18,3, -4,7, ИК (пленка неразбавленного соединения) 3500 (cл., шир. ), 2951 (ср.), 1723 (с.), 1567 (ср.), 1493 (с.), 1424 (ср.), 1289 (с.), 1259 (с.), 1122 (с.), 1006 (сл.), 829 (с.) см-1. МСВР (FAB+) m/z: вычислено для С31H48NО7Si2 (МН+) 602,2969, найдено 602,2993.
(~ 15 г) в толуоле (50 мл) добавляли дифенилфосфорилазид (2,90 мл, 13,5 ммоль, 2,0 эквив.) и получаемую суспензию нагревали при 70oС в течение 2 ч. К реакционной смеси затем добавляли бензиловый спирт (1,40 мл, 13,5 ммоль, 2,0 эквив. ), и суспензию перемешивали при 70oС в течение 1 ч. Реакционную смесь фильтровали, тщательно промывали этилацетатом (100 мл) и фильтрат концентрировали. Остаток очищали колоночной флэш-хроматографией (25% этилацетат в гексане), получая 34 в виде бледно-желтого масла (3,62 г, 89%). Rf 0,53 (25% этилацетат в гексане); 1Н ЯМР (500 МГц, СDС13) δ 7,34 (м, 5Н, CbzArH), 7,18 (с, 1Н, АrСН), 6,77 (с, 2Н, АrH), 6,14 (с(шир.) 1Н, NH), 5,13 (с, 2Н, Cbz CH2), 3,81 (с(шир.), 3Н, ОСН3), 3,75 (с, 3Н, ОСН3), 1,00 (с, 18Н, трет-бутил), 0,16 (с, 12Н, SiСН3), Z-конфигурация подвержена путем 11,6% NOE АrН при облучении NH, 13С ЯМР (100 МГц, СDС13) δ 165,8, 149,8, 144,4, 135,8, 132,5, 130,0, 128,5, 128,4, 128,2, 126,1, 123,4, 120,2, 116,4, 67,6, 60,0, 52,5, 25,7, 18,3, -4,7, ИК (пленка неразбавленного соединения) 3500 (cл., шир. ), 2951 (ср.), 1723 (с.), 1567 (ср.), 1493 (с.), 1424 (ср.), 1289 (с.), 1259 (с.), 1122 (с.), 1006 (сл.), 829 (с.) см-1. МСВР (FAB+) m/z: вычислено для С31H48NО7Si2 (МН+) 602,2969, найдено 602,2993.
Пример 16 - Защищенная аминокислота 35 (см. схему 23 в конце описания).
Раствор 34 (6,00 г, 9,98 ммоль, 1 эквив.) и Rh[(COD)R,R-DiPAMP]+BF4 - (75 мг) в смеси метилового спирта и дихлорметана (10:1 (об./об.), 110 мл) помещали в реактор Парра для высокого давления и продували газообразным водородом (5 х 3,515 атм (5 х 50 psi)). Реакционную смесь с водородом (3,515 атм) герметизировали и перемешивали при 23oС в течение 24 ч. Раствор концентрировали и остаток очищали колоночной флэш-хроматографией (2,5% этилацетат в дихлорметане), получая 35 (6,01 г, колич.) в виде бесцветного вязкого продукта. Rf 0,41 (20% этилацетат в гексане), эи (энантиомерный избыток) 96% (ВЭЖХ, ChirlPak AD, 1% изопропиловый спирт в гексанах); [α]
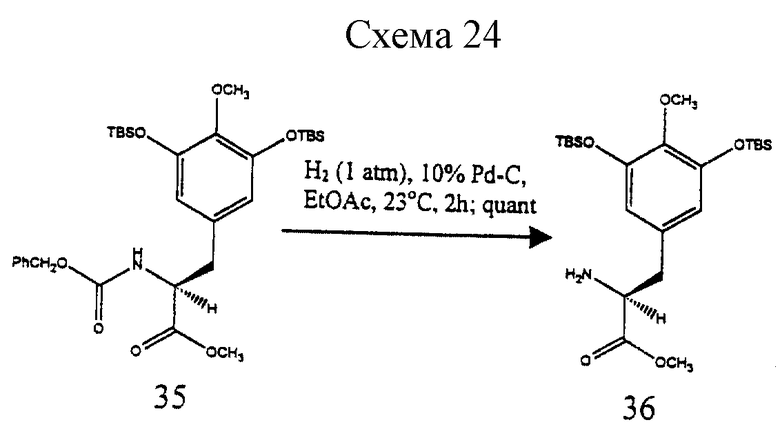
Пример 17 - Сложный аминоэфир 36 (см. схему 24 в конце описания).
Раствор 35 (1,00 г, 1,66 ммоль, 1 эквив.) и 10% палладия на активированном угле (50 мг) в этилацетате (40 мл) перемешивали при 1 атм газообразного водорода при 23oС в течение 2 ч. Реакционную смесь фильтровали под действием силы тяжести и фильтрат концентрировали, получая 36 (780 мг, колич.) в виде вязкого масла. Rf 0,38 (50% этилацетат в гексане); [α]
Пример 18 - Аллилкарбамат 7 (см. схему 25 в конце описания).
К раствору соединения 36 (780 мг, 1,66 ммоль, 1 эквив.) в пиридине (8 мл) при 0oС добавляли по каплям аллилхлорформиат (352 мл, 3,32 ммоль, 2,0 эквив. ), и реакционную смесь перемешивали при 23oС в течение 20 мин. Смесь концентрировали при 23oС и остаток распределяли между водой (50 мл) и дихлорметаном (3 х 25 мл). Объединенные органические слои сушили (сульфат натрия) и концентрировали и остаток очищали колоночной флэш-хроматографией (15% этилацетат в гексане), получая 7 (856 мг, 93%) в виде бесцветного масла. Rf 0,37 (20% этилацетат в гексане); [α]
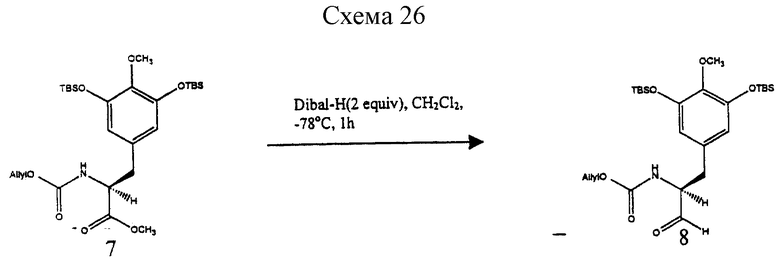
Пример 19 - Альдегид 8 (см. схему 26 в конце описания).
К раствору 7 (850 мг, 1,54 ммоль, 1 эквив.) в дихлорметане (85 мл) при -78oС добавляли диизобутилалюминийгидрид (1,5 М в толуоле, 2,05 мл, 3,08 ммоль, 2,0 эквив.) и реакционную смесь перемешивали при -78oС в течение 1 ч. Избыточный восстанавливающий агент гасили последовательным добавлением метилового спирта (700 мл), декагидрата сульфата натрия (~5 г) и целита (~2 г). Смесь перемешивали при 23oС в течение 1 ч и затем фильтровали через подушку целита. Фильтрат концентрировали и остаток растворяли в диэтиловом простом эфире (150 мл). Раствор снова фильтровали через подушку целита и фильтрат концентрировали, получая сырой альдегид 8, который использовали сразу, без дальнейшей очистки, в реакции сочетания с 6. Rf 0,33 (25% этилацетат в гексанах); 1H ЯМР сырого продукта (400 МГц, СDСl3) δ 9,61 (с, 1Н, СНО), 6,28 (с, 2Н, АrН), 5,90 (м, 1Н, Н винила), 5,30 (дд, 1Н, J=1,2, 17,2 Гц, Н винила), 5,21 (м, 2Н, Н винила, NH), 4,58 (м, 2Н, Н аллила), 4,41 (м, 1Н, СНСНО), 3,70 (с, 3Н, ОСН3), 3,01 (дд, 1Н, J=6,0, 14,4 Гц, АrСН2), 2,94 (дд, 1Н, J=6,8, 14,4 Гц, АrСН2), 0,99 (с, 18Н, Si-трет-бутил), 0,15 (с, 12Н, SiCH3).
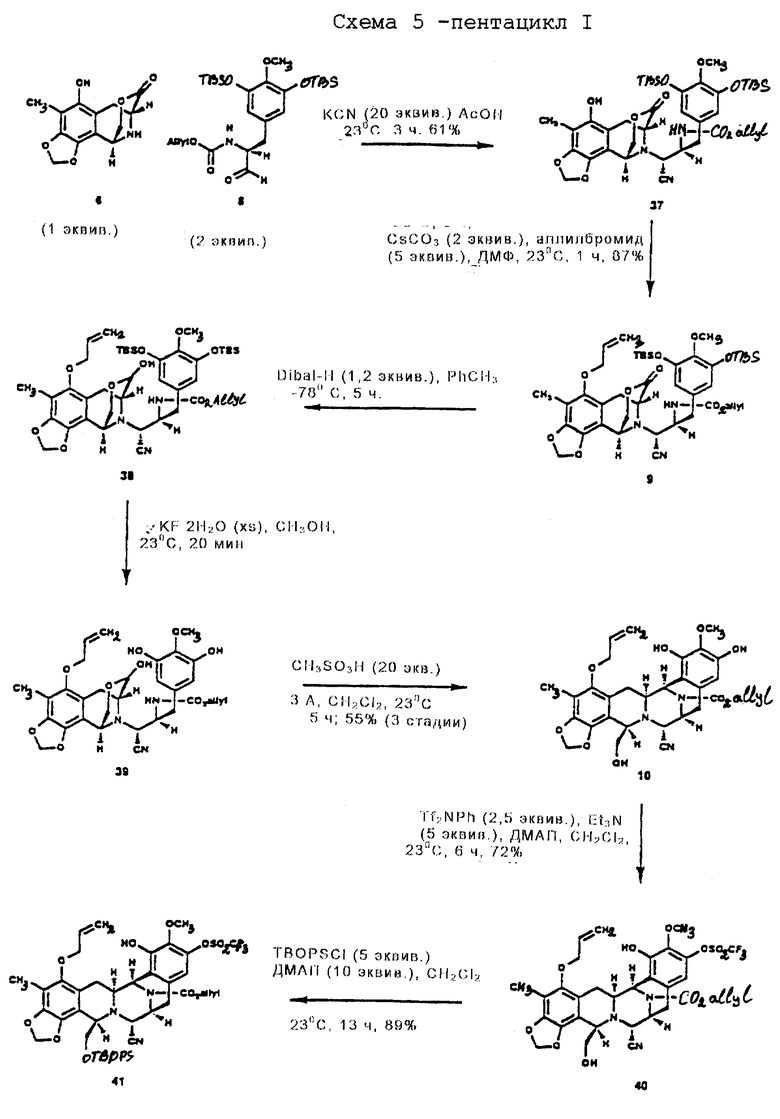
Синтез пентакцикла
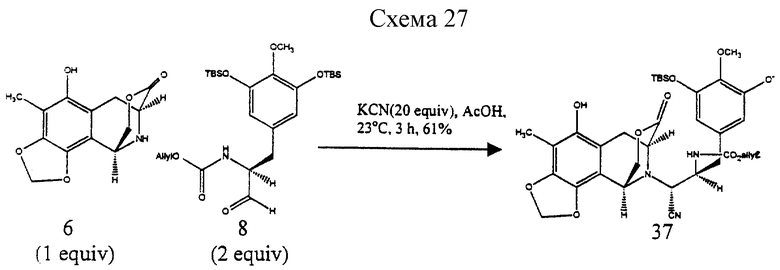
Пример 21 - Аминонитрил 37 (см. схему 27 в конце описания).
К раствору амина 6 (123 мг, 0,467 ммоль, 1 эквив.) и сырого альдегида 8 (489 мг, 0,935 ммоль, 2,0 эквив.) в ледяной уксусной кислоте (5 мл) добавляли твердый цианид калия (608 мг, 9,35 ммоль, 20 эквив.) и получаемую смесь перемешивали при 23oС в течение 1 ч. Реакционную смесь разбавляли этилацетатом (80 мл) и последовательно промывали насыщенным водным раствором бикарбоната натрия (3х60 мл) и насыщенным водным раствором хлорида натрия (60 мл). Органический слой сушили (сульфат натрия) и концентрировали и остаток очищали колоночной флэш-хроматографией (градиетное элюирование: 15%-->20% этилацетат в гексане), получая 37 (159 мг) и его аминонитрильный эпимер (67 мг) в отдельных фракциях (общий выход 61%). 37: Rf 0,19 (25% этилацетат в гексане), [α]
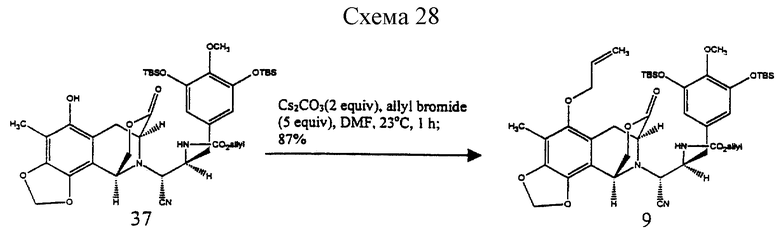
Пример 22 - Аллиловый простой эфир 9 (см. схему 28 в конце описания).
К раствору ацетонитрила 37 (986 мг, 1,24 ммоль, 1 эквив.) в ДМФ (10 мл) добавляли последовательно высушенный в пламени карбонат цезия (809 мг, 2,78 ммоль, 2,0 эквив. ) и аллилбромид (537 мл, 6,20 ммоль, 5,0 эквив.) и смесь перемешивали при 23oС в течение 1 ч. Избыточное основание нейтрализовали добавлением уксусной кислоты (4 мл) и смесь затем распределяли между насыщенным водным раствором бикарбонат натрия (100 мл) и дихлорметаном (2 х 50 мл). Водный раствор далее экстрагировали этилацетатом (2 х 50 мл). Объединенные органические слои сушили (сульфат натрия) и концентрировали и остаток очищали колоночной флэш-хроматографией (20% этилацетат в гексане), получая 9 (901 мг, 87%) в виде бесцветной пленки. Rf 0,41 (25% этилацетат в гексане); [α]
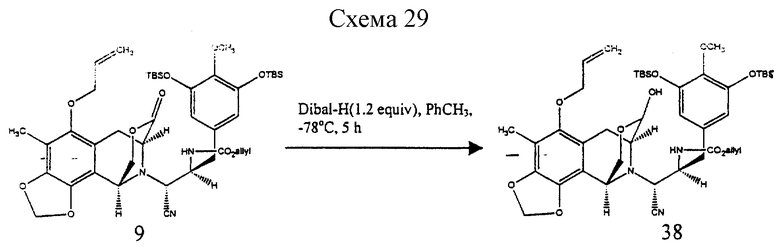
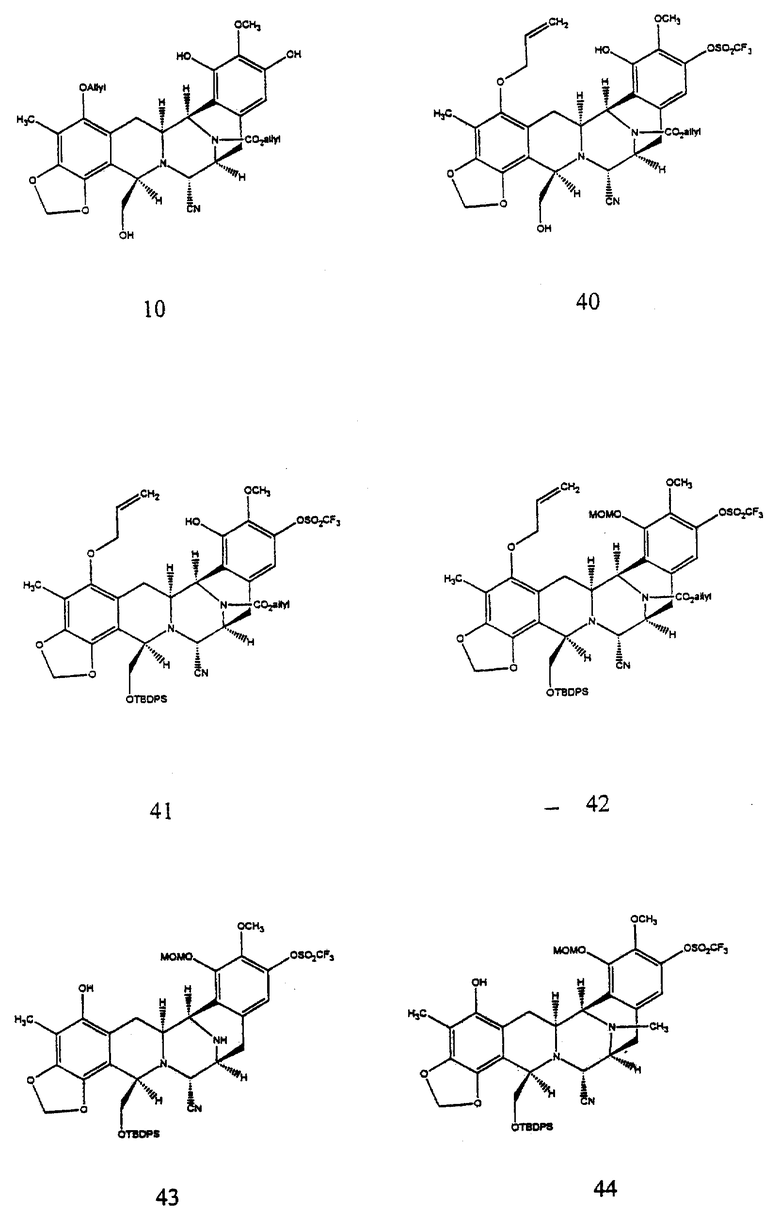
Пример 23 - Триол 10 (см. схему 29 в конце описания)
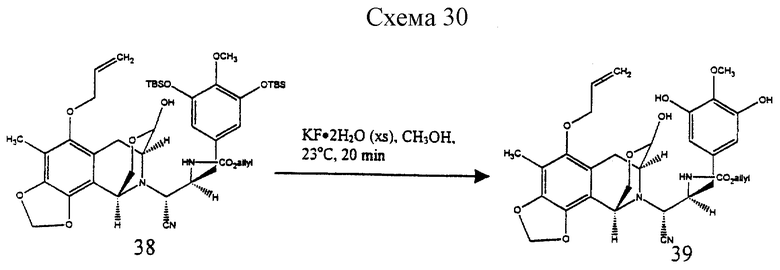
К раствору 9 (390 мг, 0,467 ммоль, 1 эквив.) в растворе толуола (50 мл) npи -78oC добавляли раствор диизобутилалюминийгидрида (1,5 М в толуоле, 374 мл, 0,560 ммоль, 1,2 эквив.) и получаемый раствор перемешивали при -78oС в течение 5 ч. Избыточный восстанавливающий агент гасили медленным последовательным добавлением метилового спирта (500 мл), декагидрата сульфата натрия (~5 г) и целита при -78oC. Суспензию перемешивали при 23oС в течение 1 ч до того, как ее фильтровали через подушку целита. Фильтрат концентрировали и остаток (38) растворяли в метиловом спирте (4 мл) (см. схему 30 в конце описания).
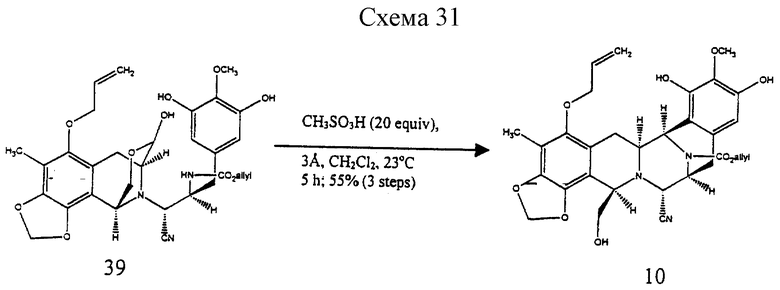
К этому раствору добавляли дигидрат фторида калия (250 мг, 2,66 ммоль, 5,7 эквив.) и реакционную смесь перемешивали при 23oС в течение 20 мин. Смесь распределяли между дихлорметаном (50 мл) и 80% насыщенным водным раствором хлорида натрия (80 мл) и водную фазу далее экстрагировали этилацетатом (2 х 50 мл). Объединенные органические слои сушили (сульфат натрия) и концентрировали и остаток (39) растворяли в дихлорметане (100 мл) (см. схему 31 в конце описания.
К этому раствору добавляли измельченные, прокаленные молекулярные сита  (6,20 г), затем метансульфоновую кислоту (531 мл, 8,21 ммоль, 20 эквив.) и суспензию перемешивали при 23oС в течение 5 ч. Избыточную кислоту гасили добавлением пиридина (1,32 мл, 16,4 ммоль, 40 эквив.) и смесь фильтровали с отсасыванием, тщательно промывали 10% изопропиловым спиртом в дихлорметане (4х20 мл). Раствор продукта промывали насыщеным водным раствором хлорида натрия (150 мл) и водный слой далее экстрагировали этилацетатом (2х100 мл). Объединенные органические слои сушили (сульфат натрия) и концентрировали и остаток очищали колоночной флеш-хромотографией (градиентной элюирование: 60-->100% этилацетат в гексане), получая триол 10 (152 мг, 55%, 3 стадии) в виде бесцветного масла. Rf 0,23 (66% этилацетат в гексане); [α]
(6,20 г), затем метансульфоновую кислоту (531 мл, 8,21 ммоль, 20 эквив.) и суспензию перемешивали при 23oС в течение 5 ч. Избыточную кислоту гасили добавлением пиридина (1,32 мл, 16,4 ммоль, 40 эквив.) и смесь фильтровали с отсасыванием, тщательно промывали 10% изопропиловым спиртом в дихлорметане (4х20 мл). Раствор продукта промывали насыщеным водным раствором хлорида натрия (150 мл) и водный слой далее экстрагировали этилацетатом (2х100 мл). Объединенные органические слои сушили (сульфат натрия) и концентрировали и остаток очищали колоночной флеш-хромотографией (градиентной элюирование: 60-->100% этилацетат в гексане), получая триол 10 (152 мг, 55%, 3 стадии) в виде бесцветного масла. Rf 0,23 (66% этилацетат в гексане); [α]
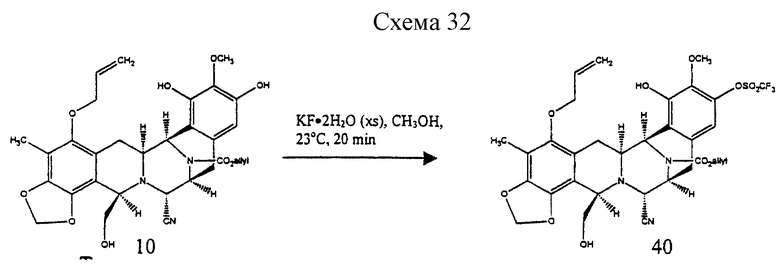
Пример 24 - Арилтрифлат 40 (см. схему 32 в конце описания).
К раствору 10 (150 мг, 0,253 ммоль, 1 эквив.) и триэтиламина (177 мл, 1,27 ммоль, 5,0 эквив. ) в дихлорметане (15 мл) последовательно добавляли N-фенилтрифлимид (227 мг, 0,634 ммоль, 2,5 эквив.) и ДМАП (1 мг) и реакционную смесь перемешивали при 23oС в течение 6,5 ч. Избыточное основание нейтрализовали добавлением уксусной кислоты (145 мл, 2,53 ммоль, 10 эквив.), затем добавляли пиридин (306 мл, 3,79 ммоль, 15 эквив.). Смесь распределяли между дихлорметаном (50 мл) и насыщенным водным раствором хлорида натрия (80 мл) и водный слой далее экстрагировали этилацетатом (2х50 мл). Объединенные органические слои сушили (сульфат натрия) и концентрировали и остаток очищали колоночной флэш-хроматографией (60% этилацетат в гексане), получая 40 (132 мг, 72%) в виде бесцветной пленки. Rf 0,44 (50% этилацетат в гексане); [α]
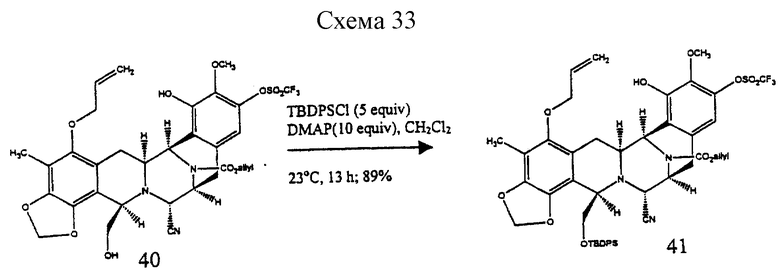
Пример 25 - Силиловый эфир 41 (см. схему 33 в конце описания.
К суспензии 41 (90 мг, 0,124 ммоль, 1 эквив.) и ДМАП (152 мг, 1,24 ммоль, 10 эквив.) и дихлорметане (10 мл) добавляли трет-бутилдифенилсилилхлорид (162 мл, 0,622 ммоль, 5,0 эквив.) и раствор перемешивали при 23oС в течение 13 ч. Избыточное основание гасили добавлением уксусной кислоты (150 мл) и смесь распределяли между водой (50 мл) и дихлорметаном (3х30 мл). Объединенные органические слои сушили (сульфат натрия) и концентрировали и остаток очищали колоночной флеш-хроматографией (градиентное элюирование: 25%-->50% этилацетат в гексане), получая 41 (106 мг, 89%) в виде бесцветного стекловидного твердого продукта. Rf 0,66 (50% этилацетат в гексане), [α]
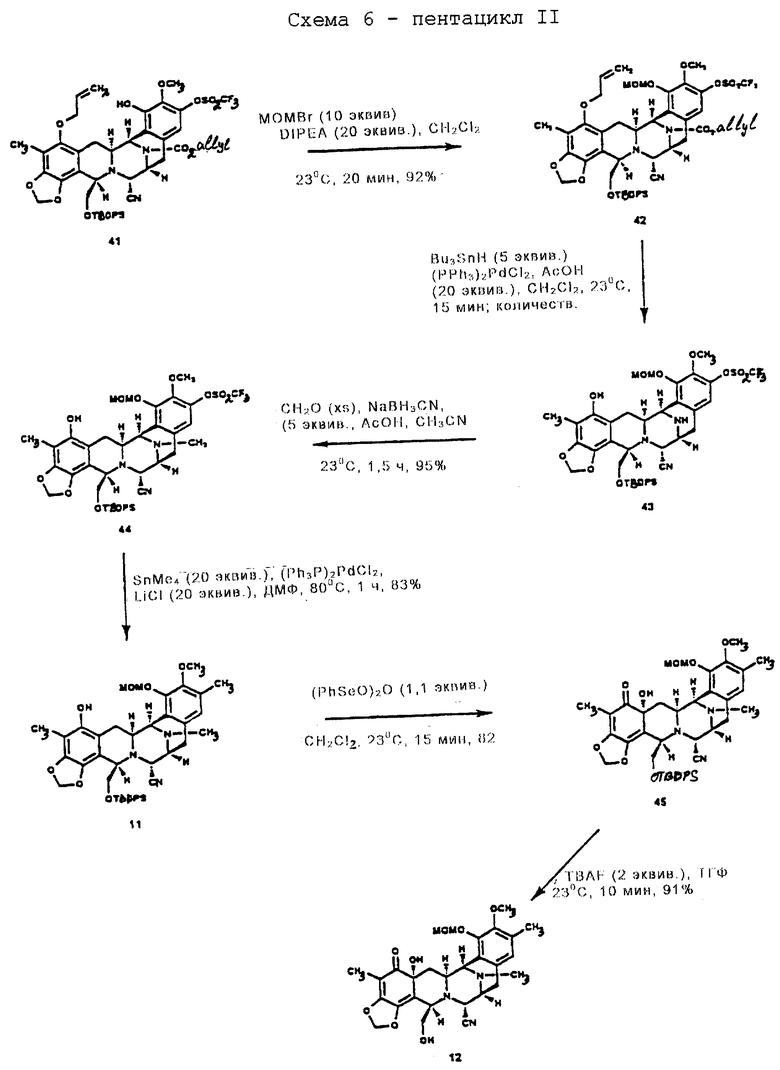
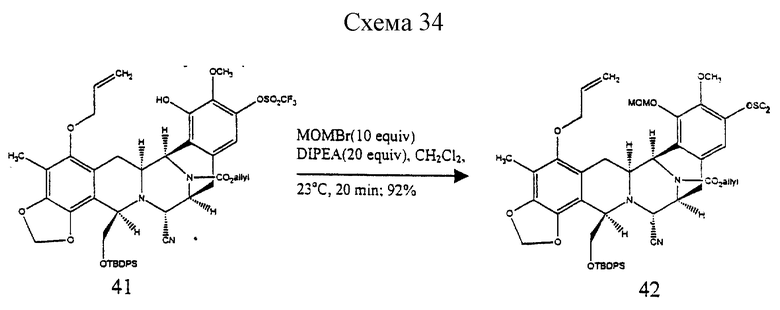
Пример 26 - Метоксиметиловый простой эфир 42 (см. схему 34 в конце описания).
К раствору 41 (94 мг, 0,0978 ммоль, 1 эквив.) и диизопропилэтиламина (340 мл, 1,96 ммоль, 20 эквив.) в дихлорметане (6 мл) при 0oС добавляли бромметилметиловый простой эфир (80 мл, 0,978 ммоль, 10 эквив.) и раствор перемешивали при 23oС в течение 20 мин. После гашения реакции метиловым спиртом (100 мл) смесь распределяли между насыщенным водным раствором бикарбоната натрия (30 мл) и дихлорметаном (2х30 мл) и объединенные органические слои сушили (сульфат натрия) и концентрировали. Остаток очищали колоночной флеш-хроматографией (25% этилацетат в гексане), получая 42 (90 мг, 92%) в виде бесцветной пленки. Rf 0,66 (50% этилацетат в гексане); [α]
5,93 (м, 1H, H винила), 5,81 (с, 1H, OCH2O), 5,65 (с, 1Н, OCH2O), 5,45-5,13 (м, H винила и ОСН2С), 4,91 (м, 1H), 4,69 (м, 1H), 4,59 (м, 2Н), 4,16 (м, 2Н), 4,07 (м, 1H, 3,87 (м, 3Н, ОСН3), 3,73-3,60 (м, 4H, ОCН3 и CHOSi), 3,4-3,2 (м, 3Н, CHOSi и АrСН), 2,97 (м, 1H, АrСН2), 2,12 (с, 3Н, АrСН3), 1,83 (м, 1H, ArCH2), 0,97 (м, 9Н, трет-бутил); 13С ЯМР (100 МГц, СDСl3) δ 154,1, 153,9, 148,5, 147,9, 144,6, 142,6, 142,4, 142,1, 139,3, 139,2, 135,7, 135,3, 134,8, 133,7, 132,9, 132,5, 132,4, 132,3, 129,8, 128,8, 328,7, 127,7, 120,3, 120,1, 118,5, 118,1, 117,5, 117,1, 116,7, 116,6, 116,5, 112,5, 112,4, 112,0, 111,8, 101,1, 99,7, 74,2, 69,2, 68,8, 67,0, 66,7, 61,1, 60,4, 60,2, 59,2, 58,4, 58,1, 56,5, 50,2, 49,3, 49,2, 48,3, 30,7, 30,1, 29,7, 26,8, 26,1, 26,0, 19,0, 9,2; ИКПФ (пленка неразбавленного соединения) 2959 (ср.), 1709 (с.), 1426 (с), 1315 (ср.), 1253 (ср.), 1213 (с.), 1140 (с. ), 1110 (с.), 1066 (с.), 1015 (c.), 987 (с.), 921 (с.), 825 (ср.) см-1; MC (FAB+) m/z: вычислено для C50H54F3N3O12SSiNa (MNa+) 1028, найдено 1028.
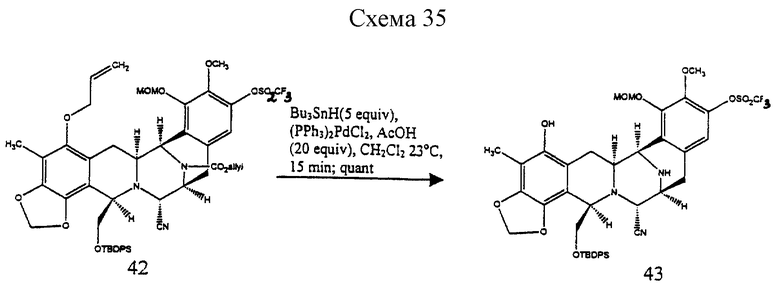
Пример 21 - Аминофенол 43 (см. схему 35 в конце описания).
К раствору 42 (90 мг, 0,0895 ммоль, 1 эквив.), уксусной кислоты (102 мл, 1,79 ммоль, 20 эквив. ) и дихлорбис(трифенилфосфин)палладия (5 мг) в дихлорметане (4 мл) добавляли трибутилоловогидрид (120 мл, 0,448 ммоль, 5,0 эквив. ) и желто/коричневый раствор перемешивали при 23oС в течение 15 мин. Смесь загружали в колонку с силикагелем и продукт очищали колоночной флэш-хроматографией (градиентное элюирование: 50% этилацетат в гексане --> 100% этилацетат), получая 43 (79 мг, колич.) в виде бесцветной пленки. Rf 0,30 (50% этилацетат в гексане); [α]
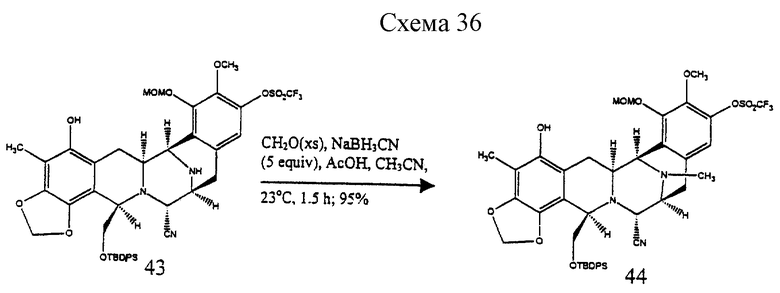
Пример 28 - Фенол 44 (см. схему 36 в конце описания).
К раствору 43 (79 мг, 0,0896 ммоль, 1 эквив.) и раствору формалина (600 мл) в ацетонитриле (6 мл) добавляли твердый цианоборогидрид натрия (17 мг, 0,269 ммоль, 5,0 эквив.) и раствор перемешивали при 23oС в течение 30 мин. Добавляли уксусную кислоту (102 мл, 1,79 ммоль, 20 эквив.) и реакционную смесь перемешивали при 23oС в течение дополнительных 1,5 ч. Смесь распределяли между насыщенным водным раствором бикарбоната натрия (40 мл) и дихлорметаном (30 мл) и водный слой далее экстрагировали этилацетатом (2 х 30 мл). Объединенные органические слои сушили (сульфат натрия) и концентрировали и остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 33-->50% этилацетат в гексане), получая 44 (76 мг, 95%) в виде бесцветной пленки. Rf 0,60 (50% этилацетат в гексане); [α]
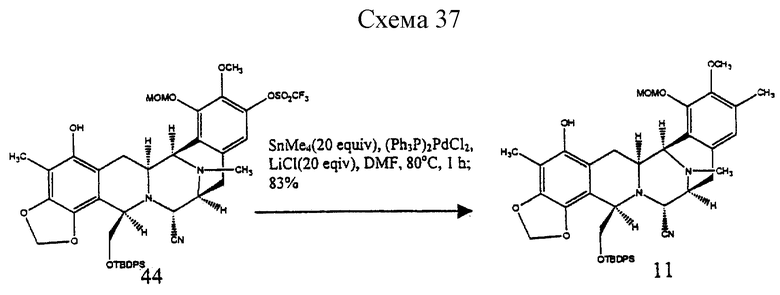
Пример 29 - Фенол 11 (см. схему 37 в конце описания).
К раствору 44 (17 мг, 0,0190 ммоль, 1 эквив.), хлорида лития (16 мг, 0,380 ммоль, 20 эквив. ) и дихлорбис(трифенилфосфин)палладия (1 мг) в ДМФ (0,5 мл) добавляли тетраметилолово (53 мл, 0,380 ммоль, 20 эквив.) и коричневый раствор перемешивали при 80oС в течение 2 ч. Реакционную смесь распределяли между водой (30 мл) и дихлорметаном (2х20 мл). Водный слой далее экстрагировали этилацетатом (2х20 мл) и объединенные органические слои сушили (сульфат натрия) и концентрировали. Продукт очищали колоночной флэш-хроматографией (градиентное элюирование: 33-->50% этилацетат в гексане), получая 11 (14 мг, 96%) в виде бесцветной пленки. Rf 0,27 (20% этилацетат в бензоле); [α]
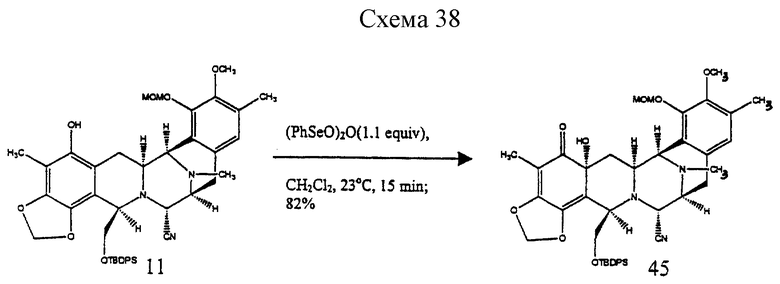
Пример 30 - Гидроксидиенон 45 (см. схему 38 в конце описания).
К раствору 11 (40 мг, 0,0525 ммоль, 1 эквив.) в дихлорметане (6 мл) добавляли бензолселениновый ангидрид (21 мг, 0,0578 ммоль, 1,1 эквив.) и темно-красный раствор перемешивали при 23oС в течение 15 мин. Смесь гасили насыщенным водным раствором бикарбоната натрия (6 мл) до того, как ее распределяли между насыщенным водным раствором бикарбоната натрия (30 мл) и дихлорметаном (2х20 мл). Водный слой далее экстрагировали этилацетатом (2х20 мл) и объединенные органические слои сушили (сульфат натрия) и концентрировали. Остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 30-->50% этилацетат в гексане), получая 45 (33 мг, 82%) в виде бесцветной пленки. Rf 0,27 (50% этилацетат в гексане); [α]
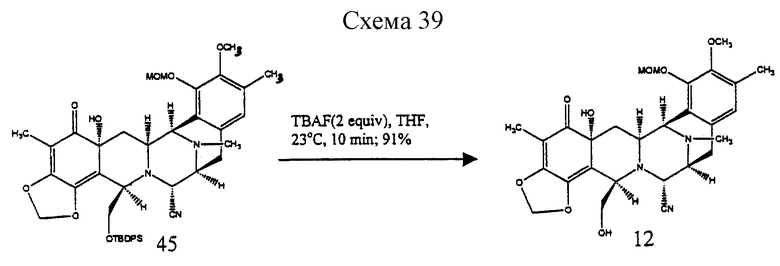
Пример 31 - Диол 12 (см. схему 39 в конце описания).
К раствору 45 (30 мг, 0,0386 ммоль, 1 эквив.) в ТГФ (4 мл) добавляли фторид тетрабутиламмония (1 М раствор в тетрагидрофуране, 77 мл, 0,0772 ммоль, 2,0 эквив.) и раствор перемешивали при 23oС в течение 10 мин. Смесь распределяли между насыщенным водным раствором хлорида натрия (30 мл) и этилацетатом (3х20 мл). Водный слой далее экстрагировали дихлорметаном (2х20 мл) и объединенные органические слои сушили (сульфат натрия) и концентрировали. Остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 75%-->100% этилацетат в гексане), получая 12 (19 мг, 91%) в виде бесцветной пленки. Rf 0,25 (75% этилацетат в гексане); [α]
Конечные стадии
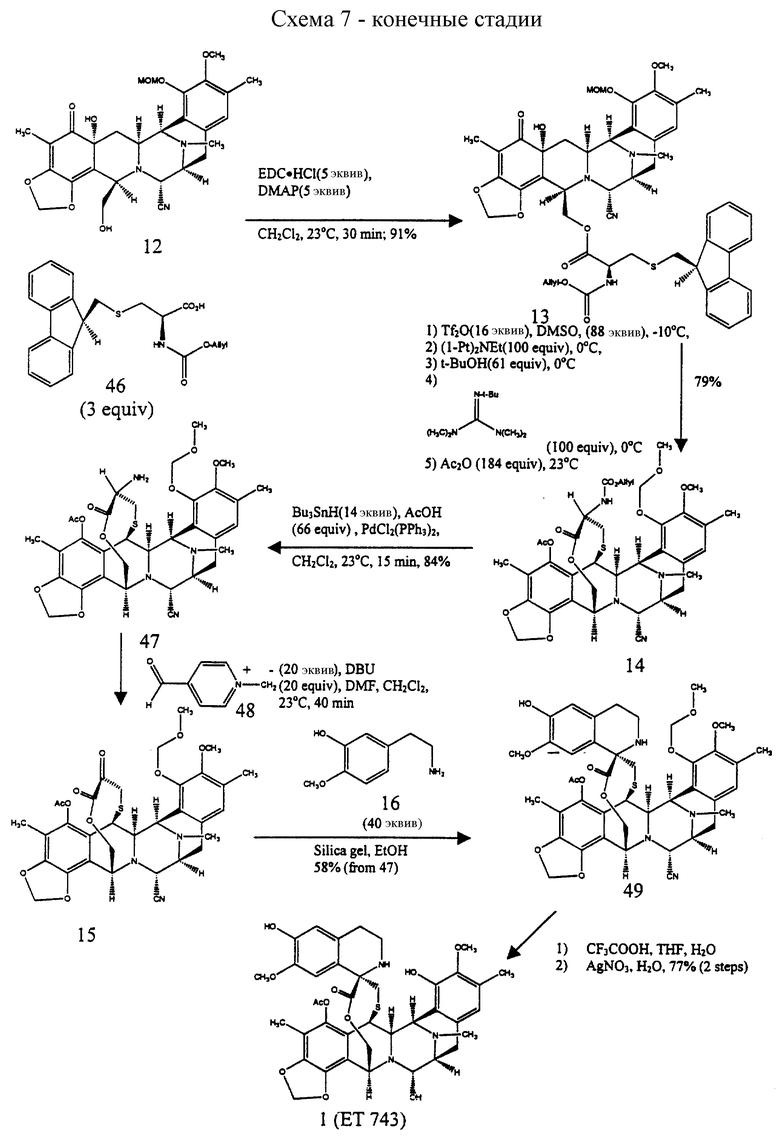
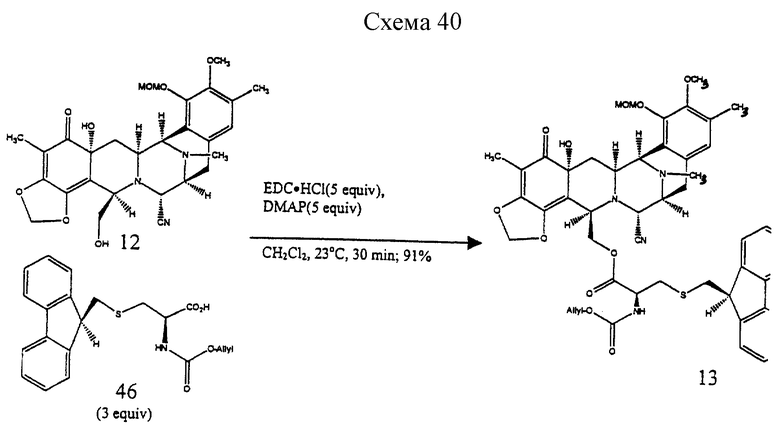
Пример 32 - Сложный эфир 13 (см. схему 40 в конце описания).
К раствору спирта 12 (9,0 мг, 0,0167 ммоль, 1 эквив.) и кислоты 46 (19 мг, 0,0501 ммоль, 3,0 эквив.) и дихлорметане (1,5 мл) добавляли ДМАП (10 мг, 0,0835 ммоль, 5,0 эквив.) и 1-(3-диметиламинопропил)-3-этилкарбодиимид•НСl (16 мг, 0,0835 ммоль, 5,0 эквив.) и получаемый раствор перемешивали при 23oС в течение 1,5 ч. Реакционную смесь распределили между насыщенным водным раствором бикарбоната натрия (30 мл) и дихлорметаном (2 х 20 мл) и водный слой далее экстрагировали этилацетатом (2 х 20 мл). Объединенные органические слои сушили (сульфат натрия) и концентрировали. Остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 50%-->60% этилацетат в гексане), получая 13 (13,7 мг, 91%). Rf 0,15 (50% этилацетат в гексане); [α]
Пример 33 - Лактон 14 (см. схему 41 в конце описания).
К раствору трифторметансульфонового ангидрида (8 мл, 0,0476 ммоль, 16,5 эквив. ) в дихлорметане (2,6 мл) при -78oС добавляли ДМСО (18 мл, 0,254 ммоль, 88 эквив.) и раствор перемешивали при -78oС в течение 15 мин. Раствор 13 (2,6 мл, 0,00287 ммоль, 1 эквив.) в дихлорметане (2,6 мл) добавляли по каплям к реакционной смеси, которую затем перемешивали при -40oС в течение 45 мин. К желто/зеленой реакционной смеси добавляли диизопропилэтиламин (51 мл, 0,288 ммоль, 100 эквив.) и желтый раствор перемешивали при 0oС в течение 45 мин до того, как избыточный реагент Сверна был погашен добавлением трет-бутилового спирта (13 мг, 0,176 ммоль, 61 эквив.) при 0oС. Трет-бутилтетраметилгуанидин (49 мл, 0,288 ммоль, 100 эквив.) добавляли к раствору, который перемешивали при 23oС в течение 1,5 ч, в течение этого времени раствор становился почти бесцветным. Добавляли уксусный ангидрид (50 мл, 0,530 ммоль, 184 эквив.) и через 1 час при 23oС реакционную смесь фильтровали через короткую колонку с силикагелем, элюируя 50% этилацетатом в гексанах. Фильтрат концентрировали и остаток очищали колоночной флэш-хроматографией (градиентное элюирование: 25-->33% этилацетат в гексане), получая 14 (1,7 мг, 79%). Rf 0,40 (50% этилацетат в гексане), [α]
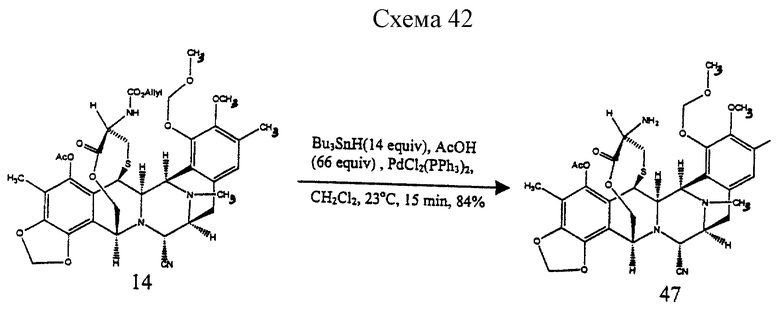
Пример 34 - Амин 47 (см. схему 42 в конце описания).
К раствору 14 (5,0 мг, 0,00666 ммоль, 1 эквив.), PdCl2 (РРh3)2 (0,5 мг) и уксусной кислоты (4 мл, 0,0666 ммоль, 10 эквив.) в дихлорметане (1 мл) добавляли трибутилоловогидрид (9 мл, 0,0333 ммоль, 5,0 эквив.) и коричневый раствор перемешивали при 23oС в течение 5 мин. Реакционную смесь сразу загружали в колонку с силикагелем и продукт очищали колоночной флеш-хроматографией (градиентное элюирование: этилацетат -->4% изопропиловый спирт в этилацетате), получая амин 47 (3,6 мг, 84%). Rf 0,25 (этилацетат); [α]
Пример 35 - Кетон 15 (см. схему 43 в конце описания).
К раствору 47 (2,9 мг, 0,00435 ммоль, 1 эквив.) в смеси ДМФ в дихлорметане (1:3 (об./об.), 640 мл) добавляли твердый 48 (22 мг, 0,0871 ммоль, 20 эквив. ) и красный раствор перемешивали при 23oС в течение 40 мин. Добавляли DBU (15 мл, 0,0871 ммоль, 20 эквив.) и черную суспензию перемешивали при 23oС в течение 15 мин до добавления насыщенного водного раствора щавелевой кислоты (0,5 мл). Желтую смесь перемешивали при 23oС в течение 30 мин до того, как ее распределяли между насыщенным водным раствором бикарбоната натрия (10 мл) и этиловым простым эфиром (30 мл). Органический слой сушили (сульфат магния) и концентрировали и фильтровали через короткую колонку силикагеля с использованием 50% этилацетата в гексанах, получая кетон 15 (2,0 мг, 70%). Rf 0,30 (50% этилацетат в гексанах); [α]
Пример 36 - Тристетрагидроизохинолин 49 (см. схему 44 в конце описания).
Кетон 15 (1,7 мг, 0,00256 ммоль, 1 эквив.) вместе с фенетиламином 16 (10 мг, 0,0599 ммоль, 23 эквив.) растеряли в абсолютном этаноле (500 мл) и к этому раствору добавляли силикагель (10 мг). Суспензию перемешивали при 23oС в течение 10 ч до того, как смесь была разбавлена этилацетатом (5 мл) и отфильтрована. Фильтрат концентрировали и остаток очищали колоночной флэш-хроматографией (5% метанол в дихлорметане), получая 49 (1,7 мг, 82%). Rf 0,32 (5% метанол в дихлорметане); [α]
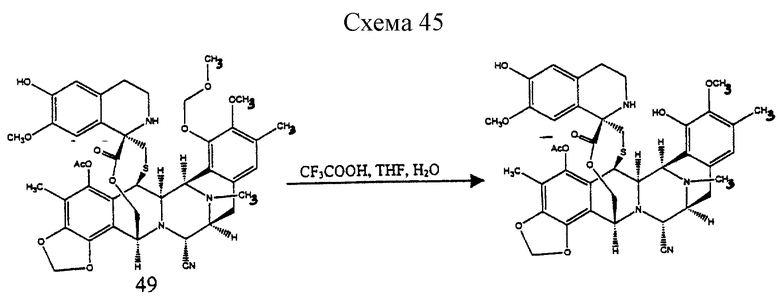
Пример 37 - Эстеинасцидин 770 (50) (см. схему 45 в конце описания).
Метоксиметиловый простой эфир 49 (2,8 мг, 0,0034 ммоль, 1 эквив.) растворяли в смеси трифторуксусная кислота:ТГФ:вода (4:1:1 (об./об.), 2,8 мл) и раствор перемешивали при 23oС в течение 9 ч. Реакционную смесь разбавляли толуолом (8 мл) и раствор концентрировали при 23oС. Все летучие компоненты удаляли в вакууме азеотропным удалением с толуолом (2х2 мл). Остаток очищали колоночной флэш-хроматографией (5% метанол в дихлорметане), получая 50 (2,2 мг, 78%). Rf 0,28 (9% метанол в дихлорметане); [α]
Пример 38 - Эстеинасцидин 743(1) (см. схему 46 в конце описания).
Эстеинасцидин 770 (50) (2,2 мг, 0,00285 ммоль, 1 эквив.) растворяли в смеси ацетонитрила и воды (3:2, (об./об.), 1,0 мл) и к этому раствору добавляли твердый нитрат серебра (15 мг, 0,088 ммоль, 30 эквив.). Суспензию перемешивали при 23oС в течение 11 ч, в это время добавляли смесь насыщенного водного раствора хлорида натрия и насыщенного водного раствора бикарбоната натрия (1:1 (об./об.), 2,0 мл). Смесь энергично перемешивали при 23oС в течение 15 мин до того, как ее распределяли между смесью насыщенного водного раствора хлорида натрия и насыщенного водного раствора бикарбоната натрия (1: 1 (об./об.), 15 мл) и дихлорметаном (3х10 мл). Объединенные органические слои сушили (сульфат натрия) и фильтровали через подушку целита. Фильтрат концентрировали, получая чистый 1 (2,0 мг, 95%), идентичный во всех отношениях с 1 аутентичного образца. ВЭЖХ (Zorbax ODS, C18, 4,6 ммх25 см, скорость потока: 1,0 мл/мин). RT 11,28 мин (соинъекция, 25% CH3CN в Н2О с 0,2% ТФК); [α]
ПРИМЕЧАНИЯ И ОБСУЖДЕНИЕ ИНФОРМАЦИИ
Следующие примечания и/или ссылки относятся к указанному выше. Авторы изобретения в соответствии с этим хотели бы процитировать публикации, которые следуют далее, как потенциальный прототип для заявленного здесь изобретения. Кроме того, публикации, цитированные ниже, таким образом, включены здесь в качестве ссылки.
1) Первое достижение в этой области обязано проф. Kenneth L. Rinehart и его группе. См. (a) Rinehart, K.L., Shield, L.S. In Topics in Parmaceutical Sciences, eds. Breimer, D.D.; Crommelin, D.J.A.; Midha, K.K. (Amsterdam Medical Press, Noordwijk, The Netherlands), 1989, pp. 613. (b) Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Keifer, P.A.; Wilson, G.R.; Perun, T.J., Jr.; Sakai, R.; Thompson, A.G.; Stroh, J.G.; Shield, L.S.; Seigler, D.S.; Li, L.H.; Martin, D.G.; Grimmelikhuijzen, C.J.P.; Gade, G.J. Nat. Prod. 1990, 53 771. (с) Rinehart, K.L.; Sakai, R. Holt, T.G.; Fregeau, N.L.; Perun, T.J., Jr.; Seigler, D. S.; Wilson, G.R., Shield, L.S. Pure Appl. Chem. 1990, 62, 1277. (d) Rinehart, K. L.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.A.; Sun F.; Li, L.H.; Martin, D.G.J. Org. Chem. 1990, 55, 4512. (e) Wright, A.E. ; Forleo, D.A.; Gunawardana, G.P.; Gunasekera, S.P.; Koehn, F.E.; MeConnell, O. J. J. Org. Chem. 1990, 55, 4508. (f) Sakai, R.; Rinehart, K.L.; Guan, Y.; Wang, H. -J. Proc. Natl. Acad. Sci. USA 1992, 89, 11456. См. также патенты США 5089273; 5149804; 5256663 и 5478932, каждый из которых включен здесь в качестве ссылки.
(2) Science, 1994, 266, 1324.
(3) Современный клинический план предусматривает введение пациенту трех доз по 0,5 мг соединения I; частное сообщение от Dr.Glynn Faircloth, PharmaMar USA, Cambridge, MA.
(4) Получен из 3,4-метилендиоксифенилметоксиметилового простого эфира в соответствии со следующей последовательностью: (1) литирование в положении С-2 (3 эквив. BuLi, 3 эквив. тетраметалендиамина в гексане при 0oС в течение 4 ч) и реакция СН3l (6 эквив. при -78-->23oС в течение 15 мин) с получением исключительно 2-метилпроизводного (87%); (2) ортолитирование (2 эквив. Buli в ТГФ при -30oС в течение 13 ч) и последующее формилирование 4 эквив. ДМФ (выход 64%); (3) отщепление защитной группы МеОСН2 (0,55 эквив. СН3SО3Н в CH2Cl2 при 0oС) и (4) обработка полученного 3-метил-4,5-метилендиоксисалицилового альдегида 1,5 эквив. NaH в ДМФ при 0oС в течение 5 мин и 2 эквив. бензилбромида при 23oС в течение 40 мин (общий выход 86%). (b) Получен из моноаллилового эфира малоновой кислоты путем превращения в смешанный ангидрид с хлорангидридом ВОР (Aldrich) и реакции с 2,2-диметоксиэтанолом.
(5) Эта стадия, которая включает полную изомеризацию в термодинамически более стабильный Z-α-ациламиноакриловый эфир, представляет собой обычно пригодный способ стереоспецифического синтеза таких соединений.
(6) Koenig, K.E. in Asymmetric Synthesis; Morrison, J.D., Ed., Academic Press. Jnc., Orlando, FL, Vol.5, 1985, p.71.
(7) Превращение 4 --> 5 демонстрирует пригодный способ регулирования стереохимии в ряду тетрагидроизохинолина.
(8) (а) Эта стадия превращает третичную гидроксильную группу 13 в О-диметилсульфониевое производное. Использование системы оксалилхлорид-ДМСО в качестве реагента неудовлетворительно вследствие препятствия, создаваемого хлоридом в последующих стадиях образования и присоединения хинонметида. (b) Эта стадия образует хинонметид, вероятно, путем циклоэлиминирования промежуточного илида оксосульфония типа Сверна.
(9) Barton, D.H.R.; Flliott, J.D.; Gero, S.D.J. Chem. Soc. Perkin Trans. 1, 1982, 2085.
(10) Получен от Prof. K.L. Rinehart and PharmaMar, USA.
(11) Для предыдущих работ по синтезу сафрамицинов см:
(a) Fukuyama, Т.; Sachleben, R.A.J. Am. Chem. Soc. 1982, 104, 4957. (b) Fukuyama, Т. ; Yang, L.; Ajeck, K.L.; Sachleben, R.A.J. Am.Chem.Soc. 1990, 112, 3712. © Saito, N.; Yamauchi, R.; Nisnioka, H.; Ida, S.; Kubo, A.J. Org. Chem. 1989, 54, 5391.
(12) Still, W.C.; Kahn, M., Mitra, A.J. Org. Chem., 1978, 43, 2923.
(13) Kofron, W.G.; Baclawski, L.M.J. Org. Chem. 1976, 41, 1979.








Данное изобретение относится к синтетическому способу получения соединений ряда эстеинасцидина и относящихся к ним структур, таких, как сафрамицины, в частности к получению эстеинасцидина 743 (1), чрезвычайно сильнодействующего и редкого противоопухолевого средства морского происхождения. Указанный способ является энантио- и стереорегулируемым, конвергентным и коротким. Описываются также промежуточные продукты способа, которые можно использовать не только для полного синтеза эстеинасцидина 743, но также других известных соединений ряда эстеинасцидина, включая их производные и аналоги. 5 с. и 1 з.п. ф-лы.
с образованием эстенасцидина 770, представленного структурной формулой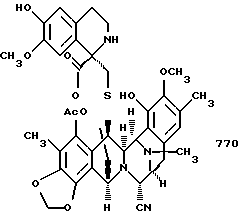
и необязательное замещение в полученном эстеинасцидине 770 группы CN на группу ОН с получением эстеинасцидина 743, представленного структурной формулой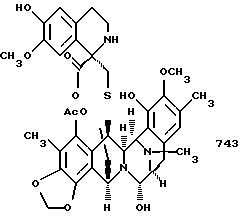
2. Способ по п. 1, отличающийся тем, что включает следующие стадии: (а) взаимодействие 2-бензилокси-3-метил-4,5-метилендиоксибензальдегида и аллил-2,2-диметоксиэтилмалоната с образованием α,β-ненасыщенного эфира малоновой кислоты формулы 2 в виде смеси Е и Z изомеров, представленного структурной формулой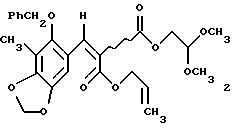
(b) стереоспецифическое преобразование соединения формулы 2 в соединение формулы 3, представленное структурной формулой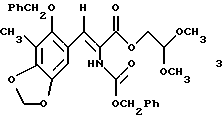
с помощью стадий селективного отщепления аллиловой сложноэфирной группы, перегруппировки Курциуса и взаимодействия промежуточного изоцианата с бензиловым спиртом; (с) преобразование соединения формулы 3 в соединение формулы 4, представленное структурной формулой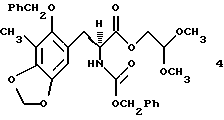
путем каталитического гидрирования над Rh[(COD)R, R-DIPAMP] +BF4 -; (d) преобразование соединения формулы 4 в соединение формулы 5, представленное структурной формулой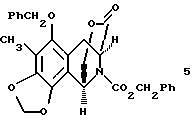
путем отщепления ацетальной группы, где выделение и обработка полученного альдегида с помощью BF3 -Et2O и молекулярными ситами  дает производное мостикового лактона формулы 5; (е) преобразования производного мостикового лактона формулы 5 в производное свободного аминофенола формулы 6, представленое структурной формулой
дает производное мостикового лактона формулы 5; (е) преобразования производного мостикового лактона формулы 5 в производное свободного аминофенола формулы 6, представленое структурной формулой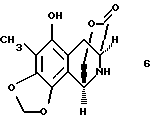
путем гидрирования над 10% Pd-C; (f) взаимодействие 3,5-бис-трет-бутилдиметилсилилокси-4-метоксибензальдегида и метилгидромалоната с получением производного защищенного сложного α-аминоэфира формулы 7, представленного структурной формулой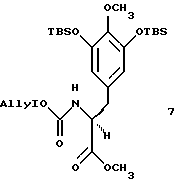
(g) восстановление производного защищенного сложного α-аминоэфира формула 7 до хирального альдегида 8, представленного cтруктурной формулой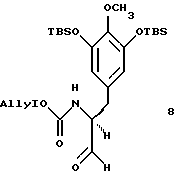
(h) взаимодействие соединений формул 6 и 8 с получением связанного фенольного α-аминонитрила с последующим O-аллилированием с получением производного аллилового эфира формулы 9, представленного структурной формулой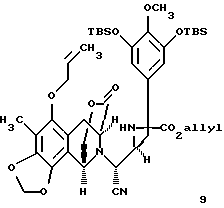
взаимодействие соединения формулы 9 с диизобутилалюминийгидридом для селективного преобразования лактоновой функциональной группы в соединении формулы 9 в лактол; десилилирование производного лактола и циклизацию десилилированного соединения внутренним бисаннелированием Манниха с получением мономостикового пентациклического производного формулы 10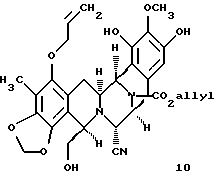
(i) преобразование пентациклического производного формулы 10 в соединение формулы 11, представленного структурной формулой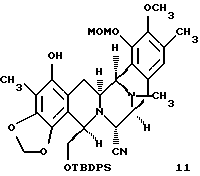
путем селективного трифторметансульфирования по меньшей мере стерически затрудненного фенольного гидроксила с последующим (1) селоктивным силилированием первичного гидроксила; (2) защитой оставшейся фенольной группы в виде метоксиметилового эфира; (3) двойным деаллилированием; (4) восстановительным N-метилированием и (5) заменой СF3SО3 на СН3; (j) окисление фенольного производного формулы 11, осуществляемого селективным по положению ангулярным гидроксилированием с получением после десилилирования дигидроскидиенового производного формулы 12, представленного структурной формулой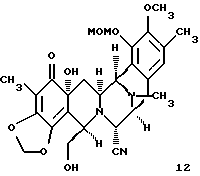
(k) этерификация первичной гидроксильной функциональной группы соединения 12 с помощью (S)-N-аллилоксикарбонил-S-(9-флуоренилметил)цистеина с получением соединения формулы 13, представленного cтруктурной формулой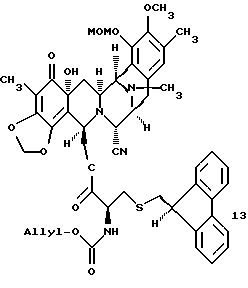
(l) преобразование соединения формулы 13 путем (1) сначала взаимодействия соединения формулы 13 с образованным in situ реагента Сверна; (2) последующего образования эксэндохинонметида; (3) разрушения избыточного реагента Сверна; (4) добавления избытка N-трет-бутил-N', N''-тетраметилгуанидина с образованием 10-членного лактонового мостика и (5) добавление избытка Ас2О для ацетилирования образовавшейся феноксидной группы и получения мостикового лактонового производного формулы 14, представленного структурной формулой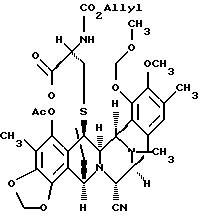
(m) отщепление N-аллилоксикарбонильной группы у соединения формулы 14 и окисления образовавшегося α-аминолактона в соответствующий α-кетолактон путем переаминирования с образованием, таким образом, соединения формулы 15, представленного cтруктурной формулой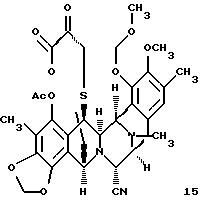
(n) взаимодействие соединения формулы 15 с 2-[3-гидрокси-4-метоксифенил] этиламином для стереоспецифического образования спиро-тетрагидроизохинолинового производного 49, представленного структурной формулой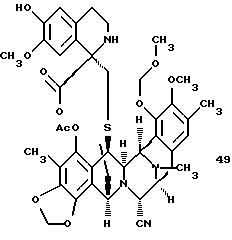
(о) отщепление метоксиметильной группы у соединения формулы 49, представленного структурной формулой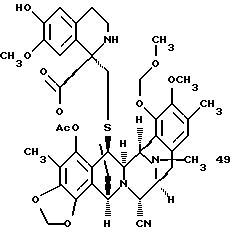
с образованием эстеинасцидина 770, представленного структурной формулой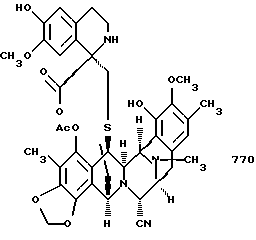
и необязательное замещение в эстеинасцидине 770 группы CN на группу НО с получением эстеинасцидина 743, представленного структорной формулой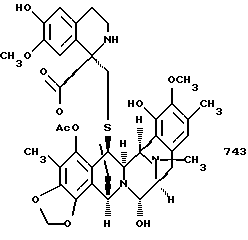
3. Эстеинасцидин 743, полученный способом по п. 1 или 2.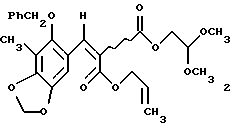
производное бензилкарбамата формулы 3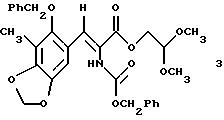
защищенное аминокислотное производное формулы 4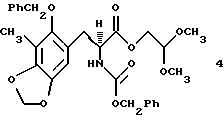
производное лактона формулы 5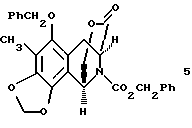
производное аминофенола формулы 6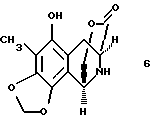
производное аминонитрила формулы 37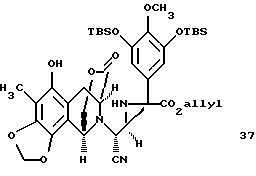
производное аллилового эфира формулы 9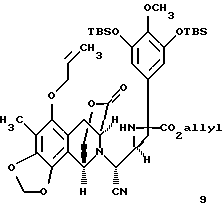
соединение формулы 38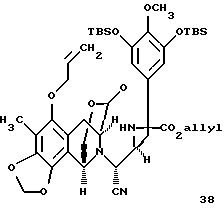
соединение формулы 39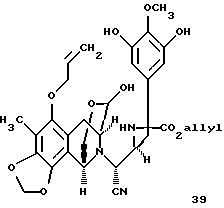
производное триола формулы 10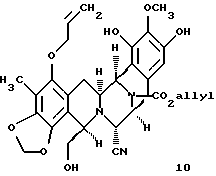
производное арилтрифлата формулы 40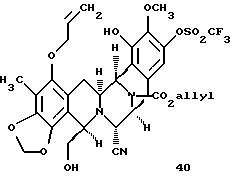
производное силилового эфира формулы 41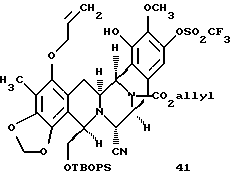
производное метоксиметилового эфира формулы 42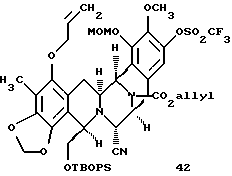
производное аминофенола формулы 43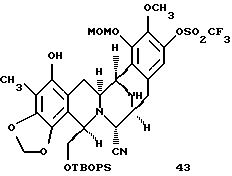
производное фенола формулы 44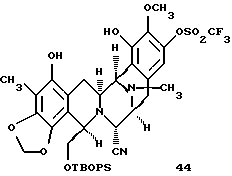
производное фенола формулы 11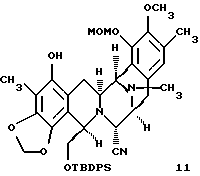
производное гидроксидиенона формулы 45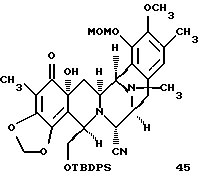
производное диола формулы 12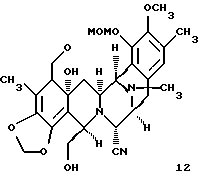
производное сложного эфира формулы 13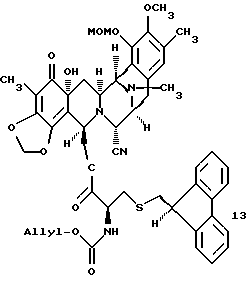
производное лактона формулы 14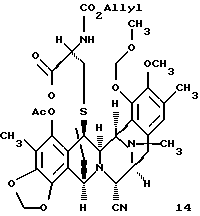
производное амина формулы 47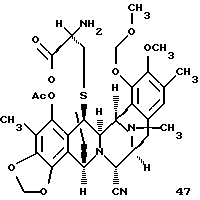
производное кетона формулы 15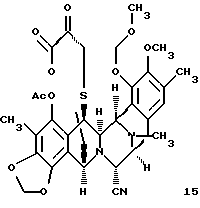
производное тристетрагидроизохинолина формулы 48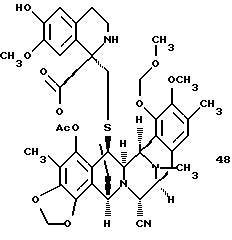
или производное спиротетрагидроизохинолина 49, представленное структурной формулой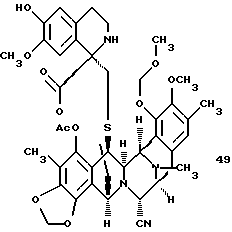
6. Способ получения промежуточных продуктов по п. 5, который включает проведение одной или нескольких стадий от (а) до (n) по п. 2.
| US 5089273, 1992 | |||
| US 5149804, 1992 | |||
| US 5478932, 1995 | |||
| US 5265663, 1993 | |||
| МАШКОВСКИЙ М.Д | |||
| Лекарственные средства | |||
| Харьков: Торсинг, 1998, ч.2, с.263. |
