Область изобретения
Изобретение относится к новым соединениям с азотом в кольце, способу их получения, их применению и фармацевтическим композициям, содержащим эти новые соединения. Новые соединения можно использовать в терапии, особенно для лечения боли.
Предпосылки создания изобретения и известный уровень техники
δ-Рецептор был идентифицирован как играющий роль во многих функциях организма, таких как сердечно-сосудистая и болевая системы. Лиганды для δ-рецептора могут, следовательно, найти потенциальное использование в качестве анальгезирующих и/или антигипертензивных средств. Было также обнаружено, что лиганды для δ-рецептора обладают иммуномодулирующей активностью.
В настоящее время проведена идентификация, по меньшей мере, трех разных популяций опиоидных рецепторов (μ,δ и κ), и все три обнаруживаются как в центральной, так и периферической нервных системах многих видов животных, включая человека. Анальгезию наблюдали на различных моделях животных, когда были активированы один или несколько из этих рецепторов.
За редкими исключениями, доступные в настоящее время селективные опиоидные δ-лиганды являются пептидами по природе и не пригодны для введения системными путями. Некоторые непептидные δ-антагонисты стали пригодны в течение некоторого последнего времени (для обзора см. Takemori and Portoghese, 1992, Ann. Rev. Pharmacol, Tox., 32:239-269). Эти соединения, например налтриндол, характеризуются весьма слабой (т. е. слабее в 10 раз) селективностью для связывания δ-рецептора по сравнению с μ-рецептором и не проявляют анальгезирующую активность, факт, который подразумевает потребность в разработке высоко селективных непептидных δ-лигандов.
Таким образом, проблемой, лежащей в основе данного изобретения, было нахождение новых анальгетиков, имеющих повышенное анальгезирующее действие, а также улучшенный профиль побочного действия по сравнению с современными μ-агонистами и потенциальную пероральную эффективность.
Анальгетики, которые были идентифицированы и известны из предшествующего уровня техники, имеют много недостатков, проявляющихся в том, что они имеют плохую фармакокинетику и не проявляют анальгезирующее действие при введении системными путями. Кроме того, было подтверждено документами, что предпочтительные соединения, описанные в известном уровне техники, проявляют также значительное судорожное действие при системном введении.
Проблема, указанная выше, была разрешена путем разработки новых соединений, которые имеют кольцо пиперидина, которое может быть 5-членным, 6-членным или 7-членным азотсодержащим кольцом, как будет описано ниже.
Описание изобретения
Новые соединения по настоящему изобретению определяются общей формулой (I)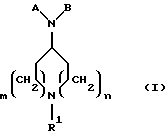
где m равно 0 или 1;
n равно 1 или 2;
R1 выбирают из водорода, разветвленного или неразветвленного C1-С6-алкила; C3-С8-циклоалкила; C4-С8-(алкилциклоалкила), где алкил представляет C1-С2-алкил и циклоалкил представляет C3-С6-циклоалкил; бензила;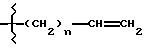 ;
; ,
,
где G представляет гидроароматическую или гетеро ароматическую группу, имеющую 5 или 6 атомов, и где гетероатомы выбирают из О, S и N; ,
,
где n = 0 или 1;
C6-С10-арила или гетероарила, имеющего от 5 до 10 атомов, выбранных из любого атома С, S, N и О, где арил или гетероарил может быть необязательно и независимо замещен 1 или 2 заместителями, независимо выбранными из водорода, СН3, (СН2)рСF3, галогена, CONR5R4, COOR5, COR5, (CH2)pNR5 R4 , (СН2)рСНз(СН2)рSОR5R4, (CH2)pSO2R5 и (СН2)pSO2NR5, где R4 и R5, каждый независимо, такие, как определено выше для R1, и р равно 0, 1 или 2;
(C1-С2-алкил) - (C1-С10-арил) или (C1-С2-алкил)гетероарил, причем гетероарильные фрагменты имеют от 5 до 10 атомов, выбранных из любого атома С, S, N и О, и где арил или гетероарил может быть необязательно и независимо замещен 1 или 2 заместителями, независимо выбранными из любого заместителя: водорода, СН3, CONR5R4, COOR5, COR5, (CH2)qNR5R4, (СH2)qСH3 (CH2)qSОR5R4,
(CH2)qSO2R5, (CH2)qSO2NR5 и (CH2)qOR4, где R4 и R5, каждый независимо, такие, как определено выше для R1, и q равно 0, 1 или 2;
А представляет
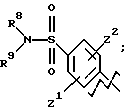


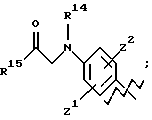

и
где R6, R7, R8, R9, R10, R11, R12, R13, R14, R15, R16 R17 и R18, каждый независимо, такие, как определено выше для R1, и где кольцо фенила каждого заместителя А может быть необязательно и независимо замещено 1 или 2 заместителями Z1 и Z2, которые, каждый независимо, выбирают из водорода, СН3, (СН2)rСF3, галогена, CONR2R3, CO2R2, COR2, (CH2)rNR2R3, (СН2)rСН3(СН2)rSОR2,
(CH2)rSO2R2 и (СН2) rSO2NR2RЗ, где R2 и R3, каждый независимо, такие, как определено выше для R1, и где r равно 0, 1 или 2; X представляет О, S или NR19, где R19 такой, как определено для R1.
В представляет замещенный или незамещенный ароматический, гетероароматический, гидроароматический или гетерогидроароматический фрагмент, имеющий от 5 до 10 атомов, выбранных из любого атома С, S, N и О, необязательно и независимо замещенный 1 или 2 заместителями, независимо выбранными из водорода, СН3, (СН2)tСF3, галогена, (СН2) tCONR5R4, (CH2)tNR5R4, (CH2)tCOR5,
(CH2)tCOOR5, OR5, (CH2)tSOR5, (CH2)tSO2R5 и (CH2)tSO2NR5R4, где R4 и R5, каждый независимо, такие, как определено выше для R1, и t равно 0, 1, 2 или 3;
В объем данного изобретения включаются также фармацевтически приемлемые соли соединений формулы (I), а также их изомеры, гидраты, изоформы и пролекарства.
Предпочтительные соединения по изобретению представляют собой соединения формулы (I), где
R1 выбирают из бензила: ;
; ,
,
где G представляет гидроароматическую или гетероароматическую группу, имеющую 5 или 6 атомов и где гетероатомы выбирают из О, S и N; и ;
;
и где n = 0 или 1;
А выбирают из любого одного из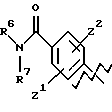

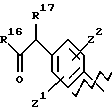
и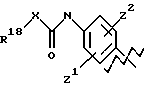
где R6, R7, R8, R9, R16, R17 и R18, каждый независимо, такие, как определено выше для R1, и Z1, Z2 и X, каждый независимо, такие, как определено выше;
В выбирают из фенила, нафтила, индолила, бензофуранила, дигидробензофуранила, бензотиофенила, пиррила, фуранила, хинолинила, изохинолинила, циклогексила, циклогексенила, циклопентила, циклопентенила, инданила, инденила, тетрагидронафтила, тетрагидрохинила, тетрагидроизохинолинила, тетрагидрофуранила, пирролидинила и индазолинила, каждый из которых необязательно и независимо замещен 1 или 2 заместителями, независимо выбранными из водорода, СН3, СF3, галогена, -(СН2)tCONR5R4, -(СН2)tNR5R4, -(СН2)tCOR5,
-(СН2)tCO2R5 и -OR5, где t равно 0 или 1, и где R4 и R5 такие, как определено выше.
Особенно предпочтительные соединения представляют собой соединения формулы (I), где
R1 представляет (С1-С2-алкил)фенил и водород;
А представляет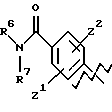
или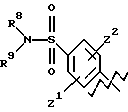
где R6, R7, R8, R9, каждый, представляют этиленовую группу, и Z1 и Z2 такие, как определено выше;
В представляет фенил или нафталин и
m и n, каждый, равны 1 или m равно 1 и n равно 0.
Заместители А и В, соответственно, могут быть необязательно замещены в любом положении кольца.
Термином "галоген" обозначается хлор, фтор, бром и иод.
Термином "арил" обозначается ароматическое кольцо, имеющее от 6 до 10 атомов углерода, такое как фенил и нафтил.
Термином "гетероарил" обозначается ароматическое кольцо, в котором один или несколько из 5-10 атомов в кольце представляют собой элементы, другие чем углерод, такие как N, S и О.
Термином "гидроароматический" обозначается структура частично или полностью насыщенного ароматического кольца, имеющая 5-10 атомов углерода в кольце.
Термином "гетерогидроароматический" обозначается структура частично или полностью насыщенного ароматического кольца, в которой один или несколько из 5-10 атомов в кольце являются элементами, другими, чем углерод, такими как N, S и О.
Термином "изомеры" обозначаются соединения формулы (I), которые отличаются положением их функциональной группы и/или ориентацией. Термином "ориентация" обозначаются стереоизомеры, диастереоизомеры, региоизомеры и энантиомеры.
Термином "изоформы" обозначаются соединения формулы (I), которые отличаются по их кристаллической решетке, такие как кристаллическое соединение и аморфные соединения.
Термином "пролекарство" обозначаются фармакологически приемлемые производные, например сложные эфиры и амиды, так чтобы образуемый продукт биопревращения такого производного был активным лекарственным средством. Ссылка на публикацию Goodman and Gilmans, The Pharmacological basis of Therapeutics, 8th ed., McGraw-Hill, Jnt. Ed. 1992, "Biotransformation of Drugs, p. 13-15, описывающая пролекарства в общем, включается здесь таким образом.
Новые соединения данного изобретения можно использовать в терапии, особенно для лечения различных болезненных состояний, таких как хроническая боль, острая боль, раковая боль, боль, вызванная ревматоидным артритом, мигрень, висцеральная боль и так далее. Этот список, однако, не должен истолковываться как исчерпывающий.
Соединения изобретения пригодны в качестве иммуномодуляторов, особенно для аутоиммунных болезней, таких как артрит, для полнослойных кожных лоскутов, органов-трансплантатов и аналогичных хирургических потребностей, для коллагеновых болезней, различных аллергий, для использования в качестве противоопухолевых средств и антивирусных средств.
Соединения изобретения можно использовать при болезненных состояниях, где имеет место дегенерация или дисфункция опиоидных рецепторов или опиоидные рецепторы принимают участие в этой парадигме. Она может включать использование меченных изотопами вариантов соединений изобретения в диагностических методиках и в применениях для получения изображений, таких как позитронная эмиссионная томография (ПЭТ).
Соединения изобретения можно использовать для лечения диареи, депрессии, недержания мочи, различных психических болезней, кашля, отека легких, различных желудочно-кишечных нарушений, спинального повреждения и привыкания к чрезмерному употреблению лекарственных средств, включая лечение от злоупотребления алкоголем, никотином, опиоидами и другими лекарственными средствами, и нарушений симпатической нервной системы, например гипертензии.
Соединения изобретения можно использовать в качестве анальгезирующего агента для применения во время общей анестезии и для оказания помощи контролируемой анестезией. Комбинации агентов с разными свойствами часто используют для достижения баланса действий, требуемых для поддержания анестезирующего состояния (например, амнезии, анальгезии, мышечной релаксации и седативного эффекта). В эту комбинацию включают вводимые ингаляцией анестетики, снотворные средства, анксиолитики, нейромышечные блокаторы и опиоиды.
Соединения данного изобретения в меченной изотопом форме можно использовать в качестве диагностического средства.
В объем данного изобретения включается также применение любого из соединений по приведенной выше формуле (I) для изготовления лекарственного средства для лечения любого из состояний, обсуждаемых выше.
Следующим аспектом изобретения является способ лечения субъекта, страдающего любым из обсуждаемых выше состояний, посредством которого эффективное количество соединения по указанной выше формуле (I) вводят пациенту, нуждающемуся в таком лечении.
Способы получения
Соединения формулы (I), как описано выше, можно получить арилированием амина формулы (II)
где R1, m и n такие, как определено выше в формуле (I), и W представляет А или В, как определено выше в формуле (I), арилирующим агентом формулы (III)
W-Z (III)
где W представляет А или В, как определено выше в формуле (I), и Z представляет подходящий заместитель, т. е. реакционноспособный компонент, подходящий для использования в определенном способе, который должен быть знаком специалисту в данной области, предпочтительно галоген, трифлат (СF3SО3-), мезилат (СН3SО3-), тозилат (СН3(С6Н4)SO3-), трибутилолово, триацетоксисвинец, диарилвисмут, борат (В(ОН)2), купрат или другие группы, известные в данной области. Арилирование можно катализировать металлами, предпочтительно Сu, Ni, Pd или их подходящими солями, комплексами, оксидами или гидроксидами. 4-Аминопиперидин вышеприведенной формулы (II) можно превратить полностью или частично в его соответствующий анион обработкой основаниями, предпочтительно триэтиламином, 4-диметиламинопиридином, К2СО3, NaOH, NaH, диизопропиламидом лития, трет-бутоксидом натрия или тому подобное, до или во время процесса арилирования. Реакцию можно проводить в присутствии комплексообразующих реагентов, предпочтительно трифенилфосфина, трифениларсина, дибензилиденацетона, 2,2'-бис(дифенилфосфино)-1,1'-бинафтила, 1,1'-бис(дифенилфосфино)ферроцена, кислорода или других таких соединений, известных в данной области. Реакцию можно необязательно проводить в присутствии одного или нескольких растворителей, таких как толуол, дихлорметан, тетрагидрофуран, диметилформамид, диоксан, ацетонитрил или диметилсульфоксид или в смеси растворителей.
R1 и заместители на А и В соединений формулы (I), как определенно выше, можно модифицировать после или во время получения (I) из (II) и (III) способами, известными в данной области, например восстановлением, окислением и алкилированием.
Амин формулы (II) можно получить восстановительным аминированием кетона формулы (IV)
где R1, R2, R3, m и n такие, как определено выше в формуле (I), замещенным ариламином (V)
W-NH2 (V)
где W такой, как определено выше в формуле (II).
Восстановительное аминирование можно проводить одно- или двухстадийным способом, включающим использование кислоты Бронстедта или Льюиса и восстанавливающего агента. Подходящими кислотами являются серная кислота, полифосфорная кислота, 4-толуолсульфоновая кислота, изопропоксид титана, трихлорид алюминия, диэтиловый эфират трифторида бора или тому подобное. Подходящими восстанавливающими агентами являются водород в присутствии катализатора, предпочтительно Pd, Pd-C, Pd(OH)2, PtO2, Rh-C или никель Ренея, борогидрид натрия, цианоборогидрид натрия, литийалюминийгидрид, диборан, диизобутилалюминийгидрид или тому подобное. Реакцию можно проводить в присутствии одного или нескольких растворителей, которые могут быть органическими или неорганическими, таких как толуол, дихлорметан, простые эфиры, спирты, уксусная кислота, вода или смеси растворителей.
R1 и заместители на W соединения (II), как определено выше, можно модифицировать после или во время получения (I) из (II) и (III) способами, известными в данной области, например восстановлением, окислением или алкилированием, после или во время получения (II) из (IV) и (V).
Соединения формулы (III), (IV) и (V) могут быть коммерчески доступны, могут быть получены по литературным методикам или могут быть получены способами, известными в данной области.
Изобретение теперь будет описываться более подробно посредством следующих примеров, которые не должны никоим образом рассматриваться, как ограничение изобретения.
ПРИМЕРЫ
ПРИМЕР 1
(i) Получение 4-[N-(1-бензилпиперидин-4-ил)амино] -N,N-диэтилбензамида (соединение 1)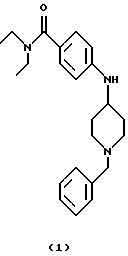
Ti(Oi-Pr)4 (14,8 мл, 50 ммоль) добавляли к смеси 4-амино-(N,N-диэтил)бензамида (4,81 г, 25 ммоль) и 1-бензил-4-пиперидона (6,95 мл, 37,5 ммоль) при комнатной температуре. Смесь обрабатывали ультразвуком при 40oС в течение 2 час и перемешивали при 60oС в течение 15 час. Смесь охлаждали на ледяной бане и добавляли EtOH (100 мл) и гранулы NaBH4 (3,5 г, 91 ммоль). После перемешивания в течение 1 час при 0oС и 20 час при комнатной температуре, добавляли 1 М NH4OH (50 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, разбавляли СН2Сl2 (100 мл) и фильтровали через слой целита®. Слои в фильтрате разделяли, водный слой экстрагировали CH2Cl2 (100 мл) и объединенные органические фазы промывали NаНСО3 (водный, насыщенный, 100 мл) и сушили над К2СО3. Смесь фильтровали, концентрировали и остаток очищали хроматографией (градиент от PhMe до Ме2СО) и кристаллизацией (PhMe), получая указанное в заголовке соединение 1 (7,48 г, 82%) в виде твердого материала бежевого цвета.
ИК (КВr): 3343, 2939, 1608, 1528, 1459, 1422, 1339, 1285, 1174, 1091, 981, 827, 735 см-1.
1H ЯМР(СDС1з): 7,32 (д, 4Н), 7,26 (т, 1Н), 7,23 (д, 2Н), 6,54 (д, 2Н), 3,72 (шир. с, 1Н), 3,53 (с, 2Н), 3,42 (шир. д, 4Н), 3,32 (шир. с, IH), 2,84 (д, 2Н), 2,15 (т, 2Н), 2,02 (д, 2Н), 1,48 (к, 2Н), 1,17 (т, 6Н).
13С ЯМР (СDСl3): 171,7, 148,1 138,3, 129,1, 128,5, 128,2, 127,0, 125,3, 112,2, 63,1, 52,2, 49,8, 41,5 (шир.), 32,4, 13,6 (шир.).
Аналитический образец получали перекристаллизацией из PhMe.
Анализ. Вычислено для С23Н31N3О:
С 75,58; Н 8,55; N 11,50.
Найдено: С 75,58; Н 8,63; N 11,31.
(ii) Получение 4-[N-(1-бензилпиперидин-4-ил)анилино]-N,N-диэтилбензамида (соединение 2)
Смесь 4- [N- (1-бензилпиперидин-4-ил) амино] -N, N-диэтилбензамида (соединение 1) (0,58 г, 1,59 ммоль), Ph3Bi (0,84 г, 1,90 ммоль) и Cu(OAc)2 (0,43 г, 2,38 ммоль) в PhMe (25 мл) нагревали при 110oС в течение 15 час. Добавляли Рh3Вi (0,84 г, 1,90 ммоль) и Сu(ОАс)2 (0,43 г, 2,38 ммоль) и смесь перемешивали при кипячении с обратным холодильником в течение 6 час. Добавляли Ph3Bi (0,84 г, 1,90 ммоль) и Сu(ОАс)2 (0,43 г, 2,38 ммоль) и смесь перемешивали при кипячении с обратным холодильником в течение 15 час, оставляли для охлаждения и гасили 1 М NH4OH (5 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, разбавляли EtOAc (25 мл) и фильтровали через слой целита®. Слои в фильтрате разделяли, темно-синий водный слой экстрагировали EtOAc (25 мл) и объединенные органические фазы промывали Н2O (50 мл) и насыщенным солевым раствором (25 мл) и сушили над К2СО3. Смесь фильтровали, концентрировали и остаток очищали хроматографией (градиент, от PhMe до Ме2O), получая указанное в заголовке соединение 2 (0,33 г, 47%) в виде бесцветного масла.
1H ЯМР (CDCl3): 7,53 (т, 2Н), 7,29-7,18 (м, 8Н), 7,01 (д, 2Н), 6,58 (д, 2Н), 3,85 (т, IH), 3,49 (с, 2Н), 3,42 (д, 4Н), 2,95 (д, 2Н), 2,11 (т, 2Н), 1,92 (д, 2Н), 1,51 (к, 2Н), 1,17 (т, 6Н).
13Н ЯМР (CDCl3): 171,5, 149,0, 143,6, 138,2, 129,5, 129,1, 128,7, 128,2, 128,1, 127,0, 126,7, 125,4, 116,0, 63,1, 55,5, 53,3, 40 (шир.), 30,7, 13 (шир.).
Аналитический образец получали в виде гидрохлорида добавлением раствора свободного основания в смеси простой эфир/EtOH к охлажденному льдом разбавленному простым эфиром НСl.
ИК (КВr): 3423, 2975, 2934, 2529, 1606, 1458, 1285, 1094, 750, 705 см-1.
Анализ. Вычислено для С29Н35N3О•НСl•Н2O:
С 70,21; Н 7,72; N 8,47.
Найдено: С 70,02; Н 7,61; N 8,35.
ПРИМЕР 2
(i) Получение 4-[N-(1-бензилпиперидин-4-ил)-4-метиланилино] -N, N-диэтилбензамида (соединение 3)
Смесь 4- [N- (1-бензилпиперидин-4-ил) амино] -N, N-диэтилбензамида (соединение 1) (0,37 г, 1,00 ммоль), три-4-толилвисмута (1,59 г, 3,30 ммоль) и Сu(ОАс)2 (0,54 г, 3,00 ммоль) в PhMe (20 мл) нагревали при кипячении с обратным холодильником в течение 16 ч. Смесь оставляли для охлаждения и гасили Н2O (2 мл). Смесь перемешивали в течение 1 час, разбавляли EtOAc (25 мл) и фильтровали через слой целита®. Слои в фильтрате разделяли, водный слой экстрагировали EtOAc (25 мл) и объединенные органические фазы промывали Н2О (50 мл) и насыщенным солевым раствором (25 мл) и сушили над MgSO4. Смесь фильтровали, концентрировали и остаток очищали хроматографией (градиент, от CH2Cl2 до 8% MeOH/CH2Cl2), получая указанное в заголовке соединение 3 (0,09 г, 20%) в виде бесцветного масла.
1H ЯМР (CDCl3,): 7,30-7,16 (м, 9Н), 6,94 (д, 2Н), 6,52 (д, 2Н), 3,83 (т, 1Н), 3,48 (с, 2Н), 3,42 (д, 4Н), 2,93 (д, 2Н), 2,36 (с, 3Н), 2,07 (т, 2Н), 1,90 (д, 2Н), 1,50 (к, 2Н), 1,16 (т, 6Н).
13С ЯМР (CDC13): 171,6, 149,7, 140,4, 138,2, 136,1, 130,2, 130,2, 129,0, 128,1, 128,0, 126,9, 125,3, 113,8, 63,0, 55,4, 53,3, 41 (шир.), 30,5, 21,0, 13 (шир.).
Аналитический образец получали в виде гидрохлорида добавлением раствора свободного основания в простом эфире к охлажденному льдом разбавленному простым эфиром НС1.
ИК (КВr): 2936, 2528, 1605, 1510, 1457, 1428, 1284, 1094, 952, 742, 701 см-1.
Анализ. Вычислено для С30Н37N3О•НСl•0,5 Н2О:
С 71,91; Н 7,84; N 8,39.
Найдено: С 71,75; Н 7,83; N 8,32.
ПРИМЕР 3
Получение 4-[N-(1-бензилпиперидин-4-ил)-1-нафтиламино] -N, N-диэтилбензамида (соединение 4)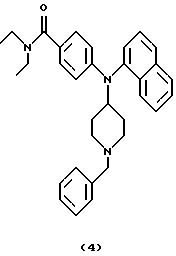
Смесь 4-[N-(1-бензилпиперидин-4-ил)амино]-N,N-диэтилбензамида (соединение 1) (0,37 г, 1,00 ммоль), три-1-нафтилвисмута (0,53 г, 1,20 ммоль) и Сu(ОАс)2 (0,27 г, 1,50 ммоль) в PhMe (20 мл) нагревали при кипячении с обратным холодильником в течение 17 ч. Три-1-нафтилвисмут (0,53 г, 1,20 ммоль) и Cu(OAc)2 (0,27 г, 1,50 ммоль) добавляли при комнатной температуре. Смесь перемешивали при кипячении с обратным холодильником в течение 22 час, оставляли для охлаждения и гасили 1 М NH4OH (5 мл). Смесь перемешивали в течение 30 мин, разбавляли EtOAc (25 мл) и фильтровали через слой целита®. Слои в фильтрате разделяли, темно-синий водный слой экстрагировали EtOAc (25 мл) и объединенные органические фазы промывали Н2О (50 мл) и насыщенным солевым раствором (25 мл) и сушили над К2СО3. Смесь фильтровали, концентрировали и остаток очищали хроматографией (градиент, от PhMe до Ме2СО), получая указанное в заголовке соединение 4 (0,41 г, 83%) в виде коричневатого твердого материала.
ИК (КВr): 2939, 2796, 1619, 1511, 1456, 1420, 1346, 1282, 1180, 1099, 783 см-1.
1H ЯМР (CDCI3): 7,87 (т, 2Н), 7,79 (д, 1Н), 7,51 (т, 1Н), 7,46 (т, 1Н), 7,37 (т, 1Н), 7,31 (д, 1Н), 7,28-7,19 (м, 5Н), 7,17 (д, 2Н), 6,41 (д, 2Н), 4,08 (т, 1Н), 3,46 (с, 2Н), 3,40 (д, 4Н), 2,89 (д, 2Н), 2,11 (т, 2Н), 2,08 (шир. с, 2Н), 1,50 (шир. с, 2Н). 1,14 (т, 6Н).
13С ЯМР (CDCl3): 171,7, 149,8, 138,9, 138,1, 135,0, 133,4, 129,1, 129,0, 128,3, 128,2, 128,1, 128,0, 127,0, 126,4, 126,2, 126,1, 124,6, 124,5, 112,2, 63,0, 56,5, 53,3, 40 (шир.), 30,4, 13,5 (шир.).
Аналитический образец получали перекристаллизацией из EtOH.
Анализ. Вычислено для C33H37N3O:
С 80,61; Н 7,59; N 8,55.
Найдено: С 80,48; Н 7,41; N 8,52.
ПРИМЕР 4
Получение 4-[N-(1-бензилпиперидин-4-ил)-2-нафтиламино] -N, N-диэтилбензамида (соединение 5)
Смесь 4-[N-(1-бензилпиперидин-4-ил)амино]-N,N-диэтилбензамида (соединение 1) (0,67 г, 1,83 ммоль), три-2-нафтилвисмута (0,97 г, 2,20 ммоль) и Сu(ОАс)2 (0,50 г, 2,75 ммоль) в PhMe (35 мл) нагревали при кипячении с обратным холодильником в течение 15 ч. Добавляли три-2-нафтилвисмут (0,97 г, 2,20 ммоль) и Сu(ОАс)2 (0,50 г, 2,75 ммоль) и смесь перемешивали при кипячении с обратным холодильником в течение 22 час. Добавляли три-2-нафтилвисмут (0,97 г, 2,20 ммоль) и Cu(OAc)2 (0,50 г, 2,75 ммоль). После кипячения с обратным холодильником в течение 70 час смесь оставляли для охлаждения и гасили 1 М NH4OH (10 мл). Смесь перемешивали в течение 30 мин, разбавляли EtOAc (35 мл) и фильтровали через слой целита®. Слои в фильтрате разделяли, темно-синий водный слой экстрагировали EtOAc (35 мл) и объединенные органические фазы промывали H2O (75 мл) и насыщенным солевым раствором (35 мл) и сушили над MgSO4. Смесь фильтровали, концентрировали и остаток очищали хроматографией (градиент, от РhМе до Ме2СО), получая указанное в заголовке соединение 5 (0,63 г, 70%) в виде коричневого масла, которое отверждалось при стоянии.
ИК (КВr): 2935, 2807, 1614, 1510, 1424, 1284 см-1.
1H ЯМР (CDCl3): 7,83-7,78 (м, 2Н), 7,74 (д, 1Н), 7,48-7,42 (м, 3Н), 7,27-7,21 (м, 7Н), 7,10 (дд, 1Н), 6,66 (д, 2Н), 3,94 (т, 1Н), 3,48 (с, 2Н), 3,43 (шир. с, 4Н), 2,94 (д, 2Н), 2,14 (т, 2Н), 1,99 (с, 2Н), 1,57 (м, 2Н), 1,17 (т, 6Н).
13С ЯМР (CDCl3): 171,4, 148,8, 141,3, 138,1, 134,2, 131,2, 129,2, 129,0, 128,1, 128,0, 127,5, 127,4, 127,2, 126,9, 126,2, 125,4, 125,3, 116,8, 63,0, 55,8, 53,3, 41 (шир.), 30,7, 13,6 (шир.).
Аналитический образец получали из МеОН.
Анализ. Вычислено для С33Н37N3О:
С 80,61; Н 7,59; N 8,55.
Найдено: С 80,35; Н 7,59; N 8,46.
ПРИМЕР 5
Получение N, N-диэтил-4-(N-пиперидин-4-иланилино) бензамида (соединение 6)
Раствор 4-[N-(1-бензилпиперидин-4-ил)анилино] -N, N-диэтилбензамида (соединение 2) (1,21 г, 2,74 ммоль) в МеОН (25 мл) гидрировали при 4,219 атм (60 пси) в течение 4 дней в присутствии каталитического количества Pd(OH)2 на угле. Смесь фильтровали через слой целита®, концентрировали и остаток очищали хроматографией (градиент, от СН2Сl2 до CH2Cl2/MeOH (9:1) и до CH2Cl2/MeOH/NH4OH (водный, концентрированный) (80:18:2)), получая указанное в заголовке соединение 6 (0,62 г, 64%) в виде бесцветного масла.
1ЯМР (CDCl3): 7,37 (т, 2Н), 7,25-7,22 (м, 3Н), 7,01 (д, 2Н), 6,61 (д, 2Н), 3,98 (т, 1Н), 3,42 (шир. д, 4Н), 3,17 (д, 2Н), 2,78 (т, 2Н), 2,00 (д, 2Н), 1,71 (шир. с, 1Н), 1,41 (к, 2Н), 1,18 (т, 6Н).
13С ЯМР (СDСl3): 171,4, 148,2, 143,1, 129,8, 128,0, 127,9, 127,7, 125,7, 116,9, 53,5, 44,4, 41 (шир.), 28,7, 13,5 (шир.).
Аналитический образец получали в виде гидрохлорида добавлением раствора свободного основания в простом эфире к охлажденному льдом разбавленному простым эфиром НС1.
ИК (КBr): 3426, 3359, 2936, 2722, 1603, 1473, 1281, 1091, 708, 503 см-1.
Анализ. Вычислено для C22H29N3O•HCl•H2O:
С 65,09; Н 7,95; N 10,35.
Найдено: С 65,37; Н 7,94; N 10,38.
ПРИМЕР 6
Получение N, N-диэтил-4- [ (N-пиперидин-4-ил]-1-нафтиламино)бензамида (соединение 7)
Раствор 4-[N-(1-бензилпиперидин-4-ил)-1-нафтиламино] -N, N-диэтилбензамида (соединение 4) (0,22 г, 0,45 ммоль) в EtOH (20 мл) гидрировали при 4,219 атм (60 пси) в течение 64 час в присутствии каталитического количества Pd(OH)2 на угле. Смесь фильтровали через слой целита®, концентрировали и остаток очищали хроматографией (градиент, от CH2Cl2 до СН2Сl2/МеОН (9:1) до CH2Cl2/MeOH/NH4OH (водный, конц.) (80:18:2)), получая указанное в заголовке соединение 7 (0,12 г, 67%) в виде бесцветного масла, которое отверждалось при стоянии.
ИК (КВr): 2942, 1609, 1512, 1448, 1280 см-1.
1H ЯМР (СDСl3): 7,86 (т, 2Н), 7,80 (д, 1Н), 7,50 (т, 1Н), 7,45 (т, 1Н), 7,36 (т, 1Н), 7,30 (д, 1Н), 7,18 (д, 2Н), 6,42 (д, 2Н), 4,20 (т, 1Н), 3,40 (шир. , д, 4Н), 3,06 (д, 2Н), 2,79-2,63 (м, 3Н), 2,03 (шир. с, 2Н), 1,39 (шир. с, 2Н), 1,14 (т, 6Н).
13С ЯМР (CDCl3): 171,6, 149,4, 138,7, 134,8, 133,3, 128,7, 128,2, 128,1, 127,9, 126,3, 126,1, 126,0, 124,4, 124,2, 112,1, 56,1, 46,0, 41 (шир.), 31,3, 13,4 (шир.).
Анализ. Вычислено для С26Н31N3О•1,25 Н2O:
С 73,64; Н 7,96; N 9,91.
Найдено: С 73,77; Н 7,54; N 9,96.
ПРИМЕР 7
Получение N, N-диэтил-4-[(N-пиперидин-4-ил] -2-нафтиламино)бензамида (соединение 8)
(1-Хлорэтил)хлорформиат (58 мкл, 0,53 ммоль) добавляли к раствору 4-[N-(1-бензилпиперидин-4-ил)-2-нафтиламино] -N, N-диэтилбензамида (соединение 5) (105 мг, 0,21 ммоль) в дихлорэтане (2,5 мл) при комнатной температуре. Смесь нагревали при кипячении с обратным холодильником в течение 17 час и затем оставляли для охлаждения до комнатной температуры и концентрировали. Добавляли метанол (2,5 мл) и смесь нагревали при кипячении с обратным холодильником в течение 2,5 час, оставляли для охлаждения и концентрировали. Остаток распределяли между CH2Cl2 (10 мл) и 1 М NH4OH (10 мл). Слои разделяли и органическую фазу промывали H2O (10 мл) и насыщенным солевым раствором (10 мл) и сушили над К2СО3. Смесь фильтровали, концентрировали и остаток очищали хроматографией (градиент, от CH2Cl2 до CH2Cl2/MeOH/NH4OH (водный, концентр. ) (85:13,5:1,5) и ВЭЖХ (LiChroPrep RP-18, элюирование с увеличивающимися количествами 0,1% ТФУ/MeCN в 0,1% ТФУ/Н2О), получая указанное в заголовке соединение 8 (30 мг) в виде трифторацетата.
ИК (неразбавленный): 3420, 1680, 1600 см-1.
1H ЯМР (CDCl3, соль ТФУ) d: 1,10 (6Н, м), 1,78 (2Н, м,), 2,10 (2Н, м, 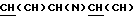 ), 3,00 (2Н, м,
), 3,00 (2Н, м,  ), 3,35 (6Н, м), 4,15 (1Н, м, ), 6,65 (2Н, м), 7,00 (1Н, м), 7,20 (2Н, м), 7,40 (3Н, м), 7,75 (3Н, м,
), 3,35 (6Н, м), 4,15 (1Н, м, ), 6,65 (2Н, м), 7,00 (1Н, м), 7,20 (2Н, м), 7,40 (3Н, м), 7,75 (3Н, м, 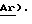
ПРИМЕР 8
Получение 4-[(N-(1-[2-фенилэтил] пиперидин-4-ил)анилино]-N,N-диэтилбензамида (соединение 9)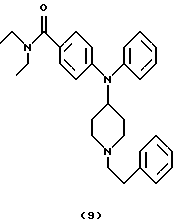
(2-Бромэтил)бензол (0,18 мл, 1,30 ммоль) добавляли при перемешивании к охлажденному льдом раствору N,N-диэтил-4-(N-пиперидин-4-иланилино)бензамида (соединение 6) (0,21 г, 0,59 ммоль), Et3N (0,10 мл, 0,75 ммоль) и каталитического количества 4-диметиламинопиридина в СН2Сl2 (5 мл). Перемешиваемую смесь выдерживали до достижения комнатной температуры в течение 5 час, нагревали при кипячении с обратным холодильником в течение 16 час, оставляли для охлаждения до комнатной температуры, разбавляли СН2Сl2 (10 мл) и промывали Н2O (15 мл), насыщенным солевым раствором (15 мл) и сушили над К2СО3. Смесь фильтровали, концентрировали и остаток очищали хроматографией (градиент, от РhМе до PhMe/Me2CO (1:2)), получая указанное в заголовке соединение 9 (0,11 г, 40%) в виде бесцветного масла, которое отверждалось при стоянии.
ИК (КВr): 2928, 1612, 1504, 1437, 1280, 1980, 754 см-1.
1H ЯМР (CDCl3): 7,36 (т, 2Н), 7,30-7,15 (м, 8Н), 7,01 (д, 2Н), 6,61 (д, 2Н), 3,88 (т, 1Н), 3,42 (шир. д, 4Н), 3,08 (д, 2Н), 2,76 (м, 2Н), 2,57 (м, 2Н), 2,17 (т, 2Н), 1,97 (д, 2Н), 1,55 (к, 2Н), 1,18 (т, 6Н).
13С ЯМР (СDСlз): 171,5, 149,0, 143,5, 140,3, 129,6, 128,6, 128,6, 128,4, 128,1, 126,8, 126,1, 125,4, 116,1, 60,6, 55,5, 53,5, 41 (шир.), 33,9, 30,7, 13,6 (шир.).
Анализ. Вычислено для С30Н37N3О•0,2 С3Н6О:
С 78,66; Н 8,24; N 8,99.
Найдено: C 78,55; Н 7,75; N 8,91.
ПРИМЕР 9
(i) Получение 3-[N-(1-бензилпиперидин-4-ил)амино] -N,N-диэтилбензамида (соединение 10)
Ti(Oi-Pr)4 (0,70 мл, 2,37 ммоль) добавляли к смеси 3-амино-(N,N-диэтил)бензамида (150 мг, 0,78 ммоль) и 1-бензил-4-пиперидона (0,18 мл, 0,97 ммоль) при комнатной температуре. Смесь обрабатывали ультразвуком при 40oС в течение 6 час и перемешивали при комнатной температуре в течение 15 час. Смесь охлаждали на ледяной бане и добавляли EtOH (5 мл) и NaBH4 (75 мг, 1,98 ммоль). После перемешивания в течение 1 час при 0oС и 2 дня при комнатной температуре добавляли 2 М NH4OH (5 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, разбавляли СН2Сl2 (10 мл) и фильтровали через целит®. Слои в фильтрате разделяли, водный слой экстрагировали CH2Cl2 (3•10 мл). Объединенные органические фазы промывали 10% НСl (2•15 мл). рН объединенных органических экстрактов устанавливали до 10 при помощи 2 н. NaOH и экстрагировали СН2Сl2 (3•10 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали, концентрировали и остаток очищали хроматографией
(EtOAc: гептан: Et3N, 9:1:0,1), получая указанное в заголовке соединение 10 (173 мг, 61%) в виде бледно-желтого густого масла.
ИК (неразбавленный): 3333, 1612 см-1.
1H ЯМР (СDСlз): 7,40-7,10 (7Н, м), 6,55 (2Н, м), 3,50 (4Н, м), 3,22 (4Н, м), 2,80 (2Н, м), 2,12 (2Н, м), 2,00 (2Н, м), 1,43 (2Н, м), 1,20 (3Н, м), 1,05 (3Н, м).
13С ЯМР (СDСl3): 171,6, 147,1, 138,3, 138,2, 129,1, 129,0, 128,1, 126,9, 114,6, 113,8, 110,6, 63,0, 52,2, 49,8, 43,1, 38,9, 32,4, 14,1, 12,8.
Анализ. Вычислено для С23Н31N3О• НСl•2,1 Н2О:
С 63,07; Н 8,28; N 9,59.
Найдено: С 63,19; Н 7,94; N 9,25.
(ii) Получение 3-[N-(1-бензилпиперидин-4-ил)анилино]-N,N-диэтилбензамида (соединение 11)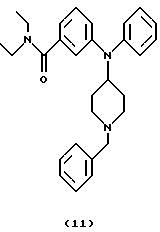
Смесь 3-[N-(1-бензилпиперидин-4-ил)амино]-N,N-диэтилбензамида (соединение 10) (360 мг, 0,98 ммоль), Ph3Bi (1,10 г, 2,50 ммоль) и Cu(OAc)2 (0,45 г, 2,48 ммоль) в толуоле (10 мл) нагревали при 110oС в течение 12 час. Добавляли другую порцию Сu(ОАс)2 (0,45 г) и смесь нагревали при 110 oС в течение дополнительных 12 час и оставляли для охлаждения до комнатной температуры. Добавляли воду (10 мл) и смесь фильтровали через целит®. Слои в фильтрате разделяли и органическую фазу промывали водой, насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Хроматография остатка (EtOAc/гептан, 9:1) давала указанное в заголовке соединение 11 (255 мг, 59%) в виде бледно-желтого густого масла.
ИК (неразбавленный) 3056, 3010, 2937, 2810, 1629 см-1.
1H ЯМР (CDCl3): 7,40-6,60 (14Н, м), 3,80 (1Н, м), 3,43 (4Н, шир. с), 3,20 (2Н, шир. с), 2,90 (2Н, м), 2,05 (2Н, м), 1,90 (2Н, м), 1,48 (2Н, м), 1,20 (3Н, шир.), 1,00 (3Н, шир., с).
13С ЯМР (CDCl3): 171,3, 147,3, 144,4, 138,1, 129,3, 129,0, 128,0, 126,9, 126,4, 123,9, 119,7, 117,5, 116,5, 62,9, 55,3, 53,2, 43,0, 39,0, 30,6, 14,0, 12,7.
Анализ. Вычислено для C29H35N3O•1,25 HC1•0,5 Н2О:
С 70,15; Н 7,41; N 8,47; Cl 8,94.
Найдено: С 69,69; Н 7,34; N 8,25; Cl 8,96.
ПРИМЕР 10
Получение N, N-диэтил-3-(N-пиперидин-4-иланилино)бензамида (соединение 12)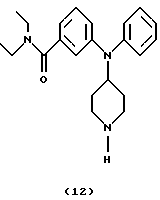
Раствор 3-[N-(1-бензилпиперидин-4-ил)анилино] -N, N-диэтилбензамида (соединение 11) (102 мг, 0,23 ммоль) в EtOH (15 мл) гидрировали при 2,812 атм (40 пси) в присутствии каталитического количества Pd(OH)2 на угле в течение 2 час. Смесь фильтровали через целит® и концентрировали. Остаток очищали хроматографией (EtOAc/гептан/Et3N, 9: 1:0,5), получая указанное в заголовке соединение 12 (50 мг, 81%) в виде бледно-желтого вязкого масла.
ИК (соль НСl, неразбавленная): 3421, 1597, 1494 см-1.
1H ЯМР (СDСl3): 7,80-6,50 (9Н, м), 4,00 (1Н, м), 3,21 (2Н, шир. с), 3,30 (2Н, м), 3,20 (2Н, шир. с), 2,90 (2Н, м), 2,05 (2Н, м), 1,70 (2Н, м), 1,17 (3Н, шир. с), 1,00 (3Н, шир. с).
13С ЯМР (СDСl3): 171,0, 143,7, 140,7, 138,0, 129,5, 129,2, 128,2, 124,5, 119,7, 117,9, 116,5, 53,2, 43,1, 41,1, 39,0, 28,7, 14,0, 8,9.
Аналитический образец получали в виде гидрохлорида добавлением раствора свободного основания в простом эфире к охлажденному льдом разбавленному простым эфиром НС1.
Анализ. Вычислено для С28Н33N3О•НСl•1,3 Н2O:
С 62,30; Н 8,08; N 9,91.
Найдено: С 62,40; Н 7,67; N 9,80.
ПРИМЕР 11
(i) Получение 4-[N-(1-бензилпиперидин-3-ил)амино] -N,N-диэтилбензамида (соединение 13)
Ti(Oi-Pr)4 (2,2 ил, 7,45 ммоль) добавляли к смеси 4-амино-(N,N-диэтил)бензамида (0,36 г, 1,87 ммоль) и 1-бензил-3-пиперидона (0,70 г, 3,69 ммоль) при комнатной температуре. Смесь обрабатывали ультразвуком при 40oС в течение 1 час и перемешивали при комнатной температуре в течение 15 час. Смесь охлаждали на ледяной бане и добавляли EtOH (15 мл) и NaBH4 (0,21 г, 5,55 ммоль). После перемешивания в течение 16 час при комнатной температуре добавляли 2 М NH4OH (15 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, разбавляли СН2Сl2 (10 мл) и фильтровали через слой целита®. Слои в фильтрате разделяли, водный слой экстрагировали СН2Сl2 (3•15 мл). Объединенные органические фазы промывали 10% НС1 (2•20 мл). рН объединенных органических экстрактов устанавливали до 10 при помощи 2 н. NaOH и экстрагировали СН2Сl2 (3•10 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали хроматографией (EtOAc/гептан: Et3N, 9: 1:0,1), получая указанное в заголовке соединение 13 (0,32 г, 47%) в виде бледно-желтого густого масла.
ИК (неразбавленный): 3320, 1736, 1608 см-1.
1H ЯМР (СDСl3): 7,20 (7Н, м), 6,50 (2Н, м), 4,22 (1Н, шир. с), 3,60-3,30 (7Н, м), 2,60 (1Н, м), 2,35 (3Н, м), 1,65 (2Н, м), 1,50 (2Н, м), 1,10 (6Н, м).
13С ЯМР (CDCl3): 171,7, 147,9, 138,1, 128,7, 128,4, 128,1, 126,9, 124,9, 112,1, 63,0, 58,6, 53,5, 48,3, 41,4, 28,6, 22,3, 13,4.
Анализ. Вычислено для С23Н31N3О•НСl•2,1 Н2O:
С 62,81; Н 8,30; N 9,55.
Найдено: С 62,75; Н 7,94; N 9,63.
(ii) Получение 4-[N-(1-бензилпиперидин-3-ил)анилино]-N,N-диэтилбензамида (соединение 14)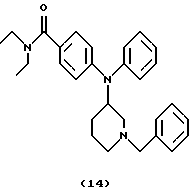
Смесь 4-[N-(1-бензилпиперидин-3-ил)амино]-N,N-диэтилбензамида (соединение 13) (0,29 мг, 0,79 ммоль), Ph3Bi (0,87 г, 1,98 ммоль) и Cu(OAc)2 (0,36 г, 1,98 ммоль) в толуоле (5 мл) нагревали при 110oС в течение 12 час и оставляли для охлаждения до комнатной температуры. Добавляли воду (5 мл) и смесь фильтровали через целит®. Слои в фильтрате разделяли и органическую фазу промывали водой, насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Хроматография остатка (EtOAc/гептан, 9:1) давала указанное в заголовке соединение 14 (0,24 г, 67%) в виде бледно-желтого вязкого масла.
ИК (неразбавленный): 3056, 3012, 2938, 2800, 1613, 1492 см-1.
1H ЯМР (СDСl3): 7,40-7,10 (10 Н, м), 6,95 (2Н, д), 6,55 (2Н, д), 4,05 (1Н, м), 3,45 (2Н, с), 3,38 (4Н, шир. с), 3,19 (1Н), 2,75 (1Н, м), 1,90 (1H, м), 1,80-1,60 (4Н, м), 1,12 (6Н, м).
Аналитический образец получали в виде гидрохлорида добавлением раствора свободного основания в простом эфире к охлажденному льдом разбавленному простым эфиром НС1.
13С ЯМР(СDСl3): 171,4, 148,8, 143,4, 138,2, 129,3, 128,7, 128,6, 128,4, 128,0, 127,8, 126,8, 125,3, 123,9, 115,7, 62,9, 57,2, 54,4, 53,2, 42 (b), 29,8, 25,9, 13,4 (b).
Анализ. Вычислено для С29Н35N3О•1,25 НСl•0,5 H2O:
С 70,15; Н 7,41; N 8,47; Cl 8,94.
Найдено: С 70,30; Н 7,30; N 8,43; Cl 8,34.
ПРИМЕР 12
Получение N, N-диэтил-4-(N-пиперидин-3-иланилино)бензамида (соединение 15)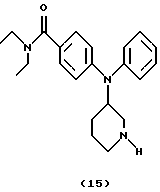
Раствор 4-[N-(1-бензилпиперидин-3-ил)анилино] -N, N-диэтилбензамида (соединение 14) (0,28 г, 0,63 ммоль) в EtOH (10 мл) гидрировали при 2,109 атм (30 пси) в присутствии каталитического количества Pd(OH)2 на угле в течение 6 час. Смесь фильтровали через целит®, концентрировали и остаток очищали хроматографией (градиент, EtOAc/гептан/Et3N, от 9:1:0 до 9:1:0,5), получая указанное в заголовке соединение 15 (80 мг, 36%) в виде бледно-желтого вязкого масла.
ИК (неразбавленный): 3300-3500, 1609, 1464 см-1.
1H ЯМР (СDСl3): 7,35 (2Н, м), 7,18 (3Н, м), 6,95 (2Н, м), 6,56 (2Н, м), 4,30 (1Н, шир. с), 4,0 (1Н, м), 3,38 (4Н, шир. с), 2,95 (1Н, м), 2,35 (2Н, м), 1,95 (1Н, м), 1,70 (2Н, т), 1,15 (2Н, м), 1,10 (6Н, м).
13С ЯМР (CDCl3): 171,3, 148,5, 142,8, 129,3, 128,5, 127,7, 126,4, 125,5, 115,4, 54,1, 49,3, 45,4, 44-38 (b), 29,9, 25,6, 13,3(b).
Анализ. Вычислено для С22Н29N3О•НСl•0,4 H2O:
С 66,87; Н 7,86; N 10,63.
Найдено: С 66,85; Н 7,68; N 10,44.
ПРИМЕР 13
(i) Получение 3-[N-(1-бензилпиперидин-3-ил)амино] -N,N-диэтилбензамида (соединение 16)
Ti(Oi-Pr)4 [6,2 мл, 21,0 ммоль) добавляли к смеси 3-амино-(N,N-диэтил)бензамида (1,0 г, 5,2 ммоль) и 1-бензил-3-пиперидона (2,0 г, 10,6 ммоль) при комнатной температуре. Смесь обрабатывали ультразвуком при 40oС в течение 2 час и перемешивали при комнатной температуре в течение 16 час. Смесь охлаждали на ледяной бане и добавляли EtOH (30 мл) и NaBH4 (0,60 г, 15,9 ммоль). После перемешивания в течение 16 час при комнатной температуре добавляли 2 М NH4OH (25 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, разбавляли CH2Cl2 (25 мл) и фильтровали через слой целита®. Слои в фильтрате разделяли и водный слой экстрагировали CH2Cl2 (3•25 мл). Объединенные органические фазы промывали 10% НСl (2•25 мл). рН объединенных водных экстрактов устанавливали до 10 при помощи 2 н. NaOH и экстрагировали CH2Cl2 (3•25 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали хроматографией (EtOAc/гептан/Et3N, 9: 1: 0,1), получая указанное в заголовке соединение 16 (1,10 г, 58%) в виде бледно-желтого вязкого масла.
ИК (неразбавленный): 3327, 1606, 1440 см-1.
1H ЯМР (СDСl3): 7,40-7,00 (7Н, м), 6,60-6,40 (2Н, м), 4,20 (1Н, шир. с), 3,50 (4Н, м), 3,20 (2Н, шир. с), 2,60 (1Н, м), 2,40-2,20 (3Н, м), 1,70 (2Н, м), 1,50 (2Н, м), 1,20 (3Н, шир. с), 1,00 (3Н, шир. с).
13С ЯМР (СDСl3): 171,4, 146,9, 138,1, 137,9, 128,9, 128,6, 127,9, 126,7, 114,1, 113,5, 110,4, 62,8, 58,5, 53,3, 48,3, 42,9, 38,7, 28,6, 22,2, 13,9, 12,6.
(ii) Получение 3-[N-(1-бензилпиперидин-3-ил)анилино] -N,N-диэтилбензамида(соединение 17)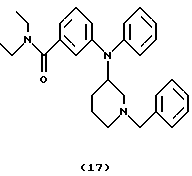
Смесь 3-[N-(1-бензилпиперидин-3-ил)амино]-N,N-диэтилбензамида (соединение 16) (0,25 мг, 0,68 ммоль), Ph3Bi (0,75 г, 1,70 ммоль) и Сu(ОАс)2 (0,31 мг, 1,70 ммоль) в толуоле (5 мл) нагревали при 110oС в течение 14 час и оставляли для охлаждения до комнатной температуры. Добавляли воду (5 мл) и смесь фильтровали через целит®. Слои в фильтрате разделяли и органическую фазу промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали. Хроматография остатка (EtОAc/гептан, 9:1) давала указанное в заголовке соединение 17 (0,16 г, 52%) в виде бледно-желтого вязкого масла.
ИК (неразбавленный): 3010, 2930, 1630, 1610 см-1.
1H ЯМР (СDСl3): 7,40-6,60 (14Н, м), 4,05 (1Н, м), 3,45 (4Н, шир. с), 3,18 (2Н, м), 2,75 (1Н, м), 1,90 (1Н, м), 1,80-1,60 (4Н, м), 1,18 (3Н, шир. с), 1,00 (3Н, шир. с).
13С ЯМР (СDСl3): 171,3, 147,1, 144,5, 138,3, 138,1, 129,3, 129,1, 128,8, 128,1, 126,9, 125,9, 123,7, 119,8, 117,8, 116,9, 63,0, 57,5, 54,4, 53,2, 43,1, 39,0, 29,9, 25,0, 14,1, 12,8.
Анализ. Вычислено для С29Н35N3О195>1,4 HCl•0,5 H2O:
С 69,38; Н 7,46; N 8,37; Cl 9,91.
Найдено С 69,11; Н 7,14; N 8,08; Cl 10,12.
ПРИМЕР 14
Получение N, N-диэтил-3-(N-пиперидин-3-иланилино)бензамида (соединение 18)
Раствор 3-[N-(1-бензилпиперидин-3-ил)анилино] -N, N-диэтилбензамида (соединение 17) (50 мг, 0,11 ммоль) в EtOH (5 мл) гидрировали при 2,109 атм (30 пси) в присутствии каталитического количества Pd(OH)2 на угле в течение 6 час. Смесь фильтровали через целит®, концентрировали и остаток очищали хроматографией (градиент, EtOAc/гептан/Et3N, от 9:1:0 до 9:1:0,5), получая указанное в заголовке соединение 18 (15 мг, 36%) в виде бледно-желтого вязкого масла.
1H ЯМР (CDCl3): 7,40-6,60 (9Н, м), 4,40 (1Н, м), 3,60 (2Н, м),), 3,40 (2Н, шир. с), 3,15 (3Н, м), 2,50 (2Н, м), 1,80 (2Н), 1,20 (2Н, м), 1,18 (3Н, шир. с), 0,95 (3Н, шир. с).
13С ЯМР (CDCl3): 171,2, 146,9, 144,0, 138,0, 129,4, 126,4, 124,3, 119,7, 117,8, 116,5, 54,1, 45,1, 43,1, 39,0, 30,0, 14,0, 12,7.
Аналитический образец получали в виде гидрохлорида добавлением раствора свободного основания в простом эфире к охлажденному льдом разбавленному простым эфиром НСl.
ИК (неразбавленный): 3412, 1598, 1493 см-1.
Анализ. Вычислено для C22H29N3O•НСl•H2O:
С 65,09; Н 7,95; N 10,35.
Найдено: С 65,03; Н 7,80; N 10,02.
ПРИМЕР 15
(i) Получение 4-[N-(1-бензилпирролидин-3-ил)aмино]-N,N-диэтилбензамида (соединение 19)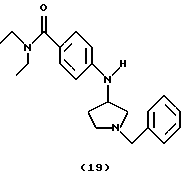
Ti(Oi-Pr)4 (3,1 мл, 10,4 ммоль) добавляли к смеси 4-амино-(N,N-диэтил)бенэамида (0,50 г, 2,51 ммоль) и 1-бензил- 3-пирролидона (0,85 мл, 5,30 ммоль) при комнатной температуре. Смесь обрабатывали ультразвуком при 40oС в течение 3 час и перемешивали при комнатной температуре в течение 16 час. Смесь охлаждали на ледяной бане и добавляли EtOH (30 мл) и NaBH4 (0,30 г, 8,00 ммоль). После перемешивания в течение 16 час при комнатной температуре добавляли 2 М NH4OH (20 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, разбавляли CH2Cl2 (20 мл) и фильтровали через слой целита®. Слои в фильтрате разделяли, водный слой экстрагировали СН2С12 (3•20 мл). Объединенные органические фазы промывали 10% НС1 (2•20 мл). рН объединенных водных экстрактов устанавливали до 10 при помощи 2 н. NaOH и экстрагировали СН2Сl2 (3•20 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали, концентрировали и остаток очищали хроматографией (градиент, ЕtOАс/гептан: Еt3N, от 9:1:0,5 до 9:0:1), получая указанное в заголовке соединение 19 (0,40 г, 44%) в виде бледно-желтого вязкого масла.
ИК (неразбавленный): 3322, 1609, 1527, 1455 см-1.
1H ЯМР (СDСl3): 7,40-7,10 (7Н, м), 6,60-6,40 (2Н, м), 4,20 (1Н, м), 3,95 (1Н, м), 3,55 (2Н, с), 3,35 (4Н, шир. с), 2,70 (2Н, м), 2,50-2,35 (2Н, м), 2,25 (1Н, м), 1,60 (1Н, м), 1,15 (6Н, шир. т).
13С ЯМР (СDСl3): 171,4, 148,2, 138,4, 128,5, 128,2, 128,1, 126,7, 126,1, 113,8, 112,1, 60,5, 59,9, 52,5, 51,9, 41 (b), 32,2, 13,3.
(ii) Получение 4-[N-(1-бензилпирролидин-3-ил)анилино]-N,N-диэтилбензамида (соединение 20)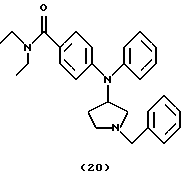
Смесь 4- [N- (1-бензилпирролидин-3-ил) амино] -N, N-диэтилбензамида (соединение 19) (0,40 мг, 1,14 ммоль), Ph3Bi (1,25 г, 2,84 ммоль) и Cu(OAc)2 (0,52 мг, 2,86 ммоль) в толуоле (10 мл) нагревали при 110oС в течение 16 час и оставляли для охлаждения до комнатной температуры. Добавляли воду (5 мл) и смесь фильтровали через целит®. Фильтрат промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали. Хроматография остатка (EtOAc/MeOH, 95: 5) давала указанное в заголовке соединение 20 (0,19 г, 40%) в виде бледно-желтого вязкого масла.
1H ЯМР (СDСl3): 7,40-7,18 (10Н, м), 7,05 (2Н, м), 6,70 (2Н, м), 4,57 (1Н, м), 3,60 (1Н, шир. с), 3,40 (5Н, м), 2,80 (1Н, м), 2,60 (1Н, м), 2,58 (2Н, м), 2,20 (1Н, м), 1,90 (1Н, м), 1,18 (6Н, шир. с).
13С ЯМР (СDСl3): 171,4, 149,3, 145,2, 138,9, 129,4, 128,4, 128,1, 128,0, 127,7, 127,2, 126,7, 125,0, 117,1, 60,3, 58,3, 57,8, 53,0, 41 (b), 29,5, 13,4.
Аналитический образец получали в виде гидрохлорида добавлением раствора свободного основания в простом эфире к охлажденному льдом разбавленному простым эфиром НСl.
ИК (неразбавленный): 3430, 1610, 1457 см-1.
Анализ. Вычислено для C28H33N3O•НСl•1,3 Н2O:
С 68,99; Н 7,24; N 8,62.
Найдено: С 68,99; Н 7,57; N 8,62.
ПРИМЕР 16
Получение N, N-диэтил-3-(N-пирролидин-3-иланилино)бензамида (соединение 21)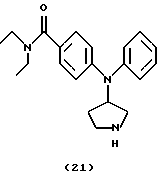
Смесь соединения 20 (90 мг, 0,2105 ммоль), NH4O2CH (27 мг, 0,4282 ммоль) и каталитического количества 10% Pd/C в МеОН (5 мл) энергично перемешивали при комнатной температуре в течение ночи. Катализатор удаляли при помощи целита и фильтрат концентрировали в вакууме, получая сырой образец, который очищали посредством MPLC (СН2С12:МеОН(10% ТФУ), от 100:0 до 9:1, на силикагеле 60), получая указанное в заголовке соединение 21 (30 мг, 42%) в виде бледно-желтого густого масла.
ИК (соль НСl, пленка) v: 3428 (NH), 1607 (CONEt2) см-1.
1H ЯМР (свободный амин, 400 МГц, CDCl3) δ: 1,06 (6Н, м, 2 х  1,90 (1Н, м,
1,90 (1Н, м, 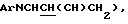 2,30 (1Н, м,
2,30 (1Н, м,  2,58 (2Н, м, ArNCHCH2CH2N), 2,95 (1Н, м,
2,58 (2Н, м, ArNCHCH2CH2N), 2,95 (1Н, м, 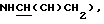 3,23 (1Н, м,
3,23 (1Н, м, 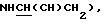 3,40 (4Н, шир. с,
3,40 (4Н, шир. с, 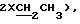 4,70 (1Н, м,
4,70 (1Н, м, 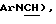 6,68 (2Н, м, Аr), 7,02 (2Н, м, Аr), 7,22 (3Н, м, Аг), 7,38 (2Н, м, Аr).
6,68 (2Н, м, Аr), 7,02 (2Н, м, Аr), 7,22 (3Н, м, Аг), 7,38 (2Н, м, Аr).
13С ЯМР (свободный амин, 100 Гц, СDСl3) d: 13,4, 29,7, 41,9, 54,6, 57,9, 59,3, 117,5, 125,5, 127,9, 128,0, 129,7, 144,8, 149,2, 171,2.
Элементный анализ. Вычислено для C21H29N3OCl2•1,5 Н2O:
С 57,66; Н 7,37; N 9,61.
Найдено: С 57,86; Н 7,38; N 9,03.
ПРИМЕР 17
(i) Получение 4-[N-(1-бензилпиперидин-4-ил)амино] -N, N-диэтилбензолсульфонамида (соединение 22)
Ti(Oi-Pr)4 (2,10 мл, 7,10 ммоль) добавляли к смеси 4-амино-(N,N-диэтил)бензолсульфонамида (0,81 г, 3,55 ммоль) и 1-бензил-4-пиперидона (0,99 мл, 5,32 ммоль) при комнатной температуре. Смесь обрабатывали ультразвуком при 40oС в течение 40 мин и перемешивали при 60oС в течение 18 час. Темную смесь охлаждали на ледяной бане и добавляли EtOH (15 мл), затем гранулы NaBH4 (0,5 г, 13,2 ммоль). После перемешивания в течение 1 час при 0oС и 20 ч при комнатной температуре добавляли 1 М NH4OH (5 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, разбавляли CH2Cl2 (20 мл) и фильтровали через слой целита®. Слои в фильтрате разделяли, водный слой экстрагировали CH2Cl2 (15 мл) и объединенные органические фазы промывали NаНСО3 (водный, насыщенный, 25 мл) и сушили над К2СО3. Смесь фильтровали, концентрировали и остаток очищали хроматографией (градиент от РhМе до Ме2СО), получая указанное в заголовке соединение 22 (0,91 г, 46%) в виде рыжевато-коричневого твердого материала.
ИК (КВr): 2942, 1560, 1520, 1321, 1146, 920 см-1.
1H ЯМР (СDСl3): 7,55 (д, 2Н), 7,34-7,23 (м, 5Н), 6,54 (д, 2Н), 4,08 (д, 1Н), 3,53 (с, 2Н), 3,29 (шир. с, 1Н), 3,17 (к, 4Н), 2,85 (д, 2Н), 2,16 (т, 2Н), 2,01 (д, 2Н), 1,51 (к, 2Н), 1,11 (т, 6Н).
13С ЯМР (СDСlз): 150,2, 138,2, 129,1, 129,0, 128,2, 127,0, 126,8, 63,0, 52,1, 49,6, 41,9, 32,2, 14,1.
(ii) Получение 4-[N-(1-бензилпиперидин-4-ил)анилино]-N,N-диэтилбензолсульфонамида (соединение 23)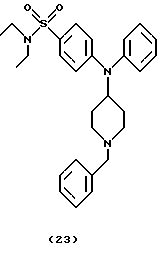
Смесь 4-[N-(1-бензилпиперидин-4-ил)амино] -N,N-диэтилбензолсульфонамида (соединение 22) (0,44 мг, 1,10 ммоль), Рh3Вi (0,58 г, 1,31 ммоль) и Cu(OAc)2 (0,30 мг, 1,64 ммоль) в PhMe (10 мл) нагревали при кипячении с обратным холодильником в течение 24 час. Добавляли Ph3Bi (0,58 г, 1,31 ммоль) и Cu(OAc)2 (0,30 г, 1,64 ммоль). Смесь перемешивали при кипячении с обратным холодильником в течение 24 час и добавляли Ph3Bi (0,58 г, 1,31 ммоль) и Cu(OAc)2, (0,30 г, 1,64 ммоль). После кипячения с обратным холодильником в течение 24 час смесь оставляли для охлаждения и гасили 1 М NH4OH (5 мл). Смесь перемешивали при комнатной температуре в течение 30 мин, разбавляли EtOAc (25 мл) и фильтровали через слой целита®. Слои в фильтрате разделяли, темно-синий водный слой экстрагировали EtOAc (25 мл) и объединенные органические фазы промывали H2O (50 мл) и насыщенным солевым раствором (25 мл) и сушили над К2СО3. Смесь фильтровали, концентрировали и остаток очищали хроматографией (градиент, от РhМе до Ме2СО), получая указанное в заголовке соединение 23 (50 мг, 10%) в виде коричневого масла.
1H ЯМР (CDCl3): 7,51 (д, 2Н), 7,43 (т, 2Н), 7,35 (т, 1Н), 7,30-7,22 (м, 5Н), 7,07 (д, 2Н), 6,48 (д, 2Н), 3,86 (т, 1Н), 3,48 (с, 2Н), 3,18 (к, 4Н), 2,94 (д, 2Н), 2,11 (т, 2Н), 1,91 (д, 2Н), 1,50 (к, 2Н), 1,11 (т, 6Н).
13С ЯМР (CDCl3): 151,8, 141,5, 138,1, 131,1, 129,9, 129,1, 128,6, 128,1, 127,5, 127,0, 125,9, 112,7, 63,0, 55,8, 53,1, 42,0, 30,6, 14,2.
Очистка ВЭЖХ (LiChroPrep RP-18, элюирование с повышающимся содержанием 0,1% ТФУ/MeCN в 1% ТФУ/Н2О) давала аналитический образец в виде белого твердого материала.
ИК (КВr): 3433, 1677, 1586, 1496, 1324, 1196, 1148, 719 см-1.
Анализ. Вычислено для С28Н35N3O2S•1,25 СF3СООН:
С 59,07; Н 5,89; N 6,78.
Найдено: С 59,00; Н 6,01; N 7,01.
ПРИМЕР 18
Получение N, N-диэтил-4-(N-пиперидин-4-иланилино)бензолсульфонамида (соединение 24)
(1-Хлорэтил)хлорформиат (10 мкл, 0,1 ммоль) добавляли к раствору 4-[N-(1-бензилпиперидин-4-ил)анилино] -N, N-диэтилбензолсульфонамида (соединение 23) (19 мг, 40 мкмоль) в толуоле (1 мл) при комнатной температуре. Смесь нагревали при кипячении с обратным холодильником в течение 16 час, оставляли для охлаждения до комнатной температуры и концентрировали. Добавляли метанол (1 мл) и смесь нагревали при кипячении с обратным холодильником в течение 4 час, оставляли для охлаждения и концентрировали. Остаток распределяли между CH2Cl2 (5 мл) и 1 М NH4OH (5 мл). Слои разделяли и органическую фазу промывали Н2О (5 мл) и насыщенным солевым раствором (5 мл) и сушили над К2СО3. Смесь фильтровали, концентрировали и остаток очищали ВЭЖХ (LiChroPrep RP-18, элюирование с повышающимся содержанием 0,1% ТФУ/MeCN в 0,1% ТФУ/Н2О), получая указанное в заголовке соединение 24 (13 мг, 84%) в виде трифторацетата.
ИК (КВr): 3420, 1658, 1199, 1146, 714 см-1.
1H ЯМР (СD3ОD) δ: 7,66-7,63 (м, 4Н), 7,55 (т, 1Н), 7,28 (д, 2Н), 6,76 (д, 2Н), 4,50 (т, 1Н), 3,54 (д, 2Н), 3,35-3,23 (м, 6Н), 2,34 (д, 2Н), 1,73 (к, 2Н), 1,19 (т, 6Н).
13С ЯМР (СD3ОD) δ: 153,4, 142,4, 132,8, 131,7, 130,1, 129,7, 128,8, 114,4, 53,6, 45,2, 43,6, 29,2, 14,9.
Анализ. Вычислено для C28H35N3O2S•2 СF3СООН•1,5 Н2O:
С 46,73; Н 5,33; N 6,54.
Найдено: С 46,54; Н 5,01; N 6,71.
Лучший способ выполнения изобретения, известный в настоящее время, должен использовать соединения примеров 1, 2, 3, 4, 5, 6, 7, 17 и 18.
Фармацевтические композиции
Новые соединения по настоящему изобретению можно вводить перорально, внутримышечно, подкожно, локально, интраназально, внутрибрюшинно, интраторакально, внутривенно, эпидурально, внутриоболочечно, интрацеребровентрикулярно и путем инъекции в суставы.
Предпочтительный путь введения пероральный, внутривенный или внутримышечный.
Дозировка будет зависеть от пути введения, тяжести болезни, возраста и массы пациента и других факторов, обычно рассматриваемых штатным врачом больницы, когда определение индивидуальной схемы приема лекарственного средства и уровня дозы наилучшим образом подходит конкретному пациенту.
Для получения фармацевтических композиций из соединений данного изобретения инертные, фармацевтически приемлемые носители могут быть либо твердыми, либо жидкими. Препараты в твердой форме включают порошки, таблетки, диспергируемые гранулы, капсулы, саше и суппозитории.
Твердым носителем может быть одно или несколько веществ, которые могут действовать также в качестве разбавителей, корригентов, солюбилизаторов, смазывающих веществ, суспендирующих агентов, связующих или агентов, дезинтегрирующих таблетки; им может быть также капсулирующий материал.
В порошках носитель представляет собой тонкоизмельченный твердый материал, который находится в смеси с тонкоизмельченным активным компонентов. В таблетках активный компонент смешивают с носителем, обладающим необходимыми связующими свойствами, в подходящих пропорциях и прессуют для получения необходимой формы и размера.
Для получения композиций в форме суппозиториев низкоплавящийся воск, такой как смесь глицеридов жирных кислот и какао-масла, сначала расплавляют и в нем диспергируют активный ингредиент, например, путем перемешивания. Расплавленную гомогенную смесь затем выливают в формы общепринятого размера и оставляют для охлаждения и затвердевания.
Подходящими носителями являются карбонат магния, стеарат магния, тальк, лактоза, сахар, пектин, декстрин, крахмал, трагакант, метилцеллюлоза, натрийкароксиметилцеллюлоза, низкоплавкий воск, какао-масло и тому подобное.
Фармацевтически приемлемые соли представляют собой ацетат, бензолсульфонат, бензоат, бикарбонат, битартрат, бромид, кальцийацетат, камсилат, карбонат, хлорид, цитрат, дигидрохлорид, этилендиаминтетраацетат, эдисилат, эстолат, эзилат, фумарат, глюкаптат, глюконат, глутамат, гликоллиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, иодид, изотионат, лактат, лактобионат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, памоат (эмбонат), пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, субацетат (основная уксуснокислая соль), сукцинат, сульфат, таннат, тартрат, теоклат, триэтиодид, соль бензатина, хлорпрокаина, холина, диэтаноламина, этилендиамина, меглумина, прокаина, алюминия, кальция, лития, магния, калия, натрия и цинка.
Предпочтительными фармацевтически приемлемыми солями являются гидрохлориды, трифторацетаты и битартраты.
Термин композиция включает готовую форму активного компонента с капсулирующим материалом в качестве носителя, обеспечивающего образование капсулы, в которой активный компонент (с другими носителями или без них) окружается носителем, который, таким образом, находится в ассоциации с ним. Аналогичным образом, включаются саше.
В качестве твердых лекарственных форм, подходящих для перорального введения, можно использовать таблетки, порошки, саше и капсулы.
Композиции в жидкой форме включают растворы, суспензии и эмульсии. В качестве примера жидких препаратов, подходящих для парентерального введения, можно упомянуть растворы активных соединений в стерильной воде или смеси вода-пропиленгликоль. Жидкие композиции можно также изготовить в растворе водного полиэтиленгликоля.
Водные растворы для перорального введения можно получить растворением активного компонента в воде и добавлением подходящих красителей, корригентов, стабилизаторов и загустителей, если необходимо. Водные суспензии для перорального использования можно приготовить диспергированием тонкоизмельченного активного компонента в воде вместе с вязким материалом, таким как синтетические камеди, смолы, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, и другими суспендирующими агентами, известными в области фармацевтических готовых препаративных форм.
Предпочтительно фармацевтические композиции находятся в виде единичной дозы. В такой форме композиция разделена на единичные дозы, содержащие подходящее количество активного компонента. Единичная доза может быть упакованным препаратом, причем упаковка содержит дискретные количества препаратов, например, упакованные таблетки, капсулы и порошки в пузырьках или ампулах. Единичная доза может также представлять собой капсулу, саше или таблетку или она может содержать подходящее число любой из этих упакованных форы.
БИОЛОГИЧЕСКАЯ ОЦЕНКА
A) МОДЕЛЬ IN VITRO
Клеточная культура
Клетки 293S человека, экспрессирующие клонированные рецепторы человека μ, δ, κ и устойчивые в неомицину, выращивали в суспензии при 37oС и атмосфере с 5% СO2 во встряхиваемых колбах, содержащих DMEM (модифицированную по способу Дульбекко среду Игла) без кальция, 10% ФТС, 5% BCS, 0,1% плюроник F-68 и 600 мкг/мл генетицина.
Препарат мембран
Клетки собирали в виде осадка после центрифугирования и ресуспендировали в буфере для лизиса (50 мМ Трис, рН 7,0, 2,5 мМ ЭДТУ с ФМСФ, добавленным непосредственно перед использованием до 0,1 мM из 0,1 М исходного раствора в этаноле), инкубировали на льду в течение 15 мин, затем гомогенизировали политроном в течение 30 сек. Суспензию центрифугировали при 1000 g (max) в течение 10 мин при 4oС. Супернатант сохраняли на льду и осадок ресуспендировали и центрифугировали, как указано ранее. Супернатанты из обоих центрифугирований объединяли и центрифугировали при 46000 g (mах) в течение 30 мин. Осадок ресуспендировали в холодном буфере Трис (50 мМ Трис/Cl, рН 7,0) и центрифугировали снова. Конечный осадок ресуспендировали в буфере для мембран (50 мМ Трис, 0,32 М сахароза, рН 7,0). Аликвоты (1 мл) в полиэтиленовых пробирках замораживали в системе сухой лед/этанол и сохраняли при -70oС до использования. Концентрации белка определяли модифицированным анализом Lowry c ДСН.
Анализы связывания
Мембраны оттаивали при 37oС, охлаждали на льду, пропускали 3 раза через иглу 25 G и разводили в буфере для связывания (50 мМ Трис, 3 мМ MgCl2, 1 мг/мл АБС (Sigma A-7888), рН 7,4, который сохраняли при 4oС после фильтрования через фильтр 22 m и к которому только что было добавлено 5 мкг/мл апротинина, 10 мкМ бестатина, 10 мкМ дипротина А без ДТТ). В охлажденные на льду полипропиленовые пробирки размером 12•75 мм, содержащие 100 мкл подходящего радиоактивного лиганда и 100 мкл испытуемых пептидов при различных концентрациях, добавляли аликвоты 100 мкл.
Общее (ОС) и неспецифическое (НС) связывание определяли в отсутствие и в присутствии 10 мкМ налоксона соответственно. Пробирки встряхивали вортексом и инкубировали при 25oС в течение 60-75 мин, после этого времени содержимое быстро фильтровали в вакууме и промывали около 12 мл/пробирку охлажденного льдом буфера для промывки (50 мМ Трис, рН 7,0, 3 мМ МgСl2) через фильтры GF/B (Ватман), предварительно вымоченные в течение, по меньшей мере, 2 час в 0,1% полиэтиленимине. Радиоактивность (число распадов в минуту), сохранившуюся на фильтрах, измеряли бета-счетчиком после вымачивания фильтров в течение, по меньшей мере, 12 час в мини-склянках, содержащих 6 - 7 мл сцинтилляционной жидкости. Если анализ проводили в планшетах с 96 глубокими лунками, фильтрование выполняли через 96 одинаковых, предварительно вымоченных в ПЭИ фильтров, которые промывали 3•1 мл буфером для промывания и сушили в сушильном шкафу при 55o С в течение 2 час. Радиоактивность пластин фильтров подсчитывали в TopCount (Packard) после добавления 50 мкл сцинтилляционной жидкости MS-20/лунку.
Анализ данных
Специфическое связывание (СС) вычисляли как ОС-НС и СС в присутствии различных испытуемых пептидов выражали как процент СС от контроля. Величины IC50 и коэффициент Хилла (nн) для лигандов в вытеснении специфически связанного радиоактивного лиганда вычисляли из логистических диаграмм или программ для построения кривых, таких как Ligand, GraphPad Prism, SigmaPlot или RecepyorFit. Величины Ki вычисляли по уравнению Cheng-Prussoff. Величины средних значений ± среднеквадратичная ошибка для IC50, Ki и nн приводились для испытуемых лигандов, по меньшей мере, в трех кривых вытеснения.
Эксперименты по насыщению рецепторов
Величины Kδ радиоактивного лиганда определяли путем проведения анализов связывания на клеточных мембранах с подходящими радиоактивными лигандами при концентрациях в диапазоне от 0,2 до 5-кратных концентраций оцененного Kδ (вплоть до 10-кратного, если возможны такие количества требуемого радиоактивного лиганда). Специфическое связывание радиоактивного лиганда выражали как пмоль/мг мембранного белка. Величины Kδ и Вmах из отдельных экспериментов получали из нелинейной зависимости специфически связанного (В) лиганда от нМ свободного (F) радиоактивного лиганда из отдельного эксперимента согласно односайтовой модели.
В) БИОЛОГИЧЕСКАЯ МОДЕЛЬ (МОДЕЛЬ IN VIVO)
МЕХАНОАЛЛОДИНИЯ У КРЫСЫ, ИНДУЦИРОВАННАЯ ПОЛНЫМ АДЪЮВАНТОМ ФРЕЙНДА (ПАФ) И МАНЖЕТКОЙ, ПОМЕЩЕННОЙ НА СЕДАЛИЩНЫЙ НЕРВ
Животные
Использовали самцов крыс Sprague-Dawley (Charles River, St-Constant, Canada), весящих 175-200 г во время хирургической операции. Их поселяли по группам из трех особей в боксах, в которых термостатически поддерживали 20oС и устанавливали суточный цикл освещение/темнота 12:12, причем крысам обеспечивали доступ к корму и воде. После помещения в боксы животных оставляли для акклиматизации в течение, по меньшей мере, 2 дней до хирургической операции. Эксперименты были одобрены Medical Ethical Committee для исследования животных.
ЭКСПЕРИМЕНТАЛЬНАЯ МЕТОДИКА
ПОЛНЫЙ АДЪЮВАНТ ФРЕЙНДА
Крыс сначала анестезировали в камере галотаном, после чего в дорсальную область левой ступни, между вторым и третьим наружными пальцами, подкожно инъецировали 10 мкл ПАФ. Животных затем оставляли для восстановления от анестезии при наблюдении за ними в клетке-камере.
МАНЖЕТКА ДЛЯ СЕДАЛИЩНОГО НЕРВА
Животных готовили по методу, описанному Mosconi and Kruger (1996). Крыс анестезировали внутрибрюшинно смесью кетамин/ксилазин (2 мл/кг) и помещали на их правый бок и делали разрез поперек и вдоль оси боковой стороны левого бедра. Мышцы верхних четырехглавых мышц были препарированы по отдельности при помощи иглы для высвобождения седалищного нерва, вокруг которого помещали пластиковую манжетку (трубка РЕ-60, длина 2 мм). Рану затем зашивали в двух слоях 3-0 викриловыми и шелковыми шовными материалами.
ОПРЕДЕЛЕНИЕ МЕХАНОАЛЛОДИНИИ С ИСПОЛЬЗОВАНИЕМ ИСПЫТАНИЯ ФОН ФРЕЯ
Испытание проводили между 08:00 и 16:00 часами, используя способ, описанный Chaplan et al. (1994). Крыс помещали в клетки из плексигласа на верх основания из проволочной сетки, которая обеспечивала доступ к лапке, и оставляли для привыкания на 10-15 мин. Испытуемой площадью была среднеплантарная часть левой задней лапки, что позволяло избежать
использования менее чувствительных подушечек ступней. Лапку трогали волосками из группы с 8 волосками фон Фрея с логарифмически увеличивающейся жесткостью (0,41, 0,69, 1,20, 2,04, 3,63, 5,50, 8,51 и 15,14 грамм; Stoelting, III, USA). Волосок фон Фрея применяли снизу сетчатого пола перпендикулярно плантарной поверхности с усилием, достаточным для вызывания слабого сгибания волоска относительно лапки, и это усилие поддерживали в течение приблизительно 6-8 секунд. Положительную ответную реакцию отмечали, если лапа была резко отдернута. Вздрагивание сразу после удаления волоска также рассматривали как положительную ответную реакцию. Перемещение крысы рассматривали как неясную реакцию и в таких случаях стимул повторяли.
ПРОТОКОЛ ИСПЫТАНИЯ
Животных испытывали в послеоперационный день 1 для ПАФ-обработанной группы и в послеоперационный день 7 для группы с манжеткой седалищного нерва. 50% порог отдергивания определяли с использованием метода Диксона "вверх-вниз" (1980). Испытание начинали с волоска 2,04 г в середине группы. Стимулы всегда предоставляли последовательным путем, вне зависимости от того, были ли они повышающимися или понижающимися. При отсутствии ответной реакции отдергивания лапы на первоначально выбранный волосок давали более сильный стимул; в случае отдергивания лапы выбирали следующий, более слабый стимул. Вычисление оптимального порога этим способом требовало 6 ответных реакций в непосредственной близости от 50% порога, и подсчет этих 6 ответных реакций начинали, когда имело место первое изменение в ответной реакции, например, когда порог был впервые пересечен. В случаях, когда пороги находились вне пределов стимулов, были назначены величины 15,14 (нормальная чувствительность) или 0,41 (максимальная аллодиния) соответственно. Получаемые результаты положительных и отрицательных ответных реакций были представлены в виде таблицы с использованием стандартного метода, Х - без отдергивания; О - отдергивание, и 50% порог отдергивания интерполировали с использованием формулы
50% порог, г = 10(Xf+kδ)/10000,
где Xf - величина последнего использованного волоска фон Фрея (log-единицы); k - табличная величина (из Chaplan et al. (1994)) для результатов положительных/отрицательных ответных реакций и δ = средняя разность между стимулами (log-единицы). Здесь δ - 0,224.
Пороги фон Фрея были превращены в процент максимально возможного эффекта (% МВЭ) по Chaptan et al., 1944. Для вычисления% МВЭ использовали следующее уравнение:
ВВЕДЕНИЕ ИСПЫТУЕМОГО ВЕЩЕСТВА
Крысам вводили инъекцией (подкожно, внутрибрюшинно или вводили перорально) испытуемое соединение до испытания по методу фон Фрея, время между введением испытуемого соединения и испытания по способу фон Фрея варьировалось в зависимости от природы испытуемого соединения.
В) БИОЛОГИЧЕСКАЯ МОДЕЛЬ (МОДЕЛЬ IN VIVO)
ПОЛНЫЙ АДЪЮВАНТ ФРЕЙНДА (FCA) И ИНДУЦИРОВАННАЯ МАНЖЕТОЙ СЕДАЛИЩНОГО НЕРВА MECHANO-ALLODYNIA У КРЫС
Животные.
Использовали самцов крыс Sprague Dawley (Charles River, St-Constant, Canada) весом 175-200 г во время хирургической операции. Их разделяли на группы по три особи в каждой и помещали в комнаты с поддерживаемой при помощи термостата температурой 20oС, при цикле свет/темнота 12:12 часов и при свободном доступе к пище и воде. По прибытии животным давали акклиматизироваться в течение, по меньшей мере, 2 дней перед хирургической операцией. Эксперименты были одобрены соответствующим Комитетом по медицинской этике для исследования животных.
ЭКСПЕРИМЕНТАЛЬНАЯ ПРОЦЕДУРА
ПОЛНЫЙ АДЪЮВАНТ ФРЕЙНДА
Крыс сначала анестезировали в галотановой камере, после чего им вводили 10 мкл FCA путем подкожной инъекции в тыльную поверхность левой лапы между вторым и третьим внешними пальцами. Затем животным давали отойти от анестезии, наблюдая их в их клетках.
МАНЖЕТА СЕДАЛИЩНОГО НЕРВА
Животных готовили в соответствии с методом, описанным Mosconi and Kruger (1996). Крыс анестезировали смесью кетамина/ксилазина (2 мл/кг, внутрибрюшинно), клали на правый бок и делали продольный разрез по все тыльной стороне левого бедра. Мышцы в верхней четверти отодвигали, раскрывая седалищный нерв, вокруг которого располагали пластмассовую манжету (трубка из ПЭ-60 длиной 2 мм). Затем рану закрывали при помощи наложения 3-0 викрилового и шелкового хирургических швов в два слоя.
ОПРЕДЕЛЕНИЕ MECHANO-ALLODYNIA МЕТОДОМ VON FREY
Испытание проводили между 08:00 и 16:00 часами, используя метод, описанный Chaplan et al. (1994). Крыс помещали в клетки из плексигласа на пол из проволочной сетки, что обеспечивало доступ к лапе, и оставляли их на 10-15 минут. Испытываемым участком была середина подошвы левой задней лапы, избегая при этом менее чувствительные подушечки на лапе. До лапы дотрагивались поочередно 8 волосками фон Фрея (Von Frey) с логарифмически увеличивающейся жесткостью (0,41, 0,69, 1,20, 2,04, 3,63, 5,50, 8,51 и 15,14 грамм; Stoelting, I11, USA). Волосок фон Фрея прикладывали снизу через проволочный пол перпендикулярно поверхности подушки лапы с достаточной силой до небольшой вмятины в лапе и держали примерно 6-8 секунд. Позитивная реакция была отмечена, если было резкое отдергивание лапы. Вздрагивание сразу же после удаления волоска также рассматривалось как позитивная реакция. Двигательную способность рассматривали как сомнительный ответ, и в таких случаях стимулирование повторяли.
ПРОТОКОЛ ИСПЫТАНИЯ
Животных испытывали на первый день после операции (группа, получавшая FCA) и на 7 день после операции (группа с манжетой седалищного нерва). 50% пороговое значение одергивания лапы определяли, используя метод вверх-вниз Диксона (Dixon) (1980). Испытание начинали с использованием волоска 2,04 г, среднего из этой серии. Стимуляцию использовали всегда последовательно, либо по степени возрастания, либо убывания. В отсутствие ответной реакции отдергивания лапы на начально используемый волосок применяли более сильный раздражитель; в случае отдергивания лапы, использовали затем следующий более слабый раздражитель. Для расчета оптимального порогового значения при помощи этого метода требуется 6 ответных реакций, непосредственно приближающихся к 50% уровню, и подсчет этих 6 ответов начинали, когда имело место первое изменение в ответной реакции, например, когда впервые был пересечен пороговый уровень. В тех случаях, когда пороговые значения оставались за пределами диапазона раздражающих стимулов, соответственно брали значения 15,14 (нормальная чувствительность) или 0,41 (максимально allodynic). Полученный образец позитивных и негативных ответов сводили в таблицу, используя преобразование, Х - отсутствие отдергивания; 0 - отдергивание, и 50% пороговое значение отдергивания интерполировали с использованием формулы
50% δ пороговое значение =10(Xfk5)/10000,
где Xf - значение последнего используемого волоска фон Фрея (log единицы); k - табличное значение (из Chaplan et al.(1994)) для модели позитивного/негативного ответов и δ = средняя разница между стимулами (log единицы). Здесь δ=0,224.
Пороговые значения фон Фрея были преобразованы в процентные значения максимально возможного эффекта (%МРЕ), в соответствии с Chaplan et а1., 1994. Для расчета% МРЕ использовали следующее уравнение:
ВВЕДЕНИЕ ИСПЫТЫВАЕМОГО СРЕДСТВА
Крысам инъецировали (подкожно, внутрибрюшинно или вводили перорально) испытываемое соединение до проведения испытания фон Фрея, при этом время между введением испытываемого соединения и испытанием фон Фрея менялось в зависимости от природы испытываемого соединения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2193029C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА ИЛИ ПИПЕРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2194702C2 |
| СПОСОБ ВОССТАНОВЛЕНИЯ НАРУШЕННЫХ ФУНКЦИЙ ОПИОИДНЫХ РЕЦЕПТОРОВ | 1996 |
|
RU2307833C2 |
| ПРОИЗВОДНЫЕ 4-МЕРКАПТОПИРРОЛИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФАРНЕЗИЛТРАНСФЕРАЗЫ | 1996 |
|
RU2191773C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА | 2004 |
|
RU2359971C2 |
| МАКРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2798235C2 |
| ЗАМЕЩЕННЫЕ ДИАЛКИЛ (ОКСИДО)-Λ-СУЛЬФАНИЛИДЕН НИКОТИНАМИД ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2014 |
|
RU2711749C2 |
| АМИДОПРОИЗВОДНЫЕ КАК БЛОКАТОРЫ TTX-S | 2013 |
|
RU2632899C2 |
| НОВЫЕ АКТИВАТОРЫ РАСТВОРИМОЙ ГУАНИЛАТЦИКЛАЗЫ И ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗЫ С ДВОЙНЫМ МЕХАНИЗМОМ ДЕЙСТВИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2758373C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛИДИНОНА В КАЧЕСТВЕ АГЕНТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2002 |
|
RU2288919C2 |




Изобретение относится к азотсодержащим гетероциклическим соединениям ф-лы (I) или их фармацевтически приемлемым солям, или изомерам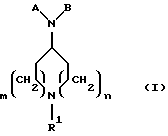
где m равно 0 или 1, n равно 0,
R1 - водород, бензил, (С1-С2-алкил)-(С6-С10-арил), гетероарил-С1 - C2-алкил,
А представляет группу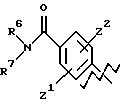
или
где R6, R7, R8, R9 - С1-С6-алкил, Z1 и Z2 - водород, В представляет С6-С10-арил, который может быть замещен СН3 или ОН. Соединения ф-лы (I) обладают аналгезирующей активностью и могут быть использованы в медицине. 4 c. и 6 з.п. ф-лы, 1 табл.

где m равно 0 или 1
n равно 1
R1 выбирают из водорода, бензила или (С1-С2-алкил)-(С6-С10-арила),
А представляет группу гетероарил-С1-2 алкил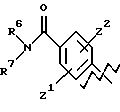
или
где R6, R7, R8 и R9 представляют С1-С6-алкил,
Z1 и Z2 представляют водород,
В представляет С6-С10-арил, который может быть замещен 1 или 2 группами СН3, OH или их фармацевтически приемлемые соли или изомеры.
или
где R6, R7, R8 и R9 представляют С1-С6-алкил,
Z1 и Z2 представляют водород,
В выбирают из фенила, нафтила, которые могут быть замещены 1 или 2 группами СН3, OH.
или
где R6, R7, R8 и R9 представляют C1-С6-алкил,
Z1 и Z2 представляют водород.
где R1, m и n имеют определенные в п. 1 значения, подвергают восстановительному аминированию ариламином формулы (V)
W - NH2 (V),
где W представляет А или В, значения которых определены в п. 1, необязательно в присутствии растворителя, с получением соединения формулы (II)
где R1, m и n имеют определенные в п. 1 значения, (ii) соединение формулы (II), полученное на стадии (i) подвергают арилированию соединением формулы (III)
W-Z (III),
где W представляет А или В, значения которых определены в п. 1,
Z представляет удаляемую группу, необязательно в присутствии катализатора, с получением соединения формулы (I) по п. 1.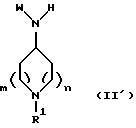
где W представляет А, значения которого определены в п. 1,
R1, m и n имеют определенные в п. 1 значения.
| US 4460586 А, 17.07.1984 | |||
| US 4680296 А, 14.07.1987 | |||
| ПРОИЗВОДНОЕ N-ЗАМЕЩЕННОГО 1-ГЕКСИЛ-4-ФЕНИЛ-4-ПИПЕРИДИНКАРБОКСАМИДА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2039043C1 |

