Настоящее изобретение относится к использованию макроциклических комплексов металлических лигандов, применяемых в качестве отбеливающих катализаторов, в частности к комплексам переходных металлов макроциклических тетраамидов лигандов как катализаторов с целью улучшения протекания реакций окислительного отбеливания.
Соединенные Штаты Америки и Канада являются основными мировыми производителями древесной пульпы, применяемой для изготовления бумаги и картона. В 1983 г. Соединенные Штаты Америки изготовили свыше 41 миллиона метрических тонн пульпы - 39% мирового ее производства. Мировой уровень производства существенно увеличился с тех пор. Пульпа, которая изготавливается либо механическим способом, либо с использованием химических процессов из древесины содержит 1) целлюлозу - линейный полимер d-глюкозы с формулой -(С6Н10О5)-; 2) лигнин - аморфные неоднородные трехмерные молекулы, содержащие в своем составе в основном С9Н8,83O2,37(ОСН3)0,96 и 3) экстрактивные вещества, компоненты древесной пульпы, которые обычно предварительно экстрагируются для использования пульпы при производстве бумаги. См. W.G.Glasser и S.Sarkanen eds. "LIGNIN PROPERTIES AND MATERIALS", American Chemical Society Symposium, Series 397.
Желаемые свойства бумаги характеризуются прочностью, белизной и степенью белизны. Прочность бумаги определяется вязкостью пульпы в процессе производства, которая в свою очередь зависит от свойств целлюлозы после завершения операций по приготовлению пульпы. Молекулярная целлюлоза, как это рассматривалось выше, содержит линейные цепи d-глюкозы, имеющие естественную форму длинных волокон. Увеличение длины отдельных волокон целлюлозы повышает вязкость пульпы и, в свою очередь, обеспечивает высокую прочность бумаги. Поэтому целесообразно исключить возможность проведения процессов обработки полимеров целлюлозы в аппаратах малого объема.
Белизна определяется по внешнему виду путем рассмотрения образца бумаги и оценивается субъективно. Степень белизны измеряется степенью отражения света с длиной волн 475 нм. Чем большая часть падающего на бумагу света отражается от ее поверхности по сравнению с поглощаемой его частью, тем степень белизны бумаги выше.
Степень белизны достигается отбеливанием. Отбеливание пульпы определяется как очистка волокон целлюлозы с использованием химических реагентов для увеличения степени белизны. Отбеливающие реагенты увеличивают степень белизны посредством удаления и обесцвечивания лигнина, содержащегося в составе пульпы. Лигнин может иметь окраску от желтоватой до темно-коричневой в зависимости от вида древесины.
Наиболее часто используемыми отбеливающими химическими реагентами являются такие окислители, как хлор, гипохлорит и диоксид хлора. Кроме того, для этой цели может быть использован газообразный кислород совместно с NaOH, однако такая технология требует дорогостоящего оборудования и используется только в установках большой производительности. Применение кислорода приводит также к потере прочности пульпы вследствие разрушения свободных радикалов, входящих в состав полимеров целлюлозы.
Хлор и гипохлорит могут привести к потере прочностных характеристик при неправильном их использовании, но, как правило, они являются более эффективными и более простыми в использовании окислителями. Применение диоксида хлора позволяет получить бумагу с высоким уровнем степень белизны без разрушения компонентов, входящих в состав пульпы. Однако при использовании всех окислителей на основе хлора образуются побочные хлорсодержащие продукты, оказывающие вредное влияние на окружающую среду и здоровье людей. Кроме того, хлор, например, может легко реагировать с горючими материалами. При реакции с H2S, CO и SO2 происходит образование токсичных и коррозионных газов; в жидкой форме хлор может служить причиной возгорания, образования вздутий и разрушения тканей. Присутствие газообразного хлора вызывает раздражение глаз, носовых каналов и дыхательных путей. Присутствие высоких концентрации хлора может привести к летальному исходу.
Отбеливатели на основе диоксида хлора разлагаются с образованием Cl2, который обладает высокой токсичностью и коррозионными свойствами.
Несмотря на опасность для окружающей среды, хлорсодержащие окислители широко используются для отбеливания пульпы в Соединенных Штатах Америки. Промышленные процессы отбеливания пульпы и бумаги в настоящее время являются сочетанием нескольких методов. Один из часто используемых процессов отбеливания предусматривает в начальной стадии использование хлорирования, затем экстрагирования с применением NaOH, очистку с использованием диоксида хлора, повторное экстрагирование с NaOH и последующую очистку с применением диоксида хлора. Модификация этого процесса предусматривает проведение дополнительных процессов окисления с использованием гипохлорита на этапах между первым экстрагированием NaOH и первичной очисткой с применением диоксида хлора. В другом варианте исключается вторичная экстракция NaOH и повторная очистка диоксидом хлора.
Альтернативным решением отбеливания с использованием хлорсодержащих окислителей является применение перекиси водорода. H2O2 окисляет и делает более белым лигнин и обеспечивает высокую производительность при обработке пульпы. Этот процесс прост в применении и не требует дорогостоящего оборудования. В процессе реагирования H2O2 диссоциирует, образуя ионы пергидроксилов, ООН-, которые обесцвечивают лигнин и не взаимодействуют с целлюлозой. Однако при разложении H2O2 образуются свободные радикалы кислорода •O2 - и гидроксид •ОН, приводящие к измельчению лигнина и разрушению целлюлозы. Несмотря на то, что сама перекись водорода является сильным окислителем и может привести к ожогам кожи и слизистой оболочки, это соединение при низких концентрациях (ниже 8%) не представляет серьезной опасности и его применение не приводит к нарушению состояния окружающей среды. Основным недостатком при использовании H2O2 в качестве окислителя при отбеливании пульпы и бумаги является малая скорость реакции и высокая стоимость. Хотя H2O2 обладает рядом существенных преимуществ с точки зрения охраны окружающей среды, малая скорость отбеливания и высокая стоимость ассоциируется с трудностями ее использования в промышленных масштабах. В промышленных установках обычно используются процессы отбеливания с применением хлора и/или диоксида хлора, применяемые также для очистки стоков.
Некоторые хелаты переходных металлов были исследованы с целью их применения для других целей. Например, комплексы переходных металлов с высокой окислительной способностью известны в качестве окислителей в различных биологических процессах, связанных с их влиянием на протеиновые формы, и в последние годы вызывают интерес при исследовании механизма их взаимодействия и реакционной способности в некоторых процессах монооксидного катализа.
Экспериментальная программа описана в работе Collins T.J., "Designing Ligands for Oxidizing Complexes" Accounts of Chemical Research, 279, Vol. 27, No. 9 (1994). Эта работа посвящена вопросам ориентированного подхода к созданию лигандов, устойчивых к окислительному разрушению, в случае использования центров окисления металлов. Некоторые диамидо-N-дифеноксид- и диамидо-N-алкоксидакриловые хелатные соединения и макроциклические тетраамидо-N-хелатные соединения описаны в статье Collins Accounts of Chemical Research.
Азиды являются основой реакции синтеза при получении макроциклических тетраамидолигандов, o чем приводится в Uffelman E.S. Ph.D. Thesis, California Institute of Technology (1992). Кроме того, синтез арила, соединенного мостиковой связью с тетраамидолигандом, благодаря направленной реакции азидов может протекать при использовании ароматических диаминов в качестве исходного материала.
Однако до сих пор не было установлено, что какие-либо макроциклические тетраамидолиганды могут быть новыми и высокоэффективными отбеливающими активаторами для пероксидных соединений. Кроме того, не изучены, не обнаружены и не рассмотрены возможности использования этих типов соединений для отбеливания пульпы и бумаги.
Настоящее изобретение предусматривает отбеливающий состав, включающий:
(а) окислительный стабильный активатор для процессов отбеливания, имеющий следующую структуру: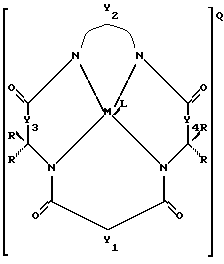
где Y1, Y3 и Y4 представляют собой соединенные мостиковыми связями группы, имеющие ноль, один, два или три узла, содержащие углерод, способные к замещению, a Y2 - группа с мостиковыми связями, имеющая по крайней мере один узел, содержащий углерод, для замещения, причем каждый из этих узлов содержит C(R), C(R1) (R2) или C(R)2 единиц, а каждая замещающая группа R является такой же или отличается от оставшихся R замещенных групп и выбирается из групп, содержащих Н, алкил, циклоалкил, циклоалкенил, алкенил, арил, алкинил, алкиларил, галоген, алкокси- или феноксигруппы, ССН2СF3, СF3 и их комбинации или формы замещенных или незамещенных бензольных колец, в которых два атома углерода в кольце образуют узел в общей точке Y, или совместно с парами R образуют замещающие связи с тем же углеродным атомом, образуя циклоалкильное кольцо, которое может включать другой атом, кроме углеродного, например кольцо циклопентила или циклогексила; М - переходной металл, способный к окислению, выбранный из элементов, образующих периоды I, II, III, IV, V, VI, VII или VIII, или выбранных из групп 6, 7, 8, 9, 10 или 11 Периодической таблицы элементов; Q - любой противоположно заряженный ион, который обеспечивает баланс зарядов соединения на стехиометрическом уровне; L - любой лабильный лиганд; и
(b) эффективное количество источников окислителя.
Пассиваторы, стабилизаторы и другие стандартные широко известные отбеливающие компоненты для пульпы и бумаги могут добавляться в состав этих смесей.
Предпочтительными отбеливающими активаторами являются макроциклические тетраамидные соединения. Среди них могут быть соединения, имеющие замещенные ароматические заместители, непосредственно введенные в лиганды циклической структуры.
Например, одна из предлагаемых структур может иметь вид: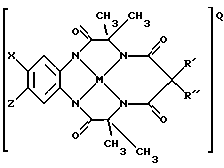
где Х и Z могут быть представлены Н, группами электронодоноров или акцепторов электронов, a R' и R" - любыми комбинациями Н, алкила, циклоалкила, циклоалкенила, алкенила, арила, алкинила, алкиларила, галогена, алкокси- или феноксизаместителей или включать сочетания колец циклоалкила или циклоалкенила, которые могут содержать по крайней мере один неуглеродный атом; М - переходной металл, способный к окислению, из элементов, образующих периоды I, II, III, IV, V, VI, VII или VIII, или выбранный из групп 6, 7, 8, 9, 10 или 11 Периодической таблицы элементов; Q - любой противоположно заряженный ион, который обеспечивает баланс зарядов соединения на стехиометрическом уровне.
Быстрый рост производства пульпы и бумаги и увеличение надежности процессов химического отбеливания для производства осветленных, прочных бумажных изделий приводит к увеличению сбросов хлорсодержащих побочных продуктов в окружающую среду. Промышленность нуждается в безопасной альтернативе хлорсодержащим окислителям для отбеливания. Так, необходим способ отбеливания пульпы, обеспечивающий снижение выбросов токсичных соединений в окружающую среду. Кроме того, необходима простая технология с использованием нетоксичных методов, предусматривающая возможность изготовления осветленной, прочной бумаги.
Каталитический активатор, в соответствии с настоящим изобретением, как это было определено, соответствует этим специфическим требованиям. Этот активатор, как это показано, значительно увеличивает скорость отбеливания щелочного лигнина благодаря введению перекиси водорода. Более того, активатор в соответствии с описываемым изобретением, как это показано, обладает высокой стабильностью.
Существует потребность в новом отбеливающем активаторе, который бы удовлетворял требованиям стабильности катализатора в буферных растворах.
Кроме того, необходим также новый отбеливающий активатор, который может быть использован в стехиометрических количествах по отношению к окисляемым соединениям.
Настоящее изобретение предусматривает использование способа отбеливания пульпы, бумаги и других материалов на основе целлюлозы, включающий этапы контактирования таких материалов в непрерывных или периодических процессах с источником окислителя, предпочтительно с пероксидными соединениями, а более предпочтительно с перекисью водорода и/или продуктами ее диссоциации и катализаторами, или близко к стехиометрическим количествам отбеливающего активатора из соединений, рассмотренных ранее. Этот способ также предусматривает возможность введения пассиватора для защиты пероксидных соединений от взаимодействия с малыми количествами металлов, которые могут создать возможность ненужного разрушения этих смесей.
Предлагаемый способ может быть использован при различных температурах, однако предпочтительно, чтобы температуры находились в интервале от окружающей до 80oС, более предпочтительно в интервале от окружающей до 40oС. Однако температура процесса не является критической величиной. Возможно проведение предлагаемого процесса в широком диапазоне температур.
Предпочтительно, чтобы интервал рН находился в пределах от 7 до 11, а предпочтительнее в интервале от 9 до 11.
Предлагаемый активатор может быть использован в других процессах в качестве улучшенного активатора окислительных реакций, протекающих в растворах вообще и, в частности, как активатор для проведения процесса с использованием окислителей с прочными связями О-атомов, таких как перекись водорода, трет-бутилгидропероксид, гидроперекись кумола, гипохлорит и перкислоты; предпочтительное использование предлагаемого способа предусматривает применение в качестве активатора пероксидных соединений, а более предпочтительно применение активатора в виде перекиси водорода при отбеливании пульпы и бумаги. Предлагаемый состав улучшает окислительную способность перекиси водорода путем эффективного использования этого улучшенного промышленного окислителя, оказывающего незначительное влияние на окружающую среду.
Значение применения новой технологии для защиты окружающей среды трудно переоценить. Многие тысячи тонн вредных для окружающей среды и высокотоксичных соединений, таких как мутагенные и канцерогенные побочные продукты, не образуются при этом процессе. Способ, предлагаемый в настоящем патенте, полностью или частично исключает использование хлорсодержащих отбеливающих окислителей и образование токсичных побочных продуктов, возникающих при их использовании.
Фиг. 1 представляет график, подтверждающий активность соединений, предлагаемых в настоящем патенте, в случае их введения совместно с перекисью водорода в образцы лигнина по сравнению с вариантом использования только перекиси водорода.
Фиг. 2 представляет спектр поглощения щелочного лигнина под воздействием ультрафиолетового излучения и при непосредственном наблюдении.
Фиг. 3 представляет протекание процесса синтеза макроциклических тетраамидолигандов с использованием реакции азидов.
Фиг. 4 представляет протекание процесса синтеза макроциклических тетраамидолигандов в соответствии с настоящим изобретением с применением реакции азида и использованием в качестве исходного материала ароматического диамина.
Фиг. 5 представляет собой график, позволяющий сравнить степень стабильности катализатора в соответствии с предлагаемым изобретением и контрольного состава.
Изобретение предлагает отбеливающую композицию, которая включает:
(а) окислительный стабильный отбеливающий активатор, имеющий химическую структуру: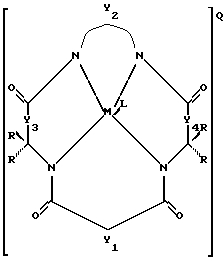
где Y1, Y3 и Y4 представляют собой соединенные мостиковыми связями группы, имеющие ноль, один, два или три узла, содержащие углерод, способные к замещению, a Y2 - группа с мостиковыми связями, имеющая по крайней мере один узел, содержащий углерод, для замещения, причем каждый из этих узлов содержит C(R), C(R1) (R2) или C(R)2 единиц, а каждая замещающая группа R является такой же или отличается от оставшихся R замещенных групп и выбирается из групп, содержащих Н, алкил, циклоалкил, циклоалкенил, алкенил, арил, алкинил, алкиларил, галоген, алкокси- или феноксигруппы, СН2СF3, СF3 и их комбинации или формы замещенного или незамещенного бензольного кольца, в котором два атома углерода в кольце образуют узел в общей точке Y, или совместно с парой R образуют замещающие связи с тем же углеродным атомом, образуя циклоалкильное или циклоалкенильное кольцо, которое может включать другой атом, кроме углеродного, например кольцо циклопентила или циклогексила; М - переходной металл, способный к окислению из элементов, образующих периоды I, II, III, IV, V, VI, VII или VIII, или выбранный из групп 6, 7, 8, 9, 10 или 11 Периодической таблицы элементов; Q - любой противоположно заряженный ион, который обеспечивает баланс зарядов соединения на стехиометрическом уровне; L - факультативно присутствующий какой-нибудь лабильный лиганд,
(b) достаточное количество источников окислителя.
Среди таких структур предпочтение предлагается отдать макроциклическим тетраамидолигандам, которые высоко эффективны в различных группах, обеспечивающих улучшенные характеристики отбеливающих активаторов.
Эти лиганды приготавливаются в соответствии с процедурой, представленной на фиг. 3 или 4, и предпочтительно включают разновидности лигандов, описанных подробно в патентной заявке Соединенных Штатов Америки Serial No 08/681237, представленной Collins et al. 22 июля 1996 г. под названием LONG-LIVED HOMOGENOUS OXIDATION CATALYSTS и используемой в этом патенте в качестве ссылки.
1. Макроциклические тетраамидолиганды
Патентуемые химические соединения имеют структуру: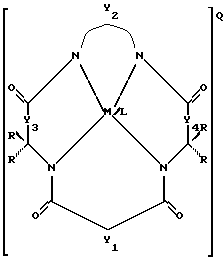
где Y1, Y3 и Y4 представляют собой соединенные мостиковыми связями группы, имеющие ноль, один, два или три узла, содержащие углерод, способный к замещению, a Y2 - группа с мостиковыми связями, имеющая по крайней мере один узел, содержащий углерод, для замещения, причем каждый из этих узлов содержит C(R), C(R1) (R2) или C(R)2 единиц, а каждая замещающая группа R является такой же или отличается от оставшихся R замещенных групп и выбирается из групп, содержащих Н, алкил, циклоалкил, циклоалкенил, алкенил, арил, алкинил, алкиларил, галоген, алкокси- или феноксигруппы, СН2СF3, СF3 и их комбинации или формы замещенного или незамещенного бензольного кольца, в котором два атома углерода в кольце образуют узел в общей точке Y, или совместно с парой R образуют замещающие связи с тем же углеродным атомом, образуя циклоалкильное или циклоалкенильное кольцо, которое может включать другие атомы, кроме углеродных, например кольца циклопентила или циклогексила; М - переходной металл, способный к окислению и состоящий из элементов, образующих периоды I, II, III, IV, V, VI, VII или VIII или выбранных из групп 6, 7, 8, 9, 10 или 11 Периодической таблицы элементов; Q - любой противоположно заряженный ион, который обеспечивает баланс зарядов соединения на стехиометрическом уровне; L - любой лабильный лиганд.
Особенно предпочтительная разновидность структуры предлагаемого макроциклического тетраамидного соединения в соответствии с изобретением подставлена ниже: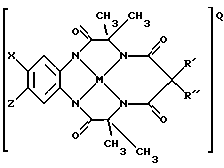
где Х и Z могут быть представлены Н, электронодонорной группой или группой акцепторов электронов, a R' и R" - любыми комбинациями Н, алкила, циклоалкила, циклоалкенила, алкенила, арила, алкинила, алкиларила, галогена, алкокси- или феноксизаместителей или включать сочетания кольца циклоалкила или циклоалкенила, которое может содержать по крайней мере один неуглеродный атом; М - переходной металл, способный к окислению из элементов, образующих периоды I, II, III, IV, V, VI, VII или VIII, или выбранный из групп 6, 7, 8, 9, 10 или 11 Периодической таблицы элементов; Q - любой противоположно заряженный ион, который обеспечивает баланс зарядов соединения на стехиометрическом уровне.
Х и Z группы могут быть Н или электронодонорными группами, или группами акцепторов электронов. В группу акцепторов электронов входят галогены, такие как Вr, I и предпочтительно Cl-. Кроме того, к соответствующим группам относятся SO3 -, OSO3 -, OSО3R (R может включать без ограничений такие составляющие, как Н, алкил, арил, алкиларил) и NO2 -. В электронодонорную группу входят алкоксисоединения (без ограничений, метокси-, этокси-, пропокси- и бутоксисоединения), алкилы (без ограничений, метил, этил, пропил, н-бутил и трет-бутил), а также водород. Эти группы изменяют плотность электронов комплексов металлических лигандов и влияют на их реакционную способность.
R' и R" должны способствовать поддержанию каталитической стабильности рассматриваемых макроциклических тетраамидолигандов. Хотя каждый из них может выбираться индивидуально из числа Н, алкилов, алкенилов, арилов, алкинилов, галогенов, алкокси- или феноксизамещающих групп, однако предпочтение следует отдавать алкилам с короткими цепями. Наиболее предпочтительно, когда R' и R" являются теми же самыми и являются этилом или метилом или когда R' и R" комбинируются, образуя кольцо циклоалкила и циклоалкенила, в частности циклопентила или циклогексила. Циклоалкильное кольцо может включать по крайней мере один атом, отличающийся от углеродных, такой как, без ограничений, N, О или S. Наиболее предпочтительными и устойчивыми являются разновидности соединений, где R' и R" совпадают и выбраны из групп, содержащих метил, CF3, водород, галоген, и пять членов кольца образуют совместную структуру с атомом углерода, непосредственно связанным с этим атомом. Эти последние группы являются интерактивными, образуя прочные связи с углеродом кольца, пространственно-затрудненные и/или конформационно-затрудненные таким образом, что ограничена внутримолекулярная реакция окислительного разрушения.
Металл М является переходным металлом, способным к окислению из периодов I, II, III, IV, V, VI, VI, VII или VIII или может быть выбран из групп 6 (Cr, Mo, W), 7 (Мn, Тc, Re), 8 (Fe, Ru, Os), 9 (Co, Rh, Ir), 10 (Ni, Pd, Pt) и 11 (Сu, Аg, Аu). Наиболее предпочтительно выбирать такой металл из группы, включающей Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn (Группа 12), Mo и W.
Q - любой противоположно заряженный ион, который обеспечивает баланс зарядов структуры на стехиометрическом уровне. Могут быть использованы как положительно, так и отрицательно противоположно заряженные ионы. Предпочтительно, но не обязательно, выбирать противоположно заряженные положительные ионы из ряда противоположно заряженных щелочных металлов (напр. К, Li, Na), NR*4 и PR*4, где каждый R* индивидуально выбирается из Н, алкилов, арилов, алкиларилов, алкенилов или может соединяться вместе, образуя кольца с циклоалкилами или циклоалкенилами или арилами, причем эти кольца могут содержать по крайней мере один неуглеродный атом. В основном, но не обязательно, отрицательно заряженный противоион выбирается из числа BF4 -1 и PF6 -1.
L - любой лабильный лиганд, способный присоединяться к М. К таким лигандам относятся, в основном, но не обязательно, H2O, Cl и С=N.
В связи со сложной природой таких соединений, в данном случае они не называются, однако для удобства указывается на возможность их использования из числа перечисленных. Вышеуказанная структура, например, может быть обозначена как 5,6-(4,5-Di-Х-бензо)-3,8,11,13-тетраоксо-2,2,9,9-тетраметил-12,12-диэтил-1,4,7,10-тетраазациклотридекан (или тетраметил-диэтил di-X-бензол (TMDE-DXB, где X = Cl, H, Me, OMe)). Так, для удобства, указанная выше структура, в состав которой входят две метиловые группы, связанные с аминами лиганда, и две этиловые группы, обозначенные как R' и R", относится к соединению TMDE. Если R' и R" - метиловые группы, то это соединение отнесено к TMDM. Когда обе группы Х и Z являются хлором, такое соединение относится к DCB. Предпочтительным переходным металлом лигандов является железо, так что такое соединение может быть отнесено к группе Fe-DCB.
Обычно способ отбеливания с применением перекиси водорода предусматривает проведение этого процесса при рН в пределах от 11 до 9 и в интервале температур от 30 до 80oС, но предпочтительно в интервале от 50 до 70oС. См. Charles, J.E. et al., 1980, TAPPI Pulping Conference Proceedings, TAPPI Press (1980). При использовании активатора в соответствии с настоящим изобретением температура реакции может быть понижена до окружающей температуры. Хотя этот каталитический активатор может быть применен и при более высоких температурах реакции, он также может успешно применяться и при температурах 35oС и 40oС. Известно, что при изменении температуры приблизительно на каждые 10oС, скорость реакции изменяется почти вдвое. Поэтому скорость реакции значительно возрастает с повышением температуры. Однако, если применяется отбеливание пульпы с использованием рассматриваемого активатора, скорость окисления с применением H2O2 значительно увеличивается по сравнению с ранее достигнутой и обеспечивается проведение реакции при более низких температурах, благодаря чему достигается существенная экономия энергии на проведение этого процесса и увеличивается производительность процесса вследствие того, что он протекает при более низкой температуре, чем это осуществлялось ранее. Поэтому предпочтительно вести процесс при температурах в пределах от окружающей до 80oС, а лучше в интервале от окружающей до 70oС и еще лучше от окружающей до 40oС. Рассматриваемая система отбеливания обеспечивает возможность эффективной работы даже при температурах ниже окружающей. Широкий интервал температур, в котором каталитический активатор может быть использован, позволяет применять его на существующем оборудовании и в связи с другими процессами отбеливания пульпы и бумаги и использовать его без специальных изменений в температурном режиме для пероксидных отбеливателей на промышленных линиях без внесения изменений, связанных с понижением температуры.
Уровень рН окислительной реакции также может быть понижен при использовании рассматриваемого активатора. Эксперименты по проведению процесса отбеливания при рН 7 с использованием H2O2 и рассматриваемым каталитическим активатором позволяют надеяться на возможность отбеливания лигнина с повышенной скоростью по сравнению с проведением этого процесса с применением H2O2, однако не с наилучшей скоростью, возможной для данного активатора. Дальнейшее увеличение скорости и получения удовлетворительных результатов было достигнуто при увеличении рН до 10. Так, обычный диапазон рН от 11 до 9 не может быть изменен при добавлении рассматриваемого каталитического активатора, но может быть использован при необходимости с целью снижения степени разложения H2O2, которая, как известно, протекает при высоких рН. Степень разложения может быть также связана с присутствием переходных металлов в отбеливающем растворе, содержащем пероксидные соединения. Пассиваторы и другие известные стабилизаторы используются для снижения вероятности разложения, вызванного присутствием переходных металлов. Эксперименты, приведенные ниже, показали, что возможно также использование пассиваторов совместно с рассматриваемым каталитическим активатором.
Следует надеяться, что отбеливание с использованием предлагаемого способа будет способствовать благоприятному числу каппа - меры, используемой в промышленных процессах отбеливания пульпы и бумаги и определяющей количество остаточного лигнина после отбеливания. Число каппа, которое должно быть по возможности минимальным, представляет собой отношение разницы между (1) полным окислительным эквивалентом, необходимым для 100% удаления лигнина и (2) разностью между действительно достигнутым уровнем окисления и полным эквивалентом окисления. Он определяется путем исследований с применением марганцовокислого калия в соответствии с известной методикой, используемой в промышленности при производстве пульпы и бумаги.
В связи с тем, что рассматриваемый макроциклический тетраамидолиганд действительно является катализатором или каталитическим активатором, количество этого добавляемого соединения обычно поддерживается на близком стехиометрическом уровне. Однако предпочтительно, хотя и не обязательно, вводить добавки в количестве от 0,0001 до 999,999 частей на миллион (ppm), a более предпочтительно от 0,001 до 100000 ppm, в состав рассматриваемого соединения.
Экспериментальный раздел, приводимый ниже, описывает некоторые предпочтительные виды синтеза рассматриваемого макроциклического тетраамидного соединения. Кроме того, были выполнены дополнительные испытания с целью продемонстрировать отбеливание лигнина и определить каталитическую активность рассматриваемого макроциклического лиганда.
2. Окислительные соединения
Окислительные соединения, такие как О-передающие атомы, в основном пероксидные соединения, могут быть органическими или неорганическими соединениями, содержащими -О-О-пероксидные связи. Примерами таких соединений могут быть перекись водорода, аддукты перекиси водорода, соединения, способные образовывать перекись водорода в водных растворах, органические перекиси, персульфаты, перфосфаты и персиликаты. Аддукты перекиси водорода включают щелочные металлы (т. е. натрий, литий, калий), карбонаты пероксигидратов и пероксид мочевины. Вещества, способные образовывать перекись водорода в водных растворах, включают щелочные металлы (натрий, калий, литий), пербораты (моно- и тетрагидраты). Пербораты производятся в коммерческих масштабах и могут быть получены от таких фирм, как Akzo N.V. и FMC Corporation. Соответственно, фермент спиртовой оксидазы и подходящий спиртовой субстрат могут быть использованы в качестве источника перекиси водорода. Органические пероксиды включают, без ограничений, бензоил и гидроперекись кумола. Персульфаты включают пероксимоносульфат калия (торговое название Oxone®, E.I. duPont de Nemours) и Caro's кислоту.
Эффективное количество пероксидных соединений - такое их количество, которое способно выделить по крайней мере 0,001 ppm активного кислорода (А.О. ). Предпочтительно, но не обязательно, чтобы количество получаемого А.О. находилось где-то в пределах от 0,001 до 1,000 ppm A.O. Для промышленного отбеливания предпочтительно использовать концентрации в пределах от приблизительно 0,01 до 50 ppm A.O. Описание и объяснение A.O. измерений приводится в статье Sheldon N. Lewis, "Peracid and Peroxide Oxidation" в Oxidation, 1969, pp. 213-258, которая используется здесь в качестве ссылки.
3. Составы
Рассматриваемый макроциклический тетраамидолиганд при необходимости может быть смешан с различными добавками или основой, которая включает основной компонент и поверхностно-активные вещества, выбираемые из группы, состоящей из анионных, неионных, катионных, амфотерных, цвиттерионных поверхностно-активных веществ, а также их смесей. Кроме указанных веществ могут быть использованы другие виды соединений. Эти материалы могут быть применены в жидкой форме для отбеливания твердых или других видов поверхностей. Такие соединения могут использоваться в процессах отбеливания пульп и текстильных материалов. Каждая из этих смесей и соответствующие составы, пригодные для использования, рассматриваются ниже.
а. Компоненты
В качестве основного компонента обычно используются щелочные материалы, т. е. такие, которые в составе водных растворов имеют рН от 7 до 14, предпочтительно от 9 до 12. Примерами неорганических компонентов могут служить карбонаты щелочных металлов и аммония (включая двойные кислые и средние углекислые соли и бикарбонаты), фосфаты (включая ортофосфаты, триполифосфаты и тетрапирофосфаты), алюмосиликаты (как природные, так и синтетические цеолиты) и их смеси. Предпочтительно рекомендуется использовать карбонаты для применения в составе рассматриваемого изобретения из-за их высокий щелочности и эффективности при удалении ионов жесткости, которые могут присутствовать в составе используемой воды, и учитывая их невысокую цену. В качестве предпочтительной основы может быть рекомендовано использование карбонатов. Кроме того, могут быть использованы силикаты (Na2O: SiO2 с модулем от 4: 1 до 1:1, а более предпочтительно с модулем от 3:1 до 1:1). Силикаты, рекомендуются к использованию в указанных составах благодаря их высокий растворимости в воде и способности образовывать стекловидные структуры, обеспечивающие связывание компонентов смеси.
Органические компоненты также могут быть использованы для указанной цели и выбираются из групп, содержащих сульфосукцинат щелочного металла и аммония, полиакрилаты, полималеаты, сополимеры акриловой и малеиновой кислот или малеиновой кислоты или малеинового ангидрида, цитраты и их смеси.
b. Наполнители/разбавители
Наполнители для отбеливающих составов служат для создания соответствующего количества или дозирования осветляющей активности при их использовании. Предпочтительно использовать соли, такие как NaCl, Na2SО4 и буру. Также могут быть использованы органические разбавители, такие как сахароза. В случае применения жидких форм могут быть использованы растворители (без ограничения, такие как алканолы, гликоли, гликолевые эфиры, углеводороды, кетоны и карбоновые кислоты), жидкие поверхностно-активные вещества и вода, применяемые также в качестве разбавителей.
с. Поверхностно-активные вещества
Поверхностно-активные вещества обычно добавляются в отбеливатель с целью удаления примесей частиц почвы, например неионные поверхностно-активные вещества обеспечивают очистку замасленного субстрата, а анионные поверхностно-активные вещества используются для удаления макрочастиц субстратов. Хотя, в общем случае, окислительный отбеливающий состав может содержать малое количество или вообще не содержать поверхностно-активные вещества.
Наиболее эффективными из поверхностно-активных веществ следует считать анионные поверхностно-активные вещества. Примерами таких веществ могут служить аммонийные, замещенные аммонийные (например, моно-, ди- и триэтаноламмоний), и соли щелочных, щелочноземельных металлов и С6-C20 жирных и смоляных кислот, линейные и разветвленные алкилбензолсульфонаты, алкилсульфонаты, алкилсульфоэфиры, алкансульфонаты, сульфоолефины, сульфонаты оксиалканов, сульфаты моноглицеридов жирных кислот, сернокислые эфиры алкилглицерина, ацилсаркозинаты и ацил-N-метилтауриды. Предпочтительно использовать поверхностно-активные вещества, содержащие алкиларилсульфонаты, такие как алкилбензолсульфонаты.
Другими предпочтительно применяемыми поверхностно-активными веществами являются линейные оксиэтилированные спирты, такие как выпускаемые Shell Chemical Company с торговым наименованием NEODOL. Другие соответствующие неионные поверхностно-активные вещества могут включать другие линейные оксиэтилированные спирты со средней длиной от 6 до 16 углеродных атомов и содержащие в среднем от 2 до 20 молей окиси этилена на моль спиртов; линейные и разветвленные первичные и вторичные оксиэтилированные и оксипропилированные спирты со средней длиной от 2 до 6 углеродных атомов и содержащие в среднем 0-10 молей окиси этилена и от примерно 1 до 10 молей окиси пропилена на моль спиртов; линейные или разветвленные алкилфенокси-(полиоксиэтилированные) спирты, известные также как оксиэтилированные алкилфенолы со средней длиной цепи от 8 до 16 углеродных атомов и в среднем содержащие от 1,5 до 30 молей окиси этилена на моль спиртов, а также их смеси. Еще один вид неионных поверхностно-активных веществ может включать полиоксиэтиленовые эфиры карбоновых кислот, глицериновые эфиры жирной кислоты, жирные кислоты и оксиэтилированные алканоламиды жирных кислот, некоторые блок-сополимеры окиси пропилена и окиси этилена, а также блок-полимеры окиси пропилена и окиси этилена с оксипропилированным этилендиамином. В состав указанных поверхностно-активных веществ могут быть также включены такие полуполярные неионные поверхностно-активные вещества, как аминоксиды, окиси фосфинов, сульфоксиды и другие оксиэтилированные производные.
Соответствующие катионные поверхностно-активные вещества могут включать четвертичные аммониевые соединения, типичным представителем которых являются те группы, которых связанны с атомом азота, содержащие C12-С18 алкильные группы, в то время как три другие группы являются алкильными группами с короткой цепью, которые могут быть замещены фенильными группами.
Далее, соответствующие амфотерные и цвиттерионные поверхностно-активные вещества, которые содержат анионные водорастворимые группы, катионные группы и гидрофобные органические группы, могут включать аминокарбоновые кислоты и их соли, аминодикарбоновые кислоты и их соли, алкилбетаины, алкиламинопропилбетаины, сульфобетаины, производные алкилимидазолинов, некоторые четвертичные аммониевые соединения, некоторые четвертичные фосфониевые соединения и некоторые третичные сульфониевые соединения.
Дальнейшие примеры анионных, неионных, катионных и амфотерных поверхностно-активных веществ, которые могут быть использованы для целей этого изобретения, описаны в Kirk-Othmer, Encyclopedia of Chemical Technology. Third Edition, Volume 22, pages 347 - 387, и в McCutcheon's Detergents and Emulsifiers, North American Edition, 1983, которые являются ссылками настоящего документа.
d. Хелатобразующие соединения
В некоторые составы (композиции), перечисленные ниже, специально желательно вводить хелатобразующие соединения, предпочтительно аминополифосфонаты. Эти хелатные хелатобразующие соединения обеспечивают стабильность растворов окислителей с целью получения оптимальных условий проведения реакции. Эти вещества воздействуют на свободные ионы тяжелых металлов хелатных соединений. Такие хелатобразующие соединения выбираются из числа известных соединений, эффективных при образовании хелатных соединений свободных ионов тяжелых металлов. Хелатобразующие соединения должны быть устойчивы к гидролизу и быстрому окислению окислителями. Предпочтительно, чтобы их коэффициент кислотной диссоциации (рКа) находился в пределах 1-9 и позволял оценить степень диссоциации при низкой величине рН с целью определения величины прочности связей с катионами металлов. Наиболее предпочтительным хелатобразующими соединением являются аминополифосфонаты, выпускаемые под торговым названием DEQUEST фирмой Monsanto Company. Примерами таких соединений могут служить DEQUEST 2000, 2041, 2060 и 2066. Могут также использоваться полифосфонаты, такие как DEQUEST 2010. Другие хелатобразующие соединения, такие как этилендиаминтетрауксусная кислота (EDTA) и нитрилотриуксусная кислота (NTA), также могут быть использованы в рассматриваемом процессе. Еще одним предпочтительным новым хелатобразующим соединением являются новые пропилендиаминтетраацетаты, такие как Hampshire 1,3 PDTA, изготавливаемые W.R.Grace, и Chel DTPA 100#F, изготавливаемые Ciba-Geigy A.G. Могут также использоваться смеси указанных выше соединений. Эффективное количество хелатобразующих соединений обычно составляет от 1 до 1000, предпочтительно от 5 до 500 и более предпочтительно от 10 до 100 ppm, хелатобразующих соединений в моющем растворе.
е. Другие компоненты
Стандартные компоненты окислительных отбеливающих составов могут быть включены в настоящее изобретение.
В их числе ферменты, являющиеся особенно желательными для использования в составе окислительных отбеливателей. Однако в их число следует включить стабилизаторы ферментов.
Протеазы являются одним из особенно предпочтительных классов ферментов. Они выбираются из кислотных, нейтральных или щелочных протеаз. Термины "кислотные", "нейтральные" и "щелочные" относятся к рН, при котором активность ферментов оптимальна. Примерами нейтральных протеаз являются MILEZYME (получаемая от Miles Laboratory) и трипсин - протеаза природного происхождения. Щелочные виды протеаз могут быть получены от различных производителей; они обычно получаются в результате жизнедеятельности различных микроорганизмов (например, Bacillis subtilis). Типичным примером щелочных протеаз являются MEXATASE и MEXACAL, получаемые от International BioSynthetics, ALCALASE, SAVINASE и ESPERASE, получаемые от Novo Industry A/S. См. также Stanislowski et al., US патент No 4511490, используемый здесь в качестве ссылки.
Другими пригодными ферментами служат амилазы, являющиеся углевод-гидролизными ферментами. Они также могут считаться предпочтительными для включения в состав смесей амилаз и протеаз. Соответствующими амилазами могут быть RAPIDASE, получаемые от Society Rapidase, MILEZYME от Miles Laboratory и MAXAMYL из International BioSynthetics.
Еще одним подходящим видом ферментов являются липазы, такие как описанные в Silver US Patent No 3950277 и Thom et al., US Patent No 4707291, используемые здесь в качестве ссылки.
Еще одним ферментом, представляющим интерес, являются пероксидазы, такие как пероксидаза из хрена, и включенные в International Patent Publication WO 93/24628, представленные здесь в качестве ссылки. Желательно также использовать смеси любых указанных гидролаз (гидролитических ферментов) и особенно смеси протеаз и амилаз.
Кроме того, в составе основы могут быть использованы такие компоненты как красители, например Monastral blue и антрахиноновый краситель (такой как описанный в Zielske US Patent No 4661293 и US Patent N 4746461).
Могут быть также использованы соответствующие красители, которые выбираются без ограничений из ряда соединений, содержащих диоксид титана, синий ультрамарин (см. также Chang et al., US Patent N 4708816) и окрашенные алюмосиликаты.
Флуоресцентные отбеливатели являются еще одним желательным компонентом. В их число входят стильбен, стирол и производные нафталинов, которые под воздействием ультрафиолетового излучения флуоресцируют в видимой области спектра.
Дополнительные органические активаторы отбеливания могут быть введены в состав без ограничений, такие как сложные эфиры (см. Fong et al., US Patent No 4778618, и Rowland et al., US Patent No 5182045), кетоны, имиды (см. Kaaret, US Patent No 5478569) и нитрилы, указанные патенты используются здесь в качестве ссылки.
Добавки могут присутствовать в количествах от 0-50%, а более предпочтительно в количестве 0-30% и лучше 0-10%. В некоторых случаях могут быть применены индивидуальные виды добавок других типов. Однако настоящее изобретение предусматривает возможность использования каждого из составов с целью получения различных свойств для конкретных категорий отбеливателей.
Экспериментальная часть
Синтез устойчивых к окислению тетраамидолигандов
Материалы. Все использовавшиеся растворители и реактивы имели реагентную чистоту (Aldrich, Aldrich Sure-Seal, Fisher) и использовались в исходном состоянии, как они были получены. Микроанализы были выполнены Midwest Microlabs, Indianapolis, IN.
Масс-спектрометрия. Масс-спектрометр с ионизационным электрораспылением был приобретен у FINNIGAN-MAT SSQ700 (San Jose, СА) и оснащен устройством для электрораспыления ANALYTICA OF BRAN FORD. Напряжение на электрораспылителе составляло 2400 - 3400 В. Пробы растворялись в ацетонириле или в дихлорметане с концентрацией примерно 10 рmоl/μl и данные о пробах вводились в систему ESI по сбору информации при непосредственном введении в поток со скоростью 1 л/мин, данные о чем также вводилась в систему сбора информации. Масс-спектрометрические исследования выполнялись на квадрупольном масс-спектрометре FINNIGAN-MAT 4615 с использованием ударной ионизации (70 eV) положительно заряженных ионов, причем масс-спектрометр был связан с системой сбора данных INCOS. Температура источника ионов составляла 150oС и в камере измерения 100oС. Пробы вводились или из газового хроматографа или непосредственно. Масс-спектр, полученный при бомбардировке быстрыми положительно заряженными ионами атомов, воспринимался магнитным сектором FINNIGAN-MAT 212, подключенным к системе сбора информации INCOS. Напряжение ускорения составляло 3 кВ, а температура источника ионов была на уровне 70oС. При этом использовался ION TECH источник поля ускорения атомов с применением ксенона при 8 кэВ. В FAB матрице использовался тиоглицерин. Эксперименты с применением ионизации положительно заряженных ионов при соударении электронов (70 eV MS/mS) были выполнены с применением сдвоенного квадрупольного масс-спектрометра FINNIGAN-MAT TSQ/700. Образцы проб вводились в прибор непосредственно. Температура источника ионов составляла 150oС и в камере измерения 70oС. Диссоциация, возникающая под воздействием соударений (CID), достигалась посредством введения аргона только в центр камеры октаполя до тех пор, пока давление не достигало величины 0,9-2,5•10-6 торр (мм вод. ст.). Номинальная энергия для CID генерации ионов была <35 эВ (по данным лаборатории). Данные с высокой разрешающей способностью были получены на масс-спектрометре с двойной фокусировкой JEOL JMS АХ-505Н с конфигурацией ЕВ при разрешающей способности 7500. Пробы вводились посредством газового хроматографа или непосредственно. При проведении масс-спектрометрических экспериментов в источник ионов через тепловой вход вводился перфторкеросин. Точные данные массового состава были получены с применением интерполяции на компьютере при проведении массового анализа перфторкеросина. GC/MS (т.е. газовый хроматограф/масс-спектрометр) условия: колонка 20 м • 0,25 мм DB-1701 (J & W Scientific); газ-носитель - гелий с линейной скоростью 40 см/с; инжектор 125oС; температура колонки 35oС в течение 3 мин с последующим повышением температуры со скоростью 10oС/мин до 100oС; инжекция, степень разделения примерно 50:1.
Спектроскопические методы. Были получены спектры ядерно-магнитного резонанса 300 МГц 1H и 75 МГц 13С с применением прибора IBM AF300, использующего сверхпроводящую магнитную систему OXFORD, и программного обеспечения контроля BRUKER. Инфракрасный участок спектра исследовался на спектрометре MATTSON GALAXY серии 5000 FTIR, управляемом компьютером MACINTOSH II. Ультрафиолетовый и видимый участки спектра были получены с применением спектрофотометра HEWLETT PACKARD 8452A, управляемым компьютером ZENITH Z-425/SX. Обычный Х-диапазон EPR спектра был записан с применением BRUKER ER300 спектрометра, укомплектованного проточным гелиевым криостатом OXFORD ESR-900. Мессбауэровский спектр был получен с применением прибора с постоянным ускорением, и изомерное смещение оценивалось по стандарту металлического железа при 298К. С целью устранения ориентации поликристаллических образцов под воздействием магнитного поля, пробы были подготовлены в форме замороженных суспензий с применением nujol.
Синтез макроциклических тетраамидо-N-донорных лигандов
Основную схему реакции см. в конце описания.
Приведенное ниже описание представляет собой основную последовательность синтеза макроциклических тетраамидолигандов в соответствии с изобретением.
α-Аминокарбоновая кислота смешивается с активированным малонатом в пиридине при температуре ниже 70oС. После окончательного завершения двухступенчатой реакции, которая длится от 72 до 144 часов, MACRO LINKER (A-L-A) изолируются. На втором этапе диамин, предпочтительно о-фенилендиамин, добавляется к пиридиновому раствору MACRO LINKER в присутствии агента сочетания, предпочтительно РСl3 или триметилацетилхлорида. Реакция образования кольцевых структур (с двойными связями) завершается с использованием орошения в течение 48-110 часов, после чего полученные макроциклические тетраамиды изолируются в достаточном количестве.
В приводимых ниже примерах 1-25 представлены различные этапы реакции. Примеры 26-39 иллюстрируют детали процесса и, в соответствии с изобретением, использование реакции окисления, включая процессы отбеливания лигнина и красителей.
Пример 1
Синтез связанных промежуточных (A-L-A) макроструктур из α-метилаланина и диэтилмалонилдихлорида (тетраметилдиэтилзамещенное промежуточное соединение).
Двугорлая колба (1 л), соединенная с затвором (250 мл) для выравнивания давления и мембраной, заполняется под N2 α-аминоизомасляной кислотой (т.е. α-метилаланином) (20,62 г, 0,2 моль) и осушенный пиридин (250 мл), полученный при сухом просеивании на сите с размером 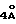 мол, помещается в колбу и нагревается до 60-70oС при перемешивании, после чего в дополнительный затвор добавляется диэтилмеланилдихлорид (23,23 мл, 0,135 моль), растворенный в осушенном пиридине (100мл осушенной фракции, полученной на сите
мол, помещается в колбу и нагревается до 60-70oС при перемешивании, после чего в дополнительный затвор добавляется диэтилмеланилдихлорид (23,23 мл, 0,135 моль), растворенный в осушенном пиридине (100мл осушенной фракции, полученной на сите 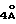 мол). Содержимое из дополнительного затвора добавляется каплями (в течение одного часа) в реакцию, и ацилирование протекает (60-70oС, 30-36 ч) под азотом или с присоединением к осушительной трубке. После завершения реакции ацилирования производится быстрое охлаждение путем добавления H2O (30 мл) и перемешивания (60-70oС, 24 ч). Объем растворителя уменьшается с использованием роторного испарителя с добавлением масла и затем HCl (конц., прибл. 25 мл) для получения конечной величины рН от 2 до 3. Горячий раствор помещается в холодильник (4oС, 15ч) и желтовато-коричневый продукт извлекается путем фильтрации фритты и промывается ацетонитрилом (2х100 мл). Просушенный на воздухе белый продукт (16,5-19,8 г, 50-60% выход) должен храниться в эксикаторе. Этот продукт обычно достаточно чистый для проведения реакций с замкнутыми кольцевыми структурами, хотя, возможно, потребуется его перекристаллизация.
мол). Содержимое из дополнительного затвора добавляется каплями (в течение одного часа) в реакцию, и ацилирование протекает (60-70oС, 30-36 ч) под азотом или с присоединением к осушительной трубке. После завершения реакции ацилирования производится быстрое охлаждение путем добавления H2O (30 мл) и перемешивания (60-70oС, 24 ч). Объем растворителя уменьшается с использованием роторного испарителя с добавлением масла и затем HCl (конц., прибл. 25 мл) для получения конечной величины рН от 2 до 3. Горячий раствор помещается в холодильник (4oС, 15ч) и желтовато-коричневый продукт извлекается путем фильтрации фритты и промывается ацетонитрилом (2х100 мл). Просушенный на воздухе белый продукт (16,5-19,8 г, 50-60% выход) должен храниться в эксикаторе. Этот продукт обычно достаточно чистый для проведения реакций с замкнутыми кольцевыми структурами, хотя, возможно, потребуется его перекристаллизация.
Свойства
1H ЯМР (ядерно-магнитный резонанс) спектр (d2 - пиридин) δ [ppm]: 8,9 (s, 2H, NH амид); 2,2 (q, 4Н); 1,8 (s, 12H); 1,2 (t, 6H). ИК-спектр (инфракрасный) (Nujol mull) ν [см-1]: 3310 (амид NH); 1721 (карбоксильная СО); 1623 (амид СО). Anal. Calcd (сокращение от "аналитический расчет") для С15Н21N2О6: С 54,53; Н 7,93; N, 8,48. Найдено: С 54,48; Н 7,88; N 8,47.
Пример 2
Увеличенная производительность, промежуточный (A-L-A) синтез связанных макроструктур из α-метилаланина и диэтилмалонилдихлорида (TMDE с промежуточным замещением).
Двугорлая колба (2 л, RB+Claisen), соединенная с затвором (250 мл) для выравнивания давления и мембраной, заполняются под N2 α-аминоизомасляной кислотой (т. е. α-метилаланином) (90,3 г, 0,9 моль), и обезвоженный пиридин (1,4 л, хранение в герметическом сосуде) помещается в затвор и реакционная смесь нагревается до 45-55oС при перемешивании. Пиридин (100мл, хранение в герметическом сосуде) и затем диэтилмалонилдихлорид (104,4 мл, 0,61 моль) помещается в дополнительный затвор. Содержимое из дополнительного затвора добавляется каплями (в течение 3-4 ч) в реакцию, затем дополнительный затвор снимается и реакция ацилирования продолжается (55-65oС, 120-130 ч) под N2. После завершения реакции ацилирования производится быстрое охлаждение путем добавления H2O (100 мл) и перемешивания (60-70oС, 24-36 ч).
Объем растворителя уменьшается с использованием роторного испарителя с добавлением масла и затем НС1 (конц., прибл. 110 мл) для получения конечной величины рН от 2 до 3. Горячий раствор помещается в холодильник (4oС, 15 ч) и желтовато-коричневый продукт извлекается путем фильтрования и промывается ацетонитрилом (700 мл, 150 мл) при перемешивании в колбе Эрленмейера. Просушенный на воздухе белый продукт (87,9 г, 60% выход) измельчается пестиком в ступке и затем хранится в эксикаторе. Полученный по технологии с увеличенной производительностью промежуточный, содержащий амиды продукт чаще требует перекристаллизации перед использованием для проведения реакций с замкнутыми кольцевыми структурами.
Пример 3
Перекристаллизация промежуточного замещенного неочищенного TMDE из примера 2 (50,4 г, 0,153 моль) осуществляется растворением промежуточного TMDE в H2O (500 мл, деионизированная) с медленным добавлением Na2CO3 (16,2 г, 0,153 моль) в трех аликвотах и тщательно избегая чрезмерного пенообразования при интенсивном перемешивании с одновременным медленным нагреванием. Раствор доводится до кипения, фильтруется и подкисляется HCl (конц., 30 мл, 0,36 моль). Раствор охлаждается (в течение ночи, 4oС) и белый осадок отфильтровывается и промывается в ацетонитриле (250 мл). Осушенный на воздухе продукт (38,8-45,4 г, перекрист., выход 77-90%) должен храниться в эксикаторе.
Пример 4
Промежуточный (A-L-A) гексаметил (НМ)
Синтез промежуточного НМ идентичен синтезу промежуточного TMDE в примере 2 со следующими изменениями: диметилмалонилдихлорид (17,8 мл, 0,135 моль) заменяется на диэтилмалонил дихлорид и температура реакции должна быть снижена до 55-65oС вследствие более низкой температуры кипения ацилирующего агента. Выход промежуточного гексаметила составляет 45-60%.
Свойства
1H ЯМР (d5-пиридин, δ [ppm]): 9,2-9,8 br s, 2 H (карбоксильный ОН), 8,23 s, 2 Н (амид), 1,87 s 12 Н (СН3), 1,74 s 6 H (СН3). ИК (nujol/NaCl) ν [см-1] : 3317,0 (амид NH); 1717,9 (карбоксильная СО); 1625,7 (амид СО). Anal. (осушка при 100oС) Calcd. для C13H22N2O6: С 51,63, Н 7,34, N 9,27. Найдено: С 51,64, Н 7,35, N 9,33.
Пример 5
Перекристаллизация промежуточного НМ
Неочищенный промежуточный гексаметил (НМ) был подвергнут перекристаллизации таким же способом, как и промежуточный TMDE амид. Вследствие несколько более высокой растворимости в воде промежуточного НМ амида в этом процессе должно использоваться несколько меньшее количество H2O.
Пример 6
Промежуточный Di CyHex Di этил (диэтилдициклогексил)
Колба с полусферическим дном (500 мл) загружается 1-амино-1-циклогексанкарбоновой кислотой (15 г, 0,1 моль), затем соединяется с дополнительным затвором (40 мл) для выравнивания давления, накрывается мембраной и продувается азотом. Обезвоженный пиридин (300 мл) помещается в реакционную колбу через дополнительный затвор и 20 мл помещается в дополнительный затвор. Начинают нагрев системы и устанавливают температуру на уровне 60oС. Когда температура 60oС достигнута, одна треть полного количества диэтилмалонилдихлорида, используемого для проведения реакции (т.е. 6 мл, 0,033 моль), добавляется с помощью шприца в дополнительную колбу. Смесь пиридина и диэтилмалонилдихлорида добавляется по каплям в реакцию и процесс ацилирования длится 12 часов. Вторая (6 мл, 0,033 моль) и третья (6 мл, 0,033 моль) аликвоты (части) добавляются с 12- часовыми интервалами. После того, как весь ацилирующий агент был введен и прореагировал (полное время реакции составляет 48-56 ч), 20 мл воды добавляется по каплям в реакцию. Реакция проводится с подогревом в течение еще 24 часов для открытия колец моно- и бис- промежуточных оксазалонов и выхода диамида дикарбоновой кислоты. Выделившийся пиридин удаляется с применением роторного выпаривания продукта, имеющего слабую желтовато-коричневую окраску, и подкисляется для получения рН 2 с использованием HCl (конц. ). Получаемый неочищенный продукт отфильтровывается, промывается ацетонитрилом и осушивается воздухом с целью получения белого промежуточного DiCyHexDE амида (16 г, 74%).
Свойства
1Н ЯМР (d5-пиридин) δ [ppm] : 8,30 (s, 2H, NH амид), 2,60 (m, 4 H, cyhex), 2,25 (q, 4 Н, этил CH2), 2,15 (m, 4 Н, циклогексил), 1,8-1,5 (m, 10 Н, циклогексил), 1,25 (m, 2H, cyhex), 1,20 (t, 6 Н, этил СН3). 13С ЯМР широкодиапазонный с развязывающими контурами (d5-пиридин) δ [ppm]: 178,0, (карбоксильный СО), 174,3 (амид СО), 60,5 (циклогексил), 59,4 (малонил quat), 33,0 (циклогексил α CH2), 30,3 (этил CH2), 26,0 (циклогексил γ CH2), 22,3 (циклогексил β CH2), 9,9 (этил СН3). ИК (nujol/NаCl) ν [см-1]: 3307 (амид NH); 3150 (sh, br, m, амид NH/карбоксильный ОН), 3057 (s, str, Н связанный амид NH/ карбоксильный ОН), 1717 (s, str, карбоксильный СО); 1621 (s, str, амид СО). Anal. Calcd. для C21H34N2O6: С 61,44; Н 8,35; N 6,82. Найдено: С 61,41; Н 8,38, N 6,90%.
Пример 7
Di CyHex диэтил монооксазалон (диэтилбициклогексилмонооксазалон)
Недостатком процесса охлаждения реакции промежуточного Di CyHex Di этила (с нагревом и водой, см. выше) при стехиометрическом соотношении 1,35 диэтилмалонилдихлорида; 2-циклогексиламинокислоты, является перемешивание промежуточного DiCyHexDE-амида (дициклогексилдиамида) и получаемого монооксазалона. Образующийся продукт DiCyHexDE монооксазалон имеет среднюю растворимость в кипящем циклогексане, в то время, как промежуточный циклогексиламид не растворяется в нем, благодаря чему достигается возможность простого выделения продукта из смеси, прибл. 10 г смеси промежуточного амида и монооксазалона, содержащей остаточное количество CH2Cl2, кипятили с интенсивным перемешиванием в 400-500 мл циклогексана. Полученный нерастворимый промежуточный продукт DiCyHexDE-амид выделялся при гравитационной фильтрации в нагретом состоянии, в то время как полученный продукт монооксазолон выкристаллизовывался под действием гравитационных сил при охлаждении и испарении раствора циклогексана. Выход продукционного амида составил около 6 г, выход монооксазолона - примерно 4 г.
Свойства монооксазолона
1H ЯМР (d5-пиридин) δ [ppm]: 9,7 (s, 1H, амид NH), 2,7-1,6 (нерастворимые Су Нех-циклогексингруппы), 1,05 (t, 6 Н, этил СН3). ИК (нуогель/NaCl) [см-1]: 3309 (sh, w, амид NH), 3229 (s, str, Н связанный амид NH/карбоксильный ОН), 3166 (s, str, H связанный амид NH/карбоксильный ОН), 3083 (s, str, Н связанный амид NH/ карбоксильный ОН), 1834 (s, str, оксаза С= O), 1809 (s, m, H связанный с оксазой С=O), 1743 (s, str, карбоксильный СО), 1663 (s, str, оксаза C=N), 1639 (s, br, str, амид СО). Anal. Calcd. для C21H32N2O5•(C6H12) 0,25: С 65,35; Н 8,53; N 6,77. Найдено: С, 65,07; Н 8,67: N, 6,68%. Присутствие сольвата циклогексана подтверждается посредством 13С ЯМР.
Реакции макроциклизации
Примеры проведения некоторых реакций синтеза для получения макроциклических тетраамидолигандов приводятся ниже.
Присоединение трихлорида фосфора
Процесс присоединения трихлорида фосфора, содержащего промежуточные (A-L-A) амиды, к ароматическим 1,2-диаминам позволяет получить макроциклические тетраамиды безопасно, с малыми затратами и с высоким выходом продукта. Могут быть использованы два различных возможных способа присоединения PCl3, причем различие состоит в порядке присоединения и выборе используемых реагентов. Эти способы пригодны для приготовления различных классов макроциклических соединений с различным замещением электронов в мостах диаминов или пространственной структуре промежуточных амидов прежде всего вследствие объединения параллельных типов макросвязей промежуточных амидов при синтезе.
Пример 8
А. Макроциклический синтез с присоединением РС13
В длинногорлую колбу (250 мл) помещались промежуточные амиды из примеров 2-7 (10 ммоль), перемешивались стеклянной палочкой и затем колба помещалась в печь (80-100oС, 30-45 минут). Горячая колба продувалась N2, арилдиамин (10 ммоль) добавлялся в колбу и вводился обезвоженный пиридин (50 мл, хранение в герметической посуде). Колба нагревалась (50-60oС) и РС13 (d=1,574 г/мл, 1,72 мл, 20 ммоль) сразу же вводился в колбу с помощью шприца без использования чрезмерного орошения. Рассматриваемая реакция относится к экзотермическим и требует соблюдения предосторожностей. Затем температура увеличивалась при орошении или становилась несколько ниже (100-115oС) и реакция продолжалась под N2 (48 ч). После завершения реакции ацилирования содержимое колбы подкислялось HCl (1 экв., прибл. 60 мл) до рН 2. Содержимое переливалось в колбу Эрленмейера (вода использовалась для ополаскивания колбы) и перемешивалось с CH2Cl2 (300 мл, 2-3ч), затем экстрагировалось с добавлением CH2Cl2 (2 х 150 мл). Комбинированные органические слои промывались разбавленной HCl (0,1М, 2 х 100 мл) и затем разбавленным водным раствором Nа2СО3 (2 х 5 г/100мл). Органические растворители удалялись на роторном испарителе до получения неочищенного продукта (30%). Вес неочищенного продукта обычно эквивалентен первоначальному весу диамина.
В. Макроциклический синтез с присоединением РС13
В длинногорлую колбу (250 мл) помещались MgSО4 (5 г), стеклянная палочка для перемешивания, диаминарил (10 ммоль) и пиридин (50 мл, осушенный на ситах 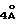 ), затем под N2 с помощью шприца добавлялся РС13 (d=1,754 г/мл, 1,72 мл, 20 ммоль) смесь находилась под орошением 30 мин, при этом образовывался оранжево-желтый осадок. Смесь несколько охлаждалась, добавлялся промежуточный амид (10 ммоль) и затем смесь находилась под орошением под азотом (115oС, 48 ч). После завершения реакции ацилирования содержимое колбы подкислялось HCl (1 экв., прибл. 60 мл) до рН 2. Содержимое переливалось в колбу Эрленмейера и перемешивалось с CH2Cl2 (2 х 150 мл). Комбинированные органические слои промывались разбавленной HCl (0,1 М, 2 х 100 мл) и затем разбавленным водным раствором Nа2СО3 (2 х 5 г/100мл). Органические растворители удалялись на роторном испарителе до получения неочищенного продукта (30%). Вес неочищенного продукта обычно эквивалентен первоначальному весу диамина.
), затем под N2 с помощью шприца добавлялся РС13 (d=1,754 г/мл, 1,72 мл, 20 ммоль) смесь находилась под орошением 30 мин, при этом образовывался оранжево-желтый осадок. Смесь несколько охлаждалась, добавлялся промежуточный амид (10 ммоль) и затем смесь находилась под орошением под азотом (115oС, 48 ч). После завершения реакции ацилирования содержимое колбы подкислялось HCl (1 экв., прибл. 60 мл) до рН 2. Содержимое переливалось в колбу Эрленмейера и перемешивалось с CH2Cl2 (2 х 150 мл). Комбинированные органические слои промывались разбавленной HCl (0,1 М, 2 х 100 мл) и затем разбавленным водным раствором Nа2СО3 (2 х 5 г/100мл). Органические растворители удалялись на роторном испарителе до получения неочищенного продукта (30%). Вес неочищенного продукта обычно эквивалентен первоначальному весу диамина.
Примечание. Для проведения макроциклических реакций в больших объемах время замыкания кольцевых структур увеличивается до 4-5 дней при орошении и большая часть пиридина, присутствующего при окончании реакции, удаляется с использованием роторного испарителя перед подкислением.
Пример 9
TMDE-DCB из промежуточного TMDE + DCB диамина
1,2-Диамино-4,5-дихлорбензол (1,77 г, 10 ммоль) был использован в качестве диаминарила с промежуточным TMDE амидом (3,3 г, 10 ммоль) в РС13 с применением способов А или В макроциклических реакций. Неочищенный макроциклический продукт (2,7 г) был подвергнут перекристаллизации из минимального количества нагретого 95% EtOH (этанола) путем выпаривания чистого продукта TMDE-DCB (1,5 г, 32%).
Свойства
1H ЯМР (CD2Cl2) δ [ppm]: 7,65 (s, 1 H, ArH), 7,35 (s, 2Н, амид NH), 6,45 (s, 2H, амид NH), 1,90 (q, 4 Н, этил CH2), 1,57 (s, 12 H, RСН3), 0,85 (t, 6H, этил СН3). ИК (nujol/NaCI) ν [см-1]: 3454 (следы ROH), 3346 (br, амид NH), 1706& 1688& 1645 (амид СО). Anal. Calcd. для С21Н28Сl2N4О4: С 53,51; Н 5,99; N 11,89. Найдено: С 53,58; Н 6,09; N 11,89.
Пример 10
TMDE-B из промежуточного TMDE + В диамина
1,2-Диаминобензол (т. е. о-фенилендиамин) (1,08 г, 10 ммоль) был использован в качестве арилдиамина с промежуточным TMDE амидом (3,3 г, 10 ммоль) в РС13 по методу А или В реакции макрокристаллизации. Неочищенный макроциклический продукт (1,5 г) был подвергнут перекристаллизации из минимального количества нагретого 95% (этанола) методом выпаривания для получения чистого TMDE-B (25% из диамина).
Свойства
1H ЯМР (CDCl3) δ [ppm]: 7,55 (m, 2 H, ArH), 7,48 (s, br, 2 H, арил амид NH), 7,17 (m, 2 Н, ArH), 6,46 (s, br, 2 Н, алкиламид NH), 2,07 (m, br, 4 Н, этил СН2), 1,60 (s, 12 Н, RСН3), 0,89 (t, 6 Н, этил СН3). ИК (nujol/NaCl) ν [см-1]: 3395&3363 (амид NH), 1702&1680&1652&1635 (амид СО). Anal. Calcd. для C21H10N4O4•H2O: С 59,98; Н 7,67; N 13,32. Найдено: С 60,18; Н 7,20; N 13,18.
Пример 11
TMDE-DMB из промежуточного TMDE + DMB диамина
1,2-Диамино-4,5-диметилбензол (1,36 г, 10 ммоль) был использован в качестве арилдиамина с промежуточным тетраметилдиэтиламидом (3,3 г, 10 ммоль) в РС13 по методу А или В реакции макрокристаллизации. Неочищенный макроциклический продукт (1,6 г) был подвергнут перекристаллизации из минимального количества нагретого 95% EtOH (этанола) методом выпаривания для получения чистого TMDE-DMB (25% из диамина).
Свойства
1H ЯМР (DMSO-d6) δ [ррm]: 8,00 (s, 2 Н, амид NH), 7,67 (s, 2 Н, амид NH), 7,28 (s, 2 H, ArH), 2,17 (s, 6 Н, арил СН3), 1,99 (q, 4 Н, этил CH2), 1,46 (s, 12 Н, RСН3), 0,75(t, 6 Н, этил СН3). ИК (nujol/NaCl) ν [см-1]: 3446 (s, m, следы ROH), 3362 (s, str, амид NH), 3348 (sh, m, амид NH), 3332 (s, str, Н амид NH), 1696 (амид СО), 1679 (амид СО), 1651 (амид СО), 1641 (амид СО), 1584 (s, m/w, кольцевой арил/амид). Anal. Calcd. для C23H34N4О4: С 64,16; Н 7,96; N 13,01. Найдено: С 64,09; 64,28; Н 8,04; 7,92; N 12,86; 13,04.
Пример 12
TMDE-DMOB из промежуточного амида TMDE + DMOB диамина
1,2-Диамино-4,5 диметоксибензол. HBr (5,0 г, 15 ммоль) были приготовлены, как это рассматривалось выше, и использованы как диаминарил непосредственно с промежуточным тетраметилдиэтиламидом (5,0 г, 15 ммоль) в 1,5 раза увеличенным количеством РСl3 по методу А или В реакции макрокристаллизации. Неочищенный макроциклический продукт (3,75 г) был получен перекристаллизацией нагретого 80-85% EtOH (1 г/40 мл) посредством испарения для получения чистого продукта TMDE-DMOB (30 % из диамина).
Свойства
1H ЯМР (CD2Cl2) δ [ppm]: 7,26 (s, 2 Н, амид NH), 7,01 (s, 2 H, ArH), 6,41 (s, 2 Н, амид NH), 3,80 (s, 6H, арил ОСН3), 2,07 (q, br, 4 Н, этил CH2), 1,54 (s, 12 Н, РСН3), 0,90 (t, 6 Н, этил СН3). ИК (nujol/NaCl) ν [см-1] : 3451 (s, m, Н связанный с H2O), 3391&3347 (амид NH), 1695&1670&1655 (амид СО). Anal. Calcd. для С23Н34N4О6, (Н2О)0,33: С 58,96; Н 7,46; N 11,96. Найдено (ESU): С 58,90; Н 7,26; N 11,76. Было обнаружено присутствие сольвата H2O при 1Н ЯМР- и ИК-анализах.
Пример 13
TMDE-Nap из промежуточного TMDE + Nap диамина
4,5-Диаминонафталин (1,68 г, 10 ммоль) был использован в качестве арилдиамина с промежуточным тетраметилдиэтиламидом (3,3 г, 10 ммоль) в РС13 по методу А или В реакции макрокристаллизации. Неоптимизированный выход составил 15-20% из диамина.
1H ЯМР (CDCl3) δ [ppm]: 8,05 (s, 2 Н, арилН α кольцо), 7,75 (m, 2Н, арилН β кольцо), 7,55 (s, 2 Н, ариламид NH), 7,35 (m, 2H, ArH' β кольцо), 6,45 (s, 2 Н алкиламид NH), 2,15 (m, br, 4 Н, этил CH2,), 1,65 (s, 12 Н, RСН3), 0,90 (t, 6 Н, этил СН3).
Пример 14
HM-DCB из промежуточного НМ + DCB диамина
1,2-Диамино-4,5-дихлорбензол (1,77 г, 10 ммоль) был использован как диамин с промежуточным гексаметиламидом (3,02 г, 10 ммоль) в РС13 по методу А или В реакции макрокристаллизации. Неочищенный макроциклический продукт (1,33 г, 30%) был получен перекристаллизацией из минимального количества нагретого н-пропанола путем испарения, выход первой порции перекристаллизированного материала составил 60%.
Свойства
1H ЯМР δ [ppm]: 7,69(s, 2 H, ArH), 7,39 (s, 2 Н, амид NH), 6,44 (s, 2 Н, амид NH), 1,58 (s, 12 Н, фиксированные метилы), 1,53 (s, 6 Н, метилмалонаты), отмечен незначительный пик н-пропанола. ИК (nujol/NaCl) ν [см-1]: 3503 (s, br, m-w, н-пропанол ОН, 3381 (sh, m, амид NH), 3338 (s, str, амид NH), 1689 (s, str, амид CO), 1643 (s, str, амид CO). Anal. Calcd. для С19H24N4О4 Cl2• (С3Н8О)0,2: С 51,70; Н 5,57, N 12,30%. Найдено: С 51,69; Н 5,63; N 12,33%.
Пример 15
HM-DMOB и НМ-В из промежуточного НМ + DMOB или В диамина
Промежуточный НМ также был использован для синтеза НМ-В и HM-DMOB по тому же самому методу и с подобными результатами, какие были получены в примере 14 для производных дихлоридов.
1H ЯМР данные для HM-DMOB в DCCl3 δ [ppm]: 7,65(s, 2 Н, амид NH), 7,21 (s, 2 Н, арил СН), 6,72 (s, 2 Н, амид NH), 4,00 (s 6 Н, метокси СН3), 1,76 (s, 12 Н, фиксированные метилы), 1,58 (s, 6 Н, метилмалонаты).
1H ЯМР данные для НМ-В в d5 пиридине δ [ppm]: 8.55 (s, 2 Н, амид NH), 8,40 (s, 2 Н, амид NH), 7,81 (m, 2H, ArH aa'bb'), 7,10 (m, 2 Н, ArH aa'bb'), 1,77 (s, 12 Н фиксированные метилы), 1,73 (s, 6 Н, метилмалонаты).
Пики амидов имеют тенденцию к изменению в несколько десятков раз величины, выраженной в ppm, в присутствии специфических примесей, таких как вода, кислоты и др.
Пример 16
DiCyHexDE DCB из промежуточного DiCyHexDE + DCB диамина
1,2-Диамино-4,5-дихлорбензол (1,77 г, 10 ммоль) был использован в качестве арилдиамина с промежуточным Di Cy Hex диэтиламидом (дициклогексилдиэтиламидом) (3,3г, 10 ммоль) в РС13, по методу А или В реакции макрокристаллизации. Вследствие увеличения пространственных затруднений рекомендуется увеличение времени реакции замыкания колец (3-4 дня по сравнению с обычными 48 часами). Су Hex оксазалоны (циклогексилоксазалоны), образующиеся при проведении реакции в качестве побочного продукта, не удаляются при воздействии кислоты на конечном этапе, так, что оказывается необходимым измельчать/промывать первоначально выделенный растворимый продукт в CH2Cl2 с использованием пентана для удаления оксазалонов. Выпаривание промывочного пентана обеспечивает возможность возвращения оксазалонов на повторную обработку. Нерастворимый в неочищенном пентане продукт подвергался перекристаллизации при растворении в CH2Cl2 или СНС13 с добавлением циклогексана до слабого помутнения и затем выпаривался на воздухе (1-2 дня) до получения белого микрокристаллического DiCyHexDE-DCB конечного продукта, который получался фильтрованием (1,38г, 25% из диамина). Возможна также перекристаллизация из нагретого чистого толуола с использованием процесса выпаривания.
Свойства
1H ЯМР (CDCl3) δ [ррm]: 7,70 (s, 2 Н, ArH), 7,45 (s, 2 Н, амид NH), 6,45 (s, 2 Н амид NH), 2,35 (m, br, 4 Н, циклогексил), 2,00 (m, br, ≈ 8 Н, циклогексил/этил CH2), 1,7 (m, br, ≈ 8 Н, циклогексил), 1,30 (m, br, ≈ 4 Н, циклогексил), 0,90 (t, 6 Н, этил СН3). Anal. (осушка при 100oС) Calcd. для С27Н36Сl2N4О4• (C6H12)0,2: С 59,60; Н 6,81; N, 986, Найдено: С 59,60; Н 6,77; N 9,77. Было обнаружено присутствие циклогексанового растворителя при анализе 1H и 13С ЯМР.
Пример 17
DiCyHexDE-B из промежуточного DiCyHexDE + В диамина
1,2-Диаминбензол (орто-фенилендиамин, 1,08 г, 10 ммоль) был использован в качестве арилдиамина при приготовлении аналогов DiCyHexDE-DCB с получением DiCyHexDE-B (1,25 г, 26% из диамина).
Свойства
1H ЯМР (DC3CN) δ [ppm]: 7,62 (s, 2 Н, ариламид NH), 7,51 (m, 2 Н, арилН), 7,18 (m, 2 Н, арилН), 6,71 (s, 2 Н алкиламид NH), 2,12 (m, 6 Н, циклогексил), 1,85 (q&m, этил CH2 & циклогексил), 1,62 (m, циклогексил), 1,37 (m, циклогексил), 0,90 (t, 6 Н этил СН3), 0,85 (m, циклогексил). ИК (nujol/NaCl) ν [см-1] : 3750 (s, m, H20), 3385 (s, str, амид NH), 314 (s, str, амид NH), 3258 (s, m, br, Н связанный амид NH), 1694 (s, str, амид CO), 1651 (s, str, амид CO), 1594 (s, m, кольцо арила/амид).
Пример 18
DiCyHex диэтил-бис-оксазалон
(диэтилдициклогексил-бис-оксазалон)
Это соединение было получено в качестве побочного продукта РС13, в реакции макрокристаллизации промежуточного DiCyHex Di этиламида с о-фенилендиамином. Бисоксазалон не удаляется из продуктов реакции при воздействии кислоты (это нейтральная молекула и легко растворяется в органических растворителях). Промывка неочищенных макроциклических соединений/оксазалона с использованием экстракции пентаном приводит к переходу бисоксазалона в пентан. Выпаривание на воздухе слоев полученного пентана приводит к образованию кристаллов чистого бисоксазалона в форме больших прозрачных призм (1 см • 1 см • 0,5 см). В связи с большим объемом гидрофобных СуНех групп этот оксазалон значительно более устойчив к гидролизу, чем соответствующие производные метила.
Свойства бисоксазалона
1H ЯМР (DС3СN) δ [ррm]: 2,05 (q, 4 Н, этил CH2), 1,8-1,4 (нерастворимые СуНех группы), 0,88 (t. t H, этил СН3). 13С ЯМР с расширенным диапазоном (СD3СN) δ [ррm]: 181,0 (оксаза (С=0), 162,7 (оксаза С=N), 69,0 (оксаза cyhex quat), 49,0 (малонат quat), 34,3 (cyhex α метилены), 25,5 (cyhex γ метилены), 24,9 (малонат метиленов), 21,8 (cyhex β метилены), 8,3 (этил СН3). ИК (nujol/NaCl) ν [см-1]: 1822 (s, str, br, оксаза С=0), 1662 (s, str, оксаза C= N). Anal. (осушка при 50oС). Calcd. для C21H30N204: С 67,36; Н 8,07; N 7,48, Найдено: С 67,26; Н 8,15; N 7,64.
Синтез хелатных комплексов
Пример 19
[Et4N] 2 и [Et4N] 3 [соли тетраэтиламмонийхлорид железа (III) TMDE-DCB моноанион и соответственно железа (III) водных TMDE-DCB моноанион]
Исходные макроциклические тетраамиды из любых приведенных выше примеров 10-18 (525 мг, 1,1 ммоль) растворяются в тетрагидрофуране (40 мл, Aldrich) под N2. Используя специальную технику, трет-бутиллитий (2,6 мл, 4,4 ммоль, 1,7 М в 2,4-диметилпентане, Aldrich) добавлялся в раствор под N2 при -108oС. Затем вводился хлорид железа (обезвоженный, 155 мг, 1,2 ммоль, Alfa) и раствор нагревался до комнатной температуры при перемешивании (16 ч), до выделения оливково-зеленого осадка, содержащего комплекс FeII, вступающий в реакцию с воздухом. Воздух удалялся через дренажную трубку (2 ч) и оранжевая твердая фаза выделялась и промывалась CH2Cl2 (2 х 10 мл). Полученный оранжевый порошок осушивался под пониженным давлением. Выход 595 мг (≈93%). Благодаря изменению сольватации и ограниченной растворимости соль лития переводилась в соль тетраэтиламмония и использовалась в дальнейшем. Соль лития (595 мг) в СН3ОН (50 мл) помещалась в ионообменную колонку (Dowex® 50Х2-100, 25г, 2 см х 12,5 см), которая предварительно насыщалась [Et4N]+ катионами и оранжевая область вымывалась с применением СН3ОН (100 мл). Растворитель удалялся при пониженном давлении. Остаток переводился в суспензию в CH2Cl2 (20 мл) и полученная смесь фильтровалась. Растворитель удалялся из маточного раствора при пониженном давлении с выделением оранжевого гигроскопического стекловидного осадка [Еt4N] 2, который использовался в дальнейшем без дополнительной очистки.
ИК (Nujol/NaCl, см-1): 1619 (ν (CO) амид), 1575 (ν (CO) амид), 1534 (ν (CO) амид).
Тщательная очистка исходного железа (III) более эффективна при использовании аксиального водного комплекса моноанионов (одноатомных анионов), чем с использованием такого же аксиального хлорсодержащего дианионного комплекса. [Et4N] 2 (550 мг, прибл. 0,7 ммоль) был растворен в СН3СN (50 мл). Тетрафторборат серебра (140 мг, 0,7 ммоль) был растворен в CH3CN (2 мл) и добавлялся в раствор при перемешивании (1 ч). Осадок АgСl отфильтровывался и растворитель удалялся при пониженном давлении. Полученный [Et4N] 3 дополнительно очищался вымыванием в колонке с силикагелем (8% МеОН в CH2Cl2). Растворитель удалялся при пониженном давлении и полученный продукт подвергался перекристаллизации из H2O. Выход: 360 мг (≈77% различная степень сольватации с водой была обнаружена в различных макрокристаллических пробах).
ИК (Nujol/NaCl, см-1): 1590 (ν (CO) амид), 1565 (ν (СО) амид), 1535 (ν (СО) амид). Anal. Calcd. для C29H46N5FeO5Cl2• (Н2О): С 50,52; Н 7,02; N 10,16; Cl 10,28. Найдено: С 50,24; Н 6,84; N 9,82; Cl 10,32.
ESIMS (отрицательный ион): m/z 522,2, [3-H2O]1- (100%); m/z 269,7, [3-Н+]2- (18%).
Пример 20
[Et4N] 4 [соли тетраэтиламмонийхлорид железа (IV) TMDE-DCB моноанион]
[Et4N] 2 (500 мг, прибл. 0,6 ммоль) был растворен в CH2Cl2 (30 мл). Нитратоцерий-аммоний (IV) (10,3 г, 18,3 ммоль) добавлялся в раствор и смесь перемешивалась (2 ч). Твердые соли церия извлекались из раствора фильтрованием. Фиолетовый продукт был получен путем удаления растворителя под пониженным давлением и осушался под вакуумом. Выход 400 мг (≈95%). Фиолетовые кристаллы были получены путем перекристаллизации из CH2Cl2 /Et2O.
ИК (Nujol/NaCl, см-1): 1688 (ν (СО) амид), 1611 (ν (СО) амид), 1582 (ν (СО) амид).
ESIMS (отрицательный ион): m/z 557 [4] -1 (100%); m/z 522, [4-Cl] 1- (65%).
Пример 21
Синтез [Ph4P] 5 [соли тетрафенилфосфонийцианид железа (IV) TMDE-DCB моноанион] из [Еt4N] 4 [соль тетраэтиламмонийхлорид железа (IV) TMDE-DCB моноанион] и NaCN.
[Et4N] 4 [соли железа (IV) тетраэтиламмония хлор TMDE-DCB моноанион] (225 мг, 0,33 ммоль) был приготовлен в форме суспензии в H2O (10 мл). Цианид натрия (140 мг, 2,85 ммоль) был растворен в H2O (10 мл) и добавлен в суспензию, после чего смесь была подвергнута звуковой (ультразвуковой) обработке (Branson 1200, 0,5ч). Фиолетовой цвет суспензии изменился на темно-синий цвет раствора и почти вся твердая фаза растворилась. Смесь была отфильтрована и голубой продукт был осажден посредством добавления PPh4Cl [хлорид тетрафенилфосфония], растворенного в воде (600 мг, 1,6 ммоль, 10 мл, Aldrich). Голубой осадок был собран и промыт Н2O (2 х 10 мл). Выход 250 мг (0,28 ммоль, ≈85%). Этот материал (120 мг) был подвергнут дальнейшей очистке посредством тонкослойной хроматографии (TCL) (слой силикагеля, GF, 20 см х 20 см х 1000 мкм, 10:1 CH2Cl2:CH3CN). Голубой продукт был экстрагирован из силикагеля с использованием CH3CN: CH2Cl2 (1:1, 60 мл). Растворитель был удален при пониженном давлении и осадок был растворен в CH2Cl2 (3 мл) и отфильтрован. При добавлении пентана (150 мл) образовался голубой порошок (90 мг, 0,10 ммоль). Выход на очистку 75%.
ИК (Nujol/NaCl, см-1): 2129 (ν (CN)), 1659 (ν (СО) амид), 1598 (ν (СО) амид), 1571 (ν (СО) амид). Anal. Calcd. для C46H44N5FeOCl2P: С 62,18; Н 4,99; N 7,88; Cl 7,98. Найдено: С 61,96; Н 5,04; N 7,84; Cl 8,06. ESIMS (отрицательный ион): m/z 548,2 [5]-l (100%); m/z 522,1, [5-CN]1- (20%). Для 13С-меченого цианида: m/z 549,2, [5]1- (100%); m/z 522,1, [5-13CN]1- (8%).
Пример 22
Синтез [Рh4Р] 5 [соль тетрафенилфосфонийцианид железа (IV) TMDE-DCB моноанион] из нитрилцианидых соединений.
[Рh4Р] 5 [соль тетрафенилфосфонийцианид железа (IV) TMDE-DCB моноанион] может быть образован в присутствии или при отсутствии основания. В случае отсутствия основания голубой цвет обесцвечивается и превращается в желто-оранжевый по мере удаления растворителя при окончании процедуры. Тем не менее, при выделении продукта достигается получение твердой фазы голубого цвета с лучшим выходом, при проведении реакции в присутствии добавляемого основания при рН в пределах 9-10. Конечный продукт реакции [Рh4Р] 5 получался с использованием каждого из соединений СН3СN, СD3СN, СН3СН2СN и (СН3)2СНСN, применяемых в качестве растворителя нижнего слоя. Основание не добавлялось в рассматриваемую каталитическую реакцию. Было установлено, что голубое соединение является эффективным каталитическим исходным веществом посредством добавления выделенного [Ph4P] 5 к раствору ацетонитрила ТВНР (третичного бутилгидропероксида) в качестве растворителя и окислителя, причем расход этих веществ показывает, что хотя [Рh4Р] 5 образуется как конечный продукт процесса каталитического окисления, этот процесс не дезактивирует форму катализатора.
Пример 23
Синтез [Ph4P] 5 в присутствии основы
[Et4N] 3 (160 мг, 0,23 ммоль) был растворен в выбранном нитрильном растворителе (6 мл), см. пример 19. Затем был добавлен гидроксид тетраметиламмония в качестве основания (20 вес. %, 0,370 мл, 0,52 ммоль, Aldrich), после чего был добавлен по каплям с перемешиванием (20 мин) гидропероксид трет-бутила (90%, 0,605 мл, 5,4 ммоль, Aldrich) и в результате был получен голубой раствор. Оставшийся нитрил был удален под пониженным давлением и образовавшийся маслянистый голубой остаток, который был растворен в H2O (15 мл), был отфильтрован. Голубой продукт был осажден из фильтрата добавлением водного раствора РРh4С1 (800 мг, 2,1 ммоль, Aldrich, 10 мл). Голубой осадок был собран и промыт в H2O (2х10 мл). Выход 130, 0,15 ммоль (65%). Последующая очистка была выполнена, как это описано в разделе [Ph4P] 5, пример 25.
Пример 24
Рентгеновская структура кристаллов и очистка [Еt4N] 3 H2O
C29H48Cl2FeN5O6, М= 689,47, Triclinic, Пространственная группа Р-1, а= 9,899(2); b=11,771 (2); 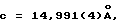 =95,33(2); в=100,09(2); γ = 92,31(2)o,
=95,33(2); в=100,09(2); γ = 92,31(2)o, 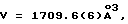 Dobs= 1.33 г см-3, D caicd (Z=2) = 1.339 г см-3, Т=293К,
Dobs= 1.33 г см-3, D caicd (Z=2) = 1.339 г см-3, Т=293К, 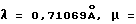 0,64 мм-1, коэф. передачи 0,87-1,00. Дифракционные данные были получены при комнатной температуре на дифрактометре Enraff-Nonius CAD-4, используя монохроматическое Мо-Kα излучение графита. Для получения информации были исследованы три отраженных пучка и были обнаружены только случайные отклонения интенсивности флуктуации (колебаний). Структура была определена прямыми методами. Связь водородных и углеродных атомов учитывалась при расчете положения исходя из того, что длина С/Н связи составляет
0,64 мм-1, коэф. передачи 0,87-1,00. Дифракционные данные были получены при комнатной температуре на дифрактометре Enraff-Nonius CAD-4, используя монохроматическое Мо-Kα излучение графита. Для получения информации были исследованы три отраженных пучка и были обнаружены только случайные отклонения интенсивности флуктуации (колебаний). Структура была определена прямыми методами. Связь водородных и углеродных атомов учитывалась при расчете положения исходя из того, что длина С/Н связи составляет 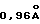 , и расчеты были выделены с использованием модели с термическим коэффициентом, на 20% большим, чем для исходного углерода. Атомы водорода молекул воды были локализованы по таблицам разности плотностей электронов и их координаты позволили выявить температурные параметры, зафиксированные на уровне выше 20%, по сравнению с теми же параметрами для кислорода. Расчеты выполнялись с использованием матричного метода наименьших квадратов F2 с учетом коэффициента рассеивания, который принимался на основании Международных Таблиц. Определение всех неводородных атомов проводилось с учетом параметров температурной анизотропии. Полученная сводка результатов была типичной. Расчеты позволили определить R=0,053, wR 2=0,112 с весовой функцией 1,0/[σ2 Fo 2) + {0,0652 (Fo 2 + 2Fc 2/3}2] для 2262 обнаруженных отражений.
, и расчеты были выделены с использованием модели с термическим коэффициентом, на 20% большим, чем для исходного углерода. Атомы водорода молекул воды были локализованы по таблицам разности плотностей электронов и их координаты позволили выявить температурные параметры, зафиксированные на уровне выше 20%, по сравнению с теми же параметрами для кислорода. Расчеты выполнялись с использованием матричного метода наименьших квадратов F2 с учетом коэффициента рассеивания, который принимался на основании Международных Таблиц. Определение всех неводородных атомов проводилось с учетом параметров температурной анизотропии. Полученная сводка результатов была типичной. Расчеты позволили определить R=0,053, wR 2=0,112 с весовой функцией 1,0/[σ2 Fo 2) + {0,0652 (Fo 2 + 2Fc 2/3}2] для 2262 обнаруженных отражений.
Пример 25
Результаты рентгеновской структуры кристаллов и очистка для [Et4N] 4
Единичные кристаллы [Еt4N] 4 при 20±1oС являются моноклинными, пространственная группа P21/c-C52h (No. 14) с 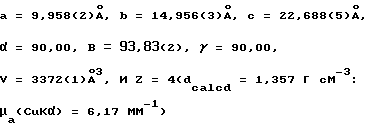
Полная информация о 4626 независимых абсорбционно-скорректированных отражениях, имеющих 2θ(CuKα) < 115,0o, была получена, используя θ-2θ сканирование и Ni-отфильтрованное CuKα излучение. Структура была рассчитана с использованием техники "прямого метода" с применением пакета программного обеспечения NICOLET SHELXTL, модифицированного Crystalytics Company. Полученные параметры структуры были ограничены сходимостью R1 (без учета веса, основанного на F) = 0,037 для 2680 независимых отражений, имеющих 2θ(CuKα) < 115,0o и I = 3σ (I). Были выделены десять метильных групп устойчиво обеспечивающих sр3-гибридную геометрию и длиной связей С-Н на уровне  Первоначальная ориентация каждой метильной группы была определена дифференциальным методом Фурье для водородных атомов. Окончательная ориентация каждой метильной группы была определена с учетом трех параметров поворота. Установленное положение для жестких вращающихся групп метила, имеющих С-С-Н угол, определено в пределах от 103-118o. Остальные водородные атомы были включены в структуру в соответствии с расчетным показателем для идеальных атомов (предполагая sp2- или sp3- гибридизацию углеродных атомов и длину связи С-Н равной
Первоначальная ориентация каждой метильной группы была определена дифференциальным методом Фурье для водородных атомов. Окончательная ориентация каждой метильной группы была определена с учетом трех параметров поворота. Установленное положение для жестких вращающихся групп метила, имеющих С-С-Н угол, определено в пределах от 103-118o. Остальные водородные атомы были включены в структуру в соответствии с расчетным показателем для идеальных атомов (предполагая sp2- или sp3- гибридизацию углеродных атомов и длину связи С-Н равной  , связанных с соответствующими атомами углерода. Изотропический температурный показатель каждого водородного атома был установлен на уровне 1,2 по отношению к эквивалентному изотопическому температурному показателю углерода, с которым он был связан ковалентной связью.
, связанных с соответствующими атомами углерода. Изотропический температурный показатель каждого водородного атома был установлен на уровне 1,2 по отношению к эквивалентному изотопическому температурному показателю углерода, с которым он был связан ковалентной связью.
Пример 26
Отбеливание лигнина с использованием перекиси водорода и Fe-DCB при рН 10
В односантиметровую удлиненную термостатированную при 25oС кварцевую кювету, содержащую 3,0 мл 0,1М NаНСО3/Nа2СО3 (рН 10), было введено 60 мкл насыщенного раствора щелочного лигнина и 300 мкл исходного каталитического раствора (1,24 х 10-4 М Fe-DCB, где в качестве R' и R" был метил) (все в воде). Этот раствор был перемешан и в него добавлено 3,8 мкл 30% H2O2. Изменение абсорбции при 350, 376, 400, 426, 450 и 476 нм проводилось с использованием УФ/визуального спектрофотометра Hewlett-Packard, действующего по программе определения кинетики в одинарной ячейке. После добавления H2O2 абсорбирование резко увеличилось на всех длинах волн и затем резко снижалось. После 15 мин абсорбирование на каждой из длин волн было ниже первоначальной величины, свидетельствуя, что произошло отбеливание лигнина. При повторном добавлении 60 мкл лигнина наблюдалось быстрое увеличение абсорбции, как это происходило и раньше, и затем происходило снижение первоначального подъема, хотя и более медленно, чем раньше. Во время эксперимента наблюдалось образование пузырьков.
После 30 мин вводилось дополнительно 3,8 мкл H2O2. Ход реакции был аналогичным предыдущей. Происходило быстрое усиление абсорбции и последующее ее затухание.
Пример 27
Отбеливание лигнина без Fe-DCB при рН 10
Были повторены все этапы, указанные в примере 26, за исключением введения катализатора. В односантиметровую удлиненную термостатированную при 25oС кварцевую кювету, содержащую 3,0 мл 0,1М NaHCO3/Na2CO3 (рН 10) было добавлено 60 мкл насыщенного раствора щелочного лигнина и смесь была перемешена. После короткого промежутка времени реакция была инициирована, было добавлено 3,8 мкл 30% H2O2.
Изменение абсорбции проводилось при тех же параметрах, как и в примере 26.
После добавления H2O2 наблюдалось увеличение абсорбции на всех шести длинах волн. Подъем не был столь быстрым и не имел ярко выраженного пика, как при каталитической реакции. Абсорбция затем постепенно начинала снижаться, но этот процесс происходил в замедленном темпе. Образование пузырьков в смеси не наблюдалось в течение 15 мин. После одного часа наблюдалось начало образования пузырьков.
Сравнение с предыдущими экспериментами в примерах 26 и 27 показывает, что добавление активатора в соответствии с настоящим изобретением увеличивает скорость реакции, при которой происходит отбеливание лигнина с использованием H2O2.
Пример 28
Отбеливание лигнина с использованием перекиси водорода, стабилизатора и без Fe-DCB при рН 10
Были повторены все этапы примера 27 с введением стабилизатора, DEQUEST 2066, 2 мкл, хелатобразующего агента для свободных ионов металлов. Введение Н2О2 приводит к резкому увеличению и снижению параметров, как это видно из примера 27.
Пример 29
Отбеливание лигнина с использованием перекиси водорода, стабилизатора и без использования Fe-DCB при рН 7
Были повторены все этапы примера 27 при рН 7 c применением 0,0087 мольного KH2P04/0,030, мольного Nа2НРО4 буферного раствора. В кювету было добавлено 2 мкл хелатобразующего агента DEQUEST 2066. В течение одночасового периода проведения эксперимента не наблюдалось видимого отбеливания. Хотя наблюдалась минимальная активность следов процесса абсорбции на длине волн 350 нм, изменение этого параметра следует отнести к возможным шумам, сопровождающим процесс измерения.
Пример 30
Отбеливание лигнина с использованием перекиси водорода, FE-DCB и стабилизатора при рН 10
В кювете, оснащенной устройством для перемешивания, было смешано 1 эквивалент катализатора из примера 26 (300 мкл исходного раствора Fe-DCB), 60 мкл насыщенного буферного раствора лигнина, как это описывалось раньше, и 2 мкл хелатобразующего DEQUEST. Абсорбция измерялась с использованием тех же самых параметров, как описано в примерах 26 и 27. После 1-2 мин в кювету было введено 1000 эквивалентов 30% H2O2 (3,8 мкл). Это привело к быстрому росту абсорбции и дальнейшему быстрому ее снижению, как это было описано в примере 26.
После 20 мин в кювету было добавлено еще 60 мкл лигнина. Абсорбция на всех длинах волн возрастала значительно медленнее и затем снижалась также медленнее, чем в случае добавления H2O2.
После 30 мин в кювету был добавлен эквивалент (300 мкл) катализатора (Fe-DCB). При этом никаких существенных изменений не наблюдалось.
После 40 мин в кювету было введено дополнительно 3,8 мкл H2O2. Это привело к существенному ослаблению абсорбции на всех длинах волн, свидетельствующему, что вновь происходит отбеливание лигнина.
Пример 31
Отбеливание лигнина с использованием перекиси водорода, FE-DCB и стабилизатора при рН 7 в процессе изменяющейся абсорбции.
Пример 29 был повторен, но с добавлением 300 мкл катализатора. После проведения некоторого количества циклов было добавлено 3,8 мкл 30% H2O2. После введения H2O2 абсорбция возросла на всех шести длинах волн, как и в примере 26, однако не столь быстро. Абсорбция продолжала медленно усиливаться первые 15 мин, без резких пиков и затем начала снижаться на всех шести длинах волн. После одного часа абсорбция была выше, чем первоначальная абсорбция.
Пример 32
Подтверждение активности катализатора
В односантиметровую удлиненную термостатированную при 25oС кварцевую кювету, содержащую 3,0 мл 0,1М NaHCO3/Na2CO3 (pH 10) было добавлено 60 мкл насыщенного раствора щелочного лигнина и 300 мкл (12, 4 мкМ) исходного каталитического раствора (1,24 х 10-4 М Fe-DCB) и 2 мкл Dequest 2066 (все в воде). Смесь была перемешана при соблюдении условий, указанных в примере 26, и затем было добавлено 19 мкл (5000 эквивалентов) 30% H2O2. После первоначального быстрого увеличения скорости абсорбции и последующего быстрого ее снижения, каждые 15 мин. в реакцию вводились аликвоты, содержащие 60 мкл насыщенного щелочного раствора лигнина и 19 мкл (5000 эквивалентов) 30% H2O2.
Полученные результаты при мониторинге на длине волн 476 нм показаны сплошной линией на графике, фиг. 1. Аналогичные результаты были получены при мониторинге на других длинах волн. Добавки лигнина и Н2О2 приведены в примечании.
Для сравнения были выполнены измерения абсорбции в специально приготовленной кювете с насыщенным раствором лигнина, хелатобразующего соединения и H2O2, но без использования катализатора. Результаты показаны пунктирной линией на фиг. 1.
Пример 33
Подтверждение стабильности катализатора
В соответствии с фиг. 5 сравнивалась долговечность катализатора двух модификаций согласно изобретению в опытах с красителями. Соединение 1 содержало заместители R' и R", в качестве которых использовались СН3, и соединение 2 с заместителями R' и R", в качестве которых использовались -СН2СН3. Регулирование добавок катализатора не проводилось.
Условия проведения исследования: рН 9, комнатная температура (21,1oС), использование буферного раствора NаНСО3/Nа2СО3. В качестве окислителя использовалась H2O2 4 мМ (30%). Как указывается звездочкой, 12 мкМ хлорида пиноцианола добавлялось в качестве красителя.
Как видно из графика, фиг. 5, каждое добавление красителя, когда присутствовал состав 1, приводило к непосредственному обесцвечиванию. Состав 2, содержащий диэтиловое соединение, обеспечивает более длительную декомпозицию. Исследования показывают только очень низкую скорость обесцвечивания.
| название | год | авторы | номер документа |
|---|---|---|---|
| МЕТАЛЛ-ЛИГАНДСОДЕРЖАЩИЕ ОТБЕЛИВАЮЩИЕ КОМПОЗИЦИИ | 1999 |
|
RU2234528C2 |
| ОТБЕЛИВАЮЩИЙ СОСТАВ И СПОСОБЫ ДЛЯ ЕГО ИСПОЛЬЗОВАНИЯ | 1997 |
|
RU2193050C2 |
| КОМПЛЕКСНОЕ СОЕДИНЕНИЕ, СОДЕРЖАЩЕЕ МАКРОЦИКЛИЧЕСКИЙ ТЕТРАДЕНТАТНЫЙ ЛИГАНД, ХЕЛАТНЫЙ КОМПЛЕКС И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ МАКРОЦИКЛИЧЕСКИХ ТЕТРАДЕНТАТНЫХ СОЕДИНЕНИЙ | 1997 |
|
RU2173322C2 |
| ПАКЕТНЫЙ КОММУТАТОР, ПАМЯТЬ N ПОРТОВ И СИСТЕМА ДЛЯ КОММУТАЦИИ ПАКЕТОВ (ВАРИАНТЫ) | 1992 |
|
RU2115254C1 |
| ЛИГАНД И КОМПЛЕКС ДЛЯ КАТАЛИТИЧЕСКОГО ОТБЕЛИВАНИЯ СУБСТРАТА | 2001 |
|
RU2283342C2 |
| ОТБЕЛИВАЮЩАЯ КОМПОЗИЦИЯ, НЕ СОДЕРЖАЩАЯ МЕТАЛЛ | 2011 |
|
RU2570902C2 |
| ФЛУОРЕСЦЕНТНЫЕ ОТБЕЛИВАЮЩИЕ СРЕДСТВА | 1998 |
|
RU2198168C2 |
| КАТАЛИТИЧЕСКИЙ СПОСОБ ОБРАБОТКИ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2003 |
|
RU2342997C2 |
| КРИПТОФИЦИНОВОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ ПРОЛИФЕРАЦИИ КЛЕТОК И СПОСОБ СМЯГЧЕНИЯ ПАТОЛОГИЧЕСКОГО СОСТОЯНИЯ | 1996 |
|
RU2195458C2 |
| КАТАЛИЗАТОР ДЛЯ ОБРАБОТКИ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2003 |
|
RU2343977C2 |
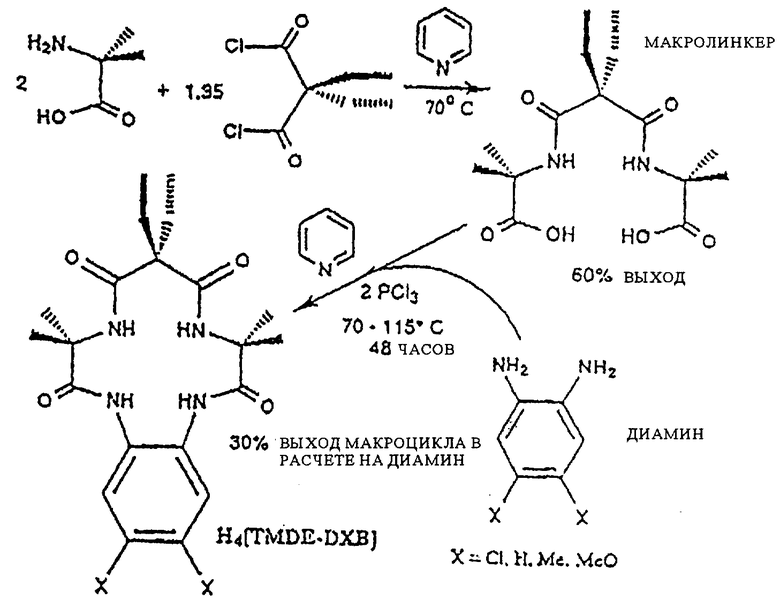
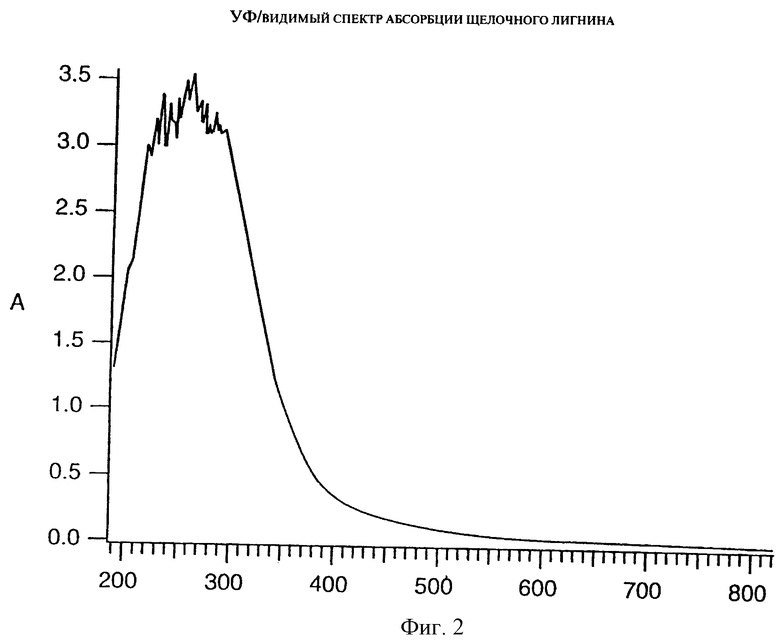
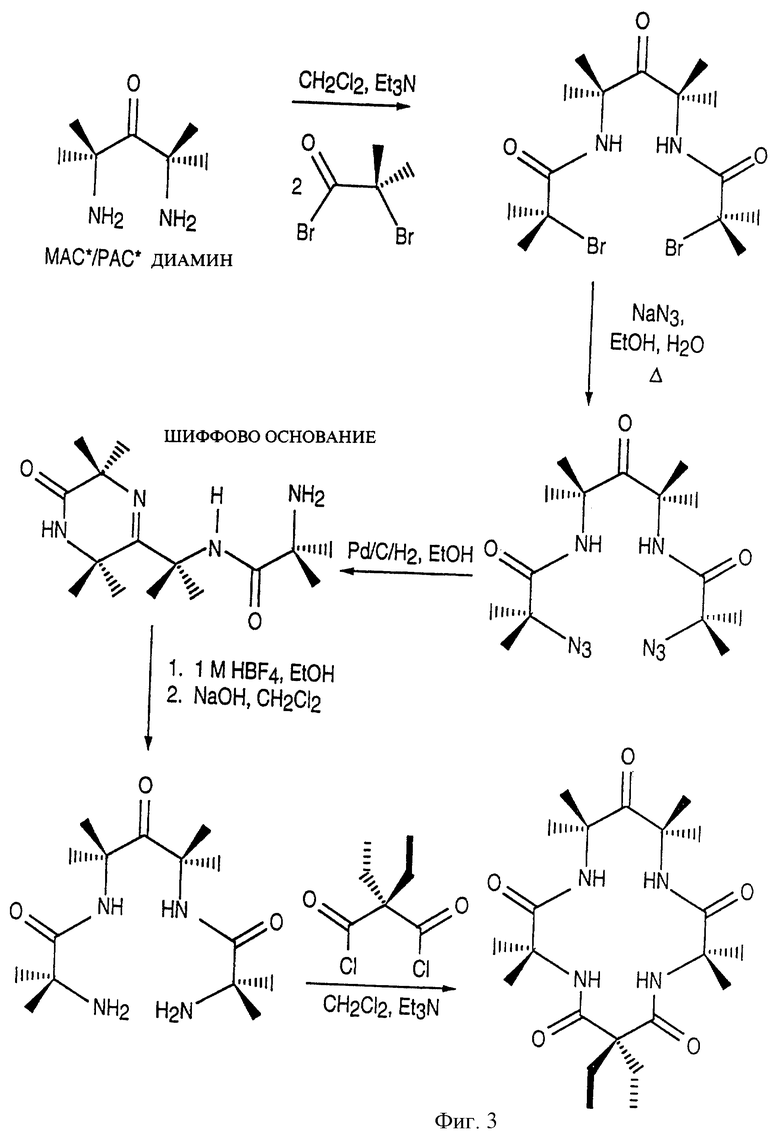
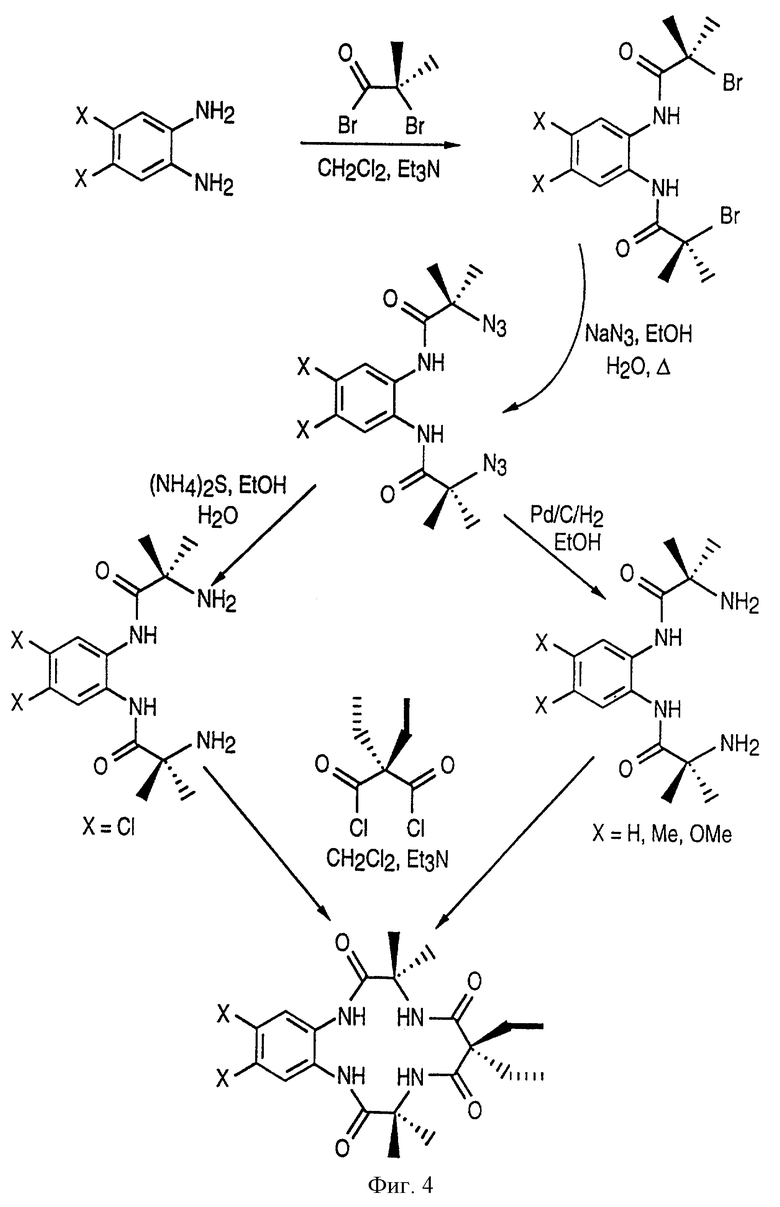
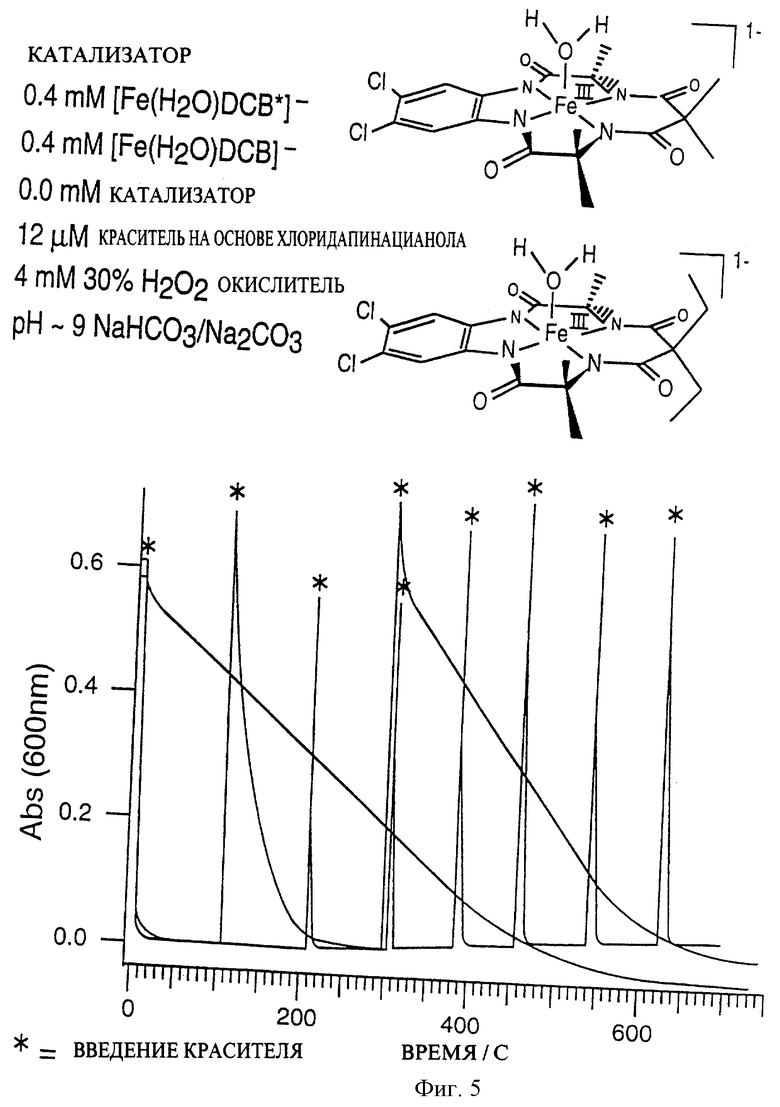
Изобретение относится к макроциклическим комплексам металлических лигандов, используемых в качестве активаторов отбеливателя. Описывается отбеливающий состав для отбеливания материалов на основе лигнина, содержащий эффективное количество источника окисляющего соединения и активатор отбеливателя, представляющий собой устойчивый к окислению макроциклический тетраамидолиганд приведенной формулы, где Х и Z - атом водорода, электронодонорная или электроноацепторная группа; R' и R'' - алкил и циклоалкил; М - переходной металл со степенью окисления I - VIII или выбирается из 3 - 11 группы Периодической таблицы элементов, система IU РАС; Q - любой противоион, который сбалансировал бы заряд соединения на стехиометрической основе; L - факультативно присутствующий лабильный лиганд, а когда присутствует в водном растворе, L представляет собой воду. Описывается также способ отбеливания лигнинсодержащих материалов, способ окисления и отбеливания лигнина. Изобретение позволяет повысить степень отбеливания лигнинсодержащих материалов за счет использования указанного активатора отбеливателя. 5 с. и 21 з.п.ф-лы, 5 ил.
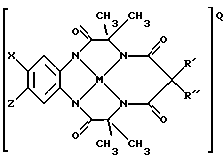
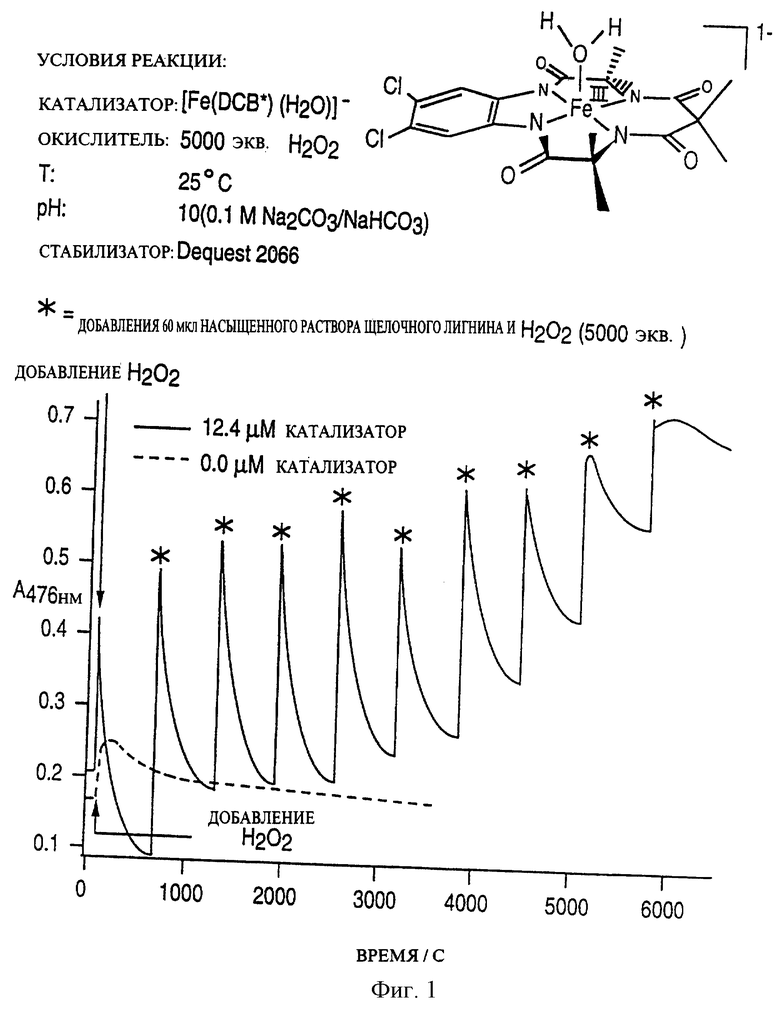
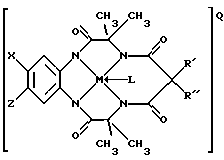
где Х и Z могут быть представлены Н, группами доноров или акцепторов электронов;
R' и R" алкил, циклоалкил;
М - переходный металл со степенями окисления I-VIII, или выбранными из групп 3-11 Периодической таблицы элементов, система IUPAC;
Q - любой противоположно заряженный ион, который обеспечивает баланс зарядов соединения на стехиометрической основе;
L - факультативно присутствующий лабильный лиганд, и когда он присутствует в водном растворе, то представляет собой воду.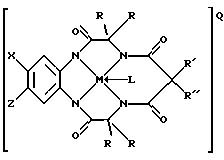
где X и Z могут быть представлены Н, группами доноров и акцепторов электронов;
R, R' и R" - алкил, циклоалкил;
М - переходный металл со степенями окисления I-VIII или выбранными из групп 3-11 Периодической таблицы элементов, система IUPAC;
Q - любой противоположно заряженный ион, который обеспечивает баланс зарядов соединения на стехиометрической основе;
L - факультативно присутствующий лабильный лиганд, и когда он присутствует в водном растворе, то представляет собой воду.
Приоритет по пунктам:
24.02.1997 по пп. 6, 19, 22, 23, 25 и 26;
22.07.1996 - остальные пункты формулы.
| US 4590005 А, 20.05.1986 | |||
| US 5288746 А, 22.02.1994 | |||
| SU 1689477 A1, 07.11.1991. |

