Заявление, касающееся федерально финансируемого исследования
Эта работа поддерживалась Национальным научным фондом, грантом СНЕ9319505, и Национальным институтом здравоохранения, грантом GM-44867. Правительство Соединенных Штатов может иметь определенные права в отношении этой заявки.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Данное изобретение относится к применению макроциклических металл-лигандных комплексов в качестве отбеливающих катализаторов и более конкретно к комплексам с металлами переходного ряда макроциклических тетраамидных лигандов в качестве катализаторов для усиления реакций окислительного отбеливания.
Соединенные Штаты и Канада являются ведущими мировыми производителями древесного волокна, используемого для производства бумаги и картона. В 1994 году Соединенные Штаты производили более 58 миллионов тонн древесного волокна. Древесное волокно, которое изготовлено либо механически, либо химически из древесины, содержит: 1) целлюлозу, гомополисахаридный линейный полимер d-глюкозы формулы -(С6Н10О5)-; 2) лигнин, неоднородную трехмерную молекулу, имеющую следующий общий состав -С9Н8,83О2,37(ОСН3)0,96, и 3) гемицеллюлозу, гетерополисахаридный полимер. См., в общем, W.G. Glasser and S. Sarkanen, eds. "LIGNIN PROPERTIES AND MATERIALS", American Chemical Society Symposium, Series 397.
Желательные качества для бумаги включают в себя прочность, белизну и яркость. Прочность бумаги связана с вязкостью древесного волокна, используемого в ее производстве, которая, в свою очередь, связана с состоянием целлюлозы после операции варки (превращения в волокнистую массу). Молекулярная целлюлоза, как указано выше, представляет собой линейную цепь d-глюкозы, которая природно образует длинные волокна. Чем длиннее отдельные цепи целлюлозного полимера, тем выше вязкость древесного волокна и тем выше, в свою очередь, прочность бумаги. Таким образом, во время обработки наиболее желательно избегать расщепления целлюлозных полимеров на более мелкие единицы.
Белизна основана на виде бумаги для наблюдателей, и, следовательно, ее мера является субъективной. Яркость (степень белизны) является термином, используемым для описания белизны древесного волокна по шкале от 0% (абсолютно черная) до 100% (относительно МgО-стандарта, который имеет абсолютную яркость приблизительно 96%) при помощи коэффициента отражения синего света (457 нм) от бумаги, изготовленной из этого древесного волокна. Чем больше падающий свет отражается, а не поглощается, тем ярче бумага.
Яркость достигается отбеливанием. Отбеливание древесного волокна определяется как обработка целлюлозных волокон химикалиями для увеличения яркости. Отбеливающие химикалии увеличивают яркость путем удаления и обесцвечивания лигнина в древесном волокне. Лигнин обнаруживает цвет от желтоватого до темно-коричневого в зависимости от типа древесины.
Наиболее обычными отбеливающими химикалиями являются окислители: хлор, источник гипохлоритного иона и диоксид хлора. Газообразный кислород в сочетании с NaOH также может быть использован, но он требует дорогостоящего оборудования и должен быть использован в больших количествах. Кроме того, кислород приводит к потере прочности древесного волокна, обусловленной вызываемым свободными радикалами повреждением целлюлозных полимеров, в частности, в тех случаях, когда содержание лигнина в древесном волокне является низким.
Хлор и гипохлорит могут приводить к потере прочности при неправильном применении, но в общем являются эффективными и относительно легкими в использовании окислителями. Гипохлорит является агрессивным окислителем, который имеет тенденцию атаковать (разрушать) целлюлозу, особенно при неоптимальном применении. Диоксид хлора дает высокий уровень яркости без деструкции древесного волокна. Однако он представляет собой дорогостоящий окислитель и склонен к взрывному разложению. Все окислители на основе хлора производят вытекающие (сточные) побочные продукты, которые являются опасными для окружающей среды и для здоровья. Кроме того, сточные воды, которые содержат хлор в любой химической форме, не могут быть сожжены в регенерирующем котле целлюлозного завода. Хлор производит коррозию регенерирующего котла. Кроме того, как отмечается ниже, сгорание хлорсодержащих частиц может приводить к образованию полихлорированных диоксинов и дибензофуранов, 17 из которых считаются токсичными и канцерогенными. Кроме того, хлор может, например, взаимодействовать сильно с горючими материалами. Он взаимодействует с H2S, CO и SO2 с образованием токсичных и коррозионных (агрессивных) газов и в жидкой форме вызывает ожоги, образование волдырей и деструкцию тканей. В газообразной форме он вызывает серьезное раздражение глаз, проходов носа и респираторной ткани. В высоких дозах он может быть летальным. Содержащий диоксид хлора отбеливатель разрушается до Сl2, который является токсичным и агрессивным.
Полихлорированные ароматические соединения являются загрязняющими веществами окружающей среды. Наиболее хорошо известными примерами являются ДДТ, полихлорированные фенолы, диоксины, дибензофураны и полихлорированные бифенилы (РСВ). Эти типы соединений могут быть образованы при экспонировании соответствующих органических соединений хлорсодержащим окислителем. Горение органического материала в присутствии хлора в любой форме может производить диоксин. Даже хотя диоксины и РСВ больше не производятся, существуют химические процессы, в которых образуются эти соединения из полихлорированных фенольных предшественников. Существует необходимость в предотвращении нежелательного образования полихлорированных ароматических соединений и превращении в безвредные подобных соединений в случае их присутствия в окружающей среде.
В целлюлозно-бумажной промышленности хлорированные органические вещества (монохлорированные и полихлорированные), называемые вместе "абсорбируемым или адсорбируемым органическим галогеном" или АОХ, образуются при отбеливании древесного волокна окислителями на основе хлора. Одним из таких соединений является 2,4,6-трихлорфенол (ТХФ), который образуется, например, во время процесса отбеливания при применении хлора в качестве отбеливающего агента. ТХФ попадает в конце концов в поток отработанных вод, уходящих с завода.
Вопреки опасности для окружающей среды окислители на основе хлора наиболее широко применяются для отбеливания древесного волокна в Соединенных. Штатах. Коммерческое оборудование для отбеливания древесного волокна и бумаги использует в действительности комбинацию нескольких способов. Одна широко используемая последовательность отбеливания начинается с хлорирования с последующими экстракцией при помощи NaOH, обработкой диоксидом хлора, еще одной экстракцией при помощи NaOH и затем еще одной обработкой диоксидом хлора. Модификация этой последовательности добавляет стадию окисления гипохлоритом между первой экстракцией NaOH и первой обработкой диоксидом хлора. В другой последовательности вторая экстракция при помощи NaOH и вторая обработка диоксидом хлора исключены.
14 ноября 1997 года Управление по охране окружающей среды Соединенных Штатов подписало Правила (Cluster Rule), требующие, чтобы целлюлозно-бумажная промышленность уменьшила производство хлорированных органических материалов. Для удовлетворения требований уменьшения отработанной воды эта промышленность прежде всего расширяет применение отбеливания, называемого "не содержащим элементного хлора" (ECF) отбеливанием, причем этот термин используют прежде всего для отбеливания диоксидом хлора. Важным моментом является то, что отбеливание диоксидом хлора производит значительно менее токсичную отработанную воду, чем это делает отбеливание элементным хлором (Cl2). Тем не менее некоторое количество АОХ образуется, и дополнительным недостатком является то, что сточные воды отбеливающего оборудования не могут сжигаться в регенерирующем котле, как отмечалось выше. Кроме того, промышленность разработала отбеливание, называемое "вообще не содержащим хлора" (TCF-отбеливанием). Основными окислителями TCF-отбеливания являются кислород и пероксид водорода, хотя озон также используется. Пероксид водорода окисляет и осветляет лигнин и производит высокие выходы древесной целлюлозы. Его легче использовать, чем кислород, и он не требует дорогостоящего оборудования, которое является одним из больших недостатков отбеливания кислородом. Обычно считается, что при использовании Н2О2 диссоциирует с образованием пергидроксильного иона ООН-, который обесцвечивает лигнин и не разрушает целлюлозу. Однако при разрушении Н2O2 производит свободные радикалы, которые фрагментируют лигнин, как это желательно, но также разрушают целлюлозу. Основным агрессивным радикалом является гидроксильный радикал НО-, который является крайне неселективным. Поскольку Н-O-связь воды является такой сильной (приблизительно 119 ккал·моль-1) радикал НО- будет извлекать атомы Н быстро из большого разнообразия органических соединений и фактически из большинства источников атома Н. По этой причине древесное волокно обычно обрабатывают пассивирующим агентом перед обработкой пероксидом. Целью пассивирующего агента является удаление ионов металлов, которые разрушают пероксисоединение, продуцирующее радикалы. Кроме того, способы отбеливания пероксидом водорода часто включают в себя добавление дополнительного пассивирующего агента, опять для защиты пероксисоединения от экспонирования следовым количеством металла, которое может разрушать пероксисоединение нежелательным образом и снижать его селективность. Хотя сам пероксид водорода является сильным окислителем, который может обжигать кожу и слизистые оболочки, он не является серьезно опасным в низких концентрациях (<8%). Наиболее важно то, что его применение не вводит в окружающую среду элементной токсичности. Пероксид является превосходным отбеливающим агентом. Основным недостатком применения Н2O2 в качестве окислителя для отбеливания древесного волокна и бумаги является то, что он не является таким селективным при делигнификации древесного волокна, как диоксид хлора. Процесс протекает относительно медленно, так что древесное волокно и пероксид должны быть подвергнуты нагреванию. Исторически пероксид был сравнительно дорогим окислителем. Однако цены на пероксид упали и заводы имеют свободу выбора в отношении окислителей нового поколения. Хотя Н2О2 был бы явно предпочтительным вследствие его благоприятных для окружающей среды свойств, факторы селективности и технологические расходы, связанные с его применением, способствуют уменьшению его коммерческой предпочтительности. При коммерческом применении его использовали прежде всего в качестве увеличивающего яркость агента для механического древесного волокна, например, в газетной бумаге, когда не требуется долгосрочная стабильность цвета, и лигнин прежде всего скорее обесцвечивается, чем по существу удаляется, или в качестве вспомогательного средства для хлорирования и/или отбеливания диоксидом хлора и/или отбеливания кислородом или для окисления вытекающего потока сточных вод.
В публикациях WO 98/03625 и WO 98/03626 описаны отбеливающие композиции, состоящие из макроциклического тетрадентатного активатора отбеливания и окислителя. В последней публикации утверждается, что эта отбеливающая композиция применима для отбеливания древесного волокна и лигнина. В патенте США №5032286 описан способ нагревания сточных вод целлюлозной фабрики под давлением для изменения химического состава с последующей обработкой кислым раствором для удаления хромофоров из этого раствора.
Воздействие на окружающую среду сточных вод, производимых обработкой древесного волокна и бумаги, были в центре значительных исследовательских работ на протяжении последних 30 лет. Традиционными областями обеспокоенности были потребность в кислороде, суспендированные твердые вещества и острая токсичность. Усовершенствования в стратегиях контроля на заводах, в технологии варки и отбеливания и системах вторичной обработки в большой степени справились с этими вопросами. В настоящее время центр внимания сместился в сторону потенциальной подострой токсичности (например, репродуктивных эффектов), остаточных питательных элементов/эутрофикации и неподатливых компонентов (рекальцитрантов), т.е. материалов, не поддающихся обработке, в частности красящих веществ и органического хлора. Снижения нагрузок масс красящих веществ и абсорбируемого органического хлора (хлорорганических соединений) после биологической обработки могут составлять в среднем 10 и 40% соответственно. В некоторых случаях могут происходить значительные увеличения уровней красящего вещества. Приблизительно 50% потребности растворимого химического кислорода в отбеленных сточных водах крафт-целлюлозного завода (ВКМЕ) остается также после вторичной обработки и, по-видимому, состоит из не поддающегося обработке (рекальцитрантного) высокомолекулярного материала (НММ). Компоненты более высокой молекулярной массы (MW > 1000 дальтон) в ВКМЕ состоят, в первую очередь, из высокодеградированных хлорированных продуктов деструкции лигнина с некоторым количеством остаточных полисахаридных компонентов. В ВКМЕ этот материал может составлять 40-90% общего органического материала, приблизительно 80% содержания АОХ и 60-100% загрязнений красящих веществ из данного завода. Мало информации доступно о химической природе или массовых расходах красящего вещества и НММ, выбрасываемых из других операций завода (например, механической варки).
Исследования в Скандинавии и Северной Америке показали, что эти не поддающиеся обработке (рекальцитрантные), хромофорные, галогенированные и углеродистые компоненты являются очень стойкими в системах сырой воды и могут быть детектированы в десятках километров от источника слива. Окрашенный материал оказывает явное действие на эстетику получающей сточные воды воды, а также снижает глубину проникновения света в водяном столбе и, следовательно, доступном месте обитания для макрофитов и источниках планктонных и бентосных продуктов питания. Ранее считали, что НММ является небионакапливающимся, нетоксичным и инертным вследствие его высокого молекулярного размера и водорастворимости. Более недавние исследования в настоящее время показывают, что бионакопление части этого материала может происходить в экспонируемых с ним организмах, и было сделано предположение, что НММ является основным токсикантом, ингибирующим способность оплодотворения у некоторых морских видов (например, иглокожих). НММ может также абсорбировать низкомолекулярные липофильные экотоксиканты, такие как хлорфенольные соединения. Эта ассоциация может существенно влиять на рассеяние и перемещение этих экотоксикантов и изменять их биодоступность для реципиентных организмов.
В свете этой обеспокоенности целлюлозно-бумажная промышленность находится под значительным давлением требований эффективного удаления компонентов сточных вод. Существует большое количество усовершенствованных технологий, в различных стадиях развития, которые имеют потенциальное применение для защиты окружающей среды. Они включают в себя ультрафильтрацию; флокуляцию; электротехнологии, такие как озонолиз, фотолиз и мокрое окисление; прокаливание и плазмолиз. Существенный потенциал существует в отношении применения ряда различных технологий для получения желательной обработки, таких как применение усовершенствованной обработки в сочетании с биологической и/или мембранной технологией разделения.
Усовершенствованные окислительные технологии, например, с использованием озона или пероксида, показали конкретную перспективу в качестве высокоэффективного способа удаления органического хлора или красящего вещества из сточных вод целлюлозно-бумажных заводов. Однако высокие уровни пероксида, требующиеся в созданных до настоящего времени обработках, по-видимому, делают эту технологию такой же запретительно дорогой, как и другие способы.
Некоторые хелаты металлов переходного ряда были подвергнуты поиску для не связанных с этим вопросом целей. Например, известно, что комплексы металлов переходного ряда с высокой степенью окисления функционируют в качестве окислителей в многочисленных биологических реакциях под влиянием белкового матрикса, и в последние годы возник широко распространенный интерес в понимании механизма действия и реакционной способности некоторых монооксигеназных катализаторов. Примерная программа описана в Collins T.J., "Designing Ligands for Oxidizing Complexes", Accounts of Chemical Research, 279, vol. 27, No. 9 (1994). Эта статья представляет ориентированный на конструирование подход для получения лигандов, которые являются устойчивыми к окислительной деструкции при координации с центрами, содержащими металл с высокой степенью окисления. Несколько диамидо-N-дифеноксидо- и диамидо-N-алкоксидо-ациклических хелатных соединений и макроциклических тетраамидо-N-хелатных соединений описаны в этой статье Collins (Accounts of Chemical Research).
Синтетический путь на основе азида к макроциклическим тетраамидо-металл-лигандным комплексам описан Uffelman E.S., Ph.D. Thesis, California Institute of Technology (1992). Кроме того, синтез имеющего арильный мостик тетраамидо-лиганда через путь на основе азида может происходить с использованием ароматического диамина в качестве исходного материала.
Однако в данной области еще не является признанным, что некоторые макроциклические тетраамидо-металл-лигандные комплексы будут обеспечивать новые и необычайно эффективные активаторы отбеливания для пероксисоединений. Кроме того, не было сообщений, описанных или предполагаемых, что эти типы соединений будут необычайно полезными в областях отбеливания древесного волокна и бумаги.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение включает в себя способ обесцвечивания хромофоров в сточных водах целлюлозно-бумажного производства, предусматривающий контактирование сточных вод с (а) устойчивым к окислению активатором, имеющим структуру

где Y1, Y3 и Y4 каждый обозначают мостиковую группу, имеющую нуль, один, два или три углеродсодержащих узла для замещения, a Y2 является мостиковой группой, имеющей по меньшей мере один углеродсодержащий узел для замещения, причем каждый указанный узел содержит единицу C(R), C(R1)(R2) или C(R)2 и каждый заместитель R является таким же или отличающимся от остальных заместителей R и (i) выбран из группы, состоящей из алкила, циклоалкила, циклоалкенила, алкенила, арила, алкинила, алкиларила, галогена, алкокси или фенокси, СН2СF3, СF3 и их комбинаций, или (ii) образуют замещенное или незамещенное бензольное кольцо, причем два атома углерода в этом кольце образуют узлы в единице Y, или (iii) вместе со спаренным заместителем R, связанным с тем же самым атомом углерода, образуют циклоалкильное или циклоалкенильное кольцо, которое может включать в себя атом, иной чем углерод, например циклопропильное, циклобутильное, циклопентильное или циклогексильное кольцо;
М обозначает металл переходного ряда со степенями окисления I, II, III, IV, V, VI, VII или VIII или выбран из групп 3, 4, 5, 6, 7, 8, 9, 10 и 11 Периодической системы элементов;
Q обозначает любой противоион, который уравновешивает заряд соединения на стехиометрической основе, и
(b) количеством источника окислителя, эффективным для окисления компонентов сточных вод. Окисление может проводиться с целью отбеливания субстрата, такого как ткань или древесное волокно, или бумага и другие целлюлозные материалы, окисления лигнина, обесцвечивания лигнина, делигнификации древесного волокна, обесцвечивания хромофоров, таких как хромофоры лигнина или происходящие из лигнина хромофоры, или для окисления адсорбируемых или абсорбируемых органических галогенов и углеродистых компонентов в сточных водах промышленной операции, такой как операция обработки древесного волокна и бумаги. Побочные продукты процесса производства целлюлозы и бумаги также включают в себя абсорбируемые разновидности органических галогенов, такие как ароматические соединения. Ароматические соединения включают в себя хлорированные фенолы, диоксины, дибензофураны, бифенилы и их комбинации.
Могут быть добавлены пассиваторы, стабилизаторы и другие стандартные вспомогательные вещества для отбеливания древесного волокна и бумаги, хорошо известные специалистам с квалификацией в области отбеливания древесного волокна и бумаги.
Предпочтительными активаторами окислителей являются макроциклические тетраамидо-металл-лигандные комплексы. Из них особенно предпочтительными являются комплексы, имеющие замещенный ароматический заместитель, конденсированный непосредственно в циклическую структуру лиганда.
Например, предпочтительное соединение имеет структуру
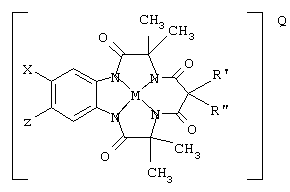
где Х и Z могут быть Н, электронодонорными или электроноакцепторными группами и R' и R” могут быть любой комбинацией заместителей Н, алкила, циклоалкила, циклоалкенила, алкенила, арила, алкинила, алкиларила, галогена, алкокси или фенокси, или объединяться с образованием циклоалкильного или циклоалкенильного кольца, которое может содержать по меньшей мере один атом, который не является углеродом; М обозначает металл переходного ряда со степенями окисления I, II, III, IV, V, VI, VII или VIII или выбран из групп 3, 4, 5, 6, 7, 8, 9, 10 и 11 Периодической системы элементов; Q обозначает любой противоион, который уравновешивает заряд соединения на стехиометрической основе.
Быстрое развитие целлюлозно-бумажной промышленности и увеличение доверия к химическим процессам отбеливания для обеспечения ярких, прочных бумажных продуктов вызывает обязательно высвобождение хлорированных побочных продуктов в окружающую среду. Промышленность нуждается в безопасной альтернативе окислителям на основе хлора для отбеливания. Любая такая технология отбеливания будет еще более желательной, если она выполняет обесцвечивание окрашенных сточных вод из любого процесса на заводе. Эта технология будет еще более желательной, если окисляющая система будет атаковать и разрушать НММ, АОХ и ВКМЕ, а также сточные воды из любого другого типа завода.
Соединения, используемые в способе данного изобретения, значительно улучшают производительность пероксида для отбеливающих древесное волокно приложений с существенными снижениями потребностей в химических веществах. Это соединение и эта окислительная композиция могут быть использованы для обработки красящих, содержащих органический хлор и не поддающихся обработке (рекальцитрантных) углеродистых материалов в древесном волокне (древесной целлюлозе).
Существует потребность в способе отбеливания древесного волокна, который значительно уменьшает выход токсических веществ в окружающую среду либо при приготовлении древесного волокна, либо при обработке сточных вод, либо в обоих процессах. Кроме того, существует большая потребность в нетоксичном для окружающей среды способе, который является легким в применении и который будет производить яркую, прочную бумагу.
Было определено, что способ данного изобретения является особенно хорошо пригодным для решения таких задач. Было показано, что способ данного изобретения быстро увеличивает скорость обесцвечивания лигнина при добавлении пероксида водорода. Кроме того, было показано, что соединение данного изобретения является очень стабильным в условиях каталитического окисления, в том числе при отбеливании древесного волокна или окислении сточных вод.
Данное изобретение обеспечивает способ окислительной деструкции полихлорированных фенолов и обесцвечивания хромофоров в сточных водах предприятий целлюлозно-бумажной промышленности. Этот способ предусматривает обычно стадии контактирования сточных вод с источником окислителя, предпочтительно пероксисоединения и более предпочтительно пероксида водорода и/или продуктов его диссоциации, и каталитических или субстехиометрических количеств активатора описанного выше состава. Кроме того, этот способ предусматривает добавление пассиватора для защиты пероксисоединения от экспонирования следовым количеством ионов металла, которые могут нежелательным образом разрушать его.
Способ может проводиться при различных температурах, но предпочтительно в диапазоне от температуры окружающей среды до приблизительно 130°С и более предпочтительно от температуры окружающей среды до 90°С. Могут быть с успехом использованы также диапазоны от температуры окружающей среды до приблизительно 40°С. Однако температура, по-видимому, не является решающей. Пригоден широкий диапазон температур. Специалистам в данной области будет понятно, что давление в этой системе должно увеличиваться при более высоких температурах.
Предпочтительным диапазоном рН является диапазон 7-12 и более предпочтительным является диапазон между 9 и 11.
Хотя в других применениях было показано, что активатор, используемый в способе данного изобретения, является превосходным активатором для окислительных реакций в растворе, в общем и в частности, в качестве активатора для активации окислителей переноса атома кислорода, таких как пероксид водорода, трет-бутилгидропероксид, кумилгидропероксид, гипохлорит и перкислоты, предпочтительным применением в способе данного изобретения является применение в качестве активатора пероксисоединений и наиболее предпочтителен в качестве активатора пероксид водорода, с кислородом или без кислорода, в отбеливании древесного волокна и бумаги. Способ данного изобретения повышает окислительные способности пероксида водорода, повышая тем самым коммерческую применимость этого благоприятного для окружающей среды окислителя.
Тысячи за тысячами метрических тонн нежелательных для окружающей среды и даже высокотоксичных, мутагенных или канцерогенных побочных продуктов теперь не должны больше образовываться. Способ данного изобретения может существенно уменьшить, если не заменить вообще, применение отбеливающих окислителей на основе хлора и токсичных побочных продуктов, которые генерирует их применение. Способ данного изобретения может быть использован для обработки крафт-целлюлозы в ранней стадии отбеливания с последующим применением диоксида хлора для дополнительного отбеливания и осветления и/или с применением некатализируемого пероксида для дополнительного отбеливания и осветления. Есть основания ожидать, что способ данного изобретения может быть также использован для усиления кислородной обработки целлюлозы путем добавления активатора и пероксида к циклам отбеливания кислородом. Кроме того, есть основания ожидать, что способ данного изобретения может быть использован для осветления целлюлозы в конце или незадолго до конца последовательности отбеливания со множественными обработками.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 является графиком, показывающим длительную активирующую стабильность предпочтительного соединения данного изобретения при добавлении с пероксидом водорода к пробе лигнина в сравнении с контролем, использующим только пероксид водорода.
Фиг.2 представляет спектр поглощения УФ/видимого света щелочного лигнина.
Фиг.3 изображает синтетический путь получения макроциклических тетраамидо-металл-лигандных комплексов данного изобретения через азидный путь.
Фиг.4 изображает синтетический путь получения макроциклических тетраамидо-металл-лигандных комплексов данного изобретения через азидный путь с использованием ароматического диамина в качестве исходного материала.
Фиг.5 является графиком, сравнивающим длительные стабильности катализаторов предпочтительных вариантов данного изобретения в сравнении с контролем.
Фиг.6 показывает продукты, идентифицированные из окисления ТХФ при помощи FePcS и H2O2 при рН 7.
Фиг.7 является графиком, показывающим изменения, которые происходят со спектром УФ/видимого света ТХФ после добавления H2O2. Жирная линия является спектром непрореагировавшего ТХФ, а пунктирная линия является спектром H2O2.
Фиг.8 является графиком, показывающим изменения поглощения при трех длинах волн, показанных на фиг.7.
Фиг.9 является графиком, показывающим множественные окисления ТХФ с использованием системы [Fe(H2O)DCB*]-/H2O2 в условиях, показанных на фиг.9. Добавление H2O2 показано * и добавление ТХФ показано #.
Фиг.10 представляет серию из трех графиков, показывающих окисление ТХФ при рН 6,8, рН 7,4 и рН 10 с использованием системы [Fe(H2O)DCB*]-/Н2О2. Все рабочие условия являются идентичными в этих трех случаях, кроме рН. ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ
Данное изобретение относится к композиции, содержащей (а) устойчивый к окислению активатор, имеющий структуру
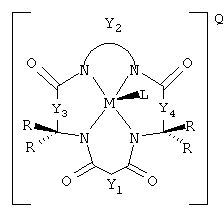
где Y1, Y3 и Y4 каждый представляют мостиковую группу, имеющую нуль, один, два или три углеродсодержащих узла для замещения, а Y2 является мостиковой группой, имеющей по меньшей мере один углеродсодержащий узел для замещения, причем каждый указанный узел содержит единицу C(R), C(R1)(R2) или С(R3) и каждый заместитель R является таким же или отличающимся от остальных заместителей R и выбран из группы, состоящей из алкила, циклоалкила, циклоалкенила, алкенила, арила, алкинила, алкиларила, галогена, алкокси или фенокси, СН2СF3, СF3 и их комбинаций, или образуют замещенное или незамещенное бензольное кольцо, причем два атома углерода в этом кольце образуют узлы в единице Y или вместе со спаренным заместителем R, связанным с тем же самым атомом углерода, образуют циклоалкильное или циклоалкенильное кольцо, которое может включать в себя атом, иной чем углерод, например циклопропильное, циклобутильное, циклопентильное или циклогексильное кольцо; М обозначает металл переходного ряда со степенями окисления I, II, III, IV, V, VI, VII или VIII, или выбран из групп 3, 4, 5, 6, 7, 8, 9, 10 и 11 Периодической системы элементов; Q обозначает любой противоион, который уравновешивает заряд соединения на стехиометрической основе, и
(b) количество источника окислителя, эффективное для окисления мишени. Окисление может проводиться с целью отбеливания субстрата, такого как ткань или древесное волокно, или бумага и другие целлюлозные материалы, окисления лигнина, обесцвечивания лигнина, делигнификации древесного волокна, обесцвечивания хромофоров, таких как хромофоры лигнина или происходящие из лигнина хромофоры, или для окисления адсорбируемых органических галогенов (АОХ) и углеродистых компонентов в сточных водах промышленной операции, такой как операция обработки древесного волокна и бумаги.
Показано, что из них предпочтительные макроциклические тетраамидо-металл-лигандные комплексы являются удивительно эффективными в разнообразной группе характеристик производительности для активаторов окислителей.
Эти лиганды получают в соответствии с процедурами, показанными на фиг.3 или 4, и описанными в патенте США №6051704, озаглавленном SYNTHESIS OF MACROCYCLIC TETRAAMIDO-N LIGANDS, включенном здесь в качестве ссылки, и включают в себя, кроме описанных здесь соединений, лиганды, описанные подробно в WO 98/03263, озаглавленном LONG-LIVED HOMOGENOUS OXIDATION CATALYSTS, который включен здесь в качестве ссылки.
1. Макроциклические тетраамидо-металл-лигандные комплексы
Соединения данного изобретения имеют структуру
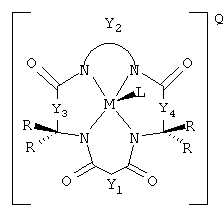
где Y1, Y3 и Y4 каждый представляют мостиковую группу, имеющую нуль, один, два или три углеродсодержащих узла для замещения, а Y2 является мостиковой группой, имеющей по меньшей мере один углеродсодержащий узел для замещения, причем каждый указанный узел содержит единицу C(R), C(R1)(R2) или С (R3) и каждый заместитель R является таким же или отличающимся от остальных заместителей R и выбран из группы, состоящей из алкила, циклоалкила, циклоалкенила, алкенила, арила, алкинила, алкиларила, галогена, алкокси или фенокси, СН2СF3, СF3 и их комбинаций, или образуют замещенное или незамещенное бензольное кольцо, причем два атома углерода в этом кольце образуют узлы в единице Y, или вместе со спаренным заместителем R, связанным с тем же самым атомом углерода, образуют циклоалкильное или циклоалкенильное кольцо, которое может включать в себя атом, иной чем углерод, например циклопропильное, циклобутильное, циклопентильное или циклогексильное кольцо; М обозначает металл переходного ряда со степенями окисления I, II, III, IV, V, VI, VII или VIII, или выбран из групп 3, 4, 5, 6, 7, 8, 9, 10 и 11 Периодической системы элементов; Q обозначает любой противоион, который уравновешивает заряд соединения на стехиометрической основе; L присутствует необязательно и может быть любым лабильным лигандом.
Особенно предпочтительный вариант этих соединений данного изобретения представлен структурой макроциклического тетраамидо-металл-лигандного комплекса
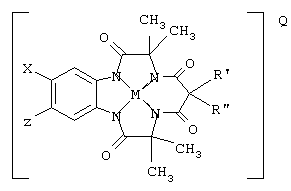
где Х и Z могут быть Н, электронодонорными или электроноакцепторными группами и R' и R" могут быть любой комбинацией заместителей Н, алкила, циклоалкила, циклоалкенила, алкенила, арила, алкинила, алкиларила, галогена, алкокси или фенокси, или объединяться с образованием циклоалкильного или циклоалкенильного кольца, которое может содержать по меньшей мере один атом, который не является углеродом; М обозначает металл переходного ряда со степенями окисления I, II, III, IV, V, VI, VII или VIII или выбран из групп 3, 4, 5, 6, 7, 8, 9, 10 и 11 Периодической системы элементов; Q обозначает любой противоион, который уравновешивает заряд соединения на стехиометрической основе, L присутствует необязательно и может быть любым лабильным лигандом.
Группы Х и Z могут быть Н или либо электронодонорными, либо электроноакцепторными группами. Электроноакцепторные группы включают в себя галогены, такие как Вr, I и наиболее предпочтительно Сl-. Кроме того, пригодными группами являются SO
R' и R", по-видимому, влияют на длительную каталитическую стабильность макроциклических тетраамидо-лигандов данного изобретения. Хотя каждый из этих заместителей может быть индивидуально выбран из заместителей Н, алкила, алкенила, арила, алкинила, галогена, алкокси или фенокси, предпочтительным, по-видимому, является алкил с короткой цепью. Особенно предпочтительно, когда R' и R" являются одинаковыми и выбраны из этила и метила или когда R' и R" объединяются с образованием циклоалкильного или циклоалкенильного кольца, в частности циклопропильного, циклобутильного, циклопентильного или циклогексильного кольца. Циклоалкильное кольцо может включать в себя по меньшей мере один атом, иной чем углерод, такой как, без ограничений, N, О или S. Наиболее предпочтительными и наиболее сильными вариантами являются варианты, в которых R' и R" являются одинаковыми и выбраны из группы, состоящей из метила, СF3, водорода, галогена и 4-членного кольца, образованного вместе с атомом углерода, с которым они оба связаны. Эти последние группы являются или нереакционноспособными, образуют сильные связи с углеродом кольца, являются стерически (пространственно) затрудненными и/или являются конформационно затрудненными, так что внутримолекулярная окислительная деструкция затруднена.
Металл М является металлом переходного ряда со степенями окисления I, II, III, IV, V, VI, VII или VIII или может быть выбран из Группы 3 (Sc, Y, лантаниды и актиниды), Группы 4 (Ti, Zr, Hf), Группы 5 (V, Nb, Та), Группы 6 (Cr, Mo, W), Группы 7 (Mn, Тс, Re), Группы 8 (Fe, Ru, Os), Группы 9 (Со, Rh, Ir), Группы 10 (Ni, Pd, Pt) и Группы 11 (Сu, Аg, Аu). Предпочтительно он выбран из группы, состоящей из Se, Ti, V, Cr, Mn, Fe, Со, Ni, Сu, Zn (Группа 12), Mo и W.
Q является любым противоионом, который уравновешивал бы заряд этого соединения на стехиометрической основе. Могут применяться как отрицательные, так и положительные противоионы. Обычно положительно заряженный противоион предпочтительно, но без ограничений, выбран из противоионов, являющихся щелочными металлами (например, К, Li, Na), [NR*4]+ и [PR*4]+, где каждый R* выбран индивидуально из Н, алкила, арила, алкиларила, алкенила или они могут быть конденсированы вместе с образованием циклоалкильного или циклоалкенильного, или арильного кольца, которое может содержать по меньшей мере один атом, иной чем углерод. Обычно отрицательно заряженный противоион предпочтительно, но без ограничения, выбран из [BF4]-1 и [PF6]-1.
L является любым лабильным лигандом, который может присоединяться к М. Эти лиганды включают в себя предпочтительно, но без ограничений, Н2O, Сl- и CN-.
Вследствие комплексной природы этих соединений они не имеют названий в данном описании, но для удобства их называют по заместителям, присутствующим в них. Например, представленная выше структура может быть названа 5,6-(4,5-ди-Х-бензо)-3,8,11,13-тетраоксо-2,2,9,9-тетраметил-12,12-диэтил-1,4,7,10-тетраазациклотридекан (или тетраметилдиэтилди-Х-бензол (TMDE-DXB, где Х = Cl, H, Me, ОМе)). Таким образом, для удобства, в приведенной выше структуре, где имеются две метильные группы, каждая на углероде α относительно амидного донора этого лиганда, и имеются две этильные группы, действующие в качестве R' и R", соединение называют TMDE-DXB. В том случае, когда R' и R" являются метильными группами, соединение называют TMDM-DXB. Когда группы Х и Z обе представляют собой хлор, соединение называют TMDE-DCB или TMDM-DCB. Используется дополнительное укорочение названия этих соединений во всем описании, где, например, TMDE-DCB называют DCB, а TMDM-DCB называют DCB*, причем * особо обозначает, что R' и R" представляют собой метил. Предпочтительным металлом переходного ряда этого лиганда является железо, так что соединение с Fe(III) и аксиальным лигандом Н2О может быть названо [Fe (H2O)DCB]-.
Общепринятые способы отбеливания пероксидом водорода практикуются при рН в диапазоне 11-9 и при температуре в диапазоне 30-80°С и наиболее часто при 50-70°С. См. Charles J.E. et al., 1980, TAPPI Pulping Conference Proceedings, TAPPI Press (1980). При использовании одного из активаторов данного изобретения температура этой реакции может быть снижена до температуры окружающей среды. Хотя эти активаторы-катализаторы могут быть использованы при более высоких обычных температурах реакции, они хорошо работают также при 35 и 40°С. В некоторых применениях могут быть предпочтительными более высокие температуры реакции, например температура до приблизительно 130°С и предпочтительно в диапазоне от температуры окружающей среды до 90°С. Известно, что при изменении на каждые десять градусов скорость реакции изменяется приблизительно в 2 раза. Таким образом, скорость реакции является гораздо более высокой при более высоких температурах. Однако при отбеливании древесного волокна с активатором данного изобретения скорости окисления H2O2, которые являются значительно лучшими, чем возможные до сих пор скорости, могут быть получены с температурами, гораздо более низкими, чем это было возможно до сих пор, что экономит энергетические ресурсы и увеличивает пропускную способность завода в случаях, когда другие характеристики этого завода делают это возможным. Таким образом, предпочтительные диапазоны температур находятся между температурой окружающей среды и 130°С, предпочтительно между температурой окружающей среды и 90°С и наиболее предпочтительно между температурой окружающей среды и 60°С. Для некоторых применений предпочтительный диапазон находится между приблизительно температурой окружающей среды и 90°С. Система отбеливания данного изобретения будет функционировать эффективно даже при температурах ниже температуры окружающей среды. Широкий диапазон температур, на протяжении которого активатор будет функционировать, позволяет использовать способ данного изобретения в существующих установках и в сочетании с другими процессами отбеливания целлюлозы и бумаги без необходимости специальной коррекции температур для части отбеливания пероксидом коммерческой технологической линии, а не производить обычно выгодное изменение снижения температуры.
рН реакции окисления также может быть понижен при использовании активатора данного изобретения. Эксперименты по отбеливанию, проводимые при рН 7 с H2О2 и активатором окислителя данного изобретения, обесцвечивали лигнин при скорости, которую считают усовершенствованием в сравнении с обычной скоростью отбеливания посредством Н2О2, но не при наилучшей скорости, возможной для этого активатора. Гораздо более быстрые и удовлетворительные скорости получали с использованием рН 10. Таким образом, нет необходимости изменения общепринятого диапазона рН 11-9 путем добавления активатора-катализатора данного изобретения, но это может быть необходимо, если требуется избежать разложения H2O2, которое, как известно, имеет место при высоком рН. Разложение может также вызываться присутствием следовых металлов в отбеливающем растворе с пероксисоединением. Пассиваторы и другие известные стабилизаторы используют для уменьшения вероятности разложения, вызываемого присутствием следовых металлов. Эксперименты, приведенные ниже, показывают, что пассиваторы могут быть также использованы с катализатором-активатором данного изобретения.
Кроме того, авторы изобретения считают, что способ данного изобретения будет давать очень благоприятные каппа-числа, меру, используемую в целлюлозно-бумажной промышленности, чтобы показать количество остаточного лигнина после отбеливания. Каппа-число, которое должно быть как можно более низким, представляет собой отношение разности между (1) общим эквивалентом окисления, необходимым для 100% удаления лигнина, и (2) разности между действительным достигаемым окислением и общим эквивалентом окисления. Его получают с использованием испытания с перманганатом калия в соответствии с процедурами, хорошо известными в целлюлозно-бумажной промышленности.
Отбеливающая композиция данного изобретения может быть использована для эффективного удаления или существенного уменьшения органического хлора или окраски, обусловленной хромофорами лигнина, из сточных вод целлюлозно-бумажного производства. Комбинация окислителя и вещества-активатора могла бы обесцвечивать хромофоры в сточных водах целлюлозно-бумажного предприятия для удаления коричневой окраски. С учетом окружающей среды окислитель в обесцвечивающей сточные воды композиции является предпочтительно пероксисоединением или озоном. Прежние попытки применения пероксидной обработки, катализируемой ионом двухвалентного железа, для удаления абсорбируемого органического галогена, оказались непригодными вследствие очень высоких уровней требующегося пероксида и запретительно высоких расходов, связанных с использованием больших количеств пероксида. Было показано, что добавление соединения-активатора данного изобретения к пероксисоединениям значительно снижает уровень пероксида, требующегося для реакций окисления. Таким образом, авторы считают, что композиция активатор/окислитель данного изобретения вполне пригодна для обработки окраски (красящего вещества), органического хлора и не поддающихся обработке углеродистых материалов (рекальцитрантов) в сточных водах целлюлозно-бумажных предприятий. Авторы считают что использование при низких уровнях потоков сточных вод либо в месте выхода, либо в месте повторного использования, является выгодным. Обработка объединенных сточных вод в местах концов трубопровода имеет то преимущество, что большая часть деградируемого органического материала в отработанной воде удалена, и остались только не поддающиеся удалению материалы (рекальцитранты), на которые должна быть нацелена композиция данного изобретения. Обработка на более ранней стадии выше по ходу потока от конца трубопровода имеет то преимущество, что она способна подвергать более высокую концентрацию соединений-мишеней действию композиции данного изобретения.
Здесь представлены эксперименты, которые демонстрируют пригодность композиции данного изобретения для окисления 2,4,6-трихлорфенола, полихлорированного ароматического соединения и загрязнителя окружающей среды, образующегося во время процесса отбеливания при использовании хлора в качестве отбеливающего агента в целлюлозно-бумажном производстве. Авторы изобретения считают, что композиция данного изобретения является эффективной для окисления также других полихлорированных ароматических соединений, таких как ДДТ, другие полихлорированные фенолы, диоксины и полихлорированные бифенилы (РСВ).
Поскольку макроциклические тетраамидо-металл-лигандные комплексы действуют как катализаторы, их количество, добавляемое к отбеливающим композициям, обычно является субстехиометрическим. Однако предпочтительно без ограничения добавлять приблизительно 0,0001 - приблизительно 999999 миллионных долей (м.д.), более предпочтительно 0,001-100000 м.д., к композициям данного изобретения.
В экспериментальном разделе ниже описаны выбранные синтезы предпочтительных макроциклических тетраамидо-металл-лигандных комплексов. Кроме того, были проведены испытания для демонстрации способности обесцвечивания лигнина и длительной каталитической активности комплексов металлов этих макроциклических лигандов данного изобретения.
2. Соединения-окислители
Соединения-окислители, такие как атомы переноса О, предпочтительно пероксисоединения, могут быть органическим или неорганическим соединением, содержащим -O-O-пероксидную связь. Примеры таких соединений включают в себя пероксид водорода, аддукты пероксида водорода, соединения, способные образовывать пероксид водорода в водном растворе, органические пероксиды, персульфаты, перфосфаты и персиликаты. Аддукты пероксида водорода включают в себя пероксигидрат карбоната щелочного металла (например, натрия, лития, калия) и пероксид мочевины, которые могут высвобождать пероксид водорода в раствор. Соединения, способные образовывать пероксид водорода в водном растворе, включают в себя перборат (моно- и тетрагидрат) щелочного металла (натрия, калия, лития). Пербораты коммерчески доступны из таких источников, как Akzo N.V. и FMC Corporation. Альтернативно в качестве источника пероксида водорода могут быть использованы фермент алкогольоксидаза и его подходящий спиртовой субстрат. Органические пероксиды включают в себя без ограничения гидропероксиды бензоила и кумена. Персульфаты включают в себя пероксимоносульфат калия (продаваемый как Oxone®, E.I. duPont de Nemours) и кислоту Каро.
Эффективным количеством пероксисоединения является количество, достаточное для генерирования по меньшей мере 0,001 м.д. активного кислорода (А.О.). Хотя это не является ограничением, предпочтительным является образование от приблизительно 0,001 до приблизительно 1000 м.д. А.О. Для отбеливания тканей предпочтительным является образование от приблизительно 0,01 до приблизительно 50 м.д. А.О. Описание и объяснение измерения А. О. можно найти в статье Sheldon N. Lewis, "Peracid and Peroxide Oxidations", In: Oxidation, 1969, pp. 213-258, которая включена здесь в качестве ссылки.
3. Вспомогательные вещества
Макроциклические тетраамидо-металл-лигандные комплексы данного изобретения, если желательно, могут быть объединены со вспомогательным веществом или основой, содержащей основные компоненты раствора и поверхностно-активные вещества, выбранные из группы, состоящей из анионогенных, катионогенных, амфотерных, цвиттерионных поверхностно-активных веществ и их смесей. Могут присутствовать другие вспомогательные материалы. Эти соединения могут быть также представлены в жидкой основе, для проведения отбеливания твердой поверхности или иной поверхности. Эти соединения могут быть применимы для отбеливающей обработки целлюлозы и текстиля. Каждое из этих соединений и вспомогательные материалы, пригодные для применения здесь, дополнительно обсуждаются ниже.
а) Основные компоненты раствора
Основными компонентами раствора являются обычно щелочные основные компоненты раствора, т.е. такие, которые в водном растворе будут давать рН 7-14, предпочтительно 9-12. Примеры неорганических основных компонентов раствора включают в себя карбонаты щелочных металлов и аммония (в том числе сесквикарбонаты и бикарбонаты), фосфаты (в том числе ортофосфаты, триполифосфаты и тетрапирофосфаты), алюминосиликаты (как природные, так и синтетические цеолиты) и их смеси. Карбонаты являются особенно желательными для применения в данном изобретении вследствие их высокой щелочности и эффективности в удалении жестких ионов, которые могут присутствовать в жесткой воде, а также их низкой стоимости. Карбонаты могут быть использованы в качестве преобладающего основного компонента раствора. Могут быть также использованы силикаты (Na2O:SiO2, модуль 4:1-1:1, наиболее предпочтительно приблизительно 3:1-1:1). Силикаты, вследствие их растворимости в воде и способности образовывать стекловидный матрикс, могут быть также выгодно использованы в качестве связующего вещества.
Органические основные компоненты также пригодны для применения и выбраны из группы, состоящей из сульфосукцинатов, полиакрилатов, полималеатов щелочных металлов и аммония, сополимеров акриловой кислоты и малеиновой кислоты или малеинового ангидрида, цитратов и их смесей.
b) Наполнители/разбавители
Наполнители для отбеливающей композиции используют для гарантии того, что на одно применение подается точное количество или точная доза очищающих активностей. Предпочтительными являются соли, такие как NaCl, Na2SO4 и бура. Возможны органические разбавители, такие как сахар. В жидком выполнении в качестве разбавителей могли бы использоваться растворители (такие как, без ограничения, алканолы, гликоли, эфиры гликолей, углеводороды, кетоны и карбоновые кислоты), жидкие поверхностно-активные вещества и вода.
с) Поверхностно-активные вещества
Поверхностно-активные вещества обычно добавляют к отбеливающему раствору для удаления конкретных загрязнений-мишеней, например, неионогенные поверхностно-активные вещества нацелены на масляные субстраты, а анионогенные поверхностно-активные вещества нацелены на состоящие из частичек субстраты. Однако, вообще говоря, окислительные отбеливающие композиции могут содержать небольшое количество поверхностно-активного вещества или вообще могут не содержать поверхностно-активных веществ.
Особенно эффективными поверхностно-активными веществами, по-видимому, являются анионогенные поверхностно-активные вещества. Примеры таких поверхностно-активных веществ могут включать в себя соли аммония, замещенного аммония (например, моно-, ди- и триэтаноламмония), щелочных металлов и щелочноземельных металлов С6-С20-жирных кислот и кислот канифоли, алкил(линейный или разветвленный)бензолсульфонаты, алкилсульфаты, сульфаты неполных алкиловых простых эфиров, алкансульфонаты, олефинсульфонаты, гидроксиалкансульфонаты, сульфаты моноглицеридов жирных кислот, сульфаты алкилглицериловых простых эфиров, ацилсаркозинаты и ацил-N-метилтауриды. Предпочтительными являются алкиларилсульфонатные поверхностно-активные вещества, такие как алкилбензолсульфонаты.
Другие применимые предпочтительные поверхностно-активные вещества включают в себя линейные этоксилированные спирты, такие как продаваемые Shell Chemical Company под товарным названием NEODOL. Другие пригодные неионогенные поверхностно-активные вещества могут включать в себя другие линейные этоксилированные спирты со средней длиной 6-16 атомов углерода и в среднем с приблизительно 2-20 моль этиленоксида на 1 моль спирта; линейные и разветвленные, первичные и вторичные этоксилированные, пропоксилированные спирты со средней длиной приблизительно 6-16 атомов углерода и в среднем с 0-10 моль этиленоксида и приблизительно 1-10 моль пропиленоксида на 1 моль спирта; линейные и разветвленные алкилфенокси(полиэтокси)спирты, известные также как этоксилированные алкилфенолы, со средней длиной цепи 8-16 атомов углерода и в среднем с 1,5-30 моль этиленоксида на 1 моль спирта, и их смеси.
Далее, пригодные неионогенные поверхностно-активные вещества могут включать в себя эфиры полиоксиэтилена и карбоновой кислоты, эфиры жирной кислоты и глицерина, алканоламиды жирных кислот и этоксилированных жирных кислот, некоторые блок-сополимеры пропиленоксида и этиленоксида и блок-сополимеры пропиленоксида и этиленоксида с пропоксилированным этилендиамином. Также включены такие семиполярные неионогенные поверхностно-активные вещества, как аминоксиды, фосфиноксиды, сульфоксиды и их этоксилированные производные.
Пригодные катионогенные поверхностно-активные вещества могут включать в себя соединения четвертичного аммония, в которых обычно одна из групп, связанная с атомом азота, является С12-С18-алкильной группой, в другие три группы являются алкильными группами с короткими цепями, которые могут нести заместители, как фенильные группы.
Далее, пригодные амфотерные и цвиттерионные поверхностно-активные вещества, которые содержат анионную солюбилизирующую в воде группу, катионную группу и гидрофобную органическую группу, могут включать в себя аминокарбоновые кислоты и их соли, аминодикарбоновые кислоты и их соли, алкилбетаины, алкиламинопропилбетаины, сульфобетаины, производные алкилимидазолиния, некоторые соединения четвертичного аммония, некоторые соединения четвертичного фосфония и некоторые соединения третичного сульфония.
Другие примеры анионогенных, неионогенных, катионогенных и амфотерных поверхностно-активных веществ, которые могут быть пригодны для применения в данном изобретении, представлены в Kirk-Othmer, Encyclopedia of Chemical Technology, Third Edition, Volume 22, pages 347-387, и McCutcheon's, Detergents and Emulsifiers, North American Edition, 1983, которые включены здесь в качестве ссылки.
d) Хелатообразователи
В некоторые описанные здесь композиции особенно предпочтительно включать хелатообразователь, наиболее предпочтительны в применениях для отбеливания белья - аминополифосфонат, а в применениях для отбеливания целлюлозы поликарбоксилат. Эти хелатообразователи способствуют поддержанию стабильности в растворе окислителя для достижения оптимальной производительности. Таким образом они действуют путем образования хелатов свободных ионов тяжелых металлов. Хелатообразователь выбран из ряда известных агентов, которые являются эффективными в образовании хелатов свободных ионов тяжелых металлов. Хелатообразователь должен быть устойчивым к гидролизу и быстрому окислению окислителями. Предпочтительно он должен иметь константу диссоциации в кислоте (рКа) 1-9, указывающую, что он диссоциирует при низких рН для усиления связывания с катионами металлов. Наиболее предпочтительным хелатообразователем для применений в отбеливании белья является аминополифосфонат, который коммерчески доступен под товарным названием DEQUEST из Monsanto Company. Примерами его являются DEQUEST 2000, 2041, 2060 и 2066. Полифосфонат, такой как DEQUEST 2010 является также пригодным для применения. Другие хелатообразователи, такие как этилендиаминтетрауксусная кислота (ЭДТА) и нитрилотриуксусная кислота (NTA), являются предпочтительными для применений в отбеливании древесного волокна. Другими новыми предпочтительными хелатообразователями являются новые пропилендиаминтетраацетаты, такие как Hampshire 1,3 PDTA от W.R. Grace и Chel DTPA 100#F от Ciba-Geigy A.G. Могут быть пригодными смеси перечисленных хелатообразователей. Эффективные количества хелатообразователя находятся в диапазоне 1-1000, более предпочтительно 5-500, наиболее предпочтительно 10-100 м.д. хелатообразователя в промывном растворе.
е. Другие вспомогательные вещества
Стандартные вспомогательные вещества окислительного отбеливания могут быть включены в данное изобретение. Те, которые включают в себя ферменты, являются особенно предпочтительными вспомогательными материалами в продуктах окислительного отбеливания. Однако может быть предпочтительным включение стабилизатора фермента.
Особенно предпочтительным классом ферментов являются протеазы. Их выбирают из кислых, нейтральных и щелочных протеаз. Термины "кислые", "нейтральные" и "щелочные" относится к рН, при котором активность ферментов является оптимальной. Примеры нейтральных протеаз включают MILEZYME (доступный из Miles Laboratory) и трипсин, природно встречающуюся протеазу. Щелочные протеазы доступны из большого круга источников и обычно их получают из различных микроорганизмов (например, Bacillus subtilis). Типичные примеры щелочных протеаз включают MAXATASE и MAXACAL, из International BioSynthetics, ALCALASE, SAVINASE И ESPERASE, все доступные из Novo Industri A/S. См. также Stanislowski et al., патент США №4511490, включенный здесь в качестве ссылки.
Другими подходящими ферментами являются амилазы, которые являются гидролизующими углеводы ферментами. Предпочтительно включать смеси амилаз и протеаз. Подходящие амилазы включают в себя RAPIDASE, из Societe Rapidase, MILEZYME из Miles Laboratory и MAXAMYL из International BioSynthetics.
Другими подходящими ферментами являются липазы, такие как описанные в Silver, патент США №3950277, и Thom et al., патент США №4707291, оба включены здесь в качестве ссылки.
Другими представляющими здесь интерес ферментами являются пероксидазы, такие как пероксидаза хрена и пероксидазы, описанные в Международной патентной публикации WO 93/24628, включенной здесь в качестве ссылки. Предпочтительными являются смеси любых из предыдущих гидролаз, в частности смеси протеазы и амилазы.
Кроме того, необязательные вспомогательные вещества включают красители, такие как "монастрал синий" (фталоцианиновый органический пигмент) и антрахиноновые красители (такие как описанные в Zielske, патент США №4661293 и патент США №4746461).
Пигменты, которые являются также пригодными красителями, могут быть выбраны, без ограничения, из диоксида титана, ультрамарина синего (см. также Chang et al., патент США №4708816) и окрашенных алюминосиликатов.
Другими желательными вспомогательными веществами являются флуоресцентные белящие агенты. Они включают производные стильбена, стирола и нафталина, которые при освещении ультрафиолетовым светом испускают или флуоресцируют свет с длиной волны видимого света.
Могут быть добавлены дополнительные органические активаторы отбеливания, в том числе, но не ограничивающиеся ими, эфиры (см. Fong et al., патент США №4778618, и Rowland et al., патент США №5182045), кетоны, имиды (см. Kaaret, патент США №5478569) и нитрилы, все включены здесь в качестве ссылки.
Эти добавки могут присутствовать в количествах в диапазоне 0-50%, более предпочтительно 0-30% и наиболее предпочтительно 0-10%. В определенных случаях некоторые из индивидуальных вспомогательных веществ могут перекрываться в других категориях. Однако данное изобретение рассматривает каждое из вспомогательных веществ как обеспечивающее различные выгодные для производительности свойства в их различных категориях.
ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
Синтез окислительно сильных тетраамидо-лигандов
Материалы. Все растворители и реагенты были требуемой для реагентов чистоты (Aldrich, Aldrich Sure-Seal, Fisher) и их использовали в том виде, в каком их получали. Микроанализы выполнялись Midwest Microlabs, Indianopolis, IN.
Масс-спектрометрия. Масс-спектры с использованием электрораспылительной ионизации получали на масс-спектрометре FINNIGAN-MAT SSQ700 (San Jose, CA), оборудованном электрораспылительным интерфейсом ANALYTICA OF BRANDFORD. Использовали напряжения электрораспыления 2400-3400 В. Пробы растворяли в ацетонитриле или дихлорметане при концентрациях приблизительно 10 пмоль/мл и вводили в ESI-интерфейс перед получением данных прямым вливанием при скорости потока 1 л/мин и вводили перед получением данных. МС-эксперименты с ионизацией электронным ударом положительных ионов (70 эВ) проводили на квадрупольном масс-спектрометре FINNIGAN-MAT 4615, соединенном с системой данных INCOS. Температура источника электронов была 150°С и температура распределительной камеры была 100°С. Пробы вводили посредством газового хроматографа или зондом прямого введения. Масс-спектры положительных ионов с бомбардировкой ускоренными атомами получали на приборе с магнитным сектором FINNIGAN-MAT 212 в комбинации с системой данных INCOS. Ускоряющее напряжение было 3 кВ и температура источника электронов была приблизительно 70°С. Использовали пушку ускоренных атомов ION TECH saddle field с ксеноном при 8 кэВ. В качестве матрикса FAB (бомбардировки ускоренными атомами) использовали тиоглицерин. МС/МС-эксперименты с ионизацией положительных ионов с электронным ударом (70 эВ) проводили на последовательном квадрупольном масс-спектрометре FINNIGAN-MAT TSQ/700. Пробы вводили с использованием зонда для прямого введения. Источник ионов поддерживали при 150°С, а распределительную камеру поддерживали при 70°С. Индуцированную соударениями диссоциацию (CID) получали введением аргона в центральный октаполюс только rf-столкновений до тех пор, пока давление в распределительной камере не достигало 0,9-2,5×10-6 Торр. Номинальная кинетическая энергия ионов для ионов продуктов CID была <35 эВ (лабораторная ссылка). Данные высокого разрешения получали на масс-спектрометре с двойной фокусировкой JEOL JMS AX-505H в ЕВ-конфигурации с использованием разрешения 7500. Пробы вводили при помощи газового хроматографа или зонда для прямого введения. Во время получения масс-спектральных данных в источник ионов вводили перфторкеросин посредством нагреваемого впускного отверстия. Точные масс-спектральные соотнесения получали компьютерной интерполяцией из масс перфторкеросина. ГХ/МС-условия: колонка 20 м × 0,25 мм DB-1701 (J & W Scientific); газ-носитель - гелий с линейной скоростью 40 см/сек; инжектор, 125°С; температура колонки 35°С в течение 3 минут, затем увеличение при 10°С/мин до 100°С; инжекция, разделенный режим, отношение приблизительно 50:1.
Спектроскопические способы. 1H-ЯМР-спектры (300 МГц) и 13С-ЯМР-спектры (75 МГц) получали на приборе IBM AF300 с использованием сверхпроводящей магнитной системы OXFORD, получение данных управлялось программным обеспечением BRUKER. Инфракрасные спектры получали на спектрофотометре MATTSON GALAXY Series 5000 FTIR, регулируемом компьютером MACINTOSH II. Спектры УФ/видимый свет получали на спектрофотометре HEWLETT PACKARD 8452A, управляемым компьютером ZENITH Z-425/SX. Обычные спектры Х-полосного ЭПР (электронного парамагнитного резонанса) регистрировали на спектрофотометре BRUKER ER300, оборудованном гелиевым проточным криостатом OXFORD ESR-900. Мессбауэровские спектры получали на приборах с постоянным ускорением и изомерные смещения даются относительно железного металлического стандарта при 298 К. Во избежание ориентации поликристаллических проб приложенным магнитным полем эти пробы суспендировали в замороженном нуджоле (очищенном парафиновом масле).
Синтезы макроциклических тетраамидо-N-донор-лигандов
Общая схема реакций
Ниже изображена предпочтительная последовательность реакций для синтеза макроциклических тетраамидо-металл-лигандных комплексов
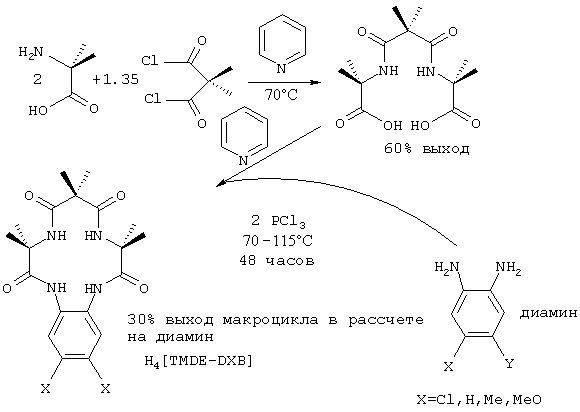
α-Аминокарбоновую кислоту смешивают с активированным малонатом в пиридине при температурах ниже 70°С. После завершения реакции селективного двойного связывания, 72-144 ч, выделяют MACRO LINKER (A-L-A). Во второй стадии диамин, предпочтительно о-фенилендиамин, добавляют к раствору в пиридине этого MACRO LINKER в присутствии связующего агента, предпочтительно РСl3 или пивалоилхлорида. Реакции замыкания цикла (двойное связывание) дают протекать при дефлегмации (с обратным холодильником) в течение 48-110 часов и затем целевой макроциклический тетраамид выделяют с хорошим выходом.
В нижеследующих примерах 1-25 представлены различные части этих реакционных стадий. Примеры 26-39 демонстрируют особенности выполнения и преимущества данного изобретения для окислительных реакций, включающих в себя обесцвечивание (отбеливание) лигнина и обесцвечивание красящего вещества.
Пример 1
Синтез промежуточного продукта макролинкера (A-L-A) из α-метилаланина и диэтилмалонилдихлорида (тетраметилдиэтилзамещенного промежуточного продукта)
Двугорлую колбу (1 л), снабженную капельной уравнительной воронкой (для уравнивания давления) (250 мл) и разделительной мембраной, помещают под N2. В эту колбу добавляют α-аминоизомасляную кислоту (т.е. α-метилаланин) (20,62 г, 0,2 моль) и сухой пиридин (250 мл, высушенный над молекулярными ситами 4  ) и нагревают при 60-70°С при перемешивании, затем в делительную воронку добавляют диэтилмалонилдихлорид (23,23 мл; 0,135 моль), растворенный в сухом пиридине (100 мл, высушенном над молекулярными ситами 4
) и нагревают при 60-70°С при перемешивании, затем в делительную воронку добавляют диэтилмалонилдихлорид (23,23 мл; 0,135 моль), растворенный в сухом пиридине (100 мл, высушенном над молекулярными ситами 4  ). Содержимое капельной воронки добавляют (по каплям, 1 ч) к реакции и ацилированию дают протекать (60-70°С, 30-36 ч) под N2 или с использованием сушильной трубки, которой снабжают колбу. По завершении ацилирования реакцию гасят добавлением Н2О (30 мл) и перемешиванием (60-70°С, 24 ч). Объем растворителя уменьшают на роторном испарителе с получением масла, затем добавляют НСl (концентрированную, приблизительно 25 мл) до конечного рН 2-3. Горячий раствор ставят в холодильник (4°С, 15 ч) и полученный рыжевато-коричневый продукт собирают фильтрованием через фритту и промывают основательно ацетонитрилом (2×100 мл). Высушенный на воздухе белый продукт (16,5-19,8 г, выход 50-60%) должен храниться в эксикаторе. Этот продукт обычно является достаточно чистым для реакции замыкания кольца, но иногда может потребоваться перекристаллизация.
). Содержимое капельной воронки добавляют (по каплям, 1 ч) к реакции и ацилированию дают протекать (60-70°С, 30-36 ч) под N2 или с использованием сушильной трубки, которой снабжают колбу. По завершении ацилирования реакцию гасят добавлением Н2О (30 мл) и перемешиванием (60-70°С, 24 ч). Объем растворителя уменьшают на роторном испарителе с получением масла, затем добавляют НСl (концентрированную, приблизительно 25 мл) до конечного рН 2-3. Горячий раствор ставят в холодильник (4°С, 15 ч) и полученный рыжевато-коричневый продукт собирают фильтрованием через фритту и промывают основательно ацетонитрилом (2×100 мл). Высушенный на воздухе белый продукт (16,5-19,8 г, выход 50-60%) должен храниться в эксикаторе. Этот продукт обычно является достаточно чистым для реакции замыкания кольца, но иногда может потребоваться перекристаллизация.
1H-ЯМР-спектр (d5-пиридин) δ [м.д.]: 8,9 (с, 2Н, амидный NH); 2,2 (к, 4Н); 1,8 (с, 12Н); 1,2 (т, 6Н). ИК (нуджол mull): n [см-1] = 3310 (амидный NH); 1721 (карбоксильный СО); 1623 (амидный СО). Анал., рассчит. для C15H21N2O6, %; С 54,53; Н 7,93; N 8,48. Найдено, %: С 54,48; Н 7,88; N 8,47.
Пример 2
Большой масштаб, синтез промежуточного продукта макролинкера (A-L-A) из α-метилаланина и диэтилмалонилдихлорида (TMDE-замещенного промежуточного продукта)
Двугорлую колбу (2л, RB + колба Кляйзена), снабженную капельной уравнительной воронкой (для уравнивания давления) (250 мл) и мембранами помещают под N2. В эту колбу добавляют α-аминоизомасляную кислоту (т.е. α-метилаланин) (90,3 г, 0,9 моль), канюлируют безводный пиридин (1,4 л, Aldrich Sure seal) и эту реакционную смесь нагревают до 45-55°С и перемешивают. Пиридин (100 мл, Aldrich Sure seal) и затем диэтилмалонилдихлорид (104,4 мл, 0,61 моль) канюлируют в делительную воронку. Содержимое делительной воронки добавляют (по каплям, 3-4 ч) к реакции, затем делительную воронку удаляют и ацилированию дают протекать (55-65°С, 120-130 ч) под N2. По завершении ацилирования реакцию гасят добавлением Н2O (100 мл) и перемешиванием (60-70°С, 24-36 ч). Объем растворителя уменьшают на роторном испарителе с получением масла, затем добавляют НСl (конц., приблизительно 110 мл) до конечного рН 2-3. Горячий раствор помещают в холодильник (4°С, 15 ч) и полученный рыжевато-коричневый продукт собирают фильтрованием через фритту и промывают основательно ацетонитрилом (700 мл, 150 мл) перемешиванием в колбе Эрленмейера. Высушенный на воздухе белый продукт (87,9 г, выход 60%) измельчают пестиком в ступке и хранят в эксикаторе. Этот промежуточный амидный продукт реакции большого масштаба, более вероятно, требует перекристаллизации перед применением его в реакции замыкания кольца.
Пример 3
Перекристаллизация TMDE-замещенного промежуточного продукта
Неочищенный TMDE-промежуточный продукт из примера 2 (50,4 г, 0,153 моль) растворяют в Н2О (500 мл, деионизованная вода) добавлением Nа2СО3 (16,2 г, 0,153 моль) в трех аликвотах медленно и осторожно во избежание избыточного пенообразования, с хорошим перемешиванием и небольшим нагреванием. Этот раствор доводят до кипения, фильтруют и подкисляют НСl (конц., 30 мл, 0,36 моль). Раствору дают остыть (в течение ночи, 4°С) и белый осадок отфильтровывают и промывают ацетонитрилом (250 мл). Высушенный на воздухе продукт (38,8-45,4 г, перекристаллизованный, выход 77-90%) должен храниться в эксикаторе.
Пример 4
TMDM-замещенный промежуточный продукт (A-L-A)
Синтез TMDM-замещенного промежуточного продукта идентичен синтезу TMDE-замещенного промежуточного продукта в примере 2 со следующими исключениями: диэтилмалонилдихлорид заменяют диметилмалонилдихлоридом (17,8 мл, 0,135 моль) и температура реакции должна быть уменьшена до 55-65°С из-за более низкой точки кипения этого ацилирующего агента. Выход TMDM-промежуточного продукта равен 45-60%.
1H-ЯМР-спектр (d5-пиридин) δ [м.д.]: 9,2-9,8 (шс, 2Н, карбоксильный ОН), 8,23 (с, 2Н, амид), 1,87 (с, 12Н, СН3), 1,74 (с, 6Н, СН3). ИК (нуджол/NaCl) ν [см-1]: 3317,0 (амидный NH); 1717,9 (карбоксильный СО); 1625,7 (амидный СО). Анал. (высушенный при 100°С) рассчит. для C13H22N2O6, %: С 51,63, Н 7,34, N 9,27. Найдено, %: С 51,64, Н 7,35, N 9,33.
Пример 5
Перекристаллизация TMDM-замещенного промежуточного продукта
Неочищенный TMDM-промежуточный продукт перекристаллизовывали таким же образом, что и TMDE-замещенный промежуточный продукт. Вследствие несколько более высокой растворимости в воде для TMDM-замещенного промежуточного продукта следует использовать немного меньшее количество H2O.
Пример 6
Di CyHex Di Ethyl (DiCyHexDE)-замещенный промежуточный продукт
Круглодонную колбу (500 мл) загружают 1-амино-1-циклогексанкарбоновой кислотой (15 г, 0,1 моль), затем подсоединяют уравнительную капельную воронку (40 мл), закрытую разделительной мембраной, и продувают азотом. Безводный пиридин (300 мл) канюлируют в эту реакционную колбу через капельную воронку и 20 мл в капельную воронку. Начинают нагревание системы и стабилизируют температуру при 60°С. По достижении 60°С одну треть всего диэтилмалонилхлорида, который должен использоваться в этой реакции (т.е. 6 мл, 0,033 моль), добавляют через шприц в капельную воронку. Смесь пиридин/диэтилмалонилдихлорид добавляют по каплям к реакции и ацилированию дают протекать в течение 12 часов. Вторую аликвоту (6 мл, 0,033 моль) и третью аликвоту (6 мл, 0,033 моль) добавляют с интервалами 12 часов. После добавления всего количества ацилирующего агента и протекания реакции (общее время реакции 48-56 ч) к реакции добавляют по каплям 20 мл воды. Реакцию нагревают в течение дополнительных 24 часов для раскрытия кольца промежуточных продуктов моно- и бис-оксазалона и получения диамиддикарбоновой кислоты. Удаление пиридина на роторном испарителе дает бледный желтовато-рыжеватый осадок (шлам), который подкисляют до рН 2 НСl (концентрированной). Неочищенный продукт собирают фильтрованием, промывают ацетонитрилом и сушат на воздухе с получением белого DiCyHexDE-замещенного промежуточного продукта (16 г, 74%).
1H-ЯМР-спектр (d5-пиридин) δ [м.д.]: 8,30 (с, 2Н, амидный NH), 2,60 (м, 4Н, cyhex), 2,25 (к, 4Н, СН2 этила), 2,15 (м, 4Н, cyhex), 1,8-1,5 (м, 10Н, cyhex), 1,25 (м, 2Н, cyhex), 1,20 (т, 6Н, СН3 этила). 13С-ЯМР со спиновой развязкой широкой полосы (d5-пиридин) δ [м.д.]: 178,0 (СО карбоксила), 174,3 (СО амида), 60,5 (cyhex кват), 59,4 (малонил кват), 33,0 (CH2 cyhex а), 30,3 (CH2 этила), 26,0 (СН2 cyhex g), 22,3 (CH2 cyhex b), 9,9 (СН3 этила). ИК (нуджол/NaCl) ν [см-1]: 3307 (амидный NH); 3150 (sh плечо), шм, амидный NH/карбоксильный ОН), 3057 (с, str (сильный), Н связанный амидный NH/карбоксильный ОН), 1717 (с, str, карбоксильный СО); 1621 (с, str, амидный СО). Анал. рассчит. для C21H34N2O6, %: С, 61,44; Н 8,35; N 6,82. Найдено, %: С 61,41, Н 8,38; N 6,90%.
Пример 7
Di CyHex Diethyl монооксазалон
Невозможность погасить реакцию Di CyHex Diethyl-промежуточного продукта ( нагреванием и водой, см. выше) при стехиометрии 1,35 диэтилмалонилдихлорид : 2 СуНех-аминокислота приводит к смеси Di CyHex Diethyl-замещенного промежуточного продукта и продукта монооксазалона. Di CyHex Diethyl-монооксазалоновый продукт является умеренно растворимым в кипящем циклогексане, тогда как циклогексиламидный промежуточный продукт является нерастворимым, что позволяет выполнить простое разделение этой смеси продуктов. Приблизительно 10 г смеси амидного промежуточного продукта и монооксазалона, содержащей небольшое количество остаточного CH2Cl2, кипятили при интенсивном перемешивании в 400-500 мл циклогексана. Нерастворимый Di CyHex Diethyl-замещенный промежуточный продукт собирали горячим гравитационным фильтрованием, тогда как монооксазалоновый продукт кристаллизовался постепенно по мере охлаждения и упаривания раствора циклогексана. Выход амидного производного равен приблизительно 6 г, выход монооксазалона равен приблизительно 4 г. Характеристика монооксазалона: 1H-ЯМР-спектр (d5-пиридин) δ [м.д.]: 9,7 (с, 1Н, амидный NH), 2,7-1,6 (неразделенные группы СyHes), 1,05 (т, 6Н, СН3 этила). ИК (нуджол/NaCl) ν [см-1]: 3309 (sh, w, амидный NH), 3229 (с, str, H связанный амидный NH/карбоксильный ОН), 3166 (с, str, Н связанный амидный NH/карбоксильный ОН), 3083 (с, str, H связанный амидный NH/карбоксильный ОН), 1834 (с, str, С=О оксаз), 1809 (с, m, H связанный С=О оксаз), 1743 (с, str, карбоксильный СО), 1663 (с, str, C=N оксаз), 1639 (с, шир, str, амидный СО). Анал. рассчит. для C21H32N2O5 (C6H12), %: 0,25; С 65,35; H 8,53; N 6,77. Найдено, %: С 65,07; H 8,67; N 6,68%. Присутствие сольватированного циклогексана было подтверждено посредством 13С-ЯМР.
Реакции макроциклизации
Далее описаны примеры нескольких синтетических путей для получения макроциклических тетраамидо-металл-лигандных комплексов.
Связывание посредством трихлорида фосфора
Связывание посредством трихлорида фосфора амидсодержащего промежуточного продукта (A-L-A) с ароматическим 1,2-диамином дает надежно, дешево и с высоким выходом макроциклические тетраамиды. Применимы два различных варианта способа РСl3-связывания, различия между ними относятся к порядку добавления и выбору используемых реагентов. Эти способы применимы для получения большого разнообразия различных макроциклов с различными электронными заместителями, присутствующими на диаминовом мостике, или стерическими заместителями, присутствующими на амидном промежуточном продукте, прежде всего вследствие параллельного включения макролинкерного типа амидных промежуточных продуктов во все эти синтезы.
Пример 8
А. Синтез макроциклов через РСl3-связывание
Длинногорлую колбу (250 мл) загружают амидным промежуточным продуктом примеров 2-7 (10 ммоль), стержнем-мешалкой и затем обжигают в сушильной камере (80-100°С, 30-45 минут). Горячую колбу помещают под N2, добавляют арилдиамин (10 ммоль) и вводят через канюлю безводный пиридин (50 мл, Aldrich Sure seal). Колбу нагревают (50-60°С) и вводят через шприц РСl3 (d=1,574 г/мл, 1,72 мл, 20 ммоль) насколько возможно быстро без излишней дефлегмации. Реакция является экзотермической, так что следует соблюдать осторожность. Затем температуру увеличивают до температуры дефлегмации или чуть ниже температуры дефлегмации (100-115°С) и реакции дают протекать под N2 (48 ч). После завершения ацилирования содержимое колбы подкисляют НСl (1 экв., приблизительно 60 мл) до конечного рН 2. Смесь переносят в колбу Эрленмейера (для ополаскивания колбы используют воду) и перемешивают с CH2Cl2 (300 мл, 2-3 ч), затем экстрагируют дополнительным количеством CH2Cl2 (2×150 мл). Объединенные органические слои промывают разбавленной НСl (0,1 М, 2×100 мл), затем разбавленным водным Nа2СО3 (2×5 г/100 мл). Органические растворители удаляют на роторном испарителе с получением неочищенного продукта (30%). Вес неочищенного продукта обычно эквивалентен первоначальному весу диамина.
В. Синтез макроциклов через РСl3-связывание
Длинногорлую колбу (250 мл) загружают МgSO4 (5 г), стержнем-мешалкой, арилдиамином (10 ммоль) и пиридином (50 мл, высушенным над молекулярными ситами 4  ), затем помещают под N2. РСl3 (d=1,754 г/мл, 1,72 мл, 20 ммоль) добавляют через шприц и смесь доводят до дефлегмации в течение 30 минут, причем образуется оранжево-желтый осадок. Смесь немного охлаждают, добавляют амидный промежуточный продукт (10 ммоль), затем содержимое колбы нагревают с обратным холодильником при дефлегмации под N2 (115°С, 48 ч). После завершения ацилирования содержимое колбы подкисляют НСl (1 экв., приблизительно 60 мл) до конечного рН 2. Смесь переносят в колбу Эрленмейера и перемешивают с CH2Cl2 (2×150 мл). Объединенные органические слои промывают разбавленной НСl (0,1 М, 2×100 мл), затем разбавленным водным Nа2СО3 (2×5 г/100 мл). Органические растворители удаляют на роторном испарителе с получением неочищенного продукта (30%). Вес неочищенного продукта обычно эквивалентен первоначальному весу диамина.
), затем помещают под N2. РСl3 (d=1,754 г/мл, 1,72 мл, 20 ммоль) добавляют через шприц и смесь доводят до дефлегмации в течение 30 минут, причем образуется оранжево-желтый осадок. Смесь немного охлаждают, добавляют амидный промежуточный продукт (10 ммоль), затем содержимое колбы нагревают с обратным холодильником при дефлегмации под N2 (115°С, 48 ч). После завершения ацилирования содержимое колбы подкисляют НСl (1 экв., приблизительно 60 мл) до конечного рН 2. Смесь переносят в колбу Эрленмейера и перемешивают с CH2Cl2 (2×150 мл). Объединенные органические слои промывают разбавленной НСl (0,1 М, 2×100 мл), затем разбавленным водным Nа2СО3 (2×5 г/100 мл). Органические растворители удаляют на роторном испарителе с получением неочищенного продукта (30%). Вес неочищенного продукта обычно эквивалентен первоначальному весу диамина.
Замечание. Для реакций макроциклизации большого масштаба периоды времени для замыкания кольца увеличивают до 4-5 дней при дефлегмации и большую часть пиридина, присутствующего в конце реакции, удаляют на роторном испарителе перед подкислением.
Пример 9
TMDE-DCB из TMDE-промежуточного продукта + DCB-диамина
1,2-Диамино-4,5-дихлорбензол (1,77 г, 10 ммоль) использовали в качестве арилдиамина с TMDE-амидным промежуточным продуктом (3,3 г, 10 ммоль) в реакции макроциклизации А или В РСl3-способа. Неочищенный макроциклический продукт (2,7 г) перекристаллизовывали из минимального количества горячего 95% EtOH упариванием с получением чистого TMDE-DCB (1,5 г, 32%).
1H-ЯМР (CD2Cl2) δ [м.д.]: 7,65 (с, 1Н, АrН), 7,35 (с, 2Н, амидный NH), 6,45 (с, 2Н, амидный NH), 1,90 (к, 4Н, СН2 этила), 1,57 (с, 12Н, RСН3), 0,85 (т, 6Н, СН3 этила). ИК (нуджол/NaCl) ν [см-1]: 3454 (следы ROH), 3346 (шир, амидный NH), 1706 и 1688 и 1645 (амидный СО). Анал. рассчит. для C21H28Cl2N4O4, %: С 53,51; Н 5,99; N 11,89. Найдено, %: С 53,58; Н 6,09; N 11,89.
Пример 10
TMDE-B из TMDE-промежуточного продукта + В-диамина
1,2-диаминобензол (т.е. о-фенилендиамин) (1,08 г, 10 ммоль) использовали в качестве арилдиамина с TMDE-амидным промежуточным продуктом (3,3 г, 10 ммоль) в реакции макроциклизации А и В РСl3-способа. Неочищенный макроциклический продукт (1,5 г) перекристаллизовывали из минимального количества горячего 95% EtOH упариванием с получением чистого TMDE-B (25% в расчете на диамин).
1H-ЯМР (CDCl3) δ [м.д.]: 7,55 (м, 2Н, АrН), 7,48 (с, шир, 2Н, ариламидный NH), 7,17 (м, 2Н, АrН), 6,46 (с, шир, 2Н, алкиламидный NH), 2,07 (м, шир, 4Н, СН2 этила), 1,60 (с, 12Н, RСН3), 0,89 (т, 6Н, СН3 этила). ИК (нуджол/NaCl) ν [см-1]: 3395, 3363 (амидный NH), 1702, 1680, 1652, 1635 (амидный СО). Анал. рассчит. для C21H10N4O4·Н2О, %: С 59,98; Н 7,67; N 13,32. Найдено, %: С 60,18; Н 7,20; N 13,18.
Пример 11
TMDE-DMB из TMDE-промежуточного продукта + DMB-диамина
1,2-Диамино-4,5-диметилбензол (1,36 г, 10 ммоль) использовали в качестве арилдиамина с TMDE-промежуточным продуктом (3,3 г, 10 ммоль) в реакции макроциклизации А и В РСl3-способа. Неочищенный макроциклический продукт (1,6 г) перекристаллизовывали из минимального количества горячего 95% EtOH упариванием с получением чистого TMDE-DMB (25% в расчете на диамин).
1H-ЯМР (DMSO-d6) δ [м.д.]: 8,00 (с, 2Н, амидный NH), 7,67 (с, 2Н, амидный NH), 7,28 (с, 2Н, АrН), 2,17 (с, 6Н, СН3 арила), 1,99 (к, 4Н, СН2 этила), 1,46 (с, 12Н, RСН3), 0,75 (т, 6Н, СН3 этила). ИК (нуджол/NaCl) ν [см-1]: 3446 (с, м, следы ROH), 3362 (с, str, амидный NH), 3348 (sh, м, амидный NH), 3332 (с, str, Н, амидный NH), 1696 (СО амида), 1679 (СО амида), 1651 (СО амида), 1641 (СО амида), 1584 (с, m/w (средне-слабый), арильное кольцо/амид). Анал. рассчит. для C23H34N4O4, %: С 64,16; Н 7,96; N 13,01. Найдено, %: С 64,09, 64,28; Н 8,04, 7,92; N 12,86, 13,04.
Пример 12
TMDE-DMOB из TMDE-промежуточного продукта + DMOB-диамина
1,2-Диамино-4,5-диметоксмбензол·2 HBr (5,0 г, 15 ммоль), полученный, как описано выше, использовали в качестве арилдиамина непосредственно с TMDE-промежуточным продуктом (5,0 г, 15 ммоль) в реакции макроциклизации А или В 1,5-масштабного РСl3-способа. Неочищенный макроциклический продукт (3,57 г) перекристаллизовывали из минимального количества горячего 80-85% EtOH (1 г/40 мл) упариванием с получением чистого TMDE-DMOB (30% в расчете на диамин).
1H-ЯМР (CD2Cl2) δ [м.д.]: 7,26 (с, 2Н, амидный NH), 7,01 (с, 2Н, АrН), 6,41 (с, 2Н, амидный NH), 3,80 (с, 6Н, ОСН3 арила), 2,07 (шк, 4Н, СН3 этила), 1,54 (с, 12Н, RСН3), 0,90 (т, 6Н, СН3 этила). ИК (нуджол/NaCl) ν [см-1]: 3451 (с, м, Н-связанная Н2О), 3391, 3347 (амидный NH), 1695, 1670, 1655 (амидный СО). Анал. рассчит. для C23H34N4O6·(Н2O)0,33, %: С 58,96; Н 7,46; N 11,96. Найдено (ESU), %: С 58,90; Н 7,26; N 11,76. Присутствие сольватированной Н2О подтверждалось 1H-ЯМР и ИК.
Пример 13
TMDE-нафталин из TMDE-промежуточного продукта + нафталиндиамина
4,5-Диаминонафталин (1,68 г, 10 ммоль) использовали в качестве арилдиамина с TMDE-промежуточным продуктом (3,3 г, 10 ммоль) в реакции макроциклизации А или В РСl3-способа. Неоптимизированный выход составлял 15-20% в расчете на диамин. 1H-ЯМР (СDСl3) δ [м.д.]: 8,05 (с, 2Н, кольцо АrН а), 7,75 (м, 2Н, кольцо АrН b), 7,55 (с, 2Н, амидный NH Аr), 7,35 (м, 2Н, кольцо АrН b), 6,45 (с, 2Н, NH алкиламида), 2,15 (шм, 4Н, СН2 этила), 1,65 (с, 12Н, RСН3), 0,90 (е, 6Н, СН3 этила).
Пример 14
TMDM-DCB из TMDM-промежуточного продукта + DCB-диамина
1,2-Диамино-4,5-дихлорбензол (1,77 г, 10 ммоль) использовали в качестве диамина с промежуточным продуктом TMDM-амидом (3,02 г, 10 ммоль) в реакции макроциклизации А или В РСl3-способа. Неочищенный макроцикл (1,33 г, 30%) перекристаллизовывали из минимального количества горячего н-пропанола упариванием, выход первой перекристаллизации был 60%.
1H-ЯМР δ [м.д.]: 7,69 (с, 2Н, АrН), 7,39 (с, 2Н, амидный NH), 6,44 (с, 2Н, амидный NH), 1,58 (с, 12Н, плеч. метилы), 1,53 (с, 6Н, метилы малоната), наблюдали небольшие пики н-пропанола. ИК (нуджол/NaCl) ν [см-1]: 3503 (с, шир, m-w, ОН н-пропанола), 3381 (sh, м, амидный NH), 3338 с, str, амидный NH), 1689 (с, str, амидный СО), (с, str, амидный СО). Анал. рассчит. для C19H24N4O4Cl2·(С3Н8О)0,2, %: С 51,70; Н 5,57; N 12,30. Найдено, %: С 51,69; Н 5,63; N 12,33.
Пример 15
TMDM-DMOB и TMDM-B из TMDM-промежуточного продукта + DMOB или В-диамина
TMDM-промежуточный продукт использовали также для синтеза TMDM-B и TMDM-DMOB в соответствии с тем же самым способом и с результатами, подобными полученным в примере 14 для дихлорпроизводного. Данные 1H-ЯМР для TMDM-DMOB в CDCl3, δ [м.д.]: 7,65 (с, 2Н, амидный NH), 7,21 (с, 2Н, СН арила), 6,72 (с, 2Н, амидный NH), 4,00 (с, 6Н, СН3 метокси), 1,76 (с, 12Н, плеч. метилы), 1,58 (с, 6Н, метилы малоната). Данные 1H-ЯМР для TMDM-B в d5-пиридине, δ [м.д.]: 8,55 (с, 2Н, амидный NH), 8,40 (с, 2Н, амидный NH), 7,81 (м, 2Н, ArH aa'bb'), 7,10 (м, 2Н, ArH aa'bb'), 1,77 (с, 12Н, плеч. метилы), 1,73 (с, 6Н, метилы малоната). Амидные пики склонны смещаться на несколько десятых м.д. в присутствии загрязнителей, таких как вода, кислоты и т.д.
Пример 16
DiCyHexDE-DCB из DiCyHexDE-промежуточного продукта + DCB-диамина
1,2-Диамино-4,5-дихлорбензол (1,77 г, 10 ммоль) использовали в качестве арилдиамина с Di Cy Hex DE-промежуточным продуктом (3,3 г, 10 ммоль) в реакции макроциклизации А или В РСl3-способа. Вследствие увеличенного пространственного затруднения рекомендуется увеличенное время реакции замыкания кольца (3-4 дня против обычных 48 часов). Су Hex Оксазалоны, образующиеся в качестве побочного продукта во время этой реакции, не удаляются основной кислотной обработкой, так что необходимо растирать/промывать исходно выделенный СН2Сl2-растворимый продукт с пентаном для удаления этих оксазалонов. Упаривание пентановых промывок делает возможным повторное использование оксазолонов. Неочищенный нерастворимый в пентане продукт перекристаллизовывали растворением в СН2Сl2 или СНСl3 с добавлением циклогексана до легкого помутнения и последующим упариванием на воздухе (1-2 дня) с получением белого микрокристаллического DiCyHexDE-DCB-продукта, который собирали фильтрованием (1,38 г, 25% в расчете на диамин). Перекристаллизация из горячего толуола с упариванием также кажется перспективной.
1H-ЯМР (CDCl3) δ [м.д.]: 7,70 (с, 2Н, АrН), 7,45 (с, 2Н, амидный NH), 6,45 (с, 2Н, амидный NH), 2,35 (шм, 4Н, cyhex), 2,00 (шм, >>8Н, cyhex/CH2 этила), 1,70 (шм, >>8Н, cyhex), 1,30 (шм, >> 4Н, cyhex), 0,90 (т, 6Н, СН3 этила). Анал. (высушенный при 100°С), рассчит. для С27Н36Сl2N4O4·(C6H12)0,2, %: С 59,60; Н 6,81; N 9,86. Найдено, %: С 59,60; Н 6,77; N 9,77. Присутствие сольватированного циклогексана подтверждалось 1H-и 13С-ЯМР.
Пример 17
DiCyHexDE-B из DiCyHexDE-промежуточного продукта + В-диамина
1,2-Диаминобензол (орто-фенилендиамин, 1,08 г, 10 ммоль) использовали в качестве арилдиамина в получении, аналогичном получению DiCyHexDE-DCB с получением DiCyHexDE-B (1,25 г, 26% в расчете на диамин).
1H-ЯМР (DC3CN) δ [м.д.]: 7,62 (с, 2Н, ариламидный NH), 7,51 (м, 2Н, АrН), 7,18 (м, 2Н, АrН), 6,71 (с, 2Н, алкиламидный NH), 2,12 (м, 6Н, Cyhex), 1,85 (к + м, СН2 этила + cyhex), 1,62 (м, cyhex), 1,37 (м, cyhex), 0,90 (т, 6Н, СН3 этила), 0,85 (м, cyhex). ИК (нуджол/NaCl) ν [см-1]: 3750 (с, м, Н2O), 3385 (с, str, амидный NH), 314 (с, str, амидный NH), 3258 (с, м, шир, Н связанный амидный NH), 1694 (с, str, амидный СО), 1651 (с, str, амидный СО), 1594 (с, м, арильное кольцо/амид).
Пример 18
DiCyHexDE-бис-оксазалон
Этот продукт получали в качестве побочного продукта реакции макроциклизации РСl3-способа DiCyHexDE-амидного промежуточного продукта с о-фенилендиамином. Бис-оксазалон не удаляется кислотно-основной обработкой (он является нейтральной молекулой и очень хорошо растворим в органических растворителях). Промывание неочищенного макроциклического/оксазалонового продукта пентаном экстрагирует большую часть этого бис-оксазалона в пентан. Упаривание на воздухе пентанового слоя дает чистый бис-оксазалон в виде больших (1 см × 1 см × 0,5 см) прозрачных призм. Из-за объемных гидрофобных СуНех-групп этот оксазалон является гораздо более устойчивым к гидролизу, чем соответствующее метилсодержащее производное. Характеристика бис-оксазалона: 1H-ЯМР (DС3СN) δ [м.д.]: 2,05 (к, 4Н, СН2 этила), 1,8-1,4 (неразделенные Су Hex-группы), 0,88 (т, тН, СН3 этила). 13С-ЯМР со спиновой развязкой широкой полосы (СD3СN) δ [м.д.]: 181,0 (С=O оксаз.), 162,7 (C=N оксаз.), 69,0 (СуНех оксаз. кват), 49,0 (малонат кват), 34,3 (метилены cyhex а), 25,5 метилены cyhex g), 24,9 (метилены малоната), 21,8 (метилены cyhex b), 8,3 (СН3 этила). ИК (нуджол/NaCl) ν [см-1]: 1822 (с, str, шир, С=O оксаз.), 1662 (с, str, C=N оксаз.). Анал. (высушенный при 50°С), рассчитано для С21Н30N2O4, %: С 67,36; Н 8,07; N 7,48. Найдено, %: С 67,26; Н 8,15; N 7, 64.
Синтезы хелатных комплексов
Пример 19
[Et4N]2 и [Et4N]3, [тетраэтиламмониевые соли дианиона железо(III)-хлор-TMDE-DCB [Fe(Cl)DCB]2- и моноаниона железо(III)-акво-TMDE-DCB [Fe(H2O)DCB]- соответственно].
Исходный макроциклический тетраамид любого из примеров 10-18, приведенных выше (525 мг, 1,1 ммоль), растворяют в тетрагидрофуране (40 мл, Aldrich) в атмосфере азота. С использованием способов Шленка к раствору добавляли трет-бутиллитий (2,6 мл, 4,4 ммоль, 1,7 М в 2,4-диметилпентане, Aldrich) в атмосфере азота при -108°С. Затем добавляли хлорид железа(II) (безводный, 155 мг, 1,2 ммоль, Alfa) и раствор нагревали до комнатной температуры при перемешивании (16 ч) с получением оливково-зеленого осадка, чувствительного к воздуху комплекса Fe(II). Воздух впускали через сушительную трубку (2 ч) и оранжевое твердое вещество собирали и промывали СН2Сl2 (2×10 мл). Полученный оранжевый порошок сушили при пониженном давлении. Выход 595 мг (>>93%). Вследствие вариабельной сольватации в ограниченной растворимости эту соль лития превращали и тетраэтиламмониевую соль для дальнейшего использования. Соль лития (595 мг) в СН3ОН (50 мл) наносили на ионообменную колонку (Dowex® 50Х2-100, 25 г, 2 см × 12,5 см), которая была предварительно насыщена катионами [Et4N]+, и оранжевую полосу элюировали СН3ОН (100 мл). Растворитель удаляли при пониженном давлении.
Остаток суспендировали в CH2Cl2 (20 мл) и смесь фильтровали. Растворитель удаляли из маточного раствора при пониженном давлении с получением оранжевого гигроскопичного стекловидного остатка [Et4N]2, который использовали без дополнительной очистки. ИК (нуджол/NaCl) ν [см-1]: 1619 (ν CO амида), 1575 (ν СО амида), 1534 (ν CO амида). Более удобным подходом для тщательной очистки исходного железо(III)-материала было использование аксиального аква-моноанионного комплекса, чем этого аксиального хлор-дианионного комплекса. [Et4N]2 (550 мг, приблизительно 0,7 ммоль) растворяли в СН3СN (50 мл). Тетрафторборат серебра (140 мг, 0,7 ммоль) растворяли в СН3СN (2 мл) и добавляли к этому раствору при перемешивании (1 ч). AgCl-осадок отфильтровывали и растворитель удаляли при пониженном давлении. Полученный [Et4N]3 дополнительно очищали элюцией через колонку силикагеля (8% МеОН в СН3Сl2). Растворитель удаляли при пониженном давлении и продукт перекристаллизовывали из Н2О. Выход 360 мг (>>77% вариабельная сольватация с водой была обнаружена в различных микрокристаллических пробах). ИК (нуджол/NaCl) [см-1]: 1590 (ν СО амида), 1565 (ν CO амида), 1535 (n СО амида). Анал. рассчит. для C29H46N5FeO5Cl2·(Н2О), %: С 50,52; Н 7,02; N 10,16; Cl 10,28. Найдено, %: С 50,24; Н 6,84; N 9,82; Cl 10,32. ESIMS (отрицательный ион): m/z 552/2, [3-Н2О]1- (100%); m/z 269,7, [3-Н+]2- (18%).
Пример 20
[Et4N]4, [тетраэтиламмониевая соль моноаниона железо(IV)-хлор-TMDE-DCB]
[Et4N]2 (500 мг, приблизительно 0,6 ммоль) растворяли в CH2Cl2 (30 мл). К раствору добавляли нитрат аммония-церия(IV) (10,3 г, 18,3 ммоль) и смесь перемешивали (2 ч). Твердые соли церия удаляли фильтрованием. Пурпурный продукт получали удалением растворителя при пониженном давлении и сушкой под вакуумом. Выход 400 мг (>>95%). Пурпурные кристаллы получали перекристаллизацией из смеси CH2Cl2/Et2O. ИК (нуджол/NaCl) [см-1]: 1688 (ν СО амида), 1611 (ν СО амида), 1582 (ν CO амида). ESIMS (отрицательный ион): m/z 557 [4]-1 (100%); m/z 522 [4-Сl]1- (65%).
Пример 21
Синтез [Ph4P]5 [тетрафенилфосфониевой соли моноаниона железо(IV)-циано-TMDE-DCB] из [Et4N]4 [тетраэтиламмониевой соли моноаниона железо(IV)-хлор-TMDE-DCB] и NaCN
[Et4N]4 [тетраэтиламмониевую соль моноаниона железо(IV)-хлор-TMDE-DCB] (225 мг, 0,33 ммоль) суспендировали в Н2O (10 мл). Цианид натрия (140 мг, 2,85 ммоль) растворяли в Н2O (10 мл), добавляли к этой суспензии и эту смесь обрабатывали ультразвуком (Branson 1200, 0,5 ч). Пурпурная суспензия изменялась, превращаясь в темно-синий раствор, и почти весь твердый материал растворялся. Смесь фильтровали и синий продукт осаждали добавлением PPh4Cl [тетрафенилфосфонийхлорида], растворенного в воде (600 мг, 1,6 ммоль, 10 мл, Aldrich). Синий осадок собирали и промывали Н2О (2×10 мл). Выход 250 мг (0,28 ммоль, >>85%). Этот материал (120 мг) дополнительно очищали тонкослойной хроматографией (ТСХ) (пластинка силикагеля, GF, 20 см × 20 см × 1000 мм, 10:1 СН2Сl2 : СН3СN). Синий материал экстрагировали из этого силикагеля смесью СН3СN :СН2Сl2 (1:1, 60 мл). Растворитель удаляли при пониженном давлении и остаток растворяли в СН2Сl2 (3 мл) и фильтровали. Добавление пентана (150 мл) давало синий порошок (90 мг, 0,10 ммоль). Выход после очистки 75%. ИК (нуджол/NaCl) [см-1]: 2119 (ν CN), 1659 (ν СО амида), 1598 (ν СО амида), 1571 (ν СО амида). Анал., рассчит. для C46H44N5FeOCl2P, %: С 62,18; Н 4,99; N 7,88; Cl 7,98. Найдено, %: С 61,96; Н 5,04; N 7,84; Cl 8,06. ESIMS (отрицательный ион): m/z 548,2, [5]1- (100%); m/z 522,1, [5-CN]1- (20%). Для 13С-меченого цианида: m/z 549,2, [5]1- (100%); m/z 522,1, [5-13CN]1- (8%).
Пример 22
Синтез [Ph4P]5 [тетрафенилфосфониевой соли моноаниона железо(IV)-циано-TMDE-DCB] из источников нитрилов (цианидов)
[Ph4P]5 [тетрафенилфосфониевая соль моноаниона железо(IV)-циано-TMDE-DCB] может быть образован в присутствии или в отсутствие основания. В отсутствие основания синий цвет превращается в желто-оранжевый по мере удаления растворителя в процедурах обработки. Таким образом, выделение продукта для получения синего твердого вещества лучше всего проводить в присутствии добавленного основания в диапазоне рН 9-10. Последующая реакция дает [Ph4P]5 с каждым из СН3СN, CD3CN, СН3СН2СN и (СН3)2СНСN в качестве субстратов-растворителей. Основание не добавляли к описанным каталитическим реакциям. Было определено, что синее соединение является эффективным предшественником катализатора, добавлением выделенного [Ph4P]5 к раствору в ацетонитриле ТВНР (трет-бутилгидропероксида) как растворитель, так и окислитель потреблялись, что указывало на то, что, хотя [Ph4P]5образуется в качестве конечного продукта каталитического окислительного процесса, он не является дезактивированной формой этого катализатора.
Пример 23
Синтез [Ph4P]5 в присутствии основания
[Et4N]3 (160 мг, 0,23 ммоль) растворяли в выбранном нитрильном растворителе (6 мл), см. пример 19. Основание - гидроксид тетраэтиламмония - добавляли (20 мас.%, 0,370 мл, 0,52 ммоль, Aldrich), затем добавляли по каплям при перемешивании (20 мин) трет-бутилгидропероксид (90%, 0,605 мл, 5,4 ммоль, Aldrich) с получением синего раствора. Остаточный нитрил удаляли при пониженном давлении с получением маслянистого синего остатка, который растворяли в Н2O (15 мл) и фильтровали. Синий материал осаждали из фильтрата добавлением водного раствора PPh4Cl (800 мг, 2,1 ммоль, Aldrich, 10 мл). Синий осадок собирали и промывали H2O (2×10 мл). Выход 130 г, 0,15 ммоль (65%). Дополнительную очистку проводили, как описано в разделе [РН4Р]5, в примере 25.
Пример 24
Данные рентгеновской (кристаллической) структуры и уточнение для [Et4N]3-H2O
C29H48Cl2FeN5O6, М = 689,47, триклинный, пространственная группа Р-1, а = 9,899(2); b = 11,771(2); с = 14,991(4)  = 95,33(2); = 100,09(2); g = 92,31(2)°, V = 1709,6(6)
= 95,33(2); = 100,09(2); g = 92,31(2)°, V = 1709,6(6)  3, Dнабл = 1,33 г·см-3, Dpaccч(Z = 2) = 1,339 г·см-3, Т = 293 К, 1 = 0,71069
3, Dнабл = 1,33 г·см-3, Dpaccч(Z = 2) = 1,339 г·см-3, Т = 293 К, 1 = 0,71069  , m=0,64 мм-1, коэффициент переноса 0,87-1,00. Данные диффракции собирали при комнатной температуре на диффрактометре Enraff-Nonius CAD-4 с использованием монохроматизированной графитом Мо-Ка-радиации. Три отражения подвергали мониторингу во время всего сбора данных, причем наблюдались только случайные флуктуации в интенсивности. Структуру определяли прямыми методами. Атомы водорода, связанные с углеродом, включали в рассчитанные положения с длиной связи С/Н 0,96
, m=0,64 мм-1, коэффициент переноса 0,87-1,00. Данные диффракции собирали при комнатной температуре на диффрактометре Enraff-Nonius CAD-4 с использованием монохроматизированной графитом Мо-Ка-радиации. Три отражения подвергали мониторингу во время всего сбора данных, причем наблюдались только случайные флуктуации в интенсивности. Структуру определяли прямыми методами. Атомы водорода, связанные с углеродом, включали в рассчитанные положения с длиной связи С/Н 0,96  и уточняли с использованием модели (riding model) с термическим параметром на 20% более высоким, чем у исходного углерода. Местоположения атомов водорода молекулы воды были определены из разностных карт (диаграмм) распределения электронной плотности, и их координаты позволили производить уточнения с термическим параметром, фиксированным при величине, на 20% большей, чем эта величина у кислорода. Уточнение проводили по методу наименьших квадратов с полной матрицей на F2 с факторами рассеяния, взятыми из Международных таблиц. Все не являющиеся водородом атомы уточняли с анизотропными термическими параметрами. Конечные разностные карты были невыразительными. Уточнение сходилось к R=0,053, wR2 = 0,112 с весами 1,0/[s2F
и уточняли с использованием модели (riding model) с термическим параметром на 20% более высоким, чем у исходного углерода. Местоположения атомов водорода молекулы воды были определены из разностных карт (диаграмм) распределения электронной плотности, и их координаты позволили производить уточнения с термическим параметром, фиксированным при величине, на 20% большей, чем эта величина у кислорода. Уточнение проводили по методу наименьших квадратов с полной матрицей на F2 с факторами рассеяния, взятыми из Международных таблиц. Все не являющиеся водородом атомы уточняли с анизотропными термическими параметрами. Конечные разностные карты были невыразительными. Уточнение сходилось к R=0,053, wR2 = 0,112 с весами 1,0/[s2F
Пример 25
Данные рентгеновской (кристаллической) структуры и уточнение для [Et4N]4
Отдельные кристаллы [Et4N]4 при 20±1°С являются моноклинными, пространственная группа P21/c-C , b = 14,956(3)
, b = 14,956(3)  , с = 22,688(5)
, с = 22,688(5)  , а = 90,00, _ = 93,83(2), g = 90,00, V= 3372(1)
, а = 90,00, _ = 93,83(2), g = 90,00, V= 3372(1)  3 и Z = 4 (dрассч = 1,357 г·см-3: ma(CuKa)- = 6,17 мм-1). Всего 4626 независимых отражений с поправкой на адсорбцию, имеющих 2q(CuKa) < 115,0°, собирали с использованием Q-2Q сканов и Ni-фильтрованного Сu-Ка-излучения. Структуру определяли с использованием способов "Прямых методов" с пакетом программного обеспечения NICOLET SHELXTL, модифицированным в Crystalytics Company. Полученные структурные параметры оганичивались сходимостью R1 (невзвешенной, на основе F) = 0,037 для 2680 независимых отражений, имеющих 2Q(CuKa) < 115,0° и I > 3s (I). Десять метильных групп были уточнены как жесткие вращающиеся части с sр3-гибридизованной геометрией и длиной связи С - Н 0,96
3 и Z = 4 (dрассч = 1,357 г·см-3: ma(CuKa)- = 6,17 мм-1). Всего 4626 независимых отражений с поправкой на адсорбцию, имеющих 2q(CuKa) < 115,0°, собирали с использованием Q-2Q сканов и Ni-фильтрованного Сu-Ка-излучения. Структуру определяли с использованием способов "Прямых методов" с пакетом программного обеспечения NICOLET SHELXTL, модифицированным в Crystalytics Company. Полученные структурные параметры оганичивались сходимостью R1 (невзвешенной, на основе F) = 0,037 для 2680 независимых отражений, имеющих 2Q(CuKa) < 115,0° и I > 3s (I). Десять метильных групп были уточнены как жесткие вращающиеся части с sр3-гибридизованной геометрией и длиной связи С - Н 0,96  . Первоначальную ориентацию каждой метильной группы определяли из разностных положений Фурье для атомов водорода. Конечную ориентацию каждой метильной группы определяли тремя ротационными параметрами. Уточненные положения для жестких вращающихся метильных групп имеют С-С-Н-углы, которые имеют диапазон от 103° до 118°. Остальные атомы водорода были включены в расчеты показателя структуры как идеализированные атомы (с предположением sp2- или sр3-гибридизации атомов углерода и длины связи С - Н 0,96
. Первоначальную ориентацию каждой метильной группы определяли из разностных положений Фурье для атомов водорода. Конечную ориентацию каждой метильной группы определяли тремя ротационными параметрами. Уточненные положения для жестких вращающихся метильных групп имеют С-С-Н-углы, которые имеют диапазон от 103° до 118°. Остальные атомы водорода были включены в расчеты показателя структуры как идеализированные атомы (с предположением sp2- или sр3-гибридизации атомов углерода и длины связи С - Н 0,96  ), несомые на их соответствующих атомах углерода. Изотропный термический параметр каждого атома водорода был фиксированным при величине в 1,2 раза большей, чем эквивалентный изотропный термический параметр углерода, с которым он ковалентно связан.
), несомые на их соответствующих атомах углерода. Изотропный термический параметр каждого атома водорода был фиксированным при величине в 1,2 раза большей, чем эквивалентный изотропный термический параметр углерода, с которым он ковалентно связан.
Пример 26
Отбеливание лигнина с пероксидом водорода и [Fe (H2O)DCB*]-при рН 10
В кварцевую кювету с длиной пути света 1 см, содержащую 3,0 мл смеси 0,1 М NаНСО3/Nа2СО3 (рН 10), термостатируемую при 25°С, добавляли 60 мкл насыщенного раствора щелочного лигнина и 300 мкл исходного раствора катализатора (1,24×10-4 М [Fe (H2O) DCB* ]- (где R' и R" представляют собой метил, что подразумевается звездочкой *, а противоионом является катион тетраэтиламмония), все в воде. Этот раствор перемешивали и добавляли 3,8 мкл 30% H2O2. Изменения поглощения при 350, 376, 400, 426, 450 и 476 нм измеряли с использованием спектрофотометра Hewlett-Packard UV/Vis, работающего в режиме Кинетики одной ячейки. При добавлении H2O2 поглощение быстро увеличивается при всех длинах волн и затем быстро снижается. После 15 минут поглощение при каждой длине волны было ниже исходной величины, что указывало на то, что имело место обесцвечивание лигнина. Производили второе добавление 60 мкл лигнина, которое вызывало быстрое повышение поглощений, как и прежде, и затем следующее снижение исходного повышения происходило более медленно, чем прежде. Во всем эксперименте образовывались пузырьки.
Спустя 30 минут добавляли дополнительные 3,8 мкл H2O2. Поведение было сходным с поведением, наблюдаемым ранее. За быстрым увеличением поглощения следовало снижение.
Пример 27
Отбеливание лигнина без [Fe(H2O)DCB*]- при рН 10
Стадии примера 26 повторяли с исключением катализатора. В кварцевую кювету с длиной пути света 1 см, содержащую 3,0 мл смеси 0,1 М NaHCO3/Na2CO3 (pH 10), термостатируемую при 25°С, добавляли 60 мкл насыщенного раствора щелочного лигнина и смесь перемешивали. Спустя короткий период времени после начала получения данных добавляли 3,8 мкл 30% Н2О2.
Измерения поглощения производили с использованием тех же самых параметров, которые описаны в примере 26.
При добавлении Н2О2 все шесть длин волн обнаружили повышение поглощения. Это повышение не было быстрым и не было таким острым, как в катализируемой реакции. Поглощение постепенно начинало опускаться вниз, но это происходило очень медленно. Пузырьков не наблюдали в смеси в пределах первых 15 минут. В конце часа начинали появляться пузырьки.
Сравнение предварительных экспериментов в примерах 26 и 27 показывает, что добавление активатора данного изобретения увеличивает скорость, с которой Н2О2 обесцвечивает (отбеливает) лигнин.
Пример 28
Отбеливание лигнина с пероксидом водорода, пассиватором и без [Fe(H2O)DCB*]- при pH 10
Стадии примера 27 повторяли с добавлением пассиватора DEQUEST 2066, 2 мкл, хелатообразователя для ионов свободного металла. Добавление H2O2 давало картину постепенного повышения и снижения, подобную картине, наблюдаемой в примере 27.
Пример 29
Отбеливание лигнина с пероксидом водорода, пассиватором и без [Fe(H2O)DCB*]- при рН 7
Стадии примера 27 повторяли при рН 7 с использованием буфера 0,0087 моляльный КН2РO4/0,030 моляльный Na2HPO4. В кювету добавляли 2 мкл хелатообразователя DEQUEST 2066. Не наблюдали поддающегося оценке отбеливания в рамках времени 1 часа этого эксперимента. Минимальную активность наблюдали в следах поглощения при 350 нм, но ее отнесли к шуму.
Пример 30
Отбеливание лигнина с пероксидом водорода, [Fe(H2O)DCB*]- и пассиватором при рН 10
В кювете, снабженной перемешивающим стержнем, смешивали 1 эквивалент катализатора примера 26 (300 мкл исходного раствора [Fe(H2O)DCB*]-, 60 мкл насыщенного раствора щелочного лигнина, забуференного, как описано прежде, и 2 мкл хелатообразователя DEQUEST 2066. Поглощение измеряли с использованием тех параметров, которые описаны в примерах 26 и 27.
Спустя 1-2 минуты в кювету добавляли 1000 эквивалентов 30% Н2О2 (3,8 мкл). Это вызывало быстрое увеличение в поглощении с последующим быстрым уменьшением, как описано в примере 26.
Спустя 20 минут дополнительные 60 мкл лигнина добавляли в кювету. Поглощение при всех длинах волн повышалось более медленно и затем снижалось более медленно, чем после добавления H2O2.
Спустя 30 минут добавляли дополнительный эквивалент (300 мкл) катализатора [Fe(H2O)DCB*]-. Не наблюдали значимых изменений.
Спустя 40 минут дополнительные 3,8 мкл H2O2 добавляли в кювету. Это вызывало значимое уменьшение поглощения при всех длинах волн, что указывало на то, что опять имело место обесцвечивание лигнина.
Пример 31
Отбеливание лигнина с пероксидом водорода, [Fe(H2O)DCB*]- и пассиватором при рН 7
Пример 29 повторяли, но с добавлением 300 мкл катализатора. 3,8 мкл 30% Н2О2 добавляли после нескольких циклов. При добавлении H2O2 поглощение повышалось при каждой из шести длин волн, использованных в примере 26, но не драматично. Поглощения продолжали медленно повышаться в течение первых 15 минут, выходили на плато и затем начинали падать для всех шести длин волн. Спустя один час поглощение было более высоким, чем первоначальное поглощение.
Пример 32
Длительная активность катализатора
В кварцевую кювету с длиной пути света 1 см, содержащую 3,0 мл смеси 0,1 М NaHCO3/Na2CO3 (рН 10), термостатируемую при 25°С, добавляли 60 мкл насыщенного раствора щелочного лигнина, 300 мкл (12,4 мкМ) исходного раствора катализатора (1,24×10-4 М [Fe(H2O)DCB*]-) и 2 мкл DEQUEST 2066, все в воде. Смесь перемешивали, получение данных начинали, как в примере 26, и затем добавляли 19 мкл (5000 эквивалентов) 30% H2O2. После первого быстрого повышения поглощения с последующим быстрым снижением, каждые 15 минут добавляли аликвоты 60 мкл насыщенного раствора щелочного лигнина и 19 мкл (5000 эквивалентов) 30% H2O2.
Результаты, полученные при подвергаемой мониторингу длине волны 476 нм, показаны сплошной линией на графике на фиг.1. Такие же результаты получали при других подвергаемых мониторингу длинах волн. Добавления лигнина и Н2О2 указаны звездочками.
Для сравнения готовили кювету насыщенного раствора лигнина, хелатообразователя и Н2O2 без катализатора и измеряли поглощение. Эти результаты показаны пунктирной линией на фиг.1.
Пример 33
Длительная стабильность катализатора
На фиг.5 показано сравнение каталитической "долговечности" двух вариантов данного изобретения в тесте с красителем. Соединение 1 имеет заместители R' и R", каждый из которых представляет собой СН3, тогда как соединение 2 имеет заместители R' и R", каждый из которых представляет собой -СН2СН3. В контроле катализатор не добавляли.
Условиями были: рН 9, комнатная температура (21,1°С), буферная система NаНСО3/Nа2СО3. Окислителем был 4 мМ (30%) H2O2. В каждой из звездочек добавляли 12 мкМ пинацианолхлоридный краситель.
Как можно видеть из графика на фиг.5, каждое добавление красителя в том варианте, где присутствовало соединение 1, приводило к почти немедленному обесцвечиванию. Соединение 2, диэтилсоединение, показало более постепенное обесцвечивание. Контроль показал лишь очень постепенную скорость обесцвечивания.
Пример 34
Окисление 2,4,6-трихлорфенола (ТХФ)
ТХФ мог быстро окисляться при различных условиях в воде посредством H2O2 с использованием комплекса железа [Fe(H2O)DCB*]-, показанного на фиг.1, в качестве активатора H2O2. Недавняя работа по окислению ТХФ была проведена с использованием либо Н2О2, либо KHSO5 в качестве окислителя и водорастворимого комплекса железа 2,9,16,23-тетрасульфофталоцианина (FePcS) в качестве активатора окислителя. См. Sorokin A., Seris J.-L., Meunier В., Science, vol. 268, рр. 1163-1166 (1995). В этих исследованиях было обнаружено, что ТХФ более эффективно расщеплялся до молекулярных разновидностей, показанных на фиг.6, когда окислителем был KHSO5. Однако с практической точки зрения желательно использовать в качестве окислителя Н2O2 вследствие его легкой доступности при низкой стоимости. В статье Sorokin et al., Science, цитируемой выше, сообщалось, что соединения, названные "связанными продуктами" на фиг.6, дают пурпурный раствор. Эти связанные продукты не окисляются далее системой FePcS/H2O2. См. Sorokin A., De Suzzoni-Dezard S.; Poullain D., Noel J.-P., Meunier B. J. Am. Chem. Soc., vol. 118, pp. 7410-7411 (1996). Поскольку эти связанные продукты являются также полихлорированными ароматическими соединениями, существует вероятность того, что они являются нежелательными с точки зрения защиты окружающей среды. Здесь обеспечено доказательство, что [Fe (H2O)DCB*]- окисляет далее некоторые из связанных продуктов окисления ТХФ или все эти связанные продукты.
На фиг.7, показаны изменения в спектре ультрафиолет/видимый свет (UV/vis), которые имеют место при окислении ТХФ в фосфатном буфере с рН 7,4 1 мкМ [Fe(Н2О) DCB*]- в присутствии 5,4 мМ Н2О2. В этом эксперименте [Fe(Н2О)DCB*]- и ТХФ объединяют в буфере с рН 7,4, добавляют Н2O2 и затем подвергают мониторингу спектральные изменения. Максимумы поглощения ТХФ наблюдаются при 220, 256 и 316 нм. Длины волн, выбранные для кинетического анализа окислительного процесса, показаны на фиг.7. Результаты этого анализа показаны на фиг.8 (для ясности показаны только изменения поглощения для трех длин волн). В отсутствие [Fe(H2O)DCB*]- не было уменьшений в величинах поглощения для полос, возникающих из ТХФ.
Из фиг.8 видно, что поглощение для полосы при 316 нм уменьшается, появляется новое поглощение при 516 нм, которое увеличивается по интенсивности в течение приблизительно 100 секунд, но затем опять уменьшается до его начальной величины. В экспериментах, в которых проводили визуальный мониторинг этой реакции, быстрое образование пурпурного раствора происходило после добавления Н2О2 и затем этот раствор постепенно обесцвечивался, давая в конце концов бесцветный раствор. Пурпурную окраску относили к связанным продуктам, подобным продуктам на фиг.6. Тот факт, что этот раствор становится бесцветным, указывает на то, что система [Fe(H2O)DCB*]-/H2O2 способна окислять эти связанные продукты.
Система [Fe(H2O)DCB*]-/Н2О2 способна выполнять множественные окисления ТХФ, как показано на фиг.9. Экспериментальные процедуры были такими же, что и использованные на фиг.8 (однако обратите внимание на более высокую температуру для фиг.9), но после первого цикла окисления ТХФ добавляли дополнительные порции 45 мкМ ТХФ, отмеченные #, когда поглощение при 516 нм возвращалось по существу к его исходной величине. Первая часть ТХФ окисляется в пределах 100 секунд, тогда как пятая порция требует более 400 секунд для ее окисления. Снижение окислительного процесса возникает, возможно, из комбинации разложения [Fe(H2O)DCB*]- и дополнительного окисления некоторой части продуктов окисления ТХФ посредством [Fe(H2O)DCB*]-.
Система [Fe(H2O)DCB*]-/H2O2 окисляет ТХФ при различных рН. На фиг.10 показаны кинетические линии для окисления ТХФ в условиях, указанных на фиг.10. Эти данные показывают, что окисление ТХФ является наиболее быстрым при рН 10. Однако, в противоположность результатам, показанным на фиг.9, множественные циклы окисления ТХФ не достигаются одной загрузкой [Fe(H2O)DCB*]-. Кроме того, изменения поглощения при 300 и 316 нм являются более сложными при рН 10, свидетельствуя о повышенной сложности этого процесса окисления. Таким образом, из трех величин рН, испытанных в этом эксперименте, оптимальными величинами в условиях данного эксперимента в отношении времени и долговечности активатора, являются, по-видимому, величины при рН 7,4. Это очень важный результат, так как выход сточных вод в озера и реки должен иметь место в области рН 7.
Пример 35
Обесцвечивание сточных вод
Две пробы по 25 мл сточных вод, имеющие сильную грязно-черную/коричневую окраску, полученные из целлюлозно-бумажного обрабатывающего предприятия в Тасмании, Новой Зеландии, готовили в растворе рН 11. К одному из этих растворов добавляли 25 мг Н2O2 и 12,5 мкг активатора, [Fe(Н2O)DCB*]-. Спустя четыре часа величину поглощения этой пробы измеряли при 465 нм при помощи спектрофотометра видимого света. Добавленные вещества показаны в таблице.
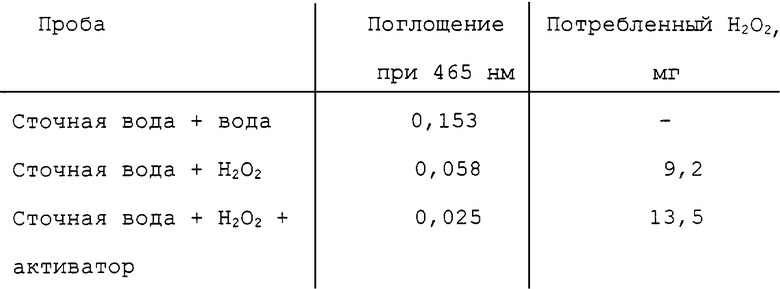
Проба, содержащая активатор, была слабого желтого цвета после обработки. Визуальное обследование раствора, содержащего активатор, показало, что превращение черного/коричневого раствора в бледно-желтый фактически завершалось в пределах первого часа обработки, при приблизительно 30-60 минутах. Сравнимые изменения не наблюдали для раствора, содержащего только H2O2.
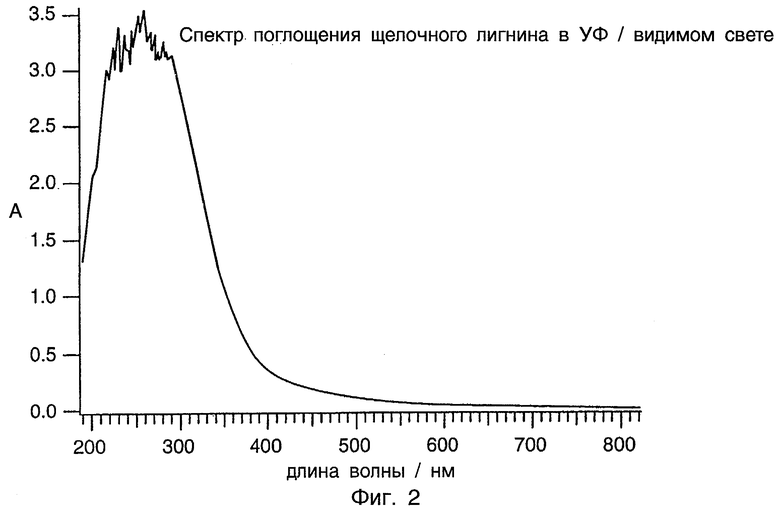
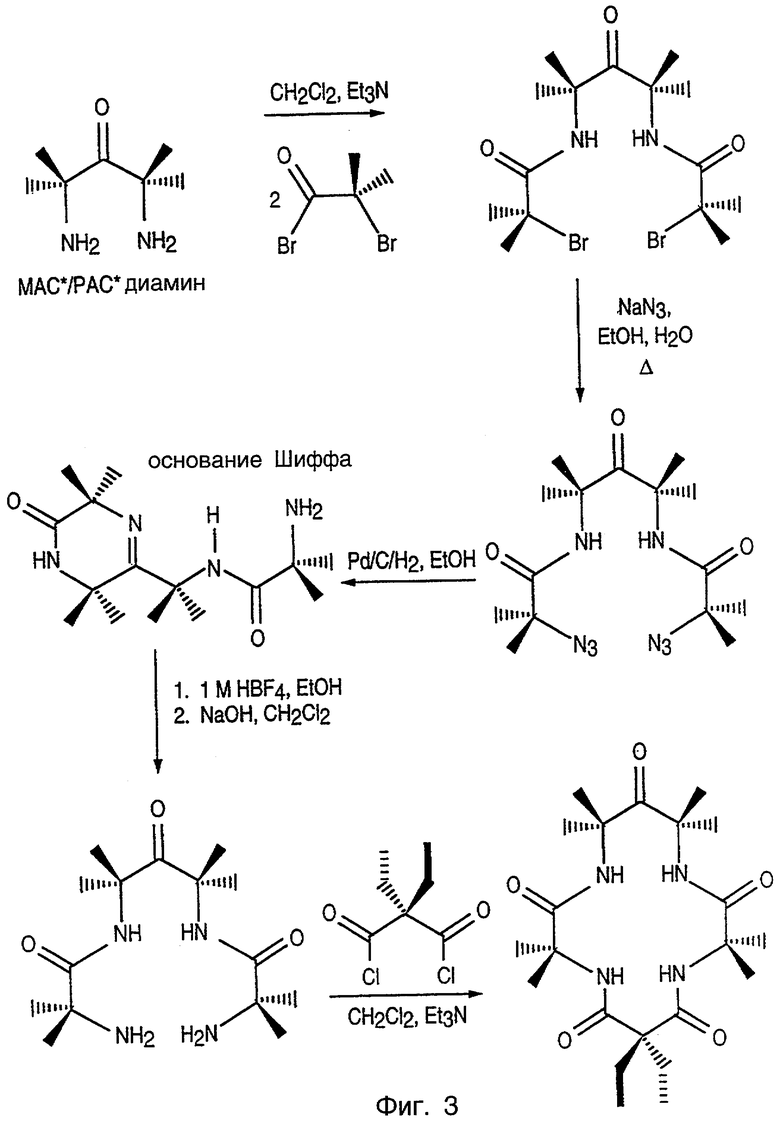
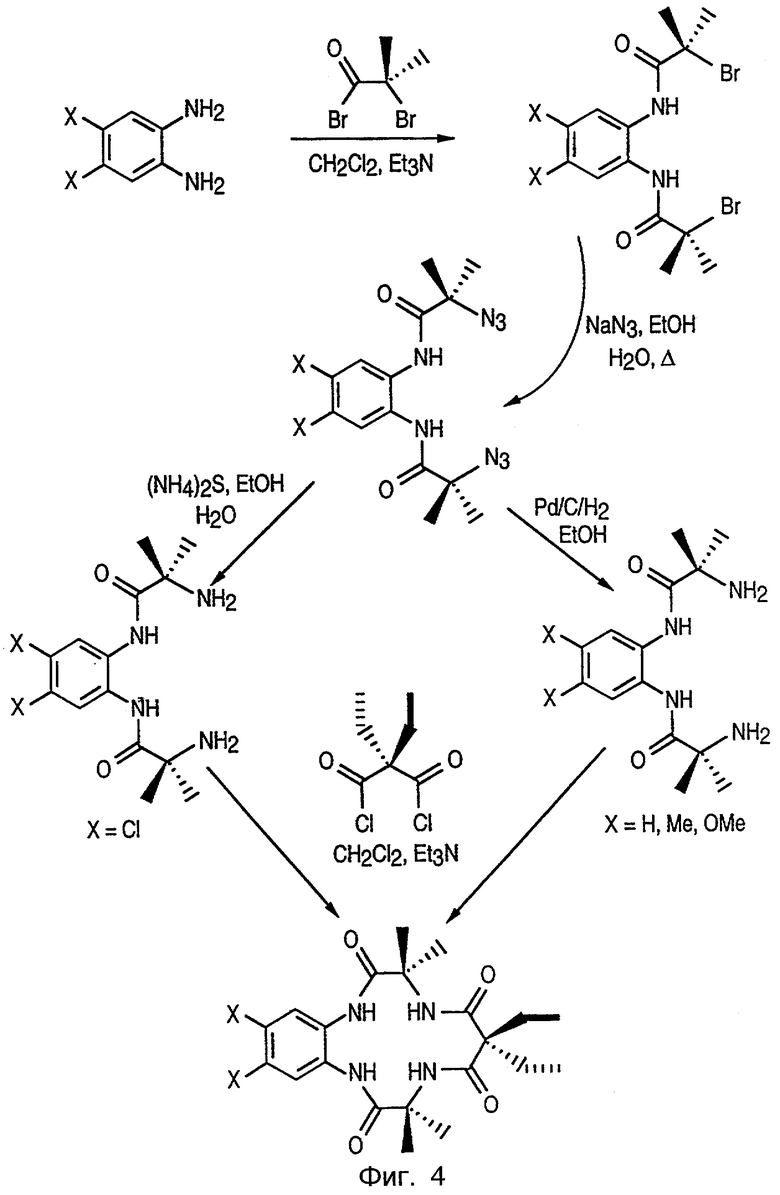
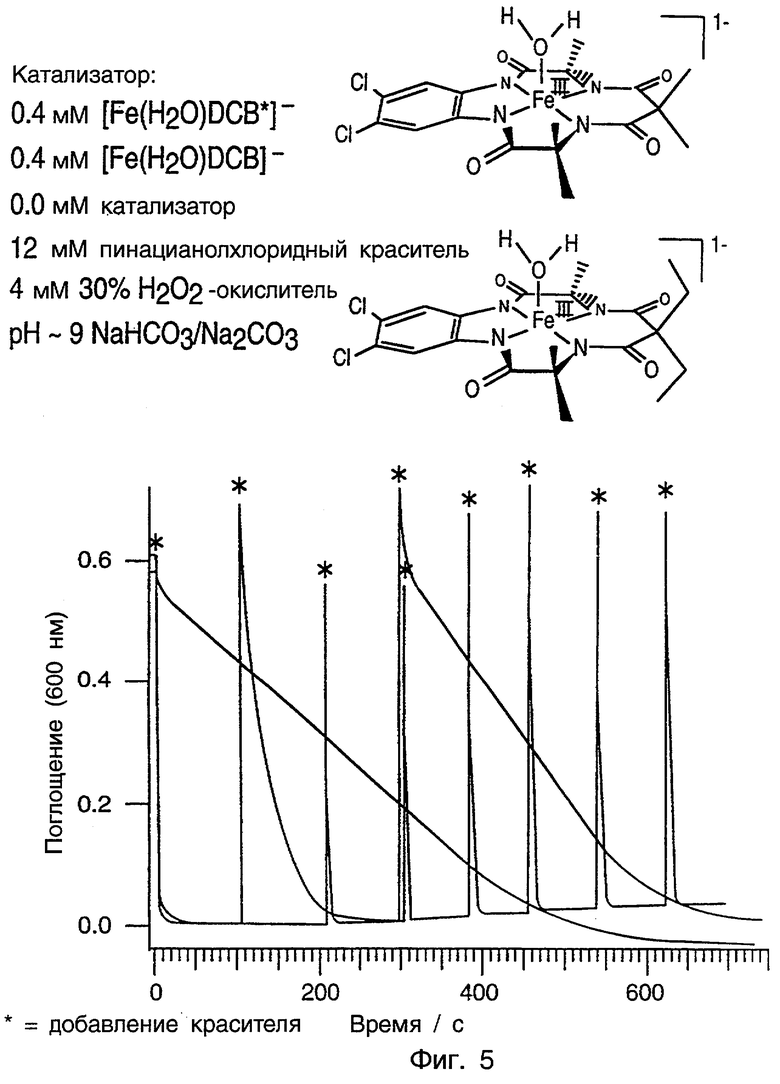
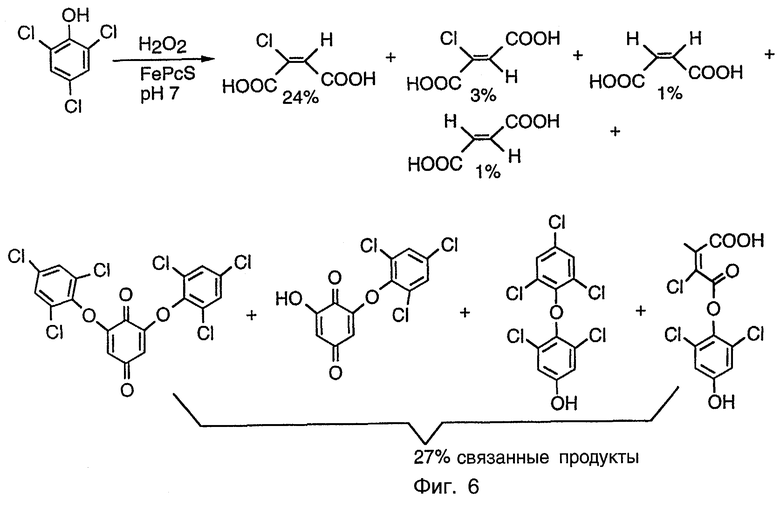
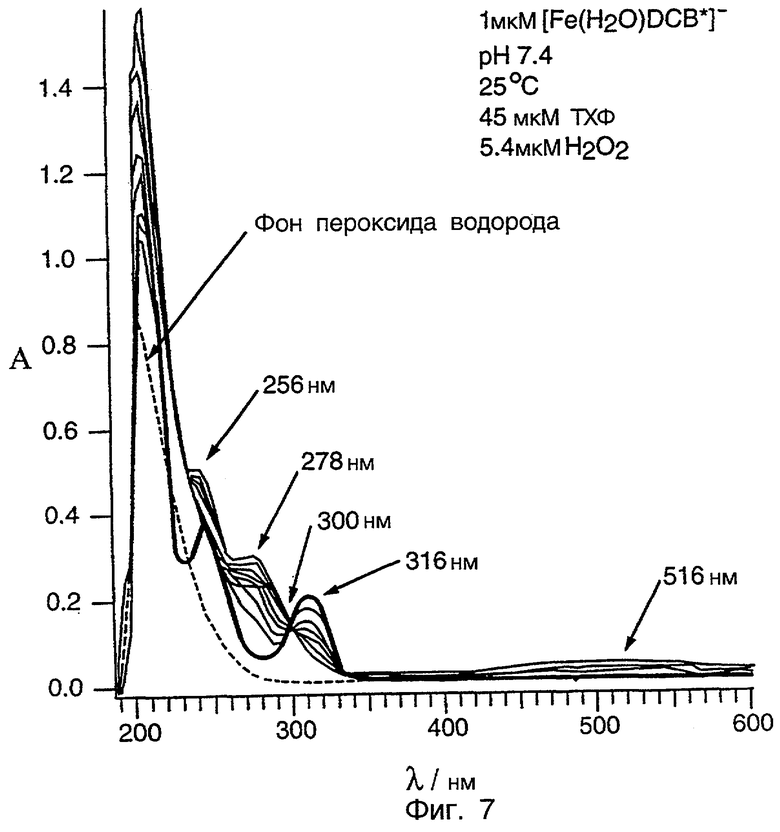
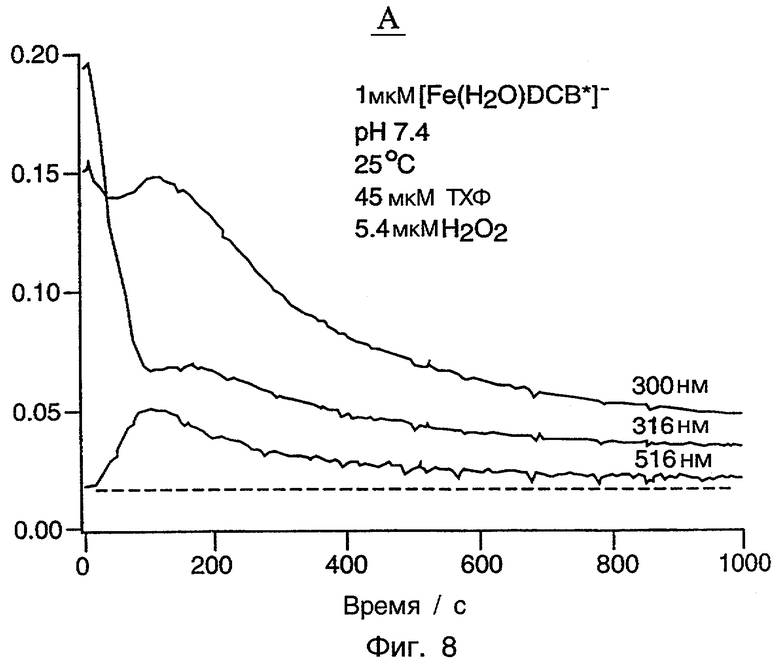
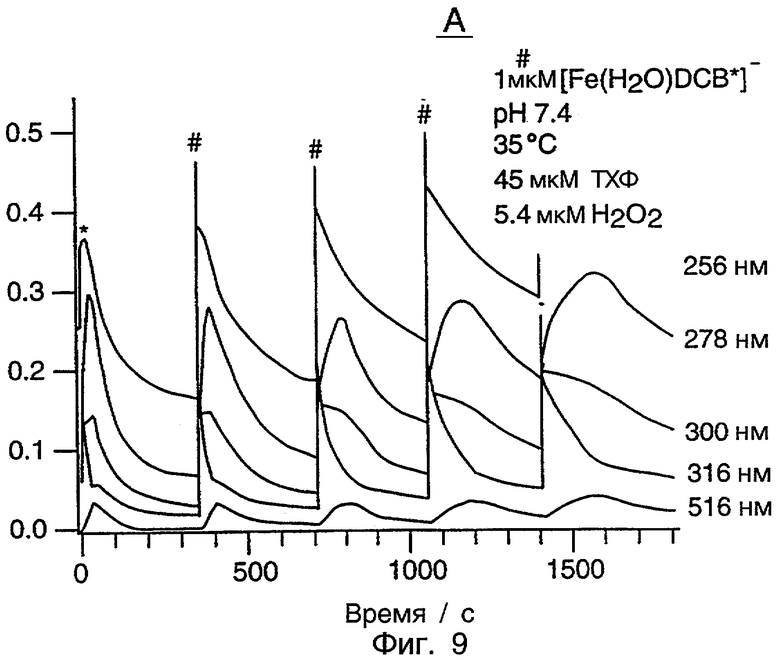
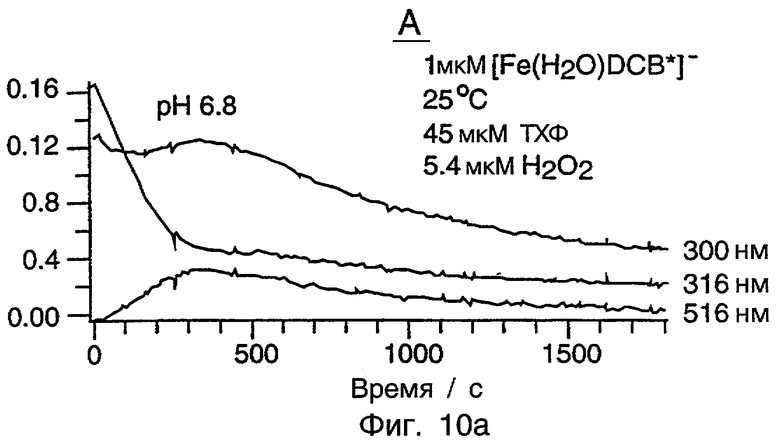
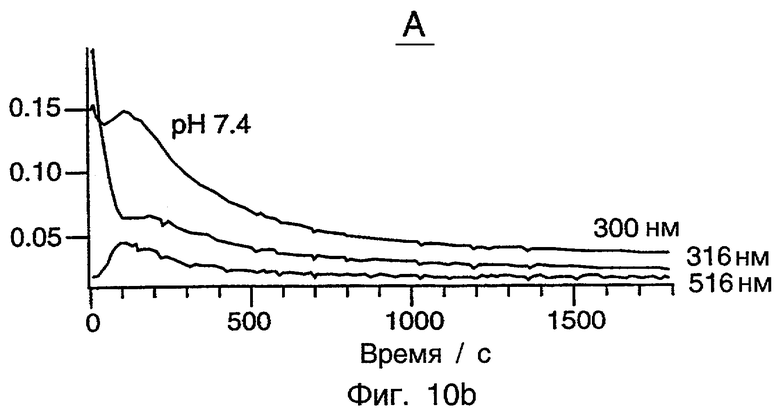
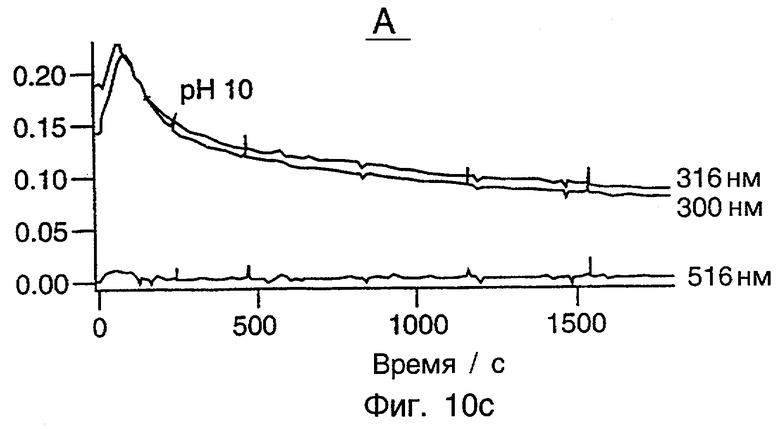
Изобретение относится к макроциклическим металлокомплексным лигандам, используемым в качестве катализаторов окислительного отбеливания. Описывается способ обесцвечивания хромофоров в сточных водах производства целлюлозы и бумаги, который предусматривает использование композиции, состоящей из (а) источника окислителя в количестве, эффективном для отбеливания субстрата, и (b) устойчивого к окислению активатора окислителя, имеющего структуру (I)
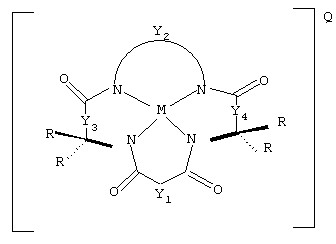
где Y1, Y3 и Y4 каждый представляют мостиковую группу, имеющую нуль, один, два или три углеродсодержащих звена для замещения, a Y2 является мостиковой группой, имеющей по меньшей мере одно углеродсодержащее звено для замещения, причем каждое указанное звено содержит группу C(R), C(R1)(R2) или C(R)2 и каждый заместитель R является таким же или отличающимся от остальных заместителей R; М обозначает металл переходного ряда со степенями окисления I, II, III, IV, V, VI, VII или VIII или выбран из групп 3, 4, 5, 6, 7, 8, 9, 10 и 11 Периодической системы элементов и Q обозначает любой противоион, который уравновешивает заряд соединения на стехиометрической основе. Способ позволяет повысить эффективность отбеливания указанных субстратов. 2 c. и 9 з.п. ф-лы, 10 ил.
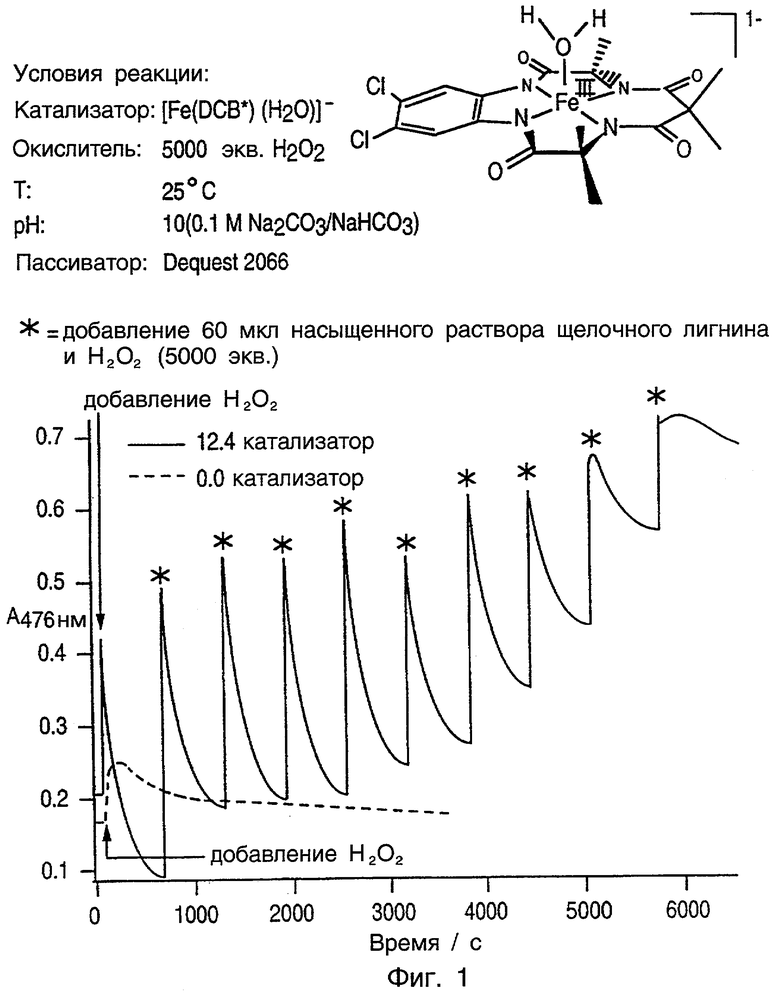
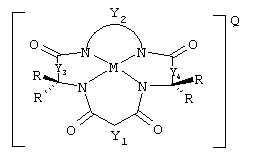
где Y1, Y3 и Y4, каждый, представляют мостиковую группу, имеющую нуль, один, два или три углеродсодержащих узла для замещения;
Y2 является мостиковой группой, имеющей по меньшей мере один углеродсодержащий узел для замещения, причем каждый указанный узел содержит группу С(R), C(R1)(R2) или C(R)2 и где R, R1, R2 являются такими же или отличающимися и (i) выбран из группы, состоящей из алкила, алкенила, циклоалкила, циклоалкенила, арила, алкиларила, галогена, алкокси или фенокси, CH2CF3, CF3, или (ii) образуют замещенное или незамещенное бензольное кольцо, причем два атома углерода в этом кольце образуют узлы мостиковой группы Y, или (iii) вместе с заместителем R, связанным с тем же самым атомом углерода, образуют циклоалкильное или циклоалкенильное кольцо, которое может включать в себя атом, иной чем углерод;
М обозначает металл переходного ряда со степенями окисления I, II, III, IV, V, VI, VII или VIII, или выбран из групп 3, 4, 5, 6, 7, 8, 9, 10 и 11 Периодической таблицы элементов (система ИОПАК);
Q обозначает любой противоион, который уравновешивает заряд соединения на стехиометрической основе,
и (b) с источником окислителя в количестве, эффективном для отбеливания субстрата.
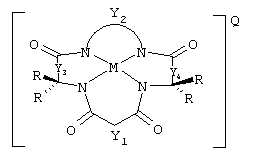
где Y1, Y3 и Y4, каждый, представляют мостиковую группу, имеющую нуль, один, два или три углеродсодержащих узла для замещения;
Y2 является мостиковой группой, имеющей по меньшей мере один углеродсодержащий узел для замещения, причем каждый указанный узел содержит группу С (R), C(R1)(R2) или С(R)2 и где R, R1, R2 являются такими же или отличающимися и (i) выбран из группы, состоящей из алкила, алкенила, циклоалкила, циклоалкенила, арила, алкиларила, галогена, алкокси или фенокси, CH2CF3, CF3, или (ii) образуют замещенное или незамещенное бензольное кольцо, причем два атома углерода в этом кольце образуют узлы мостиковой группы Y, или (iii) вместе с заместителем R, связанным с тем же самым атомом углерода, образуют циклоалкильное или циклоалкенильное кольцо, которое может включать в себя атом, иной чем углерод;
М обозначает металл переходного ряда со степенями окисления I, II, III, IV, V, VI, VII или VIII, или выбран из групп 3, 4, 5, 6, 7, 8, 9, 10 и 11 Периодической таблицы элементов (система ИОПАК);
Q обозначает любой противоион, который уравновешивает заряд соединения на стехиометрической основе,
и (b) с источником окислителя в количестве, эффективном для окисления указанных углеродистых веществ.
| US 5032286 A, 16.07.1991 | |||
| WO 9803626 A, 22.12.1998 | |||
| WO 9803625 A, 29.12.1998 | |||
| SU 1689477 A1, 07.11.1991. |

