Настоящая работа осуществлена при поддержке Национального института здоровья, программа GM-44867 и Национального научного фонда, программа CHE9319505. Правительство США имеет права на данное изобретение.
Обоснование изобретения
Область изобретения
Настоящее изобретение относится к хелатным комплексным соединениям металлов, предназначенным для получения катализаторов окисления, и, более конкретно, к долговечным макроциклическим соединениям, служащим катализаторами окисления, которые способны участвовать в реакциях окисления пероксидами и другими первичными окислителями.
Описание известного уровня техники
Хотя основным источником окислителей и в химических, и в биологических процессах являются вещества, основанные на переходных металлах, процесс окисления намного лучше изучен именно в биологии, т.е. многие сложные селективные реакции окисления, которые осуществляются в биологических процессах, не удается осуществить с применением гомогенных синтетических систем. Это отличие более заметно в области химии окисления, чем в любых других областях химических взаимодействий. Так, по сравнению с химией восстановительных процессов, или образованием связей "углерод-углерод", химия окисления использует очень ограниченное число технологий невысокого качества для осуществления стехиометрических или каталитических процессов.
Относительная дороговизна хороших гомогенных окислительных систем и катализаторов обусловлена окислительной деградацией катализаторов. Обладающие высокой окисляющей способностью комплексные соединения переходных металлов, находящихся посредине ряда переходных металлов и в конце него, аналогичны соединениям, которые функционируют в качестве активных промежуточных соединений в многочисленных реакциях ферментного окисления, и эти соединения трудно было синтезировать, поскольку в таких соединениях наблюдается тенденция к быстрому разложению лигандов.
В работе Collins, T. J., "Designing Ligands for Oxidizing Complexes," Accounts of Chemical Research, 279, Vol. 27, N 9 (1994), синтетические основанные на металлах окислители разделяют на два основных класса - соединения, обладающие окислительно-восстановительной активностью в отношении металлов, и окислители металлов матричного типа. В системах, обладающих окислительно-восстановительным действием в отношении металлов, окислительная составляющая содержит ион металла, который находится в непосредственном контакте с лигандами. В результате эти системы ограничены небольшим набором лигандов, которые совместимы с окислительными ионами металлов. Окислители матричного типа не подвержены таким ограничениям, поскольку окисляющая составляющая находится дальше от иона металла, но матричные системы пригодны только для мягкого окисления, в отличие от строгого окисления, для которого требуются высокоактивные металлоокислители. Окислители в виде ионов металлов в оксигеназных ферментах часто являются катализаторами строгого окисления, такого как взаимодействие метан-монооксигеназы, т.е. окисление метана до метанола кислородом в качестве первичного окислителя. Роли металлоокислителей в таких ферментах относятся к типу окислителей-восстановителей. Основная проблема при перенесении этого предполагаемого ферментного механизма в искусственные системы состоит в том, чтобы разработать устойчивые системы лигандов, которые могут выдерживать воздействие исключительно сильных окислителей - ионов металлов, вызывающих отщепление атома.
В статье "Accounts" Коллинз описывает "конструкционно-ориентированный" подход к получению лигандов и хелатных комплексных соединений металлов, которые устойчивы к окислительной деградации. В статье "Accounts" также описаны несколько диамидо-N-дифеноксидо ациклических и тетраамидо-N- макроциклических лигандов, которым придали устойчивость к окислительной деградации, наряду с комплексными соединениями переходных металлов, находящихся посредине ряда переходных металлов и в конце него, причем ионы металлов находятся в таком состоянии, что обладают беспрецедентно высокой окисляющей способностью, которой удалось добиться при помощи макроциклических лигандов.
Хотя достаточно получить вышеописанные ионы с очень высокой валентностью в стабильной форме, включая сильные окислители, осуществляющие перенос электронов, тем не менее приведенный в статье "Accounts" набор правил является недостаточным для решения задачи инкапсулирования особенно мощного металлоксоокислителя, аналогично тому, как это происходит в монооксигеназных ферментах, так чтобы окислитель имел достаточный срок службы для того, чтобы осуществить процессы бимолекулярного окисления. Решение этой задачи заключается в структуре лиганда, описанного в настоящей заявке.
Краткое описание изобретения
Требуемой стабильности лиганда и производного комплексного соединения добивались при помощи соединений с макроциклическим тетрадентатным лигандом по настоящему изобретению. Соединения по изобретению имеют общую формулу: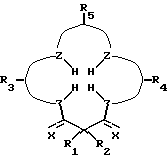
где R1 и R2 имеют одинаковые или различные значения, являются связанными или несвязанными, и каждый из них выбирают из заместителей, которые являются нереакционноспособными, внутримолекулярно образуют сильные связи с R1 и R2 и с циклическим углеродом, являются пространственно затрудненными и конформационно затрудненными, так что окислительная деградация металлокомплекса соединения является ограниченной, когда металлокомплекс находится в присутствии окисляющей среды. Затруднение предотвращает присоединение конформеров, которые приводят к внутримолекулярной окислительной деградации.
Z представляет собой донорный атом, предпочтительно, устойчивый к окислению атом, образующий металлокомплекс, такой как N или O, при необходимости несущий водород;
X представляет собой функциональную группу, предпочтительно, устойчивую к окислению функциональную группу, такую как O или N Rs, где Rs представляет собой метил, фенил, гидроксил, оксильную группу, -CF3 или -CH2CF3;
R3 представляет собой фрагмент, соединяющий соседние атомы Z и состоящий из: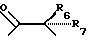
или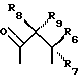
R4 представляет собой фрагмент, соединяющий соседние атомы Z и состоящий из: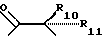
или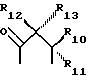
где R6 и R7, R8 и R9, а также R10 и R11, R12 и R13, попарно и совокупно имеют одинаковые или различные значения и каждый выбирают из группы, в которую входят алкил, арил, галогены и CF3, а также
R5 представляет собой фрагмент, соединяющий соседние атомы Z, и состоящий из: (i)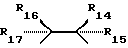
и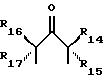
где R14 - R17 имеют одинаковые или различные значения и каждый представляет собой алкил, арил, галоген, или CF3, а также (ii) арильной группы, включая моно-, ди-, три- и тетразамещенный арил и гетероарильные заместители: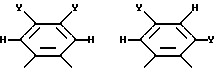
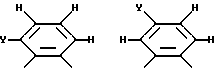
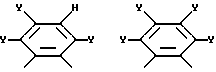
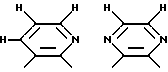
где каждый из Y может представлять собой любой заместитель или заместители, но, предпочтительно, галоген, водород, алкил, арил, амино, замещенный амино, нитро, алкокси, арилокси и их сочетания. Арильная группа заменяет собой четыре заместителя, а также атомы углерода, к которым они присоединены.
Настоящее изобретение относится к новым изменениям в структуре макроцикла, которые повышают устойчивость тетра-аза-макроциклических лигандов, благодаря которым можно получить систему лигандов, которая поддерживает катализ, основанный на преднамеренно высокоактивных промежуточных металлоксосоединениях, аналогичную системе лигандов для моноксигеназ. Механизм разложения, в котором потребовались вышеописанные изменения, оказался совершенно неожиданным. Важнее всего то, что новые системы, описанные здесь, поддерживают катализ с крайне желательными окислителями, осуществляющими перенос O-атома, особенно пероксидами, что позволяет использовать эти окислители для широкого круга технологических окислительных процессов, где можно применять химически эффективный и экономичный катализ.
Комплексные соединения переходных металлов с макроциклическими лигандами в прошлом использовались в качестве катализаторов окисления. Патентованные системы включают порфирины и фталоцианины, галогенированные порфирины и лиганды, относящиеся к порфиринам, а также замещенный трициклоазанонан и соответствующие макроциклы. Все эти системы фундаментальным образом отличаются от системы по настоящему изобретению. Во-первых, макроциклические тетраамиды являются тетраанионными и сильными донорами, так что лиганды по настоящему изобретению делают доступными такие формы металлов, при которых они обладают высокой валентностью и реакционной способностью, причем эффективность лигандов по изобретению гораздо выше, чем любых других макроциклических соединений, применявшихся для этой цели. Во-вторых, макроциклические соединения по настоящему изобретению можно получить в виде соединений, сильно защищенных без обращения к галогенным заместителям (или с ними) - негалогенированные соединения причиняют меньше ущерба окружающей среде. В-третьих, комплексы макроциклических тетраамидов по настоящему изобретению обладают выраженной устойчивостью к гидролизу, что позволяет использовать их в протолитической среде, такой как вода, в которой растворяются соли, содержащие различные ионы металлов.
Тетрадентатное макроциклическое соединение по настоящему изобретению предназначено для образования комплексного соединения с металлом, предпочтительно с переходным металлом, а лучше всего с переходным металлом, выбранным из группы VI (группа Cr), VII (группа Mn), VIII (группа Fe), IX (группа Co), X (группа Ni), XI (группа Cu) Периодической таблицы элементов, для получения соответствующего хелатного комплекса.
Таким образом, настоящее изобретение включает хелатный комплекс формулы: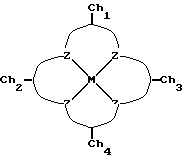
где M представляет собой металл, Z представляет собой донорный атом, такой как устойчивый к окислению атом, образующий комплекс с металлом, который описан выше как макроциклическое тетрадентатное соединение по настоящему изобретению, и Ch1, Ch2, Ch3 и Ch4 представляют собой устойчивые к окислению компоненты хелатной системы, которые являются одинаковыми или различными и которые образуют пяти-, шестичленные кольца с соседними атомами ZMZ.
В предпочтительном варианте осуществления изобретения осевой лиганд L1 связывается с металлом. Лиганд L1 является лабильным, поскольку он занимает свое положение относительно металла до тех пор, пока хелатную систему не введут в раствор, содержащий окислитель. Лабильный лиганд диссоциирует в растворе и будет замещен окислителем, обычно агентом переноса O-атома, но может быть замещен любым окислителем, который может служить для того, чтобы активировать ион металла для осуществления функций катализатора. Предпочтительные лабильные лиганды включают Cl-анион, ионы галидов в целом, CN-, ROH, NH3, или любой амин, карбоксилат, фенол или феноксид, нитрил, пиридин, простой эфир, сульфоксид, кетон или карбонат.
Было обнаружено, что местами окисления в комплексных соединениях железа, представляющих собой макроциклы, содержащие ароматические кольца (один электрон окислен над FeIII) можно манипулировать, выбирая осевые лиганды, а также при помощи заместителей в виде ароматических колец. Сильный s-донорный анионный осевой лиганд (CN-) способствует окислению металла в центре, т.е. FeIV, в то время как более слабые доноры (Cl-) способствуют локализованному на лиганде окислению. Оксо-соединение является промежуточной формой системы хелатного комплекса и считается, что в некоторых случаях оно функционирует как реальный катализатор. В других случаях хелатная система может быть единственным местом окисления, или место окисления может быть смешанным - хелатная система, металл и любой другой лиганд, присоединенный к металлу.
Хелатная группа Ch1, предпочтительно представляет собой радикал, описанный выше как Rs в макроциклическом тетрадентатном соединении. Ch2 и Ch3 соответствуют радикалам R3 и R4 макроциклического тетрадентатного соединения, описанного выше.
Ch4, предпочтительно, представляет собой связующую часть общей формулы X= CC(R>>)2'C=X, где (R>>)2 эквивалентно R1 и R2, описанным выше, а X представляет собой устойчивую к окислению функциональную группу, описанную выше.
R1 и R2 представляют собой ключевые заместители в конструкции устойчивого хелатного комплекса и катализаторов по настоящему изобретению. R1 и R2, предпочтительно, представляет собой метил, CF3, водород или галоген, или могут образовывать, вместе с атомом углерода, к которому оба они присоединены, кольцо, такое как четырех-, пяти- или шестичленное кольцо. Считается, что внутримолекулярные взаимодействия между заместителями R1 и R2 в известных ранее комплексах и оксолиганде в действующей каталитической системе способствуют быстрой деградации хелатного лиганда, как об этом свидетельствует опыт. См. Фиг. 1, где изображен предполагаемый механизм окислительного разложения катализатора. Например, было отмечено (и эти данные соответствуют данным по фиг. 1), что известные каталитические соединения, имеющие диэтиловые заместители в положениях R1 и R2, чувствительны к окислительному воздействию, так что пока можно было наблюдать каталитическое окисление, система лигандов одновременно претерпевала медленное разложение окислением. Все тетраамидные макроциклические соединения, описанные в работе Collins Accounts of Chemical Research, которую мы цитировали выше, включают диэтильные заместители в положениях R1 и R2. Таким образом, до настоящего времени ни одно комплексное соединение переходного металла, имеющее макроциклический тетраамидный лиганд, не могло выполнять функции катализатора окисления в течение продолжительного периода времени, необходимого для каталитической реакции.
Описание чертежей
Фиг. 1 схематично иллюстрирует предложенный путь окислительной деградации каталитической системы, состоящей из соединения II и пероксидов, вследствие внутримолекулярных взаимодействий между диэтильной составляющей и оксоаксиальным лигандом.
Фиг. 2 иллюстрирует способ, при помощи которого конформационные ограничители предотвращают окислительную деградацию оксогруппы.
Фиг. 3 (а) и (б) иллюстрируют две возможные структуры макроциклического тетраамидного лиганда по настоящему изобретению, где показаны компоненты соединения - ответвление, линкер и мостик.
Фиг. 4 представляет собой вид рециркулируемой системы металлоокислителя.
Фиг. 5 представляет собой схематичное изображение аминозависимого макроциклического комплекса металла, ковалентно связанного с подлежащей поверхностью.
Фиг. 6 иллюстрирует несколько хелатных комплексов, полученных из макроциклических лигандов по изобретению.
Подробное описание предпочтительных вариантов осуществления изобретения
Предпочтительным вариантом осуществления тетрадентатного макроциклического соединения по настоящему изобретению является следующее: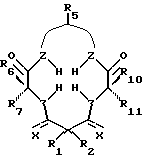
в котором R1 и R2 имеют одинаковые или различные значения и каждый из них выбирают из группы заместителей, которые являются нереакционноспособными, образуют сильные межмолекулярные связи с R1 и R2 и с циклическим углеродом, являются пространственно затрудненными и конформационно затрудненными, так что окислительная деградация комплекса металла соединения ограничена, когда комплекс находится в присутствии окислительной среды. Низкая конформационная свобода определенных видов соединений предотвращает приобретение конформеров, которые могут приводить к внутримолекулярной окислительной деградации. Z обозначает донорный атом, предпочтительно устойчивый к окислению атом, образующий комплекс с металлом, более предпочтительно, N или O, несущий, при необходимости H. Предпочтительно, чтобы по меньшей мере три Z представляли собой N. X представляет собой функциональную группу, предпочтительно устойчивую к окислению функциональную группу, а более предпочтительно O или NRs, где Rs представляет собой метил, фенил, гидроксил, оксильную группу, -CF3 или -CH2CF3.
R6, R7, R10 и R11 имеют одинаковые или различные значения и каждое выбирают из группы, в которую входят алкил, арил, галогены и CF3. R5 представляет собой фрагмент, соединяющий соседние Z атомы, и его выбирают из группы, в которую входят (i)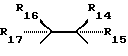
и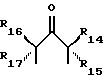
где радикалы с R14 по R17 имеют одинаковые или различные значения и обозначают алкил, арил, водород, галоген, CF3 или их сочетания, и (ii) арильная группа, включая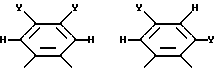

где Y представляет собой галоген, водород, алкил, арил, аминогруппу, замещенную аминогруппу, нитрогруппу, алкокси, арилокси и их сочетания.
Соединения по настоящему изобретению образуют устойчивые долговечные катализаторы окисления и предкатализаторы. Для удобства термин "катализатор" в настоящем тексте включает предкатализаторы и реальные каталитические комплексы, причем последние представляют собой такие катализаторы, которые осуществляют окисление. Во многих случаях точный механизм каталитического действия является неизвестным и поэтому точная роль хелатной системы по настоящему изобретению в той или иной реакции окисления не известна. В тексте настоящей заявки термин "устойчивый катализатор окисления" означает, что когда катализатор добавляют в растворитель в присутствии окислителя, такого как пероксид, период полураспада активированной формы комплекса металла составляет 30 секунд или более. Период полураспада - это время, в течение которого половина металлокомплекса разлагается или деградирует.
Было обнаружено, что в соответствии с наиболее предпочтительным вариантом изобретения новые устойчивые соединения отличаются от известных соединений только по одной составляющей. После замены диэтиловых заместителей R1, R2 в известных тетраамидных соединениях на диметиловые заместители, бывшие хрупкие недолговечные хелатные комплексы превращались в стабильные долговечные комплексы, которые очень устойчивы к окислительной деградации. То, что казалось незначительным изменением в структуре, фактически стало ключом к новому классу устойчивых долговечных катализаторов окисления. Прочность связи C-H метилового заместителя примерно на 3 Ккал•мол-1 выше, чем прочность C-H связи соответствующего этилового заместителя. Было обнаружено, что любые заместители R1, R2, которые являются нереакционноспособными или которые образуют сильные связи с циклическим углеродом, или которые являются пространственно или конформационно затрудненными, так что они не могут вступать во внутримолекулярное взаимодействие с осевым оксолигандом, также будут образовывать устойчивые катализаторы, или предкатализаторы по настоящему изобретению.
Важность прочности связи и/или конформационного ограничения можно увидеть из следующего.
Чтобы создать носитель для катализатора окисления, каждый компонент лигандной системы должен быть устойчивым к окислительной деградации. Ключевой фактор стабильности групп R1 и R2 обнаружили, наблюдая один наиболее показательный случай. Как показано на фиг. 1 аквакомплексы железа (III) взаимодействуют с гидропероксидами до получения указанного оксокомплекса, который, как было показано, обладает каталитическими свойствами в отношении окисления нитрилов, содержащих C-H связи, до получения цианогруппы. Однако в процессе каталитической реакции лигандная система медленно разлагается и предполагается, что такая деградация происходит при отделении атома H от метиленовой группы этильного заместителя в R1 положении, что соответствует структуре гидантоинового кольца, содержащего продукт деградации, помеченный III (фиг. 1). Молекулярные модели показывают, что сильно напряженная конформация хелатного кольца, содержащего Ch4, требуется для того, чтобы подвести отделяемый H-атом близко к отделяющему O-атому. Соединение III однозначно характеризовалось различными анализами, включая масс-спектрометрию, 1H и 13C ЯМР, ИК, элементный анализ. Одновременно с наблюдаемой деградацией система осуществляет каталитическое окисление наиболее слабой связи C-H в серии нитрилов [(CH3)2CHCN, CH3CH2CN, CH3CN, CD3CN], которые применяются в качестве растворителей. Продукты представляют собой смеси продуктов окисления нитрилов. Так, если трет-бутилгидропероксид является первичным окислителем, то смесь продуктов с (CH3)2CHCN субстратом содержит (CH3)2C(OH)CN, (CH3)2(CN)COOC(CH3)3, (CH3)2(CN)COOCH3, (CH3)2C=O, (CH3)3COH. Было также показано, что хотя данная смесь продуктов предполагает, что происходит самоокисление свободных радикалов, где роль комплекса железа II (фиг. 1), будет состоять в инициации процесса, самоокисление свободных радикалов не может быть доминантным механизмом. Так, когда окисление осуществляют в 18O2 (1 атм. >98%), выход продуктов, маркированных 18O2, является слишком низким для того, чтобы механизм реакции соответствовал процессу самоокисления свободных радикалов. Путем замены CH3- на CH3CH2 - в положениях R1 и R2, деградация лигандов сильно подавляется, так что окисление нитрила само по себе является доминирующим в окислительной способности. Такое подавление деградации лигандов путем замены CH3- на CH3CH2- можно признать результатом повышенной прочности связи C-H в CH3- относительно CH3CH2-, приблизительно 3ккал/мол-1, посредством чего скорость отделения H-атома при помощи оксолиганда замедляется приблизительно в три раза. Поскольку очевидно, что отделение является крайне важным фактором деградации, то ориентация отделяемого H-атома относительно оксолиганда также играет важную роль, поскольку такая ориентация определяет расстояние приближения, а реакции отделения очень сильно зависят от расстояния. Молекулярные модели показывают, что если циклопентильный фрагмент применяется для замены этильных групп R1 и R2, то метиленовая группа C-H, эквивалентная группе, отделяемой от этильной C-H группы, не может достичь оксолиганда без того, чтобы образовалось значительно более сильное напряжение кольца, чем то, которое обнаруживается в случае этила. Таким образом, конформационное ограничение приводит к сильному возрастанию сопротивления замещенного таким образом хелатного комплекса до окислительной деградации.
В структуре, показанной на фиг. 2, оксогруппа и метиленовый H ограничены от такого близкого приближения, которое может происходить в случае этила, поскольку метиленовая группа циклопентильного заместителя не может свободно вращаться, чтобы приблизить две группы настолько близко друг к другу.
Соединения по настоящему изобретению являются макроциклическими, состоят из четырех анионных донорных лигандов, в результате чего образуется практически плоская тетрадентатная платформа, которая может образовать комплекс с металлом и осевым лигандом до получения хелатно/каталитической системы по настоящему изобретению. Предпочтительная структура устойчивых лигандов - макроциклический тетраамидный лиганд, не имеющий водородных атомов на α-N-амидных донорных группах. При координировании с ионами металла пяти- и шестичленные хелатные кольца являются наиболее стабильными. Заместители могут быть самыми различными, при условии, что они удовлетворяют вышеуказанным требованиям. Это особенно важно в отношении заместителей R1 и R2.
Основанный на азиде синтетический путь получения макроциклических тетраамидных лигандов описан в работе Uffelman, E.S., Ph.D. Thesis, California Institute of Technology, (1992). Иначе (и предпочтительно) соединения по изобретению можно синтезировать новыми путями.
Новый способ синтеза позволяет осуществлять синтез вариантов, которые невозможно синтезировать с помощью известного способа синтеза на основе азида. Однако при изменении макроцикла важно сохранить общую схему соединения. Макроцикл будет состоять из 5- и 6-членных колец, в виде схемы 5,5,5,6, схемы 5,6,5,6, схемы 5,6,6,6 или схемы 6,6,6,6, о чем более подробно будет сказано ниже.
Новый метод синтеза обычно идет как показано в схеме 1 и 2. Конкретные примеры применения нового метода для синтеза некоторых конкретных макроциклических тетраамидов показаны на схеме 3. Для удобства классификации исходные материалы, которые состоят из диаминных функциональных групп, иногда называют "мостами" (В), исходные материалы, состоящие из двухосновных функциональных групп, иногда называют "линкерами" (L), а исходные материалы, состоящие из аминных/кислотных функциональных групп иногда называют "ответвлениями" (A). См. Фиг. 3(а) и (б). Ответвления макроциклического соединения являются более устойчивыми, чем линкеры, они противостоят деградации.
Схема 1 (см. в конце описания) представляет собой обобщенную схему синтеза макроциклических тетраамидов, имеющих конфигурацию (B-A-L-A-), из α-аминокарбоновых кислот посредством нового способа синтеза. Промежуточное соединение, содержащее диамид-дикарбоксил, которое иногда называют "макролинкерным промежуточным соединением", или просто промежуточным соединением (A-L-A), предварительно получают без использования защитных групп посредством реакции селективного двойного присоединения, в которой α-аминокарбоновую кислоту (выступающую в роли "ответвления", A) и активированное производное малоновой кислоты (выступающее в роли линкера, L), в растворителе нагревают до получения макролинкерного промежуточного соединения. Макролинкерное промежуточное соединение затем соединяют с диамином, выступающим в роли моста (В) в процессе другой реакции селективного двойного присоединения, в ходе которой применяют растворитель, агент присоединения и нагрев. Методология синтеза очень упрощена и позволяет использовать широкий круг функциональных групп. Широкий круг макроциклических тетраамидов, имеющих различные электронные или пространственные заместители, получали таким способом и при высоком выходе продукта.
Схема 2 (см. в конце описания) представляет собой обобщенную схему синтеза макроциклических тетраамидов, имеющих конфигурацию (B-A-L-A-), из β -аминокарбоновых кислот посредством модифицированного варианта основного или первичного способа синтеза. К выбору β -аминокарбоновых кислот в качестве исходных материалов применим практически тот же подход, что и к выбору α -аминокарбоновых кислот в качестве исходных материалов. Для некоторых β аминокарбоновых кислот использование защитной группы может быть желательным, как показано на Схеме 2. Макролинкерное промежуточное соединение (A-L- A) получают предварительно посредством реакции селективного двойного присоединения, в ходе которой сложный эфир β -аминокарбоновой кислоты в качестве ответвления (A) и активированное производное малоновой кислоты в качестве линкера (L) в растворителе нагревают до получения промежуточного вещества, которое после удаления защитной группы можно присоединить к диаминовому мосту (В) в ходе еще одной реакции селективного присоединения до получения широкого круга замещенных макроциклических тетраамидов с расширенным размером кольца по сравнению с тетраамидами, которые были получены из α - аминокарбоновых кислот.
Макролинкерное промежуточное соединение (A-L-A) можно получать в больших количествах в ходе периодического или непрерывного процесса посредством прямого взаимодействия замещенного малонил- дигалида с раствором (предпочтительно пиридиновым раствором) α или β -аминокарбоновой кислоты или ее сложного эфира. Множество реакций давали хорошие выходы без защитных групп при температурах, предпочтительно, ниже или равных около 70oC. Некоторые примеры требуют использования защитных групп, и эти реакции обычно также дают хороший выход. Промежуточное соединение можно разделить на порции и каждую отдельную порцию затем взаимодействуют с широким кругом диаминовых соединений, образующих мосты, имеющие разные пространственные или электронные заместители, в присутствии агента присоединения. Для α -аминокарбоновых кислот этап закрытия кольца занимает от 48 до 120 часов и в идеале должен проходить практически без влаги (см. Схему 3 в конце описания). Широкий круг тетраамидных макроциклов, имеющих тщательно подобранные электронные свойства, можно синтезировать при значительной экономии расходов в сопоставлении с известным азидным способом.
Схема 3 представляет собой конкретный пример получения макроциклического тетраамида, имеющего конфигурацию (В-A-L-A-), из α-аминокарбоновой кислоты в качестве исходного материала, α-аминокарбоновую кислоту смешивают с активированным малонатом в пиридине при температурах ниже 70oC. После того как реакция селективного двойного присоединения завершена, через 72-144 часа, выделяют макролинкерное промежуточное соединение (A-L-A). На втором этапе диамин, предпочтительно о-фенилендиамин, добавляют в пиридиновый раствор макролинкерного промежуточного соединения в присутствии агента, вызывающего присоединение, предпочтительно PCl3, или пивалоилхлорида. Реакция, приводящая к закрытию кольца, реакция двойного присоединения, протекает при перегонке в течение 48-110 часов, а затем нужный макроциклический тетраамид изолируют при хорошем выходе продукта реакции.
Синтез устойчивых к окислению макроциклических тетраамидов требует, чтобы все атомы H, от α до донорных атомов были замещены более устойчивыми к окислению группами, такими как алкил, галоген, арил или гетероциклические заместители.
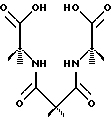
Структура 1 показывает ключевое промежуточное соединение при получении катализатора по настоящему изобретению - устойчивого к окислению макролинкера (ответвление-линкер-ответвление Arm- Linker-Arm). Эту молекулу можно легко синтезировать в ходе одного этапа без использования защитных групп посредством прямого ацилирования а-метилаланина с использованием диметилмалонил- дихлорида.
В альтернативном варианте осуществления изобретения в способе по настоящему изобретению используется последовательное присоединение/удаление защитной группы для создания защищенной формы макролинкерного промежуточного соединения. После удаления защитной группы промежуточное соединение присоединяют посредством реакции двойного присоединения, описанной выше, для создания тетраамидного макроцикла. Аналогичным образом последовательное присоединение/удаление защитной группы можно применять в отношении заместителей, присутствующих на мостиковом фрагменте для расширения круга мостиковых заместителей, которые могут применяться для реакции макроциклизации.
Оба варианта осуществления способа по настоящему изобретению основаны на использовании в качестве исходных материалов аминов или карбоновых кислот, которые перечислены в Таблице 1. В Таблице 1 перечислены исходные материалы в нескольких формах, содержащие аминогруппы и группы карбоновых кислот в виде родственных групп, защитных/активированных групп, и скрытых групп. В Таблице 2 используются эти же категории в связи с ограничениями размеров хелатного кольца (5- и 6-членные хелатные кольца являются предпочтительными), чтобы идентифицировать исходные материалы, пригодные для синтеза хелатных макроциклических тетраамидных соединений, имеющих требуемое пяти- или шестичленное кольцо.
В тексте настоящей заявки термином "родственные группы" (указанные курсивом в Таблице 1) обозначаются предпочтительные синтезированные функциональные группы. Термин "защитная/активированная группа" относится к тем группам, которые содержат легко различимую часть родственной группы. Термин "скрытые группы" относится к таким группам, которые не должны содержать легко распознаваемую часть родственной группы, но которые могут легко быть преобразованы в родственную группу или в родственную группу, содержащую защитную/активированную группу. Более подробные примеры можно легко найти в работе "Green and Green, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York (1981). Полный список защитных/активирующих групп находится в работе G. A. Fletcher and J. H. Jones, "A List of Amino-Acid Derivatives Which are Useful in Peptide Synthesis", Int. J. Peptide Protein Res. 4, (1972), P.347-371.
Структура 2 приведена ниже для того, чтобы разъяснить краткое обозначение по Таблице 2 и Таблице 3, где указаны размеры хелатного кольца (включая ион металла), которое сформировалось, когда данный макроциклический лиганд скоординирован относительно центрального переходного металла.
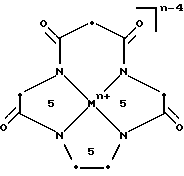
Амин будет обозначаться "a", а карбоксилат "c".
Черточками (-) обозначены амидные связи. Каждая черточка должна соединять следующий "a" с ведущим "c", или наоборот, а конечная черточка возвращает к началу. Структура 2 иллюстрирует макроциклический лиганд (5,5,6,5), показанный в металлокоординированной форме с указанными размерами хелатного кольца (включая ион металла). Идя против часовой стрелки, конкретная использованная макромолекула представляет собой 5aa-5ca-6cc-5ac- (или любую циклическую перестановку).
Родственные формы (=) функциональных групп для каждого исходного материала показаны графически в Таблице 2 ниже, а возможные комбинации защитных/активированных групп (p/а групп) или скрытые формы (h) каждого исходного материала показаны в виде табличек. Переменные позиции отмечены точкой (•). Подчеркнутые сочетания букв и цифр сбоку представляют собой сокращенные схемы и относятся к размерам хелатного кольца, образованного, когда конкретный исходный материал вводится в макроцикл и координируется относительно металла в центре (см. Структуру 2).
Полный диапазон макроциклических тетраамидных соединений, которые можно синтезировать из исходных материалов, описанных в Таблице 2, в общих чертах приведен в Таблице 3. Каждое уникальное сочетание указано графически и помечено сокращением как в Структуре 2, приведенной выше.
Отдельные исходные материалы для мостика, ответвления и линкера можно либо закупать, либо синтезировать обычными способами. Примеры синтеза немногих исходных материалов, которых нет в продаже, приведены в настоящем тексте в Экспериментальной части. Эффективный альтернативный способ получения замещенных и незамещенных малонатов описан в работе A.P.Krapcho, E.G.E. Jahngen, Jr. and D. S. Kashdan. α-carbalkoxylations of carboxylic acids. A general synthetic route to monoesters ofmalonic acids", Tet. Lett. 32, p. 2721-2723 (1974). Устойчивые к окислению макроциклические тетраамиды, показанные в Таблице 3, могут использоваться с синтезированными, причем не приходится прибегать к использованию тех видов, которые содержат сильные связи N-N, таких как оксиды, гидразины и азосоставляющие.
Схемы 4-6 (см. в конце описания) графически изображают замещение в различных положениях, показанных (•) в таблице 3. В оставшейся части настоящего раздела обсуждается в целом, как выбирать заместители R, и в виде таблиц перечислены некоторые конкретные примеры замещенных исходных материалов мостика, ответвления и линкера.
Замещение на одном узле
Исходные материалы, содержащие только одно изменяемое положение, замещают атомом углерода, несущим две R группы, фрагмент -C(Ra)(Rb)- (в данном случае черточкой (-) обозначена одинарная связь в отличие от амидных связей).
В схеме 4 одно изменяемое положение всегда заменяют фрагментом -C(Ra)(Rb)-.
Чтобы произвести замещение в любом одном изменяемом положении R-группы, на фрагменте -C(Ra)(Rb)- могут быть одинаковыми или различными и их выбирают из группы, состоящей из углеводородов и углеводородов, замещенных гетероатомом (напр. , галогеном, N, O, Si, P, S). Конкретный выбор R-групп, помимо R1 и R2, осуществляют из следующих типов/подтипов групп либо по отдельности, либо в сочетании (напр., для R= арилсилиловый эфир, перечислены только арил, сложные эфиры и силоксаны): H, кетоны, альдегиды, карбоновые кислоты, карбоновые кислоты, имеющие скрытые или защитные/активированные группы (см. Таблицу 1), сложные эфиры, простые эфиры, амины, амины, имеющие скрытые или защитные/активированные группы (см. Таблицу 1), имины, амиды, нитрогруппы, сульфонилы, сульфаты, фосфорилы, фосфаты, силил, силоксаны, алкил, алкенил, алкинил, гелогены, арил и соединения, выбираемые из биологических систем, напр. , природные или искусственные аминокислотные боковые цепи, гетероциклические кольца, лактамы, лактоны, алкалоиды, терпены (стероиды, изопреноиды), липиды или фосфолипидные цепи.
Для замещения на одном узле слияние групп Ra и Rb в положении, которое не является тем местом, где происходит замещение, но к месту замещения дает виды, связанные двойной связью с узлом, такие как оксо (=О), имин (=NRa), или замещенную виниловую группу (=C(RaRb). Образование иминов или замещенных виниловых групп представляет собой вид миграции на узле. Если первоначальные группы Ra и Rb слиты в месте, которое не является местом замещения и не к месту замещения, тогда образуется циклическая кольцевая структура. Если образуются такие циклические группы, то дополнительные R заместители на циклических группах выбирают так же, как для нормального замещения на единственном узле или на нескольких узлах (включая возможность дальнейших слияний R групп в одном или нескольких узлах до получения дополнительных оксогрупп, иминов, замещенных виниловых групп, или спиро-колец, бензольных, замещенных бензольных, гетероциклических, замещенных гетероциклических, циклоалкильных, замещенных циклоалкильных, циклоалкенильных или замещенных циклоалкенильных колец). Предпочитаемыми размерами спиро/циклических колец являются четырех-, пяти- или шестичленные кольца.
Замещение на нескольких узлах см. в схеме 5.
В схеме 5 замена на двух изменяемых положениях может быть осуществлена двумя фрагментами -C(Ra)(Rb)-, или же два изменяемых положения можно объединить, получая часть арильного или гетероциклического кольца.
Для проведения замещения на нескольких узлах, замещения на индивидуальных положениях -C(Ra)(Rb)- производят так же, как замещение на единственном узле. Помимо типов замещений для единственных узлов можно объединить или связать множество узлов вместе посредством конденсации R групп, расположенных на различных узлах в местах, которые либо являются (сочетание), либо не являются (соединение) местами присоединения. Сочетание мест, которые соседствуют друг с другом, ведет к появлению этиленовых фрагментов (-C(Ra)= C(Rb)-) формы устранения R групп. Соединение узлов через конденсацию R групп в местах, которые не являются точками присоединения, или сочетание мест, которые не соседствуют друг с другом, ведет к появлению циклических структур, таких как спиро-колец, бензольных, замещенных бензольных, гетероциклических, замещенных гетероциклических, циклоалкильных, замещенных циклоалкильных, циклоалкенильных или замещенных циклоалкенильных колец. Пяти- и шестичленные кольца являются предпочтительными.
Если образовались циклические группы или если имеются остаточные R группы, оставшиеся после сочетания соседних мест, то остаточные R группы и заместители на циклических группах выбирают так же, как для обычного замещения на единственном узле или на нескольких узлах (включая возможность последующей конденсации R групп до получения дополнительных спиро- колец, бензольных, замещенных бензольных, гетероциклических, замещенных гетероциклических, циклоалкильных, замещенных циклоалкильных, циклоалкенильных или замещенных циклоалкенильных колец).
Важный момент состоит в том, что замещение и единственного узла, и множества узлов может идти в виде рекурсивной функции, напр., замещенный o-фенилендиамин ⇒ замещенный гетероциклический о-фенилендиамин ⇒ замещенный спироциклоалкил гетероциклический о-фенилендиамин и т.д.
На схеме 6 показано, что замена на трех изменяемых положениях может быть осуществлена или тремя фрагментами -C(Ra)(Rb)-, или же два изменяемых положения можно объединить, получая часть арильного или гетероциклического кольца, где третье положение замещено фрагментом -C(Ra)(Rb)-, или же три изменяемых положения можно объединить до образования части конденсированного диарила, конденсированного арильного гетероциклического кольца или конденсированного дигетероциклического кольца.
Некоторые примеры выпускаемых промышленностью и/или синтезируемых исходных материалов для линкеров, ответвлений и мостиков даны в Таблицах 4, 5 и 6 соответственно. Макроциклическое тетраамидное соединение, имеющее нужную конфигурацию хелатного кольца, показанную в Таблице 3, т.е. 556, 5566, 5656, 5666 или 6666, и их варианты, можно получить при использовании общих принципов выбора и комбинации исходных материалов для различных хелатных конфигураций, показанных в Таблице 2, т.е. родственных, защитных/активированных или скрытых групп, с последующим выбором конкретных исходных материалов из Таблиц 4, 5 и 6. Использование таких функциональных групп и аналогичных исходных материалов в новом способе синтеза позволит получить макроциклические тетраамидные соединения, имеющие хелатную конфигурацию колец и совокупность заместителей, пригодных для конкретного конечного использования. Значком * в таблицах отмечены заместители, которые обладают сравнительной устойчивостью к окислению. Значком  в таблицах обозначены заместители, которые очень устойчивы к окислению.
в таблицах обозначены заместители, которые очень устойчивы к окислению.
В таблице 4 указаны некоторые представители производных малонатов дикарбоновых кислот, т.е. линкеры, которые используют для получения макроциклических тетраамидов, находятся либо в форме родственных, либо в форме скрытых групп или защитных/активированных групп.
Таблица 5 идентифицирует некоторые типичные α- и β-аминокарбоновые кислоты, т. е. ответвления, представляющие интерес для получения макроциклических тетраамидов в виде материнских, скрытых или защищенных/активированных групп.
Таблица 6 идентифицирует некоторые типичные диамины, т.е. мостики, представляющие интерес при получении макроциклических тетраамидов, как в виде материнских, скрытых или защищенных/активированных групп. Аминные и защищенные/активированные или скрытые аминогруппы используются взаимозаменяемо.
Список n,n- + 2-диаминов значительно короче, чем список других производных, в значительной степени это объясняется тем, что синтез требуемых n,n + 2-диаминов более сложен, чем n,n + 1-диаминов.
Некоторые конкретные примеры исходных материалов для мостика, ответвления и линкера приведены в Таблице 7. В каждом случае амидные связи разлагались в ходе ретросинтеза до образования аминного эквивалента (амина, нитро, азид, изоцианат и т. п. , см. Таблицу 1) и эквивалента карбоновой кислоты (кислота, сложный эфир, ацилхлорид, нитрил и т.п., см. Таблицу 1).
Мостики и линкеры из Таблицы 7 сохраняют местную двухкратную симметрию, в то время как все ответвления приводят к пятичленным хелатным кольцам.
Некоторые конкретные примеры мостика (Bridge=В), ответвления (Arm=A) и линкера (Linker=L)
Поскольку R группы не принимают участия в реакции синтеза, то возможны многочисленные варианты. Однако, как говорилось выше, чтобы получить устойчивое к окислению соединение и катализатор, к R группам необходимо применить определенные ограничения. Достаточно точно известно, что удаление атома водорода происходит между R-заместителями линкера и аксиальным лигандом, связанным с центральным атомом металла конечной хелатной системы, Такое удаление затем приводит к окислительному разложению, как показано на Фиг. 1. Молекулярные модели показали, что при конформации "ванны" шестичленного кольца линкера в макроциклическом комплексе, атомы H метилена этильных групп могут достигать атомов кислорода в комплексе Fe-оксо. Этот факт, а также другие данные подтверждают механизм, изображенный на Фиг. 1, и объясняют параметры заместителей R1 и R2. Чтобы избежать удаления H атома и последующей деградации, R-группы предпочтительных макроциклических соединений должны быть такими, чтобы они снижали реакцию отделения H-атома и посредством этого замедляли окислительную деградацию. Чтобы осуществить это, группы R1 и R2 соединения по настоящему изобретению должны представлять собой такие группы, которые обладают высокой прочностью связей, не вступают в реакции, или которые недоступны для осевого лиганда, такие как пространственно или конформационно затрудненные группы. Можно использовать любое такое соединение или любую комбинацию таких признаков. Последнего можно достичь путем ограничения конформационной свободы групп R1 и R2, так что они будут просто недостаточно близки друг к другу, чтобы началась реакция. В данном тексте высокая прочность связи C-H означает свыше 94 ккал•моль-1 или свыше 85 ккал•моль-1 для пространственно недоступных C-H связей.
Малонатная часть линкера является наиболее чувствительной частью макроциклического лиганда. Предпочитаемые R группы на линкере включают метил, галоген, водород, CF3 и спироциклопентильное или спироциклогексильное кольцо на месте R1 и R2.
Значительно менее ограничен выбор R-заместителей для ответвлений, чем для линкера из-за устойчивости этой части соединения, которая может быть связана с неспособностью пятичленного кольца изменяться так, чтобы его могли окислять C-H группы в контакте с осевым оксолигандом. Так, R группы α- и β-аминокарбоновой кислоты также можно выбрать с тем, чтобы подобрать заместители для конечной молекулы и обеспечить определенные свойства этой молекулы. Макроцикл может быть симметричным и асимметричным. Для асимметричных макроциклов используют две различных аминокислоты в качестве исходных материалов, и полученные макроциклы представляют собой смесь симметричных и асимметричных вариантов. Два варианта можно разделять известными способами разделения. Несколько примеров соединений по настоящему изобретению показаны ниже.
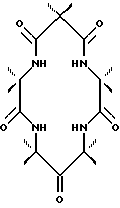
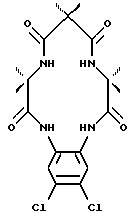
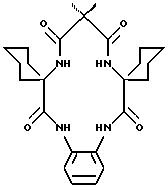
После того как макроциклический тетрадентатный лиганд был получен, макроциклическое соединение можно преобразовать в комплексное соединение с самыми различными ионами металла, предпочтительно переходного металла, и большинство предпочтительных переходных металлов выбирают из металлов VI, VII, VIII, IX, X или XI группы Периодической таблицы для образования хелатного комплекса формулы: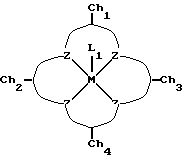
где М представляет собой металл, Z представляет собой устойчивый к окислению атом, образующий комплекс с металлом, такой как N или O, L1 представляет собой любой лабильный лиганд, Ch1, Ch2, Ch3 и Ch4 представляют собой устойчивые к окислению компоненты хелатной системы, описанной выше, которые могут быть одинаковыми или различными и которые образуют пяти- или шестичленные кольца с соседними атомами ZMZ.
Комплексообразования добиваются следующим способом. Макроциклический лиганд растворяют в растворителе, который является носителем, обычно в ТГФ, производят удаление протонов обработкой основанием, предпочтительно бис-триметилсилиламидом лития, диизопропиламидом лития, трет-бутилом лития, n-бутилом лития или фенилом лития. Подойдет любое основание, с помощью которого можно удалять протоны в месте образования комплекса с металлом, т.е. амидные N-H протоны тетраамидного соединения. Предпочтительными являются некоординирующие органические растворимые основания. После того как из лиганда удалили протоны, добавляют ионы металла. Полученное промежуточное соединение, лиганд металла с относительно низкой валентностью, затем подвергают окислению. Этап окисления, предпочтительно, осуществляют при помощи воздуха, хлора, брома или пероксида бензоила до получения хелатного комплекса металла, обычно в виде литиевой соли. Реакция обмена полученного комплекса до получения тетраалкиламмониевой, тетрафенилпосфониевой или бис(трифенилфосфоранилиден)аммониевой (PPN) соли приводит к получению хелатных комплексов металлов, которые легче поддаются очистке, по сравнению с комплексами, содержащими ион лития. Очищенный хелатный комплекс металла можно затем использовать в качестве катализатора реакций окисления.
Если комплекс затем соединяют с сильным окислителем, передающим атом O, предпочтительно, пероксидом, таким как перекись водорода, трет- бутилгидропероксид, кумилгидропероксид, или надкислотой, то получают оксо-промежуточное соединение лиганда металла IV, V или VI группы. Когда устойчивые к окислению заместители использовали для получения каркаса лиганда, то в качестве промежуточных соединений получали устойчивые содержащие оксогруппы соединения. Такие содержащие оксогруппы соединения с высокой валентностью являются активными агентами переноса в ряде каталитических реакций окисления.
Когда металлы с низкой валентностью подвергают воздействию пероксида или другого окислителя, содержащего [O], то металл притягивает и связывает кислород из окислителя. В зависимости от металла связь между металлом и кислородом может быть очень сильной, или она может быть достаточно сильной для того, чтобы удалить кислород из окислителя для последующего переноса к другой составляющей.
Если металл представляет собой ион металла III группы, то полученное оксосоединение в целом будет представлять собой ион металла V группы. Если металл представляет собой ион металла IV группы, то полученное оксосоединение обычно будет содержать ион металла VI группы или комплексное соединение металла V группы со вторым участком окисления на лиганде, т.е. радикал лиганд/катион. Комбинированный стабилизирующий эффект макроциклического лиганда и участие числа d электронов в центре, которым является металл, в процессе изменения степени связывания с оксолигандом, позволяет получать комплексные соединения с переходными металлами (с малыми номерами групп), которые образуют очень сильные связи между кислородом и металлом, благодаря чему получают стабильные оксиды. Переходные металлы, которые относятся к группам со средними или большими номерами, имеют тенденцию удалять кислород из окислителя и связывать оксолиганд до получения реакционноспособного промежуточного соединения. В системе лиганд-металл, полученной способом по изобретению, переходные металлы, которые относятся к группам со средними или большими номерами, имеют тенденцию способствовать переносу кислорода.
Помимо стабилизирующего действия лиганд также влияет на свойства металла. Контролируя выбор металла, плотность электронов в макроцикле, заряд комплекса и прочность связи/порядок связи с координированным оксолигандом, комплекс лиганд/металла можно точно настроить таким образом, чтобы реализовать все возможности по передаче кислорода от стабильных оксидов к катализаторам окисления с высокой валентностью.
В предпочтительном варианте осуществления осевой лиганд L1 является лабильным, поскольку он занимает свое положение относительно металла до тех пор, пока хелатная система не введена в раствор, содержащий окислитель. Лабильный лиганд будет диссоциировать и заменяться окислителем, чаще всего агентом, переносящим атом O, но может заменяться также любым другим окислителем, который можно использовать для активации иона металла для осуществления катализа. Предпочтительные лабильные лиганды включают, в числе прочих анион Cl-, ионы галогенидов в целом, CN-, H2O, OH-, ROH, NH3 или амин, карбоксилат, фенол или феноксид, пиридин, простой эфир, сульфоксид, кетон или карбонат. Участок окисления можно выбирать по осевым лигандам, а также по заместителям на кольце.
Получали макроциклические соединения со спироциклическими заместителями, и было обнаружено, что такие заместители делают макроциклическое соединение очень гидрофобным и, что примечательно, растворимым в пентане и других легких насыщенных алифатических растворителях. Длинноцепочечные заместители, такие как додецильные цепочки или фосфолипидные цепочки, делают макроциклическое соединение растворимым в мембранах.
Спироциклогексильное производное является пространственно затрудненным и дает замедленную реакцию по сравнению с другими предпочтительными заместителями, так что изменяется нормальный синтез амидного промежуточного соединения по первому этапу способа по изобретению.
Синтез бис-спироциклогексильного макролинкерного промежуточного соединения осуществляли путем добавки по капле ацилирующего реагента в нескольких аликвотных количествах, предпочтительно в трех, вводимых через определенные промежутки времени. Наилучшие результаты получали, когда после двенадцатичасовых интервалов следовали длительные периоды взаимодействия. Если период взаимодействия был не очень длительным, то выход был ниже. Последовательность реакций показана на схеме, приведенной ниже. Циклогексан можно использовать для отделения оксазалоновой формы макролинкера от других продуктов реакции, или можно добавить воду, чтобы осуществить гидролиз оксазалона на месте. Гидролиз промежуточных оксазалонов дает повышенный выход искомого бис-циклогексильного макролинкера.
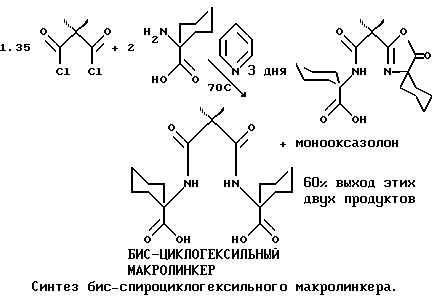
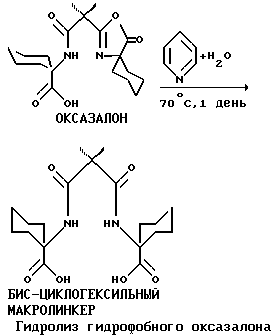
Содержащий циклогексил макролинкер затем готов для стадии закрывания кольца; это осуществляют так же, как для других промежуточных соединений по изобретению. Однако вследствие повышенной стабильности промежуточных макроциклических соединений, содержащих спироциклогексил, отделение макроциклических соединений от побочных продуктов реакции отличается от других предпочтительных способов закрывания колец. Обычно неочищенные продукты реакции, представляющие собой макроциклические соединения, экстрагируют в органический растворитель, такой как CH2Cl2. Раствор CH2Cl2 промывают кислотами и основаниями для удаления примесей и побочных продуктов, которые содержат кислотные или основные функциональные группы, и для осуществления гидролиза любых промежуточных соединений, содержащих оксазалон. Циклогексил-тетраамидное макроциклическое соединение не очень поддается обычной очистке путем промывания кислотами и основаниями, поэтому в результате очистки получают смесь приблизительно 1: 1 бисциклогексилоксазалона и бис- циклогексил-тетраамидного макроциклического соединения. Экстракция пентана в смеси дает чистое разделение. Макроциклическое соединение является нерастворимым и его выделяют в виде порошка, в то время как растворимая в пентане фракция может выпариваться до получения крупных кристаллов бис-циклогексил-оксазалона.
Было замечено, что добавка замещенного малонилдихлорида в избыточных количествах повышает выход макролинкера при оптимальном отношении около 2 моль аминокислоты на 1,35 - 1,5 моль замещенного малонилдихлорида. Смесь продуктов включает макролинкер и монооксазалоновую форму макролинкера, которую можно легко подвергнуть гидролизу до получения дополнительного продукта. Выход продукта, получаемого способом по изобретению, значительно повышается, если из реакционного раствора исключить воду в ходе реакций по закрыванию кольца.
Можно использовать также пиридиндиамины. Известный способ синтеза азидов, который включает в себя этап восстановления, в ходе которого восстанавливается также пиридиновое кольцо, не позволяет получать макроциклическое соединение, имеющее пиридиновый мостик. Варианты соединений, содержащих дополнительную аминогруппу, также довольно сложно синтезировать при помощи известных способов синтеза. Варианты соединений, содержащих дополнительную аминогруппу, представляют значительный интерес, поскольку они позволяют связывать макроциклическое соединение или металлокомплекс с основой, такой как полимер или песок, или с другими молекулами или субстратами, имеющими функциональные группы, которые ковалентно связаны с амином. Группы, которые ковалентно связаны с аминами, хорошо известны и включают (в форме комплексов), например, алкиламины, амиды, сульфонамиды, имины и другие скрытые или защитные/активированные формы, см. Таблицу 1.
Синтез вариантов соединений, содержащих дополнительную аминогруппу, обычно происходит так, как показано на Схемах реакций 7 и 8 (см. в конце описания).
Последовательность реакций включает стратегическое и селективное введение защитной аминогруппы (ацетамида) на арилдиаминовую группу (Мостик). Защищенная форма мостика, ацетамидо-диамин, после этого пригодна для проведения реакции закрытия кольца через стандартный синтез диамин + промежуточный линкер, как описано здесь. Более длительный период реакции закрытия кольца требуется для того, чтобы достичь макроциклизации и это объясняют тем, что между присоединенным оксазалоном и ацетамидной группой образуется нежелательная водородная связь, которая замедляет требуемую реакцию макроциклизации.
После того как было синтезировано макроциклическое соединение, содержащее дополнительную защитную аминогруппу, как показано на Схеме реакций 5, это соединение можно металлировать кобальтом. Удаление ацетильной защитной группы позволяет получить комплексное макроциклическое соединение кобальта, которое легко присоединить к носителю. Наилучшие результаты получали путем реацилирования дополнительной аминогруппы акрилоил-хлоридом до получения макроциклического соединения с дополнительной винильной группой, соединенной с амидной группой.
Затем это соединение можно сополимеризовать с двенадцатикратным избытком различных акрилоиловых мономеров до получения акрилового полимера, который содержит макроциклическое комплексное соединение кобальта в качестве боковой цепи приблизительно на каждых 20 остатков, что схематично показано на Фиг. 5.
Путем присоединения макроциклического комплексного соединения кобальта к полимеру или другому носителю, металл можно восстановить и рециркулировать с помощью системы, схематично показанной на Фиг. 4. Экологически токсичные металлы, например CrVI, можно заместить более экологически безвредными реагентами окисления, такими как CoIV или CoIIILI, где LI относится к окислению, в центре которого находится лиганд.
Из Фиг. 4 видно, что после требуемого процесса окисления прикрепленный окислитель можно рециркулировать, для чего его собирают и повторно окисляют первичным окислителем, таким как гипохлорит, бром или с применением электролиза. Используя прикрепленные макроциклические соединения металлов, как ожидается, можно будет получить действенный способ значительного снижения уровней захоронения отработавших токсичных соединений металлов в окружающей среде.
Экспериментальная часть
Синтез устойчивых к окислению тетраамидных лигандов.
Материалы: Все растворители и реагенты представляли собой вещества химического класса (Aldrich, Aldrich Sure-Seal, Fisher) и использовались по мере получения. Микроанализы проводили в Midwest Microlabs, Indianapolis, Indiana.
Электронные измерения: напряжение синхронизации измеряли в атмосфере N2 в камере с тремя отделениями, при использовании стекловидного угольного дискового рабочего электрода (A ~ 0,0078 см2 или 0,071 см2), противоэлектрода из платиновой проволоки, и электрода из насыщенной хлоридом натрия каломели (SSCE) в качестве справочного электрода. В качестве растворителей применяли CH2Cl2 (Aldrich Sureseal) или CH3CN (высушенный над CaH2), причем в качестве электролита использовали [Bu4N][ClO4](0,1 М, Fluka, высушенный под вакуумом при 24oC) или [Bu4N][PF6](0,1 М, Fluka, puriss). Измерения проводили прибором Princeton Applied Research Model 273 Potentiostat/Galvanostat под управлением компьютера "Compudyne 486DX", а кривые силы тока/напряжения были получены с применением двухкоординатного самописца Graphtec Model WX1200, или при помощи регулятора напряжения/цифрового кулонометра Princeton Applied Research Model 173/179, оснащенного ИК компенсацией с положительной обратной связью, универсального программатора, и двухкоординатного самописца Houston Instruments Model 2000. В некоторых экспериментах добавляли ферроцен (Fc) в качестве внутреннего стандарта потенциала, применимого для выводов. Формальные потенциалы вычисляли как средние значения анодных и катодных пиковых потенциалов и их сопоставляли с NHE. Разделение от пика к пику пары Fc+/Fc во всех случаях соответствовало аналогичному показателю для пар соединения железа. Графики пиков силы тока в зависимости от квадратного корня скорости сканирования в диапазоне 20-500 мВ с-1 для всех пар носили характер линейной зависимости.
Масс-спектрометрия. Спектры электрораспылительной масс-спектрометрии получали с помощью масс-спектрометра Finnigan-MAT SSQ700 (Сан- Хосе, Калифорния), оснащенного электрораспылительным интерфейсом Analytica of Branford. Использовали напряжение электрораспыления в 2400-3400 В. Образцы растворяли либо в ацетонитриле, либо в дихлорметане при концентрациях приблизительно 10 пмоль/мкл и вводили в интрфейс ESI перед сбором данных путем прямого вливания при скорости потока 1 мкл/мин. Эксперименты по масс-спектрометрии электронной ударной ионизации положительными ионами (70 эB) осуществляли при использовании квадрупольного масс-спектрометра Finnigan-MAT 4615 в сочетании с системой сбора данных INCOS. Температура источника ионов составляла 150oC и температура в камере с несколькими отделениями составляла 100oC. Введение образцов производили при помощи газхроматографа или путем прямого введения зонда. Спектры в результате бомбардировки быстрыми атомами (положительными ионами) получали при помощи прибора Finnigan-MAT 212 в сочетании с системой сбора данных INCOS. Ускоряющее напряжение составляло 3 кВт и температура источника ионов составляла приблизительно 70oC. Использовали пушку для седловидного поля, полученного по ионной технологии с использованием быстрых атомов, причем ксенон был в 8 кэВ. В качестве матрицы бомбардировки быстрыми атомами использовали тиоглицерин. Эксперименты по ударной электронной ионизации положительными ионами (70 эВ) MS/MS проводили на тандемном квадрупольном масс-спектрометре Finnigan-MAT TSQ/700. Введение образцов осуществляли при помощи зонда прямого введения. Температуру источника ионов поддерживали в 150oC, а в камере с несколькими отделениями - в 70oC. Диссоциацию, вызванную столкновениями атомов, получали при введении аргона в центр только-рч столкновений восьмиполюсника до тех пор, пока давление в камере не достигло 0,9-2,5•10-6 торр. Номинальная кинетическая энергия ионов, являющихся продуктами диссоциации, вызванной столкновениями атомов, составляла < 35 эВ (лабораторные данные). Данные приборов с высоким разрешением были получены при помощи масс-спектрометра с двойной фокусировкой JEOL JMS АХ-505H конфигурации EB с использованием разрешения 7500. Введение образцов осуществляли при помощи газ-хроматографа или зонда для непосредственного введения. В процессе получения масс-спектров перфторкеросин вводили в источник ионов через нагретое отверстие. Точные массы получали при помощи компьютерной интерполяции из масс перфторкеросина. Условия ГХ/МС: колонна 20 м х 0,25 мм DB-1701 (J&W Scientific); газ-носитель - гелий с линейной скоростью 40 см/сек; инжектор, 125oC; температура колонны 35oC в течение 3 мин, затем поднимается с интенсивностью 10oC/мин до 100oC; впрыскивание, разделительное, примерно при отношении 50:1.
Спектроскопические методы: 300 МГц 1H ЯМР спектры и 75 МГц 13C ЯМР спектры получали на приборе IBM AF300 при помощи магнитной системы Oxford Superconducting, сбор данных контролировался программным обеспечением Bruker. Инфракрасные спектры получали на спектрометре Mattson Galaxy Series 5000 FTIR под управлением компьютера Macintosh II. Спектры УФ/vis получали на спектрофотометре Hewlett Packard 8452A под управлением компьютера Zenith Z- 425/SX. Обычные спектры ЭПР X-полоса были записаны на спектрометре Bruker ER300, оснащенном криостатом гелиевого потока Oxford ESR-900. Спектры Moessbauer были получены на приборах с постоянным ускорением, а изомерические сдвиги отмечали относительно металлического железа в качестве стандарта при 298 К. Чтобы избежать ориентации поликристаллических образцов под действием магнитного поля, образцы суспендировали в замороженном нуйоле (nujole).
Синтез диаминов, не выпускаемых промышленностью
Пример 1
A. Синтез 1.2-диамино-4.5-диметоксибензола из 1.2- диметоксибензола (вератролa) 1,2-динитро-4.5-диметоксибензол: Вератрол дважды нитрировали в соответствии со способом по работе Drake et al, in "Synthetic Antimalarials, Some Derivatives of 8- Aminoquinoline", J. Amer. Chem. Soc., 1536, Vol. 68 (1946). Азотную кислоту (68,3 г, конц.) добавили (по капле, 1 час) в хорошо перемешанный раствор вератрола (48,3 г, 350 ммоль, d = 1,084) в ледяной уксусной кислоте (1450 мл), изначально охлажденной до 15oC. Смесь необходимо выдерживать при температуре ниже 40oC, но выше 10oC путем охлаждения и регулирования скорости добавки кислоты. Отделяется значительное количество мононитровератрола. Перемешивание продолжали и добавили дополнительное количество азотной кислоты (212,7 мл, дымящая) (добавку производили по капле в течение 1 часа), в то время как температуру раствора поддерживали на уровне ниже 30oC. По мере того, как шло второе нитрирование, мононитровератрол растворялся и, когда добавили всю кислоту, раствор становился прозрачным. Смесь оставили на два часа и затем вылили на примерно 1,5 л ледяной воды. Выпавшее в осадок динитросоединение отфильтровывали, обильно промывали водой до полного удаления кислоты (pH > 5) и рекристаллизовывали непосредственно из минимального количества горячего EtOH (600 мл). Выход 1,2-диметокси-4,5-динитробензола составил 69,0 г (87%). Характеристики: темп. плавления 129,5-130,5oC. 1H ЯМР (CDCl3) δ [ppm] : 7.35 (s, 2H, ArH), 4.02 (s, 6H, OCH3). ИК nujol ν [см-1]: 3124 (s, w, арил CH), 3073 (s, w, арил CH), 1592 (s, str, распрямление арильного кольца), 1535 & 1518 (s, str, ArNO2).Расчетные данные для C8H8N2O6: C, 42.11; H, 3.53; N, 12.28. Обнаружено: C, 42.12; H, 3,54; N 12,33.
1.2-диамино-4.5- диметоксибензол: 1,2-диметокси-4,5-динитробензол (10 г, 43,8 ммоль) восстанавливали до 1,2-диметокси-4,5-диаминобензола в кислом MeOH (175 мл + 2 экв. неорганической кислоты (т.е. 10 мл конц. HBr)) путем каталитического гидрирования при использовании 10% Pd/C катализатора (24-36 часов, 20-22 фунтов на кв.дюйм H2 были поглощены из резервуара). Если вначале добавляли более 2 экв. HBr, то действие Pd/C катализатора подавлялось. После завершения гидрирования добавляли еще 4-5 экв. конц. неорганической кислоты, чтобы защитить материал от окисления воздухом и смесь выпаривали в роторном испарителе до получения красного/фиолетового масла. Неочищенный материал очищали добавкой небольшого объема абс. EtOH6 затем выливали шлам на 600 мл ледяного Et2O и оставляли в морозильнике на ночь.
Красно-фиолетовый продукт собирали фильтрацией, быстро высушивали воздухом, затем хранили в эксикаторе до завершения процесса сушки. Продолжительное воздействие на соль диамина воздуха/воды вызывает позеленение, которое свидетельствует о необратимом окислении. В результате гидрирования выход составил около 90%. Характеристики красно-фиолетового 1,2- диметокси-4,5- диаминобензола (гидрата дигидробромидной соли): 1H ЯМР (d5пиридин) δ [ppm]: 10.35 (s, br, 7.5 H, H2O/py.HBr/R-NH2 быстрый обмен), 7.35 (s, 2 H, ArH), 3.60 (s, 6 H, ArOCH3) ИК (nujol/NaCl) ν [cm-1: 3085 (br, OH), 2557 (s, str, ArNH3 +), 1623 (s, w, асимметричн. NH3 + сгиб/растяжение арильного кольца), 1539, 1519 (s, m. симметр. MH3 + сгиб). (Расчетные данные для C8H12N2O2) (HBr)2, (H2O)0,66; C, 28.09; H, 4.52: N, 8.19. Обнаружено: C, 27.82; H, 4.18; N, 8.37. Независимое подтверждение гидрирования было получено из результатов ИК и ЯМР спектроскопии.
Получение безводной сульфатной соли 1,2-диамино-4,5-диметоксибензола было описано в работе Nakamura, M. et al. in "Fluorimetric Determination of Aromatic Aldehydes with 4,5- Dimethoxy-l,2-Diaminobenzene". Anal.Chim. Acta. (1982), 134, p.39-45: 1,2-диамино-4,5-диметоксибензол (2 г) растворяли в EtOH (20 мл) и смешивали с H2SO4 (конц., около 2 мл). Продукт рекристаллизовывали из EtOH, получая почти бесцветные иглы (выход около 2 г). Расчетные данные для C8H14O6N2S: C, 36.1; H, 5.3; N, 10.5. Обнаружено: C, 35.85; H, 5.6; 20 N, 10.4.
Б. Получение 1.2-диамино-4-ацетамидобензола из 1.4-диамино-2-нитробензола (2-нитро-1.4-фенилендиамин).
1-амино-2-нитро-4-ацетамидобензол: 1,4- диамино-2-нитробензол (2-нитро-1,4- фенилендиамин) селективно ацетилировали по способу, описанному в работе McFarlane et. al, J. Chem. Soc. Perkin Trans., 691 (1988), которая упомянута здесь для сведения. Аминогруппы от мета- до нитрогрупп легко подвергаются ацетилированию при помощи уксусного ангидрида в ацетоне (аминогруппы от орто до нитродезактивируются). Выход 1-амино-2-нитро-4- ацетамидобензола (2-нитро-4-ацетамидоанилина) составил > 90%. Характеристики: 1H ЯМР (CD3OD) δ [ppm]: 8,3 (m, 1, H, ArH), 7.5 (M, 1H, ArH), 6.9 (M, 1H, ArH), 2.1 (s, 3H, ацетил CH3, что хорошо согласуется с McFarlane. ИК (nujol/NaCl) ν [cm-1] : 3470 (s, str, HOAc), 3340- 3150(m, m/str, ацетамид ArNH + ArMH2), 1661 (s, str, ацетамид CO), 1643 (s, str, H связанный ацетамид CO), 1592 (s, m/w, растяж.арильн.), 1547 (s, str, ArNO2,) & 1512 (s, m ArNO2). Анал. (Высушено при 80oC) Расчетные данные для: C8H9N3O3; C 49.23; H, 4.65; N, 21.53. Обнаружено: C, 49.36; H, 4.55; N, 21.31.
1.2-диамино-4-ацетамидобензол: 1 -амино-2-нитро-4-ацетамидобензол восстанавливали до 1,2-диамино-4-ацетамидобензола в уксусной кислоте (HOAc)/MeOH, используя каталитическое гидрирование на 10% Pd/C катализаторе. Материал выделяли в виде дигидрохлоридной соли. Выход > 90%. Характеристики:
1H ЯМР (CD3OD) δ [ppm]: 6.94 (т, 1 H, ArH), 6.68 (т, 1 H, ArH), 6.62 (т, 1 H, ArH), 2.1
(s, 3H, ацетил CH3). ИК (nujol/NaCl) ν [cm-1]: 3348 (s, str, ацетамид ArNH), 3226- 3100
(m, m, ArNH2), 2588 (s,br, str, ArNH3 +), 1649 (s, str, ацетамид CO), 1623 (s, str, H - связанный ацетамид CO). Анал. (Высушено при 80oC) Расчетные данные для C8H13N3OCl2. (HCl/H2O)0,1: C, 39.45; H, 5.50; N, 17.25; Cl, 30,57. Обнаружено: C, 39.39;
H, 5.53; N, 17.32; Cl, 30.37. Присутствие сольвата HCl/H2O подтверждалось ИК, и соответствовало постоянно кипящей 36,5-38% HCl, используемой для получения гидрохлоридной соли.
В. Получение 2.4-диамино-2.4-диметилпентона из 2.4-диметилпентанона.
2.4-дибромо-2.4-диметилпентанон: в 2,4-диметилпентанон (85 мл, 68,5 г, 0,60 моль) в
CCl4 или 1,2-дихлорэтане (1 л) добавили N-бромосукцинимид (NBS, 240 г, 1,35 моль,
2,26 эквив.). Смесь нагревали с обратным холодильником и добавляли бензоилпероксид (около 20 мг). Пока раствор нагревали с обратным холодильником (24 часа), светло-оранжевое твердое вещество (сукцинимид) всплывало к поверхности галогенированного растворителя, в то время как непрореагировавший N-бромосукцинимид оставался на дне. Бензоилпероксид несколько раз добавляли в смесь при перегонке (около 20 мг; 12-24 часовые интервалы) до тех пор, пока никакого N-бромосукцинимида не будет видно, обычно реакция завершалась через 24 часа. Когда реакция завершалась, твердые вещества собирали фильтрацией и передавали в отходы, галогенированный растворитель/Br2 удаляли из маточного раствора при пониженном давлении, получая светло-желтое масло. Чтобы удалить остатки галогенированного растворителя добавляли 95% EtOH (100 мл), растворитель снова удаляли при пониженном давлении, и получали желтое слегка загрязненное масло (159,99 г, 0,59 моль, 98%). 1H ЯМР (CDCl3): 2.1 (s). ИК(чист./NaCl) ν [cm-1]: 3375 (s, w, загрязнение OH), 3014, 2978, 2933 (s, str, CH), 2858 (s, w, CH), 1701 (s, str, кетон CO).
2.4-диазидо-2.4-диметилпентанон: Раствор 2,4-дибромо-2,4- диметилпентанона получали вышеописанным способом или закупали у фирмы Lancaster Synthesis (89,8 г, 0,33 моль) в EtOH (1,2 л, 95%) добавили в раствор NaN3 (Внимание! 47,2 г, 0,726 моль, 2,2 эквив.) в воде (0,6 л). Раствор нагревали с обратным холодильником (16 ч) до получения светло-оранжевого раствора. EtOH удаляли при пониженном давлении до тех пор, пока раствор не становился мутным. Мутный водный раствор три раза экстрагировали, все еще теплым, пентаном (500 мл), и соединенные экстракты высушивали над Na2SO4 и концентрировали до 300 мл при пониженном давлении. Затем добавляли ледяную уксусную кислоту (100 мл) и оставшийся пентан удаляли при пониженном давлении. Эта обработка нужна для того, чтобы удалить избыток NaN3, поскольку продукт на следующем этапе подвергается воздействию Pd/C и следует избегать образования азидов тяжелых металлов (из-за опасности взрыва). Растворитель удаляли из небольшого образца при пониженном давлении до получения чистого масла (< 20 мг), которое использовали для проведения спектроскопии: 1H ЯМР (CDCl3):1,54 (s). ИК (чист.) ν [см-1]: 2115 (RN3), 1720 (кетон CO). Следует учитывать, с точки зрения безопасности, что органические азиды, получаемые в ходе данной реакции и синтеза на основе азидов, никогда не выделяются в концентрированном виде или в виде твердых веществ в количествах, превышающих 20 мг.
2.4-диамино-2.4-диметилпентан-3- он: Ледяную уксусную кислоту (50 мл) добавляли в HOAc раствор диалкилазида, полученного на предыдущем этапе, и этот раствор добавляли в 10% Pd/C (2,7 r). Смесь гидрировали при давлении 50 фунтов на кв.дюйм (344,738 кПа) (1 неделю) в гидрогенераторе Парра. Поскольку в ходе реакции на каждую абсорбированную молекулу H2 получается одна молекула N2, бомбу извлекали и повторно 10 раз увеличивали давление H2 до 50 фунтов на кв.дюйм (344,738 кПа). (H2 из резервуара высокого давления не достаточно эффективно поглощается). Уголь удаляли фильтрацией, а HOAc удаляли при пониженном давлении. После добавки HBr (48%, 76 мл) смесь растворяли в EtOH. Летучие соединения удаляли при пониженном давлении до получения твердого рыжевато-коричневого вещества, которое промывали смесью (200 мл) ТГФ (50%(, EtOH (45%), и конц. HBr (5%), или смесью ТГФ (95%) и конц. HBr (5%). Полученный белый порошок представлял собой дигидробромидную соль 2,4-диамино-2,4- диметилпентан- -она (56,2 г, 48% из 2,4-дибромо-2,4- диметилпентанона). Дополнительный продукт можно собрать из промывной воды, которую сливают из нескольких операций получения веществ. Продукт нужно хранить в виде дигидробромидной или дигидрохлоридной соли, чтобы защитить амины от окислительной деградации.
Характеристики: 1H ЯМР(CDCl3/DMSO-d6) 2,4-диамино-2,4-диметилпентан-3-он. 2 HBr: 8.62 (6H, s, br. MH3), 1.77 (12 H, s, Me). ИК (свободное основание, nujol mull) ν [см-1: 3460-3160 (RNH2), 1690 (кетон CO). Анал. (Высушено при 80oC). Расчетные данные для C7H16N2O.(HBr)2: C, 27.47; H, 5.93; N, 9.15; Br, 52.22. Обнаружено C, 27.43; H, 5.91; N, 9. 11;Br, 52.46.
Синтез макроцикличестких тетраамидо-N доноров лигандов
Пример 2
Синтез макролинкерного промежуточного соединения (A-L-A) из α-метилаланина и диметилмалонилдихлорида (тетраметилдиметил-замещенное промежуточное соединение).
Гексаметильное промежуточное соединение (НМ)
Помещают двухгорлую колбу (1 л), оснащенную воронкой, которая позволяет уравновешивать давление (250 мл), и перегородкой в атмосферу N2. Добавляют α\-аминоизомасляную кислоту (т. е. α -метилаланин) (20,62 г, 0,2 моль) и сухой пиридин (250 мл, высушенный на ситах 4 А моль) в колбу и нагревают до 55-65oC при перемешивании, затем добавляют диметилмалонилдихлорид (17,8 мл, 0,135 моль), растворенный в сухом пиридине (100 мл, высушен на ситах 4 А моль)в воронку.
Добавляют содержимое воронки (по капле, 1 час) в реакционную смесь и оставляют смесь для осуществления ацилирования (60-70oC, 30-36 часов) в атмосфере N2, или используют сушильную трубку. Когда ацилирование завершено, реакцию прекращают путем добавки H2O (30 мл) при перемешивании (60-70oC, 24 часа).
Уменьшают объем растворителя выпариванием на роторном испарителе до получения масла, затем добавляют HCl (конц., около 25 мл) до окончательного значения pH в 2-3. Ставят горячий раствор в холодильник (4oC, 15 час.) и собирают полученный продукт фильтрацией на фритте, и тщательно промывают ацетонитрилом (2х100 мл). Высушенный воздухом белый продукт (16,5 -19,8 г, выход 45-60%) нужно хранить в эксекаторе. Этот продукт обычно является достаточно чистым для того, чтобы происходила реакция закрытия колец, но иногда может потребоваться рекристаллизация. Характеристики: 1H ЯМР (d5 пиридин, δ [ppm] ); 9/2-9.8 br s, 2H (карбоксильн. OH), 8.23 s, 2H (амид), 1.87 s 12H (CH3), 1.74 s 6H (CH3). ИK(nujol/NaCl) ν [см-1]: 3317.0 (амид NH); 1717.9 (карбоксильн. CO); 1625.7 (амид CO). Анал. (высушено при 100oC). Расчетн. данные для C13H22N2O6: C 51.63, H 7.34.N 9.27. Обнаруж.: C 51.64, H 7.35, N 9.33.
Пример 3
Крупномасштабный синтез макролинкерного промежуточного вещества из α -метилаланина и диэтилмалонилдихлорида (тетраметилдиэтил-замещенное промежуточное соединение)
Если нужно осуществить крупномасштабный синтез, то двухгорлая колба (2 л, RB+Claisen) должна быть оснащена воронкой уравновешивающей давление (250 мл) и перегородкой, и эту колбу помещают в атмосферу N2. Добавляют α -аминоизомасляную кислоту (т.е. α -метилаланин) (90,3 г, 0,9 моль)(или любой описанный выше α или β -амино), через трубочку вводят безводный пиридин (1,4 л, sure seal) в колбу и нагревают до 45- 55oC при перемешивании, Через трубочку вводят пиридин (100 мл, sure seal), а затем диметилмалонилдихлорид (104,4 мл, 0,61 моль) в воронку. Добавляют содержимое воронки (по капле, 3-4 часа) в реакционную смесь, удаляют воронку, оставляют колбу для осуществления ацилирования (55-65oC, 120-130 часов) в атмосфере N2. После того, как ацилирование завершено, реакцию прекращают путем добавки H2O (100 мл) при перемешивании (60-70oC, 24-36 часов). Уменьшают объем растворителя выпариванием на роторном испарителе до получения масла, затем добавляют HCl (конц., около 110 мл) до окончательного значения pH в 2-3. Ставят горячий раствор в холодильник (4oC, 15 час. ) и собирают полученный продукт фильтрацией на фритте, и тщательно промывают ацетонитрилом (700 мл, 150 мл) при перемешивании в колбе Эрленмейера. Толкут высушенный на воздухе белый продукт (87,9 г, выход 60%) в ступке и хранят в эксикаторе. Промежуточное амидное вещество, полученное в результате крупномасштабной реакции, перед использованием в реакции закрытия колец, скорее всего, необходимо будет подвергнуть рекристаллизации.
Пример 4
Рекристаллизация гексаметильного промежуточного соединения.
Неочищенное промежуточное соединение из Примера 2 или 3 (50,4 г, 0,153 моль) в H2O (немного менее 500 мл, деионизиров.) растворяют путем добавления Na2CO3 (16,2 г, 0,153 моль) в три части, добавку производят медленно и осторожно, чтобы избежать чрезмерного пенообразования. Перемешивать нужно медленно, а нагревать понемногу. Раствор доводят до кипения, фильтруют и подкисляют HCl (конц., 30 мл, 0,36 моль). Оставляют раствор остывать (на ночь, 4oC), затем отфильтровывают осадок и промывают ацетонитрилом (250 мл). Продукт, высушенный воздухом (38,8-45,4 г, выход 77-90%) нужно хранить в эксикаторе.
Реакции макрокристаллизации.
Разработано несколько путей синтеза для получения макроциклических тетраамидолигандов. Способ, основанный на органических азидах, описан в работе Uffelman, E. S. , Ph.D. Thesis, California Institute of Technology (1992) and Kostka, K. L, Ph.D. Thesis Carnegie Mellon University (1993). Примеры нескольких путей синтеза для получения макроциклических тетраамидолигандов при использовании нового способа синтеза, приведены ниже.
Присоединение трихлорида фосфора.
Способ PCl3 присоединения амидного промежуточного продукта реакции к ароматическим 1,2- диаминам позволяет безопасно и дешево получать макроциклические тетраамиды при высоком выходе продукта. Можно использовать два различных варианта метода PCl3 присоединения, различия между которыми относятся к последовательности введения добавок и к выбору реагентов. Эти способы можно применять для получения широкого круга различных макроциклов с различными заместителями на уровне электронов, присутствующими на мостиковом диамине, или с пространственными заместителями, присутствующими на амидном промежуточном соединении, главным образом из-за параллельного включения амидных производных макролинкерного типа во все виды синтеза.
Пример 5
A. Синтез макроциклических соединений посредством PCl3 присоединения.
В колбу с длинным горлом (250 мл) помещают амидное промежуточное соединение из Примеров 2-4 (10 ммоль) и мешалку, а затем колбу прокаливают в печи (80-100oC, 30- 45 мин). Горячую колбу помещают в атмосферу N2, добавляют арилдиамин (10 ммоль) и безводный пиридин (50 мл, sure seal). Колбу нагревают (50-60oC) и через шприц впрыскивают PCl3 (d=1.574 г/мл, 1,72 мл, 20 ммоль); впрыскивание производят как можно быстрее без перегонки. Происходит экзотермическая реакция, поэтому следует принять меры предосторожности. Затем температуру повышают до температуры перегонки или чуть ниже нее (100- 115oC) оставляют смесь для осуществления реакции в атмосфере N2 (48 часов). После завершения ацилирования содержимое колбы подкисляют HCl (1 экв., около 60 мл) до итогового значения  . Смесь перемещают в колбу Эрленмейера (для промывки колбы используют воду) и перемешивают с CH2Cl2 (300 мл, 2-3 часа), затем экстрагируют дополнительным количеством CH2Cl2 (2 х 150 мл). Объединенные органические слои промывают разбавленной HCl (0,1 М, 2х100 мл), а затем разбавленным водным Na2CO3 (2 х 5 г/100 мл). Органические растворители удаляют на роторном испарителе до получения неочищенного продукта (30%). Масса неочищенного продукта обычно эквивалентна начальной массе диамина.
. Смесь перемещают в колбу Эрленмейера (для промывки колбы используют воду) и перемешивают с CH2Cl2 (300 мл, 2-3 часа), затем экстрагируют дополнительным количеством CH2Cl2 (2 х 150 мл). Объединенные органические слои промывают разбавленной HCl (0,1 М, 2х100 мл), а затем разбавленным водным Na2CO3 (2 х 5 г/100 мл). Органические растворители удаляют на роторном испарителе до получения неочищенного продукта (30%). Масса неочищенного продукта обычно эквивалентна начальной массе диамина.
Б. Синтез макроциклических соединений посредством PCl3 присоединения
В колбу с длинным горлом (250 мл) помещали MgSO4 (5 г), мешалку, арилдиамин (10 ммоль) и пиридин 50 мл (высушен на ситах 4 А моль), затем помещали в атмосферу N2. Через шприц добавляли PCl3 (d=1.754 г/мл, 1,72 мл, 20 ммоль) и смесь перегоняли в течение 30 мин, до образования оранжево-желтого осадка. Затем смесь немного охлаждали, добавляли амидное промежуточное соединение (10 ммоль), после чего смесь перегоняли в атмосфере N2 (115oC, 48 часов). После завершения ацилирования содержимое колбы подкисляли HCl (1 экв., около 60 мл) до итогового значения pH ≈ 2. Смесь перемещали в колбу Эрленмейера и перемешивали с CH2Cl2 (300 мл, 2- 3 часа), затем экстрагировали дополнительным количеством CH2Cl2 (2 х 150 мл). Объединенные органические слои промывают разбавленной HCl (0,1 М, 2х100 мл), а затем разбавленным водным Na2CO3 (2х5 г/100 мл). Органические растворители удаляют на роторном испарителе до получения неочищенного продукта (30%). Масса неочищенного продукта обычно эквивалентна начальной массе диамина.
Примечание: для крупномасштабных реакций макроциклизации время взаимодействия увеличивали до 4-5 дней при перегонке смеси, и большую часть пиридина, присутствовавшего в конце взаимодействия, удаляли при помощи роторного испарителя перед подкислением.
Пример 6
Получение гексаметил-дихлорбензола из гексаметильного промежуточного соединения + дихлорбензолдиамин 1.2-диамино-4.5- дихлорбензол (1,77 г, 10 ммоль) использовали в качестве диамина с гексаметиламидным промежуточным соединением (3,02 г, 10 ммоль) в PCl3 способе A или Б проведения реакции макроциклизации. Неочищенное макроциклическое соединение (1,33 г, 30%) рекристаллизовывали из минимального количества горячего н- пропанола выпариванием. После первой рекристаллизации получали выход 60%. Характеристики: 1H ЯМР δ [ppm] : 7.69 (s, 2H, ArH), 7.39 (s, 2H, амид NH), (s, 2H, амид NH), 1.58 (s, 12H, метильные ответвления), 1.53 (s, 6H, малонатметил), отмечали небольшие пики n-пропанола. ИК (nujol/NaCl) ν [cm-1]: 3503 (s, br, m-w, n-пропанол OH), 3381 (sh, m, амид NH), 3338 (s, str, амид NH), 1689 (s, str, амид CO), 1643 (s, str, амид CO). Анализ. Расчетн. данные для C19H24N4O4Cl2• (C3H8O)0,2: C, 51.70; H, 5.57, N 12.30%. Обнаружено C, 51.69; H, 5.63; N, 12.33%.
Реакции присоединения посредством оксазалона
Оксазалоновое присоединение амидного промежуточного соединения к ароматическим диаминам также позволяет получать макроциклические тетраамиды безопасным и дешевым способом, причем выход продукта реакции будет высоким, но при этом наблюдается меньшая чувствительность к дополнительным функциональным группам. Макроциклы, способные образовываться посредством PCl3 присоединения, можно получать также посредством присоединения через оксазалон. Кроме того, меньшая чувствительность к дополнительным функциональным группам позволяет получать лиганды с дополнительными функциональными группами, которые придают новые свойства получаемым комплексным соединениям металлов. Конкретные примеры включают введение реакционноспособных групп (таких как амин или винил), присоединенных в виде дополнений к арильному кольцу макроцикла, что позволяет ковалентное присоединение предварительно полученных макроциклов к некоторым полимерным основам.
Пример 7
Синтез макроциклических соединений с использованием оксазалонового способа.
В колбу с длинным горлом (250 мл) помещали амидное промежуточное соединение (3,3 г, 10 ммоль), мешалку, а затем колбу прокаливали в печи (80-100oC, 30- 45 мин). Горячую колбу оснащали перегородкой и помещали в атмосферу N2. Через трубочку вводили безводный пиридин (50 мл, sure seal) и продолжали нагрев, одновременно добавляя через шприц триметилацетилхлорид (т. е. пивалоилхлорид) (22-24 ммоль). Температуру повысили до перегонки или немного ниже температуры перегонки (110-115oC) и позволили реакции протекать в атмосфере N2 (22-26 часов), при этом тщательно избегали загрязнения продуктами других реакций с N2. Реакционная смесь меняет цвет с прозрачного светло-желтого до желто-коричневого. После того как образование оксазалона завершается (примечание: *Откачивание равного количества и повторное растворение в сухом d5 пиридине позволяло получить доминантный вид (> 80% бис-оксазалона через 24 ч перегонки), имеющий следующие характеристики: 1H ЯМР d [ppm] : 2,10 (q.4H, метилен CH2's), 1,38 (s, 12H, RCH3), 0,85 (t, 6H, этил CH3's). После добавки воды в образец для ЯМР, восстанавливался нормальный спектр для амидного промежуточного соединения через примерно 20 часов при КТ), добавляют арилдиамин (8-10 ммоль) либо в виде твердого вещества, либо в виде суспензии в безводном пиридине, вводимой через трубку с большим отверстием, либо в виде раствора в безводном пиридине (sure seal), из которого в атмосфере N2 отведены газы, если ограничения, накладываемые растворимостью и наличием пространства в верхней части колбы, позволяют это сделать. Реакция закрытия колец происходит при перегонке в течение последующих 48-72 часов (более длительное взаимодействие потребуется при крупномасштабных реакциях) в атмосфере N2, без взаимного загрязнения продуктами других реакций. Смесь обычно становится черно-коричневой. После того как ацилирование завершено, реакцию останавливают добавкой H2O (30 мл) и перемешивают при перегонке (100oC, 22-26 часов). Смесь охлаждают и перемещают в колбу с круглым дном (500 мл) используя минимальное количество H2O, чтобы промыть длинногорлую колбу. Растворитель удаляют посредством роторного испарителя до получения смеси неочищенного продукта в виде масляного рыжевато-коричневого или черно-коричневого твердого вещества. Следует отметить, что если позволяют функциональные группы, то смесь неочищенных продуктов может растворяться в CH2Cl2 и ее можно промывать разбавленной водной HCl и разбавленным водным Na2CO3. Удаление органического растворителя при пониженном давлении дает нормальное макроциклическое соединение, знакомое из реакция PCl3 присоединения и пригодное для непосредственной рекристаллизации, в результате которой получают чистое макроциклическое соединение.
Пример 8
Получение TMDE-AcB из тетраметилдиэтил- промежуточного соединения + АсВ диамин через оксазалон.
Это макроциклическое соединение представляет собой защищенную форму макроциклического соединения, содержащего дополнительные аминогруппы, которое может быть присоединено к различным носителям благодаря образованию амидной связи между основой и дополнительной аминогруппой. Благодаря образованию нежелательной водородной связи (как предполагают) реакция закрытия колец требует длительной перегонки, чтобы добиться образования макроциклов, 1,2-диамино-4- ацетамидобензол-дигидрохлорид (9 ммоль) использовали в качестве диамина в реакции закрытия оксазалонового кольца. Время макроциклизации увеличивали (при перегонке 5 дней), после чего реакцию останавливали обычным способом и проводили кислотно- основную обработку до получения смеси триамидного соединения, содержащего макроциклический имидазол, и искомого тетраамидного макроциклического соединения. Дальнейшая очистка осуществлялась с применением хроматографии на силикагеле (1 >> х 4-5 >>), при использовании ацетонитрила в качестве элюента. В альтернативном варианте неочищенный продукт можно очистить рекристаллизацией из горячего этанола, хлороформа или дихлорэтана. Выход из диамина составил 15-20%. Характеристики: 1H ЯМР (CD3CN) δ [ppm]): 8.31 (s, 1H, арилацетамид NH), 7.72 (m, 1H, ArH), 7.55 (s, 1H, ариламид NH), 7.44 (s, 1H, ариламид NH), 7.30 (m, 2H, ArH), 6.86 (s, 2H, алкиламид NH), 2.05 (q, 4H, этил CH2's), 2.01 (s, 3H, ацетил CH3, 1.49 (d, 12H, RCH3's), 0.82 (1, 6H, этил CH3's). ИК (nujol/NaCl) ν [см-1]: 3368 (s, m, амид NH), 3319 (s, m, амид NH), 3291 (sh, m, амид NH), 3268 (s, str, амид NH), 1678 (sh, m, амид CO), 1667 (s, str, амид CO), 1656 (s, str, амид CO), 1639 (sh, m, амид CO), 1608 (s, m, арильное кольцо/амид). Анал. расчетн. данные для C23H33N5O5: (H2O)1,25: C, 57.31 H, 7.42 N, 14.53 Обнаружено: C, 57.02; H, 7.15; N, 14.33. Присутствие сольвата H2O было подтверждено 1H ЯМР и ИК.
Пример 9
Синтез пералкилированных макроциклических соединений (MAC*) или тетраметилдиметил - диметилпентана из тетраметилдиметил промежуточного соединения + 2.4-диамино-2.4-диметил-пентан-3-она (DMP) через оксазалон
PCl3 способ для получения H4[MAC*](_тетраметилдиметил диметилпентана) не позволил получить приемлемые количества макроциклического соединения; предположили, что это вызвано образованием нежелательного комплекса между функциональной группой диаминкетона и фосфорным реагентом. В отличие от PCl3 способа, который использует гетерогенный раствор, оксазалоновый способ получения H4[MAC*] является способом, где используется гомогенный раствор, что упрощает применение диагностических методик, таких как 1H ЯМР, для определения причин неудачи синтеза. Взаимодействие тетраметилдиметил-бис-оксазалона с диметилпентан-диамином в сухом пиридине не позволяет получить амиды (по анализу ЯМР). Поскольку оксазалоновый способ нечувствителен к кетонным функциональным группам, то неудачу при получении амидов отнесли за счет образования солей кислот на алкиламинной функциональной группе, причем алкилдиамин на 3-4 pKa единиц более щелочной, чем пиридин, в то время как арилдиамины имеют значения pKa, близкие к пиридину. Поэтому более щелочной высококипящий растворитель (триэтиламин, трипропиламин, диэтиланилин) можно использовать для увеличения количества образующегося амида. Для аминсодержащих растворителей присутствие воды и аминных загрязнителей вызывает проблемы, учитывая низкую растворимость реагентов. Добавка льюисовой кислоты в качестве высушивающего агента приносит большую пользу. Заметный выход H4[MAC*] можно получить (неоптимизированный выход макроциклического соединения составляет 2-3%) в результате взаимодействия (1 этап) тетраметилдиметил-бис-оксазалона с диметилпентан-алкилдиамином в трипропиламин+CaO, при перегонке. Выделение продукта следует осуществлять путем фракционной рекристаллизации из толуола в сочетании с 1H ЯМР.
Наиболее высокий выход H4[MAC*] из алкилдиамина посредством известного способа Uffelman (4 этапа из алкилдиамина) составляет 8-10%. Заметный выход H4[MAC*] можно получить посредством оксазалонового способа.
Синтез хелатных комплексов
Соединения под номерами 2, 3, 4 и 5 в приведенных ниже примерах являются диметильными копиями соединений по Фиг. 6.
Пример 10.
[Et4N] 2 и [Et4N]3. [тетраэтиламмониевые соли хлоро тетраметилдиметил-дихлорбензол-моноаниона железа(III) и акво - тетраметилдиметил-дихлорбензол-моноаниона железа (III) соответственно.
Исходный макроциклический тетраамид по любому из приведенных ниже примеров (525 мг, 1,1 ммоль) растворяют в тетрагидрофуране (40 мл, Альдрих) в атмосфере N2. Трет-бутиллитий в атмосфере N2 (2,6 мл, 4,4 ммоль, 1,7 М в 2,4-диметилпентане, Альдрих) добавляют в раствор в атмосфере N2 при -108oC. Добавляют хлорид железа (безводный, 155 мг, 1,2 ммоль, Альфа) и раствор прогревают до комнатной температуры при перемешивании (16 часов) до получения осадка, - чувствительного к воздуху комплексного соединения FeIIL. Через сушильную трубку пропускают воздух (2 часа) и собирают твердое вещество, промывают его CH2Cl2 (2х10мл). Полученный порошок высушивают при пониженном давлении. Выход: 595 мг (около 93%). Из-за различного сольватирования и ограниченной растворимости литиевую соль необходимо преобразовать в тетраэтиламмониевую соль для последующего использования. Литиевую соль (595 мг) в CH3OH (50 мл) помещают в ионообменную колонну Dowex® 50Х2-100, 25 г, 2 см х 12,5 см), которая предварительно насыщена катионами [Et4N]+ и полосу элюируют CH3OH (100 мл). Растворитель удаляют при пониженном давлении. Остаток суспендируют в CH2Cl2 (20 мл) и смесь фильтруют. Растворитель удаляют из маточного раствора при пониженном давлении до получения гигроскопичного стекловидного остатка [Et4N] 2, который можно использовать без последующей очистки. ИК (nujol/NaCl, см-1): 1619 ν (CO)амид), 1575 ν (CO)амид), 1534 ν (CO)амид). Тщательную очистку исходного материала железа (III) удобнее всего осуществить при помощи аксиального аквамоноанионного комплекса, а не при помощи данного аксиального хлородианионного комплекса. [Et4N]2 (550 мг, около 0,7 ммоль) растворяли в CH3CN (2 мл) и добавляли в раствор, который при этом перемешивали (1 час). Осадок AgCl отфильтровывали и растворитель выпаривали при пониженном давлении. Полученный [Et4N]3 далее очищали, элюируя через колонну с силикагелем (8% MeOH в CH2Cl2). Растворитель удаляли при пониженном давлении и продукт рекристаллизовывали из H2O.
Пример 11
[Et4N] (тетраэтиламмониевая соль хлоро- тетраметилдиметил-дихлорбензол-моноаниона железа (IV)
[Et4N]2 (500 мг, около 0,6 ммоль) растворяли в CH2Cl2 (30 мл). В раствор добавляли нитрат аммонийцерия (IV) (10,3 г, 18,3 ммоль) и смесь перемешивали (2 часа). Твердые соли церия удаляли фильтрацией. Получали продукт путем удаления растворителя при пониженном давлении и высушивания под вакуумом.
Пример 12
Синтез [Ph4P] 5[тетрафенилфосфониевой соли циано- тетраметилдиметил-дихлорбензол-моноаниона железа (IV)
[Et4N] 4 (тетраэтиламмониевая соль хлоро-_тетраметилдиметил-дихлорбензол-моноаниона железа (IV)) (225 мг, 0,33 ммоль) суспендируют в H2O (10 мл). Цианид натрия (140 мг, 2,85 ммоль) растворили в H2O (10 мл) и добавили в суспензию, после чего смесь подвергали ультразвуковой обработке (Branson 1200, 0,5 часа). Затем смесь фильтровали и вызывали осаждение голубого продукта реакции при помощи добавки PPh4Cl [тетрафенилфосфонийхлорида], растворенного в воде (600 мл, 1,6 ммоль, 10 мл, Альдрих). Осадок собирали и промывали H2O (2х10 мл). Материал нужно экстрагировать из силикагеля при помощи CH3CN: CH2Cl2 (1:1,60 мл). Растворитель удаляли при пониженном давлении и осадок растворяли в CH2Cl2 (3 мл) и отфильтровывали. После добавки пентана (150 мл) получали порошок (90 мг, 0,10 ммоль).
Пример 13
Синтез [Ph4P] 5[тетрафенилфосфониевая соль железо (IV) циано-тетраметилдиметил-дихлорбензол-моноанион] из исходных материалов, содержащих нитрилцианид.
[Ph4] 5[тетрафенилфосфониевая соль железо (IV) циано -_тетраметилдиметил-дихлорбензол моноанион] может быть получен в присутствии основания или без него. При отсутствии основания цвет исчезает по мере того, как растворитель удаляют в процессе обработки. Поэтому выделение продукта для получения твердого вещества лучше всего производить в присутствии добавленного основания при pH в диапазоне 9-10. Нижеследующая реакция позволит получить 5 при использовании каждого из CH3CN, CD3CN,CH3CH2CN и (CH3)2CHCN в качестве растворяющей среды. Основание в вышеописанную реакционную среду не добавляют.
Пример 14
Синтез [Ph4P]5 в присутствии основания.
[Et4N] 3 (160 мг, 0,23 ммоль) растворяют в выбранном нитриловом растворителе (6 мл). См. Пример 13. Добавляют тетраэтиламмония гидроксид в качестве основания (20 мас.%, 0,370 мл, 0,52 ммоль, Альдрих), затем трет- бутилгидропероксид (90%, 0,605 мл, 5,4 ммоль, Альдрих), добавку производят по капле при перемешивании (20 мин) в результате чего получают синий раствор. Оставшийся нитрил удаляют при пониженном давлении, получая масляный осадок, который растворяют в H2O (15 мл) и фильтруют. Материал осаждают из фильтрата путем добавки водного раствора PPh4Cl (800 мг, 2,1 ммоль, Альдрих, 10 мл). Синий осадок собирают и промывают H2O (2х10 мл). Выход: 130 мг, 0,15 ммоль (65%). Последующую очистку проводили как описано в разделе по [Ph4P]5, Пример 12.
Пример 15
1-[2-((E)-2-бутенил-2-этиламидо)- 2-метилпропанамидо] -2-[5.5-диметилгидантоин]-4.5-дихлорбензол (т.е. продукт разложения лиганда)
[Et4N] 2 (130 мг, 0,13 ммоль) растворяют в CH3CN (5 мл, Альдрих). Медленно (в течение 3 минут) добавляют 90% раствор трет-бутилгидропероксида (0,445 мл, 4 ммоль, Альдрих). Реакционную смесь перемешивают (25 мин) а затем все жидкости удаляют при пониженном давлении. Остаток растворяют в CH2Cl2, а затем загружают на пластинку для тонкослойной препаративной хроматографии (TLC) (Silica gel GF, 1000 мм, 20 см x 20 см) и элюируют смесью растворителей 15% CH3CN/85% CH2Cl2. Полосу обнаружили при УФ облучении при показателе Rf 0.3. Часть кремнезема, содержащую продукт, удаляют с пластины, и продукт подлежит экстрагированию CH2Cl2:CH3CN (1:1). Раствор фильтруют и растворитель удаляют при пониженном давлении. Твердые частицы получают путем растворения остатка в CH2Cl2 (3 мл), после чего добавляют пентан (150 мл). Твердое вещество собирают фильтрацией и промывают пентаном (2х10 мл).
Некоторые примеры конкретных вариантов осуществления изобретения, т.е. макроциклических соединений по изобретению, раскрыты в заявке на патент США N 08/684670 на имя Т. Collins et al., entitled "Metal Ligand Containing Bleaching Compositions".
| название | год | авторы | номер документа |
|---|---|---|---|
| ОТБЕЛИВАЮЩИЙ СОСТАВ, СОДЕРЖАЩИЙ МЕТАЛЛИЧЕСКИЙ ЛИГАНД, И СПОСОБ ОТБЕЛИВАНИЯ | 1997 |
|
RU2193049C2 |
| ОТБЕЛИВАЮЩИЙ СОСТАВ И СПОСОБЫ ДЛЯ ЕГО ИСПОЛЬЗОВАНИЯ | 1997 |
|
RU2193050C2 |
| МЕТАЛЛ-ЛИГАНДСОДЕРЖАЩИЕ ОТБЕЛИВАЮЩИЕ КОМПОЗИЦИИ | 1999 |
|
RU2234528C2 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ГИСТОНДЕАЦЕТИЛАЗ | 2011 |
|
RU2565076C2 |
| КОМПЛЕКСНОЕ СОЕДИНЕНИЕ И СПОСОБ ИЗВЛЕЧЕНИЯ ЖЕЛАЕМЫХ ИОНОВ ИЗ РАСТВОРА С ИСПОЛЬЗОВАНИЕМ ЭТИХ СОЕДИНЕНИЙ | 1993 |
|
RU2116828C1 |
| МАКРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2020 |
|
RU2818822C1 |
| КАТАЛИЗАТОР ДЛЯ ОБРАБОТКИ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2003 |
|
RU2343977C2 |
| СПОСОБ ПОЛУЧЕНИЯ МАКРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2006 |
|
RU2456296C2 |
| ПИРИДИЛДИАМИДНЫЕ КОМПЛЕКСЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2009 |
|
RU2514405C2 |
| КОНТРАСТНЫЕ АГЕНТЫ | 2016 |
|
RU2743167C2 |

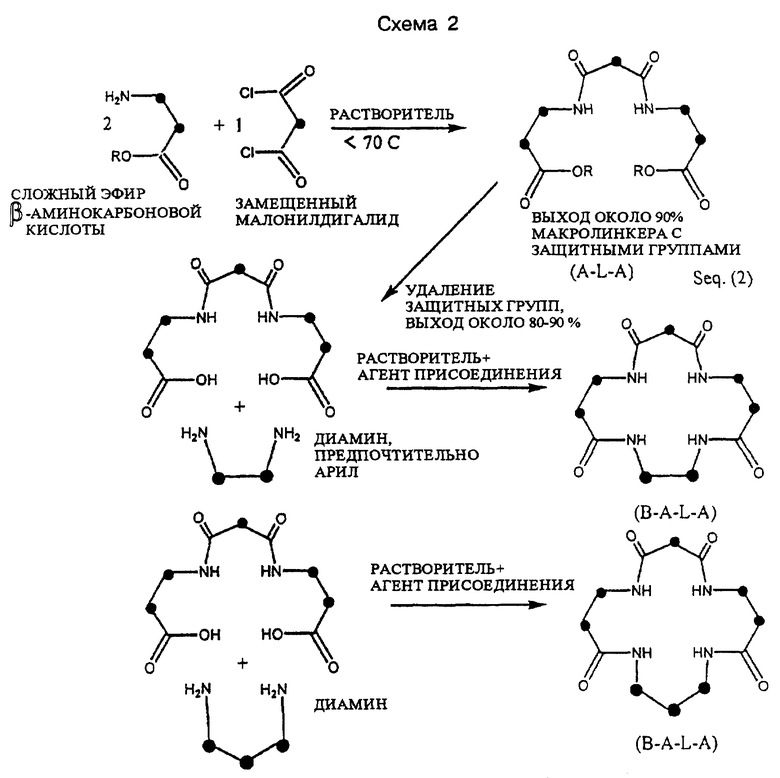
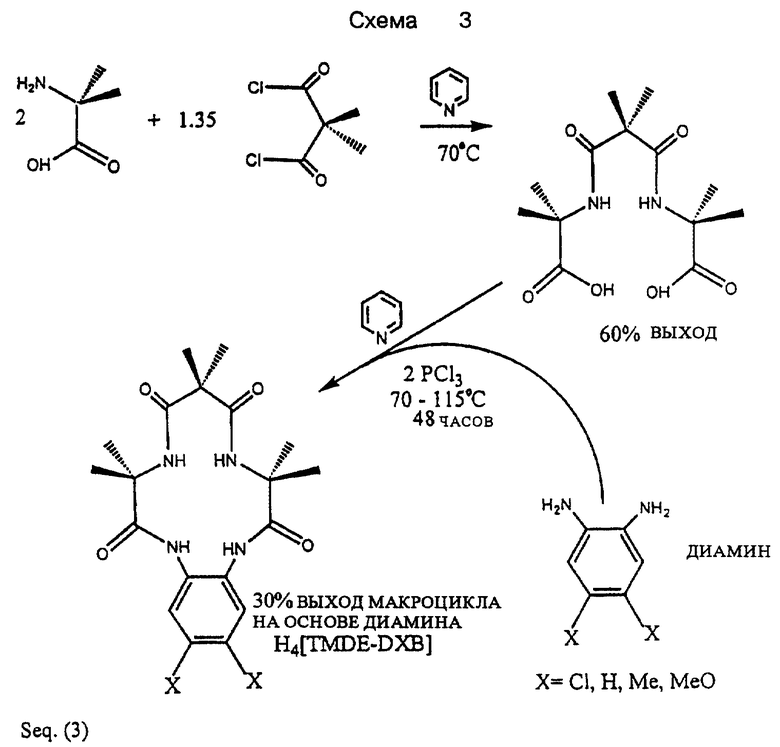
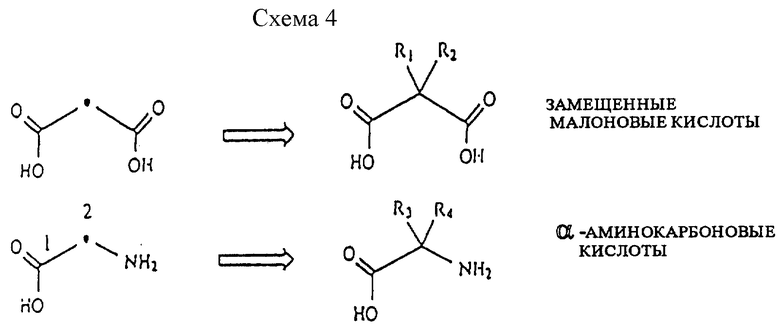
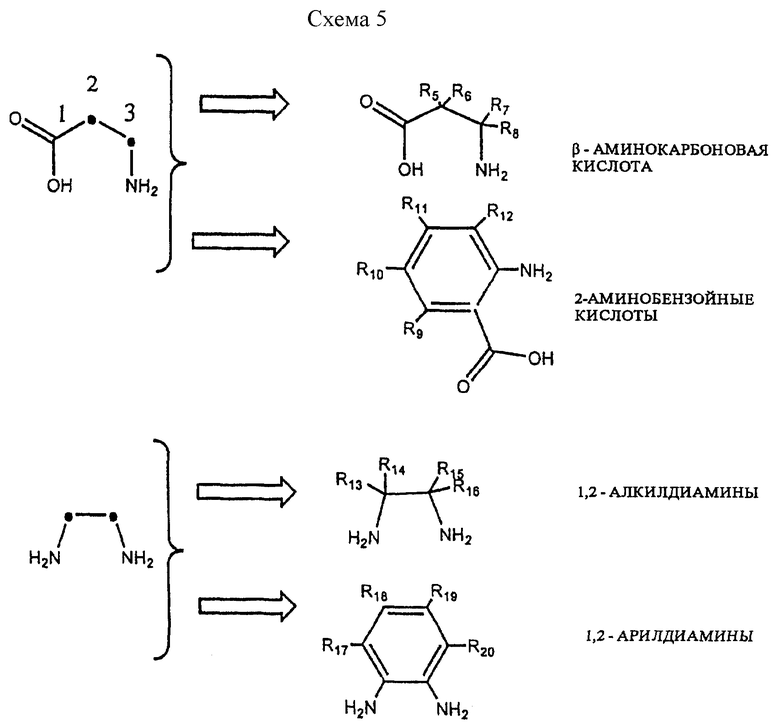

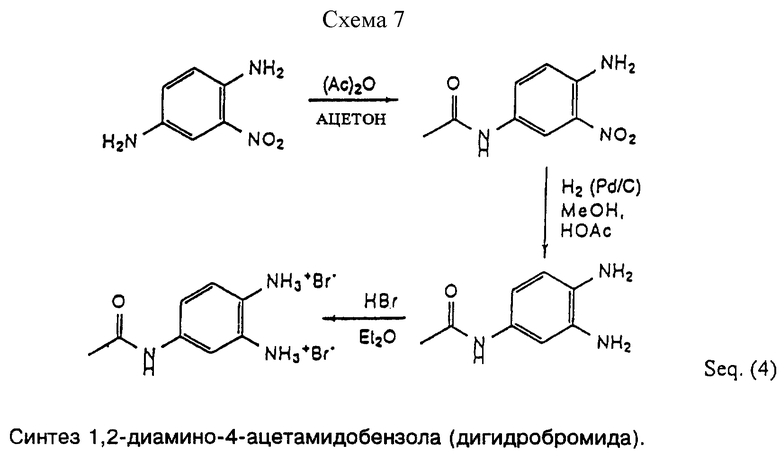
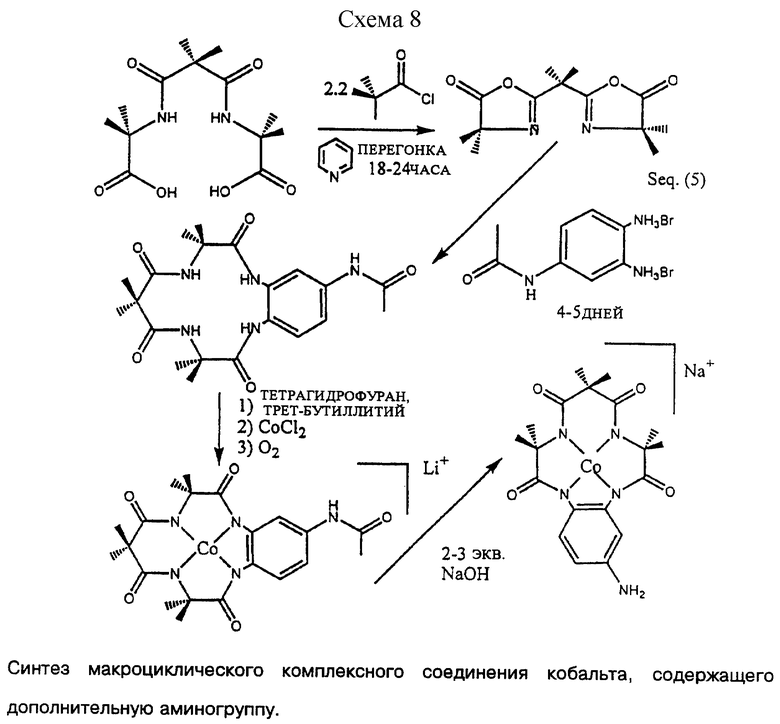
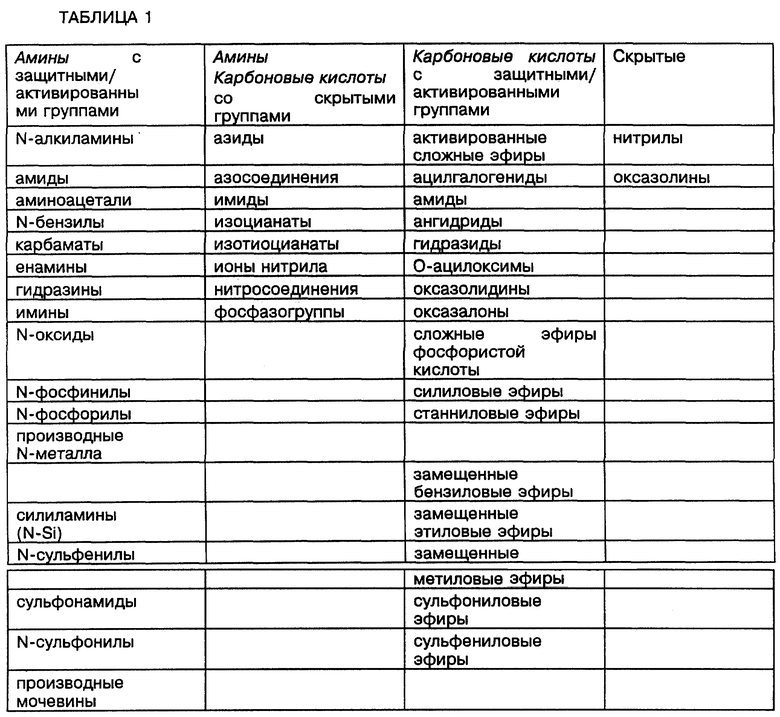
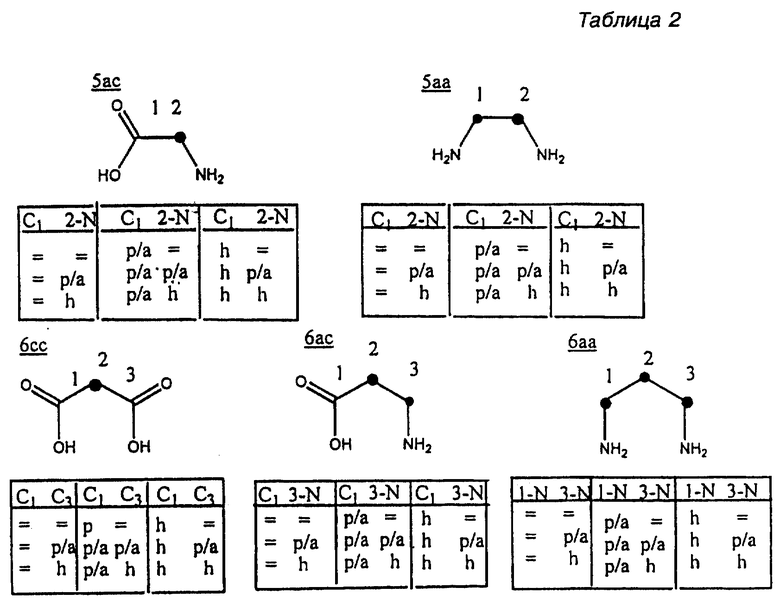
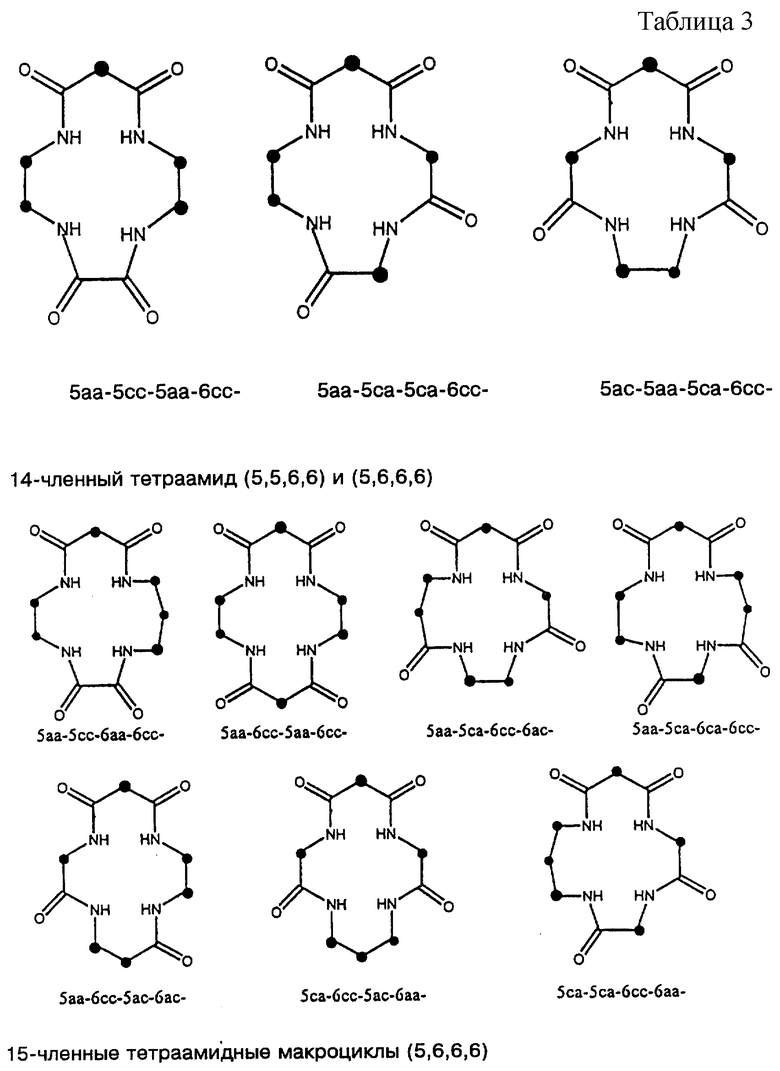
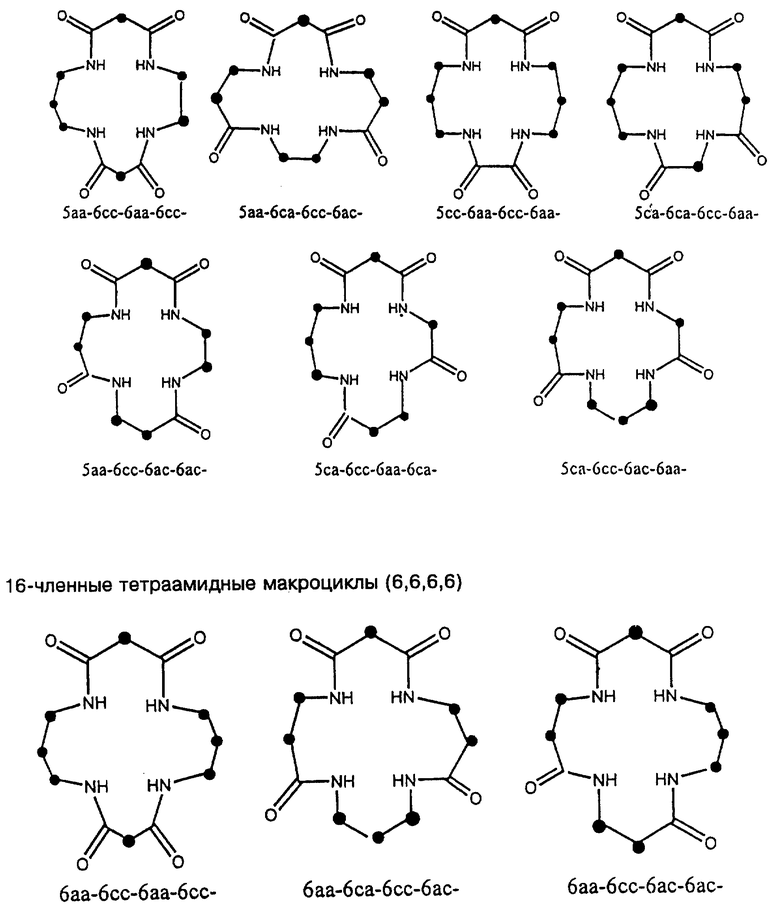
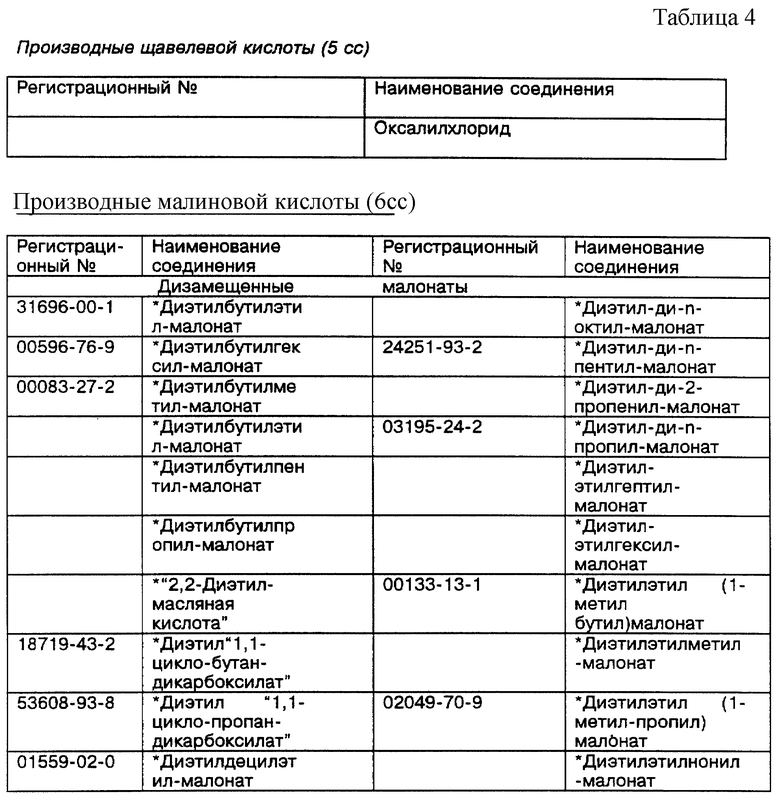

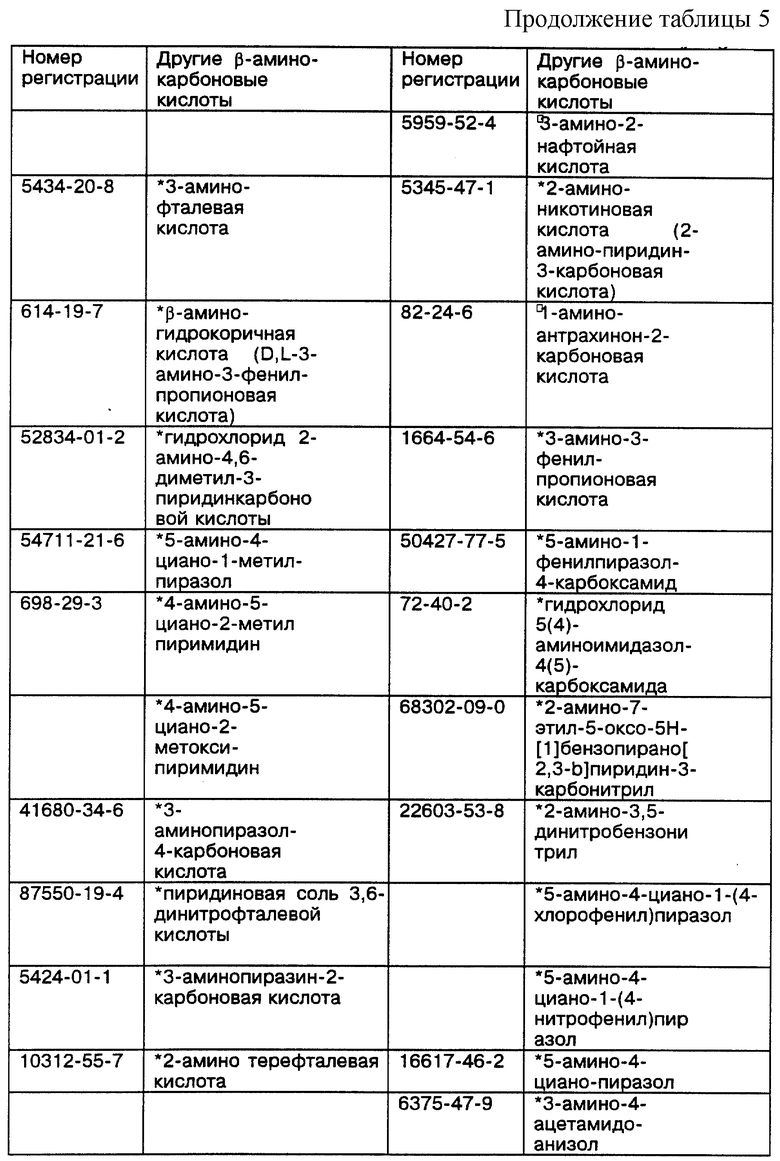
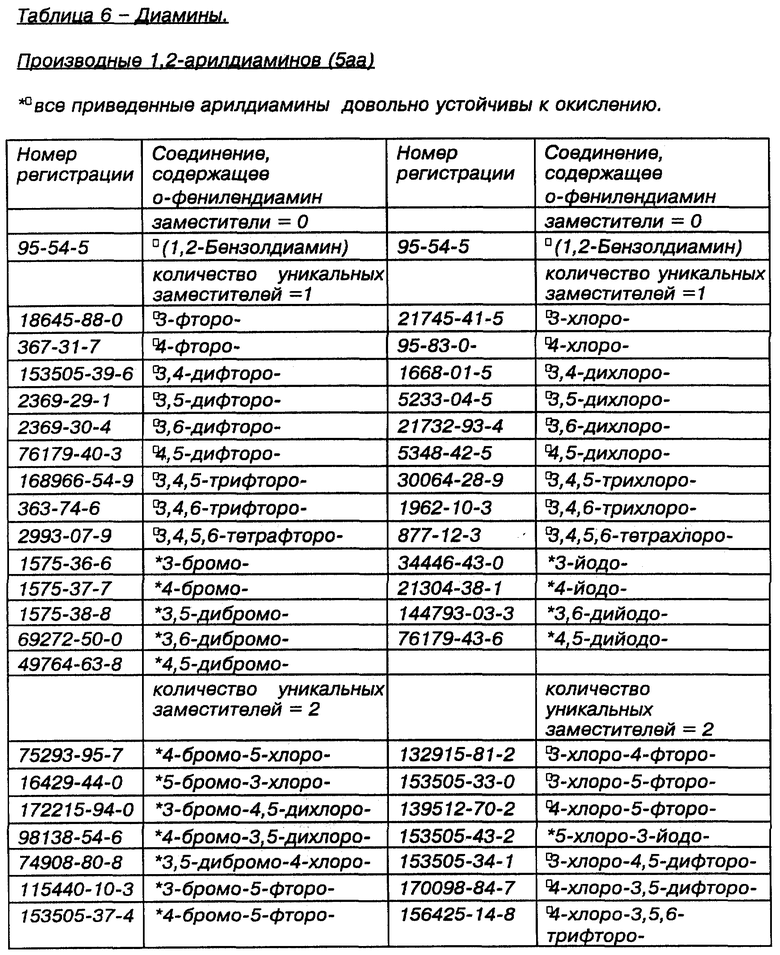
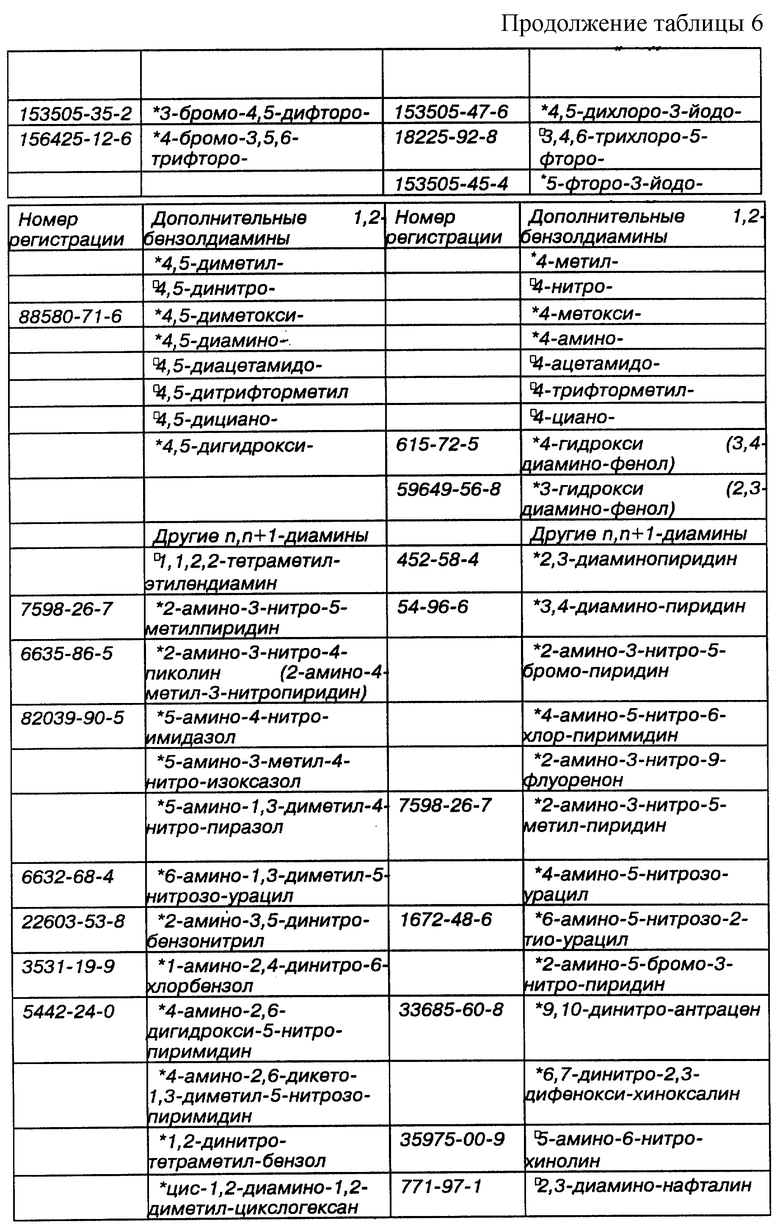
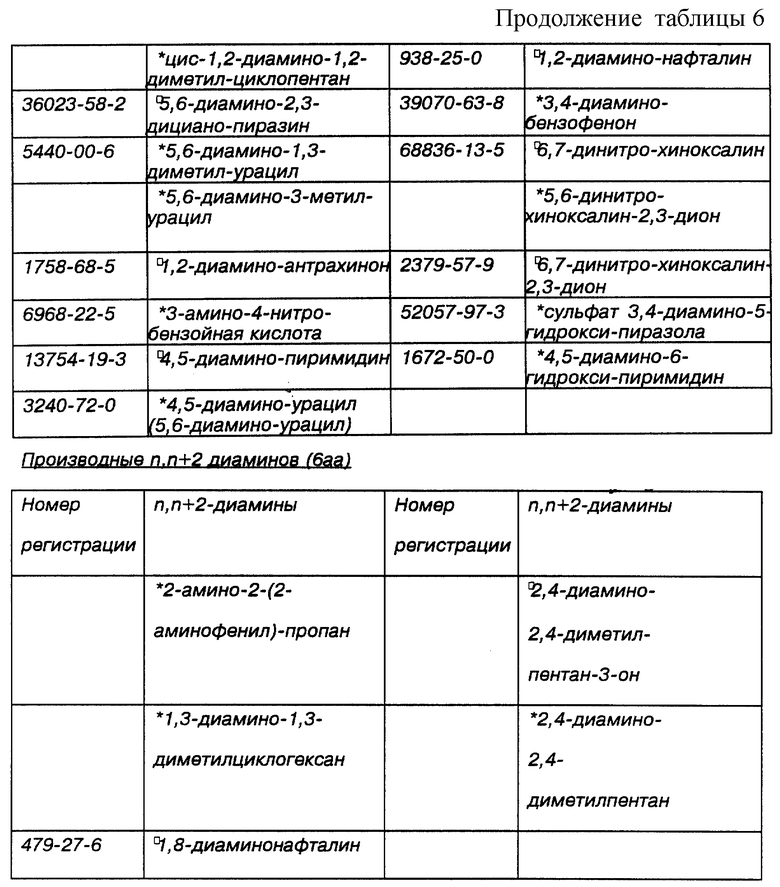
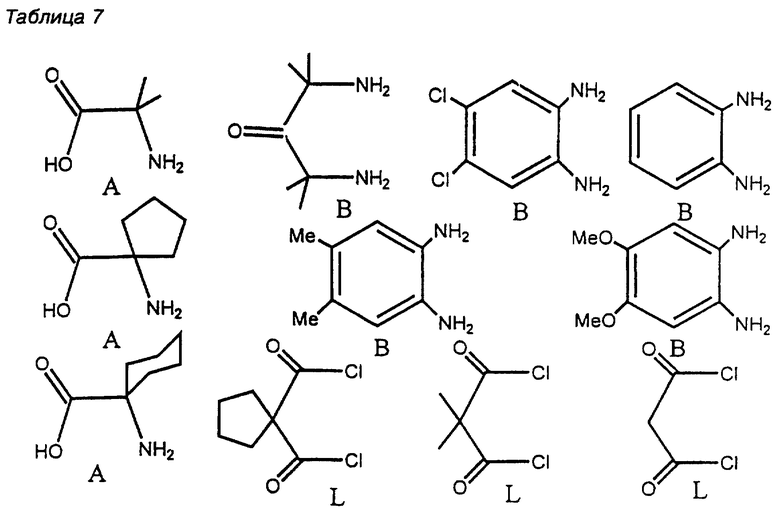
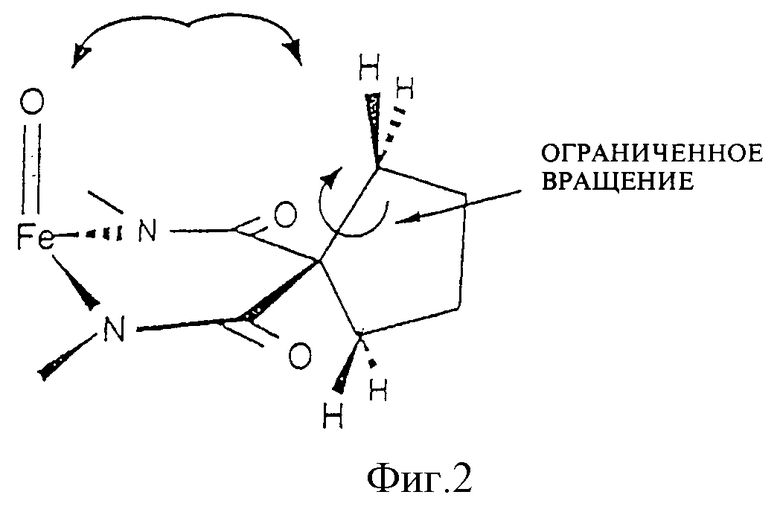
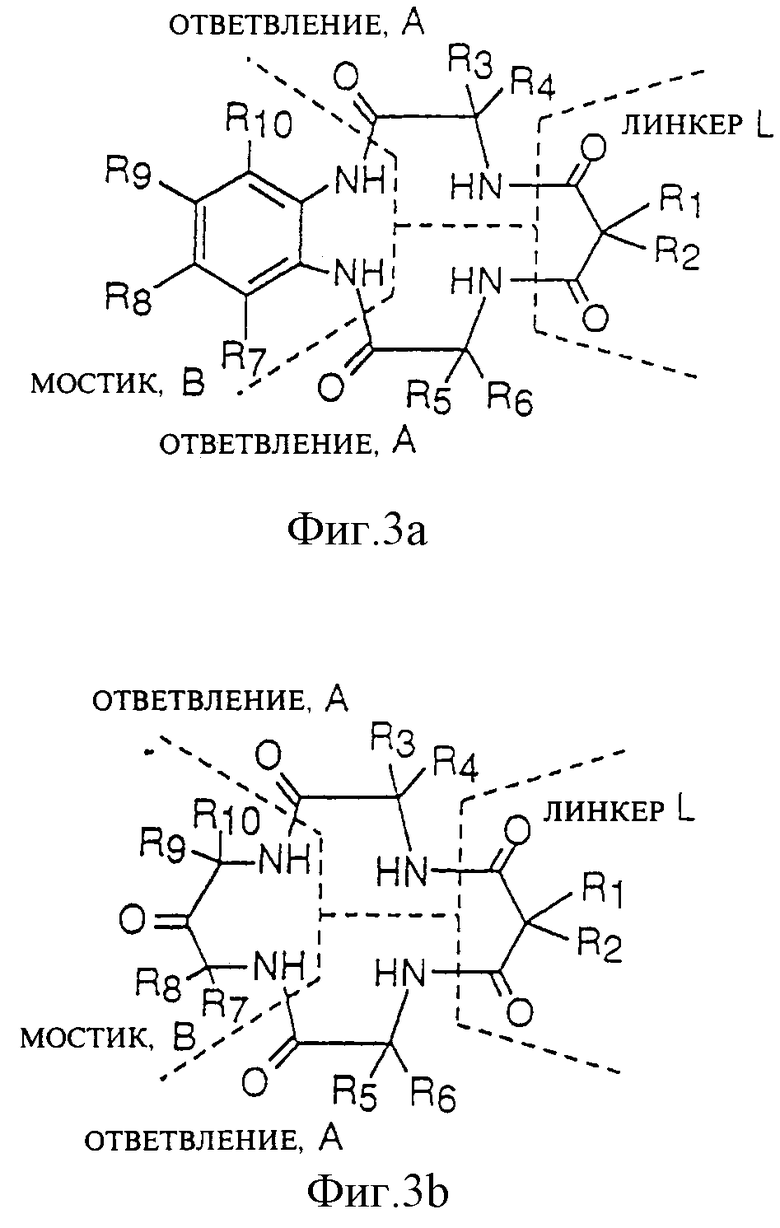
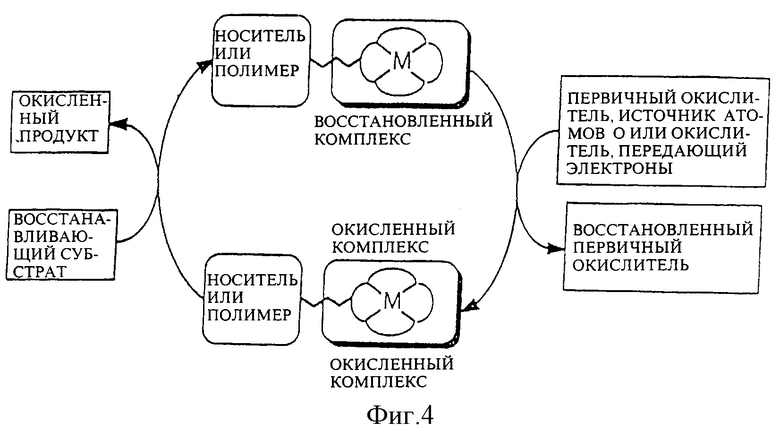
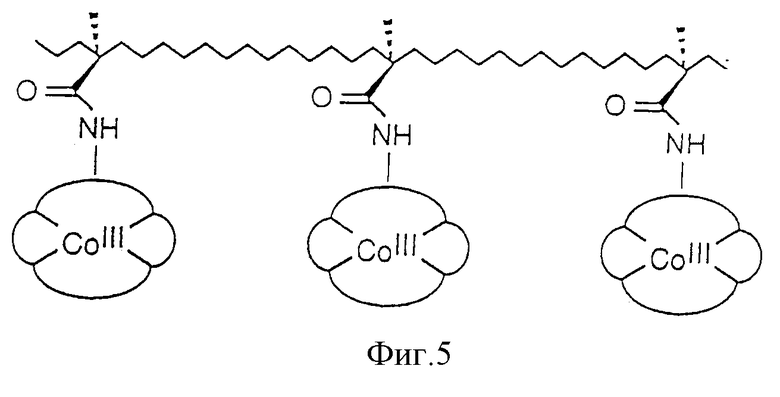
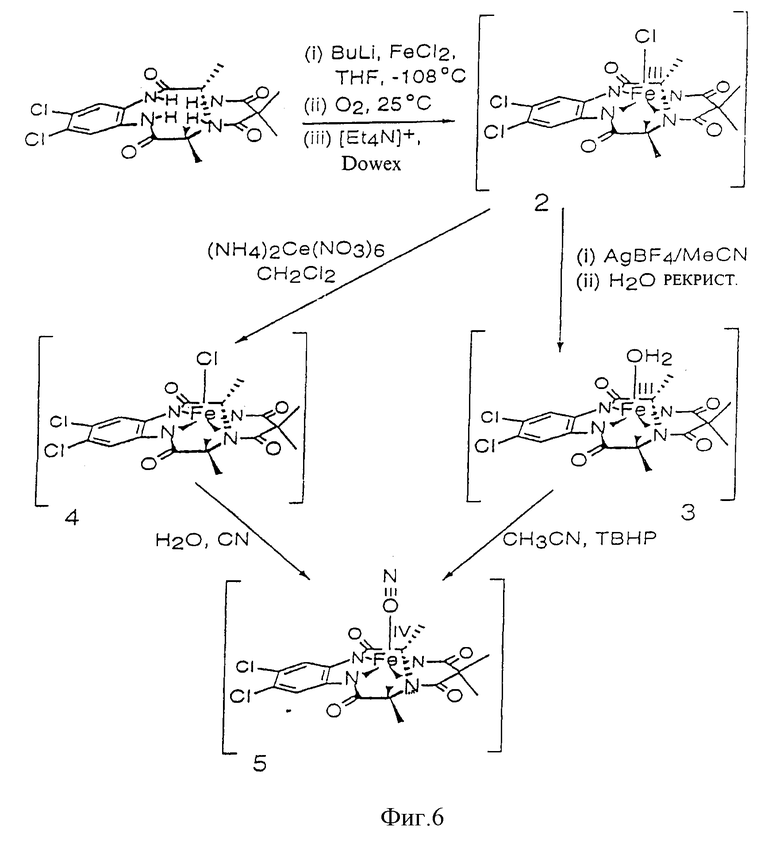
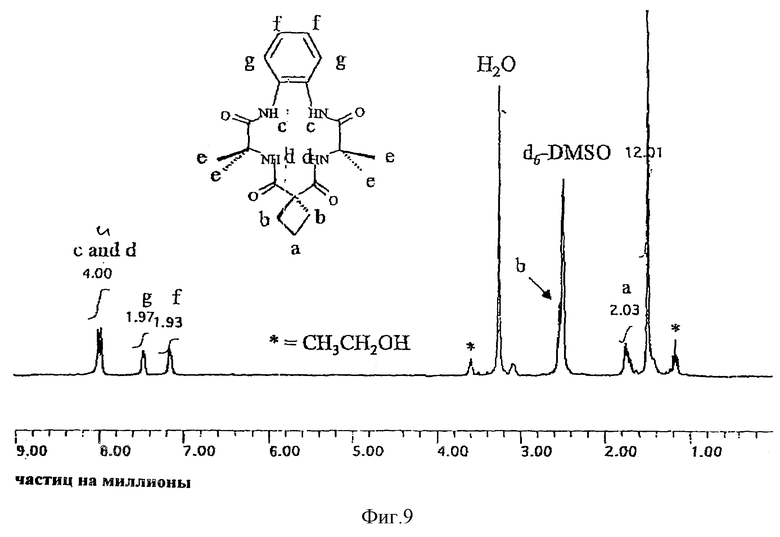
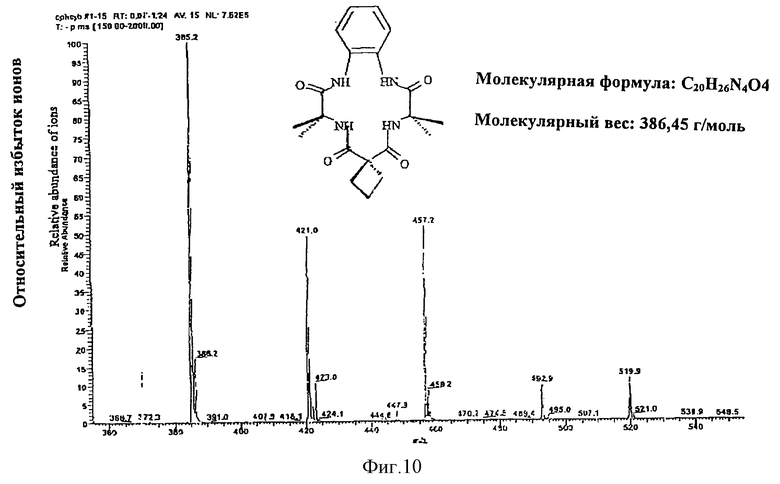
Изобретение относится к новому устойчивому комплексному соединению, содержащему макроциклический тетрадентатный лиганд, имеющий структуру формулы I, где R1 и R2 имеют одинаковые или различные значения, являются связанными или несвязанными и каждый выбирается из группы, состоящей из водорода, галогена, метила, CF3 и, если они связаны, циклопропила, циклобутила, циклопентила или циклогексила, являются пространственно и конфармационно затрудненными, так что окислительная деградация комплекса металла в соединении ограничена, когда комплекс находится в присутствии окисляющей среды, Z представляет собой устойчивый к окислению атом, являющийся металлокомплексообразователем, выбираемый из азота и кислорода, Х представляет собой устойчивую к окислению функциональную группу, выбираемую из О или NRs, где Rs представляет собой метил, фенил, гидроксил, оксильную группу, CF3 или CH2CF3, R3, R4, R5 представляют собой фрагменты, соединяющие соседние атомы Z, содержащие структуры, описанные в формуле изобретения. Также раскрыты хелатный комплекс и промежуточное соединение для получения комплексного соединения, содержащего макроциклический тетрадентатный лиганд. Изобретение позволяет получить стабильный долговечный катализатор окисления или промотор катализатора, используемые в химических или биологических процессах. 3 с. и 8 з.п.ф-лы, 10 ил., 7 табл.
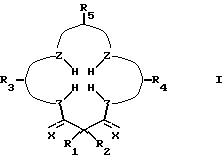
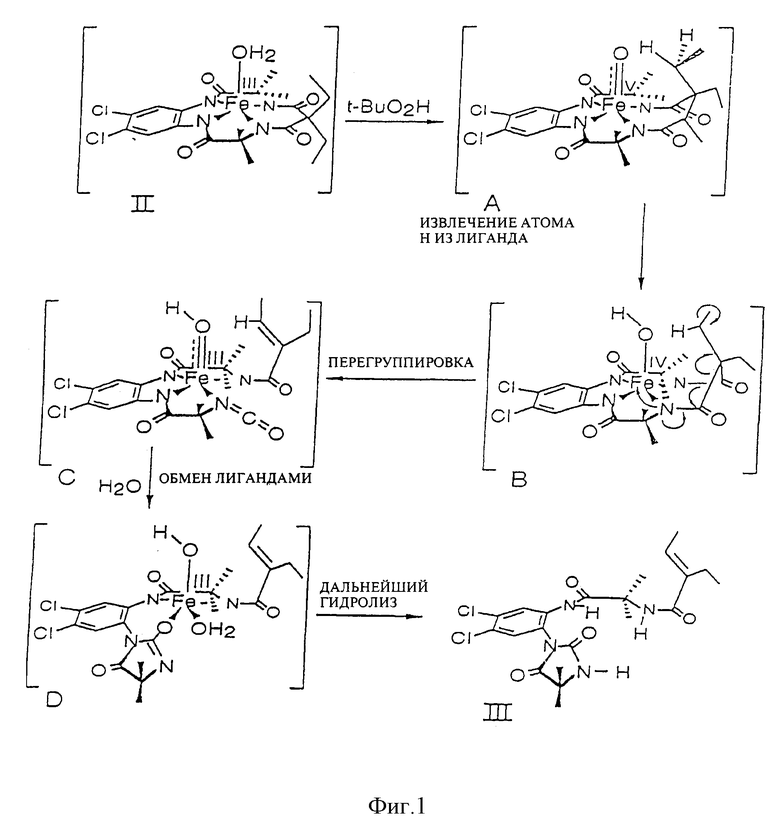

где R1 и R2 имеют одинаковые или различные значения, являются связанными или несвязанными и каждый выбирается из группы, состоящей из водорода, галогена, метила, CF3 и, если они связаны, циклопропила, циклобутила, циклопентила или циклогексила, являются пространственно затрудненными и конформационно затрудненными, так что окислительная деградация комплекса металла в соединении ограничена, когда комплекс находится в присутствии окисляющей среды;
Z представляет собой устойчивый к окислению атом, являющийся металлокомплексообразователем, выбираемый из группы, состоящей из азота и кислорода;
Х представляет собой устойчивую к окислению функциональную группу, выбранную из О или NRs, где Rs представляет собой метил, фенил, гидроксил, оксильную группу, CF3 или CH2CF3;
R3 представляет собой фрагмент, соединяющий соседние атомы Z, содержащий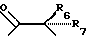
и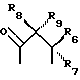
где R6, R7, R8 и R9 попарно и совокупно имеют одинаковые или различные значения и каждый выбирается из группы, состоящей из алкила, фенила, галогена и CF3;
R4 представляет собой фрагмент, соединяющий соседние атомы Z, содержащий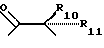
или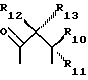
где R10, R11, R12 и R13 попарно и совокупно имеют одинаковые или различные значения и каждый выбирается из группы, состоящей из алкила, фенила, галогена и CF3;
R5 представляет собой фрагмент, соединяющий соседние атомы Z, выбранный из группы, состоящей из: (i)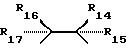
и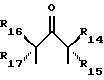
где R14, R15, R16 и R17 имеют одинаковые или различные значения и каждый представляет собой алкил, фенил, галоген или CF3,
и (ii) моно-, ди-, три- и тетразамещенных арильных и гетероарильных заместителей, содержащих
где каждый из Y имеет одинаковые или различные значения и содержит галоген, водород, алкил, фенил, амино, замещенный амино, нитро, алкокси, фенилокси и их сочетания.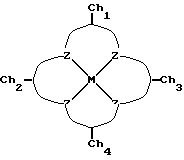
где М представляет собой переходный металл, выбираемый из группы VIII Периодической таблицы;
Z представляет собой донорный атом N или О;
Ch1, Ch2 и Ch3 представляют собой устойчивые к окислению хелатные группы, которые имеют одинаковые или различные значения и которые образуют 5- или 6-членные кольца с указанным металлом, причем Ch1 представляет собой фрагмент, соединяющий соседние атомы, Z выбранный из группы, состоящей из: (i)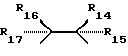
и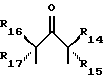
где R14, R15, R16 и R17 имеют одинаковые или различные значения и каждый представляет собой алкил, фенил, галоген или CF3,
и (ii) моно-, ди-, три- и тетразамещенных арильных и гетероарильных заместителей, содержащих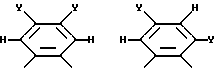
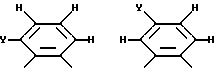
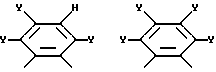
где каждый из Y имеет одинаковые или различные значения и содержат галоген, водород, алкил, фенил, амино, замещенный амино, нитро, алкокси, фенилокси и их сочетания;
Ch2 представляет собой фрагмент, соединяющий соседние атомы Z, содержащий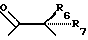
или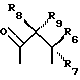
где R6, R7, R8 и R9 попарно или совокупно имеют одинаковые или различные значения и каждый выбирается из группы, состоящей из алкила, фенила, галогена и CF3;
Ch3 представляет собой фрагмент, соединяющий соседние атомы Z, содержащий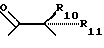
или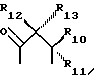
где R10, R11, R12 и R13 попарно и совокупно имеют одинаковые или различные значения и каждый выбирается из группы, состоящей из алкила, фенила, галогена и CF3;
Ch4 представляет собой хелатную группу формулы
где R1 и R2 имеют одинаковые или различные значения, являются связанными или несвязанными и каждый выбирается из группы, состоящей из водорода, галогена, метила, CF3 и, если они связаны, циклопропила, циклобутила, циклопентила или циклогексила, являются пространственно затрудненными и конформационно затрудненными, так что окислительная деградация комплекса металла в соединении ограничена, когда комплекс находится в присутствии окисляющей среды.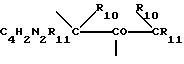 где Y представляет собой галоген, водород, алкил, CH3, NH2 или СНО, а R10 и R11 имеют одинаковые или различные значения и каждый представляет собой алкил, фенил, водород, галоген или CF3.
где Y представляет собой галоген, водород, алкил, CH3, NH2 или СНО, а R10 и R11 имеют одинаковые или различные значения и каждый представляет собой алкил, фенил, водород, галоген или CF3.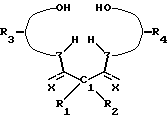
где Z представляет собой устойчивый к окислению атом, являющийся металлокомплексообразователем, выбираемый из группы, состоящей из азота и кислорода;
R1 и R2 имеют одинаковые или различные значения, являются связанными или несвязанными и каждый выбирается из группы, состоящей из водорода, галогена, метила, CF3 и, если они связаны, циклопропила, циклобутила, циклопентила или циклогексила, являются пространственно затрудненными и конформационно затрудненными, и их сочетания достаточны для того, чтобы не дать заместителю подвергнуться окислительной деградации, когда промежуточное соединение находится в присутствии окисляющей среды;
Х представляет собой устойчивую к окислению функциональную группу, выбираемую из О или NRs, где Rs - это метил, фенил, гидроксил, оксильная группа, CF3 или CH2CF3;
R3 представляет собой фрагмент, соединяющий соседние атомы Z, содержащий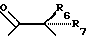
или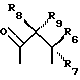
где R6, R7, R8 и R9 попарно и совокупно имеют одинаковые или различные значения и каждый выбирается из группы, состоящей из алкила, фенила, водорода, галогена, галогенированных алкилов, галогенированных арилов и CF3;
R4 представляет собой фрагмент, соединяющий соседние Z атомы, содержащий
или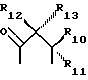
где R10, R11, R12 и R13 попарно и совокупно имеют одинаковые или различные значения и каждый выбирается из группы, состоящей из алкила, фенила, водорода, галогена, галогенированных алкилов, галогенированных арилов и CF3.
| Дисульфокислота 29,31-металл28,5:14,19-ди(дитиациклогексено) ( )-7,12:21,26-дибензо ( ) -тетраазопорфина как катализатор окисления тиоловых соединений | 1976 |
|
SU595334A1 |
| US 4758682 A, 19.07.1988 | |||
| US 4577042 A, 18.03.1986 | |||
| US 4952289 A, 28.08.1990 | |||
| US 5021595 A, 04.06.1991. | |||

