Настоящее изобретение относится к новым пиразолиновым производным общей формулы (I) и к их физиологически приемлемым солям, к способам их получения, к их применению в качестве лекарственных средств для лечения человека и/или животных и к содержащим их фармацевтическим составам.
Новые соединения по настоящему изобретению могут использоваться в фармацевтической промышленности в качестве промежуточных продуктов и для получения лекарственных средств.
Уровень техники
Нестероидные противовоспалительные препараты (NSAIDS) традиционно классифицируются как противовоспалительные, жаропонижающие и болеутоляющие агенты для симптоматического облегчения воспаления, лихорадки и легкой до умеренной боли. Основными показаниями для применения этих препаратов являются остеоартриты, ревматоидные артриты и другие воспалительные заболевания суставов, а также лечение воспалений, связанных с небольшими поражениями, и в качестве анальгетиков широкого спектра. По существу NSAIDS являются ингибиторами острой воспалительной реакции, однако, при ревматических поражениях они оказывают слабое воздействие на основные дегенеративные изменения, наблюдаемые в тканях.
Открытие основного механизма действия NSAIDS путем ингибирования циклооксигеназы (СОХ) (J.R.Vane, Nature, 1971, 231, 232) дало удовлетворительное объяснение их терапевтического действия и доказало важность того факта, что некоторые простагландины являются медиаторами при воспалительных заболеваниях (R.J.Flower, J.R.Vane, Biochem. Pharm., 1974, 23, 1439; J.R.Vane, R.M.Botting, Postgrad. Med. J., 1990, 66 (Suppl. 4), S2). Желудочно-кишечная токсичность классических NSAIDS, a также их полезные эффекты связаны с подавлением синтеза простагландинов путем ингибирования фермента СОХ. Несмотря на использование некоторых приемов (энтеросолюбильное покрытие для предотвращения всасывания в желудке, парентеральное введение, пролонгированная лекарственная форма и т.д.) для ослабления поражения желудочно-кишечного тракта, провоцируемого NSAIDS, ни одна из этих модификаций не дала значительного эффекта в случае серьезных побочных реакций, таких как прободение и кровоизлияние.
Открытие индуцированной простагландинсинтетазы, названной циклооксигеназой-2 (СОХ-2), отличающейся от основного фермента, называемого в настоящее время циклооксигеназой-1 (COX-1) (J.Sirois, J.R.Richards, J.Biol. Chem., 1992, 267, 6382), возобновило интерес к созданию новых противовоспалительных препаратов. Идентификация изоформы СОХ-2 привела к гипотезе о том, что она может быть ответственна за секрецию простагландинов в участках воспаления. В итоге селективное подавление этого изофермента уменьшило бы воспаление без проявления побочных эффектов желудочной и почечной токсичности. По существу изофермент СОХ-1 экспрессируется в большинстве тканей для синтеза простагландинов, которые регулируют нормальную клеточную активность. С другой стороны, изофермент СОХ-2 обычно присутствует в клетках, но при хроническом воспалении уровни белка СОХ-2 повышаются параллельно с усилением секреции простагландинов (J.R.Vane, R.M.Botting, Inflamm. Res., 1995, 44, 1). Следовательно, селективный ингибитор СОХ-2 имеет такие же противовоспалительные, жаропонижающие и болеутоляющие свойства, как и обычный нестероидный противовоспалительный агент, и ингибирует также маточные сокращения, индуцированные гормонами, и обладает потенциальным антиканцерогенным действием и полезными свойствами в предупреждении развития болезни Альцгеймера. С другой стороны, селективный ингибитор СОХ-2 уменьшает потенциальную желудочно-кишечную токсичность, снижает потенциальные побочные почечные эффекты и время кровотечения.
Трехмерная структура СОХ-1 определена с помощью рентгеноструктурного анализа (D.Picot, P.J.Loll, R.M.Garavito, Nature, 1994, 367, 243). Три спирали структуры образуют вход в циклооксигеназный канал, и его включение в мембрану позволяет арахидоновой кислоте достигать активного центра с внутренней стороны бислоя. Активный центр циклооксигеназы представляет собой большой гидрофобный канал, и авторы полагают, что NSAIDS ингибируют СОХ-1 путем вытеснения арахидоновой кислоты из верхней части канала. Недавно была описана (R.S.Service, Science, 1996, 273, 1660) трехмерная структура СОХ-2, которая позволяет сравнить схожесть и различия между двумя изоформами и, следовательно, исследовать новые препараты, которые селективно ингибируют СОХ-2. Из структур СОХ-1 и СОХ-2 следует, что сайты связывания противовоспалительных агентов с ферментами очень схожи, но существует отличие, по меньшей мере, в одной важной аминокислоте. Объемистый изолейцин, присутствующий в активном центре СОХ-1, заменен валином в СОХ-2. Изолейцин закрывает боковую полость, которая отделена от основной связи обоих изоферментов. Закрытая полость СОХ-1 не препятствует связыванию классических NSAIDS, a ингибитор, которому необходима дополнительная точка опоры, предоставляемая боковой полостью, будет связываться с СОХ-2 легче, чем с СОХ-1. В результате прототипом для нового поколения противовоспалительных препаратов являются ингибиторы циклооксигеназы, которые имеют большее сродство к боковой полости СОХ-2.
В химической литературе описаны производные пятичленных азотсодержащих ароматических гетероциклов с ингибирующей активностью в отношении СОХ-2. Производные азолов в них являются пирролами (W.W.Wilkerson et al, J.Med. Chem., 1994, 37, 988; W.W.Wilkerson et al, J.Med. Chem., 1995, 38, 3895; I.K.Khanna et al, J.Med. Chem., 1997, 40, 1619), пиразолами (T.D.Penning et al, J.Med. Chem., 1997, 40, 1347; К.Tsuji, et al, Chem. Pharm. Bull., 1997, 45, 987; К.Tsuji et al, Chem. Pharm. Bull., 1997, 45, 1475) или имидазолами (Khanna et al, J.Med. Chem., 1997, 40, 1634).
Заявители открыли, что новые соединения на основе пирролов, имеющие общую формулу (I), обладают интересными биологическими свойствами, что делает их особенно полезными для применения при лечении человека и/или в ветеринарии. Соединения, являющиеся объектом данного изобретения, применимы в качестве агентов с противовоспалительной активностью и для лечения других заболеваний, в развитии которых принимает участие циклооксигеназа-2, и не обладают желудочной и почечной токсичностью, свойственной классическим NSAIDS.
Сущность изобретения
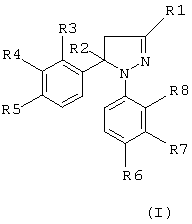
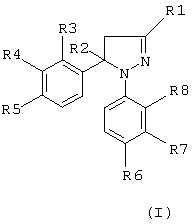
Настоящее изобретение обеспечивает новые пиразолины, которые ингибируют фермент циклооксигеназу-2, с применением их при лечении человека и/или животных в качестве противовоспалительных средств и в случае других заболеваний, в развитии которых принимает участие циклооксигеназа-2, при этом они обладают низкой или не обладают вовсе желудочной и почечной токсичностью. Следовательно, эти противовоспалительные агенты имеют лучшие показатели безопасности. Новые соединения, являющиеся объектом настоящего изобретения, представляют собой производные Δ2-пиразолинов, известные также как 4,5-дигидро-1H-пиразолы. Таким образом, они являются неароматическими азотсодержащими гетероциклическими соединениями. Отсюда следует, что пиразолиновые кольца не являются планарными в отличие от описанных ранее азолов. Соединения, являющиеся объектом настоящего изобретения, имеют следующую общую формулу (I).
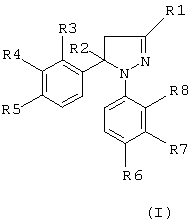
где R1 означает атом водорода, метил, фторметил, дифторметил, трифторметил, карбоксил, низший алкил карбоксилат с 1-4 атомами углерода, карбоксамидную или цианогруппу;
R2 означает атом водорода или метальную группу;
R3, R4, R5 и R8, идентичные или разные, означают атом водорода, хлора, фтора, метильную, трифторметильную или метоксигруппу; один из R5 и R6 означает атом водорода, хлора или фтора, метильную, трифторметильную, метокси- или трифторметоксигруппу, а другой из R5 и R6 означает метилсульфонильную, аминосульфонильную или ацетиламиносульфонильную группу, при условии, что когда R1 означает метильную группу, тогда R2 означает атом водорода или метальную группу;
R3 и R8, идентичные или разные, означают атом водорода, хлора, или фтора, метильную или трифторметильную группу,
R4 означает атом водорода или фтора, метильную, трифторметильную или метокси группу,
R5 означает атом фтора, трифторметильную, трифторметокси-, метилсульфонильную или аминосульфонильную группу;
R6 означает атом водорода, хлора, фтора, метильную, трифторметильную, метокси- или трифторметокси-, метилсульфонильную или аминосульфонильную группу, при условии, что один из R5 и R6 означает метилсульфонильную или аминосульфонильную группу, и
R7 означает атом водорода, хлора или фтора, метильную, трифторметильную или метоксигруппу.
Новые соединения общей формулы (I) имеют асимметрический атом углерода и поэтому могут быть получены в энантиомерно чистой форме или в виде рацематов. Рацематы соединений (I) могут быть разделены на оптические изомеры с помощью общеизвестных методов, таких как, например, хроматография с хиральной стационарной фазой, или путем фракционной кристаллизации их диастереоизомерных солей, которые могут образовываться при взаимодействии соединений (I) с энантиомерно чистыми кислотами. Подобным образом они могут быть получены путем энантиоселективного синтеза с использованием энантиомерно чистых хиральных предшественников.
Настоящее изобретение относится также к физиологически приемлемым солям соединений общей формулы (I), в частности к солям, образующимся путем присоединения минеральных кислот, таких как хлористоводородная, бромистоводородная, фосфорная, серная, азотная кислота и т.д., и органических кислот, таких как лимонная, малеиновая, фумаровая кислота, винные кислоты или их производные, n-толуолсульфоновая, метансульфоновая, камфосульфоновая кислота и т.д.
Новые производные общей формулы (I) могут применяться для млекопитающих, включая человека, в качестве противовоспалительных агентов для лечения воспаления и для лечения других нарушений, связанных с воспалением, в качестве анальгетиков при болях и мигрени и в качестве антипиретиков при лихорадке. Например, новые производные общей формулы (I) могут применяться в лечении артритов, включая, но не ограничиваясь лечением ревматоидных артритов, спондилоартропатий, подагрического артрита, системной красной волчанки, остеоартритов и ювенильных артритов. Новые производные общей формулы (I) могут применяться в лечении астмы, бронхита, нарушений менструального цикла, тендинита, бурсита и разнообразных состояний, которые воздействуют на кожу, таких как псориаз, экзема, ожоги и дерматиты. Новые производные общей формулы (I) могут также применяться в лечении желудочно-кишечных заболеваний, таких как синдром воспаленного кишечника, болезнь Крона, гастрит, синдром воспаленной толстой кишки и язвенный колит.
Новые производные общей формулы (I) могут быть получены следующими методами по изобретению, которые приведены ниже.
Метод А
Получение соединений общей формулы (I) осуществляют путем взаимодействия соединения общей формулы (II)
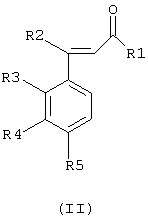
где R1 означает атом водорода, метильную, фторметильную, дифторметильную, трифторметильную или карбоксильную группу;
R2, R3, R4 и R5 имеют значения, приведенные для общей формулы (I), с фенилгидразином общей формулы (III) в виде основания или соли
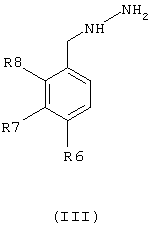
где R6, R7 и R8 имеют значения, приведенные ранее для общей формулы (I).
Реакцию проводят в присутствии соответствующего растворителя, такого как; например, спирты, такие как метанол и этанол, эфиры, такие как диоксан, тетрагидрофуран или их смеси или других растворителей. Реакция проходит в кислой среде, которая может быть органической кислотой, такой как, например, уксусная кислота, или неорганической кислотой, такой как, например, хлористоводородная кислота, или смесью двух кислот, или в основной среде, такой как, например, пиперидин, пиперазин, гидроксид натрия, гидроксид калия, метоксид натрия либо этоксид натрия, или их смеси. Кислая или основная среда сама по себе может играть роль растворителя. Наиболее подходящие температуры варьируют между комнатной температурой и температурой кипения растворителя, а продолжительность реакции может быть от нескольких часов до нескольких суток.
Метод Б
Получение соединений общей формулы (I), где R1 означает алкилкарбоксилат низшего алкила с 1-4 атомами углерода, и R2, R3, R4, R5, R6, R7 и R8 имеют те же значения, которые приведены выше, осуществляют путем взаимодействия соединения общей формулы (I), где R1 означает карбоксильную группу (СООН) и R2, R3, R4, R5, R6, R7 и R8 имеют те же значения, которые приведены выше, с соответствующим реагентом для получения хлорангидрида кислоты, таким как, например, тионилхлорид или оксалилхлорид, проводя затем реакцию этерификации с алифатическим спиртом, содержащим 1-4 атома углерода, в присутствии органического основания, такого как триэтиламин или пиридин, или путем прямой реакции карбоновой кислоты с соответствующим безводным спиртом, насыщенным газообразным хлористым водородом. Реакцию проводят в реагенте, который одновременно является и растворителем, или в других подходящих растворителях, таких как галоидированные углеводороды, такие как дихлорметан, хлороформ или четыреххлористый углерод, в эфирах, таких как диоксан, тетрагидрофуран, этиловый эфир или диметоксиэтан. Наиболее подходящие температуры варьируют между 0°С и температурой кипения растворителя, и продолжительность реакции находится в интервале от 10 минут до 24 часов.
Метод В
Получение соединений общей формулы (I), где R1 означает карбоксамидную группу и R2, R3, R4, R5, R6, R7 и R8 имеют те же значения, которые приведены выше, осуществляют путем взаимодействия соединения общей формулы (I), где R1 означает карбоксильную группу (СООН) и R2, R3, R4, R5, R6, R7 и R8 имеют те же значения, которые приведены выше, с соответствующим реагентом для получения хлорангидрида кислоты, например, с тионилхлоридом или оксалилхлоридом, проводя затем реакцию с аммиаком, который может быть в виде концентрированного водного раствора или раствора в соответствующем растворителе. Реакцию проводят в соответствующем растворителе, таком как, например, эфиры, такие как диоксан, тетрагидрофуран, этиловый эфир или диметоксиэтан. Наиболее подходящие температуры варьируют между 0°С и температурой кипения растворителя, и продолжительность реакции находится в интервале 1-24 часа.
Метод Г
Получение соединений общей формулы (I), где R1 означает цианогруппу и R2, R3, R4, R5, R6, R7 и R8 имеют те же значения, которые приведены выше, осуществляют путем взаимодействия соединения общей формулы (I), где R1 означает карбоксамидную группу и R2, R3, R4, R5, R6, R7 и R8 имеют те же значения, которые приведены выше, с соответствующим реагентом, таким как, например, смешанный диметилформамидтионилхлорид или метансульфонилхлорид. Реакцию проводят в соответствующем растворителе, таком как, например, диметилформамид или пиридин. Наиболее подходящие температуры варьируют между 0°С и температурой кипения растворителя, и продолжительность реакции изменяется в интервале между 15 минутами и 24 часами.
Метод Д
Соединения общей формулы (II), являющиеся промежуточными при получении соединений общей формулы (I), коммерчески доступны или могут быть получены с использованием различных известных методов, среди которых найдены следующие.
Метод Д-1
Получение соединения общей формулы (II), где R1 означает моно-, ди- или трифторметильную группу, R2 означает атом водорода, a R3, R4 и R5 имеют значения, приведенные выше для соединений общей формулы (I), осуществляют с помощью реакции бензальдегида общей формулы (IV)
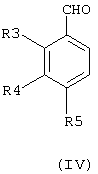
где R3, R4 и R5 имеют значения, приведенные выше для соединений общей формулы (I), с N-фенил(моно-, ди- или трифтор)ацетимидоилхлоридом в присутствии диалкилфосфоната, такого как диэтилметилфосфонат, и сильного органического основания, такого как LDA (диизопропиламид лития), или по реакции Виттига с моно-, ди- или трифторацетилметилентрифенилфосфораном и основанием, таким как карбонат натрия или карбонат калия. Реакцию проводят в соответствующем растворителе, таком как, например, дихлорметан, хлороформ или бензол, или в эфире, таком как тетрагидрофуран, этиловый эфир, диметоксиэтан или диоксан. Наиболее подходящая температура варьирует между -70°С и температурой кипения растворителя, и продолжительность реакции находится в интервале между 15 минутами и 20 часами.
Метод Д-2
Получение соединений общей формулы (II), в которой R1 означает метильную или трифторметильную группу, R2 означает метильную группу и R3, R4 и R5 имеют значения, приведенные выше для соединений общей формулы (I), проводили путем взаимодействия соединения общей формулы (V)
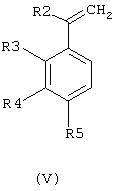
где R2 означает метальную группу и R3, R4 и R5 имеют значения, приведенные выше для соединений общей формулы (I), с моно-, ди- или трифторуксусным ангидридом в присутствии комплекса диметилсульфид-трифторид бора. Реакцию проводят в соответствующем растворителе, таком как, например, галоидированные углеводороды, такие как дихлорметан, хлороформ или четыреххлористый углерод, или в эфире, таком как диоксан, тетрагидрофуран, этиловый эфир или диметоксиэтан. Наиболее подходящая температура варьирует между -70°С и температурой кипения растворителя, и продолжительность реакции находится в интервале между 20 минутами и 20 часами.
Метод Д-3
Получение соединений общей формулы (II), в которой R1 означает метильную или трифторметильную группу, R2 означает атом водорода и R3, R4 и R5 имеют значения, приведенные ранее для соединений общей формулы (I), проводили с помощью различных методов, среди которых может быть упомянута реакция Кляйзена-Шмидта между бензальдегидом общей формулы (IV) и ацетоном или 1,1,1-трифторацетоном в присутствии водного раствора гидроксида щелочного металла, такого как гидроксид натрия или гидроксид калия, или уксусной кислоты и пиперидина; реакция Виттига-Хорнера между бензальдегидом общей формулы (IV) и 2-оксоалкилфосфонатом в присутствии водного раствора основания, такого как, например, карбонат калия или бикарбонат калия; реакция бензальдегида общей формулы (IV) с α,α-бис(триметилсилил)-трет-бутилкетимином в присутствии кислоты Льюиса, такой как, например, бромид цинка, или реакция соединения общей формулы (VI)
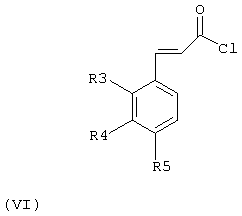
где R3, R4 и R5 имеют значения, приведенные выше для соединений общей формулы (I), с триметилалюминием в присутствии трихлорида алюминия. Реакцию проводят в подходящем растворителе, таком как, например, спирт, такой, как метанол или этанол; галоидированный углеводород, такой как четыреххлористый углерод, хлороформ или дихлорметан; эфир, такой как тетрагидрофуран, этиловый эфир, диоксан или диметоксиэтан; вода, или их смеси. Температура реакции может изменяться в интервале между -60°С и температурой кипения растворителя, и продолжительность реакции находится в интервале между 2 часами и несколькими днями.
Метод Д-4
Получение соединений общей формулы (II), в которой R1 и R2 означают атом водорода и R3, R4 и R5 имеют значения, приведенные ранее для соединений общей формулы (I), проводили, используя различные методы, среди которых может быть упомянута, например, реакция Виттига-Хорнера с бензальдегидом общей формулы (IV) с последующим восстановлением ненасыщенного α,β-эфира с гидридом металла, таким как диизобутилалюминийгидрид (Dibal); с помощью реакции бензальдегида общей формулы (IV) с α,α-бис(триметилсилил)-трет-бутилацетальдимином в присутствии кислоты Льюиса, такой как дибромид цинка, или путем конденсации бензальдегида общей формулы (IV) с ацетальдегидом в присутствии гидроксида щелочного металла, такого как гидроксид натрия или гидроксид калия.
Метод Е
Получение соединений общей формулы (I), где R1, R2, R3, R4, R7 и R8 имеют значения, описанные выше, и один из R5 и R6 означает атом водорода, хлора или фтора, метильную, трифторметильную, метокси- или трифторметоксигруппу, а другой из R5 и R6 означает ацетиламиносульфонильную группу, проводили путем взаимодействия соединения общей формулы (I), где R1, R2, R3, R4, R7 и R8 имеют значения, описанные выше, и один из R5 и R6 означает атом водорода, хлора, фтора, метильную, трифторметильную, метокси- или трифторметоксигруппу, а другой из R5 и R6 означает ацетиламиносульфонильную группу, с соответствующим реагентом, таким как, например, ацетилхлорид или уксусный ангидрид. Реакцию проводили в отсутствие растворителя или в соответствующем растворителе, таком как, например, диметилформамид или пиридин. Наиболее подходящие температуры варьировали между 0°С и температурой кипения растворителя, и продолжительность реакции изменяли в интервале от 15 минут до 14 часов.
Изобретение обеспечивает фармацевтические составы, которые, помимо фармацевтически приемлемого наполнителя, включают, по меньшей мере, одно соединение общей формулы (I) или его физиологически приемлемую соль. Изобретение относится также к применению соединения общей формулы (I) и его физиологически приемлемых солей при получении препаратов для лечения воспаления и/или для лечения других нарушений, связанных с воспалением. В последующих примерах приведено получение новых соединений по изобретению. Раскрыты также некоторые типичные формы применения их в различных областях, а также фармацевтические рецептуры, применимые к соединениям, которые являются объектом изобретения. Примеры, приведенные ниже в качестве иллюстрации, никоим образом не должны ограничивать область изобретения.
Сведения, подтверждающие возможность осуществления изобретения
Пример 1 (позиция 1 в таблицах)
1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(4-метилфенил)-3-трифторметил-1H-пиразол
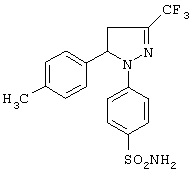
Получение (E)-1,1,1-трифтор-4-(4-метилфенил)-3-бутен-2-она (метод Д-1)
В колбу с сухой инертной атмосферой помещали 15 мл безводного ТГФ (тетрагидрофурана), колбу охлаждали до -70°С. Прибавляли раствор 2М LDA в смеси ТГФ-гексан (5 мл, 10 ммоль) и диэтилметилфосфонат (0.75 мл, 5 ммоль), растворенный в 5 мл ТГФ, колбу встряхивали в течение 30 минут. Затем прибавляли по каплям N-фенилтрифторацетимидоилхлорид (1.04 г, 5 ммоль) (полученный в соответствии с Tamura К., Mizukami H., et al. J.Org. Chem., 1993, 58, 32-35), продолжая встряхивание в тех же условиях в течение 1 часа. Прибавляли n-толуальдегид (0.6 г, 5 ммоль), охлаждающую баню убирали, колбу продолжали встряхивать при комнатной температуре в течение 16 часов. Прибавляли 10 мл 2 н. НСl при перемешивании в течение последующих 4 часов. Удаляли ТГФ на роторном испарителе, смесь экстрагировали этиловым эфиром (3×20 мл), объединенные органические экстракты промывали 5%-ным раствором бикарбоната натрия и насыщенным раствором хлористого натрия до рН~6. Смесь сушили над безводным сульфатом натрия и упаривали. Полученное неочищенное масло освобождали от примесей с помощью колоночной хроматографии на силикагеле под давлением (элюирование смесью этилацетат-петролейный эфир, 1:9), получая (E)-1,1,1-трифтор-4-(4-метилфенил)-3-бутен-2-он (0.8 г, выход 75%) в виде прозрачного масла. ИК (пленка, см-1): 1715, 1601, 1201, 1183, 1145, 1056, 811, 703. 1Н-ЯМР (CDCl3, δ): 2.4 (с, 3Н), 6.97 (д, J=18 Гц, 1H), 7.25 (д, J=9 Гц, 2Н), 7.54 (д, J=9 Гц, 2Н), 7.95 (д, J=18 Гц, 1Н). Тонкослойная хроматография (ТСХ, петролейный эфир): Rf 0.16.
Получение 1-(4-аминосульфонилфенил)-4,5-дигидро-5-(4-метилфенил)-3-трифторметил-1H-пиразола (метод А)
Раствор гидрохлорида 4-(аминосульфонил)гидразина (0.82 г, 3.69 ммоль) и (Е)-1,1,1-трифтор-4-(4-метилфенил)-3-бутен-2-она (0.79 г, 3.69 ммоль) в 15 мл уксусной кислоты нагревали при кипении в течение 3 часов в атмосфере азота. Раствор охлаждали, выливали в воду и экстрагировали этилацетатом. Органический раствор промывали водой, сушили над безводным сульфатом натрия и упаривали досуха в вакууме. Полученный таким образом неочищенный продукт кристаллизовали из смеси этанол-петролейный эфир, получая 1-(4-аминосульфонилфенил)-4,5-дигидро-5-(4-метилфенил)-3-трифторметил-1H-пиразол (0.65 г, выход 45%). Т. пл. 140-143°С. ИК (КВr, см-1): 3356, 3268, 1594, 1326, 1170, 1139, 1120, 1097. 1Н-ЯМР (CDCl3, δ): 2.34 (с, 3Н), 2.99-3.06 (дд, J=6.9 и 14 Гц, 1H), 3.66-3.73 (дд, J=12.6 и 14 Гц, 1H), 4.69 (уш. с, 2Н), 5.38-5.45 (дд, J=6.9 и 12.6 Гц, 1H), 7.04-7.11 (2д, J=8.1 и 9.3 Гц, 4Н), 7.17 (д, J=8.1 Гц, 2Н), 7.70 (д, J=9.3 Гц, 2Н). 13C-ЯМР (CDCl3, δ): 20.9, 41.2, 64.5, 113.4, 120.5 (кв, J=268 Гц), 125.3, 127.6, 130.1, 133.2, 136.7,138.3, 138. (кв, J=38 Гц), 146.0. ТСХ (этилацетат): Rf 0.89.
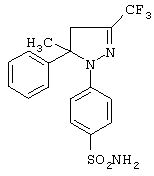
Пример 2 (позиция 2 в таблицах)
1-(4-Аминосульфонилфенил)-4,5-дигидро-5-фенил-5-метил-3-трифторметил-1H-пиразол
Получение (E)-1,1,1-трифтор-4-метил-4-фенил-3-бутен-2-она (метод Д-2)
К раствору комплекса бортрифторид-диметилсульфид (3.9 г, 30 ммоль) в 75 мл дихлорметана, охлажденного до -60°С, медленно прибавляли трифторуксусный ангидрид (6.3 г, 30 ммоль). Смесь встряхивали в течение 10 минут и медленно прибавляли раствор α-метилстирола (3.54 г, 30 ммоль) в 15 мл дихлорметана, поддерживая температуру -60°С. Затем давали температуре подняться до -50°С и выдерживали при этом значении в течение 15 минут, затем давали температуре подняться до 0°С и в этих условиях смесь встряхивали в течение 30 минут. Прибавляли 50 мл этилового эфира и 50 мл водного раствора 10%-ного бикарбоната натрия. Фазы разделяли, водную фазу дополнительно промывали эфиром. Объединенные эфирные фазы промывали водой, сушили над безводным сульфатом натрия и упаривали досуха на роторном испарителе. Полученный таким образом неочищенный продукт очищали от примесей, используя колоночную хроматографию на силикагеле под давлением, элюируя петролейным эфиром. Возвращали 2.0 г (51%) непрореагировавшего исходного α-метилстирола, получали 2.35 г (E)-1,1,1-трифтор-4-метил-4-фенил-3-бутен-2-она (выход 75%) в виде бесцветного масла. ИК (пленка, см-1): 1709, 1596, 1204, 1142, 1072. 1Н-ЯМР (CDCl3, δ): 2.71 (с, 3Н), 6.8 (с, 1Н), 7.45 (м, 3Н), 7.6 (м, 2Н).
Получение 1-(4-аминосульфонилфенил)-4,5-дигидро-5-фенил-5-метил-3-трифторметил-1H-пиразола (метод А)
В колбу с инертной атмосферой прибавляли (Е)-1,1,1 -трифтор-4-метил-4-(4-метилфенил)-3-бутен-2-он (1.75 г, 8.2 ммоль), гидрохлорид 4-(аминосульфонил)фенилгидразина (2 г, 9 ммоль) и пиперидин (0.85 г, 10 ммоль), растворенные в 100 мл этанола, и нагревали при кипении в течение 5.5 часа. Смесь охлаждали, растворитель удаляли на роторном испарителе, к остатку прибавляли воду и раствор экстрагировали этилацетатом. Органическую фазу промывали водой, сушили над безводным сульфатом натрия и упаривали досуха. Неочищенный продукт освобождали от примесей, используя колоночную хроматографию на силикагеле под давлением, элюируя смесью этилацетат-петролейный эфир (4:6) и получая 1-(4-аминосульфонилфенил)-4,5-дигидро-5-фенил-5-метил-3-трифторметил-1H-пиразол в виде белого твердого вещества (1.46 г, выход 47%), т. пл. 60-66°С. ИК (КВr, см-1): 3384, 3266, 1593, 1498, 1327, 1151, 1099, 703. 1Н-ЯМР (CDCl3, δ): 1.6 (с, 3Н), 2.8 (м, 1Н), 3.1 (м, 1H), 4.5 (уш. с, 2Н), 7.2 (м, 3Н), 7.4-7.55 (м, 4Н), 7.7 (д, 2Н). 13C-ЯМР (CDCl3): 27.6, 54.2, 63.1, 114.6, 124.0 (кв, J=268 Гц), 125.6, 127.4, 127.8, 129.1. 131.0, 142.0 (кв, J=38 Гц), 142.6, 147.5.
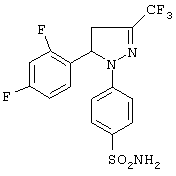
Пример 3 (позиция 3 в таблицах)
1-(4-Аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
Получение (E)-1,1,1-трифтор-4-(2,4-дифторфенил)-3-бутен-2-она (метод Д-3)
В колбе растворяли 2,4-дифторбензальдегид (20 г, 0.14 моль), ледяную уксусную кислоту (12.2 г, 0.2 моль) и пиперидин (12.2 г, 0.14 моль) в ТГФ (300 мл). Раствор охлаждали до 5-10°С и барботировали через него трифторацетон (СF3СОСН3) (8 г, 0.07 моль). Убирали охлаждающую баню, температуру поднимали до комнатной и выдерживали при этой температуре в течение 1.5 часа при непрерывном встряхивании. Снова прибавляли трифторацетон (5 г, 0.045 моль), смесь оставляли на 1.5 часа при встряхивании. Снова прибавляли 5 г, и смесь продолжали встряхивать в течение 1.5 часа. Операцию повторяли до тех пор, пока не было прибавлено всего 35 г (0.31 моль) трифторацетона. Добавляли 20% раствора (50 мл), растворитель удаляли при пониженном давлении. Прибавляли 50 мл воды, раствор экстрагировали этилацетатом. Органическую фазу промывали водой, 5% серной кислотой, водой, смесь сушили над безводным сульфатом натрия. Раствор фильтровали и упаривали. Полученный неочищенный продукт перегоняли, получая 18.1 г (Е)-1,1,1-трифтор-4-(2,4-дифторфенил)-3-бутен-2-она, т. пл. 50-51°С. ИК (КВr, см-1): 1717, 1602, 1583, 1277, 1146, 1059. 706. 1Н-ЯМР (CDCl3, δ): 6.9 (м, 2Н), 7.05 (д, J=16 Гц, 1Н), 7.6 (м, 1H), 8.0 (д, J=16 Гц, 1Н).
Получение 1-(4-аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразола (метод А)
Раствор гидрохлорида 4-(аминосульфонил)фенилгидразина (47.8 г, 0.21 моль) и (E)-1,1,1-трифтор-4-(2,4-дифторфенил)-3-бутен-2-она (53.1 г 95%-ного, 0.21 моль) в 315 мл уксусной кислоты нагревали при кипении в течение 24 часов в атмосфере азота. Смесь охлаждали, выливали в воду и фильтровали. Смесь промывали толуолом, полученный таким образом неочищенный продукт кристаллизовали из изопропанола, получали 46.2 г. Водный маточный раствор от кристаллизации концентрировали, получая дополнительно 12.6 г продукта. Общий выход 1-(4-аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразола 58.8 г (68%), т. пл. 160-162°С.
Может быть использована также следующая методика: в колбу с инертной атмосферой вносили этилат натрия (0.53 г, 7.72 ммоль), растворенный в 45 мл этанола, 1,1,1-трифтор-4-(2,4-дифторфенил)-3-бутен-2-он (полученный по методу Д-1) (0.913 г, 3.86 ммоль) и гидрохлорид 4-(аминосульфонил)фенилгидразина (0.87 г, 3.87 ммоль), смесь нагревали при кипении в течение 16 часов. Смесь охлаждали, упаривали досуха, прибавляли холодную воду, смесь подкисляли путем добавления уксусной кислоты, осажденное твердое вещество отделяли фильтрованием. Твердое вещество растворяли в эфире, обрабатывали активированным углем, раствор фильтровали и удаляли растворитель на роторном испарителе. Полученный остаток кристаллизовали из смеси этиловый эфир-петролейный эфир (50:50), получая 1-(4-аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол (1.02 г, выход 65%) в виде твердого вещества, т. пл. 160-162°С. ИК (КВr, см-1): 3315, 3232, 1617, 1593, 1506, 1326, 1179, 1099, 1067. 1Н-ЯМР (CDCl3, δ): 3.0 (дд, J=6.3 и 11.4 Гц, 1Н), 3.80 (дд, J=11.4 и 12.6 Гц, 1Н), 4.79 (уш. с, 2Н), 5.70 (дд, J-6.3 и 12.6 Гц, 1Н), 6.8-6.95 (м, 2Н), 7.01-7.09 (м, 3Н), 7.74 (д, J=8.7 Гц, 2Н).
Пример 4 (позиция 4 в таблицах)
4,5-дигидро-1-(4-метилфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол (метод А)
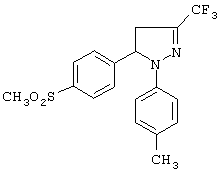
В колбе с инертной атмосферой растворяли (Е)-1,1,1-трифтор-4-(4-метилсульфонилфенил)-3-бутен-2-он (полученный по методу Д-1) (1.83 г, 6.58 ммоль) и гидрохлорид 4-метилфенилгидразина (1.04 г, 6.58 ммоль) в 50 мл этанола. Прибавляли несколько капель соляной кислоты, смесь нагревали при кипении в инертной атмосфере в течение 4 суток. Смесь охлаждали, продукт кристаллизовали. Раствор отделяли фильтрованием, продукт перекристаллизовывали из этанола. Получали 4,5-дигидро-1-(4-метилфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол (0.8 г, выход 32%) в виде твердого вещества, т. пл. 140-143°С. ИК (КВr, см-1): 1516, 1310, 1148, 1131, 1060, 774. 1Н-ЯМР (CDCl3, δ): 2.2 (с, 3Н), 2.9 (дд, J=7.8 и 17.1 Гц, 1Н), 3.05 (с, 3Н), 3.7 (дд, J=12.9 и 17.1 Гц, 1Н), 5.45 (дд, J=7.8 и 12.9 Гц, 1H), 6.8 (д, J=8.4 Гц, 2Н), 7.0 (д, J=8.4 Гц, 2Н), 7.45 (д, J=8.4 Гц, 2Н), 7.9 (д, J=8.4 Гц, 2Н).
Пример 5 (позиция 39 в таблицах)
Метил-4,5-дигидро-5-(4-метилфенил)1-(4-метилсульфонилфенил)-1H-пиразол-3-карбоксилат (метод Б)
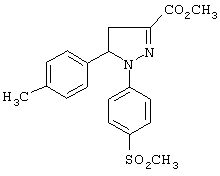
Растворяли 4,5-дигидро-5-(4-метилфенил)-1-(4-метилсульфонилфенил)-1H-пиразол-3-карбоновую кислоту (6.9 г, 19.3 ммоль) и тионилхлорид (3.5 мл; 48 ммоль) в 50 мл тетрагидрофурана, смесь встряхивали при комнатной температуре в течение 16 часов. Смесь упаривали досуха на роторном испарителе, полученный таким образом неочищенный хлорангидрид растворяли в 150 мл метанола в колбе с инертной атмосферой, прибавляли 8 мл (58 ммоль) триэтиламина, смесь встряхивали при комнатной температуре в течение 2 часов. Прибавляли воду, осадок отделяли фильтрованием, промывали тщательно водой и метанолом. Таким образом получали целевой метиловый эфир (5.8 г, выход 82%) в виде твердого вещества кремового цвета, т. пл. 155-160°С. ИК (КВr, см-1): 1741, 1561, 1260, 1226, 1135, 1089. 1Н-ЯМР (CDCl3, δ): 2.3 (с, 3Н), 3.0 (с, 3Н), 3.1 (дд, J=6 и 18.3 Гц, 1Н), 3.75 (дд, J=12.6 и 18.3 Гц, 1Н), 5.4 (дд, J=6 и 12.6 Гц, 1Н), 7-7,25 (д,J=8.7 Гц, 2Н).
Пример 6 (позиция 41 в таблицах)
Получение 1-(4-аминосульфонилфенил)-4,5-дигидро-5-(4-метилфенил)-1H-пиразол-3-карбоксамида (метод В)
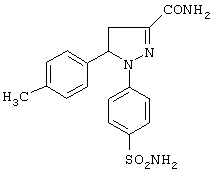
Раствор 1-(4-аминосульфонилфенил)-4,5-дигидро-5-(4-метилфенил)-1H-пиразол-3-карбоновой кислоты (3.7 г, 10.3 ммоль) и тионилхлорида (3 г, 25.8 ммоль) в 70 мл тетрагидрофурана встряхивали при комнатной температуре в течение 16 часов. Смесь упаривали досуха на роторном испарителе, полученный таким образом неочищенный хлорангидрид растворяли в 30 мл метанола в инертной атмосфере и охлаждали до 0°С. Прибавляли 9 мл концентрированного раствора гидроксида аммония, растворенного в 20 мл ТГФ. Смесь встряхивали при комнатной температуре в течение 16 часов, растворитель удаляли на роторном испарителе. К остатку прибавляли воду, смесь экстрагировали этилацетатом, органический слой промывали водой, сушили над безводным сульфатом натрия и упаривали досуха. Полученный таким образом неочищенный остаток кристаллизовали из смеси этилацетат-петролейный эфир, получая 2.6 г (выход 72%) целевого соединения, т. пл. 210-215°С. ИК (КВr, см-1): 3450, 3337, 1656, 1596, 1345, 1141. 1Н-ЯМР (СD3ОD), δ: 2.4 (с, 3Н), 3.05 (дд, J=6 и 17.7 Гц, 1Н), 3.8 (дд, J=12.9 и 17.7 Гц, 1Н), 5.6 (дд, J=6 и 12.9 Гц, 1Н), 7.2-7.3 (м, 6Н), 7.75 (д, J=8.7 Гц, 2Н).
Пример 7 (позиция 34 в таблицах)
Получение 3-циан-4,5-дигидро-5-(4-метилфенил)-1-(4-метилсульфонилфенил)-1H-пиразола (метод Г)
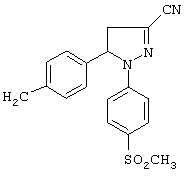
В колбу с инертной атмосферой вносили 6.3 мл безводного диметилформамида, охлаждали до 0°С и медленно прибавляли 2.1 мл тионилхлорида. Колбу встряхивали в течение 2 часов в тех же условиях. Прибавляли раствор 4,5-дигидро-5-(4-метилфенил)-1-(4-метилсульфонилфенил)-1H-пиразол-3-карбоксамида (3.8 г, 10.6 ммоль) в 30 мл диметилформамида, смесь встряхивали в течение 5 часов при 0°С, затем в течение 16 часов при комнатной температуре. Содержимое колбы выливали на лед, выпавший осадок отделяли фильтрованием. Получали 3.35 г (выход 93%) неочищенного продукта, который кристаллизовали из этилацетата, получая желтое твердое вещество, т. пл. 162-164°С. ИК (КВr, см-1): 2220, 1593, 1500, 1389, 1296, 1143. 1Н-ЯМР (CDCl3, δ): 2.3 (с, 3Н), 3-3.1 (с+дд, 4Н), 3.75 (дд, J=12.6 и 18 Гц, 1Н), 5.5 (дд, J=6.3 и 12.6 Гц, 1Н), 7-7.2 (м, 6Н), 7.7 (д, J=8.7 Гц, 2Н).
Пример 8 (позиция 64 в таблицах)
1-(4-Ацетиламиносульфонилфенил)-5-(2,4-дифторфенил)-4.5-дигидро-3-трифторметил-1H-пиразол (метод Е)
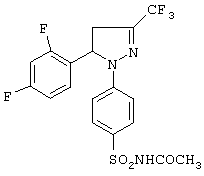
Смесь, состоящую из 0.58 г (1.43 ммоль) 1-(4-аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразола и 2 мл ацетилхлорида, нагревали при кипении в течение 2 часов. Смесь охлаждали, упаривали досуха при пониженном давлении, полученный остаток растворяли в этилацетате, раствор промывали водой, сушили над сульфатом натрия и упаривали досуха. Получали 0.49 г (76%) 1-(4-ацетиламиносульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразола в виде белого твердого вещества, т. пл. 172-174°С. ИК (КВr, см-1): 3302, 1723, 1593, 1506, 1337, 1165. 1Н-ЯМР (CDCl3, δ): 2.0 (с, 3Н), 3.0 (дд, J=6.6 и 18.0 Гц, 1Н), 3.8 (дд, J=12.9 и 18.0 Гц, 1H), 5.7 (дд, J=6.6 и 12.9 Гц, 1Н), 6.9 (м, 2Н), 7.05 (м+д, 3Н), 7.85 (д, J=8.7 Гц, 2Н), 8.1 (с,1Н).
Примеры 9 и 10 (позиции 75 и 76 в таблицах)
(+)-1-(4-Аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол и (-)-1-(4-аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
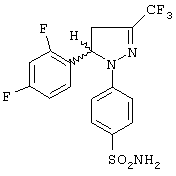
Рацемическую смесь (±)-1-(4-аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразола разделяли на энантиомеры с помощью высокоэффективной жидкостной хроматографии, используя колонку CHIRALPAK AS с частицами 10 мкм и размерами 25×2 см (Daicel), мобильная фаза 0.1% диэтиламин в метаноле, скорость потока 8 мл/мин. При времени удерживания 7.4 минуты получали (+)-1-(4-аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол в виде белого твердого вещества, т. пл. 173-174°С, энантиомерная чистота 99.9%, [α]D+189.3 (с 1, метанол). При времени удерживания 9.2 минуты получали (-)-1-(4-аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол в виде белого твердого вещества, т. пл. 173-174°С, энантиомерная чистота >99.9%, [α]D=-189.4 (с 1, метанол). С использованием такой же методики получали соединения, соответствующие позициям 77 и 78 в таблицах.
В таблице 1 приведены некоторые примеры, относящиеся к общей формуле (I), в таблице 2 приведены данные идентификации этих соединений. Соединения примеров 1-36, 44-63 и 65-74 получены по методу А, примеров 37-39 - по методу Б, примеров 40-42 - по методу В, примера 64 - по методу Е, энантиомерно чистые соединения 75-78 получены разделением рацемической смеси.
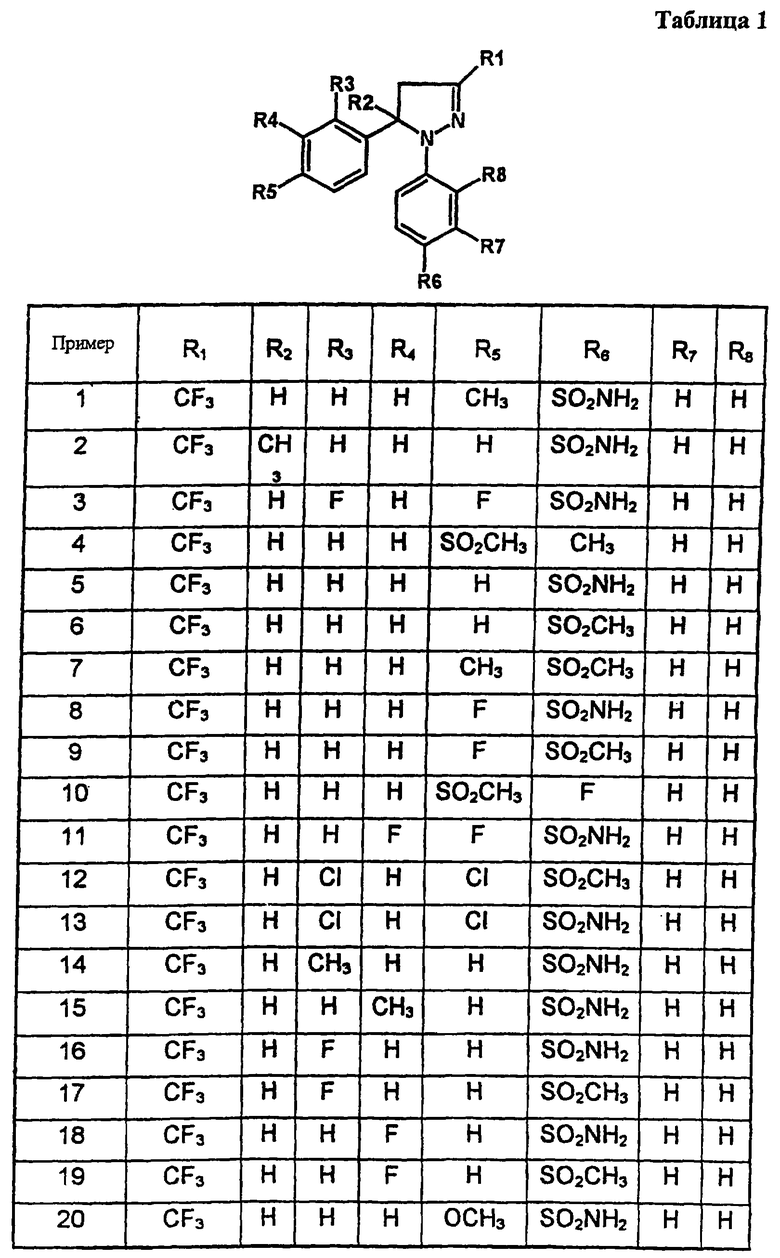
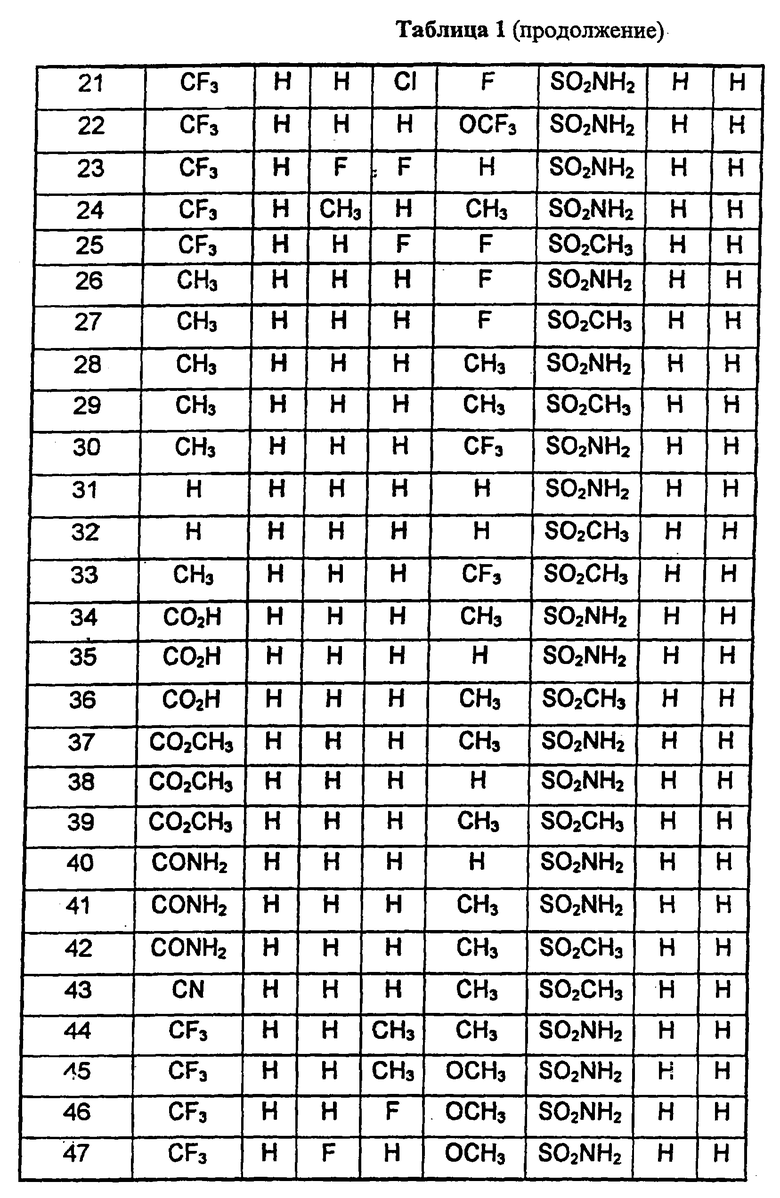
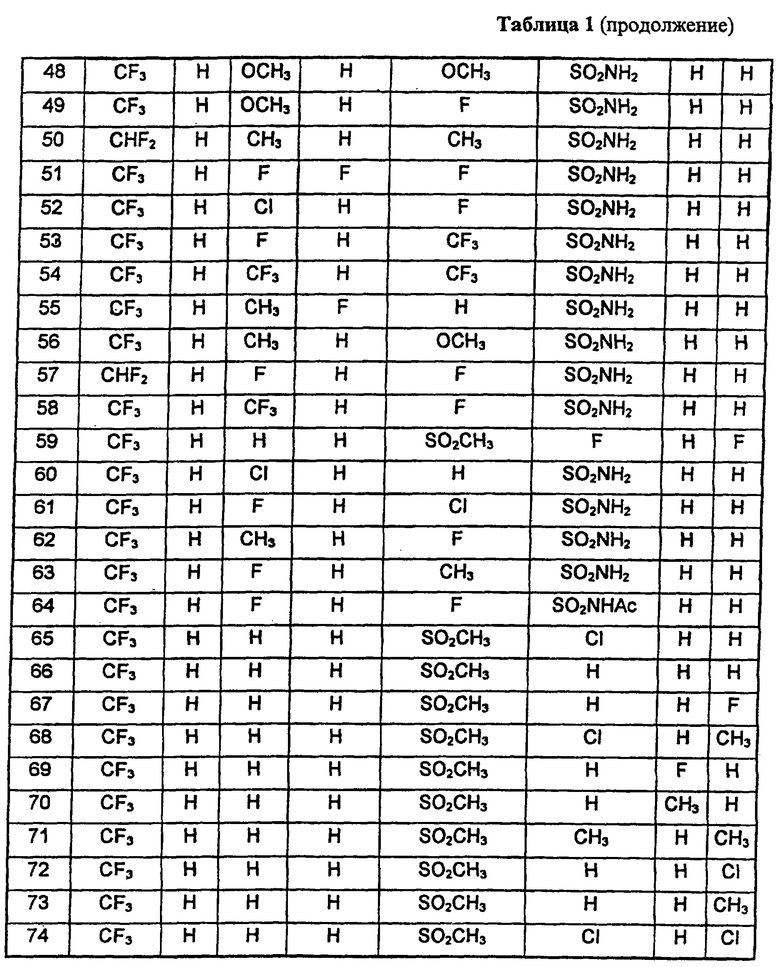
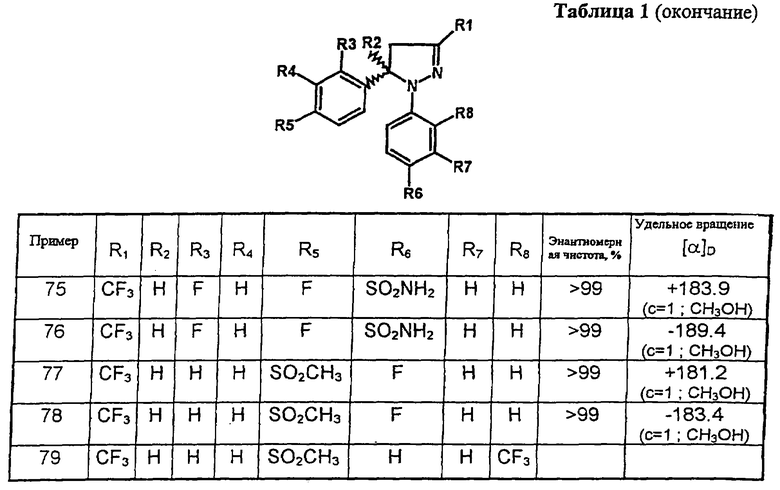
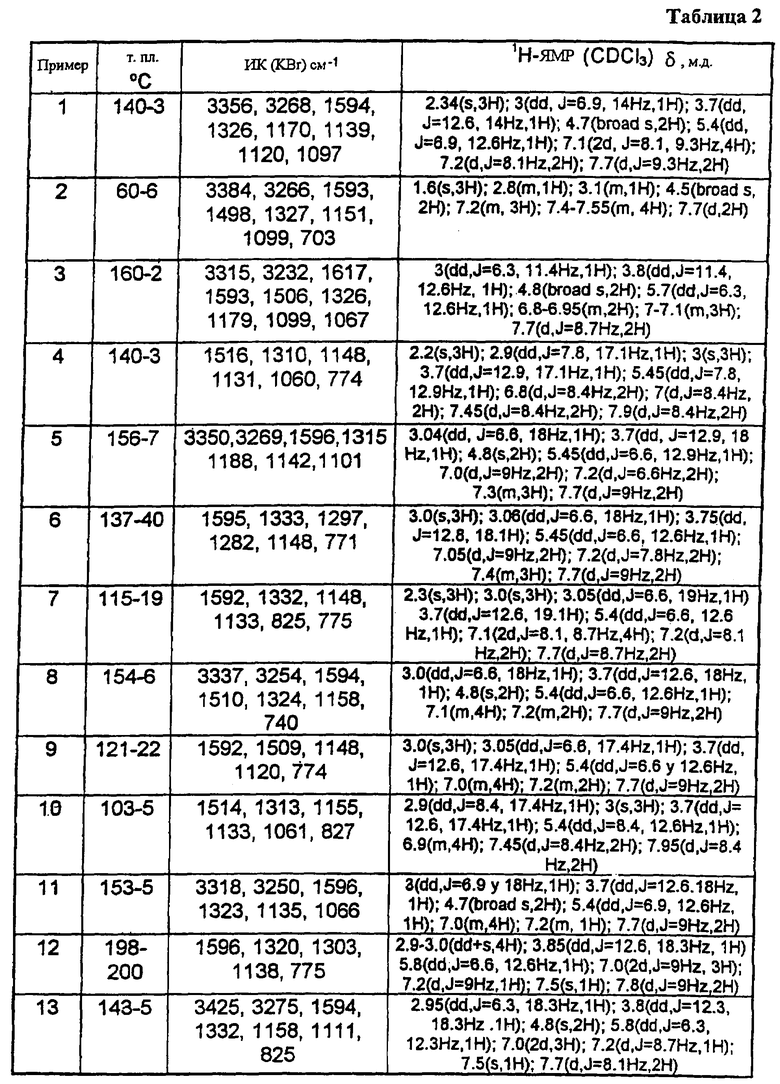
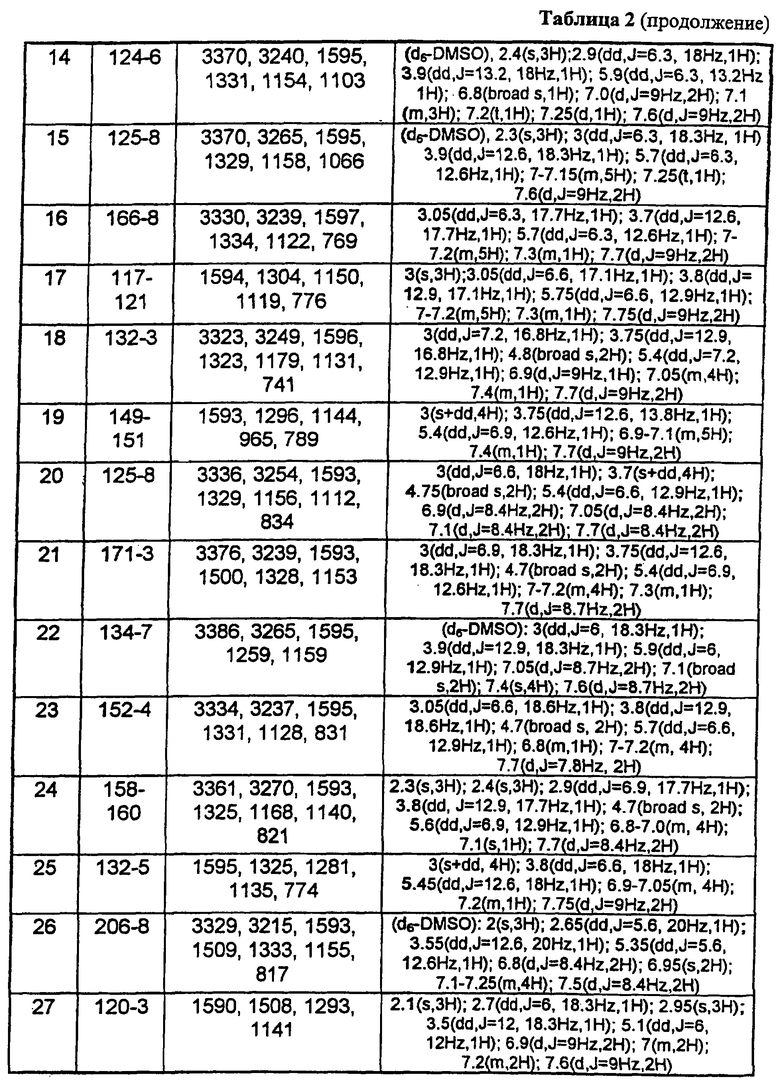
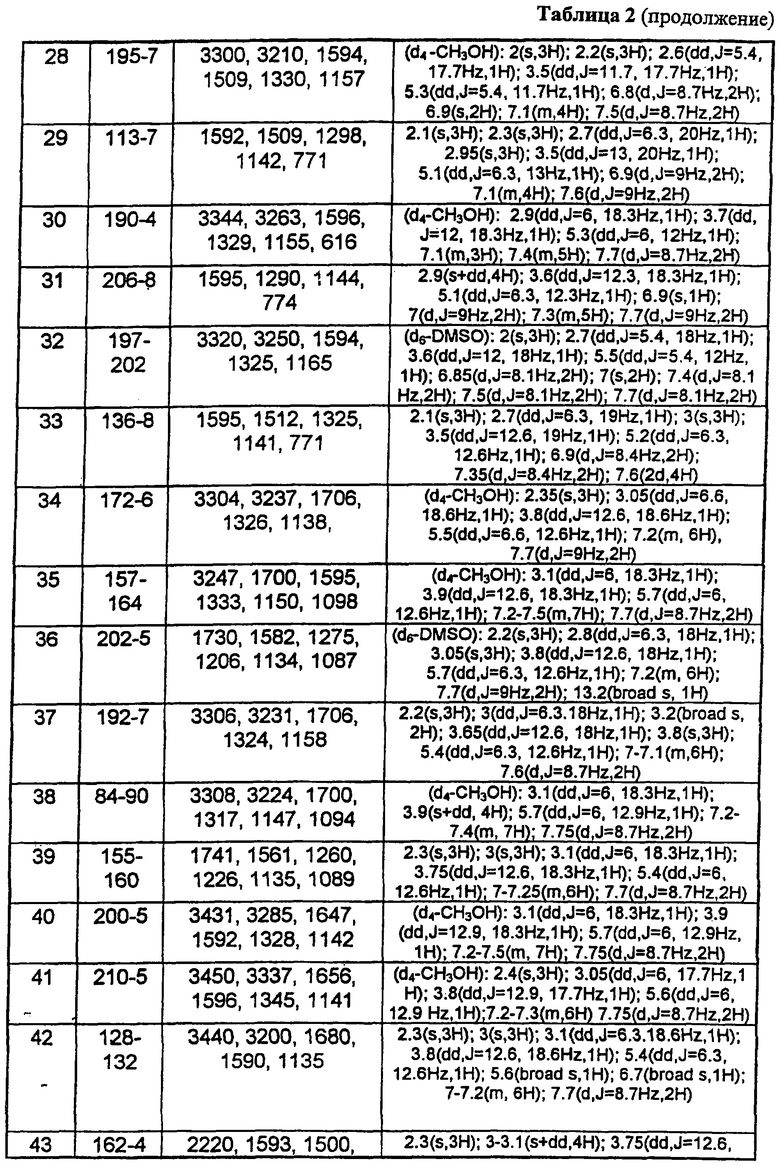
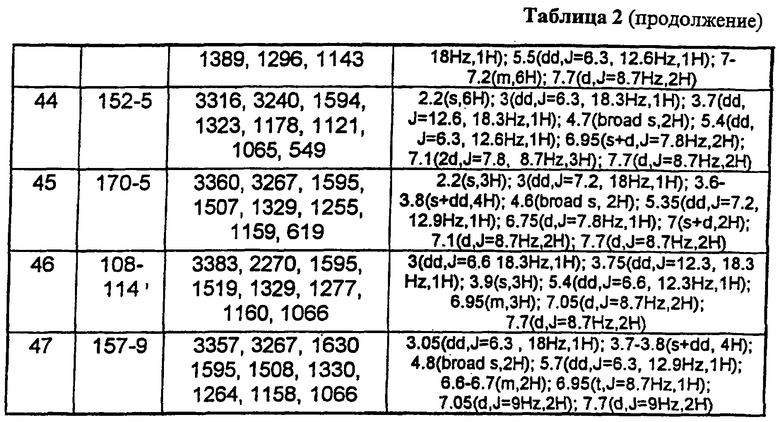
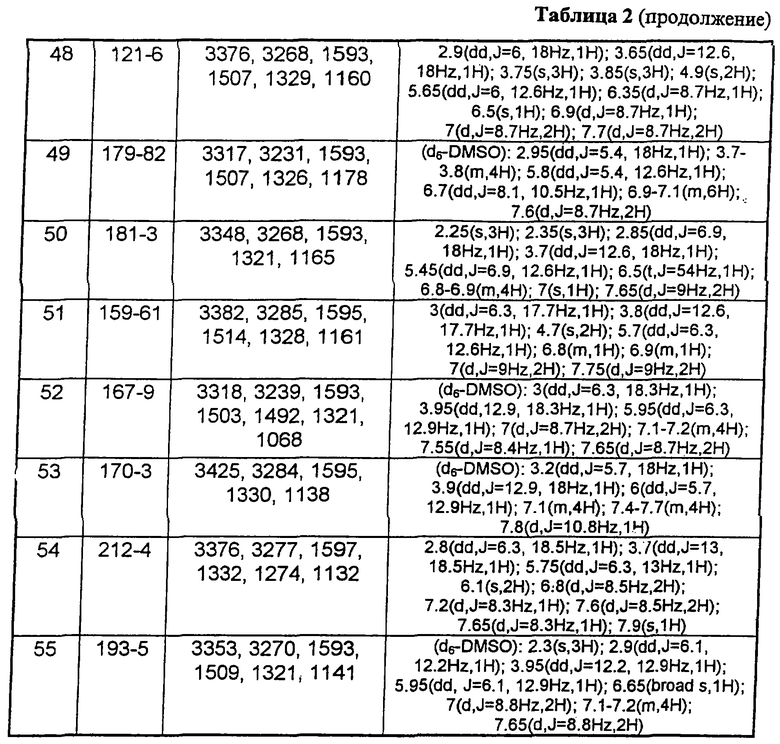
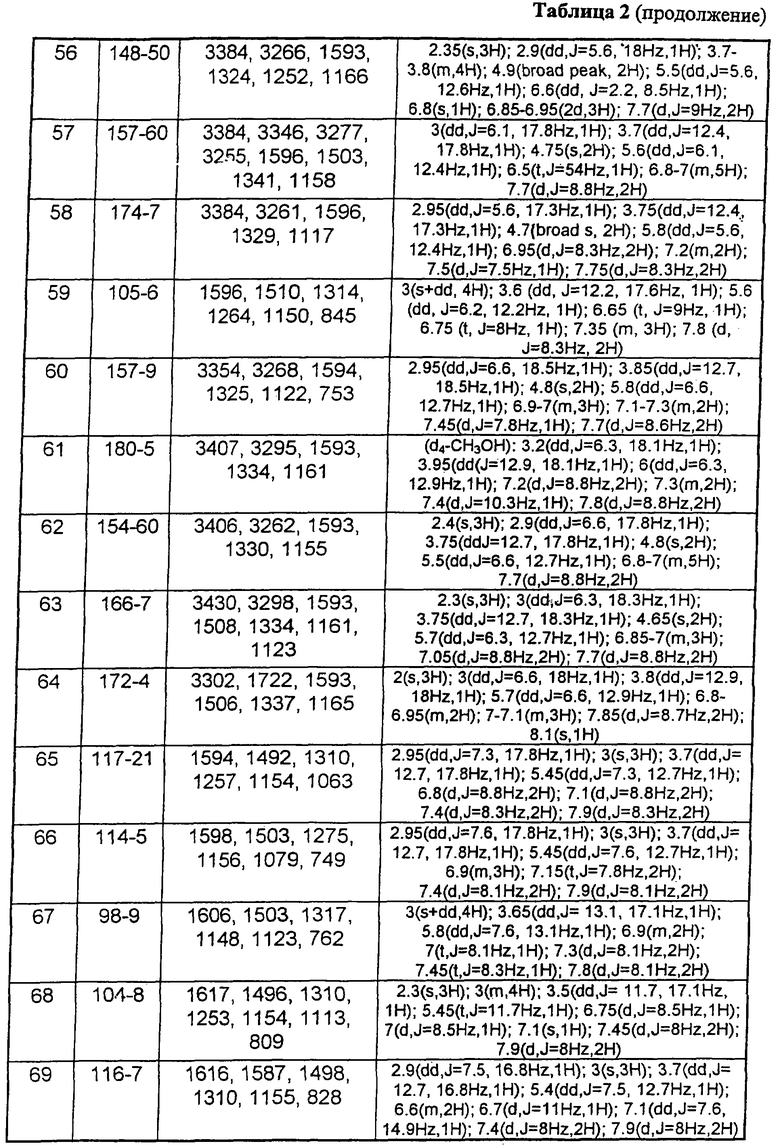
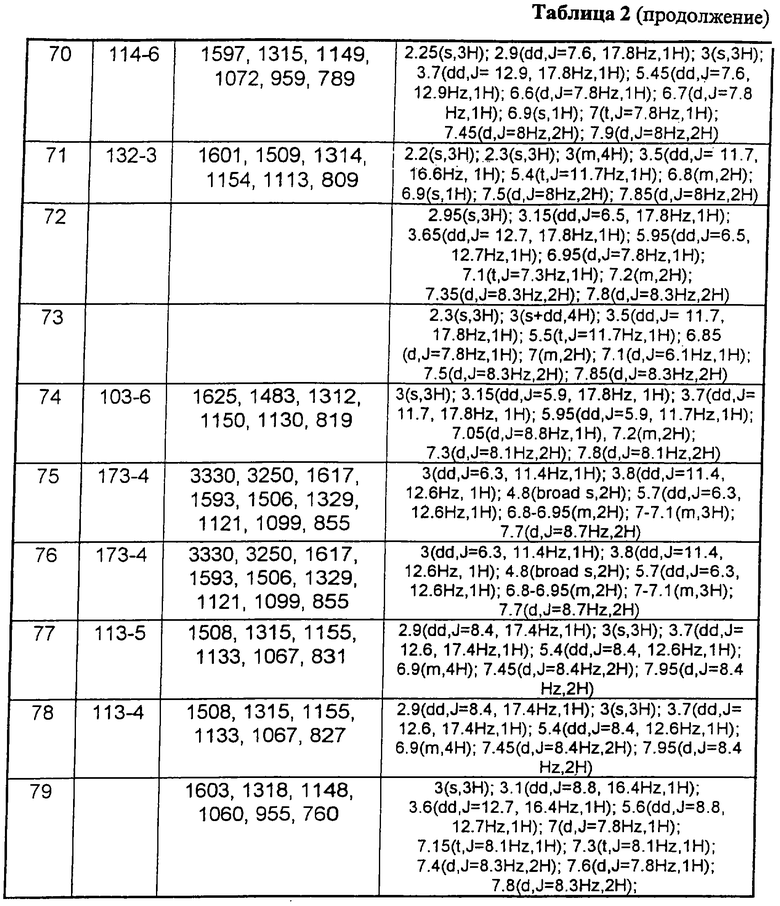
В таблице 2 при описании спектров 1Н-ЯМР использована следующая аббревиатура: s - синглет, broad s - уширенный синглет, d - дублет, dd - дублет дублетов m - мультиплет, t - триплет, J - константа спин-спинового взаимодействия, Hz - Герц (Гц).
Продукты, являющиеся объектом изобретения, представляют собой эффективные, активные при пероральном применении противовоспалительные агенты и селективные ингибиторы СОХ-2 со значительной обезболивающей активностью, лишенные ульцерогенного действия, и очень активные в тестах с экспериментальными артритами. С целью демонстрации этих видов активности приведены примеры некоторых фармакологических тестов.
Ингибирование синтеза простагландинов в воспалительных экссудатах и слизистой оболочке крыс
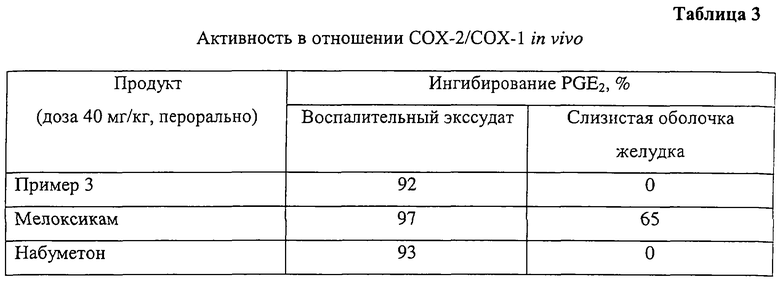
В этом опыте, помимо демонстрации селективного ингибирования СОХ-2, показана также противовоспалительная активность наряду с отсутствием влияния на простагландины желудка после перорального введения. Опыт проводили с использованием модификации метода, описанного О. Tofanetti et al. (Med. Sci. Res. 1989, 17, 745-746). Исследуемые соединения вводили перорально в начальной скрининговой дозе 40 мг/кг. Через час после введения крысам давали наркоз и имплантировали подкожно в межлопаточную область тампоны, смоченные в каррагенане. Через шесть часов после имплантации крыс умерщвляли и экстрагировали межлопаточные тампоны, а также слизистую оболочку желудка. Далее в каждом образце определяли простагландин PGE2 с помощью иммуноанализа как в экссудате тампона, так и в слизистой оболочке желудка. Ингибирование РGЕ2 в воспалительном экссудате свидетельствует о противовоспалительной активности как ингибиторов СОХ-2, так и ингибиторов СОХ-1, тогда как ингибирование PGE2 в слизистой оболочке желудка, как принято считать, является эффектом ингибирования СОХ-1.
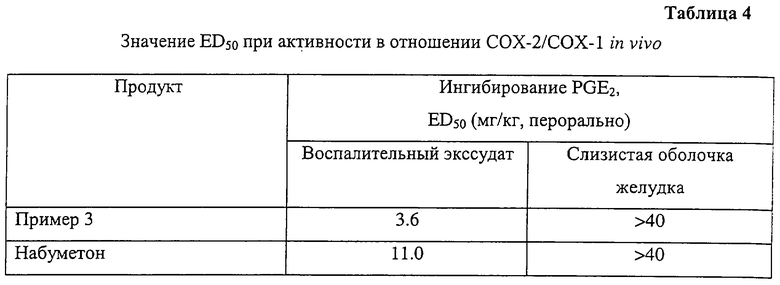
В таблице 3 суммированы результаты, полученные с соединением примера 3, в таблице 4 приведены значения ED50 (50%-ной эффективной дозы), а также селективности. Соединение обладает более выраженной противовоспалительной активностью, чем соединение сравнения.
Болеутоляющая активность в отношении “гипералгезии”, вызванной тепловым раздражителем предвоспаленной лапы крысы
В этом исследовании болеутоляющую активность у крыс контролировали с помощью метода, описанного К.Hargreaves et al. (Pain, 1988, 32, 77-78). Первоначально каждой крысе с помощью инъекции в правую заднюю лапу вводили суспензию каррагенана. Через два часа вводили перорально исследуемые соединения в скрининговой дозе 40 мг/кг. Через два часа после обработки тепловой источник прижимали к подошве стопы каждой задней лапы крыс и измеряли время, которое им потребовалось для того, чтобы отдернуть контролируемую лапу. Гиперальгезию определяли путем сравнения процента альгезии инъецированной каррагенаном лапы с другой задней лапой. Болеутоляющую активность рассчитывали, сопоставляя эти значения гиперальгезии для группы животных, обработанных соединением, со значениями для группы, получавшей только наполнитель.
В таблице 5 представлены суммированные результаты, полученные с соединением примера 3, в таблице 6 приведены значения ED50, свидетельствующие о том, что это соединение более активно, чем другие селективные ингибиторы СОХ-2, при изучении активности в опыте с тепловой гиперальгезией.


Желудочно-кишечные эффекты (GI): индукция язв у крыс, подвергнутых стрессу путем охлаждения
В этом исследовании определяли возможные ульцерогенные эффекты на желудочно-кишечном уровне после перорального введения. С этой целью использовали модификацию метода, описанного K.D.Rainsford (Agents and Actions, 1975, 5, 553-558). Сначала крысы получали перорально исследуемые соединения в различных дозах. По истечении двух часов крыс помещали в нагрудный холодильник при -15°С на 1 час. После этого их выдерживали в течение 1 часа при комнатной температуре. Затем животных умерщвляли и извлекали желудки. Желудок выдерживали в физиологическом растворе в течение 15 минут. После этого определяли процент площади поверхности желудка с язвами с использованием для каждого желудка анализатора изображения Project C.S.V. vs 1.2. Для каждого соединения определяли максимальную дозу, которая не приводила к ульцерогенезу, с помощью линейного регрессионного анализа данных доза-ответ.
Результаты, полученные с соединением примера 3, суммированы в таблице 7. Было показано, что оно не обладает ульцерогенным действием даже в очень высоких дозах, как и следовало ожидать в случае селективного ингибитора СОХ-2. С другой стороны, как диклофенак, так и пироксикам, селективные ингибиторы СОХ-1, обладали ульцерогенным действием в очень низких дозах.

Противоартритная активность у крыс
В этом опыте изучали противоартритную активность соединения примера 3 у крыс. С этой целью использовали метод, описанный B.J.Jaffee et al. (Agents and Actions, 1989, 27, 344-346). Сначала вводили адъювант Фрейда (суспензию Mycobacterium butiricum в масле соевых бобов) путем инъекции в подошву стопы задней левой лапы крысы. Через 14 дней, когда в неинъецированной лапе развилось вторичное воспаление, которое считается экспериментальным артритом, начинали лечение путем введения исследуемого соединения или наполнителя контрольной группе. Соединение примера 3 вводили перорально в дозе 10 мг/кг/день в течение 11 дней. Измеряли объем лапы с вторичным воспалением в последние дни лечения. Противоартритную активность рассчитывали путем сравнения среднего объема лапы с вторичным воспалением для группы, получавшей соединение из примера 3, и контрольной группы в течение 5 дней.
Полученные результаты свидетельствуют о том, что соединение из примера 3 обладает высокой противоартритной активностью, поскольку пероральное введение дозы 10 мг/кг/день привело к подавлению вторичного воспаления на 71%.
С учетом хороших фармакодинамических показателей производные пиразолинов по изобретению могут быть удобными для применения в лечении человека и животных, в частности, в качестве противовоспалительных агентов для лечения воспаления и других нарушений, связанных с воспалением, например, в качестве противоартритных агентов, обезболивающих агентов для лечения боли и мигрени или в качестве жаропонижающих средств при лечении лихорадки.
При лечении человека вводимая доза соединения по настоящему изобретению изменяется в зависимости от тяжести переносимого недуга. Обычно доза будет находиться между 100 и 400 мг/кг/день. Соединения по изобретению будут вводиться, например, в форме капсул, таблеток либо инъекционных растворов или суспензий.
Ниже приведены в качестве примера два фармацевтических состава, содержащие соединения, являющиеся объектом настоящего изобретения (см. табл.8).

| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ ПИРАЗОЛИНА ПРИ ПОЛУЧЕНИИ ЛЕЧЕБНОГО СРЕДСТВА ДЛЯ ПРЕДУПРЕЖДЕНИЯ И/ИЛИ ЛЕЧЕНИЯ БОЛЕЗНЕЙ, СВЯЗАННЫХ С ПРОЛИФЕРАЦИЕЙ КЛЕТОК | 2002 |
|
RU2305545C2 |
| НОВЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ, И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2001 |
|
RU2288225C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАЦЕМИЧЕСКИХ И ЭНАНТИОМЕРНО ЧИСТЫХ ПРОИЗВОДНЫХ 1,5-ДИАРИЛ-3-ТРИФТОРМЕТИЛ-Δ-ПИРАЗОЛИНОВ | 2002 |
|
RU2288915C2 |
| СПОСОБ ЛЕЧЕНИЯ ВОСПАЛЕНИЯ ИЛИ СВЯЗАННОГО С ВОСПАЛЕНИЕМ ЗАБОЛЕВАНИЯ У СОБАК | 1996 |
|
RU2253456C2 |
| НИТРООКСИПРОИЗВОДНЫЕ ИНГИБИТОРОВ ЦИКЛООКСИГЕНАЗЫ-2 | 2003 |
|
RU2339617C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРИМЕНЕНИЯ В ТЕРАПИИ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ, ВКЛЮЧАЮЩАЯ КОМБИНАЦИЮ БИФОСФОНАТА, ИНГИБИТОРА СОХ-2 И ТАКСОЛА | 2002 |
|
RU2317819C2 |
| СПОСОБ ПРИМЕНЕНИЯ ИНГИБИТОРОВ ЦИКЛООКСИГЕНАЗЫ-2 ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ НЕОПЛАЗИИ | 1997 |
|
RU2239429C2 |
| НОВЫЕ 1,2-БИС-СУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ КАК МОДУЛЯТОРЫ ХЕМОКИНОВОГО РЕЦЕПТОРА | 2011 |
|
RU2654213C9 |
| ПИРАЗОЛИЛЗАМЕЩЕННЫЙ БЕНЗОЛСУЛЬФОНАМИД ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ ОТ ВОСПАЛЕНИЯ ИЛИ СВЯЗАННОГО С ВОСПАЛЕНИЕМ ЗАБОЛЕВАНИЯ | 1994 |
|
RU2139281C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ 4,5-ДИГИДРО-1H-ПИРАЗОЛА, ИМЕЮЩИЕ CB-АНТАГОНИСТИЧЕСКУЮ АКТИВНОСТЬ | 2002 |
|
RU2299199C2 |
Описывается новое производное пиразолина общей формулы (I)
 ,
,
где R1 означает атом водорода, метил, фторметил, дифторметил, трифторметил, карбоксил, низший алкилкарбоксилат, карбоксамидную или цианогруппу, R2 означает атом водорода или метильную группу, R3, R4, R7 и R8, идентичные или разные, означают атом водорода, хлора, фтора, метильную, трифторметильную или метоксильную группу, один из R5 и R6 означает атом водорода, хлора или фтора, метильную, трифторметильную, метокси- или трифторметоксигруппу, а другой из R5 и R6 означает метилсульфонильную, аминосульфонильную или ацетиламиносульфонильную группу, в форме рацемата или энантиомера или его физиологически приемлемые соли. Описываются 5 способов получения соединения общей формулы (I), способы получения энантиомерно чистого соединения формулы (I) и его физиологически приемлемой соли, а также фармацевтическая композиция на его основе. Технический результат – соединения обладают противовоспалительной, анальгетической и жаропонижающей активностью. 9 с. и 7 з.п. ф-лы, 8 табл.
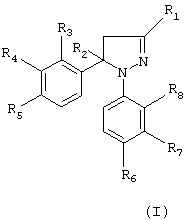
где R1 означает атом водорода, метил, фторметил, дифторметил, трифторметил, карбоксил, низший алкилкарбоксилат с 1-4 атомами углерода, карбоксамидную или цианогруппу;
R2 означает атом водорода или метильную группу;
R3, R4, R7 и R8, идентичные или разные, означают атом водорода, хлора, фтора, метильную, трифторметильную или метоксильную группу;
один из R5 и R6 означает атом водорода, хлора или фтора, метильную, трифторметильную, метокси или трифторметокси группу, а другой из R5 и R6 означает метилсульфонильную, аминосульфонильную или ацетиламиносульфонильную группу при условии, что когда R1 означает метильную группу, тогда R2 означает атом водорода или метильную группу;
R3 и R8, идентичные или разные, означают атом водорода, хлора или фтора, метильную или трифторметильную группу,
R4 означает атом водорода или фтора, метильную, трифторметильную или метокси группу,
R5 означает атом фтора, трифторметильную, трифторметокси, метилсульфонильную или аминосульфонильную группу;
R6 означает атом водорода, хлора, фтора, метильную, трифторметильную, метокси или трифторметокси, метилсульфонильную или аминосульфонильную группу при условии, что один из R5 и R6 означает метилсульфонильную или аминосульфонильную группу;
R7 означает атом водорода, хлора или фтора, метильную, трифторметильную или метоксигруппу,
в форме рацемата или энантиомера или его физиологически приемлемые соли.
[1] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(4-метилфенил)-3-трифторметил-1H-пиразол
[2] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-метил-5-(4-метилфенил)-3-трифторметил-1H-пиразол
[3] 1-(4-Аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
[4] 4,5-Дигидро-1-(4-метилфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[5] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-фенил-3-трифторметил-1H-пиразол
[6] 4,5-Дигидро-5-фенил-1-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[7] 4,5-Дигидро-5-(4-метилфенил)-1-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[8] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(4-фторфенил)-3-трифторметил-1H-пиразол
[9] 4,5-Дигидро-5-(4-фторфенил)-1-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[10] 4,5-Дигидро-1-(4-фторфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[11] 1-(4-Аминосульфонилфенил)-5-(3,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
[12] 5-(2,4-Дихлорфенил)-4,5-дигидро-1-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[13] 1-(4-Аминосульфонилфенил)-5-(2,4-дихлорфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
[14] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(2-метилфенил)-3-трифторметил-1H-пиразол
[15] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(3-метилфенил)-3-трифторметил-1H-пиразол
[16] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(2-фторфенил)-3-трифторметил-1H-пиразол
[17] 4,5-Дигидро-5-(2-фторфенил)-1-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[18] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(3-фторфенил)-3-трифторметил-1H-пиразол
[19] 4,5-Дигидро-5-(3-фторфенил)-1-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[20] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(4-метоксифенил)-3-трифторметил-1H-пиразол
[21] 1-(4-Аминосульфонилфенил)-5-(3-хлор-4-фторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
[22] 1-(4-Аминосульфонилфенил)-4,5-дигидро-3-трифторметил-5-(4-трифторметоксифенил)-1H-пиразол
[23] 1-(4-Аминосульфонилфенил)-5-(2,3-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
[24] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(2,4-диметилфенил)-3-трифторметил-1H-пиразол
[25] 5-(3,4-Дифторфенил)-4,5-дигидро-1-(4-метилсульфонилметил)-3-трифторметил-1H-пиразол
[26] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(4-фторфенил)-3-метил-1H-пиразол
[27] 4,5-Дигидро-5-(4-фторфенил)-3-метил-1-(4-метилсульфонилфенил)-1H-пиразол
[28] 1-(4-Аминосульфонилфенил)-4,5-дигидро-3-метил-5-(4-метилфенил)-1H-пиразол
[29] 4,5-Дигидро-3-метил-5-(4-метилфенил)-1-(4-метилсульфонилфенил)-1H-пиразол
[30] 1-(4-Аминосульфонилфенил)-4,5-дигидро-3-метил-5-(4-трифторметилфенил)-1H-пиразол
[31] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-фенил-1H-пиразол
[32] 4,5-Дигидро-5-фенил-1-(4-метилсульфонилфенил)-1H-пиразол
[33] 4,5-Дигидро-3-метил-1-(4-метилсульфонилфенил)-5-(4-трифторметилфенил)-1H-пиразол
[34] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(4-метилфенил)-1H-пиразол-3-карбоновая кислота
[35] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-фенил-1H-пиразол-3-карбоновая кислота
[36] 4,5-Дигидро-5-(4-метилфенил)-1-(4-метилсульфонилфенил)-1H-пиразол-3-карбоновая кислота
[37] Метил-1-(4-аминосульфонилфенил)-4,5-дигидро-5-(4-метилфенил)-1H-пиразол-3-карбоксилат
[38] Метил-1-(4-аминосульфонилфенил)-4,5-дигидро-5-фенил-1H-пиразол-3-карбоксилат
[39] Метил-4,5-дигидро-5-(4-метилфенил)-1-(4-метилсульфонилфенил)-1H-пиразол-3-карбоксилат
[40] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-фенил-1H-пиразол-3-карбоксамид
[41] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(4-метилфенил)-1H-пиразол-3-карбоксамид
[42] 4,5-Дигидро-5-(4-метилфенил)-1-(4-метилсульфонилфенил)-1H-пиразол-3-карбоксамид
[43] 3-Циан-4,5-дигидро-5-(4-метилфенил)-1-(4-метилсульфонилфенил)-1H-пиразол
[44] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(3,4-диметилфенил)-3-трифторметил-1H-пиразол
[45] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(3-метил-4-метоксифенил)-3-трифторметил-1H-пиразол
[46] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(3-фтор-4-метоксифенил)-3-трифторметил-1H-пиразол
[47] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(2-фтор-4-метоксифенил)-3-трифторметил-1H-пиразол
[48] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(2,4-диметоксифенил)-3-трифторметил-1H-пиразол
[49] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(4-фтор-2-метоксифенил)-3-трифторметил-1H-пиразол
[50] 1-(4-Аминосульфонилфенил)-3-дифторметил-4,5-дигидро-5-(2,4-диметилфенил)-1H-пиразол
[51] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(2,3,4-трифторфенил)-3-трифторметил-1H-пиразол
[52] 1-(4-Аминосульфонилфенил)-5-(2-хлор-4-фторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
[53] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(2-фтор-4-трифторметилфенил)-3-трифторметил-1H-пиразол
[54] 1-(4-Аминосульфонилфенил)-5-[2,4-(бистрифторметил)фенил]-4,5-дигидро-3-трифторметил-1Н-пиразол
[55] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(2-метил-3-фторфенил)-3-трифторметил-1H-пиразол
[56] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(2-метил-4-метоксифенил)-3-трифторметил-1H-пиразол
[57] 1-(4-Аминосульфонилфенил)-5-(2,4-дифторфенил)-3-дифторметил-4,5-дигидро-1H-пиразол
[58] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(4-фтор-2-трифторметилфенил)-3-трифторметил-1H-пиразол
[59] 1-(2,4-Дифторфенил)-4,5-дигидро-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[60] 1-(4-Аминосульфонилфенил)-5-(2-хлорфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
[61] 1-(4-Аминосульфонилфенил)-5-(4-хлор-2-фторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
[62] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(4-фтор-2-метилфенил)-3-трифторметил-1H-пиразол
[63] 1-(4-Аминосульфонилфенил)-4,5-дигидро-5-(2-фтор-4-метилфенил)-3-трифторметил-1H-пиразол
[64] 1-(4-Ацетиламиносульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
[65] 1-(4-Хлорфенил)-4,5-дигидро-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[66] 4,5-Дигидро-1-фенил-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[67] 4,5-Дигидро-1-(2-фторфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[68] 1-(4-Хлор-2-метилфенил)-4,5-дигидро-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[69] 4,5-Дигидро-1-(3-фторфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1Н-пиразол
[70] 4,5-Дигидро-1-(3-метилфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[71] 4,5-Дигидро-1-(2,4-диметилфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[72] 1-(2-Хлорфенил)-4,5-дигидро-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[73] 4,5-Дигидро-1-(2-метилфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[74] 1-(2,4-Дихлорфенил)-4,5-дигидро-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[75] (+)-1-(4-Аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
[76] (-)-1-(4-Аминосульфонилфенил)-5-(2,4-дифторфенил)-4,5-дигидро-3-трифторметил-1H-пиразол
[77] (+)-4,5-Дигидро-1-(4-фторфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол
[78] (-)-4,5-Дигидро-1-(4-фторфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-пиразол;
[79] 4,5-Дигидро-5-(4-метилсульфонилфенил)-3-трифторметил-1-(2-трифторметилфенил)-1H-пиразол,
или его физиологически приемлемые соли.
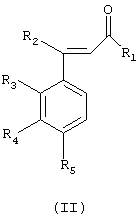
где R1 означает атом водорода, метильную, фторметильную, дифторметильную, трифторметильную или карбоксильную группу;
R2, R3, R4 и R5 имеют значения, приведенные для общей формулы (I),
с фенилгидразином общей формулы (III) в виде основания или соли
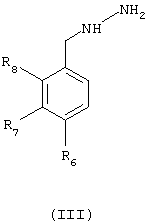
где R6, R7 и R8 имеют значения по п.1,
при условии, что когда R1 означает фторметильную группу, гидразин должен быть в виде соли.
взаимодействием соединения общей формулы (I), где R1 означает карбоксильную группу (СООН), a R2 - R8 имеют значения по п.1, с соответствующим реагентом с целью получения хлорангидрида кислоты, с таким, как, например, тионилхлорид или оксалилхлорид, с последующей реакцией этерификации действием алифатического спирта с 1-4 атомами углерода в присутствии органического основания, такого, как триэтиламин или пиридин, или прямым взаимодействием карбоновой кислоты с соответствующим безводным спиртом, насыщенным газообразным хлористым водородом.
| US 4425179 A, 10.01.1984 | |||
| WO 8806583 A, 07.09.1988 | |||
| Indian Journal of Chemistry | |||
| Vol | |||
| Прибор для равномерного смешения зерна и одновременного отбирания нескольких одинаковых по объему проб | 1921 |
|
SU23A1 |
| RU 2059622 C1, 10.05.1996. | |||

