Предпосылки создания изобретения
a) Область техники, к которой относится изобретение
Настоящее изобретение относится к новым соединениям, классифицированным в данной области химии как поли(монопероксикарбонаты) структуры А: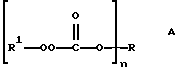
(определения n, R и R1 даны в разделе Краткое изложение сущности изобретения), к способам их получения и их применению и к промежуточным соединениям, используемым в способах получения.
В промышленности полимеров существует потребность в эффективных свободно-радикальных инициаторах для полимеризации этиленоненасыщенных (с двойными связями) мономеров, таких, как стирол с более высокой производительностью при сохранении молекулярной массы полимера и физических свойств полимера, например механических свойств при растяжении. Обычно применение более активных свободно-радикальных инициаторов и увеличение температур полимеризации для повышения темпов производства полимеров (например, полистирола) приводят к требуемому повышению производительности, но также влекут за собой нежелательные последствия, такие, как снижение молекулярной массы полимера и ухудшение физических свойств полимера. Снижение температур полимеризации, уменьшение используемого количества инициатора и применение менее активных инициаторов обычно обеспечивают увеличение молекулярной массы полимера, но при этом снижается производительность. В 80-х годах имел место прогресс в области полимеризации стирола. Применение дипероксикеталей, таких, как 1/1-бис(трет-бутилперокси)циклогексан, в качестве инициаторов вместо обычных инициаторов, таких, как дибензоилпероксид и трет-бутилпероксибензоат, в промышленных процессах полимеризации стирола привело к повышению молекулярной массы и росту производства полистирола. Данные заявители способствовали дальнейшему развитию этой области техники и нашли, что новые поли(монопероксикарбонаты) структуры А по настоящему изобретению можно использовать в качестве инициаторов для полимеризации этиленоненасыщенных мономеров с получением полимеров (например, полистирола), имеющих значительно более высокую молекулярную массу при одновременном сохранении или повышении скоростей полимеризации, или с получением полимеров при значительно более высоких скоростях полимеризации с сохранением молекулярной массы и что соединения по настоящему изобретению лучше, чем дипероксикетали, такие, как 1,1-бис(трет-бутилперокси)циклогексан. Таким образом, новые поли(монопероксикарбонаты) по настоящему изобретению способны удовлетворить потребности промышленности полимеров, относящихся к процессам полимеризации.
В производстве сложных полиэфиров тоже существует потребность в свободно-радикальных инициаторах, отверждающих ненасыщенные полиэфирные смолы быстрее и/или при более низких температурах. Новые поли(монопероксикарбонаты) по настоящему изобретению способны также удовлетворить эту потребность промышленности полимеров.
b) Описание известного уровня техники
В патенте США 3652631 (выдан PPG 28 марта 1972 г.) раскрыты бис(монопероксикарбонаты) 1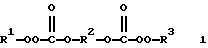
(где R1 и R3 представляют алкил, имеющий до 10 углеродных атомов, необязательно замещенный галогеном или нитрогруппами, и R2 представляет двухвалентный остаток органического диола, содержащий до 12 углеродных атомов и до трех простых эфирных связей), полученные из трет-бутилгидропероксида, трет-амилгидропероксида, или трет-гексилгидропероксида и бис(хлорформиатов) и применение этих соединений для полимеризации мономеров, таких, как стирол. Патент США 3652631 охватывает такой бис(монопероксикарбонат), как 1,5-бис(трет-бутилпероксикарбонилокси)-3-окса-пентан. Заявители настоящего изобретения нашли, что поли(монопероксикарбонаты) структуры А являются новыми и лучшими инициаторами полимеризации стирола, чем 1,5-бис(трет-бутилпероксикарбонилокси)-3-оксапентан, так как они давали полистиролы со значительно более высокими молекулярными массами при одних и тех же условиях полимеризации.
В патенте США 4136105 (выдан Pennwalt Corp. 23 января 1979 г.) раскрыты О-алкил ОО-трет-окстилмонопероксикарбонаты 2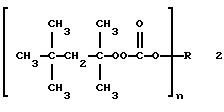
(где n представляет целое число от 1 до 4, предпочтительно 1; когда n = 1, R выбран из алкила с 1-16 углеродными атомами, циклоалкила с 5-12 углеродными атомами, арила с 6-14 углеродными атомами, аралкила с 7-14 углеродными атомами, алкенила с 3-10 углеродными атомами, циклоалкенила с 5-10 углеродными атомами и алкинила с 3-14 углеродными атомами; когда n = 2, R выбран из алкилена с 2-12 углеродными атомами, циклоалкилена с 4-12 углеродными атомами, арилена с 6-14 углеродными атомами, алкенилена с 2-12 углеродными атомами, алкинилена с 4-12 углеродными атомами, метиленфенилметилена, метиленциклогексилметилена, -R1 XR1 и -R2YR2-, где R1 представляет алкилен с 2-6 углеродными атомами, R2 представляет фенилен, Х представляет -О- или -S- и Y представляет -О-, -S-, -CH2- или С(СН3)2-; когда n = 3, R представляет R3 С(СН2-)3, -CH(CH2-)2 и -СН2CН(-)СН2СН2СН2СН2-, где R3 представляет алкил с 1-5 углеродными атомами; и когда n = 4, R представляет С(СН2-)4) и применение этих соединений для инициирования полимеризации виниловых мономеров и для отверждения ненасыщенных сложных полиэфиров. Этот патент охватывает трис- и тетракис(моно-трет-октилпероксикарбонаты), полученные из трет-октилгидропероксида, но не раскрывает новых поли-(монопероксикарбонатов) по настоящему изобретению, которые получены из трет-бутил- и трет-амилгидропероксидов.
В патенте США 5314970 (выдан Elf Atochem 24 мая 1994 г.) раскрыты ОО-трет-алкил О-поликапролактонмонопероксикарбонаты, то есть поликапролактоны с блокированными концевыми ОО-трет-алкилпероксикарбонатными группами 3, полученные из трет-алкилгидропероксидов и хлорформиатов
(A-X-)m-R-(-X'-B)n
где А представляет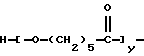
В представляет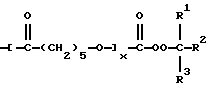
m представляет целое число от 0 до 3, n представляет целое число от 1 до 4, m+n представляет целое число от 1 до 4, R1 и R2 являются одинаковыми или разными и представляют алкил с 1-4 углеродными атомами, R3 представляет алкил с 1-12 углеродными атомами или алкинил с 2-12 углеродными атомами, у представляет целое число от 0 до примерно 10000, х представляет целое число от 4 до примерно 22000, (y)(m)+(х)(n) представляет целое число от 4 до примерно 22000, Х и X' независимо выбраны из -О- или -N(R4-), причем R4 представляет водород, замещенный или незамещенный алифатический с 1-20 углеродными атомами, замещенный или незамещенный алициклический с 5-18 углеродными атомами, замещенный или незамещенный ароматический с 6-14 углеродными атомами и замещенный или незамещенный аралифатический с 7-22 углеродными атомами, и R представляет замещенный или незамещенный алифатический, алициклический, ароматический или аралифатический радикал, бирадикал, трирадикал или тетрарадикал),
поликарболактоны с концевыми гидроксигруппами и применение этих соединений для инициирования полимеризации виниловых мономеров, для отверждения ненасыщенных сложных полиэфиров, для получения поликапролактоновых блок-сополимеров, для сшивания полиолефинов, для отверждения эластомеров, для модифицирования полипропилена, для прививки поликарболактоновых блоков на полиолефинах, для получения взаимопроникающих полимерных сеток и для получения привитых полиолов.
Единственными монопероксикарбонатами, которые были раскрыты в примерах, были бис(трет-бутилмонопероксикарбонаты) и бис(трет-амилмонопероксикарбонаты), полученные из диолов. Единственная полезность, раскрытая в примерах, и полезность, подчеркнутая в реферате, описании и формуле изобретения, заключалась в применении бис(монопероксикарбонатов) для получения поликапролактон-полистироловых блок-сополимеров и привитых сополимеров для использования в качестве агента, улучшающего совместимость, для смесей полимеров. Поскольку наиболее эффективными блок-сополимерами для совмещающихся смесей полимеров были блок-сополимеры с более крупными блочными сегментами, то наиболее предпочтительными поли(е-капролактонами) были поли(е-капролактоны) с дигидроксильными концевыми группами с молекулярной массой примерно 3000-15000 (патент США 5314970, колонка 12, строки 30-33). Исходные материалы по настоящему изобретению, относящиеся к поли(е-капролактонам) с гидроксильными концевыми группами, ограничены поли(е-капролактонами) с концевыми полигидроксигруппами, кроме специальных случаев, когда новые пероксидзамещенные бис(монопероксикарбонаты) получают путем осуществления взаимодействия бис(галогенформиатов) поли(е-капролактонов), имеющих бис-гидроксильные концевые группы, с 1,1,4-триметил-4-(трет-бутилперокси)пентилгидропероксидом или с 1,1,4-триметил-4-(трет-амилперокси)пентилгидропероксидом. Кроме того, полигидроксильные исходные материалы (т.е. диолы, триолы или высшие полиолы) для соединений по настоящему изобретению должны иметь молекулярные массы менее чем примерно 1000, менее чем примерно 1000 и менее чем примерно 1300 соответственно.
Патент США 5314970 не предполагает продвижения вперед в области полимеризации стирола при использовании предлагаемых в патенте бис(монопероксикарбонатов). Предлагаемым в патенте США 5314970 соединением является бис(трет-бутилмонопероксикарбонат, полученный из TONE® 200. Заявители настоящего изобретения нашли, что новые поли(монопероксикарбонаты) структуры А были лучшими инициаторами полимеризации стирола, так как они давали полистиролы со значительно более высокими молекулярными массами при одних и тех же условиях полимеризации, чем у полученных с использованием бис(трет-бутилмоноперокси-карбоната) из TONE® 200.
В патенте США 5455321 (выдан The Dow Chemical Company 3 октября 1995 г.) раскрыт способ получения моновинилиденового ароматического полимера (например, полистирола), имеющего молекулярную массу выше 275000, который (способ) включает полимеризацию моновинилиденового ароматического мономера (например, стирола) в присутствии а) 10-2000 частей на миллион (по массе) по крайней мере одного инициатора свободно-радикальной полимеризации с разветвлением, имеющего структуру:
R'((CO)nOOR)m,
где n представляет 0 или 1, m представляет 3-6, R' представляет многофункциональный органический радикал, имеющий до 25 неводородных атомов, и R представляет C1-15 третичные алкильные или C7-15 третичные аралкильные группы, и
b) 10-2000 частей на миллион одного или нескольких органических уменьшающих гелеобразование агентов, выбранных из группы, состоящей из i) меркаптанов, терпенов, галогенуглеродов и галогенуглеводородов, имеющих до 20 углеродных атомов, ii) рецикловой жидкости, полученной путем удаления летучих продуктов из полимеризуемой смеси полимеров, и iii) смеси органических агентов, уменьшающих гелеобразование, из i) и ii). Предпочтительным инициатором свободно-радикальной полимеризации с разветвлением был 2,2-бис(4,4-ди-трет-бутилпероксициклогексил)-пропан: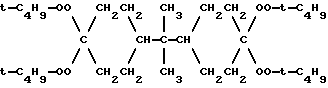
Другими инициаторами свободно-радикальной полимеризации с разветвлением, раскрытыми в этом патенте, были сложные три-трет-алкиловые эфиры 1,3,5-бензолтрикарбоперкислоты, сложные тетра-трет-алкиловые эфиры 1,2,4,5-бензолтетракарбоперкислоты и 2,4,6-три-трет-алкилперокси-1,3,5-триазины, 2-(4-изопропенилфенил)-2-пропил трет-алкилпероксиды, трет-алкил 4-изопропенилпероксибензоаты, ди-трет-алкилдипероксималеинаты и дипероксифумараты и ОО-трет-алкил О-алкилмонопероксималеинаты и монопероксифумараты. В патенте США 5455321 не раскрыты ни новые поли(монопероксикарбонаты) по настоящему изобретению, ни новые способы их применения в производстве полимеров.
В патенте США 5266603 (выдан Huels Aktiengesellschaft 30 ноября 1993 г.) раскрыт способ получения вспенивающихся гомополимеров и сополимеров стирола путем а) приготовление водной суспензии стирольного мономера и пероксидной инициирующей системы, содержащей по крайней мере один алифатический или циклоалифатический дипероксикеталь (например, 2,2-бис(трет-бутилперокси)бутан или 1,1-бис(трет-бутилперокси)циклогексан) или монопероксикарбонатный инициатор (например, ОО-трет-бутил О-(2-этилгексил)монопероксикарбонат или ОО-трет-амил О-(2-этилгексил)монопероксикарбонат) и пероксидный инициатор, имеющий более короткий период полураспада, чем алифатический или циклоалифатический дипероксикеталевый или монопероксикарбонатный инициатор (например, дибензоилпероксид), b) нагревания перемешиваемой суспензии при температуре от 80 до 100oС в течение первого периода времени для осуществления начальной полимеризации, с) добавления к перемешиваемой суспензии С3-6 углеводородного диспергатора, d) повышение температуры полученной суспензии до температуры в пределах от 100 до 130oС во втором периоде для осуществления окончательной полимеризации и получения вспенивающегося полистирола. В этом патенте не используют ни бис-, ни трис-, ни более высокие поли(монопероксикарбонаты), а используют лишь моно-(монопероксикарбонаты), такие, как ОО-трет-бутил О-(2-этилгексил)монопероксикарбонат или ОО-трет-амил О-(2-этилгексил)монопероксикарбонат.
В общем в описанных выше материалах поли(монопероксикарбонаты) структуры A не раскрыты.
с) Определения
Диол определяется как структура R(-ОН)2, где R представляет бирадикал, например R(-)2. Триол определяется как структура R(-ОН)3, где R представляет трирадикал, например R(-)3. Полиол определяется как структура R(-OH)n, где R представляет полирадикал, например R(-)n, и n представляет целое число ≥2. Тетраол определяется как структура R(-OH)4, где R представляет тетрарадикал, например R(-)4.
Когда в общей формуле или структуре обобщенная функциональная группа или индекс, такие, как R, R1, R2, х, n и т.д., появляется более чем один раз, их значения не зависят друг от друга.
Краткое изложение сущности изобретения
В соответствии с настоящим изобретением в первом его аспекте, касающемся соединения, предлагается поли(монопероксикарбонат) структуры А: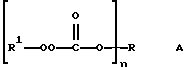
где n предоставляет целое число от 3 до 8, R1 выбран из группы, состоящей из трет-алкильных радикалов с 4-12 углеродными атомами, 1,1,4-триметил-4(трет-бутилперокси) пентильного радикала, 1,1,4-триметил-4(трет-амилперокси) пентильного радикала, трет-циклоалкильных радикалов с 6-10 углеродными атомами, трет-аралкильных радикалов с 9-13 углеродными атомами и 3-метил-1-бутин-3-ила и 3-метил-1-пентин-3-ила, при условии, что:
когда R1 выбран из 1,1,4-триметил-4(трет-бутилперокси) пентильного радикала и 1,1,4-триметил-4(трет-амилперокси) пентильного радикала, n может иметь также значение 2;
когда n = 2, R представляет бирадикал, выбранный из алкилена с 2-12 углеродными атомами, алкенилена с 4-8 углеродными атомами и бирадикальных структур (n) и (о)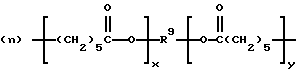
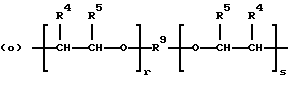
где R9 представляет алкиленовый бирадикал с 2-8 углеродными атомами;
когда n = 3, R представляет трирадикал, выбранный из 1, 3, 5-циклогекстриила, R2C(CH2-)3, -CHR2H (-)СН2- и структур (а), (b), (с), (d) и (е),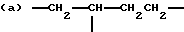
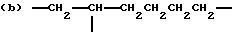
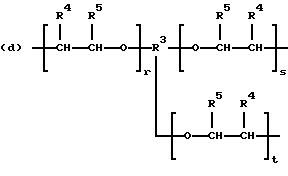
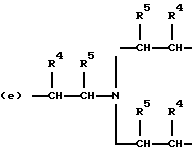
где R2 выбран из водорода и алкильного радикала с 1-6 углеродными атомами, R3 представляет трирадикал, выбранный из группы, состоящей из R2С(CH2-)3, -CHR2CH(-)CH2- и структур (а) и (b), R4 и R5 являются одинаковыми или разными и выбраны из водорода и алкильных радикалов с 1-4 углеродными атомами, х, у и z представляют целые числа от 0 до 5 с условием, что сумма х, у и z составляет от 2 до 8, и r, s и t представляют целые числа от 0 до 6 с условием, что сумма r, s и t составляет от 3 до 18, и когда n = 4-8, R представляет полирадикал, выбранный из C(CH2-)4 и структур (f), (g), (h), (i), (j), (k) и (l),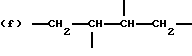
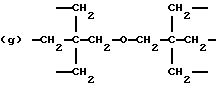
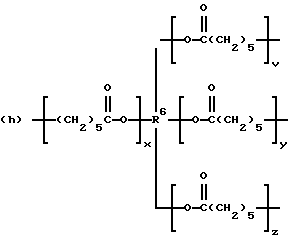
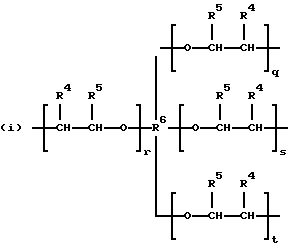
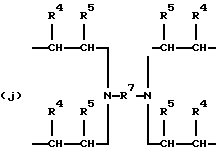
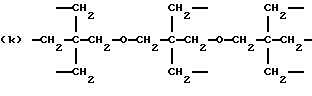
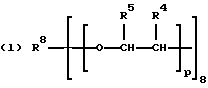
где R6 представляет тетрарадикал, выбранный из C(CH2-)4 и структуры (f), R7 представляет бирадикал, выбранный из алкилена с 2-6 углеродными атомами и 1,2-, 1,3-и 1,4-фенилена, R8 представляет октарадикал на основе сахарозы, имеющий структуру (m)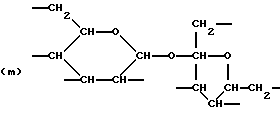
р представляет целое число от 1 до 3, v представляет целое число от 0 до 5 с условием, что сумма v, х, у и z составляет от 3 до 10, и q представляет целое число от 0 до 4 с условием, что сумма q, r, s и t составляет от 2 до 16, и с дополнительным условием, что когда R представляет R3C(CH2-)3, структуру (b) или С(СН2-)4, то R1 не является трет-октилом; причем такой новый поли(монопероксикарбонат) структуры A синтезирован из диола, триола или высшего полиола структуры АА: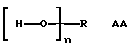
имеющего молекулярную массу менее примерно 1000, менее примерно 1000 или менее примерно 1300 соответственно.
В соответствии с настоящим изобретением в подродовом аспекте, касающемся соединения, предлагается соединение структуры А', в котором в структуре А, когда n = 3, R представляет трирадикал, выбранный из 1,3,5-циклогекстриила, R2C(СH2-)3, -CHR2CH(-)CH2 - и структур (а), (б), (d) и (е), определенных выше.
В соответствии с настоящим изобретением во втором подродовом аспекте, касающемся соединения, предлагается соединение структуры А", в котором в структуре А, когда n = 3, R - такой, как определенный выше для первого подродового аспекта настоящего изобретения, и когда n = 4-8, R представляет полирадикал, выбранный из С(СН2-)4 и структур (f), (g), (i), (j), (k) и (1), определенных выше.
Соединения по первому аспекту настоящего изобретения имеют физические свойства, присущие аморфным твердым веществам или вязким жидкостям, причем указанные твердые вещества имеют цвет от белого до цвета светлой соломы, а указанные жидкости окрашены в цвет светлой соломы. Твердые вещества имеют пределы плавления, и все соединения имеют инфракрасные спектры и содержание активного кислорода пероксида, которые убедительно подтверждают необходимость патентования указанных структур.
Соединения по первому аспекту настоящего изобретения по своей природе пригодны для прикладного применения в качестве инициаторов для полимеризации этиленоненасыщенных мономеров, в частности стирола, и для модификации молекулярной массы полимеров, таких, как ненасыщенные сложные полиэфиры, термопластические полимеры, эластомерные полимеры и смеси таких полимеров.
В соответствии с настоящим изобретением в первом его аспекте, касающемся способа, предлагается способ свободно-радикального инициированного модифицирования субстрата, выбранного из группы, состоящей из этиленоненасыщенных мономеров и полимеров, восприимчивых к свободно-радикальному индуцированному модифицированию молекулярной массы, который включает обработку указанных субстратов при условиях, эффективных для инициирования свободно-радикального индуцированного модифицирования указанных субстратов, одним или несколькими соединениями структуры (А) в эффективных инициирующих количествах.
Конкретно можно назвать следующие процессы свободно-радикального индуцированного модифицирования молекулярной массы:
а) полимеризация этиленоненасыщенных мономеров (таких, как стирол, этилен, аллилдигликолькарбонат (ADC, АДК) и тому подобное, которые известны в данной области техники как соединения, восприимчивые к такой полимеризации), необязательно в присутствии ненасыщенного эластомера (такого, как полибутадиен, полиизопрен и тому подобное, полезного, как известно в данной области техники, в случае присутствия в таких процессах полимеризации);
b) отверждение ненасыщенных сложных полиэфиров;
с) сшивание и отверждение термопластических и эластомерных полимеров; и
е) модифицирование молекулярной массы полиолефинов.
В соответствии с настоящим изобретением во втором его аспекте, касающемся способа, предлагается способ свободно-радикальной инициированной полимеризации этиленоненасыщенных мономеров (таких, как стирол, этилен, аллилдигликолькарбонат (ADC, АДК) и тому подобное), которые известны в данной области техники как соединения, восприимчивые к такой полимеризации необязательно в присутствии ненасыщенного эластомера (такого, как полибутадиен, полиизопрен и тому подобное), полезного, как известно, в данной области техники, в случае присутствия в таких процессах полимеризации, при условиях, эффективных для инициирования свободно-радикальной индуцированной полимеризации, с одним или несколькими соединениями структуры А в сочетании с другими инициаторами свободно-радикальной полимеризации, выбранными из группы, состоящей из монопероксидов и дипероксидов (таких, как диацилпероксиды, дипероксикетали, сложные пероксиэфиры, монопероксикарбонаты и диалкилпероксиды), в эффективных инициирующих количествах.
Подробное описание предпочтительных вариантов
Новые поли(монопероксикарбонаты) структуры А - Способы получения
Новые поли(монопероксикарбонаты) структуры А могут быть получены путем осуществления взаимодействия одного или более трет-алкилгидропероксидов структуры B
R1-OOH
c поли(галогенформиатом) структуры С при температуре от -30 до 50oС,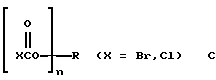
где R, R1 и n такие, как определенные для структуры A, необязательно в присутствии неорганического или органического основания и необязательно в присутствии одного или нескольких растворителей.
Неограничивающие примеры подходящих необязательных оснований включают триэтиламин, трибутиламин, N, N-диизопропилэтиламин, 2,2,6,6-тетраметилпиперидин, N, N-диметиланилин, N,N-диметиламинопиридин, 2,4,6-колидин, мочевина, тетраметилмочевина, гидроксид натрия, карбонат натрия, гидрокарбонат натрия, гидроксид калия, карбонат калия, гидрокарбонат калия, гидроксид кальция, гидроксид магния, гидроксид бария, карбонат кальция и тринатрийфосфат.
Неограничивающие примеры подходящих необязательных растворителей включают пентан, гексаны, гептаны, додеканы, смеси уайт-спиритов без запаха, толуол, ксилолы, кумен, метиленхлорид, этилацетат, 2-этилгексилацетат, изобутил-изобутират, диметиладипат, диметилсукцинат, диметилглутарат (или его смеси), диметилфталат, дибутилфталат, бензилбутилфталат, диэтиловый эфир, метил трет-бутиловый эфир (МТВЕ, МТВЭ), 2-метоксиэтилацетат, тетрагидрофуран (THF, ТГФ) и другие.
Подходящие гидропероксиды структуры В, которые могут быть подвергнуты взаимодействию с поли(галогенформиатами) структуры C, включают трет-бутилгидропероксид, трет-амилгидропероксид, 2-метил-2-пентилгидропероксид, 3-метил-3-пентилгидропероксид, 3-метил-1-бутин-3-илгидропероксид, 3-метил-1-пентин-3-илгидропероксид, 2-метил-2-гексилгидропероксид, 1,1,3,3-тетраметилбутилгидропероксид, 1,1,4-триметил-4(трет-бутилперокси)пентилгидропероксид, 1,1,4-триметил-4(трет-амилперокси)пентилгидропероксид, 1-метил-1-циклогексилгидропероксид, параментангидропероксид, α-кумилгидропероксид, 4-метил-α-кумилгидропероксид, 3-метил-α-кумилгидропероксид и диизопропилбензолмоногидропероксид.
Неограничивающие примеры подходящих поли (галогенформиатов) структуры C, которые могут быть подвергнуты взаимодействию с гидропероксидами структуры В, включают 1,1,1-трис(хлоркарбонилоксиметил)этан, 1,1,1-трис (хлор-карбонилоксиметил)-пропан, 1,1,1-трис(хлоркарбонилоксиметил)бутан, 1,2,3-трис(хлоркарбонилокси)пропан, 1,2,3-трис(хлоркарбонилокси)гексан, 1,2,3-трис(хлоркарбонилокси)гептан, 1,2,4-трис(хлоркарбонилокси)бутан, 1,2,6-трис(хлоркарбонилокси)гексан, 1,3,5-трис(хлоркарбонилокси) циклогексан, тетракис(хлоркарбонилоксиметил)метан, 1,2,3,4-тетракис(хлоркарбонилокси)бутан, 1,1,1,5,5,5-гекса(хлоркарбонилоксиметил)-3-оксапентан и 1,1,1,5,5,9,9,9-окта (хлоркарбонилоксиметил)-3,7-диоксанонан.
Подходящие поли(галогенформиаты) структуры C включают также трис- и тетракис(хлорформиаты) структур D и Е: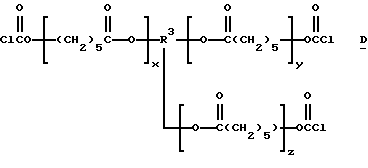
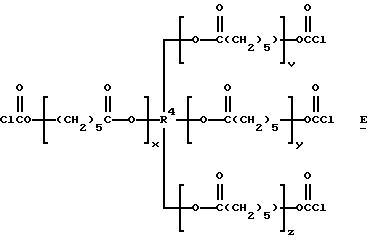
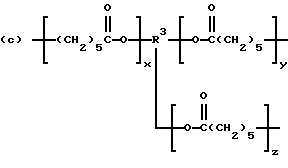
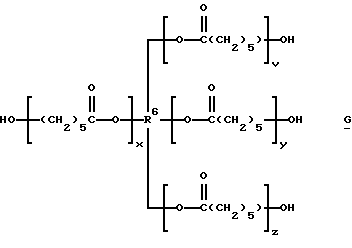
которые являются производными от поликапролактонтриолов и -тетраолов (структуры F и G соответственно):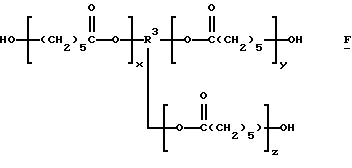
таких, как те, что производит ф. Union Carbide Corporation и продает под торговым знаком TONE®, например TONE® 0301, TONE® 1303, TONE® 0305, TONE® 0310 и TONE® 4411, и трис- и тетракис(хлорформиаты) структур H и I: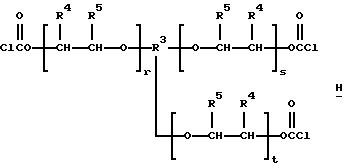
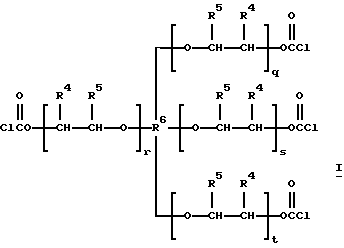
которые являются производными от полиэфиртриолов и -тетраолов (структуры J и K соответственно):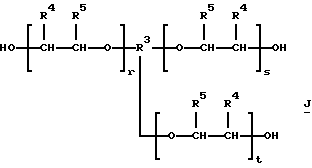
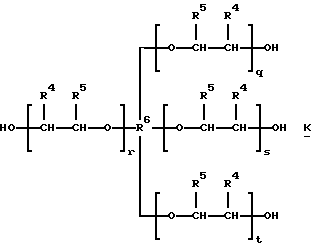
некоторые из которых производит BASF Corporation под торговым знаком PLURACOL®; где R4 представляет метил и R5 представляет водород, например PLURACOL® GP-730, PLURACOL® ТР-740, PLURACOL® PeP 450, PLURACOL®PeP 550 и PLURACOL® PeP 650, и другие, которые производит Dow Chemical Company под торговым знаком VORANOL®, такие, как структура J, где R4 и R5 представляют водород, например VORANOL® 234-630, и еще некоторые другие, которые производит Arco Chemical Company под торговым знаком ARCOL®, такие, как структура J, где R4 представляет метил и R3 представляет водород, например ARCOL® LG-650 и ARCOL® LHT-240. Молекулярные массы полиолов TONE®, PLURACOL®, VORANOL® и ARCOL® указаны производителями и даны в табл.I.
Когда R1 структуры А представляет 1,1,4-триметил-4(трет-бутилперокси)пентильный радикал или 1,1,4-триметил-4(трет-амилперокси)пентильный радикал и n = 2, для получения полипероксидов структуры А можно использовать бис(галогенформиаты), являющиеся производными диолов. Неограничивающие примеры диоловых предшественников бис(галогенформиатов) включают этиленгликоль, 1,2 и 1,3-пропиленгликоли, 2,2-диметил-1,3-пропандиол, 1,4-бутандиол, диэтиленгликоль, триэтиленгликоль, дипропиленгликоль, 1,4-циклогександиметанол, диолы TONE и другие.
Определения R3, R6, q, r, s, t, v, х, у и z даны в разделе Краткое изложение сущности изобретения.
Вышеуказанные поли(галогенформиаты) могут быть получены путем осуществления взаимодействия 0-100% избыточных карбонилдигалогенидов (таких, как дибромид или дихлорид, т.е. фосген) с соответствующим полиолом, т.е. (HO)nR, в присутствии или в отсутствии тетраалкилмочевины (например, тетраметилмочевины) и в присутствии или в отсутствии растворителя до полного завершения реакции. Избыточный карбонилдибромид или фосген удаляют путем отпаривания или путем перегонки. Неограничивающие примеры подходящих полиолов, взаимодействующих с карбонилдигалогенидами с образованием три- и поли(галогенформиатов) структуры С включают 1,1,1-трис(гидроксиметил)этан, 1,1,1-трис(гидроксиметил)пропан, 1,1,1-трис(гидроксиметил)бутан, глицерин, 1,2,3-тригидроксигексан, 1,2,3-тригидроксигептан, 1,2,4-тригидроксибутан, 1,2,6-тригидроксигексан, 1,3,5-тригидроксициклогексан, пентаэритриол, 1,2,3,4-тетрагидроксибутан, 1,1,1,5,5,5-гекса(гидроксиметил)-3-оксапентан, 1,1,1,5,5,9,9,9-окта(гидроксиметил)-3,7-диоксанонан и поликапролактонтриолы и -тетраолы структур F и G соответственно, и полиэфиртриолы и -тетраолы структур J и К соответственно.
В соответствии с другим вариантом новые поли(монопероксикарбонаты) структуры А могут быть получены путем осуществления взаимодействия трет-алкил-пероксигалогенформиатов структуры L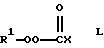
(где X = Br или Cl)
с полиолом, т.е. (HO)nR, в присутствии неорганического или органического основания и необязательно в присутствии одного или нескольких растворителей. Трет-алкилпероксигалогенформиаты структуры L могут быть получены путем осуществления взаимодействия трет-алкилгидропероксида структуры В с избыточным карбонилдигалогенидом (карбонилдибромидом или фосгеном) и удаления избыточного карбонилдигалогенида путем отпарки или перегонки.
Неограничивающие примеры неорганических или органических оснований, необязательных растворителей, полиолов и трет-алкилгидропероксидов перечислены выше. Неограничивающие примеры подходящих трет-алкилпероксигалогенформиатов структуры, L включают трет-бутилпероксихлорформиат, трет-амилпероксихлорформиат, 2-метил-2-пентилпероксихлорформиат, 3-метил-3-пентил-пероксихлорформиат и 3-метил-1-бутин-3-илпероксихлорформкат.
Новые пероксидзамещенные бис(монопероксикарбонаты) структуры А, где R1 выбран из 1,1,4-триметил-4(трет-бутилперокси)-пентильного радикала и 1,1,4-триметил-4(трет-амилперокси)пентильного радикала и n = 2, могут быть получены путем осуществления взаимодействия гидропероксида, выбранного из 1,1,4-триметил-4(трет-бутилперокси)пентилгидропероксида и 1,1,4-триметил-4(трет-амилперокси)пентилгидропероксида, с бис (галогенформиатом) структуры С (где n = 2) при температуре от -30 до 50oС необязательно в присутствии неорганического или органического основания и необязательно в присутствии одного или нескольких растворителей.
Неограничивающие примеры подходящих бис(галоген-формиатов) структуры C (где n = 2), которые могут быть подвергнуты взаимодействию с 1,1,4-триметил-4(трет-бутилперокси)пентилгидропероксидом или 1,1,4-три-метил-4(трет-амилперокси)пентилгидропероксидом, включают 1,2-бис(хлоркарбонилокси)этан, 1,2 и 1,3-бис(хлоркарбонилокси) пропаны, 2,2-диметил-1,3-бис(хлоркарбонилокси)пропан, 1,6-бис(хлоркарбонилокси)гексан, 1,5-бис-(хлоркарбонилокси)-3-оксапентан, 1,4-бис(хлоркарбонилокси)-2-бутен и бис (монопероксикарбонаты) структур НН и II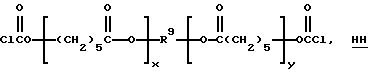
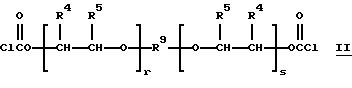
Вышеуказанные бис(галогенформиаты) могут быть получены путем осуществления взаимодействия 0-100% избыточных карбонилдигалогенидов (таких, как дибромид или дихлорид, т.е. фосген) с соответствующим диолом в присутствии или в отсутствии тетраалкилмочевины (например, тетраметилмочевины) и в присутствии или в отсутствие растворителя до полного завершения реакции. Избыточный карбонилдибромид или фосген удаляют путем отпаривания или путем перегонки.
Неограничивающие примеры подходящих диолов, взаимодействующих с карбонилдигалогенидами с образованием бис(галогенформиатов) структуры С (где n = 2), включают 1,2-этандиол, 1,2 и 1, 3-пропандиолы, 1,2-, 1,3 и 1,4-бутандиолы, 2-бутен-1,4-диол, 2 -этил-1,3 гександиол, 2, 2, 4-триметил-1,3-пентандиол, 1,6-гександиол, диэтилен-гликоль, дипропиленгликоль и поликапролактондиолы структуры JJ (диолы TONE®: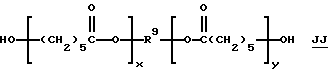
такие, как TONE® 200 и TONEH 210, производства Union Carbide Corporation) и полиалкиленгликоля структуры КК: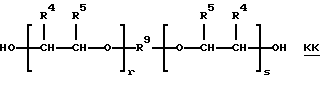
Новые поли(монопероксикарбонаты) структуры А - Иллюстративные примеры
Неограничивающие примеры новых поли(монопероксикарбонатов) структуры А, кроме соединений в рабочих примерах, включают следующие:
1,1,1-трис(трет-амилпероксикарбонилоксиметил)этан,
1,1,1-трис(трет-амилпероксикарбонилоксиметил)пропан,
1,1,1-трис(трет-амилпероксикарбонилоксиметил)бутан,
1,1-бис[2-(трет-амилпероксикарбонилокси)этоксиметил] -1-[2-(трет-бутилпероксикарбонилокси)этоксиметил]пропан,
1-[2-(трет-амилпероксикарбонилокси)этоксиметил] -1,1-бис[2-(трет-бутилпероксикарбонилокси)этоксиметил]пропан,
1,2,3-трис(трет-амилпероксикарбонилокси)пропан, 1,2,3-трис(трет-бутилпероксикарбонилокси)гексан, 1,2,3-трис(трет-бутилпероксикарбонилокси)гептан, 1,2,4-трис(трет-бутилпероксикарбонилокси)бутан, 1,2,6-трис (С трет- бутилпероксикарбонилокси)гексан, 1,3,5-трис(трет-бутилпероксикарбонилокси)циклогексан, тетракис(трет-амилпероксикарбонилоксиметил)метан, 1,2,3,4-тетракис(трет-амилпероксикарбонилокси)бутан, 1,1,1,5,5,5-гекса(трет-бутилпероксикарбонилоксиметил)-3-оксапентан, 1,5-бис[1,1,4-триметил-4-(трет-амилперокси) пентилпероксикарбонилокси 3-3-оксапентан, 1,1,1-трис[1,1,4-триметил-4(трет-бутилперокси)пентилпероксикарбонилоксиметил] пропан, 1,1,1,5,5,9,9,9-окта(трет-бутилпероксикарбонилокси-метил)-3,7-диоксанонан и трис- и тетракис(трет-алкилмонопероксикарбонаты) поликапролактонтриолов и -тетраолов и полиэфиртриолов и -тетраолов, т.е. соединений структур М, N, О и Р соответственно: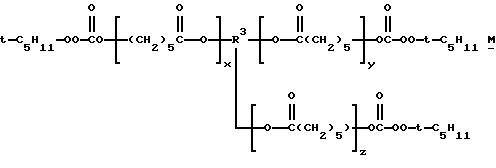
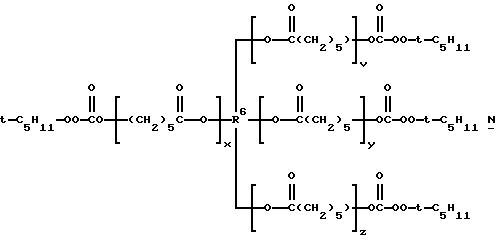
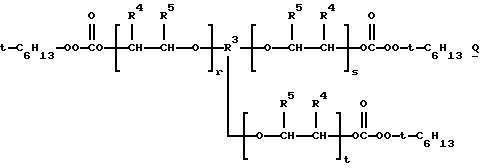
где t-C5H11 представляет трет-амил, t-C6H13 представляет 2-метил-2-пентил или 3-метил-3-пентил и t-C8H17 представляет 2-метил-2-гептил или 1,1,3,3-тетраметилбутил.
Новые поли(монопроксикарбонаты) структуры А - Полезность
А. Полимеризация этиленоненасыщенных мономеров
Как было установлено, в процессах свободно-радикальной полимеризации этиленоненасыщенных мономеров при подходящих температурах и давлениях новые пероксидные соединения структуры А по настоящему изобретению являются эффективными инициаторами в отношении рентабельности (пониженные потребности в инициаторе и т. д.). Этиленоненасыщенные мономеры включают олефины, такие, как этилен, пропилен, стирол, альфа-метилстирол, n-метилстирол, хлорстиролы, бромстиролы, винилбензилхлорид, винилпиридин и дивинилбензол; диолефины, такие, как 1,3-бутадиен, изопрен и хлоропрен; сложные виниловые эфиры, такие, как винилацетат, винилпропионат, виниллаурат, винилбензоат и дивинилкарбонат; ненасыщенные нитрилы, такие, как акрилнитрил и метакрилонитрил; акриловую и метакриловую кислоты и их ангидриды, сложные эфиры и амиды, такие, как ангидрид акриловой кислоты, аллил-, метил-, этил-, н-бутил-, 2-гидроксиэтил, глицидил-, лаурил- и 2-этилгексилакрилаты и метакрилаты и акриламиды и метакриламиды; малеиновый и итаконовый ангидриды; малеиновую, итаконовую и фумаровую кислоты и их эфиры; винилгалогеновые и винилидендигалогеновые соединения, такие, как винилхлорид, винилбромид, винилфторид, винилиденхлорид и винилиденфторид; пергалогенолефины, такие, как тетрафторэтилен, гексафторпропилен и хлортрифторэтилен; простые виниловые эфиры, такие, как метилвиниловый эфир, этилвиниловый эфир и н-бутилвиниловый эфир; сложные аллиловые эфиры, такие, как аллилацетат, аллилбензоат, аллилэтилкарбонат, триаллилфосфат, диаллилфталат, диаллил-фумарат, диаллилглутарат, диаллиладипат, диаллилкарбонат, бис(аллилкарбонат) диэтиленгликоля (т.е. ADC); акролеин; метилвинилкетон или их смеси.
При свободно-радикальной полимеризации с прививкой на полимеры этиленоненасыщенных мономеров при подходящих температурах и давлениях новые пероксидные соединения структуры А также являются эффективными инициаторами в отношении эффективности прививки. Этиленоненасыщенные мономеры включают стироловые мономеры, такие, как стирол, альфа-метилстирол, n-метилстирол, хлорстиролы, бромстиролы и винилбензилхлорид; ненасыщенные нитрилы, такие, как акрилонитрил и метакрилонитрил; сложные эфиры акриловой и метакриловой кислот, такие, как аллил-, метил-, этил-, н-бутил-, 2-гидроксиэтил-, глицидил-, лаурили 2-этилгексилакрилаты и метакрилаты; и малеиновый ангидрид. Прививаемые полимеры включают полибутадиен и полиизопрен. Двумя важными полимерными соединениями, которые получают путем прививки этиленоненасыщенных мономеров на главные цепи полимеров, являются ударопрочный полистирол (HIPS) и акрилонитрил-бутадиен-стирол (ABS, АБС). HIPS получают путем свободно-радикальной прививки стирола на полибутадиен, а ABS получают путем свободно-радикальной прививки акрилонитрила и стирола на полибутадиен. Такие полибутадиен-модифицированные соединения имеют более высокую ударную прочность, чем немодифицированные полимеры.
В традиционный процессах полимеризации и сополимеризации этиленоненасыщенных мономеров и при прививке этиленоненасыщенных мономеров на главные цепи полимеров обычно используют температуры в пределах от 0 до 190oС, предпочтительно от 20 до 175oС, а более предпочтительно от 30 до 160oС, и содержания поли(монопероксикарбонатов) структуры А (в чистом виде) от 0,002 до 10% или более, предпочтительно от 0,005 до 2%, а более предпочтительно от 0,01 до 1% по массе мономера. Новые пероксиды по настоящему изобретению могут быть использованы в сочетании с другими инициаторами свободно-радикальной полимеризации, такими, как 1,5-ди(трет-бутилпероксикарбонилокси)-3-оксапентан, 2,5-диметил-2,5-ди(2-этилгексаноилперокси)гексан, 2,5-диметил-2,5-ди(изопропоксикарбонилперокси)гексан, 2,5-диметил-2-(2-этилгексоксикарбонилперокси)-5-(трет-бутилперокси)гексан, трет-бутилпероксибензоат, трет-амилпероксибензоат, ди-трет-бутилдипероксифталат и некоторые из тех, что перечислены внизу колонки 4 и вверху колонки 5 описания к патенту США 4525308 (выдан Pennwalt Corporation 25 июня 1985 г.). Использование пероксидных соединений по настоящему изобретению в сочетании с указанными инициаторами добавляет гибкости процессам, используемым производителями полимеров, и позволяет им осуществлять "тонкую отладку" процессов полимеризации.
В. Отверждение ненасыщенных сложных полиэфиров
При отверждении ненасыщенных полиэфирных смол путем нагревания при подходящих температурах отверждения в присутствии свободно-радикальных отверждающих агентов новые поли(монопероксикарбонаты) структуры А по настоящему изобретению проявляют повышенную отверждающую активность в отверждаемых ненасыщенных сложных полиэфирах. Ненасыщенные полиэфирные смолы, которые могут быть отверждены новыми поли(монопероксикарбонатами) по настоящему изобретению, обычно содержат ненасыщенный сложный полиэфир и один или несколько этиленоненасыщенных мономеров.
Ненасыщенные сложные полиэфиры представляют собой, например, полиэфиры, полученные путем этерификации по крайней мере одной этиленоненасыщенной ди- или высшей поликарбоновой кислоты (или ангидрида или галогенангидрида кислоты), такой, как малеиновая кислота, фумаровая кислота, глутаконовая кислота, итаконовая кислота, мезаконовая кислота, цитраконовая кислота, аллилмалоновая кислота, тетрагидрофталевая кислота и другие, насыщенными и ненасыщенными ди- или высшими полиолами, такими, как этиленгликоль, диэтиленгликоль, триэтиленгликоль, 1,2 и 1,3-пропандиолы, 1,2-, 1,3- и 1,4-бутандиолы, 2,2-диметил-1,3-пропандиол, 2-гидроксиметил-2-метил-1,3-пропандиол, 2-бутен-1,4-диол, 2-бутин-1,4-диол, 2,4,4-триметил-1,3-пентандиол, глицерин, пентаэритритол, маннит и другие. Можно также использовать смеси таких ди- и высших поликислот и/или смеси таких ди- или высших полиолов. Этиленоненасыщенные ди- или высшие поликарбоновые кислоты могут быть частично заменены насыщенными ди- или поликарбоновыми кислотами, такими, как адипиновая кислота, янтарная кислота, себациновая кислота и другие, и/или ароматическими ди- или высшими поликарбоновыми кислотами, такими, как фталевая кислота, тримеллитовая кислота, пиромеллитовая кислота, изофталевая кислота и терефталевая кислота. Используемые кислоты могут иметь замещающие группы, такие, как галоген. Примерами таких подходящих галогензамещенных кислот являются, например, тетрахлорфталевая кислота, тетрабромфталевая кислота, 5,6-дикарбокси-1,2,3,4,7,7-гексахлорбицикло(2,2,1)-2-гептен и другие.
Другим компонентом ненасыщенной полиэфирной смолы (полимеризуемым мономером или мономерами) могут предпочтительно быть этиленоненасыщенные мономеры, такие, как стирол, альфа-метилстирол, n-метилстирол, хлорстиролы, бромстиролы, винилбензилхлорид, дивинилбензол, диаллилмалеат, дибутилфумарат, триаллилфосфат, триаллилцианурат, диаллилфталат, диаллилфумарат, метилакрилат, метилметакрилат, н-бутилакрилат, н-бутилметакрилат, этилакрилат и другие или их смеси, которые известны в данной области техники как сополимеризуемые с указанными ненасыщенными полиэфирами. Предпочтительная ненасыщенная полиэфирная смола содержит в качестве ненасыщенного полиэфирного компонента продукт этерификации 1,2-пропандиола (полиола), малеинового ангидрида (ангидрида ненасыщенной поликарбоновой кислоты) и фталевого ангидрида (ангидрида ароматической дикарбоновой кислоты), а также мономерный компонент - стирол.
Другие типы ненасыщенных полиэфирных смол могут быть отверждены с использованием новых пероксидных соединений по настоящему изобретению в качестве катализатора отверждения. Эти смолы, называемые ненасыщенными полимерами сложных виниловых эфиров, состоят из винилэфирной смолы и одного или нескольких способных полимеризоваться мономерных компонентов. Винилэфирный полимерный компонент может быть получен путем осуществления взаимодействия хлорэпоксида, такого, как эпихлоргидрин, с соответствующим количеством бисфенола, такого, как Бисфенол А (т.е. 2,2-(4-гидроксифенил)пропан), в присутствии основания, такого, как гидроксид натрия, с получением продукта конденсации, имеющего концевые эпоксигруппы, произведенные от хлорэпоксида. Последующее взаимодействие продукта конденсации с полимеризуемыми ненасыщенными карбоновыми кислотами, такими, как акриловая и метакриловая кислоты, в присутствии или в отсутствие кислотных или основных катализаторов приводит к образованию винилэфирного полимерного компонента. Для завершения получения ненасыщенного полимера сложных виниловых эфиров обычно добавляют стирол в качестве полимеризуемого мономерного компонента.
Обычно используют температуры в пределах примерно от 20 до 200oС и содержания новых поли(монопероксикарбонатов) структуры А примерно от 0,05 до 5% или более предпочтительно от 0,10 до 4%, а более предпочтительно от 0,25 до 3% по массе отверждаемой ненасыщенной полиэфирной смолы.
Описанные выше ненасыщенные полиэфирные смолы могут быть наполнены различными материалами, такими, как сера, стекло, углеродные и борные волокна, углеродные сажи, кремнеземы, силикаты металлов, глины, карбонаты металлов, антиоксиданты (АО), стабилизаторы против действия тепла, ультрафиолетового излучения (УФ) и света, сенсибилизаторы, красители, пигменты, ускорители, оксиды металлов, такие, как оксид цинка, порообразователи, зародышеобразователи и другие.
С. Отверждение аллилдигликолькарбонатных (ADC, АДК) смол
При отверждении или полимеризации бис(аллилкарбоната) диэтиленгликоля (ADC)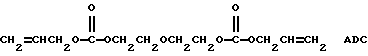
путем нагревания ADC-мономера при подходящих температурах отверждения в присутствии свободно-радикальных отверждающих агентов новые поли(монопероксикарбонаты) структуры А по настоящему изобретению проявляют повышенную отверждающую или полимеризующую активность по отношению к ADC-мономерам. Промышленное производство ADC осуществляет Pittsburgh Plate Glass Company (PPG) в виде мономера CR-39 (CAS per. N 142-22-3), который получают путем осуществления взаимодействия бис(хлорформиата) диэтиленгликоля с аллиловым спиртом в присутствии щелочи (R. Dowbenco, Издат. J.I. Kroschwitz and M. Howe-Grant, Kirk-Othmer Encyclopedia of Chemical Technology, "Allyl Monomers and Polymers", четвертое издание, том 2, Willey-Interscience Publication, John Willey & Sons, Inc., Нью-Йорк, 1992, с. 163-168). ADC-мономер может быть отвержден или полимеризован один или с другими сомономерами, такими, как сложные эфиры акриловой кислоты, сложные эфиры метакриловой кислоты, сложные аллиловые эфиры, диаллилдикарбоксилаты (например, ди-аллилфталат), малеиновый ангидрид и другие мономеры, для получения светлых отливок или линз, прозрачных, ударно-вязких, прочных на разрыв и стойких к растворителям. Отверждение или полимеризацию ADC-мономеров осуществляют в массе (без растворителя). Обычно отверждение или полимеризацию ADC-мономеров для формования литых листов или линз осуществляют в две стадии. Первая стадия включает основную часть полимеризации и проводится в присутствии отверждающего инициатора при температуре от 35 до 150oС. Время отверждения или полимеризации на первой стадии колеблется в пределах от примерно 5 до 50 ч. Вторая стадия отверждения или полимеризации ADC-мономеров включает доотверждение или отжиг ADC-смолы в течение одного или нескольких часов при 100-170oС.
Новые поли(монопероксикарбонаты) структуры А обычно используют в количестве примерно от 1 до 6% или более, предпочтительно 2-5%, более предпочтительно 2,5-4% по массе отвержденного или полимеризуемого ADC-мономера.
Описанные выше ADC-смолы могут быть наполнены различными материалами, такими, как антиоксиданты (АО), стабилизаторы против действия тепла, ультрафиолетового излучения (УФ) и света, тональные пигменты, фотохромные добавки и красители. Кроме того, ADC-смолы могут содержать такие добавки, как акриловые полимеры и противоусадочные низкомолекулярные акриловые смолы, раскрытые в патенте США 4217433 (выдан Pecnnwalt Corporation 12 августа 1980 г. ). Такие противоусадочные добавки используют для противодействия усадке, имеющей место при полимеризации ADC-мономера.
D. Отверждение эластомеров и сшивание термопластичных полимеров
При отверждении эластомерных соединений и сшивании полимерных соединений путем нагревания при подходящих температурах отверждения и сшивания в присутствии свободно-радикальных отверждающих и сшивающих агентов новые поли(монопероксикарбонаты) структуры А по настоящему изобретению проявляют отверждающую и сшивающую активности.
Эластомерные смолы, которые могут быть отверждены с помощью новых поли(монопероксикарбонатов) по настоящему изобретению, включают эластомеры, такие, как этилен-пропиленовые сополимеры (EPR), этилен-пропилен-диеновые терполимеры (EPDM), полибутадиен (PBD), силоксановый каучук (SR), нитрильный каучук (NR), неопрен, фторэластомеры и этилен-винилацетатный сополимер (EVA).
Полимерные соединения, которые могут быть сшиты новыми поли(монопероксикарбонатами) по настоящему изобретению, включают олефиновые термопласты, такие, как хлорированный полиэтилен (СРЕ), полиэтилен низкой плотности (LDPE), линейный полиэтилен низкой плотности (LLDPE) и полиэтилен высокой плотности (HDPE). Другие сшиваемые термопластические полимеры включают поливинилхлорид (PVC), полистирол, поли(винилацетат), полиакрилаты, сложные полиэфиры, поликарбонаты и т.д.
Обычно используют температуры в пределах примерно от 80 до 310oС и содержания новых поли(монопероксикарбонатов) примерно от 0,1 до 10%, предпочтительно от 0,5 до 5%, а более предпочтительно от 0,5 до 3% по массе отверждаемой эластомерной смолы или сшиваемого олефинового полимера.
Отверждаемая эластомерная смола или сшиваемый полимер могут быть, хотя и необязательно, наполнены материалами, перечисленными выше для применения с традиционными ненасыщенными сложными полиэфирами.
Е. Модифицирование полиолефинов и других полимеров
В процессах модифицирования полиолефинов (например, выгодная деструкция полипропилена (РР) путем уменьшения молекулярной массы полимера и уменьшения молекулярно-массового распределения РР и улучшения молекулярно-массовых и пленкообразующих свойств линейного полиэтилена низкой плотности (LLDPE)) и сополимеров новые поли-(монопероксикарбонаты) структуры А по настоящему изобретению проявляют модифицирующую по отношению к полиолефинам активность. Другие полимеры, которые могут быть модифицированы трис- и поли(монопероксикарбонатами), включают полиэтилен высокой плотности (HDPE), этилен-пропиленовый сополимер и т.д.
Обычно используют температуры в пределах примерно от 140 до 340oС и содержания поли(монопероксикарбонатов) примерно от 0,001 до 1,0%, предпочтительно от 0,01 до 1,0%, а более предпочтительно от 0,01 до 0,5% по массе модифицируемых полиолефинов или сополимеров. В качестве сокатализатора модифицирования можно, хотя и не обязательно, использовать молекулярный кислород в количестве до 1% по массе.
Новые поли(монопероксикарбонаты) структуры А - Примеры получения и полезности
Следующие далее примеры дополнительно иллюстрируют наилучшие способы осуществления настоящего изобретения и представлены для подробного описания способов получения и полезности предлагаемых соединений и их не следует считать ограничивающими объем изобретения.
Пример 1. Получение 1,1,1-трис(трет-бутилпероксикарбонилоксиметил)этана (I-1)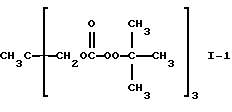
В этом примере продукт получали в две стадии синтеза. На первой стадии 1,1,1-трис(гидроксиметил)этан (0,15 моль) подвергали взаимодействию с избыточным фосгеном (0,85 моль) в 175 мл 1,4-диоксана при 0-8oС. Для подавления образования циклического карбоната прибавляли 1,1,3,3-тетраметилмочевину (0,4 г). По окончании реакции избыточный фосген и растворитель отгоняли из продукта при 15-30oС и пониженном давлении с получением 1,1,1-трис(хлоркарбонилоксиметил)этана в виде жидкости с содержанием продукта по результатам анализа 89,3% и скорректированным выходом 74,6%.
На второй стадии 1,1,1-трис(хлоркарбонилоксиметил)этан подвергали взаимодействию с трет-бутилгидропероксидом в присутствии водного раствора гидроксида калия и поверхностно-активного вещества и в результате получили продукт так, как описано ниже:
В реактор емкостью 300 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 70,0 г (0,25 моль) 20% водного раствора гидроксида калия, 25 г (0,25 моль) 90,2% трет-бутилгидропероксида и 10 капель TERGITOL® NP-10 [смесь поверхностно-активных веществ, содержащих поли(окси-1,2-этандиил), α-(4-нонилфенил)-ω-гидрокси-; 26027-38-3 в регистре САS, и поли (окси-1,2-этандиил), α-гидро-ω-гидрокси-; 25322-68-3 в регистре САS; производство Union Carbide], и полученный раствор перемешивали при 25oС в течение 10 мин. К перемешанной смеси при 22-29oС медленно прибавляли 17,2 г (0,05 моль) 89,3% 1,1,1-трис(хлоркарбонилоксиметил)этана в течение 25 мин. По окончании прибавления реакционную массу перемешивали 3 ч при 30-35oС, после чего прибавляли 150 мл МТВЕ (метил трет-бутиловый эфир), и реакционную массу перемешивали одну минуту при 30-35oС. Затем нижний водный слой отделяли, и органический слой охлаждали до 17oС и промывали 100 мл 10% водного раствора гидроксида калия. Затем органический слой промывали три раза 50 мл порциями 10% водного раствора гидросульфита натрия, потом 100 мл 10% водного раствора гидроксида натрия и затем насыщенным водным раствором сульфата натрия до рН 7-8. Продуктный раствор сушили над 5% по массе безводного МgS04 и, отфильтровав отработанный осушитель, удаляли в вакууме растворитель с получением в остатке 7,4 г (31,6% теоретически, без поправки) белого твердого вещества, т.пл. = 55-60oС. Инфракрасный (ИК) спектр продукта содержал основную полосу поглощения карбонила монопероксикарбоната при 1790 см-1 и основную карбонильную полосу карбоната примерно при 1755 см-1. В ИК-спектре не было полосы поглощения гидроксида (ОН). Продукт содержал 9,42% активного кислорода(теоретически 10,25%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 91,9%, и скорректированный выход составлял 29,1%.
Согласно способу получения, данным выхода и данных ИК-спектра, продукт, полученный при этой реакции, был указанным в заголовке целевым продуктом.
Пример 2. Получение 1,1,1-трис(трет-бутилпероксикарбонилоксиметил)пропана (I-2)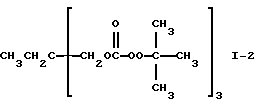
В этом примере продукт получали в две стадии синтеза. На первой стадии 1,1,1-трис(гидроксиметил)пропан (0,10 моль) подвергали взаимодействию с избыточным фосгеном (0,60 моль) в 200 мл 1,4-диоксана при 2-8oС. Для подавления образования циклического карбоната прибавляли 1,1,3,3-тетраметилмочевину (0,3 г). По окончании реакции избыточный фосген и растворитель отгоняли из продукта при 20-30oС и пониженном давлении с получением 1,1,1-трис(хлоркарбонилоксиметил) пропана в виде жидкости с содержанием продукта по результатам анализа 87,7% и скорректированным выходом 95,6%.
На второй стадии 1,1,1-трис(хлоркарбонилоксиметил)пропан подвергали взаимодействию с трет-бутилгидропероксидом в присутствии водного раствора гидроксида калия и поверхностно-активного вещества, и в результате получили продукт так, как описано ниже:
В реактор емкостью 300 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 56,0 г (0,20 моль) 20% водного раствора гидроксида калия и 19,5 г (0,20 моль) 92% трет-бутилгидропероксида, и полученный раствор перемешивали при примерно 25oС. К перемешанной смеси при 23-31oС медленно прибавляли раствор 18,3 г (0,05 моль) 87,7% 1,1,1-трис(хлоркарбонилоксиметил)пропана и 50 мл МТВЕ в течение 30 мин. По окончании прибавления реакционную массу перемешивали 3 ч при 30-32oС, после чего прибавляли 50 мл МТВЕ, и реакционную массу перемешивали одну минуту при 30-32oС. Затем нижний водный слой отделяли, и органический слой охлаждали до 12oС и промывали 50 мл 10% водного раствора гидросульфита натрия. Затем полученный органический слой промывали два раза 50 мл порциями 3% водного раствора гидрокарбоната натрия. Продуктный раствор сушили над 5% по массе безводного МgS04 и, отфильтровав отработанный осушитель, удаляли в вакууме растворитель с получением в остатке 10,8 г (44,8% теоретически, без поправки) прозрачной бесцветной жидкости. ИК-спектр продукта содержал основную полосу поглощения карбонила монопероксикарбоната при 1792 см-1 и основную карбонильную полосу карбоната примерно при 1767 см-1. В ИК-спектре был лишь след полосы поглощения ОН. Продукт содержал 8,65% активного кислорода (теоретически 9,95%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 86,9%, и скорректированный выход составлял 38,9%.
Согласно способу получения, данным выхода и данным ИК-спектра, продукт, полученный при этой реакции, был указанным в заголовке целевым продуктом.
Пример 3. Получение поликапролактон-трис(моно-трет-бутилпероксикарбоната) (I-3)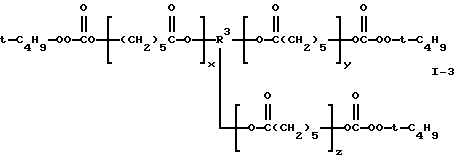
где сумма х, у и z равна примерно 2 и R3 представляет трирадикал.
В этом примере продукт получали в две стадии синтеза. На первой стадии 0,12 моль поликапролактонтриола (С-1) (TONE 0301; молекулярная масса = 300; производство Union Cabride Corp.):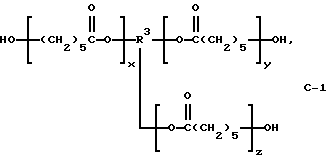
где сумма х, у и z равна примерно 2 и R3 представляет трирадикал, подвергали взаимодействию с избыточным фосгеном (0,60 моль) при 5-10oС. По окончании реакции избыточный фосген отгоняли из продукта при 15-25oС и пониженном давлении с получением поликапролактон-трис(хлорформиата) в виде светло-розовой вязкой жидкости с содержанием продукта по результатам анализа 91,0% и скорректированным выходом 84,2%.
На второй стадии поликапролактон-трис(хлорформиат) подвергали взаимодействию с трет-бутилгидропероксидом в присутствии водного раствора гидроксида калия, и в результате получили продукт так, как описано ниже:
В реактор емкостью 250 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 28,0 г (0,10 моль) 20% водного раствора гидроксида калия, 12,9 г (0,10 моль) водного 70% трет-бутилгидропероксида и 3 капли (приблизительно 0,1 г) TERGITOL NP-10 при 20-30oС. Полученный раствор перемешивали при примерно 25oС. К перемешанному раствору при 23-29oС медленно прибавляли 16,1 г (0,03 моль) 91,0% поликапролактон-трис(хлорформиата) в течение 20 мин. Для обеспечения хорошего перемешивания прибавляли примерно 50 мл МТВЕ. По окончании прибавления реакционную массу перемешивали 3 часа при 30oС, еще прибавляя при этом МТВЕ (50-60 мл). Затем реакционной массе давали разделиться на жидкие фазы. После этого нижний водный слой отделяли, и оставшийся органический слой охлаждали до 15oС и промывали 50 мл 10% водного раствора гидросульфита натрия, а потом промывали 50 мл 10% водного раствора гидроксида калия и 50 мл порциями насыщенного водного раствора сульфата натрия до получения рН 7-8. Продуктный раствор сушили над 5% по массе безводного MgSO4 и, отфильтровав отработанный осушитель, удаляли в вакууме растворитель с получением в остатке 19,6 г (приблизительно 100% теоретически, без поправки) бесцветной жидкости. ИК-спектр продукта содержал основную полосу поглощения карбонила монопероксикарбоната при 1785 см-1 и основную карбонильную полосу карбоната или сложного эфира примерно при 1740 см-1. В ИК-спектре не было полосы поглощения ОН. Продукт содержал 6,69% активного кислорода (теоретически 7,40%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 90,4%, и скорректированный выход составлял 91,3%.
Пример 4. Получение поликапролактон-трис(моно-трет-бутилпероксикарбоната) (I-4)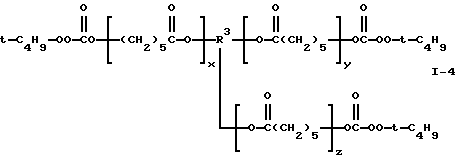
где сумма х, у и z равна примерно 4 и R3 представляет трирадикал.
В этом примере продукт получали в две стадии синтеза. На первой стадии 0,05 моль поликапролактонтриола (С-2) (TONE 0305; молекулярная масса = 540; производство Union Carbide Corp.)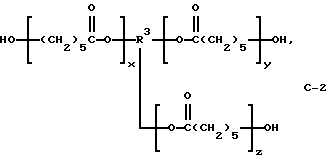
где сумма х, у и z равна примерно 4 и R3 представляет трирадикал, подвергали взаимодействию с избыточным фосгеном (0,45 моль) при 3-7oС. По окончании реакции избыточный фосген отгоняли из продукта при 15-25oС и пониженном давлении с получением поликапролактон-трис(хлорформиата) в виде светло-розовой жидкости с содержанием продукта по результатам анализа 97,9% и скорректированным выходом 93,3%.
На второй стадии поликапролактон-трис(хлорформиат) подвергали взаимодействию с трет-бутилгидропероксидом в присутствии водного раствора гидроксида калия, и в результате получили продукт так, как описано ниже.
В реактор емкостью 200 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 15,7 г (0,07 моль) 25% водного раствора гидроксида калия и 9,0 г (0,07 моль) 70% водного раствора трет-бутилгидропероксида. Полученный раствор перемешивали при примерно 25oС. К перемешанному раствору при 24-28oС медленно прибавляли 14,8 г (0,02 моль) 97,9% поликапролактон-трис(хлорформиата) в течение 15 мин. По окончании прибавления реакционную массу перемешивали 3,5 ч при 28-32oС, после чего прибавляли 80 мл МТВЕ, и реакционную массу перемешивали одну минуту при 28-32oС и затем давали разделиться. После этого нижний водный слой отделяли и оставшийся органический слой охлаждали до 15oС и промывали 25 мл 10% водного раствора гидросульфита натрия. Затем полученный органический слой промывали 25 мл 10% водного раствора гидроксида калия и 50 мл порциями насыщенного водного раствора сульфата натрия до получения рН 7-8. Продуктный раствор сушили над 5% по массе безводного MgSO4 и, отфильтровав отработанный осушитель, удаляли в вакууме растворитель с получением в остатке 17,4 г (98% теоретически, без поправки) бесцветной жидкости. ИК-спектр продукта содержал основную полосу поглощения карбонила монопероксикарбоната при 1785 см-1 и основную карбонильную полосу карбоната или сложного эфира примерно при 1730 см-1. В ИК-спектре был лишь след полосы поглощения ОН. Продукт содержал 5,00% активного кислорода (теоретически 5,40%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 92,6%, и скорректированный выход составлял 90,7%.
Согласно способу получения, данным выхода и данным ИК-спектра, продукт, полученный при этой реакции, был указанным в заголовке целевым продуктом.
Пример 5. Получение 1,1,1-трис[2-(трет-бутилпероксикарбонилокси)этоксиметил]пропан (I-5)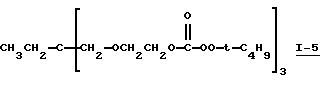
В этом примере продукт (т.е. 1,1,1-трис[2-(трет-бутилпероксикарбонилокси)этоксиметил] пропан I-5) получали в две стадии синтеза. На первой стадии 0,15 моль простого полиэфиртриола (т.е. 1,1,1-трис[2-(гидроксиэтоксиметил] пропана, С-3) (коммерческий триоловый продукт VORANOL 234-630; молекулярная масса = 267; производство Dow Chemical)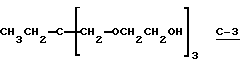
подвергали взаимодействию с избыточным фосгеном (0,65 моль) при 3-7oС. Затем реакционную смесь перемешивали 4 ч при 0-10oС и давали отстояться в течение ночи при 20-25oС. Избыточный фосген отгоняли из продукта при 20-25oС и пониженном давлении в течение 5 ч с получением полиэфир-трис (хлорформиата) в виде прозрачной вязкой жидкости с содержанием продукта по результатам анализа 97,4% и скорректированным выходом 94,8%.
На второй стадии полиэфир-трис(хлорформиат) подвергали взаимодействию с трет-бутилгидропероксидом в присутствии водного раствора гидроксида калия, и в результате получили продукт так, как описано ниже:
В реактор емкостью 200 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 29,4 г (0,105 моль) 20% водного раствора гидроксида калия, 13,5 г (0,105 моль) водного 70% трет-бутилгидропероксида и 3 капли (приблизительно 0,1 г) TERGITOL NP-10 при 20-30oС. Полученный раствор перемешивали при примерно 25oС. К перемешанному раствору при 23-29oС медленно прибавляли раствор, состоявший из 14,0 г (0,03 моль) 97,4% полиэфир-трис(хлорформиата) и 20 мл МТВЕ в течение 15 мин. По окончании прибавления реакционную массу перемешивали 2,5 ч при 30oС, после чего прибавляли 80-90 мл МТВЕ, и реакционную массу перемешивали одну минуту при 30oС и затем давали разделиться на жидкие фазы. После этого нижний водный слой отделяли, и оставшийся органический слой охлаждали до 15oС и промывали 50 мл 10% водного раствора гидроксида калия. Неочищенный продуктный раствор промывали затем 50 мл 10% водного раствора гидросульфита натрия. Полученный органический слой потом промывали 50 мл насыщенного водного раствора гидрокарбоната калия. Далее органический слой промывали 50 мл насыщенного водного раствора сульфата натрия до получения рН, равного примерно 7. Продуктный раствор сушили над 5% по массе безводного MgSO4 и, отфильтровав отработанный осушитель, удаляли в вакууме растворитель с получением в остатке 18,3 г (приблизительно 100% теоретически, без поправки) бесцветного жидкого продукта. ИК-спектр продукта содержал основную полосу поглощения карбонила монопероксикарбоната при 1785 см-1 и основную полосу поглощения карбоната примерно при 1735 см-1. В ИК-спектре не было полосы поглощения ОН. Продукт содержал 7,52% активного кислорода (теоретически 7,81%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 94,2%, и скорректированный выход составлял 93,7%.
Согласно способу получения, данным выхода и данным ИК-спектра продукт, полученный при этой реакции, был указанным в заголовке целевым продуктом.
Пример 6. Получение поликапролактон-тетракис(моно-трет-бутилпероксикарбоната) (I-6)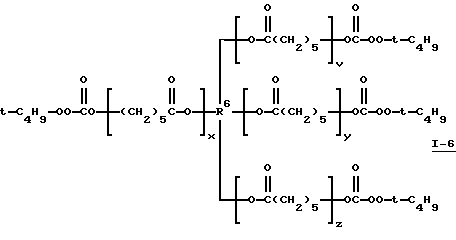
где сумма v, x, у и z равна примерно 8 и R6 представляет тетрарадикал.
В этом примере продукт получали в две стадии синтеза. На первой стадии 0,03 моль поликапролактонтетраола (С-4) (экспериментальный капролактоновый олигомерный тетраол TONE 4411; молекулярная масса = 1006; производство Union Carbide Corp.)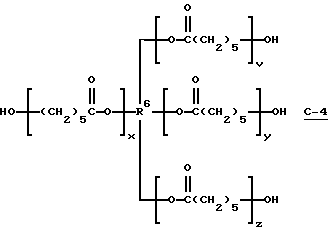
где сумма v, х, у и z равна примерно 8 и R6 представляет тетрарадикал, подвергали взаимодействию с избыточным фосгеном (0,35 моль) при 3-7oС. Реакционную смесь затем перемешивали 5 ч при 10-20oС и давали ей отстояться в течение ночи при 20-25oС. Затем избыточный фосген отгоняли из продукта при 20-25oС и пониженном давлении в течение 5 часов с получением поликапролактон-тетракис(хлорформиата) в виде прозрачной вязкой жидкости с содержанием продукта по результатам анализа 97,3% и скорректированным выходом 91,9%.
На второй стадии поликапролактон-тетракис(хлорформиат) подвергали взаимодействию с трет-бутилгидропероксидом в присутствии водного раствора гидроксида калия, и в результате получили продукт так, как описано ниже:
В реактор емкостью 200 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 11,2 г (0,05 моль) 25% водного раствора гидроксида калия и 6,4 г (0,05 моль) водного 70% трет-бутилгидропероксида при 20-30oС. Полученный раствор перемешивали при примерно 25oС. К перемешанному раствору при 24-31oС медленно прибавляли раствор, состоявший из 12,9 г (0,01 моль) 97,3% поликапролактон-тетракис(хлорформиата) и 30 мл МТВЕ в течение 15 мин. По окончании прибавления реакционную массу перемешивали 3 ч при 30-35oС, после чего прибавляли 70 мл МТВЕ, и реакционную массу перемешивали одну минуту при 30-35oС и затем давали ей разделиться. После этого нижний водный слой отделяли, и органический слой охлаждали до 15oС и промывали 50 мл 10% водного раствора гидроксида калия. Затем неочищенный продуктный раствор промывали 50 мл 10% водного раствора гидросульфита натрия. Далее полученный органический слой промывали 10% водным раствором гидрокарбоната калия до получения рН, равного примерно 7. Полученный раствор сушили над 5% по массе безводного МgSO4 и, отфильтровав отработанный осушитель, удаляли в вакууме растворитель с получением в остатке 14,9 г (приблизительно 100% теоретически, без поправки) вязкой бесцветной жидкости. ИК-спектр продукта содержал основную полосу поглощения карбонила монопероксикарбоната при 1785 см-1 и основную карбонильную полосу карбоната или сложного эфира примерно при 1730 см-1. В ИК-спектре не было полосы поглощения ОН. Продукт содержал 3,73% активного кислорода (теоретически 4,35%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 85,7%, и скорректированный выход составлял 86,9%.
Согласно способу получения, данным выхода и данным ИК-спектра, продукт, полученный при этой реакции, был указанным в заголовке целевым продуктом.
Пример 7. Получение полиэфир-тетракис(моно-трет-бутилпероксикарбоната) (I-7)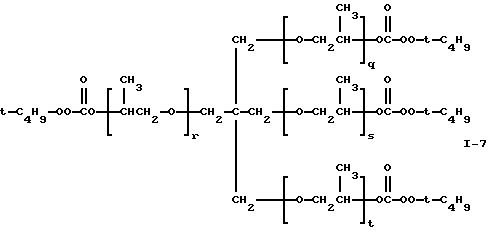
где сумма q, r, s и t равна примерно 6-7.
В этом примере продукт получали в две стадии синтеза. На первой стадии 0,075 моль полиэфиртетраола (С-5) (PLURACOL РеР 550; молекулярная масса = 500; производство BASF Corporation)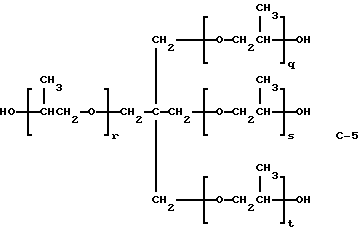
где сумма q, r, s и t равна примерно 6-7, подвергали взаимодействию с избыточным фосгеном (0,60 моль) при 3-7oС. Затем реакционную смесь перемешивали 2-3 ч при 1-20oС и давали ей отстояться всю ночь при 20-25oС. Далее избыточный фосген отгоняли из продукта при 20-30oС и пониженном давлении с получением полиэфир-тетракис(хлорформиата) в виде прозрачной жидкости с содержанием продукта по результатам анализа 100% и скорректированным выходом 97,4%.
На второй стадии полиэфир-тетракис(хлорформиат) подвергали взаимодействию с трет-бутилгидропероксидом в присутствии водного раствора гидроксида калия и в результате получили продукт так, как описано ниже.
В реактор емкостью 250 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 29,2 (0,13 моль) 25% водного раствора гидроксида калия и 16,7 г (0,13 моль) 70% водного раствора трет-бутилгидропероксида при 22-29oС. Полученный раствор перемешивали при примерно 25oС. К перемешанному раствору при 23-28oС медленно прибавляли 18,8 г (0,025 моль) 100% полиэфир-тетракис(хлорформиата) в течение 15 мин. По окончании прибавления реакционную массу перемешивали 3 ч при 25-30oС, после чего прибавляли 100 мл МТВЕ, и реакционную массу перемешивали одну минуту при примерно 30oС и затем давали разделиться на жидкие фазы. После этого нижний водный слой отделяли, и оставшийся органический слой охлаждали до 12oС и промывали 50 мл 10% водного раствора гидросульфита натрия, а затем 50 мл 10% водного раствора гидроксида калия и 50 мл порциями насыщенного водного раствора сульфата натрия до получения рН 7-8. Продуктный раствор сушили над 5% по массе безводного МgSO4 и, отфильтровав отработанный осушитель, удаляли в вакууме растворитель с получением в остатке 22,4 г (92,9% теоретически, без поправки) бесцветной жидкости. ИК-спектр продукта содержал основную полосу поглощения карбонила монопероксикарбоната при 1785 см-1 и основную карбонильную полосу карбоната или сложного эфира примерно при 1752 см-1. В ИК-спектре был лишь след полосы поглощения ОН. Продукт содержал 6,16% активного кислорода (теоретически 6,64%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 92,8%, и скорректированный выход составлял 86,3%.
Согласно способу получения, данным выхода и данным ИК-спектра, продукт, полученный при этой реакции, был указанным в заголовке целевым продуктом.
Пример 8. Получение поликапролактон-бис(моно-трет-бутилпероксикарбоната) (А-1)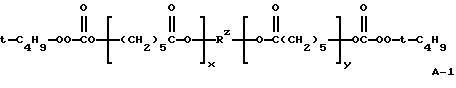
где сумма х и у равна примерно 4 и R2 представляет бирадикал.
В этом примере продукт получали в две стадии синтеза. На первой стадии 0,03 моль поликапролактодиола (С-6) (диол TONE® 0200; молекулярная масса = 530; производство Union Carbide Corp.)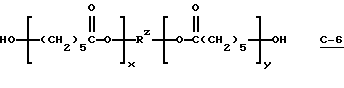
где сумма х и у равна примерно 4 и R3 представляет бирадикал, подвергали взаимодействию с избыточным фосгеном в соответствии с описанным выше процессом. В результате получили поликапролактон-бис(хлорформиат) в виде розовой вязкой жидкости с содержанием продукта по результатам анализа 100%.
На второй стадии поликапролактон-бис(хлорформиат) подвергали взаимодействию с трет-бутилгидропероксидом в присутствии водного раствора гидроксида калия и в результате получили продукт так, как описано ниже:
В реактор емкостью 400 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 14,9 г (0,12 моль) 45% водного раствора гидроксида калия, 10,0 г воды и 14,1 г (0,11 моль) водного 70% трет-бутилгидропероксида при 20-30oС. Полученный раствор перемешивали при примерно 25oС. К перемешанному раствору при 23-31oС медленно прибавляли 32,7 г (0,05 моль) 100% поликапролактон-бис(хлорформиата) в течение 25 мин. По окончании прибавления прибавляли 75 мл МТВЕ, и реакционную массу перемешивали примерно 2 ч при 30±2oС, после чего прибавляли еще 125 мл МТВЕ, и реакционную массу перемешивали одну минуту при 30oС и затем давали разделиться на жидкие фазы. После этого нижний водный слой отделяли и оставшийся органический слой охлаждали до 15oС и промывали 50 мл 10% водного раствора гидросульфита натрия при 15-25oС. Разделение полученной массы на две жидкие фазы было очень медленным. Прибавление сульфита натрия ускорило разделение на фазы. Нижнюю водную фазу отделяли и выливали. После этого верхний органический раствор промывали два раза 50 мл порциями 20% водного раствора гидроксида калия при 20-30oС. Полученный органический слой промывали затем насыщенным водным раствором сульфата натрия до получения рН, равного примерно 7. Органический продуктный раствор сушили над 5% по массе безводного MgS04 и, отфильтровав отработанный осушитель, удаляли в вакууме растворитель с получением в остатке 33,8 г (приблизительно 89% теоретически, без поправки) вязкой бесцветной жидкости. ИК-спектр продукта содержал основную полосу поглощения карбонила монопероксикарбоната при 1785 см-1 и основную карбонильную полосу карбоната или сложного эфира примерно при 1731 см-1. В ИК-спектре не было полосы поглощения ОН. Продукт содержал 3,97% активного кислорода (теоретически 4,20%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 94,5%, и скорректированный выход составлял 84,1%.
Согласно способу получения, данным выхода и данным ИК-спектра, продукт, полученный при этой реакции, был указанным в заголовке целевым продуктом.
Пример 9. Получение 1,1,1-трис[2-(трет-амилперокси-карбонилокси)этоксиметил]пропана (I-8)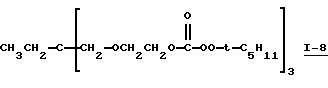
В этом примере продукт (т.е. 1,1,1-трис[2-(трет-амилпероксикарбонилокси)этоксиметил] пропан I-8) получали путем осуществления взаимодействия полиэфир-трис(хлорформиата) из VORANOL 234-630 (пример 5), трет-амилгидропероксида и водного раствора гидроксида калия так, как описано ниже:
В реактор емкостью 200 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 19,6 (0,070 моль) 20% водного раствора гидроксида калия, 8,0 г (0,070 моль) 91% трет-амилгидропероксида и 3 капли (приблизительно 0,1 г) TERGITOL NP-10 при примерно 20-25oС. Полученный раствор перемешивали при температуре 25oС. К перемешанному раствору при 24-32oС медленно прибавляли 9,3 г (0,020 моль) 98,7% полиэфир-трис(хлорформиата) (из VORANOL 234-630) в течение 15 мин. Во время прибавления добавляли 50 мл МТВЕ. Затем реакционную массу перемешивали 3,0 часа при примерно 30oС. В конце периода реакции прибавляли еще 50 мл МТВЕ, и реакционную массу перемешивали еще две минуты и затем давали ей разделиться на жидкие фазы. После этого нижний водный слой отделяли, и оставшийся органический слой охлаждали до 20oС и промывали 50 мл 20% водного раствора гидроксида калия. Неочищенный продуктный раствор промывали затем 50 мл 15% водного раствора гидросульфита натрия. Полученный органический слой потом промывали насыщенным водным раствором гидрокарбоната натрия до получения рН, равного примерно 7. Продуктный раствор сушили над 5% по массе безводного МgS04 и, отфильтровав отработанный осушитель, удаляли в вакууме растворитель с получением в остатке 10,8 г (приблизительно 82,2% теоретически, без поправки) бесцветного жидкого продукта. ИК-спектр продукта содержал основную полосу поглощения карбонила монопероксикарбоната при 1785 см-1 и основную полосу поглощения карбоната при 1753 см-1. В ИК-спектре была небольшая полоса поглощения ОН. Продукт содержал 7,52% активного кислорода (теоретически 7,31%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 79,9%, и скорректированный выход составлял 65,7%.
Согласно способу получения, данным выхода и данным ИК-спектра, продукт, полученный при этой реакции, был указанным в заголовке целевым продуктом.
Пример 10. Получение полиэфир-тетракис(моно-трет-амилпероксикарбоната) (I-9)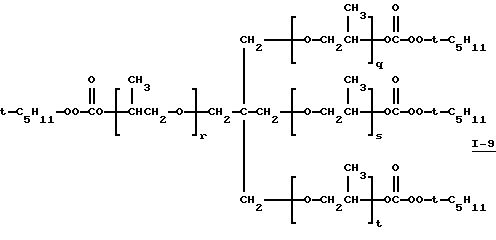
(где сумма q, r, s и t равна примерно 6-7).
В этом примере продукт получали в две стадии синтеза. На первой стадии полиэфиртетраол (С-5) (PLURACOL РеР 550) подвергали взаимодействию с избыточным фосгеном, получив в результате полиэфир-тетракис(хлорформиат) примера 7.
На второй стадии полиэфир-тетракис(хлорформиат) подвергали взаимодействию с трет-амилгидропероксидом в присутствии водного раствора гидроксида калия, и в результате получили продукт так, как описано ниже:
В реактор емкостью 250 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 28,1 (0,10 моль) 20% водного раствора гидроксида калия, 10,1 г (0,09 моль) 92,6% трет-амилгидропероксида и 2 капли (приблизительно 0,1 г) ALIQUAT® 336 (трикаприлилметиламмонийхлорид), производство Henkel Corporation), и полученный раствор перемешивали при примерно 25oС. К перемешанному раствору при 43-45oС медленно прибавляли 15,2 г (0,020 моль) 100% полиэфир-тетракис(хлорформиата) в течение 10 мин. По окончании прибавления реакционную массу перемешивали 5 ч примерно при 35-40oС, после чего прибавляли 75 мл МТВЕ, и реакционную массу охлаждали до 25oС, перемешивали одну минуту и затем давали ей разделиться на жидкие фазы. После этого нижний водный слой отделяли, и оставшийся органический слой промывали 50 мл 20% водного раствора гидроксида калия и затем 50 г водного раствора сульфита натрия с буферными свойствами (приготовленного путем растворения 1,2 г уксусной кислоты, 2,5 г ацетата натрия и 4,3 г сульфита натрия в 42,0 г воды). Водный слой выливали, а органический слой промывали 100 г насыщенного раствора хлорида натрия. Продуктный раствор сушили над 5% по массе безводного MgSO4 и, отфильтровав отработанный осушитель, удаляли в вакууме растворитель с получением в остатке 18,0 г (88,2% теоретически, без поправки) бесцветной жидкости. Продукт содержал 5,56% активного кислорода (теоретически 6,27%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 88,7%,и скорректированный выход составлял 80,0%.
Согласно способу получения и данным выхода, продукт, полученный при этой реакции, был указанным в заголовке целевым продуктом.
Пример 11. Получение полиэфир-трис (моно-трет-бутилпероксикарбоната), I-10, из PLURACOL® ТР-740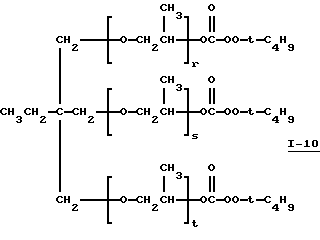
где сумма г, s и t равна примерно 6-7.
В этом примере продукт получали в две стадии синтеза. На первой стадии 0,06 моль полиэфиртриола (С-7) (PLURACOL ТР-740; молекулярная масса = 730, производство BASF Corporation)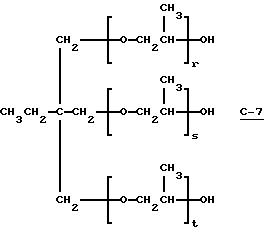
где сумма r, s и t равна примерно 10-11, подвергали взаимодействию с избыточным фосгеном (0,28 моль) при 3-7oС. Затем реакционную смесь перемешивали 2-3 ч при 10-20oС и давали ей отстояться всю ночь при 20-25oС. Далее избыточный фосген отгоняли из продукта при 20-30oС и пониженном давлении с получением полиэфир-трис(хлорформиата) А в виде прозрачной жидкости с содержанием продукта по результатам анализа 100% и скорректированным выходом 93,8%.
На второй стадии полиэфир-трис(хлорформиат) А подвергали взаимодействию с трет-бутилгидропероксидом в присутствии водного раствора гидроксида калия и в результате получили продукт так, как описано ниже:
В реактор емкостью 250 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 19,6 г (0,07 моль) 25% водного раствора гидроксида калия и 9,0 г (0,07 моль) 70% водного раствора трет-бутилгидропероксида при 22-29oС. Полученный раствор перемешивали при примерно 25oС. К перемешанному раствору при 33-40oС медленно прибавляли 18,3 г (0,02 моль) 100% полиэфир-трис (хлорформиата) А в течение 15 мин. По окончании прибавления реакционную массу перемешивали 1,5 часа при 40oС, после чего прибавляли 17 г этилбензола (ЕВ), и реакционную массу перемешивали две минуты при примерно 30oС и затем давали разделиться на жидкие фазы. После этого нижний водный слой отделяли и оставшийся органический слой охлаждали до 25oC и промывали 50 мл 20% водного раствора гидрксида калия, а затем 50 г водного раствора сульфита натрия с буферными свойствами (приготовленного путем растворения 1,2 г уксусной кислоты, 2,5 г ацетата натрия и 4,3 г сульфита натрия в 42,0 г воды) и 50 г насыщенного раствора хлорида натрия. Продуктный раствор сушили над 1,7 г безводного МgSO4 и, отфильтровав отработанный осушитель, получили 35,7 г бесцветной жидкости. Продуктный раствор содержал 2,49% активного кислорода (теоретически 4,45%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 55,9%, и скорректированный выход составлял 92,4%.
Согласно способу получения и данным выхода, продукт, полученный при этой реакции, был указанным в заголовке целевым продуктом в виде 55,9% раствора в этилбензоле.
Пример 12. Получение полиэфир-трис(моно-трет-бутил-пероксикарбоната), I-11, из PLUORACOL® GP-730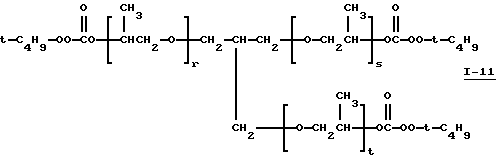
где сумма r, s и t равна примерно 10-11.
В этом примере продукт получали в две стадии синтеза. На первой стадии 0,05 моль полиэфиртриола (С-8) (PLURACOL GP-730; молекулярная масса = 730, производство BASF Corporation)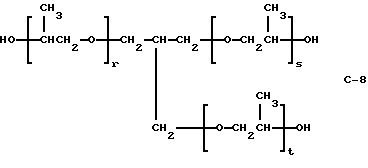
где сумма r, s и t равна примерно 10-11, подвергали взаимодействию с избыточным фосгеном (0,40 моль) при 3-7oС. Затем реакционную смесь перемешивали 2-3 ч при 10-20oС и давали ей отстояться всю ночь при 20-25oС. Далее избыточный фосген отгоняли из продукта при 20-30oС и пониженном давлении с получением полиэфир-трис(хлорформиата) В в виде прозрачной жидкости с содержанием продукта по результатам анализа 100% и скорректированным выходом 96,3%.
На второй стадии полиэфир-трис(хлорформиат) В подвергали взаимодействию с трет-бутилгидропероксидом в присутствии водного раствора гидроксида калия и в результате получили продукт так, как описано ниже:
В реактор емкостью 250 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 19,6 (0,07 моль) 25% водного раствора гидроксида калия и 9,0 г (,07 моль) 70% водного раствора трет-бутилгидропероксида при 22-29oС. Полученный раствор перемешивали при примерно 25oС. К перемешанному раствору при 23-28oС медленно прибавляли 18,3 г (0,02 моль) 100% полиэфир-трис(хлорформиата) В в течение 15 мин. По окончании прибавления реакционную массу перемешивали 3 ч при 25-30oС, после чего прибавляли 100 мл МТВЕ, и реакционную массу перемешивали одну минуту при примерно 30oС и затем давали разделиться на жидкие фазы. После этого нижний водный слой отделяли, и оставшийся органический слой охлаждали до 12oС и промывали 50 мл 10% водного раствора гидросульфита натрия, а затем 50 мл 10% водного раствора гидроксида калия и 50 мл порциями насыщенного водного раствора сульфата натрия до получения рН 7-8. Продуктный раствор сушили над 5% по массе безводного МgS04 и, отфильтровав отработанный осушитель, получали в остатке 20,3 г (94% теоретически, без поправки) бесцветной жидкости. Продукт содержал 4,23% активного кислорода (теоретически 4,45%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 95,1%, и скорректированный выход составлял 89,3%.
Согласно способу получения и данным выхода продукт, полученный при этой реакции, был указанным в заголовке целевым продуктом.
Пример 13. Получение 1,5-бис(1,1,4-триметил-4-(трет-бутилперокси)пентилпероксикарбонилокси)-3-оксапентана, I-12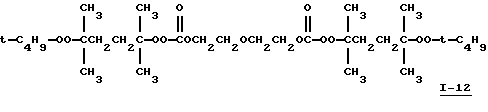
В этом примере продукт получали путем осуществления взаимодействия диэтиленгликоль-бис(хлорформиата) (С-9)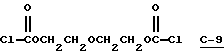
с 1,1,4-триметил-4-(трет-бутилперокси)пентилгидро-пероксидом (С-10)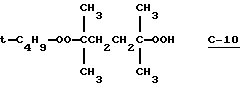
и водным гидроксидом калия с получением продукта так, как описано ниже:
В реактор емкостью 250 мл с водяной рубашкой, оборудованный механической мешалкой, термометром и капельной воронкой, загружали 8,0 (0,05 моль) 25% водного раствора гидроксида натрия и 11,0 г (0,043 моль) 91% 1,1,4-триметил-4-(трет-бутилперокси)пентилгидропероксида при 22-29oС. Полученный раствор перемешивали при примерно 25oС. К перемешанному раствору при 23-28oС медленно прибавляли 5,8 г (0,025 моль) 99% диэтиленгликоль-бис(хлорформиата) (С-9) в течение 15 мин. По окончании прибавления реакционную массу перемешивали 3,5 часа при 30-35oС, после чего прибавляли 50 мл МТВЕ, и реакционную массу перемешивали одну минуту при примерно 30oС и затем давали ей разделиться на жидкие фазы. После этого нижний водный слой отделяли, и оставшийся органический слой охлаждали до 17oC и промывали 50 мл 10% водного раствора гидросульфита натрия, а затем 50 мл 20% водного раствора гидроксида натрия и 50 мл порциями
насыщенного водного раствора сульфата натрия до получения рН 7-8. Продуктный раствор сушили над 5% по массе безводного MgS04 и, отфильтровав отработанный осушитель, получили в остатке 14,7 г (88,6% теоретически, без поправки) бесцветной жидкости. ИК-спектр продукта содержал основную полосу поглощения карбонила монопероксикарбоната при 1785 см-1 и основную карбонильную полосу карбоната или сложного эфира примерно при 1752 см-1. В ИК-спектре был лишь след полосы поглощения ОН. Продукт содержал 4,48% активного кислорода (теоретически 5,10%) согласно пероксиэфирному методу определения содержания активного кислорода, следовательно, содержание продукта по результатам анализа составляло 87,0%, и скорректированный выход составлял 77,0%.
Согласно способу получения, данным выхода и данным ИК-спектра, продукт, полученный при этой реакции, был указанным в заголовке целевым продуктом.
Пример 14. Данные экзотермического эффекта при 280oF (138oС) по методике SPI для поликапролактон-трис(моно-трет-бутилпероксикарбоната), I-4
Ненасыщенная полиэфирная смола в этом примере представляла собой смесь ненасыщенного сложного полиэфира и стирольного мономера. Ненасыщенным полиэфиром была алкидная смола, полученная путем этерификации следующих компонентов:
Компонент - Количество (моль)
Малеиновый ангидрид - 1,0
Фталевый ангидрид - 1,0
Пропиленгликоль - 2,2
К полученной смоле добавляли 0,013% (по массе) гидрохинонового ингибитора. Алкидная смола имела кислотное число 45-50. Семь (7) частей (по массе) указанного выше ненасыщенного полиэфира алкида разбавляли тремя (3) массовыми частями стирольного мономера. Полученная ненасыщенная полиэфирная смола имела следующие свойства:
Вязкость (по Брукфилду 2 при 20 об/мин) - 13,0 П
Относительная плотность - 1,14
Гелеобразующие и отверждающие свойства трет-бутилпероксибензоата (А-2) (коммерческий пероксидный продукт, используемый для отверждения ненасыщенной полиэфирной смолы) и поликапролактон-трис(моно-трет-бутилпероксикарбоната) 1-4 (новый поли(монопероксикарбонат) по настоящему изобретению) определяли по стандартной методике Общества промышленности пластмасс (SPI) для определения экзотермического эффекта (предложенная SPI методика, касающаяся рабочих экзотерм для полиэфирных смол, опубликована в сигнальном экземпляре материалов 24-й ежегодной технической конференции - Reinforced Plastics/Composites Division, Society of the Plastics Industry, Inc., 1969). По этой методике при 280oF (138oС) была произведена сравнительная оценка А-2 и I-4. Количество использованного I-4 было эквивалентно по содержанию активного кислорода 1,0 г чистого А-2 на 100 г ненасыщенной полиэфирной смолы.
Результаты этого исследования даны в таблице к примеру 14 и показывают, что I-4 желатинизировал и отверждал смолу быстрее, чем А-2, поскольку I-4 был более активным в отверждении ненасыщенной полиэфирной смолы, чем коммерческий пероксидный катализатор А-2.
Пример 15. Улучшенные процессы полимеризации стирола с использованием новых поли(монопероксикарбонатов) в качестве свободно-радикальных инициаторов
Полимеризацию стирола осуществляли с использованием мономерного раствора, содержащего 95% стирола и 5% этилбензола (ЕВ). В качестве инициаторов использовали 1,1-ди(трет-бутилперокси)циклогексан (А-3), т.е. Lupersol 331 (производство Elf Atochem North America, Inc: коммерческий инициатор, используемый в настоящее время для получения высокомолекулярного полистирола при повышенных скоростях полимеризации), 1,5-бис-(трет-бутилпероксикарбонилокси)-5-оксапентан (А-4; известный бис(монопероксикарбонат); патент США 3652631), поликапролактон-бис(моно-трет-бутил-пероксикарбонат) (А-1) (известный бис-(монопероксикарбонат); патент США 5314970) и несколько поли(монопероксикарбонатов) по настоящему изобретению, а именно: 1,1,1-трис(трет- бутилпероксикарбонилоксиметил) этан (I-1), 1,1,1-трис (трет-бутилпероксикарбонилоксиметил)пропан (I-2), поликапролактон-трис(моно-трет-бутилпероксикарбонат) (I-3), поликапролактон-трис(монотрет-бутилпероксикарбонат) (I-4), 1,1,1-трис[2-(трет-бутилпероксикарбонилокси)этокси-метил] пропан (I-5) и поликапролактон-тетракис(моно-трет-бутилпероксикарбонат) (I-6).
Приготовление растворов стирола и инициатора
К растворам 95% стирола и 5% этилбензола при комнатной температуре добавляли свободно-радикальные инициаторы в количествах, эквивалентных 0,00277 моль активного кислорода на 1000 г стирольного раствора (или 0,00252 моль активного кислорода на литр стирольного раствора). Полученные стирольные растворы продували перед запаиванием в стеклянных ампулах (наружный диаметр 10 мм, внутренний диаметр 8 мм) азотом.
Полимеризация стирола
Ампулы, содержащие стирольные растворы (несколько для каждого раствора), погружали в циркуляционную масляную ванну, температуру в которой регулировали с помощью программного регулятора температуры. Температуру образцов изменяли по линейному закону от 100 до 151oС с запрограммированной скоростью 0,17oС/минуту (5-часовая программа). Образцы каждого раствора извлекали из ванны с 1-часовыми интервалами в течение 5-часовой программы и охлаждали путем погружения в водяную баню со льдом. Затем стирольные растворы извлекали из ампул и анализировали для определения среднемассовой молекулярной массы (Mw) полистирола и остаточного содержания стирольного мономера.
Peзультаты
Характеристики трис(моно-трет-бутилпероксикарбонатов) I-1,I-2, I-3, I-4 и I-5 и тетракис(моно-трет-бутилпероксикарбоната) 1-6 сравнивали с известными соединениями А-1, А-3 и А-4 в соответствии с вышеописанной методикой. Полученные результаты сведены в таблицу к примеру 15.
Судя по результатам, касающимся среднемассовой молекулярной массы (Mw) полистирола, применение трис- и поли(монопероксикарбонатов) по настоящему изобретению, т. е. I-1, I-2, I-3, I-4, I-5 и I-6 в качестве инициаторов полимеризации стирола дало значительно более высокие значения Mw (330000-375000) после 5-часовой программы полимеризации, чем те, что были получены с известными соединениями А-1 (Mw равна приблизительно 290000), А-3 (Mw равна приблизительно 280000) и А-4 (Mw равна приблизительно 300000). Известный бис(монопероксикарбонат) А-1 был значительно менее эффективен в увеличении молекулярной массы полистирола (максимальное значение Mw было равно приблизительно 290000), чем трис- и поли(монопероксикарбонаты) по настоящему изобретению (Mw равна примерно 330000-375000). Таким образом, трис- и поли(монопероксикарбонаты) по настоящему изобретению являются существенным шагом вперед в области полимеризации этиленоненасыщенных мономеров, таких, как стирол.
Пример 16. Улучшенные процессы полимеризации стирола с использованием поли (монопероксикарбонатов) в качестве свободно-радикальных инициаторов
Полимеризацию стирола осуществляли по методике, описанной в примере 15. Оценивали в качестве свободно-радикальных инициаторов в сравнении с 1,1-ди(трет-бутилперокси)циклогексаном (А-3) несколько других поли(монопероксикарбонатов) структуры А: полиэфир-тетракис(моно-трет-бутилпероксикарбонат) (I-7), полиэфир-тетракис(моно-трет-амилпероксикарбонат) (I-9), полиэфир-трис(моно-трет-бутилпероксикарбонат) (I-10) из PLURACOL ТР-740, полиэфир-трис(моно-трет-бутилпероксикарбонат) (I-11) из PLURACOL GP-730 и 1,5-бис(1,1,4-триметил-4-(трет-бутил-перокси)пентилпероксикарбонилокси)-3-оксапентан (I-12). В этом примере использовали свободно-радикальные инициаторы в количествах, эквивалентных 0,00277 моль активного кислорода на 1000 г стирольного раствора (или 0,00252 моль активного кислорода на литр стирольного раствора).
Полимеризация стирола
Ампулы, содержавшие стирольные растворы (несколько для каждого раствора), погружали в циркуляционную масляную ванну, температуру в которой регулировали с помощью программного регулятора температуры. Температуру образцов изменяли по линейному закону от 100 до 151oС с запрограммированной скоростью 0,17oС/минуту (5-часовая программа). Образцы каждого раствора извлекали из ванны с 1-часовыми интервалами в течение 5-часовой программы и охлаждали путем погружения в водяную баню со льдом. Затем стирольные растворы извлекали из ампул и анализировали для определения среднемассовой молекулярной массы (Mw) полистирола и остаточного содержания стирольного мономера.
Результаты
Характеристики поли(монопероксикарбонатов) I-7, I-9, I-10, I-11 и I-12 сравнивали с известным соединением А-3 в соответствии с вышеописанной методикой. Полученные результаты сведены в таблицу к примеру 16.
Судя по результатам, касающимся среднемассовой молекулярной массы (Mw) полистирола, применение поли(монопероксикарбонатов) по настоящему изобретению, т. е. I-7, I-9, I-10, I-11 и I-12, в качестве инициаторов полимеризации стирола дало значительно более высокие значения Mw (300000-390000) после 5-часовой программы полимеризации, чем те, что были получены с известным соединением А-3 (Mw равна приблизительно 280000). Кроме того, по окончании 5-часового периода остаточные содержания стирола в полистиролах, полученных с использованием новых поли(монопероксикарбонатов) по настоящему изобретению, были значительно ниже, чем в полистироле, полученном с использованием А-3 (5-10% остаточного стирола против 17-18% остаточного стирола). Особенно привлекательным в этом отношении было соединение I-12. Эти результаты показывают, что поли(монопероксикарбонаты) по настоящему изобретению являются существенным шагом вперед в области полимеризации этиленоненасыщенных мономеров, таких, как стирол.
Пример 17. Улучшенные процессы полимеризации стирола с использованием поли(монопероксикарбонатов) в сочетании с 1,1-ди(трет-бутилперокси)циклогексаном (А-3)
Полимеризацию стирола осуществляли с использованием мономерного раствора, содержащего 95% стирола и 5% этилбензола (ЕВ). В этом примере использовали модификацию методики, описанной в примере 15. Были использованы комбинации двух свободно-радикальных инициаторов, причем один из инициаторов комбинации был новый поли(монопероксикарбонат) по настоящему изобретению, а именно 1,1,1-трис[2-(трет-бутилпероксикарбонилокси)-этоксиметил]пропан (I-5) или полиэфир-тетракис (моно-трет-бутилпероксикарбонат ) (I-7). Вторым инициатором комбинаций был 1,1-ди(трет-бутилперокси) циклогексан (А-3), уже известное соединение.
Приготовление растворов стирола и инициатора
Содержания комбинаций свободно-радикальных инициаторов в этом примере были эквивалентны суммарно 0,00230 моль активного кислорода на 1000 г стирольного раствора (или 0,00209 моль активного кислорода на литр стирольного раствора).
Полимеризация стирола
Ампулы, содержащие стирольные растворы (несколько для каждого раствора), погружали в циркуляционную масляную ванну, температуру в которой регулировали с помощью программного регулятора температуры. Температуру образцов изменяли по линейному закону от 100 до 145,6oС с запрограммированной скоростью 0,19oС/минуту (4-часовая программа). По окончании 4-часового периода образцы раствора извлекали из ванны и охлаждали путем погружения в водяную баню со льдом. Затем стирольные растворы извлекали из ампул и анализировали для определения среднемассовой молекулярной массы (Mw) полистирола.
Результаты
В таблице к примеру 17 представлены значения среднемассовых молекулярных масс, полученных при использовании комбинации А (1-5 и А-3) и комбинации В (I-7 и А-3) инициаторов в качестве свободно-радикальных инициирующих систем.
Результаты показывают, что среднемассовую молекулярную массу можно регулировать в сторону увеличения путем замены некоторой части инициатора А-3 инициатором I-5 или I-7 или в сторону уменьшения путем замены некоторой части инициатора I-5 или I-7 инициатором А-3. Таким образом, производители полистирола могут использовать новые поли-(монопероксикарбонаты) по настоящему изобретению в сочетании с другими свободно-радикальными инициаторами для регулирования молекулярной массы полистирола и, следовательно, регулирования физических свойств полистирола.
Пример 18. Улучшенные процессы полимеризации стирола с использованием полиэфир-тетракис(моно-трет-бутилпероксикарбоната) (I-7) в сочетании с трет-бутилпероксибензоатом (А-2).
Полимеризацию стирола осуществляли с использованием мономерного раствора, содержащего 95% стирола и 5% этилбензола (ЕВ). В этом примере использовали модификацию методики, описанной в примере 15. Были использованы комбинации двух свободно-радикальных инициаторов, причем одним из инициаторов комбинации был новый поли(монопероксикарбонат) по настоящему изобретению, а именно полиэфир-тетракис(моно-трет-бутилпероксикарбонат) (I-7), а вторым инициатором комбинаций был известный монопероксид, а именно трет-бутилпероксибензоат (А-2).
Приготовление растворов стирола и инициатора
Суммарные содержания свободно-радикальных инициаторов, использованных в этом примере, были эквивалентны суммарно 0,00277 моль активного кислорода на 1000 г стирольного раствора (или 0,00230 моль активного кислорода на литр стирольного раствора).
Полимеризация стирола
Ампулы, содержавшие стирольные растворы (несколько для каждого раствора), погружали в циркуляционную масляную ванну, температуру в которой регулировали с помощью программного регулятора температуры. Температуру образцов изменяли по линейному закону от 100 до 151oС с запрограммированной скоростью 0,17oС/минуту (5-часовая программа). По окончании 5-часового периода образцы раствора извлекали из ванны и охлаждали путем погружения в водяную баню со льдом. Затем стирольные растворы извлекали из ампул и анализировали для определения среднемассовой молекулярной массы (Mw) полистирола.
Результаты
В таблице к примеру 18 представлены значения среднемассовых молекулярных масс, полученных при использовании комбинации С (I-7 и А-2) инициаторов в качестве свободно-радикальной инициирующей системы.
Результаты показывают, что среднемассовую молекулярную массу можно регулировать в сторону увеличения путем замены некоторой части известного монопероксида А-2 инициатором I-7 или в сторону уменьшения путем замены некоторой части инициатора I-7 известным монопероксидом А-2. Таким образом, производители полистирола могут использовать новые поли (монопероксикарбонаты) по настоящему изобретению в сочетании с другими монопероксидными инициаторами для регулирования молекулярной массы полистирола и, следовательно, регулирования физических свойств полистирола.
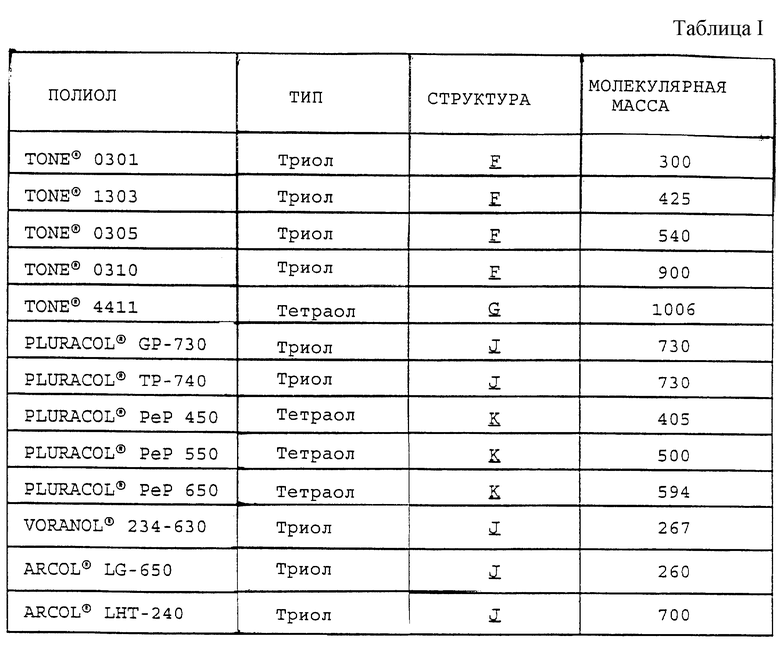
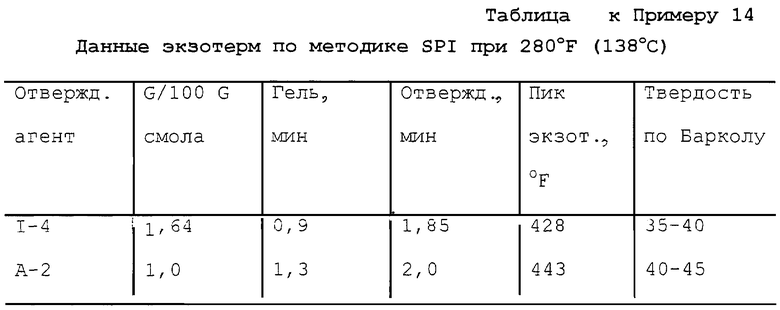
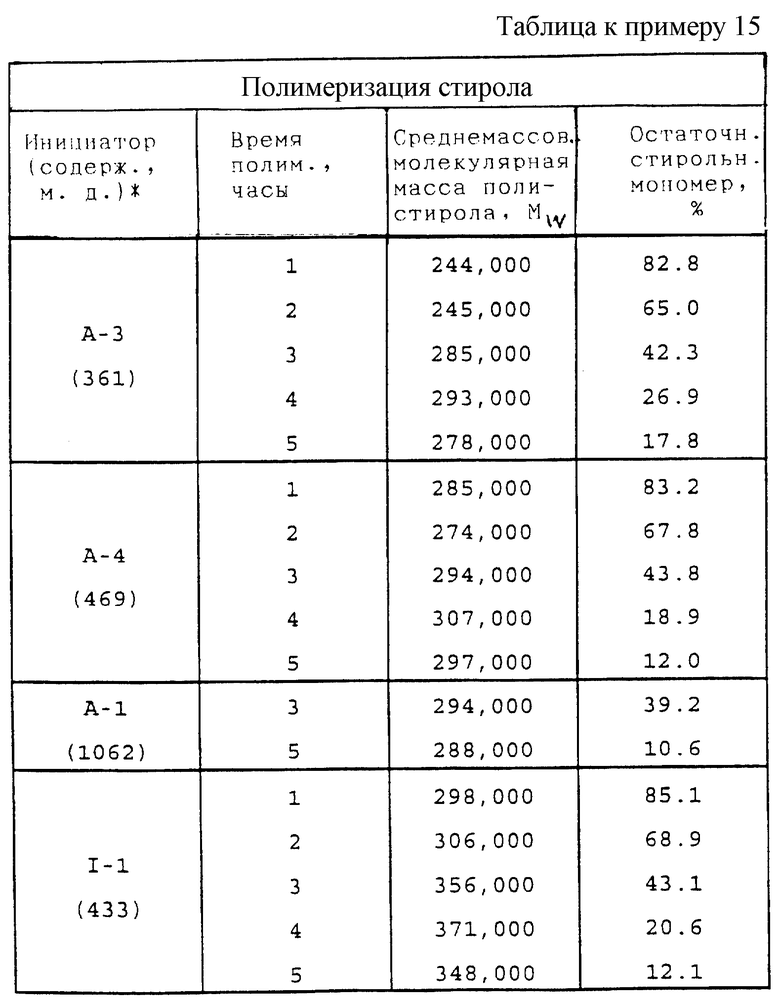
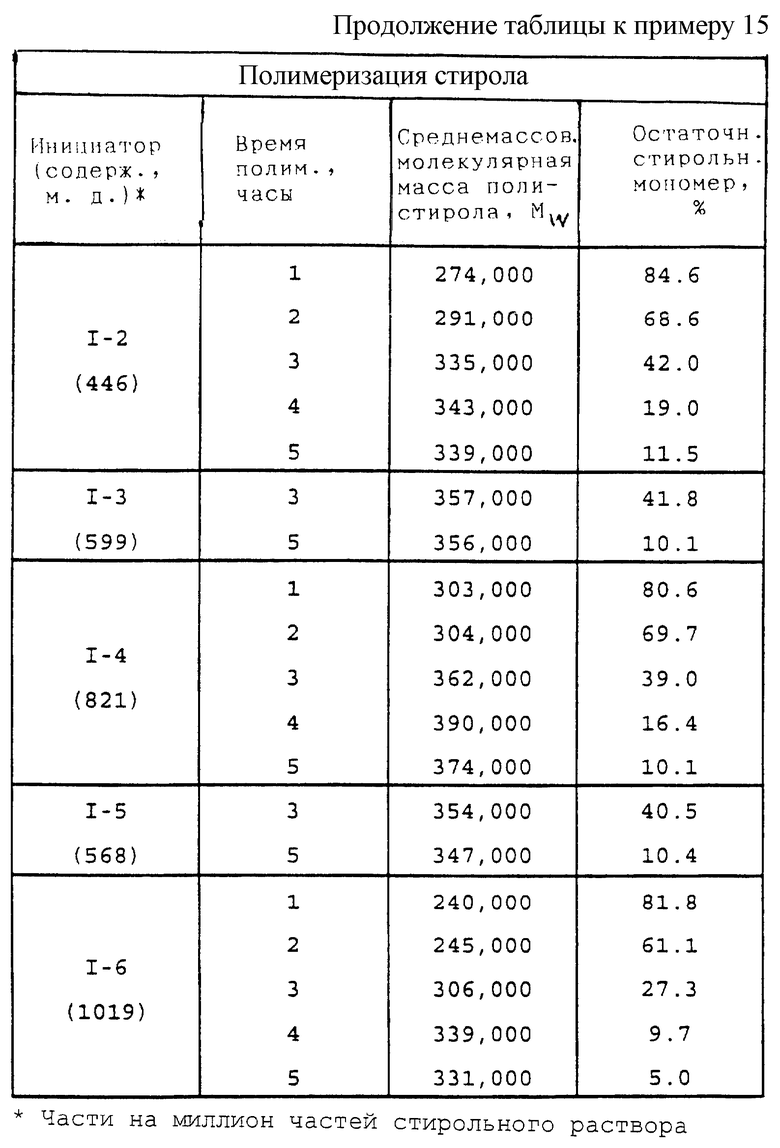
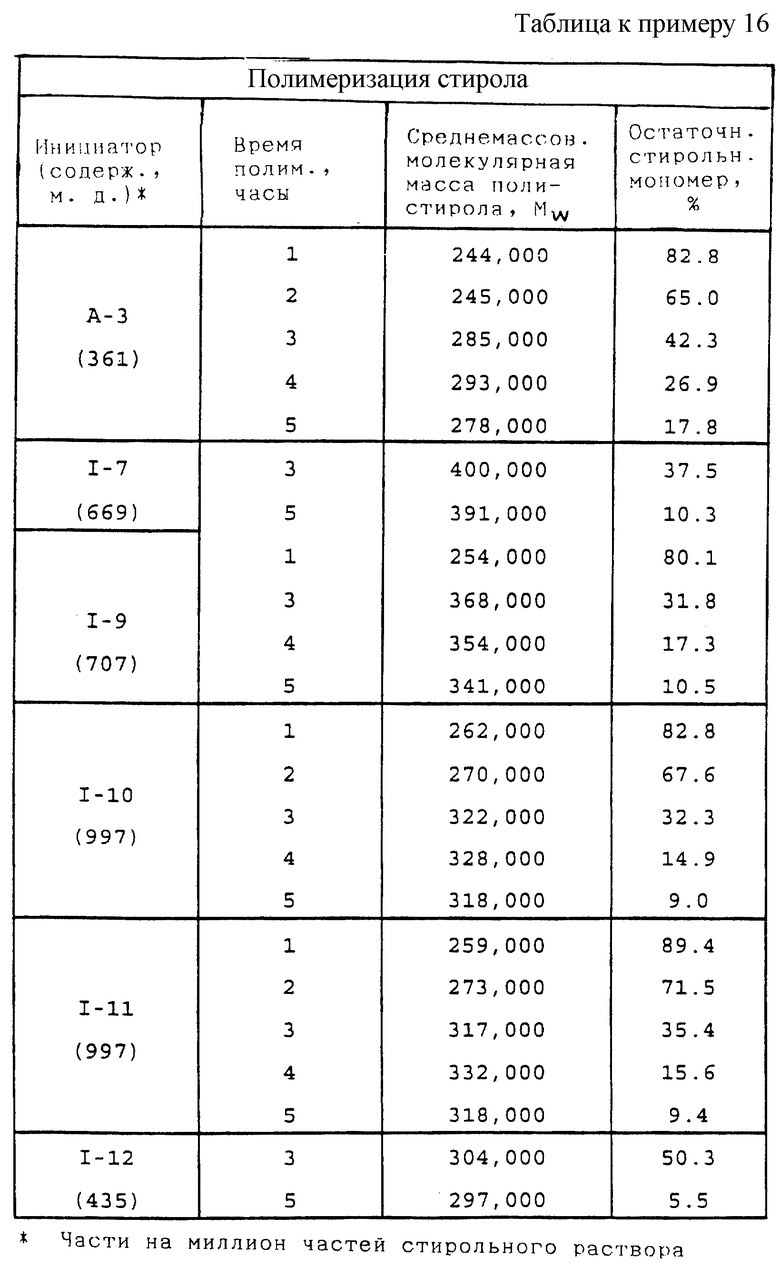
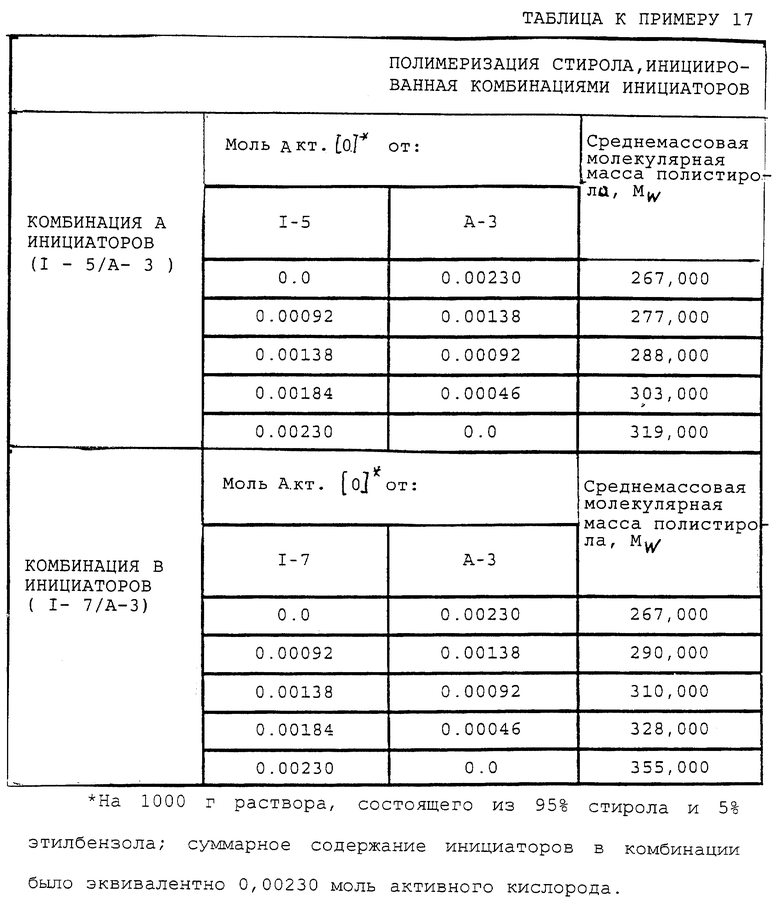
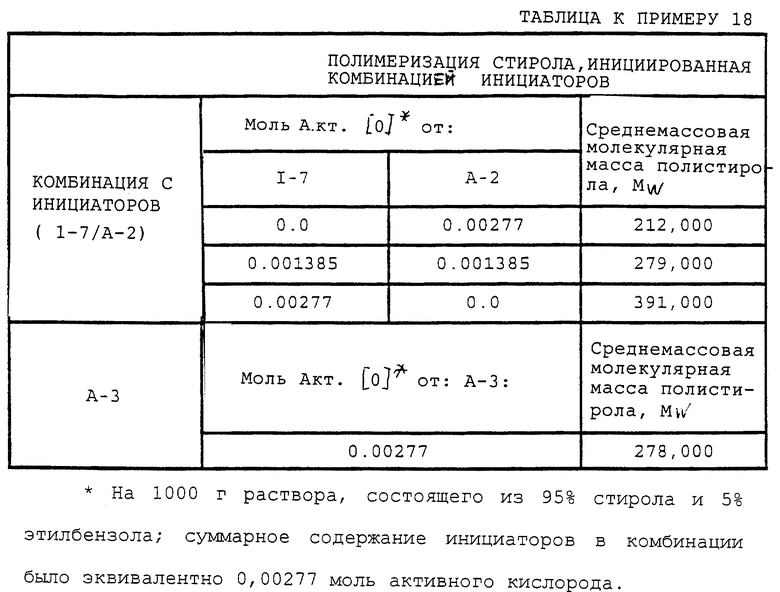
Изобретение относится к новым соединениям, таким как поли(монопероксикарбонаты) общей структуры А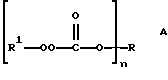
где R, R1 и n определены в кратком изложении сущности изобретения, такие, как 1,1,1-трис(трет-бутилпероксикарбонилоксиметил)этан, промежуточные соединения для их получения, а также способы их получения и применения. Монопероксикарбонаты полезны для инициирования полимеризации этиленоненасыщенных мономеров, в частности стирола, отверждения ненасыщенных сложных полиэфиров и модифицирования молекулярной массы полимеров путем сшивания или регулируемой деструкции цепи. 2 с. и 16 з.п.ф-лы, 6 табл.
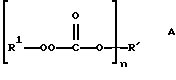
где n представляет целое число от 2 до 8;
R1 выбран из трет-алкильных радикалов с 4-12 углеродными атомами, 1,1,4-триметил-4(трет-бутилперокси)пентильного радикала, 1,1,4-триметил-4(трет-амилперокси)пентильного радикала, трет-циклоалкильных радикалов с 6-10 углеродными атомами, трет-аралкильных радикалов с 9-13 углеродными атомами и 3-метил-1-бутил-3-ила и 3-метил-1-пентин-3-ила, при условии, что когда n= 2, R1 выбран из 1,1,4-триметил-4(трет-бутилперокси)пентильного радикала и 1,1,4-триметил-4-(трет-амилперокси)пентильного радикала;
i) когда n= 2, R представляет бирадикал, выбранный из алкилена с 2-12 углеродными атомами, алкенилена с 4-8 углеродными атомами и бирадикальных структур (n) и (о)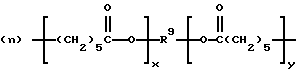
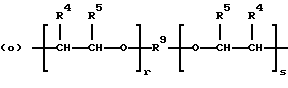
где R9 представляет алкиленовый бирадикал с 2-8 углеродными атомами;
ii) когда n= 3, R представляет трирадикал, выбранный из 1,3,5-циклогекстриила, R2C(CH2-)3, -CHR2CH(-)CH2- и структур (а), (b), (с), (d) и (е),
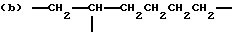
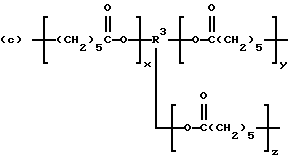
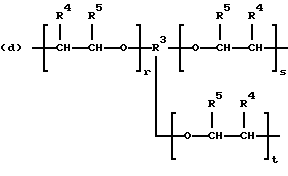
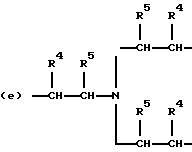
где R2 выбран из водорода и алкильных радикалов с 1-6 углеродными атомами;
R3 представляет трирадикал, выбранный из R2С(CН2-)3, -CHR2CH(-)СН2- и структур (а) и (b);
R4 и R5 являются одинаковыми или разными и выбраны из водорода и алкильных радикалов с 1-4 углеродными атомами;
х, y и z представляют целые числа от 0 до 5, сумма х, y и z составляет от 2 до 8;
r, s и t представляют целые числа от 0 до 6, сумма r, s и t составляет от 3 до 18,
при условии, что когда трирадикал R представляет собой R2(CН2)3- или структуру (b), то R1 не является трет-октилом;
iii) и когда n= 4-8, R представляет полирадикал, выбранный из С(СН2-)4 и структур (f), (g), (h), (i), (j), (k) и (1)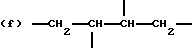
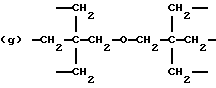
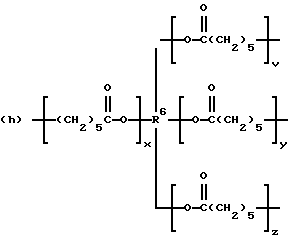
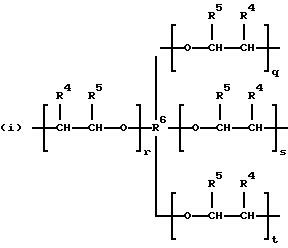
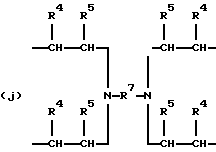
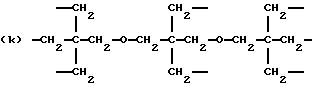
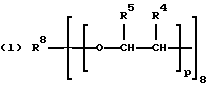
где R6 представляет тетрарадикал, выбранный из С(СН2-)4 и структуры (f);
R7 представляет бирадикал, выбранный из алкилена с 2-6 углеродными атомами и 1,2-, 1,3- и 1,4-фенилена;
R8 представляет октарадикал на основе сахарозы, имеющий структуру (m)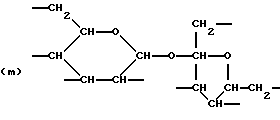
р представляет целое число от 1 до 3;
v представляет целое число от 0 до 5; сумма v, х, y и z составляет от 3 до 10;
q представляет целое число от 0 до 4; сумма q, r, s и t составляет от 2 до 16,
и с условием, что когда R представляет С(СН2-)4, то R1 не является трет-октилом.
Приоритет по пунктам и признакам:
23.08.1996 по пп. 1-18;
08.08.1997 по пп. 1-18 (уточнение признаков).
| Chem | |||
| abstr., ТОМ 92, №164324, МАЙ 1980, (Columbus, ОН | |||
| USA), C.2 | |||
| Chem | |||
| abstr., TOM 71, №123928, сентябрь 1969, (Columbus, OH | |||
| US) | |||
| US 5245075, 14.09.1993 | |||
| ЕР 0717035 A1, 13.12.1995 | |||
| СПОСОБ ПОЛУЧЕНИЯ ДИЭФИРОВ ПЕРОКСИДИКАРБОНОВЫХКИСЛОТ | 0 |
|
SU323398A1 |
| 0 |
|
SU189859A1 | |

