Изобретение относится к некоторым соединениям хиназолина и к их фармацевтически приемлемым солям. Соединения настоящего изобретения ингибируют действие определенных протеинтирозинкиназ (РТК), являющихся рецепторами факторов роста, и таким образом подавляют аномальный рост некоторых типов клеток. Поэтому соединения настоящего изобретения являются противораковыми средствами и могут быть использованы для лечения рака у млекопитающих. Кроме того, настоящее изобретение относится к получению указанных хиназолинов, к их использованию для лечения рака, и к фармацевтическим препаратам, содержащим указанные соединения.
Протеинтирозинкиназы представляют собой класс ферментов, которые катализируют перенос фосфатной группы от АТФ на тирозиновый остаток, присутствующий в белковом субстрате. Очевидно, что протеинтирозинкиназы играют важную роль в росте нормальных клеток. Многие белки, являющиеся рецепторами факторов роста, функционируют как тирозинкиназы, осуществляя таким образом передачу сигнала. Взаимодействие факторов роста с этими рецепторами является необходимым процессом для нормальной регуляции клеточного роста. Однако при определенных условиях, в результате мутации или усиленной экспрессии, эти рецепторы могут становиться нерегулируемыми, что приводит к неконтролируемой пролиферации клеток, вызывающей рост опухолей, и в конечном счете, к развитию заболевания, известного как рак (Wilks A.F., Adv. Cancer Res., 60, 43 (1993) и Parsons J.T.; Parsons S.J., Important Advances in Oncology, DeVita V. T. Ed. , J.B. Lippincott Co., Phila (1993). Одним из идентифицированных киназных рецепторов фактора роста и их протоонкогенов, которые являются целью для соединений настоящего изобретения, является киназный рецептор эпидермального фактора роста (EGF-R-киназа, белковый продукт онкогена erbB) и продукт, продуцируемый онкогеном erbB-2 (называемым также neu или НЕR2). Поскольку процесс фосфорилирования является необходимым сигналом для деления клеток и поскольку сверхэкспрессия или мутация киназ связана с возникновением рака, то ингибитор фосфорилирования, то есть ингибитор протеинтирозинкиназы может быть использован в качестве терапевтического средства для лечения рака и других заболеваний, которые характеризуются неконтролируемым или аномальным ростом клеток. Так, например, сверхпродуцирование рецепторного киназного продукта онкогена erbB-2 связано с раком молочной железы и яичников (Slamon, D. J. et al., Science 244, 707 (1989) и Science 235, 1146 (1987)). Отсутствие регулирования ЕGF-R-киназы связано с опухолями кожного покрова (Reiss, M. , et al., Cancer Res., 51, 6254 (1991)), раком молочной железы (Macias, A. , et al. , Anticancer Res., 7, 459 (1987)), а также с опухолями других важных органов (Gullick, W.J., Brit. Med. Bull., 47, 87 (1991)). Поскольку негулируемые киназные рецепторы играют важную роль в патогенезе рака, то многие исследования, проводимые за последнее время, были направлены на разработку специфических ингибиторов РТК, которые можно было бы использовать в качестве противораковых терапевтических средств (см. некоторые последние работы: Burke T.P., Drugs Future, 17, 119 (1992) и Chang, C.J., Geahlen, P.Z., J. Nat. Prod. 55, 1529 (1992)).
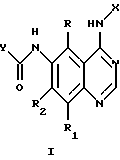
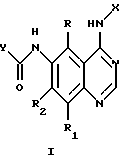
Соединения настоящего изобретения представляют собой определенные 4-анилинохиназолины. В данном изобретении система хиназолиновых колец пронумерована следующим образом: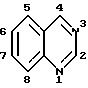
Было отмечено, что другие 4-анилинохиназолины, которые по своей природе и по расположению заместителей в положениях 5-8 отличаются от соединений настоящего изобретения, обладают РТК-ингибирующей активностью. В Европейской патентной заявке 520 722 А1 описаны некоторые 4-анилинохиназолины, которые в положениях 5-8 содержат водород, хлор, трифторметил или нитрозаместители. При этом ни одно из соединений, описанных в вышеуказанной патентной заявке, не имеет той уникальной комбинации заместителей, которая присутствует в соединениях настоящего изобретения. Кроме того, следует отметить, что, хотя в вышеуказанной патентной заявке заявлено использование описываемых в ней соединений в качестве противораковых средств, однако в этой заявке не приводится какой-либо иллюстрации противоракового эффекта in vivo. В Европейской патентной заявке 566226 А1 раскрываются некоторые 4-анилинохиназолины, которые необязательно содержат ряд заместителей в положениях 5-8. Однако ни одно из соединений, описанных в вышеуказанной патентной заявке, не имеет той уникальной комбинации заместителей, которая присутствует в соединениях настоящего изобретения. Кроме того, следует отметить, что, хотя в вышеуказанной патентной заявке заявлено использование раскрываемых в ней соединений в качестве противораковых средств, однако в этой заявке не приводится какой-либо иллюстрации их противоракового действия in vivo, в указанной Европейской заявке проиллюстрирована лишь in vivo - активность, направленная на ингибирование TGF-α-стимулируемого роста гепатоцитов крыс. В заявке на европейский патент 635 498 А1 раскрываются некоторые 4-анилинохиназолины, которые необязательно имеют ряд заместителей в положении 6 и обязательно содержат галоген в положении 7. Однако ни одно из соединений, описанных в указанной заявке, не имеет той уникальной комбинации заместителей, которая присутствует в соединениях настоящего изобретения. Кроме того, следует отметить, что, хотя в вышеуказанной патентной заявке заявлено использование раскрытых в ней соединений в качестве противораковых средств, однако в этой заявке не приводится какой-либо иллюстрации их противоракового действия in vivo. В указанной заявке на европейский патент проиллюстрирована лишь in vivo-активность, направленная на ингибирование TGF-α-стимулируемого роста гепатоцитов у крыс. Кроме того, известны некоторые хиназолиновые ингибиторы, которые не имеют 4-анилиновой группы. В заявке на европейский патент 602 851 А1 раскрыты некоторые хиназолины, которые не содержат анилиновой группы в 4-положении и которые необязательно содержат ряд заместителей в положениях 5-8. Однако ни одно из соединений, описанных в этой заявке, не имеет той уникальной комбинации заместителей, которую имеют соединения настоящего изобретения. Кроме того, следует отметить, что, хотя в вышеуказанной патентной заявке заявлено использование раскрываемых в ней соединений в качестве противораковых средств, однако какой-либо иллюстрации их противоракового действия in vivo в этой заявке не приводится. В указанной заявке на европейский патент показана лишь in vivo-активность, направленная на ингибирование TGF-альфа-стимулируемого роста гепатоцитов крыс. В патентной заявке WO 95/19774 раскрываются некоторые гетероциклы, которые ингибируют РТК и которые имеют пиримидиновое кольцо, подобное кольцу 4-анилинохиназолов настоящего изобретения. Однако в этой заявке отсутствуют какие-либо упоминания о 4-анилинохиназолинах или об уникальной комбинации заместителей, присутствующей в соединениях настоящего изобретения. Кроме того, следует отметить, что хотя в вышеуказанной патентной заявке заявлено использование раскрытых в ней соединений в качестве противораковых средств, однако какой-либо иллюстрации их противоракового действия in vivo в этой заявке не приводится. В патентной заявке WO 95/157581 описаны некоторые хиназолины, которые необязательно содержат ряд заместителей в положениях 5-7. Но ни одно из этих соединений не имеет той уникальной комбинации заместителей, которая присутствует в соединениях настоящего изобретения. Кроме того, следует отметить, что хотя в вышеуказанной патентной заявке заявлено использование раскрытых в ней соединений в качестве противораковых средств, однако какой-либо иллюстрации их противоракового действия in vivo в этой заявке не приводится.
Помимо вышеуказанных патентных заявок, имеется ряд публикаций, в которых описаны 4-анилинохиназолины, а именно: Fry, D.W., et al., Science, 265, 1093 (1994), Rewcastle G.W., et al., J. Med. Chem., 38, 3482 (1995); и Bridges, A. J., et al., J. Med. Chem., 39, 267, (1996). Однако ни одно из соединений, описанных в указанных публикациях, не имеет уникальной комбинации заместителей, присутствующей в соединениях настоящего изобретения. Кроме того, следует отметить, что в этих публикациях не приводится описания, иллюстрирующего противоопухолевое действие этих соединений in vivo.
РТК (протеинтирозинкиназа) катализирует перенос фосфатной группы от молекулы АТФ на тирозиновый остаток, присутствующий в белковом субстрате. Все известные в настоящее время ингибиторы обычно конкурируют либо с АТФ, либо с белковым субстратом киназы. Некоторые из этих ингибиторов, так называемые смешанные конкурентные ингибиторы, могут конкурировать как с АТФ, так и с субстратом одновременно, причем все такие конкурентные ингибиторы являются обратимо действующими ингибиторами. Известные в настоящее время 4-анилинохиназолины являются обратимо действующими ингибиторами, конкурирующими с АТР (Fry, D.W., et al., Science, 265, 1093 (1994)). Поскольку концентрация АТФ в клетке в нормальном состоянии очень высока (миллимоль), то соединения, которые конкурируют с АТФ, могут не обладать in vivo-активностью, так как нежелательно, чтобы концентрации указанных соединений увеличивались до значений, необходимых для вытеснения АТФ из ее сайтов связывания. Было показано, что хиназолиновые ингибиторы настоящего изобретения обладают уникальной способностью необратимо ингибировать РТК, а поэтому не конкурируют с АТФ или с белковым субстратом. Соединения настоящего изобретения являются необратимо действующими ингибиторами благодаря тому, что они могут образовывать ковалентные связи с аминокислотными остатками, присутствующими в активном центре фермента. Как показано ниже, это является результатом того, что соединения настоящего изобретения обладают повышенной терапевтической полезностью по сравнению с ингибиторами обратимого типа. Было показано, в частности, что именно уникальная природа и комбинация заместителей, присутствующих в соединениях настоящего изобретения, приводит к необратимому связыванию этих соединений-ингибиторов с ферментом. Указанные уникальные свойства соединений настоящего изобретения обуславливают их способность ингибировать рост опухолей человека в in vivo-модели рака.
Описание изобретения
Настоящее изобретение относится к соединению формулы 1: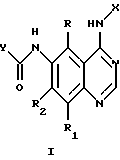
где X представляет собой фенил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, включающей в себя: галоген, алкил с 1-6 атомами углерода, алкокси с 1-6 атомами углерода, гидрокси, трифторметил, циано, нитро, карбокси, карбоалкокси с 2-7 атомами углерода, карбоалкил с 2-7 атомами углерода, амино, и алканоиламино с 1-6 атомами углерода;
R и R1 независимо представляют собой водород, галоген, алкил с 1-6 атомами углерода, алкокси с 1-6 атомами углерода, гидрокси или трифторметил;
R2 представляет собой водород, алкил с 1-6 атомами углерода, алкокси с 1-6 атомами углерода, гидрокси или трифторметил;
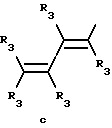
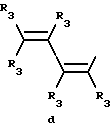
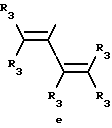
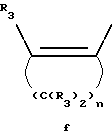
Y представляет собой радикал, выбранный из группы, включающей в себя: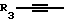
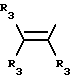
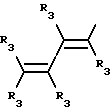
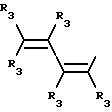
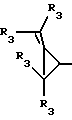
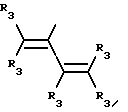
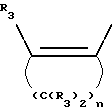
R3 независимо представляет собой водород, алкил с 1-6 атомами углерода, карбокси, карбоалкокси с 1-6 атомами углерода, фенил или карбоалкил с 2-7 атомами углерода;
n=2-4;
или к их фармацевтически приемлемой соли при условии, что все R3 в Y могут быть одинаковыми или различными.
Фармацевтически приемлемыми солями являются соли, полученные с органическими и неорганическими кислотами, такими как уксусная, молочная, лимонная, винная, янтарная, малеиновая, малоновая, глюконовая, хлористоводородная, бромистоводородная, фосфорная, азотная, серная метансульфоновая кислота или с подобными известными фармацевтически приемлемыми кислотами.
Алкильная часть алкильного, алкокси-, карбоалкокси-, карбоалкильного и алканоиламино-заместителей представляет собой прямую или разветвленную углеродную цепь. "Карбокси" означает радикал -СО2Н. "Карбоалкокси с 2-7 атомами углерода" означает радикал -CO2R", где R" является алкильным радикалом с 1-6 атомами углерода. "Карбоалкил" означает радикал -COR", где R" означает алкильный радикал с 1-6 атомами углерода. В случае, когда Х является замещенным, то он предпочтительно является моно-, ди- или тризамещенным, а наиболее предпочтительно монозамещенным. Если соединение настоящего изобретения имеет ассиметрический центр, то настоящее изобретение включает в себя отдельные R- и S-энантиомеры указанного соединения, а также их рацемическую смесь.
Из соединений настоящего изобретения предпочтительными являются соединения, в которых R, R1 и R2 являются водородом; и соединения, в которых R, R1 и R2 являются водородом, а Х - либо не замещен, либо монозамещен галогеном или алкилом с 1-6 атомами углерода.
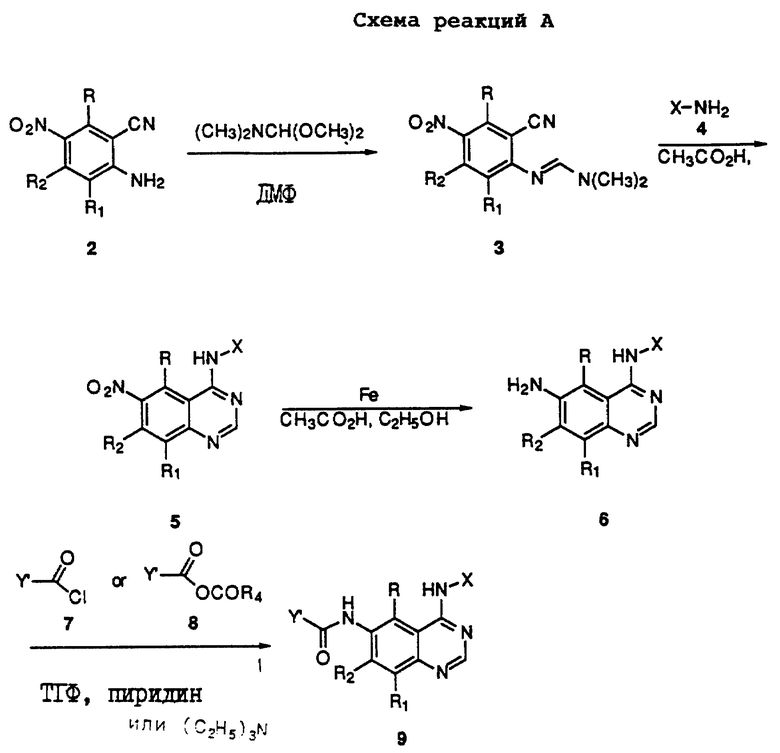
Получение соединений настоящего изобретения Формулы 9 описано ниже в схеме реакций А (см. в конце текста), где R, R1, R2, R3, Х и n являются такими, как они были определены выше, а R4 представляет собой алкил с 1-6 атомами углерода (предпочтительно изобутил). Y' представляет собой радикал, выбранный из группы, состоящей из: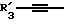
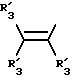
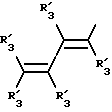
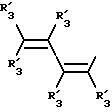
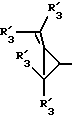
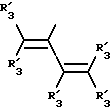
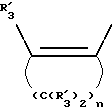
где каждый из R'3 независимо представляет собой водород, алкил с 1-6 атомами углерода, карбокси, карбоалкокси с 1-6 атомами углерода, фенил или карбоалкил с 2-7 атомами углерода. В соответствии с приведенной ниже схемой реакций А: 5-нитроантранилонитрил формулы 2 нагревают при температуре около 100oС в отсутствие или в присутствии растворителя, содержащего избыточное количество диметилацеталя диметилформамида, в результате чего получают амидин Формулы 3. Нагревание раствора амидина 3 и анилина 4 в уксусной кислоте в течение 1-5 часов дает 6-нитро-4-анилинохиназолины Формулы 5. В результате восстановления нитрогруппы с помощью восстановителя, такого как железо в смеси уксусной кислоты и спирта, при повышенной температуре получают 6-амино-4-анилинохиназолины Формулы 6. После ацилирования соединения 6 с использованием либо хлорангидрида Формулы 7, либо смешанного ангидрида Формулы 8 (который получают из соответствующей карбоновой кислоты) в инертном растворителе, таком как тетрагидрофуран (ТГФ) в присутствии органического основания, такого как пиридин или триэтиламин, получают соединения настоящего изобретения, представленные Формулой 9. В случаях, когда соединения Формул 7 или 8 имеют ассиметрический атом углерода, они могут быть использованы в качестве рацемата или отдельных R- или S-энантиомеров, причем в указанном случае соединения настоящего изобретения будут находиться в рацемической или в оптически активной R- и S-форме соответственно. 5-нитро-антранилонитрилы Формулы 2, необходимые для получения соединений настоящего изобретения, являются либо уже известными соединениями, либо они могут быть получены известными способами, подробно описанными в следующих работах: Baudet, Recl. Trav. Chim. Pay-Bas, 43, 710 (1924); Hartmans, Recl. Trav. Chim. Pays-Bas, 65, 468, 469 (1946); Taylor et al., J. Am. Chem. Soc., 82. 6058, 6063 (1960); Taylor et al., J. Am. Chem. Soc., 82, 3152, 3154 (1960); Deshpande; Seshadri, Indian J. Chem., 11, 538 (1973); Katritzky, Alan P.; Laurenzo, Kathleen S. , J. Org. Chem., 51 (1986); Niclas, Hans-Joachim; Bohle, Matthias; Rick, Jens-Detlev, Zeuner, Frank; Zoelch, Lothar, Z. Chem., 25(4), 137-138 (1985).
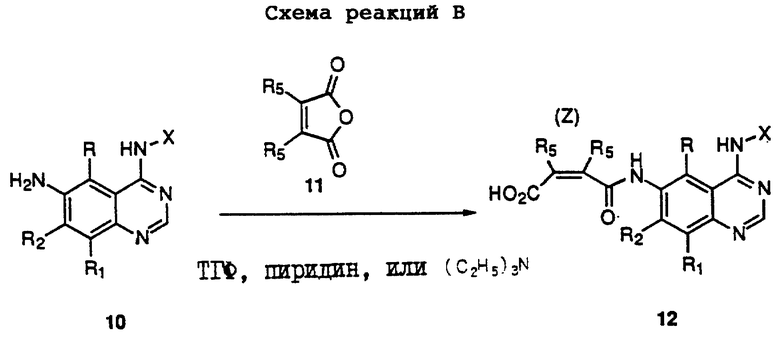
Получение соединений настоящего изобретения, представленных Формулой 12, описано ниже в схеме реакций В (см. в конце текста), где R, R1, R2, X и n определены выше. Каждый из R5 независимо представляет собой водород, фенил или алкил с 1-6 атомами углерода. В соответствии со схемой реакций В 6-амино-4-анилинохиназолины Формулы 10 (полученные, как описано в схеме реакций А) ацилируют циклическим ангидридом формулы 11 в инертном растворителе, таком как тетрагидрофуран в присутствии основного катализатора, такого как пиридин или триэтиламин.
Характерные соединения настоящего изобретения оценивали по нескольким стандартным фармакологическим тестам, которые показали, что эти соединения обладают значительной активностью в качестве ингибиторов протеинтирозинкиназ и являются антипролиферативными агентами. Как показали стандартные фармакологические тесты, соединения настоящего изобретения являются активными ингибиторами, а поэтому они могут быть использованы в качестве противоопухолевых агентов. Методики проведения этих тестов и полученные результаты описаны ниже.
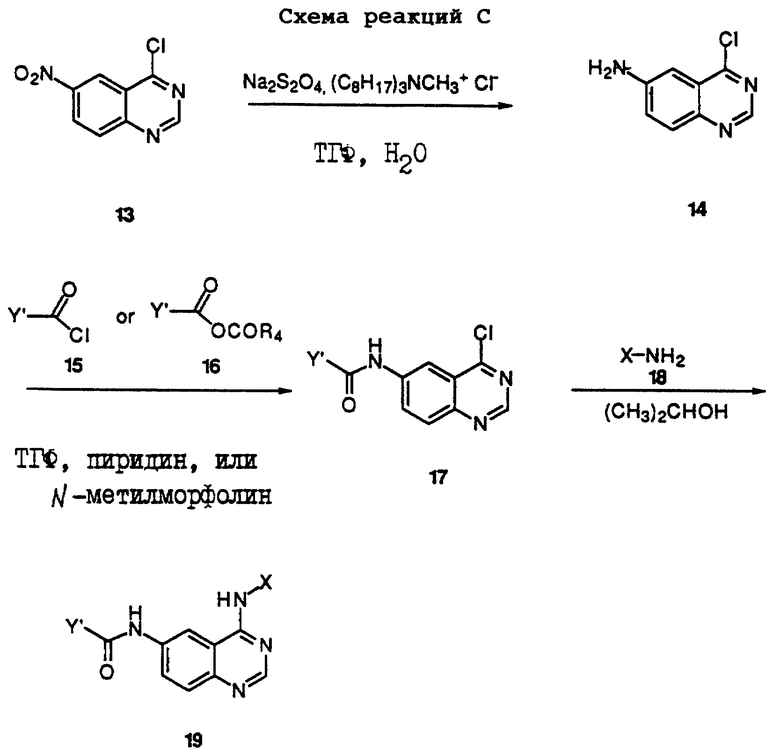
Получение соединений настоящего изобретения, представленных Формулой 19, описано ниже в схеме реакций С (см. в конце текста), где Y', R4, и Х определены выше. В соответствии с реакциями, проиллюстрированными в схеме реакций С, 4-хлор-6-нитрохиназолин Формулы 13 (Morley, J.S., and Simpson J. Chem. Soc., 360 (1948)) восстанавливают до 6-амино-4-хлорхиназолина Формулы 14 с использованием восстановителя, такого как гидросульфит натрия в смеси растворителей, содержащей инертный растворитель и воду, предпочтительно в двухфазной системе, состоящей из тетрагидрофурана и воды, в присутствии небольшого количества катализатора фазового переноса. В результате ацилирования соединения Формулы 14 с использованием либо хлорангидрида Формулы 15, либо смешанного ангидрида Формулы 16 (полученного из соответствующей карбоновой кислоты) в инертном растворителе, таком как тетрагидрофуран (ТГФ) в присутствии основания амина, например органического основания, такого как пиридин или N-метилморфолин, получают соединения Формулы 17. В тех же случаях, когда соединения Формул 15 и 16 имеют ассиметрический атом углерода, они могут быть использованы в виде рацемата или в виде отдельных R- или S-энантиомеров; причем в этом случае соединения настоящего изобретения будут находиться в рацемической или в оптически активной R- и S-форме соответственно. После нагревания соединения Формулы 17 с анилином Формулы 18 в инертном растворителе, таком как изопропанол, получают соединения настоящего изобретения, представленные Формулой 19.
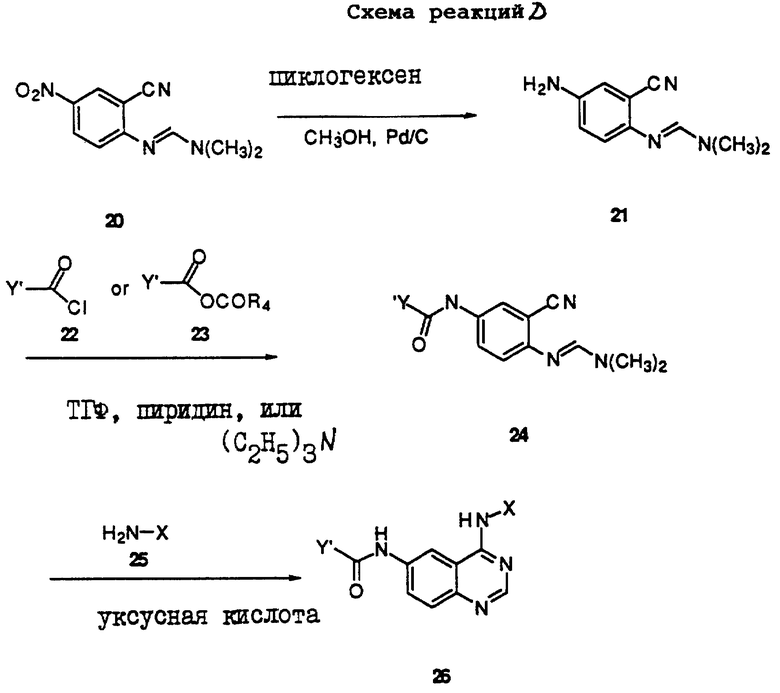
Получение соединений настоящего изобретения, представленных Формулой 26, описано ниже в схеме реакций D (см. в конце текста), где Y', R4 и Х определены выше. В соответствии с реакциями, проиллюстрированными в схеме Реакций D, нитрогруппу соединения Формулы 20 (полученного, как описано в схеме реакций А) восстанавливают с получением соответствующего аминосоединения Формулы 21, используя палладиевый катализатор и источник водорода, который может быть непосредственно самим водородом или циклогексеном. После ацилирования соединения Формулы 21 с использованием либо хлорангидрида Формулы 22, либо смешанного ангидрида Формулы 23 (полученного из соответствующей карбоновой кислоты) в инертном растворителе, таком как тетрагидрофуран (ТГФ) в присутствии основания амина, например органического основания, такого как пиридин или N-метилморфолин, получают соединения Формулы 24. В тех случаях, когда соединения Формул 22 или 23 имеют ассиметрический атом углерода, они могут быть использованы в виде рацемата или в виде отдельных R- или S-энантиомеров, причем в этом случае соединения настоящего изобретения будут находиться в рацемической или в оптически активной R- или S-энантиомерной форме соответственно. После нагревания соединения Формулы 24с анилином Формулы 25 в инертном растворителе, таком как уксусная кислота, получают соединения настоящего изобретения, представленные Формулой 26.
Ингибирование киназного рецептора эпидермального фактора роста (EGF-R)
Тестируемые соединения оценивали на их способность ингибировать фосфорилирование тирозинового остатка пептидного субстрата, катализируемое рецептором эпидермального фактора роста, обладающим ферментативной киназной активностью. Пептидный субстрат (RR-SRC) имеет последовательность
arg-arg-leu-ile-glu-asp-ala-glu-tyr-ala-ala-arg-gly
Фермент получали в виде мембранного экстракта клеток А431 (Американская коллекция типовых культур, Роквилл, МD). Клетки А431 культивировали в колбах Т175 до состояния 80%-ной сплошности. Затем клетки два раза промывали забуференным фосфатным физиологическим раствором (РВS), не содержащим СА2+. После этого колбы с клетками в 20 мл РВS, содержащем 1,0 мМ этилендиаминтетрауксусной кислоты (EDTА), вращали в течение 1,5 часа при комнатной температуре, а затем центрифугировали при скорости 600 об/мин в течение 10 минут. Клетки солюбилизировали в 1 мл на 5•106 клеток охлажденного буфера для лизиса {10 мМ 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота (НЕРЕS), рН 7,6, 10 мМ NaCl, 2 мМ EDTA, 1 мМ фенилметилсульфонилфторид (PMSF), 10 мг/мл апротинина, 10 мг/мл лейпептина, 0,1 мМ ортованадат натрия} в гомогенизаторе Даунса с 10 ходами при охлаждении льдом. Лизат центрифугировали при 600 об/мин в течение 10 минут до осветления клеточного дебриса, а затем надосадочную жидкость центрифугировали при 100000 об/мин в течение 30 минут при 4oС. После этого мембранный осадок суспендировали в 1,5 мл буфера HNG (50 мМ НЕРЕS, рН 7,6, 125 мМ NaCl, 10%-ный глицерин). Мембранный экстракт разделяли на аликвоты, после чего сразу замораживали в жидком азоте и хранили при -70oС.
Испытуемые соединения приготавливали в виде концентрированных (10 мг/мл) растворов для хранения в 100% диметилсульфоксиде (ДМСО). Перед проведением эксперимента эти концентрированные растворы для хранения разводили до 500 мМ буфером (30 мМ Нереs, рН 7,4), а затем подвергали серийному разбавлению до нужной концентрации.
Аликвоту мембранного экстракта А431 (10 мг/мл) разбавляли в 30 мМ НЕРЕS (рН 7,4), получая концентрацию белка 50 мкг/мл. К 4 мкл ферментного препарата добавляли EGF (1 мкл при концентрации 12 мкг/мл) и инкубировали на льду в течение 10 минут, после чего добавляли 4 мкл испытуемого соединения или буфера и полученную смесь инкубировали на льду в течение 30 минут. Затем к этой смеси добавляли 32Р-АТФ (10 мКюри/мл, разбавленный 1:10 в буфере для анализа только пептидом-субстратом в концентрации 0,5 мМ (в контрольных реакциях испытуемое соединение отсутствовало) и полученную смесь оставляли на 30 минут при 30oС для прохождения реакции. Реакцию прекращали добавлением 10% ТСА, а затем выдерживали на льду, по крайней мере, 10 минут, после чего пробирки микроцентрифугировали на полной скорости в течение 15 минут. Часть надосадочных жидкостей наносили каплями на фосфоцеллюлозные (Р81) диски и два раза промывали 1%-ной уксусной кислотой, а затем водой каждый раз в течение 5 минут, после чего проводили подсчет импульсов. Данные по ингибированию, полученные для характерных соединений настоящего изобретения, представлены ниже в Таблице 1. Величина IС50 представляет собой концентрацию испытуемого соединения, необходимую для уменьшения общего количества фосфорилированного сусбрата на 50%. Процент (%) ингибирования для испытуемого соединения определяли, по крайней мере, для трех различных концентраций, а величину IС50 определяли по кривой зависимости "доза-ответ". Процент ингибирования вычисляли по следующей формуле:
% ингибирования= 100 [имп. /мин (лекарственное соединение) /имп./мин (контроль)] • 100
где "имп. /мин (лекарственное соединение)" обозначает число импульсов в минуту, которое соответствует количеству радиоактивно меченной АТФ (g-33Р), включенному в пептидный субстрат RR-SRC под действием фермента в присутствии испытуемого соединения при 30oС за 30 минут; причем измеренное путем подсчета импульсов в жидкой фазе "имп./мин (контроль)" означает число импульсов в минуту, которое соответствует количеству радиоактивно меченной АТФ (g-33Р), включенному в пептидный сусбтрат RR-SRC под действием фермента при 30oС за 30 минут в отсутствии испытуемого соединения; измеренное путем подсчета импульсов в жидкой фазе. В полученные значения импульсов были внесены поправки на фоновое число импульсов, продуцируемое АТФ при отсутствии ферментативной реакции. Величины 1С50, представленные в Таблице 1, представляют собой средние значения, полученные из нескольких экспериментов.
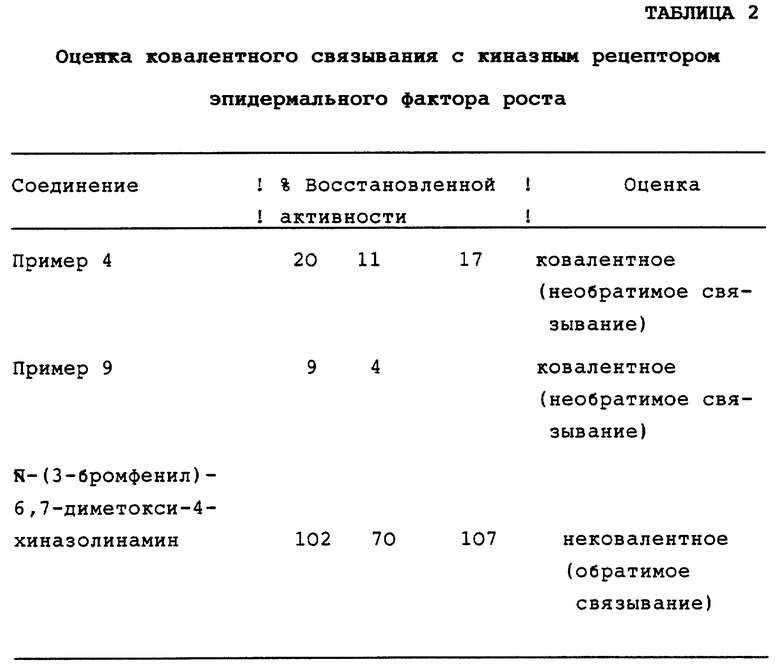
Оценка ковалентного связывания испытуемого соединения с киназным рецептором эпидермального фактора роста
Аликвоту ферментного экстракта А431 (полученного как описано выше) разбавляли до концентрации 50-100 мкг/мл буфером (30 мМ) Hepes при рН 7,4, содержащим EGF в концентрации 12 мкг/мл, так, чтобы в стандартных экспериментальных условиях имела место приблизительно 2%-ная реакция, а конечная концентрация EGF составляла 2,4 мкг/мл (как в стандартном анализе, описанном выше). Перед использованием полученную смесь инкубировали в течение, по крайней мере, 10 минут при 4oС. Этот ферментный препарат использовали в описанных ниже тестах, проведенных методом диализа.
К 60 мкл ферментного препарата добавляли 48 мкл испытуемого соединения, растворенного в 5%-ном диметилсульфоксиде (ДМСО) (или только 48 мкл 5%-ного ДМСО для контроля). Для полной гарантии 80-90%-ного ингибирования концентрации испытуемых соединений выбирали так, чтобы они в 20-100 раз превышали величины 1C50. Раствор, содержащий фермент и ингибитор, инкубировали в течение 45-60 минут при 4oС. Для недиализованного контроля 9 мкл-аликвоту раствора фермента и ингибитора оценивали в соответствии со стандартной схемой, описанной выше. Для теста, проводимого методом диализа, 60 мкл-аликвоту раствора фермента и ингибитора помещали в ячейку микродиализатора (Pierce Microdialyser System 100) и диализовали при 4oС против 30 мМ буфера Hepes, содержащего 1,25 мкг/мл EGF, в течение 24 часов с двумя заменами буфера (перед каждой заменой диализ проводили в течение минимум 3 часов). При этом использовали мембраны с пропускной способностью по молекулярной массе 8000. 9 мкл-аликвоту (по крайней мере в двойном повторе) диализованного раствора оценивали на активность в соответствии со стандартной схемой, описанной выше. После диализа фермент, к которому не добавляли испытуемое соединение, сохранял 50-90% от своей первоначальной активности. Диализованные растворы испытуемых соединений, в которые не был добавлен фермент, также оценивали для того, чтобы убедиться в диализуемости испытуемых соединений.
Если после диализа ферментативная активность не восстанавливается, то это указывает на ковалентное связывание испытуемых соединений с ферментом (необратимое ингибирование). Если после диализа наблюдается высокая степень восстановления ферментативной активности, то это свидетельствует о том, что испытуемое соединение связывается с ферментом нековалентно (обратимое ингибирование). Ковалентное связывание может быть определено как % восстановленной активности, вычисленный по следующей ниже формуле, исходя из данных % ингибирования, полученных до и после диализа:
% восстановленной активности=[(% ингибировании (до диализа) - % ингибирования (после диализа)] % ингибирования (до диализа)] • 100.
Величина % восстановленной активности, близкая к 100%, свидетельствует о нековалентном связывании (обратимое ингибирование). Величина % восстановленной активности, являющаяся намного меньше 100%, свидетельствует о ковалентном связывании (необратимое ингибирование). Результаты, полученные для ковалентного связывания характерных соединений настоящего изобретения с EGF-R-киназой, представлены ниже в таблице 2. Для сравнения, в Таблице 2 также приводятся данные связывания для N-(3-бромфенил)-6,7-диметокси-4-хиназолинамина. Этот хиназолиновый ингибитор был идентифицирован как сильный ингибитор EGF-R-киназы (Fry, D.W., et al., Science, 265, 1093 (1994); Rewcastle G. W., et al., J. Med. Chem., 38, 3482 (1995), и Bridges, A.J., et al., J. Med. Chem. 39, 267, (1996)) и описан в заявке на европейский патент 566266 A1. Результаты многократных независимых оценок для каждого исследуемого соединения представлены в Таблице 2.
Результаты, представленные в Таблице 2, указывают на то, что соединения настоящего изобретения являются необратимыми ингибиторами ЕGF-R-киназы, действующими путем образования ковалентной связи с аминокислотным остатком, присутствующим в ферменте. Отсюда можно сделать вывод, что соединения настоящего изобретения явно отличаются от обычных 4-анилинохиназолинов, таких как N-(3-бромфенил)-6,7-диметокси-4-хиназолинамин, который действует путем обратимого связывания. Как будет показано ниже, такое отличие связывающей способности соединений настоящего изобретения от связывающей способности известных хиназолиновых ингибиторов приводит к значительному увеличению биологической активности соединений настоящего изобретения, что придает им большую терапевтическую ценность.
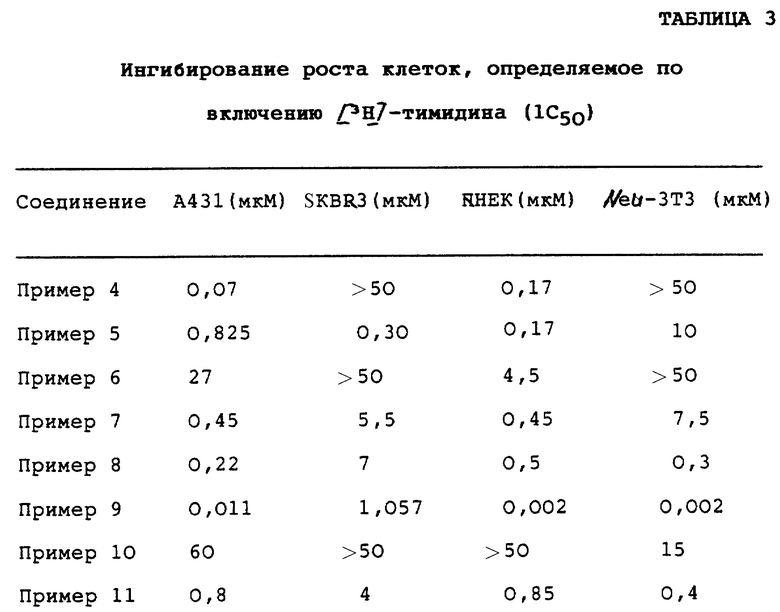
Ингибирование клеточного роста, измеренное с помощью включения [3H]-тимидина
Характерные соединения настоящего изобретения оценивали на их способность к in vitro-ингибированию роста клеточных линий, описанных ниже. Количественную оценку ингибирования проводили измерением снижения включения радиоактивно меченного тимидина при культивировании клеток в присутствии ингибитора. Клеточные линии А431 и SKBP3 были получены из Американской коллекции типовых культур (Rockville, MD). Клетки Neu-3Т3 были получены путем трансфекции мышиных фибробластов NIH 3Т3 активированным крысиным Neu онкогеном. Клетки NHEK были получены из Clonetcs (San Diego, СА). Клетки культивировали традиционным способом во влажной камере в атмосфере воздуха с 5% СО2. Указанные клеточные линии зависят от факторов роста, являющихся лигандами для рецепторов тирозинкиназы, которые, в свою очередь, являются мишенями для соединений настоящего изобретения; при этом указанные клеточные линии имеют следующие характеристики:
А431: клетки карциномы кожного покрова человека, сверхэкспрессирующие EGFR
Neu-3Т3: клетки N1Н 3Т3, трансфецированные активированным Neu онкогеном
NНЕК: EGF-зависимые нормальные кератиноциты эпидермиса человека
SKBR3: раковые клетки молочной железы человека, сверхэкспрессирующие ErbB2 ген.
Клеточные линии культивировали в следующих соответствующих средах:
А431: модифицированная по способу Дульбекко среда Игла с высоким содержанием глюкозы, BR1/Gibco (10% фетальная бычья сыворотка (FВS), глутамин, пенициллин-стрептомицин) Dulbecco, R., Freeman, G. Virology 8, 396 (1959).
Nue-3Т3: модифицированная по способу Дульбекко среда Игла с высоким содержанием глюкозы (10% фетальная бычья сыворотка, глутамин, пенициллин-стрептомицин).
SKBR3: Roswell Park Memorial Institute 1640 W/GLU (10% FBS, глутамин, пенициллин-стрептомицин) Moore, G. E., Gerner, R.E. & Franklin, H.A. А.М.А 199, 516 (1967).
NHEK: среда для культивирования кератиноцитов, Clonetics, Boyce, S.T. & Ham, R.G. In vitro 17, 239 (Abstract 159) (1981).
Клетки засевали при плотности 10 000 клеток на ячейку в 96-ячеечные планшеты в полную среду и культивировали до логарифмической фазы роста (лог-фазы). На этой стадии полную среду заменяли средой, содержащей 0,5% FВS (для клеток, растущих в 10% FBS), или средой, не содержащей эпидермального фактора роста (EGF) (для клеток, растущих в бессывороточной среде). После инкубирования в течение ночи в средах с низким содержанием сыворотки (или в средах, не содержащих EGF) к клеткам добавляли испытуемые соединения, после чего клетки выдерживали в присутствии соединений в течение 48-72 часов. Затем среду с испытуемым соединением удаляли и снова добавляли полную среду. После этого клетки культивировали в течение 18 часов. Затем клетки инкубирования в [3Н]-тимидине (1 мКюри/мл в средах, содержащих сыворотку/EGF) в течение 4 часов. Лизис клеток проводили в 0,5 М NaOH в течение, по крайней мере, 30 минут при 37oС, после чего делали анализ на радиоактивность.
Данные ингибирования роста клеток представлены в Таблице 3. Величина 1C50 представляет собой концентрацию испытуемого соединения, необходимую для уменьшения количества включения [3H]-тимидина на 50%. Процент (%) ингибирования испытуемым соединением определяли, по крайней мере, для трех различных концентраций, а величину 1C50 определяли по кривой зависимости "доза-ответ". Процент ингибирования вычисляли по следующей формуле:
% ингибирования = 100-[имп. /мин (лекарственное соединение)/ имп./мин (контроль )] • 100
где "имп. /мин (лекарственное соединение)" означает число импульсов в минуту, которое соответствует количеству [3H]-тимидина, включенного в ДНК при культивировании клеток в присутствии испытуемого соединения; причем указанное число измеряли путем подсчета импульсов в жидкой фазе;
"имп./мин (контроль)" означает число импульсов в минуту, которое соответствует количеству [3H] -тимидина, включенного в ДНК при культивировании клеток в отсутствие испытуемого соединения; причем указанное число измеряли путем подсчета импульсов в жидкой фазе.
In vivo - Ингибирование роста опухолей эпидермиса человека (А431)
Для проведения стандартных фармакологических тестов in vivo были использованы самки "голых" мышей (nu/nu) BALB/c (Charles River, Walmington, MA).
Клетки эпидермоидной карциномы человека А-431 (Американская коллекция типовых культур, Rockville, Maryland, CRL-155) культивировали in vitro, как описано выше. Порцию 5•106 клеток подкожно (S.с.) инъецировали мышам. Когда опухоли достигали массы 100-150 мг, мышей произвольно распределяли на обрабатываемые группы (день 0). В дни 1, 5 или 9 либо в дни 1-10 после начала эксперимента (т. е. со дня 0) мышам обрабатываемой группы вводили один раз в день внутрибрюшинно (i. р. ) соединения в дозах 80, 40 или 20 мг/кг в 0,2% Klucel. Контрольным животным лекарственные соединения не вводили. Массу опухоли определяли каждые 7 дней [(длина х ширина2)/2] в течение 28 дней после начала эксперимента. Величину относительного роста опухоли (средняя масса опухоли в дни 7, 14, 21 и 28, деленная на среднюю массу опухоли в день 0) определяли для каждой обрабатываемой группы. % Т/С (опухоль/контроль) определяли делением величины относительного роста опухоли для обрабатываемой группы на величину относительного роста опухоли для контрольной группы (обработанной плацебо) и умножением на 100. Испытуемое соединение считается активным, если % Т/С составляет ≤42%.
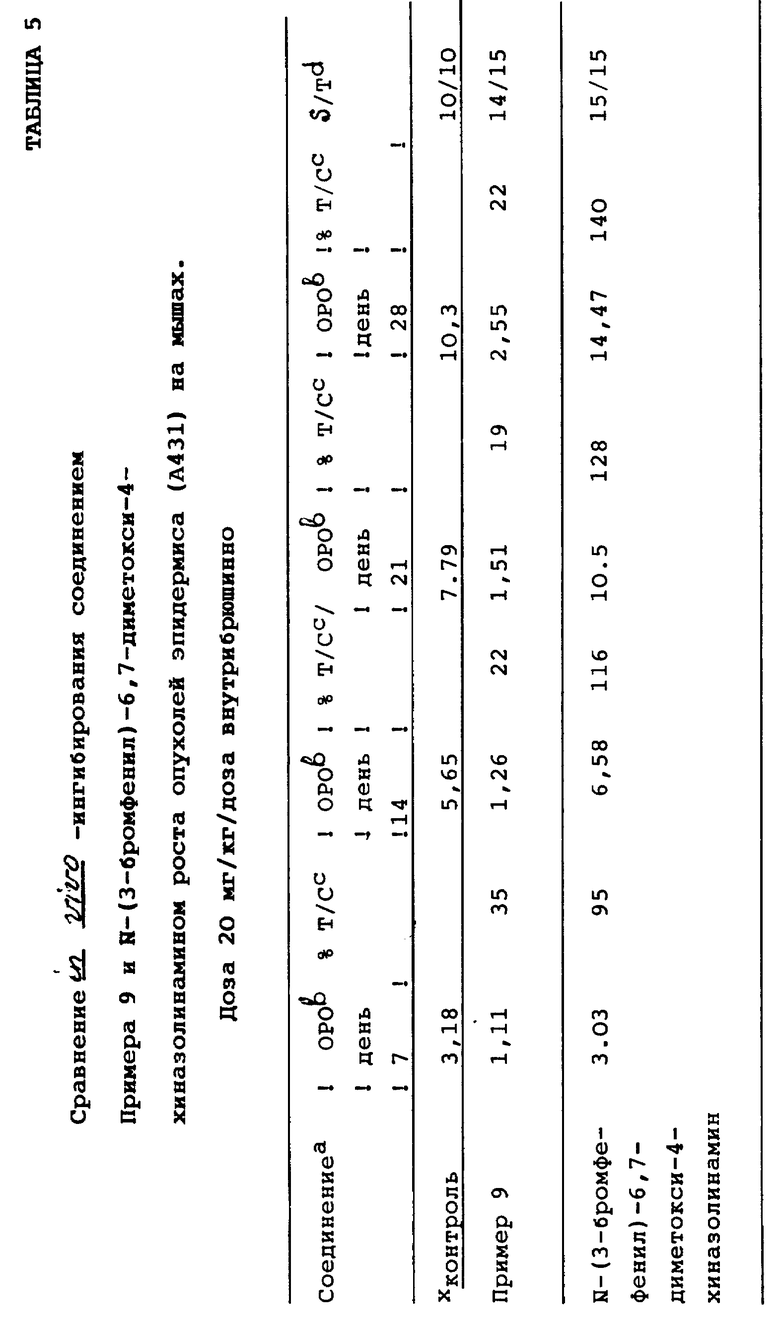
Данные ингибирования, полученные для соединения Примера 9, представлены ниже в Таблице 4.
Сравнение способности соединения Примера 9 и N-(3-бромфенил)-6,7-диметокси-4-хиназолинамина ингибировать рост опухолей эпидермиса человека (А431) in vivo показано ниже в Таблице 5. N-(3-бромфенил)-6,7-диметокси-4-хиназолинамин был выбран в качестве соединения для сравнения потому, что это соединение хиназолина было идентифицировано как сильный ингибитор EGF-R-киназы (Fry, D. W. , et al., Science, 265, 1093 (1994); Rewcastle G.W., et al., J. Med. Chem., 38, 3482 (1995); Bridges, A.J., et. al., J. Med. Chem., 39, 267 (1996)), который был описан в заявке на европейский патент 566266 А1.
Как показано в Таблицах 4-5, соединения настоящего изобретения ингибируют рост опухолей человека у млекопитающих, а поэтому они могут быть использованы в качестве противоопухолевых средств. В этой связи следует отметить, что соединения настоящего изобретения существенно отличаются от известных 4-анилинохиназолинов, таких как N-(3-бромфенил)-6,7-диметокси-4-хиназолинамин, который не обладает противоопухолевой активностью.
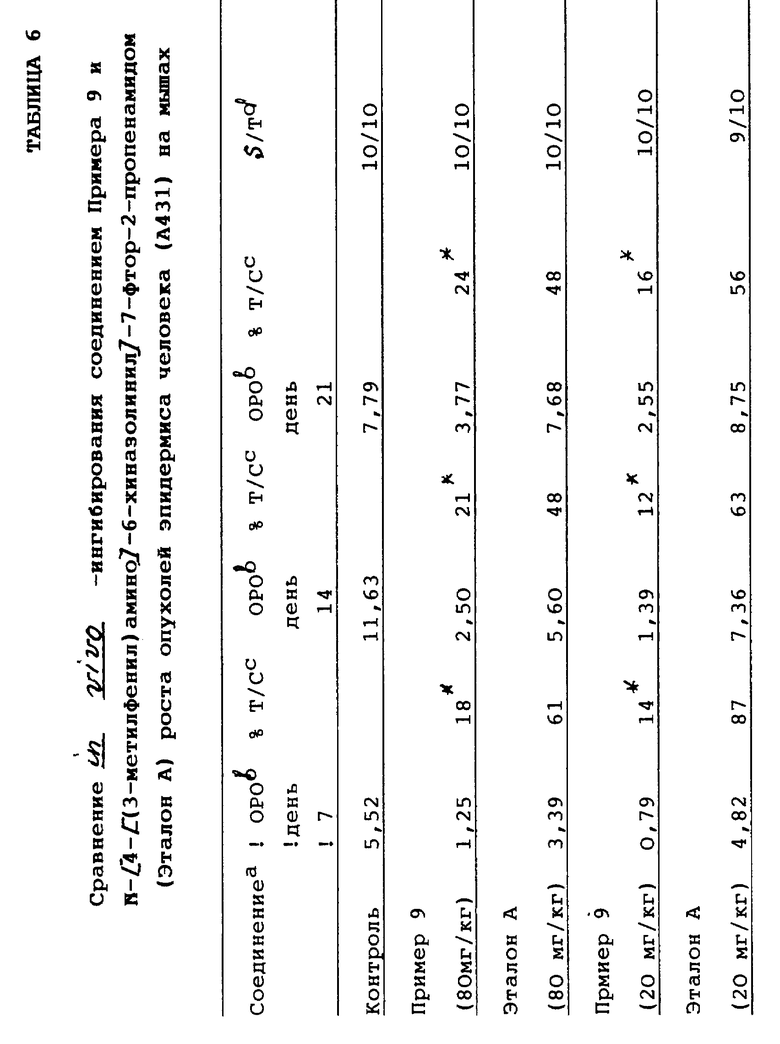
Способность соединения Примера 9 ингибировать рост эпидермоидных опухолей человека (А431) in vivo сравнивали со способностью двух структурно родственных соединений N-[4-(3-метилфенил)амино]-6-хиназолинил]-7-фтор-2-пропенамид (обозначаемый далее эталоном А) и N-[4-[(3-бромфенил)амино]-6-хиназолинил]-бутанамид (обозначаемый далее Эталоном В), которые были описаны в заявках на европейские патенты 635488 A1 и 566226 A1 соответственно. Результаты этого сравнения показаны в Таблицах 6 и 7.
Результаты, представленные в Таблицах 6 и 7, свидетельствуют о том, что соединение Примера 9, являющееся характерным соединением настоящего изобретения, значительно (р<0,01) ингибирует in vivo-рост опухоли эпидермиса человека. При этом наиболее структурно близкие соединения, описанные в заявках на европейские патенты 635488 (Эталон А) и 566226 А1 (Эталон В), обнаруживают значительно меньшую активность, чем соединение Примера 9, и кроме того, оба эти соединения оказались не способными к значительному снижению роста опухоли при обеих испытуемых дозах.
Исходя из результатов, полученных для характерных соединений настоящего изобретения, можно сделать вывод, что эти соединения могут быть использованы для лечения опухолевых заболеваний, ингибирования роста опухолей или уничтожения опухолей. В частности, соединения настоящего изобретения могут быть использованы для лечения, ингибирования роста или уничтожения опухолей, экспрессирующих рецептор EGF (EGF-R), таких как опухоли молочной железы, почек, мочевого пузыря, полости рта, гортани, пищевода, желудка, толстой кишки, яичников или легких.
Соединения настоящего изобретения могут быть введены в чистом виде либо они могут быть введены в сочетании с одним или несколькими фармацевтически приемлемыми носителями, например растворителями, разбавителями и т.п. Соединения настоящего изобретения могут быть введены перорально в форме таблеток; капсул; диспергируемых порошков; гранул или суспензий, содержащих, например, от около 0,05 до 5% суспендирующего агента; сиропов, содержащих, например, от около 10 до 50% сахара; и эликсиров, содержащих, например, от около 20 до 50% этанола, и т.п.; или парентерально в форме растворов для инъекций или суспензий, содержащих от около 0,05 до 5% суспендирующего агента в изотонической среде. Такие фармацевтические препараты могут содержать, например, от около 0,05 до около 90% активного ингредиента в сочетании с носителем, а в основном, примерно 5-60 мас.% активного ингредиента.
Используемая эффективная доза активного ингредиента может варьироваться в зависимости от конкретно используемого соединения, способа его введения и тяжести заболевания. Однако, в основном, удовлетворительные результаты могут быть получены при введении суточной дозы соединения, составляющей от около 0,5 до около 1000 мг на кг массы тела животного, причем указанная доза может быть разделена, но необязательно, на 2-4 приема в день либо она может быть введена в виде лекарственной формы с пролонгированным высвобождением. Для наиболее крупных млекопитающих суточная доза составляет от около 1 до 1000 мг, а предпочтительно от около 2 до 500 мг. Лекарственные формы, пригодные для внутреннего употребления, содержат от около 0,5 до 1000 мг активного соединения в однородной смеси с твердым или жидким фармацевтически приемлемым носителем. Указанная схема приема лекарственного средства может быть скорректирована для получения оптимального терапевтического эффекта. Так, например, суточная доза может быть введена в виде нескольких дробных доз либо она может быть пропорционально снижена, если это необходимо для данного режима лечения.
Указанные активные соединения могут быть введены перорально, а также внутривенно, внутримышечно или подкожно. Твердыми носителями, соответствующими природе активного ингредиента и подходящими для данной формы введения, являются крахмал, лактоза, дикальцийфосфат, микрокристаллическая целлюлоза, сахароза и каолин, а подходящими жидкими носителями являются стерильная вода, полиэтиленгликоли, неионогенные поверхностно-активные вещества и пищевые масла, такие как арахисовое и кунжутное масло. Кроме того, могут быть также использованы добавки, широко применяемые при изготовлении фармацевтических композиций, например, такие как ароматизаторы, красители, консерванты и антиоксиданты, а в частности, витамин Е, аскорбиновая кислота, ВНТ и ВНА.
С точки зрения простоты изготовления и введения предпочтительными фармацевтическими композициями являются твердые композиции, а в частности, таблетки и капсулы с твердым или жидким наполнителем. При этом предпочтительным способом введения соединений является пероральное введение.
В некоторых случаях может оказаться предпочтительным вводить соединения настоящего изобретения непосредственно через дыхательные пути в виде аэрозолей.
Указанные активные соединения могут быть также введены парентерально или интраперитонально. Растворы или суспензии, содержащие указанные активные соединения в виде свободного основания или фармакологически приемлемой соли, могут быть получены в воде, соответствующим образом смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии могут быть также получены в глицерине, жидком полиэтиленгликоле или в их смесях в масле. При стандартных условиях хранения и использования эти препараты содержат консервант для предупреждения роста микроорганизмов.
Фармацевтическими формами, подходящими для введения путем инъекции, являются стерильные водные растворы или дисперсии, а также стерильные порошки для быстрого приготовления стерильных растворов для инъекций или дисперсий непосредственно перед их использованием. В любом случае лекарственная форма для инъекций должна быть стерильной и должна быть достаточно жидкой, такой чтобы ее можно было легко вводить с помощью шприца. Кроме того, указанная лекарственная форма должна быть стабильной в условиях изготовления и хранения и должна быть защищена от заражения микроорганизмами, такими как бактерии и грибки. В качестве носителя может быть использован растворитель или дисперсионная среда, содержащая, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла.
Для лечения рака соединения настоящего изобретения могут быть использованы в сочетании с другими противоопухолевыми средствами или в сочетании с облучением. Эти другие противоопухолевые средства или лучевая терапия могут быть использованы в разное время или одновременно с использованием соединений настоящего изобретения. Такая комплексная терапия может иметь синергическое действие, в результате которого может быть получен лучший терапевтический эффект. Так, например, соединения настоящего изобретения могут быть использованы в сочетании с митотическими ингибиторами, такими как таксол или винбластин; алкилирующими агентами, такими как цисплатин или циклофозамид; антиметаболитами, такими как 5-фторурацил или гидроксимочевина; интеркаляторами ДНК, такими как адриамицин или блеомицин; ингибиторами топоизомеразы, такими как этопозид или камптотецин; и антиэстрогенами, такими как тамоксифен.
В нижеприведенных примерах описано получение характерных соединений настоящего изобретения.
ПРИМЕР 1
N'-(2-Циано-4-нитрофенил)-N,N-диметилформамидин
40,8 г 5-нитроантранилонитрила и 40 мл N-диметилформамидодиметилацеталя нагревали на паровой бане в течение двух часов. После удаления растворителей при пониженном давлении остаток растворяли в метиленхлориде. Полученный раствор пропускали через Magnesоl и растворитель удаляли. После промывки эфиром получали 50,8 г N'-(2-циано-4-нитрофенил)-N,N-диметилформамидина.
ПРИМЕР 2
N'-(3-Бромфенил)-6-нитро-4-хиназолинамин
Раствор 23,74 мл 3-броманилина и 40,5 г N-(2-циано-4-нитрофенил)-N,N-диметилформамидина в 100 мл ледяной уксусной кислоты перемешивали и нагревали на масляной бане в течение 1,5 часа при температуре 148oС. После охлаждения и фильтрования было получено твердое вещество N-(3-бромфенил)-6-нитро-4-хиназолинамин с количественным выходом; т. пл. 267-270oС; Массс-спектр (m/e): 345.
ПРИМЕР 3
N-(3-Бромфенил)-4,6-хиназолиндиамин
Смесь, содержащую 34,5 г N-(3-бромфенил)-6-нитро-4-хиназолинамина и 16,8 г порошкообразного железа в 150 мл этанола и 150 мл ледяной уксусной кислоты, нагревали на масляной бане в течение двух часов при температуре 120oС. Полученное твердое вещество фильтровали и к фильтрату добавляли твердый карбонат натрия, в результате чего образовывалось твердое вещество. После фильтрации твердое вещество экстрагировали метанолом. Экстракты обрабатывали активированным углем и выпаривали с получением твердого вещества. После промывки твердого вещества эфиром получали 27,5 г N-(3-бромфенил)-4,6-хиназолиндиамина; Масс-спектр (m/е): 315.
ПРИМЕР 4
4-[[4-[(3-Бромфенил)амино] -6-хиназолинил] амино]-4-оксо-(Z)-2-бутеновая кислота
К 1,6 г N-(3-бромфенил)-4,6-хиназолиндиамина и 0,6 г малеинового ангидрида добавляли 15 мл пиридина. После перемешивания в течение ночи растворители удаляли на роторном испарителе. Твердое вещество растворяли приблизительно в 400 мл горячего этанола, а нерастворившееся вещество отфильтровали, в результате чего было получено 0,33 г 4-[[4-[(3-бромфенил)амино]-6-хиназолинил]амино]-4-оксо-(Z)-2-бутеновой кислоты; Масс-спектр (m/е): М+Н: 413, 415.
ПРИМЕР 5
Этиловый эфир 4-[[4-[(3-бромфенил)амино]-6-хиназолинил]-амино]-4-оксо-(Е)-2-бутеновой кислоты
Раствор N-(3-бромфенил)-4,6-хиназолиндиамина в 15 мл пиридина охлаждали на ледяной бане, а затем по капле добавляли раствор 1,22 г этилфумарилхлорида в 10 л метиленхлорида. После перемешивания в течение полутора часов реакционную смесь оставляли для нагревания до комнатной температуры, затем растворители удаляли при пониженном давлении, а остаток обрабатывали водой. Полученное красное твердое вещество отфильтровывали и экстрагировали горячим ацетоном. После фильтрования нерастворившегося вещества фильтрат концентрировали, в результате чего было получено 0,45 г этилового эфира 4-[[4-[(3-бромфенил)амино]-6-хиназолинил]амино]-4-оксо-(Е)-2-бутеновой кислоты; т. пл. 259-263oС: Масс-спектр (m/е): М+Н; 441, 443.
ПРИМЕР 6
N-[4-[(3-Бромфенил)амино]-6-хиназолинил]-3-метил-2-бутенамид
Раствор, содержащий 1,58 г N-(3-бромфенил)-4,6-хиназолиндиамина и 15 мл пиридина, охлаждали на ледяной бане, а затем по капле добавляли раствор, содержащий 0,67 мл 3,3-диметилакрилоилхлорида и 7 мл эфира. После перемешивания и охлаждения в течение 2 часов растворители удаляли при пониженном давлении. Остаток обрабатывали водой и полученное твердое вещество перекристаллизовывали из метилцеллюлозы, в результате чего получали 0,97 г N-[4-[(3-бромфенил)амино] -6-хиназолинил] -3-метил-2-бутенамида; т. пл. 300-301oС; Масс-спектр (m/е): 396, 398.
ПРИМЕР 7
N-[4-(3-Бромфенил)амино]-6-хиназолинид]-(E)-бутенамид
Раствор, содержащий 1,6 г N-(3-бромфенил)-4,6-хиназолиндиамина и 15 мл пиридина, охлаждали на ледяной бане, а затем по капле добавляли раствор, содержащий 0,57 мл транс-кротоноилхлорида и 6 мл эфира. После перемешивания и охлаждения в течение 2 часов растворители удаляли при пониженном давлении. Образовавшийся остаток обрабатывали водой, а затем твердое вещество перекристаллизовывали из н-бутанола, в результате чего получали 0,69 г N-[4-[(3-бромфенил)амино] -6-хиназолинил] -(Е)-2-бутенамида; т. пл. 153-160oС; Масс-спектр (m/e): M+H 383, 385.
ПРИМЕР 8
N-[4-(3-Бромфенил)амино]-6-хиназолинил]-2-метил-2-пропенамид
Раствор, содержащий 1,6 г N-(3-бромфенил)-4,6-хиназолиндиамина и 15 мл пиридина, охлаждали на ледяной бане, а затем по каплям добавляли раствор, содержащий 0,59 мл метакриоилхлорида и 6 мл эфира. После перемешивания и охлаждения в течение двух часов растворители удаляли при пониженном давлении. Остаток обрабатывали водой и полученное твердое вещество растворяли в нагретом н-бутаноле. Этот раствор охлаждали добавлением эфира, в результате чего получали 0,44 г N-[4-[(3-бромфенил)амино]-6-хиназиолинил]-2-метил-2-пропенамида; т. пл. 40-245oС, Масс-спектр (m/e): M+H 383, 385.
ПРИМЕР 9
N-[4-[(3-Бромфенил)амино]-6-хиназолинил]-2-бутинамид
Раствор 0,50 г 2-бутиновой кислоты в 10 мл тетрагидрофурана охлаждали на ледяной бане. К этому раствору добавляли изобутилхлороформат (0,79 мл), а затем N-метилморфолин (0,66 мл). После выдерживания в течение приблизительно одной минуты к смеси добавляли раствор 1,6 г N-(3-бромфенил)-4,6-хиназолиндиамина в 10 л пиридина. Полученную реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Растворители удаляли при пониженном давлении и образовавшееся твердое вещество перекристаллизовывали из н-бутанола, в результате чего получали 1,07 г N[4-[(3-бромфенил)амино]-6-хиназолинил]-2-бутинамида; масс-спектр (m/е): 381, 383.
ПРИМЕР 10
4-[[4-[(3-Бромфенил)амино] -6-хиназолинил] амино]-4-оксо-(Е)-2-бутеновая кислота
К 2,3 г этилового эфира 4-[[4-[(3-бромфенил)амино]-6-хиназолинил]амино] -4-оксо-(Е)-2-бутеновой кислоты (Пример 5) в 25 мл этанола добавляли 2,5 мл водного раствора 10 н. гидроксида натрия. После перемешивания в течение одного часа к раствору добавляли 2,1 мл концентрированной соляной кислоты и эту реакционную смесь перемешивали еще 2 часа. Полученное твердое вещество перекристаллизовывали из н-бутанола, в результате чего получали 0,97 г 4-[[4-[(3-бромфенил)амино] -6-хиназолинил] амино] -4-оксо-(Е)-2-бутеновой кислоты; масс-спектр (m/е): М+Н 413.
ПРИМЕР 11
N-[4-[(3-Бромфенил)амино]-6-хиназолинил]-2,4-гексадиенамид
Раствор 2,4-гексадиеновой кислоты (0,67 г) в 10 мл тетрагидрофурана охлаждали на ледяной бане. К раствору добавляли изобутилхлороформат (0,79 мл), а затем N-метилморфолин (0,66 мл). После выдерживания приблизительно в течение 1 минуты к смеси добавляли раствор 1,6 г N-(3-бромфенил)-4,6-хиназолиндиамина в 10 мл пиридина. Полученную реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. После удаления растворителей при пониженном давлении твердое вещество перекристаллизовывали и получали 1,0 г N-[4-[(3-бромфенил)-амино]-6-хиназолинил]-2,4-гексадиенамида; т. пл. 258-260oС.
ПРИМЕР 12
N-[4-[(3-Бромфенил)амино]-6-хиназолинил]-2-циклопентенамид
Раствор 0,43 г 2-циклопентеновой кислоты в 5 мл тетрагидрофурана охлаждали на ледяной бане. К этому раствору добавляли изобутилхлороформат (0,49 мл), а затем N-метилморфолин (0,41 мл). Приблизительно через одну минуту к смеси добавляли раствор 1,0 г N-(3-бромфенил)-4,6-хиназолиндиамина в 10 мл пиридина. Полученную реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Затем добавляли еще 0,5 эквивалента смешанного ангидрида. Полученную смесь перемешивали в течение пяти часов. После удаления растворителей при пониженном давлении твердое вещество очищали с помощью хроматографии на силикагеле, в результате чего получали 0,30 г N-[4-[(3-бромфенил)амино] -6-хиназолинил] -2-циклопентенамида; масс-спектр (m/е): 409 (М+Н, EI).
ПРИМЕР 13
N-[4-[(3-Бромфенил)амино]-6-хиназолинил]-2-пропенамид
Раствор 2,0 г N-(3-бромфенил)-4,6-хиназолиндиамина в 10 мл пиридина охлаждали на ледяной бане, а затем при температуре 0oС по каплям добавляли раствор 0,61 мл акриоилхлорида в 30 мл эфира. После перемешивания в течение 3,5 часа при комнатной температуре растворители удаляли при пониженном давлении. Остаток очищали с помощью хроматографии и получали 0,2 г N-[4-[(3-бромфенил)амино] -6-хиназолинил] -2-пропенамида; масс-спектр (m/е): М+Н 369.
ПРИМЕР 14
N-[4-[(3-Бромфенил)амино]-6-хиназолинил]-(3-фенил-2-пропинамид)
Раствор 0,93 г 3-фенил-2-пропиновой кислоты в 10 мл тетрагидрофурана охлаждали на ледяной бане. К этому раствору добавляли изобутилхлороформат (0,82 мл), а затем N-метилморфолин (0,69 мл). После выдерживания приблизительно в течение 1 минуты к смеси добавляли раствор 1,0 г N-(3-бромфенил)-4,6-хиназолиндиамина в 7 мл пиридина. Реакция протекала в течение одного часа при температуре 0oС. Растворители удаляли при пониженном давлении. Твердое вещество очищали с помощью хроматографии на силикагеле, в результате чего получали 0,01 г N-[4-[(3-бромфенил)амино]-6-хиназолинил]-(3-фенил-2-пропинамида); масс-спектр (m/е): 443,2, 445,2 (М+Н, электронапыление).
ПРИМЕР 15
6-Амино-4-хлорхиназолин
Смесь, содержащую 3,25 г 4-хлор-6-нитрохиназолина, 10,8 г бисульфита натрия и 0,3 г катализатора фазового переноса (C8H17)3NCH3 +Сl- в 97 мл тетрагидрофурана и 32 мл воды, быстро перемешивали в течение двух часов, эту смесь разбавляли эфиром и органический слой отделяли. Органический раствор промывали солевым раствором, а затем сушили над сульфатом магния. Полученный раствор пропускали через небольшую колонку с силикагелем. Растворитель удаляли при пониженном давлении при температуре 30oС, в результате чего получали 6-амино-4-хлорхиназолин, который использовали на следующей стадии без дополнительной очистки.
ПРИМЕР 16
[4-Хлор-6-хиназолинил]-2-бутинамид
Раствор 1,64 г 2-бутиновой кислоты в 46 мл тетрагидрофурана охлаждали на ледяной бане. К этому раствору добавляли изобутилхлороформат (2,34 мл), а затем N-метилморфолин (4,13 мл). Приблизительно через 10 минут смесь выливали в раствор 6-амино-4-хлорхиназолина в 46 мл тетрагидрофурана. Эту смесь перемешивали при комнатной температуре в течение 2 часов. Полученную смесь выливали в смесь солевого раствора и насыщенного бикарбоната натрия, а затем экстрагировали эфиром. Эфирный раствор сушили сульфатом магния и фильтровали. После удаления растворителя получали [4-хлор-6-хиназолинил]-2-бутинамид в виде бесцветного маслообразного вещества, которое использовали на следующей стадии без дополнительной очистки.
ПРИМЕР 17
N-[4-[(3-Бромфенил)амино]-6-хиназолинил]-2-бутинамид
Раствор, содержащий 1,76 [4-хлор-6-хиназолинил]-2-бутинамида и 1,23 г 3-броманилина, нагревали с обратным холодильником в 23 мл изопропанола в течение 40 минут в атмосфере инертного газа. Полученную смесь охлаждали до комнатной температуры, а затем добавляли 200 мл эфира и получали 0,4 г N-[4-[(3-бромфенил)амино] -6-хиназолинил] -2-бутинамида в виде соли с соляной кислотой. После нейтрализации раствором бикарбоната натрия, экстрагирования этилацетатом, удаления растворителя и перекристаллизации из 1-бутанола был получен N-[4-[(3-бромфенил)амино]-6-хиназолинил]-2-бутинамид в виде свободного основания.
ПРИМЕР 18
N'-(4-Амино-2-цианофенил)-N,N-диметилформамидин
Раствор, содержащий 6,0 г (27,5 ммоль) N'-(2-циано-4-нитрофенил)-N,N-диметилформамидина, 33,9 г (41,8 мл, 412,4 ммоль) циклогексена и 0,6 г 10% палладия-на-угле в 360 мл метанола, нагревали с обратным холодильником в течение 4 часов. Горячую смесь фильтровали через целит. После удаления растворителя остаток перекристаллизовывали из хлороформа/тетрахлорметана, в результате чего получали 4,9 г (95%) целевого соединения в виде светло-серого кристаллического твердого вещества; масс-спектр (m/е): 188,9 (М+Н, электронапыление).
ПРИМЕР 19
N-[3-Циано-4-[[(диметиламино)метилен]амино]фенил]-2-бутинамид
Раствор, содержащий 2,01 г (23,9 ммоль) 2-бутиновой кислоты и 2,9 мл (22,3 ммоль) изобутилхлороформата в 30 мл тетрагидрофурана, перемешивали в атмосфере азота при температуре 0oС, а затем к этой смеси в течение 3 минут добавляли 2,42 г (2,63 мл, 22,3 ммоль) N-метилморфолина. После 15-минутного перемешивания к смеси в течение 4 минут добавляли раствор N'-(4-амино-2-цианофенил)-N, N-диметилформамидина и 1,6 г (1,75 мл, 15,9 миллимоль) N-метилморфолина в 25 мл тетрагидрофурана. Смесь перемешивали в течение 30 минут при температуре 0oС, а затем в течение 30 минут при комнатной температуре. Полученную смесь разбавляли 70 мл этилацетата и выливали в смесь солевого раствора и насыщенного бикарбоната натрия. Органический слой сушили сульфатом магния и фильтровали через слой силикагеля. После удаления растворителя остаток перемешивали с 50 мл эфира. Суспендированное твердое вещество собирали и получали 3,61 г (89%) беловатого твердого продукта: масс-спектр (m/е): 255,0 (М+Н, электронапыление).
ПРИМЕР 20
N-[4-[(3-Бромфенил)амино]-6-хиназолинил]-2-бутинамид
Раствор, содержащий 3,0 г (11,8 ммоль) N-[3-циано-4-[[(диметиламино)метилен] амино] фенил] -2-бутинамида и 2,23 г (12,98 ммоль) 3-броманилина в 18 мл уксусной кислоты, мягко нагревали с обратным холодильником при перемешивании в атмосфере азота в течение 1 часа и 15 минут. Полученную смесь охлаждали на ледяной бане, в результате чего образовывалось твердое вещество. Это твердое вещество собирали фильтрованием и промывали эфиром/ацетонитрилом (1:1), в результате чего получали желтое твердое вещество, которое перекристаллизовывали из этанола с получением 2,51 г N-[4-[(3-бромфенил)амино] -6-хиназолинил] -2-бутинамида; масс-спектр (m/е): 381, 383.






Изобретение относится к производным хиназолина формулы I, где Х представляет собой фенил, который является необязательно замещенным галогеном; R, R1 и R2 представляют собой водород, Y представляет собой радикал, выбранный из группы, включающей группы формул (а-f); R3 независимо представляет собой водород, алкил с 1-6 атомами углерода, карбокси, карбоалкокси с 1-6 атомами углерода или фенил; n=2-4; или их фармацевтически приемлемым солям, при условии, что каждый R3 в группе Y может быть одинаковым или различным. Соединения формулы I ингибируют действие определенных протеинтирозинкиназ, являющихся рецепторами факторов роста и могут быть использованы для лечения рака у млекопитающих. Кроме того, настоящее изобретение относится к способам получения указанных хиназолинов, способу ингибирования биологического действия нерегулируемой протеинтирозинкиназы у млекопитающего, способу лечения или ингибирования роста опухолей у млекопитающих и фармацевтической композиции на основе указанных соединений. 8 с. и 13 з.п.ф-лы, 7 табл.
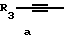
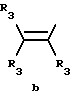
где X представляет собой фенил, необязательно замещенный галогеном;
R, R1 и R2 представляют собой водород;
Y представляет собой радикал, выбранный из группы, включающей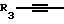
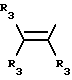
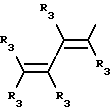
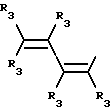
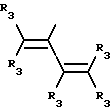
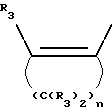
где R3 независимо представляет собой водород, алкил с 1-6 атомами углерода, карбокси, карбоалкокси с 1-6 атомами углерода или фенил;
n= 2-4,
или их фармацевтически приемлемые соли, при условии, что каждый R3 в группе Y может быть одинаковым или различным.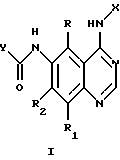
где Х представляет собой фенил, необязательно замещенный галогеном;
R, R1 и R2 представляют собой водород;
Y представляет собой радикал, выбранный из группы, включающей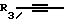
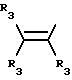
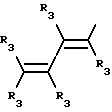
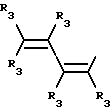
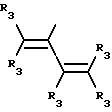
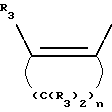
где R3 независимо представляет собой водород, алкил с 1-6 атомами углерода, карбокси, карбоалкокси с 1-6 атомами углерода или фенил;
n= 2-4,
или его фармацевтически приемлемой соли, при условии, что каждый R3 в группе Y может быть одинаковым или различным.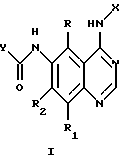
где X представляет собой фенил, необязательно замещенный галогеном;
R, R1 и R2 представляют собой водород;
Y представляет собой радикал, выбранный из группы, включающей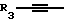
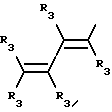
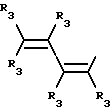
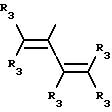
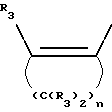
где R3 независимо представляет собой водород, алкил с 1-6 атомами углерода, карбокси, карбоалкокси с 1-6 атомами углерода или фенил;
n= 2-4,
или его фармацевтически приемлемой соли, при условии, что каждый R3 в группе Y может быть одинаковым или различным.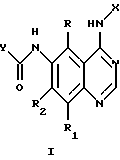
где Х представляет собой фенил, необязательно замещенный галогеном;
R, R1 и R2 представляют собой водород;
Y представляет собой радикал, выбранный из группы, включающей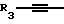
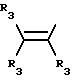
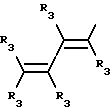

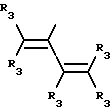
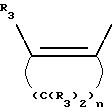
где R3 независимо представляет собой водород, алкил с 1-6 атомами углерода, карбокси, карбоалкокси с 1-6 атомами углерода или фенил;
n= 2-4,
или его фармацевтически приемлемой соли, при условии, что каждый R3 в группе Y может быть одинаковым или различным.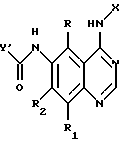
где R, R1, R2 и Х имеют значения, определенные в п. 1;
Y' представляет собой радикал, выбранный из группы, включающей
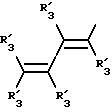
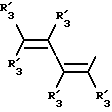
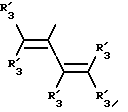
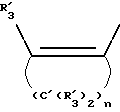
где каждый R'3 независимо представляет собой водород, алкил с 1-6 атомами углерода, карбокси, карбоалкокси с 1-6 атомами углерода, фенил;
n равно целому числу от 2 до 4,
отличающийся тем, что антранилонитрил формулы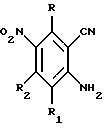
обрабатывают диметилацеталем диметилформамида в присутствии или отсутствии растворителя с получением соединения формулы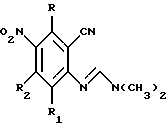
указанное соединение нагревают с анилином формулы
X-NH2
в кислотном органическом растворителе с получением 6-нитрохиназолина формулы
указанное соединение обрабатывают восстанавливающим агентом с получением 6-амино-хиназолина формулы
и указанное соединение ацилируют хлорангидридом или смешанным ангидридом формул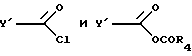
где R4 представляет собой алкил с 1-6 атомами углерода.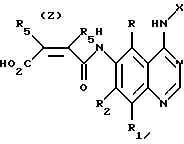
где R, R1, R2 и Х имеют значения, определенные в п. 1;
R5 независимо представляет водород, фенил или алкил с 1-6 атомами углерода;
(Z) означает конфигурацию двойной связи,
отличающийся тем, что соединение формулы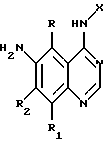
ацилируют циклическим ангидридом формулы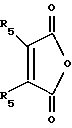
в инертном растворителе в присутствии органического основания.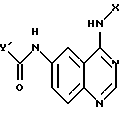
где Х имеет значения, определенные в п. 1;
Y' имеет значения, определенные в п. 18,
отличающийся тем, что соединение формулы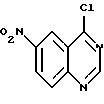
восстанавливают бисульфитом натрия в присутствии катализатора фазового переноса в смеси растворителей, содержащей инертный органический растворитель и воду, с получением соединения формулы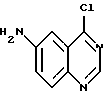
указанное соединение ацилируют хлорангидридом или смешанным ангидридом формулы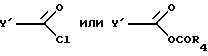
где R4 представляет собой алкил с 1-6 атомами углерода,
в присутствии основания амина в инертном растворителе с получением соединения формулы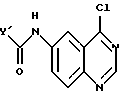
и указанное соединение нагревают с анилином формулы
X-NH2
в инертном растворителе.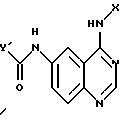
где Х имеет значения, определенные в п. 1;
Y' имеет значения, определенные в п. 18,
отличающийся тем, что соединение формулы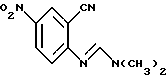
восстанавливают в присутствии палладиевого катализатора и источника водорода в инертном растворителе с получением аминосоединения формулы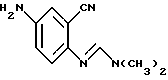
указанное соединение ацилируют хлорангидридом или смешанным ангидридом формул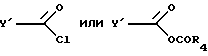
где R4 представляет собой алкил с 1-6 атомами углерода,
в присутствии основания амина в инертном растворителе с получением соединения формулы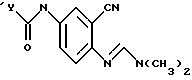
и указанное соединение нагревают с анилином, имеющим формулу
X-NH2
в кислотном растворителе.
| RU 93004423 A1, 20.05.1995 | |||
| Устройство для передачи сигналов времени и кодовой информации о текущем времени в составе телевизионных сигналов | 1975 |
|
SU520722A1 |
| Ультразвуковой искатель | 1975 |
|
SU602851A1 |
| US 5196446 А, 23.03.1993. | |||

