Изобретение относится к новой группе производных имидазола, к способам их получения, к использованию их при лечении опосредованных цитокинами заболеваний и к фармацевтическим композициям для использования в таком лечении.
Предпосылки изобретения
Интерлейкин 1 (ИЛ-1) и фактор некроза опухоли (ФИО) являются биологическими веществами, производимыми различными клетками, такими как моноциты или макрофаги. ИЛ-1, как показано, опосредует множество видов биологической активности и, как предполагается, является важным при иммунорегуляции и других физиологических состояниях, таких как воспаление [см., например, Dinarello et al., Rev. Infect. Disease, 6, 51 (1984)]. Множество известных видов биологической активности ИЛ-1 включают активацию Т-клеток-хелперов, возбуждение лихорадки, стимуляцию продуцирования простагландина и коллагеназы, хемотаксиса нейтрофилов, активацию белков острой фазы и понижение уровней железа в плазме крови.
Существует много болезненных состояний, при которых избыточное или нерегулируемое производство ИЛ-1 вызывает обострение и/или начало заболевания. Эти заболевания включают ревматоидный артрит, остеоартрит, эндотоксемию и/или синдром токсического шока, другие острые или хронические воспалительные болезненные состояния, такие как воспалительная реакция, вызванная эндотоксином, или воспаление кишечника; туберкулез, атеросклероз, дегенерация мышц, кахексия, псориатический артрит, синдром Рейтера, ревматоидный артрит, подагра, травматический артрит, артрит при краснухе и острый синовит. Недавно полученные факты также связывают активность ИЛ-1 с диабетом и бета-клетками поджелудочной железы.
Dinarello, J. Clinical Immunology, 5(5), 287-297 (1985) делает обзор видов биологической активности, которые связывают с ИЛ-1. Необходимо заметить, что некоторые из этих эффектов были описаны другими авторами как непрямые эффекты ИЛ-1.
Избыточное или нерегулируемое продуцирование ФНО подразумевается при опосредовании или обострении ряда заболеваний, включая ревматоидный артрит, ревматоидный спондилит, остеоартрит, артрит при краснухе и другие состояния с артритом; сепсис, септический шок, эндотоксический шок, грамотрицательный сепсис, синдром токсического шока, синдром недомогания у взрослых при простуде, церебральную малярию, хроническое воспаление легких, силикоз, саркоидоз легких, заболевания резорбции костных тканей, реперфузионные повреждения, реакцию на прививку, отторжение трансплантатов, лихорадку и боли в мышцах, вызванные инфекцией, такой как грипп, вторичную кахецию после инфекции или злокачественного заболевания, кахецию после синдрома приобретенного иммунодефицита (СПИД), СПИД, КОС (комплекс, обусловленный СПИДом), образование келоидной ткани, образование рубцовой ткани, болезнь Крона, язвенный колит или лихорадка.
СПИД возникает в результате инфицирования Т-лимфоцитов вирусом иммунодефицита человека (ВИЧ). Идентифицированы, по крайней мере, три типа или штамма ВИЧ, то есть ВИЧ-1, ВИЧ-2 и ВИЧ-3. Как следствие ВИЧ инфекции, Т-клетка, опосредующая иммунитет, ослабляется, и инфицированные индивидуумы демонстрируют тяжелые случайные инфекции и/или необычные новообразования. Внедрение ВИЧ в Т-лимфоцит требует активации Т-лимфоцита. Другие вирусы, такие как ВИЧ-1, ВИЧ-2, инфицируют Т-лимфоциты после активации Т-клетки, и такая экспрессия и/или репликация белка вируса поддерживается или опосредуется с помощью такой активации Т-клетки. Как только активированный Т-лимфоцит инфицируется ВИЧ, Т-лимфоцит должен продолжать находиться в активированном состоянии, чтобы сделать возможной экспрессию гена ВИЧ и/или репликацию ВИЧ. Монокины, в частности ФНО, как предполагается, участвуют в опосредованной активированной Т-клеткой экспрессии белка ВИЧ и/или репликации вируса, играя свою роль в поддержании активации Т-лимфоцита. По этой причине влияние на активность монокина, такую как ингибирование продукции монокина, в частности ФНО, у ВИЧ инфицированного индивидуума помогает при ограничении поддержания активации Т-клетки, тем самым уменьшая развитие ВИЧ инфицирования ранее не инфицированных клеток, что дает в результате замедление или устранение распространения иммунной дисфункции, вызванной инфекцией ВИЧ. Моноциты, макрофаги и родственные клетки, такие как kupffer и глиальные клетки, также, как предполагается, поддерживают ВИЧ инфекцию. Эти клетки, подобно Т-клеткам, являются мишенями для репликации вируса, и уровень репликации вируса зависит от активированного состояния клеток [см., Rosenberg et al., The Immunopathogenesis of HIV Infection, Advances in Immunology, Vol. 57 (1989)]. Монокины, такие как ФНО, как показано, активируют репликацию ВИЧ в моноцитах и/или макрофагах [см. , Poli, et al., Proc. Natl. Acad. Sci., 87:782-784 (1990)] , следовательно, ингибирование продукции или активности монокинов помогает при ограничении распространения ВИЧ, как показано выше для Т-клеток.
ФНО также, как показано, играет разнообразные роли с другими вирусными инфекциями, такими как цитомегаловирус (ЦМВ), вирус гриппа и вирус герпеса, по тем же причинам, которые уже были отмечены.
Интерлейкин-8 (ИЛ-8) является хемотаксическим фактором, впервые идентифицированным и охарактеризованным в 1987 году. ИЛ-8 продуцируется несколькими видами клеток, включая моноядерные клетки, фибробласты, эндотелиальные клетки и кератиноциты. Его продуцирование эндотелиальными клетками индуцируется ИЛ-1, ФНО или липополисахаридом (ЛПС). ИЛ-8 человека, как показано, действует на нейтрофилы мыши, морской свинки, крысы и кролика. По отношению к ИЛ-8 применялось множество различных названий, таких как белок-1 привлечения/активации нейтрофилов (БАН-1), хемотаксический фактор нейтрофилов, полученный из моноцитов (ХФНМ), активирующий фактор нейтрофилов (АФН) и хемотаксический фактор лимфоцитов Т-клеток.
ИЛ-8 стимулирует ряд функций in vitro. Как показано, он имеет свойства хемоаттрактанта для нейтрофилов, Т-лимфоцитов и базофилов. Кроме того, он индуцирует высвобождение гистамина из базофилов как у нормальных, так и у аллергических индивидуумов, а также высвобождение лизосомальных ферментов и быстрое выделение при респираторных заболеваниях из нейтрофилов. ИЛ-8 также, как показано, увеличивает поверхностную экспрессию Mac-1 (CD11b/CD18) на нейтрофилах без синтеза белков de novo, что может вносить вклад в повышенную адгезию нейтрофилов к клеткам эндотелия сосудов. Многие заболевания характеризуются массированной инфильтрацией нейтрофилов. Состояния, связанные с увеличенным производством ИЛ-8 (который ответственен за хемотаксис нейтрофилов в области воспаления) могли бы быть облегчены с помощью соединений, которые являются суппрессорами производства ИЛ-8.
ИЛ-1 и ФНО воздействуют на широкое разнообразие клеток и тканей, и эти цитокины, наряду с другими цитокинами из лейкоцитов, являются важными и критичными медиаторами воспаления в большом разнообразии болезненных состояний и симптомов. Ингибирование этих цитокинов дает выигрыш при контролировании, уменьшении и облегчении многих из этих болезненных состояний.
В этой области при лечении остается необходимость в соединениях, которые являются противовоспалительными лекарственными средствами для суппрессии цитокинов, то есть соединений, которые способны ингибировать цитокины, такие как ИЛ-1, ИЛ-6, ИЛ-8 и ФНО.
Краткое описание изобретения
Данное изобретение относится к новым соединениям формулы (I) и (II) и к фармацевтическим композициям, содержащим соединения формулы (I) или (II) и фармацевтически приемлемый разбавитель или связующее вещество.
Данное изобретение также относится к способу ингибирования цитокинов и к лечению заболеваний, опосредованных цитокином, у млекопитающего, нуждающегося в этом, который включает введение указанному млекопитающему эффективного количества соединения формулы (I) или (II).
Данное изобретение, более конкретно, относится к способу ингибирования продуцирования ИЛ-1 у млекопитающего, нуждающегося в этом, который включает введение указанному млекопитающему эффективного количества соединения формулы (I) или (II).
Данное изобретение, более конкретно, относится к способу ингибирования продуцирования ИЛ-8 у млекопитающего, нуждающегося в этом, который включает введение указанному млекопитающему эффективного количества соединения формулы (I) или (II).
Данное изобретение, более конкретно, относится к способу ингибирования производства ФНО у млекопитающего, нуждающегося в этом, который включает введение указанному млекопитающему эффективного количества соединения формулы (I) или (II).
Соответственно, настоящее изобретение касается соединения формулы (I)
где R1 представляет 4-пиридил, пиримидинил, хинолил, изохинолил, хиназолин-4-ил, 1-имидазолил или 1-бензимидазолил, где гетероарильное кольцо замещено N(R10)C(O)Ra или галогензамещенным моно- или ди-С1-6-алкилзамещенным амино и это кольцо дополнительно необязательно замещено С1-4алкилом, галогеном, гидроксилом, С1-4алкокси, С1-4алкилтио, С1-4алкилсульфинилом, CH2OR12, амино, моно- и ди-С1-6алкилзамещенным амино или N-гетероциклильным кольцом, которое имеет от 5 до 7 членов NR15;
R4 представляет фенил, нафт-1-ил или нафт-2-ил, или гетероарил, который необязательно замещен одним или двумя заместителями, каждый из которых независимо выбран, и который для заместителя 4-фенила, 4-нафт-1-ила, 5-нафт-2-ила или 6-нафт-2-ила, представляет галоген, циано, нитро, -С (Z)NR7R17, -C(Z)OR16, - (CR10R20)vCOR12,
-SR5, -SOR5, -OR12, галогензамещенный-С1-4алкил, С1-4алкил,
-ZC(Z)R12, -NR10C(Z)R16 или - (CR10R20) vNR10R20 и который для других положений замещения представляет галоген, циано,
-C(Z)NR13R14, -C(Z)OR3, - (CR10R20)m''-COR3, -S(O)mR3, -ОR3,
-OR12, галогензамещенный-С1-4алкил, С1-4алкил,
- (CR10R20)m''NR10C(Z)R3, -NR10S(O)m'R8, -NR10S(O)m'NR7R17,
-ZC(Z)R3, -ZC(Z)R12 или - (CR10R20)m''NR13R14;
v равно 0 или целому числу, имеющему значение 1 или 2;
m равно 0 или целому числу 1 или 2;
m' равно целому числу, имеющему значение 1 или 2;
m'' равно 0 или целому числу, имеющему значение от 1 до 5;
R2 представляет C1-10алкил N3, -(CR10R20)n'OR9, гетероциклил, гетероциклилС1-10алкил, C1-10алкил, галогензамещенный-C1-10-алкил, С2-10алкенил, С2-10алкинил, С3-7циклоалкил, С3-7циклоалкилС1-10алкил, С5-7циклоалкенил, С5-7циклоалкенил-C1-10алкил, арил, арилС1-10алкил, гетероарил, гетероарил-C1-10алкил, (CR10R20)nOR11, (CR10R20)nS(O)mR18, (CR10R20)nNHS(O)2R18,
(CR10R20)nNR13R14, (CR10R20)nNO2, (CR10R20)nCN, (CR10R20)n'SO2R18,
(CR10R20)nS(O)m'NR13R14, (CR10R20)nC(Z)R11, (CR10R20)nOC(Z)R11, (CR10R20)nC(Z)OR11, (CR10R20)nC(Z)NR13R14, (CR10R20)nC(Z)NR11ОR9,
(CR10R20)nNR10C (Z) R11, (CR10R20)nNR10C(Z)R13R14, (CR10R20)nN(OR6)С(Z)R13R14, (CR10R20)nN(OR6)С(Z)R11, (CR10R20)nC(= NOR6)R11, (CR10R20)nNR10C(=NR19) NR13R14, (CR10R20)nOC(Z)NR13R14, (CR10R20)nNR10C(Z)NR13R14, (CR10R20)nNR10C(Z)OR10, 5-(R18)-1, 2, 4-оксадиазол-3-ил или 4-(R12)-5-(R18R19)-4, 5-дигидро-1, 2, 4-оксадиазол-3-ил; где арильная, арилалкильная, гетероарильная, гетероарилалкильная, циклоалкильная, циклоалкилалкильная, гетероциклическая и гетероциклическая алкильная группы могут быть необязательно замещены;
n равно целому числу, имеющему значение от 1 до 10;
n' равно 0 или целому числу, имеющему значение от 1 до 10;
Z является кислородом или серой;
Ra представляет водород, C1-6алкил, С3-7циклоалкил, арил, арилС1-4алкил, гетероарил, гетероарилС1-4алкил, гетероциклил, или гетероциклилС1-4алкил;
R3 представляет гетероциклил, гетероциклилС1-10алкил или R8;
R5 представляет водород, С1-4алкил, С2-4алкенил, С2-4алкинил или NR7R17, за исключением остатков -SR5, являющегося -SNR7R17, и -S(O)R5, являющегося -SОН;
R6 представляет водород, фармацевтически приемлемый катион, C1-10алкил, С3-7циклоалкил, арил, арилС1-4алкил, гетероарил, гетероарилС1-10алкил, гетероциклил, ароил, или C1-10алканоил;
R7 и R17, каждый, независимо выбраны из водорода или С1-4алкила, или R7 и R17 вместе с азотом, к которому они присоединены, образуют 5-7-членное гетероциклическое кольцо, которое необязательно содержит дополнительный гетероатом, выбранный из кислорода, серы или NR15;
R8 представляет C1-10 алкил, галогензамещенный- С1-10алкил, С2-10алкенил, С2-10алкинил, С3-7циклоалкил, С5-7циклоалкенил, арил, арилС1-10 алкил, гетероарил, гетероарилС1-10алкил, (CR10R20)nOR11, (CR10R20)nS(O)mR18, (CR10R20)nNHS(O)2R18, (CR10R20)nNR13R14, где арил, арилалкил, гетероарил, гетероарилалкил могут быть необязательно замещенными;
R9 представляет водород, -C(Z)R11, необязательно замещенный C1-10алкил, S(O)2R18, необязательно замещенный арил или необязательно замещенный арилС1-4алкил;
R10 и R20, каждый независимо, выбраны из водорода или С1-4алкила;
R11 представляет водород, C1-10алкил, С3-7циклоалкил, гетероциклил, гетероциклилС1-10алкил, арил, арилС1-10-алкил, гетероарил или гетероарилС1-10алкил;
R12 представляет водород или R16;
R13 и R14, каждый независимо, выбраны из водорода или необязательно замещенного С1-4алкила, необязательно замещенного арила или необязательно замещенного арилС1-4-алкила или вместе с азотом, к которому они присоединены, образуют 5-7-членное гетероциклическое кольцо, которое необязательно включает дополнительный гетероатом, выбранный из кислорода, серы или NR9;
R15 представляет R10 или С(Z) -С1-4алкил;
R16 представляет С1-4алкил, галогензамещенныйС1-4алкил или С3-7циклоалкил;
R18 представляет C1-10алкил, С3-7циклоалкил, гетероциклил, арил, арилС1-10алкил, гетероциклил, гетероциклилС1-10алкил, гетероарил, или гетероарилС1-10алкил;
R19 представляет водород, циано, С1-4алкил, С3-7циклоалкил или арил;
или его фармацевтически приемлемой соли.
Подробное описание изобретения
Новые соединения формулы (I) могут быть также использованы в связи с ветеринарным лечением млекопитающих, отличных от людей, нуждающихся в ингибировании производства или ингибирования цитокинов. В частности, опосредованные цитокинами заболевания у животных, подлежащие терапевтическому или профилактическому лечению, включают болезненные состояния, такие как те, которые указаны здесь в разделе способов лечения, но в особенности, вирусные инфекции. Примеры таких вирусов включают, но не ограничиваясь ими, инфекции лентивирусов, таких как вирус инфекционной анемии лошади, вирус козлиного артрита, visna или maedi вирус, или ретровирусные инфекции, такие как, но не ограничиваясь ими, вирус иммунодефицита кошек (ВИК), вирус иммунодефицита быка или вирус иммунодефицита собаки, или другие ретровирусные инфекции.
Другим аспектом настоящего изобретения является предпочтительный выбор соединения формулы
где R1 представляет 4-пиридил или пиримидиниловое кольцо, замещенное С1-4алкиламиногруппой, это кольцо может быть дополнительно необязательно замещено С1-4алкилом, галогеном, гидроксилом, С1-4алкокси, С1-4алкилтио, C1-4aлкилсульфинилом, CH2OR12, амино, моно- и ди-С1-6алкилзамещенным амино или N-гетероциклильным кольцом, и это кольцо имеет от 5 до 7 членов и необязательно включает дополнительный гетероатом, выбранный из кислорода, серы или NR15;
R4 представляет фенил, нафт-1-ил или нафт-2-ил или гетероарил, который необязательно замещен одним или двумя заместителями, каждый из которых выбран независимо, и который для заместителя 4-фенила, 4-нафт-1-ила, 5-нафт-2-ила или 6-нафт-2-ила представляет галоген, циано, нитро, -C(Z)NR7R17, -C(Z)OR16, - (CR10R20)vCOR12, -SR5, -SOR5, -OR12, галогензамещенный-С1-4алкил, С1-4алкил, -ZC(Z)R12, -NR10C(Z)R16 или - (CR10R20)vNR10R20, и который для других положений замещения представляет галоген, циано -С (Z) NR13R14, -C(Z)OR3, -(CR10R20) m''COR3, -S(O)mR3, -ОR3, -OR12, галогензамещенный-С1-4алкил, С1-4алкил, -(CR10R20)m''NR10C(Z)R3, -NR10S(O)m'R8, -NR10S(O)m'NR7R17, -ZC(Z)R3, -ZC(Z)R12 или -(CR10R20)m''NR13R14;
v равно 0 или целому числу, имеющему значение 1 или 2;
m равно 0 или целому числу 1 или 2;
m' равно целому числу, имеющему значение 1 или 2;
m'' равно 0 или целому числу, имеющему значение от 1 до 5;
R2 представляет C1-10алкил N3, - (CR10R20) n'OR9, гетероциклил, гетероциклилС1-10алкил, C1-10алкил, галогензамещенный-C1-10-алкил, С2-10алкенил, С2-10алкинил, С3-7циклоалкил, С3-7циклоалкилС1-10алкил, С5-7циклоалкенил, С5-7циклоалкенил-C1-10алкил, арил, арилС1-10алкил, гетероарил, гетероарил-C1-10алкил, (CR10R20)nOR11, (CR10R20)nS(O)mR18, (CR10R20)nNHS(O)2R18,
(CR10R20)nNR13R14, (CR10R20)nNO2, (CR10R20)nCN, (CR10R20)n'SO2R18,
(CR10R20)nS(O)m'NR13R14, (CR10R20)nC(Z)R11, (CR10R20)nOC(Z)R11, (CR10R20)nC(Z)OR11, (CR10R20)nC(Z)NR13R14, (CR10R20)nC(Z)NR11ОR9,
(CR10R20)nNR10C(Z)R11, (CR10R20)nNR10C(Z)R13R14, (CR10R20)nN(OR6)С(Z)R13R14, (CR10R20)nN(OR6)С(Z)R11, (CR10R20)nC(= NOR6)R11, (CR10R20)nNR10C(=NR19)NR13R14, (CR10R20)nOC(Z)NR13R14, (CR10R20)nNR10C(Z)NR13R14, (CR10R20)nNR10C(Z)OR10, 5-(R18)-1,2,4-оксадиазол-3-ил или 4-(R12)-5-(R18R19)-4,5-дигидро-1,2,4-оксадиазол-3-ил; где арильная, арилалкильная, гетероарильная, гетероарилалкильная, циклоалкильная, циклоалкилалкильная, гетероциклическая и гетероциклическая алкильная группы могут быть необязательно замещены;
n равно целому числу, имеющему значение от 1 до 10;
n' равно 0 или целому числу, имеющему значение от 1 до 10;
Z является кислородом или серой;
Ra представляет водород, C1-6алкил, С3-7циклоалкил, арил, арилС1-4алкил, гетероарил, гетероарилС1-4алкил, гетероциклил или гетероциклилС1-4алкил;
R3 представляет гетероциклил, гетероциклилС1-10алкил или R8;
R5 представляет водород, С1-4алкил, С2-4алкенил, С2-4алкинил или NR7R17, за исключением радикалов -SR5, являющихся -SNR7R17 и -S(O)R5, являющегося -SОН;
R6 представляет водород, фармацевтически приемлемый катион, C1-10алкил, С3-7циклоалкил, арил, арилС1-4алкил, гетероарил, гетероарилС1-10алкил, гетероциклил, ароил, или C1-10алканоил;
R7 и R17, каждый независимо, выбраны из водорода или С1-4алкила, или R7 и R17 вместе с азотом, к которому они присоединены, образуют 5-7-членное гетероциклическое кольцо, которое необязательно включает дополнительный гетероатом, выбранный из кислорода, серы или NR15;
R8 представляет C1-10алкил, галогензамещенный C1-10алкил, С2-10алкенил, С2-10алкинил, С3-7циклоалкил, С5-7циклоалкенил, арил, арилС1-10алкил, гетероарил, гетероарил-C1-10алкил, (CR10R20)nOR11, (CR10R20)nS(O)mR18, (CR10R20)nNHS(O)2R18, (CR10R20)nNR13R14, где арил, арилалкил, гетероарил, гетероарилалкил могут быть необязательно замещенными;
R9 представляет водород, -C(Z)R11, необязательно замещенный C1-10алкил, S(O)2R18, необязательно замещенный арил или необязательно замещенный арилС1-4алкил;
R10 и R20, каждый независимо, выбраны из водорода или С1-4алкила;
R11 представляет водород, C1-10алкил, С3-7циклоалкил, гетероциклил, гетероциклилС1-10алкил, арил, арилС1-10-алкил, гетероарил или гетероарилС1-10алкил;
R12 представляет водород или R16;
R13 и R14, каждый, независимо выбраны из водорода или необязательно замещенного С1-4алкила, необязательно замещенного арила или необязательно замещенного арилС1-4-алкила, или вместе с азотом, к которому они присоединены, образуют 5-7-членное гетероциклическое кольцо, которое необязательно включает дополнительный гетероатом, выбранный из кислорода, серы или NR9;
R15 представляет R10 или С(Z)-С1-4алкил;
R16 представляет С1-4алкил, галогензамещенный-С1-4алкил или С3-7циклоалкил;
R18 представляет C1-10алкил, С3-7циклоалкил, гетероциклил, арил, арилС1-10алкил, гетероциклил, гетероциклилС1-10алкил, гетероарил или гетероарилС1-10алкил;
R19 представляет водород, циано, С1-4алкил, С3-7циклоалкил или арил;
или их фармацевтически приемлемую соль.
В формуле (I) соответствующие радикалы R1 включают 4-пиридил, 4-пиримидинил, 4-хинолил, 6-изохинолил, 4-хиназолинил, 1-имидазолил и 1-бензимидазолил, причем 4-пиридил, 4-пиримидинил и 4-хинолил являются предпочтительными. Более предпочтительным является 4-пиримидинильный или 4-пиридильный радикал и наиболее предпочтительным является 4-пиримидинил.
Гетероарильное кольцо R1 является замещенным N(R10)C(O)Ra или галогензамещенным моно- или ди-С1-6алкилзамещенным амино. Когда заместитель R1 является N(R10) C(O)Ra, Ra представляет водород, C1-6алкил, С3-7циклоалкил, арил, арилС1-4алкил, гетероарил, гетероарилС1-4алкил, гетероциклил или гетероциклилС1-4алкил, С1-4алкил, Ra предпочтительно является C1-6алкилом; предпочтительно R10 является водородом. Также наблюдается, что радикалы Ra, в частности C1-6алкильная группа, могут быть необязательно замещены, предпочтительно от одного до трех раз, предпочтительно галогеном, таким как фтор, как в трифторметиле или трифторэтиле.
Когда заместитель R1 является галогензамещенным моно- и диС1-6алкилзамещенным амино, предпочтительно таким, где аминогруппа является монозамещенной, более предпочтительно метилом. Алкильная группа в моно- и диС1-6алкилзамещенном аминорадикале является галогензамещенной, такой как трифтор-, то есть трифторметилом или трифторэтилом.
Гетероарильное кольцо R1 может содержать дополнительную замещающую группу, такую как С1-4алкил, галоген, ОН, С1-4алкокси, С1-4алкилтио, С1-4алкилсульфинил, CH2OR12, амино, моно- и диС1-6алкилзамещенная амино или N-гетероциклическое кольцо, которое состоит из 5-7 членов и необязательно содержит дополнительный гетероатом, выбранный из кислорода, серы или NR15.
Предпочтительным положением в кольце заместителя R1 у 4-пиридильного производного является положение 2, такое как 2-метил-4-пиридил. Предпочтительное положение в кольце у 4-пиримидинилового кольца является также положение 2, такое как 2-метилпиримидинил, 2-аминопиримидинил или 2-метиламинопиримидинил.
Соответственно, для соединений формулы (II) R1 является 4-пиридилом или пиримидиниловым кольцом, замещенным С1-4алкил-аминогруппой. С1-4алкиламиногруппа является, соответственно, метиламино, этиламино, изопропиламино, н-бутиламино или трет-бутиламино. Предпочтительно, кольцо является 4-пиримидиниловым кольцом. Предпочтительным положением в кольце у 4-пиримидинилового кольца является положение 2, такое как в 2-метиламинопиримидиниле. Предпочтительное положение в кольце заместителя R1 у 4-пиридилового производного является положение 2, такое как 2-метиламино-4-пиридил. Пиридильное или пиримидинильное кольцо могут содержать дополнительную замещающую группу, такую как С1-4алкил, галоген, гидроксил, С1-4алкокси, С1-4алкилтио, С1-4алкилсульфинил, CH2OR12, амино, моно- и диС1-6алкилзамещенная амино или N-гетероциклическое кольцо, которое имеет 5-7 членов, и необязательно содержит дополнительный гетероатом, выбранный из кислорода, серы или NR15.
Соответственно, R4 для соединений формулы (I) и (II) является фенилом, нафт-1-илом или нафт-2-илом или гетероарилом, который необязательно замещен одним или двумя заместителями. Более предпочтительно R4 является фенильным или нафтильным кольцом. Подходящими заместителями R4, когда он является 4-фенильным, 4-нафт-1-ильным, 5-нафт-2-ильным или 6-нафт-2-ильным радикалом, являются один или два заместителя, каждый из которых является независимо выбранным из галогена, -SR5, -SOR5, -OR12, СF3 или - (CR10R20)vNR10R20, и для других положений замещения этих колец предпочтительным заместителем является галоген, -S(O)mR3, -ОR3, СF3, -(CR10R20)nS(O)m'NR13R14, -NR10C(Z)R3 и NR10S(O)m'R8. Предпочтительные заместители для 4-положения в фениле и нафт-1-иле и для положения 5 в нафт-2-иле включают галоген, главным образом, фтор и хлор и -SR5 и -SOR5, где R5 является предпочтительно C1-2 алкилом, более предпочтительно метилом; из которых фтор и хлор являются более предпочтительными и наиболее предпочтительным является фтор. Предпочтительные заместители для положения 3 в фенильном и нафт-1-ильном кольце включают: галоген, главным образом, фтор и хлор; -ОR3, главным образом, С1-4алкокси; СF3; NR10R20, такой как амино; -NR10C(Z)R3, главным образом, -NHCO-(C1-10алкил); -NR10S(O)m'R8, главным образом, -NHSO2 (C1-10алкил); и -SR3 и SOR3, где R3 является предпочтительно С1-2алкилом, более предпочтительно метилом. Когда фенильное кольцо является дизамещенным, предпочтительно эти заместители представляют собой два независимых галогеновых радикала, такие как фтор или хлор, предпочтительно ди-хлор и более предпочтительно в положении 3, 4. Также предпочтительно, что в положении 3 как для -ОR3, так и для -ZC(Z)R3 радикалах R3 может также включать водород.
Предпочтительно радикал R4 является незамещенным или замещенным фенильным радикалом. Более предпочтительно R4 является фенилом, замещенным в положении 4 фтором и/или замещенным в положении 3 фтором, хлором, С1-4алкокси, метансульфонамидо или ацетамидо, или R4 является фенилом, ди-замещенным в положениях 3 и 4, независимо, хлором или фтором, более предпочтительно хлором. Более предпочтительно R4 является 4-фторфенилом.
Соответственно, для соединений формулы (I) и (II) Z является кислородом или серой, предпочтительно кислородом.
Соответственно, для соединений формулы (I) и (II) R2 представляет C1-10алкил N3, -(CR10R20)n'OR9, гетероциклил, гетероциклилС1-10алкил, C1-10алкил, галогензaмещенный C1-10-алкил, С2-10алкенил, С2-10алкинил, С3-7циклоалкил, С3-7циклоалкилС1-10алкил, С5-7циклоалкенил, С5-7циклоалкенилС1-10алкил, арил, арилС1-10алкил, гетероарил, гетероарилС1-10алкил, (CR10R20)nOR11, (CR10R20)nS(O)mR18, (CR10R20)nNHS(O)2R18,
(CR10R20)nNR13R14, (CR10R20)nNO2, (CR10R20)nCN, (CR10R20)n'SO2R18,
(CR10R20)nS(O)m'NR13R14, (CR10R20)nC(Z)R11, (CR10R20)nOC(Z)R11, (CR10R20)nC(Z)OR11, (CR10R20)nC(Z)NR13R14, (CR10R20)nC(Z)NR11OR9,
(CR10R20)nNR10C(Z)R11, (CR10R20)nNR10C(Z)R13R14, (CR10R20)nN(OR6)С(Z)R13R14, (CR10R20)nN(OR6)С(Z)R11, (CR10R20)nC(= NOR6)R11, (CR10R20)nNR10C(=NR19)NR13R14, (CR10R20)nOC(Z)NR13R14, (CR10R20)nNR10C(Z)NR13R14, (CR10R20)nNR10C(Z)OR10, 5-(R18)-1,2,4-оксадиазол-3-ил или 4-(R12)-5-(R18R19)-4,5-дигидро-1,2,4-оксадиазол-3-ил; где циклоалкильный, циклоалкилалкильный, арильный, арилалкильный, гетероарильный, гетероарилалкильный, гетероциклический и гетероциклический алкильный радикалы могут быть необязательно замещены; где n является целым числом, имеющим значения от 1 до 10, m равно 0 или целому числу 1 или 2; n' равно 0 или целому числу, имеющему значения от 1 до 10; и m' равно 1 или 2. Предпочтительно n равно 1-4.
Предпочтительно R2 является необязательно замещенным гетероциклильным кольцом и необязательно замещенным гетероциклилС1-10алкилом, необязательно замещенным C1-10алкилом, необязательно замещенным С3-7циклоалкилом, необязательно замещенным С3-7циклоалкилС1-10алкилом, группой (CR10R20)nC(Z)OR11, (CR10R20)nNR13R14, (CR10R20)nNHS(O)2R18, (CR10R20)nS(O)mR18, необязательно замещенным арилом; необязательно замещенным арил-C1-10алкилом, (CR10R20)nOR11, (CR10R20)nC(Z)R11 или группой (CR10R20)nC(= NOR6)R11. Более предпочтительно R2 является необязательно замещенным гетероциклильным кольцом и необязательно замещенным гетероциклилС1-10алкилом, необязательно замещенным С3-7циклоалкилом, необязательно замещенным С3-7циклоалкил- C1-10алкилом, необязательно замещенным арилом, группой (CR10R20)nNR13R14 или (CR10R20)nC(Z)OR11.
Когда R2 является необязательно замещенным гетероциклилом, кольцо является предпочтительно морфолино, пирролидинильной или пиперидинильной группой. Когда кольцо является необязательно замещенным, заместители могут быть непосредственно присоединены к свободному азоту, такому как в пиперидинильной группе или в пиррольном кольце, или на самом кольце. Предпочтительно кольцо является пиперидином или пирролом, более предпочтительно пиперидином. Гетероциклильное кольцо может быть необязательно замещено один - четыре раза, независимо, галогеном; C1-4 алкилом; галозамещенным C1-4алкилом, таким как трифторметил или трифторэтил; арилом, таким как фенил; арилалкилом, таким как бензил, где арильный или арилалкильный радикалы сами могут быть необязательно замещены (как в разделе определений, ниже); С(О)OR11, таким как радикалы C(O)С1-4алкил или C(O)ОН; С(O)Н; C(O)С1-4алкилом, гидроксизамещенным С1-4алкилом, С1-4алкокси, S(O)mС1-4алкилом (где m равно 0, 1 или 2), NR10R20 (где R10 и R20 являются независимо водородом или С1-4алкилом).
Предпочтительно, если кольцо является пиперидином, кольцо присоединено к имидазолу в положении 4, и заместители находятся непосредственно на доступном азоте, то есть образуют 1-формил-4-пиперидин, 1-бензил-4-пиперидин, 1-метил-4-пиперидин, 1-этокси-карбонил-4-пиперидин, 2,2,2-трифтор-этил-4-пиперидин или 1-трифторацетил-4-пиперидин. Если кольцо является замещенным алкильной группой и кольцо присоединено в положении 4, предпочтительным является замещение в положении 2 или 6 или в обоих, такие как 2,2,6,6-тетраметил-4-пиперидин. Также, если кольцо является пирролом, кольцо присоединяется к имидазолу в положении 3 и все заместители находятся непосредственно на доступном азоте.
Когда R2 является необязательно замещенной гетероциклилС1-10алкильной группой, кольцо является предпочтительно морфолино, пирролидинильной или пиперидинильной группой. Предпочтительно этих алкильных радикалов имеется от 1 до 4, более предпочтительно 3 или 4 и наиболее предпочтительно 3, как в пропильной группе. Предпочтительные гетероциклические алкильные группы включают, но не являются ограниченными ими, морфолиноэтильный, морфолинопропильный, пирролидинилпропильный, и пиперидинилпропильный радикалы. Гетероциклическое кольцо здесь является также необязательно замещенным подобным же образом, как и выше, для прямого присоединения гетероциклила.
Когда R2 является необязательно замещенным С3-7циклоалкилом или необязательно замещенным С3-7циклоалкилС1-10алкилом, циклоалкильная группа является предпочтительно C5-C6 кольцом, причем это кольцо может быть необязательно замещенным 1 или более раз, независимо, галогеном, таким как фтор, хлор, бром или йод; гидрокси; C1-10алкокси, таким как метокси и этокси; S(O)mалкилом, где m равно 0, 1 или 2, таким как метилтио, метилсульфинилом или метилсульфонилом; амино, моно- и ди-замещенным амино, таким как в группе NR7R17; или где R7R17 могут быть циклизованы вместе с азотом, к которому они присоединены, с образованием 5-7-членного кольца, которое необязательно включает дополнительный гетероатом, выбранный из O/N/S; C1-10алкилом, таким как метил, этил, пропил, изопропил или трет-бутил; галогензамещенным алкилом, таким как СF3 или трифторэтил; гидроксизамещенным C1-10алкилом; С(O)ОR11, таким как свободная кислота или производное сложного метилового эфира; необязательно замещенным арилом, таким как фенил; необязательно замещенным арилалкилом, таким как бензил или фенэтил; и далее, где эти арильные радикалы могут также быть замещены один или два раза галогеном; гидрокси; C1-10алкокси; S(O)mалкилом; амино, моно- или ди-замещенным амино, таким как группа NR7R17; алкилом или галогензамещенным алкилом.
Когда R2 представляет (CR10R20) nNR13R14, R13 и R14 являются такими, как определены в формуле (I), то есть R13 и R14 являются, каждый независимо, выбранными из водорода, необязательного замещенного С1-4алкила, необязательно замещенного арила или необязательно замещенного арилС1-10алкила, или вместе с азотом, к которому они присоединены, образуют 5-7-членное гетероциклическое кольцо, необязательно включающее дополнительный гетероатом, выбранный из кислорода, серы или NR9. Наблюдается, что в некоторых случаях это может давать тот же самый радикал как и гетероциклический C1-10алкильный радикал, отмеченный выше, который также является пригодным в качестве R2. Если кольцо NR13R14 циклизировано, то он может быть необязательно замещенным как здесь определено. Предпочтительно R13 и R14 являются, независимо, водородом, С1-4алкилом, предпочтительно метилом или бензилом. Значение n предпочтительно равно от 1 до 4, более предпочтительно равно 3 или 4 и наиболее предпочтительно равно 3, так как в пропильной группе. Предпочтительные группы включают, но не ограничиваются ими, аминопропил, (N-метил-N-бензил)аминопропил, (N-фенилметил)амино-1-пропил или диэтиламинопропил.
Когда R2 является группой (CR10R20)nC(Z)OR11, R11 является, соответственно, водородом, С1-4алкилом, главным образом метилом. Значение n предпочтительно равно от 1 до 4, более предпочтительно равно 2 или 3, как в пропильной группе. Предпочтительные группы включают, но не ограничиваются ими, карбоксиметил-1-бутил, карбокси-1-пропил или 2-ацетоксиэтил.
Когда R2 является группой (CR10R20)nS(О)mR18, m равно 0, 1 или 2 и R18 предпочтительно является арилом, главным образом фенилом или C1-10алкилом, главным образом метилом. Значение n предпочтительно равно от 1 до 4, более предпочтительно равно 2 или 3, так как в этильной или пропильной группе.
Когда R2 является группой (CR10R20)nOR11, R11 является, соответственно, водородом, арилом, главным образом фенилом, или C1-10алкилом, главным образом метилом или этилом. Значение n предпочтительно равно от 1 до 4, более предпочтительно равно 2 или 3, так как в этильной или пропильной группе.
Когда R2 является группой (CR10R20)nNHS(O)2R18, R18 является, соответственно, алкилом, главным образом метилом. Значение n предпочтительно равно от 1 до 4, более предпочтительно равно 2 или 3, так как в этильной или пропильной группе.
Когда R2 является необязательно замещенным арилом, арил предпочтительно является фенилом. Арильное кольцо может быть необязательно замещенным один или несколько раз, предпочтительно одним или двумя заместителями, независимо выбранными из С1-4алкила, галогензамещенного С1-4алкила, такого как трифтрометил или трифторэтил, галогена, главным образом фтора или хлора, (CR10R20)tOR11, -(CR10R20)tNR10R20, главным образом амино или моно- или ди-алкиламино -(CR10R20)nS(О)mR18, где m равно 0, 1 или 2; -SH-, -(CR10R20)nNR13R14, -NR10C(Z)R3 (такой как -NНСО(С1-10алкил)); -NR10S(O)mR8 (такой как -NHSO2 (C1-10алкил)); и t равно 0 или целому числу от 1 до 4. Предпочтительно фенил является замещенным в 3 или 4 положении - (CR10R20)tS(О)mR18, и R18 предпочтительно является C1-10алкилом, главным образом метилом.
Когда R2 является необязательно замещенным гетероарильной или гетероарилалкильной группой, кольцо может быть необязательно замещенным один или несколько раз, предпочтительно одним или двумя заместителями, независимо выбирая из одного или нескольких раз, С1-4алкилом, галогензамещенным С1-4алкилом, таким как трифторметил или фторэтил, галогеном, главным образом фтором или хлором, (CR10R20) tOR11, -(CR10R20)tNR10R20, главным образом амино или моно- или ди-алкиламино -(CR10R20)nS(O)mR18, где m равно 0, 1 или 2; -SH-, -(CR10R20)nNR13R14, -NR10C(Z)R3 (такой как -NHCO (C1-10алкил)); -NR10S(O)mR8 (такой как -NHSO2 (C1-10алкил)); и t равно 0 или целому числу от 1 до 4.
Специалист в данной области легко заметит, что, когда R2 является радикалом (CR10R20)nOC(Z)R11 или (CR10R20)nOC(Z)R13R14, или любой другой подобным образом замещенной группой, n предпочтительно равно не менее 2, что позволяет осуществить синтез стабильных соединений.
Предпочтительно R2 представляет С1-4алкил (разветвленный и неразветвленный), главным образом метил, метилтиопропил, метилсульфинилпропил, аминопропил, N-метил-N-бензиламинопропильную группу, диэтиламинопропил, циклопропилметил, морфолинилбутил, морфолинилпропил, морфолинилэтил, пиперидин или замещенный пиперидин. Более предпочтительно R2 представляет метил, изопропил, бутил, трет-бутил, н-пропил, метилтиопропил или метилсульфинилпропил, морфолинпропил, морфолинилбутил, фенил, замещенный галогеном, тиоалкилом или сульфинилалкилом, такой как метилтио, метилсульфинильный или метилсульфонильный радикал; пиперидинил, 1-формил-4-пиперидин, 1-бензил-4-пиперидин, 1-метил-4-пиперидин, 1-этокси-карбонил-4-пиперидин, 2,2,2-трифторэтил-4-пиперидин или 1-трифторацетил-4-пиперидин.
Во всех случаях там, где существует алкенил или алкиниловый радикал в качестве замещающей группы, ненасыщенная связь, то есть виниленовая или ацетиленовая связь предпочтительно не является связанной непосредственно с азотным, кислородным или серным радикалами, например в ОR3 или для некоторых радикалов R2.
Как здесь используется, термин "необязательно замещенный", если он не определяется специально, должен означать такие группы, как фтор, хлор, бром или йод; гидрокси; гидроксизамещенный C1-10алкил; C1-10алкокси, такой как метокси или этокси; S(O)mалкил, где m равно 0, 1 или 2, такой как метилтио, метилсульфинил или метилсульфонил; амино, моно- и ди-замещенный амино, такой как в группе NR7R17; или где R7R17 могут вместе с азотом, к которому они присоединены, циклизироваться с образованием 5-7-членного кольца, которое необязательно включает дополнительный гетероатом, выбранный из O/N/S; C1-10алкил, циклоалкил или циклоалкилалкильная группа, такая как метил, этил, пропил, изопропил, трет-бутил и тому подобное или циклопропилметил; галогензамещенный C1-10алкил, такой как СF3; или трифторэтил, необязательно замещенный арил, такой как фенил, или необязательно замещенный арилалкил, такой как бензил или фенэтил, где эти арильные радикалы могут также быть замещены один или два раза галогеном; гидрокси; гидроксизамещенный алкил; C1-10алкокси; S(O)mалкил; амино, моно- и ди-замещенный амино, такой как в группе NR7R17; алкил или СF3.
В предпочтительной подгруппе соединений формулы (I) R2 представляет морфолинилпропил, аминопропил, пиперидинил, N-бензил-4-пиперидинил, N-метил-4-пиперидинил или 2,2,6,6-тетраметилпиперидин-4-ил, 2,2,2-трифторэтил-4-пиперидин или 1-трифторацетил-4-пиперидин; и R4 представляет фенил или фенил, замещенный один или два раза фтором, хлором, C1-4aлкокси, S(O)mалкил, метансульфонамид или ацетамид.
В предпочтительной подгруппе соединений формулы (II) R1 представляет 2-метиламино-4-пиримидинил или 2-метиламино-4-пиридил; R2 представляет морфолинилпропил, аминопропил, пиперидинил, N-бензил-4-пиперидинил, N-метил-4-пиперидинил или 2,2,6,6-тетраметилпиперидин-4-ил; и R4 представляет фенил или фенил, замещенный один или два раза фтором, хлором, С1-4алкокси, S(О)mалкилом, метансульфонамид или ацетамид.
Соответствующие фармацевтически приемлемые соли хорошо известны специалистам в данной области и включают основные соли неорганических и органических кислот, таких как хлористо-водородная кислота, бромисто-водородная кислота, серная кислота, фосфорная кислота, метансульфоновая кислота, этансульфоновая кислота, уксусная кислота, яблочная кислота, винная кислота, лимонная кислота, молочная кислота, щавелевая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, бензойная кислота, салициловая кислота, фенилуксусная кислота и миндальная кислота. В дополнение, фармацевтически приемлемые соли соединений формулы (I) могут быть также образованы с фармацевтически приемлемым катионом, например, если замещающая группа включает карбоксирадикал. Соответствующие фармацевтически приемлемые катионы хорошо известны специалистам в данной области и включают катионы щелочных металлов, щелочно-земельных металлов, аммония и четвертичного аммония.
Следующие термины, как они здесь используются, относятся к:
"гало" или "галогены" включают галогены: хлор, фтор, бром и йод.
"C1-10алкил" или "алкил" - радикалы как с прямой, так и с разветвленной цепью из от 1 до 10 атомов углерода, если эта длина цепи не ограничена другим образом, включают, но не ограничиваются ими, метил, этил, н-пропил, изо-пропил, н-бутил, втор-бутил, изо-бутил, трет-бутил, н-пентил и тому подобное.
Термин "циклоалкил" используют здесь для обозначения циклических радикалов предпочтительно с 3-8 углеродами, которые включают, но не ограничиваются ими, циклопропил, циклопентил, циклогексил и тому подобное.
Термин "циклоалкенил" используют здесь для обозначения циклических радикалов предпочтительно с 5-8 углеродами, которые имеют хотя бы одну связь, включая, но не ограничиваясь ими, циклопентенил, циклогекенсил и тому подобное.
Термин "алкенил" используют здесь во всех случаях для обозначения радикалов с прямой или разветвленной цепью с 2-10 атомами углерода, если эта длина цепи не ограничена другим образом, включающих, но не ограничивающихся ими, этенил, 1-пропенил, 2-пропенил, 2-метил-1-пропенил, 1-бутенил, 2-бутенил и тому подобное.
"Арил" - фенил и нафтил.
"Гетероарил" (сам по себе или в любом сочетании, в таком как "гетероарилокси" или "гетероарилалкил") - 5-10-членная ароматическая кольцевая система, в которой одно или несколько колец включают один или несколько гетероатомов, выбранных из группы, включающей N, О или S, такие как, но не ограничиваясь ими, пиррол, пиразол, фуран, тиофен, хинолин, изохинолин, хиназолинил, пиридин, пиримидин, оксазол, тиазол, тиадиазол, триазол, имидазол или бензимидазол.
"Гетероциклический" (сам по себе или в любом сочетании, в таком как "гетероциклилалкил") - насыщенная или частично ненасыщенная 4-10-членная кольцевая система, в которой одно или несколько колец включают один или несколько гетероатомов, выбранных из группы, включающей N, О или S, такие как, но не ограничиваясь ими, пирролидин, пиперазин, морфолин, тетрагидропиран или имидазолидин.
Термин "аралкил" или "гетероарилалкил", или "гетероциклический алкил" используют здесь для обозначения C1-4 алкила, как определено выше, присоединенного к арилу, гетероарилу или гетероциклическому радикалу, как также определено здесь, если не указано другого.
"Сульфинил" - оксид S(O) соответствующего сульфида, термин "тио" относится к сульфидам, а термин "сульфонил" относится к полностью окисленному S(O)2 радикалу.
"Ароил" - С(O)Аr, где Аr представляет производное фенила, нафтила или арилалкила, такое как определены выше, такая группа включает, но не ограничивается ими, бензил и фенэтил.
"Алканоил" - С(О) C1-10алкил, где алкил является таким, как определено выше.
Для целей настоящего изобретения "ядро" 4-пиримидинил радикала для R1 или R2 упоминается как формула
Соединения по настоящему изобретению могут включать один или несколько асимметричных атомов углерода и могут существовать в рацемических и оптически активных формах. Все эти соединения входят в объем настоящего изобретения.
Примеры соединений формулы (I) включают:
5-(2-ацетамидо-4-пиримидинил)-4- (4-фторфенил)-1-(4-морфолино-3-пропил)имидазол;
5-(2-ацетамидо-4-пиримидинил)-4- (4-фторфенил)-1-(1-метил-4-пиперидинил)имидазол.
Примеры соединений формулы (II) включают:
5-[4-(2-метиламино)пиримидинил] -4- (4-фторфенил)-1-(1-метил-пиперидин-4-ил)имидазол;
5-[4-(2-метиламино)пиримидинил] -4- (4-фторфенил)-1-(4-морфолин-3-пропил)имидазол;
5-[4-(2-метиламино)пиримидинил] -4- (4-фторфенил)-1-(пиперидин-4-ил)имидазол;
5-[(2-этиламино)пиримидин-4-ил] -4- (4-фторфенил)-1-(1-метилпиперидин-4-ил)имидазол;
4-(4-фторфенил)-5-[2-(изопропил)аминопиримидинил -4-ил] -1-(1-метилпиперидин-4-ил)имидазол;
5-[4-(2-метиламино-4-пиримидинил)] -4- (4-фторфенил)-1-(2,2,6,6-тетраметилпиперидин-4-ил)имидазол;
5-(2-метиламино-4-пиримидинил)-4- (4-фторфенил)-1-(2-цианоэтил)имидазол;
5-(2-метиламино-4-пиримидинил)-4- (4-фторфенил)-1-[1-(2,2,2-трифторфенил)-4-пиперидинил]имидазол;
5-(2-амино-4-пиримидинил)-4- (4-фторфенил)-1-[1-(2,2,2-трифторэтил)-4 -пиперидинил]имидазол;
5-(2-амино-4-пиримидинил]-4- (4-фторфенил)-1-[(1-трифторацетил) -4-пиперидинил]имидазол.
Предпочтительные соединения формулы (II) включают:
5-[4-(2-метиламино)пиримидинил] -4- (4-фторфенил)-1-(4-морфолино-3-пропил)имидазол;
5-[4-(2-метиламино)пиримидинил] -4- (4-фторфенил)-1-(1-метилпиперидин-4-ил)имидазол;
5-[4-(2-метиламино)пиримидинил]-4- (4-фторфенил)-1-(4-пиперидин)имидазол.
Настоящее изобретение, кроме того, включает новые соединения, приведенные ниже:
5-[4-(2-метилтио)пиримидинил]-4- (4-фторфенил)-1-(4-пиперидин)имидазол;
4-(фторфенил)-1-(1-метилпиперидин- 4-ил)-5-(2-метилтио-4-пиримидинил)имидазол;
4-(фторфенил)-1-(1-метилпиперидин- 4-ил)-5-(2-метилсульфинил-4-пиримидинил)имидазол;
1-трет-бутил-4-(4-фторфенил)-5-(2 -метилсульфинил-4-пиримидинил)имидазол;
5-(2-амино-4-пиримидинил)-4-(4 -фторфенил)-1-(тетрагидро-4-тиопиранил)имидазол;
5-(2-амино-4-пиримидинил)-4-(4- фторфенил)-1-(тетрагидро-4-пиранил)имидазол;
5-(2-амино-4-пиримидинил)-4-(4- фторфенил)-1-(тетрагидро-4-сульфинилпиранил)имидазол;
5-(2-амино-4-пиримидинил)-4-(4- фторфенил)-1-(тетрагидро-4-сульфонилпиранил)имидазол;
5-(2-амино-4-пиримидинил)-4-(4- фторфенил)-1-(1-трифторацетил-пиперидин-4-ил)имидазол;
5-(4-пиридил)-4-(4-фторфенил)-1- (4-пиперидинил) имидазол;
5-(4-пиридил)-4-(4-фторфенил)-1- (1-трет-бутоксикарбонил-4-пиперидинил)имидазол.
Для целей настоящего изобретения области доз, детали приготовления препаратов и способы получения соединений формулы (II) являются аналогичными тем, которые относятся к соединениям формулы (I). Кроме того, дозы и подробности приготовления препаратов с использованием соединений формулы (А) в качестве ингибиторов СОХ-2 и PGHS являются аналогичными тем, которые относятся к соединениям формулы (I), как описано здесь.
В дальнейшем аспекте настоящее изобретение касается способа синтеза соединений формулы (I) и (II), как иллюстрируется выше и далее, соединений формулы (А), где R1 представляет 4-пиридил, пиримидинил, хинолил, изохинолинил, хиназолин-4-ил, 1-имидазолил или 1-бензимидазолил, где гетероарильное кольцо необязательно замещено одним или двумя заместителями, каждый из которых, независимо, выбран из C1-4алкила, галогена, гидроксила, С1-4алкокси, С1-4алкилтио, С1-4алкилсульфинила, CH2OR12, амино, моно- или ди-С1-6алкилзамещенного амино, N(R10)C(O)Ra или N-гетероциклильного кольца, причем кольцо имеет от 5 до 7 членов и необязательно содержит дополнительный гетероатом, выбранный из кислорода, серы или NR15; и где все замещающие группы в R1, R2 и R4 являются такими же, как в формулах (I) и (II). Для использования в разделе описания синтеза синтез "соединений формулы (I)" будет также относиться к этому более широкому описанию R1.
В дальнейшем аспекте настоящее изобретение касается соединения формулы (IIа), имеющего структуру
где р равно 0 или 2; R4 является таким, как определено для формулы (I), и Аr представляет необязательно замещенный арил, как здесь определено. Соответственно, Аr представляет фенил необязательно замещенный С1-4алкилом, С1-4алкокси или галогеном. Предпочтительно Аr представляет фенил или 4-метилфенил, то есть тозильное производное.
Соединения формулы (I) и (II) могут быть получены способами синтеза, некоторые из которых иллюстрируются на схемах I-XI (см. в конце описания). Синтез, предусмотренный на этих схемах, может быть применен для получения соединений формулы (I), имеющих множество различных групп R1, R2 и R4, которые взаимодействуют с использованием необязательных заместителей, соответствующим образом защищенных в целях совместимости с реакциями, приведенными здесь. Последующее снятие защиты в этих случаях затем дает соединения с обычно описанными свойствами. Как только образуется имидазольное ядре, другие соединения формулы (I) и (II) могут быть получены стандартными способами взаимного превращения функциональных групп, хорошо известными в данной области.
Например: -C(O)NR13R14 из СО2СН3 путем нагревания без или с каталитическим цианидом металла, например NaCN, и HNR13R14 в СН3ОН; ОС(O)R3 из -ОН, например, с СlС(O)R3 в пиридине; -NR10-C (S) NR13R14 из -NHR10 с алкилизотиоцианатом или тиоциановой кислотой; NR6C(O)OR6 из -NНR6 с алкилхлорформиатом; -NR10C(О)NR13R14 из -NHR10 путем обработки изоцианатом, например, HN= C= O или R10N=C=O; -NR10C(O)R8 из -NHR10 путем обработки СlС(O)R3 в пиридине; -С(= NR10)NR13R14 из -С(NR13R14)SR3 с Н3NR3 +OАс- путем нагревания в спирте; -С(NR13R14)SR3 из -С(S)NR13R14 с R6-I в инертном растворителе, например ацетоне; -C(S)NR13R14 (где R13 или R14 не является водородом) из -C(S)NH2 с HNR13R14-C(=NCN)-NR13R14 из -С(=NR13R14)-SR3 с NH2CN путем нагревания в безводном спирте, альтернативно из -C(=NH)-NR13R14 путем обработки BrCN и NaOEt в EtOH; -NR10-C (=NCN) SR8 из -NHR10 путем обработки (R8S)2C= NCN; -NR10SО2R3 из -NHR10 путем обработки СlSO2R3 путем нагревания в пиридине; -NR10C(S)R3 из NR10C(O)R8 путем обработки реагентом Лавессона (Lawesson's reagent) [2,4-бис-(4-метоксифенил)-1,3,2,4-дитиа-дифосфетан-2,4-дисульфид] ; -NR10SО2СF3 из -NHR6 с ангидридом трифторметансульфоновой кислоты и снованием, где R3, R6, R10, R13 и R14 такие, как определены здесь для формулы (I).
Предшественники групп R1, R2 и R4 могут быть другими группами R1, R2 и R4, которые могут быть взаимно преобразованы друг в друга стандартными способами взаимного превращения функциональных групп. Например, соединение формулы (I), где R2 представляет галогензамещенный C1-10алкил, может быть преобразовано в соответствующее С1-10алкилN3 производное путем взаимодействия с соответствующей азидной солью, а после этого, если желательно, может быть восстановлено до соответствующего С1-10алкилNН2 соединения, которое, в свою очередь, может взаимодействовать с R18S(O)2X, где Х представляет галоген (например, хлор), с получением соответствующего соединения С1-10алкилNНS (O)2R18.
Альтернативно соединение формулы (I), где R2 представляет галогензамещенный C1-10алкил, может взаимодействовать с aминR13R14NH с получением соответствующего соединения С1-10алкилNR13R14 или может взаимодействовать с солью щелочного металла R18SH с получением соответствующего соединения С1-10алкилSR8.
Относительно схемы I, соединения формулы (I) соответствующим образом получают путем взаимодействия соединения формулы (IIа) с соединением формулы (III), где р равно 0 или 2, R1, R2 и R4 такие, как определены здесь для формулы (I), или являются предшественниками групп R1, R2 и R4, и Аr представляет необязательно замещенную фенильную группу, и после этого, если необходимо, путем преобразования предшественника R1, R2 и R4 в группу R1, R2 и R4. Как обнаружено, R2NH2, который взаимодействует с R1CHO с образованием имина, радикала R2 формулы (III), когда он содержит реакционноспособную функциональную группу, такую как первичный или вторичный амин, спирт или тиольное соединение, эта группа должна быть соответствующим образом защищена. Соответствующие защитные группы можно найти в Protecting Groups Organic Synthesis, Green T.W., Wiley-Interscience, New York, 1981, описание которого включено здесь в качестве ссылки. Например, когда R2 представляет гетероциклическое кольцо, такое как пиперидиновое кольцо, азот защищен группами, такими как трет-Вос, CO2R18 или замещенный арилалкильный радикал.
Реакцию удобно проводить при температуре окружающей среды или при охлаждении (например, от -50o до 10o), или при нагревании в инертном растворителе, таком как метиленхлорид, ДМФ, тетрагидрофуран, толуол, ацетонитрил или диметоксиэтан, в присутствии подходящего основания, такого как К2СО3, трет-бутилNН2, 1,8-диазабицикло[5,4,0] ундец-7-ен (ДБУ) или гуанидинового основания, такого как 1,5,7-триазабицикло[4,4,0]дец-5-ен (ТБД). Промежуточные соединения формулы (II), как обнаружено, являются очень стабильными и пригодны для хранения в течение длительного времени. Предпочтительно, р равно 2.
Взаимодействие соединения формулы (IIа), где р=2, с соединением формулы (III), схема I, дает существенно более высокие выходы соединений формулы (I), чем там, где р=0. Кроме того, взаимодействие соединений формулы (IIа), где р= 2, является более привлекательным с точки зрения окружающей среды и экономики. Когда р=0, предпочтительным используемым растворителем является метиленхлорид, который является непривлекательным с точки зрения окружающей среды для производства в промышленном масштабе, а предпочтительное основание ТБД, является также более дорогим и дает некоторые побочные продукты и примеси, чем тогда, когда используют коммерчески привлекательный синтез (р=2), как описано далее.
Как отмечено, по схеме I используют 1,3-диполярные циклодобавления аниона замещенного арилтиометилизицианида (когда р=0) к имину. Более конкретно, это взаимодействие требует сильного основания, такого как аминовое основание, для использования на стадии депротонирования. Коммерчески доступный ТБД является предпочтительным, хотя могут быть использованы также трет-бутоксид, Li+ или Na+, или К+ гексаметилдисилазид. Хотя метиленхлорид является предпочтительным растворителем, могут быть использованы и другие галогенированные растворители, такие как хлороформ или четыреххлористый углерод; эфиры, такие как ТГФ, ДМЭ, ДМФ, диэтиловый эфир, трет-бутилметиловый эфир; а также ацетонитрил, толуол или их смеси. Реакция может иметь место при от около -20oС до около 40oС, предпочтительно от около 0oС до 20oС, более предпочтительно от около 0oС до около 10oС и наиболее предпочтительно около 4oС, для реакций, включающих R1 группу пиримидина. Для соединений, где R1 является пиридином, обнаружено, что могут быть необходимы изменения условий реакции, как температуры, так и растворителя, такие как уменьшение температур до около -50oС или замена растворителя на ТГФ.
В дальнейшем процессе соединения формулы (I) могут быть получены путем присоединения соответствующего производного соединения формулы (IX)
где T1 представляет водород, а Т4 представляет R4 или альтернативно T1 представляет R1 и Т4 представляет Н, в которой R1, R2 и R4 такие, как определены выше; к соединению, представляющему собой: (i) когда T1 представляет водород, соответствующее производное гетероарильного кольца R1H, в условиях связывания кольца, для осуществления связывания гетероарильного кольца R1 с имидазольным ядром в положении 5; (ii) когда Т4 представляет водород, соответствующее производное арильного кольца R4H, в условиях связывания кольца для осуществления связывания арильного кольца R4 с имидазольным ядром в положении 4.
Такие реакции присоединения к арилу/гетероарилу хорошо известны специалистам в данной области. Обычно металлоорганический синтетический эквивалент аниона одного компонента связывают с реакционноспособным производным второго компонента в присутствии соответствующего катализатора. Анионный эквивалент может быть образован либо из имидазола формулы (IX), в этом случае арильное/гетероарильное соединение обеспечивает реакционноспособное производное, или из арильного/гетероарильного соединения, в этом случае имидазол обеспечивает реакционноспособное производное. Соответственно, подходящие производные соединения формулы (IX) или арильные/гетероарильные кольца включают металлоорганические производные, такие как магнийорганические, цинкорганические, оловоорганические и производные борной кислоты, и подходящие реакционноспособные производные включают производные с бромом, йодом, фторсульфонатом и трифторметансульфонатом. Соответствующие способы описаны в WO 91/19497, описание которого включено здесь в качестве ссылки.
Соответствующие магнийорганические и цинкорганические производные соединения формулы I могут взаимодействовать с галогеновым, фторсульфонатным или трифлатным производным гетероарильного или арильного кольца в присутствии катализатора связывания кольца, такого как палладиевый (0) или палладиевый (II) катализатор, следуя способу Kumada et al., Tetrahedron Letters, 22, 5319 (1981). Удобный такой катализатор включает тетракис(трифенилфосфин)палладий и PdCl2[1,4-бис(дифенилфосфин)бутан], необязательно в присутствии хлорида лития и основания, такого как триэтиламин. Кроме того, никелевый (II) катализатор, такой как Ni (II) Сl2 (1, 2-бифенилфосфин) этан может также быть использован для связывания арильного кольца, следуя способу Pridgen et al. , J. Org. Chem, 1982, 47, 4319. Подходящие реакционные растворители включают гексаметилфосфорамид. Когда гетероарильное кольцо представляет собой 4-пиридил, соответствующие производные включают 4-бром- и 4-йод-пиридин и фторсульфонатные и трифлатные сложные эфиры 4-гидроксипиридина. Подобным же образом соответствующие производные в случае, когда арильное кольцо является фенилом, включают бром, фторсульфонат, трифлат и предпочтительно йодпроизводные. Соответствующие магнийорганические и цинкорганические производные могут быть получены путем обработки соединения формулы (IX) или его бромпроизводного алкиллитиевым соединением с получением соответствующего литиевого реагента путем депротонирования или трансметаллирования соответственно. Литиевое промежуточное соединение затем может быть обработано избытком галогенпроизводного магния или цинка с получением соответствующего металлоорганического реагента.
Производное триалкилолова соединения формулы (IX) может быть обработано бром-, фторсульфон-, трифлат- или предпочтительно йодпроизводным арильного или гетероарильного кольца в инертном растворителе, таком как тетрагидрофуран, предпочтительно содержащем 10% гексаметилфосфорамида, в присутствии соответствующего катализатора присоединения, такого как палладиевый (0) катализатор, например тетракис-(трифенилфосфин)палладия, с помощью способа, описанного в Stille, J. Amer. Soc, 1987, 109, 5478, в патентах США 4719218 и 5002942, или путем использования палладиевого (II) катализатора в присутствии хлорида лития, необязательно с добавлением основания, такого как триэтиламин, в инертном растворителе, таком как диметилформамид. Производные триалкилолова могут быть удобно получены путем металлирования -соответствующего соединения формулы (IX) соответствующим агентом для присоединения лития, таким как втор-бутиллитий или н-бутиллитий, в эфирном растворителе, таком как терагидрофуран, или путем обработки бромпроизводного соответствующего соединения формулы (IX) алкиллитием с последующей, в каждом случае, обработкой галогенидом триалкилолова. Альтернативно бромпроизводное соединения формулы (IX) может быть обработано соответствующим гетероарильным или арильным производным триалкилолова в присутствии катализатора, такого как тетракис(трифенилфосфин)палладий, в условиях, подобных условиям, описанным выше.
Производные борной кислоты также являются пригодными для использования. Следовательно, соответствующее производное соединения формулы (IX), такое как производное бром, йод, трифлат или фторсульфонатпроизводное, может взаимодействовать с гетероарил- или арилборной кислотой в присутствии палладиевого катализатора, такого как тетракис(трифенилфосфин)палладий или PdCl2[1,4-бис(дифенилфосфин)бутан] , в присутствии основания, такого как бикарбонат натрия, в условиях нагревания с обратным холодильником, в растворителе, таком как диметоксиэтан (смотри Fisher and Haviniga, Rec. Trav. Chim. Pays Bas, 84, 439, 1965, Snieckus., Tetrahedron Lett., 29, 2135, 1988 and Terashimia, M. , Vhem. Pharm. Bull., 11, 4755, 1985). Также могут быть использованы неводные условия, например растворитель, такой как ДМФ, при температуре около 100oС в присутствии Pd(II) катализатора (смотри Thompson W. J. et al., J. Org. Chem, 49, 5237, 1984). Подходящие производные борной кислоты могут быть получены путем обработки магний или литийпроизводного с эфиром триалкилборной кислоты, таким как триэтил, триизопропил или трибутилборат, в соответствии со стандартными способами.
В таких реакциях присоединения легко заметить, что необходимо учитывать наличие функциональных групп в соединениях формулы (IX). Таким образом, как правило, заместители с аминогруппой или с серой должны быть неокисленными или защищенными.
Соединения формулы (IX) являются имидазолами и могут быть получены с помощью любого из способов, описанных ранее для получения соединений формулы (I). В частности, α-галогенкетон или другие соответственно активированные кетоны R4COCH2Hal (для соединений формулы (IX), где T1 представляет водород), или R1COCH2Hal (для соединений формулы (IX), где Т4 представляет водород), могут взаимодействовать с амидином формулы R2NH-C=NH, где R2 является таким, как определен в формуле (I), или с его солью в инертном растворителе, таком как галогенированный углеводородный растворитель, например хлороформ, при умеренно повышенной температуре, и, если необходимо, в присутствии соответствующего конденсирующего агента, такого как основание. Получение соответствующих α-галогенкетонов описано в WO 91/19497. Соответствующие реакционноспособные сложные эфиры включают эфиры сильных органических кислот, таких как низшая алкансульфоновая или арилсульфоновая кислота, например метан- или п-толуолсульфоновая кислота. Амидин предпочтительно используют в виде соли, удобным образом, гидрохлоридной соли, которая затем может быть преобразована в свободный амидин in situ путем использования двухфазной системы, в которой реакционноспособный сложный эфир находится в инертном органическом растворителе, таком как хлороформ, а соль находится в водной фазе, к которой медленно добавляют водный раствор основания в димолярном количестве и при энергичном перемешивании. Соответствующие амидины могут быть получены с помощью стандартных способов, смотри, например, Garigipati R., Tetrahedron Letters, 190, 31, 1989.
Соединения формулы (I) также могут быть получены с помощью способа, который включает взаимодействие соединения формулы (IX), где T1 представляет водород, с N-ацилгетероарильной солью в соответствии со способом, описанным в патенте США 4803279, патенте США 4719218 и патенте США 5002942, с получением промежуточного соединения, в котором гетероарильное кольцо присоединено к имидазольному ядру и присутствует как его 1,4-дигидропроизводное, причем это производное может быть затем подвергнуто условиям окисления-деацилирования (схема II). Гетероарильная соль, например пиридиниевая соль, может быть либо получена предварительно, либо более предпочтительно получена in situ путем добавления замещенного карбонилгалогенида (такого как ацилгалогенид, ароилгалогенид, арилалкилгалогенидформиатный эфир или предпочтительно алкилгалогенформиатный эфир, такой как ацетилбромид, бензоилхлорид, бензилхлорформиат или предпочтительно этилхлорформиат) к раствору соединения формулы (IX) в гетероарильном соединении R1H или в инертном растворителе, таком как метиленхлорид, к которому добавляют гетероарильное соединение. Соответствующие условия деацилирования и окисления описаны в патентах США 4803279, 4719218 и 5002942, эти ссылки тем самым включены во всей их полноте. Соответствующие системы окислителей включают серу в инертном растворителе или в смеси растворителей, таких как декалин, декалин и диглим, п-цимен, ксилол или мезитилен, при нагревании с обратным холодильником, или предпочтительно трет-бутоксид калия в трет-бутаноле с сухим воздухом или кислородом.
В дальнейшем процессе, проиллюстрированном на схеме III, соединения формулы (I) могут быть получены путем обработки соединения формулы (X) термически или с помощью циклирующего агента, такого как оксихлорид фосфора или пентахлорид фосфора (смотри Engel and Steglich, Leibigs Ann. Chem, 1978, 1916 и Strzybny et al., J. Org. Chem, 1963, 28, 3381). Соединения формулы (X) могут быть получены, например, путем ацилирования соответствующего α-кетоамина активированным производным формиата, таким как соответствующий ангидрид, в обычных условиях ацилирования с последующим образованием имина с R2NH2. Аминокетон может быть получен из основного кетона путем оксаминирования и восстановления, а требуемый кетон может, в свою очередь, быть получен декарбоксилированием бета-кетоэфира, полученного при конденсации арил(гетероарил)уксусного эфира с реагентом R1-СОХ.
На схеме IV проиллюстрированы далее два (2) различных способа, в которых используются кетон (формула XI) для получения соединения формулы (I). Гетероциклический кетон (XI) получают путем добавления аниона алкилгетероцикла, такого как 4-метил-хинолин (получают путем его обработки алкиллитием, таким как н-бутиллитий) к N-алкил-О-алкоксибензамиду, сложному эфиру и другому соответственным образом активированному производному в том же окисленном состоянии. Альтернативно анион может быть конденсирован с бензальдегидом с получением спирта, который затем окисляют до кетона (XI).
В следующем способе N-замещенные производные формулы (I) могут быть получены путем обработки аниона амида формулы (XII)
R1CH2NR2COH (ХII)
где R1 и R2:
(a) нитрилом формулы (XIII)
R4CN (ХIII)
где R4 определен выше, или
(b) избытком ацилгалида, например ацилхлорида, формулы (XIV)
R4COHal (XIV)
где R4 определен выше и Hal представляет галоген, или соответствующим ангидридом с получением бис-ацилированного промежуточного продукта, который затем обрабатывают источником аммония, таким как ацетат аммония.
Одна из разновидностей этого подхода проиллюстрирована на схеме V выше. Первичный амин (R2NH2) обрабатывают галогенметилгетероциклом формулы R1CH2X с получением вторичного амина, который затем преобразуют в амид обычным способом. Альтернативно амид может быть получен так, как показано на схеме V, путем алкилирования формамида R1CH2X. Депротонирование этого амида сильным амидным основанием, таким как диизопропиламид лития или бис(триметилсилил)амид натрия, с последующим добавлением избытка ароилхлорида дает бисацилированное соединение, которое затем замыкают до имидазольного соединения формулы (I) путем нагревания в уксусной кислоте, включающей ацетат аммония. Альтернативно анион амида может взаимодействовать с замещенным арилнитрилом с получением имидазола формулы (I) непосредственно.
Последующее описание и схемы являются дальнейшими примерами способа, первоначально описанного выше в схеме I. Различные пиримидинальдегидные производные 6, 7 и 8, как показано в схеме VI, могут быть получены путем модификации способов Bredereck et al. (Chem. Ber 1964, 97, 3407), это описание включено здесь в качестве ссылки. Эти пиримидинальдегиды затем используют в качестве промежуточных соединений в синтезе, как далее здесь описано. Незащищенное аминоальдегидное производное, например, 8 может быть несколько нестабильным. Использование способа ацетолиза, как описано в схеме VI, где альдегид 7 выделяют в виде ацетамидного производного (соединение 3 преобразовывают в 7 через промежуточный продукт 4), ведет к более стабильному соединению для использования в реакции прибавлении цикла с получением соединений формулы (I).
Для такой реакции используют обычные условия ацетолиза, и они являются хорошо известными специалистам в данной области. Соответствующие условия представлены, например, в примере 83. Более детально, в реакции используют нагревание 2-аминопиримидиндиалкоксиацеталя с уксусным ангидридом в присутствии каталитического количества концентрированной серной кислоты, которая одновременно ацетилирует амин и ведет к замене одной из алкоксигрупп на ацетоксигруппу. Полученное в результате соединение преобразуют в альдегид путем деацетилирования с каталитическим количеством алкоксидной соли в соответствующем спиртовом растворителе, например Na+ метоксид и метанол. Альтернативно более высокие выходы могут быть получены путем сначала ацетилирования амина уксусным ангидридом, а затем осуществления обмена путем последующего добавления концентрированной серной кислоты.
О взаимодействии иминов с тозилметилизонитрилами было впервые сообщено van Leusen (van Leusen, et al., J. Org. Chem. 1977, 42, 1153). Были сообщены следующие условия: трет-бутиламин(трет-ВuNН2) в диметоксиэтане (ДМЭ), К2СО3 в МеОН и NaH в ДМЭ. При проверке этих условий, как может обнаружить любой, получаются низкие выходы. Желаемый продукт, например, 5-[(2-(1-метиламино)пиримидин-4-ил] -4-(4-фторфенил)-1-(1-метилпиперидин-4-ил)имидазол, выделяют с выходами менее 50%, используя трет-BuNH2 в ДМЭ при комнатной температуре, но можно использовать также и второй путь реакции, включающий аминный обмен с получением трет-бутилимина с последующим взаимодействием с изоцианидом 1 с получением трет-Вuимидазола. Вероятно, это можно осуществить, используя любой первичный амин в качестве основания. Вторичные амины, хотя и не являются предпочтительными, могут быть использованы, но могут они также медленно разлагать изонитрил. Вероятно, для реакции требуется около 3 эквивалентов амина, чтобы дойти до завершения, с получением примерно 50%-ных выходов выделенных продуктов. Вторичные амины со стерическими ограничениями (диизопропиламин), хотя и могут быть использованы, реагируют очень медленными и обычно не слишком эффективны. Использование третичных и ароматических аминов, таких как пиридин и триэтиламин, не дает взаимодействия в некоторых исследуемых условиях, но более щелочные виды, такие как ДБУ и 4-диметиламинопиридин (ДМАП), хотя и реагируют медленно, дают некоторые выходы и поэтому могут быть пригодными здесь для использования.
Как указано в схемах VII и VIII, пиримидинальдегиды по схеме VI могут быть конденсированы с первичным амином с получением имина, который может быть выделен соответствующим образом, или может взаимодействовать in situ с желаемым изонитрилом в присутствии разнообразных соответствующих оснований и растворителей, как здесь описано, давая 5-(4- пиримидинил)имидазолы, где R2 и R4 являются такими, как здесь описано для соединений формулы (I).
Предпочтительный способ получения соединений формулы (I) представлен на схеме VII. Имины, полученные и выделенные на отдельной стадии, часто являются смолами, с которыми трудно обращаться. Черный цвет часто переносится также на конечный продукт. Выход при получении иминов изменяется, и при их получении часто используют менее приемлемые с точки зрения окружающей среды растворители, такие как СН2Сl2.
Для этой реакции, где р=2, требуется соответствующее основание для осуществления реакции. Для реакции требуется основание, достаточно сильное для депротонирования изонитрила. Соответствующие основания включают реагенты на основе амина, карбоната, гидрида или алкил- или ариллития или их смеси. Основания включают, но не ограничиваются ими, карбонат калия, карбонат натрия, первичные и вторичные амины, такие как трет-бутиламин, диизопропиламин, морфолин, пиперидин, пирролидин, и другие ненуклеофильные основания, такие как ДБУ, ДМАП и 1,4-диазабицикло[2,2,2]октан (ДАБЦО).
Соответствующие растворители для использования здесь включают, но не ограничиваются ими, N, N-диметилформамид (ДМФ), MeCN, галогенированные растворители, такие как метиленхлорид или хлороформ, тетрагидофуран (ТГФ), диметилсульфоксид (ДМСО), спирты, такие как метанол или этанол, бензол, толуол, ДМЭ или EtOAc. Предпочтительно растворителем является ДМФ, ДМЭ, ТГФ или MeCN, более предпочтительно ДМЭ. Выделение продукта обычно достигается путем добавления воды и фильтрования продукта в виде прозрачного вещества.
Хотя и не очень удобное для работы в промышленном масштабе, добавление NaH вместо трет-бутиламина к изонитрилу при температурах, возможно, ниже 25oС (в ТГФ) является, вероятно, необходимым. Кроме того, BuLi также, как сообщается, является эффективным основанием для депротонирования тозилбензилизонитрилов при -50oС. (DiSanto, R. ; Costi, R; Massa, S.; Artico, M. Synt. Commun. 1995, 25, 795).
Могут быть использованы различные температурные условия в зависимости от предпочтительного основания, например, используя трет-BuNH2/DME и К2СО3/МеОН, пробуют проводить реакции при 0, комнатной температуре, 40, около 64 и 80oС. При температурах выше 40oС выходы могут упасть до около 20%, хотя не наблюдается особой разницы между 0oС и комнатной температурой. Используя К2СО3 в ДМФ, пытаются проводить взаимодействия при 0oС и 25oС без видимой разницы в продукте, качестве или выходе. Следовательно, диапазоны температур ниже 0oС и выше 80oС, как предполагается, также находятся в рамках настоящего изобретения. Предпочтительный диапазон температур составляет от около 0o до около 25oС. Для настоящих целей комнатная температура считается равной 25oС, но, как наблюдается, она может изменяться от 20oС до 30oС.
Как показано на схеме VIII, имин предпочтительно образуется in situ в растворителе. Этот предпочтительный синтез является способом, который осуществляется как синтез в одном сосуде. Удобно, когда первичный амин используют в виде соли, такой как дигидрохлоридная соль, например, реакция может, кроме того, включать основание, такое как карбонат калия, перед добавлением изонитрила. Альтернативно азот пиперидина может оказаться необходимым защитить (PG), как показано ниже, удобно, когда PG является ВОС или C(O)2R, где R представляет предпочтительно алкильный, арильный, арилалкильный радикалы, хорошо известные специалистам в данной области. Условия реакции, такие как растворители, основания, температуры и тому подобное, подобны тем, которые иллюстрируются и обсуждаются выше для выделения имина, как показано на схеме VII. Специалист в данной области легко обнаружит, что при некоторых обстоятельствах образование имина in situ может потребовать условий дегидрирования или может потребовать кислотного катализа.
Другой способ получения соединений формулы (I) представлен ниже на схеме VIIIa. Чтобы избежать сложностей, связанных с выделением пиримидинальдегида 8, является возможным гидролиз ацеталя 3 до альдегида 8, как описано в примере 3, часть (b). Альдегид 8, полученный in situ, может быть обработан последовательно первичным амином, этилацетатом и NаНСО3 с получением соответствующего имина in situ, который экстрагируют этилацетатом. Добавление изонитрила, карбонатного основания и ДМФ делает возможным образование 5-(4- пиримидинил)имидазолов, где R2 и R4 являются такими, как здесь определено для соединений формулы (А).
Предпочтительный способ синтеза соединений формулы (I) также предусматривает соответствующий и надежный способ введения S(О)mалкильного радикала в пиримидин (R1 группа) с использованием, например, 2-метилтиопиримидинальдегидого производного, как описано в секции примеров. Ниже в схеме IX соединение 1 (X=S метил), хотя и является конечным продуктом, может быть также использовано в качестве предшественника, как отмечено ранее, для получения дальнейших соединений формулы (I). В этом конкретном случае метилтиорадикал окисляют до метилсульфинильного радикала, который дополнительно может быть далее модифицирован до замещенной аминогруппы.
Другим воплощением настоящего изобретения является новый способ гидролиза 2-тиометилпиримидин ацеталя до 2-тиометилпиримидин альдегида, как показано в схеме X. Гидролиз ацеталя до альдегида с использованием различных известных условий реакции, таких как муравьиная кислота, не дают удовлетворительного выхода альдегида, получают <13%. Один из способов синтеза включает использование АсОН (свежий) в качестве растворителя и концентрированную H2SO4 в условиях нагревания, предпочтительно с каталитическим количеством серной кислоты. Условия нагревания включают температуры от около 60o до 85oС, предпочтительно от около 70o до около 80oС, поскольку более высокие температуры дают потемнение реакционной смеси. После завершения реакции смесь охлаждают
примерно до комнатной температуры и удаляют уксусную кислоту. Более предпочтительный способ включает нагревание ацеталя в 3 н. HCl при 40oС в течение 18 ч, охлаждение и экстрагирование нейтрализованного бикарбонатом раствора EtOAc. Примеры этих двух способов описаны здесь в примерах 6 (b) и 25.
Целевые производные 2-аминопиримидин-4-илимидазола формулы (I), а также подобные пиридинсодержащие соединения могут быть получены с помощью одного из трех способов: 1) непосредственное взаимодействие 2-аминопиримидинимина с изонитрилом; 2) конденсация 2-ацетамидпиримидинимина с изонитрилом с последующим удалением ацетамидогруппы; и 3) окисление 2-метилтиопиримидинового производного до соответствующего сульфоксида с последующим замещением желаемым амином.
Хотя эти схемы представлены, например, с необязательно замещенным пиперидиновым радикалом для конечного положения R2 или 4-фторфенилом для R4, любой соответствующий радикал R2 или R4 может быть добавлен таким способом, если он получается на первичном амине. Подобным образом, любой подходящий R4 может быть добавлен в способе с использованием изонитрила.
Соединения формулы (IIа) на схеме I могут быть получены с помощью способов van Leusen et al., выше. Например, соединение формулы (IIа) может быть получено путем дегидрирования соединения формулы (IV), схема I, где Ar, R4 и р являются такими, как здесь описано.
Соответствующие дегидрирующие агенты включают оксихлорид фосфора, оксалилхлорид, тионилхлорид, фосген или тозилхлорид в присутствии соответствующего основания, такого как триэтиламин или диизопропилэтиламин, или подобных оснований и так далее, таких как пиридин. Соответствующими растворителями являются диметоксиэфир, тетрагидрофуран или галогенированные растворители, предпочтительно ТГФ. Реакция является наиболее эффективной, когда температуру реакции поддерживают между -10oС и 0oС. При более низких температурах происходит незавершенная реакция, а при более высоких температурах раствор делается темным и выход продукта падает.
Соединения формулы (IV), схема I, могут быть получены путем взаимодействия соединения формулы (V), схема I, R4CHO, где R4 определен здесь, с ArS(O)pH и формамидом с удалением воды или без удаления, предпочтительно в условиях дегидрирования при комнатной или повышенной температуре, например от 30o до 150oС, удобно с обратным холодильником, необязательно в присутствии кислотного катализатора. Альтернативно, вместо кислотного катализатора может быть использован триметилсилилхлорид. Примеры кислотных катализаторов включают камфар-10-сульфоновую кислоту, муравьиную кислоту, п-толуолсульфоновую кислоту, хлористый водород и серную кислоту.
Оптимальный способ получения изонитрила формулы (IIа) проиллюстрирован в схеме XI и в разделе примеров, в его примере 10.
Преобразование замещенного альдегида в тозилбензил-формамид может быть достигнуто путем нагревания альдегида, 1 - схема XI, с кислотой, такой как п-толуолсульфоновая кислота, муравьиная кислота или камфарсульфоновая кислота; с формамидом и п-толуолсульфиновой кислотой [в условиях реакции около 60oС в течение около 24 ч]. Предпочтительным является не использовать никакого растворителя. Реакция может давать низкие выходы (<30%), когда используют растворители, такие как ДМФ, ДМСО, толуол, ацетонитрил или избыток формамида. Температуры менее 60oС обычно являются плохими при получении желаемого продукта, а температуры существенно выше 60oС могут давать продукт, который разлагается, или давать бензильный бис-формамид 2 - схема XI. В примере 23 (а) описан синтез 4-фторфенилтозил-метилформамида, соединения формулы (IV), схема I, где р=2, описанный WO 95/02591 Adams et al. Этот способ отличается от того, который описывается здесь в примере 10, следующими условиями: использованием натриевой соли толуолсульфиновой кислоты, что приводит к неравномерному нагреву, более низким выходам и более низкой воспроизводимости, чем по настоящему изобретению, как здесь описывается, по которому используют сульфиновую кислоту и получают возможность использования неводных условий.
Условия для получения α-(п-толуолсульфонил)-4-фторбензилизонитрила, как описано в примере 23(b), описанном в WO 95/02591 Adams et al., в качестве растворителя для экстрагирования продукта используют МеCl и ДМЭ в качестве растворителя. Настоящее изобретение усовершенствует этот способ путем использования менее дорогостоящих растворителей, таких как ТГФ или EtOAc, для экстрагирования. Еще более высокие выходы получают путем повторной кристаллизации в спирте, таком как 1-пропанол, хотя другие спирты, такие как метанол, этанол и бутанолы, являются приемлемыми. Ранее соединения частично очищали хроматографическими методами, а также опасными растворителями для дополнительной очистки.
Другим воплощением настоящего изобретения является синтез тозилбензилформамидного производного, который осуществляют путем взаимодействия бис-формамидного промежуточного продукта 2 - схема XI с п-толуолсульфиновой кислотой. В этом предпочтительном способе получение бис-формамида из альдегида достигается путем нагревания альдегида с формамидом в соответствующем растворителе с кислотным катализом. Соответствующими растворителями являются толуол, ацетонитрил, ДМФ и ДМСО или их смеси. Кислотные катализаторы представляют собой катализаторы, хорошо известные в данной области, и включают, но не ограничиваются ими, хлористо-водородную кислоту, п-толуолсульфоновую кислоту, камфарсульфоновую кислоту и другие безводные кислоты. Реакцию проводят при температуре в диапазоне от около 25oС до 110oС, предпочтительно около 50oС, лучше всего в течение от около 4 до около 5 ч, являются приемлемыми также более длинные времена реакции. При более высоких температурах (>70oС) и при более длительном времени реакции могут наблюдаться разложение продукта и более низкие выходы. Полное превращение продукта обычно требует удаления воды из реакционной смеси.
Предпочтительные условия преобразования бисформамидного производного в тозилбензилформамид достигаются путем нагревания бисформамида в соответствующем растворителе с кислотным катализатором и п-толуолосульфоновой кислотой. Растворители для использования в этой реакции включают, но не являются ограниченными ими, толуол и ацетонитрил и их смеси. Кроме того, также могут быть использованы смеси этих растворителей с ДМФ или ДМСО, но они могут приводить к более низким выходам. Температура может изменяться от около 30oС до около 100oС. Температура, более низкая чем 40oС и более высокая чем 60oС, не является предпочтительной, поскольку уменьшаются выход и скорость реакции. Предпочтительно диапазон составляет от около 40oС до 60oС, наиболее предпочтительно около 50oС. Оптимальное время составляет от около 4 до 5 ч, хотя оно может быть более продолжительным. Предпочтительно используемые кислоты включают, но не ограничены ими, толуолсульфоновую кислоту, камфарсульфоновую кислоту и хлористо-водородную кислоту, а также другие безводные кислоты. Наиболее предпочтительно бис-формамид нагревают в смеси толуол:ацетонитрил в отношении 1:1 с п-толуолсульфиновой кислотой и хлористым водородом.
Другим воплощением настоящего изобретения является предпочтительный способ получения тозилбензилформамидного соединения, который осуществляют проведением процесса в одном сосуде. Этим способом сначала преобразуют альдегид в бис-формамидное производное, а затем осуществляют взаимодействие бисформамидного производного с толуолсульфиновой кислотой. Этот способ объединяет оптимизированные условия в едином эффективном процессе. Таким способом могут быть достигнуты высокие выходы, >90%, арил(тозил)бензилформамида.
Предпочтительные условия реакции включают катализатор, такой как триметилсилилхлорид (ТМСCl), в предпочтительном растворителе, смеси толуол:ацетонитрил, предпочтительно при соотношении 1:1. Предпочтительным является реагент, такой как ТМСС1, который взаимодействует с водой, образующейся в процессе, и в то же самое время образует хлористый водород для катализа реакции. Также предпочтительным является использование хлористого водорода и п-толуолсульфоновой кислоты. Следовательно, три соответствующих условия реакции, используемые здесь, включают 1) использование дегидрирующего агента, который также поставляет хлористый водород, такого как ТМССl или п-толуолсульфиновая кислота; или 2) использование соответствующего дегидрирующего агента и соответствующего источника кислоты, источника, такого как, но не ограничивающегося им, камфарсульфооновая кислота, хлористо-водородная кислота или п-толуолсульфоновая кислота; и 3) альтернативные условия дегидрирования, такие как азеотропное удаление воды и использование кислотного катализатора и п-толуолсульфиновой кислоты.
Соединения формулы (IIа), где р=2, могут также быть получены путем взаимодействия в присутствии сильного основания соединения формулы (VI), схема I, R4CH2NC, с соединением формулы (VII), схема I, ArSO2L1, где R4 и Аr являются такими, как здесь определены, и L1 является уходящей группой, такой как галоген, например фтор. Соответствующие сильные основания включают, но не ограничиваются ими, алкиллитиевые соединения, такие как бутиллитий или литийдиизопропиламид (Van Leusen et al., Tetrahedron Letters No. 23, 2367-68 (1972)).
Соединения формулы (VI), схема I, могут быть получены путем взаимодействия соединения формулы (VIII), схема I, R4CH2NH2, с алкилформиатом (например, этилформиатом) с получением промежуточного амида, который может быть преобразован в желаемый изонитрил путем взаимодействия с хорошо известным дегидрирующим агентом, таким как, но не ограничиваясь им, оксалилхлорид, фосфороксихлорид или тозилхлорид, в присутствии соответствующего основания, такого как триэтиламин.
Альтернативно, соединение формулы (VIII), схема I, может быть преобразовано в соединение формулы (VI), схема I, путем взаимодействия с хлороформом и гидроксидом натрия в водном растворе дихлорметана с катализом с переносом фазы.
Соединения формулы (III), схема I, могут быть получены путем взаимодействия соединения формулы R1CHO с первичным амином R2NH2.
Аминосоединения формулы (VIII), схема I, являются известными или могут быть получены из соответствующих спиртов, оксимов или амидов с использованием стандартных взаимных преобразований функциональных групп.
Соответствующие защитные группы для использования с гидроксильными группами и азотом имидазола хорошо известны в данной области и описаны во многих ссылках, например, Protecting Groups in Organic Synthesis, Greene T. W. , Wiley-Interscience, New York, 1981. Соответствующие примеры гидроксилзащитных групп включают силильные эфиры, такие как трет-бутилдиметил или трет-бутилдифенил, и алкильные эфиры, такие как метил, соединенный в виде алкильной цепи переменной длины, (CR10R20)n. Соответствующие примеры защитных групп для азота имидазола включают тетрагидропиранил.
Фармацевтические соли добавления кислот соединений формулы (I) могут быть получены известным способом, например путем их обработки соответствующим количеством кислоты в присутствии соответствующего растворителя.
Способы лечения
Соединения формулы (I) или (II) или их фармацевтически приемлемая соль может быть использована при получении лекарственного средства для профилактического или терапевтического лечения любого болезненного состояния у человека или другого млекопитающего, которое обостряется или вызывается избыточной или нерегулируемой продукцией цитокинов такими клетками млекопитающих, такими как, но не ограничиваясь ими, моноциты и/или макрофаги.
Соединения формулы (I) или (II) способны ингибировать про-воспалительные цитокины, такие как ИЛ-1, ИЛ-6, ИЛ-8 и ФНО, и поэтому пригодны для использования в терапии. ИЛ-1, ИЛ-6, ИЛ-8 и ФНО оказывают влияние на широкое разнообразие клеток и тканей, и эти цитокины, а также другие цитокины, продуцированные лейкоцитами, являются важными и критическими медиаторами воспаления широкого разнообразия болезненных состояний и симптомов. Ингибирование этих про-воспалительных цитокинов может использоваться при контроле, уменьшении и ослаблении многих из этих болезненных состояний.
Соответственно, настоящее изобретение относится к способу лечения заболеваний, опосредованных цитокинами, который включает введение эффективно воздействующего на цитокины количества соединения формулы (I) или (II), или их фармацевтически приемлемой соли.
В частности, соединения формулы (I) или (II) или их фармацевтически приемлемая соль могут использоваться при профилактике или терапии болезненного состояния у людей или у других млекопитающих, которое обостряется или вызывается избыточным или нерегулируемым продуцированием ИЛ-1, ИЛ-8 или ФНО, такими клетками млекопитающих, такими как, но не ограничиваясь ими, моноциты и/или макрофаги.
Другой аспект настоящего изобретения заключается в том, что соединения формулы (А), формулы (I) и формулы (II) способны ингибировать индуцируемые про-воспалительные белки, такие СОХ-2, также упоминаемые под многими другими названиями, такими как простагландин, эндопероксидсинтаза-2 (ПГЭС-2) и по этой причине могут использоваться в терапии. Эти про-воспалительные липидные медиаторы циклооксигеназного (ЦО) пути продуцируются индуцируемым ферментом СОХ-2. Следовательно, регуляция СОХ-2, который является ответственным за эти продукты, производные от арахидоновой кислоты, такие как простагландины, оказывают воздействие на широкое разнообразие важных клеток и тканей, и на критические медиаторы воспаления широкого разнообразия болезненных состояний и симптомов. Экспрессия СОХ-1 не подвержена воздействию соединений формулы (I). Селективное ингибирование СОХ-2 может ослабить или уменьшить склонность к образованию язв, связанную с ингибированием СОХ-1, тем самым ингибируя простагландины, наиболее важные для воздействия по защите клеток. Такое ингибирование этих про-воспалительных медиаторов является полезным при контроле, уменьшении и ослаблении многих из этих болезненных состояний. Более заметно такие медиаторы воспаления, в частности простагландины, являются существенными при боли, такой как при обострении чувствительности болевых рецепторов или отек. Следовательно, этот аспект управления болью включает лечение нейромышечной боли, головной боли, боли при раке и боли при артрите. Соединения формулы (I) или их фармацевтически приемлемая соль могут использоваться при профилактике или лечении человека или другого млекопитающего путем ингибирования синтеза фермента СОХ-2.
Соответственно, настоящее изобретение относится к способу ингибирования синтеза СОХ-2, который заключается во введении эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Настоящее изобретение также относится к способу профилактического лечения человека или другого млекопитающего путем ингибирования синтеза фермента СОХ-2.
Соответственно, в другом аспекте это изобретение относится к способу ингибирования продукции ИЛ-1 у млекопитающего, которое в этом нуждается, который включает введение указанному млекопитающему эффективного количества соединения формулы (I) или (II) или их фармацевтически приемлемых солей.
Существует много болезненных состояний, в которых избыточная или нерегулируемая продукция ИЛ-1 участвует в обострении и/или вызове болезни. Они включают ревматоидный артрит, остеоартрит, раздражение, эндотоксемию и/или синдром токсического шока, другие острые или хронические воспалительные болезненные состояния, такие как воспалительная реакция, вызванная эндотоксином, или воспаление кишечника, туберкулез, атеросклероз, дегенерация мышц, множественный склероз, кахеция, резорбция костной ткани, псориатический артрит, синдром Рейтера, ревматоидный артрит, подагра, травматический артрит, артрит при краснухе и острый синовит. Последние данные также связывают активность ИЛ-1 с диабетом, β-клетками поджелудочной железы и болезнью Альцхаймера.
В дальнейшем аспекте настоящее изобретение относится к способу ингибирования продукции ФНО у млекопитающего, которое в этом нуждается, который включает введение указанному млекопитающему эффективного количества соединения формулы (I) или (II) или его фармацевтически приемлемой соли.
Избыточная или нерегулируемая продукция ФНО участвует в опосредовании или обострении ряда заболеваний, включая ревматоидный артрит, ревматоидный спондилит, остеоартрит, артрит при подагре и другие артритные состояния, сепсис, септический шок, эндотоксический шок, грамотрицательный сепсис, синдром токсического шока, синдром недомогания при простуде у взрослых, раздражение, церебральную малярию, хроническое воспаление легких, силикоз, саркоидоз легких, заболевания резорбции костной ткани, такие как остеопороз, реперфузионное повреждение, реакцию на прививку, отторжение аллотрансплантата, лихорадку и судороги, вызванные инфекцией, такой как грипп, вторичную кахецию после инфекции или злокачественного заболевания, вторичную кахецию в результате синдрома приобретенного иммунодефицита (СПИД), СПИД, ВСК (комплекс, вызванный СПИДом), образование келоидной ткани, образование рубцовой ткани, болезнь Крона, язвенный колит и повышенная температура.
Соединения формулы (I) могут также использоваться при лечении вирусных инфекций, где такие вирусы являются чувствительными к увеличению их уровня путем регулирования с помощью ФНО или устанавливают производство ФНО in vivo. Вирусы, предназначающиеся здесь для лечения, являются вирусами, которые производят ФНО в результате инфекции, или вирусами, которые являются чувствительными к ингибированию, например, с помощью пониженной репликации, прямо или косвенно, с помощью ингибирующих ФНО соединений формулы (I) или (II). Такие вирусы включают, но не ограничиваются ими, ВИЧ-1, ВИЧ-2 и ВИЧ-3, цитомегаловирус (ЦМВ), грипп, аденовирус и вирусы группы Herpes, такие как, но не ограничиваются ими, Herpes Zoster и Негрез Simplex. Соответственно, в дальнейшем аспекте настоящее изобретение относится к способу лечения млекопитающего, страдающего от вируса иммунодефицита человека (ВИЧ), который включает введение такому млекопитающему эффективно ингибирующего ФНО количества соединения формулы (I) или (II), или их фармацевтически приемлемой соли.
Соединения формулы (I) или (II) могут также быть использованы в связи с ветеринарным лечением млекопитающих, но не людей, нуждающихся в ингибировании производства ФНО. Заболевания, опосредуемые ФНО и предполагающиеся для терапевтического или профилактического лечения у животных, включают болезненные состояния, такие как те, которые отмечены выше, но в особенности вирусные инфекции. Примеры таких вирусов включают, но не являются ограниченными ими, лентивирусные инфекции, такие как вирус инфекционной анемии лошади, вирус артрита козла, visna или maedi вирус или ретровирусные инфекции, такие как, но не ограничиваясь ими, вирус иммунодефицита кошачьих (ВИК), вирус иммунодефицита быка или вирус иммунодефицита собаки или другие ретровирусные инфекции.
Соединения формулы (I) или (II) также могут быть использованы местно при лечении или профилактике местных болезненных состояний, опосредуемых или обостряемых избыточным производством цитокинов, таких как ИЛ-1 или ФНО соответственно, таких как воспаление связок, экзема, псориаз и другие воспалительные состояния кожи, такие как солнечный ожог; воспалительные состояния глаз, включая коньюктивит; повышенной температуры, боли и других симптомов, связанных с воспалением.
Соединения формулы (I) или (II), как показано, также ингибируют продуцирование ИЛ-8 (Интерлейкин-8, БАН). Соответственно, в следующем аспекте настоящее изобретение относится к способу ингибирования продукции ИЛ-8 у млекопитающего, нуждающегося в этом, который включает введение указанному млекопитающему эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Существует много болезненных состояний, в которых избыточное или нерегулируемое продуцирование ИЛ-8 участвует в обострении и/или является причиной заболевания. Эти заболевания характеризуются массированной инфильтрацией нейтрофилов, такие как псориаз, воспаление кишечника, астма, сердечное и почечное реперфузионное повреждение, синдром недомогания при простуде у взрослых, тромбоз и гломерулонефрит. Все эти заболевания связаны с повышенным производством ИЛ-8, который является ответственным за хемотаксис нейтрофилов в области воспаления. В противоположность другим воспалительным цитокинам (ИЛ-1, ФНО и ИЛ-6), ИЛ-8 обладают уникальным свойством промотора хемотаксиса и активации нейтрофилов. Следовательно, ингибирование производства ИЛ-8 вело бы к прямому уменьшению инфильтрации нейтрофилов.
Соединения формулы (I) или (II) вводят в количестве, достаточном для ингибирования продукции цитокина, в частности ИЛ-1, ИЛ-6, ИЛ-8 или ФНО, так что она понижается при регулировании до нормальных уровней или, в некоторых случаях, до субнормальных уровней, так чтобы ослабить или предотвратить болезненное состояние. Ненормальные уровни ИЛ-1, ИЛ-6, ИЛ-8 или ФНО, например, в контексте настоящего изобретения составляют: (i) уровни свободных (не связанных в клетках) ИЛ-1, ИЛ-6, ИЛ-8 или ФНО, превышающие или равные 1 пикограмм на мл; (ii) любое количество ИЛ-1, ИЛ-6, ИЛ-8 или ФНО, связанное с клетками; или (iii) присутствие мРНК ИЛ-1, ИЛ-6, ИЛ-8 или ФНО при уровнях, выше чем базальные, в клетках или тканях, в которых производятся ИЛ-1, ИЛ-6, ИЛ-8 или ФНО, соответственно.
Обнаружение того, что соединения формулы (I) или (II) являются ингибиторами цитокинов, в частности ИЛ-1, ИЛ-6, ИЛ-8 и ФНО, основывается на воздействии соединений формулы (I) на производство ИЛ-1, ИЛ-8 и ФНО в исследованиях in vitro, которые здесь описаны.
Как здесь используется, термин "ингибирование продуцирования ИЛ-1 (ИЛ-6, ИЛ-8 или ФНО)" относится к следующим явлениям:
a) уменьшение избыточных уровней in vivo цитокина (ИЛ-1, ИЛ-6, ИЛ-8 или ФНО) у человека до нормальных или субнормальных уровней путем ингибирования высвобождения in vivo цитокина из всех клеток, включая, но не ограничиваясь ими, моноциты или макрофаги;
b) регулируемое понижение при генетически обусловленном уровне избыточных уровней цитокина (ИЛ-1, ИЛ-6, ИЛ-8 или ФНО) in vivo у человека до нормальных или субнормальных уровней;
c) регулируемое понижение путем ингибирования прямого синтеза цитокина (ИЛ-1, ИЛ-6, ИЛ-8 или ФНО) как посттрансляционного события; или
d) регулируемое понижение при трансляционном уровне избыточных уровней in vivo цитокина (ИЛ-1, ИЛ-6, ИЛ-8 или ФНО) у человека до нормальных или субнормальных уровней.
Как здесь используется, термин "вызванное ФНО заболевание или болезненное состояние" относится к любому и ко всем болезненным состояниям, в которых ФНО играет какую-либо роль, либо путем продуцирования самого ФНО, либо обусловленного ФНО высвобождения другого монокина, такого как, но не ограничиваясь ими, ИЛ-1, ИЛ-6, ИЛ-8. Болезненное состояние, в котором, например, ИЛ-1 является главным компонентом, и продуцирование или действие которого обостряется или выделяется в ответ на ФНО, должен, следовательно, рассматриваться как болезненное состояние, опосредуемое ФНО.
Как здесь используется, термин "цитокин" относится к любому секретируемому полипептиду, который воздействует на функции клетки и является молекулой, которая модулирует взаимодействия между клетками при иммунном, воспалительном или гематопоэтическом отклике. Цитокин включает, но не ограничивается этим, монокины или лимфокины, независимо от того, какие клетки их производят. Например, обычно упоминается, что монокин продуцируется и выделяется моноядерной клеткой, такой как макрофаг и/или моноцит. Однако множество других клеток также продуцирует монокины, такие как природные клетки-киллеры, фибробласты, базофилы, нейтрофилы, эндотелиальные клетки, астроциты мозга, стромальные клетки костного мозга, эпидермальные кератиноциты и B-лимфоциты. Лимфокины, как обычно упоминается, продуцируются клетками-лимфоцитами. Примеры цитокинов включают, но не ограничиваются ими, интерлейкин-1 (ИЛ-1), интерлейкин-6 (ИЛ-6), интерлейкин-8 (ИЛ-8), фактор некроза опухоли α(ФНО-α) и фактор некроза опухоли β(ФНО-β).
Как здесь используется, термин "влияющее на цитокины" или "подавляющее цитокины количество" относится к эффективному количеству соединения формулы (I) или (II), которое вызывает уменьшение уровней цитокинов in vivo до нормальных или субнормальных уровней, когда его дают пациенту для профилактики или лечения болезненного состояния, которое обостряется или вызывается избыточным или нерегулируемым производством цитокинов.
Как здесь используется, цитокин, упоминаемый в фразе "ингибирование цитокина, для использования при лечении ВИЧ-инфицированного человека", представляет собой цитокин, который участвует в (а) инициировании/поддержании активации Т-клетки и/или экспрессии и/или репликации гена ВИЧ, активированной Т-клеткой, и/или (b) в проблеме, связанной с любым опосредованным цитокином заболеванием, таким как кахеция или дегенерация мышц.
Поскольку ФНО-β (также известный как лимфотоксин) имеет схожую структурную гомологию с ФНО-α (также известный как кахектин), и поскольку каждый из них вызывает подобные биологические отклики и связывается с одним и тем же клеточным рецептором, как ФНО-α, так и ФНО-β, ингибируются соединениями настоящего изобретения, и, следовательно, упоминаются здесь вместе как "ФHО", если специально не указано иного.
Новый член семейства MAP киназ, альтернативно называемый CSBP, р38 или RK, был недавно идентифицирован независимо несколькими лабораториями. Активацию этой новой протеинкиназы через двойное фосфорилирование наблюдали в различных клеточных системах при стимуляции с помощью широкого спектра стимулов, таких как физико-химический стресс и обработка липополисахаридом или про-воспалительными цитокинами, такими как интерлейкин-1 и фактор некроза опухоли. Ингибиторы биосинтеза цитокинов настоящего изобретения, соединения формулы (I), (II) и (А), как определено, являются сильнодействующими и селективными ингибиторами активности CSBP/p38/RK киназ. Эти ингибиторы могут использоваться при определении сигнальных путей, участвующих в воспалительных откликах. В частности, в первый раз определенный путь передачи сигнала может быть приписан действию липополисахарида на производство цитокинов в макрофагах.
Ингибиторы цитокинов впоследствии исследовались на противовоспалительную активность на ряде модельных животных. Модельные системы выбирали так, что они были относительно нечувствительными к ингибиторам циклооксигеназы в порядке получения активности только агентов, суппрессивных по отношению к цитокинам. Ингибиторы проявили значительную активность во многих таких исследованиях in vivo. Наиболее примечательными являются их активность на модели коллагениндуцированного артрита и ингибирование производства ФНО в модели эндотоксического шока. В последнем исследовании уменьшение уровня ФНО в плазме коррелирует с выживанием и защитой от смертности, связанной с эндотоксическим шоком. Также большую важность имеет эффективность соединений при ингибировании ресорбции костной ткани в системе культуры дифференцированных клеток трубчатой кости зародыша крысы. Griswold et al. (1988) Arthritis Rhum. 31: 1406-1412; Badger, et al. (1989) Circ. Shock 27, 51-61; Votta et al. (1994) in vitro. Bone 15, 533-538: Lee et al. (1993). В Ann. N.Y. Acad. Sci. 696, 149-170.
Для использования соединения формулы (I), (II) или (А) или их фармацевтически приемлемых солей при лечении, их обычно включают в фармацевтическую композицию в соответствии с обычной фармацевтической практикой. Настоящее изобретение, следовательно, также относится к фармацевтической композиции, содержащей эффективное нетоксическое количество соединения формулы (I) или (II) и фармацевтически приемлемые связующее вещество или разбавитель. Для целей настоящего изобретения соединения формулы (II) и (А) включены и представлены в разделе способов лечения с помощью использования термина формула (I).
Соединения формулы (I), их фармацевтически приемлемые соли и фармацевтические композиции, включающие их, могут удобно вводиться с помощью любого из способов, обычно используемых для введения лекарственных средств, например, перорально, местным образом, парентерально или путем ингаляции. Соединения формулы (I) могут вводиться в обычных формах дозировки, полученных путем объединения соединения формулы (I) со стандартными фармацевтическими связующими материалами в соответствии с обычными способами. Соединения формулы (I) могут также быть введены в обычных дозировках в сочетании с известным вторым терапевтически активным соединением. Эти способы могут включать перемешивание, гранулирование и прессование или растворение ингредиентов в согласии с требованиями желаемого препарата. Необходимо заметить, что форма и характер фармацевтически приемлемых связующего вещества или разбавителя диктуется количеством активного ингредиента, с которым они должны быть объединены, способом введения и другими хорошо известными параметрами. Связующее вещество (связующие вещества) должно быть "приемлемым" в смысле того, чтобы быть совместимым с другими ингредиентами препарата и не быть вредным для принимающего его.
Используемое фармацевтическое связующее вещество может быть, например, либо твердым, либо жидким. Примерами твердых связующих веществ являются лактоза, белая земля, сахароза, тальк, желатин, агар, пектин, аравийская камедь, стеарат магния, стеариновая кислота и тому подобное. Примерами жидких связующих веществ являются сироп, арахисовое масло, оливковое масло, вода и тому подобное. Подобным же образом, связующее вещество или разбавитель могут содержать материал для увеличения времени высвобождения, хорошо известный в данной области, такой как глицерилмоностеарат или глицерилдистеарат, сам по себе или с воском.
Может быть использовано широкое разнообразие фармацевтических форм. Так, если используют твердое связующее вещество, препарат может быть приготовлен в виде таблеток, помещен в твердую желатиновую капсулу в порошкообразной или гранулированной форме, или в форме драже или лепешек. Количество твердого связующего вещества может изменяться в широких пределах, но предпочтительно составляет от около 25 мг до около 1 г. Когда используют жидкое связующее вещество, препарат может быть в виде сиропа, эмульсии, мягкой желатиновой капсулы, стерильной жидкости для инъекций, такой как ампула или неводная жидкая суспензия.
Соединения формулы (I) могут вводиться местным образом, то есть путем несистемного введения. Этот способ включает нанесение соединения формулы (I) наружно на эпидермис или в ротовую полость и закапывания такого соединения в ухо, глаз и нос, так чтобы соединение не вводилось бы в значительных количествах в поток крови. В противоположность этому, системное введение относится к пероральному, внутривенному, внутрибрюшинному и внутримышечному введению.
Препараты, пригодные для местного введения, включают жидкие или полужидкие препараты, пригодные для проникновения через кожу к области воспаления, такие как жидкие мази, примочки, кремы, консистентные мази или пасты и капли, пригодные для введения в глаз, ухо или нос. Активный ингредиент может составлять при местном введении от 0,001% до 10% мас./мас., например от 1% до 2% от массы препарата. Он может, однако, содержать вплоть до 10% мас./мас., но предпочтительно содержит менее чем 5% мас./мас., более предпочтительно от 0,1% до 1% мас./мас. препарата.
Примочки по настоящему изобретению включают примочки, пригодные для нанесения на кожу или глаз. Примочка для глаз может содержать стерильный водный раствор, необязательно содержащий бактерицид, и может быть получена с помощью способов, подобных способам для приготовления капель. Примочки или жидкие мази для нанесения на кожу могут также включать агент для ускорения высыхания или охлаждения кожи, такой как спирт или ацетон, и/или увлажнитель, такой как глицерин, или масло, такое как касторовое масло или арахисовое масло.
Кремы, консистентные мази или пасты по настоящему изобретению являются полутвердыми препаратами активного ингредиента для наружного нанесения. Они могут быть получены путем смешивания активного ингредиента в тонко измельченной или порошкообразной форме, самого по себе или в растворе или суспензии в водной или неводной жидкости на жировой или нежировой основе с помощью соответствующих технических средств. Основа может включать углеводороды, такие как твердый, мягкий или жидкий парафин, глицерин, пчелиный воск или мыло на основе металла; клейкое вещество; масло природного происхождения, такое как миндальное, кукурузное, арахисовое, касторовое или оливковое масло, жир из шерсти или его производные или жирную кислоту, такую как стеариновую или олеиновую кислоту вместе со спиртом, таким как пропиленгликоль, или макрогель. Препарат может включать любой соответствующий поверхностно-активный агент, такой как анионное, катионное или неионное поверхностно-активное вещество, такое как сорбитановый эфир или его полиоксиэтиленовое производное. Также могут быть включены суспендирующие агенты, такие как натуральные смолы, производные целлюлозы или неорганические материалы, такие как различные формы окиси кремния и другие ингредиенты, такие как ланолин.
Капли по настоящему изобретению могут включать стерильные водные или масляные растворы или суспензии, и могут быть приготовлены путем растворения активного ингредиента в соответствующем водном растворе бактерицидного и/или фунгицидного агента и/или любого другого соответствующего консерванта и предпочтительно включать поверхностно-активный агент. Полученный раствор может затем быть очищен до прозрачности путем фильтрования, помещен в соответствующий сосуд, который затем герметизуют и стерилизуют в автоклаве или выдерживая при 98-100oС в течение получаса. Альтернативно, раствор может быть стерилизован путем фильтрования и помещен в сосуд с помощью любой асептической методики. Примерами бактерицидных и фунгицидных агентов, пригодных для включения в капли, являются нитрат- или ацетатфенил ртути (0,002%), бензалконийхлорид (0,01%) и хлоргексидинацетат (0,01%). Соответствующие растворители для приготовления масляного раствора включают глицерин, разбавленный спирт и пропиленгликоль.
Соединения формулы (I) могут вводиться парентерально, то есть путем внутривенного, внутримышечного, подкожного, интраназального, ректального, вагинального или внутрибрюшинного введения. Подкожная и внутримышечная формы парентерального введения обычно являются предпочтительными. Соответствующие дозированные формы для такого введения могут быть приготовлены с помощью обычных методик. Соединения формулы (I) могут также вводиться с помощью ингаляции, то есть путем интраназального или перорального ингаляционного введения. Соответствующие дозированные формы для такого введения, такие как аэрозольный препарат или ингалятор с фиксированной дозой, могут быть приготовлены с помощью обычных методик.
Для всех способов использования, описанных здесь для соединений формулы (I), дневной режим пероральной дозировки составляет предпочтительно от около 0,01 до около 30 мг/кг общей массы тела, предпочтительно от около 0,01 мг/кг до 10 мг/кг, более предпочтительно от около 0,01 мг до 5 мг. Дневной режим парентеральной дозировки составляет от около 0,001 до около 30 мг/кг общей массы тела, предпочтительно от около 0,01 до около 10 мг/кг, а более предпочтительно от 0,01 мг до 5 мг/кг. Дневной режим дозировки при местном применении составляет предпочтительно от 0,1 мг до 150 мг, наносимых от одного до четырех, предпочтительно от двух до трех раз в день. Дневной режим дозировки при ингаляции составляет предпочтительно от около 0,01 мг/кг до около 1 мг/кг в день. Специалист в данной области также заметит, что оптимальное количество и промежутки между индивидуальными дозировками соединения формулы (I) или ее фармацевтически приемлемой соли определяются природой и степенью состояния, подлежащего лечению, формой, способом и областью введения и конкретным пациентом, подвергающемуся лечению, и что такие оптимумы могут определяться с помощью обычных методик. Специалист в данной области также заметит, что оптимальный курс лечения, то есть число доз соединений формулы (I) или ее фармацевтически приемлемой соли, даваемых за день в течение определенного числа дней, может быть определено специалистом в данной области с использованием обычных исследований для установления курса лечения.
Далее изобретение будет описано со ссылками на последующие биологические примеры, которые являются чисто иллюстративными и не предназначены для ограничения рамок настоящего изобретения.
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
Эффекты ингибирования цитокинов соединений настоящего изобретения определяют с помощью следующих анализов in vitro:
Интерлейкин-1 (ИЛ-1): Моноциты из периферической крови человека выделяют и очищают либо из свежих препаратов крови от добровольных доноров, либо из светлого слоя хранимого запаса крови, в соответствии со способом Colotta et al. J. Immunol., 132, 936 (1984). Эти моноциты (1•106) размещают на 24-луночных планшетах при концентрации 1-2 миллион/мл на лунку. Клеткам дают возможность прилипнуть в течение 2 ч, после этого времени неприлипшие клетки удаляют с помощью осторожной промывки. Затем к клеткам добавляют исследуемые соединения в течение 1 ч перед добавлением липополисахарида (50 нг/мл), и культуры инкубируют при 37oС в течение дополнительных 24 ч. В конце этого периода супернатанты культур удаляют и очищают до прозрачности от клеток и всех остатков. Супернатанты культур затем непосредственно оценивают на биологическую активность ИЛ-1 либо с помощью способа Simon et al., J. Immunol. Methods, 84, 85 (1985) (на основе способности ИЛ-1 стимулировать линию клеток, производящих интерлейкин-2 (EL-4) к выделению ИЛ-2 во взаимодействии с ионофором А23187), либо методом Lee et al., J. Immuno Therapy, 6(1), 1-12 (1990) (анализ ELISA). Соединения формулы (I), как доказывают примеры 1-24, являются, как показано, ингибиторами ИЛ-1, продуцированного in vitro моноцитами человека.
Фактор некроза опухоли (ФНО): Моноциты из периферической крови человека выделяют и очищают либо из светлых слоев крови из хранимых запасов, либо из остатков от тромбоцитофереза, в соответствии со способом Colotta, R. et al., J. Immunol., 132(2), 936 (1984). Моноциты помещают при плотности 1x106 клеток/мл среды/лунка в 24-луночные планшеты. Клеткам дают возможность прилипнуть в течение 1 ч, после этого времени супернатант отсасывают и добавляют свежую среду (1 мл, RPMI-1640, Whitaker Biomedical Products, Whitaker, CA, USA), содержащую 1% фетальной сыворотки теленка плюс пенициллин и стрептомицин (10 единиц/мл). Клетки инкубируют в течение 45 минут в присутствии или в отсутствие исследуемого соединения в диапазоне доз 1 нМ - 10 мМ (соединения растворяют в диметилсульфоксиде/этаноле, так что конечная концентрация растворителя в культурной среде составляет 0,5% диметил-сульфоксида/0,5% этанола). Затем добавляют бактериальный липополисахарид (E.coli 055: В5[ЛПС] от Sigma Chemical Co.) (100 нг/мл в 10 мл солевого раствора с фосфатным буфером), и культуры инкубируют в течение 16-18 ч при 37oС в инкубаторе с 5% СO2. В конце периода инкубации супернатанты культур отделяют от клеток, центрифугируют при 3000 об/мин для удаления остатков клеток. Затем супернатант оценивают на активность ФНО с использованием либо радиоиммунного анализа, либо анализа ELISA, как описано в WO 92/10190 и Becker et al., J. Immunol. , 1991, 147, 4307. Соединение формулы (I), как доказывают примеры 1-24, являются, как показано, ингибиторами ФНО, произведенного in vitro моноцитами человека.
Ингибирующая активность ИЛ-1 и ФНО, по-видимому, не коррелирует со свойством соединений формулы (I) при опосредовании ингибирования метоболизма арахидоновой кислоты. Кроме того, способность ингибировать производство простагландина и/или синтез лейкотриена нестероидными противовоспалительными лекарственными средствами с сильной ингибиторной активностью по отношению к циклооксигеназе и/или липоксигеназе не означает, что соединение будет обязательно ингибировать продуцирование ФНО или ИЛ-1 в нетоксичных дозах.
Интерлейкин-8 (ИЛ-8): Первичные эндотелиальные клетки пуповины человека (ПЭКПЧ) (Cell Systems, Kirland, Wa) выдерживают в культурной среде с добавленными 15% фетальной сывороткой теленка и 1% контрасуппрессивного-HBGF, состоящего из аФХФ и гепарина. Затем клетки 20-кратно разбавляют перед размещением (250 мкл) на желатинированных 96-луночных планшетах. Перед использованием культурную среду заменяют свежей средой (200 мкл). Буфер или исследуемое соединение (25 мкл в концентрациях от 1 до 10 мкМ) затем добавляют к каждой лунке по 4 лунки за раз и планшеты инкубируют в течение 6 ч в увлажняемом инкубаторе при 37oС в атмосфере с 5% СO2. В конце инкубационного периода супернатант удаляют и оценивают концентрацию ИЛ-8 с использованием набора ELISA для ИЛ-8, полученного от R&D Systems (Minneapolis, MN). Все данные представлены как среднее значение (нг/мл) для множества образцов на основе стандартной кривой отклонений. Значения IC50, там где они видны, получают с помощью нелинейного регрессионного анализа.
Анализ специфического связывания белков цитокинами
Был разработан анализ радиоконкурентного связывания для обеспечения первичного скрининга с высокой воспроизводимостью для исследований зависимости структура - активность. Этот анализ обеспечивает ряд преимуществ по сравнению с обычными биологическими анализами, в которых используют свежевыделенные
моноциты человека в качестве источника цитокинов и анализы ELISA для получения количественных данных. Кроме того, что этот анализ является гораздо более легким, предложенный анализ связывания был широко проверен и показал высокую степень корреляции с результатами биологического анализа. Специфичный и воспроизводимый анализ ингибирования связывания цитокина разработали с использованием растворимой цистосолической фракции из клеток ТНР.1 и радиоактивно меченного соединения. Заявка на патент США USSN 08/123175 Lee et al., зарегистрированная в сентябре 1993; Lee et al., PCT 94/10529, зарегистрированная 16 сентября 1994 и Lee et al., Nature 300, p(72), 739-746 (Dec. 1994), описание которых включены сюда в качестве ссылки во всей их полноте, описывают указанный выше способ скрининга лекарственных средств для идентификации соединений, которые взаимодействуют и связываются с белком, специфически связывающим цитокин (здесь и далее БССЦ). Однако для целей настоящего изобретения связывающий белок может находиться в выделенном виде в растворе или в иммобилизованном виде, или может с помощью генной инженерии быть экспрессирован на поверхность рекомбинантных клеток-хозяев, таких как система проявления фага или слитые белки. Альтернативно целые клетки или цистосолические фракции, содержащие БССЦ, могут быть использованы в протоколе скрининга. Независимо от вида связывающего белка, множество соединений контактирует со связывающим белком при условиях, достаточных для образования комплекса соединение/связывающий белок, и определяются соединения, способные к формированию, увеличению или взаимодействию с указанными комплексами.
Показанные нулевые соединения формулы (I), примеры 3-27, все демонстрируют положительную ингибирующую активность, такую как IC50 связывания от около 0,18 до 5 мкМ, в этом анализе связывания, за исключением примера 12, где соединение не исследовалось.
Анализ с помощью простагландинэндопероксидсинтазы-2 (ПГЭС-2):
Последующий анализ описывает способ определения ингибиторных воздействий соединений формулы (I) на экспрессию белка человека ПГЭС-2 в стимулированных ЛПС моноцитах человека.
Метод: Моноциты периферической крови человека выделяют из светлых слоев путем центирифугирования в градиенте Ficoll и Percoll. Клетки высевают по 2•106/лунка в 24-луночные планшеты и позволяют прилипнуть в течение 1 ч в RPMI, с добавлением 1% сыворотки АВ человека, 20 мМ L-глютамина, пенициллина-стрептомицина и 10 мМ HEPES. Соединения добавляют при различных концентрациях и инкубируют при 37oС в течение 10 минут. ЛПС добавляют при 50 нг/лунка (чтобы вызвать экспрессию фермента) и инкубируют в течение ночи при 37oС. Супернатант удаляют и клетки промывают один раз холодным ЗФР. Клетки лизируют в 100 мкл холодного лизирующего буфера (50 мМ Трис/НСl рН 7,5, 150 мМ NaCl, 1% NP40, 0,5% деоксихолата натрия, 0,1% НДС, 300 мкг/мл ДНКазы, 0,1% TRITON X-10, 1 мМ PMSF, 1 мМ лейпептина, 1 мМ пепстатина). Лизат центрифугируют (10000 • g в течение 10 минут при 4oС) для удаления остатков клеток и растворимую фракцию подвергают анализу на НДС с помощью ПАГЭФ (12% гель). Белок, отделенный на геле, переносят на нитроцеллюлозную мембрану посредством электрофореза в течение 2 ч при 60 В. Мембрану предварительно обрабатывают в течение 1 ч ЗФР/0,1% Tween 20 с 5% обезжиренным сухим молоком. После трехкратной промывки в буфере ЗФР/Tween мембрану инкубируют с разбавленной в отношении 1:2000 моноспецифичной сыворотке антител к ПГЭС-2 или разбавленной в отношении 1:1000 сыворотке антител к ПГЭС-1 ЗФР/Tween с 1% BSA в течение 1 ч при непрерывном встряхивании. Мембрану трижды промывают в ЗФР/Tween, а затем инкубируют с разбавленным в отношении 1:3000 конъюгатом сыворотки антител осла к Ig кролика (Amersham) и пероксидазы хрена в ЗФР/Tween с 1% BSA в течение 1 ч при непрерывном встряхивании. Мембрану затем промывают трижды в ЗФР/Tween, и используют иммуннодетектирующую систему ECL (Amersham) для детектирования уровня экспрессии простагландинэндопероксидсинтаз-2.
РЕЗУЛЬТАТЫ: Следующие соединения были исследованы, и, как обнаружено, они являются активными (ингибировали индуцированную ЛПС экспрессию белка PGHS-2 в степени, заметной для ранжирования, подобной той, которая отмечается при ингибировании производства цитокинов в указанных анализах):
1-[3-(4-Морфолинил)пропил] 4-(4-фторфенил)-5-(4-пиридил)имидазол, показательное соединение формулы (I); 6-(4-Фторфенил)-2,3-дигидро-5-(4-пиридинил)[2,1-b]тиазол; Дексаметазон
Несколько соединений были исследованы и оказались неактивными (до 10 мкМ);
2-(4-Метилсульфинилфенил)-3-(4-пиридил)-6,7-дигидро-(5Н)-пирроло[1,2-а] имидазол ролипрам; фенидон и NDGA. Ни одно из исследуемых соединений, как обнаружено, не ингибирует уровни белков ПГЭС-1 или cPLA2 в подобных экспериментах.
Примеры синтеза
Далее изобретение описано со ссылками на последующие примеры, которые являются только иллюстративными и не предназначены для ограничения объема настоящего изобретения. Все температуры даны в градусах Цельсия, все растворители имеют наивысшую доступную степень чистоты, и все реакции проходят в безводных условиях в атмосфере аргона, если не указано иного.
В примерах все температуры даны в градусах Цельсия (oС). Масс-спектры получают с помощью масс-спектрометра VG Zab, с использованием бомбардировки быстрыми атомами, если не указано иного. Спектры 1Н-ЯМР (далее ЯМР) записывают при 250 МГц с использованием спектрометра Bruker AM 250 или Am 400. Мультиплетности указывают следующим образом: с=синглет, д=дублет, т=триплет, кв=квартет, м=мультиплет и ушир. указывает на уширенный сигнал. Насыщ. означает насыщенный раствор, экв. означает пропорцию молярного эквивалента реагента по отношению к главному участнику реакции. Флэш-хроматография проводится на Merck Silica gel 60 (230-400 меш.).
Пример 1
1-[3-(4-Морфолинил)пропил]-4-(4-фторфенил)-5-(4-пиридил)-имидазол
а) 4-Фторфенил-толилтиометилформамид
Раствор п-фторбензальдегида (13,1 миллилитра (далее мл), 122 миллимоля (далее ммоль)), тиокрезола (16,64 грамма (далее г)), формамида (15,0 мл, 445 ммоль) и толуола (300 мл) объединяют и нагревают до температуры кипения толуола с азеотропным удалением Н2О в течение 18 ч. Охлажденную реакционную смесь разбавляют EtOAc (500 мл) и промывают насыщенным водным раствором Nа2СО3 (3•100 мл), насыщенным водным раствором NaCl (100 мл), сушат (Nа2SO4) и концентрируют. Остаток растирают с петролейным эфиром, фильтруют и сушат в вакууме с получением 28,50 г указанного в заголовке соединения в виде белого твердого продукта (85%). Температура плавления (далее т.пл.) = 119-120oС.
b) 4-Фторфенил-толилтиометилизоцианид
Соединение примера 1(а) (25 г, 91 ммоль) в СН2Cl2 (300 мл) охлаждают до -30oС и при механическом перемешивании добавляют по каплям РОСl3 (11 мл, 110 ммоль) с последующим добавлением по каплям Et3N (45 мл, 320 ммоль), поддерживая температуру ниже -30oС. Перемешивают при -30oС в течение 30 мин и при 5oС в течение 2 ч, разбавляют CH2Cl2 (300 мл) и промывают 5% водным раствором Na2CO3 (3•100 мл), сушат (Na2SO4) и концентрируют до 500 мл. Этот раствор фильтруют через цилиндрический слой кварца 12 х 16 см в большой тигель (воронку) с фильтром из спеченного стекла с CH2Cl2 с получением 12,5 г (53%) очищенного изонитрила в виде светло-коричневого, смолообразного твердого продукта. ИК (CH2Cl2) 2130 см-1.
c) Пиридин-4-карбоксальдегид[4-морфолинилпроп-3-ил] имин
Пиридин-4-карбоксальдегид (2,14 г, 20 ммоль), 4-(3-аминопропил)морфолин (2,88 г, 20 ммоль), толуол (50 мл) и МgSO4 (2 г) объединяют и перемешивают в атмосфере аргона в течение 18 ч. МgSO4 отфильтровывают и фильтрат концентрируют, а остаток повторно концентрируют из CH2Cl2 с получением 4,52 г (97%) указанного в заголовке соединения в виде желтого масла, включающего менее чем 5% альдегида, основываясь на 1H-ЯМР. 1H-ЯМР (CD3Cl): δ 8,69 (д, J=4,5 Гц, 2Н), 8,28 (с, 1Н), 7,58 (д, J=4,5 Гц, 2Н), 3,84 (м, 6Н), 2,44 (м, 6Н), 1,91 (м, 2Н).
d) 1-[3-(4-Морфолинил]-4-(4-фторфенил)-5-(4-пиридил)имидазол
Соединение примера 1(b) (1,41 г, 5,5 ммоль) и соединение примера 1(с) (1,17 г, 5,0 ммоль), и CH2Cl2 (10 мл) охлаждают до 5oС. Добавляют 1,5,7-триазабицикло[4,4,0] дец-5-ен, далее упоминаемый как (ТБД) (0,71 г, 5,0 ммоль) и реакционную смесь выдерживают при 5oС в течение 16 ч, разбавляют EtOAc (80 мл) и промывают насыщенным водным раствором Na2CO3 (2•15 мл). EtOAc экстрагируют 1 н. HCl (3•15 мл) и кислотные фазы промывают EtOAc (2•25 мл), наслаивают EtOAc (25 мл) и делают щелочными путем добавления твердого К2СО3 до рН 8,0, а затем - 10% NaOH до рН 10. Фазы отделяют и водную экстрагируют дополнительным EtOAc (3•25 мл). Экстракты сушат (К2СО3), концентрируют и остаток кристаллизуют из ацетона/гексана с получением 0,94 г (51%) указанного в заголовке соединения. Т. пл. = 149-150oС.
Пример 2
5-(2-Аминопиримидин-4-ил)-4-(4-фторфенил)-1-[3-(4-морфолинил)пропил]имидазол
a) 2-Аминопиримидин-4-карбоксальдегид диметилацеталь
Диметилформамид диметилацеталь (55 мл, 0,41 моль) и пировиноградный альдегид диметилацеталь (50 мл, 0,41 моль) объединяют и нагревают до 100oС в течение 18 ч. Метанол удаляют в вакууме с получением масла. Раствор NaOH (18 г, 0,45 моль) в Н2O (50 мл) добавляют к гуанидин HCl (43 г, 0,45 моль) в Н2О (100 мл) и полученный раствор добавляют к описанному выше маслу. Полученную смесь перемешивают при 23oС в течение 48 ч. Фильтрование дает 25 г (50%) указанного в заголовке соединения.
b) 2-Аминопиримидин-4-карбоксальдегид
Соединение предыдущей стадии (1,69 г, 10 ммоль) и 3 н. НС1 (7,3 мл, 22 ммоль) объединяют и нагревают до 48oС в течение 14 ч, охлаждают, наслаивают EtOAc (50 мл) и нейтрализуют путем добавления NaHCO3 (2,1 г, 25 ммоль) маленькими порциями. Водную фазу экстрагируют EtOAc (5•50 мл), и экстракты сушат (Na2SO4) и концентрируют с получением 0,793 г (64%) указанного в заголовке соединения.
c) 2-Аминопиримидин-4-карбоксальдегид [3-(4-морфолинил)-пропил]имин
Соединение предыдущей стадии и 4-(3-аминопропил)морфолин взаимодействуют по способу примера 1(с), выше, с получением указанного в заголовке соединения в виде желтого масла.
d) 5-(2-Аминопиримидин-4-ил)-4-(4-фторфенил)-1-[3-(4-морфолинил)пропил] имидазол
По способу примера 1(d), выше, исключая использование соединения предыдущей стадии в качестве имина, получают указанное в заголовке соединение в виде белого твердого продукта. 1H-ЯМР (СD3Сl): δ 8,15 (д, J=5,4 Гц, 1Н), 7,62 (с, 1Н), 7,46 (дд, 2Н), 7,00 (т, J=8,6 Гц, 2Н), 6,50 (д, J=5,4 Гц, 1Н), 5,09 (ушир. с, 2Н), 4,34 (т, J=7,0 Гц, 2Н), 3,69 (м, 4Н), 2,35 (ушир. с, 4Н), 2,24 (т, J=4,6 Гц, 2Н), 1,84 (м, 2Н).
Пример 3
5-[4-(2-Метиламино)пиримидинил] -4-(4-фторфенил)-1-(4-морфолино-3-пропил)имидазол
а) 2-Метиламинопиримидин-4-диметилацеталь
Натрий (3,27 г, 142 ммоль) растворяют в абсолютном этиловом спирте (425 мл). Добавляют 1-метилгуанидингидрохлорид (15,5 г, 142 ммоль) и полученную суспензию перемешивают в течение около 10 мин. Добавляют 1,1-диметокси-2-оксо-4-диметиламино-3-бутен (142 ммоль), растворенный в этаноле (20 мл), и смесь перемешивают при нагревании с обратным холодильником в течение 24 ч. Смесь охлаждают и фильтруют. Этанол выпаривают и полученный остаток растирают с горячим EtOAc. Промывочные жидкости EtOAc объединяют и растворитель выпаривают с получением указанного в заголовке соединения (23,5 г, 91%) в виде желтого масла. 1H-ЯМР (СDСl3): δ 8,35 (д, J=4,5 Гц, 1Н), 6,74 (д, 1Н), 5,10 (с, 1Н), 3,40 (с, 1Н), 3,00 (д, 3Н).
b) 2-Метиламинопиримидин-4-карбоксальдегид
Следуя способу примера 2 (b), выше, исключая использование соединения предыдущей стадии (11,75 г, 64,6 ммоль), получают указанное в заголовке соединение в виде желтой пены (7,3 г, выход 82,7%). 1H-ЯМР (CDCl3): δ 9,85 (д, J=4,5 Гц, 1Н), 8,52 (с, 1Н), 7,03 (д, 1Н), 5,52 (с, 1Н), 3,10 (д, 1Н).
c) 5-[4-(2-Метиламино)пиримидинил]-4-(4-фторфенил)-1-(4-N-морфолино-1-пропил)имидазол
Соединение предыдущей стадии (5,0 г, 36,5 ммоль) и 4-(3-аминопропил)морфолин (5,3 мл, 36,5 ммоль) перемешивают в СН2Сl2 (180 мл). По истечении около 16 ч смесь охлаждают до 0oС. Добавляют соединение примера 1(b) (11,3 г, 43,8 ммоль) и ТБД (8,4 г, 61,32 ммоль). Смесь оставляют стоять в течение около 3 дней при около 5oС. Продукт фильтруют и перетирают в порошок с горячим EtOH с получением указанного в заголовке соединения (6,06 г, выход 41,9%) в виде бледно-желтого твердого продукта. Т. пл. = 203-305oС. 1H-ЯМР (CDCl3/MeOD): δ 8,01 (д, J=4,5 Гц, 1Н), 7,60 (с, 1H), 7,37 (кв, 2H), 6,95 (т, 2Н), 6,29 (д, 1Н), 4,32 (с, 1Н), 3,63 (т, 4Н), 3,57 (м, 2Н), 2,95 (с, 3Н), 2,33 (м, 4Н), 2,23 (т, 2Н), 1,82 (т, 2Н).
Пример 4
5-[4-(2-Метиламино)пиримидинил] -4-(4-фторфенил)-1-(1-метил-пиперидин-4-ил)имидазол
а) Соединения примера 75(а) (4,25 г, 37,2 ммоль), полученное как описано в WO 95/02591, Adams et al. , описание которых включено сюда в качестве ссылки во всей полноте, и примера 3(b) (5,1 г, 37,2 ммоль), полученного так же, как и выше, объединяют в CH2Cl2 (150 мл). Эту смесь перемешивают в течение около 16 ч при комнатной температуре и охлаждают до 0oС. Добавляют соединение примера 1(b), выше, и ТБД, и полученную смесь перемешивают при комнатной температуре в течение около 3 дней. Смесь очищают непосредственно на колонке с силикагелем и очищают путем флэш-хроматографии, элюируя 0%-5% MeOH/CH2Cl2. Полученное масло промывают в ацетоне/гексане и осадок фильтруют, промывают ацетоном с получением указанного в заголовке соединения (1,36 г, выход 10%) в виде бледно-желтого твердого продукта. Т. пл.=209-210oС. 1H-ЯМР (СDСl3): δ 8,16 (д, J=4,5 Гц, 1Н), 7,77 (с, 1Н), 7,45 (кв, 2Н), 6,98 (т, 2Н), 6,41 (д, 1Н), 5,20 (д, 1Н), 4,66 (с, 1Н), 3,05 (д, 3Н), 2,98 (д, 2Н), 2,32 (с, 3Н), 2,14 (м, 2Н), 2,01 (м, 4Н).
Отмеченные выше условия используют соединение формулы (IIа) - схема I, где р= 0. В способе, аналогичном способу части (а), но используя соединение формулы (IIа) - схема I, где р=2, и изолированный имин, используют по таблице.
При способах, аналогичных описанным выше, может быть получено следующее соединение.
Пример 5
5-[4-(2-Метиламино)пиримидинил]-4-(4-фторфенил)-1-(4-пиперидин)имидазол
а) 5-[4-(2-Метиламино)пиримидинил] -4-(4-фторфенил)-1-(4-N-Вос-пиперидин)имидазол
Раствор 2-метиламино-4-пиримидинкарбоксальдегида (2,47 г, 17,99 ммоль) и трет-бутил 4-амино-1-пиперидинкарбоксилата (как описано в примере 46(а) в WO 95/02591, Adams et al.) (3,96 г, 19,79 ммоль) в 36 мл ДМФ перемешивают при около 25oС в течение около 5-6 ч. После охлаждения до около 0oС добавляют изонитрил стадии (b) примера 10(b), ниже (6,24 г, 21,60 ммоль) и порошкообразный К2СО3 (2,98 г, 21,60 ммоль). Раствор постепенно нагревают до около 25oС в течение около 3 ч. По истечении около 16 ч добавляют 100 мл Н2О и полученную смесь фильтруют, промывают 20 мл Н2О и 50 мл трет-бутилметилового сложного эфира. После сушки получают 6,85 г (84%) указанного в заголовке соединения в виде белого порошка. 1H-ЯМР (300 МГц, CDCl3): δ 8,15 (1Н, д, J= 5,0 Гц), 7,72 (1Н, с), 7,45 (2Н, м), 6,99 (2Н, т, J=8,7 Гц), 6,40 (1Н, д, J= 5,1 Гц), 5,20 (1Н, м), 4,80 (1Н, м), 4,28 (2Н, м), 3,03 (3Н, д, J=5,0 Гц), 2,76 (2Н, т, J=12,2 Гц), 2,17 (2Н, д, J=12,2 Гц), 1,86 (2Н, дкв, J=4,3, 12,4 Гц), 1,48 (9Н, с).
b) 5-[4-(2-Метиламино)пиримидинил] -4-(4-фторфенил)-1-(4-пиперидин)имидазол
К перемешиваемой суспензии N-BOC производного стадии (а) выше (31 г, 68 ммоль) в этилацетате (310 мл, 10 объемов) добавляют 3 н. HCl (160 мл, 476 ммоль, 7 экв. ) при 25oС. Полученный мутно-желтый раствор перемешивают при 25oС в течение 2 ч. рН реакционной смеси доводят до 12-13 путем медленного добавления 50% водного раствора NaOH. Фазы отделяют, и водную фазу экстрагируют дважды метиленхлоридом (200 мл каждый раз). Объединенные органические экстракты промывают водой, сушат над MgSO4 и выпаривают в роторном испарителе досуха. Полученный светло-желтый остаток суспендируют в горячем этилацетате/метиленхлориде (200 мл, смесь 9:1) и позволяют охладиться до 25oС. Продукт собирают путем фильтрования отсасыванием и промывают этилацетатом (25 мл). Белый твердый продукт сушат до постоянного веса при 50oС/<1 мм с получением 19 г (54 ммоль) описанного в заголовке продукта, получая выход 79%. 1H-ЯМР (300 МГц, CDCl3): δ 8,15 (1H, д, J=5,0 Гц), 7,77 (1H, с), 7,45 (2Н, м), 6,99 (2Н, т, J=8,7 Гц), 6,40 (1H, д, J=5,1 Гц), 5,23 (1H, м), 4,76 (1H, м), 3,22 (2Н, д, J=12,4 Гц), 3,05 (3Н, д, J=5,1 Гц), 2,67 (2Н, дт, J=2,0, 12,3 Гц), 2,16 (2Н, д, J=11,8 Гц), 1,86 (2Н, дкв, J=3,9, 12,2 Гц).
В альтернативном синтезе указанное в заголовке соединение может быть получено следующим образом:
5-[4-(2-Метиламино)пиримидинил] -4-(4-фторфенил)-1-(пиперидин 4- ил)имидазол
i) 5-[4-(2-Метиламино)пиримидинил]-4-(4-фторфенил)-1-(4-N- карбоксиэтилпиперидин)имидазол
Раствор 2-метиламинопиримидина-4-карбоксальдегид диметилацеталя (1,88 г, 10,3 ммоль) в 10 мл 3 н. HCl нагревают при 47oС в течение 14 ч до тех пор, пока по данным ВЭЖХ не исчезнет исходный материал. Реакционную смесь охлаждают до 25o и добавляют последовательно этил 4-амино-1-пиперидин-карбоксилат (1,95 г, 11,3 ммоль), этилацетат (30 мл) и NаНСО3 (3,45 г, 41,1 ммоль). Через 7 ч добавляют ДМФ (5 мл), изонитрил стадии (b), примера 10(b) (2,97 г, 10,3 ммоль) и порошкообразный К2СО3 (1,56 г, 11,3 ммоль). Реакционную смесь перемешивают в течение 14 ч, разбавляют 50 мл EtOAc, промывают водой (2•50 мл), насыщенным раствором К2СО3 (50 мл) и солевым раствором (30 мл), и органическую фазу концентрируют. Продукт перекристаллизовывают из этилацетата с получением указанного в заголовке соединения (1,96 г 45%). 1H-ЯМР (300 МГц, СDСl3): δ 8,16 (1Н, д, J=5,0 Гц), 7,72 (1H, с), 7,45 (2Н, м), 7,00 (2Н, т, J=8,7 Гц), 6,41 (1H, д, J=5,0 Гц), 5,19 (1H, м), 4,84 (1H, м), 4,35 (2Н, м), 4,16 (2Н, кв, J=7,1 Гц), 3,04 (3Н, д, J=5,0 Гц), 2,82 (2Н, м), 2,20 (2Н, м), 1,88 (2Н, дкв, J=4,4, 12,5 Гц), 1,28 (3Н, т, J=7,1 Гц).
ii) 5-[4-(2-Метиламино)пиримидинил] -4-(4-фторфенил)-1-(пиперидин-4-ил)имидазол
К перемешиваемому раствору соединения, полученного выше (2,1 г, 4,64 ммоль), в EtOH (40 мл) и воде (20 мл) добавляют NaOH (1,48 г, 37,2 ммоль), и раствор нагревают с обратным холодильником в течение 36 ч. Раствор охлаждают до комнатной температуры и добавляют толуол (20 мл). Раствор концентрируют в вакууме, добавляют толуол (20 мл) и повторно концентрируют в вакууме. Добавляют воду (30 мл) и толуол (30 мл), и белый твердый продукт, который образовался, фильтруют и сушат с получением указанного в заголовке соединения (1,26 г, 77%). 1H-ЯМР (300 МГц, CDCl3): δ 8,15 (1H, д, J=5,0 Гц), 7,77 (1H, с), 7,45 (2Н, м), 6,99 (2Н, т, J=8,7 Гц), 6,40 (1H, д, J=5,1 Гц), 5,23 (1H, м), 4,76 (1H, м), 3,22 (2Н, д, J=12,4 Гц), 3,05 (3Н, д, J=5,1 Гц), 2,67 (2Н, дт, J= 2,0, 12,3 Гц), 2,16 (2Н, д, J=11,8 Гц), 1,86 (2Н, дкв, J=3,9, 12,2 Гц).
Еще в одном альтернативном воплощении настоящего изобретения защитная группа заменяется с трет-ВОС на карбоксиэтил и синтезируется с использованием условий, подобных условиям частей (а) и (b) выше:
5-[4-(2-N-Метиламино)пиримидинил] -4-(4-фторфенил)-1-(4-N-карбоксиэтилпиперидин)имидазол
Раствор 2-метиламино-4-пиримидинкарбоксальдегида (2,47 г, 17,99 ммоль) и этил 4-амино-1-пиперидинкарбоксилата (3,25 г, 18,9 ммоль) в 36 мл ДМФ перемешивают при комнатной температуре в течение около 3,3 ч. Добавляют изонитрил стадии (b) примера 10(b) (6,0 г, 20,7 ммоль) и порошкообразный К2СО3 (3,11 г, 22,5 ммоль). Через приблизительно 16 ч добавляют 250 мл Н2O, 30 мл ТБМЭ и 30 мл EtOAc, и полученную смесь фильтруют, промывают 200 мл Н2О и ТБМЭ (2•100 мл). Изолированный материал повторно кристаллизуют из EtOAc/Et2O с получением 5,3 г (65%) указанного в заголовке соединения в виде белого порошка. Т. пл. 205-206oС; 1H-ЯМР (300 МГц, CDCl3): δ 8,16 (1Н, д, J=5,0 Гц), 7,72 (1H, с), 7,45 (2Н, м), 7,00 (2Н, т, J=8,7 Гц), 6,41 (1H, д, J=5,0 Гц), 5,19 (1H, м), 4,84 (1H, м), 4,35 (2Н, м), 4,16 (2Н, д, J=7,1 Гц), 3,04 (3Н, д, J= 5,0 Гц), 2,82 (2Н, м), 2,20 (2Н, м), 1,88 (2Н, дкв, J=4,4, 12,5 Гц), 1,28 (3Н, т, J=7,1 Гц).
Еще в одном альтернативном воплощении настоящего изобретения использование кислотного гидролиза этилкарбомата дает указанное в заголовке соединение:
5-[4-(2-Метиламино)пиримидинил]-4-(4-фторфенил)-1-(4-пиперидин)-имидазол
Концентрированную соляную кислоту (12 мл) добавляют к 4[[1-(1-этоксикарбонил)-4-пиперидинил)] -4-(4-фторфенил)-1Н-имидазол-5-ил] -N-метил-2-пиримидинамину (25 г, 0,059 моль) и нагревают с обратным холодильником в течение 18 ч. Реакционную смесь охлаждают до 0oС и нейтрализуют 50% водным раствором гидроксида натрия. Полученный осадок собирают путем фильтрования, промывают водой, сушат на воздухе и сушат в вакууме при 40oС с получением указанного в заголовке соединения в виде белого твердого продукта с выходом 62%.
Пример 6
5-[(2-Этиламино)пиримидин-4-ил] -4-(4-фторфенил)-1-(1-метилпиперидин-4-ил)имидазол
a) 2-Метилтиопиримидин-4-карбоксальдегид диметилацеталь
Пировиноградный альдегид диметилацеталь (19,2 мл, 159,1 ммоль) и N,N-диметилформамид диметилацеталь (21,12 мл, 159,1 ммоль) объединяют в 500 мл колбе и нагревают до 100oС. Через 4,5 ч колбу убирают с нагревателя, добавляют тиомочевину (11,0 г, 144,5 ммоль), NaOMe (25 мас. % раствор в МеОН, 39,7 мл, 173 ммоль) и 30 мл МеОН, и нагревание продолжают при 65oС. Через 18 ч раствор охлаждают до 25oС и добавляют МеI (10,8 мл, 173 ммоль) в течение 5 мин (экзотерм.) Через 3 ч раствор разбавляют 250 мл Н2О и экстрагируют EtOAc (3 х 100 мл). Органические фазы объединяют, сушат над Na2SO4 и концентрируют с получением указанного в заголовке соединения (26,8 г, 93%) в виде коричневого масла.
b) 2-Метилтиопиримидин-4-карбоксальдегид
2-Метилтиопиримидин-4-карбоксальдегид диметилацеталь (30,0 г, 150 ммоль) растворяют в 300 мл ледяном АсОН и 3 мл конц. H2SO4, и нагревают при около 70-80oС. Через 10 ч раствор охлаждают до 25oС, и АсОН удаляют в вакууме, оставляя остаток в виде коричневого масла. Этот остаток разбавляют 200 мл СН2Сl2 и промывают насыщенным NаНСО3 (3•50 мл), Н2О (50 мл) и солевым раствором (50 мл). Органическую фазу сушат над МgSO4 и концентрируют с получением 22,1 г (96%) указанного в заголовке соединения в виде коричневого масла.
c) 2-Метилтиопиримидин-4-карбоксальдегид (1-метилпиперидин-4-ил)имин
2-Метилтиопиримидин-4-карбоксальдегид (5,6 г, 36 ммоль) и 4-амино-1-метилпиперидин дигидрохлорид (6,73 г, 36 ммоль) растворяют в 200 мл СН2Сl2 и добавляют NаНСО3 (10,6 г, 126 ммоль). Через 20 ч раствор фильтруют и концентрируют с получением 8,9 г (98%) указанного в заголовке соединения в виде коричневого масла.
d) 4-(Фторфенил)-1-(1-метилпиперидин-4-ил)-5-(2-метилтио-4-пиримидинил)имидазол
Трет-BuNH2 (3,90 мл, 37,08 ммоль) быстро добавляют к раствору 2-метилтиопиримидин-4-карбоксальдегид (1-метилпиперидин-4-ил)имина (3,71 г, 14,83 ммоль) и 4-фторфенилтозилметилизоцианида (5,15 г, 17,8 ммоль), растворенным в 50 мл ДМЭ, при 25oС. Через 14 ч раствор разбавляют 50 мл EtOAc и промывают 50 мл насыщ. NаНСО3 25 мл солевого раствора. Органические фазы сушат над Na2SO4 и концентрируют. Кристаллизация из сырого остатка с использованием EtOAc/гексана дает 2,85 г (50%) продукта в виде светло-коричневых кристаллов. 1H-ЯМР (300 МГц, CDCl3): δ 8,31 (1H, д, J=5,1 Гц), 7,78 (1Н, с), 7,40 (2Н, м), 6,99 (2Н, т, J=8,7 Гц), 6,76 (1Н, д, J=5,2 Гц), 4,67 (1H, м), 2,97 (2Н, м), 2,58 (3Н, с), 2,31 (3Н, с), 2,06 (6Н, м).
Альтернативно, для синтеза в части (d) выше используют дополнительные условия реакции с другим аминовыми основаниями и растворителями:
а) трет-бутиламин и ТГФ; b) диизопропиламин и ТГФ; с) пирролидин и ТГФ; d) карбонат калия и этанол.
e) 4-(Фторфенил)-1-(1-метилпиперидин-4-ил)-5-(2-метил-суль финил-4-пиримидинил)имидазол
Персульфат калия (3,2 г, 7,0 ммоль) в воде (75 мл) добавляют к раствору 4-(фторфенил)-1-(1-метилпиперидин -4-ил)-5-(2-метилтио-4-пиримидинил)имидазола (2,7 г, 7,0 ммоль) в ледяном АсОН (150 мл). После перемешивания при температуре окружающей среды в течение 72 ч реакционную смесь нейтрализуют путем добавления по частям концентрированным водным раствором NH4OH и экстрагируют CH2Cl2. Органический экстракт промывают солевым раствором, сушат (MgSO4) и концентрируют. Остаток растирают с этиловым эфиром с получением указанного в заголовке соединения в виде не совсем белого твердого продукта; выход 2,3 г (83%).
f) 5-[(2-Этиламино)пиримидин-4-ил] -4-(4-фторфенил)-1- (1-метилпиперидин-4-ил)имидазол
4-(Фторфенил)-1-(1-метилпиперидин-4-ил)-5-(2-метилсульфинил-4-пиримидинил) имидазол (0,25 г, 0,65 ммоль) и 70% водный раствор этиламина (2,5 мл) нагревают до 120oС в герметичном реакционном сосуде в течение 18 ч. После охлаждения до температуры окружающей среды летучие продукты выпаривают и остаток растирают с этиловым эфиром с получением указанного в заголовке соединения в виде белого твердого продукта; выход 0,13 г (53%): ES(+) MS m/e= 381 (MH+).
Пример 7
4-(4-Фторфенил)-5-[2-(изопропил)аминопиримидинил-4-ил] -1-(1-метилпиперидин-4-ил)имидазол
По способу примера 6, стадии (F), исключая замещение изопропиламина, получают указанного в заголовке соединения в виде желтовато-киричневого твердого продукта с выходом 20%: ES(+) MS m/e=395 (MH+).
Пример 8
5-(2-Амино-4-пиримидинил)-4-(4-фторфенил)-1-(1-метил-4- пиперидинил)имидазол
а) 2-Ацетамидопиримидин-4-карбоксальдегид монометил моноацетоксиацеталь
Смесь 2-аминопиримидин-4-карбоксальдегид диметилацеталя (9,0 г, 53 ммоль) и уксусного ангидрида (25 мл) нагревают до 60oС в течение 18 ч. Добавляют концентрированную H2SO4 (10 капель), и раствор нагревают до 100oС в течение 10 ч. После охлаждения до температуры окружающей среды летучие продукты выпаривают и остаток фильтруют с помощью вакуума через пад из силикагеля, элюируя 4% МеОН в CH2Cl2. Выпаривание фильтрата с последующим перетиранием остатка со сложным эфиром дает указанное в заголовке соединение в виде белого твердого продукта; выход 8,6 г (68%).
b) 2-Ацетамидопиримидин-4-карбоксальдегид
Метоксид натрия (0,056 г, 1,0 ммоль) добавляют к раствору 2-ацетамидо-пиримидин-4-карбоксальдегидмонометилмоноацетоксиацеталя (5,0 г, 21 ммоль) в МеОН (25 мл) при температуре окружающей среды. После перемешивания при этой температуре в течение 3 ч реакционную смесь нейтрализуют добавлением 3 н. HCl. Полученный раствор концентрируют, и остаток нагревают с CH2Cl2. Оставшийся твердый продукт удаляют путем фильтрования, и растворитель выпаривают с получением указанного в заголовке соединения в виде желтого твердого продукта; выход 3,2 г (92%).
c) 2-Ацетамидопиримидин-4-карбоксальдегид (1-метилпиперидин-4-ил)имин
Следуя способу примера 75(b), как описано в WO 95/02591, Adams et al., исключая замещение 2-ацетамидо-пиримидин-4-карбоксальдегида, получают указанное в заголовке соединение в виде не совсем белого твердого продукта с выходом 75%.
d) 5-(2-Ацетамидо-4-пиримидинил)-4-(4-фторфенил)-1-(1-метил-4-пиперидинил)имидазол
Следуя способу примера 6(d), исключая замещение 2-ацетамидопиримидин-4-карбоксальдегид (1-метилпиперидин-4-ил)имина, получают указанное в заголовке соединение в виде желтого твердого продукта с выходом 51%.
e) 5-(2-Амино-4-пиримидинил)-4-4(4-фторфенил)-1-(1-метил-4-пиперидинил)имидазол
Раствор 5-(2-ацетамидо-4-пиримиди-нил)-4-(4-фторфенил)-1-(1-метил-4-пиперидинил)имидазола (4,0 г, 0,010 моль) в 40 мл 3 н. HCl нагревают до 75oС в течение 18 ч. После охлаждения до температуры окружающей среды реакционную смесь нейтрализуют твердым гидрокарбонатом натрия. Полученный осадок выделяют путем фильтрования, промывают водой и сушат на воздухе с получением указанного в заголовке соединения в виде белого твердого продукта с количественным выходом.
Пример 9
5-(2-Ацетамидо-4-пиримидинил)-4-(4-фторфенил)-1-(4-морфолино-3-пропил)имидазол
Раствор 5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-(4-морфолино-3-пропил)имидазола (0,50 г, 1,3 ммоль) в уксусном ангидриде (10 мл) нагревают с обратным холодильником в течение 18 ч. После охлаждения до температуры окружающей среды избыток уксусного ангидрида выпаривают и остаток распределяют между насыщенным водным раствором NаНСО3 и этилацетатом. Слои разделяют и органическую фазу концентрируют. Остаток растворяют в МеОН (10 мл) и добавляют 2,5 н. NaOH (1 мл). После перемешивания при температуре окружающей среды в течение 2 ч раствор частично выпаривают и полученный осадок собирают путем фильтрования, промывают водой и сушат на воздухе с получением указанного в заголовке соединения в виде белого твердого продукта; выход 0,28 г (51%): ES(+) MS m/e=425 (MH+).
Пример 10
а) α-(п-Толуолсульфонил)-4-фторбензилформамид
К перемешиваемому раствору 4-фторбензальдегида (124 г, 979 ммоль) в ацетонитриле (620 мл, 5 объемов) и толуола (620 мл, 5 объемов) добавляют формамид (110 г, 2,45 ммоль, 2,5 экв.), затем хлортриметилсилан (119 г, 1,07 моль, 1,1 экв. ). Реакционную смесь нагревают до 50oС в атмосфере азота в течение 5 ч. К полученной белой суспензии добавляют п-толуолсульфиновую кислоту (230 г, 1,47 моль, 1,5 экв.), и реакционную смесь нагревают до 50oС в течение еще 5 ч, затем охлаждают до температуры окружающей среды. Добавляют метанол (250 мл) и трет-бутилметиловый эфир (620 мл). Через 15 минут реакционную смесь выливают в воду (3 л), предварительно охлажденную до 0oС. После перемешивания в течение 30 минут при 0oС продукт собирают путем фильтрования отсасыванием и промывают с трет-бутилметиловым эфиром (250 мл). Продукт, белое, кристаллическое, твердое вещество, сушат до постоянного веса при 40oС/<1 мм рт.ст. с получением 270 г (879 ммоль) описанного продукта (выход 90%). 1H-ЯМР (300 МГц, СD3СN): δ 7,99 (1Н, с), 7,92 (1H, м), 7,71 (2Н, д, J= 8,3 Гц), 7,49 (2Н, дд, J=5,3, 8,8 Гц), 7,39 (2Н, д, J=8,1 Гц), 7,16 (2Н, т, J=8,8 Гц), (1Н, м), 6,31 (1H, д, J=10,6 Гц), 2,42 (3Н, с).
Условия, альтернативные используемым выше в части (а), используют:
а) толуол при около 50oС; b) ацетонитрил при около 50oС; и с) используют те же условия, что и выше, но при температурах 30, 40, 50, 60 и 70oС.
b) α-(п-Толуолсульфонил)-4-фторбензилизонитрил
Перемешиваемую суспензию α-(п-толуолсульфонил)-4-фтор-бензилформамида, полученную выше в стадии (а) (100 г, 325 ммоль) в ТГФ (650 мл, 6,5 объемов) охлаждают до 0oС и добавляют РОCl3 (46 мл, 487 ммоль, 1,5 экв.). Наблюдают скачок температуры в 1oС, обусловленный экзотермичностыо реакции. Через 15 минут при 0oС белую суспензию охлаждают до -5oС. Триэтиламин (166 г, 1,62 моль, 5 экв. ) добавляют по каплям к суспензии в течение 45 минут с такой скоростью, чтобы поддерживать температуру реакции ниже 0oС, но выше -5oС. Необходимо соблюдать осторожность при начале добавления, так как реакция имеет тенденцию к экзотермическому скачку температуры. После окончания добавления желтую суспензию перемешивают в течение 30 минут при 0oС. Реакционная суспензия имеет тенденцию к потемнению в течение периода перемешивания. Реакционную суспензию выливают в смесь насыщенного водного раствора бикарбоната натрия (1 л) и этилацетата (1 л), обе предварительно охлаждают до 0oС. Органическую фазу позже промывают водой, затем солевым раствором. Органическую фазу концентрируют в вакууме с помощью выпаривания на роторном испарителе до тех пор, пока не останется 10% от начального объема. Добавляют 1-пропанол (200 мл) и снова концентрируют в вакууме при 35oС до тех пор, пока не останется 10% от начального объема. Этот процесс повторяют со свежим 1-пропанолом (200 мл). По окончании наблюдают мелкодисперсный желтый осадок. Осадок охлаждают до 0oС, и продукт собирают путем фильтрования отсасыванием и промывают 1-пропанолом (50 мл). Не совсем белый твердый продукт сушат до постоянного веса при 40oС/<1 мм рт. ст. с получением 65,7 г (227 ммоль) описанного продукта, получая выход 70%. 1H-ЯМР (300 МГц, CDCl3): δ 7,62 (2Н, д, J=6,7 Гц), 7,46 (4Н, м), 7,08 (2Н, т, J=8,6 Гц), 5,62 (1Н, с), 2,46 (3Н, с).
Используют альтернативные условия по отношению к используемым выше в части (b), такие как а) различные растворители: ДМЭ, ДМЭ/ацетонитрил (10:1) с использованием таких же реакционных условий; b) используют реакционные условия, указанные выше, но в диапазоне температур от -30, -15, -10 и 0oС; с) используя реакционную температуру 0oС и -10oС; d) ряд дегидрирующих агентов, включая трифторуксусный ангидрид, тионилхлорид и оксалилхлорид.
Пример 11
5-(2-Ацетамидо-4-пиримидинил)-4-(4-фторфенил)-1-(1-метилпиперидин-4-ил)имидазол
К раствору 2-ацетиламидопиримидинил-4-карбоксальдегида (0,84 г, 5,08 ммоль) и 1-метилпиперидин-4 -иламинодигидрохлоридной соли (1,04 г, 5,59 ммоль) в 21 мл ДМФ добавляют порошкообразный K2СО3 (1,54 г, 11,2 ммоль). Через примерно 6 ч добавляют α-(п-толуолсульфонил)-4-фторбензилизонитрил, продукт стадии (b) примера 10, выше, (1,76 г, 6,10 ммоль) и порошкообразный К2СО3 (0,84 г, 6,10 ммоль) и стенки колбы промывают 5 мл ДМФ. Через 16 ч добавляют 300 мл Н2О к реакционной смеси, и раствор экстрагируют EtOAc (3•100 мл). Объединенные органические фазы промывают H2O (3•50 мл), сушат над Na2SO4 и концентрируют. Чистое соединение, указанное в заголовке (0,75 г, 38%), перекристаллизовывают из EtOAc в виде светло-желтых кристаллов. 1H-ЯМР (300 МГц, CDCl3): δ 8,71 (1Н, с), 8,39 (1H, д, J=5,2 Гц), 7,81 (1Н, с), 7,39 (2Н, м), 7,13 (2Н, т, J=8,7 Гц), 6,81 (1H, д, J=5,2 Гц), 4,88 (1H, м), 2,94 (2Н, д, J=10,1 Гц), 2,47 (3Н, с), 2,32 (3Н, с), 2,07 (6Н, м).
Пример 12
5-[4-(2-Метилтиоамино)пиримидинил] -4-(4-фторфенил)-1-(4-пиперидин)имидазол
К раствору 2-метилтиоамино-4-пиримидинкарбоксальдегида (3,4 г, 22,07 ммоль) и 4-амино-1- метилпиперидиндигидрохлоридной соли (4,54 г, 24,3 ммоль) в 44 мл ДМФ добавляют порошкообразный К2СО3 (7,02 г, 50,8 ммоль). По истечении около 6 ч раствор охлаждают до около 0oС и добавляют изонитрил примера 10, стадии (b), выше, (7,68 г 26,5 ммоль) и К2СО3 (3,57 г, 25,38 ммоль), и перемешивают с постепенным нагревом до около 25oС. Через примерно 16 ч реакционную смесь разбавляют 200 мл EtOAc и промывают 200 мл Н2O. Водный слой экстрагируют EtOAc (2•100 мл), и объединенные органические фазы промывают Н2O (3•100 мл). Органические фазы сушат над Na2SO4 и концентрируют, и указанное в заголовке соединение перекристаллизовывают из EtOAc/Hex с получением 5,12 г (61%) светло-желтых кристаллов. 1H-ЯМР (300 МГц, CDCl3): δ 8,33 (1Н, д, J=5,3 Гц), 7,79 (1H, с), 7,41 (2Н, м), 7,01 (2Н, т, J=8,7 Гц), 6,77 (2Н, д, J= 5,2 Гц), 4,68 (1H, м), 2,98 (2Н, м), 2,59 (3Н, с), 2,32 (3Н, с), 2,07 (6Н, м).
Пример 13
5-[4-(2-Метиламино)пиримидинил] -4-(4-фторфенил)-1-(1-метилпиперидин-4-ил)имидазол
К раствору 2-метиламино-4-пиримидинкарбоксальдегида (2,79 г, 20,37 ммоль) и 4-амино-1- метилпиперидиндигидрохлоридной соли (4,19 г, 22,41 ммоль) в 41 мл ДМФ добавляют порошкообразный К2СО3 (6,19 г, 44,82 ммоль). Смесь перемешивают при комнатной температуре в течение около 6 ч. Раствор охлаждают до около 0oС и добавляют изонитрил примера 85, стадии (b) (7,07 г, 24,44 ммоль) и порошкообразный К2СО3 (3,10 г, 22,41 ммоль). Перемешивают при около 0oС в течение около 3 ч, затем медленно нагревают до комнатной температуры в течение 2 ч. Прибавляют 100 мл Н2О и перемешивают около 15 минут. Фильтруют и промывают 50 мл Н2О и 50 мл ТБМЭ. После сушки 5,56 г (74%) указанного в заголовке соединения выделяют в виде не совсем белого порошка. 1H-ЯМР (300 МГц, CDCl3): δ 8,15 (1H, д, J=4,9 Гц), 7,76 (1H, с), 7,45 (2Н, м), 6,99 (2Н, т, J=8,7 Гц), 6,40 (1H, д, J=5,1 Гц), 5,29 (1H, м), 4,65 (1H, ушир. с), 3,04 (3Н, д, J=5,1 Гц), 2,97 (2Н, м), 2,31 (3Н, с), 2,13-1,98 (6Н, м).
Альтернативно, реакция, описанная выше, проводится при комнатной температуре.
Пример 14
5-[4-(2-Аминопиримидинил)-4-(4-фторфенил)-1-(2,2,6,6-тетраметил-4-пиперидинил)имидазол
a) 2-Аминопиримидинин-4-карбоксальдегид 4-(2,2, 6,6-тетра-метилпиперидинил) имин
С соединением примера 2(b) (0,752 г 6,1 ммоль), выше, объединяют 4-амино-2,2,6,6-тетраметил-пиперидин (1,00 г, 6,42 ммоль), СН2Сl2 (90 мл) и СН3ОН (1 мл), перемешивают в течение ночи и концентрируют с получением указанного в заголовке соединения в виде желтого твердого продукта.
b) 5-[4-(2-Амино)пиримидинил)-4-(4-фторфенил)-1-(2,2,6,6-тетраметил-4-пиперидинил)имидазол
Продукт из (а), выше, и продукт примера 4(b), выше, (1,86 г, 6,42 ммоль), К2СО3 (0,842 г, 6,1 ммоль), и ДМФ (12 мл) объединяют и перемешивают в течение 3 дней. Выливают в Н2O (25 мл) и экстрагируют EtOAc (4•25 мл), сушат (Na2SO4) и концентрируют до масла. Флэш-хроматография (0-10% МеОН в CH2Cl2) дает 0,837 г (35%) указанного в заголовке соединения. Т. пл. 227-230 (разл.).
Пример 15
При способах, аналогичных способам, описанным выше, исключая использование соединения примера 3(b) в качестве альдегидного предшественника имина, может быть получено следующее соединение:
5-(2-Метиламино-4-пиримидинил)-4-(4-фторфенил)-1-(2,2,6,6-тетраметил-4-пиперидин)имидазол; т. пл.=184-185oС.
При способах, аналогичных способам, описанным в примере 1, исключая использование продукта примера 2(b) или продукта примера 3(b) в качестве альдегида и соответствующего амина для получения промежуточного имина, могут быть получены следующие соединения:
Пример 16
5-(2-Амино-4-пиримидинил)-4-(4-фторфенил)-1-(тетрагидро-4-тиопиранил)имидазол. Т. пл. 228-230oС.
Пример 17
5-(2-Амино-4-пиримидинил)-4-(4-фторфенил)-1-(тетрагидро-4-пиранил)имидазол. Т. пл. 222-223oС.
Пример 18
5-(2-Метиламино-4-пиримидинил)-4-(4-фторфенил)-1-(2-цианоэтил)имидазол. Т. пл. 193-194oС.
При способах, аналогичных тем, что описаны в примере 14 WO 95/02591 Adams et al. (Attorney Docket P50172-1), исключая использование продукта примера 16 в качестве исходного продукта, могут быть получены следующие соединения:
Пример 19
5-(2-Амино-4-пиримидинил)-4-(4-фторфенил)-1-(тетрагидро-4-сульфинилпиранил)имидазол. Т. пл. 255-265oС (разл.).
Пример 20
5-(2-Амино-4-пиримидинил)-4-(4-фторфенил)-1-(тетрагидро-4-сульфонилпиранил)имидазол
Продукт примера 16, выше (213 мг, 0,6 ммоль), СН2Сl2 (2,25 мл), СН3ОН (0,75 мл) и ТФА (92 мл, 1,2 ммоль) охлаждают до 4oС и добавляют МЦПБА (около 80%) (387 мг), нагревают до 23oС в течение 20 мин, выливают в EtOAc (50 мл) и промывают 5% водн. Nа2СО3, сушат (Na2SO4), концентрируют, фильтруют через слой кремнезема (0-4% МеОН), получая чистый 5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-(тетрагидро-4-сульфонилпиранил)имидазол (80 мг, 34%). Т. пл. =228-230oС.
Пример 21
5-(2-Метиламино-4-пиримидинил)-4-(4-фторфенил)-1-[1-(2,2,2-трифторэтил)пиперидин-4-ил]имидазол
a) 2-N-Метиламинопиримидин-4-карбоксальдегид диметилацеталь
Пировиноградный альдегид диметилацеталь (277 мл, 2,3 моль) и N,N-диметилформамид диметилацеталь (304 мл, 2,3 моль) перемешивают вместе при 100oС в течение 18 ч. Смесь концентрируют до коричневого масла.
Металлический натрий (25 г) растворяют в EtOH (3 л). Добавляют метилгуанидин гидрохлорид (112 г), и смесь перемешивают 5 ч. Добавляют описанное выше масло (1,15 ммоль), и смесь нагревают с обратным холодильником в течение 24 ч, охлаждают, фильтруют и концентрируют. Полученный остаток растирают с горячим EtOAc и фильтруют через целит. Фильтрат концентрируют, получая указанное в заголовке соединение в виде коричневого масла. 1H-ЯМР (CDCl3): δ 8,33 (д, 1Н), 6,75 (д, 1H), 5,10 (с, 1Н), 3,40 (с, 6Н), 3,00 (с, 3Н).
b) 2-N-Метиламинопиримидин-4-карбоксальдегид
Смесь соединения примера 21(а) гидролизуют по способу примера 26(е) с получением соединения в виде желтой пены. 1H-ЯМР (CDCl3): δ 9,88 (с, 1H), 7,13 (д, 1Н), 7,01 (д, 1Н), 2,05 (с, 3Н).
c) 2-N-Метиламинопиримидин-4-карбоксальдегид (1- [1- (2, 2, 2-трифторэтил)аминопиперидин]имин
Продукт примера 26(с) и продукт примера 21(b) взаимодействуют по способу примера 1(f) с получением указанного в заголовке соединения в виде желтого масла. 1H-ЯМР (CDCl3): δ 8,34 (д, 1), 8,14 (с, 1), 7,13 (д, 1), 5,2 (м, 1), 3,33 (м, 1), 3,05 (м, 7), 2,55 (м, 2), 1,88 (м, 2), 1,75 (м, 2).
d) 5-(2-Метиламино-4-пиримидинил)-4-(4-фторфенил)-1-[1-(2,2,2-трифторэтил)пиперидин-4-ил]имидазол
Продукт примера 21(с) и продукт примера 26 (h) взаимодействуют по способу примера 26(i) с получением указанного в заголовке соединения. Кристаллы из ацетона/гексана. Т. пл. 189-191oС.
Пример 22
5-(2-Амино-4-пиримидинил)-4-(4-фторфенил)-1-[(1-трифтор-ацетил)-4-пиперидинил]имидазол
Продукт примера 46 (с) (500 мг, 1,12 ммоль), описанный в WO 95/02591 Adams et al. (Attorney Docket P50172-1), суспендируют в СН2Сl2 (50 мл) и добавляют Et3N (585 мл, 4,2 ммоль), а через 30 с добавляют трифторуксусный ангидрид (160 мл, 1,12 ммоль). Через 1 ч нерастворимый материал отфильтровывают и фильтрат концентрируют. Полученный белый порошок фильтруют через слой кварца (1-2% СН3ОН в CH2Cl2) с
получением 350 мг (72%) указанного в заголовке соединения. Т. пл.=249-250oС.
Пример 23
5-(4-Пиридил)-4-(4-фторфенил)-1-(4-пиперидинил)имидазол
a)Пиридин-4-карбоксальдегид(этил-4-аминопиперидин-карбоксилат)имин
Следуя способу примера 26(с), исключая замещение этил-4-аминопиперидинкарбоксилата 4-(3-аминопропил)морфолином, получают указанное в заголовке соединение с количественным выходом.
b) 1-[(1-Этоксикарбонил)пиперидин-4-ил] -4-(4-фторфенил)-5-(4-пиридил)имидазол
Следуя способу примера 11, исключая замещение пиридин-4-карбоксальдегид (этил-4-аминопиперидинкарбоксилат)имина 2-N-метиламино-4-карбоксальдегид(4-этиленкетальциклогексил)-имином, получают указанное в заголовке соединение в виде светло-желтого твердого продукта с выходом 71%.
с) 4-(4-Фторфенил)-5-(4-пиридил)-1-(4-пиперидинил)имидазол
Концентрированную соляную кислоту (40 мл) добавляют к 1-[(1-этоксикарбонил)пиперидин-4-ил] -4-(4-фторфенил)-5-(4-пиридил)имидазолу (9,4 г, 24 ммоль) и смесь нагревают с обратным холодильником в течение 18 ч. Полученный желтый раствор охлаждают до температуры окружающей среды и нейтрализуют 10% водным раствором гидроксида натрия. Осадок собирают, промывают водой и сушат на воздухе с получением указанного в заголовке соединения в виде белого твердого продукта с выходом 71%. ESMS=323 [М+Н]; т. пл. 185-187,0oС.
Пример 24
5-(4-Пиридил)-4-(4-фторфенил)-1-(1-трет-бутоксикарбонил-4-пиперидинил)имидазол
Следуя способу примера 11, исключая замещение пиридин-4-карбоксальдегид (трет-бутокси-4 -аминопиперидинкарбоксилат)-имина 2-N-метиламино -4-карбоксальдегид(4-этиленкетальциклогексил)имином, получают указанное в заголовке соединение в виде светло-желтого твердого продукта с выходом 42%. ESMS=423 [М+Н].
Пример 25
2-Метилтиопиримидин-4-карбоксальдегид
Смесь 2-метилтиопиримидин-4-карбоксальдегид диметил-ацеталя (3 г, 15,5 ммоль) в 3 н. НСl (12 мл) перемешивают при 40oС в течение 18 ч. После охлаждения до 0oС смесь разбавляют EtOAc и нейтрализуют добавлением твердого NaHCO3. Водную фазу отделяют и экстрагируют EtOAc (4•25 мл). Объединенные органические экстракты сушат (Na2SO4) и концентрируют. Полученный остаток очищают флэш-хроматографией (силикагель, CH2Cl2) и перекристаллизовывают в гексане с получением указанного в заголовке соединения в виде иголок кремового цвета (1,12 г, выход 48%). 1H-ЯМР (CDCl3): δ 9,95 (с, 1Н), 8,77 (д, 1H), 7,43 (д, 1Н), 2,63 (с, 3Н).
Пример 26 (см. схему XII в конце описания)
5-(2-Амино-4-пиримидинил)-4-(4-фторфенил)-1-[1-(2,2,2-трифторэтил)-4-пиперидинил]имидазол
a) 1-Трифторацетил-4-(1,2-диоксиэтил)пиперидин
4-(1,2-Диоксиэтил)пиперидин (7,15 г, 50 ммоль), CH2Cl2 (50 мл), Et3N (8,35 мл, 60 ммоль) и ДМАП (0,61 г, 5 ммоль) объединяют и добавляют по каплям трифторуксусный ангидрид (11,03 г, 52,5 ммоль) в CH2Cl2 (50 мл), и поддерживают при температуре <30o с внешним охлаждением. После затухания начальной реакции реакционную смесь перемешивают при 23oС в течение 16 ч, промывают 1 н. HCl (2•50 мл), сушат (Na2SO4) и концентрируют с получением 13,38 г (100%) 1-трифторацетил-4-(1,2-диоксиэтил)пиперидина в виде белого твердого продукта. IR 1693 см-1.
b) 1-(2,2,2-Трифторэтил)-4-пиперидинон
Продукт предыдущей стадии (1,19 г, 5,0 ммоль) в ТГФ (5 мл) добавляют к 1 М борану в ТГФ (10 мл) при 4-10oС и нагревают с обратным холодильником, и перемешивают 5 ч, охлаждают до 4oС, и добавляют 6 н. HCl (1,5 мл). Смесь концентрируют, подщелачивают (рН>12) с 50% водн. NaOH и экстрагируют Et2O (3•40 мл). Экстракт сушат (К2СО3) и концентрируют до бесцветного масла.
Полученное выше масло растворяют в 1 н. HCl (15 мл) и раствор нагревают с обратным холодильником в течение 1 ч, охлаждают до 23oС и экстрагируют Et2O (3•20 мл), сушат (Na2SO4) и концентрируют с получением 0,84 г (93%) 1-три-фторэтил-4-пиперидинона в виде светло-желтого масла. IR 1719 см-1.
с) Гидрохлорид 1-(2,2,2-трифторэтил)-4-аминопиперидина
Продукт предыдущей стадии (3,14 г, 17,35 ммоль), Н2О (29 мл) и H2NOH НС1 (4,82 г, 69,4 ммоль) растворяют вместе и добавляют маленькими порциями Na2CO3 (4,82 г). Смесь перемешивают при 23oС в течение 2 ч, устанавливают рН>10 50% с помощью водн. NaOH, экстрагируют Et2O (5•50 мл) и концентрируют до белой пены.
Полученный выше остаток растворяют в EtOH (абс.) и добавляют Raney Ni (5 мл суспензии в EtOH), и смесь восстанавливают в Н2 (50 фунт/дюйм2) (3100 г/см2) в течение 3,5 ч. Катализатор отфильтровывают и промывают EtOH. Добавляют эфирный раствор НСl (1 М) (40 мл), и растворитель удаляют в вакууме с получением масла. При добавлении Et2O (300 мл) осаждается белый твердый осадок. Твердый продукт отфильтровывают, промывают дополнительным количеством Et2O и сушат в вакууме с получением 1,71 г (39%) указанного в заголовке соединения. 1H-ЯМР (CD3OD): δ 4,1 (м, 2), 3,7 (м, 2), 3,5 (м, 1), 3,3 (м, 2), 2,3 (м, 2), 2,1 (м, 2).
d) 2-Аминопиримидин-4-карбоксальдегид диметилацеталь
Диметилформамид диметилацеталь (55 мл, 0,41 моль) и пировиноградный альдегид диметилацеталь (50 мл, 0,41 моль) объединяют и нагревают до 100oС в течение 18 ч. Метанол удаляют в вакууме с получением масла. Раствор NaOH (18 г, 0,45 моль) в Н2О (50 мл) добавляют к гуанидин НСl (43 г, 0,45 моль) в H2O (100 мл), и полученный раствор добавляют к описанному выше маслу. Полученную смесь перемешивают при 23oС в течение 48 ч. Фильтрование дает 25 г (50%) указанного в заголовке соединения.
e) 2-Аминопиримидин-4-карбоксальдегид
Соединение предыдущей стадии (1,69 г, 10 ммоль) и 3 н. HCl (7,3 мл, 22 ммоль) объединяют и нагревают до 48oС в течение около 14 ч, охлаждают, наслаивают EtOAc (50 мл) и нейтрализуют путем добавления NаНСО3 (2,1 г, 25 ммоль) маленькими порциями. Водную фазу экстрагируют EtOAc (5•50 мл), и экстракты сушат (Na2SO4) и концентрируют с получением 0,793 г (64%) указанного в заголовке соединения.
f) 2-Аминопиримидин-4-карбоксальдегид(1-трифторэтил-4-аминопиперидин)имин
Продукт примера 26(с) (1,71 г, 6,79 ммоль), H2O (16 мл), К2СО3 (1,21 г, 8,77 ммоль) и продукт примера 1(е) (0,79 г, 6,45 ммоль), и CH2Cl2 (100 мл) объединяют и перемешивают в атмосфере Аr в течение 14 ч. Фазы отделяют и водную фазу экстрагируют дополнительной порцией CH2Cl2. Объединенные органические фазы сушат (К2СО3) и концентрируют с получением указанного в заголовке соединения в виде желтого масла. 1Н-ЯМР (CDCl3): δ 8,36 (д, 1), 8,13 (с, 1), 7,19 (д, 1), 5,2 (м, 2), 3,35 (м, 1), 3,0 (м, 4), 2,55 (м, 2), 1,90 (м, 2), 1,75 (м, 2).
g) 4-Фторфенил-толилсульфонометилформамид
К суспензии натриевой соли п-толуолсульфоновой кислоты (30 г) в Н2O (100 мл) добавляют ТБМЭ (50 мл), затем по каплям добавляют конц. НСl (15 мл). После перемешивания в течение 5 мин органическую фазу удаляют и водную фазу экстрагируют ТБМЭ. Органическую фазу сушат (Na2SO4) и концентрируют почти досуха. Добавляют гексан и фильтруют свободную кислоту.
Свободную кислоту (22 г, 140,6 ммоль), п-фторбензальдегид (22 мл, 206 ммоль), формамид (20 мл, 503 ммоль) и камфарсульфоновую кислоту объединяют и перемешивают при 60oС в течение 18 ч. Полученный твердый продукт раскрашивают и перемешивают со смесью МеОН (35 мл) и гексана (82 мл), затем фильтруют. Твердый продукт повторно суспендируют в МеОН/гексан (1:3, 200 мл) и энергично перемешивают до разрушения оставшихся кусков. Фильтрация дает указанное в заголовке соединение (27 г, выход 62%). 1H-ЯМР (400 МГц, СDСl3): δ 8,13 (с, 1Н), 7,71 (д, 2Н), 7,43 (дд, 2Н), 7,32 (д, 2Н), 7,08 (т, 2Н), 6,34 (д, 1Н), 2,45 (с, 3Н).
h) 4-Фторфенил-толилсульфонометилизоцианид
Соединение примера 1(f) (2,01 г, 6,25 ммоль) в ДМЭ (32 мл) охлаждают до -10oС. Добавляют РОС13 (1,52 мл, 16,3 ммоль), затем по каплям добавляют триэтиламин (4,6 мл, 32,6 ммоль) в ДМЭ (3 мл), поддерживая внутреннюю температуру ниже -5oС. Смесь постепенно нагревают в течение 1 ч, гасят в Н2O и экстрагируют EtOAc. Органическую фазу промывают насыщенным водным раствором NaHCO3, сушат (Na2SO4) и концентрируют. Полученный остаток растирают с петролейным эфиром и фильтруют с получением указанного в заголовке соединения (1,7 г, выход 90%). 1H-ЯМР (CDCl3): δ 7,63 (д, 2Н), 7,33 (м, 4Н), 7,10 (т, 2Н), 5,60 (с, 1Н), 2,50 (с, 3Н).
i) 5-(2-Амино-4-пиримидинил)-4-(4-фторфенил)-1-[1-(2,2,2-трифторэтил)-4-пиперидинил]имидазол
Продукт примера 1(h) (1,96 г, 6,79 ммоль), продукт примера 26(f) (<6,45 ммоль) и К2СО3 (0,89 г, 6,45 ммоль), и ДМФ (14 мл) объединяют и перемешивают в атмосфере Аr в течение 2 дней. Полученную смесь разбавляют Et2O (100 мл) и фильтруют. Фильтрат концентрируют до сухого остатка в вакууме. Полученный твердый продукт растирают с Et2O, фильтруют и промывают Et2O. Полученный твердый продукт кристаллизуют из ацетона/гексана с получением 0,751 г (28% от продукта примера 1(е)) 5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-[1-(2,2,2-трифторэтил)-4-пиперидинил]имидазола. Т. пл.=229-230oС.
Пример 27
5-(2-Метилтио-4-пиримидинил)-4-(4-фторфенил)-1-(4-пиперидинил)имидазол
a) 1-трет-Бутоксикарбонил-4-аминопиперидин 1-трет-
Бутоксикарбонил-4-аминопиперидин -4-он (коммерчески доступный от Lancaster Chem) преобразуют в свободное основание указанного в заголовке соединения по способу примера 26 (с) с исключением стадии обработки НСl.
b) 2-Метилтиопиримидин-4-карбоксальдегид [1-трет-бутокси-карбонил-4-аминопиперидин]имин
2-Метилтиопиримидин-4-карбоксальдегид [Bredereck et al. Chem. Ber. 1964, 3407] (1,51 г, 9,8 ммоль), продукт примера 27 (а) (2,1 г, 10,5 ммоль), MgSO4 (около 2 г) и CH2Cl2 (75 мл) объединяют и перемешивают при 23oС в течение 16 ч. Фильтрование и концентрация фильтрата дает указанное в заголовке соединение в виде желтого масла. 1H-ЯМР (CDCl3): δ 8,57 (д, 1), 8,27 (с, 1), 7,58 (д, 1), 4,05 (м, 2), 3,55 (м, 1), 3,00 (м, 2), 2,60 (с, 3), 1,75 (м, 4), 1,48 (с, 9).
c) 5-(2-Метилтио-4-пиримидинил)-4-(4-фторфенил)-1-[(1-трет-бутоксикарбонил)-4-пиперидинил)имидазол
Следуя способу примера 26(i), исключая использование продукта примера 27(b) в качестве имина, получают масло из эфирной фазы. Флэш-хроматография в 0-1% МеОН в CH2Cl2, концентрирование и растирание остатка с гексаном дает 2,32 г (50% от 2-метилтиопиримидин-4-карбоксальдегида) указанного в заголовке соединения в виде коричневого твердого продукта. ESP+MS m/z = 470 (MH+).
d) 5-(2-Метилтио-4-пиримидинил)-4-(4-фторфенил)-1-(4-пиперидинил)имидазол
Продукт предшествующего примера (469 мг, 1,0 ммоль) суспендируют в СН3ОН (2 мл), охлаждают до 0oС в атмосфере Аr и добавляют 4 М НСl в диоксане. Полученный светло-желтый твердый продукт нагревают до 23oС, перемешивают в течение 4 ч и разбавляют Et2O. Отделяют смолообразный твердый продукт, который отверждают, растирая с Et2O в течение 20 мин. Твердый продукт отфильтровывают, повторно растворяют в Н2O, промывают EtOAc, наслаивают вторую порцию EtOAc, и водную фазу подщелачивают путем добавления 50% водного раствора NaOH. Экстрагирование EtOAc (3х), сушка объединенных экстрактов (К2СО3) и концентрирование дает твердый продукт, который растирают с Et2O и фильтруют с получением 168 мг (46%) указанного в заголовке соединения в виде беловатого твердого продукта. Т. пл.=182-183oС.
Указанное выше описание полностью описывает настоящее изобретение, включая его предпочтительные воплощения. Модификации и усовершенствования воплощений, конкретно описанных здесь, находятся в рамках последующей формулы изобретения. Без дальнейшего изложения предполагается, что специалист в данной области может, используя предыдущее описание, использовать настоящее изобретение наиболее полным образом. По этой причине приведенные примеры предполагаются в качестве исключительно иллюстративных и не представляющих какого-либо ограничения рамок настоящего изобретения никаким образом. Воплощения настоящего изобретения, в которых сформулировано исключительное право и привилегия, определены следующим образом.
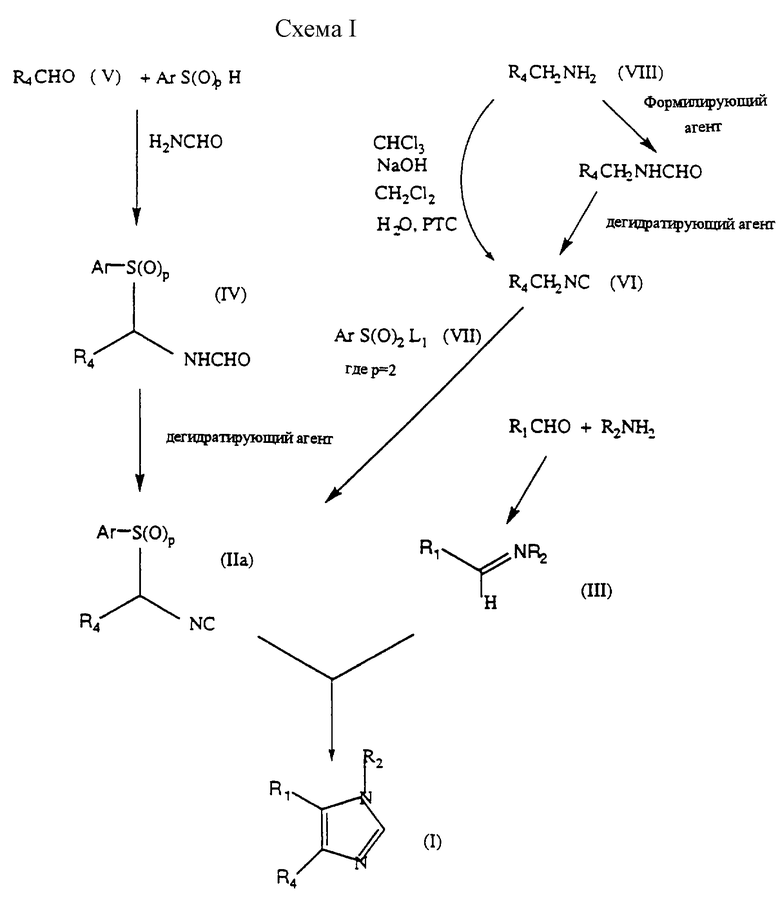

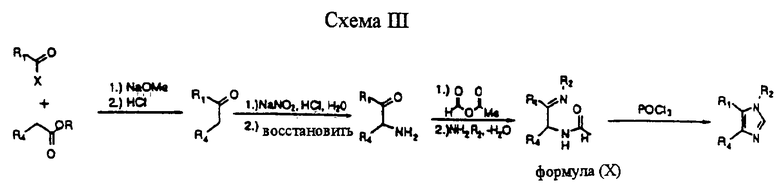










Изобретение относится к способу получения производных имидазола формулы А, где R1 представляет собой замещенный гетероцикл, R4 - фенил, необязательно замещенный, R2 представляет собой алкилN3, -(CR10R20)nOR9 и дальше как указано в описании. Способ включает взаимодействие соединения формулы II с фоединением формулы III. Также изобретение включает в себя способы получения а) соединения формулы IV путем взаимодействия R4-CHO с формамидом, кислотным катализатором и дегидрирующим агентом, б) соединения формулы IV, заключающийся во взаимодействии соединения формулы I с п-толуолсульфиновой кислотой, кислотным катализатором, органическим растворителем. Соединения по п. 1 обладают ингибирующей активностью в отношении цитокинов и поэтому могут использоваться в качестве активного соединения в фармацевтической композиции. 7 с. и 23 з.п. ф-лы, 1 табл.






где R1 представляет собой 4-пиридил, 4-пиримидинил, где гетероарильное кольцо необязательно замещено одним или двумя заместителями, каждый из которых независимо выбран из С1-4алкила, галогена, гидроксила, С1-4алкокси, С1-4алкилтио, C1-4алкилсульфинила, амино, CH2OR12, моно- или ди-С1-6алкилзамещенного амино, N(R10)C(O)Ra или N-гетероциклического кольца, где кольцо включает от 5 до 7 членов и необязательно содержит дополнительный гетероатом, выбранный из кислорода или NR15;
R4 представляет собой фенил, необязательно замещенный одним или несколькими заместителями, такими, как галоген, S(С1-С6)алкил, циано, нитро, карбокси;
R2 представляет собой С1-10алкилN3, -(CR10R20)n'OR9, гетероцикл, который представляет собой насыщенную или частично ненасыщенную 5 - 7-членную кольцевую систему, содержащую один или несколько гетероатомов, выбранных из группы, включающей N или О; C1-6алкил, галогензамещенный C1-6алкил, С2-6алкенил, гетероарил-С1-6алкил, (CR10R20)nOR11, (CR10R20)nS(O)mR18, (CR10R20)nNR13R14, (CR10R20)nCN, (CR10R20)n′SO2R18, (CR10R20)nS(O)m′NR13R14, (CR10R20)nC(Z)R11, (CR10R20)nOC(Z)R11, (CR10R20)nC(Z)OR11, (CR10R20)nC(Z)NR13R14, (CR10R20)nC(Z)NR11OR9,
(CR10R20)nNR10C(Z)R11, (CR10R20)nNR10C(Z)R13R14, (CR10R20)nNR10C(= NR19)NR13R14, (CR10R20)nOC(Z)NR13R14, (CR10R20)nNR10C(Z)NR13R14, (CR10R20)nNR10C(Z)OR10;
m' равно целому числу 1 или 2;
n равно целому числу, имеющему значения от 1 до 10;
n' равно 0 или целому числу, имеющему значения от 1 до 10;
Z является кислородом или серой;
Ra представляет собой водород или С1-6алкил;
R6 представляет собой водород, С1-6алкил, морфолин;
R9 представляет собой водород или C1-6алкил;
R10 и R20 каждый независимо выбраны из водорода или С1-4 алкила;
R11 представляет собой водород, C1-6алкил, гетероцикл, который представляет собой насыщенную или частично ненасыщенную 5 - 6-членную кольцевую систему, содержащую один или два атома азота;
R12 представляет собой водород или R16;
R13 и R14 каждый независимо выбраны из водорода или необязательно замещенного С1-4алкила, необязательно замещенного фенила или необязательно замещенного фенилС1-4алкила, или вместе с азотом, к которому они присоединены, образуют 5 - 6-членное гетероциклическое кольцо, которое необязательно содержит дополнительный гетероатом, выбранный из кислорода или NR9;
R15 представляет собой R10 или С(Z)-С1-4алкил;
R16 представляет собой С1-4алкил;
R18 представляет собой С1-4алкил, гетероцикл, который представляет собой насыщенную или частично ненасыщенную 5 - 6-членную кольцевую систему, содержащую один или два гетероатома, выбранных из кислорода или азота;
R19 представляет собой водород, циано, С1-4алкил,
или его фармацевтически приемлемую соль, отличающийся тем, что включает взаимодействие соединения формулы IIа
с соединением формулы III
где р равно 2;
и основанием, достаточно сильным для депротонирования изонитрильного радикала формулы IIа и где имин формулы III образуется in situ перед реакцией с соединением формулы IIа; R1, R2 и R4 являются такими, как определено для формулы А, и Аr является необязательно замещенной фенильной группой.
R1CHO,
где R1 является таким, как определен для формулы А,
с первичным амином формулы
R2NH2,
где R2 является таким, как определен для формулы А;
R2 может быть соответствующим образом защищен так, как это необходимо.
где Х и X1 определены как заместитель R1 в формуле А по п. 1,
с получением соединения формулы А или его фармацевтически приемлемой соли.
где Х определен как заместитель R1 в формуле А по п. 1,
с получением соединения формулы А или его фармацевтически приемлемой соли.
R1CH(ORa)2,
где R1 определен в формуле А;
Ra представляет собой алкил, арил, арилалкил, гетероарил, гетероарилалкил.
1-трет-бутил-4-(4-фторфенил)-5-(2-метилсульфинил-4-пиримидинил)имидазол;
5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-(тетрагидро-4-тиопиранил)имидазол;
5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-(тетрагидро-4-пиранил)имидазол;
5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-(тетрагидро-4-сульфинилпиранил)имидазол;
5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-(тетрагидро-4-сульфонилпиранил)имидазол;
5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-(1-трифторацетил-4-пиперидинил)имидазол;
5-(2-амино-4-пиримидинил)-4-(4-фторфенил)-1-[1-(2,2,2-трифторэтил)-4-пиперидинил] имидазол или
5-(4-пиридил)-4-(4-фторфенил)-1-(4-пиперидинил)имидазол,
или его фармацевтически приемлемая соль.
где р равно 0 или 2;
Аr представляет собой необязательно замещенный фенил или нафтил;
R4 является таким, как определено для соединений формулы А по п. 1,
отличающийся тем, что осуществляют взаимодействие альдегида формулы
R4-CHO,
где R4 является таким, как определено для соединений формулы А,
и соединения формулы
Ar-S(O)p-H,
где Аr указан выше,
с формамидом, кислотным катализатором и необязательно дегидрирующим агентом.
где R4 является таким, как определено для соединений формулы А в п. 1.
где R4 является таким, как определено для соединений формулы А в п. 1, за исключением того, что не является незамещенным фенилом.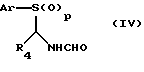
где р равно 2;
Аr представляет собой необязательно замещенный фенил или нафтил;
R4 является таким, как определено для соединений формулы А в п. 1,
заключающийся в том, что осуществляют взаимодействие соединения формулы
где R4 является таким, как определено для формулы А,
с соединением формулы
Ar-S(O)p-H,
где Аr указан выше,
в присутствии кислотного катализатора и органического растворителя.
Приоритетов по пунктам:
09.01.1995 по п. 1;
07.06.1995 по пп. 2-24 - уточнение признаков по указанным пунктам.
| Способ получения производных тризамещенных имидазолов или их солей с основанием | 1982 |
|
SU1169534A3 |
| US 5166400 A1, 24.11.1992 | |||
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| US 4461770 A1, 24.07.1984 | |||
| US 4822805 A1, 18.04.1989 | |||
| Д.Р.ЛОУРЕНС и др | |||
| Клиническая фармакология | |||
| - М.: Медицина, 1993, стр.87-114, 469-505. | |||

