Данное изобретение относится к пиримидиновым производным, или их фармацевтически приемлемым солям, или сложным эфирам, способным подвергаться гидролизу in vivo, которые обладают ингибиторной активностью по отношению к клеточному циклу и являются полезными вследствие их антипролиферативной активности (такой, как противораковая) и, следовательно, могут применяться в способах лечения человека или животных. Изобретение также относится к способам промышленного получения упомянутых пиримидиновых производных, к содержащим их фармацевтическим композициям и к их применению в производстве лекарственных препаратов, применение которых основано на получении антипролиферативного эффекта у теплокровного животного, такого как человек.
Семейство внутриклеточных белков, называемых циклинами, играет центральную роль в клеточном цикле. Синтез и деградация циклинов строго регулируется таким образом, что уровень их экспрессии колеблется в процессе клеточного цикла. Циклины связываются с циклинзависимыми серин/треониновыми киназами (CDKs) и такое связывание является необходимым для проявления активности CDK (такой как CDK1, CDK2, CDK4 и/или CDK6) в клетке. Хотя точные подробности того, как каждый из данных факторов объединяется для регуляции активности CDK, не вполне понятны, равновесие между двумя из них определяет, действительно ли клетка будет развиваться в процессе клеточного цикла.
В последнее время в результате сближения исследований онкогена и гена супрессии опухолей было установлено, что регуляция вступления в клеточный цикл является ключевой контрольной точкой мутагенеза в опухолях. Более того, оказалось, что CDK6 находятся внизу ряда онкогенных сигнальных путей. Нарушение регуляции активности CDK в результате повышающей регуляции (upregulation) циклинов и/или удаления эндогенных ингибиторов является важным связующим звеном между митогенными сигнальными путями и пролиферацией опухолевых клеток.
Соответственно, было установлено, что ингибитор киназ клеточного цикла, особенно относящийся к ингибиторам CDK2, CDK4 и/или CDK6 (действующих в S-фазе, G1-S и G1-S фазе, соответственно), должен иметь ценность как селективный ингибитор клеточной пролиферации, такой как рост раковых клеток млекопитающих.
Настоящее изобретение основано на открытии, что некоторые пиримидиновые соединения неожиданно ингибируют действие киназ клеточного цикла, демонстрируя селективность в отношении CDK2, CDK4 и CDK6, и, следовательно, обладают антипролиферативными свойствами. Ожидается, что такие свойства должны иметь ценность при лечении болезненных состояний, связанных с аберрантными клеточными циклами и клеточной пролиферацией, таких как раковые заболевания (твердые опухоли и лейкемии), фибропролиферативные и дифференционные нарушения, псориаз, ревматоидный артрит, саркома Капоши, гемангиома, острая и хроническая нефропатии, атерома, атеросклероз, артериальный повторный стеноз, аутоиммунные заболевания, острое и хроническое воспаление, костные заболевания и глазные заболевания с пролиферацией сосудов клетчатки.
Соответственно, настоящее изобретение относится к соединению формулы (I):
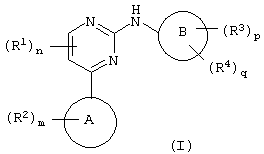
где цикл А представляет собой имидазо[1,2а]пирид-3-ил или пиразол[2,3а]пирид-3-ил;
R2 присоединен к циклическому атому углерода и выбран из галогена, нитро, циано, гидрокси, трифторметила, трифторметокси, амино, карбокси, карбамоила, меркапто, сульфамоила, C1-6алкила, С2-6алкенила, С2-6алкинила, C1-6алкокси, C1-6алканоила, C1-6алканоилокси, N-(C1-6алкил) амино, N,N-(C1-6алкил)2амино, C1-6алканоиламино, N-(C1-6алкил) карбамоила, N,N-(C1-6алкил)2карбамоила, C1-6алкилS(О)а, где а равно от 0 до 2, C1-6алоксикарбонила, N-(C1-2алкил) сульфамоила, N,N-(C1-6алкил)2сульфамоила, фенила, гетероциклической группы, фенилтио или (гетероциклическая группа) тио; где любой С1-2акил, С2-6алкенил, С2-6алкинил, фенил или гетероциклическая группа может быть необязательно замещенным по атому углерода одним или несколькими G; и где, если упомянутая гетероциклическая группа содержит -NH-фрагмент, то азот может быть необязательно замещен группой, выбранной из Q;
m равно 0-5; при этом значения R2 могут быть одинаковыми или разными;
R1 обозначает галоген, нитро, циано, гидрокси, трифторметил, трифторметокси, амино, карбокси, карбамоил, меркапто, сульфамоил, С1-3алкил, C2-3алкенил, C2-3алкинил, С1-3алкокси, C1-6алканоил, N-(C1-3алкил) амино, N,N-(С1-2алкил)2амино, C1-3алканоиламино, N-(C1-3алкил) карбамоил, N,N-(С1-2алкил)2карбамоил, C1-3алкил(О)а, где а равно от 0 до 2, N-(C1-3алкил)сульфамоил или N,N-(C1-3алкил)2сульфамоил; где любой C1-2алкил, C1-3алкил, С2-3алкенил или С2-3алкинил может быть необязательно замещен по атому углерода одним или несколькими J;
n равно от 0 до 2, при этом значения R1 могут быть одинаковыми или разными;
Цикл В представляет собой фенил или фенил, конденсированный с С5-7циклоалкильным циклом;
R3 обозначает галоген, нитро, циано, гидрокси, амино, карбокси, карбамоил, меркапто, сульфамоил, С2-6алкенил или С2-6алкинил;
р равно 0-4; при этом значения R3 могут быть одинаковыми или разными;
R4 обозначает группу А-Е-; где
А выбран из C1-6лкила, фенила, гетероциклической группы, С3-8циклоалкила, фенилС1-6алкила, (гетероциклическая группа)-C1-6алкила или С3-8циклоалкилС1-6циклоалкила; причем C1-6лкил, фенил, гетероциклическая группа, С3-8циклоалкил, фенилС1-6алкил, (гетероциклическая группа) C1-6алкил или С3-8циклоалкилС1-6циклоалкил могут быть необязательно замещены по атому углерода одним или несколькими D; и где, если упомянутая гетероциклическая группа содержит -NH-фрагмент, то азот может быть необязательно замещен группой, выбранной из R;
Е обозначает прямую связь или -О-, -С(О)-, -ОС(О)-, -С (О) О-, -N(Ra)C(O)-, -C(О)N(Ra)-, -N(Ra)-, -S(О)r-, SO2N(Ra)- или -N(Ra)SО2-; где Ra обозначает водород или С1-6лкил, необязательно замещенный одним или несколькими D, а r равно 0-2;
D независимо выбран из оксо, галогена, нитро, циано, гидрокси, трифторметила, трифторметокси, амино, карбокси, карбамоила, меркапто, сульфамоила, C1-6алкила, С2-6алкенила, С2-6алкинила, C1-6алкокси, С2-6алканоила, C1-6алканоилокси, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, C1-6алканоиламино, N-(C1-6алкил)карбамоила, N,N-(C1-6алкил)2карбамоила, C1-6алкилS(О)а, где а равно от 0 до 2, C1-6алкоксикарбонила, С1-6алкоксикарбониламино, бензилоксикарбониламино, N-(C1-6алкил)сульфамоила и N,N-(C1-6алкил)2сульфамоила; где любой C1-6алкил, С2-6алкенил, С2-6алкинил или фенил может быть необязательно замещен по атому углерода одним или несколькими К;
q равно 0-2; при этом значения R4 могут быть одинаковыми или разными; и где р+q≤ 5;
G, J и К независимо выбраны из галогена, нитро, циано, гидрокси, трифторметокси, трифторметила, амино, карбокси, карбамоила, меркапто, сульфамоила, метила, этила, метокси, этокси, ацетила, ацетокси, метиламино, этиламино, диметиламино, диэтиламино, N-метил-N-этиламино, ацетиламино, N-метилкарбамоила, N-этилкарбамоила, N,N-диметилкарбамоила, N,N-диэтилкарбамоила, N-метил-N-этилкарбамоила, метилтио, этилтио, метилсульфинила, этилсульфинила, мезила, этилсульфонила, метоксикарбонила, этоксикарбонила, N-метилсульфамоила, N-этилсульфамоила, N,N-диметилсульфамоила, N,N-диэтилсульфамоила или N-метил-N-этилсульфамоила; и
Q и R независимо выбраны из С1-4алкила, С1-4алканоила, С1-4алкилсульфонила, С1-4алкоксикарбонила, карбамоила, N-(C1-4алкил) карбамоила, N,N-(С1-4алкил) карбамоила, бензила, бензилоксикарбонила, бензоила и фенилсульфонила; или его фармацевтически приемлемую соль, или сложный эфир, способный подвергаться гидролизу in vivo.
Во избежание неопределенности, фраза "где любой С1-6алкил необязательно замещен", а также другие подобные фразы также включают возможность необязательного замещения по другим группам, содержащим С1-6алкильную группу, например, таким, как C1-6алкокси, C1-6алканоил, C1-6алканоилокси, N-(C1-6алкил)амино, N,N-(С1-6алкил)2амино, C1-6алканоиламино, N-(C1-6алкил)карбамоил, N,N-(C1-6алкил)2карбамоил, С1-6алкилS(О)а, где а равно от 0 до 2, C1-6алкоксикарбонил, C1-6алкоксикарбониламино, N-(C1-6алкил)-сульфамоил или N,N-(С1-6алкил)2сульфамоил.
В данном описании термин "алкил" включает алкильные группы как с прямой, так и с разветвленной цепью, но указания на индивидуальные алкильные группы, такие как "пропил", относятся только к вариантам с прямой цепью. Например, "C1-6алкил" включает C1-4алкил, C1-3алкил, С1-2алкил, пропил, изопропил и трет-бутил. Однако указания на индивидуальные алкильные группы, такие как "пропил", относятся только к вариантам с прямой цепью, а указания на индивидуальные алкильные группы с разветвленной цепью, такие как "изопропил", относятся только к вариантам с разветвленной цепью. Подобное правило применяется к другим радикалам, например "фенилС1-6алкил" включает фенилС1-4-алкил, бензил, 1-фенилэтил и 2-фенилэтил. Термин "галоген" относится к фтору, хлору, брому и иоду.
Если необязательные заместители выбраны из "одной или нескольких" групп, следует понимать, что данное определение включает все заместители, выбранные из одной из описанных групп, или заместители, выбранные из двух или более из описанных групп.
"Гетероциклическая группа" представляет собой насыщенную, частично насыщенную или ненасыщенную моно- или бициклическую структуру, содержащую 4-12 атомов, из которых по меньшей мере один атом выбран из азота, серы или кислорода, который может быть связан, если не оговорено иначе, с углеродом или азотом, где группа -СН2-может быть необязательно заменена на -С(О)-, циклический атом азота может необязательно нести C1-6алкильную группу и образовывать четвертичное соединение, или циклический атом азота и/или серы необязательно может быть окислен с образованием N-оксида и/или S-оксидов. Примерами и подходящими значениями термина "гетероциклическая группа" являются морфолин, пиперидил, пиридил, пиранил, пирролил, изотиазолил, индолил, хинолил, тиенил, 1,3-бензодиоксолил, тиадиазолил, пиперазинил, тиазолидинил, пирролидинил, тиоморфолин, пирролинил, гомопиперазинил, 3,5-диоксапиперидинил, тетрагидропиранил, имидазолил, пиримидил, пиразинил, пиридазинил, изоксазолил, N-метилпирролил, 4-пиридон, 1-изохинолон, 2-пирролидон, 4-тиазолидон, пиридин-N-оксид и хинолин-N-оксид. Предпочтительно, "гетероциклическая группа" представляет собой насыщенную, частично насыщенную или ненасыщенную моно- или бициклическую структуру, содержащую 5 или 6 атомов, из которых по меньшей мере один атом выбран из азота, серы или кислорода, который может быть связан, если не оговорено иначе, с углеродом или азотом, где группа –СН2-может быть необязательно заменена на -С(О)-, циклический атом азота может необязательно нести C1-6алкильную группу и образовывать четвертичное соединение, или циклический атом азота и/или серы необязательно может быть окислен с образованием N-оксида и/или S-оксидов.
Подходящим значением для фенила, конденсированного с С5-7 циклоалкильным циклом, является инданил или тетралинил.
Примером "C1-6алканоилокси" является ацетокси. Примеры "C1-6алкоксикарбонила" включают С1-4алкоксикарбонил, метоксикарбонил, этоксикарбонил, н- и трет-бутоксикарбонил. Примеры "C1-6алкокси" включают C1-3алкокси, метокси, этокси и пропокси. Примеры "C1-6алканоиламино" включают C1-3алканоиламино, формамидо, ацетамидо и пропиониламино. Примеры "C1-6алкилS(О)а, где а равно от 0 до 2", включают C1-4алкилсульфонил, C1-3алкилS(О)а, метилтио, этилтио, метилсульфинил, этилсульфинил, мезил и этилсульфонил. Примеры "C1-6алканоила" включают C1-4алканоил, C1-3алканоил, пропионил и ацетил. Примеры "N-C1-6алкиламино" включают N-(С1-3алкил)амино, метиламино и этиламино.
Примеры "N,N-(С1-6алкил)2амино" включают N,N-(С1-2алкил)2амино, ди-N-метиламино", ди-(N-этил) амино и N-этил-N-метиламино. Примерами "С2-6алкенила" являются С2-3алкенил, винил, аллил и 1-пропенил. Примерами "С2-6алкинил" являются С2-3алкинил, этинил, 1-пропинил и 2-пропинил. Примерами "N-(C1-6алкил) сульфамоила" являются N-(C1-3алкил) сульфамоил, N-(метил) сульфамоил и N-(этил) сульфамоил. Примерами "N-(C1-6алкил)2сульфамоила" являются N,N-(C1-3алкил)2сульфамоил, N,N-(диметил)сульфамоил и N-(метил)-N-(этил)сульфамоил. Примерами "N-(C1-6алкил) карбамоила" являются N-(C1-4алкил)карбамоил, N-(C1-3алкил)карбамоил, метиламинокарбонил и этиламинокарбонил. Примерами "N,N-(C1-6алкил)2-карбамоила" являются N,N-(С1-4алкил)2карбамоил, N,N-(С1-2алкил)2-карбамоил, диметиламинокарбонил и метилэтиламинокарбонил. Примерами "C5-7циклоалкила" являются циклопропил и циклогексил. Примеры "(гетероциклическая группа) C1-6алкила" включают пиридилметил, 3-морфолинпропил и 2-пиримид-2-илэтил. Примеры "(гетероциклическая группа)тио" включают тиенилтио и пиридилтио. Примеры "С3-8циклоалкила" включают циклопропил и циклогексил. Примеры "С3-8циклоалкил С1-6циклоалкила" включают циклопропилметил и 2-циклогексилпропил. Примеры "C1-6алкоксикарбониламино" включают метоксикарбониламино и трет-бутоксикарбониламино.
Подходящей фармацевтически приемлемой солью соединения данного изобретения является, например, кислотно-аддитивная соль соединения данного изобретения, которое является достаточно основным, например, кислотно-аддитивная соль, например, с неорганической или органической кислотой, например хлористоводородной, бромистоводородной, серной, фосфорной, трифторуксусной, лимонной или малеиновой кислотой. Кроме того, подходящей фармацевтически приемлемой солью соединения данного изобретения, которое является достаточно кислым, является соль щелочного металла, например натриевая или калийная соль, соль щелочноземельного металла, например кальциевая или магниевая соль, аммонийная соль, или соль с органическим основанием, которое дает физиологически приемлемый катион, например, соль с метиламином, диметиламином, триметиламином, пиперидином, морфолином или трис-(2-гидроксиэтил)амином.
Соединения формулы (I) могут быть введены в виде пролекарственной формы, которая разрушается в организме человека или животного с образованием соединения формулы (I). Примеры пролекарственных форм включают сложные эфиры соединения формулы (I), которые могут подвергаться гидролизу in vivo.
Способным подвергаться гидролизу in vivo сложным эфиром соединения формулы (I), содержащего карбоксильную или гидроксильную группу, является, например, фармацевтически приемлемый эфир, который гидролизуется в организме человека или животного с образованием исходной кислоты или спирта. Подходящие фармацевтически приемлемые сложные эфиры, образуемые карбоксильной группой, включают C1-6алкоксиметиловые эфиры, например, метоксиметиловый, C1-6алканоилоксиметиловые эфиры, например, пивалоилоксиметиловый, фталидиловый эфиры, С3-8циклоалкоксикарбонилокси С1-6алкиловые эфиры, например, 1-циклогексилкарбонилоксиэтил; 1,3-диоксолен-2-онилметиловые эфиры, например, 5-метил-1,3-диоксолен-2-онилметиловый; и C1-6алкоксикарбонилоксиэтиловые эфиры, например, 1-метоксикарбонилоксиэтиловый, и могут быть образованы по любой карбоксильной группе соединений данного изобретения.
Способные подвергаться гидролизу in vivo сложные эфиры соединений формулы (I), содержащих гидроксильную группу, включают неорганические эфиры, такие как эфиры фосфорной кислоты, и α -ацилоксиалкиловые эфиры, а также родственные соединения, которые в результате гидролиза сложного эфира in vivo распадаются с образованием исходной гидроксильной группы. Примеры α -ацилоксиалкиловых эфиров включают ацетоксиметоксильные и 2,2-диметилпропионилоксиметоксильные. Группы, образующие с гидроксильными группами сложноэфирные группы, способные к гидролизу in vivo, включают алканоил, бензоил, фенилацетил, а также замещенный бензоил и фенилацетил, алкоксикарбонил (образующий алкилкарбонатные эфиры), диалкилкарбамоил и N-(диалкиламиноэтил)-N-алкилкарбамоил (образующий карбаматы), диалкиламиноацетил и карбоксиацетил. Примеры заместителей бензоильной группы включают морфолин и пиперазин, атом азота которых связан с положением 3 или 4 бензоильного кольца через метиленовую группу.
Некоторые соединения формулы (I) могут иметь хиральные центры, и/или образуют геометрические изомеры (Е- и Z-изомеры), и следует понимать, что изобретение охватывает все такие оптические и геометрические изомеры, а также диастереомеры, обладающие ингибиторной активностью по отношению к CDK.
Изобретение относится ко всем таутомерным формам соединений формулы (I) и к каждой из них, которые обладают ингибиторной активностью по отношению к CDK.
Следует также понимать, что некоторые соединения формулы (I) могут существовать как в несольватированном виде, так и в сольватированном виде, например в виде гидратов. Следует понимать, что изобретение охватывает все такие сольватированные формы, которые обладают ингибиторной активностью по отношению к CDK.
В другом аспекте изобретения предлагается соединение формулы (I) (изображенной выше), в котором:
Цикл А представляет собой имидазо[1,2а]пирид-3-ил или пиразол[2,3а]пирид-3-ил;
R2 присоединен к циклическому атому углерода и выбран из галогена, нитро, циано, гидрокси, трифторметила, трифторметокси, амино, карбокси, карбамоила, меркапто, сульфамоила, C1-6алкила, С2-6алкенила, С2-6алкинила, C1-8алкокси, C1-6алканоила, C1-6алканоилокси, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, C1-6алканоиламино, N-(C1-6алкил) карбамоила, N,N-(C1-6алкил)2-карбамоила, С1-6алкилS(О)а, где а равно от 0 до 2, C1-6алкоксикарбонила, N-(C1-6алкил) сульфамоила и N,N-(C1-6алкил)2-cульфамоила;
m равно 0-5; при этом значения R2 могут быть одинаковыми или разными;
R1 обозначает галоген, нитро, циано, гидрокси, трифторметил, трифторметокси, амино, карбокси, карбамоил, меркапто, сульфамоил, C1-3алкил, С2-3алкенил, С2-3алкинил, C1-3алкокси, C1-3алканоил, N-(C1-3алкил) амино, N,N-(C1-2алкил)2амино, C1-3алканоиламино, N-(C1-3алкил) карбамоил, N,N-(С1-2алкил)2 карбамоил, C1-3алкилS(O)а, где а равно от 0 до 2, N-(C1-3алкил) сульфамоил или N,N-(C1-3алкил)2сульфамоил;
n равно от 0 до 2, при этом значения R1 могут быть одинаковыми или разными;
Цикл В представляет собой фенил или фенил, конденсированный с C5-7циклоалкильным циклом;
R3 обозначает галоген, нитро, циано, гидрокси, амино, карбокси, карбамоил, меркапто, сульфамоил, С2-6алкенил или С2-6алкинил;
р равно 0-4; при этом значения R3 могут быть одинаковыми или разными;
R4 обозначает группу А-Е-; где
А необязательно замещен по атому углерода одним или несколькими D и выбран из C1-6алкила, фенила, гетероциклической группы, фенилC1-6алкила или (гетероциклическая группа) C1-6алкила;
Е обозначает прямую связь или -О-, -С(О)-, -ОС(О)-, -С(O)O-, -N(Ra)C(O)-, -C(O)N(Ra)-, -N(Ra)-, -S(O)r-, -SO2N(Ra)- или -N(Ra)SO2-; где R3 обозначает водород или C1-6алкил, необязательно замещенный одним или несколькими D, а r равно 0-2;
D независимо выбран из оксо, галогена, нитро, циано, гидрокси, трифторметила, трифторметокси, амино, карбокси, карбамоила, меркапто, сульфамоила, C1-6алкила, С2-6алкенила, C2-6алкинила, C1-6алкокси, C1-6алканоила, С1-6алканоилокси, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, C1-6алканоиламино, N-(C1-6алкил) карбамоила, N,N-(C1-6алкил)2карбамоила, C1-6алкилS(О)а, где а равно от 0 до 2, C1-6алкоксикарбонила, N-(C1-6алкил) сульфамоила и N,N-(С1-6алкил)2сульфамоила; и
q равно 0-2; при этом значения R4 могут быть одинаковыми или разными; и где р+q<5;
или его фармацевтически приемлемая соль, или сложный эфир, способный подвергаться гидролизу in vivo.
Предпочтительные значения R1, R2, R3, R4, n, m, р, q, цикла А и цикла В указаны ниже. Такие значения могут применяться в случае соответствия любому из определений, пунктов формулы изобретения или воплощений, определенных в описании выше или ниже.
В одном аспекте изобретения цикл А предпочтительно представляет собой имидазо[1,2а]пирид-3-ил.
В другом аспекте изобретения цикл А предпочтительно представляет собой пиразол[2,3а]пирид-3-ил.
Предпочтительно, R2 присоединен к циклическому атому углерода и выбран из галогена, нитро, циано, гидрокси, трифторметила, трифторметокси, амино, карбокси, карбамоила, сульфамоила, C1-3алкила, С2-3алкенила, C1-3алкокси, C1-3алканоила, C1-3алканоилокси, N-(C1-3алкил) амино, N,N-(C1-2-алкил)2амино, C1-3алканоиламино, N-(C1-3алкил) карбамоила, N,N-(С1-2алкил)2карбамоила, C1-3алкилS(О)а, где а равно от 0 до 2, N-(С1-3алкил) сульфамоила и N,N-(C1-3алкил)2сульфамоила.
Более предпочтительно, R2 присоединен к циклическому атому углерода и обозначает C1-3алкил.
В частности, R2 присоединен к циклическому атому углерода и обозначает метил.
В другом аспекте изобретения предпочтительно R2 присоединен к циклическому атому углерода и выбран из галогена, нитро, циано, гидрокси, трифторметила, трифторметокси, амино, карбокси, карбамоила, меркапто, сульфамоила, C1-3алкила, С2-3алкенила, С2-3алкинила, C1-3алкокси, C1-3алканоила, C1-3алканоилокси, N-(C1-3алкил) амино, N,N-(С1-3алкил)2амино, C1-3алканоиламино, N-(C1-3алкил) карбамоила, N,N-(C1-3алкил)2карбамоила, C1-3алкилS(O)а, где а равно от 0 до 2, C1-3алкоксикарбонила, N-(C1-3алкил) сульфамоила, N,N-(C1-3алкил)2сульфамоила, фенила, гетероциклической группы, фенилтио или (гетероциклическая группа) тио; где любой C1-3алкил, С2-3алкенил, С2-3алкинил, фенил или гетероциклическая группа может быть необязательно замещенным по атому углерода одним или несколькими G; и где, если упомянутая гетероциклическая группа содержит -NH-фрагмент, то азот может быть необязательно замещен группой, выбранной из Q.
В другом аспекте изобретения более предпочтительно R2 присоединен к циклическому атому углерода и выбран из галогена, циано, C1-6алкила, C1-6алкокси, C1-6алкилS(О)а, где а равно О фенила, фенилтио или (гетероциклическая группа)тио; где любой C1-6алкил, фенил или гетероциклическая группа может быть необязательно замещенным по атому углерода одним или несколькими G; где G выбран из гидрокси и диметиламино.
В другом аспекте изобретения особенно предпочтительно R2 присоединен к циклическому атому углерода и выбран из брома, циано, метила, метокси, этилтио, 2-гидроксиэтилтио, 2-диметиламиноэтилтио, фенил, фенилтио или тиен-2-илтио.
В другом аспекте изобретения еще более предпочтительно R2 присоединен к циклическому атому углерода и выбран из брома, циано, метила, метокси, этилтио, 2-гидроксиэтилтио или 2-диметиламиноэтилтио.
В другом аспекте изобретения особенно предпочтительно R2 присоединен к циклическому атому углерода и выбран из 2-гидроксиэтилтио.
В следующем аспекте изобретения предпочтительно R2 присоединен к циклическому атому углерода и выбран из фтора, хлора, брома, циано, метила, метокси, этилтио, 2-гидроксиэтилтио или 2-диметиламиноэтилтио.
Предпочтительно, m равно 0-2; при этом значения R2 могут быть одинаковыми или разными.
В одном аспекте изобретения предпочтительно m равно 0.
В другом аспекте изобретения предпочтительно m равно 1.
В следующем аспекте изобретения предпочтительно m равно 2; при этом значения R2 могут быть одинаковыми или разными.
В следующем аспекте изобретения предпочтительно R2 присоединен к циклическому атому углерода и выбран из фтора, хлора, брома, циано, метила, метокси, этилтио, 2-гидроксиэтилтио или 2-диметиламиноэтилтио и m равно 0-2; при этом значения R2 могут быть одинаковыми или разными.
Предпочтительно, R2 присоединен к циклическому атому углерода и выбран из брома, циано, метила, метокси, этилтио, 2-гидроксиэтилтио или 2-диметиламиноэтилтио и m равно 0-2; при этом значения R2 могут быть одинаковыми или разными.
Предпочтительно, цикл А и (R2)m вместе образуют имидазо[1,2а]пирид-3-ил, пиразол[2,3а]пирид-3-ил, 2-метил-имидазо[1,2а]пирид-3-ил, 2-метилпиразол[2,3а]пирид-3-ил или 2,5-диметилимидазо[1,2а]пирид-3-ил.
В другом аспекте изобретения предпочтительно цикл А и (R2)m вместе образуют имидаэо[1,2 а]пирид-3-ил,
пиразол[2,3а]пирид-3-ил, 2-метилимидазо[1,2 а]пирид-3-ил,
2-метилпиразол[2,3а]пирид-3-ил,
2,5-диметилимидазо[1,2 а]пирид-3-ил,
6-фенилимидазо[1,2а]пирид-3-ил,
2-метил-6-метоксиимидазо[1,2а]пирид-3-ил,
5-бромимидазо[1,2а]пирид-3-ил,
5-фенилтиоимидазо[1,2а]пирид-3-ил,
5-этилтиоимидазо[1,2а]пирид-3-ил,
5-(2-гидроксиэтилтио)имидазо[1,2а]пирид-3-ил,
5-тиен-2-илтиоимидазо[1,2а]пирид-3-ил,
5-цианоимидазо[1,2а]пирид-3-ил или
5-(2-диметиламиноэтилтио)имидазо[1,2а]пирид-3-ил.
В другом аспекте изобретения более предпочтительно цикл А и (R2)m вместе образуют имидазо[1,2а]пирид-3-ил или 5-(2-гидроксиэтилтио)имидазо[1,2а]пирид-3-ил.
Предпочтительно, n равно 0 или 1, и, если n равно 1, предпочтительно R1 присоединен по положению 5 пиримидинового цикла.
Более предпочтительно, n равно 0.
Предпочтительно, R1 обозначает галоген или C1-3алкилS(О)а, где а равно 0; где C1-3алкильная группа может быть необязательно замещена по атому углерода одним или несколькими J; где J обозначает гидрокси.
Более предпочтительно, R1 обозначает бром или 2-гидроксиэтилтио.
Особенно предпочтительно, R1 обозначает бром или 2-гидроксиэтилтио и n равно 0-1.
Предпочтительно, цикл В представляет собой фенил или инданил.
Более предпочтительно, цикл В представляет собой фенил или индан-5-ил.
Особенно предпочтительно, цикл В представляет собой фенил.
Предпочтительно, R3 представляет собой галоген или сульфамоил.
Более предпочтительно, R3 представляет собой фтор, хлор, бром или сульфамоил.
Особенно предпочтительно, R3 представляет собой хлор или сульфамоил.
Еще более предпочтительно, R3 обозначает сульфамоил.
Предпочтительно, р равно 0-2; при этом значения R3 могут быть одинаковыми или разными.
В одном аспекте изобретения предпочтительно р равно 0.
В другом аспекте изобретения предпочтительно р равно 1.
В следующем аспекте изобретения предпочтительно р равно 2; при этом значения R3 могут быть одинаковыми или разными.
Предпочтительно, R3 обозначает фтор, хлор, бром или сульфамоил; и р равно 1.
В следующем аспекте изобретения, если цикл В представляет собой фенил и р равно 1, предпочтительно, R3 присоединен в мета-положении по отношению к фрагменту -NH-формулы (I).
Предпочтительно, Ra обозначает водород.
Предпочтительно, Е обозначает -NHSO2-.
Предпочтительно, R4 обозначает группу А-Е-; где А является необязательно замещенным по атому углерода одним или несколькими D и выбран из С1-4алкила, фенила, гетероциклической группы или фенил С1-4алкила;
Е обозначает прямую связь или -О-, -С (О)-, -N(Ra)C(O)-, -S(O)r- или –N(Ra)SO2-; где Ra обозначает водород, метил или этил, а r равно 0-2;
D обозначает оксо, циано, гидрокси, амино, карбокси, карбамоил, сульфамоил, C1-3алкил, С2-3алкенил, С2-3алкинил, C1-3алкокси, C1-3алканоил, N-(C1-3алкил)амино, N,N-(C1-2алкил)2амино, C1-3алканоиламино, N-(C1-3алкил) карбамоил, N,N-(C1-3алкил)2карбамоил, C1-3алкилS(О)а, где а равно от 0 до 2, N-(C1-3алкил)сульфамоил или N,N-(C1-3алкил)2сульфамоил.
Более предпочтительно, R4 обозначает группу А-Е-; где А является необязательно замещенным по атому углерода одним или несколькими D и выбран из С1-4алкила, фенила, гетероциклической группы или фенилС1-4алкила;
Е обозначает прямую связь или -О-, -С(О)-или -S(O)r; где r равно 0-2;
D обозначает гидрокси или N,N-(С1-2алкил)2амино.
Особенно предпочтительно, R4 обозначает метил, этил, метокси, метилтио, мезил, ацетил, 3-N,N-диметиламино-2-гидроксипропокси, 2-N,N-диэтиламиноэтокси, бензилокси, анилиносульфонил, пиримидин-2-иламиносульфонил, фенокси, 3,5-диоксапиперидин-1-илсульфонил.
В другом аспекте изобретения предпочтительно R4 обозначает группу А-Е-; где
А выбран из C1-6алкила, фенила, гетероциклической группы, С3-8циклоалкила, фенилС1-6алкила, (гетероциклическая группа) C1-6-алкила или С3-8циклоалкил С1-6циклоалкила; указанные C1-6алкил, фенил, гетероциклическая группа, С3-8циклоалкил, фенилС1-6алкил, (гетероциклическая группа) С1-6алкил или С3-8циклоалкил С1-6циклоалкил могут быть необязательно замещены по атому углерода одним или несколькими D; и, если упомянутая гетероциклическая группа содержит фрагмент -NH-, то азот может быть необязательно замещен группой, выбранной из R;
Е обозначает прямую связь или -О-, -С (О)-, -N(Ra)C(O)-, -S(O)r-или –N(Ra)SO2-; где R3 обозначает водород или C1-6алкил, а r равно 0-2;
D независимо выбран из гидрокси, амино, C1-6алкокси, N-(C1-6алкил) амино, N,N-(C1-6алкил)2амино, C1-6алкоксикарбониламино или бензилоксикарбониламино; где любой C1-6алкил может быть необязательно замещен по атому углерода одним или несколькими К;
К выбран из гидрокси или диэтиламино; и
R обозначает С1-6алкил.
В другом аспекте изобретения более предпочтительно R4 обозначает группу А-Е-; где
А выбран из метила, этила, пропила, пентила, фенила, пиримидила, 3,5-диоксапиперидин-1-ила, циклопропила, бензила, пирролидин-1-илэтила, пиперидин-1-илэтила, пирролидин-2-ил-этила, 3-(2-оксо-пирролидин-1-ил)пропила, 3-имидазол-1-ил-пропила, 2-морфолинэтила, 3-морфолинпропила, 2-имидазол-4-илэтила, 2-пиперазин-1-илэтила, 3-пиперазин-1-илпропила или циклопропилметила; где А может быть необязательно замещен по атому углерода одним или несколькими D; и где пирролидин-2-ил, имидазол-4-ил или пиперазин-1-ил может быть необязательно замещен по атому азота группой, выбранной из R;
Е обозначает прямую связь или -О-, -С(О)-, -N(Ra)C(O)-, -S(O)r-или -N(Ra)SO2-; где R4 обозначает водород или метил, а r равно 0-2;
D независимо выбран из гидрокси, амино, метокси, метиламино, этиламино, изопропиламино, N,N-диметиламино, N,N-диэтиламино, трет-бутоксикарбониламино или бензилоксикарбониламино; где любой из метила, этила, изопропила или трет-бутила может быть необязательно замещен по атому углерода одним или несколькими К;
К выбран из гидрокси или диэтиламино; и
R обозначает метил.
В другом аспекте изобретения особенно предпочтительно R4 обозначает метил, этил, метокси, метилтио, ацетил, бензилокси,
мезил, N,N-диэтиламиноэтокси,
3-N,N-диметиламино-2-гидроксипропокси, фенокси,
N-метилкарбамоил, N,N-диметилкарбамоил,
N-(3-имидазол-1-илпропил)карбамоил,
N-[3-(2-оксопирролидин-1-ил)пропил]карбамоил,
3,5-диоксапиперидин-1-илсульфонил,
N-циклопропилсульфамоил, N-циклопропилметилсульфамоил,
анилиносульфонил, N-пиримидин-2-илсульфамоил,
N-метилсульфамоил, N-пропилсульфамоил,
N-(2-метоксиэтил)сульфамоил,
N-(2-метиламиноэтил)сульфамоил,
N-(2-изопропиламиноэтил)сульфамоил,
N-(2-диметиламиноэтил)сульфамоил,
N-(2-диэтиламиноэтил)сульфамоил,
N-[2-(гидроксиэтиламино)этил]сульфамоил,
N-[2-(диэтиламиноэтил)этил]сульфамоил,
N-(пирролидин-1-илэтил)сульфамоил,
N-[2-(1-метилпирролидин-2-ил)этил]сульфамоил,
N-(2-пиперидин-1-илэтил)сульфамоил,
N-(2-пиперазин-1-илэтил)сульфамоил,
N-(2-морфолинэтил)сульфамоил,
N-(2-имидазол-4-илэтил)сульфамоил,
N-(3-гидроксипропил)сульфамоил,
N-(2,3-дигидроксипропил)сульфамоил,
N-(3-метоксипропил)сульфамоил,
N-(3-аминопропил)сульфамоил,
N-(3-метиламинопропил)сульфамоил,
N-(3-диметиламинопропил)сульфамоил,
N-(3-диэтиламинопропил)сульфамоил,
N-(3-изопропиламинопропил)сульфамоил,
N-(3-трет-бутоксикарбониламинопропил)сульфамоил,
N-(3-бензилоксикарбониламинопропил)сульфамоил,
N-[3-(2-оксопирролидин-1-ил)пропил]сульфамоил,
N-(3-морфолинпропил)сульфамоил,
N-[3-(4-метилпиперазин-1-ил)пропил]сульфамоил,
N-(3-имидазол-1-илпропил)сульфамоил или
N-(5-гидроксипентил)сульфамоил.
В другом аспекте изобретения, еще более предпочтительно R4 обозначает N-метилсульфамоил, N-(2-метоксиэтил)сульфамоил,
N-(2-метиламиноэтил)сульфамоил,
N-(2-диметиламиноэтил)сульфамоил,
N-(3-метоксипропил)сульфамоил,
N-(3-диметиламинопропил)сульфамоил или
N-(3-изопропиламинопропил)сульфамоил.
Предпочтительно, R4 обозначает метил, этил, метокси, метилтио, ацетил, бензилокси, мезил, N,N-диэтиламиноэтокси,
3-N,N-диметиламино-2-гидроксипропокси, фенокси,
N-метилкарбамоил, N,N-диметилкарбамоил,
N-(3-имидазол-1-илпропил)карбамоил,
N-[3-(2-оксо-пирролидин-1-ил)пропил]карбамоил,
3,5-диоксапиперидин-1-илсульфонил,
N-циклопропилсульфамоил, N-циклопропилметилсульфамоил,
анилиносульфонил, N-пиримидин-2-илсульфамоил,
N-метилсульфамоил, N-пропилсульфамоил,
N-(2-метоксиэтил)сульфамоил,
N-(2-метиламиноэтил)сульфамоил,
N-(2-изопропиламиноэтил)сульфамоил,
N-(2-диметиламиноэтил)сульфамоил,
N-(2-диэтиламиноэтил)сульфамоил,
N-[2-(гидроксиэтиламино)этил]сульфамоил,
N-[2-(диэтиламиноэтил)этил]сульфамоил,
N-(пирролидин-1-илэтил)сульфамоил,
N-[2-(1-метилпирролидин-2-ил)этил]сульфамоил,
N-(2-пиперидин-1-илэтил)сульфамоил,
N-(2-пиперазин-1-илэтил)сульфамоил,
N-(2-морфолинэтил)сульфамоил,
N-(2-имидазол-4-илэтил)сульфамоил,
N-(3-гидроксипропил)сульфамоил,
N-(2,3-дигидроксипропил)сульфамоил,
N-(3-метоксипропил)сульфамоил,
N-(3-аминопропил)сульфамоил,
N-(3-метиламинопропил)сульфамоил,
N-(3-диметиламинопропил)сульфамоил,
N-(3-диэтиламинопропил)сульфамоил,
N-(3-изопропиламинопропил)сульфамоил,
N-(3-трет-бутоксикарбониламинопропил)сульфамоил,
N-(3-бензилоксикарбониламинопропил)сульфамоил,
N-[3-(2-оксопирролидин-1-ил)пропил]сульфамоил,
N-(3-морфолинпропил)сульфамоил,
N-[3-(4-метилпиперазин-1-ил)пропил]сульфамоил,
N-(3-имидазол-1-илпропил)сульфамоил или
N-(5-гидроксипентил)сульфамоил; и q равно 1.
Предпочтительно, q равно 0-1.
В одном аспекте изобретения q равно 0.
В другом аспекте изобретения q равно 1.
В следующем аспекте изобретения, если цикл В представляет собой фенил и q равно 1, предпочтительно R4 присоединен в пара-положение по отношению к -NH-фрагменту формулы (I).
Предпочтительно, цикл В, (R3)p(R4)q и вместе образуют фенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил, 3-бромфенил, 3-метилфенил, 4-метилфенил, 3-этилфенил, 3-метоксифенил, 4-метоксифенил, 3-метилтиофенил, 4-метилтиофенил, 4-мезилфенил, 3-сульфамоилфенил, 4-сульфамоилфенил, 3-ацетилфенил, 3,4-дихлорфенил, 3-хлор-4-фторфенил, 2-хлор-4-метилфенил, 4-(3-N,N-диметиламино-2-гидроксипропокси)-фенил, 4-бензилоксифенил, 4-анилиносульфонилфенил, 4-(пиримидин-2-илсульфонил)фенил, 4-феноксифенил, 4-(2-N,N-диэтиламиноэтокси)фенил, 4-(3,5-диоксапиперидин-1-илсульфонил)фенил или инданил.
В другом аспекте изобретения, более предпочтительно цикл В, (R3)p(R4)q и вместе образуют фенил, 2-хлорфенил, 2-фторфенил, 3-фторфенил, 3-хлорфенил, 3-бромфенил, 3-метилфенил, 3-метоксифенил, 3-метилтиофенил, 3-ацетилфенил, 3-этилфенил, 3-сульфамоилфенил, 4-фторфенил, 4-хлорфенил, 4-метилфенил, 4-метоксифенил, 4-сульфамоилфенил, 3-метил-4-сульфамоилфенил,
4-(N-метилкарбамоил)фенил, 4-(N,N-диметилкарбамоил)фенил,
4-метилтиофенил, 4-мезилфенил,
4-(N-метилсульфамоил)фенил, 4-(N-пропилсульфамоил)фенил,
3-хлор-4-(N-пропилсульфамоил)фенил,
4-(N,N-диэтиламиноэтокси)фенил, 4-бензилоксифенил,
4-феноксифенил, 4-(N-циклопропилсульфамоил)фенил,
4-(N-циклопропилметилсульфамоил)фенил,
4-(3,5-диоксапиперидин-1-илсульфонил)фенил,
4-анилиносульфонилфенил,
4-(N-пиримидин-2-илсульфамоил)фенил,
4-[N-(2-метоксиэтил)сульфамоил]фенил,
3-хлор-4-[N-(2-метоксиэтил)сульфамоил]фенил,
3-метил-4-[N-(2-метоксиэтил)сульфамоил]фенил,
4-[N-(3-диэтиламинопропил)сульфамоил]фенил,
4-{N-[2-(1-метилпирролидин-2-ил)этил]сульфамоил]}фенил,
3-хлор-4-фторфенил, 3,4-дихлорфенил, 2-хлор-4-метилфенил,
4-(3-N,N-диметиламино-2-гидроксипропокси) фенил,
4-[N-(3-гидроксипропил)сульфамоил]фенил,
4-{N-[3-(2-оксопирролидин-1-ил)пропил]сульфамоил}фенил,
4-[N-(5-гидроксипентил)сульфамоил]фенил,
4-[N-(3-метоксипропил)сульфамоил]фенил, индан-5-иламино,
4-[N-(3-изопропиламинопропил)сульфамоил]фенил,
4-[N-(2-изопропиламиноэтил)сульфамоил]фенил,
4-[N-(З-имидазол-1-илпропил)сульфамоил]фенил,
4-[N-(3-диметиламинопропил)сульфамоил]фенил,
4-[N-(3-морфолинпропил)сульфамоил]фенил,
3-метил-4-[N-(3-морфолинпропил)сульфамоил]фенил,
4-[N-(3-аминопропил)сульфамоил]фенил,
4-{N-[2-(гидроксиэтиламино)этил]сульфамоил}фенил,
4-[N-(2-имидазол-4-илэтил)сульфамоил]фенил,
4-[N-(3-метиламинопропил)сульфамоил]фенил,
4-[N-(2-пиперазин-1-илэтил)сульфамоил]фенил,
4-[N-[3-(4-метилпиперазин-1-ил)пропил]сульфамоил]фенил,
4-[N-(2,3-дигидроксипропил)сульфамоил]фенил,
4-[N-(3-имидазол-1-илпропил)карбамоил]фенил,
4-[N-[2-(диэтиламиноэтил)этил]сульфамоил]фенил,
4-[N-[3-(2-оксо-пирролидин-1-ил)пропил]карбамоил]фенил,
4-[N-(2-диметиламиноэтил)сульфамоил]фенил,
4-[N-(2-морфолинэтил)сульфамоил]фенил,
3-метил-4-[N-(2-морфолинэтил)сульфамоил]фенил,
4-[N-(пирролидин-1-илэтил)сульфамоил]фенил,
4-[N-(2-метиламиноэтил)сульфамоил]фенил,
4-[N-(2-пиперидин-1-илэтил)сульфамоил]фенил,
4-[N-(2-диэтиламиноэтил)сульфамоил]фенил,
4-[N-(3-трет-бутоксикарбониламинопропил)сульфамоил]фенил,
4-[N-(3-бензилоксикарбониламинопропил)сульфамоил]фенил или
4-[N-(3-диэтиламинопропил)сульфамоил]фенил.
В другом аспекте изобретения, особенно предпочтительно цикл В, (R3)p(R4)q и вместе образуют 4-сульфамоилфенил,
4-(N-метилсульфамоил)фенил,
4-[N-(2-метоксиэтил)сульфамоил]фенил,
4-[N-(3-метоксипропил)сульфамоил]фенил,
4-[N-(3-изопропиламинопропил)сульфамоил]фенил,
4-[N-(3-диметиламинопропил)сульфамоил]фенил,
4-[N-(2-диметиламиноэтил)сульфамоил]фенил или
4-[N-(2-метиламиноэтил)сульфамоил]фенил.
Таким образом, в одном аспекте изобретения предлагается соединение формулы (I), изображенной выше, где:
Цикл А представляет собой имидазо[1,2а]пирид-3-ил или пиразол[2,3а]пирид-3-ил;
R2 присоединен к циклическому атому углерода и выбран из галогена, нитро, циано, гидрокси, трифторметила, трифторметокси, амино, карбокси, карбамоила, сульфамоила, C1-3алкила, С2-3алкенила, C1-3алкокси, C1-3алканоила, C1-3алканоилокси, N-(C1-3алкил) амино, N,N-(C1-2алкил)2амино, C1-3алканоиламино, N-(C1-3алкил) карбамоила, N,N-(С1-2алкил)-карбамоила, C1-3алкилS(О)а где а равно от 0 до 2, N-(C1-3-алкил) сульфамоила и N,N-(C1-3алкил)2сульфамоила;
m равно 0-2; при этом значения R2 могут быть одинаковыми или разными;
n равно 0;
цикл В представляет собой фенил или инданил;
R3 представляет собой галоген или сульфамоил;
R4 обозначает группу А-Е-; где
А необязательно замещен по атому углерода одним или несколькими D и выбран из С1-4алкила, фенила, гетероциклической группы или фенил С1-4алкила;
Е обозначает прямую связь или -О-, -С (О)-, -N(Ra)C(O)-, -S(O)r- или N(Ra) SO2-; где Ra обозначает водород, метил или этил, а r равно 0-2;
р равно 0-2; при этом значения R3 могут быть одинаковыми или разными;
D обозначает оксо, циано, гидрокси, амино, карбокси, карбамоил, сульфамоил, C1-3алкил, С2-3алкенил, С2-3алкинил, C1-3алкокси, C1-3алканоил, N-(C1-3алкил) амино, N,N-(С1-2алкил)2-амино, C1-3алканоиламино, N-(C1-3алкил) карбамоил, N,N-(C1-2-алкил)2карбамоил, С1-3алкилS(О)а где а равно от 0 до 2, N-(C1-3алкил) сульфамоил или N,N-(C1-3алкил)2сульфамоил;
q равно 0-1; при этом значения R4 могут быть одинаковыми или разными;
или его фармацевтически приемлемая соль, или его сложный эфир, способный подвергаться гидролизу in vivo.
Таким образом, в следующем аспекте изобретения предлагается соединение формулы (I), изображенной выше, где:
Цикл А представляет собой имидазо[1,2а]пирид-3-ил или пиразол[2,3а]пирид-3-ил;
R2 присоединен к циклическому атому углерода и обозначает C1-3алкил;
m равно 0-2; при этом значения R2 могут быть одинаковыми или разными;
n равно 0;
Цикл В представляет собой фенил или индан-5-ил;
R3 обозначает фтор, хлор, бром или сульфамоил;
р равно 0-2; при этом значения R3 могут быть одинаковыми или разными;
R4 обозначает метил, этил, метокси, метилтио, мезил, ацетил, 3-N,N-диметиламино-2-гидроксипропокси, 2-N,N-диэтиламиноэтокси, бензилокси, анилиносульфонил, пиримидин-2-ил-аминосульфонил, фенокси, 3,5-диоксапиперидин-1-илсульфонил.
q равно 0-1; при этом значения R4 могут быть одинаковыми или разными;
или его фармацевтически приемлемая соль, или сложный эфир, способный подвергаться гидролизу in vivo.
Таким образом, в дополнительном аспекте изобретения предлагается соединение формулы (I), изображенной выше, где:
Цикл А и (R2)m вместе образуют имидазо[1,2 а]пирид-3-ил, пиразол[2,3а]пирид-3-ил, 2-метилимидазо[1,2а]пирид-3-ил, 2-метилпиразол[2,3а]пирид-3-ил или 2,5-диметилимидазо[1,2а]пирид-3-ил;
n равно 0;
Цикл В, (R3)p и (R4)q вместе образуют фенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил, 3-бромфенил, 3-метилфенил, 4-метилфенил, 3-этилфенил, 3-метоксифенил, 4-метоксифенил, 3-метилтиофенил, 4-метилтиофенил, 4-мезилфенил, 3-сульфамоилфенил, 4-сульфамоилфенил, 3-ацетилфенил, 3,4-дихлорфенил, 3-хлор-4-фторфенил, 2-хлор-4-метилфенил, 4-(3-N,N-диметиламино-2-гидроксипропокси)фенил, 4-бензилоксифенил, 4-анилиносульфонилфенил, 4-(пиримидин-2-ил-сульфонил)фенил, 4-феноксифенил, 4-(2-N,N-диэтиламиноэтокси)-фенил, 4-(3,5-диоксапиперидин-1-илсульфонил)фенил или инданил; или его фармацевтически приемлемая соль, или его сложный эфир, способный подвергаться гидролизу in vivo.
Таким образом, в следующем дополнительном аспекте изобретения предлагается соединение формулы (I), изображенной выше, где:
Цикл А представляет собой имидазо[1,2а]пирид-3-ил или пиразол[2,3а]пирид-3-ил;
R1 представляет собой галоген или C1-3алкилS(О)а, где а равно 0; где C1-3алкильная группа может быть необязательно замещена по атому углерода одним или несколькими J; где J обозначает гидрокси.
n равно 0-1;
R2 присоединен к циклическому атому углерода и выбран из галогена, циано, C1-6алкила, C1-6алкокси, C1-6алкилS(О)а, где а равно 0, фенила, фенилтио или (гетероциклическая группа)тио; где любой C1-6алкил, фенил или гетероциклическая группа могут быть необязательно замещены по атому углерода одним или несколькими G; где G выбран из гидрокси и диметиламино.
m равно 0-2; при этом значения R2 могут быть одинаковыми или разными;
Цикл В представляет собой фенил или индан-5-ил;
R3 обозначает фтор, хлор/бром или сульфамоил;
р равно 0-1;
R4 обозначает группу А-Е-; где
А выбран из C1-6алкила, фенила, гетероциклической группы, С3-8циклоалкила, фенилС1-6алкила, (гетероциклическая группа) C1-6-алкила или С3-6циклоалкил С1-6циклоалкила; причем C1-6алкил, фенил, гетероциклическая группа, С3-8цинклоалкил, фенил С1-6алкил, (гетероциклическая группа) C1-6алкил или С3-8циклоалкил С1-6циклоалкил могут быть необязательно замещены по атому углерода одним или несколькими D; и где, если упомянутая гетероциклическая группа содержит -NH-фрагмент, то азот может быть необязательно замещен группой, выбранной из R;
Е обозначает прямую связь или -О-, -С(О)-, -N(Ra)C(O)-, -S(О)r- или -N(Ra)SO2-, где R8 обозначает водород или C1-6алкил, а r равно 0-2;
D независимо выбран из гидрокси, амино, C1-6алкокси, N-(C1-6алкил) амино, N,N-(C1-6алкил)2амино, C1-6алкоксикарбониламино или бензилоксикарбониламино; где любой C1-6алкил может быть необязательно замещен по атому углерода одним или несколькими К;
К выбран из гидрокси или диэтиламино; и
R обозначает С1-4алкил; а
q равно 0-1;
или его фармацевтически приемлемая соль, или его сложный эфир, способный подвергаться гидролизу in vivo.
Таким образом, в другом дополнительном аспекте изобретения предлагается соединение формулы (I), изображенной выше, где:
Цикл А представляет собой имидазо[1,2а]пирид-3-ил;
n равно 0;
Цикл В представляет собой фенил;
R3 обозначает сульфамоил;
р равно 0-1;
R4 обозначает N-метилсульфамоил, N-(2-метоксиэтил)-сульфамоил, N-(2-метиламиноэтил)сульфамоил, N-(2-диметиламиноэтил)сульфамоил, N-(3-метоксипропил)сульфамоил, N-(3-диметиламинопропил)сульфамоил или N-(3-изопропиламинопропил)-сульфамоил; и
q равно 0-1;
или его фармацевтически приемлемая соль, или его сложный эфир, способный подвергаться гидролизу in vivo.
В другом аспекте изобретения к предпочтительным соединениям изобретения относится любое соединение примеров 1-38 или его фармацевтически приемлемая соль, или его сложный эфир, способный подвергаться гидролизу in vivo.
В другом аспекте изобретения к предпочтительным соединениям изобретения относится любое соединение примеров 1-98 или его фармацевтически приемлемая соль, или его сложный эфир, способный подвергаться гидролизу in vivo.
В следующем аспекте изобретения к предпочтительным соединениям изобретения относятся соединения примеров 7, 39, 40, 52, 53, 55, 65, 68 и 86 или их фармацевтически приемлемые соли, или их сложные эфиры, способные подвергаться гидролизу in vivo.
Предпочтительными аспектами изобретения являются такие аспекты, которые относятся к соединению формулы (I) или к его фармацевтически приемлемой соли.
В другом аспекте настоящего изобретения предлагается способ получения соединения формулы (I) или его фармацевтически приемлемой соли, или его сложного эфира, способного подвергаться гидролизу in vivo, данный способ (где R1, R2, R3, R4, цикл А, цикл В, m, р, q и n, если не указано иначе, являются такими, как определено в формуле (I)), включает:
а) взаимодействие пиримидина формулы (II):
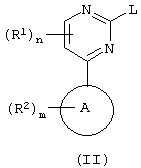
где L является уходящей группой;
с амином формулы (III):
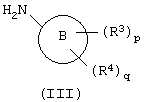
b)взаимодействие пиримидина формулы (IV)
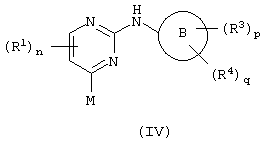
c соединением формулы (V):
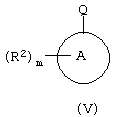
где один из М и Q является способной к замещению группой X, а другой является металлическим реагентом Y; или
с) взаимодействие соединений формулы (VI):
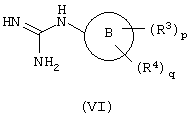
с соединением формулы (VII):
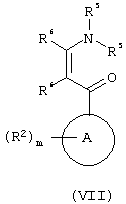
где R5 обозначает C1-6алкил, a R6 обозначает водород или R1;
и затем, при необходимости:
i) превращение соединения формулы (I) в другое соединение формулы (I);
ii) удаление всех защитных групп;
iii) получение фармацевтически приемлемой соли или сложного эфира, способного подвергаться гидролизу In vivo.
L представляет собой уходящую группу, подходящими значениями для L являются, например галоген или сульфонилоксильная группа, например хлор, бром, метансульфонилоксильная или толуол-4-сульфонилоксильная группа.
Хорошей уходящей группой Х является, например, галоген или сульфонильная группа, например бром, иод или трифторметилсульфонильная группа.
Подходящей металлической группой Y является, например, медь, литий, бор органический реагент, такой как -В(ОН)2, -В(ОРri)2 или -B(Et)2, или оловоорганическое соединение, такое как SnBu3, кремнийорганическое соединение, такое как Si(Me)F2, цирконий органическое соединение, такое как ZrCl2, алюминийорганическое соединение, такое как AlEt2, магнийорганическое соединение, такое как МgВr, цинкорганическое соединение, такое как ZnCl, или ртутьорганическое соединение, такое как НgВr. Конкретные условия реакции для указанных выше взаимодействий приведены ниже.
а) Пиримидины формулы (II) и амины формулы (III) могут взаимодействовать: i) в присутствии подходящего растворителя, например, кетона, такого как ацетон, или спирта, такого как этанол или бутанол, или ароматического углеводорода, такого как толуол, или N-метилпирролидина, необязательно, в присутствии подходящей кислоты, например, неорганической кислоты, такой как хлористоводородная или серная кислота, или органической кислоты, такой как уксусная или муравьиная кислота (или подходящая кислота Льюиса), и при температуре от 0° С до температуры дефлегмации, предпочтительно, при температуре дефлегмации; или ii) в стандартных условиях Бухвальда (Buchwald) (например, см. J.Am.Chem.Soc., 118, 7215; J.Am. Chem.Soc., 119, 8451; J.Org.Chem., 62, 1568 и 6066), например, в присутствии ацетата палладия, в подходящем растворителе, например, в ароматическом растворителе, таком как толуол, бензол или ксилол, с подходящим основанием, например, неорганическим основанием, таким как карбонат цезия, или органическим основанием, таким как трет-бутоксид калия, в присутствии подходящего лиганда, такого как 2,2’-бис(дифенилфосфин)-1,1’-бинафтил, и при температуре в интервале от 25 до 80° С.
Пиримидины формулы (II) могут быть получены по схеме I

где один из М и Q является уходящей группой X, как определено выше, а другой представляет собой металлический реагент Y, как определено выше.
Условия перекрестного сочетания хорошо известны из уровня техники. Подходящие условия включают, например, условия, описанные ниже в пункте b).
Если цикл А представляет собой имидазо[1,2а]пирид-3-ил, то соединения формулы (II) могут быть также получены по схеме II.
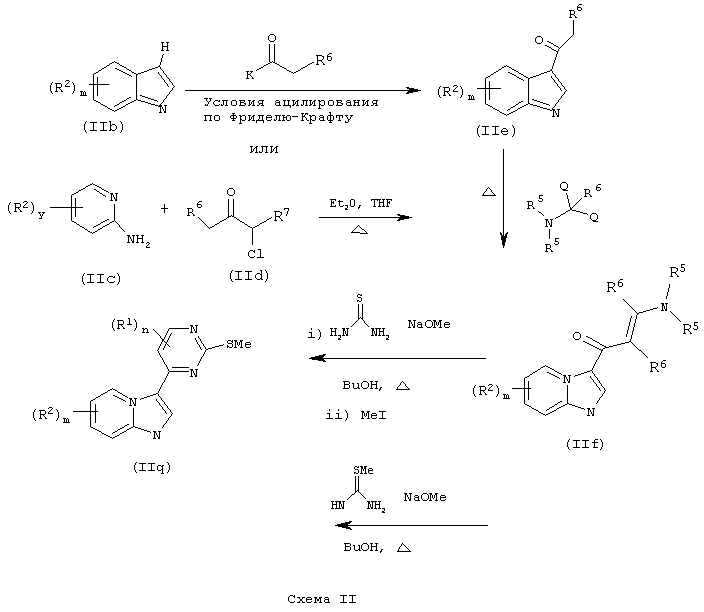
К обозначает хорошую уходящую группу (например, C1-6-алканоилокси), R6 такой, как определено выше, у равен 0-4, R7 обозначает водород или R2; Q обозначает хорошую уходящую группу (например, C1-6алкокси) и R5 является таким, как определено выше.
Если цикл А представляет собой пиразол[2,3а]пирид-3-ил, то соединения формулы (II) могут быть также получены по схеме III.
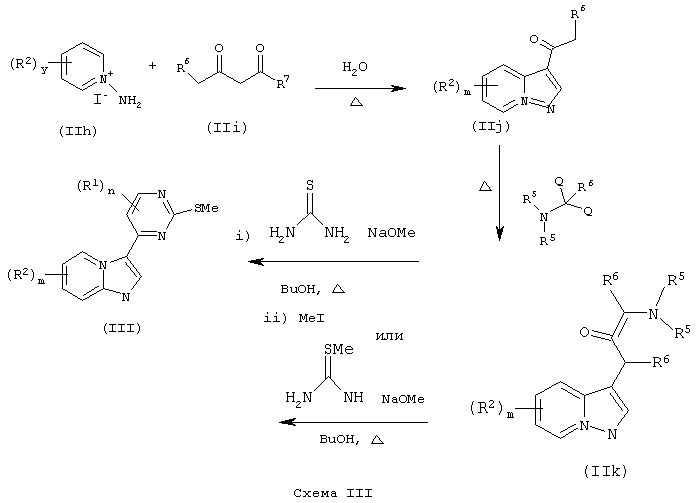
где R5, R6 и R7 такие, как определены выше.
Соединения формулы (IIf) или (IIk) могут быть далее модифицированы с получением соединений формулы (IIn):
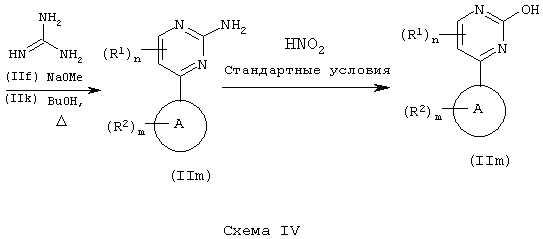
Специалисту в данной области должно быть понятно, что соединения формулы (IIn) могут быть дополнительно модифицированы с помощью стандартных реакций модификации функциональных групп, известных из уровня техники, с получением соединений формулы (II), где L представляет собой другую уходящую группу, например хлор, бром, тозил и мезил.
Соединения формул (IIа), (IIb), (IIc), (IId), (IIh), (IIi) и (III) представляют собой коммерчески доступные или известные из литературы соединения, или они могут быть получены с помощью стандартных способов, известных из уровня техники.
b) Соединения формулы (IV) и соединения формулы (V) могут взаимодействовать друг с другом в стандартных условиях перекрестного сочетания. Примерами таких условий являются присутствие катализатора, например, металлического катализатора, такого как тетракис(трифенилфосфин)палладий(0), хлорид палладия(II), бромид палладия(II), хлорид никеля(II), бромид никеля(II) или хлорид бис(трифенилфосфин)никеля(II), присутствие подходящего инертного растворителя или разбавителя, например тетрагидрофурана, 1,4-диоксана, 1,2-диметоксиэтана, бензола, толуола, ксилола, метанола или этанола. Реакцию предпочтительно проводят в присутствии подходящего основания, такого как, например, карбонат натрия или карбонат калия, пиридин, 4-диметиламинопиридин, триэтиламин или морфолин, и обычно при температуре, находящейся в интервале, например, от 10 до 250° С, предпочтительно в интервале от 60 до 120° С.
Соединения формулы (IV) могут быть получены по схеме V

Соединения формулы (V) представляют собой коммерчески доступные или известные из литературы соединения, или они могут быть получены с помощью стандартных способов, известных из уровня техники.
c) соединения формулы (VI) и соединения формулы (VII) взаимодействуют друг с другом в подходящем растворителе, таком как N-метилпирролидинон или бутанол при температуре, находящейся в интервале 100-200° С, предпочтительно в интервале 150-170° С. Реакцию предпочтительно проводят в присутствии подходящего основания, такого как, например, метоксид натрия или карбонат калия.
Соединения формул (VI) и (VII) представляют собой коммерчески доступные или известные из литературы соединения, или они могут быть получены с помощью стандартных способов, известных из уровня техники, или соединения формулы (VII) могут быть получены способом, подобным описанному выше для (IIf) и (IIk).
Следует понимать, что некоторые из различных циклических заместителей в соединениях настоящего изобретения могут быть введены с помощью стандартных реакций ароматического замещения, или могут быть образованы путем общепринятых модификаций функциональных групп, либо до, либо сразу после проведения упомянутых выше способов, и, как таковые, включены в аспект способа получения в соответствии с настоящим изобретением. Такие реакции и модификации включают, например, введение заместителя с помощью реакции ароматического замещения, восстановления заместителей, алкилирования заместителей и окисления заместителей. Реагенты и условия реакций для таких процедур хорошо известны в химии. Конкретные примеры реакций ароматического замещения включают введение нитрогруппы с использованием концентрированной азотной кислоты, введение ацильной группы с использованием, например, галогенангидрида кислоты и кислоты Льюиса (такой как трихлорид алюминия) в условиях Фриделя-Крафтса; введение алкильной группы с использованием алкилгалогенида и кислоты Льюиса (такой как трихлорид алюминия) в условиях Фриделя-Крафтса; а также введение галогенной группы. Конкретные примеры модификаций включают восстановление нитрогруппы до аминогруппы, например, путем каталитического гидрирования, с использованием никелевого катализатора или обработки железом в присутствии хлористоводородной кислоты при нагревании; окисление алкилтио до алкилсульфинила или алкилсульфонила.
Следует также понимать, что в некоторых упомянутых здесь реакциях может быть необходимо/желательно защищать в соединениях чувствительные группы. Случаи, в которых защита является необходимой или желательной, а также подходящие методы защиты известны специалистам в данной области. Традиционные защитные группы могут использоваться в соответствии со стандартными практическими руководствами (например, см. T.W.Green, Protective Groups in Organic Synthesis, John Wiley and Sons, 1991). Таким образом, если реагенты включают такие группы, как амино, карбокси или гидрокси, защита такой группы в некоторых из упомянутых реакций может являться желательной.
Подходящей защитной группой для аминогруппы или алкиламиногруппы является, например, ацильная группа, например, алканоильная группа, такая, как ацетил, алкоксикарбонильная группа, например, метоксикарбонильная, этоксикарбонильная или трет-бутоксикарбонильная группа, арилметоксикарбонильная группа, например, бензилоксикарбонильная, или ароильная группа, например, бензоильная. Условия, в которых проводят удаление вышеуказанных защитных групп, необходимо варьировать в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или алкоксикарбонильная группа, или ароильная группа может быть удалена, например, путем гидролиза в присутствии подходящего основания, такого как гидроксид щелочного металла, например гидроксид лития или натрия. Альтернативно, ацильная группа, такая как трет-бутоксикарбонильная группа, может быть удалена, например, путем обработки подходящей кислотой, такой как хлористоводородная, серная или фосфорная кислота, или трифторуксусная кислота, и арилметоксикарбонильная группа, такая как бензилоксикарбонильная группа, может быть удалена, например, путем гидрирования над таким катализатором, как палладий на угле, или путем обработки кислотой Льюиса, например, трис-трифторацетатом бора. Подходящей альтернативной защитной группой для первичной аминогруппы является, например, фталоильная группа, которая может быть удалена путем обработки алкиламином, например диметиламинопропиламином, или гидразином.
Подходящей защитной группой для гидроксильной группы является, например, ацильная група, например, алканоильная группа, такая как ацетильная, ароильная группа, например, бензоильная, или арилметильная группа, например, бензильная. Условия, в которых проводят удаление вышеуказанных защитных групп, необходимо варьировать в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или ароильная группа может быть удалена, например, путем гидролиза в присутствии подходящего основания, такого как гидроксид щелочного металла, например гидроксид лития или натрия. Альтернативно, арилметильная группа, такая как бензильная группа, может быть удалена, например, путем гидрирования над таким катализатором, как палладий на угле.
Подходящей защитной группой для карбоксильной группы является, например, этерифицирующая группа, например, метильная или этильная группа, которая может быть удалена, например, путем гидролиза в присутствии основания, такого как гидроксид натрия, или, например, трет-бутильная группа, которая может быть удалена, например, путем обработки кислотой, например органической кислотой, такой как трифторуксусная кислота, или, например, бензильная группа, которая может быть удалена, например, путем гидрирования над таким катализатором, как палладий на угле.
Защитные группы могут быть удалены на любой подходящей стадии синтеза с помощью традиционных методов, хорошо известных в области химии.
Как указано ранее, соединения, определенные в настоящем изобретении, обладают активностью против пролиферации клеток, такой как противораковая активность, которая, как полагают, является следствием CDK-ингибиторной активности соединения. Данные свойства могут быть оценены, например, с помощью приведенной ниже процедуры:
Анализ
Используемые аббревиатуры:
HEPES обозначает N-[2-гидроксиэтил]пиперазин-N’-[2-этансульфоновую кислоту]
DTT обозначает дитиотретиол
PMSF обозначает фенилметилсульфонилфторид.
Соединения тестируют в киназном анализе in vitro в 96-луночном формате, используя сцинтилляционный проксимальный анализ (Scintillation Proximity Assay) (SPA получают от Amersham) для измерения включения [γ -33-Р]-аденозинтрифосфата в исследуемый субстрат (GST-ретинобластомный белок; GST-Rb). В каждую лунку помещают тестируемое соединение (разбавленное в DMSO и воде до нужных концентраций) и в контрольные лунки помещают либо росковитин, как контроль ингибирования, либо DMSO, как положительный контроль.
Приблизительно 0,2 мкл частично очищенного фермента Е (количество зависит от активности фермента), разбавленного 25 мкл инкубационного буфера, добавляют в каждую лунку, затем добавляют 20 мкл смеси GST-Rb/ATP/АТР33 (содержащей 0,5 мкг GST-Rb и 0,2 мкМ АТР и 0,14 мкКи [у-33-Р]-аденозинтрифосфата в инкубационном буфере), полученную смесь осторожно встряхивают, после чего инкубируют при комнатной температуре в течение 60 минут.
В каждую лунку затем добавляют 150 мкл останавливающего раствора (содержащего 0,8 мг/лунку гранул белка A-PVT SPA (Amersham)), 20 пМ/лунку кроличьих IgG против глутатионтрансферазы (полученных от Molecular Probes), 61 мМ EDTA и 50 мМ HEPES, pH 7,5, содержащего 0,05% азида натрия.
Планшеты закрывают приспособлением для заклеивания планшетов Topseal-S, оставляют на два часа, затем центрифугируют при 2500 об/мин, 1124× g, в течение 5 минут. Планшеты прочитывают на Topcount в течение 30 секунд на лунку.
Инкубационный буфер, используемый для разбавления смесей фермента и субстрата, содержит 50 мМ HEPES, рН 7,5, 10 мМ МnСl2, 1 мМ DTT, 100 мкМ ванадата натрия, 100 мкМ NaF, 10 мМ глицерофосфата натрия, BSA (конечная концентрация 1 мг/мл).
Аналитический субстрат
В данном анализе используют только часть ретинобластомного белка (Science, 1987 Mar.13; 235(4794):1394-1399; Lee W.H., Bookstein R., Hong F., Young L.J., Shew J.Y., Lee E.Y.), гибридизованного с GST-маркером. Проводят PCR ретинобластомного гена, кодирующего аминокислоты 379-928 (полученного из ретинобластомной плазмиды АТСС pLRbRNL), и последовательность клонируют в гибридном векторе pGEX 2T (Smith D.B. and Johnson, K.S. Gene, 67, 31 (1988); который содержит tac промотор для индукции экспрессии, внутренний lac Iq ген для применения в любом хозяине E.coli, и кодирующий участок для расщепления тромбином - полученный от Pharmacia Biotech), который используют для амплификации аминокислот 792-928. Данную последовательность снова клонируют в векторе pGEX 2T.
Полученную таким образом ретинобластомную последовательность 792-928 экспрессируют в E.coli (клетки BL21 (DE3) pLysS), используя стандартные методы индукции экспрессии, и очищают, как описано ниже.
Пасту E.coli. ресуспендируют в 10 мл/г буфера NETN (50 мМ Tris, рН 7,5, 120 мМ NaCl, 1 мМ EDTA, 0,5% об/об NP-40, 1 мМ PMSF, 1 мкг/мл лейпептина, 1 мкг/мл апротинина и 1 мкг/мл пепстатина) и обрабатывают ультразвуком в течение 2× 45 секунд на 100 мл гомогената. После центрифугирования супернатант загружают на колонку с 10 мл глутатион-сефарозы (Pharmacia Biotech, Herts, UK) и промывают буфером NETN. После промывания киназным буфером (50 мМ HEPES, рН 7,5,10 мМ MgCl2, 1 мМ DTT, 1 mm PMSF, 1 мкг/мл лейпептина, 1 мкг/мл апротинина и 1 мкг/мл пепстатина) белок элюируют 50 мМ раствором восстановленного глутатиона в киназном буфере. Фракции, содержащие GST-Rb(792-927) объединяют и диализуют в течение ночи против киназного буфера. Конечный продукт анализируют методом SDS (додецилсульфат натрия)-PAGE (полиакриламидный гель), используя 8-16% трис-глициновые гели (Novex, San Diego, USA).
CDK2 и циклин Е
Открытые рамки считывания CDK2 и циклина Е выделяют методом PCR с обратной транскриптазой, используя клетки HeLa, a также мРНК активированных Т-клеток в качестве матрицы, и клонируют в экспрессионном векторе насекомых pVL1393 (полученном от Invitrogen, номер по каталогу 1995: V1392-20). Затем проводят двойную экспрессию CDK2 и циклина Е [используя стандартную методику соинфицирования вируса Baculogold] в клеточной системе насекомых SF21 (клетки Spodoptera Frugiperda, полученные из ткани яичника Fall Army Worm - коммерчески доступные).
Пример получения Циклина E/CDK2
В следующем примере приведены детали получения циклина E/CDK2 в клетках SF21 (в ТС100+10% FBS(TCS)+0,2% Pluronic), подвергавшихся двойному инфицированию с MOI (множественность заражения) 3 для каждого вируса с циклином Е и CDK2.
Клетки SF21, выращенные в среде в роллер-флаконе до концентрации 2,33× 106 клеток/мл, используют для инокуляции 10× 500 мл роллер-флаконов при 0,2× 10Е6 клеток/мл. Роллер-флаконы инкубируют на роллерной установке при 28° С.
Через 3 дня (72 ч) клетки считают и среднее для 2 флаконов определяют как 1,86× 10Е6 клеток/мл (99% жизнеспособных). Затем культуры подвергают повторному инфицированию вирусами с MOI3 для каждого вируса.
Вирусы смешивают вместе перед добавлением к культурам и культуры возвращают на роллерную установку при 28° С.
Через 2 дня (48 ч) после инфицирования собирают 5 литров культуры. Общий клеточный счет в момент сбора составляет 1,58× 10Е6 клеток/мл (99% жизнеспособных). Клетки центрифугируют при 2500 об/мин, 30 мин, 4° С, в Heraeus Omnifuge 2,0 RS порциями объемом 250 ml. Супернатант отбрасывают.
Частичная совместная очистка Cdk2 и циклина Е
Клетки Sf21 ресуспендируют в буфере для лизиса (50 мМ Tris рН 8,2, 10 мМ MgCl2, 1 мМ DTT, 10 мМ глицерофосфата, 1,1 мМ ортованадата натрия, 0,1 мМ NaF, 1 мМ PMSF, 1 мкг/мл лейпептина и 1 мкг/мл апротинина) и гомогенизируют в течение 2 минут в гомогенизаторе Dounce объемом 10 мл. После центрифугирования супернатант загружают на анионообменную колонку Poros HQ/M 1,4/100 (РЕ Biosystems, Hertford, UK). Cdk2 и циклин Е совместно элюируются в начале градиента 0-1М NaCl (градиент осуществляют в буфере для лизиса, не содержащем ингибиторов протеаз) на протяжении 20 объемов колонки. Совместное элюирование контролируют с помощью вестерн-блоттинга, используя антитела против Cdk2 и против циклина Е (Santa Cruz Biotechnology, California, US).
По аналогии можно провести анализ ингибирования CDK4 и CDK6. CDK2 (EMBL № по каталогу Х62071) может использоваться вместе с циклином А или циклином Е (см. EMBL № по каталогу М73812), более подробно такие анализы описаны в международной публикации РСТ №WO 99/21845, разделы которой, относящиеся к разделам биохимических и биологических анализов, включены в данный документ в качестве ссылки.
Хотя фармакологические свойства соединений формулы (I) варьируются в зависимости от структурных изменений, в основном активность соединений формулы (I) может проявляться при концентрациях IC50 или дозах в интервале от 250 мкМ до 1 нМ.
При тестировании в описанном выше анализе CDK2-ингибиторной активности in vitro было обнаружено, что для соединения примера 11 IC50=0,19 мкМ, а для соединения примера 12 IC50=0,17 мкМ.
Активность соединений настоящего изобретения in vivo может быть оценена с помощью стандартных методов, например путем измерения ингибирования клеточного роста и оценки цитотоксичности.
Ингибирование клеточного роста может быть измерено путем окрашивания клеток Сульфородамином С (SRB), флуоресцентным красителем, который окрашивает белки и, следовательно, позволяет оценить количество белка (т.е. клеток) в лунке (см. Boyd, M.R. (1989) Status of the NCl preclinical antitumour drug discovery screen, Prin.Prac.Oncol., 10:1-12). Таким образом, далее приводятся детали измерения ингибирования клеточного роста.
Клетки помещают в соответствующей среде объемом 100 мл в 96-луночные планшеты; для MCF-7, SK-UT-1B и SK-UT-1 в качестве среды используют модифицированную по Дульбекко среду Игла. Клетки оставляют прикрепляться в течение ночи, затем добавляют ингибиторные соединения в различных концентрациях с максимальной концентрацией DMSO 1% (об./об.). Анализируя контрольный планшет, получают значение для клеток перед получением дозы ингибиторного соединения. Клетки ингибируют при 37° С (5% СO2) в течение 3 дней.
По окончании трех дней к планшетам добавляют ТСА до конечной концентрации 16% (об./об.). Затем планшеты инкубируют при 4° С в течение 1 часа, супернатант удаляют и планшеты промывают водопроводной водой. После просушивания добавляют 100 мл красителя SRB (0,4% SRB в 1% уксусной кислоте) на 30 минут при 37° С. Избыток SRB удаляют и планшеты промывают 1% уксусной кислотой. Связанный с белком SRB солюбилизируют в 10 мМ Tris pH 7,5 и встряхивают в течение 30 минут при комнатной температуре. Значения оптической плотности прочитывают при 540 нм и концентрацию ингибитора, вызывающую 50% ингибирование роста, определяют из полулогарифмической зависимости поглощения от концентрации ингибитора. Концентрация соединения, которая уменьшает оптическую плотность ниже значения, полученного для клеток, помещенных в планшет в начале эксперимента, дает значение токсичности.
Обычно значения IC50 для соединений данного изобретения при тестировании в SRB анализе находятся в интервале от 1 мМ до 1 нМ.
В соответствии со следующим аспектом данного изобретения предлагается фармацевтическая композиция, которая включает пиримидиновое производное формулы (I) или его фармацевтически приемлемую соль, или сложный эфир, способный к гидролизу in vivo, как указано выше, в сочетании с фармацевтически приемлемым разбавителем или носителем.
В соответствии со следующим аспектом данного изобретения предлагается фармацевтическая композиция, которая включает соединение формулы (I) или его фармацевтически приемлемую соль, или сложный эфир, способный к гидролизу in vivo, как указано выше, в сочетании с фармацевтически приемлемым разбавителем или носителем.
Композиция может быть представлена в форме, подходящей для перорального введения, например, в виде таблетки или капсулы, для парентеральной инъекции (включая внутривенную, подкожную, внутримышечную, внутрисосудистую, или вливание) в виде стерильного раствора, суспензии или эмульсии, для местного введения в виде мази или крема, или для ректального введения в виде свечи.
В основном, вышеупомянутые композиции могут быть получены традиционным способом, с использованием традиционных эксципиентов.
Соединение формулы (I) обычно вводят теплокровному животному в единичной дозе, находящейся в интервале 5-5000 мг на квадратный метр площади тела животного, т.е. приблизительно 0,1-100 мг/кг, данная доза обычно является терапевтически эффективной. Разовая лекарственная форма, такая как таблетка или капсула, обычно содержит, например, 1-250 мг активного ингредиента. Предпочтительно используют дневную дозу, находящуюся в интервале 1-50 мг/кг. Однако дневную дозу необходимо варьировать в зависимости от хозяина, подлежащего лечению, конкретного способа введения, а также тяжести заболевания, подлежащего лечению. Соответственно, оптимальный дозировочный режим может быть установлен врачом, лечащим конкретного пациента.
В соответствии со следующим аспектом данного изобретения предлагается соединение формулы (I) или его фармацевтически приемлемая соль, или сложный эфир, способный к гидролизу in vivo, как указано выше, для применения в терапевтическом способе лечения организма человека или животного.
Авторы данного изобретения обнаружили, что соединения, определенные в настоящем изобретении, или их фармацевтически приемлемые соли, или сложные эфиры, способные к гидролизу in vivo, являются эффективными ингибиторами клеточного цикла (антипролиферационными агентами), полагают, что данное свойство является следствием их ингибиторной активности по отношению к CDK. Соответственно ожидают, что соединения в соответствии с изобретением могут быть полезными при лечении заболеваний или медицинских состояний, опосредованных только или частично ферментами CDK, т.е. данные соединения могут использоваться для достижения ингибиторного эффекта по отношению к CDK у теплокровных животных, нуждающихся в таком лечении. Таким образом, соединения в соответствии с изобретением обеспечивают способ лечения пролиферации злокачественных клеток, характеризующийся ингибированием ферментов CDK, т.е. данные соединения могут использоваться для продуцирования антипролиферативного эффекта, опосредованного только, или частично ингибированием CDKs. Полагают, что такое соединение в соответствии с изобретением обладает широким диапазоном противораковых свойств, так как CDKs участвуют во многих распространенных раковых заболеваниях человека, таких как лейкемия, а также рак груди, легкого, ободочной кишки, прямой кишки, желудка, простаты, мочевого пузыря, поджелудочной железы и яичников. Таким образом, полагают, что соединение в соответствии с изобретением обладает противораковой активностью по отношению к данным раковым заболеваниям. Кроме того, полагают, что соединение в соответствии с изобретением может обладать активностью против ряда лейкемий, лимфоидных злокачественных заболеваний и твердых опухолей, таких как карциномы и саркомы, в таких тканях, как печень, почки, простата и поджелудочная железа. Полагают, что такие соединения в соответствии с изобретением существенно замедляют рост первичных и рецидивных твердых опухолей, например, ободочной кишки, груди, простаты, легких и кожи. Более конкретно, полагают, что такие соединения в соответствии с изобретением или их фармацевтически приемлемые соли, или сложные эфиры, способные к гидролизу in vivo, ингибируют рост таких первичных и рецидивных твердых опухолей, которые связаны с CDKs, особенно тех опухолей, рост и распространение которых в значительной степени зависят от CDKs, включая, например, некоторые опухоли ободочной кишки, груди, простаты, легких, вульвы и кожи.
Далее полагают, что соединение в соответствии с настоящим изобретением может обладать активностью против других пролиферационных заболеваний в широком диапазоне других болезненных состояний, включающих лейкемии, фибропролиферативные и дифференционные нарушения, псориаз, ревматоидный артрит, саркому Калоши, гемангиому, острые и хронические нефропатии, атерому, атеросклероз, артериальный повторный стеноз, аутоиммунные заболевания, острое и хроническое воспаление, костные болезни и глазные болезни с пролиферацией сосудов сетчатки.
Таким образом, в соответствии с данным аспектом изобретения предлагается соединение формулы (I) или его фармацевтически приемлемая соль, или сложный эфир, способный к гидролизу in vivo, как определено выше, для применения в качестве лекарственного препарата; а также применение соединения формулы (I) или его фармацевтически приемлемой соли, или сложного эфира, способного к гидролизу in vivo, как определено выше, в промышленном получении лекарственного препарата, предназначенного для применения при получении ингибиторного эффекта по отношению к клеточному циклу (антипролиферативного эффекта) у теплокровного животного, такого, как человек. В особенности, ингибиторный эффект достигается путем предотвращения входа в S-фазу или развития S-фазы, в результате ингибирования CDK2, CDK4 и/или CDK6, особенно CDK2.
В соответствии со следующим аспектом данного изобретения предлагается применение соединения формулы (I) или его фармацевтически приемлемой соли, или сложного эфира, способного к гидролизу in vivo, как определено выше, в промышленном получении лекарственного препарата, предназначенного для применения при лечении раковых заболеваний (твердых опухолей и лейкемий), фибропролиферативных и дифференционных нарушений, псориаза, ревматоидного артрита, саркомы Капоши, гемангиомы, острых и хронических нефропатий, атеромы, атеросклероза, артериального повторного стеноза, аутоиммунных заболеваний, острого и хронического воспаления, костных болезней и глазных болезней с пролиферацией сосудов сетчатки.
В соответствии со следующим аспектом изобретения, предлагается способ получения ингибиторного эффекта по отношению к клеточному циклу (антипролиферативного эффекта) у теплокровного животного, такого, как человек, нуждающегося в таком лечении, который включает введение упомянутому животному эффективного количества соединения, как определено непосредственно перед данным абзацем. В особенности, ингибиторный эффект достигается путем предотвращения входа в S-фазу или развития S-фазы, в результате ингибирования CDK2, CDK4 и/или CDK6, особенно, CDK2.
Как указано выше, размер дозы, необходимой для лечения или профилактики конкретного клеточно-пролиферативного заболевания, необходимо варьировать в зависимости от подлежащего лечению хозяина, способа введения и тяжести заболевания, подлежащего лечению. Предусмотренная разовая доза находится в интервале, например, 1-100 мг/кг, предпочтительно 1-50 мг/кг.
CDK-ингибиторная активность, определенная выше, может применяться как самостоятельная терапия, или может включать, кроме соединения данного изобретения, одно или несколько других веществ и/или лечений. Такое объединенное лечение может быть достигнуто путем одновременного, последовательного или раздельного введения индивидуальных компонентов лечения. В области медицинской онкологии обычной практикой является применение сочетания различных форм лечения для каждого пациента с раковым заболеванием. В медицинской онкологии другим(и) компонентом(компонентами) такого объединенного лечения, в добавление к лечению ингибиторами клеточного цикла, определенными выше, могут являться: хирургия, радиотерапия или химиотерапия. Такая химиотерапия может охватывать три основные категории терапевтических агентов:
(i) другие агенты, ингибирующие клеточный цикл, механизмы действия которых совпадают с механизмами, определенными выше, или отличаются от них;
(ii) цитостатические агенты, такие как антиэстрогены (например, тамоксифен, торемифен, ралоксифен, дролоксифен, иодоксифен), прогестогены (например, ацетат мегестрола), ингибиторы ароматаз (например, анастрозол, летразол, воразол, экземестан), антипрогестогены, антиандрогены (например, флютамид, нилютамид, бикалютамид, ацетат ципротерона), агонисты и антагонисты LHRH (например, ацетат госерелина, люпролид), ингибиторы тестостерон-5α -дигидроредуктазы (например, финастерид), антиинвазийные агенты (например, ингибиторы металлопротеиназ, такие как маримастат, и ингибиторы функции рецептора урокиназного активатора плазминогена) и ингибиторы функции факторов роста (такие факторы роста включают, например, тромбоцитарный фактор роста и гепатоцитарный фактор роста, а такие ингибиторы включают антитела против фактора роста, антитела против рецептора фактора роста, ингибиторы тирозинкиназы и ингибиторы серин/треонинкиназы); и
(iii) антипролиферативные/антинеопластические лекарственные препараты и их сочетания, применяющиеся в медицинской онкологии, такие как антиметаболиты (например, антифолаты, такие как метотрексат, фторпиримидины, такие как 5-фторурацил, пурин и аналоги аденозина, арабинозид цитозина); противоопухолевые антибиотики (например, антрациклины, такие как доксорубицин, дауномицин, эпирубицин и идарубицин, митомицин-С, дактиномицин, митрамицин); производные платины (например, цисплатин, карбоплатин); алкилирующие агенты (например, азотистый иприт, мелфалан, хлорамбуцил, бусульфан, циклофосфамид, ифосфамид, нитрозамочевины, тиотепа); антимитотические агенты (например, алкалоиды vinca, такие как винкристин, и таксоиды, такие как таксол, таксотер); ингибиторы топоизомеразы (например, эпиподофиллотоксины, такие как этопозид и тенипозид, амсакрин, топотекан). В соответствии с данным аспектом изобретения предлагается фармацевтический продукт, включающий соединение формулы (I), как определено выше, и добавочное противоопухолевое вещество, как определено выше, для консолидированного лечения ракового заболевания.
Кроме применения в терапевтической медицине, соединения формулы (I) и их фармацевтически приемлемые соли могут также использоваться в качестве фармакологических инструментов в разработке и стандартизации аналитических систем in vitro и in vivo для оценки эффектов ингибиторов активности клеточного цикла у лабораторных животных, таких как кошки, собаки, кролики, обезьяны, крысы и мыши, как часть изыскания новых терапевтических агентов.
К другим, описанным выше фармацевтическим композициям, процессам, способам применения и промышленного производства лекарственных препаратов также применимы альтернативные и предпочтительные воплощения соединений данного изобретения, описанные в данном документе.
Примеры
Далее данное изобретение иллюстрируется с помощью нижеследующих не ограничивающих примеров, в которых, если не указано обратное:
(i) температуры приведены в градусах Цельсия (° С); операции проводят при комнатной температуре или температуре окружающей среды, то есть при температуре в интервале 18-25° С;
(ii) органические растворы сушат над безводным сульфатом магния; упаривание растворителя осуществляют с помощью роторного испарителя при пониженном давлении (600-4000 Па; 4,5-30 мм рт.ст.) и температуре бани не выше 60° С;
(iii) хроматография означает флэш-хроматографию на силикагеле; тонкослойную хроматографию (ТСХ) проводят на пластинах с силикагелем; если упоминается колонка с оксидом кремния Bond Elut, то имеют в виду колонку, содержащую 10 г или 20 г частиц оксида кремния размером 40 микрон, причем оксид кремния содержится в одноразовом шприце объемом 60 мл и поддерживается пористым диском, шприц получают от Varian, Harbor City, California, USA под названием "Mega Bond Elut SI", "Mega Bond Elut" является торговой маркой;
(iv) в основном протекание реакций отслеживают с помощью ТСХ, а время реакций приводят только для иллюстрации;
(v) конечные продукты имеют удовлетворительные спектры протонного ядерного магнитного резонанса (ЯМР) и/или масс-спектральные данные;
(vi) выходы приведены только для иллюстрации и не являются в точности такими, какие могут быть получены в результате тщательно проведенного процесса; получение повторяют, если требуется большее количество вещества;
(vii) если присутствуют данные ЯМР, они приводятся в виде значений дельта для основных характеристических протонов, в миллионных долях (м.д.) по отношению к внутреннему стандарту тетраметилсилану (TMS), значения определяют при 300 МГц, используя пердейтерий-диметилсульфоксид (DMSO-d6) в качестве растворителя, если не указано иначе;
(viii) химические символы имеют обычные значения; используются единицы и символы системы СИ;
(ix) соотношения растворителей приводятся как объем: объем (об./об.); и
(х) масс-спектры снимают при энергии электронов, равной 70 электрон-вольт, в режиме химической ионизации (CI), используя систему прямого ввода; где указано, ионизацию вызывают электронным ударом (EI), бомбардировкой быстрыми атомами (FAB) или электрораспылением (ESP); приводятся значения m/z; обычно, приводятся только ионы, которые определяют исходную массу;
(xi) если не оговорено иначе, соединения, содержащие асимметрично замещенный атом углерода и/или серы, не разделяют;
(xii) если синтез описывают как аналогичный описанному в предыдущем примере, используемые количества являются в миллимолярном отношении эквивалентными количествам, приведенным в предыдущем примере;
(xvi) используются следующие сокращения:
NMP 1-метил-2-пирролидинон;
DMF N,N-диметилформамид;
DMFDMA N,N-диметилформамиддиметилацетил;
DMSO диметилсульфоксид;
THF тетрагидрофуран; и
ЕА элементный анализ.
ПРИМЕР 1. 2-(3-Хлоранилин)-4-(2-метилимидазо[1,2а]пирид-3-ил)пиримидин
Гидрид натрия (236 мг, 60% суспензии в минеральном масле, 5,9 ммоль) добавляют к раствору 3-хлоранилина (496 мл, 4,7 ммоль) в NMP (10 мл) в атмосфере азота. Смесь перемешивают в течение 30 минут при температуре окружающей среды и добавляют раствор 4-(2-метилимидазо[1,2а]пирид-3-ил)-2-метилтиопиримидина (способ 1) (600 мг, 2,3 ммоль) в NMP (2 мл). Смесь нагревают при 150° С в течение 3 часов. Реакционную смесь оставляют охлаждаться, разбавляют водой и экстрагируют этилацетатом. Объединенные экстракты сушат и летучие вещества удаляют упариванием. Остаток очищают хроматографией, элюируя смесью этилацетат/гексан (1:1), увеличивая полярность до этилацетат/метанол (97:3). Очищенный продукт растирают с эфиром и гексаном, отделяют фильтрацией и сушат, получая указанное в заголовке соединение (159 мг, 21%).
ЯМР: 2,62(с, 3Н), 6,98-7,04(м, 2Н), 7,12(д, 1Н), 7,25 (дд, 1Н), 7,42(дд, 1Н), 7,59-7,64(м, 2Н), 8,02(с, 1Н), 8,55(д, 1Н), 9,72(д, 1Н), 9,84(с, 1Н).
ПРИМЕРЫ 2-12
Следуя способу примера 1 и используя соответствующие исходные вещества, получают следующие соединения.
ПРИМЕР 13. 2-[4-(3-Димеламино-2-гидроксипропокси)-анилин]-4-(имидазо[1,2а]пирид-3-ил)пиримидин
Смесь 4-(3-диметиламино-2-гидроксипропокси)анилина (497 мг, 1,76 ммоль) (способ 11) и цианамида (185 мг, 4,4 ммоль) в NMP (1 мл) нагревают при 160° С в течение 30 минут. Затем добавляют смесь 3-(3-диметиламинопроп-2-ен-1-оил)имидазо[1,2а]-пиридина (способ 5) (400 мг, 1,76 ммоль) и метоксида натрия (183 мг, 3,5 ммоль) в 1-бутаноле (10 мл) и полученную смесь кипятят с обратным холодильником в течение 3 часов. Смесь оставляют охлаждаться и остаток очищают хроматографией, элюируя смесью этилацетат/метанол (97:3, увеличивая полярность до 90:10), получая указанное в заголовке соединение (30 мг, 4%). ЯМР: 2,35(с,6Н), 2,40-2,63(м, 2Н), 3,82-4,02(м, 3Н), 6,90(д, 2Н), 7,06(дд, 1Н), 7,30(д, 1Н), 7,50(дд, 1Н), 7,59(с, 2Н), 7,74 (д, 1Н), 8,38(д, 1Н), 8,58(с, 1Н), 9,42(с, 1Н).
m/z: 405 [МН]+
ПРИМЕРЫ 14-15
Следуя способу примера 13 и используя соответствующие исходные вещества, получают следующие соединения.
2Продукт очищают хроматографией, элюируя смесью дихлорметан/метанол/триэтиламин (96:4:0,5), и перекристаллизо-вывают из смеси ацетонитрил/метанол.
ПРИМЕРЫ 16-36
Следующие примеры соединений получают, очищают и характеризуют с помощью нижеприведенного общего способа:
Бис(триметилсилил)амид натрия (2,05 мл 1М раствора в THF, 2,05 ммоль) добавляют к раствору анилина (1,65 ммоль) в NMP (1,5 мл) в атмосфере азота. Смесь перемешивают в течение 30 минут при температуре окружающей среды и добавляют раствор 4-(имидазо[1,2а]пирид-3-ил)-2-метилтиопиримидина (способ 4) (200 мг, 0,83 ммоль) в NMP (1 мл). Реакционную смесь нагревают при 150° С в течение 2,5 часов. Растворитель и летучие вещества удаляют упариванием и остаток очищают хроматографией, элюируя этилацетатом, затем смесью этилацетат/метанол (97:3) и в конце смесью этилацетат/метанол (97:3). Продукты реакции характеризуют методом ВЭЖХ на колонке 4,6 мм × 10 см Hichrom RPB 100А, элюируя смесью вода/ацетонитрил/муравьиная кислота (95:5:0,1 в течение 1,5 минут, затем в условиях 10-минутного градиента доводят до 5:95:0,1) со скоростью потока 1,0 мл/минуту, детектируют при 254 нм (ширина полосы 10 нм).
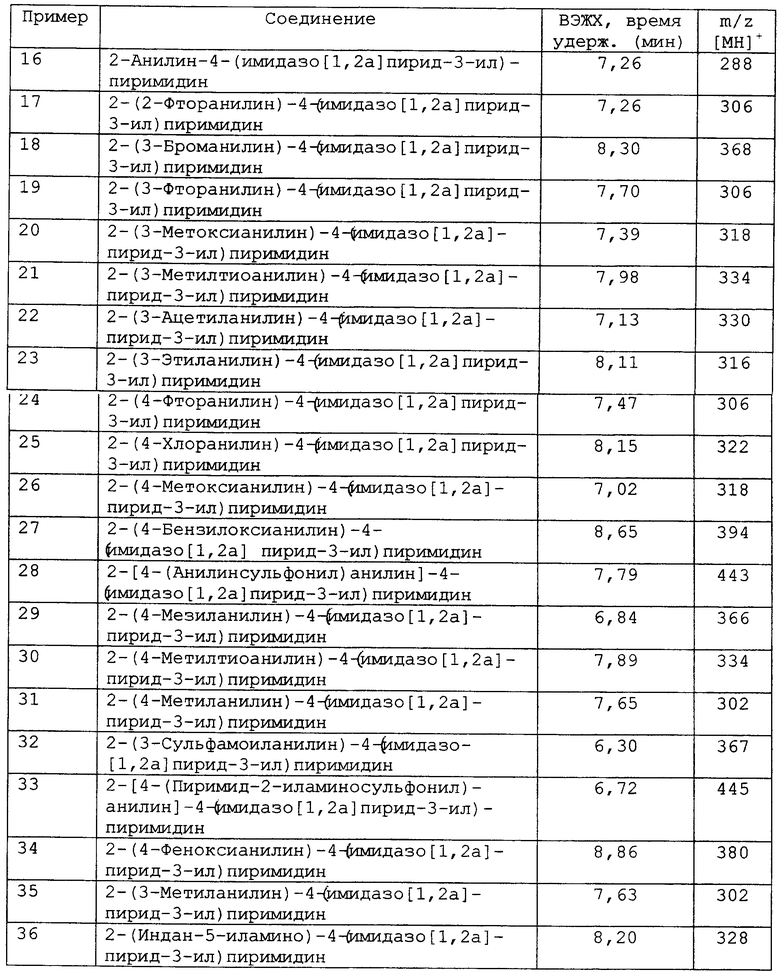
ПРИМЕР 37
2-(3-Хлоранилин)-4-(2,5-диметилимидазо-[1,2а]пирид-3-ил)пиримидин
2-Метилтио-4-(2,5-диметилимидазо[1,2а]пирид-3-ил)пиримидин (способ 14) (200 мг, 0,74 ммоль) добавляют к раствору 3-хлоранилина (0,16 мл, 1,48 ммоль) и гидрида натрия (60 мг, 1,48 ммоль) в NMP (1 мл) в атмосфере азота. Смесь нагревают при 150° С в течение 4 часов и затем оставляют охлаждаться. Неочищенную реакционную смесь загружают на колонку Bond Elut, элюируют дихлорметаном для удаления NMP и затем смесью дихлорметан/метанол/метиламин (75:20:5) элюируют продукт. Продукт дополнительно очищают хроматографией, элюируя смесью этилацетат/гексан (8:2) и затем этилацетатом, получая указанное в заголовке соединение (22 мг, 9%). ЯМР: 2,27 (с, 3Н), 2,61 (с, 3Н), 7,01(д, 1Н), 7,12(д, 1Н), 7,30(м, 2Н), 7,56(д, 1Н), 7,62(д, 1H), 8,57(д, 1H), 9,41(c, 1H), 9,83(c, 1H); m/z: 350 [MH]+.
ПРИМЕР 38
Следуя способу примера 37 и используя соответствующие исходные вещества, получают следующие соединения.
пиразол [2,3а]пирид-3-ил)-
Пиримидин
ПРИМЕР 39. 2 -[4-N-Метилсульфамоил)анилин]-4 -(имидазо-[1,2а]пирид-3-ил)пиримидин
Толуол (4 мл) добавляют к смеси трис(дибензиденацетон)-дипалладия(0) (24 мг, 0,026 ммоль), 2,2’-бис(дифенилфосфин)-1,1’-бинафтила (21 мг, 0,034 ммоль), 2-хлор-4-(имидазо-[1,2 а]пирид-3-ил)пиримидина (способ 20; 150 мг, 0,652 ммоль) и 4-(N-метилсульфамоил)анилина (способ 23; 135 мг, 0,725 ммоль) в атмосфере азота. Колбу вакуумируют и заполняют азотом, добавляют трет-бутоксид натрия (140 мг, 1,46 ммоль) и колбу снова вакуумируют и заполняют азотом. Смесь нагревают при 100° С в течение 3 часов и затем оставляют охлаждаться. Смесь разбавляют этилацетатом и промывают водой. Органическую фазу отделяют, сушат и летучие вещества удаляют упариванием. Остаток очищают хроматографией, элюируя смесью этилацетат/метанол (100:0, с увеличением полярности до 97:3), получая 15 мг указанного в заголовке соединения, (60%). ЯМР: 2,42(д, 3Н), 7,25-7,10(м, 2Н), 7,62-7,45(м, 2Н), 7,79-7,70(м, 3Н), 7,98(д, 2Н), 8,50(д, 1H), 8,62(с, 1H); m/z: 381 [МН]+.
ПРИМЕРЫ 40-44
Следуя способу примера 39 и используя соответствующие исходные вещества, получают следующие соединения.
[МН]+
[м-н]
2Продукт очищают хроматографией, элюируя смесью гексан/этилацетат (50:50) с увеличением полярности до этилацетат/метанол (95:5).
3Продукт очищают хроматографией, элюируя смесью гексан/этилацетат (80:20) с увеличением полярности до этилацетат/метанол (90:10).
ПРИМЕР 45
2-{4-[N-(3-Гидроксипропил)сульфамоил]анилин}-4-(имидазо[1,2а]пирид-3-ил) пиримидин
2-Анилин-4 -(имидазо[1,2 а]пирид-3-ил)пиримидин (пример 16; 100 мг, 0,347 ммоль) растворяют в тионилхлориде (4 мл) и смесь охлаждают до 5° С. Добавляют хлорсульфоновую кислоту (0,06 мл, 0,90 ммоль) и смесь перемешивают при 5° С в течение 30 минут, затем оставляют нагреваться до температуры окружающей среды и перемешивают в течение 60 минут. Затем смесь кипятят с обратным холодильником в течение 90 минут. Летучие вещества удаляют упариванием и остаток подвергают азеотропной перегонке с толуолом. К остатку добавляют 3-аминопропанол (3 мл) и смесь перемешивают при температуре окружающей среды в течение 30 минут. Смесь очищают хроматографией, элюируя смесью гексан/этилацетат (50:50) с увеличением полярности до этилацетат/метанол (85:15). 60 мг (41%). ЯМР: 1,45-1,56(м, 2Н), 2,79(кв, 2Н), 3,35(кв, 2Н), 4,39(т, 1Н), 7,15(дд, 1Н), 7,31(т, 1Н), 7,45-7,54(м, 2Н), 7,70-7,79(м, 3Н), 7,95(д, 2Н), 8,50(д, 1Н), 8,62(с, 1Н); m/z: 423 [М-Н]-.
ПРИМЕРЫ 46-50
Следуя способу примера 45 и используя соответствующие исходные вещества, получают следующие соединения.
2Продукт очищают хроматографией, элюируя смесью гексан/этилацетат (50:50) с увеличением полярности до этилацетат/метанол (95:5).
3Продукт очищают хроматографией, элюируя смесью гексан/этилацетат (80:20) с увеличением полярности до этилацетат/метанол (90:10).
ПРИМЕР 51
2-{4-(N-[3-(2-оксопирролидин-1-ил)пропил]-су льфамоил}анилин)-4-(имидазо[1,2а]пирид-3-ил)пиримидин
2-Анилин-4-(имидазо[1,2 а]пирид-3-ил)пиримидин (пример 16; 100 мг, 0,347 ммоль) растворяют в тионилхлориде (3 мл) и смесь охлаждают до 5° С. Добавляют хлорсульфоновую кислоту (0,06 мл, 0,90 ммоль) и смесь перемешивают 30 минут при 5° С, оставляют нагреваться до температуры окружающей среды и перемешивают 60 минут. Затем смесь кипятят с обратным холодильником в течение 90 минут. Летучие вещества удаляют упариванием и остаток подвергают азеотропной перегонке с толуолом. К остатку добавляют пиридин (3 мл) и 3-(2-оксопирролидин-1-ил)пропиламин (3 мл), смесь перемешивают при температуре окружающей среды в течение одного часа. Смесь очищают хроматографией, элюируя смесью гексан/этилацетат (50:50) с увеличением полярности до этилацетат/метанол (80:20); 60 мг (36%). ЯМР: 1,51-1,60(м, 2Н), 1,80-1,90(м, 2Н), 2,13(т, 2Н), 2,70(т, 2Н), 3,10(т, 2Н), 3,20 (т, 2Н), 7,16(дд, 1Н), 1,48-7,55(м, 2Н), 7,70-7,80(м, 3Н), 7,95(д, 2Н), 8,50(д, 1Н), 8,62 (с, 1Н); m/z: 492 [МН]+.
ПРИМЕРЫ 52-70
Следуя способу примера 45 и используя соответствующие исходные вещества, получают следующие соединения.

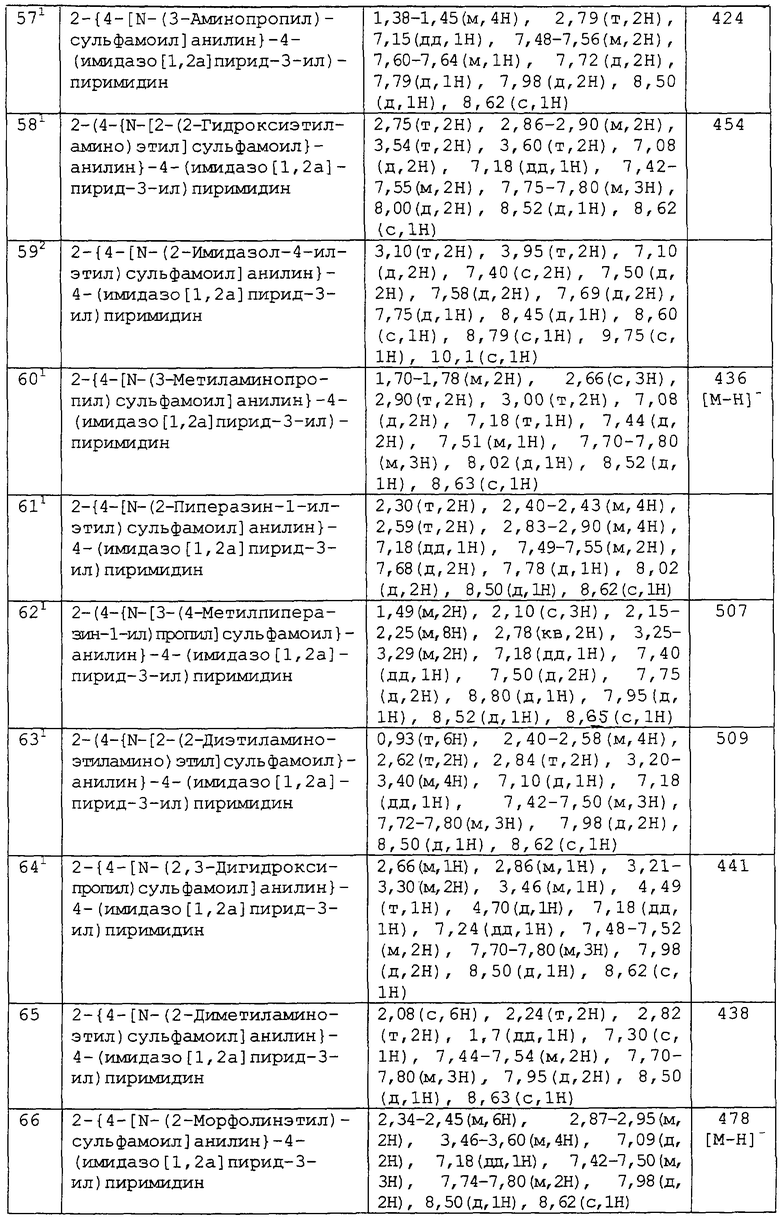
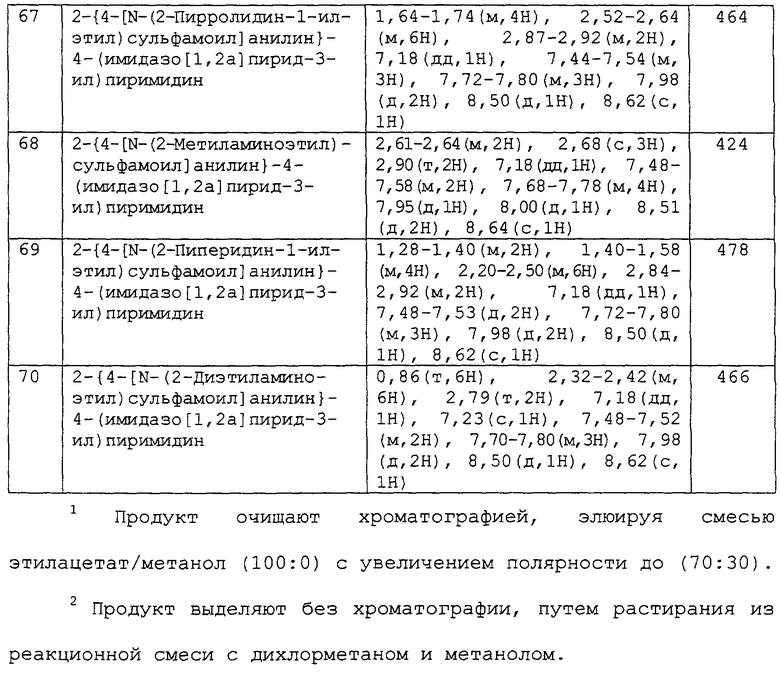
ПРИМЕР 71
2-{4-[N-(3-Имидазол-1-илпропил)карбамоил]-анилино}-4-(ииидазо[1,2а]пирид-3-ил)пиримидин.
К смеси 2-амино-4-(имидазо[1,2а]пирид-3-ил)пиримидина (способ 22; 200 мг, 0,95 ммоль), 1-[3-(4-бромбензоиламино)-пропил]имидазола (способ 27; 350 мг, 1,14 ммоль), трис(дибензиденацетон)дипалладия(0) (43 мг, 0,047 ммоль) и 2,2’-бис(дифенилфосфин)-1,1’-бинафтила (28 мг, 0,046 ммоль) добавляют толуол (10 мл) в атмосфере азота. Добавляют трет-бутоксид натрия (218 мг, 0,0023 ммоль), реакционную смесь тщательно продувают азотом и затем нагревают при 100° С в течение 24 часов. Летучие вещества удаляют упариванием и остаток очищают хроматографией, элюируя смесью гексан/этилацетат (50:50) с увеличением полярности до этилацетат/метанол (95:5), получая указанное в заголовке соединение, 99 мг (24%). ЯМР: 1,90-2,00(м, 2Н), 3,22(кв, 2Н), 4,02 (т, 2Н), 6,86 (с, 1Н), 7,16(дд, 1Н), 7,21 (с, 1Н), 7,42-7,55(м, 2Н), 6,80 (с, 3Н), 7,78(д, 1Н), 7,83(д, 4Н), 8,38 (т, 1Н), 8,48(д, 1Н), 8,62(с, 1Н), 9,92 (с, 1Н); m/z: 439 [МН]+.
ПРИМЕРЫ 72-74
Следуя способу примера 71 и используя соответствующие исходные вещества, получают следующие соединения.
[МН]
2Исходным веществом является 2,4-дихлор-1-(2-метоксиэтил-сульфамоил)бензол (способ 29)
3Исходным веществом является 2,4-дихлор-1-(1-пропил-сульфамоил)бензол (способ 30)
ПРИМЕР 75
2-(3-Метил-4-сульфамоиланилин)-4-(имидазо-[1, 2а] пирид-3-ил) пиримидин
2-(3-Метиланилин)-4-(имидазо[1,2а]пирид-3-ил)пиримидин (пример 35; 80 мг, 0,266 ммоль) обрабатывают, как описано в примере 45, но используют 2М этанольный раствор аммиака, получая указанное в заголовке соединение (6 мг, 17%).
ЯМР: 2,60(с, 3Н), 6,95-7,20(м, 4Н), 7,46-7,50(м, 2Н), 7,70-7,80(м, 4Н), 8,50(д, 1Н), 8,62(с, 1Н), 9,87(с, 1Н); m/z: 381 [МН]+.
ПРИМЕРЫ 76-78
Следуя способу примера 75 и используя соответствующие исходные вещества, получают следующие соединения.
Пример 79
5-Бром-2-(4-сульфамоиланилин)-4-(имидазо-[1,2а]пирид-3-ил)пиримидин
2-Анилин-5-бром-4-(имидазо[1,2 а]пирид-3-ил)пиримидин (пример 97; 73 мг, 0,2 ммоль) обрабатывают, как описано в примере 45, но используют 2М этанольный раствор аммиака, получая указанное в заголовке соединение (18 мг, 21%). ЯМР: 7,12(дд, 1Н), 7,19(с, 2Н), 7,53(дд, 2Н), 7,72(д, 2Н), 7,79(д, 1Н), 7,84(д, 2Н), 8,76(с, 1Н), 8,78(с, 1Н), 9,62 (с, 1Н); m/z: 445 [MH]+.
ПРИМЕРЫ 80-81
Следуя способу примера 79 и используя соответствующие исходные вещества, получают следующие соединения.
ПРИМЕР 83
2-(4-[N-(2-Метоксиэтил)сульфамоил]анилин}-4-(5-бромимидазо [1, 2а] пирид-3-ил) пиримидин.
2-Анилин-4-(5-бромимидазо[1,2а]пирид-3-ил)пиримидин (пример 98; 70 мг, 0,2 ммоль) обрабатывают 2-метоксиэтиламином в условиях, описанных в примере 51, получая указанное в заголовке соединение, 23 мг (25%). 1H ЯМР: 2,90(кв, 2Н), 3,18 (с, 3Н), 3,26-3,29(м, 2Н), 7,49-7,54(м, 2Н), 7,60(дд, 1Н), 7,74-7,78(м, 3Н), 7,90(д, 1Н), 8,54(д, 1Н), 8,62(с, 1Н); m/z: 503 [МН]+.
ПРИМЕР 84
2-{4-[Н-(2-Метоксиэтил)сульфамоил]анилин}-4-(5-фенилтиоимидазо[1,2а]пирид-3-ил)пиримидин.
Гидрид натрия (80 мг 60% суспензии в минеральном масле, 2,0 ммоль) добавляют к раствору тиофенола (0,102 мл, 1,0 ммоль) в NMP (4 мл) и смесь перемешивают в течение 30 минут. Добавляют 2-[4-(N-(2-метоксиэтил)сульфамоил)анилин]-4-(5-бромимидазо[1,2а]-пирид-3-ил)пиримидин (пример 83; 100 мг, 0,19 ммоль) в NMP (1 мл) и смесь нагревают при 150° С в течение 18 часов. Смесь оставляют охлаждаться, разбавляют водой и экстрагируют этилацетатом. Экстракты промывают водой, сушат и летучие вещества удаляют упариванием. Остаток растирают с эфиром и отделяют фильтрацией, получая указанное в заголовке соединение, 20 мг (20%). ЯМР: 2,85(кв, 2Н), 3,15(с, 3Н), 3,24(кв, 2Н), 7,10-7,30(м, 5Н), 7,38(д, 1Н), 7,46(дд, 1Н), 7,52(д, 1Н), 7,75 (д, 2Н), 7,79(д, 1Н), 7,92(д, 2Н), 8,54(д, 1Н), 8,66(с, 1Н);m/z: 533 [МН]+.
ПРИМЕРЫ 85-88
Следуя способу примера 84 и используя соответствующие исходные вещества, получают следующие соединения.
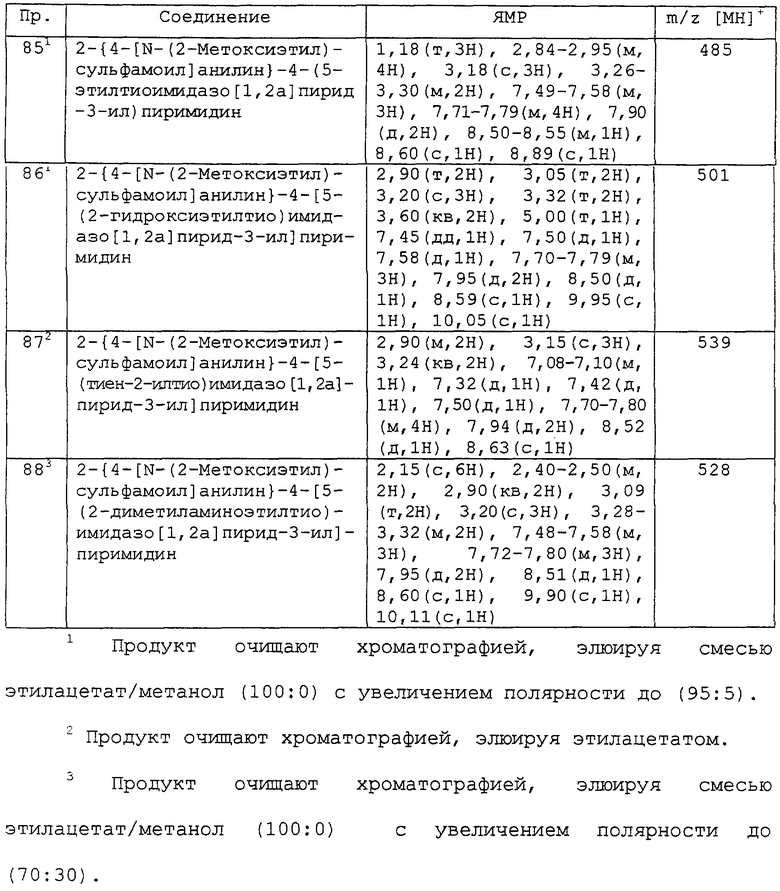
ПРИМЕР 89
2-{4-[N-(2-Метоксиэтил)сульфамоил]анилин}-4-(5-цианоимидазо[1,2 а]пирид-3-ил)пиримидин
2-{4-[N-(2-Метоксиэтил)сульфамоил]анилин}-4-(5-бромимидазо[1,2а]пирид-3-ил)пиримидин (пример 83; 87 мг, 0,17 ммоль), цианид тетраэтиламмония (27 мг, 0,17 ммоль), дифенилфосфинферроцен (23 мг, 0,03 ммоль), цианид меди (I) (62 мг, 0,7 ммоль) и трис(дибензиденацетон)дипалладий(0) (7 мг, 0,008 ммоль) в безводном диоксане (6 мл) тщательно продувают азотом и кипятят с обратным холодильником в течение 48 часов. Летучие вещества удаляют упариванием и остаток очищают хроматографией, элюируя смесью гексан/этилацетат (50:50) с увеличением полярности до (0:100), получая указанное в заголовке соединение, 16 мг (21%). ЯМР: 2,90(кв, 2Н), 3,15(с, 3Н), 3,25-3,30(м, 2Н), 7,42(дд, 1Н), 7,58(д, 1Н), 7,72-7,78 (м, 3Н), 7,90-7,98 (м, 3Н), 8,59(д, 1Н), 8,40(с, 1Н), 10,23(с, 1Н), 10,53 (с, 1Н); m/z: 447[МН]+.
ПРИМЕР 90.
2-{4-[N-(3-Диметиламинопропил)сульфамоил]анилин}-4-(5-бромимидазо[1,2а1пирид-3-ил)пиримидин
2-Анилин-4-(5-бромимидазо[1,2а]пирид-3-ил)пиримидин (пример 98; 200 мг, 0,52 ммоль) обрабатывают, как описано в примере 45, но используют 3-диметиламинопропиламин, получая указанное в заголовке соединение (92 мг, 34%). ЯМР: 1,48-1,58(м, 2Н), 2,10(с, 6Н), 2,20-2,28 (м, 2Н), 2,72-2,80(м, 2Н), 7,08(д, 1Н), 7,40-7,48 (м, 2Н), 7,51(д, 1Н), 7,61(дд, 1Н), 7,71-7,78(м, 3Н), 7,90(д, 2Н), 8,55(д, 1Н), 8,64(с, 1Н); m/z: 530 [МН]+.
ПРИМЕР 91.
5-(2-Гидроксиэтилтио)-2-{(4-[N-(2-метоксиэтил)-сульфамоил]анилин}-4-имидазо[1,2а]пирид-3-ил)-пиримидин.
Гидрид натрия (158 мг 60% суспензии в минеральном масле, 4,0 ммоль) добавляют к 2-меркаптоэтанолу (0,139 мл, 2,0 ммоль) в NMP (4 мл) и смесь перемешивают в течение 30 минут. Добавляют 5-бром-2-{4-[N-(2-метоксиэтил) сульфамоил] анилин}-4-(имидазо [1, 2а]-пирид-3-ил) пиримидин (пример 80; 100 мг, 0,19 ммоль) в NMP (1 мл) и смесь нагревают при 120° С в течение 3 часов. Смесь оставляют охлаждаться, разбавляют водой, нейтрализуют 2М хлористоводородной кислотой и экстрагируют этилацетатом. Экстракты промывают водой и насыщенным соляным раствором, сушат и летучие вещества удаляют упариванием. Остаток очищают хроматографией, элюируя смесью гексан/этилацетат (50:50) с увеличением полярности до этилацетат/метанол (95:5), получая указанное в заголовке соединение, 39 мг (20%). ЯМР: 2,85-2,98(м, 4Н), 3,15(с, 3Н), 3,24-3,30(м, 2Н), 3,51(кв, 2Н), 4,82 (т, 1Н), 7,10(дд, 1Н), 7,45-7,54(м, 2Н), 7,70(д, 2Н), 7,78(д, 1Н), 7,90(д, 2Н), 8,70(с, 1Н), 8,85(с, 1Н), 9,72(д, 1Н), 10,18 (с, 1Н); m/z: 501 [MH]+.
ПРИМЕР 92
2-4-{N-[3-(трет-Бутоксикарбониламино)пропил]-сульфамоил}анилин)-4-(имидазо[1,2а]пирид-3-ил) пиримидин
2-Анилин-4-(имидазо[1,2а]пирид-3-ил)пиримидин (пример 16; 290 мг, 1,0 ммоль) растворяют в тионилхлориде (6 мл) и смесь охлаждают до 0° С. Медленно добавляют хлорсульфоновую кислоту (0,266 мл, 4,0 ммоль) и смесь перемешивают при 0° С 30 минут, оставляют нагреваться до температуры окружающей среды, перемешивают два часа и затем кипятят с обратным холодильником в течение одного часа. Летучие вещества удаляют упариванием. Остаток растворяют в сухом пиридине (5 мл) и полученный раствор медленно добавляют к раствору 3-(трет-бутоксикарбониламино)пропиламина (0,209 мл, 1,2 ммоль) и диэтилметиламина (1,21 мл, 10 ммоль) в пиридине (10 мл) и охлаждают до 0° С в атмосфере азота. Смесь перемешивают при 0° С в течение одного часа, затем при температуре окружающей среды в течение двух часов. Летучие вещества удаляют упариванием и остаток подвергают азеотропной перегонке с водой. Остаток растирают с водой, собирают фильтрацией и затем очищают хроматографией, элюируя смесью дихлорметан/метанол (95:5) с увеличением полярности до (90:10), получая указанное в заголовке соединение, 207 мг (40%). ЯМР: 1,30(с, 9Н), 1,50(квнт, 2Н), 2,67(м, 2Н), 2,85(м, 2Н), 7,38(м, 2Н), 7,58(д, 1Н), 7,68(д, 1Н), 7,70(д, 2Н), 7,89(д, 1Н), 7,95(д, 2Н), 8,58 (д, 1Н), 8,80(с, 1Н),; m/z: 524 [МН]+.
ПРИМЕР 93
2-(4-{N-[3-(Бензилоксикарбониламино)пропил]-сульфамоил}анилин)-4-(имидазо[1,2а]пирид-3-ил)пиримидин
2-Анилин-4 -(имидазо[1,2а]пирид-3-ил)пиримидин (пример 16; 290 мг, 1,0 ммоль) и 3-(бензилоксикарбониламино)пропиламин (0,294 мл, 1,2 ммоль) обрабатывают, как описано в примере 92, получая указанное в заголовке соединение, 212 мг (38%). ЯМР: 1,50(квнт, 2Н), 2,70(кв, 2Н), 2,98(дд, 2Н), 4,98(с, 2Н), 7,12-7,15(м, 4Н), 7,18(т, 2Н), 7,19(т, 1Н), 7,75(д, 2Н), 7,79(д, 1Н), 7,90(д, 2Н), 8,50(д, 1Н), 8,60(с, 1Н); m/z: 558 [МН]+.
ПРИМЕР 94
2-[4-(2-Диэтиламиноэтокси)анилин]-4-(6-фенилимидазо[1,2a] пирид-3-ил)пиримидин
3-(3-Диметиламинопроп-2-ен-1-оил)-6-фенилимидазо[1,2а]-пиридин (способ 38; 50 мг, 0,17 ммоль) добавляют к раствору 4-(2-диэтиламиноэтокси)фенилгуанидина (способ 42; 60 мг, 0,19 ммоль) и метоксида натрия (11 мг, 0,21 ммоль) в н-бутаноле (1,5 мл) и смесь нагревают при 115° С в течение 15 часов. Летучие вещества удаляют упариванием и остаток очищают хроматографией, элюируя смесью гексан/этилацетат (50:50) с увеличением полярности до этилацетат/метанол (80:20), получая указанное в заголовке соединение (5 мг, 6%). ЯМР: 1,07(т, 6Н), 2,64(кв, 4Н), 2,92 (т, 2Н), 4,10 (т, 2Н), 6,98(д, 2Н), 7,08(м, 2Н), 7,15 (д, 1Н), 1,37-7,60 (м, 4Н), 7,70(д, 2Н), 7,92(с, 1Н), 8,30(с, 1Н), 8,35(д, 1Н), 9,80(д, 1Н); m/z: 479 [MH]+.
ПРИМЕР 95
4-(6-Метокси-2-метилимидазол[1,2а]пирид-3-ил)-2-(4-сульфамоиланилин) пиримидин
3-(3-Диметиламинопроп-2-ен-1-оил)-2-метил-6-метоксиимидазо-[1,2а]пиридин (способ 39; 862 мг, 3,51 ммоль) добавляют к раствору 4-сульфамоилфенилгуанидина (способ 41; 1,5 г, 7,0 ммоль) и метоксида натрия (758 мг, 14 ммоль) в н-бутаноле (4 мл) и смесь кипятят с обратным холодильником в течение 24 часов. Смесь оставляют охлаждаться, полученный осадок отделяют фильтрацией и очищают хроматографией, элюируя смесью гексан/этилацетат (50:50) с увеличением полярности до этилацетат/метанол (90:10), получая указанное в заголовке соединение. ЯМР: 2,60(с, 3Н), 3,88(с, 3Н), 6,70(дд, 1Н), 7,03 (Д, 1Н), 7,12(д, 1Н), 7,18(с, 2Н), 7,75(д, 2Н), 7,90(д, 2Н), 8,52(д, 1Н), 9,68(д, 1Н), 9,97 (с, 1Н); m/z: 411 [МН]+.
ПРИМЕР 96
2-(3-Хлоранилин)-4-(пиранол[2,3а]пирид-3-ил)пиримидин.
Безводный н-бутанол (6,0 мл) добавляют к смеси 3-(3-диметиламинопроп-2-ен-1-оил)-2-метилпиразол[2,3а]пиридина (способ 18; 180 мг, 0,84 ммоль), 3-хлорфенилгуанидина (142 мг, 0,84 ммоль) и гидрида натрия (67 мг 60% дисперсии в минеральном масле, 1,67 ммоль) и смесь нагревают в атмосфере азота при 125° С в течение 7 часов. Летучие вещества удаляют упариванием и остаток растирают со смесью эфира и дистиллированной воды. Осажденное твердое вещество отделяют фильтрацией, промывают эфиром и дистиллированной водой и сушат, получая указанное в заголовке соединение (78 мг, 29%). ЯМР: 7,00(д, 1Н), 7,10(т, 1Н), 7,35(м, 2Н), 7,50(т, 1Н), 7,60(д, 2Н), 8,08(с, 1Н), 8,43 (д, 1Н), 8,70(д, 1Н), 8,82(д, 2Н), 9,68 (с, 1Н); m/z: 322 [МН]+.
ПРИМЕР 97
2-Анилин-5-бром-4-(имидазо[1,2а]пирид-3-ил)пиримидин
2-Амино-5-бром-4-(имидазо[1,2а]пирид-3-ил)пиримидин (способ 31; 200 мг, 0,67 ммоль) и бромбензол (0,08 мл, 0,76 ммоль) обрабатывают, как описано в примере 71, и продукт очищают хроматографией, элюируя смесью гексан/этилацетат (50:50) с увеличением полярности до (0:100), получая указанное в заголовке соединение. ЯМР: 6,98-7,10(м, 2Н), 7,30(дд, 2Н), 7,50(дд, 1Н), 7,66(д, 2Н), 7,78(д, 1Н), 8,64(с, 2Н), 8,72(с, 1Н), 9,01(д, 1Н), 9,82(с, 1Н).
ПРИМЕР 98
2-Анилин-4-(5-бромимидазо[1,2а]пирид-3-ил)пиримидин
2-Амино-4-(5-бромимидазо[1,2а]пирид-3-ил)пиримидин (способ 35; 1,0 г, 3,4 ммоль) и бромбензол (4,36 мл, 4,1 ммоль) обрабатывают, как описано в примере 71, и продукт очищают хроматографией, элюируя смесью этилацетат/метанол (98:2) с увеличением полярности до (90:10), получая указанное в заголовке соединение, 70 мг (6%). ЯМР: 7,00(дд, 1Н), 7,30-7,40(м, 4Н), 7,59(д, 1Н), 7,65-7,75(м, 3Н), 8,42(д, 1Н), 8,60 (с, 1Н), 9,70(с, 1Н); m/z: 364 [М-Н]-.
Получение исходных веществ
Исходные вещества для описанных выше примеров либо являются коммерчески доступными, либо могут быть легко получены с помощью стандартных способов из известных веществ. Например, нижеследующие реакции являются иллюстрациями, но не ограничениями способов получения некоторых исходных веществ, используемых в описанных выше реакциях.
СПОСОБ 1. 4-(2-Метилимидаао[1,2а]пирид-3-ил)-2-метилтиопиримидин.
Смесь 3-(3-диметиламинопроп-2-ен-1-оил)-2-метилимидазо-[1,2а]пиридина (способ 2) (20 г, 87 ммоль), тиомочевины (6,52 г, 86 ммоль) и метоксида натрия (1,19 г, 22 ммоль) в бутаноле (220 мл) нагревают при 85° С в течение двух часов в атмосфере азота. Добавляют метилиодид (2 мл, 32 ммоль) и смесь нагревают при 85° С в течение еще 1 часа. Добавляют метанол и летучие вещества удаляют упариванием. Остаток очищают хроматографией, элюируя смесью этилацетат/метанол (100:0 с увеличением полярности до 97:3), получая указанное в заголовке соединение (16 г, 71%). ЯМР: 2,59 (с, 1Н), 2,62 (с, 3Н), 7,10(дд, 1Н), 7,40 (дд, 1Н), 7,42(д, 1Н), 7,63(д, 1Н), 8, 62 (с, 1Н), 9,54(д, 1Н); m/z: 257 [МН]+.
СПОСОБ 2. 3-(3-Диметиламинопроп-2-ен-1-оил)-2-метилимидазо[1,2а]пиридин.
Смесь 3-ацетил-2-метилимидазо[1,2 а]пиридина (способ 3) (40 г, 0,23 моль) и DMFDMA (200 мл) кипятят с обратным холодильником в атмосфере азота в течение 4 дней. Летучие вещества удаляют упариванием, остаток растирают с горячим эфиром и твердый продукт собирают фильтрацией, получая указанное в заголовке соединение (21 г, 40%). ЯМР: 2,64(с, 3Н), 3,29 (с, 6Н), 5,50(д, 1Н), 7,00(дд, 1Н), 7,38(дд, 1Н), 7,54 (д, 1Н), 7,70(д, 1Н), 9,55(д, 1Н); m/z: 230 [МН]+.
СПОСОБ 3. 3-Ацетил-2-метилимида[1,2а]пиридин
Смесь 2-аминопиридина (60 г, 0,64 моль) и 3-хлор-2,4-пентандиона (101,4 г, 0,75 моль) в эфире (450 мл) и THF (750 мл) кипятят с обратным холодильником в течение 12 часов, затем оставляют стоять 18 часов при температуре окружающей среды. Растворитель удаляют упариванием и остаток очищают хроматографией, элюируя смесью дихлорметан/гексан (1:1) с увеличением полярности до дихлорметан/метанол (98:2). Очищенный продукт растирают с гексаном, получая указанное в заголовке соединение (46,2 г, 40%). ЯМР: 2,55 (с, 3Н), 2,68 (с, 3Н), 7,15 (дц, 1Н), 7,56(дд, 1Н), 7,64(д, 1Н), 9,58(д, 1Н); m/z: 175 [МН]+.
СПОСОБ 4. 4-Имида[1,2а] пирид-3-ил)-2-метилтиопиримидин
Смесь 3-(3-диметиламинопроп-2-ен-1-оил) имидазо [1,2а]пиридина (способ 5) (0,90 г, 4,2 ммоль), тиомочевины (0,32 г, 4,2 ммоль) и метоксида натрия (0,34 г, 6,3 ммоль) нагревают при 85° С в н-бутаноле (10 мл) в течение 2 часов. Смесь оставляют охлаждаться до 30° С, добавляют по каплям метилиодид (0,6 мл, 9,6 ммоль) и перемешивание продолжают в течение еще 3 часов. Летучие вещества удаляют упариванием и остаток очищают хроматографией, элюируя смесью этилацетат/метанол (100:0 с увеличением полярности до 97:3), получая указанное в заголовке соединение (0,94 г, 93%). ЯМР: 2, 61 (с, 3Н), 7,22(дд, 1Н), 7,54(дд, 1Н), 7,72(д, 1Н), 7,77 (д, 1Н), 8,56(д, 1Н), 8, 66 (с, 1Н), 9, 83 (д, 1Н); m/z: 243 [МН]+.
СПОСОБ 5. 3-3-Диметиламинопроп-2-ен-1-оил)имидазо-[1,2а]пиридин.
Смесь неочищенного 3-ацетилимидазо[1,2,а]пиридина (способ 6) (3,3 г, 19,1 ммоль) и DMFDMA (40 мл) кипятят с обратным холодильником в течение 60 часов. Смесь оставляют охлаждаться, летучие вещества удаляют упариванием и остаток растирают с горячим эфиром. Твердый продукт собирают фильтрацией, получая указанное в заголовке соединение, 2,29 г, 52%. ЯМР: 2,90 (шир.с, 3Н), 3,10(шир.с, 3Н), 5,81(д, 1Н), 7,09(дд, 1Н), 7,42 (дд, 1Н), 7,65(д, 1Н), 7,70(д, 1Н), 8,43 (с, 1Н), 9,72(д, 1Н); m/z: 216 [MH]+.
СПОСОБ 6. 3-Ацетилимидазо[1,2]пиридин
Хлорид алюминия (20,4 г, 153,2 ммоль) добавляют маленькими порциями к раствору имидазо[1,2 а]пиридина (8,9 г, 75,7 ммоль) в охлажденном дихлорметане (150 мл) при 5° С. Затем смесь оставляют нагреваться до температуры окружающей среды и перемешивают 1 час, после чего кипятят с обратным холодильником. Затем медленно в течение 30 минут добавляют уксусный ангидрид (5,1 мл, 53,9 ммоль) и смесь кипятят с обратным холодильником в течение еще 90 минут. Смесь оставляют охлаждаться, растворитель удаляют упариванием и к остатку добавляют воду со льдом. Водную смесь подщелачивают 2М водным раствором гидроксида натрия и экстрагируют этилацетатом. Объединенные экстракты сушат и летучие вещества удаляют упариванием, получая коричневое масло. Показано, что данное масло приблизительно на 35% состоит из указанного в заголовке соединения, остальное вещество представляет собой имидазо[1,2,а]пиридин. Данную смесь используют без дополнительной очистки. ЯМР: 2,57(с, 3Н), 7,22(дд, 1Н), 7,61 (дд, 1Н), 7,79(д, 1Н), 8,60 (с, 1Н), 9,52(д, 1Н).
СПОСОБ 7. 4-(3,5-Диоксапиперидин-1-ил)сульфониланилин
Смесь 1-(3,5-диоксапиперидин-1-ил)сульфонил-4-нитробензола (способ 8) (500 мг, 1,82 ммоль) и катализатора 10% палладия на угле (150 мг) в этаноле (25 мл) и этилацетате (25 мл) перемешивают в атмосфере водорода в течение 3 часов. Катализатор удаляют фильтрацией через диатомовую землю и фильтровальную подушку промывают этанолом и этилацетатом. Летучие вещества удаляют из фильтрата упариванием и остаток растирают с эфиром и гексаном, получая указанное в заголовке соединение (395 мг, 88%). ЯМР: 4,90(с, 2Н), 5,10(с, 4Н), 6,02(с, 2Н), 6,58(д, 2Н), 7,50(д, 2Н).
СПОСОБ 8. 1-(3,5-Диоксапиперидин-1-ил)сульфонил-4-нитробензол
4-Нитробензолсульфонамид (2,02 г, 10 ммоль) добавляют к раствору 1,3,5-триоксана (1,96 г, 20 ммоль) в уксусной кислоте (5 мл). Смесь перемешивают в течение 5 минут и медленно добавляют метансульфоновую кислоту (10 мл). Затем смесь перемешивают при 35° С 20 минут, охлаждают до 0° С, разбавляют водой и экстрагируют этилацетатом. Объединенные экстракты промывают дважды водой и дважды 5% водным раствором гидрокарбоната натрия, затем сушат и летучие вещества удаляют упариванием. Остаток перекристаллизовывают из этанола, получая указанное в заголовке соединение (955 мг, 35%). ЯМР: 4,87(с, 2Н), 5,30(с, 4Н), 8,20(д, 2Н), 8,42(д, 2Н).
СПОСОБ 9. 4-(2-Диэтиламиноэтокси)анилин
Смесь 4-(2-диэтиламиноэтокси)-1-нитробензола (способ 10) (1,0 г, 4,2 ммоль) и катализатора 10% палладия на угле (200 мг) в этаноле (30 мл) перемешивают в атмосфере водорода в течение 3 часов. Катализатор удаляют фильтрацией через диатомовую землю и фильтровальную подушку промывают метанолом. Летучие вещества удаляют из фильтрата упариванием, получая указанное в заголовке соединение (400 мг, 46%) в виде масла. M/z: 209 [МН]+.
СПОСОБ 10. 4-(2-Диэтиламиноэтокси)-1-нитробензол
К смеси 4-нитрофеноксида натрия (10,5 г, 65 ммоль), гидрохлорида 2-(диэтиламино)этилхлорида (8,6 г, 50 ммоль) и карбоната калия (10,4 г, 75 ммоль) добавляют воду (8 мл) и ксилол (35 мл) и полученную смесь кипятят с обратным холодильником в течение 2 часов. Затем присоединяют насадку Дина-Старка и удаляют воду. Органический раствор оставляют охлаждаться до температуры окружающей среды и оставляют стоять на 18 часов. С осажденного твердого вещества декантируют раствор и летучие вещества удаляют из декантированного раствора упариванием, получая указанное в заголовке соединение (8,0 г, 52%) в виде масла. ЯМР: 0,90(т, 6Н), 2,50(кв, 2Н), 2,89(т, 2Н), 4,15(т, 2Н), 7,15(д, 2Н), 8,18(д, 2Н); m/z: 239 [МН]+.
СПОСОБ 11. 4-[3-(N,N-Диметил)амино-2-тадрокаьтропокси]-анилин
3-N,N-Диметиламино-2-гидрокси-3-(4-нитрофенокси)пропан (способ 12) (3,75 г) растворяют в этаноле (40 мл). В атмосфере азота добавляют 10% палладий на угле (0,4 г). Атмосферу азота заменяют водородом и реакционную смесь перемешивают в течение ночи. Катализатор удаляют фильтрацией через диатомовую землю и фильтрат упаривают досуха. Остаток растворяют в диэтиловом эфире, содержащем небольшое количество изопропанола, и добавляют раствор хлористого водорода (1М в эфире, 16 мл). Эфир упаривают и твердый остаток суспендируют в изопропаноле. Данную смесь нагревают на паровой бане в течение нескольких минут, затем оставляют охлаждаться до температуры окружающей среды. Полученный порошок отделяют фильтрацией, промывают изопропанолом, эфиром и сушат (3,04 г, 72,4%). ЯМР: 2,80(с, 6Н), 3,15(м, 2Н), 3,88(м, 2Н), 4,25(м, 1Н), 5,93(шир.с, 1Н), 6,88(м, 4Н); m/z: 211 [МН]+.
ЕА: для C11H18N2О2·1,6HCl
требуется: С: 49,2, Н: 7,4, N: 10,4, Cl: 21,7%;
обнаружено: С: 49,2, Н: 7,2, N: 10,1, Cl: 19,1%.
СПОСОБ 12. 3-N,N-Диметиламино-2-гидрокси-1-(4-нитрофенокси)пропан
1-(4-Нитрофенокси)-2,3-эпоксипропан (способ 13) (4,3 г) растворяют в метаноле (30 мл) и DMF (10 мл). Добавляют диметиламин (2М раствор в метаноле, 17 мл) и смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь упаривают досуха и остаток растворяют в насыщенном растворе гидрокарбоната натрия и этилацетате. Этилацетатный слой отделяют и промывают дважды насыщенным соляным раствором, сушат над безводным сульфатом натрия, фильтруют и упаривают, получая масло, которое медленно кристаллизуется в высоком вакууме (4,79 г, 89,9%). ЯМР (CDCl3): 2,33(с, 6Н), 2,98(м, 1Н), 2,54(м, 1Н), 4,00(м, 3Н), 7,00(д, 2Н), 8,20(д, 2Н); m/z: 241 [МН]+.
СПОСОБ 13. 1-(4-Нирофенокси)-2,3-эпоксипропан
1-(4-Нитрофенокси)-2,3-эпоксипропан получают по способу, аналогичному описанному Zhen-Zhong Lui et al. в Synthetic Communications (1994), 24, 833-838.
4-Нитрофенол (4,0 г), безводный карбонат калия (8,0 г) и бромид тетрабутиламмония (0,4 г) смешивают с эпибромгидрином (10 мл). Реакционную смесь нагревают при 100° С в течение 1 часа. После охлаждения до температуры окружающей среды реакционную смесь разбавляют этилацетатом и фильтруют. Фильтрат упаривают досуха и остаток дважды перегоняют с толуолом. Полученное масло очищают с помощью колоночной хроматографии, элюируют смесью этанол (1,0%):дихлорметан, получая после упаривания масло, которое кристаллизуется (4,36 г, 77,7%).
ЯМР (CDCl3): 2,78(м, 1Н), 2,95(м, 1Н), 3,38(м, 1Н), 4,02 (дц, 1Н), 4,38(дд, 1Н), 7,00(д, 2Н), 8,20(д, 2Н); m/z: 196 [MH]+.
СПОСОБ 14. 2-Метилтио-4-(2,5-диметилимидазо[1,2а]пирид-3-ил)пиримидин
Смесь 3-(3-диметиламинопроп-2-ен-1-оил)-2,5-диметилимидазо-[1,2а]пиридина (способ 15) (3,50 г, 14,4 ммоль), тиомочевины (1,09 г, 14,4 ммоль) и метоксида натрия (1,01 г, 18,7 ммоль) нагревают при 85° С в 1-бутаноле (50 мл) в течение 2 часов. Смесь оставляют охлаждаться до 30° С, добавляют по каплям метилиодид (1,8 мл, 28,8 ммоль) и смесь перемешивают еще 3 часа. Летучие вещества удаляют упариванием и остаток очищают хроматографией, элюируя смесью этилацетат/метанол (100:0 с увеличением полярности до 97:3), получая указанное в заголовке соединение (2,37 г, 61%). ЯМР: 2,41 (с, 3Н), 2,60 (с, 3Н), 2,70(с, 3Н), 7,56 (д, 1Н), 7,88(д, 1Н), 7,92(д, 1Н), 8,81(д, 1Н), 9,39 (с, 1Н); m/z: 271 [МН]+.
СПОСОБ 15. 3-(3-Диметиламинопроп-2-ен-1-оил)-2,5-диметилимидазо[1,2а]пиридин
Раствор 3-ацетил-2,5-диметилимидазо[1,2а]пиридина (способ 16) (3,60 г, 19,1 ммоль) в DMFDMA (20 мл) кипятят с обратным холодильником в течение 60 часов. Смесь оставляют охлаждаться и растворитель удаляют упариванием. Остаток растирают с горячим эфиром, твердое вещество собирают фильтрацией и сушат, получая указанное в заголовке соединение (3,61 г, 84%). ЯМР: 2,30 (с,3Н), 2,62(с, 3Н), 2,90(шир.с, 3Н), 3,10(шир.с, 3Н), 5,48(д, 1Н), 7,22(дд, 1Н), 7,44(д, 1Н), 7,68(д, 1Н), 9,39 (дд, 1Н).
СПОСОБ 16. 3-Ацетил-2,5-диметилимидазо [1,2 а] пиридин
3-Хлор-2,4-пентандион (6,5 мл, 54,4 ммоль) добавляют к суспензии 2-амино-4-метилпиридина (5,00 г, 46,3 ммоль) и иодида натрия (10 мг) в THF (60 мл) и смесь кипятят с обратным холодильником в течение 16 часов. Реакционную смесь оставляют охлаждаться и растворитель удаляют упариванием. Полученный твердый остаток растирают с горячим гексаном, собирают фильтрацией и сушат, получая указанное в заголовке соединение (3,69 г, 43%). ЯМР: 2,35(с, 3Н), 2,75(с 3Н), 7,41(дд, 1Н), 7,57(д, 1Н), 9,40(д, 1Н); m/z: 189 [МН]+.
СПОСОБ 17. 4-(2-Метилпиразол[2,3а]пирид-3-ил)-2-метилтиопиримидин
Смесь 3-(3-диметиламинопроп-2-ен-1-оил)-2-метил-пиразол-[2,3а]пиридина (способ 18) (3,89 г, 17 ммоль), тиомочевины (1,27 г, 17 ммоль) и метоксида натрия (0,929 г, 17 ммоль) в бутаноле (45 мл) нагревают при 85° С в течение двух часов в атмосфере азота. Добавляют метилиодид (1,05 мл, 17 ммоль) и смесь нагревают при 85° С еще 2 часа. Летучие вещества удаляют упариванием и остаток очищают хроматографией, элюируя смесью этилацетат/метанол (100:0 с увеличением полярности до 97:3), получая указанное в заголовке соединение (3,1 г, 68%).
ЯМР: 2,58 (с, 1Н), 2,68 (с, 3Н), 7,04(дц, 1Н), 7,39(дд, 1Н), 7,48 (д, 1H), 8,35(д, 1Н), 8,50(д, 1H), 8,72(д, 1Н); m/z: 257 [MH]+.
СПОСОБ 18. 3-(3-Диметаминопроп-2-ен-1-оил)-2-метилпиразол[2,3а]пиридин
Смесь 3-ацетил-2-метилпиразол[2,3а]пиридина (способ 19) (2 г, 11,5 ммоль) и DMFDMA (10 мл) нагревают при 110° С в атмосфере азота в течение 48 часов. Летучие вещества удаляют упариванием, остаток растирают с горячим эфиром и твердый продукт отделяют фильтрацией, получая указанное в заголовке соединение (1,98 г, 75%). ЯМР: 2,60(с, 3Н), 3,30(с, 6Н), 5,49(д, 1Н), 6,95(дд, 1Н), 7,38(дд, 1Н), 7,62(д, 1Н), 8,10(д, 1Н), 8,62(д, 1Н); m/z: 230 [МН]+.
СПОСОБ 19. 3-Ацетил-2-метилпиразол [2, 3а] пиридин
Карбонат калия (53,8 г, 0,39 моль) и затем 2,4-пентандион (24,8 г, 0,25 моль) добавляют к раствору иодида 1-аминопиридина (26,9 г, 0,12 моль) в воде (336 мл) и смесь нагревают при 80° С в течение 2 часов, оставляют охлаждаться до температуры окружающей среды и оставляют стоять на 18 часов. Добавляют воду и смесь экстрагируют этилацетатом. Объединенные экстракты сушат и летучие вещества удаляют упариванием. Остаток перекристаллизовывают из горячего гексана и продукт отделяют фильтрацией. Растворитель удаляют из фильтрата упариванием и добавляют к нерастворимому остатку от перекристаллизации. Неочищенную смесь очищают хроматографией, элюируя смесью дихлорметан/гексан (1:1) с увеличением полярности до дихлорметан/метанол (97:3). Полученный продукт растирают с гексаном и добавляют к продукту, полученному в результате первичной перекристаллизации, получая указанное в заголовке соединение (9,6 г, 33%). ЯМР: 2,50(с, 2Н), 2,62(с, 3Н), 7,09 (дц, 1Н), 7,55(дц, 1Н), 8,12(д, 1Н), 8,72(д, 1Н); m/z: 175 [MH]+.
СПОСОБ 20. 2-Хлор-4-(имидазо[1,2пирид-3-ил) пиримидин
Суспензию 2-гидрокси-4-(имидазо[1,2а]пирид-3-ил)пиримидина (способ 21; 9,92 г, 46%) в фосфорилхлориде (200 мл) и пентахлорид фосфора (11 г, 53%) кипятят с обратным холодильником в атмосфере азота 24 часа. Избыток фосфорилхлорида удаляют упариванием, добавляют ледяную воду и смесь нейтрализуют 2М водным раствором гидроксида натрия. Водную смесь экстрагируют этилацетатом, сушат и упаривают, получая указанное в заголовке соединение, 7,42 г (69%). ЯМР: 7,15(дд, 1Н), 7,59(дд, 1Н), 7,80(д, 1Н), 8,05(д, 1Н), 8,64(д, 1Н), 8,79(с, 1Н), 9,72(д, 1Н); m/z: 231 [МН]+.
СПОСОБ 21. 2-Гидрокси-4-(имидазо[1,2а]пирид-3-ил)пиримидин
Раствор нитрата натрия (11,04 г, 0,16 моль) в воде (100 мл) добавляют к раствору 2-амино-4-(имидазо[1,2а]пирид-3-ил)пиримидина (способ 22; 11,27 г, 0,053 моль) в 70% уксусной кислоте (330 мл) при 60° С. Смесь нагревают при 60° С в течение 3 часов, оставляют охлаждаться и нейтрализуют 5М водным раствором гидроксида натрия, полученный осадок отделяют фильтрацией, быстро промывают холодной водой и сушат в вакуумном шкафу при 50° С, получая указанное в заголовке соединение, 9,95 г (89%). ЯМР: 6,98(д, 1Н), 7,12(дд, 1Н), 7,55(дд, 1Н), 7,80(д, 1Н), 7,82(д, 1Н), 8,70 (с, 1Н); m/z: 213 [МН]+.
СПОСОБ 22. 2-Амино-4-(имидазо[1,2а]пирид-3-ил)пиримидин
Смесь 3-(3-диметиламинопроп-2-ен-1-оил)имидазо[1,2а]-пиридина (способ 5; 20 г, 0,093 моль), метоксида натрия (20,1 г, 0,372 моль) и гидрохлорида гуанидина (22,09 г, 0,233 моль) в н-бутаноле (1500 мл) и метаноле (1000 мл) кипятят с обратным холодильником в течение 60 часов. Полученный раствор декантируют с нерастворимого осадка, летучие вещества удаляют упариванием и остаток очищают хроматографией, элюируя смесью дихлорметан/метанол (97:3), получая указанное в заголовке соединение, 13 г (67%). 1H ЯМР: 6,78 (с, 1Н), 7,15-7,05 (м, 2Н), 7,45(дд, 2Н), 7,70(д, 1Н), 8,20(д, 1Н), 8,50 (с, 1Н), 10,15(д, 1Н); m/z: 212 [MH]+.
СПОСОБ 23. 4-(N-Метилсульфамоил)анилин
К сульфанилилфториду (200 мг, 1,1 ммоль) добавляют метиламин (3 мл 33% раствора в этаноле) и затем триэтиламин (0,159 мл, 1,1 ммоль) и смесь нагревают при 80° С 6 часов, затем выдерживают при температуре окружающей среды 18 часов. Летучие вещества удаляют упариванием и остаток подвергают азеотропной перегонке с толуолом, получая указанное в заголовке соединение (160 мг, 76%). ЯМР: 2,30(с, 3Н), 5,85(с, 2Н), 6,60 (д, 2Н), 7,39(д, 2Н); m/z: 187 [МН]+.
СПОСОБ 24. 4-[N-(2-Метоксиэтил) сульфамоил]анилин
Смесь 2-метоксиэтиламина (859 мг, 11,4 ммоль), сульфанилилфторида (1,0 г, 5,71 ммоль) и триэтиламина (1,72 г, 22,9 ммоль) в н-бутаноле (15 мл) кипятят с обратным холодильником в течение 18 часов. Смесь оставляют охлаждаться и летучие вещества удаляют упариванием. Остаток очищают хроматографией, элюируя смесью этилацетат/гексан (50:50) с увеличением полярности до (70:30), получая указанное в заголовке соединение (860 мг, 65%). ЯМР: 2,78(кв, 2Н), 3,15 (с, 3Н), 3,25(т, 2Н), 5,87(с, 2Н), 6,58(д, 2Н), 7,10(т, 1Н), 7,40(д, 2Н); m/z: 231 [MH]+.
СПОСОБЫ 25-26
Следующие соединения получают, используя методику способа 24.
СПОСОБ 27. 1-[3-(4-Бромбензоиламино)пропил]имидазол
1-(3-Аминопропил)имидазол (2,39 мл, 0,02 моль) добавляют к раствору 4-бромбензоилхлорида (4,0 г, 0,018 моль) в этаноле (250 мл). Смесь перемешивают при температуре окружающей среды в течение 18 часов. Летучие вещества удаляют упариванием и остаток очищают хроматографией, элюируя смесью гексан/дихлорметан (50:50) с увеличением полярности до дихлорметан/метанол (80:20), получая указанное в заголовке соединение. ЯМР: 1,95(м, 2Н), 3,20(кв, 2Н), 4,0 (т, 2Н), 6,87 (с, 1Н), 7,19 (с, 1Н), 7,64(д, 2Н), 7,68 (с, 1Н), 7,78(д, 2Н), 8,58 (т, 1Н); т/г: 308 [МН]+.
СПОСОБ 28. 1-[3-(4-Бромбензиламино)пропил]-2-оксопирролидин
1-(3-Аминопропил)-2-оксопирролидин (3,07 мл, 14 ммоль) обрабатывают, как описано в способе 27, получая указанное в заголовке соединение. ЯМР: 1,68(квнт, 2Н), 1,90(квнт, 2Н), 2,0(т, 2Н), 3,15-3,22(М, 4Н), 3,29-3,33(м, 2Н), 7,64(д, 2Н), 7,78(д, 2Н), 8,48(т, 1Н).
СПОСОБ 29. 2,4 -Дихлор-1 -(2 -метоксиэтилсульфамоил) бензол
2,4-Дихлорбензолсульфонилхлорид (500 мг, 2,1 ммоль) и 2-метоксиэтиламин (230 мг, 3,1 ммоль) в н-бутаноле (10 мл) кипятят с обратным холодильником в течение одного часа. Летучие вещества удаляют упариванием и остаток очищают хроматографией, элюируя смесью гексан/этилацетат (50:50), получая указанное в заголовке соединение. ЯМР: 3,04(т, 2Н), 3,08(с, 3Н), 3,22(т, 2Н), 7,60(дд, 1Н), 7,82(д, 1Н), 7,92(д, 1Н), 8,0 (с, 1Н); m/z: 282 [М-Н]-.
СПОСОБ 30. 2,4-Дихлор-1-(1-пропилсульфамоил) бензол
2,4-Дихлорбензолсульфонилхлорид (500 мг 2,1 ммоль) и 1-пропиламин (0,2 мл, 2,4 ммоль) в н-бутаноле (10 мл) кипятят с обратным холодильником в течение 48 часов. Летучие вещества удаляют упариванием, остаток растирают с эфиром и продукт отделяют фильтрацией, получая указанное в заголовке соединение. ЯМР: 0,78(т, 3Н), 1,35(кв, 2Н), 2,79(т, 2Н), 7,60(дд, 1Н), 7,84(д, 1Н), 7,92(д, 2Н).
СПОСОБ 31. 2-Амино-5-бром-4-(имидазо[1,2а]пирид-3-ил)пиримидин
Бром (54 мл, 0,0011 моль) добавляют по каплям к раствору 2-амино-4-(имидазо[1,2а]пирид-3-ил)пиримидина (способ 22; 200 мг, 0,95 ммоль) в уксусной кислоте (4 мл) при температуре окружающей среды. Смесь нагревают при 65° С в течение 90 минут и оставляют охлаждаться. Полученный осадок отделяют фильтрацией, промывают гексаном и сушат, получая указанное в заголовке соединение. ЯМР: 7,44(дд, 1Н), 7,90-8,00(м, 2Н), 8,59(с, 1Н), 8,99(с, 1Н), 9,78(д, 1Н); m/z: 290 [MH]+.
СПОСОБ 32. 5-Бромимидазо[1,2а]пиридин
Раствор диэтилацетил бромацетальдегида (50 мл, 0,332 моль) в диоксане (143 мл), воде (85 мл) и концентрированной хлористоводородной кислоте (5 мл) кипятят с обратным холодильником в течение 30 минут и смесь оставляют охлаждаться. Добавляют гидрокарбонат натрия (53 г), затем раствор 5-бром-2-аминопиридина (30 г, 0,174 моль) в диоксане (230 мл) и воде (85 мл) и смесь кипятят с обратным холодильником в течение 24 часов. Смесь оставляют охлаждаться, выливают в воду и подкисляют 2М хлористоводородной кислотой. Смесь промывают этилацетатом и водный слой подщелачивают 2М водным раствором гидроксида натрия. Водную смесь экстрагируют этилацетатом. Экстракты объединяют, сушат и летучие вещества удаляют упариванием. Остаток очищают хроматографией, элюируя смесью гексан/этилацетат (50:50) с увеличением полярности до (25:50), получая указанное в заголовке соединение, 20 г (59%). ЯМР: 7,30(дд, 1Н), 7,54(д, 1Н), 7,59 (с, 1Н), 7, 90 (с, 1Н), 8,89(с, 1Н); m/z: 197 [МН]+.
СПОСОБ 33. 3-Ацетил-5-бромимидазо[1,2а]пиридин
Хлорид алюминия (10,2 г, 77 ммоль) добавляют порциями в течение 10 минут к раствору 5-бромимидазо[1,2 а]пиридина (способ 32; 5,0 г, 26 ммоль) в дихлорметане (100 мл), охлажденному до 0° С. Смесь нагревают до температуры дефлегмации и в течение 15 минут добавляют ацетилхлорид (2,54 мл, 36 ммоль). Смесь кипятят с обратным холодильником в течение 24 часов, охлаждают до 0° С и добавляют дополнительное количество хлорида алюминия (10,2 г, 77 ммоль) и затем ацетилхлорид (3,26 мл). Смесь кипятят с обратным холодильником в течение 24 часов и затем летучие вещества удаляют упариванием. Добавляют ледяную воду, смесь подщелачивают 2М водным раствором гидроксида натрия и экстрагируют этилацетатом. Экстракты промывают водой, сушат и растворитель упаривают, получая указанное в заголовке соединение, которое используют без дополнительной очистки, 4,0 г. ЯМР: 2,58 (с, 3Н), 7,74-7,82(м, 2Н), 8,62(с, 1Н), 9,62(с, 1Н); m/z: 241 [МН]+.
СПОСОБ 34. 5-Бром-3-(3-диметиламинопроп-2-ен-1-оил)-имидазо[1,2а]пиридин
3-Ацетил-5-бромимидазо[1,2а]пиридин (способ 33; 4,0 г) растворяют в DMFDMA (200 мл) и смесь кипятят с обратным холодильником в атмосфере азота в течение 72 часов. Избыток DMFDMA удаляют упариванием, остаток растирают с горячим эфиром, отделяют фильтрацией и промывают эфиром, получая указанное в заголовке соединение, 2,6 г (53%). ЯМР: 2,90(с, 3Н), 3,12(с, 1Н), 5,82(д, 1Н), 7,58(дд, 1Н), 7,64(д, 1Н), 7,70 (с, 1Н), 8,44 (с, 1Н), 9,90(с, 1Н); m/z: 294 [МН]+.
СПОСОБ 35. 2-Амино-4-(5-бромимидазо[1,2а]пирид-3-ил)-пиримидин
Смесь 5-бром-3-(3-диметиламинопроп-2-ен-1-оил)имидазо [1, 2а] -пиридина (способ 34; 2,5 г, 8,5 ммоль), гидрохлорида гуанидина (2,01 г, 21 ммоль) и метоксида натрия (1,83 г, 34 ммоль) в н-бутаноле (140 мл) и метаноле (45 мл) кипятят с обратным холодильником в течение 18 часов. Летучие вещества удаляют упариванием и остаток очищают хроматографией, элюируя смесью дихлорметан/метанол (95:5), получая указанное в заголовке соединение 1,1 г (45%). ЯМР: 6,86 (с, 2Н), 7,12 [д, 1Н), 7,57 (дд, 1Н), 7,68(д, 1Н), 8,22(д, 1Н), 8,51 (с, 1Н) m/z: 290 [MH]+.
СПОСОБ 36. 6-Фенилимидазо[1,2а]пиридин
2-Амино-4-фенилпиридин (0,90 г, 5,29 ммоль) обрабатывают, как описано в способе 32, получая указанное в заголовке соединение. ЯМР: 7,07(д, 1Н), 7,35-7,53(м, 4Н) 7,59(с, 1Н), 7,64(д, 2Н), 7,83 (с, 1Н) 8,18(д, 1Н); m/z: 195 [MH]+.
СПОСОБ 37. 3-Бром-6-фенилимидазо[1,2а]пиридин
Раствор брома (0,24 мл, 4,6 ммоль) в воде (10 мл) добавляют к раствору 6-фенилимидазо[1,2а]пиридина (способ 36; 0,85 г, 4,88 ммоль) в этаноле (15 мл) и смесь перемешивают 14 часов в темноте. Смесь подщелачивают водным раствором гидрокарбоната натрия и экстрагируют дихлорметаном. Экстракты сушат, растворитель удаляют упариванием, остаток растирают с эфиром и отделяют фильтрацией, получая указанное в заголовке соединение. ЯМР: 7,38-7,56(м, 4Н), 7,77(с, 1Н), 7,83(д, 2Н), 7,96(с, 1Н), 8,39(д, 1Н); m/z: 273 [МН]+.
СПОСОБ 38. 3-(3-Диметиламинопроп-2-ен-1-оил)-6-фенилимидазо[1,2 а]пиридин
Фенилмагнийбромид (2,7 мл 1М раствора в THF) добавляют к раствору 3-бром-6-фенилимидазо[1,2 а]пиридина (способ 37; 0,48 г, 1,76 ммоль) в THF в атмосфере азота и смесь кипятят с обратным холодильником в течение 2 часов. Смесь охлаждают до 0° С и по каплям добавляют N-метокси-N-метилацетамид (0,3 мл, 2,64 ммоль). Смесь оставляют нагреваться до температуры окружающей среды и перемешивают в течение 18 часов. Реакционную смесь разбавляют эфиром, промывают водным раствором гидрокарбоната натрия, затем соляным раствором, сушат и летучие вещества удаляют упариванием. Остаток растворяют в DMFDMA (10 мл) и смесь кипятят с обратным холодильником в атмосфере азота в течение 60 часов. Избыток DMFDMA удаляют упариванием, остаток растирают с горячим эфиром, отделяют фильтрацией и промывают эфиром, получая указанное в заголовке соединение, 170 мг (33%). ЯМР: 2,8-3,2(шир.д, 6Н), 5,85(д, 1Н), 7,38-7,58(м, 4Н), 7,67(д, 1Н), 7,86(д, 2Н), 8,00(с, 1Н), 8,48(с, 1Н), 9,76(д, 1Н); m/z: 292 [МН]+.
СПОСОБ 39. 3-(3-Диметиламинопроп-2-ен-1-оил)-2-метил-6-метоксиимидазо[1,2а]пиридин
3-Ацетил-6-метокси-2-метилимидазо[1,2а]пиридин (способ 40; 1,49 г, 7,3 ммоль) и толуолсульфоновую кислоту (5 мг) в DMFDMA (25 мл) кипятят с обратным холодильником в течение 20 часов. Избыток DMFDMA удаляют упариванием. Остаток растирают с эфиром и продукт отделяют фильтрацией, получая указанное в заголовке соединение. ЯМР: 2,69(с, 3Н), 3,28(с, 6Н), 3,82(с, 3Н), 5,44 (д, 1Н), 6,69(дд, 1Н), 6,97(д, 1Н), 7,65(д, 1Н), 9,21(д, 1Н); m/z: 260 [МН]+.
СПОСОБ 40. 3-Ацетил-6-метокси-2-метилимидазо[1,2а] пиридин
Раствор 3-хлорацетоацетона (2,86 мл) в THF (6 мл) добавляют к раствору 2-амино-4-метоксипиридина (2,71 г, 21,8 ммоль) в THF (14 мл) и смесь перемешивают при температуре окружающей среды в течение 30 минут и затем кипятят с обратным холодильником в течение 3 часов. Растворитель удаляют упариванием и остаток очищают хроматографией, элюируя смесью дихлорметан/метанол (100:0) с увеличением полярности до (97:3). Продукт перекристаллизовывают из трет-бутилметилового эфира, получая указанное в заголовке соединение (2,1 г, 47%). ЯМР: 2,05 (с, 3Н), 2,63 (с, 3Н), 3,86 (с, 3Н), 6,83 (дд, 1Н), 7,07(д, 1Н), 9,20(д, 1Н); m/z: 205 [МН]+.
СПОСОБ 41. 4-Сульфамоилфенилгуанидин
Смесь сульфаниламида (20 г, 0,166 моль), бензоилцианамида (34 г, 0,33 моль) в этаноле (60 мл) и концентрированной хлористоводородной кислоты (11 мл) нагревают на паровой бане до упаривания растворителя. Добавляют воду и смесь кипятят с обратным холодильником в течение 5 минут. Добавляют гидроксид натрия (14,4 г) и смесь кипятят с обратным холодильником. Смесь оставляют охлаждаться, доводят до рН 2 хлористоводородной кислотой и осажденное твердое вещество удаляют фильтрацией. Фильтрат нейтрализуют и растворитель удаляют упариванием. Остаток перекристаллизовывают из воды, получая указанный в заголовке неочищенный продукт; m/z: 215 [МН]+.
СПОСОБ 42. 4 -(2-Диэтиламиноэтокси) фенилгуанидин
Смесь нитрата 3,5-диметилпиразолилформидиния (0,20 г, 1 ммоль) и 4-(2-диэтиламиноэтокси) анилина (способ 9; 1,0 г, 4,8 ммоль) в воде (1 мл) кипятят с обратным холодильником в течение 3 часов. Растворитель удаляют упариванием, остаток растирают с горячим эфиром и продукт отделяют фильтрацией, получая указанное в заголовке неочищенное соединение. 1H ЯМР: 0,98(т, 6Н), 2,57(кв, 4Н), 2,79(т, 2Н), 4,00(т, 2Н), 6,99(д, 2Н), 7,15(д, 2Н); m/z: 251 [МН]+.
ПРИМЕР 99
Далее приводятся примеры типичных лекарственных дозированных форм, содержащих соединение формулы (I) или его фармацевтически приемлемую соль, или сложный эфир, способный подвергаться гидролизу in vivo (далее упоминается как соединение X), предназначенных для терапевтического или профилактического применения у людей:
Замечания
Указанные выше композиции могут быть получены с помощью традиционных методик, хорошо известных в области фармацевтики. Таблетки (а)-(с) могут иметь энтеропокрытия, нанесенные с помощью обычных способов, например, с получением покрытия из ацетатфталатцеллюлозы.
Предложено соединение формулы (I):
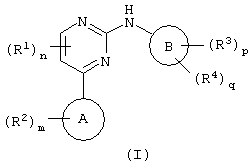
где цикл А представляет собой имидазо[1,2а]пирид-3-ил или пиразол [2, 3а]пирид-3-ил; R2 присоединен к циклическому атому углерода и выбран из галогена, циано, C1-6алкила, C1-6алкокси, C1-6алкилS (О)а, где а равно 0, фенила, фенилтио или (гетероциклическая группа)тио; где любой C1-6алкил, фенил или гетероциклическая группа могут быть необязательно замещенными по атому углерода одним или несколькими G, где гетероциклическая группа является насыщенной, частично насыщенной или ненасыщенной, моно- или бициклической структурой, содержащую 4-12 атомов, из которых по меньшей мере один атом выбран из азота, серы или кислорода, который может быть связан, если не оговорено иначе, с углеродом или азотом, где группа -СН2- может быть необязательно заменена на -С(О)-, циклический атом азота может необязательно нести C1-6алкильную группу и образовывать четвертичное соединение, или циклический атом азота и/или серы необязательно может быть окислен с образованием N-оксида и/или S-оксидов; m равно 0-2; при этом значения R2 могут быть одинаковыми или разными; R1 обозначает галоген, C1-3алкилS(О)а, где а равно 0; где любой C1-3алкил может быть необязательно замещен по атому углерода одним или несколькими J; n равно 0-1. Цикл В представляет собой фенил или фенил, конденсированный с C5-7циклоалкильным циклом; R3 обозначает галоген или сульфамоил; р равно 0-2; при этом значения R3 могут быть одинаковыми или разными; R4 обозначает группу А-Е-; где А выбран из C1-6алкила, фенила, гетероциклической группы, C3-8циклоалкила, фенилC1-6алкила, (гетероциклическая группа)-C1-6алкила или C3-8циклоалкилC1-6алкила; причем C1-6алкил, фенил, гетероциклическая группа, C3-8циклоалкил, фенилC1-6алкил, (гетероциклическая группа) C1-6алкил или C3-8циклоалкилC1-6алкил могут быть необязательно замещены по атому углерода одним или несколькими D; и где, если упомянутая гетероциклическая группа содержит -NH- фрагмент, то азот может быть необязательно замещен группой, выбранной из R; Е обозначает прямую связь или -О-, -С(О)-, -N(Ra)C(O)- или -N(Ra)SO2-, -S(O)r-; где Ra обозначает водород или C1-6алкил, а r равно 0-2; D независимо выбран из гидрокси, амино, C1-6алкокси, N-(C1-6алкил) амино, N,N-(C1-6алкил)2амино, C1-6алкоксикарбониламино и бензилоксикарбониламино; где любой C1-6алкил или фенил может быть необязательно замещен по атому углерода одним или несколькими К; q равно 0-1; G, J и К независимо выбраны из гидрокси, диметиламино, диэтиламино; и R выбран из C1-4алкила; или его фармацевтически приемлемая соль. Предложено применение пиримидиновых соединений в ингибировании действия киназ клеточного цикла CDK2, CDK4 и CDK6 и соответственно обладают антипролиферативными свойствами. Указанные свойства имеют ценность при лечении раковых заболеваний (твердые опухоли и лейкемии), фибропролиферативных и дифференционных нарушений, псориаза, ревматоидного артрита, саркомы Капоши, гемангиомы, острой и хронической нефропатии, атеромы, атеросклероза, артериального повторного стеноза, аутоиммунных заболеваний, острых и хронических воспалений, костных и глазных заболеваний с пролиферацией сосудов клетчатки. 7 н. и 15 з.п. ф-лы.
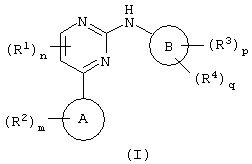
где
цикл А представляет собой имидазо[1,2а]пирид-3-ил или пиразол [2, 3а]пирид-3-ил;
R2 присоединен к циклическому атому углерода и выбран из галогена, циано, C1-6алкила, C1-6алкокси, C1-6алкилS (О)а, где а равно 0, фенила, фенилтио или (гетероциклическая группа)тио;
где любой C1-6алкил, фенил или гетероциклическая группа могут быть необязательно замещенными по атому углерода одним или несколькими G, где гетероциклическая группа является насыщенной, частично насыщенной или ненасыщенной, моно- или бициклической структурой, содержащую 4-12 атомов, из которых по меньшей мере один атом выбран из азота, серы или кислорода, который может быть связан, если не оговорено иначе, с углеродом или азотом, где группа -СН2- может быть необязательно заменена на -С(О)-, циклический атом азота может необязательно нести C1-6алкильную группу и образовывать четвертичное соединение, или циклический атом азота и/или серы необязательно может быть окислен с образованием N-оксида и/или S-оксидов;
m равно 0-2; при этом значения R2 могут быть одинаковыми или разными;
R1 обозначает галоген, C1-3алкилS(О)а, где а равно 0; где любой C1-3алкил может быть необязательно замещен по атому углерода одним или несколькими J;
n равно 0-1;
цикл В представляет собой фенил или фенил, конденсированный с C5-7циклоалкильным циклом;
R3 обозначает галоген или сульфамоил;
р равно 0-2; при этом значения R3 могут быть одинаковыми или разными;
R4 обозначает группу А-Е-; где
А выбран из C1-6алкила, фенила, гетероциклической группы, C3-8циклоалкила, фенилC1-6алкила, (гетероциклическая группа)-C1-6алкила или C3-8циклоалкилC1-6алкила; причем C1-6алкил, фенил, гетероциклическая группа, C3-8циклоалкил, фенилC1-6алкил, (гетероциклическая группа) C1-6алкил или C3-8циклоалкилC1-6алкил могут быть необязательно замещены по атому углерода одним или несколькими D; и где, если упомянутая гетероциклическая группа содержит -NH- фрагмент, то азот может быть необязательно замещен группой, выбранной из R;
Е обозначает прямую связь или -О-, -С(О)-, -N(Ra)C(O)- или -N(Ra)SO2-, -S(O)r- ; где Ra обозначает водород или C1-6алкил, а r равно 0-2;
D независимо выбран из гидрокси, амино, C1-6алкокси, N-(C1-6алкил) амино, N,N-(C1-6алкил)2амино, C1-6алкоксикарбониламино и бензилоксикарбониламино; где любой
C1-6алкил или фенил может быть необязательно замещен по атому углерода одним или несколькими К;
q равно 0-1;
G, J и К независимо выбраны из гидрокси, диметиламино, диэтиламино; и
R выбран из C1-4алкила;
или его фармацевтически приемлемая соль.
или его фармацевтически приемлемая соль.
или его фармацевтически приемлемая соль.
или его фармацевтически приемлемая соль.
или его фармацевтически приемлемая соль.
3-N,N-диметиламино-2-гидроксипропокси, фенокси,
N-метилкарбамоил, N,N-диметилкарбамоил,
N-(3-имидазол-1-илпропил)карбамоил,
N-[3-(2-оксо-пирролидин-1-ил)пропил]карбамоил,
3,5-диоксапиперидин-1-илсульфонил,
N-циклопропилсульфамоил,
N-циклопропилметилсульфамоил, анилиносульфонил,
N-пиримидин-2-илсульфамоил, N-метилсульфамоил,
N-пропилсульфамоил, N-(2-метоксиэтил)сульфамоил,
N-(2-метиламиноэтил)сульфамоил,
N-(2-изопропиламиноэтил)сульфамоил,
N-(2-диметиламиноэтил)сульфамоил,
N-(2-диэтиламиноэтил)сульфамоил,
N-[2-(гидроксиэтиламино)этил]сульфамоил,
N-[2-(диэтиламиноэтил)этил]сульфамоил,
N-(пирролидин-1-илэтил)сульфамоил,
N-[2-(1-метилпирролидин-2-ил)этил]сульфамоил,
N-(2-пиперидин-1-илэтил)сульфамоил,
N-(2-пиперазин-1-илэтил)сульфамоил,
N-(2-морфолинэтил)сульфамоил,
N-(2-имидазол-4-илэтил)сульфамоил,
N-(3-гидроксипропил)сульфамоил,
N-(2,3-дигидроксипропил)сульфамоил,
N-(3-метоксипропил)сульфамоил,
N-(3-аминопропил)сульфамоил,
N-(3-метиламинопропил)сульфамоил,
N-(3-диметиламинопропил)сульфамоил,
N-(3-диэтиламинопропил)сульфамоил,
N-(3-изопропиламинопропил)сульфамоил,
N-(3-трет-бутоксикарбониламинопропил)сульфамоил,
N-(3-бензилоксикарбониламинопропил)сульфамоил,
N-[3-(2-оксопирролидин-1-ил)пропил]сульфамоил,
N-(3-морфолинпропил)сульфамоил,
N-[3-(4-метилпиперазин-1-ил)пропил]сульфамоил,
N-(3-имидазол-1-илпропил)сульфамоил или
N-(5-гидроксипентил)сульфамоил; и
q равно 1;
или его фармацевтически приемлемая соль.
или его фармацевтически приемлемая соль.
2-(4-сульфамоиланилин)-4-(имидазо[1,2а]пирид-3-ил)пиримидина;
2-[4-(N-метилсульфамоил)анилин]-4-(имидазо[1,2а]пирид-3-ил)пиримидина;
2-{4-[N-(2-метоксиэтил)сульфамоил]анилин}-4-(имидазо[1,2а]-пирид-3-ил)пиримидина;
2-{4-[N-(3-метоксипропил)сульфамоил]анилин}-4-(имидазо-[1,2 а]пирид-3-ил)пиримидина;
2-{4-[N-(3-изопропиламинопропил)сульфамоил]анилин}-4-(имидазо[1,2а]пирид-3-ил)пиримидина;
2-{4-[N-(3-диметиламинопропил)сульфамоил]анилин}-4-(имидазо[1,2 а] пирид-3-ил)пиримидина;
2-{4-[N-(2-диметиламиноэтил)сульфамоил]анилин}-4-(имидазо-[1,2 а]пирид-3-ил)пиримидина;
2-{4-[N-(2-метиламиноэтил)сульфамоил]анилин}-4-(имидазо-
[1,2 а]пирид-3-ил)пиримидина или
2-{4-[N-(2-метоксиэтил)сульфамоил]анилин}-4-[5- (2-гидроксиэтилтио)имидазо[1,2а]пирид-3-ил]пиримидина;
или его фармацевтически приемлемая соль.
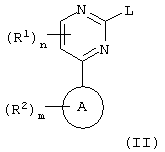
где L является способной к замещению группой, вводят в реакцию с амином формулы (III)
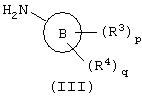
и затем, при необходимости:
i) соединения формулы (I) превращают в другое соединение формулы (I);
ii) удаляют все защитные группы;
iii) получают фармацевтически приемлемую соль.
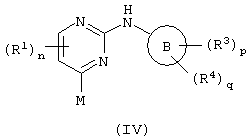
вводят в реакцию с соединением формулы (V)
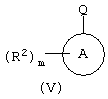
где один из М и Q является способной к замещению группой X, а
другой является металлическим реагентом Y;
и затем, при необходимости:
i) соединения формулы (I) превращают в другое соединение
формулы (I);
ii) удаляют все защитные группы;
iii) получают фармацевтически приемлемую соль.
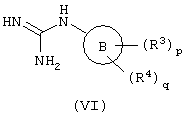
вводят в реакцию с соединением формулы (VII)
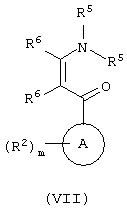
где R5 обозначает C1-6алкил, a R6 обозначает водород или R1;
и затем, при необходимости:
i) соединения формулы (I) превращают в другое соединение формулы (I);
ii) удаляют все защитные группы;
iii) получают фармацевтически приемлемую соль.
| US 5521184 A, 28.05.1996 | |||
| WO 9640143 A, 19.12.1996 | |||
| JP 63135382 A, 07.06.1988 | |||
| ИМИДАЗО-АННЕЛИРОВАННЫЕ ИЗО- И ГЕТЕРОЦИКЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2076105C1 |
| ПРОИЗВОДНЫЕ ДИФЕНИЛМЕТИЛИМИДАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1990 |
|
RU2024521C1 |

