Изобретение относится к производным 1,2,3,4-тетрагидробензофуро[3,2-с] пиридина, обладающим антагонистической активностью в отношении центральных α2-адренорецепторов. Оно также относится к их получению, содержащих их композициях, а также к их применению в качестве лекарственных средств.
Известно, что антагонисты центральных α2-адренорецепторов увеличивают высвобождение норадреналина путем блокирования пресинаптических α2-рецепторов, которые осуществляют ингибирующий контроль над высвобождением этого нейромедиатора. Благодаря повышению концентраций норадреналина, α2-антагонисты можно использовать в клинике для лечения или профилактики депрессии, когнитивных расстройств, болезни Паркинсона, сахарного диабета, сексуальной дисфункции и импотенции, повышенного внутриглазного давления и заболеваний, связанных с нарушением перистальтики кишечника, поскольку все эти состояния связаны с дефицитом норадреналина в центральной или периферической нервной системе.
Соединения настоящего изобретения являются новыми и обладают специфичной и селективной аффинностью к различным известным подтипам α2-адренорецепторов, т.е. к α2A-, α2B- и α2C-адренорецепторам.
Настоящее изобретение относится к соединениям формулы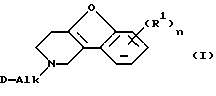
их N-оксидным формам, фармацевтически приемлемым солям присоединения и стереохимически изомерным формам, где:
каждый R1 независимо представляет водород, галоген, C1-6 алкил, нитро, гидрокси или C1-4 алкилокси;
Alk обозначает C1-6 алкандиил;
n равно 1 или 2;
D представляет 1- или 2-бензимидазолил, 2(3Н)бензоксазолон-3-ил или радикал формулы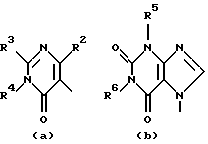
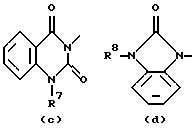
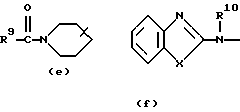
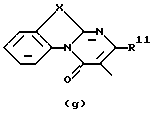
где каждый Х независимо представляет О, S или NR12;
R2 представляет водород, C1-6 алкил, арил или apил C1-6 алкил;
R3 представляет водород, C1-6 алкил, C1-6 алкилокси, C1-6 алкилтио, амино или моно- или ди(С1-6 алкил)амино;
R4, R5, R6, R7, R8, R10, R11 и R12 каждый независимо представляет водород или C1-6 алкил;
R9 представляет водород, C1-6 алкил или арил; или R3 и R4, взятые вместе, могут образовывать двухвалентный радикал -R3-R4- формулы
- CH2-CH2-CH2- (а-1);
- СН2-СН2-СН2-СН2- (а-2);
- СН=СН-СН2- (а-3);
- СН2-СН=СН- (а-4) или
- СН=СН-СН=СН (а-5);
где один или два атома водорода указанных радикалов от (а-1) до (а-5) каждый независимо могут быть заменены галогеном, С1-6 алкилом, арил С1-6 алкилом, трифторметилом, амино, гидрокси, C1-6 алкилокси или C1-10 алкилкарбонилокси; или, где возможно, два геминальных атома водорода могут быть заменены C1-6 алкилиденом или арил С1-6 алкилиденом; или
-R3-R4- может также представлять
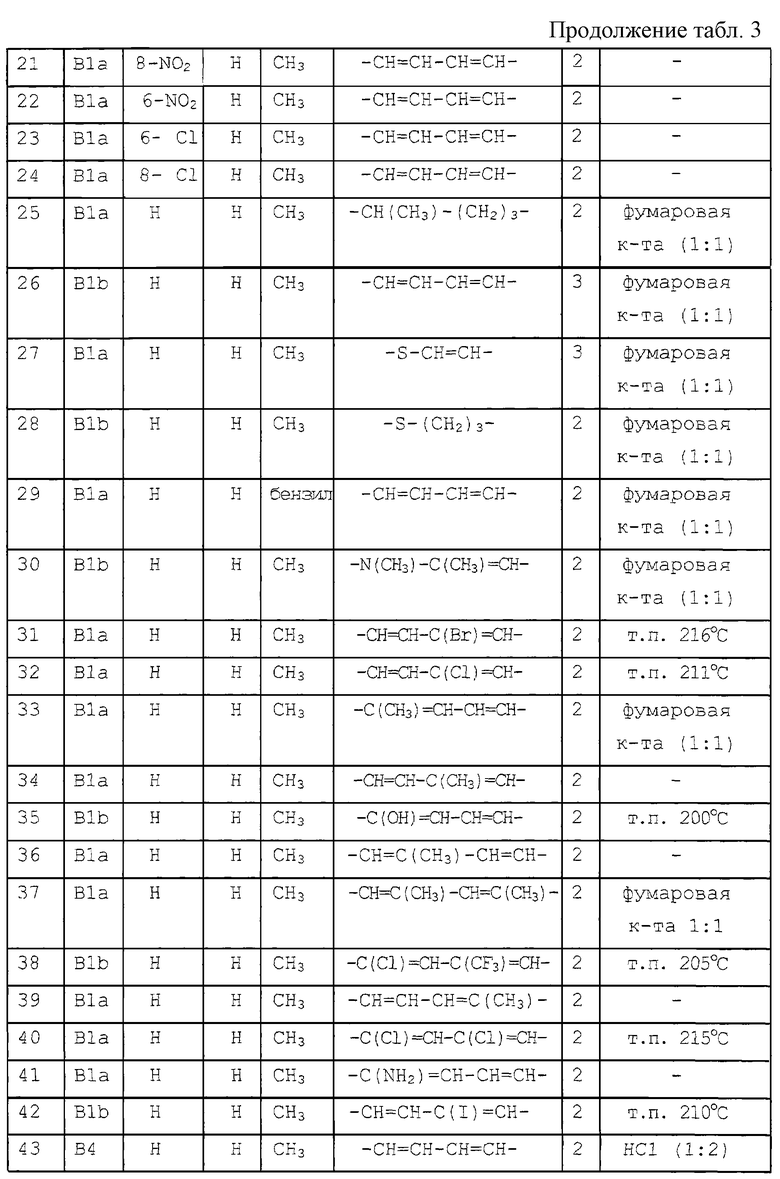
-S-CH2-CH2- (а-6);
-S-CH2-CH2-CH2- (а-7);
-S-СН=СН- (а-8);
-NH-CH2-CH2- (а-9);
-NH-CH2-CH2-CH2 (а-10);
-NH-CH=CH- (а-11);
-NH-CH=N- (a-12);
-S-CH=N (a-13) или
-СН=СН-O- (а-14),
где один или, где возможно, два или три атома водорода указанных радикалов от (а-6) до (а-14) каждый, независимо, могут быть заменены C1-6 алкилом или арилом; и арил представляет фенил, или фенил, замещенный галогеном или C1-6 алкилом.
Употребляемый в предыдущих определениях термин галоген относится к фтору, хлору, брому и иоду. Термин C1-6 алкил определяет прямые и разветвленные насыщенные углеводороды, имеющие от 1 до 6 атомов углерода, такие как, например, метил, этил, пропил, бутил, 1-метилэтил, 1,1-диметилэтил, 2-метилпропил, пентил, гексил и т.п. Термин C1-10 алкил включает C1-6 алкильные радикалы и их высшие гомологи, имеющие от 7 до 10 атомов углерода, такие как, например, гептил, октил, нонил, децил и т.п. Термин C1-6 алкандиил определяет двухвалентные прямые или разветвленные алкандиильные радикалы, имеющие от 1 до 6 атомов углерода, такие как, например, метилен, 1,2-этандиил, 1,3-пропандиил, 1,4-бутандиил, 1,5-пентандиил, 1,6-гександиил и т.п.; термин C1-6 алкилиден определяет двухвалентные прямые или разветвленные алкилиденовые радикалы, имеющие от 1 до 6 атомов углерода, такие как, например, метилен, этилиден, 1-пропилиден, 1-бутилиден, 1-пентилиден, 1-гексилиден и т.п.
Соли присоединения, указанные в настоящем документе, включают терапевтически активные соли присоединения, которые (соединения формулы (I)) способны образовывать с подходящими кислотами, такими как, например, неорганические кислоты, такие как галогенводородные кислоты, например, хлорводородная или бромводородная кислота; серная, азотная, фосфорная и т.п. кислоты; или органические кислоты, такие как, например, уксусная, пропановая, гидроксиуксусная, молочная, пировиноградная, щавелевая, малоновая, янтарная, малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памоевая и т.п. кислоты.
Фармацевтически приемлемые соли присоединения, указанные в настоящем документе, включают терапевтически активные нетоксичные соли присоединения оснований, в частности, металлов или аминов, которые способны образовывать соединения формулы (I). Указанные соли можно удобно получить действием на соединения формулы (I), содержащие кислотные водородные атомы, подходящими органическими и неорганическими основаниями, такими как, например, соли аммония, соли щелочных и щелочноземельных металлов, например, лития, натрия, калия, магния, кальция и т.п., соли с органическими основаниями, например, бензатином, N-метил-D-глюкамином, гидрабаминовые соли и соли с аминокислотами, такими как, например, аргинин, лизин и т.п.
Наоборот указанные солевые формы можно получать действием подходящего основания или кислоты на свободную кислотную или основную форму.
Термин соль присоединения, указанный в настоящем документе, также включает сольваты, которые соединения формулы (I) способны образовывать, и указанные сольваты также входят в объем настоящего изобретения. Примерами таких сольватов являются, например, гидраты, алкоголяты и т.п.
N-оксидные формы соединений формулы (I) включают такие соединения формулы (I), в которых один или несколько атомов азота окислены до так называемого N-оксида.
Термин стереохимически изомерные формы, используемый в настоящем документе, определяет все возможные изомерные формы, в которых соединения формулы (I) могут существовать. Если не указано иное, химическое обозначение соединений обозначает смесь всех возможных стереохимически изомерных форм; указанные смеси содержат все диастереоизомеры и энантиомеры основной молекулярной структуры.
Некоторые из соединений формулы (I) могут также существовать в таутомерных формах. Такие формы, хотя и не указаны ясно в вышеприведенной формуле, также входят в объем настоящего изобретения.
Где бы в настоящем описании не использовался термин соединения формулы (I), подразумевается, что он включает также N-оксидные формы, фармацевтически приемлемые соли присоединения и все стереоизомерные формы.
Особую группу соединений представляют соединения формулы (I), в которых n равно 1, a R1 представляет водород, галоген, C1-6 алкил или нитро.
Интересную группу соединений представляют соединения формулы (I), в которых n равно 1, а R1 представляет водород, хлор, фтор, метил или нитро, особенно, где R1 представляет водород; или в которых n равно 2, а R1 представляет метокси.
Еще одну интересную группу соединений представляют соединения формулы (I), в которых Alk представляет метилен, 1,2-этандиил, 1,3-пропандиил, 1,4-бутандиил или 1,5-пентандиил.
Еще одну интересную группу соединений представляют соединения формулы (I), в которых D представляет 1-бензимидазолил; 2(3Н)бензоксазолон-3-ил или D представляет радикал формулы (а), в котором R3 представляет C1-6 алкилтио, а R4 представляет C1-6 алкил; или в котором R3 и R4, взятые вместе, образуют двухвалентный радикал формулы (а-2) или (а-5), где один или два атома водорода указанных радикалов, каждый, независимо, могут быть заменены галогеном, C1-6 алкилом, арил С1-6 алкилом, трифторметилом, амино, гидрокси, C1-6 алкилокси или C1-10 алкилкарбонилокси; или, где возможно, два геминальных атома водорода могут быть заменены C1-6 алкилиденом или арил С1-6 алкилиденом; или двухвалентный радикал формулы (а-6), (а-7), (а-8), (а-11) или (а-14), где один или, где возможно, два или три атома водорода указанных радикалов, каждый независимо, могут быть заменены C1-6 алкилом или арилом; или D представляет радикал формулы (b), в котором R5 и R6 представляют C1-6 алкил; или D представляет радикал формулы (с), в котором R7 представляет водород; или D представляет радикал формулы (d), в котором R8 представляет водород или C1-6 алкил; или D представляет радикал формулы (е), в котором R9 представляет арил; или D представляет радикал формулы (f), в котором Х представляет S, а R10 представляет водород; или D представляет радикал формулы (d), в котором Х представляет S, а R11 представляет C1-6 алкил.
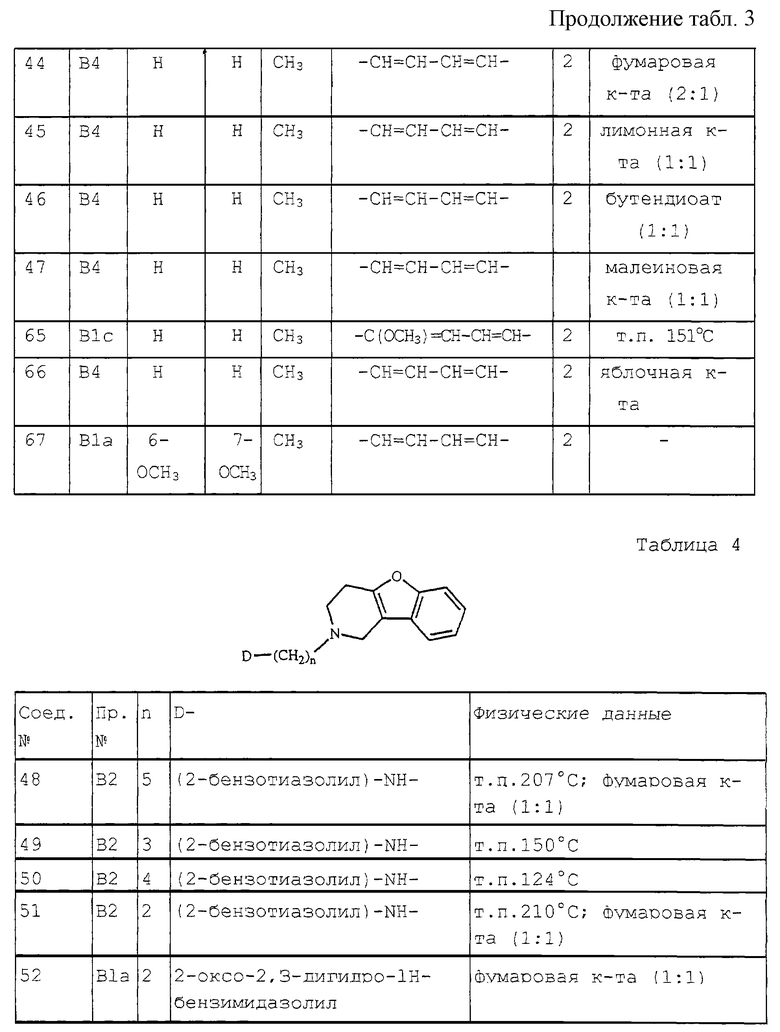
Особые соединения представляют соединения формулы (I), в которых D представляет 2(3Н)бензоксазолон-3-ил или D представляет радикал формулы (а), в котором R3 представляет метилтио, а R4 представляет метил; или в котором R3 и R4, взятые вместе, образуют двухвалентный радикал формулы (а-2) или (а-5), где один или два атома водорода, каждый, независимо, могут быть заменены галогеном, C1-6 алкилом, C1-6 алкилокси, арил С1-6 алкилом, трифторметилом, амино или гидрокси; или где два геминальных атома водорода могут быть заменены арил С1-6 алкилиденом; или в котором R3 и R4, взятые вместе, образуют двухвалентный радикал формулы (а-6), (а-7), (а-8), (а-11) или (а-14), где один или, где возможно, два или три атома водорода заменены C1-6 алкилом; или D представляет радикал формулы (b), в котором R5 и R6 представляют метил; или D представляет радикал формулы (с), в котором R7 представляет водород; или D представляет радикал формулы (d), в котором R8 представляет водород; или D представляет радикал формулы (е), в котором R9 представляет арил, присоединенный к Alk в 4 положении пиперидиновой части; или D представляет радикал формулы (f), в котором Х представляет S, a R10 представляет водород; или D представляет радикал формулы (д), в котором Х представляет S, а R11 представляет метил.
Предпочтительными являются такие соединения формулы (I), в которых n равно 1, R1 представляет водород, а D представляет радикал формулы (а), в котором R3 и R4, взятые вместе, образуют двухвалентный радикал формулы (а-2) или (а-5), где один или два атома водорода, каждый, независимо могут быть заменены галогеном, метилом, метокси, арилметилом, трифторметилом, амино или гидрокси; или где два геминальных атома водорода могут быть заменены арилметиленом; или в котором R3 и R4, взятые вместе, образуют двухвалентный радикал формулы (а-6), (а-7), (а-8), (а-11) или (а-14), где один или, где возможно, два или три атома водорода заменены метилом.
Наиболее предпочтительными являются следующие соединения:
3-[2-[3,4-дигидробензофуро[3,2-с] пиридин-2(1Н)-ил] этил]-2-метил-4Н-пиридо[1,2-а]пиримидин-4-он;
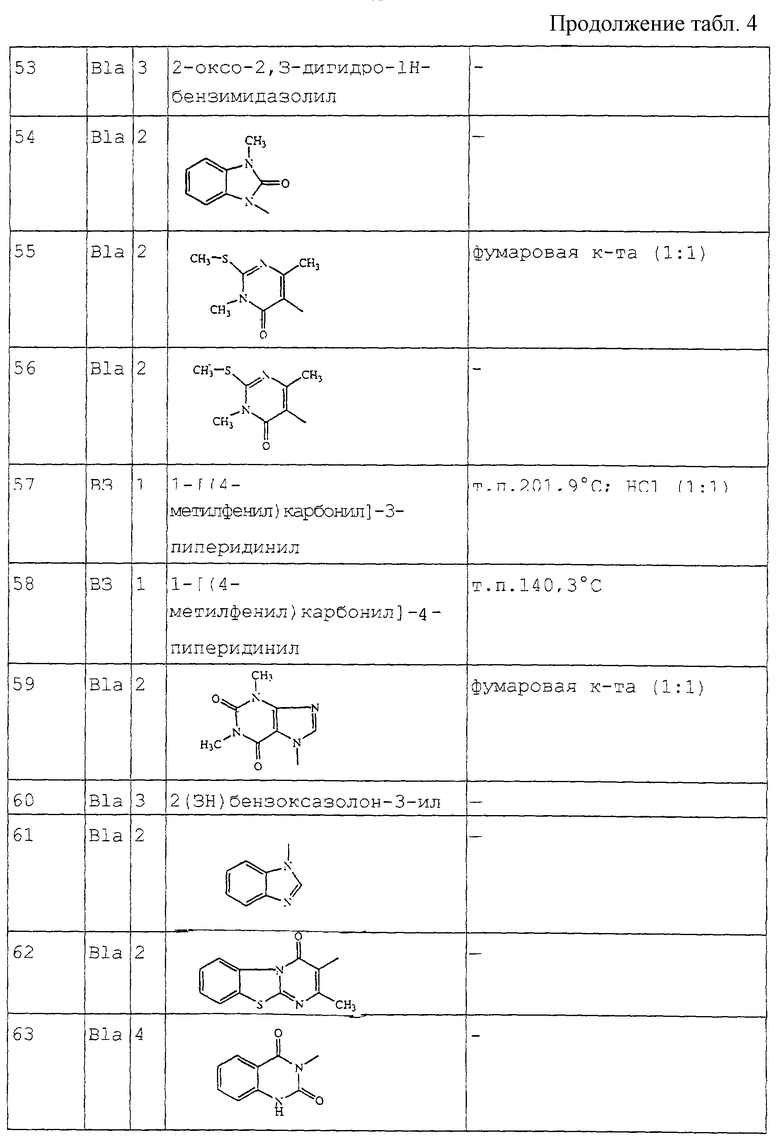
6-[[3,4-дигидробензофуро[3,2-с] пиридин-2(1Н)-ил] этил] -7-метил-3Н-тиазоло[3,2-а]пиримидин-5-он;
6-[[3,4-дигидробензофуро[3,2-е] пиридин-2(1Н)-ил] этил]-3,7-диметил-5Н-тиазоло[3,2-а]пиримидин-5-он;
3-[2-[3,4-дигидробензофуро[3,2-е] пиридин-2(1Н)-ил] этил] -2,7-диметил-4Н-пиридо[1,2-а] пиримидин-4-он; а также их N-оксиды, фармацевтически приемлемые соли присоединения и стереохимически изомерные формы.
Соединения формулы (I) обычно могут быть получены путем N-алкилирования производного 1, 2, 3, 4-тетрагидробензофурано[3,2-с]пиридина формулы (II) алкилирующим реагентом формулы (III), согласно процедуре, описанной в ЕР-А-0 037 265, ЕР-А-0 070 053, ЕР-А-0 196 132 и ЕР-А-0 378 255.
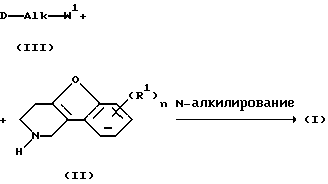
В промежуточном соединении (III) W1 представляет подходящую реакционную удаляемую группу, такую как, например, галоген, например, хлор, бром или иод; сульфонилокси, например, метансульфонилокси, 4-метилбензолсульфонилокси.
В этой и последующих реакциях продукты реакции можно выделить из реакционной среды и, если необходимо, подвергнуть дальнейшей очистке согласно известным методикам, таким как экстракция, кристаллизация, растирание и хроматография.
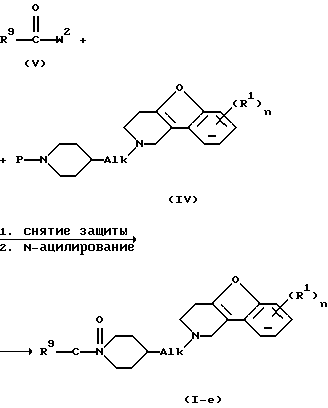
Соединения формулы (I), где D представляет радикал формулы (а), представлены формулой (1-а), могут быть получены снятием защиты с промежуточного N-защищенного вещества формулы (IV), в котором Р представляет защитную группу, такую как, например, C1-4 алкилоксикарбонил, и последующим N-ацилированием полученного промежуточного продукта ацильным производным формулы (V), в котором W2 представляет подходящую реакционную удаляемую группу, такую как, например, галоген.
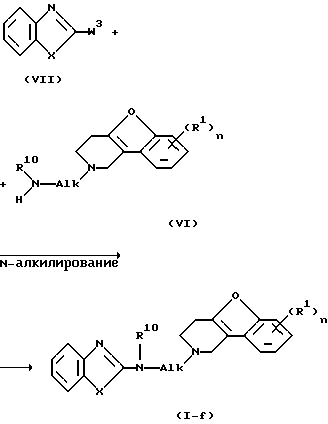
Соединения формулы (I), в которых D представляет радикал формулы (f), представленные формулой (I-f), могут быть получены N-алкилированием амина формулы (VI) промежуточным продуктом формулы (VII), где W3 представляет подходящую реакционную удаляемую группу, такую как, например, галоген.
Соединения формулы (I) могут быть преобразованы друг в друга с помощью известных способов преобразования функциональных групп.
Соединения формулы (I) могут быть также преобразованы в соответствующие N-оксидные формы посредством следующих известных способов преобразования трехвалентного азота в его N-оксидную форму. Указанная реакция N-окисления обычно может быть осуществлена путем взаимодействия исходного продукта формулы (I) с подходящим органическим или неорганическим пероксидом. Подходящие неорганические пероксиды включают, например, пероксид водорода, пероксиды щелочных или щелочноземельных металлов, например, пероксид натрия, пероксид калия; подходящие органические пероксиды могут включать пероксикислоты, такие как, например, бензолкарбопероксикислоту или галогензамещенную бензолкарбопероксикислоту, например, 3-хлорбензолкарбопероксикислоту, пероксиалкановые кислоты, например, пероксиуксусную кислоту, алкилгидропероксиды, например, трет-бутилгидропероксид. Подходящими растворителями являются, например, вода, низшие спирты, например, этанол и т.п., углеводороды, например, толуол, кетоны, например, 2-бутанон, галогенированные углеводороды, например, дихлорметан, и смеси этих растворителей.
Ряд промежуточных соединений и исходных материалов являются коммерчески доступными или являются известными соединениями, которые можно получить с помощью известных методик.
Например, некоторые из промежуточных соединений формулы (III) и их получение описаны в ЕР-А-0 037 265, ЕР-А-0 070 053, ЕР-А-0 196 132 и ЕР-А-0 378 255.
Промежуточные соединения формулы (II) можно получить по способам, описанным Cattanach С. et al. (J. Chem. Soc (С), 1971, 53-60); Карташова Т (Хим. Гетероцикл. Соедин., 1979 (9), 1178-1180) и Закусов и др. (Изобретения, 1992 (15) 247).
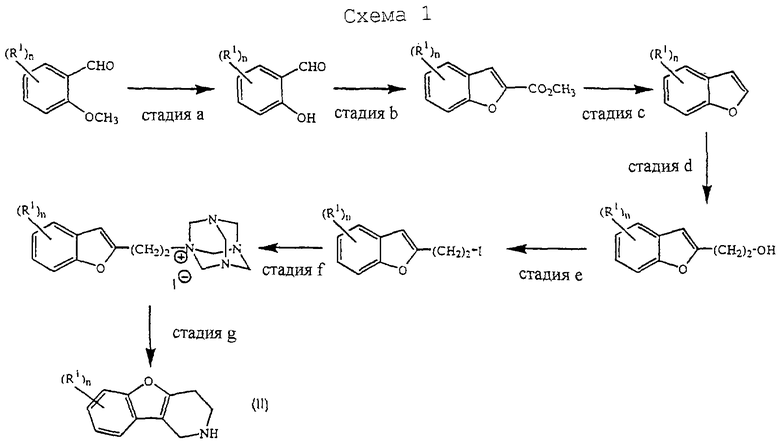
Конкретный путь синтеза для получения промежуточных соединений формулы (II) изображен на схеме 1, приведенной в конце описания.
Стадию а можно выполнить аналогично способу, описанному в Tetrahedron (1981), 37, 979-982. Бензофураны, полученные на стадии с, использовались в качестве промежуточных соединений в US 4 210 655. Следующие этапы реакции аналогичны способам, описанным в US 3 752 820.
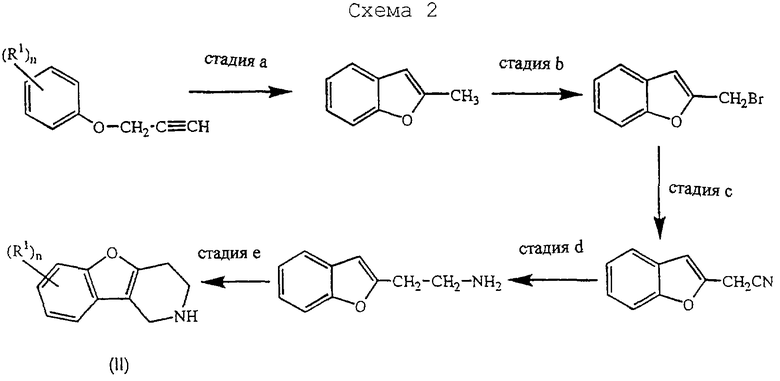
Альтернативно, промежуточные соединения формулы (II) можно получить на стадиях реакции, изображенных на схеме 2, приведенной в конце описания.
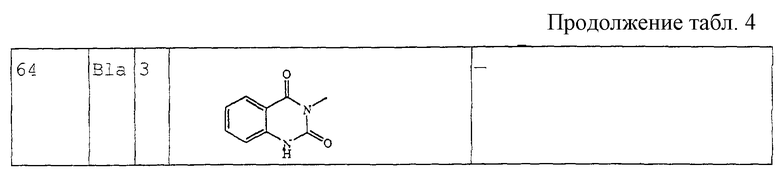
Стадию а можно проводить аналогично способу, описанному в Heterocycles (1994), 39(1), 371-380. Стадию b можно осуществлять аналогично способу, описанному в J. Med. Chem. (1986), 29(9), 1643-1650. Следующие стадии реакции можно проводить аналогично способам, описанным в J. Heterocycl. Chem. (1979), 16, 1321.
Некоторые из соединений формулы (I) и некоторые из промежуточных соединений в настоящем изобретении содержат по меньшей мере один асимметрический атом углерода. Чистые стереохимически изомерные формы указанных соединений и указанных промежуточных соединений можно получить с помощью известных методов. Например, диастереоизомеры можно разделить с помощью физических методов, таких как селективная кристаллизация или хроматография, например, противоточное распределение, жидкостная хроматография и т.п. методы. Энантиомеры можно получить из рацемических смесей путем преобразовывания сначала указанных рацемических смесей с помощью подходящих разделяющих агентов, таких как, например, хиральные кислоты, в смеси диастереоизомерных солей или соединений; затем физического разделения указанных смесей диастереоизомерных солей или соединений путем, например, селективной кристаллизации или хроматографии, например, жидкостной хроматографии и т.п. методов; и, наконец, преобразовывания указанных разделенных диастереоизомерных солей или соединений в соответствующие энантиомеры.
Чистые стереохимически изомерные формы соединений формулы (I) можно получить также из чистых стереохимически изомерных форм соответствующих промежуточных соединений и исходных материалов, при условии, что эти реакции протекают стереоспецифическим образом. Чистые и смешанные стереохимически изомерные формы соединений формулы (I) также входят в объем настоящего изобретения.
Соединения формулы (I), их N-оксидные формы, фармацевтически приемлемые соли присоединения и стереохимически изомерные формы блокируют пресинаптические аг-рецепторы на центральных норадренергических нейронах, увеличивая, таким образом, высвобождение норадреналина. Блокирование указанных рецепторов будет подавлять или облегчать множество разнообразных симптомов, связанных с дефицитом норадреналина в центральной или периферической нервной системе. Терапевтическими показаниями к применению настоящих соединений являются депрессия, когнитивные расстройства, болезнь Паркинсона, сахарный диабета, сексуальная дисфункция и импотенция, а также повышенное внутриглазное давление.
Показано также, что блокирование α2-рецепторов в центральной нервной системе усиливает высвобождение серотонина, который также может внести свой вклад в лечение депрессии (Maura et al., 1992, Naunyn-Schmiedeberg's Arch. Pharmacol., 345: 410-416).
Показано также, что блокирование α2-рецепторов с помощью настоящих соединений индуцирует повышение внеклеточной 3,4-дигидрофенилуксусной кислоты, которая является метаболитом дофамина и норадреналина.
С точки зрения пригодности соединений настоящего изобретения для лечения заболеваний, связанных с дефицитом норадреналина в центральной нервной системе, в частности, депрессии и болезни Паркинсона, настоящее изобретение относится к способу лечения теплокровных животных, страдающих этими заболеваниями, в частности, депрессией и болезнью Паркинсона; указанный способ включает системное введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли присоединения.
Настоящие соединения также потенциально пригодны для лечения болезни Альцгеймера и деменции, поскольку известно, что α2-антагонисты стимулируют высвобождение ацетилхолина (Tellez et al., 1997, J. Neurochem. 68:778-785).
В общих чертах, ожидается, что терапевтически эффективное суточное количество составит приблизительно от 0,01 до 4 мг/кг веса тела.
Настоящее изобретение, таким образом, относится также к соединениям формулы (I), определенным выше, предназначенным для применения в качестве лекарственного средства. Помимо этого, настоящее изобретение относится также к применению соединения формулы (I) для получения лекарственного средства для лечения депрессии и болезни Паркинсона.
Исследования, посвященные изучению трансдукции сигнала рецепторов и связывания рецепторов, как ex vivo, так и in vitro, и способности реверсировать индуцированное клонидином сокращение стимулированного электрическим раздражением высвобождения норадреналина из коры головного мозга кроликов, можно использовать для оценки антагонизма настоящих соединений в отношении α2-адренорецепторов. В качестве показателей блокады центральных α2-адренорецепторов in vivo можно использовать реверсирование угнетения рефлекса выпрямления, наблюдающегося у крыс после внутривенной инъекции ксилазина, и ингибирование тремора, индуцированного резерпином.
В тесте на доминирование в сообществе, в котором крысы должны конкурировать за раствор сахарозы для питья, соединения настоящего изобретения были способны усиливать конкурентное поведение подчиненных особей.
Настоящие соединения также демонстрируют воздействие на кишечник; они реверсируют противодиарейный эффект клонидина и стимулируют выделение фекалий у накормленных крыс. У собак они способны ускорять начало индуцированной магнезией диареи, а в тесте на опорожнение желудка они обладают свойством реверсировать задержку эвакуации желудочного содержимого, индуцированную α2-агонистом лидамидином. Таким образом, настоящие соединения пригодны также для лечения заболеваний, связанных с нарушением перистальтики кишечника.
Соединения настоящего изобретения также обладают способностью быстро проникать в центральную нервную систему.
Для удобства введения настоящие соединения можно помещать в различные фармацевтические композиции, включающие фармацевтически приемлемый носитель и, в качестве активного ингредиента, терапевтически эффективное количество соединения формулы (I). Для изготовления фармацевтических композиций настоящего изобретения эффективное количество конкретного соединения в виде соли присоединения или в свободном виде кислоты или основания в качестве активного ингредиента, объединяют с гомогенной смесью фармацевтически приемлемого носителя, который может принимать широкое разнообразие форм, в зависимости от желательной формы препарата для определенного пути введения. Эти фармацевтические композиции желательно изготавливать в форме стандартных лекарственных форм, удобных, предпочтительно, для перорального, чрескожного или парентерального введения. Например, при изготовлении композиции в виде лекарственной формы для перорального введения можно использовать любую из обычных фармацевтических сред, таких как, например, вода, гликоли, масла, спирты и т.п., в случае жидких препаратов для перорального введения, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие агенты, связывающие агенты, разрыхлители и т.п., в случае изготовления порошков, пилюль, капсул и таблеток. Ввиду легкости применения таблетки и капсулы представляют наиболее выгодную дозированную лекарственную форму для перорального введения, для изготовления которой, разумеется, используются твердые фармацевтические носители. Для изготовления композиций для парентерального введения носитель обычно включает стерильную воду, по меньшей мере, большую его часть, хотя можно включать в состав и другие ингредиенты, например, агенты для улучшения растворимости. Растворы для инъекций, например, можно изготавливать с включением в носитель физиологического раствора, раствора глюкозы и смеси этих растворов. Растворы для инъекций, содержащие соединения формулы (I), можно изготавливать на масляной основе, обеспечивающей пролонгированное действие. Подходящими маслами для этой цели являются, например, арахисовое масло, кунжутное масло, масло семян хлопчатника, кукурузное масло, соевое масло, синтетические сложные эфиры глицерина и длинноцепочечных жирных кислот и смеси этих и других масел. Можно изготавливать также суспензии для инъекций, в которых можно использовать подходящие жидкие носители, суспендирующие агенты и т.п. В композициях, подходящих для чрескожного введения, носитель, необязательно, включает агенты, усиливающие проникновение через кожу, и/или подходящий увлажняемый агент, необязательно, в сочетании с подходящими вспомогательными веществами различной природы в малых долях, которые не оказывают никакого значительного неблагоприятного воздействия на кожу. Указанные вспомогательные вещества могут облегчать нанесение препарата на кожу и/или могут облегчать само изготовление желаемых композиций. Эти композиции можно вводить различными путями, например, в виде чрескожного пластыря, в виде аппликации или мази. Очевидно, что для изготовления водных композиций более удобны соли присоединения (I) в силу их более высокой растворимости в воде по сравнению с соответствующими формами свободной кислоты или основания.
Особенно удобно изготавливать вышеуказанные фармацевтические композиции в виде дозированных лекарственных форм, для простоты применения и обеспечения стандартных дозировок. Дозированная лекарственная форма, указанная в описании и формуле изобретения, относится к физически дискретным единицам, подходящим в качестве стандартной дозы, каждая из которых содержит заранее определенное количество активного ингредиента, рассчитанное для обеспечения желаемого лечебного эффекта, в сочетании с требующимся фармацевтическим носителем. Примерами таких дозированных лекарственных форм являются таблетки (включая таблетки с насечками и таблетки в оболочке), капсулы, пилюли, пакетики с порошком, облатки, растворы и суспензии для инъекций, растворы с индикацией приема чайными или столовыми ложками, и содержащие их в определенном количестве упаковки.
Следующие примеры предназначены для иллюстрации настоящего изобретения.
Экспериментальная часть
Далее "КТ" будет обозначать комнатную температуру, "ТГФ" - тетрагидрофуран, "ДМФ" - N,N-диметилформамид и "ДИПЭ" - диизопропиловый эфир.
А. Получение промежуточных соединений
Пример А1
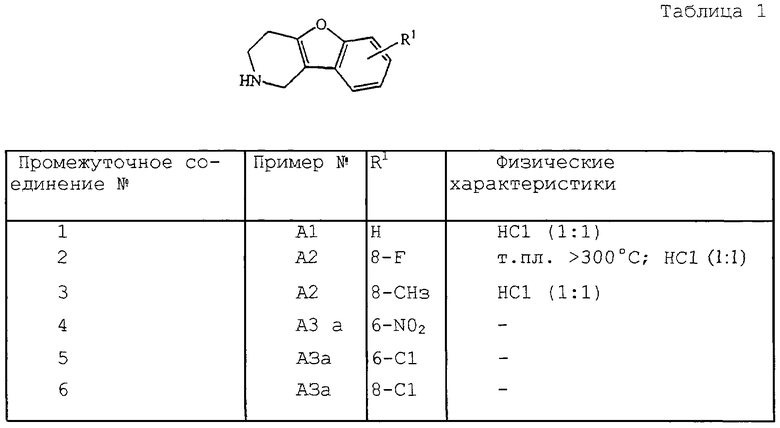
Смесь гидрохлорида О-фенилгидроксиламина (1:1) (0,625 моль) и гидрохлорида 4,4-пиперидиндиола (1:1) (0,682 моль) в 2-пропаноле (615 мл) перемешивали при 20oС. По каплям при 20oС добавляли НСl (353 мл). Реакционную смесь осторожно нагревали до температуры кипения с обратным холодильником. Реакционную смесь перемешивали и нагревали с обратным холодильником в течение 3 часов, затем охлаждали до комнатной температуры. Осадок отфильтровывали, промывали ДИПЭ и высушивали. Эту фракцию кристаллизовали из воды (1600 мл). Нужное соединение кристаллизовалось при перемешивании. Осадок отфильтровывали, промывали 2-пропанолом и ДИПЭ, затем высушивали с получением 84 г (64%) гидрохлорида 1,2,3,4-тетрагидробензофуро [3,2-е] пиридина (1: 1) (промежуточное соединение 1).
Пример А2
1,4-Диокса-8-азаспиро[4,5] декан (0,12 мл) добавляли по каплям к смеси гидрохлорида О-(4-фторфенил)гидроксиламина (1: 1) (0,1 моль) в смеси НСl и 1,1-оксибисэтана (150 мл). Реакционную смесь перемешивали и нагревали с обратным холодильником в течение 4 часов, а затем охлаждали. Осадок отфильтровывали и высушивали, затем перекристаллизовывали из воды с получением 10 г (43,9%) гидрохлорида 1,2,3,4-тетрагидро-8-фторбензофуро[3,2-с] пиридина (промежуточное соединение 2; т. пл. >300oС).
Пример A3
а) 1,2,3,4-Тетрагидро-2-метил-6-нитробензофуро[3,2-с]пиридин (0,0224 моль), полученный в соответствии со способом, описанным в J. Chem. Sоc. (С), 1971, 53-60, растворяли в 1,2-дихлорэтане (40 мл) и охлаждали до 0oС. При этой температуре добавляли по каплям (1-хлорэтил)ацетилхлорид (0,0291 моль). Эту суспензию перемешивали и нагревали с обратным холодильником в течение 2 часов. 1,2-Дихлорэтан выпаривали. Смесь растворяли в метаноле, перемешивали и нагревали с обратным холодильником в течение 2 часов, затем фильтровали. И фильтрат, и кристаллы обрабатывали 2н. Nа2СО3 и эту смесь экстрагировали CH2Cl2. Отделившийся органический слой высушивали, фильтровали, а растворитель выпаривали. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент: CH2Cl2/CH3OH 90/10). Нужные фракции собирали и растворитель выпаривали с получением 1,5 г 1,2,3,4-тетрагидро-6-нитробензофуро[3,2-с]пиридина (промежуточное соединение 4).
b) Смесь 1,2,3,4-тетрагидро-2-метил-6-нитробензофуро[3,2-с] пиридина (0,0215 моль) и триэтиламина (2 г) в ТГФ (200 мл) гидрировали с использованием палладия на угле в качестве катализатора 10% (2 г) в присутствии тиофена 4% (2 мл). После поглощения H2 (3 эквивалента) катализатор отфильтровывали и фильтрат выпаривали с получением 4,2 г 1,2,3,4-тетрагидро-6-амино-2-метилбензофуро[3,2-е]пиридина (промежуточное соединение 7).
с) Смесь промежуточного соединения (7) (0,0100 моль) в НСl (2 мл) диазотировали при -5oС с помощью NaNO2 (0,0105 моль) в воде (1,2 мл) в течение 30 мин. Раствор перемешивали в течение 30 мин при -5oС. В течение 10 мин добавляли смесь CuCl (0,010 моль) в НСl (10,6 мл). Полученную реакционную смесь перемешивали в течение 15 мин при 80oС, затем охлаждали до 20oС. После разбавления водой добавляли избыток 40% раствора К2СО3 и эту смесь экстрагировали CH2Cl2. Отделившийся органический слой высушивали, фильтровали и растворитель выпаривали с получением 1,7 г (78%) 1,2,3,4-тетрагидро-6-хлор-2-метилбензофуро[3,2-с]пиридина (промежуточное соединение 8).
Пример А4
а) Смесь промежуточного соединения (1) (0,03 моль), хлорацетонитрила (0,04 моль), йодида калия (0,1 г) и Na2CO3 (5 г) в 4-метил-2-пентаноне (180 мл) перемешивали и нагревали с обратным холодильником в течение 3 часов. Теплую смесь фильтровали и фильтрат выпаривали. Остаток очищали на силикагеле на стеклянном фильтре (элюент: СН2Сl2/СН3ОН/NН3 95/5). Нужные фракции собирали и растворитель выпаривали. Остаток кристаллизовали из ДИПЭ/петролейного эфира 1/1. Осадок отфильтровывали и высушивали с получением 5,74 г (90%) 3,4-дигидробензофуро[3,2-с]пиридин-2(1Н)-ацетонитрила (промежуточное соединение 10; т.пл. 78oС).
b) Смесь промежуточного соединения (10) (0,027 моль) в СН3ОН/NН3 (200 мл) гидрировали с использованием в качестве катализатора никеля Ренея (2 г) в присутствии тиофена 4% (1 мл). После поглощения H2 (2 эквивалента) катализатор отфильтровывали через дикалит и фильтрат выпаривали с получением 5 г (85,6%) 1,2,3,4-тетрагидро-2-(аминоэтил)бензофуро[3,2-с]пиридина (промежуточное соединение 12).
Пример А5
a) Смесь промежуточного соединения (1) (0,03 моль), этил(5-хлорпентил)карбамата (0,04 моль), иодида калия (0,1 г) и Nа2СО3 (5,7 г) в толуоле (250 мл) перемешивали и нагревали с обратным холодильником в течение ночи. Реакционную смесь охлаждали, перемешивали в воде (200 мл) и слои разделяли. Органическую фазу выпаривали. Остаток очищали посредством колоночной хроматографии на силикагеле (элюент: CH2Cl2/CH3OH 95/5). Чистые фракции собирали и растворитель выпаривали с получением 7 г этил[5-(3,4-дигидробензофуро[3,2-с]пиридин-2 (1Н)пентил] карбамата (промежуточное соединение 15).
b) Смесь промежуточного соединения (15) (0,021 моль) и гидроксида калия (12 г) в 2-пропаноле (120 мл) перемешивали и нагревали с обратным холодильником в течение 6 часов. Растворитель выпаривали. Остаток распределяли между CH2Cl2 и водой. Органический слой отделяли, высушивали, отфильтровывали и растворитель выпаривали с получением 4 г 3,4-дигидробензофуро[3,2-с]пиридин-2(1Н)-пентанамина (промежуточное соединение 16).
Пример А6
a) Смесь 3-гидроксиметилпиперидина (0,6 моль) и Na2CO3 (130 г) в СНСl3 (600 мл) перемешивали при 10oС. По каплям добавляли этилхлорформиат (115 г) (температуру поддерживали на уровне 10oС). Смесь перемешивали до тех пор, пока она не достигала комнатной температуры, и реакционную смесь перемешивали в течение ночи. Добавляли воду (500 мл). Органический слой отделяли, промывали водой, высушивали, фильтровали и растворитель выпаривали с получением 110 г (98%) (±)-этил 3-(гидроксиметил)-1-пиперидинкарбоксилата (промежуточное соединение 17).
b) Раствор метилфенилсульфонилхлорида (0,79 моль) в пиридине (200 мл) добавляли по каплям к раствору промежуточного соединения 17 (0,4 моль) в пиридине (150 мл), перемешивали при 10oС. Смесь перемешивали до тех пор, пока она не достигала комнатной температуры, и реакционную смесь перемешивали в течение ночи. При постоянном перемешивании эту смесь выливали в воду (1000 мл) и экстрагировали метилизобутилкетоном. Отделившийся органический слой промывали водой, высушивали, фильтровали и растворитель выпаривали. Остаток кристаллизовали из смеси диизопропилового эфира и петролейного эфира. Осадок отфильтровывали и высушивали с получением 96 г (70,3%) (±)-этил 3-[[[(4-метилфенил) сульфонил] окси] метил] -1-пиперидинкарбоксилата (промежуточное соединение 18).
c) Смесь промежуточного соединения (18) (0,0088 моль), свободного основания промежуточного соединения (1) (0,0080 моль) и Na2CO3 (0,016 моль) в ДМФ (25 мл) перемешивали и нагревали с обратным холодильником в течение ночи. Реакционную смесь охлаждали и растворитель выпаривали. Остаток распределяли между CH2Cl2 и 50% водным раствором NaCl. Слои разделяли. Водный слой трижды экстрагировали СН2Сl2. Объединенные органические слои высушивали, фильтровали и растворитель выпаривали. Остаток очищали посредством ВЭЖХ на силикагеле (элюент: СН2Сl2/СН3ОН 95/5). Чистые фракции собирали и растворитель выпаривали с получением 1,5 (55%) г этил 4-[[3,4-дигидробензофуро[3,2-с] пиридин-2(1Н)-ил] метил] -1-пиперидинкарбоксилата (промежуточное соединение 19).
Следующие промежуточные соединения, представленные в табл. 1 и 2, были получены аналогично одному из приведенных выше примеров.
В. Получение конечных соединений
Пример В1
а) Смесь 3-(2-хлорэтил)-2-метил-4Н-пиридо[1,2-а] пиримидин-4-она (0,0050 моль), полученного в соответствии со способами, описанными в ЕР 0 070 053, свободного основания промежуточного соединения (1) (0,0040 моль), Na2CO3 (0,008 моль) и йодида калия (0,0040 моль) в 4-метил-2-пентаноне (8 мл) перемешивали и нагревали с обратным холодильником в течение ночи. Реакционную смесь охлаждали. Добавляли 50% водный раствор NaCI и CH2Cl2. Фазы разделяли. Водный слой дважды экстрагировали CH2Cl2. Объединенные органические слои высушивали, фильтровали и растворитель выпаривали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (элюент: C2H5ОH/CH2Cl2 5/95). Нужные фракции собирали и растворитель выпаривали. Остаток растирали и обрабатывали ультразвуком в ДИПЭ, затем отфильтровывали и высушивали с получением 0,9 г (63%) 3-[2-[3,4-дигидробензофуро[3,2-с]пиридин-2(1Н)-ил]этил] -2-метил-4Н-пиридо[1,2-а]пиримидин-4-она (соединение 3; т.пл. 186,2oС).
b) 6-[[3,4-дигидробензофуро[3,2-с] пиридин-2(1Н)-ил]этил]-3,7-диметил-5Н-тиазоло[3,2-а] пиримидин-5-он (соединение 5) получали аналогично способу, описанному в примере В1а, но к реакционной смеси не добавляли йодид калия.
c) Смесь промежуточного соединения (1) (0,015 моль) и триэтиламина (4 г) в 4-метил-2-пентаноне (150 мл) перемешивали в течение 5 мин. Добавляли 9-метокси-2-метил 3-[2-[(метилсульфонил)окси] этил] -4Н-пиридо[1,2-а] пиримидин-4-он, полученный, как описано в W095/14691, и полученную реакционную смесь перемешивали и нагревали с обратным холодильником в течение 6 часов. Теплую смесь фильтровали и фильтрат перемешивали с водой (100 мл). Органический слой отделяли, высушивали, фильтровали и растворитель выпаривали. Остаток кристаллизовали из ДИПЭ и небольшого количества СН3СN. Этот продукт отфильтровывали и высушивали. Эту фракцию очищали посредством колоночной хроматографии на силикагеле (элюент: CH2Cl2/C2H5OH 92/8). Чистые фракции собирали и растворитель выпаривали. Остаток кристаллизовали из ДИПЭ. Осадок отфильтровывали и высушивали с получением 0,6 г 3-[2-[3,4-дигидробензофуро[3,2-с] пиридин-2(1Н)-ил] этил]-9-метокси-2-метил-4Н-пиридо[1,2-а]пиримидин-4-она (соединение 65; т. пл. 151oС).
Пример В2
Смесь промежуточного соединения (12) (0,0116 моль), 2-хлорбензотиазола (0,0118 моль) и NaHCO3 (2 г) в 2-этоксиэтаноле (45 мл) перемешивали и нагревали с обратным холодильником в течение 2 часов. Реакционную смесь охлаждали и добавляли при перемешивании воду (45 мл). Твердое вещество отфильтровывали с отсосом, промывали водой, перемешивали в ДИПЭ, отфильтровывали и высушивали. Эту фракцию растворяли в небольшом количестве метанола и переносили в (Е)-2-бутендиоевую кислоту (1:1) при перемешивании и нагревании. Эту смесь оставляли охлаждаться до комнатной температуры при перемешивании и полученный осадок отфильтровывали и высушивали с получением 3,4 г (63%) N-2-бензотиазол-3,4-дигидробензофуро[3,2-с]пиридин-2(1Н)-этанамин (Е)-2-бутандиоата (1:1) (соединение 51; т. пл. 210oС).
Пример В3
Гидроксид калия (0,088 моль) добавляли к горячему раствору промежуточного соединения (19) (0,0044 моль) в 2-пропаноле (50 мл) и полученную реакционную смесь перемешивали и нагревали с обратным холодильником в течение 16 часов. Растворитель выпаривали. Остаток распределяли между водой и CH2Cl2. Слои разделяли. Водную фазу трижды экстрагировали CH2Cl2. Объединенные органические фазы высушивали, фильтровали и растворитель удаляли. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (элюент: CH2Cl2/CH3OH/NH3 95/5). Нужные фракции собирали и растворитель выпаривали. Остаток растворяли в СНСl3 (15 мл). Добавляли триэтиламин (0,726 г). Добавляли 4-метилбензоилхлорид (0,0075 моль) и реакционную смесь перемешивали в течение одного часа. Добавляли 50% водный раствор NaOH. Слои разделяли. Водный слой дважды экстрагировали CH2Cl2. Объединенные органические слои высушивали, фильтровали и растворитель выпаривали. Остаток очищали посредством колоночной флэш-хроматографии на силикагеле (элюент: СН2Сl2/СН3ОН 97/3). Чистые фракции собирали и растворитель выпаривали. Остаток высушивали с получением 1,1 г (64%) 4-[(3,4-дигидробензофуро[3,2-с]пиридин-2(1Н)-ил)метил] -1-(4-метилбензоил)пиперидина (соединение 58; т. пл. 140,3oС).
Пример В4
Соединение (3) (0,0083 моль) растворяли в нагретом до температуры кипения с обратным холодильником 2-пропаноле (80 мл). По каплям добавляли смесь НСl и 2-пропанола к перемешиваемому теплому раствору до тех пор, пока он не становился кислым. Нужное соединение оставляли кристаллизоваться. Осадок отфильтровывали и высушивали с получением 3,2 г 3-[2-(3,4-дигидробензофуро[3,2-с] пиридин-2 (1Н)-ил) этил]-2-метил- 4Н-пиридо [1,2-а]пиримидин-4-она (соединение 43).
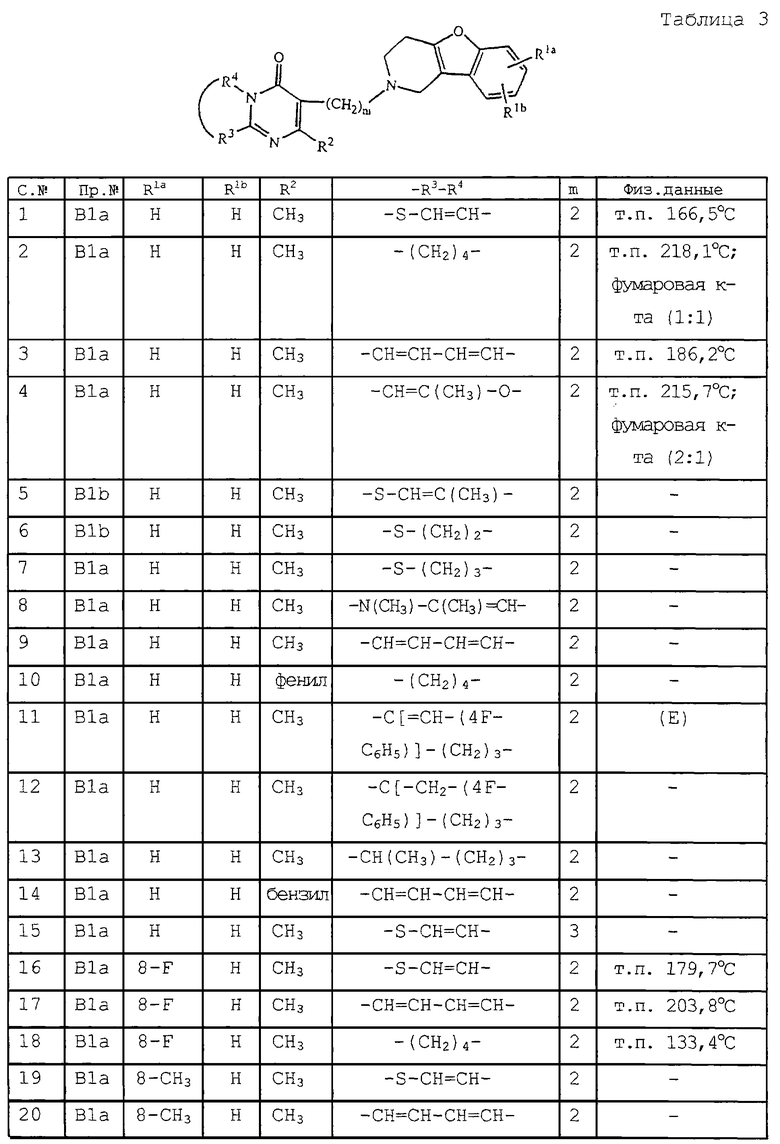
Следующие соединения, представленные в табл. 3 и 4, были получены аналогично одному из приведенных выше примеров.
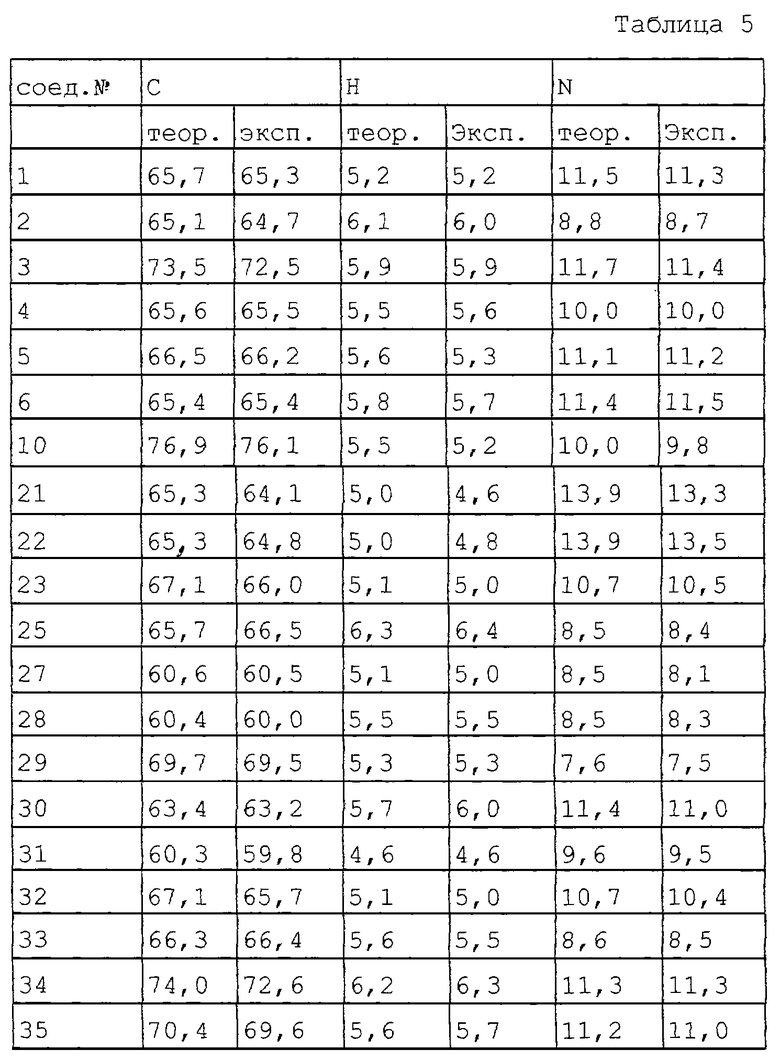
В табл. 5 перечисляются как экспериментальные (колонка "эксп"), так и теоретические (колонка "теор") величины элементного анализа на углерод, водород и азот для соединений, полученных в экспериментальной части, выше.
С. Фармакологические примеры
Пример С1: Связывающее сродство в отношении α2-рецепторов in vitro
Взаимодействие соединений формулы (I) и α2-рецепторов оценивали в экспериментах связывания радиолиганда in vitro.
Обычно, радиолиганд, обладающий высокой степенью связывающего сродства по отношению к конкретному рецептору, в низкой концентрации инкубируют с образцом препарата ткани, обогащенным конкретным рецептором, или с препаратом клеток, экспрессирующих клонированные рецепторы человека, в буферной среде. Во время инкубации радиолиганд связывается с рецептором. По достижении равновесия связывания отделяли радиоактивность, связанную с рецепторами, от несвязанной радиоактивности, и подсчитывали радиоактивность, связанную с рецепторами. Взаимодействие испытуемых соединений и рецептора оценивали в экспериментах конкурентного связывания. К инкубационной смеси, содержавшей препарат рецепторов и радиолиганд, добавляли испытуемое соединение в различных концентрациях. Связывание радиолиганда ингибируется испытуемым соединением пропорционально его связывающему сродству и концентрации.
Радиолигандом, который использовался для связывания с α2A-рецепторами, являлся 3H-раувольсцин, а препаратом рецепторов - клетки яичника китайского хомячка (СНО), экспрессирующие клонированные человеческие α2-рецепторы. Соединения под номерами 1-8, 10, 13-15, 17, 18, 23-25, 27-31, 33, 34, 36-38, 48, 49, 52, 53, 55, 56, 60, 62, 63, 65 и 66 вызывали ингибирование более чем на 50% при испытуемой концентрации 10-8 М и менее; соединения под номерами 9, 11, 12, 16, 19, 20, 22, 26, 35, 41, 44, 51, 57, 58, 59 и 64 вызывали ингибирование более чем на 50% при испытуемой концентрации от 10-8 до 10-6 M, а остальные вызывали ингибирование менее чем на 50% при испытуемой концентрации 10-6 М.
Радиолигандом, который использовался для связывания с α2B-рецепторами, являлся 3H-раувольсцин, а препаратом рецепторов - клетки СНО, экспрессирующие клонированные человеческие α2B-рецепторы. Соединения под номерами 1-8, 10, 13-15, 23, 25-28, 30, 31, 33, 34, 38-40, 48-50, 52, 53, 55, 56, 62, 63 и 66 вызывали ингибирование более чем на 50% при испытуемой концентрации 10-8 М и менее; соединения под номерами 9, 11, 12, 16-19, 24, 29, 35-37, 41, 44, 49, 51, 54-61, 64 и 65 вызывали ингибирование более чем на 50% при испытуемой концентрации от 10-8 до 10-6 М, а остальные вызывали ингибирование менее чем на 50% при испытуемой концентрации 10-6 М.
Радиолигандом, который использовался для связывания с α2C-рецепторами, являлся 3H-раувольсцин, а препаратом рецепторов - клетки СНО, экспрессирующие клонированные человеческие α2C-рецепторы. Соединения под номерами 1-6, 8, 10, 13, 14, 23, 25, 27-31, 33, 34, 36-40, 48, 50, 52, 53, 55, 58, 62, 63, 65 и 66 вызывали ингибирование более чем на 50% при испытуемой концентрации 10-8 М и менее; соединения под номерами 7, 9, 11, 12, 15-19, 22, 24, 26, 35, 41, 44, 49, 51, 56, 57, 59-61 и 64 вызывали ингибирование более чем, на 50% при испытуемой концентрации от 10-8 до 10-6 М, а остальные вызывали ингибирование менее чем 50% при испытуемой концентрации 10-6 М.
Пример С2: Ксилазин-индуцированное угнетение рефлекса выпрямления у крыс
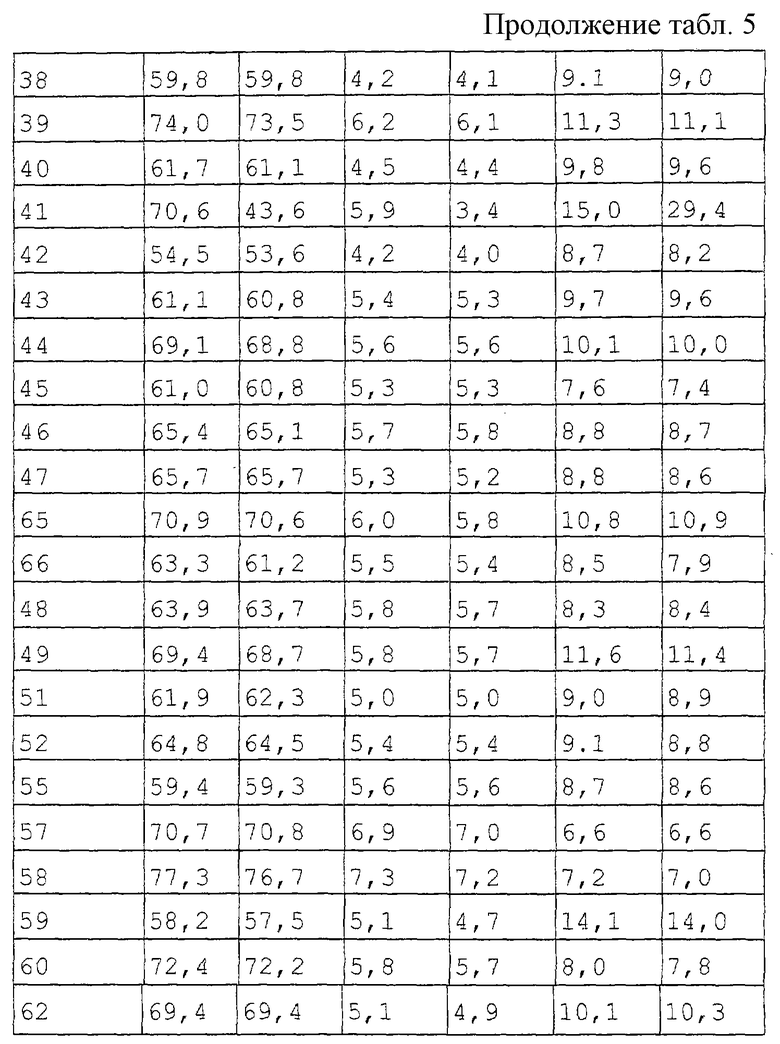
Этот тест основан на том факте, что антагонисты α2-адренорецепторов центрального действия реверсируют угнетение рефлекса выпрямления, индуцированное внутривенной инъекцией α2-агониста ксилазина. За час до инъекции ксилазина (15 мг/кг в/в) самцам крыс (200-250 г) предварительно вводили испытуемое соединение (перорально п/о) или подкожно (п/к) или только растворитель. У крыс, получивших растворитель, ксилазин-индуцированное угнетение рефлекса выпрямления отмечалось в течение периода времени до 120 мин после инъекции. Критерием активности испытуемого соединения служило отсутствие угнетения рефлекса выпрямления. Наименьшая активная доза (НАД) испытуемых соединений в отношении антагонизма к ксилазину определялась как наименьшая испытанная доза, при которой по меньшей мере 66% экспериментальных животных не демонстрировали угнетения рефлекса выпрямления. Табл. 6 приводит результаты этого изучения настоящих соединений.
D. Примеры композиций
"Активный ингредиент" (АИ), указанный в настоящих примерах, относится к соединению формулы (I), его фармацевтически приемлемой соли присоединения или стереохимически изомерной форме.
Пример D1: Капсулы
20 г АИ, 6 г лаурилсульфата натрия, 56 г крахмала, 56 г лактозы, 0,8 г коллоидного диоксида кремния и 1,2 г стеарата магния интенсивно перемешивали. Полученной смесью затем наполняли 1000 подходящих твердых желатиновых капсул, по 20 мг АИ в каждой.
Пример Р2: таблетки в оболочке
Изготовление ядра таблетки
Смесь 100 г АИ, 570 г лактозы и 200 г крахмала тщательно перемешивали, а затем увлажняли раствором 5 г додецилсульфата натрия и 10 г поливинилпирролидона приблизительно в 200 мл воды. Влажную порошкообразную смесь просеивали, высушивали и вновь просеивали. Затем добавляли 100 г микрокристаллической целлюлозы и 15 г гидрированного растительного масла. Все вместе тщательно перемешивали и прессовали в таблетки, с получением таблеток, по 10 мг активного ингредиента в каждой.
Оболочка
К раствору 10 г метилцеллюлозы в 75 мл денатурированного этанола добавляли раствор 5 г этилцеллюлозы в 150 мл дихлорметана. Затем добавляли 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. 10 г Полиэтиленгликоля расплавляли и растворяли в 75 мл дихлорметана. Последний раствор добавляли к предыдущему, а затем добавляли 2,5 г октадеканоата магния, 5 г поливинилпирролидона и 30 мл концентрированной суспензии красителя и все вместе гомогенизировали. Ядра таблеток покрывали полученной смесью в аппарате для нанесения покрытия на таблетки.
Пример D3: Раствор для перорального приема
9 г метил-4-гидроксибензоата и 1 г пропил-4-гидроксибензоата растворяли в 4 л кипящей очищенной воды. В 3 л этого раствора растворяли сначала 10 г 2,3-дигидроксибутандиоевой кислоты, а затем 20 г АИ. Последний раствор объединяли с оставшейся частью предыдущего раствора и к ним добавляли 12 л 1,2,3-пропантриола и 3 л 70% раствора сорбитола. 40 г сахарин-натрия растворяли в 0,5 л воды и добавляли 2 мл малиновой и 2 мл крыжовенной эссенции. Последний раствор добавляли к предыдущему, а затем добавляли воду до объема 20 л, с получением перорального раствора, содержащего 5 мг активного ингредиента в одной чайной ложке (5 мл). Полученным раствором наполняли подходящие контейнеры.
Пример D4: Раствор для инъекций
1,8 г метил-4-гидроксибензоата и 0,2 г пропил-4-гидроксибензоата растворяли приблизительно в 0,5 л кипящей воды для инъекций. После охлаждения приблизительно до 50oС добавляли при перемешивании 4 г молочной кислоты, 0,05 г пропиленгликоля и 4 г АИ. Раствор охлаждали до комнатной температуры и добавляли воду для инъекций до объема 1 л с получением раствора для инъекций, содержащего 4 мг/мл АИ. Этот раствор стерилизовали фильтрованием и наполняли им стерильные контейнеры.

Изобретение относится к области органической химии. Описываются производные 1,2,3,4-тетрагидробензофуро[3,2-с]пиридина общей формулы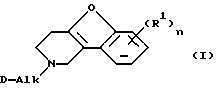
его N-оксидная форма, фармацевтически приемлемая соль присоединения или стереохимически изомерная форма, где каждый R1 независимо представляет водород, галоген, C1-6 алкил, нитрогруппу или С1-4алкилокси; Alk представляет C1-6 алкандиил; n = 1 или 2; D представляет 1- или 2-бензимидазолил, 2(3Н)бензоксазолон-3-ил или радикал формулы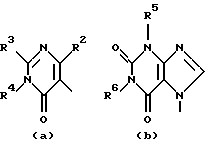
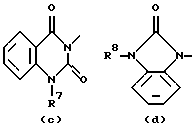
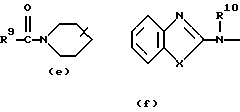
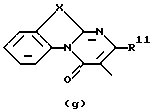
каждый X представляет S; R2 представляет C1-6алкил, арил или арилС1-6алкил; R3 представляет С1-6алкилтио; R4, R5, R6, R7, R8, R10 и R11 каждый, независимо, представляет водород или С1-6алкил; R9 представляет арил или R3 и R4, вместе взятые, могут образовывать двухвалентный радикал - R3- R4- формулы (а-2) или (а-5): -СН2-СН2-СН2-СН2- (а-2); -СН=СН-СН=СН- (а-5), где один или два атома водорода указанных радикалов каждый, независимо, могут быть заменены галогеном, С1-6алкилом, арилС1-6алкилом, трифторметилом, амино, гидрокси или С1-6алкилокси; или, где это возможно, два геминальных атома водорода могут быть заменены арилС1-6алкилиденом; или - R3-R4- также может иметь формулу (а-6), (а-7), (а-8), (а-11) или (а-14): -S-CH2-CH2- (а-6); -S-CH2-CH2-CH2- (а-7); -S-CH=CH- (а-8); -NH-CH-CH- (а-11); -СН=СН-О- (а-14), где от одного до трех атомов водорода указанных радикалов, каждый независимо, могут быть заменены C1-6алкилом и арил представляет собой фенил или фенил, замещенный С1-6алкилом. Также описываются способы получения соединений формулы (1), композиция, обладающая антагонистической активностью в отношении центральных α-адренорецепторов, и композиция, пригодная для лечения заболевания, ассоциированного с недостатком норадреналина в центральной или периферической нервной системе. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 6 с. и 6 з.п. ф-лы, 6 табл.
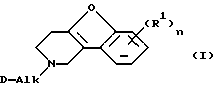
N-оксидная форма или фармацевтически приемлемая соль присоединения или их стереохимически изомерная форма,
где каждый R1 независимо представляет водород, галоген C1-6алкил, С1-4алкокси или нитрогруппу;
Alk представляет C1-6алкандиил;
n равно 1 или 2;
D представляет 1- или 2-бензимидазолил, 2 (3Н)бензоксазолон-3-ил или радикал формулы (а), (b), (с), (d), (е), (f), (g), (h)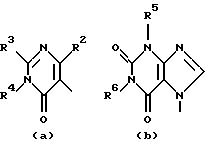
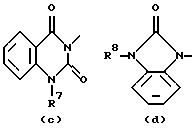
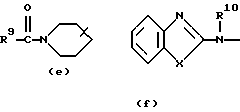
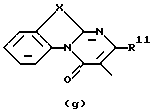
где каждый Х представляет S;
R2 представляет C1-6алкил, арил или арилС1-6алкил;
R3 представляет C1-6алкилтио;
R4, R5, R6, R7, R8, R10 и R11 каждый, независимо, представляет водород или C1-6алкил;
R9 представляет арил
или R3 и R4, взятые вместе, могут образовывать двухвалентный радикал -R3-R4- формулы (а-2) или (а-5)
-СН2-СН2-СН2-СН2- (а-2)
-СН=СН-СН=СН- (а-5)
где один или два атома водорода указанных радикалов каждый, независимо, могут быть заменены галогеном, C1-6алкилом, арилС1-6алкилом, трифторметилом, амино, гидрокси или C1-6алкилокси, или, где это возможно, два геминальных атома водорода могут быть заменены арилС1-6алкилиденом; или -R3-R4-также может иметь формулу (а-6), (а-7), (а-8), (а-11) или (а-14)
-S-CH2-CH2- (а-6)
-S-CH2-CH2-CH2- (а-7)
-S-CH=CH- (а-8)
-NH-CH=CH- (а-11)
-СН=СН-O- (а-14)
где от одного до трех атомов водорода указанных радикалов, каждый, независимо, могут быть заменены C1-6алкилом, и арил представляет собой фенил или фенил, замещенный C1-6алкилом.
6-[[3,4-дигидробензофуро[3,2-с] пиридин-2(1Н)-ил] этил] -7-метил-5Н-тиазоло[3,2-а]пиримидин-5-он;
6-[[3,4-дигидробензофуро[3,2-с] пиридин-2(1Н)-ил] этил]-3,7-диметил-5Н-тиазоло[3,2-а]пиримидин-5-он;
3-[2-[3,4-дигидробензофуро[3,2-с] пиридин-2(1Н)-ил] этил] -2,7-диметил-4Н-пиридо[1,2-а] пиримидин-4-он; его N-оксид и фармацевтически приемлемую соль присоединения или их стереоизомерная форма.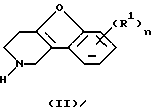
алкилирующим реагентом формулы (III)
D-Alk-W1, (III)
где W1 представляет подходящую реакционную удаляемую группу, a D, Alk, R1 и n определены в п.1,
и, если желательно, преобразование соединений формулы (I) друг в друга с помощью известных преобразований и далее, если желательно, преобразование соединений формулы (I) в терапевтически активные нетоксичные соли присоединения кислот обработкой кислотой, или в терапевтически активные нетоксичные соли присоединения оснований обработкой основанием, или, наоборот, преобразование соли присоединения кислоты в свободное основание обработкой щелочью, или преобразование соли присоединения основания в свободную кислоту обработкой кислотой и, если желательно, получение их стереоизомерных форм или N-оксидов.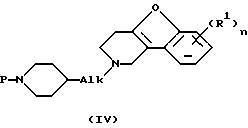
и последующее N-ацилирование полученного промежуточного соединения ацильным производным формулы (V)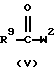
где Р представляет защитную группу, W2 представляет подходящую реакционную удаляемую группу, a R1, R9, Alk и n определены в п.1, в результате чего получается соединение формулы (1-е),
и, если желательно, преобразование соединений формулы (I) друг в друга с помощью известных преобразований и далее, если желательно, преобразование соединений формулы (I) в терапевтически активные нетоксичные соли присоединения кислот обработкой кислотой, или в терапевтически активные нетоксичные соли присоединения оснований обработкой основанием, или, наоборот, преобразование соли присоединения кислоты в свободное основание обработкой щелочью, или преобразование соли присоединения основания в свободную кислоту обработкой кислотой и, если желательно, получение их стереоизомерных форм или N-оксидов.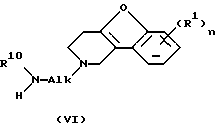
промежуточным продуктом формулы (VII)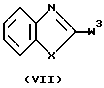
где W3 представляет подходящую реакционную удаляемую группу, а X, Alk, R1, R10 и n определены в п.1, в результате чего получается соединение формулы (I-f),
и, если желательно, преобразование соединений формулы (I) друг в друга с помощью известных преобразований и далее, если желательно, преобразование соединений формулы (I) в терапевтически активные нетоксичные соли присоединения кислот обработкой кислотой, или в терапевтически активные нетоксичные соли присоединения оснований обработкой основанием, или, наоборот, преобразование соли присоединения кислоты в свободное основание обработкой щелочью, или преобразование соли присоединения основания в свободную кислоту обработкой кислотой и, если желательно, получение их стереоизомерных форм или N-оксидов.
| US 4688803 А, 26.05.1987 | |||
| Способ формования покрышек пневматических шин | 1974 |
|
SU588137A1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕВАЛОНОЛАКТОНОВЫХ ПРОИЗВОДНЫХ | 1989 |
|
RU2045529C1 |
