Изобретение относится к новым мевалонолактонам, имеющим пиразолопиридиновое кольцо, способам их получения, фармацевтическим композициям, содержащим их, и их фармацевтическому использованию, особенно в качестве антигиперлипидемических, гиполипопротеинемических и антиатеросклеротических агентов, и к промежуточным продуктам полезным для их получения и способам получения таких промежуточных продуктов.
Известно, что некоторые метабололические продукты брожения, такие как компактин, CS-514, мевинолин или полусинтетические производные или полностью синтетические их производные являются ингибиторами HMG-CoA редуктазы, которая является ферментом, ограничивающим скорость при биосинтезе холестерина.
Клиническим путем было доказано, что С 514 и мевонолин являются потенциально полезными антигиперлипопротеинемическими агентами, и считается что они эффективны для лечения или профилактики заболеваний коронарного артериосклероза или атеросклероза.
Проведенные исследования обнаружили, что мевалонолактоновые производные, имеющие пиразолопиридиновое кольцо, соответствующие дигидрокси карбоновые кислоты и соли и их сложные эфиры имеют высокую ингибиторную активность биосинтеза холестерина, при котором HMG-CoA редуктаза действует как фермент, ограничивающий скорость реакции.
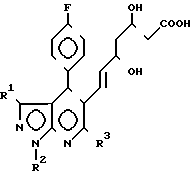
Новые мевалонолактоновые производные предлагаемого способа представлены формулой I: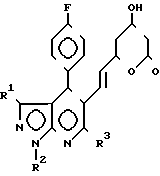 где R1 C1-6-алкил или фенил;
где R1 C1-6-алкил или фенил;
R2 С1-6-алкил;
R3 (С1-С5)-алкил, циклопропил.
С1-6-алкил включает, например, метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил, трет-бутил, н-пентил, н-гексил.
Кроме того, эти соединения, могут иметь, по крайней мере, один или два асимметричных атома углерода и могут иметь по крайней мере от двух до четырех оптических изомеров. Соединение формулы I включают все эти оптические изомеры и все их смеси.
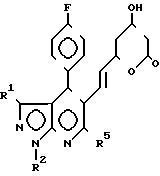
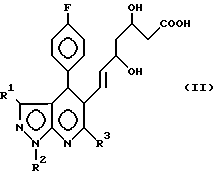
Соединения формулы I получают циклизацией соединений формулы II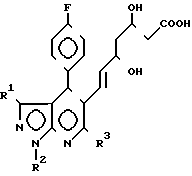 II где R1, R2 и R3 определены выше.
II где R1, R2 и R3 определены выше.
Данная реакция является реакцией образования мевалонолактона формулы I с помощью реакции дегидратации свободной гидроксикислоты II.
Реакция дегидратации может быть проведена в бензоле или толуоле при нагревании с обратным холодильником при удалении образующейся воды или с помощью добавления подходящего гидратирующего агента такого как молекулярное сито.
Кроме того, реакция дегидратации может быть проведена в сухом метиленхлориде с использованием лактонобразующего агента такого, как карбодиимид, предпочтительно водорастворимый карбодиимид такой, как пара-толуолсульфонат N-цикло- гексил-N'-(2'-)метилморфолиний(этил)карбодиимида при температуре 10 35оС, предпочтительно 20 25оС.
Исходные мевалонолактоны формулы II могут получаться восстановлением соединения формулы III
R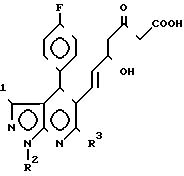 (III) где R1 R2 и R3 см. ранее.
(III) где R1 R2 и R3 см. ранее.
Реакция восстановления кетокарбоксилата формулы III, проходит с помощью различных агентов восстановления. Эта реакция включает восстановление карбонила с помощью например: боpгидрида натрия, цианобоpгидрида натрия, боргидрида цинка, диизоамилборана, диборана, трет.-бутиламиноборана, комплекса пиридин-борана, дициклогексилборана, гексилборана, 9-борабицикло (3,3,1)нонана, диизопинокамренилборана или три-втор.-бутилборгидрида лития в соответствующий дигидроксикарбоксилат формулы II.
Данная реакция может быть проведена в растворителе выбранном из углеводородов, галоидированных углеводородов, С1-4-спиртов, простых эфиров и их смесей, при температуре (-100) (50оС), предпочтительно (-78) 30оС.
Кроме того, триалкилборан такой как три-н-бутилборан или три-этилборан и боргидрид натрия используются при низкой температуре. Помимо этого, эритро-форма, имеющая биологически превосходящие активности может быть успешно получена с использованием алкоксидиалкилборана такого как метоксидиэтилборан или этоксидиэтилборан или боргидрид натрия.
Данная реакция может быть проведена с использованием смеси растворителем С1-4-спирта и тетрагидрофурана при температуре (-80) (-50)оС, предпочтительно (-72) (-68)оС.
Соединения данного изобретения проявляют высокую ингибиторную активность против биосинтеза холестерина, при котором HMG-CoA редуктаза действует как фермент, ограничивающий скорость, как показано результатами испытаний данными ниже, и таким образом способны подавлять или понижать количество холестерина в крови как липопротеин. Таким образом, соединения данного изобретения являются полезными в качестве лечебных агентов против гиперлипидемии, гиперлипопротеинемии и атеросклероза. Они могут преобразовываться в разнообразные подходящие выпускные или готовые формы препаратов в зависимости от способа назначения. Соединения могут назначаться в форме свободных кислот или в виде физиологически гидролизуемых и приемлемых сложных эфиров или лактонов, или фармацевтически приемлемых солей.
Фармацевтическая композиция изобретения предпочтительно назначается орально в форме предлагаемого соединения (самого по себе) или в виде порошков, гранул, таблеток или капсул, сформулированных с помощью смешения предлагаемого соединения с подходящим фармацевтически приемлемым носителем, включающим связующее, такое как гидроксипропилцеллюлоза, сироп, аравийская камедь, желатин, сорбит, камедь трагаканта, поливинилпирролидон или СМС-Са, эксципиент, такой как лактоза, сахар, кукурузный крахмал, фосфат кальция, сорбит, глицин или порошок кристаллической целлюлозы, смазочный агент, такой как стеарат магния, тальк, полиэтиленгликоль или двуокись кремния, и дезинтегратор, такой как картофельный крахмал.
Однако, фармацевтическая композиция данного изобретения не ограничивается таким оральным назначением, она применима и для парентерального назначения. Например, она может назначаться в форме суппозитория образованного с использованием масляного основного материала такого, как масло какао, полиэтиленгликоль, ланолин или триглицерид жирной кислоты, трансдермального терапевтического основания образованного с использованием жидкого парафина, белого вазелина, высших спиртов, мази Макроголя, гидрофильной мази или гидрогельного основного материала, инъекционной препаративной формы, приготавливаемой с помощью использования одного или более материалов, выбираемых из группы, состоящей из полиэтиленгликоля, гидрогельного основного материала, дистиллированной воды, дистиллированной воды для инъекций и эксципиента, такого как лактоза или кукурузный крахмал, или препаративной формы для назначения через слизистую оболочку, такую как слизистая оболочка глаза, носовая слизистая оболочка и слизистая оболочка рта.
Кроме того, предлагаемые соединения могут комбинироваться с основными ионообменными смолами, которые способны связывать желчные кислоты и все же не абсорбируются желудочно-кишечным трактом.
Суточная доза соединения формулы (I-III) составляет 0,05 500 мг, предпочтительно 0,5 50 мг для взрослого человека. Оно назначается для приема от одного до трех раз в день. Доза может конечно варьироваться в зависимости от возраста, веса и состояния болезни пациента.
Испытания А. Ингибирование биосинтеза холестерина из ацетата ин витро.
Раствор фермента приготавливался из печени самцов крыс Вистар, которым была вставлена канюля и выводилась желчь в течение 24 ч. Печень вырезалась в сумерки и микросомы и фракция, плавающая сверху, способная осаждаться 40 80% -ным раствором сульфата аммония (суп-фракция) приготавливались из гомогената печени в соответствии с модифицированным методом Knauss et al. Kuroda, M. et al. Biochim. Biophys. Acta, 489, 119 (1977). Для анализа биосинтеза холестерина, микросомы (0,1 мг белка и суп-фракция (1,0 мг белка) инкубировались в течение 2 ч при 37оС в 200 мкл реакционной смеси, содержащей АТФ 1 ммоль глютатиона; 6 мМ глюкоза-1-фосфата, 10 ммоль NAD; 0,25 ммоль NADP; 0,25 ммоль, СоА; 0,04 ммоль и 0,2 ммоль (2-14С) ацетат натрия (0,2 μ Ci) с 4 мкл раствора испытуемого соединения, растворенного в воде или диметилсульфоксиде. Для остановки реакции и омыления, к реакционной смеси добавлялся 1 мл 15% EtOH КОН и смесь нагревалась при 75оС в течение 1 ч. Не способные к омылению липиды экстрагировались петролейным эфиром и подсчитывалось введенная радиоактивность 14С. Ингибирующая активность соединений выражалась показателем 50 1G.
Испытание В. Ингибирование биосинтеза холестерина в клетках культуры.
Клетки Нер С2 высевались на 12-ячеистых пластин и инкубировались с модифицированной bagle средой Dulbecco (DME) содержащей 10% бычьей зародышевой сыворотки (PBS) при 37оС, 5 СО2 до тех пор, пока клетки не становились сливающимися в течение примерно 7 дней. Клетки подвергались воздействию МЕ среды, содержащей 5% сыворотки с недостаточным содержанием липопротеина (LpDS), полученной с помощью метода ультрацентрифугирования в течение почти 24 часов. Среда заменялась 0,5 мл свежей 5% LpDS, содержащей DME, перед анализом и добавлялось 10 мкл раствора испытуемого соединения, растворенного в воде или ДМСО. Через 0 ч (В-1) или 4 ч (В-2) после добавления соединений добавлялось 0,2 μ Ci (2-13С) ацетата натрия (20 мкл). После дополнительного инкубирования в течение 4 ч с (2-14С) ацетатом натрия среда удалялась и клетки промывались солевым раствором, буферированным фосфатом (PBS), охлажденным при 4оС. Клетки соскабливались резиновой палочкой и собирались в трубки с PBS и дигерировались с 0,2 мл 0,5 норм, КОН при 37оС. Некоторая доза жидкости от стадии дигерирования использовалась для анализа белка, а остальное количество омылялось 1 мл 15% EtOH-КОН при 75оС в течение 1 ч. Hеомы-ляемые липиды экстрагировались петролейным эфиром и подсчитывалась 14С радиоактивность. Результаты подсчета проверялись по клеточному белку и обозначались величиной ДРМ/мг (распадов в минуту/мг) белка. Ингибиторная активность соединений выражалась величиной 1С 50.
Испытание С. Ингибирование биосинтеза холестерина ин виво.
Самцам крыс Sprague-Dawley весом около 150 г давали корм в виде обычной еды Purina и воду по потребности и перед использованием для испытания на ингибирование ин виво биосинтеза холестерина крыс подвергли воздействию освещения по схеме 12 ч света/12 ч темноты (2:00 после полудня 2:00 до полудня темное время). Животных разделяли на группы, состоящие из пяти крыс, так, чтобы средняя величина веса тела была в каждой группе. Испытуемые соединения в дозе 0,02 0,2 мг/кг веса тела (0,4 мл/100 г веса тела) растворялись в воде или суспендировались в 0,5%-ной метилцеллюлозе и назначались орально за 2-3 ч до наступления сумерек (8:00 после полудня), когда биосинтез холестерина у крыс достигал максимума. В качестве контроля крысам назначали только воду или носитель. Через 90 мин после назначения пробы, крыс инъецировали интраперитонально 10 мк Сi (2-14С) ацетата натрия в объеме 0,2 мл на каждую. Спустя 2 ч брали пробы крови и сразу же отделяли сыворотку. Общие липиды экстрагировались в соответствии с методом Folch и др. и омылялись смесью EtОН-КОН. Неомыляемые липиды экстрагировались петролейным эфиром, подсчитывалась радиоактивность, введенная в неомыляемые липиды.
Ингибиторная активность отмечалась как процентное уменьшение числа отсчета (показатель фоновой радиации) в испытуемых группах (распады/ в минуту /2 мл сыворотки/2 ч) по сравнению с показателем в контрольной группе.
Что касается предлагаемых соединений показатели ингибиторной активности против биосинтеза холестерина, при котором HMG-CoA редуктаза служит в качестве ограничивающего скорость фермента, измерялись с помощью описанных выше испытаний А и В. Результаты показаны в табл. 1, 2, 3, 4. Дополнительно представлены также результаты измерений с помощью испытания С.
Показатели ингибиторной активности ссылочного соединения с помощью испытания А: ссылочное соединение: CS-514; 1С 50 (молярная концентрация) 1,1 х 10-8
В табл. 1 представлены показатели относительной активности в расчете на активность СS-514 по испытанию А, которая оценивалась равной 1.
Структура ссылочного соединения
CS-514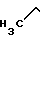
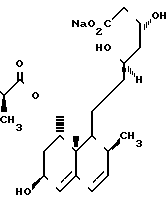
Ингибиторная активность по испытанию В-1: ссылочное соединение CS-514; 1С 50 (молярная концентрация) 1,1 х 10-8
П р и м е р 1 (Е) -7-[4'-(4''-фторфенил)-1',3'-диметил-6'-]1''-метилэтил[пиразоло(3,4-b)пириди н-5'-ил] -3,5-дигидроксигент-6- еновая кислота (соединение 1-2-1).
0,25 г (0,53 ммоль) этил-(Е)-7-[4'-(4-фторфенил)-1',3'-диметил-6']1''-метилэтил[-пиразоло(3,4-b) пиридин-5'-ил]-3,5-дигидроксигепт-6-еноата растворялось в 3 мл этанола и в раствор по каплям добавлялось 1,06 мл 0,5 н. водного раствора гидроокиси натрия. Этанол отгонялся при пониженном давлении, а затем в смесь добавлялось 3 мл дистиллированной воды. Раствор промывался диэтиловым эфиром. Водный слой осторожно нейтрализовался 1%-ной соляной кислотой и экстрагировался диэтиловым эфиром. Эфирный слой сушился над безводным сульфатом магния и упаривался при пониженном давлении, давая требуемое соединение.
Количество 0,12 г (выход 90%)
ПЯМР (ДМСЩ-d6)S млн.дол. 1,29 (д. I 7 Гц, 6Н), 1,83 (с. 3Н), 2,1-2,3 (м. 2н), 2,4-2,6 (м.1Н), 3,0-3,6 (м. 4Н), 3,96 (с. 3Н), 4,3-4,8 (м. 2Н), 5,2-5,6 (м. 1Н), 6,3-6,6 (м. 1Н), 7,2-7,4 (м. 4Н), 11,5-12,0 (шир.с, 1Н).
П р и м е р 2. Тем же способом, что описан в примере 1, были получены соединения 1-2-5. Физические свойства соединений представлены в табл.3.
ПЯМР соединения 1-2-5 (ДМСO-d6) δ млн.дол. 0,8-1,1 (м, 4Н), 1,4-1,8 (м. 2Н), 1,89 (с, 3Н), 2,1-2,6 (м. 4Н), 3,0-3,8 (м, 2Н), 3,98 (с, 3Н), 4,3-4,6 (м. 1Н), 5,3-5,7 (м, 1Н), 6,4-6,8 (м,1Н), 6,9-7,3 (м, 4Н).
П р и м е р 3. (Е)-транс-6-2'-[4''-(4'''-фторфенил-1'',3''-диметил-6'')-1'''-метилэтил] пиразоло(3,4-b)пиридин-5-ил(этинил)-4-гидрокси-3,4,5,6-тетрагидро-2Н-пиран- 2-он (соединение 1-3-1).
130 мг (0,29 ммоль) соединения 1-2-1 растворялось в 6 мл дихлорметана, и к раствору добавлялось 125 мг (0,29 ммоль) n-толуолсульфоната N-циклогексил-N'(2'- метилморфолиноэтил)-карбодиимида. Смесь перемешивалась при комнатной температуре в течение 2 ч, а затем подвергалась перегонке при пониженном давлении для удаления растворителя досуха. Остаточное масло очищалось с помощью тонкослойной хроматографии на силикагеле (элюент: смесь гексана и этилацетата 9:1 (объем/объем)), давая чистое требуемое соединение в виде бесцветного вязкого маслянистого вещества. Количество 48 мг (выход 39%).
П-ЯМР (CDCl3) δ млн.дол. 1,33 (д. I=6,8 Гц, 6н), 1,4-1,5 (м. 1Н), 1,6-1,7 (м. 2Н), 1,93 (с. 3Н), 2,5-2,6 (м. 1Н), 2,68 (дд. I=18 Гц, I=5 Гц, 1Н), 3,39 (гепталет, I= 6,8 Гц, 1Н), 4,07 (с. 3Н), 4,1-4,2 (м. 1Н), 5,1-5,2 (м. 1Н), 5,31 (дд. I= 16 Гц, I=6 Гц, 1Н), 6,61 (дд. I 16 Гц, I=1,5 Гц, 1Н), 7,1-7,3 (м. 4Н).
П р и м е р 4. Способом, описанным в примере 3, были получены соединения 1-3-5 ≈ 1-3-17. Физические данные этих соединений представлены в табл.4.
ПЯМР соединения 1-3-7 (ДМСО-d6) δ млн.дол. 1,32 (д, I=7 Гц, 6Н). 1,43-1,51 (м. 1Н), 1,65-1,70 (м. 1Н), 1,76 (с. 9Н), 1,84 (с. 3Н), 2,08 (с. 1Н), 2,59-2,65 (м. 1Н), 2,71 (д.д. I=18 Гц, I=5 Гц, 1Н), 3,41 (гепталет, I=7 Гц, 1Н), 4,23-4,29 (м. 1Н), 5,15-5,21 (м. 1Н), 5,58 (дд. I 16 Гц, I 6 Гц, 1Н), 6,67 (дд. I 10 Гц, I 1 Гц, 1Н), 7,10-7,25 (м. 1Н).
Использование: в химико-фармацевтической промышленности. Сущность изобретения: продукт-мевалолактоновые производные ф-лы I. Реагент 1: производное пиразоло-[3,4-в] -пиридин-5-ил-3.5-ди-гидрокси- гепт-6-еновой кислоты ф-лы II. Условия: циклизация в органическом растворителе в присутствии дегидратирующего агента. 4 табл. Структура соединений ф-лы I, II: 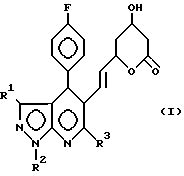
СПОСОБ ПОЛУЧЕНИЯ МЕВАЛОНОЛАКТОНОВЫХ ПРОИЗВОДНЫХ общей формулы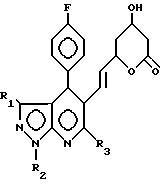
где R1 С1 С6-алкил или фенил;
R2 С1 С6-алкил;
R3 С1 С5-алкил, циклопропил,
отличающийся тем, что производное пиразоло(3,4-b)пиридин-5-ил-3,5-дигидроксигепт-6-еновой кислоты общей формулы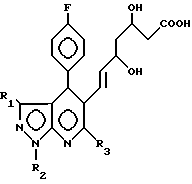
где R1 R3 имеют указанные значения,
подвергают циклизации в среде органического растворителя в присутствии дегидратирующего агента.
| 0 |
|
SU183132A1 | |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
