Изобретение относится к способу получения олефиновых олигомеров путем катионной олигомеризации олефинового сырья и может быть использовано в нефтехимической промышленности.
Получаемые по этому способу продукты могут использоваться в качестве основы синтетических олигоолефиновых масел разнообразного назначения: моторных (автомобильных, авиационных, вертолетных, тракторных, танковых); трансмиссионных, редукторных, вакуумных, компрессорных, холодильных, трансформаторных, кабельных, веретенных, медицинских, а также в качестве пластификаторов для пластмасс, каучуков, твердых ракетных топлив; сырья для получения присадок, эмульгаторов, флотореагентов, пенообразователей, компонентов смазочно-охлаждающих и гидравлических жидкостей; высокооктановых добавок к топливам и т.п.
Известны способы получения олефиновых олигомеров, включающие олигомеризацию олефинового сырья, выделение отработанного катализатора методом водно-щелочной и последующей водной отмывки, разделение очищенного олигомеризата на фракции и гидрирование выделенных целевых фракций (1. US 3780128, 18.12.1973; 2. US 3997621, 14.12.1976; 3. US 4032591, 28.06.1977; 4. US 4376222, 08.03.1983; 5. US 4225739, 30.09.1980; 6. US 4263465, 21.04.1981; 7. US 4409415, 11.10.1983; 8. US 4956512, 11.09.1990; 9. US 4436947, 13.03.1984; 10. US 4451689, 29.05.1984; 11. US 4454366, 12.06.1984; 12. US 4587368, 06.05.1986; 13. US 5254784, 20.12.1991; 14. US 5191140, 02.03.1993; 15. US 5420373, 30.05.1995; 16. US 5550307, 27.08.1996; 17. US 3725498, 03.04.1973; 18. US 3952071, 20.04.1976; 19. US 3997622, 14.12.1976; 20. US 3997623, 14.12.1976; 21. US 4006199, 01.02.1977; 22. US 4031158, 21.06.1977; 23. US 4031159, 21.06.1977; 24. US 4066715, 03.01.1978; 25. US 4113790, 12.09.1978; 26. US 4219691, 26.08.1980; 27. US 5196635, 23.03.1993; 28. US 5489721, 06.02.1996; 29. US 4214112, 22.07.1980; 30. US 3168588, 12.03.1975; 31. US 3884988, 20.05.1975; FR 2221467, 1974; 32. US 4384089; 33. US 4510342, 09.04.1985; 34. US 4579991, 01.04.1986; 35. US 4855526, 08.08.1989; 36. GB 1430497; GB 1522129; 37. SU 1073279; 38. JP 52-11350, 1977; 39. US 4952739, 28.08.1990; 40. US 4041098, 09.08.1977; 41. GB 1535324, 1535325, 1978; 42. DE 2526615, 13.06.1975; DE 2304314, 1980; 43. SU 1723101, 20.04.1989). В соответствии с этими способами олигомеризацию олефинового сырья осуществляют под действием катионных и полифункциональных каталитических систем, включающих кислоты Льюиса (основу катионных катализаторов - BF3, АlX3, RnАlX3-n, где Х - Сl, Br, I; n=1,0; 1,5; 2,0; R - СН3, С2Н5, С3Н7, i-С4Н9) и различные сокатализаторы - воду, спирты, карбоновые кислоты, кетоны, полиолы, простые и сложные эфиры, галоидалкилы, TiCl4, ZrCl4 при температурах, выбранных из интервала 20-250oС. В качестве олефинового сырья при этом используют линейные альфа-олефины (ЛАО). Наиболее часто при получении олефиновых олигомеров в качестве олефинового сырья используется децен-1. Это обусловлено тем, что выделяемые из олигомеров децена-1 тримеры децена-1 после гидрирования характеризуются уникальным комплексом физических свойств (кинематическая вязкость при 100oС равна 3,9 сСт, индекс вязкости = 130, температура застывания = минус 60oС, температура вспышки = 215-220oС). Они используются в качестве основы широко потребляемых синтетических и полусинтетических моторных (автомобильных, авиационных, вертолетных, тракторных, танковых) и других масел. Из олефинов с другим числом атомов углерода в молекуле олефиновые масла с приведенными выше характеристиками не получаются.
Все способы такого типа имеют два существенных недостатка.
1. Главным общим недостатком известных способов получения деценовых олигомеров является относительно низкая их селективность, обусловленная тем, что в результате катионной олигомеризации децена-1 под действием упомянутых каталитических систем образуются димеры децена-1 с почти линейной структурой цепи. Температура застывания таких димеров децена-1 после их гидрирования превышает минус 20oС. Это резко ограничивает возможности квалифицированного использования димеров децена-1, что приводит к повышению стоимости целевых продуктов.
2. Другим общим недостатком упомянутых способов является то, что в них в качестве исходного сырья используется только децен-1 или смеси его с некоторыми другими ЛАО. Децен-1 относится к категории дефицитного и дорогостоящего (примерно 1200 долларов США за тонну) химического сырья. Его получают только в процессах олигомеризации этилена под действием триэтилалюминия или комплексных металлов (Ti, Zr, Ni, Fe, Pd) органических катализаторов. Высокая стоимость децена-1 обусловлена тем, что содержание его в продуктах олигомеризации этилена не превышает 15 мас.% в расчете на превращенный в ЛАО этилен.
Разработан способ получения олефиновых олигомеров, в соответствии с которым для повышения селективности и улучшения технико-экономических показателей негидрированные димеры децена-1 направляют в рецикл на соолигомеризацию их с деценом-1 в широкопотребляемые три- и тетрамеры децена (44, US 4263467, 2.04.1981). Соолигомеризацию димеров децена-1 (43,8 мас.% в шихте) с деценом-1 (40 мас.% децена-1 и 15,4 мас.% декана в шихте) при этом осуществляют под действием системы ВF3/SiO2 (D=0,8-2,0 мм) + Н20 (65 м.д. в шихте) при температурах 15-30oС, давлении 0,1-0,69 МПа при расходе шихты 2,5 л в час на 1 л катализатора. Содержание димеров децена-1 на выходе из реактора уменьшается до 20,7 мас.%, а содержание тримеров децена-1 возрастает до 41,8 мас. %. Это решение позволяет квалифицированно использовать непотребляемые (застывающие при высоких температурах) димеры. Недостатком этого решения является резкое снижение производительности процесса.
Известен способ получения олефиновых олигомеров, в соответствии с которым в качестве исходного олефинового сырья используют олефины дегидрирования парафинов С10-С13 с ISOSIV процесса со статистическим (случайным) расположением двойных СН=СН связей внутри молекулы (далее "внутренние" олефины) (45, US 4167534, 11.09.1979).
Олигомеризацию указанного олефинового сырья осуществляют под действием треххлористого алюминия (1-3 мас.% в расчете на олефины) в интервале температур от 80 до 150oС. Образующиеся в этом процессе продукты (включая димеры) характеризуются высокой разветвленностью. Благодаря этому температура застывания их не превышает минус 50oС. Недостатком этого процесса является его низкая производительность, весьма сложное многостадийное технологическое оформление и как следствие низкие технико-экономические показатели.
Разработан усовершенствованный способ получения олефиновых олигомеров, в соответствии с которым в качестве исходного сырья используется смесь деценов, содержащая 50-90 мас.% децена-1 и 10-50 мас.% "внутренних" деценов (46. пат. США 4910355, 20.03.1990). Указанную смесь олефинов получают путем частичной изомеризации децена-1 под действием кислот Льюиса. Олигомеризацию смеси децена-1 и "внутренних" деценов производят под действием системы ВF3-n-С4Н9ОН в интервале температур от 10 до 80oС. Частичная изомеризация децена-1 во "внутренние" децены приводит к снижению температуры застывания олигомеров от -54 - -57 до -66 - -69oС [46].
Недостатком этого решения является то, что в процессе олигомеризации частично изомеризованного децена-1 параллельно с высоко разветвленными ди-, три- и более высокомолекулярными олигодеценами из децена-1 образуется небольшое количество почти линейных димеров децена:


После гидрирования углеводороды (1-3) превращаются в 9-метилнанодекан:
При температурах ниже минус 20oС этот углеводород образует коллоидно-дисперсные кристаллиты (наблюдаемые визуально в виде тонкой белой взвеси), которые забивают трубопроводы, фильтры и затрудняют эксплуатацию техники.
Способ [48] по технической сущности и по достигаемым результатам является ближайшим аналогом разработанного согласно настоящему изобретению способа. Поэтому мы рассматриваем его в качестве прототипа.
Задачей настоящего изобретения являлась разработка способа получения олефиновых олигомеров (в частности, ди-, три- и тетрамеров деценов), реализация которого позволила бы повысить его селективность по выходу целевых продуктов, улучшить их качество и расширить доступную сырьевую базу для их получения.
Поставленная задача решена тем, что разработан способ получения олефиновых олигомеров, включающий катионную олигомеризацию олефинового сырья, выделение отработанного катализатора из олигомеризата, разделение олигомеризата на фракции и гидрирование выделенных из олигомеризата фракций, в соответствии с которым в качестве олефинового сырья для катионной олигомеризации берут смесь цис- и трансизомеров деценов-2, -3, -4 и/или -5.
В соответствии с заявляемым способом смесь цис- и трансизомеров деценов-2, -3, -4 и -5 получают путем изомеризации децена-1. Изомеризацию децена-1 в смесь цис- и трансизомеров деценов-2, -3, -4 и -5 ведут в трубчатом реакторе вытеснения непрерывного действия в среде исходного и образующихся деценов под действием каталитической системы, включающей олеорастворимое соединение никеля (NiY2) и алкил алюминий галогенид (RnА1Х3-n) при температурах 20 (преимущественно) - 150oС, давлениях 0,1-0,5 МПа, концентрациях соединения никеля 0,0005-0,004 моль/л, концентрациях алкилалюминийгалогенида 0,01-0,08 моль/л (преимущественно 0,04-0,08 моль/л), при мольных соотношениях Al/Ni = 5-50 (преимущественно 20), временах реакции 1-60 минут (преимущественно 3-5 минут). При этом в качестве олеорастворимого соединения никеля берут ацетилацетонат никеля или безводные никелевые соли насыщенных, ненасыщенных и ароматических карбоновых кислот (от масляной, изомасляной, метакриловой и бензойной до стеариновой и олеиновой включительно), а в качестве алкилалюминийгалогенида берут RnА1Х3-n, где R - СН3, С2Н5, С3Н7, i-C4H9; Х-С1 (преимущественно), Вr или I; n=1,0 и 1,5 (преимущественно) или 2,0.
После завершения изомеризации смесь цис- и трансизомеров деценов-2, -3, -4 и -5 вместе с каталитической системой изомеризации NiY2-RnА1Х3-n направляют в реактор олигомеризации, где каталитическую систему изомеризации обрабатывают R'X (R' - трет-бутил, аллил, бензил, ацетил, бензоил) или TiCl4 для превращения ее в растворимую каталитическую систему катионной олигомеризации цис- и трансизомеров 2-, 3-, 4- и 5-деценов в ди-, три-, тетрамеры и другие олигомеры и соолигомеры упомянутых деценов.
Для упрощения технологического оформления этого способа изомеризацию децена-1 в смесь цис- и трансизомеров деценов-2, -3, -4 и -5 под действием каталитических систем NiY2-RnА1Х3-n (1) и (со)олигомеризацию всех изомеров деценов под действием систем NiY2-RnА1Х3-n - R'X или TiCl4 (2) проводят одновременно путем синхронной подачи децена-1 и всех компонентов систем (2) в реактор.
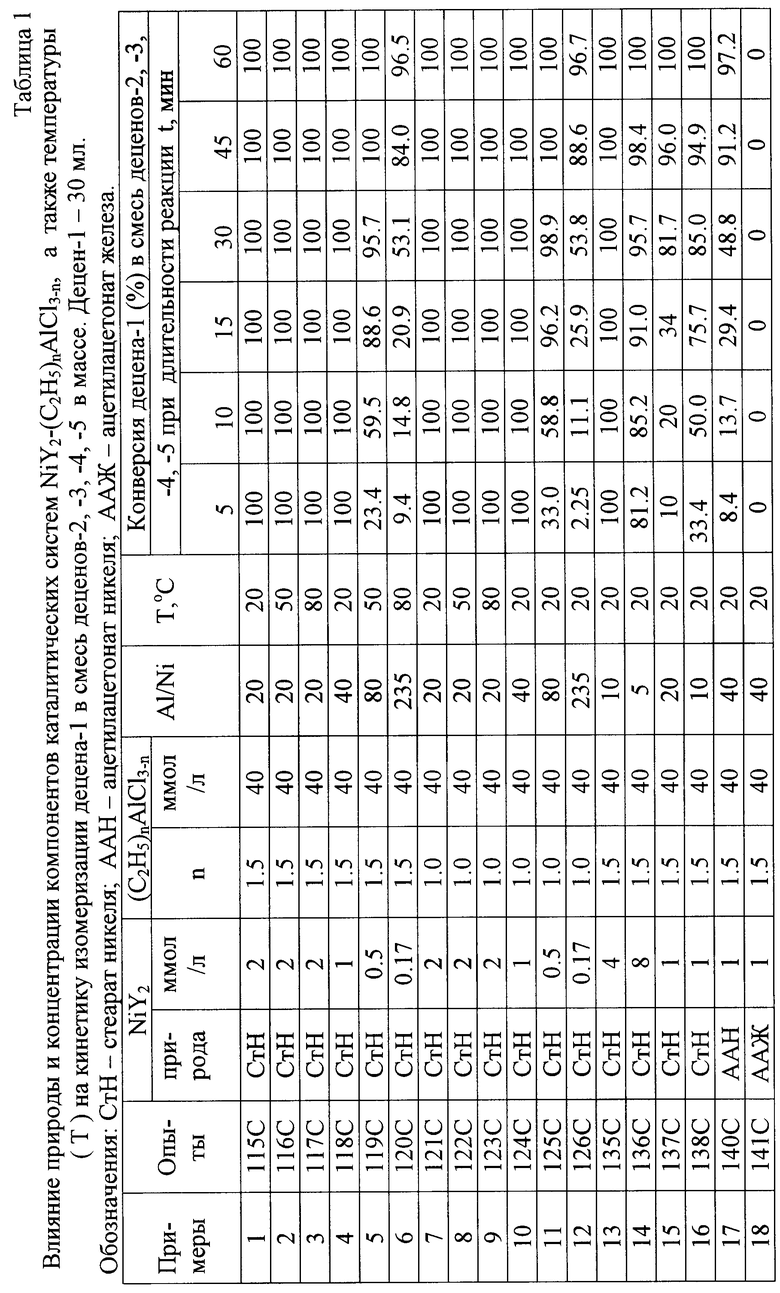
Изомеризация децена-1 под действием системы NiY2-RnА1Х3-n в указанных условиях протекает термонейтрально, селективно и с очень высокой скоростью (таблица 1).
Из таблицы 1 видно, что в лабораторных условиях под действием каталитических систем стеарат никеля (СтН)-(C2H5)1,5AlCl1,5 (ЭАСХ) и СтН - С2Н5А1С12 (ЭАДХ) при концентрациях ЭАСХ и ЭАДХ = 40 ммоль/л (оптимальная концентрация ЭАСХ и ЭАДХ в каталитических системах катионной олигомеризации олефинов) и мольных соотношениях Al/Ni = 20 при температурах 20-80oС стопроцентная изомеризация децена-1 во "внутренние" децены достигается менее чем за пять минут. Аналогичный результат в указанных выше условиях получен и при 100oС (пример 20). Однако повышение температуры до 150oС при прочих неизменных условиях приводит к снижению конверсии децена-1 во "внутренние" децены до 38,2% за 120 минут. При этом при 150oС из деценов образуется 3,9 мас.% димеров и 1,2 мас.% тримеров деценов (пример 21).
При прочих одинаковых условиях скорость изомеризации и конверсия децена-1 во "внутренние" децены снижается при уменьшении концентрации NiY2 и мольного соотношения Al/Ni (таблица 1).
Высокую активность в изомеризации децена-1 во "внутренние" децены проявила также каталитическая система ацетилацетонат никеля (ААН) - ЭАСХ (пример 17). В противоположность этому система, включающая ацетилацетонат трехвалентного железа и ЭАСХ, в оптимальных для упоминавшихся выше никельсодержащих каталитических систем условиях оказалась неактивной как катализатор изомеризации децена-1 (пример 18).
Из приведенных в таблицах 1, 2 примеров следует, что изомеризация децена-1 в смесь цис- и трансизомеров деценов-2, -3, -4 и -5 сопровождается (со)олигомеризацией деценов и, что повышение температуры и увеличение продолжительности реакции способствует повышению конверсии деценов в ди- и тримеры деценов. При низких температурах (до 70oС) и малых временах реакции (например 6 минут) дии тримеризация деценов под действием каталитических систем изомеризации NiY2-RnА1Х3-n не имеет места.
Указанные особенности изомеризации децена-1 под действием каталитических систем NiY2 - (С2Н5)nА1С13-n обеспечивают возможность проведения этой реакции в малогабаритном трубчатом реакторе вытеснения при температуре окружающей среды при временах пребывания 3-5 минут. Аналогичные результаты были получены при осуществлении изомеризации децена-1 в смесь "внутренних" деценов (а также при осуществлении совмещенного процесса изомеризации-олигомеризации) и в металлических реакторах при объеме реакционной массы 0,2 литра (примеры 19-21, 27, 28) и 5,0 литров (примеры 22, 23, 26, 29-31), а также на опытной установке непрерывного действия с трубчатым реактором вытеснения (примеры 24, 25, 32, 33).
В примерах 1-18 изомеризацию децена-1 во "внутренние" децены в лабораторных условиях осуществляли следующим образом:
Пример 1. В стеклянный термостатируемый через рубашку реактор объемом 60 мл при перемешивании с помощью электромагнитной мешалки в инертной атмосфере загружали 0,0375 г (0,06 ммоль) стеарата никеля (ММ = 624,7 г/моль), 30 мл децена-1 и 1,2 ммоля (0,1485 г) ЭАСХ. Изомеризацию проводили в изотермических условиях в течении 60 минут. По ходу процесса с помощью шприца на 5, 10, 15, 30, 45 и 60 минутах отбирали по 5 мл реакционной массы на анализы. Сразу же после отбора проб в пробирки, куда их сливали, для дезактивации катализатора изомеризации при перемешивании прибавляли 2 мл этанола. Конверсию децена-1 во "внутренние" децены определяли методом ИК-спектроскопии на приборах Specord M 80, М 82 и Nicolet по полосам 910 и 968 см-1 (СН2=СН- и транс-СН= СН- группы соответственно), а также методом ПМР на приборе Varian НА-100 в соответствии с методикой Дормана (D.E.Dorman et al. // J. Organic Chem. 1971. V. 36. N 19. P.2757-2766). Стереоконфигурацию (цис-, транс-) и положение (2, 3, 4, 5) двойных связей в молекулах изомеризованных деценов определяли методом 13С-ЯМР по интегральной интенсивности аналитических полос в спектрах образцов, отобранных из реактора при различной продолжительности процесса.
В примерах со стопроцентной конверсией децена-1 во "внутренние" децены соотношение цис-/трансизомеров обычно =1:5, соотношение СН2/СН3=5,0-5,1, а соотношение между позиционными изомерами децен-2:децен-3:децен-4:децен-5 = 1: 1:1,1:0,9, т.е. соответствовало термодинамически равновесному соотношению указанных изомеров.
В примерах 5, 6, 11, 12, 14-17 все упомянутые соотношения изменялись в широких пределах, что определялось кинетикой перемещения двойной связи от конца к центру деценовых молекул. В частности, соотношение цисизомеров к трансизомерам деценов изменялось в пределах от 1:2,9 до 1:5,8.
В соответствии с настоящим изобретением продукты изомеризации децена-1 вместе с каталитической системой изомеризации NiY2-RnА1Х3-n поступают на стадию олигомеризации, где каталитическую систему изомеризации обрабатывают R'X (R' - трет-бутил, аллил, бензил, ацетил, бензоил) или TiCl4 для превращения (инверсии) ее в растворимую катионную каталитическую систему олигомеризации цис- и трансизомеров -2, -3, -4 и деценов-5 в ди-, три-, тетра- и другие олигомеры и соолигомеры упомянутых деценов. R'X или TiCl4 подают в реактор в неразбавленном виде независимым потоком. В этом варианте процесса изомеризация и олигомеризация разделены во времени и в пространстве: они осуществляются последовательно в двух отдельных реакторах.
Принимая во внимание рассмотренные кинетические данные, для упрощения технологического оформления способа получения олефиновых олигомеров в соответствии с настоящим изобретением изомеризацию децена-1 в смесь цис- и трансизомеров деценов-2, -3, -4 и -5 под действием каталитических систем изомеризации NiY2-RnА1Х3-n (1) (со)олигомеризацию всех изомеров деценов под действием бифункциональных каталитических систем NiY2-RnА1Х3-n - R'X или TiCl4 (2) проводят одновременно путем синхронной подачи децена-1 и всех компонентов систем (2) в реактор (примеры 27-33).
Из таблицы 2 видно, что в случае осуществления способа при температурах 20-70oС непревращенный децен даже при временах пребывания 6 минут полностью изомеризуется в смесь "внутренних" деценов. Примеры 29-33 свидетельствуют о том, что скорость изомеризации децена-1 под действием системы (1) настолько высока, что существенно превышает высокую скорость (со)олигомеризации всех изомеров деценов. При изменении условий осуществления способа конверсия всех изомеров деценов в олигомеры изменяется в пределах от 67,0 до 98,7 мас.% по исходному децену-1 (примеры 27-33). Из примеров 27 и 28 следует, что повышение температуры до 100-150oС, видимо, приводит к быстрой дезактивации активных центров изомеризации, что проявляется в резком снижении степени изомеризации непревращенных деценов (до 17,8-22,7 мас.% соответственно).
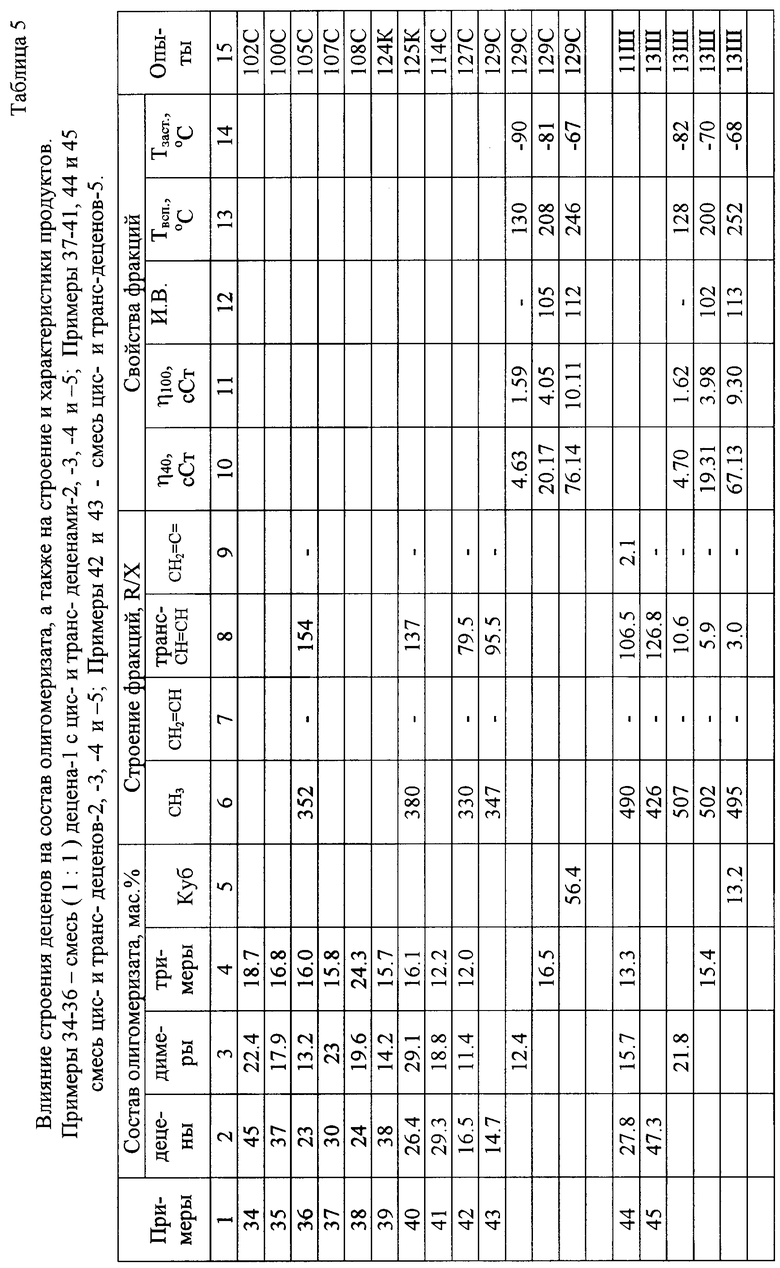
Децены с "внутренним расположением" двойных связей в виде смесей цис- и трансизомеров деценов-5 в соответствии с настоящим способом получают метатезисом (диспропорционированием) гексена-1.
Метатезис (диспропорционирование) гексена-1 в смесь цис- и трансизомеров децена-5 осуществляют в среде смесей исходного гексена-1 и образующейся смеси цис- и трансдеценов-5 при температурах 20-90oС и давлениях 0,1-0,3 МПа под действием суспендированного в реакционной среде предварительно механо- или плазмообработанного и дополнительно активированного в реакторе (в жидкой фазе) с помощью А1R3 катализатора (50-100 г/л), выбранного из группы, включающей МоО3/А12О3, MoО3/SiО2, Rе2О7/А12O3, Re2О7/SiО2 (где R - СН3 (преимущественно), С2Н5, С3Н7 или i-C4H9).
Катализаторы МоО3/А12О3 или МоО3/SiO2 и Rе2О7/Аl2О3 или Re2О7/SiО2 готовили пропиткой носителей водными растворами молибденовокислого аммония (NН4)6Мо7О24•4Н2О (ГОСТ 3765-78) и рениевокислого аммония NH4ReО4 (ТУ 6-09-04-133-75) соответственно, а затем сушили при перемешивании и (для превращения аммонийных солей в оксиды Мо и Re) прокаливали при 550oС в течение десяти часов.
В качестве носителей использовали Аl2О3 (фракция 0,1-0,5 мм) с удельной поверхностью 210-350 м2/г и силикагель SiO2 марок С-3, КСК, ШСК или Davison 955 (фракция 10-100 мкм с удельной поверхностью 210-312 м2/г и объемом пор 1,6-1,9 см3/г).
Эксперименты по диспропорционированию гексена-1 в жидкой и парогазовой фазе под действием неактивированных и плазмоактивированных нанесенных металлоксидных катализаторов МоО3/Аl2О3 или МоО3/SiO2 и Re2O7/Al2O3 или Re2О7/SiО2 проводили в трубчатом кварцевом электрообогреваемом реакторе непрерывного действия с внутренним диаметром трубы 1,4 см и длиной 35 см. В нижней части трубчатого реактора при его изготовлении была вмонтирована кварцевая решетка (типа фильтра Шотта) с диаметром пор около 0,1 мм. В реактор насыпью или в мешочке из стеклоткани загружали от 3,5 до 30 мл одного из упомянутых катализаторов. Гексен-1 (производства НКНХ, ТУ 2411-05766801-96) с помощью микродозирующего насоса непрерывно подавали в верхнюю часть вертикально установленного трубчатого реактора. Образующиеся продукты реакции (этилен и децены-5) непрерывно выводили из нижней части реактора. Скорость подачи гексена-1 в реактор варьировали в пределах от 1,0 до 100 час-1.
Эксперименты по диспропорционированию гексена-1 в жидкой фазе (в среде смесей гексена-1 и деценов-5) под действием не активированных физическими методами и механообработанных, дополнительно активированных А1R3, нанесенных металлооксидных катализаторов МоО3/Аl2О3 или МоО3/SiO2 и Re2O7/Al2O3 или Re2О7/SiО2 проводили также и в реакторе смешения периодического действия с электромагнитной мешалкой. Реактор был оснащен специальным устройством для подачи в зону реакции в любой момент времени заданного количества триалкилалюминия - AlR3. Образующийся в ходе реакции этилен непрерывно выводили из реактора через обратный холодильник в калиброванный по объему газгольдер.
Перед реакцией неактивированный катализатор в течение трех часов обрабатывали при 600oС воздухом, а затем в течение одного часа (для обеспечения деаэрации и охлаждения) продували высокочистым азотом. Охлажденный до температуры окружающей среды катализатор далее загружали в описанные реакторы, либо подвергали механо- или плазмообработке.
Предварительными опытами было установлено, что диспропорционирование гексена-1 под действием Re2O7/Al2O3 протекает при температурах 20-120oС под действием МоО3/Аl2О3 при температурах 35-150oС, а под действием WO3/Al2O3 при температурах 300-400oС. Установлено также, что диспропорционирование гексена-1 под действием МоО3/Аl2О3 протекает при более низкой температуре, чем под действием МоО3/SiO2. Из этих данных и данных о стоимости катализаторов следует, что предпочтительным является доступный (выпускаемый в промышленном масштабе) алюмомолибденовый катализатор МоО3/Аl2О3.
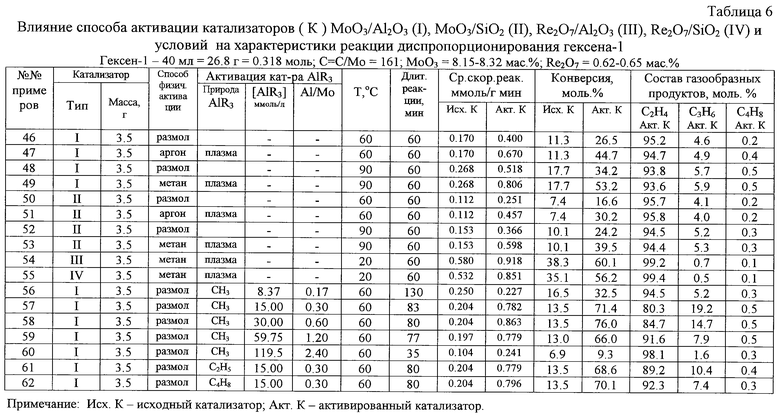
В таблице 6 показана зависимость средней скорости диспропорционирования гексена-1 от содержания МоО3 в образцахМоО3/Аl2О3, МоО3/SiO2. Из этой таблицы следует, что максимальное значение активности наблюдается при содержании в катализаторе МоО3/Аl2О3 8,15 мас. % МоО3 (в случае МоО3/SiO2 15 мас.% МоО3).
В случае неактивированных образцов рассматриваемых катализаторов наблюдался индукционный период, наличие которого свидетельствует об активации оксида переходного металла гексеном-1. Индукционный период в случае МоО3/Аl2О3 был значительно меньше, чем в случае МоО3/SiO2.
Диспропорционирование гексена-1 при температурах 20-150oС сопровождается протеканием побочных реакций изомеризации, олигомеризации и полимеризации и характеризуется падением активности алюмомолибденового катализатора. При повышенных температурах наряду с упомянутыми реакциями имеет место скелетная изомеризация, дегидрирование и расщепление углерод-углеродных связей гексена-1 и образующихся деценов-5. Снижение активности катализаторов в ходе диспропорционирования гексена-1 может быть обусловлено блокировкой поверхности частиц катализатора образующимися полимерами и/или глубоким восстановлением молибдена под действием гексена-1. Оба эти предположения подтверждаются тем, что максимальная активность дезактивированных катализаторов МоО3/Аl2О3, МоО3/SiO, Rе2О7/А12О3 и Re2О7/SiО2 восстанавливается в результате прокаливания их в токе воздуха при 550oС в течение двух часов. Дезактивация катализатора МоО3/Аl2О3 в ходе диспропорционирования гексена-1 в жидкой фазе происходит более медленно, чем в случае парофазного диспропорционирования. Это можно объяснить тем, что жидкофазное диспропорционирование протекает при более низких температурах (при которых полимеризация этилена и гексена-1 не идет или протекает с низкой скоростью), а также тем, что образующиеся полимеры, по крайней мере, частично растворяются в жидкой фазе и не блокируют активные центры диспропорционирования.
Механообработку упомянутых катализаторов проводят в шаровой мельнице при температуре окружающей среды в атмосфере аргона или метана (природного газа). Для этого в реактор из нержавеющей стали объемом 100 мл загружают 5 г катализатора (например, МоО3/SiO2) и 60-100 г стальных шаров диаметром 12 мм, реактор герметично закрывают, деаэрируют с помощью вакуумного насоса и заполняют аргоном или метаном из баллонов до достижения давления 0,1-0,3 МПа. Механообработку катализаторов осуществляют путем встряхивания заполненного реактора на механическом вибраторе с частотой 5-15 Гц и амплитудой 5-20 мм в течение 5-60 минут. Активированный катализатор из реактора механообработки перегружают в реактор метатезиса, исключая контакт его с кислородом и влагой. В процессе механообработки к частицам катализаторов в актах механического воздействия подводится энергия. В результате этого в частицах создается поле напряжений. Релаксация поля напряжений происходит несколькими путями: дроблением частиц катализатора с образованием новой дефектной высокоактивной поверхности, эмиссией электронов при раскалывании микрокристаллов, возбуждением и разрывом некоторых связей, инициированием и протеканием химических реакций в твердой фазе. Все эти процессы могут приводить к формированию потенциально активных центров метатезиса. Эти центры при контакте с гексеном-1 в жидкой или в паровой фазе превращаются в карбеновые активные центры метатезиса. Аналогичные процессы протекают и при обработке молибденоксидных и ренийоксидных катализаторов аргоновой или метановой плазмой высокочастотного (ВЧ) разряда. Обработку упомянутых катализаторов плазмой ВЧ разряда проводят при температурах 20-80oС и при давлениях аргона или метана 0,01-10,0 мм рт. ст. Для этого в кварцевый реактор-уточку (диаметр 35 мм, длина 150 мм, объем 0,2 л) загружают катализатор, реактор закрепляют в механической качалке, соединяют с вакуумной системой и с устройством для дозирования аргона или метана, реактор деаэрируют, устанавливают непрерывный поток аргона или метана (линейная скорость газов 30 см/с) и подключают с помощью внешних электродов к генератору тлеющего разряда УВЧ-66. Частота тока разряда 40,68 МГц, напряженность электрического поля 8000 В/см, плотность тока = 1,8-8,8 мА/см2, концентрацию электронов в разряде изменяют в пределах от 1013 до 1014 е/см3, мощность разряда изменяют в пределах от 20 до 70 Вт. Перемешивание осуществляют с помощью механической мешалки, а температуру в реакторе - с помощью термостата. Указанные условия и параметры обеспечивают непрерывное горение ВЧ разряда и эффективную активацию упомянутых катализаторов.
Для упрощения технологического оформления этого варианта способа метатезис (диспропорционирование) гексена-1 в смесь цис- и трансизомеров децена-5 осуществляют в паровой фазе под действием плазмообработанных однокомпонентных катализаторов МоО3/SiO2 или Rе2О7/Al2О3 при температурах 120-150oС (таблица 6).
Полученные результаты свидетельствуют о том, что предварительная механо(размалывание) и плазмообработка катализаторов обеспечивают возрастание их активности в процессе метатезиса гексена-1 как в жидкой, так и в паровой фазе (таблица 6).
Переработка гексена-1 в децены-5, а деценов-5 в основы синтетических масел разнообразного назначения позволит существенно расширить сырьевую базу для получения указанных продуктов и получить существенный экономический эффект. Последнее обеспечивается тем, что цена гексена-1 на мировых рынках (600-800 долларов США/т) существенно ниже цены децена-1 (1200 долларов США/т) и, что гексен-1 производится в больших количествах, а потребляется в меньших количествах, чем децен-1.
Смеси цис- и трансизомеров 2-, 3-, 4- и/или 5-деценов могут использоваться не только при получении олефиновых олигомеров, но и в других процессах (например, в процессах алкилирования при получении высших алкилароматических углеводородов и различных поверхностно-активных веществ на их основе, в процессах эпоксидирования и т.д.). Поэтому разработанный способ получения олефиновых олигомеров дополнительно включает выделение и очистку смеси цис- и трансизомеров 2-, 3-, 4- и/или 5-деценов соответствующими известными методами.
Согласно настоящему изобретению олефиновые олигомеры из индивидуальных или смесей цис- и трансизомеров 2-, 3-, 4- и/или 5-деценов получают в реакторах смешения или вытеснения непрерывного действия при временах пребывания от 1 до 120 минут под действием растворимой катионной каталитической системы, включающей алкилалюминийгалогенид RnAlX3-n и галоидалкил R'X или TiCl4 в среде смеси олигомеризуемого олефинового сырья и продуктов реакции в отсутствии или в присутствии соединений никеля при концентрациях RnAlX3-n 0,02-0,08 моль/л, мольных соотношениях R'X/Al=1-3 или TiCl4/Al = 0,5-1,5 в интервале температур от 20 до 150oС, при давлениях 0,1-1,0 МПа. При этом в качестве RnAlX3-n и R'X используют соединения, в которых R - СН3, С2Н5, С3Н7, i-C4H9; X-C1 (преимущественно), Вr или I; n =1,0 и 1,5 (преимущественно) или 2,0; R' - трет-бутил (преимущественно), аллил, бензил, ацетил или бензоил.
Разработанный способ позволяет осуществлять олигомеризацию олефинового сырья в присутствии парафиновых или ароматических углеводородов (таких как декан, бензол, толуол, ксилол или нафталин). Осуществление олигомеризации олефинового сырья (в том числе и деценов) приводит к образованию высокоустойчивых к термоокислительной деструкции "ароматизированных" олигомеров, функционализированных молекулами ароматических углеводородов.
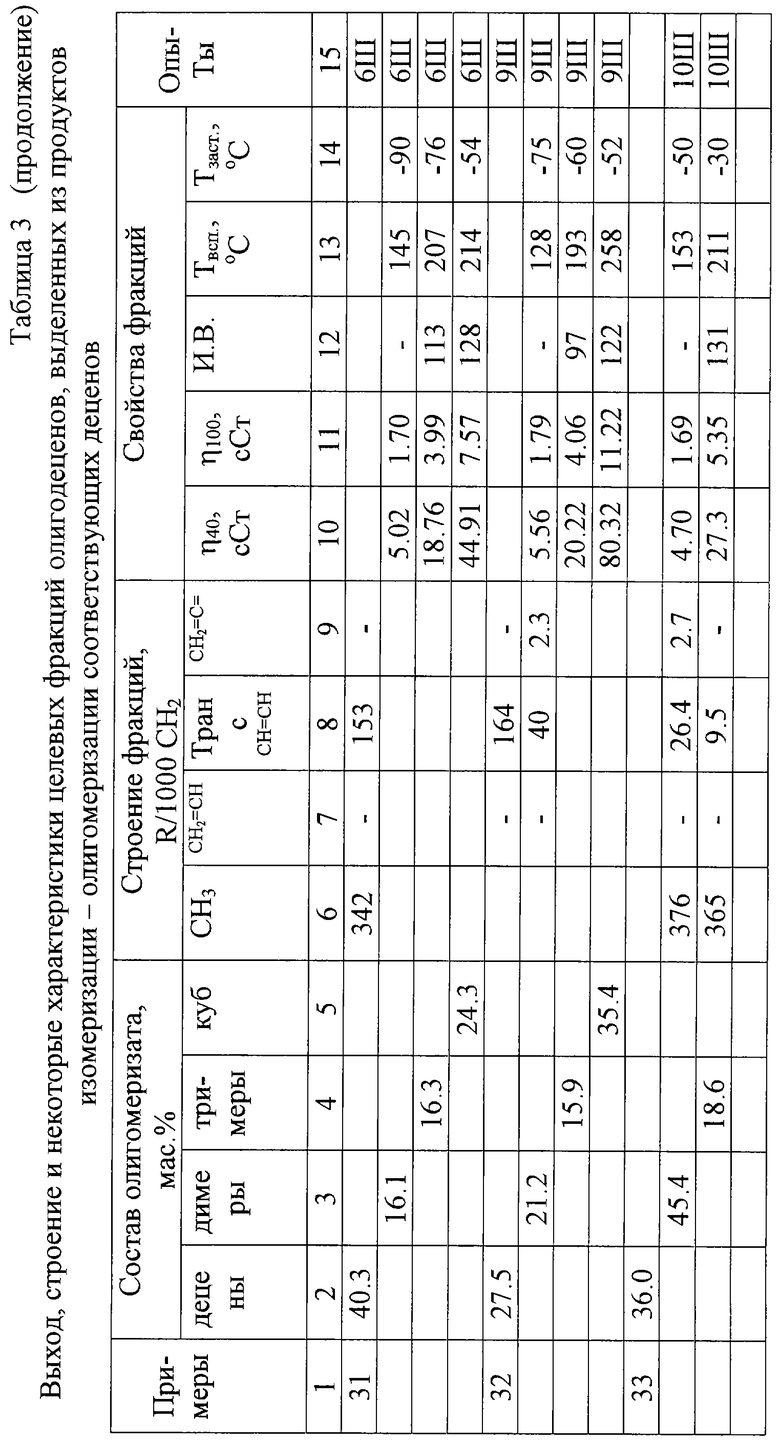
Выход, строение и некоторые характеристики целевых фракций олигодеценов, выделенных из продуктов превращения децена-1 (изомеризации олигомеризации) в соответствии с настоящим изобретением, приведены в таблице 5.
Испытание катализаторов и отработку процессов изомеризации децена-1 и катионной олигомеризации других деценов в лабораторных условиях и на опытной установке производили в металлических реакторах смешения и вытеснения (соответственно емкостного и трубчатого типа) следующим образом.
В термостатируемый очищенный и высушенный реактор емкостного типа периодического действия с мешалкой объемом 1 или 10 литров в инертной атмосфере при температуре окружающей среды (+15-+25oС) в случае осуществления изомеризации децена-1 вначале загружали соединение никеля. Вслед за этим в реактор из мерных емкостей загружали олефин (в некоторых случаях и ароматический углеводород) (степень заполнения реактора ≅ 50 об.%). Туда же с помощью калиброванного шприца при перемешивании вводили растворы галоидорганического соединения R'X и органического модификатора (ОМ), с помощью термостата разогревали реактор и его содержимое до заданной температуры и только после этого с помощью специального дозирующего устройства при перемешивании загружали необходимое количество алкилалюминийгалогенида RnAlX3-n. Сразу же после этого начиналась изомеризация и/или олигомеризация олефина. Олигомеризация обычно сопровождалась заметным повышением температуры реакционной среды (олигомеризата). Реакцию продолжали в течение запланированного времени, а затем обрывали путем введения в реактор водного раствора щелочи (NaOH) либо этанола. Полученные продукты из реактора выгружали далее в промежуточную емкость. Отработанный (дезактивированный) катализатор из продуктов реакции выделяли адсорбционным способом или методом водно-щелочной и водной отмывки.
При отработке реакций изомеризации и/или олигомеризации деценов в непрерывно действующем трубчатом реакторе использовались два типа трубчатых реакторов: трубчатый реактор, представляющий собой металлическую трубку с внутренним диаметром 10 мм и длиной один метр с рубашкой (объем реакционного пространства равнялся 78,5 мл), и трубчато-щелевой реактор длиной 1,5 м с трубчато-щелевым реакционным пространством с шириной щели между коаксиальными трубками, образующими реакционное пространство, равной 4 мм. Объем реактора этого типа равнялся 205 мл. Трубчато-щелевое реакционное пространство разогревали или охлаждали с обеих сторон щелевого зазора с помощью термостата или сетевой проточной воды.
Перед началом отработки указанных реакций в реакторах трубчатого типа на опытной установке в инертной атмосфере раздельно готовили по 30 л растворов компонентов катализатора в олефине (в частности, в децене-1). В первом аппарате (реакторе с мешалкой) готовили раствор NiY2 и/или галоидорганического соединения R'X в олефине. Во втором аппарате (реакторе с мешалкой) готовили 30 л раствора алкилалюминийгалогенида RnAlX3-n заданной концентрации (обычно 0,08 моль RnAlX3-n/л раствора в этом аппарате). После непродолжительного (5-10 мин) интенсивного перемешивания с помощью рамочной мешалки (100 об/мин) растворы компонентов катализатора заданной концентрации независимыми (одинаковыми по расходу) потоками подавались на вход упомянутых трубчатых реакторов. Температура входящих в реактор потоков соответствовала температуре окружающей среды (15-25oС). В некоторых случаях растворы компонентов катализатора перед подачей в реактор разогревались до заданной температуры. В момент смешения входящих в реактор потоков растворов компонентов катализатора с очень высокой скоростью образовывались активные центры изомеризации и/или катионные активные центры и начиналась изомеризация и/или олигомеризация олефина. Изомеризация протекала с высокой скоростью, но без выделения тепла. Олигомеризация также протекала с высокой скоростью, но с выделением большого количества тепла (ΔН)-(ΔН=22[11/Рn] ккал/моль превращенного олефина, где Рn - среднечисловая степень олигомеризации олефина). Выделяющееся тепло олигомеризации удалялось из зоны реакции хладагентом (теплоносителем) (обычно водой) через рубашки упомянутых трубчатых реакторов. Использование трубчато-щелевого реактора при отработке стадии олигомеризации деценов согласно изобретению обеспечило осуществление олигомеризации практически в изотермических условиях.
Из трубчатого реактора реакционная масса (олигомеризат) поступала в сборник-дозреватель, в адсорбер либо в промыватель.
Сборник-дозреватель представлял собой емкостной реактор с мешалкой, где изомеризация и/или олигомеризация непревращенного олефина продолжалась еще в течение 60 минут. Пребывание реакционной массы в сборнике-дозревателе способствовало повышению конверсии исходного олефина на 3-10 мас.% по сравнению со случаем, когда реакционноя масса сразу же направлялась на очистку. Из сборника-дозревателя реакционная масса направлялась в адсорбер или в промыватель.
Адсорбер представлял собой термостатируемый колонный аппарат, заполненный активированными калиброванными частицами оксида алюминия или боксита. При прохождении реакционной массы (снизу-вверх) через слой указанного адсорбента происходило фронтальное адсорбционное выделение отработанного дезактивированного катализатора и очистка реакционной массы.
Промыватель использовался для выделения отработанного катализатора методом водно-щелочной, а затем водной отмывки (очистки) реакционной массы. Он функционировал периодически и представлял собой термостатируемый (обогреваемый) реактор с мешалкой, в который из вспомогательных емкостей-мерников подавалась реакционная масса, 5%-ный водный раствор NaOH или NН3, а затем свежая деминерализованная вода. В некоторых случаях реакционная масса поступала в промыватель непосредственно из трубчатого реактора. После отбора проб для анализов очищенная реакционная масса загружалась в куб вакуумной колонны для последующей азеотропной осушки, выделения непревращенных олефинов и для разделения олигомера на фракции.
Фракционный состав продуктов олигомеризации определяли стандартным хроматографическим методом с использованием приборов ЛХМ-8 МД, Hewlett Packard Model 5710A и/либо методом фракционирования на вакуумной ректификационной колонне. Результаты этих анализов совпадали с точностью до ±2 мас.%. Строение выделенных продуктов количественно определяли методами озонолиза на приборе АДС-4М, ИКС, ПМР и 13C-ЯМР.
Для лучшего понимания данного изобретения в качестве иллюстраций в таблицах приведены примеры осуществления разработанного согласно изобретению способа получения олефиновых олигомеров и результаты определения состава, строения и свойств полученных продуктов. Эти примеры демонстрируют, но не исчерпывают возможности изобретения.
Из приведенных примеров следует, что "внутренние" децены так же, как и децен-1, могут использоваться для получения олигодеценовых олигомеров. Реакционная способность "внутренних" деценов ниже реакционной способности децена-1, что при прочих одинаковых условиях осуществления олигомеризации проявляется в некотором снижении конверсии деценов в олигодецены. Использование "внутренних" деценов в процессе получения олефиновых олигомеров согласно изобретению обеспечивает повышение управляемости процессом и позволяет получать высокоразветвленные димеры, тримеры и олигомеры деценов с рекордно низкими температурами застывания при некотором изменении комплекса и других физических свойств получаемых продуктов.
Совокупность физико-химических свойств олефиновых олигомеров, синтезированных в соответствии с настоящим изобретением, позволяет выделить их в новую группу олефиновых олигомеров.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ КАТИОННОЙ ОЛИГОМЕРИЗАЦИИ ИНДИВИДУАЛЬНЫХ ИЛИ СМЕСЕЙ ЛИНЕЙНЫХ ОЛЕФИНОВ | 2001 |
|
RU2212935C2 |
| ТРУБЧАТЫЙ РЕАКТОР | 2000 |
|
RU2201799C2 |
| КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ ОЛИГОМЕРИЗАЦИИ ОЛЕФИНОВ, СПОСОБ ЕЁ ПРИГОТОВЛЕНИЯ И СПОСОБ ОЛИГОМЕРИЗАЦИИ | 2001 |
|
RU2212936C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВЫХ ОСНОВ СИНТЕТИЧЕСКИХ МАСЕЛ | 2004 |
|
RU2287552C2 |
| КАТАЛИЗАТОР И СПОСОБ ОЛИГОМЕРИЗАЦИИ АЛЬФА-ОЛЕФИНОВ | 2011 |
|
RU2452567C1 |
| Способ получения полиальфаолефинов с кинематической вязкостью 10-25 сСт | 2018 |
|
RU2666725C1 |
| СПОСОБ ДЕЗАКТИВАЦИИ КОМПЛЕКСНОГО МЕТАЛЛООРГАНИЧЕСКОГО КАТАЛИЗАТОРА ГОМОГЕННЫХ ПРОЦЕССОВ, ТАКИХ КАК ДИМЕРИЗАЦИЯ ИЛИ ОЛИГОМЕРИЗАЦИЯ ЭТИЛЕНА В ЛИНЕЙНЫЕ АЛЬФА-ОЛЕФИНЫ, И ЕГО ВЫДЕЛЕНИЯ ИЗ РЕАКЦИОННОЙ МАССЫ | 1997 |
|
RU2123501C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛИГОМЕРОВ ВЫСШИХ ЛИНЕЙНЫХ α-ОЛЕФИНОВ | 2011 |
|
RU2487112C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛИГОМЕРОВ ВЫСШИХ ЛИНЕЙНЫХ α-ОЛЕФИНОВ | 2011 |
|
RU2483053C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛИГОМЕРОВ ВЫСШИХ ЛИНЕЙНЫХ АЛЬФА-ОЛЕФИНОВ | 2011 |
|
RU2483052C2 |




Использование: нефтехимия. Сущность: проводят катионную олигомеризацию олефинового сырья, выделение отработанного катализатора из олигомеризата, разделение олигомеризата на фракции и гидрирование выделенных из олигомеризата фракций. В качестве олефинового сырья для катионной олигомеризации берут смесь цис- и трансизомеров деценов-2, -3, -4 и/или -5 ("внутренних" деценов). Технический результат: повышение селективности процесса, улучшение качества целевого продукта. 2 с. и 10 з.п. ф-лы, 6 табл.
| US 4910355 A, 20.03.1990 | |||
| US 4263467 A, 21.04.1981 | |||
| Способ интегрального приема сигналов частотной телеграфии | 1972 |
|
SU449453A1 |
| WO 9415895 A1, 21.07.1994 | |||
| Способ получения основы синтетического смазочного масла | 1989 |
|
SU1723101A1 |

