Изобретение касается способа дезактивации комплексного металлоорганического катализатора (КМК) гомогенных процессов, таких как димеризация или олигомеризация этилена в линейные альфа-олефины (ЛАО), и его выделения из реакционной массы.
Изобретение может найти применение в химической и нефтехимической промышленности на заводах, на которых осуществляются гомогенные процессы с применением КМК, в частности на заводах по производству бутена-1 или ЛАО.
Продукт димеризации этилена - бутен-1 - используется в качестве исходного сырья для получения кристаллического полибутена-1, этилен-бутеновых и пропилен-бутеновых пластиков (в том числе - линейного полиэтилена низкой плотности) и эластомеров, олигобутеновых масел, бутилароматических углеводородов, бутадиена, альфа-окиси бутена, альфа- и бета-бутанолов, метилэтилкетона, димеров и содимеров бутена-1 с другими мономерами и других продуктов.
Продукты олигомеризации этилена - ЛАО C4 - C30 - используются в качестве исходного сырья при получении бытовых моющих препаратов, флотореагентов, эмульгаторов, компонентов смазочно-охлаждающих и бурильных жидкостей, пластификаторов, различных типов присадок, синтетических низкозастывающих масел, полимеров и сополимеров, мономеров, депрессаторов нефтей и нефтепродуктов, высших алкиламинов, высших алкилалюминийорганических соединений, теплоносителей, синтетических жирных спиртов и кислот, а также при получении компонентов различных композиций на основе ЛАО (C20-C30) - мастик, герметиков, покрытий.
В описаниях патентов США 3879485, 3911942, 3969429, Великобритании 1447811, 1447812 и ФРГ 2274583, 2462771 описан способ димеризации этилена в бутен-1 на КМК, включающем тетрабутоксид титана и триэтилалюминий в среде органического растворителя (гексана, гептана, бензина, бензола, толуола, хлористого этила, простых эфиров - диэтилового, дибутилового, диизопропилового, метил-изоамилового, метилфенилового, тетрагидрофурана и их смесей) при температурах 20 - 80oC и давлениях этилена от 0,1 до 1,6 МПа.
В соответствии с описаниями изобретений по авторскому свидетельству СССР 1042701, неакцептированной заявке Италии 2449879 и патентам ФРГ 4338414 и 4338416 олигомеризацию этилена в ЛАО C4 - C30 осуществляют в среде органического растворителя при температурах 60 - 80oC и давлениях этилена 2,0 - 4,0 МПа. В качестве реакционной среды (органического растворителя) для олигомеризации этилена используют толуол, бензол или гептан.
Олигомеризацию этилена в ЛАО в соответствии с указанными охранными документами осуществляют под действием КМК, который включает циркониевую соль органической кислоты - ClmZr(OCOR)4-m или ClmZr(OSO3R')4-m и алюминийорганическое соединение (AOC)-(C2H5)nAlCl3-n, где R и R' - алкил, алкен или фенил; m - плавно изменяется от 1 до 4; n - плавно изменяется от 1 до 2.
В оптимальных условиях процессы ди- и олигомеризации этилена от начала и до конца протекают гомофазно: осадки катализатора в рассматриваемых процессах не образуются, а растворимые продукты превращения этилена (в частности - полиэтилен) образуются в незначительных количествах.
При осуществлении процессов ди- и олигомеризации в реакторах смешения непрерывного действия реакционная масса на выходе из реактора представляет собой смесь растворителя с продуктами превращения этилена - бутеном-1 и ЛАО соответственно, в которой растворены непревращенный этилен, компоненты катализатора, активные центры ди- или олигомеризации этилена и продукты их спонтанной термической дезактивации.
В результате протекания ди- или олигомеризации этилена под действием живых активных центров вне реактора в неконтролируемых условиях (а в некоторых случаях вследствие дросселирования) концентрация этилена снижается, существенно повышается (или снижается при дросселировании) температура и резко возрастает относительная концентрация бутена-1 или ЛАО соответственно.
Наличие живых активных центров ди- или олигомеризации этилена, низкая концентрация или отсутствие этилена и относительно высокая (до 8 моль/л) концентрация бутена-1 или ЛАО в растворе обуславливают протекание вторичных реакций бутена-1 или ЛАО: в частности, в указанных условиях протекает изомеризация бутена-1 в цис- и транс-бутены-2; другие ЛАО изомеризуются в олефины с двойной связью между внутримолекулярными атомами углерода; одновременно с этим бутен-1 или ЛАО ди-, три- и олигомеризуются; превращаются в изоолефины.
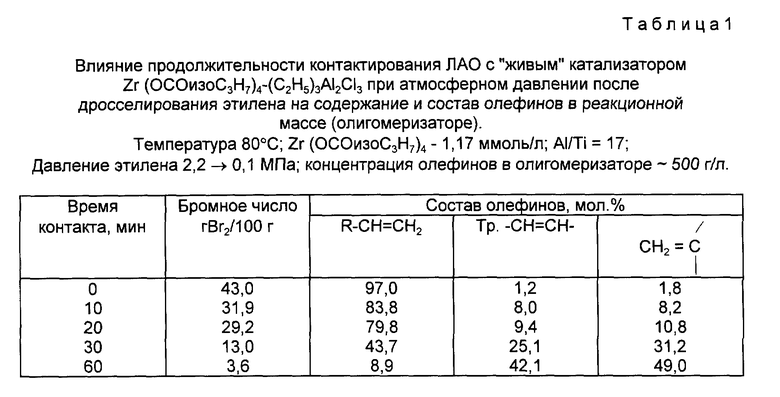
Особенно критичной оказалась продолжительность контактирования олигомеризата, содержащего ЛАО с "живым" катализатором олигомеризации этилена в отсутствие этилена. Из табл. 1 видно, что в отсутствие этилена ЛАО R-CH=CH2 под действием катализатора олигомеризуются и превращаются в изоолефины. Протекание олигомеризации ЛАО под действием "живого" катализатора подтверждается снижением общей концентрации двойных связей в реакционной массе (табл. 1) и образованием маслообразных олигомеров со среднечисловой молекулярной массой 350 - 800. Глубина превращения олефинов с винильными двойными связями возрастает с увеличением продолжительности контактирования ЛАО с "живым" катализатором.
Упомянутые катализаторы олигомеризации этилена в ЛАО содержат АОС (C2H5)nAlCl3-n, которые являются сильными кислотами Льюиса. На их основе в рассматриваемых системах легко образуются катионные активные центры. Под действием катионных активных центров ЛАО также изомеризуются, ди-, три- и олигомеризуются; алкилируют ароматический растворитель.
Замечено, что диффузионное поступление в реакционную массу, содержащую ЛАО и "живой" катализатор, микропримесей воды или спиртов приводит к резкому ускорению катионных процессов и сильному разогреву реакционной массы. В статических условиях процессы диффузии воды и спиртов, катионные реакции ЛАО и разогрев реакционной массы протекают в фронтальном режиме.
Все упомянутые вторичные реакции бутена-1 или ЛАО протекают с высокой скоростью.
Благоприятные условия для протекания вторичных реакций бутена-1 или ЛАО под действием "живого" катализатора возникают в аппаратах для дросселирования этилена, в сборниках реакционной массы и в колоннах для выделения из реакционной массы бутена-1 или узких фракций ЛАО.
Протекание вторичных реакций бутена-1 или ЛАО приводит к расходованию олефинов с винильными двойными связями и к загрязнению бутена-1 или ЛАО продуктами их превращений. В большей или в меньшей степени это приводит к снижению селективности катализатора и процесса.
Очевидно, что вторичные реакции бутена-1 или ЛАО нежелательны.
Для предотвращения протекания вторичных реакций бутена-1 или ЛАО "живой" катализатор ди- или олигомеризации этилена сразу же после выноса его из реактора необходимо дезактивировать. При этом промежуток времени между выносом "живого" катализатора из реактора и его дезактивацией должен быть сведен к минимуму.
Известны (пат. США 4486615, пат. ФРГ 4338415) способы дезактивации катализатора олигомеризации этилена в ЛАО, включающего ClmZr(OCOизоC3H7)4-m и (C2H5)nAlCl3-n, добавками стехиометрических количеств карбоновых кислот.
Недостатком способа по пат. США 4486615 является то, что в результате дезактивации катализатора олигомеризации этилена образуется значительное количество хлористого водорода, который вызывает интенсивную коррозию оборудования.
Другие недостатком этого способа является образование значительного количества химически загрязненных сточных вод, образующих при выделении дезактивированного катализатора методом противоточной водной отмывки олигомеризата.
Цирконий и алюминийсодержащие продукты дезактивированного карбоновыми кислотами катализатора по второму способу (пат. ФРГ 4338415) извлекают из реакционной массы путем пропускания ее через слой адсорбента с большой удельной площадью поверхности. В качестве адсорбентов в этом способе используют гранулированные силикагель, каолин, цеолиты, оксид алюминия, диоксид циркония, опилки.
Недостатком этого способа является то, что дезактивация указанного катализатора спиртами приводит к образованию значительных количеств хлористого водорода, который не улавливается адсорбентами, попадает в колонны для разделения олигомеризата на узкие фракции и вызывает их коррозию.
Другим недостатком рассматриваемого известного способа является наличие значительного количества химически загрязненных водных стоков, образующихся на стадии регенерации адсорбентов.
Еще одним недостатком рассматриваемого известного способа выделения дезактивированного катализатора является весьма сложное технологическое оформление этой стадии процесса, а также трудности в подборе материалов для изготовления адсорбентов, подвергающихся в процессе регенерации адсорбентов разогреву до 600oC.
Наиболее близким по технической сущности и достигаемому результату к разработанному нами техническому решению проблемы дезактивации и выделения катализаторов ди- и олигомеризации этилена является способ, в котором в качестве дезактивирующих систем используют водно-щелочные или водно-аммиачные растворы (заявка Японии N 3-220135 от 24.01.1990; РЖ Хим. 1993, 15А17П). Эти растворы используют и для выделения дезактивированного катализатора из олигомеризата. При этом вслед за водно-щелочным или водно-аммиачным выделением отработанного дезактивированного катализатора олигомеризат подвергают дополнительной отмывке деминерализованной водой.
Щелочь и аммиак включают в дезактивирующие системы в первую очередь для обеспечения нейтрализации хлористого водорода, образующегося при гидролизе хлорсодержащих продуктов реакций КМК ди- или олигомеризации этилена.
Этот способ также имеет существенные недостатки.
Применение рассматриваемого способа дезактивации и выделения КМК ди- или олигомеризации этилена водно-щелочной или водно-аммиачной отмывкой олигомеризата приводит к образованию большого количества химически загрязненных водных стоков.
С другой стороны, медленное и недостаточно эффективное смешение водной и олеофаз в аппаратах с мешалками или в широко применяемых диафрагменных смесителях приводит к образованию катионных активных центров, к протеканию катионной олигомеризации ЛАО и к алкилированию толуола линейными альфа-олефинами.
Характерной особенностью всех известных способов является то, что дезактивацию катализатора и выделение дезактивированного катализатора производят в две стадии перед разделением олигомеризата на узкие фракции.
Целью настоящего изобретения является повышение чистоты ЛАО, т.е. повышение селективности катализатора и процесса ди- или олигомеризации этилена.
Другой целью настоящего изобретения является упрощение технологического оформления стадий дезактивации катализатора ди- или олигомеризации этилена и выделения дезактивированного катализатора из реакционной массы.
Еще одной целью настоящего изобретения является полное исключение образования в процессе химически загрязненных водных стоков и обеспечение экологической чистоты процесса.
Указанные цели достигаются разработанным способом дезактивации КМК гомогенных процессов, таких как димеризация или олигомеризация этилена в ЛАО, и его выделения из органической реакционной массы путем смешивания ее с раствором гидроокиси металла в протоносодержащем растворителе и выделением дезактивированного катализатора из органической фазы в одну стадию при температуре 60 - 80oC и давлении этилена 2 - 4 МПа.
Дезактивацию "живого" катализатора в указанном интервале температур проводят потому, что процессы ди- и олигомеризации проводятся в аналогичных условиях.
В соответствии с изобретением процесс дезактивации катализатора раствором гидроокиси металла в протоносодержащем растворителе гибок в отношении температуры - она практически не оказывает влияния на скорость дезактивации и селективность процесса по ЛАО. Однако для обеспечения повышенной селективности процесса по ЛАО дезактивацию катализатора следует проводить при повышенных давлениях этилена.
Выбранный интервал давлений обеспечивает высокую скорость дезактивации КМК и высокую селективность процесса.
Скорость дезактивации катализатора и селективность процесса по ЛАО существенно зависят от эффективности смешения упомянутых растворов. Поэтому механическое смешивание органической фазы с раствором гидроокиси металла в протоносодержащем растворителе дополнительно интенсифицируют облучением получаемой смеси ультразвуком.
В качестве устройства для ультразвуковой обработки смешиваемых жидкостей используют трехмерную решетку с магнитостриктором, которая размещается в смесителе олео- и водной фаз и связывается с генератором ультразвуковых излучений УЗГ-2,5. Ультразвуковое облучение обеспечивает резкое повышение степени диспергирования капель смешиваемых жидкостей вплоть до полной гомогенизации получаемой смеси (Н.Н. Круглицкий и др. "Ультразвук в химической технологии". Киев, Укрниинти, 1970; Сборник "Термосолеустойчивость дисперсных систем", Киев, Наукова Думка, 1971). Достигаемое при этом повышение качества смешения олигомеризата с водно-щелочным раствором и повышение устойчивости получаемой дисперсии способствуют увеличению скорости дезактивации катализатора и повышению селективности процесса.
После дезактивации катализатора протоносодержащий растворитель выделяют из реакционной массы перегонкой и адсорбционной очисткой, а дезактивированный катализатор остается в органической фазе. Выделенный протоносодержащий растворитель возвращают в рецикл для приготовления раствора щелочи. Это исключает образование химически загрязненных стоков и обеспечивает экологическую чистоту процесса.
В качестве протоносодержащего растворителя для приготовления растворов гидроокиси металла, применяемых для дезактивации катализатора, используют воду, спирт или аммиак. Концентрацию гидроокиси металла в протоносодержащем растворителе варьируют в пределах от 1 до 40 мас.%.
Предпочтительными являются растворы, содержащие от 5 до 10 мас.% гидроокиси металла.
В качестве гидроокиси металла используют гидроокиси, выбранные из группы, включающей гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид аммония, гидроксид бериллия, гидроксид магния, гидроксид кальция и гидроксид алюминия. Последние соединения плохо растворимы и их используют в виде суспензии.
Несмотря на значительные усилия, направленные на изучение реакций между компонентами КМК в процессе ди- или олигомеризации этилена и в модельных условиях, точный состав продуктов этих реакций неизвестен. Это исключает возможность обоснованного и точного описания химизма процессов дезактивации "живых" катализаторов ди- или олигомеризации этилена водно-, спирто- или аммиачными растворами гидроксидов металлов. Имеющиеся данные позволяют говорить только о вероятном химизме этих процессов.
Общепризнано, что активный центр олигомеризации этилена включает цирконийорганическое соединение, содержащее сигма цирконий-углеродную связь, например 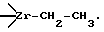 Очевидно, что в процессе водно-, спирто- или аммиачно-щелочной дезактивации "живого" КМК будет происходить гидролиз или алкоголиз именно этих связей
Очевидно, что в процессе водно-, спирто- или аммиачно-щелочной дезактивации "живого" КМК будет происходить гидролиз или алкоголиз именно этих связей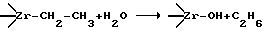
Поскольку активных центров в КМК очень мало, то основная масса дезактивирующего агента будет расходоваться в реакциях с компонентами КМК.
В качестве примера рассмотрим предполагаемый химизм водно-щелочной дезактивации КМК олигомеризации этилена, включающего Zr(OCOизоC3H7)4 и (C2H5)1,5AlCl1,5 (Al/Zr = 13). Исходя из результатов изучения гидролиза отдельных компонент этой системы водно-щелочными растворами в толуоле, химизм протекающих в дезактиваторе реакций очень приближенно можно отразить следующей схемой:
Zr(OCOизоC3H7)4 + 4NaOH ---> Zr(OH)4 + 4NaOCOизоC3H7;
13(C2H5)1,5AlCl1,5 + 19,5H2O ---> 19,5C2H6 + 13(HO)1,5AlCl1,5;
13(HO)1,5AlCl1,5 + 19,5NaOH ---> 13Al(OH)3 + 19,5NaCl;
13Al(OH)3 + 13NaOH ---> 13NaAlO2 + 26H2O.
Суммируя левые и правые части приведенных четырех уравнений и производя простые арифметические сокращения, получим балансное брутто-уравнение
Zr(OCOизоC3H7)4 + 13(C2H5)1,5AlCl1,5 + 36,5NaOH ---> Zr(OH)4 + 4NaOCOизоC3H7 + 19,5C2H6 + 19,5NaCl + 13NaAlO2 + 6,5H2O.
Это уравнение отражает особенности и стехиометрию протекающих в дезактиваторе реакций. Видно, что основным дезактивирующим агентом в рассматриваемой системе является гидроксид натрия. Для обеспечения полноты дезактивации целесообразно использовать 10 - 20%-ный избыток гидроксида натрия по отношению к стехиометрии этих реакций.
Поскольку вода в ходе дезактивации КМК не расходуется, растворять необходимое количество гидроксида натрия желательно в минимально возможном количестве воды. Это способствует снижению энергетических затрат в ходе сушки реакционной массы.
После выделения протонодонорного растворителя в олеофазе в виде высокодисперсной суспензии содержатся нерастворимые в углеводородах неизрасходованный гидроксид натрия, хлористый натрий, алюминаты и цирконаты натрия, гидроксихлориды алюминия и циркония, гидроксиды и оксиды алюминия и циркония, а также изомаслянокислый натрий. Из олеофазы их извлекают методом инерционно-гравитационного осаждения или фильтрации до разделения, либо после разделения олеофазы на индивидуальные компоненты и узкие фракции методом атмосферно-вакуумной разгонки в системе колонн при температурах 60 - 300oC.
Применение второго варианта разработанного способа стало возможным благодаря тому, что в результате целенаправленных исследований с использованием модельных и реальных объектов было установлено, что ЛАО сами по себе и в присутствии продуктов водно-, спирто- или аммиачно-щелочной дезактивации КМК ди- или олигомеризации этилена при температурах 60-300oC термостабильны и не подвергаются никаким превращениям.
По мере выделения индивидуальных компонентов и узких фракций из олеофазы в процессе ее атмосферно-вакуумной разгонки концентрация суспензии дезактивированного катализатора постепенно возрастает (в 10-20 раз). После завершения разделения олеофазы на индивидуальные компоненты и узкие фракции дезактивированный катализатор вместе со следами образовавшегося полиэтилена и с воскообразными ЛАО попадает в кубовый остаток.
Разделение упомянутых продуктов дезактивации отработанного катализатора и воскообразных ЛАО в соответствии с изобретением производят экстракционным методом.
Сущность этого решения состоит в том, что воскообразные олефины, содержащиеся в кубовом остатке, экстрагируют углеводородным, преимущественно легкокипящим растворителем, выбранным из группы, включающей изопентан, гексан, гептан, бензин, бензол, толуол, бутен-1, гексен-1 и октен-1, а упомянутые продукты дезактивации катализатора в виде шлама после промывки применяемым растворителем направляют в виде суспензии в отстойник, на фильтр или на центрифугу, где их отделяют от углеводородного растворителя.
Из экономических соображений в качестве экстрагента рекомендуется применять тот растворитель, который применяется или образуется в процессе олигомеризации. По экстрагирующей способности лучшим среди названных экстрагентов для воскообразных ЛАО является толуол.
Выделенные нерастворимые продукты дезактивации катализатора направляют далее для обжига (высокотемпературной окислительной минерализации) в распылительную огневую (пламенную) сушилку, а из нее - на утилизацию, т.е. на рекуперацию натрия, алюминия и циркония. Рекуперация циркония, в частности, металлургическим способом выгодна в том случае, когда содержание циркония в сухом шламе превышает 3 мас.%. Именно этот случай имеет место при дезактивации и выделении отработанного цирконий-алюминийсодержащего КМК олигомеризации этилена.
Воскообразные ЛАО из экстракта, объединенного с промывочным углеводородным раствором, выделяют путем отгонки углеводородного растворителя. Выделенный углеводородный растворитель возвращают в рецикл для экстракции воскообразных ЛАО из кубового остатка, а воскообразные ЛАО направляют на утилизацию или на склад.
Блок-схема стадии дезактивации "живого" КМК ди- или олигомеризации этилена и выделения образовавшихся продуктов дезактивации катализатора из олигомеризата приведена на фиг.1. Она включает следующие узлы:
1 - узел приготовления раствора гидроксида металла;
2, 3 - узел дезактивации КМК ди- или олигомеризации этилена, оснащенный устройством и генератором (3) ультразвукового облучения смешиваемых олеофазы с раствором гидроксида металла в протоносодержащем растворителе;
4 - узел выделения протоносодержащего растворителя;
5 - узел разделения олигоимеризата на индивидуальные компоненты и узкие фракции ЛАО;
6 - узел экстракционного разделения воскообразных ЛАО и продуктов дезактивации КМК ди- или олигомеризации этилена;
7 - отстойная центрифуга или друк-фильтр;
8 - узел выделения очищенных воскообразных ЛАО;
9 - узел высокотемпературной окислительной минерализации выделенного дезактивированного катализатора (распылительная огневая сушилка);
10 - циклон-пылеуловитель.
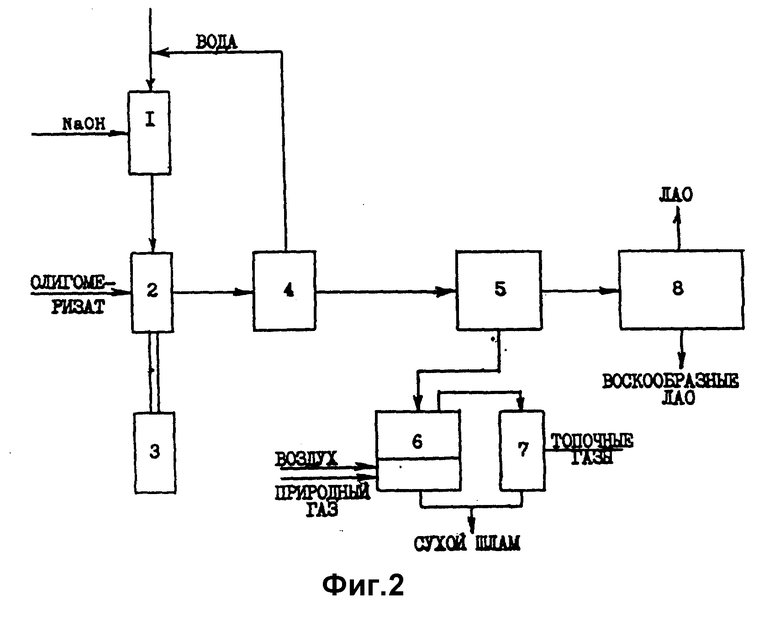
Второй альтернативный вариант технологии дезактивации и выделения дезактивированного катализатора из олеофазы включает следующие узлы (фиг.2):
1 - узел приготовления раствора гидроксида металла;
2, 3 - узел дезактивации КМК ди- или олигомеризации этилена водным, спиртовым или аммиачным (протоносодержащим) раствором гидроксида металла, оснащенный устройством и генератором (3) облучения смеси ультразвуком;
4 - узел выделения протоносодержащего растворителя;
5 - узел выделения дезактивированного катализатора и высокомолекулярного полиэтилена (отстойная центрифуга или друк-фильтр);
6 - узел высокотемпературной окислительной минерализации выделенного дезактивированного катализатора (распылительная огневая сушилка);
7 - циклон-пылеуловитель;
8 - узел разделения олеофазы на индивидуальные компоненты и узкие фракции ЛАО.
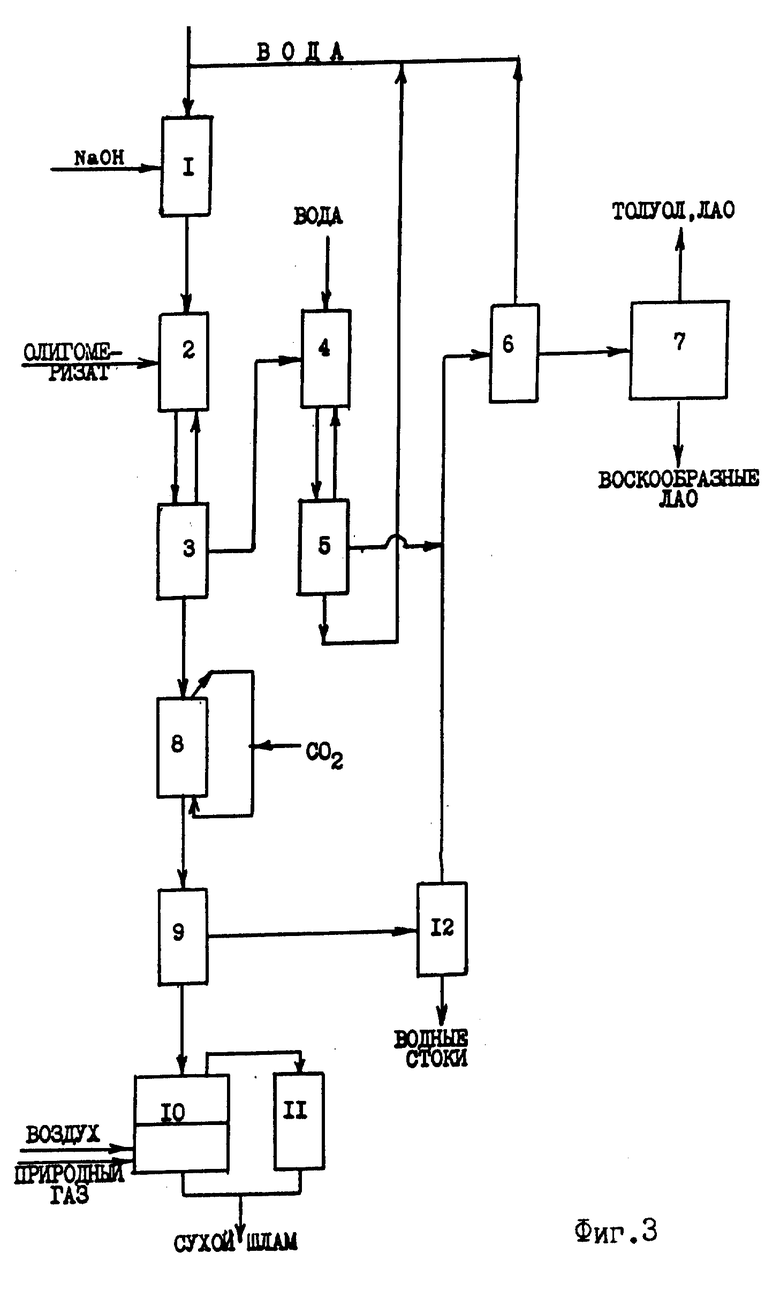
Известная технология дезактивации и выделения дезактивированного катализатора включает следующие узлы (фиг.3):
1 - узел приготовления раствора щелочи;
2, 3 - узел водно-щелочной дезактивации катализатора ди- или олигомеризации этилена, где 2 - смеситель-дезактиватор; 3 - промыватель-отстойник;
4, 5 - узел водной промывки олигомеризата, где 4 - смеситель-промыватель; 5 - промыватель-отстойник;
6 - узел азеотропной осушки олигомеризата;
7 - узел атмосферно-вакуумного разделения олигомеризата на индивидуальные компоненты и узкие фракции;
8 - узел углекислотной нейтрализации водных стоков;
9 - отстойная или фильтрующая центрифуга;
10, 11 - узел переработки катализаторного шлама, где 10 - распылительная огневая сушилка; 11 - мультициклон;
12 - узел выделения углеводородов, растворенных в сточных водах.
На схемах показаны также направления основных сырьевых и материальных потоков.
Сопоставление приведенных на фиг. 1 - 3 блок-схем стадии дезактивации и выделения отработанного катализатора ди- и олигомеризации этилена в ЛАО свидетельствует о том, что патентуемый способ имеет более простое технологическое оформление и характеризуется полным отсутствием сточных вод. Важной отличительной особенностью патентуемого способа является то, что в этом способе при дезактивации КМК используется не менее чем в 10 раз меньшее количество растворов гидроксидов металла, и то, что деминерализованная вода для промывки олигомеризата и газообразная углекислота для нейтрализации водных стоков в патентуемом способе вообще не используются.
Патентуемый способ может использоваться в гомофазных процессах ди-, олигомеризации, метатезиса, теломеризации, гидрирования, алкилирования других мономеров с использованием растворимых титан-, цирконий-, гафний-, никель-, молибден-, вольфрам- и других переходных металлсодержащих КМК.
Изобретение подтверждается и поясняется (но не исчерпывается) следующими примерами.
Пример 1.
1.1. Олигомеризацию этилена проводили на системе Zr(ОСОизоC3H7)4 (0,382 ммоль) (0,0348 г циркония) + (C2H5)1,5 AlCl1,5 (Al/Zr=13) в среде толуола (0,25 л) при 80oC и давлении этилена 2,0 МПа в течение 60 минут. В результате олигомеризации израсходовалось 187,5 г этилена и образовалось ≈ 250 мл ЛАО.
1.2. Дезактивацию катализатора в олигомеризате (≈ 500 мл) проводили водно-щелочным раствором, содержащим 15 мл воды и 3,4 г гидроксида натрия (18,5 мас. %), непосредственно в реакторе в указанных выше условиях при интенсивном перемешивании олигомеризата и дезактивирующего раствора в течение 20 минут с помощью экранированной электромагнитной мешалки и вмонтированного в днище реактора устройства для облучения смешиваемых жидкостей ультразвуком. Через 20 минут после загрузки в реактор водного раствора щелочи отбирали пробу олигомеризата для различных анализов, температуру реакционной массы снижали до 20-25oC и производили дросселирование этилена. В процессе дросселирования вместе с этиленом из реактора удалился почти весь образовавшийся бутен-1. Вслед за этим реакционную массу выгружали из реактора. После выгрузки из реактора расслаивания реакционной массы на олео- и водную фазы и выделения из нее осадков не наблюдалось.
1.3. Полученную реакционную массу (≈ 500 мл), содержащую продукты дезактивации катализатора, загружали в куб высокоэффективной колонки для атмосферно-вакуумной разгонки олигомеризата.
Вначале при температурах 60-70oC производилась азеотропная осушка олигомеризата. Из флорентины было отобрано 14,2 мл воды. Это составляет ≈ 94,6 мас.% в расчете на загруженную в реактор воду.
1.4. Вслед за осушкой производили выделение из олигомеризата гексена-1, толуола, октена-1, децена-1 и фракций ЛАО C12 - C18. После завершения разгонки олигомеризата на фракции из куба колонки в горячем состоянии было выгружено 30 г молочно-белого гомогенного кубового остатка. Выделенный кубовый остаток содержал воскообразные ЛАО, соединения циркония, алюминия, хлористый и изомаслянокислый натрий.
1.5. Полученный кубовый остаток (≈30 г) загрузили в стеклянный реактор с рубашкой для термостатирования, оснащенный винтовой мешалкой с электромеханическим приводом. Туда же загрузили 280 мл толуола. В результате перемешивания полученной смеси при 80oC в течение 20 минут образовался раствор воскообразных ЛАО и суспензия продуктов дезактивации катализатора. Осадок от раствора воскообразных ЛАО в толуоле после отстаивания отделяли декантацией. После этого к осадку добавили 100 мл свежего толуола. Полученную суспензию перемешивали 20 минут в описанных выше условиях. Осадок от промывного раствора отделяли на центрифуге, полученный фугат объединили с отделенным ранее раствором ЛАО в толуоле.
1.6. Выделенные продукты дезактивации катализатора сушили в сушильном шкафу при 300oC в воздушной атмосфере в течение 60 минут. Получено 3,82 г осадка. Для анализа на Zr взяли 0,0111 г осадка и растворили его в 3 мл стандартного аналитического раствора. Колориметрическим методом найдено, что 0,0111 г осадка содержат 95•10-6 циркония. Из этого следует, что 3,82 г осадка содержат 0,0327 г циркония (94 мас.% в расчете на исходный цирконий (0,0348 г), загруженного в реактор в виде Zr (OCOизо C3H7)4). Потери циркония составили 6 мас.%. Они обусловлены тем, что гидроксиды циркония обладают высокой адгезией и прилипают к поверхности реактора, колб и пипеток.
В результате анализов на алюминий и хлор найдено, что выделенный осадок содержит 0,13 г алюминия (97,2 мас.% в расчете на исходный алюминий (0,1342 г), введенный в реактор в виде (C2H5)1,5AlCl1,5) и 0,253 г хлора (95,6 мас.% в расчете на исходный хлор (0,266 г), загруженный в реактор в виде (C2H5)1AlCl1,5).
1.7. Объединенный раствор воскообразных ЛАО в толуоле (≈370 мл) загрузили в куб атмосферно-вакуумной ректификационной колонки и при 111oC и атмосферном давлении отогнали основную массу толуола. Остатки толуола из воскообразных олефинов удаляли в вакууме с использовании ловушки, охлаждаемой жидким азотом. Всего отогнано 361 мл (95%) толуола. Из куба выгружено 24,1 г воскообразных ЛАО (≈12,85 мас.% в расчете на израсходованный в процессе олигомеризации этилен).
1.8. В табл. 2 приведен групповой состав ЛАО, выделенных из олигомеризата, полученного при дезактивации катализатора в условиях ультразвукового облучения смешиваемых жидкостей и в контрольных условиях - без ультразвукового облучения. Сопоставление полученных данных свидетельствует о том, что ультразвуковое облучение смешиваемых олигомеризата и водного раствора гидроксида натрия способствует повышению селективности катализатора и процесса. Это проявляется в снижении содержания в фракциях ЛАО олефинов с виниловыми и винилиденовыми двойными связями и в полном исключении алкилирования.
Пример 2.
2.1. Димеризацию этилена проводили на системе Ti(OHC4H9)4-Al(C2H5)3 в среде диэтилового эфира. В реактор объемом 1,1 л загрузили 0,2 л диэтилового эфира, 0,1875 г (0,55 ммоль) тетрабутоксида титана и 3,11 г (27,28 ммоль) триэтилалюминия (Al/Ti=49,6). При 40oC и давлении этилена 8,0 ат за 250 минут израсходовалось 435 г (7,768 моль) этилена. Средняя скорость димеризации 8,5 г C2H4/(л•мин) (0,52 кг C4H8/(л•ч)). Выход бутена-1 = 2,32 кг/т Ti(OHC4H9)4 или 14100 моль бутена-1 в расчете на 1 моль Ti(OHC4H9)4. Цис- и транс-бутены-2, а также полиэтилен в продуктах отсутствовали. Наряду с бутеном-1 образовалось 9,1 г гексенов и октенов (2,1 мас.% в расчете на бутен-1).
2.2. Дезактивацию катализатора в реакционной массе (≈900 мл) проводили водно-щелочным раствором, содержащим 15 мл воды и 0,15 г (3,75 ммоль) гидроксида натрия (0,99 мас.% в расчете на воду), непосредственно в реакторе в указанных выше условиях при интенсивном перемешивании реакционной массы раствора в течение 20 минут с помощью экранированной электромагнитной мешалки и вмонтированного в днище реактора устройства для облучения образующейся смеси ультразвуком. Через 20 минут после загрузки в реактор водного раствора щелочи отбирали пробу газовой фазы для хроматографического анализа.
2.3. Полученную реакционную массу, содержащую диэтиловый эфир, бутен-1, гексены и октены, воду и продукты разложения катализатора, имеющимся в реакторе давлением этилена передавливали в куб металлической реакционной колонки, оснащенной дефлегматором и флорентийским сосудом. Вначале при 20-30oC производилось дросселирование этилена, а затем при 0-5oC - азеотропная осушка реакционной массы. Из флорентины было отобрано 11,0 мл воды, что составляет 73,3 мас.% в расчете на загруженную в реактор воду.
2.4. Вслед за осушкой производили выделение бутена-1, диэтилового эфира и гексенов. После завершения разгонки в кубе колонки остался кубовый остаток, содержащий октены, небольшое количество смолообразного неидентифицированного вещества и продукты дезактивации катализатора (предположительно гидроксиды титана, алюминия и натрия, а также алюминат натрия).
2.5. В куб колонки загрузили 100 мл н-гептана. В результате перемешивания полученной смеси при 20oC в течение 20 минут с помощью гибкой мешалки, которую вводили в куб через боковой штуцер, образовался раствор смолообразных продуктов и суспензия продуктов дезактивации катализатора. Осадок и раствор выгружали в стеклянный стакан. Углеводородный слой отделяли декантацией, а осадок промывали 40 мл н-гептана. Осадок от промывного раствора отделяли на центрифуге, полученный фугат объединили с отделенным ранее н-гептановым раствором.
2.6. Выделенные продукты дезактивации катализатора сушили и прокаливали в муфельной печи при 600oC в воздушной атмосфере в течение 60 минут. Получено 1,53 г осадка, содержащего 0,0246 г титана (92,8 мас.% в расчете на загруженный в реактор титан в виде Ti(OHC4H9)4), и 0,70 г алюминия (95,1 мас.% в расчете на загруженный в реактор алюминий в виде Al(C2H5)3).
2.7. Объединенный н-гептановый раствор (≈130 мл) загрузили в куб атмосферно-вакуумной ректификационной колонки и при 98oC и атмосферном давлении отогнали массу н-гептата. Остатки н-гептана удаляли в вакууме с использованием ловушки, охлаждаемой жидким азотом. Всего отогнано 128 мл (91,4%) н-гептана. Из куба в горячем состоянии выгружено 1,1 г маслянистого вещества (0,25 мас.% в расчете на израсходованный этилен).
Примеры 3-14.
Загрузка реагентов в реактор и условия олигомеризации этилена в ЛАО были такими же, как и в примере 1. Дезактивацию катализатора в олигомеризате проводили водными, спиртовыми и аммиачными растворами щелочи непосредственно в реакторе при механическом перемешивании смешиваемых растворов с дополнительной гомогенизацией раствора с помощью ультразвука в тех же условиях, что и в примере 1.
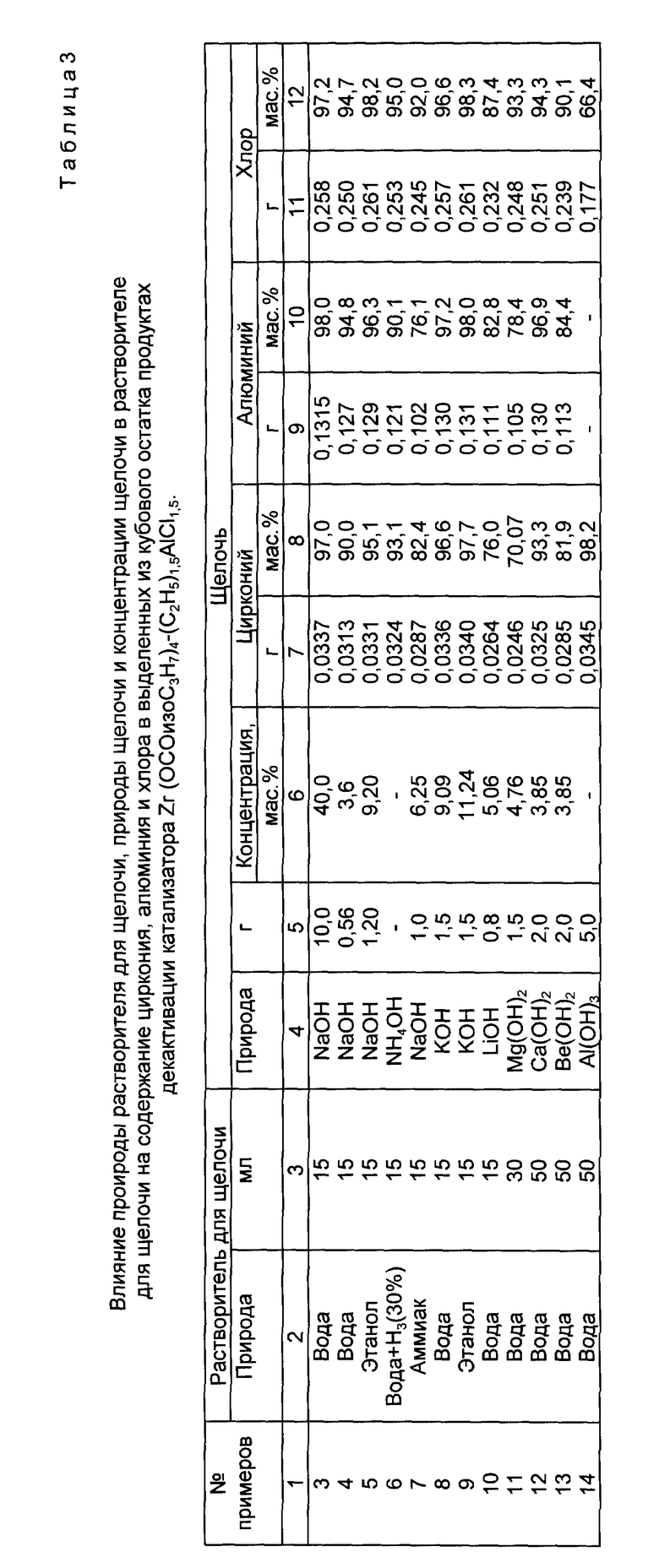
В примерах 3-14 варьировали природу растворителя для щелочи, природу и концентрацию щелочи (табл. 3). Все последующие операции разделения реакционной массы на фракции, выделения и обработки продуктов дезактивации катализатора Zr(OCOизоC3H7)4- (C2H5)1,5AlCl1,5 были такими же, как и в примере 1. Полнота выделения циркония, алюминия и хлора в расчете на исходную загрузку этих элементов в виде компонентов катализатора приведена в табл. 3. Несмотря на повышенный расход воды, гидроксиды магния, кальция и бериллия растворялись лишь частично, а гидроксид алюминия практически не растворялся. Они вводились в реактор в виде суспензии.
Полученные данные свидетельствуют о том, что предпочтительными являются водные растворы гидроксида натрия.
В совокупности эти результаты свидетельствуют о том, что патентуемый способ позволяет дезактивировать катализатор, повысить селективность процесса и практически количественно выделить дезактивированный КМК из олигомеризата, исключить образование химически загрязненных стоков, упростить технологическое оформление и обеспечить экологическую чистоту процесса.
Источники информации:
1. Патент США, 3879485.
2. Патент США, 3911942.
3. Патент США, 3969429.
4. Патент Великобритании, 1447811.
5. Патент Великобритании, 1447812.
6. Патент ФРГ, 2274583
7. Патент ФРГ, 2462771.
8. Авторское свидетельство СССР, 1042701, 19.06.78.
9. Заявка Италии, 24498.79.
10. Патент ФРГ, 4338414, C 08 F 110/02, 1993.
11. Патент ФРГ, 4338416, C 08 F 4/642, 1993.
12. Патент США, 4486615.
13. Патент ФРГ, 4338415, C 08 F 6/03, 1993.
14. Заявка Японии 3-220135, 1990.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ ОЛИГОМЕРИЗАЦИИ ЭТИЛЕНА В ЛИНЕЙНЫЕ АЛЬФА-ОЛЕФИНЫ. | 1997 |
|
RU2117012C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВЫХ ОЛИГОМЕРОВ | 2001 |
|
RU2199516C2 |
| КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ ОЛИГОМЕРИЗАЦИИ ОЛЕФИНОВ, СПОСОБ ЕЁ ПРИГОТОВЛЕНИЯ И СПОСОБ ОЛИГОМЕРИЗАЦИИ | 2001 |
|
RU2212936C2 |
| ТРУБЧАТЫЙ РЕАКТОР | 2000 |
|
RU2201799C2 |
| КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ КАТИОННОЙ ОЛИГОМЕРИЗАЦИИ ИНДИВИДУАЛЬНЫХ ИЛИ СМЕСЕЙ ЛИНЕЙНЫХ ОЛЕФИНОВ | 2001 |
|
RU2212935C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИС-(ЦИКЛОГЕКСИЛЦИКЛОПЕНТАДИЕНИЛ)ЦИРКОНИЙДИХЛОРИДА | 1989 |
|
RU1760748C |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВЫХ ОСНОВ СИНТЕТИЧЕСКИХ МАСЕЛ | 2004 |
|
RU2287552C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЛИНЕЙНЫХ АЛЬФА-ОЛЕФИНОВ | 2010 |
|
RU2497798C1 |
| ГОМОГЕННЫЙ КАТАЛИЗАТОР ДЛЯ ПОЛИМЕРИЗАЦИИ α -ОЛЕФИНОВ | 1988 |
|
RU1575391C |
| ГОМОГЕННАЯ КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ СИНТЕЗА НИЗКОМОЛЕКУЛЯРНОГО РАЗВЕТВЛЕННОГО ПОЛИЭТИЛЕНА И СПОСОБ ПОЛУЧЕНИЯ НИЗКОМОЛЕКУЛЯРНОГО РАЗВЕТВЛЕННОГО ПОЛИЭТИЛЕНА | 1999 |
|
RU2161628C1 |

Изобретение может найти применение в химической и нефтехимической промышленности, на заводах по производству бутена-1, линейных альфа-олефинов С4 - С30 (ЛАО) и других растворимых в реакционной среде продуктов с применением комплексных металлоорганических катализаторов (КМК). Способ включает дезактивацию КМК ди- или олигомеризации этилена в ЛАО путем смешивания выходящей из реактора реакционной массы, содержащей растворитель, ЛАО и "живой" катализатор, с раствором гидроокиси металла в протоносодержащем растворителе, таком как вода, спирт или аммиак, при 60-80o и давлении этилена 2-4 МПа при дополнительной интенсификации смешивания облучением получаемой смеси ультразвуком; выделение протоносодержащего растворителя из реакционной массы перегонкой и возвращение его в рецикл; последующее разделение реакционной массы на фракции и выделение дезактивированного катализатора совместно с воскообразными ЛАО из последней разделительной колонны в виде кубового остатка; выделение дезактивированного катализатора из кубового остатка путем экстракции воскообразных ЛАО углеводородным растворителем, инерционно-гравитационного осаждения дезактивированного катализатора из углеводородного экстракта и последующей высокотемпературной окислительной минерализации выделенного дезактивированного катализатора; выделение воскообразных ЛАО из экстракта, содержащего углеводородный растворитель и воскообразные ЛАО, путем отгонки углеводородного растворителя, который также возвращают в рецикл. Технический эффект данного изобретения заключается в повышении чистоты ЛАО, т. е. в повышении селективности катализатора и процесса ди- или олигомеризации этилена. 10 з.п.ф-лы, 3 табл., 3 ил.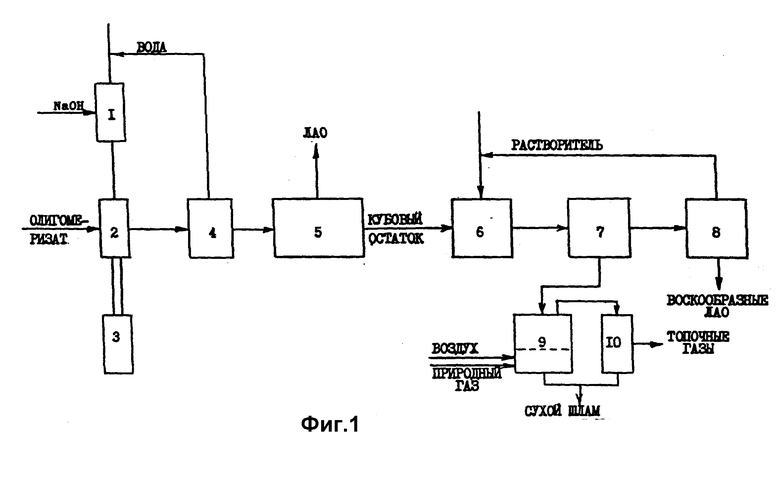
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| СПОСОБ УДАЛЕНИЯ ОСТАТКОВ | 0 |
|
SU357735A1 |
| Способ удаления остатков катализатора из полиэтилена высокой плотности | 1970 |
|
SU575033A3 |
| RU 95105686 A1, C 08 F 6/08, 10.01.97 | |||
| DE 3520103 A1, C 08 F 6/08, 11.12.86 | |||
| Преобразователь временного сдвига в код | 1973 |
|
SU505992A1 |
| US 5175247 A, C 08 F 6/08, 29.12.92 | |||
| US 5426176 A, C 08 F 6/08, 20.06.95. | |||

