Изобретение относится к способу получения соединения формулы I, [2-((8,9)-диоксо-2,6-диазобицикло-[5.2.0] нон-1(7)-ен-2-ил)этил] фосфоновой кислоты, антагонисту NMDA-рецептора, используемой в качестве противосудорожного средства и нейропротектора в случае избыточной секреции возбудительных аминокислот.
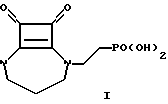
Соединение формулы I и способ его получения раскрыт в патенте США 5168103. Согласно этому способу метилфениловый эфир 3-аминопропилкарбаминовой кислоты взаимодействует с диалкиловым эфиром 2-оксоэтилфосфорной кислоты и натрийцианборгидридом с образованием промежуточного фенилметилового эфира [3-[[2-(диалкоксифосфинил}этил]амино]пропил]карбаминовой кислоты (а) с 36%-ным выходом. Взаимодействие (а) с 3,4-диалкокси (или 3,4-диарилалкокси)циклобут-3-ен-1,2-дионом, таким как 3,4-диэтоксициклобут-3-ен-1,2-дионом, приводит к фенилметиловому эфиру 3-[[2-(диалкоксифосфорил)-этил]-(2-алкокси-3,4-диоксо-1-циклобутен-1-ил)амино] пропилкарбаминовой кислоты (b) с 89% выходом. Снятие бензилокси-карбонильной защитной группы и циклизация (b) в диалкиловый эфир [2-((8,9)-диоксо-2, 6-диазабицикло[5.2.0]нон-1(7)-ен-2-ил)этил] фосфоновой кислоты (с) путем обработки (b) 10% палладием на угле и 1,4-циклогексадиеном позволяет получить (с) с выходом 62%. Обработка бициклического диэфира (с) бромтриметилсиланом дает [2-((8,9)-диоксо-2,6-диазобицикло-[5.2.0. ] нон-1(7)-ен-2-ил)этил] фосфоновую кислоту (I) с выходом 78%. Общий выход реакций в указанной последовательности составляет 15,5%.
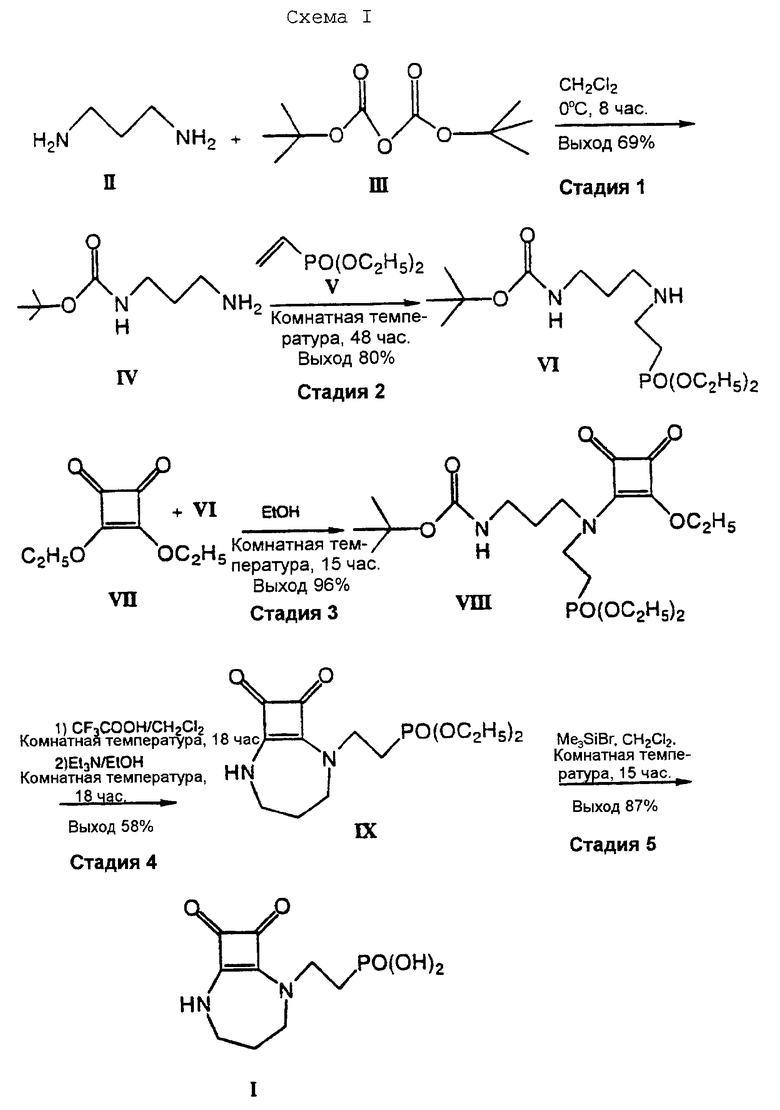
Согласно способам настоящего изобретения, как показано ниже для стадии II на схеме I, диалкиловый эфир 3-аминопропилкарбаминовой кислоты (IV) вводится во взаимодействие с диалкилвинилфосфонатом (V) с получением диалкилового эфира N-[3-(трет-бутилоксикарбониламино)пропил]-2-аминоэтилфосфоновой кислоты (VI). Дальнейшие усовершенствования согласно настоящему изобретению по сравнению со способом патента США 5168103, как будет отмечено ниже, увеличивают общий выход целевого продукта I до 38,8%.
Одним из аспектов настоящего изобретения является способ получения [2-((8,9)-диоксо-2,6-диазабицикло[5.2.0]нон-1(7)-ен-2-ил)этил]фосфоновой кислоты, который включает следующие стадии:
а) взаимодействие 3,4-ди-С1-С4-алкоксициклобут-3-ен-1,2-ди-она с ди-С1-С6-алкиловым эфиром N-[3-(трет- бутилоксикарбониламино)пропил]-2-аминоэтилфосфоновой кислоты с образованием трет-бутилового эфира 3-[[2-(ди-С1-С6-алкоксифосфорил)-этил] -(2-С1-С6-алкокси-3,4-диоксо-1-циклобутен-1-ил)амино]-пропилкарбаминовой кислоты;
b) снятие защиты с 3-аминогруппы в продукте, полученном на стадии (а);
c) циклизация продукта, полученного на стадии (b), с образованием бициклического промежуточного соединения, а именно, ди-С1-С6-алкилового эфира [2-((8,9)-диоксо-2,6-диазабицикло[5.2.0] нон-1(7)-ен-2-ил)-этил] фосфоновой кислоты; и
d) превращение ди-С1-С6-алкилового эфира, полученного на стадии (с) в соответствующую фосфоновую кислоту.
Стадию (а) проводят предпочтительно в безводном метаноле или этаноле при комнатные температурах (25-30oС). Стадии (a), (b) и (с) проводятся предпочтительно in situ. Стадию (b), предпочтительно, проводят в метиленхлориде в интервале температур от -5 до 25oС. Стадию (d) проводят, предпочтительно, в метиленхлориде или ацетонитриле при температуре около 20oС. Предпочтительным 3,4-ди-С1-С4-алкоксициклобут-3-ен-1,2-дионом на стадии (а) является 3,4-диэтоксициклобут-3-ен-1, 2-дион.
Другим аспектом настоящего изобретения является то, что ди-С1-С6-алкиловый эфир N-[3-(трет-бутилоксикарбониламино)пропил] -2-аминоэтилфосфоновой кислоты, полученный на стадии (а), получают способом, который включает взаимодействие ди-С1-С6-алкилового эфира винилфосфоната с 1,1-диметилэтиловым эфиром 3-аминопропилкарбаминовой кислоты. Предпочтительным ди-С1-С6-алкиловым эфиром винилфосфоната является диметилвинилфосфонат или диэтилвинилфосфонат, из которых наиболее предпочтителен диметилвинилфосфонат.
Другим аспектом настоящего изобретения является то, что настоящее изобретение предусматривает получение следующих соединений:
ди-С1-С6-алкиловый эфир N-[3-(трет-бутилоксикарбонил-амино)пропил]-2-аминоэтилфосфоновой кислоты;
диметиловый эфир N-[3-(трет-бутоксикарбониламино)пропил]-2-аминоэтилфосфоновой кислоты или диэтиловый эфир N-[3-(трет-бутилоксикарбониламино)пропил]-2-аминометилфосфоновой кислоты;
трет-бутиловый эфир 3-[[2-(ди-С1-С6-алкоксифосфорил)-этил] -(2-С1-С4-алкокси-3,4-диоксоциклобут-1-енил)амино]пропилкарбаминовой кислоты; и
трет-бутиловый эфир 3-[[2-(диэтоксифосфорил)этил] -(2-этокси-3,4-диоксоциклобут-1-енил)амино] пропилкарбаминовой кислоты или трет-бутиловый эфир 3-[[2-(диэтоксифосфорил)-этил] - (2-метокси-3,4-диоксоциклобут-1-енил)амино] пропилкарбаминовой кислоты.
Способ настоящего изобретения приведен ниже на схеме 1, где "алкил" представлен этильной группой и "алкокси" представлен этоксигруппой, и далее приведены следующее данные.
На стадии 1 1,3-диаминопропан (II) реагирует с дитрет-бутилкарбонатом (III) с образованием 1,1-диметилэтилового эфира 3-аминопропилкарбаминовой кислоты (IV).
На стадии 2 эфир карбаминовой кислоты IV реагирует с ди-С1-С6-алкиловым эфиром винилфосфоната (V) с образованием ди-С1-С6-алкилового эфира N-[3-(трет-бутилоксикарбониламино)пропил] -2-аминоэтилфосфоновой кислоты (VI). На приведенной схеме, где V является диэтилвинилфосфонатом, эта стадия протекает с выходом 80%. Однако и диметилвинилфосфонат обеспечивает сравнимые выходы и коммерчески предпочтителен, поскольку он более доступен при производстве больших количеств.
На стадии 3 ди-С1-С6-алкиловый эфир аминофосфоновой кислоты (VI) реагирует с 3,4-ди-С1-С4-алкоксициклобут-3-ен-1,2-дионом (VII) с образованием 1,1-диметилэтилового эфира 3-[[2-ди-С1-С6-алкоксифосфорил)этил] -(2-С1-С4-алкокси-3,4-диоксо-1-циклобутен-1-ил) амино]пропилкарбаминовой кислоты (VIII). В случае, приведенном на схеме, где алкил означает этил в обоих примерах, эта стадия протекает с 96%-ным выходом. Данную стадию проводят предпочтительно в безводных метаноле или этаноле при комнатных температурах.
На стадии 4, после снятия защиты с соединения (VIII) в трифторуксусной кислоте, в результате циклизации действием триэтиламина в качестве циклизующего агента, получают ди-C1-С6-алкиловый эфир [2-((8,9)-диоксо-2,6-диазабицикло[5.2.0] -нон-1(7)-ен-2-ил)этил] фосфоновой кислоты (IX). В случае, приведенном на схеме, выход продукта (IX) составляет 58%. Снятие защиты предпочтительно проводят в метиленхлориде при температуре от -5oС до 25oС. Циклизация, предпочтительно, проводится в метиленхлориде или ацетонитриле при температуре примерно 20oС, причем метиленхлорид является особенно предпочтительным.
На стадии 5 диэтиловый эфир фосфоновой кислоты (IX) обрабатывают бромтриметилсиланом, получая соединение I с выходом 87%. Общий выход при получении [2-((8,9)-диоксо-2,6-диазобицикло[5,2.0. ] нон-1(7)-ен-2-ил)этил] фосфоновой кислоты (I) согласно способу настоящего изобретения составляет 38,8%, что является значительно выше по сравнению с выходом 15,5%, раскрытом в патенте США 5168103.
Стадии 2, 3 и 4 предпочтительно проводятся in situ.
Для вышеприведенной последовательности реакций термин "алкил" означает C1-C6-алкил, нормальный или разветвленный, а термин "алкокси" означает С1-С6-алкоксигруппу, за исключением соединений с четырехчленным циклом VII, для которых алкил означает метил, этил или бутил. Выходы приведены для отдельных примеров, в которых "алкил" означает этил и "алкокси" означает этокси.
Преимущества способа настоящего изобретения заключаются в следующем. Во-первых, 1,3-пропандиамин является монозащищенным с превосходным выходом путем взаимодействия с ди-трет-бутилдикарбонатом, что менее безопасно, чем использование бензилхлорформиата для образования бензилоксикарбаматной защитной группы. Во-вторых, монозащищенный-1,3-пропандиамин реагирует с диэтилвинилфосфонатом с превосходным выходом без использования положительных реагентов, таких как натрийцианборгидрид и кислота, причем использование каждого из последних реагентов осуществляется не без риска, а они необходимы при применении 2-оксоэтилфосфоната, как это было раскрыто ранее, в-третьих, трет-бутилоксикарбонильная группа легко снимается при действии кислоты, в то время как удаление бензилоксикарбонильной группы требует использования палладиевого катализатора и источника водорода.
Способ согласно настоящему изобретению изображен на нижеследующей схеме I. На этой схеме "алкил" представлен этильной группой и "алкокси"-группа является этоксигруппой.
Приведены следующие конкретные примеры для иллюстрации способа настоящего изобретения, которые никоим образом не должны быть истолкованы как ограничения способа настоящего изобретения. Специалисты в данной области техники могут быть информированы еще и о других методиках или модификациях, которые могут эквивалентно использоваться для осуществления данного способа изобретения.
Пример 1
3-(трет-бутилоксикарбониламино)пропанамин
К раствору 1,3-диаминопропана (500 г, 6,75 моль; А1-drich D2, 360-2) в тетрагидрофуране (1,6 л) при температуре от -3oС до +2oС и при перемешивании добавляли по каплям раствор ди-трет-бутилдикарбоната (300 г, 1,375 моль; Aldrich 19, 913-3) в тетрагидрофуране (800 мл) в течение примерно 8 ч. Полученную белую суспензию перемешивали при 0oС и оставляли медленно нагреваться при комнатной температуре в течение ночи. Реакционную смесь упаривали в вакууме и остаток переносили в смесь этилацетата и насыщенного раствора хлорида натрия. Водный слой экстрагировали этилацетатом. Объединенные экстракты один раз промывали насыщенным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и получили масло (примерно 245 г, количественно). Масло суспендировали в воде (200 мл) и охлаждали до температуры от 0 до -5oС. Прибавляли водный раствор бромкрезола зеленого (3 мл, 0,04% в воде; Aldrich 31, 870-1), при этом получился голубой раствор. При энергичном перемешивании прибавляли по каплям 1 н. соляную кислоту (1,4 л), при этом цвет раствора изменялся от бледно-голубого до зеленовато-желтого (рН примерно 5-6, рН-индикаторная бумага). Эту суспензию экстрагировали метиленхдоридом (2•300 мл) и подщелачивали водный слой 2,5 н. раствором гидроксида натрия до рН примерно 12. Щелочной водный слой экстрагировали метиленхлоридом (5•300 мл), а органический слой промывали насыщенным раствором хлорида натрия (2•100 мл), сушили над пылеобразным безводным карбонатом калия, фильтровали через целит, упаривали и получали желаемое соединение в виде бледно-голубоватого масла, кристаллизующегося при стоянии (165 г, 69%) (очень гигроскопичное твердое вещество).
ЯМР (CDC13, 400 МГц); 1,43 (с, 9Н), 1,61 (п, 2Н), 1,59 (с, 2Н), 2,76 (т, 2Н), 3,20 (кв, 2H), 4,95 (уш. сигнал, 1Н).
Пример 2
Диэтиловый эфир N-[3-(трет-бутилоксикарбониламино)-пропил] -2-аминоэтилфосфоновой кислоты
К раствору 3-(трет-бутоксикарбониламино)пропанамина (77 г, 0,44 моль) в метаноле (500 мл) в токе азота и при выдерживании на водяной бане при примерно 20o прибавляли 97%-ный диэтилвинилфосфонат (75 г, 0,44 моль; Aldrich 11, 613-0) в течение 48 ч. Реакционную смесь упаривали в вакууме, а остаток (примерно 160 г) переносили на колонку с флоризилом (3"•6") и элюировали смесью метиленхлорид/гексан 1:1, метиленхлоридом и, наконец, 10%-ной смесью метанол/метиленхлорид, получая указанное в заглавии соединение в виде бесцветного масла (121 г, 80%).
ЯМР (CDCl3, 400 МГц): 1,32 (т, 6Н), 1,43 (с, 9Н), 1,65 (т, 2Н), 1,80 (уш. сигнал, 1Н), 1,97 (дт, 2Н), 2,67 (т, 2Н), 2,85 (дт, 2Н), 3,20 (кв, 2Н), 4,09 (м, 4Н), 5,08 (уш. сигнал, 1Н).
Пример 3
Ди-трет-бутиловый эфир 3-[[2-(диэтоксифосфорил)этил]-(2-этокси-3,4-диоксоциклобут-1-енил)амино]пропилкарбаминовой кислоты
К раствору 3,4-диэтокси-3-циклобутен-1,2-диону (45 г, 0,265 моль; Aldrich 31, 077-8) в абсолютном метаноле (1,2 л) в токе азота прибавляли по каплям раствор диэтилового эфира N-[3-(трет-бутилоксикарбониламино)пропил]-2-аминоэтилфосфоновой кислоты (80 г, 0,24 моль) в абсолютном этаноле (600 мл) и перемешивали реакционную смесь при комнатной температуре в течение 15 ч. Реакционную смесь упаривали в вакууме, а остаток переносили на колонку с силикагелем (6"•4") и элюировали сначала смесью метиленхлорид/гексан 1:1 для удаления избытка 3,4-диэтокси-3-циклобутен-1,2-диона и окончательно 10%-ной смесью метанол/метиленхлорид, получая после упаривания указанное в заглавии соединение в виде вязкого масла (107 г, 96%).
ЯМР (СDСl3, 400 МГц): 1,34 (т, 6Н), 1,43 (С, 9Н), 1,46 (т, 3Н), 1,80 (м, 2Н), 2,12 (м, 2Н), 3,14 (м, 2Н), 3,49 (т, 1Н), 3,66 (м, 1Н5, 3,73 (т, 1Н), 3,90 (м, 1Н), 4,10 (м, 4Н), 4,74 (м, 4Н), 5,05 (уш. сигнал, Н).
Пример 4
Диэтиловый эфир 2-(8,9-диоксо-2,6-диазабицикло[5.2.0] -нон-1(7)-ен-2-ил)этилфосфоновой кислоты
Раствор диэтилового эфира N-[3-(трет-бутоксикарбонил-амино)пропил]-N-[4-этокси-2,3-диоксоциклобут-1-ен-1-ил] -2-аминоэтилфосфоновой кислоты (100 г, 0,22 моль) в метиленхлориде (600 мл) охладили во льду и обработали трифторуксусной кислотой (300 мл). Реакционную смесь оставляли самопроизвольно нагреваться до комнатной температуры в течение ночи. Раствор упарили в вакууме при максимальной температуре 40oС и упаривали с толуолом (2•500 мл), получая вязкое масло (159,5 г), которое растворяли в абсолютном этаноле (1,5 л) и прибавляли по каплям в течение восьми часов к раствору триэтиламина (350 мл) в этаноле (1,5 л), затем перемешивали 8 ч при комнатной температуре. Реакционную смесь упаривали в вакууме до получения масла, которое переносили в этилацетат (1 л). Закристаллизовавшееся соединение охлаждали льдом, отфильтровывали, промывали этилацетатом и затем гексаном, получая указанное в заглавии соединение в виде белого вещества (40 г, 58%).
ЯМР (CDCl3, 400 МГц): 1,34 (т, 6Н), 2,06 (м, 2Н), 2,20 (дт, 2Н), 3,50 (м, 4Н), 4,05 (м, 2Н), 4,15 (м, 4Н), 7,87 (уш. сигнал, 1Н).
Масс-спектр (DEI) М+ м/е 316.
Анализ жидкостной хромэтографией (колонка: Microsorb-MV С18, 150•4,6 мм; элюент: 30/70 МеОН/0,01 М NН4Н2Р04 рН 4,7; скорость потока: 1мл/мин; УФ-детектор при 210 нм.
Анализ. Рассчитано для C13H21N2O5P:
С 49,36; Н 6,69; N 8,85%
Найдено: С 49,47; Н 6,74; N 8,77%
Пример 5
[2-(8,9-диоксо-2,6-диазабицикло[5.2.0] нон-1(7)-ен-2-ил)этил] фосфоновая кислота
В токе азота к раствору диэтилового эфира 2-(8,9-диоксо-2,6-диазабицикло[5.2.0] нон-1(7)-ен-ил)этил] фосфоновой кислоты (37,6 г, 0,12 моль) в метиленхлориде (350 мл) добавляли по каплям, но быстро, бромтриметилсилан (83 мл, 96,3 г, 0,63 моль; Aidrich 19, 440-9). Реакционную смесь выдерживали на водяной бане в течение 15 часов при температуре примерно 20oС. Прозрачный раствор упаривали в вакууме и пенистый остаток переносили в ацетон (600 мл) при интенсивном перемешивании, получая тонкую суспензию. Прибавляли воду (50 мл, 2,78 моль), при этом образовывался немедленно затвердевающий клейкий осадок. Суспензию интенсивно перемешивали в течение 10 мин, отфильтровывали и промывали ацетоном, получая желтое твердое вещество. Твердое вещество переносили в кипящую воду (450 мл) и горячий раствор отфильтровывали через складчатый бумажный фильтр для удаления небольших количеств нерастворившегося вещества. Прозрачный водный раствор охлаждали на льду сопровождающейся кристаллизацией. Густую массу кристаллов разбавляли, медленно прибавляя ацетон (800 мл), выдерживали при охлаждении в течение 1 ч, фильтровали и промывали ацетоном, затем гексаном, получая указанное в заглавии соединение в виде бледно-желтого твердого вещества (20,2 г). Из маточного раствора была получена вторая дополнительная порция вещества (100% чистота по ЖХ) (6,5 г), общий выход составил 87%.
ЯМР (ДМСО-d6, 400 МГц): 1,90 (м, 4Н), 3,25 (м, 2Н), 3,36 (м, 2Н), 3,84 (кв, 2Н), 8,45 (с, 1Н).
Анализ ЖХ (колонка: Nova РаК C18, 300•3,9 мм; элюент: 20/80 МеОН/0,005 М полиизоцианурата А; скорость потока: 1 мл/мин; УФ-детектор при 210 нм).
Анализ. Рассчитано для C9H13N2O5P•0,1 H2O:
С 41,26; Н 5,08; N 10,69%
Найдено: С 41,1,7; Н 5,04; N 10,42%
Анализ по Карлу-Фишеру: 0,55% H2O;
Масс-спектр: -FAM [М-Н]- м/е 259
Пример 6
Получение 3-(трет-бутоксикарбоииламино)пропанамина ("t-BOC-пропанамин")
Раствор ди-трет-бутилкарбоната (0,50 кг, 2,29 моль) в метаноле прибавляли к избытку (5 эквивалентов) 1,3- диаминопропана (0,83 кг, 11,2 моль) при 25-30oС за 4 часа. Продукт, который является 1,3-диаминопропан-трет-бутоксикарбонатом, отфильтровывали, а метанол удаляли в вакууме. Оставшееся масло переносили в этидацетат, промывали солевым раствором, прибавляли воду и доводили рН до 5,5. Слои разделяли и водную фазу сильно подщелачивали, водный щелочной раствор экстрагировали 6 раз толуолом. Объединенные толуольные экстракты сушили над сульфатом натрия и упаривали, получая t-ВОС-пропанамин (0,365 кг) с выходом 77,4%, общее количество примесей составляло 11,5%.
Пример 7
Получение диметилового эфира [2-(8,9-диоксо-2,6-диазабицикло[5.2.0] нон-1(7)-ен-2-ил]фосфоновой кислоты ("эфир диметилфосфоната")
А. Образование диметилового эфира трет-ВОС-фосфоната
Раствор t-BOC-пропанамина, 98%-ной чистоты, (0,67 кг, 3,77 моль) и диметилвинилфосфоната (0,59 кг, 4,12 моль) в безводном метаноле (2,7 л) перемешивали при комнатной температуре в течение 2-х дней с образованием раствора целевого продукта - эфира диметилфосфоната.
В. Образование t-ВОС-диметилфосфонатэтилскуарата (соответствующий фосфонат с четырехчленным циклом в молекуле)
Полученный в А раствор эфира диметилфосфоната прибавляли затем к раствору 3,4-диэтокси-3-циклобутен-1,2-диона ("этилскуарат") (0,55 кг, 3,23 моль) в безводном метаноле (3,24 л) в течение 6 часов. После перемешивания в течение ночи при 0-5oС реакционную смесь упарили отгонкой. Прибавили толуол (1 л) и повторили отгонку до конечного объема раствора трет-бутилового эфира 3-[[2-(диэтоксифосфорил)этил] -(2-этокси-3,4-диоксоциклобут-1-енил) амино] пропилкарбаминовой кислоты ("трет-ВОС-диметилфосфонатэтилскуарат") (1,6 л).
С. Образование незащищенного диметилфосфонатэтилскуарата
К полученному в В раствору t-BOC-диметилфосфонатэтилскуарата прибавили толуол (5,0 л) при 0-5oС, и далее обрабатывали трифторуксусной кислотой (4,71 кг, 41,31 моль) в течение 0,25-5 часов, поддерживая температуру в области 15oС. После 4-часового перемешивания при комнатной температуре, реакционную смесь упарили, получив неочищенный незащищенный диметилфосфонатэтилскуарат.
D. Образование эфира диметилфосфоната
К продукту реакции со стадии С после упаривания прибавляли безводный метанол (4,5 л), и полученный раствор прибавляли при комнатной температуре в течение 6 часов к раствору избытка триэтиламина (2,9 кг, 28,66 моль) в безводном метаноле. Полученную реакционную смесь упарили, затем прибавили этилацетат, которым и осадили указанный в заглавии продукт, являющийся эфиром диметилфосфоната. После фильтрования и промывания осадка на фильтре холодным этилацетатом получили с выходом 50,4% [2-(8,9-диоксо-2,6-диазабицикло[5.2.0] нон-3-(75-ен-2-ил)этил] фосфоновой кислоты ("эфир диметилфосфоната") (0,56 кг), концентрация которого составляла 97,6%, одна примесь составляла 1,05% и общее количество примесей составляло 1,76%.
Пример 8
Получение и очистка [2-(8,9-диоксо-2,6-диазабицикло-[5.2.0]нон-1(7)-ен-2-ил)этил]фосфоновой кислоты
Прибавляли при комнатной температуре бромтриметилсилан 97,6%-ной чистоты, (0,55 кг, 3,59 моль), к перемешиваемой суспензии эфира диметилфосфоната (0,46 кг, 1,56 моль) в ацетонитриле (4,1 л). Затем полученный раствор прибавляли к перемешиваемой смеси ацетонитрил-вода. Указанная в заглавии фосфоновая кислота осаждалась из раствора в виде твердого вещества кремового цвета, полученную взвесь охлаждали до 0oС и продукт выделяли фильтрованием. Влажный осадок с фильтра перемешивали в воде и добавляли 30%-ный раствор NaOH до рН 13. К полученному раствору прибавляли соляную кислоту до рН 1,0. Целевая фосфоновая кислота осаждалась из раствора в виде твердого вещества белого цвета. Продукт отфильтровали на воронке Бюхнера и промыли ледяной водой. Целевая фосфоновая кислота затем была очищена перекристаллизацией из воды. Влажный осадок с фильтра снова растворили в 12 частях воды, отфильтровали через бумажный фильтр на воронке Бюхнера и фильтрат упарили. После охлаждения из раствора выкристаллизовалась целевая фосфоновая кислота. Взвесь охладили до 0oС и собрали на воронке Бюхнера. Осадок на фильтре промывали водой и сушили в вакууме при 65oС, получив целевую очищенную фосфоновую кислоту с 86%-ным выходом, концентрация которой составляла 99,9%, с одной примесью (0,05%), общее количество примесей составляло 0,13%.
Изобретение относится к способу получения [2-((8,9)-диоксо-2,6-диазобицикло-[5.2.0] нон-1(7)-ен-2-ил)этил] фосфоновой кисло-ты (I). В способе настоящего изобретения 1,1-диметилэтиловый эфир 3-аминопропилкарбаминовой кислоты взаимодействует с диалкил-винилфосфонатом с получением диалкилового эфира N-[3-(трет-бутилоксикарбониламино)пропил] -2-аминоэтилфосфоновой кислоты (d) с 80%-ным выходом. Реакция (d) с 3,4-диалкоксициклобут-3-ен-1,2-дионом приводит к 1,1-диметиловому эфиру [3-[[2-(диалкоксифосфорил)этил]-(2-алкокси-3,4-диоксо-1,2-циклобутен-1-ил)амино] пропил]карбаминовой кислоты (е) с 96%-ным выходом. Снятие защиты и циклизация (е) в трифторуксусной кислоте приводит к диалкиловому эфиру [2-((8,9)-диоксо-2,6-диазабицикло[5.2.0] нон-1(7)-ен-2-ил)этил] фосфоновой кислоты (с) с 58%-ным выходом. Диэтиловый эфир фосфоновой кислоты (с) обрабатывают триметилбромсиланом с образованием соединения (I). Соединения I антагонисту NMDA-рецептора, применяемой в качестве антиконвульсанта и нейропротектора при избыточной секреции возбудительных аминокислот. 4 с. и 10 з.п. ф-лы.
10 Способ по одному из пп.1-9, отличающийся тем, что ди-С1-С6-алкиловый эфир N-[3-(трет-бутилоксикарбониламино)пропил]-2-аминоэтилфосфоновой кислоты является диметиловым эфиром N-[3-(трет-бутилоксикарбониламино)пропил]-2-аминоэтилфосфоновой кислоты или диэтиловым эфиром N-[3-(трет-бутилоксикарбониламино)пропил]-2-аминоэтилфосфоновой кислоты.
| Устройство для диагностики неисправностей | 1973 |
|
SU496561A1 |
| US 5168103, 01.12.1992. | |||

