Изобретение относится к замещенным пиримидинам, которые могут использоваться для лечения гипертензии. Эта возможность обусловлена оказываемым ими антагонистическим действием на ангиотенсин II, являющийся активным компонентом ренин-антиотенсинной системы.
Ангиотенсиноген превращается в ангиотенсин I под действием фермента ренина. Ангиотенсин II (AII) образуется в результате воздействия ангиотенсинконвертирующего фермента (ACE) на ангиотенсин I. A II оказывает сильное сосудосужающее действие и является одной из причин высокого кровяного давления у многих животных, включая и человека. Такая реакция, вызываемая A II, связана с воздействием им на специфические рецепторные центры. Соединения, описанные в настоящем изобретении, конкурируют с A II за эти рецепторные центры и таким образом противодействуют вызываемому им сосудосуживающему эффекту.
E. E. Allen и др. в европейском патенте 0419048 A описаны N - замещенные оксопиримидины.
E. E. Allen и др. в европейском патенте 0411766 A описаны 4-оксохиназолины.
D. A.Roberts и др. в европейском патенте 0412848 A описаны простые эфиры хинолина.
D.J.Carini и др. в патенте США 4880804 описаны N-замещенные бензимидазолы. Аналогичные по строению имидазольные производные с семизвенным гетероциклом вместо фенильного ароматического кольца описаны P.Chakravarty и др. в европейском патенте 0401030 A. Азабензимидазолы описаны P.Herold и др. в европейском патенте 0415886 A.
D. J.Cartini и др. в европейских патентах 0253310, 0324377 и патенте США 4916129 описаны N-замещенные имидазолы.
D.J.Cartini и др. в европейском патенте 0323841 описаны N-замещенные пиразолы, пирролы и триазолы. Аналогичные производные пиразола описаны T.Naka и др. в европейском патенте 0411507 A, а триазолы описаны L.L.Chang и др. в европейском патенте 0412594 A.
Авторы всех перечисленных патентов утверждают, что описанные в них соединения являются антагонистами A II.
Соединения в соответствии с настоящим изобретением отличаются от вышеупомянутых тем, что они содержат пиримидиновое кольцо, конденсированное с пирролоновым, пиридиноновым, азепиноновым или азоциноновым кольцами. За исключением первой ссылки (E.E. Allen и др. Европейский патент 0419048 A) все непептидные антагонисты A II, описанные в вышеперечисленных патентах, содержат пятизвенные азотистые кольца, индивидуальные или конденсированные с фенильными кольцами, или 6-звенные азотистые кольца, конденсированные с фенильными кольцами. Соединения, описанные в европейском патенте 0419048 A, представляют собой пиримидин-4-оны.
R. F. Shuman и др. в патенте США 5039814 описан способ получения 2-замещенных 1-/тетразол-5-ил/-бензолов, включающий прямое ортолитирование 1-/тетразол-5-ил/-бензолов и последующее взаимодействие образующегося промежуточного литиевого соединения с электрофилом. В качестве защитной группы тетразола используется трифенилметильная группа.
Отличие способа в соответствии с настоящим изобретением состоит в том, что 2-бром-1-/тетразол-2-ил/-арил, защищенный третбутильной группой, переводят в 2-/тетразол-5-ил/-арилбороновую кислоту непосредственно путем одновременного добавления Mg и триалкилбората.
Настоящее изобретение относится к замещенным пиримидинам формулы I: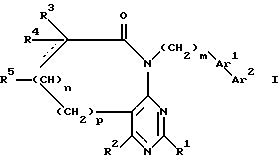
в которой
R1, R2, R3 и R4 независимо друг от друга означают H, низший алкил с 1-6 атомами углерода или перфторалкил с 1-6 атомами углерода; R5 - H или, в том случае, если n равно 1, R5 вместе с R3 означает двойную связь; n равно 0-1; p 0-2; m 0-3; Ar1 означает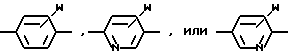
где
W означает H, низший алкил с 1-6 атомами углерода, атом галогена, окси-группу или низшую алкокси-группу с 1-6 атомами углерода; Ar2 означает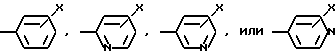
где
x означает CO2H, CN или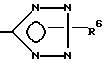
где
R6 означает H, третбутил, три-н-бутилстаннил или трифенилметил, а также к их фармацевтически приемлемым солям и N-оксидам.
Предпочтительным предметом настоящего изобретения являются соединения формулы II: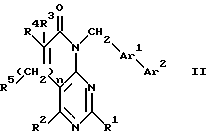
в которой
R1, R2, R3 и R4 независимо друг от друга означают H, низший алкил с 1-6 атомами углерода или перфторалкил с 1-6 атомами углерода; R5 - H или в том случае, если n равно 1, R5 вместе с R3 образует двойную связь; n равно 0-1; Ar1 означает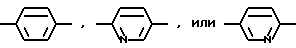
Ar2 означает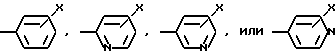
где
x означает
CO2H, CN или 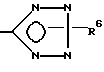 ;
;
где
R6 означает H, третбутил, три-н-бутилстаннил или трифенилметил, а также их фармацевтически приемлемые соли.
Более предпочтительным предметом настоящего изобретения являются соединения формулы III: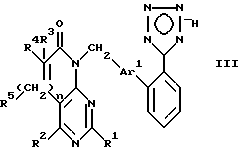
в которой
R1, R2, R3 и R4 независимо друг от друга означают H, метил или трифторметил; R5 - H или в том случае, если n равно 1, R5 вместе с R3 образует двойную связь; n равно 0-1; Ar1 означает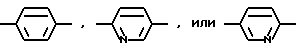
а также их фармацевтически приемлемые соли.
Наиболее предпочтительным предметом настоящего изобретения являются 2,4-диметил-5,7-дигидро-7-[[2'-/1Н-тетразол-5-ил/-[1,1-бифенил]-4-ил]метил]- 6Н-пирроло[2,3-d]-пиримидин-6-н и его фармацевтически приемлемые соли;
5,7-дигидро-2-метил-7-[[2'-/1Н-тетразол-5-ил-/[1,1-бифенил]-4-ил]-метил] -4-/трифторметил/-6Н-пирроло[2,3-d] -пиримидин-6-он и его фармацевтически приемлемые соли;
2,4-диметил-5,6,8-тригидро-8-[[2'-/1Н-тетразол-5-ил/[1,1-бифенил]-4- ил] метил]-7Н-пиридо[2,3-d]пиримидин-7-он и его фармацевтически приемлемые соли;
2-метил-4-трифторметил-5,6,8-тригидро-8-[[2'-/1Н-тетразол-5-ил/ [1,1-бифенил] -4-ил] метил] -7Н-пиридо[2,3-d]пиримидин-7-он и его фармацевтически приемлемые соли;
2,4-диметил-5,6-8-тригидро-8-[[6-[2-/1Н-тетразол-5-ил/фенил] - 3-пиридинил]метил]-7Н-пиридо[2,3-d]пиримидин-7-он и его фармацевтически приемлемые соли;
2-метил-4-трифторметил-5,6,8-тригидро-8-[[6-[2-/1Н-тетразол-5-ил/фенил] -3-пиримидинил] метил]-7Н-пиридо[2,3-d]пиримидин-7-он и его фармацевтически приемлемые соли;
2,4-диметил-5,6,8-тригидро-8-[[5-[2-/1Н-тетразол-5-ил/фенил] -2 пиридинил] метил] -7Н-пиридо[2,3-d]пиримидин-7-он и его фармацевтически приемлемые соли;
2,4,6-триметил-5,6,8-тригидро-8-[[2'-/1Н-тетразол-5-ил/ [1,1-бифенил]-4-ил] метил]-7Н-пиридо[2,3-d]пиримидин-7-он и его фармацевтически приемлемые соли;
2,4-диметил-8-[[2'-/1Н-тетразол-5-ил/[1,1-бифенил]-4-ил]метил]- 7Н-пиридо[2,3-d]пиримидин-7-он и его фармацевтически приемлемые соли.
Соединения в соответствии с настоящим изобретением могут быть получены с помощью способа, включающего:
(а) конденсацию, например, в присутствии неорганического или органического основания, пиримидина формулы: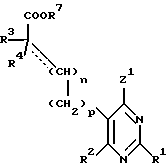
в которой
R1, R2, R3, R4, R5, n и p имеют вышеприведенные определения, Z1 означает отщепляющуюся группу, например атом галогена (в частности хлора), метансульфонилокси-группу или п-толуолсульфонилокси-группу, а R7 - низший алкил, с амином формулы:
H2N-(CH2)m-Ar1-Ar2,
в которой
m, Ar1 и Ar2 имеют вышеприведенные определения, или (b) взаимодействие бициклического соединения формулы: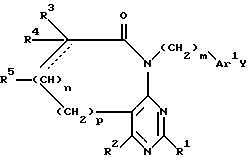
в которой
R1, R2, R3, R4, R5, n, m, p и Ar1 имеют вышеприведенные определения, а Y означает пара-бром или пара-иод, с арилбороновой кислотой (например, соединением формулы Ar2B(OH)2, в которой Ar2 имеет вышеприведенное определение) или с арилотаннатом в присутствии палладиевого катализатора, или
(c) конденсацию, например, в присутствии основания, в частности NaH, бициклического соединения формулы: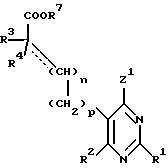
в которой
R1, R2, R3, R4, R5, n и p имеют вышеприведенные определения, с биариловым соединением формулы:
Z1-(CH2)m-Ar1-Ar2,
в которой
m, Ar1 и Ar2 имеют вышеприведенные определения, а Z1 означает отщепляющуюся группу, например атом галогена, метансульфонилокси- или n-толуолсульфонилокси-группы, или
(d) удаление защитной группы из соединения формулы I, у которого x означает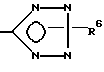
где
R6 означает третбутил, три-н-бутилстаннил или трифенилметил, например, путем кислотного или щелочного гидролиза или каталитического гидрирования с образованием в результате соединения, у которого R6 означает атом водорода, или
(e) взаимодействие соединения формулы I, у которого x означает -CN, в азидом, например триалкилкилазидом олова, или с азотисто-водородной кислотой (азид натрия/хлорид аммония) с образованием в результате соединения формулы I, у которого x означает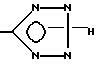
(f) получение соли соединения формулы I путем взаимодействия его с неорганической или органической кислотой или основанием, или
(g) перевод соединения формулы I или его соли в его N-оксид с помощью пероксидного соединения.
Ниже приведено более подробное описание указанных вариантов предлагаемого способа, а также получение отдельных исходных соединений для его осуществления с использованием различных схем. Исходные соединения, получение которых не описано в настоящем описании, являются известными или они могут быть получены аналогичными для получения известных соединений методами.
где
R1, R2, R3, R4, R5, n, m, p, Ar1 и Ar2 имеют вышеприведенные определения.
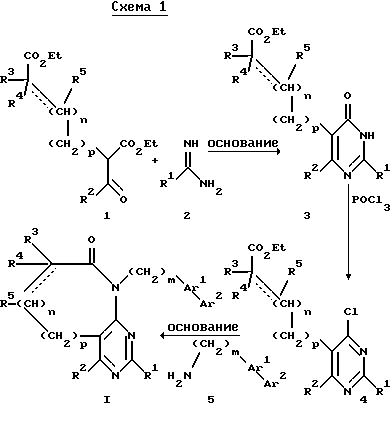
Согласно схеме 1 β -кетоэфир 1 может быть подвергнут конденсации с амидином 2 в присутствии основания, например этоксида натрия, в среде спиртового растворителя, например этанола, при температуре - от температуры окружающей среды до температуры кипения с образованием в результате пиримидона 3. Обработка пиримидона 3 оксихлоридом фосфора при температуре кипения приводит к образованию хлорпиримидина 4. Взаимодействие 4 с амином 5 в присутствии органического основания, например триэтиламина, или неорганического основания, например карбоната калия, в среде полярного растворителя, например этанола, бутанола или диметилсульфоксида, при температуре - от температуры окружающей среды до температуры кипения приводит к образованию пиримидина I. Указанный амин 5 может быть получен путем катализируемого палладием перекрестного сочетания арилстанната или арилбороновой кислоты с арилбромидом или арилиодидом и последующего перевода образующегося соединения в амин.
где
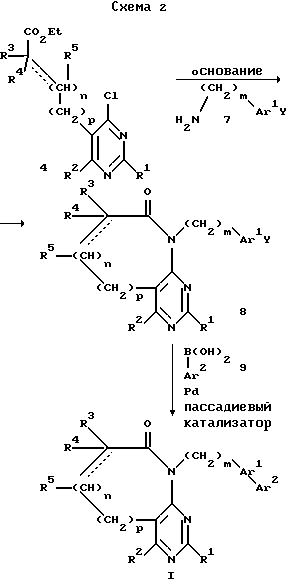
R1, R2, R3, R4, R5, n, m, p, Ar1 и Ar2 имеют вышеприведенные определения, а Y означает пара-бром или пара-иод.
При проведении процесса по схеме 2 хлорпиримидин 4 может быть подвергнут обработке амином 7 в присутствии неорганического основания, например бикарбоната натрия, в среде спиртового растворителя, например этанола или н-бутанола, например, при температуре 80-150oC с образованием в результате бициклического соединения 8. Взаимодействие 8 с арилбороновой кислотой 9 в присутствии палладиевого катализатора, например, в среде растворителя, такого как ДМФ или толуол, приводит к образованию целевого пиримидина I. В том случае, если заместителем Ar2 является нитрил, полученное соединение может быть переведено в тетразол в стандартных условиях с использованием азида.
Арилбороновая кислота 9 может быть получена следующим образом: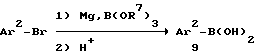
где
Ar2 имеет вышеприведенное определение, а R7 означает низший алкил с 1-6 атомами углерода.
По этой схеме арилбромид 10 может быть подвергнут обработке Mg и триалкилборатом в среде апротонного растворителя, например ТГФ, например, при температуре -20oC80oC. В результате кислотного или щелочного гидролиза образуется бороновая кислота 9.
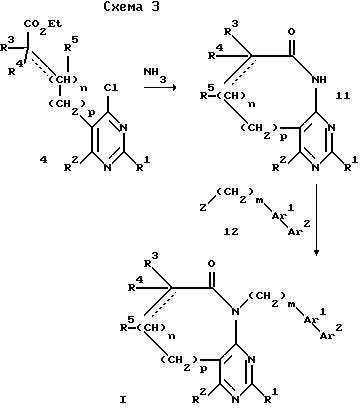
где R1, R2, R3, R4, n, m, Ar1 и Ar2 имеют вышеприведенные определения, а Z означает атом галогена.
Согласно схеме 3 хлорпиримидин 4 может быть переведен в бициклическое соединение II путем обработки аммиаком в среде спиртового растворителя, например этанола, при температуре 100-150oC. Алкилирование II биарильным соединением 12 может осуществляться, например, в присутствии основания, например NaH, в среде растворителя, например ДМФ или ТГФ. В результате образуется пиримидин I. В том случае, если заместителем Ar2 является нитрил, полученное соединение может быть переведено в тетразол в стандартных условиях с использованием азида. Биарильное соединение 12 может быть получено путем катализируемого палладием перекрестного сочетания арилбороновой кислоты с арилбромидом или арилиодидом.
В том случае, если в вышеуказанной схеме x означает тетразол, предпочтительными защитными группами этого тетразола являются группы, в которых R6 означает третбутил, три-н-бутилстанил или трифенилметил. Эти группы легко могут быть удалены путем кислотного или щелочного гидролиза или каталитического гидрирования, как это описано T.Greene в Protective Croups in Organic Synthesis, Wiley-Interscience (1980).
Соединения в соответствии с настоящим изобретением могут также образовывать соли при взаимодействии их с неорганическими или органическими кислотами и основаниями. Любые фармацевтически приемлемые соли этих соединений также охватываются настоящим изобретением. Примерами таких солей (объем изобретения, однако, не ограничивается этими примерами) являются аммониевые соли, соли щелочных металлов, например натриевые и калиевые соли, соли щелочноземельных металлов, например кальциевые соли, соли дициклогексиламина, трис-соли и соли аминокислот. Примерами солей неорганических или органических кислот являются соли, образующиеся при взаимодействии предлагаемых соединений с HCl, HBr, малеиновой и фумаровой кислотами. Соединения в соответствии с настоящим изобретением могут быть также переведены в N-оксиды путем обработки их пероксидом, например перекисью водорода, обычными способами.
Предметом настоящего изобретения являются также фармацевтические композиции, включающие заявляемое соединение и фармацевтически приемлемый носитель. Предметом его, в частности, являются гипотензивные фармацевтические композиции, включающие эффективное в отношении снижения кровяного давления количество заявляемого соединения и фармацевтически приемлемый носитель.
Предлагаемые композиции предназначены предпочтительно для орального введения. Они однако могут вводиться и другими способами, например парентерально, например, в случае пациентов, страдающих сердечной недостаточностью.
Для удобства введения предпочтительно, чтобы композиции в соответствии с настоящим изобретением готовились в виде единичных доз. Подходящими единичными дозами являются таблетки, капсулы и порошки в пакетиках или ампулах. Такие единичные дозы могут содержать 0,1-100, предпочтительно 1-50 мг, заявляемого соединения. Соединения в соответствии с настоящим изобретением могут вводиться орально в количестве примерно 0,01-100, предпочтительно 0,1-10 мг/кг. Такие композиции могут назначаться для приема 1-6, предпочтительно 1-4 раза в день. Предлагаемые соединения могут также вводиться парентерально.
Композиции в соответствии с настоящим изобретением могут содержать обычные добавки, такие как наполнители, диспергаторы, связующие, скользящие, вкусовые добавки и т. п. Они могут быть приготовлены обычным образом, в частности, как это принято при получении известных гипотензивных средств, диуретиков, β -блокирующих агентов или ингибиторов ACE.
Предметом настоящего изобретения является далее использование заявляемых соединений в качестве активных терапевтических агентов. Описанные в настоящем изобретении соединения могут, в частности, использоваться для лечения гипертензии.
Предметом настоящего изобретения является, кроме того, способ лечения гипертензии у млекопитающих, в том числе и человека, включающий введение больному млекопитающему эффективного в отношении снижения кровяного давления количества фармацевтической композиции в соответствии с настоящим изобретением.
Предпочтительными для использования в фармацевтических композициях в качестве терапевтических агентов и для лечения являются те из заявляемых соединений, в которых x означает CO2H, CN или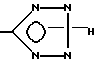
Высокое сродство заявляемых соединений к рецепторам ангиотенсина II было установлено в опытах на крысах, по связыванию их с рецепторами надпочечников, в которых определялось вытеснение меченного радиоактивной меткой ангиотенсина II из рецепторов.
Опыты проводились следующим образом. Получение мембраны.
1. Усыпляли самцов крыс породы Spraque-Dawley массой 300-400 г CO2 и умерщвляли их путем цервикального сдвига. 2. Иссекали надпочечники и хранили их в охлаждаемом льдом сахарозном буфере (0,2 М сахароза, 1 мМ ЭДТА, 10 мМ основания Trizma, pH 7,2). 3. Удаляли путем раздавливания мозговой слой. Корковый слой измельчали, промывали и гомогенизировали в охлаждаемом стеклянном гомогенизаторе тканей с 15 мл сахарозного буфера. 4. Центрифугировали гомогенизат в течение 10 мин при 3000 • g (центрифуга Sonall RCSC, ротор SS34, 6200 об/мин). Отделяли путем декантации через сетку надосадочную жидкость. 5. Центрифугировали объединенные порции надосадочной жидкости в течение 13 мин при 12000 • g (ультрацентрифуга фирмы Beckman, ротор 80 Ti, 13000 об/мин). 6. Центрифугировали надосадочную жидкость, полученную на стадии 5, в течение 60 мин при 102000 •g (ультрацентрифуга фирмы Beckman, ротор 80 Ti, 38200 об/мин). Все вышеуказанные операции проводили при 4oC. 7. Суспендировали осадок после центрифугирования в 0,5 мл в буфере, в котором проводились исследования (50 мМ трис HCl, 5 мМ MgCl2, 0,2% бычьего сывороточного альбумина, не содержащего протеазы, pH 7,4, 25oC). 8. Полученную суспензию хранили на льду. 9. Белок мембраны анализировали по методу Lowzy или Bradford с использованием бычьего сывороточного альбумина в качестве стандарта.
Испытания на связывание/анализ повторяли трижды в пластиковых пробирках 12•75 мм или в планшете с 96 лунками (суммарный объем 0,25 мл) 1. Добавляли 140 мкл буфера для проведения испытаний. 2. Добавляли 10 мкл холодного A II (до конечной концентрации 10-10 -10-7 М для построения калибровочной кривой и до 10-4 М для неспецифического связывания), испытуемые соединения (например, до конечных концентраций 25 и 100 мкМ или 1 мкМ, 10 и 100 нМ) в 50% ДМСО или 50% ДМСО в качестве контрольного раствора. 3. Добавляли 50 мкл суспензии мембраны (например, 10 мкг белка). 4. Выдерживали смесь в течение 30 мин при 25oC. 5. Добавляли 50 мкл 125I-A II, приготовление которого описано ниже (конечная концентрация 1 нМ). 6. Выдерживали смесь в течение 35 мин при 25oC 7. Инкубацию прекращали путем добавления 1 мл охлажденного на льду буфера (буфер для проведения анализа без бычьего сывороточного альбумина), фильтровали через фильтры GF/C - харвестер клеток (фильтры предварительно вымачивали в буфере для анализа, содержащем 1% полиэтиленимина). 9. Трижды ополаскивали пробирку для анализа 5 мл холодного буфера (буфер для анализа без бычьего сывороточного альбумина). 10. Разрезали фильтры на диски, помещали их в пробирки для анализа и подсчитывали число гамма-квантов за 1 мин.
Приготовление 125I-A II. Устанавливали удельную активность 125I-A II, поставленного фирмой New England Nuclear, равной 500 мкКи/нмоль, путем добавления холодного A II в воде. 1. Рассчитывали необходимые количества горячего и холодного A II и делали соответствующее разбавление. Аликвотную часть хорошо герметизировали и хранили до использования в замороженном состоянии. 2. Рассчитывали суммарную концентрацию A II (холодный + горячий) после разбавления. 3. В день анализа оттаивали замороженную аликвотную часть приготовленного препарата и доводили объем буфером для анализа (+ бычий сывороточный альбумин, не содержащий протеазы) до такой величины, чтобы концентрация равнялась 5 пмоль/мл (или 0,25 пмоль / 50 мкл). При конечной концентрации 125I-A II в пробирку для анализа добавляли 50 мкл (или 0,25 пмоль) до конечного объема 250 мкл. Результаты анализа связывания выражали в ингибирующей концентрации испытуемого соединения, необходимой для вытеснения 50% A II с радиоактивной меткой из рецептора (IC50), или в процентах вытеснения, связанного с рецептором A II (% 1) при концентрации испытуемого соединения 10-8 М. Все приведенные в настоящем описании примеры свидетельствуют о существенном ингибировании связывания A II. Как правило, для всех соединений определенная в ходе описанного анализа IC50 не превышала 50 мкМ.
Благодаря своей способности оказывать антагонистическое действие на ангиотенсин II соединения в соответствии с настоящим изобретением снижали кровяное давление у крыс, которым вводился A II. Опыты проводились следующим образом. Крыс усыпляли Dial Urethane (0,60 мл/кг парентерально) и в трахею вводили канюлю РР240. Вводили канюли РЕ 50 в одну бедренную артерию или в обе бедренные вены, или в сонную артерию и соответствующую яремную вену. В случае канюлирования яремной вены в одну вену вводили две канюли. Через небольшой срединный разрез вводили канюлю РЕ 50 в начальную часть двенадцатиперстной кишки (дистально по отношению к желудку). С помощью канюлей в артериях измеряли артериальное давление и частоту сердечных сокращений. После операции выжидали 10-15 мин для стабилизации артериального давления, затем осуществляли блокаду ганглия путем внутривенного введения мекамиламина в количестве 3 мг/кг (1 мл/кг раствора с концентрацией 3 мг/мл). Блокада ганглия приводила к снижению артериального давления примерно на 50 мм рт.ст. Мекамиламин вводили через каждые 90 мин в продолжение всего оставшегося эксперимента. После этого начинали делать вливание A II через канюлю в другой вене со скоростью 0,25 мкг/кг в минуту (9,6 мкл/мин). В результате вливания A II артериальное давление снова возвращалось к контрольному значению или несколько превышало его. После стабилизации артериального давления с помощью вливания A II определяли базовые значения среднего артериального давления (МАР) и частоты сердечных сокращений и через канюлю в двенадцатиперстной кишке вводили суспензию испытуемого соединения в метилцеллюлозе в количестве 0,1; 3 или 30 мг/кг (объем вводимой суспензии составлял 1 мл/кг). Через 15, 30, 60, 90, 120, 150, 180, 210 и 240 мин после введения испытуемого соединения определяли среднее артериальное давление и частоту сердечных сокращений. Так, например, соединение в соответствии с примером 1 при введении его внутрикожно в количестве 3 мг/кг снижало зависимость кровяного давления от A II в среднем на 41% в течение 4 ч после введения.
Как уже отмечалось, соединения в соответствии с настоящим изобретением являются эффективными антагонистами A II и поэтому могут использоваться для лечения гипертензии. Кроме того, они могут использоваться при лечении острой и хронической застойной сердечной недостаточности, первичного и вторичного легочного гиперальдостеронизма, вторичного гиперальдостеронизма, первичной и вторичной легочной гипертензии, гипертензии, вызываемой приемом оральных контрацептивов, сосудистых заболеваний, таких как мигрень, болезнь Рейно, гиперплазии и атеросклеротические процессы, болезней почек или осложнений на почки, вызываемых другими болезнями или лекарствами, таких как протеинурия, гломерулонефрит, гломерулярный склероз, склеродермия, диабетическая нефропатия, последняя стадия болезни почек, терапия при трансплантации почек и др. Предлагаемые соединения могут также использоваться при лечении нарушения функции левого желудочка, диабетической ретинопатии, болезни Альцгеймера, для улучшения познавательной способности, при лечении повышенного внутриглазного давления и для усиления ретинального кровообращения. Они могут также использоваться в качестве антидепрессантов и анксиолитиков и для профилактики или лечения рестеноза после пластических операций на сосудах. Применение заявляемых соединений для лечения этих и других аналогичных болезней очевидно для специалиста в данной области.
В нижеприведенных примерах описаны конкретные процедуры для осуществления настоящего изобретения. Эти примеры однако служат исключительно для иллюстрации изобретения и не ограничивают его объем, определяемый содержащейся в приложении формулой изобретения.
Пример 1
2,4-Диметил-5,7-дигидро-7-[[2'-(1Н-тетразол-5-ил)/1,1-бифенил-4-ил] метил]-6Н-пирроло[2,3-d]пиримидин-6-он
Стадия 1
Этил-(2,6-диметил-3Н-пиримидин-4-он-5-ил)-ацетат
Смесь NaOEt (0,060 моль) в EtOH (полученную из 1,6 г Na в 100 мл EtOH), ацетамидингидрохлорида (6,5 г, 0,069 моль) и диэтил-2-ацетилсукцината (15,0 г, 0,060 моль) кипятили с обратным холодильником в течение 16 ч. Затем ее концентрировали, остаток растворяли в воде (50 мл) и подкисляли 2 н. HCl (12 мл). Полученную таким образом водную смесь подвергали экстракции CH2Cl2, экстракты высушивали (MgSO4) и концентрировали. При растирании с эфиром получали 3,9 г (27%) продукта в виде твердого белого вещества. Температура плавления 175-177oC.
1H ЯМР (DMCO-d6): δ 1,10 (т, 3Н), 2,15 (с, 3Н), 2,25 (с, 3Н), 3,20 (с, 2Н), 4,05 (кв., 2Н), 12,20 (шир. с, 1Н)
Результаты анализа из расчета на формулу C10H14N2O3:
Рассчитано: C 57,18; H 6,71; N 13,32
Найдено: C 57,10; H 6,63; N 13,25
Стадия 2
(Тил-(4-хлор-2,6-диметилпиримидин-5-ил)-ацетат
Смесь этил-(2,6-диметил-3Н-пиримидин-4-он-5-ил)-ацетата (1,13 г, 5,4 ммоль), оксихлорида фосфора (10 мл) и N,N-диметиланилина (1,3 мл, 10,3 ммоль) кипятили в течение 3,5 ч с обратным холодильником. После этого ее концентрировали, выливали на лед и образующуюся смесь подвергали экстракции эфиром. Объединенные экстракты промывали водой, высушивали (MgSO4) и концентрировали, получая в результате 1,2 г (98%) продукта в виде маслянистой жидкости желтого цвета.
1H ЯМР (DMCO-d6): δ 1,20 (т, 3Н), 2,20 (с, 3Н), 2,60 (с, 3Н), 3,80 (с, 2Н), 4,10 (кв., 2Н).
Стадия 3
2,4-Диметил-5,7 дигидро-7-[[2'-(1Н-тетразол-5-ил)[1,1-бифенил-4-ил] метил]-6Н-пирроло[2,3-d]-пиримидин-6-он
Смесь этил-(4-хлор-2,6-диметилпиримидин-6-ил)-ацетата (0,55 г, 2,4 ммоль), 5-[4'-аминометил)[1,1-бифен-2-ил]]-1Н-тетрахолгидрохлорида (0,69 г, 2,4 ммоль), триэтиламина (0,73 г, 7,2 ммоль), ацетата натрия (0,60 г, 7,2 ммоль) и EtOH (14 мл) кипятили в течение 5 дн с обратным холодильником. После этого ее концентрировали, растворяли в воде и подвергали экстракции CHCl3. Экстракты высушивали (MgSO4), концентрировали и сырой продукт подвергали очистке с помощью флеш-хроматографии (10% MeOH/CHCl3), получая в результате твердое вещество желтого цвета. После растирания его с 10% EtOH/Et2O и перекристаллизации из EtOH получали 0,17 г (17%) продукта в виде твердого белого вещества. Температура плавления 228-229oC
1H ЯМР (DMCO-d6): δ 2,29 (с, 3Н), 2,47 (с, 3Н), 3,67 (с, 2Н), 4,83 (с, 2Н), 7,05 (д, J = 8,1 Гц, 2Н), 7,24 (д, J = 8,1 Гц, 2Н), 7,57 (м, 2Н), 7,63 (м, 2Н).
Результаты анализа из расчета на формулу C22H19N7O:
Рассчитано: C 66,48; H 4,82; N 24,67
Найдено: C 66,09; H 4,84; N не определялся.
Пример 2
5,7-Дигидро-2-метил-7-[[2'-(1Н-тетразол-5-ил)[1,1- бифенил]-4-ил]метил] -4-(трифторметил)-6Н-пирроло[2,3-d]-пиримидин-6-он
Стадия 1
Этил-(2-метил-6-трифторметил-3Н-пиримидин-1он-5-ил)ацетат
Смесь NaOEt (0,074 моль) в EtOH (полученного из 1,7 г Na и 75 мл EtOH), ацетамидингидрохлорида (3,5 г, 0,037 моль) и диэтил-2-трифторацетилсукцината (10,0 г, 0,037 моль) кипятили в течение 20 ч с обратным холодильником. После этого ее концентрировали, растворяли в воде, подкисляли до pH 4 концентрированной HCl и подвергали экстракции EtOAc. Экстракты промывали рассолом, высушивали (MgSO4) и концентрировали, получая маслянистое твердое вещество. После растирания его со смесью эфира и гексана получали 2,63 г (27%) продукта в виде белого твердого вещества.
Пробу для анализа перекристаллизовывали из смеси эфира и гексана. Температура плавления 108-110oC.
1H ЯМР (DMCO-d6): δ 1,25 (т, J = 7,2 Гц, 3Н), 2,51 (с, 3Н), 3,72 (с, 2Н), 4,17 (кв. J = 7,2 Гц, 2Н), 13,25 (шир. с, 1Н).
Результаты анализа из расчета на формулу C10H11F3N2O3
Рассчитано: C 45,46; H 4,20; N 10,60
Найдено: C 45,43; H 4,18; N 10,53.
Стадия 2
Этил-(4-хлор-2-метил-6-трифторметилпиримидин-5-ил)-ацетат
Раствор этил-(2-метил-6-трифторметил-3Н-пиримидин-4-он-5-ил)-ацетата (2,50 г, 9,46 ммоль), N,N-диметиланилина (3 капли) и оксихлорида фосфора (35 мл) кипятили в течение 4,5 ч с обратным холодильником. После этого смесь концентрировали, охлаждали и добавляли к ней ледяную воду. Для нейтрализации смеси к ней добавляли твердый KOH, после чего ее подвергали экстракции эфиром. Объединенные экстракты промывали рассолом, высушивали (MgSO4) и концентрировали, получая в результате 2,61 г (98%) продукта в виде желтой маслянистой жидкости.
1H ЯМР (DMCO-d6): δ 1,24 (т, J = 7,0 Гц, 3Н), 2,77 (с, 3Н), 3,93 (с, 2Н), 4,19 (кв., J = 7,0 Гц, 2Н).
Стадия 3
5,7-Дигидро-2 метил-7-[[2'-(1Н, тетразол-5-ил)[1,1-бифенил]-4-ил]метил] -4-/трифторметил)-6Н-пирроло[2,3-d]-пиримидин-6-он
Смесь этил-(4-хлор-2-метил-6-трифторметилпиримидин-5-ил)-ацетата (848 мг, 3,0 ммоль), 5-[(4'-аминометил)[1,1-бифен-2-ил]]-1Н- тетразолгидрохлорида (863 мг, 3,00 ммоль), триэтиламина (1,25 мл, 9,00 ммоль), ацетата натрия (738 мг, 9,00 ммоль) и EtOH (10 мл) кипятили в течение 16 ч с обратным холодильником. После этого ее концентрировали, растворяли в 1 н. NaOH и подвергали экстракции эфиром. Водную фазу подкисляли до pH 3 концентрированной HCl и собирали темно-коричневый осадок путем фильтрования. В результате очистки с помощью флеш-хроматографии (дважды, 5-10% MeOH/CH2Cl2) получали пену коричневого цвета. После кристаллизации из смеси эфира и гексана получали 109 мг (8%) продукта в виде белого твердого вещества. Температура плавления 195-197oC.
1H ЯМР (DMCO-d6): δ 2,59 (с, 3Н), 3,92 (с, 2Н), 7,05 (д, J = 8,1 Гц, 2Н), 7,28 (д, J = 8,1 Гц, 2Н), 7,52 (д, J = 6,2 Гц, 1Н), 7,57 (д, J = 7,4 Гц, 1Н), 7,66 (м, 2Н).
ИК (KBr, см-1): 1750 (С=О).
Результаты анализа из расчета на формулу C22H16F3N7O:
Рассчитано: C 58,54; H 3,57; N 21,72
Найдено: C 58,58; H 3,71; N 21,44
Пример 3
2,4-Диметил-5,6,8-тригидро-8-[[2'-(1Н-тетразол-5-ил)[1,1-бифенил] -4-ил] метил]-7Н-пирроло[2,3-d]-пиримидин-7-он
Стадия 1
Этил-3-(2,6-диметил-3Н-пиримидин-4-он-5-ил)-пропионат
Смесь NaOEt (0,069 моль) в EtOH (полученного из 1,6 г Na и 35 мл (EtOH), ацетамидингидрохлорида (3,3 г, 0,035 моль) и диэтилацетилглутарата (8,0 г, 0,035 моль) кипятили в течение 22 ч с обратным холодильником. После этого ее концентрировали, растворяли в воде, подкисляли до pH 4 концентрированной HCl и подвергали экстракции EtOAc. Экстракты промывали рассолом, высушивали (MgSO4) и концентрировали. После растворения с гексаном получали 4,0 г (51%) продукта в виде белого вещества. Пробу для анализа перекристаллизовывали из смеси эфира и гексана. Температура плавления 114-116oC.
1H ЯМР (DMCO-d6): δ 1,22 (т, J = 7,2 Гц, 3Н), 2,33 (с, 3Н), 2,40 (с, 3Н), 2,54 (т, J = 8,0 Гц, 2Н), 2,81 (т, J = 8,0 Гц, 2Н), 4,10 (кв., J = 7,2 Гц, 2Н).
Результаты анализа из расчета на формулу C11H16N2O3
Рассчитано: C 58,91; H 7,19; N 12,49
Найдено: C 59,18; H 7,26; N 12,20
Стадия 2
Этил-3-(4-хлор-2,6-диметилпиримидин-5-ил)-пропионат
Смесь этил-3-(2,6-диметил-3Н-пиримидин-4-он-5-ил)-пропионата (3,87 г, 0,017 моль) оксихлорида фосфора (40 мл) и N,N-диметиланилина (10 капель) кипятили в течение 2 ч с обратным холодильником. После этого ее концентрировали, охлаждали и добавляли к ней ледяную воду. С помощью твердого KOH устанавливали pH смеси равным 6 и подвергали ее экстракции эфиром. Экстракты высушивали (MgSO4) и концентрировали, получая в результате 2,95 г (72%) продукта в виде маслянистой жидкости коричневого цвета.
1H ЯМР (DMCO-d6): δ 1,24 (т, J = 7,2 Гц, 3Н), 2,54 (с, 3Н), 2,56 (т, J = 8,1 Гц, 2Н), 2,81 (с, 3Н), 3,04 (т, J = 8,1 Гц, 2Н), 4,14 (кв, J = 7,2 Гц, 2Н).
Стадия 3
2,4-Диметил-5,6,8-тригидро-8-[[2'-(1Н-тетразол-5-ил) [1,1-бифенил]-4-ил] метил]-7Н-пирридо[2,3-d]-пиримидин-7-он
Смесь этил-3-(4-хлор-2,6-диметилпиримидин-5-ил)пропионата, (1,27 г, 5,21 ммоль), 5-[(4'-аминометил)[1,1-бифен-2-ил]]-1Н-тетразолгидрохлорида (1,50 г, 5,21 ммоль), триэтиламина (2,2 мл, 15,64 ммоль), ацетата натрия (1,28 г, 15,64 ммоль) и EtOH (20 мл) кипятили в течение 5 дн с обратным холодильником. После этого ее охлаждали и фильтровали для удаления нерастворимого материала. Фильтрат подкисляли до pH 4 с помощью метанольного раствора HCl и концентрировали. После очистки с помощью флеш-хроматографии (10% MeOH/CH2Cl2) и кристаллизации из смеси EtOH и эфира получали 91 мг продукта в виде твердого белого вещества. Температура плавления 197-199oC.
1H ЯМР (DMCO-d6): δ 2,35 (с, 3Н), 2,42 (с, 3Н), 1,72 (т, J = 7,2 Гц, 2Н), 2,87 (т, J = 7,2 Гц, 2Н), 5,17 (с, 2Н), 6,99 (д, J = 8,2 Гц, 2Н), 7,18 (д, J = 8,2 Гц, 2Н), 7,54 (м, 2Н), 7,65 (м, 2Н).
ИК (KBr, см-1): 1710 (С=О)
Результаты анализа из расчета на формулу C23H21N7O:
Рассчитано: C 67,14; H 5,14; N 23,83
Найдено: C 67,38; H 5,39; N 24,19
Соединение 2,4-диметил-5,6,8-тригидро-8-[[2'-(1Н- тетразол-5-ил)[1,1-бифенил] -4-ил] метил] -7Н-пиридо[2,3-d] пиримидин- 7-он может быть получено также другим способом в соответствии со схемой 2.
Стадия 1
2,4-Диметил-5,6,8-тригидро-8-[(4-бромфенил)метил] -7Н- пиридо[2,3-d]пиримидин-7-он
Смесь этил-3-(4-хлор-2,6-диметилпиримидин-5-ил)пропионата (8,4 г, 0,035 моль), 4-бромбензиламингидрохлорида (8,5 г, 0,038 моль) NaHCO3 (5,8 г, 0,070 моль) и н-BuOH (75 мл) кипятили в течение 48 ч с обратным холодильником. После этого ее разбавляли EtOAc (30 мл) и промывали водой (100 мл), 10%-ным водным раствором HOAc (50 мл), водой (100 мл), насыщенным водным раствором NaHCO3 (2 • 100 мл), высушивали (MgSO4) и концентрировали, получая в результате твердый продукт желтого цвета. После растирания с гексаном (50 мл) получали 8,3 г (69%) продукта в виде не совсем белого твердого вещества. Температура плавления 123-124oC
1H ЯМР (DMCO-d6): δ 2,34 (с, 3Н), 2,42 (с, 3Н), 2,71 (т, J = 7,9 Гц, 2Н), 2,86 (т, J = 7,9 Гц, 2Н), 5,13 (с, 2Н), 7,23 (д, J = 8,3 Гц, 2Н), 7,44 (д, J = 8,3 Гц, 2Н).
Стадия 2
5-(2-бромфенил)-1Н-тетразол
Смесь 2-бромбензонитрила (10,0 г, 0,055 моль), азида натрия (3,9 г, 0,060 моль), хлорида аммония (3,2 г, 0,060 моль) и ДМФ (90 моль) нагревали в течение 18 ч при 100oC. После этого ее концентрировали, растворяли в воде и подщелачивали (pH 9) 1 н. KOH. Затем ее подвергали экстракции эфиром (сбрасывали) и подкисляли 2 н. HCl. Осадок отфильтровывали, получая в результате 9,1 г (73%) продукта в виде не совсем белого твердого вещества. Температура плавления 179-181oC.
1H ЯМР (DMCO-d6): δ 7,56 (м, 2Н), 7,69 (дв. д. J = 7,0 Гц, 2,1 Гц, 1Н), 7,86 (дв. д. J = 7,6 Гц, 1,3 Гц, 1Н).
Стадия 3
1 третбутил-5-(2-бромфенил)-1Н-тетразол
К раствору 5-(2-бромфенил)-1Н-тетразола (7,9 г, 0,035 моль) в трифторуксусной кислоте (35 мл) добавляли третBuOH (5,2 г, 0,070 моль) и H2SO4 (1,0 мл, 0,0175 моль). Через 18 ч раствор концентрировали и остаток растворяли в EtOAC. Смесь промывали водой 2,5 н. NaOH, высушивали (MgSO4) и концентрировали. После очистки с помощью флеш-хроматографии (20% EtOAc/гексан) получали 6,6 г (67%) продукта в виде светло-желтой маслянистой жидкости.
1H ЯМР (DMCO-d6): δ 1,74 (с, 9Н), 7,55 (м, 2Н), 8,06 (м, 2Н).
Стадия 4
2-[(1-Третбутил)-1Н-тетразол-5-ил]фенилбороновая кислота.
К сухим магниевым стружкам (76 мг, 3,13 ммоль) в ТГФ (1 мл) добавляли раствор 1-третбутил-5-(2-бромфенил)-1Н-тетразола (733 мг, 2,61 ммоль) в ТГФ (1,5 мл), затем 1,2-дибромэтан (20 мкл) и смесь медленно нагревали феном. Через 5 мин к ней добавляли триизопропилборат (564 мг, 3,0 ммоль) и перемешивали ее в течение 23 ч при комнатной температуре. Затем к ней добавляли лед и 0,5 н. HCl (7 мл) и перемешивали в течение 5 мин. Смесь затем подвергали экстракции эфиром и объединенные экстракты подвергали экстракции 1 н. KOH (10 мл). Водные экстракты фильтровали и подкисляли до pH 32 н. HCl. Осадок отфильтровывали, получая в результате 306 мг (48%) продукта в виде не совсем белого твердого вещества. Температура плавления 112-115oC.
1H ЯМР (DMCO-d6): δ 1,72 (с, 9Н), 7,46 (м, 2Н), 7,90 (м, 2Н).
Стадия 5
2,4-диметил-5,6,8-тригидро-8-[[2'-(1-третбутил-1Н-тетразол-5-ил)[1,1-бифенил]-4-ил]метил]-7Н-пиридо[2,3-d]-пиримидин-7-он
Смесь 2,4-диметил-5,6,8-тригидро-8-[(бромфенил)метил] -7Н-пиридо[2,3-d] пиримидин-7-она (1,08 г, 3,43 ммоль), 2-[(1-третбутил)-1Н-тетразол-5-ил] фенилбороновой кислоты (844 мг, 3,12 ммоль), триэтиламина (1,30 мл, 9,36 ммоль), тетракис (трифенилфосфин)палладия (108 мг, 0,094 ммоль) и ДМФ (15 мл) нагревали в течение 22 ч при 100oC. После этого ее концентрировали, остаток растворяли в CHCl3 и промывали раствор рассолом. Органическую фазу высушивали (MgSO4) и концентрировали. После очистки с помощью флеш-хроматографии (60% EtOAc/гексан) получали 1,05 г (66%) продукта в виде пены желтого цвета.
1H ЯМР (DMCO-d6): δ 2,35 (с, 3Н), 2,43 (с, 3Н), 2,71 (т, J = 7,1 Гц, 2Н), 2,85 (т, J = 7,1 Гц, 2Н), 5,18 (с, 2Н), 6,97 (д, J = 8,2 Гц, 2Н), 7,44 (дв., д, J = 7,3 Гц, 1,3 Гц, 1Н), 7,55 (м, 2Н), 7,76 (дв. д, J = 7,5 Гц, 1,3 Гц, 1Н).
Стадия 6
2,4 Диметил-5,6,8-тригидро-8-[[2'-(1Н-тетразол-5-ил)[1,1-бифенил]-4-мл] метил]-7Н-пиридо[2,3-d]-пиримидин-7-он
Смесь 2,4-диметил-5,6,8-тригидро-8-[[2'-(1-третбутил-1Н-тетразол-5-ил)[1,1-бифенил] -4-мл] метил] -7Н-пиридо[2,3-d] - пиримидин-7-она (200 мг, 0,0428 ммоль), метансульфоновой кислоты (280 мкл, 4,28 ммоль) и толуола (4 мл) кипятили в течение 18 ч с обратным холодильником. После этого ее концентрировали и добавляли к остатку воду (2 мл) и 1 н. KOH (4,5 мл), получая в результате раствор с pH 8. Этот раствор подвергали экстракции EtOAc для удаления непрореагировавшего исходного материала и водную фазу подкисляли до pH 51 н. HCl. В результате выпадал смолистый осадок, который экстрагировали в EtOAc и раствор высушивали (MgSO4) и концентрировали, получая в результате бесцветную маслянистую жидкость. После растирания со смесью ацетона и эфира получали 70 мг (40%) продукта в виде белого твердого вещества. Температура плавления 197-198oC
1H ЯМР-(DMCO-d6): δ 2,35 (с, 3Н), 2,42 (с, 3Н), 2,72 (т, J = 7,2 Гц, 2Н), 2,87 (т, J = 7,2 Гц, 2Н), 5,17 (с, 2Н), 6,99 (д, J = 8,2 Гц, 2Н), 7,18 (д, J = 8,2 Гц, 2Н), 7,54 (м, 2Н), 7,65 (м, 2Н).
ИК (KBr, см-1): 1710 (С=О)
Пример 4
2-Метил-4-трифторметил-5,6,8-тригидро-8-[[2'-/(1Н-тетразол-5-ил)[1,1-бифенил]-4-ил]метил]-7Н-пиридо[2,3-d]-пиримидин-7-он
Стадия 1
Диэтилтрифторацетилглутарат
К раствору NaOEt (0,217 моль) в EtOH (полученного из 5,0 г Na и 220 мл EtOH) добавляли электрифторацетоацетат (40,0 г, 0,217 моль). Через 10 мин добавляли этил-3-бромпропионат (35,0 г, 0,194 моль) и смесь кипятили в течение 24 ч с обратным холодильником. После этого ее концентрировали, остаток растворяли в эфире и промывали раствор водой. Эфирные слои высушивали (MgSO4) и концентрировали, получая в результате 50 г желтой маслянистой жидкости. После дистилляции (100-110oC 25 мм) получали 14,0 г (23%) продукта в виде бесцветной маслянистой жидкости.
1H ЯМР (DMCO-d6): δ 1,20 (м, 6Н), 2,93 (т, J = 6,4 Гц, 2Н), 3,62 (т, J = 6,4 Гц, 2Н), 4,13 (м, 5Н).
Стадия 2
Этил-3-(2-метил-6-трифторметил-3Н-пиримидин-4-он-5-ил)- пропионат.
К раствору NaOEt (0,026 моль) в EtOH (полученного из 0,60 г Na и 30 мл EtOH) добавляли диэтилтрифторацетилглутарат (3,70 г, 0,013 моль) и ацетамидингидрохлорид (1,23 г, 0,013 моль). Реакционную смесь кипятили в течение 18 ч с обратным холодильником, концентрировали и остаток растворяли в воде. Смесь подкисляли до pH 4 2н. HCl и подвергали экстракции CH2Cl2. Экстракты высушивали (MgSO4) и концентрировали, получая твердое вещество желтого цвета. После растирания его с гексаном получали 0,87 г продукта в виде белого твердого вещества.
1H ЯМР (DMCO-d6): δ 1,16 (т, J = 7,0 Гц, 3Н), 2,35 (с, 3Н), 2,45 (т, J = 6,4 Гц, 2Н), 2,70 (т, J = 6,4 Гц, 2Н), 4,06 (кв., J = 7,0 Гц, 2Н).
Стадия 3
Этил-3-(4-хлор-2 метил-6-трифторметилпиримидин-5-ил)- пропионат
Смесь этил-3-(2-метил-6-трифторметил-3Н-пиримидин-4-он-5-ил)пропионата (0,61 г, 2,20 ммоль), N,N-диэтиланилина (0,33 г, 2,2 ммоль) и POCl3 (8 мл) кипятили в течение 2 ч с обратным холодильником. После этого ее концентрировали, выливали на лед и подвергали экстракции эфиром. Объединенные экстракты высушивали (MgSO4) и концентрировали, получая в результате 0,65 г продукта в виде коричневой маслянистой жидкости.
1H ЯМР (DMCO-d6): δ 1,18 (т, J = 7,0 Гц, 3Н), 2,56 (т, J = 8,5 Гц, 2Н), 2,67 (с, 3Н), 3,06 (т, J = 8,5 Гц, 2Н), 4,10 (м, 2Н).
Стадия 4
2-Метил-4-трифторметил-5,6,8-тригидро-8-[[2'-(1Н, тетразол-5-ил)[1,1-бифенил]-4-ил]метил]-7Н, пиридо[2,3-d]-пиримидин-7-он
Смесь этил-3-(4-хлор-2 метил-6-трифторметилпиримидин-5-ил) пропионата (1,30 г, 4,40 ммоль), 5-[(4'-амминометил)[1,1-бифенил-2-ил]]-1Н-тетразолгидрохлорида (1,26 г, 4,40 ммоль), ацетата натрия (1,08 г, 13,20 ммоль) и триэтиламина (1,33 г, 31,20 ммоль) и н-BuOH (25 мл) кипятили в течение 24 ч с обратным холодильником. После этого ее концентрировали и распределяли остаток между CHCl3 и водой. Слои разделяли и органическую фазу промывали водой, высушивали (MgSO4) и концентрировали, получая в результате желтую маслянистую жидкость. После очистки с помощью флеш-хроматографии (7% MeOH/CH2Cl2) и перекристаллизации из смеси ацетона и эфира (дважды) получали 0,07 г продукта в виде белого твердого вещества. Температура плавления 219-220oC.
1H ЯМР (DMCO-d6): δ 2,53 (с, 3Н), 2,81 (т, J = 7,1 Гц, 2Н), 3,07 (т, J = 7,1 Гц, 2Н), 5,2 (с, 2Н), 7,00 (д, J = 8,1 Гц, 2Н), 7,22 (д, J = 8,1 Гц, 2Н), 7,56 (м, 2Н), 7,65 (м, 2Н).
Результаты анализа из расчета на формулу C23H18F3N7O:
Рассчитано: C 59,35; H 3,90; N 21,07
Найдено: C 59,12; H 3,85; N 21,29
Пример 5
2,4-диметил-5,6,8-тригидро-8-[[6-/2-(1Н-тетразол-5-ил)фенил] -3-пиридинил]метил]-7Н-пиридо[2,3-d]-пиримидин-7-он
Стадия 1
2,4-диметил-5,6,8-тригидро-7Н-пиридо-[2,3-d]-пиримидин-7-он
К охлажденному (-78oC) раствору этил-3-(4-хлор-2,6-диметил-пиримидин-5-ил) пропионата (27,1 г, 0,111 моль), полученного таким же образом, как это описано в примере 3, стадия 2, в EtOH (110 мл) добавляли жидкий NH3 (20 мл). Смесь нагревали в автоклаве при 150oC в течение 12 ч и затем концентрировали. После этого к ней добавляли воду (20 мл) и подвергали экстракции CH2Cl2. Экстракты высушивали (MgSO4) и концентрировали, получая в результате коричневое твердое вещество. После очистки с помощью флеш-хроматографии (2% MeOH/CH2Cl2) и растирания со смесью эфира и гексана получали 149 г (76%) продукта в виде не совсем белых кристаллов. Температура плавления 167-168oC.
1H ЯМР (DMCO-d6): δ 2,30 (с, 3Н), 2,40 (с, 3Н), 2,52 (т, J = 7,7 Гц, 2Н), 2,80 (т, J = 7,7 Гц, 2Н), 10,64 (с, 1Н),
ИК (KBr, см-1): 1690.
Результаты анализа из расчета на формулу C9H11N3O
Рассчитано: C 61,00; H 6,26; N 23,71
Найдено: C 61,04; H 6,07; N 24,10
Стадия 2
2-бром-5-оксиметилпиридин
К охлажденной (0oC) перемешиваемой суспензии 6-броминикотиновой кислоты (13,8 г, 0,068 моль), полученной по методу Campbell и др., Aust. J. Chem. 1971, 24, 277, в ТГФ (20 мл) добавляли 1,0 М BH3 в ТГФ (204 мл, 0,204 моль). Смесь перемешивали при комнатной температуре в течение 3 ч, снова охлаждали до 0oC и добавляли насыщенный водный раствор K2CO3 и воду, после чего подвергали экстракции EtOAc. Объединенные экстракты промывали водой, высушивали (MgSO4) и концентрировали, получая в результате желтую маслянистую жидкость. После очистки ее с помощью флеш-хроматографии (2% MeOH/CH2Cl2) получали 7,5 г (59%) твердого желтого вещества. Температура плавления 49-51oC.
1H ЯМР (DMCO-d6): δ 4,50 (д, J = 5,7 Гц, 2Н), 5,40 (т, J = 5,7 Гц, 1Н), 7,57 (д, J = 8,3 Гц, 1Н), 7,70 (дв, д, J = 8,3 Гц, 1,5 Гц,) 1Н), 8,35 (д, J = 1,5 Гц, 1Н).
Стадия 3
2-Бром-5-(третбутилдиметилсилилокси)метилпиридин
К перемешиваемой смеси 2-бром-5-оксиметилпиридина (4,7 г, 0,023 моль) и триэтиламина (3,4 мл, 0,024 моль) в ДМФ (30 мл) добавляли третбутилдиметилсилихлорид. Через час смесь разбавляли эфиром и промывали водой. Эфирную фазу высушивали (MgSO4) и концентрировали, получая в результате 6,8 г (97%) продукта в виде бесцветной жидкости.
1H ЯМР (DMCO-d6): δ 0,10 (с, 6Н), 0,90 (с, 3Н), 4,73 (с, 2Н), 7,65 (м, 2Н), 8,35 (д, J = 1,5 Гц, 1Н).
Стадия 4
[5-(Третбутилдиметилсилиоксиметил)пиридин-2-ил]три-н-бутилстаннан
К охлажденному (-78oC) перемешиваемому раствору 2 бром-5-(третбутилдиметилсилиокси)метилпиридина (6,8 г, 0,022 моль) в ТГФ (60 мл) добавляли 1,6 М н-BuLi в гексане (14,1 мл, 0,022 моль). Через час добавляли хлорид три-н-бутилолова (6,1 мл, 0,022 моль) и продолжали перемешивать смесь в течение еще 3 ч, после чего добавляли к ней воду, нагревали до комнатной температуры и подвергали экстракции эфиром. Объединенные экстракты высушивали (MgSO4) и концентрировали, получая в результате 11,5 г (100%) продукта в виде коричневой маслянистой жидкости.
1H ЯМР (DMCO-d6): δ 0,10 (с, 6Н), 0,80 (м, 18Н), 1,10 (м, 6Н), 1,25 (м, 6Н), 1,50 (м, 6Н), 4,73 (с, 2Н), 7,55 (м, 2Н), 8,61 (д, J = 2,2 Гц, 1Н).
Стадия 5
2-[5-(третбутилдиметилсилиоксиметил)пиридин-2-ил]бензонитрил
Смесь [5-(третбутилдиметилсилиоксиметил)пиридин-2-ил] три-н- бутилстаннана (11,5 г, 0,022 моль), 2-иодбензонитрила (5,1 г, 0,022 моль) CuI (0,43 г, 0,002 моль) и хлорида бис(трифенилфосфин)палладия (0,80 г, 0,001 моль) в ТГФ (40 мл) кипятили в течение 48 ч с обратным холодильником. После этого ее разбавляли эфиром и промывали насыщенным водным раствором NH4Cl, водным раствором NH4OH, водой и рассолом, высушивали (MgSO4) и концентрировали, получая в результате 4,9 г (67%) продукта в виде коричневой маслянистой жидкости.
1H ЯМР (DMCO-d6): δ 0,10 (с, 6Н), 0,90 (с, 9Н), 4,73 (с, 2Н), 7,45 (м, 1Н), 7,60 (м, 4Н), 7,75 (дв. д, J = 7,9 Гц, 2,2 Гц, 1Н), 8,50 (д, J = 2,2 Гц, 1Н).
Стадия 6
2-[5-(Оксиметил)пиридин-2-ил]бензонитрил
Смесь 2-[5-(третбутилдиметилсилиоксиметил)пиридин-2-ил] - бензонитрила (4,9 г, 0,021 моль) и н. Bu4NF-гидрата (8,1 г, 0,031 моль) в ТГФ (60 мл) перемешивали при комнатной температуре в течение 18 ч. Затем ее разбавляли EtOAc, промывали водой и рассолом, высушивали (MgSO4) и концентрировали, получая в результате 3,5 г (80%) продукта в виде коричневого твердого вещества. Температура плавления 152-153oC.
1H ЯМР (DMCO-d6): δ 4,61 (д, J = 5,6 Гц, 2Н), 5,42 (т, J = 5,6 Гц, 1Н), 7,57 (дв. д, J = 7,3 Гц, 1,5 Гц), 7,65 (м, 4Н), 7,80 (дв., д, J = 7,9 Гц, 2,2 Гц, 1Н), 8,52 (д, J = 2,2 Гц, 1Н).
Стадия 7
2-[5-(Хлорметил)пиридин-2-ил]бензонитрил
К охлажденному (0oC) перемешиваемому раствору 2-[5-(оксиметил)пиридин-2-ил]бензонитрила (4,3 г, 0,020 моль) и ZnCl2 0,09 г, 0,61 моль) в п-диоксане (40 мл) добавляли по каплям SOCl2 (1,50 мл, 0,020 моль). Смесь перемешивали при комнатной температуре в течение 18 ч, после чего разбавляли ее эфиром, промывали водой и рассолом, высушивали (MgSO4) и концентрировали, получая в результате 4,30 г (92%) продукта в виде коричневого твердого вещества. Температура плавления 97-98oC.
1H ЯМР (DMCO-d6): δ 4,90 (с, 2Н), 7,63 (дв. д, J = 7,7 Гц, 1,3 Гц, 1Н), 7,80 (м, 1Н), 7,85 (м, 2Н), 7,95 (д, J = 7,7 Гц, 1Н), 8,01 (дв. д, J = 8,0 Гц, 2,2 Гц, 1Н), 8,80 (д, J = 2,2 Гц, 1Н).
Стадия 8
2,4-Диметил-5,6,8-тригидро-8-[[6-(2-цианофенил)-3-пиридинил)-метил] -7Н-пиридо[2,3-d]-пиримидин-7-он
К перемешиваемой суспензии NaH (60%-ная дисперсия в минеральном масле; 0,42 г, 9,18 ммоль) в ДМФ (20 мл) добавляли 2,4-диметил-5,6,8-тригидро-7Н-пирило[2,3-d] -пиримидин-7-он (1,55 г, 8,75 ммоль). Через час добавляли 2-[5-(хлорметил)пиридин-2-ил] бензонитрил (2,00 г, 8,75 ммоль). Добавление осуществляли в несколько порций. Перемешивание продолжали в течение еще 4 ч, после чего смесь концентрировали, добавляли к ней воду и подвергали экстракции CH2Cl2. Объединенные экстракты высушивали (MgSO4) и концентрировали, получая в результате 3,20 г (100%) продукта в виде коричневого твердого вещества. Температура плавления 161-162oC.
1H ЯМР (DMCO-d6): δ 2,36 (с, 3Н), 2,45 (с, 3Н), 2,78 (т, J = 7,8 Гц, 2Н), 2,87 (т, J = 7,8 Гц, 2Н), 5,27 (с, 2Н), 7,61 м, 1Н), 7,80 (м, 4Н), 7,93 (дв. д, J = 7,7 Гц, 1,3 Гц, 1Н), 8,70 (д, J = 1,3 Гц, 1Н).
Стадия 9
2,4-Диметил-5,6,8-тригидро-8-[[6-[2-(1Н-тетразол-5-ил)фенил] -3-пирилинил]метил]-7Н-пиридо[2,3-d]-пиримидин-7-он
Смесь 2,4-диметил-5,6,8-тригидро-8-[[6-(2-цианофенил)-3-пиридинил]метил] -7Н-пиридо[2,3-d] -пиримидин-7-она (4,0 г, 10,8 ммоль), NaN3 (0,8 г, 11,9 ммоль) и хлорида три-н-бутилолова (3,9 г, 11,9 ммоль) в ксилолах (100 мл) кипятили в течение 24 ч с обратным холодильником, после чего добавляли еще 1,5 эквивалента NaN3 и хлорида три-н-бутилолова и продолжали нагревать смесь в течение еще 24 ч. После этого ее концентрировали и добавляли к ней 2н. HCl, подвергали экстракции эфиром (отбрасывали) и с помощью 50%-ного раствора NaOH устанавливали ее pH равным 5. Водную фазу подвергали экстракции CH2Cl2, экстракты промывали водой, высушивали (MgSO4) и концентрировали. После очистки остатка с помощью флеш-хроматографии (5% MeOH/CH2Cl2) и перекристаллизации из смеси EtOH и воды получали 2,8 г (63%) продукта в виде белого твердого вещества. Температура плавления 217-218oC.
1H ЯМР (DMCO-d6): δ 2,35 (с, 3Н), 2,44 (с, 3Н), т, J = 7,8 Гц, 2Н), 2,85 (т, J = 7,8 Гц, 2Н), 5,18 (с, 2Н), 7,33 (д, J = 8,3 Гц, 1Н), 7,65 (м, 5Н), 8,36 (с, 1Н).
ИК (KBr, см-1: 1690
Результаты анализа из расчета на формулу C22H20N8O:
Рассчитано: C 64,07; H 4,89; N 27,17
Найдено: C 63,87; H 4,81; N 27,56
Пример 6
2-Метил-4-трифторметил-5,6,8-тригидро-8-[[6-[2-(1Н- тетразол-5-ил)фенил] -3-пиридинил]метил]-7Н-пиридо[2,3-d]-пиримидин- 7-он
Стадия 1
2-Метил-4-трифторметил-5,6,8-тригидро-7Н-пиридо[2,3-d]- пиримидин-7-он
Раствор этил-3-(4-хлор-2-метил-6-трифторметилпиримидин-5-ил)пропионата (0,50 г, 1,70 ммоль), полученного, как это описано в примере 4, стадия 3, в насыщенном этанольном растворе аммиака (15 мл) нагревали в герметизированной трубке в течение 18 ч при 110oC. После этого смесь концентрировали, растворяли остаток в CH2Cl2 и промывали водой. Органический слой высушивали (MgSO4) и концентрировали, получая в результате 0,33 г (84%) продукта в виде белого твердого вещества. Температура плавления 147-150oC.
1H ЯМР (DMCO-d6): δ 2,53 (с, 3Н), 2,61 (т, J = 7,3 Гц, 2Н), 3,01 (т, J = 7,3 Гц, 2Н), 11,30 (с, 1Н).
Стадия 2
2-Метил-4-трифторметил-5,6,8-тригидро-8-[[6-(2-цианофенил)-3-пиридинил] метил]-7Н-пиридо[2,3-d]-7-он
К суспензии NaH (60%-ная дисперсия в минеральном масле; 33 мг, 1,40 моль) в ДМФ добавляли 2 метил-4-трифторметил-5,6,8-тригидро-7Н-пиридо[2,3-d] -пиримидин-7-он (0,32 г, 1,40 моль) при комнатной температуре. Через 30 мин добавляли 2-[5-(хлорметил)пиридин-2-ил] бензонитрил (0,32 г, 1,40 ммоль), полученный по способу в соответствии со стадией 7 примера 5. Смесь перемешивали в течение 42 ч, после чего разбавляли водой и подвергали экстракции CH2Cl2. Объединенные экстракты высушивали (MgSO4) и концентрировали, получая в результате 0,51 г (86%) продукта в виде желтой пены.
1H ЯМР (DMCO-d6): δ 2,58 (с, 3Н), 2,86 (т, J = 7,6 Гц, 2Н), 3,11 (т, J = 7,6 Гц, 2Н, 5,31 (с, 2Н), 7,62 (м, 1Н), 7,85 (м, 5Н), 8,75 (с, 1Н).
Стадия 3
2-Метил-4-трифторметил-5,6,8-тригидро-8-[[6-[2-(1Н-тетразол-5-ил) фенил] -3-пиридинил]метил]-7Н-пиридо[2,3-d]-пиримидин-7-он
Смесь 2-метил-4-трифторметил-5,6,8-тригидро-8-[[6-[2-цианофенил)-3-пиридинил] метил] -7Н-пиридо[2,3-d]-пиримидин-7-она (0,50 г, 1,23 ммоль), NaN3 (0,08 г, 1,23 ммоль) и хлорида три-н-бутилолова (0,40 г, 1,23 ммоль) в смеси ксилолов (9 мл) кипятили в течение 48 ч с обратным холодильником, после чего добавляли к ней 1 н. HCl (1,2 мл), разбавляли ее CH2Cl2, промывали водой, высушивали (MgSO4) и концентрировали. После очистки остатка с помощью флеш-хроматографии (5% MeOH/CH2Cl2) получали пену, которую растирали с 10% EtOA/эфир и в результате получали 0,145 г (25%) продукта в виде не совсем белого твердого вещества. Температура плавления 189-191oC.
1H ЯМР (DMCO-d6-): δ 2,55 (с, 3Н), 2,82 (т, J = 7,0 Гц, 2Н), 3,07 (т, J = 7,0 Гц, 2Н), 5,20 (с, 2Н), 7,31 (д, J = 8,0 Гц, 1Н,) 7,65 (м, 5Н), 8,39 (с, 1Н).
Результаты анализа из расчета на формулу C22H17F3N8O:
Рассчитано: C 56,65; H 3,67; N 24,03
Найдено: C 56,2; H 4,11; N 23,44.
Пример 7
2,4-Диметил-5,6,8-тригидро-8-[[5-[2-(1Н-тетразол-5-ил)фенил] -2-пиридинил]метил]-7Н-пиридо[2,3-d]-пиримидин-7-он
Стадия 1
(6-Метилпиридин-3-ил)-трифторметансульфонат
К перемешиваемому охлажденному (0oC) раствору 3-окси-6-метилпиридина (14,0 г, 0,128 моль) в пиридине (70 мл) добавляли по каплям ангидрид трифторметансульфоновой кислоты (39,8 г, 0,141 моль). Смесь перемешивали при комнатной температуре в течение 5 ч, после чего добавляли к ней воду и подвергали экстракции эфиром. Экстракты промывали рассолом, высушивали (MgSO4) и концентрировали, получая в результате 27,3 г (88%) продукта в виде коричневой маслянистой жидкости.
1H ЯМР (DMCO-d6): δ 2,50 (с, 3Н), 7,45 (д, J = 9,2 Гц, 1Н), 7,90 (дв. д. J = 9,2 Гц, 2,3 Гц, 1Н), 8,60 (д, J = 2,3 Гц, 1Н),
Стадия 2
N-оксид (6-метилпиридин-3-ил)-трифторметансульфоната
К перемешиваемому раствору (6-метилпиридин-3-ил)трифторметансульфоната (27,3 г, 0,113 моль) в CH2Cl2 (140 мл) добавляли порциями м-CPBA (21,5 г, 0,124 моль). Через 16 ч выпадающий осадок отфильтровывали и фильтрат концентрировали. В результате очистки с помощью флеш-хроматографии (2% MeOH/CH2Cl2) получали 25,0 г (86%) продукта в виде бесцветных кристаллов. Температура плавления 47-48oC.
1H ЯМР (DMCO-d6): δ 2,36 (с, 3Н), 7,56 (дв. д, J = 8,9 Гц, 2,3 Гц, 1Н), 7,69 (д, J = 8,9 Гц, 1Н), 8,84 (д, J = 2,3 Гц, 1Н).
Стадия 3
(6-Оксиметилпиридин-3-ил)-трифторметансульфонат
К перемешиваемому охлажденному (0oC) N-оксиду (6-метилпиридин-3-ил)-трифторметансульфонату (25,0 г, 0,097 моль) добавляли по каплям ангидрид трифторуксусной кислоты (69,0 мл, 0,487 моль). Смесь перемешивали при комнатной температуре в течение 30 мин, после чего кипятили в течение часа с обратным холодильником. После этого ее охлаждали до комнатной температуры и добавляли к ней 10%-ный водный раствор NaHCO3 (400 мл), подвергали экстракции CH2Cl2, экстракты промывали рассолом, высушивали (MgSO4) и концентрировали. После очистки остатка с помощью флеш-хроматографии (2% MeOH/CH2Cl2) получали 10,0 г (40%) продукта в виде бесцветной маслянистой жидкости.
1H ЯМР (DMCO-d6): δ 4,60 (с, 1Н), 5,60 (с, 1Н), 7,64 (д, J = 8,8 Гц, 1Н), 8,03 (дв. д, J = 8,8 Гц, 1Н), 8,67 (д, J = 2,8 Гц, 1Н).
Стадия 4
2-(2-третбутил-1Н-тетразол-5-ил)фенил-три-н-бутилстаннат
К охлажденному (-78oC) раствору 1-третбутил-5-(2-бромфенил) 1Н-тетразола (12,3 г, 0,043 моль), полученного альтернативным способом в соответствии с примером 3, стадия 3, в ТГФ (80 мл) добавляли 1,6 М н-BuLi в смеси гексанов (32,7 мл, 0,052 моль). Через час добавляли хлорид три-н-бутилолова (17,1 г, 0,052 моль) и перемешивание продолжали при -78oC в течение еще 3 ч. После этого смесь нагревали до комнатной температуры и перемешивали в течение 18 ч, затем добавляли воду и подвергали экстракции эфиром. Объединенные экстракты промывали водой, рассолом высушивали (MgSO4) и концентрировали, получая в результате коричневую маслянистую жидкость. Избыток хлорида три-н-бутилолова и непрореагировавший исходный материал удаляли путем дистилляции в высоком вакууме. В результате получали 9,6 г (45%) продукта в виде коричневой маслянистой жидкости.
1H ЯМР (DMCO-d6): δ 0,78 (т, J = 7,3 Гц, 9Н), 0,93 (т, J = 8,3 Гц, 6Н), 1,21 (м, 6Н), 1,43 (м, 6Н), 7,43 (д, J = 8,9 Гц, 1Н), 7,45 (д, J = 8,5 Гц, 1Н), 7,60 (дв. д, J = 8,9 Гц, 2,8 Гц, 1Н), 8,0 (дв. д, J = 8,5 Гц, 1Н, 2,3 Гц).
Стадия 5
2-третбутил-5-[2-[2-оксиметил)пиридин-5-ил]фенил]-тетразол
Смесь (6-оксиметилпиридин-3-ил)-трифторметансульфоната (5,0 г, 0,019 моль), 2 (2-третбутил-1Н-тетразол-5-ил)фенил-три-н-бутилстанната (9,5 г, 0,019 моль), бис(трифенилфосфин)палладий хлорида (0,70 г, 0,97 ммоль), CuI (0,37 г, 1,9 ммоль) и ДМФ (30 мл) нагревали в течение 18 ч при 100oC. После этого к ней добавляли 20%-ный водный раствор KF и перемешивали при комнатной температуре в течение 30 мин. Выпадающий осадок отфильтровывали, а фильтрат подвергали экстракции эфиром. Экстракты промывали NH4OH, насыщенным водным раствором NH4Cl, водой, рассолом, высушивали (MgSO4) и концентрировали. После очистки остатка с помощью флеш-хроматографии (5% EtOAc/гексан) получали 0,5 г (8%) продукта в виде желтой маслянистой жидкости.
1H ЯМР (DMCO-d6): δ 1,55 (с, 9Н), 4,56 (д, J = 6,0 Гц, 2Н), 5,40 (т, J = 6,0 Гц, 1Н), 7,38 (д, J = 8,8 Гц, 1Н), 7,60 (м, 4Н), 7,90 (дв. д, J = 8,8 Гц, 2,3 Гц, 1Н), 8,20 (д, J = 2,3 Гц, 1Н).
Стадия 6
2-Третбутил-5-[2-[(2-бромметил)пиридин-5-ил]-фенил]-тетразол
Смесь 2-третбутил-5-[2-[(2-оксиметил)пиридин-5-ил] фенил] - тетразола (0,50 г, 1,62 ммоль), трифенилфосфина (0,51 г, 1,94 ммоль) и CBr4 (0,64 г, 1,94 ммоль) в ТГФ (30 мл) перемешивали в течение 16 ч при комнатной температуре. Смесь затем концентрировали и остаток подвергали очистке с помощью флеш-хроматографии (20% EtOAc/гексан). В результате получали 0,40 г (66%) продукта в виде бесцветной маслянистой жидкости.
1H ЯМР (DMCO-d6): δ 1,56 (с, 9Н), 4,70 (с, 2Н), 7,60 (м, 5Н), 7,95 (дв. д, J = 8,8 Гц, 1,6 Гц, 1Н), 8,28 (д, J = 1,6 Гц, 1Н)
Стадия 7
2,4-Диметил-5,6,8-тригидро-8-[[5-[2-(1Н-тетразол-5-ил)фенил] -2-пиридинил]метил]-7Н-пиридо[2,3-d]-пиримидин-7-он
К перемешиваемой суспензии NaH (60%-ная дисперсия в минеральном масле; 41 мг, 1,03 ммоль) в ЛМФ (1 мл) добавляли 2,4-диметил-5,6,8-тригидро-7Н-пиридо[2,3-d] -пиримидин-7-он (170 мг), 0,94 моль), полученный по способу в соответствии со стадией 1 примера 5. Через 30 мин добавляли раствор 2-третбутил-5-[2-[(2-бромфенил)пиридин-5-ил] -фенил]тетразола (350 мг, 0,94 ммоль) в ДМФ (5 мл) и смесь перемешивали в течение 3 ч. Затем ее концентрировали, остаток растворяли в EtOAc и промывали водой. Органическую фазу высушивали (MgSO4), концентрировали и остаток подвергали очистке с помощью флеш-хроматографии (3% MeOH/CH2Cl2), получая в результате желтую пену. Эту пену нагревали в течение 16 ч в 6 н. HCl (2 мл), охлаждали и устанавливали pH равным 4 с помощью 50%-ного раствора NaOH. Смесь затем подвергали экстракции CHCl3, объединенные экстракты высушивали (MgSO4) и концентрировали. После растирания остатка со смесью EtOH и эфира получали 40 мг (10%) продукта в виде белого твердого вещества. Температура плавления 208-209oC
1H ЯМР (DMCO-d6): δ 2,35 (с, 3Н), 2,37 (с, 3Н), 2,77 (т, J = 7,9 Гц, 2Н), 2,90 (т, J = 7,9 Гц, 2Н), 5,30 (с, 2Н), 7,10 (д, J = 8,8 Гц, 1Н), 7,36 (дв. д. J = 8,8 Гц, 1,6 Гц, 1Н), 7,60 (м, 4Н), 8,23 (д, J = 1,6 Гц, 1Н).
ИК (KBr, см-1): 1690
Результаты анализа из расчета на формулу C22H20N8O:
Рассчитано: C 64,07; H 4,89; N 27,17
Найдено: C 63,87; H 5,16; N 27,22
Пример 8
2,4,6-Триметил-5,6,8-тригидро-8-[[2'-(1Н-тетразол-5-ил)[1,1-бифенил] -4-ил]-метил]-7Н-пиридо[2,3-d]пиримидин-7-он
Стадия 1
2,4-Диметил-5,6,8-тригидро-8[[2'-циано[1,1-бифенил] -4-ил] метил-7Н -пиридо[2,3-d]пиримидин-7-он
К перемешиваемой суспензии NaH (60%-ная дисперсия в минеральном масле; 0,115 г, 4,80 ммоль) в ДМФ (5 мл) добавляли 2,4-диметил-5,6,8-тригидро-7Н-пиридо[2,3-d] -пиримидин-7-он (0,85 г, 4,80 ммоль), полученный по способу в соответствии со стадией 1 примера 5. Через 45 мин добавляли 2-[4-(бромметил)фенил] -бензонитрил (1,00 г, 3,7 ммоль) в ДМФ (4 мл) и смесь перемешивали при комнатной температуре в течение 18 ч. После этого к ней добавляли воду и подвергали экстракции CH2Cl2. Экстракты концентрировали и сырой продукт подвергали очистке с помощью флеш-хроматографии (2% MeOH/CH2Cl2). После растирания с эфиром получали 0,80 г (60%) продукта в виде желтого твердого вещества. Температура плавления 133-137oC.
1H ЯМР (DMCO-d6): δ 2,37 (с, 2Н), 2,45 (с, 3Н), 2,76 (т, J = 7,9 Гц, 2Н), 2,88 (т, J = 7,9 Гц, 2Н), 5,27 (с, 2Н), 7,42 (д, J = 8,2 Гц, 2Н), 7,50 (д, J = 8,2 Гц, 2Н), 7,58 (м, 2Н), 7,77 (м, 1Н), 7,93 (J = 7,6 Гц, 1Н).
Стадия 2
2,4,6-Триметил-5,6,8-тригидро-8-[[2'-циано-[1,1-бифенил] -4-ил] метил]-7Н-пиридо[2,3-d]-пиримидин-7-он
К охлажденному (-78oC) раствору 2,4-диметил-5,6,8-тригидро-8-[[2'-циано[1,1-бифенил] -4-ил]метил]-7Н-пиридо-[2,3-d]-пиримидин-7-она (0,40 г, 1,10 ммоль) в ТГФ (2,0 мол) добавляли LDA (2М; 0,55 мл, 1,10 ммоль). Через 45 мин добавляли MeI (0,16 г, 1,15 ммоль), смеси давали нагреться до комнатной температуры и перемешивали ее в течение 18 ч. После этого смесь концентрировали и распределяли между водой и CH2Cl2. Органическую фазу высушивали (MgSO4) и концентрировали, получая в результате оранжевую маслянистую жидкость. После очистки ее с помощью флеш-хроматографии (60% EtOAc/гексан) получали 0,15 г (36%) продута в виде желтого твердого вещества. Температура плавления 153-156oC.
1H ЯМР (DMCO-d6): δ 1,21 (д, J = 6,3 Гц, 3Н), 2,37 (с, 3Н), 2,44 (с, 3Н), 2,60 (м, 1Н), 2,80 (м, 1Н), 3,05 (дв. д, J = 9,6 6,0 Гц, 1Н), 5,21 (д, J = 15,1 Гц, 1Н), 7,39 (д, J = 8,3 Гц, 2Н), 7,49 (д, J = 8,3 Гц, 2Н), 7,58 (м, 2Н), 7,80 (м, 1Н), 7,91 (дв. д, J = 6,9, 0,9 Гц, 1Н).
Стадия 3
2,4,6-Триметил-5,6,8-тригидро-8-[[2'-/(1Н-тетразол-5-ил) [1,1-бифенил] -4-ил]метил]-7Н-пиридо[2,3-d]пиримидин-7-он
Смесь 2,4,6-триметил-5,6,8-тригидро-8-[[2'-циано-[1,1-бифенил]-4-ил]метил] -7Н-пиридо[2,3-d] -пиримидин-7-она (0,57 г, 1,50 ммоль), NaN3 (0,13 г, 1,90 ммоль) и хлорида три-н-бутилолова (0,62 г, 1,90 ммоль) в смеси ксилолов (5 мл) кипятили в течение 48 ч с обратным холодильником. После этого смесь охлаждали и добавляли к ней 1 н. HCl (2 мл), а затем CHCl3 и воду. Органическую фазу высушивали (MgSO4) и концентрировали. После перекристаллизации остатка из EtOAc получали 0,25 г (39%) продукта в виде белого твердого вещества. Температура плавления 161-163oC.
1H ЯМР (DMCO-d6): δ 1,19 (д, J = 6,7 Гц), 2,37 (с, 3Н), 2,43 (с, 3Н), 2,59 (м, 1Н), 2,80 (м, 1Н), 3,02 (дв. д, J = 15,7, 7,0 Гц, 1Н), 5,12 (д, J = 15,0 Гц, 1Н), 5,22 (д, J = 15,0 Гц, 1Н), 7,00 (д, J = 8,0 Гц, 2Н), 7,18 (д, J = 8,0 Гц, 2Н), 7,57 (м, 2Н), 7,63 (м, 2Н).
Пример 9
2,4-Диметил-8-[[2'-(1Н-тетразол-5-ил)[1,1-бифенил]-4-ил]метил]-7Н-пиридо[2,3-d]пиримидин-7-он
Стадия 1
2,4-Диметил-8-[[2'-циано[1,1-бифенил]-4-ил]метил]-7Н-пиридо-[2,3-d]пиримидин-7-он
Таким же образом, как это описано в примере 8, стадия 1, к перемешиваемой суспензии NaH (60%-ная дисперсия в минеральном масле) в ДМФ добавляли 2,4-диметил-7Н-пиридо[2,3-d]-пиримидин-7-он, полученный по методу, описанному T.Sakarnoto и др. в Chem. Pharm. Bull., 1982, 30, 2410. Через 45 мин добавляли 2-[4-(бромметил)фенил] -бензонитрил в ДМФ и перемешивали смесь при комнатной температуре в течение 18 ч. После этого добавляли к ней воду и подвергали экстракции CH2Cl2. Экстракты концентрировали и сырой продукт подвергали очистке с помощью флеш-хроматографии (2% MeOH/CH2Cl2). После растирания с эфиром получали целевой продукт.
Стадия 2
2,4-Диметил-8-[[2'-(1Н-тетразол-5-ил)[1,1-бифенил] -4-ил]метил]-7Н-пиридо[2,3-d]пиримидин-7-он
Таким же образом, как это описано в примере 8, стадия 3, кипятили смесь 2,4-диметил-8-[[2'-циано-[1,1-бифенил] -4-ил] метил]7Н-пиридо[2,3-d]-пиримидин-7-он, NaN3 и хлорид три-н-бутилолова в смеси ксилолов в течение 48 ч с обратным холодильником. После этого смесь охлаждали и добавляли к ней 1 н. HCl (2 мл), а затем CHCl3 и воду. Органическую фазу высушивали (MgSO4) и концентрировали. После перекристаллизации остатка из EtOAc получали целевой продукт.
Пример 10
2-Этил-4-метил-8-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил] - 5,8-дигидро-6Н-пиридо[2,3-d]пиримидин-7-он
Указанное в заголовке соединение получали согласно способу, описанному в примере 3, с использованием гидрохлорида пропионамидина вместо гидрохлорида ацетамидина в стадии 1 с получением белого твердого вещества, т.пл. 231-232oC.
Пример 11
4-Этил-2-метил-8-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил] -5,8-дигидро-6Н-пиридо[2,3-d]-пиримидин-7-он
Указанное в заголовке соединение получали согласно способу, описанному в примере 3, с использованием диэтилпропионилглутарата вместо диэтилацетилглутарата в стадии 1, с получением белого твердого вещества, т.пл. 187-189oC.
Пример 12
4-Метил-2-пропил-8-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил] -5,8-дигидро-6Н-пиридо[2,3-d]пиримидин-7-он
Указанное в заголовке соединение получали согласно способу, описанному в примере 3, с использованием гидрохлорида бутириламидина вместо гидрохлорида ацетамидина в стадии 1, с получением белого твердого вещества, т.пл. 120-122oC.
Пример 13
2-Метил-4-пропил-8-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил] -5,8-дигидро-6Н-пиридо[2,3-d]пиримидин-7-он
Указанное в заголовке соединение получали согласно способу, описанному в примере 3, с использованием диэтилбутирилглутарата вместо диэтилацетилглутарата в стадии 1, с получением белого твердого вещества, т.пл. 165-167oC.
1H-ЯМР (DMCO-d6) α 0,93 (т, J = 7,3 Гц, 3Н), 1,62 (м, 2Н), 2,44 (с, 3Н), 2,63 (м, 2Н), 2,72 (м, 2Н), 5,18 (с, 2Н), 7,00 (д, J = 8,3 Гц, 2Н), 7,20 (д, J = 8,3 Гц, 2Н), 7,53 (м, 2Н), 7,64 (м, 2Н); MS (DCI) m/z 440 [M+H]+.
Анал. вычислено для C25H25N7O: C, 68,32; H, 5,73; N, 22,31
Найдено: C, 68,35; H, 5,72; N, 22,49
Пример 14
2-Изопропил-4-метил-8-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил]-5,8-дигидро-6Н-пиридо[2,3-d]пиримидин-7-он
Указанное в заголовке соединение получали согласно способу, описанному в примере 3, с использованием гидрохлорида изобутириламидина вместо гидрохлорида ацетамидина в стадии 1, с получением белого твердого вещества, т.пл. 193-195oC.
Пример 15
4-Изопропил-2-метил-8-[2'-(1Н-тетразол-5-ил)-бифенил-4-ил-метил] -5,8-дигидро-6Н-пиридо[2,3-d]пиримидин-7-он
Указанное в заголовке соединение получали согласно способу, описанному в примере 3, с использованием диэтилизобутирилглутарата вместо диэтилацетилглутарата в стадии 1, с получением твердого вещества нечистого белого цвета, т.пл. 190-191oC.
1H-ЯМР (DMCO-d6) α 1,16 (д, J = 6,6 Гц, 6Н), 2,45 (с, 3Н), 2,72 (м, 2Н), 2,93 (м, 2Н), 3,22 (м, 1Н), 5,18 (с, 3Н), 7,00 (д, J = 8,3 Гц, 2Н), 7,20 (д, J = 8,3 Гц, 2Н), 7,54 (м, 2Н), 7,65 (м, 2Н); MS (PBCI) m/z 440 [M+H]+.
Анал. вычислено для C25H25N7O: C, 68,32; H, 5,73; N, 22,31
Найдено: C, 68,38; H, 5,97; N, 21,64
Пример 16
2,4,6,6-Тетраметил-5,6,8-тригидро-8-[[2'-(1Н-тетразол-5-ил) [1,1-бифенил]-4-ил]метил]-7Н-пиридо[2,3-d]пиримидин-7-он
Стадия 1) 5,8-Дигидро-2,4,6-триметил-8[(4-бромфенил)метил]пиридо [2,3-d] пиримидин-7(6Н)он
К охлажденному раствору (-78oC) 2,4-диметил-5,6,8-тригидро-8-[(4-бромфенил)метил] -7Н-пиридо [2,3-d]пиримидин-7-она (2,50 г, 7,20 ммоль), полученного, как описано в альтернативной стадии 1 процесса примера 3, в ТГФ (25 мл) добавляли 2,0 М LDA (3,70 мл, 7,40 ммоль). Спустя 45 мин добавляли CH3I (1,03 г, 7,20 ммоль) и смеси давали нагреться до комнатной температуры в течение более 16 ч. Смесь концентрировали, и неочищенный продукт очищали флэш-хроматографией (60% EtOAc/гексан) с получением 1,40 г (54%) продукта в виде желтого твердого вещества, т.пл. 108-110oC.
1H-ЯМР (DMCO-d6) α 1,14 (д, J = 6,7 Гц, 3Н), 2,36 (с, 3Н), 2,43 (с, 3Н), 2,53 (м, 1Н), 2,78 (м, 1Н), 3,05 (дд, J = 15,7, 6,2 Гц, 1Н), 5,11 (м, 2Н), 7,22 (д, J = 8,3 Гц, 2Н), 7,45 (д, J = 8,3 Гц, 2Н).
Стадия 2) 5,8-Дигидро-2,4,6,6-тетраметил-8-[(4-бромфенил)метил] пиридо[2,3-d]пиримидин-7(6Н)-он
К охлажденному (-78oC) раствору 5,8-дигидро-2,4,6-триметил-8-[(4-бромфенил)метил] пиридо [2,3-d]пиримидин-7(6Н)-она (1,36 г, 3,80 ммоль) в ТГФ (8 мл) добавляли 2,0 М LDA (2,00 мл, 4,00 ммоль). Спустя 45 мин добавляли CH3I (0,54 г, 3,80 ммоль), смеси давали нагреться до комнатной температуры, перемешивание продолжали в течение 64 ч. Смесь концентрировали, и неочищенный продукт очищали флэш-хроматографией (60% EtOAc/гексан) с получением 1,10 г (77%) продукта в виде желтого твердого вещества; т.пл. 134-136oC;
1H-ЯМР (DMCO-d6) α 1,11 (с, 6Н), 2,37 (с, 3Н), 2,44 (с, 3Н), 2,79 (с, 2Н), 5,13 (с, 2Н), 7,20 (д, J = 8,4 Гц, 2Н), 7,47 (д, J = 8,4 Гц, 2Н).
Анал. вычислено для C18H20BrN3O: C, 57,76; H, 5,39; N, 11,23
Найдено: C, 57,84; H, 5,34; N, 11,05
Стадия 3) 2,4,6,6-Тетраметил-5,6,8-[[2'-(1Н-тетразол-5-ил)[1,1-бифенил] -4-ил]метил]-7Н-пиридо[2,3-d]пиримидин-7-он
5,8-Дигидро-2,4,6,6-тетраметил-8-[(4-бромфенил)метил] пиридо[2,3-d]пиримидин-7(6Н)-он (1,43 г, 3,82 ммоль) обрабатывали согласно процедурам альтернативных стадий процесса 5 и 6 примера 3 с получением 300 мг (19%) продукта в виде белого твердого вещества, т.пл. 208-210oC.
Анал. вычислено для C25H25N7O: C, 68,34; H, 5,69; N, 21,32
Найдено: C, 68,27; H, 5,70; N, 21,98
Пример 17
2,4-Диметил-5,6,8-тригидро-8-[3-фтор-[2'-(1Н- тетразол-5-ил)[1,1-бифенил]-4-ил]метил]-7Н-пиридо[2,3-d]пиримидин-7-он
Стадия 1) 2-Циано-4'-(бромметил)-3'-фторбифенил
Это соединение получали согласно способу, описанному в D.J.Carini, et al. J. Org. Chem. 1991, 34, 2525-2547, для 2-циано-4'-метилбифенила.
Стадия 2) 2,4-Диметил-5,6,8-тригидро-8-[2'-циано-3-фтор[1,1-бифенил]-4-ил]метил]-7Н-пиридо[2,3-d]пиримидин-7-он
К охлажденному (5oC) раствору 2,4-диметил-5,6,8-тригидро-7Н-пиридо[2,3-d] пиримидин-7 она (484 мг, 2,73 ммоль), полученного, как описано в стадии 1 примера 5, в ДМФ (3 мл) добавляли NaH (60% дисперсию в минеральном масле; 120 мг, 3,00 ммоль). Охлаждающую баню удаляли спустя 5 мин, перемешивание продолжали в течение 30 мин. Смесь повторно охлаждали, добавляли раствор 2-циано-4'-(бромметил)-3'-фторбифенила (890 мг, 2,73 ммоль) в ДМФ (93 мл), перемешивание продолжали в течение 1 ч 15 мин. Добавляли воду и смесь экстрагировали в EtOAc. Объединенные экстракты промывали рассолом, сушили и концентрировали. Очистка флэш-хроматографией (2% MeOH/CH2Cl2) дала 600 мг (57%) твердого вещества нечистого белого цвета. Перекристаллизация из смеси ацетон/эфир дала продукт в виде белого твердого вещества, т.пл. 153-155oC.
1H-ЯМР (DMCO-d6) α 2,37 (с, 3Н), 2,40 (с, 3Н), 2,77 (м, 2Н), 2,93 (т, J = 7,0 Гц, 2Н), 5,29 (с, 2Н), 7,25 (м, 2Н), 7,45 (дд, J = 11,1 1,3 Гц, 1Н), 7,59 (м, 2Н), 7,77 (м, 1Н), 7,39 (дд, J = 6,9, 0,9 Гц, 1Н).
Анал. вычислено для C23H19FN4O: C, 71,49; H, 4,96; N, 14,50
Найдено: C, 71,36; H, 5,09; N, 14,40.
Стадия 3) 2,4-Диметил-5,6,8-тригидро-8-[3-фтор-[2'-(1Н-тетразол-5-ил)[1,1-бифенил]-4-ил]метил]-7Н-пиридо [2,3-d]пиримидин-7-он
Смесь 2,4-диметил-5,6,8-тригидро-8-[2'-циано-3-фтор[1,1-бифенил]-4-ил-] метил] -7Н-пиридо[2,3-d] пиримидин-7-она (550 мг, 1,42 ммоль), nBu3SnCl (602 мг, 1,85 ммоль), NaN3 (120 мг, 1,85 ммоль) и ксилена (5 мл) нагревали с обратным холодильником в течение 48 ч, охлаждали и разбавляли эфиром. Добавляли 1 н. HCl и смесь перемешивали в течение 10 мин. Органический слой удаляли и водную фазу доводили до pH 4 2,5 н. NaOH. Осадок собирали фильтрованием и растирали с эфиром. Продукт перекристаллизовывали из фильтрата (98 мг, 16%), т.пл. 223-225oC.
1H-ЯМР (DMCO-d6) α 2,37 (с, 3Н), 2,39 (с, 3Н), 2,75 (м, 2Н), 2,90 (т, J = 7,3 Гц, 2Н), 5,21 (с, 2Н), 6,73 (дд, J = 7,9, 1,7 Гц, 1Н), 6,97 (м, 3Н), 7,56 (м, 2Н), 7,67 (м, 2Н).
Анал. вычислено для C23H20FN7O: C, 64,33; H, 4,69; N, 22,83
Найдено: C, 64,47; H, 4,88; N, 22,37.
Пример 18
Калиевая соль 4'-(2,4-диметил-7-оксо-6,7-дигидро-5Н-пиридо [2,3-d]пиримидина-8-илметил)бифенил-2-карбоновой кислоты
Стадия 1) Метил 4'-(2,4-диметил-7-оксо-6,7-дигидро-5Н-пиридо[2,3-d]пиримидин-8-илметил)бифенил-2-карбоксилат
К раствору 2,4-диметил-5,6,8-тригидро-7Н-пиридо [2,3-d]пиримидин-7-она (500 мг, 2,82 ммоль), полученного, как описано в стадии 1 примера 5, в ДМФ (5 мл) добавляли NaH (60% дисперсию в минеральном масле; 124 мг, 3,10 ммоль) при комнатной температуре. Спустя 30 мин добавляли метил 4'-(бромметил)бифенил-2-карбоксилат (80% чистоты; 1,08 г, 2,82 ммоль), полученный согласно D.J.Carini, et al., J. Org. Chem. 1991, 34, 2525-2547. Смесь перемешивали в течение 2,5 дн и затем разбавляли EtOAc, промывали рассолом, и органическую фазу сушили и концентрировали. Очистка флэш-хроматографией (1% MeOH/CH2Cl2) дала 800 мг (71%) продукта в виде коричневой пены.
1H-ЯМР (DMCO-d6) α 2,42 (с, 3Н), 2,59 (с, 3Н), 2,75 (м, 2Н), 2,85 (м, 2Н), 3,60 (с, 3Н), 5,35 (с, 2Н), 7,20 (д, J = 8,2 Гц, 2Н), 7,32 (м, 1Н), 7,37 (дд, J = 7,6, 1,3 Гц, 1Н), 7,44 (д, J = 8,2 Гц, 2Н), 7,46 (м, 1Н), 7,77 (дд, J = 7,6 1,0 Гц, 1Н).
Стадия 2) 4'-(2,4-Диметил-7-оксо-6,7-дигидро-5Н-пиридо -[2,3-d]пиримидин-8-илметил)бифенил-2-карбоновая кислота
Раствор метил 4'-(2,4-диметил-7-оксо-6,7-дигидро-5Н-пиридо[2,3-d] пиридимин-8-илметил)бифенил-2-карбоксилата (800 мг, 1,99 ммоль) и иодтриметилсилана (797 мг, 3,98 ммоль) в CH2Cl2 нагревали с обратным холодильником в течение 22 ч. Дополнительно добавляли иодтриметилсилан (797 мг, 3,98 ммоль) и нагревание продолжали в течение 20 ч. Добавляли насыщенный водный раствор NaHCO3 и EtOAc и слои разделяли. Органическую фазу экстрагировали насыщенным водным раствором NaHCO3, и объединенные водные слои обрабатывали Na2S2O3, экстрагировали CH2Cl2 (выбрасывали) и подкисляли до pH 4 концентрированной HCl. Смесь экстрагировали CH2Cl2 и объединенные экстракты сушили и концентрировали с получением 400 мг твердого вещества нечистого белого цвета. Перекристаллизация из EtOH дала 316 мг (41%) продукта в виде белого твердого вещества, т.пл. 215-216oC.
Анал. вычислено для C23H21N3O3: C, 71,30; H, 5,46; N, 10,85
Найдено: C, 71,06; H, 5,44; N, 10,60
Стадия 3) Калиевая соль 4'-(2,4-диметил-7-оксо-6,7-дигидро-5Н-пиридо[2,3-d]пиримидин-8-илметил)бифенил-2-карбоновой кислоты
4'-(2,4-Диметил-7-оксо-6,7-дигидро-5Н-пиридо[2,3-d] пиримидин- 8-илметил)бифенил-2-карбоновую кислоту превращали в калиевую соль следующим образом: смесь кислоты (300 мг), 0,774 ммоль) и 1 н. KOH (736 мкл) перемешивали в течение 10 мин. Смесь фильтровали и фильтрат концентрировали с получением 310 мг (99%) калиевой соли в виде твердого вещества нечистого белого цвета, т. пл. > 300oC.
1H-ЯМР (DMCO-d6) α 2,36 (с, 3Н), 2,48 (с, 3Н), 2,73 (м, 2Н), 2,89 (м, 2Н), 5,22 (с, 2Н), 7,12 (м, 3Н), 7,20 (м, 3Н), 7,39 (дд, J = 6,6, 1,7 Гц, 2Н).
ИК (KBr, см-1) 1680
Анал. вычислено для C23H20KN3O3 • 0,6 H2O: C, 63,31; H, 4,90; N, 9,63
Найдено: C, 63,07; H, 4,87; N, 9,72
Пример 19
Капсулы (доза 1 мг),мг:
Активное соединение 0,5% - 1
Лактоза, сухой аэрозоль 74,0% - 148
Крахмал, предварительно желатинированный 25,0% - 50
Стеарат магния 0,5% - 1 - 200
Перечисленные ингредиенты тщательно перемешивают и заполняют твердые желатиновые капсулы в указанной выше дозировке.
Пример 20
Капсулы (доза 10 мг),мг:
Активное соединение 5,0% - 10
Лактоза, сухой аэрозоль 69,5% - 139
Крахмал, предварительно желатинированный 25,0% - 50
Стеарат магния 0,5% - 1 - 200
Твердые желатиновые капсулы с дозировкой 10 мг готовят аналогично примеру 19.
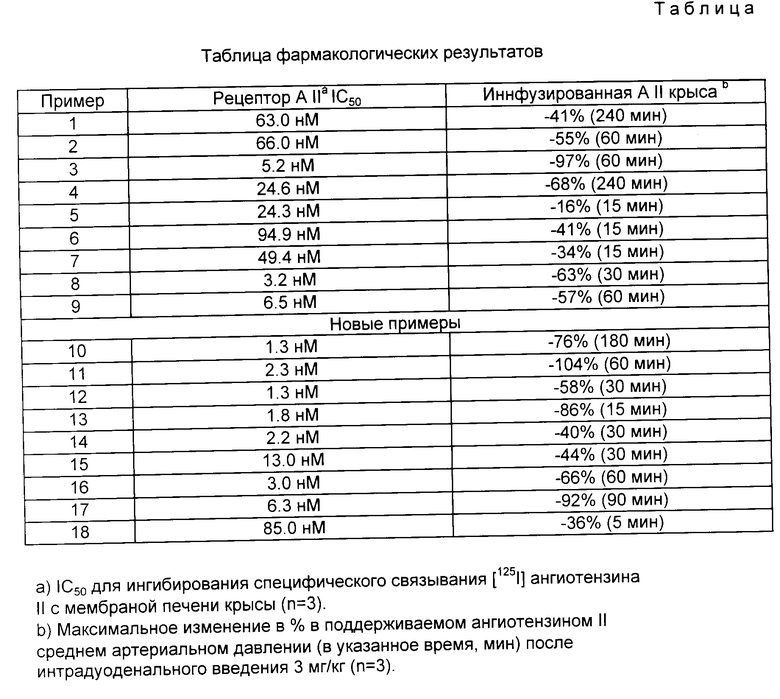
Использование: в медицине в качестве антагонистов ангиотезина II. Сущность изобретения: продукты: замещенные пиримидины ф-лы (I), где R1, R2, R3 и R4 - H, C1-C6 - алкил, C1-C6 - перфторалкил, R5 - H или в случае, если n = 1, R5 вместе с R3 образует двойную связь, n = 0-1, p = 0-2, m = 0-3, Ar1 и Ar2 - замещенные фенилен, пиридин. Реагент 1: пиримидин ф-лы (II), где R1, R2, R3, R4, R5, n и p имеют вышеуказанные значения, Z1 - отщепляемая группа, R7 - низший алкил. Реагент 2: амин ф-лы H2N - (CH2)m - Ar1 - Ar2, где m, Ar1 и Ar2 имеют вышеуказанные значения. Условия реакции: в присутствии неорганического или органического основания.
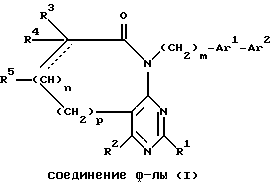
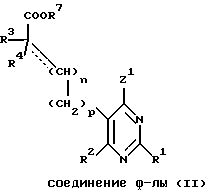 6 с. и 7 з.п. ф-лы, 1 табл.
6 с. и 7 з.п. ф-лы, 1 табл.
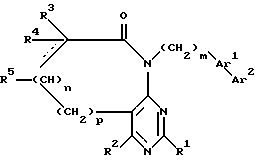
в которой R1 - R4 независимо друг от друга - H, низший алкил с 1 - 6 атомами углерода или перфторалкил с 1 - 6 атомами углерода;
R5 - H или в случае, если n = 1, R5 вместе с R3 образуют двойную связь;
n = 0 - 1;
p = 0 - 2;
m = 0 - 3;
Ar1 -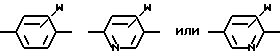
где W - H, низший алкил с 1 - 6 атомами углерода, галоген, окси- или низшая алкоксигруппа с 1 - 6 атомами углерода;
Ar2 -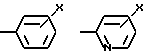
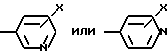
где X - CO2H, CN или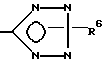
где R6 - H, трет-бутил, три-н-бутилстаннил или трифенилметил,
или их фармацевтически приемлемые соли или N-оксиды.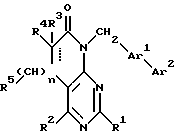
в которой R1 - R4 независимо друг от друга - H, низший алкил с 1 - 6 атомами углерода или перфторалкил с 1 - 6 атомами углерода;
R5 - H или в том случае если n = 1, R5 вместе с R3 образуют двойную связь;
n = 0 - 1;
Ar1 -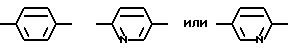
Ar2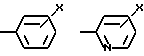
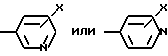
где X - CO2H, CN или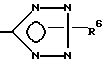
где R6 - H, трет-бутил, три-н-бутилстаннил или трифенилметил,
или их фармацевтически приемлемые соли.
4. Соединения по п.3 общей формулы III
в которой R1 - R4 независимо друг от друга - H, метил, трифторметил;
R5 - H или в случае, если n = 1, R5 вместе с R3 образуют двойную связь;
n = 0 - 1;
Ar1 -
или их фармацевтически приемлемые соли.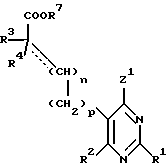
в которой R1 - R5, n и p имеют значения по п.1;
Z1 - отщепляемая группа;
R7 - низший алкил,
с амином общей формулы
H2N-(CH2)m-Ar1-Ar2,
в которой m, Ar1 и Ar2 имеют значения по п.1,
и необязательно удаляют защитную группу из соединения формулы I, у которого X -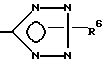
где R6 - трет-бутил, три-н-бутилстаннил или трифенилметил,
с образованием в результате соединения, у которого R6 - водород, или превращают соединение формулы I, у которого X -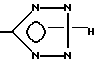
или получают соль соединения формулы I с неорганической или органической кислотой или основанием, переводят соединение формулы I или его соль в N-оксид с помощью пероксидного агента.
в которой R1 - R5, n, m, p и Ar1 имеют значения по п.1;
Y - парабром или параиод,
с арилбороновой кислотой или арилстаннаном в присутствии палладиевого катализатора, и, необязательно, удаляют защитную группу из соединения формулы I, у которого X -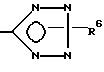
где R6 - трет-бутил, три-н-бутилстаннил или трифенилметил,
с образованием в результате соединения, у которого R6 - водород, или превращают соединение формулы I, у которого X - CN, реакцией с азидом в соединение формулы I, у которого X -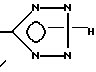
или получают соль соединения формулы I с неорганической или органической кислотой или основанием, или переводят соединение формулы I или его соль в N-оксид с помощью пероксидного агента.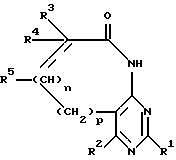
в которой R1 - R5, n и p имеют значения по п.1,
с биарильным соединением общей формулы
Z1-(CH2)m-Ar1-Ar2,
в которой Ar1, m и Ar2 имеют указанные значения;
Z1 - отщепляющаяся группа, и,
необязательно, удаляют защитную группу из соединения формулы I, у которого X -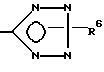
где R6 - трет-бутил, три-н-бутилстаннил или трифенилметил,
с образованием в результате соединения, у которого R6 - водород, или превращают соединение формулы I, у которого X - CN, реакцией с азидом в соединение формулы I, у которого X -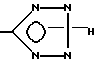
или получают соль соединения формулы I с неорганической или органической кислотой или основанием, или переводят соединение формулы I или его соль в N-оксид с помощью пероксидного агента.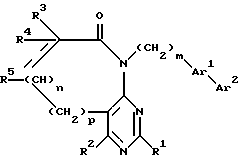
в которой R1 - R4 независимо друг от друга - H, низший алкил с 1 - 6 атомами углерода или перфторалкил с 1 - 6, атомами углерода;
R5 - H или в том случае, если n = 1, R5 вместе с R3 образуют двойную связь;
n = 0 - 1
p = 0 - 2
m = 0 - 3
Ar1 -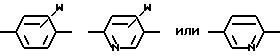
где W - H, низший алкил с 1 - 6 атомами углерода, галоген, окси- или низшая алкоксигруппа с 1 - 6 атомами углерода;
Ar2 -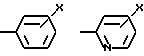
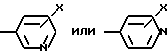
где X - CO2H, CN или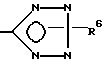
где R6 - H или трет-бутил,
и их фармацевтически приемлемых солей, отличающийся тем, что осуществляют взаимодействие соединения общей формулы I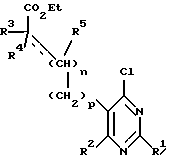
в которой R1 - R5, n и p имеют указанные значения,
с амином формулы 2
H2N-(CH2)m-Ar1Y,
где m и Ar1 имеют указанные значения
Y - парабром или параиод,
с образованием в результате соединения формулы 3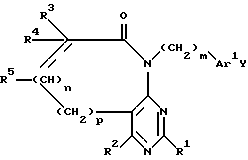
в которой R1 - R5, n, m, p, Ar1 и Y имеют указанные значения
и взаимодействие затем соединения формулы 3 с арилбороновой кислотой формулы 4
Ar2-B(OH)2,
в которой Ar2 имеют указанное значение,
в присутствии палладиевого катализатора и при желании (в том случае, если R6 - трет-бутил) удаляют эту защитную группу путем кислотного гидролиза с образованием в результате соединения общей формулы I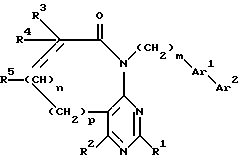
в которой R1 - R5, n, m, p, Ar1 и Ar2 имеют указанные значения.
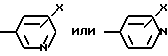
где X -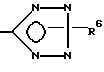
где R6 - трет-бутил,
получают путем обработки соединения формулы 5
Ar2 - Br,
в которой Ar2 имеет указанное значение, Mg и триалкилборатом, B(OR7)3, где R7 - низший алкил с 1 - 6 атомами углерода,
в среде апротонного растворителя с получением в результате (после кислотного или щелочного гидролиза) арилбороновой кислоты формулы 4 в которой Ar2 имеет указанное значение.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| EP, заявка, 0426180 A2, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| EP, заявка, 040103 0 A2, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| EP, заявка, 0419048 A2, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| EP, заявка, 0430485 A2, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

