Изобретение относится к C-22 циклическим стабилизированным рапамицин производным или их фармацевтически приемлемым солям, которые полезны для индуцирования иммунодепрессии и для лечения отторжения трансплантанта, гомологичной болезни, аутоаллергических болезней, воспалительных заболеваний, твердых опухолей, лейкоза/лимфом дифференцированных Т-клеток и гиперпролиферативных сосудистых (васкулярных) нарушений.
Рапамицин является макроциклическим триеновым антибиотиком, производимым Streptomyces hygroscopicus, который, как было найдено, обладает противогрибковой активностью, в частности против Candida albicans, как in vitro, так и in vivo [C.Vezina et al., J.Antibiot., 28, 721 (1975); S.N. Sehgal et al., J. Antibiot. , 28, 727 (1975), H. A. Backer et al., J.Antibiot., 31, 539 (1978), патент США 3.929,992 и патент США 3.993.749].
Показано, что рапамицин сам по себе (патент США 4.885.171) или в комбинации с пицибанилом (патент США, 4.401.653) обладает противоопухолевой активностью. R. Martel и др. [Can J. Physiol. Pharmacol 55, 48 (1977)] открыли, что рапамицин эффективен в экспериментальной модели аллергического энцефаломиелита, в модели множественного склероза, в модели адъювантного артрита, модели ревматоидного артрита и эффективно ингибирует образование IgE-подобных антител.
Иммуносупрессивное действие рапамицина раскрыто в FASEB 3.3411 (1989). Было также показано, что циклоспорин A и FK-506, другие макроциклические молекулы, эффективные в качестве иммуносупрессивных средств, кроме того, полезны в предотвращении отторжения трансплантата [FASEB 3, 3411 (1989), FASEB 3, 5256 (1989), R.J. Calne et al., Zancet 1183 (1978) и патент США 5,100,899].
Показано также, что рапамицин полезен в профилактике или лечении системной красной волчанки [патент США 5.078.999], воспаления легкого [патент США 5,080,899] , инсулинзависимого сахарного диабета [Fifth Int. Cont. Inflamm. Res. , Assoc. 121 (Abctract), (1990)], пролиферации клеток гладкой мышцы и интимального утолщения после васкулярной травмы [Morris, R.J Heart Zung Transplant. П (часть 2): 197 (1992)].
Показано, что моно- и диацилированные производные рапамицина (этерифицированные в положениях 28 и 43), полезны в качестве противогрибковых средств (патент США 4.316.885) и используются для изготовления водорастворимых пролекарств рапамицина (патент США 4.650.803).
Недавно была изменена условная нумерация для рапамицина, поэтому согласно номенклатуре Chemical Abstract описанные выше сложноэфирные группы должны соответствовать положениям 31- и 42-. Патент США 5.118.678 раскрывает карбаматы рапамицина, которые полезны в качестве иммуносупрессивных, противовоспалительных, противогрибковых и противоопухолевых средств. Патент США 5.100.883 раскрывает фторированные сложные эфиры рапамицина. Патент США 5.118.677 раскрывает амидоэфиры (сложные) рапамицина. Патент США 5.130.307 раскрывает сложные аминоэфиры рапамицина. Патент США 5.117.203 раскрывает сульфонаты и сульфаматы рапамицина. Патент США 5.194.447 описывает сульфонилкарбаматы рапамицина.
Данное изобретение касается C-22 замещенных рапамицин производных, которые проявляют иммуносупрессивную и/или противогрибковую и/или противоопухолевую активность in vivo и/или ингибируют тимоцитпролиферацию in vitro. Кроме того, эти соединения полезны для лечения Candida albicans инфекций, воспалительных заболеваний и аутоаллергической болезни отторжения трансплантата, включая волчанку, ревматоидный артрит, сахарный диабет, множественный склероз и т.д.
Более конкретно, данное изобретение касается производных рапамицина, стабилизированных замещением в C-22 положении, и всех их фармацевтически приемлемых солей. Такие производные имеют структуру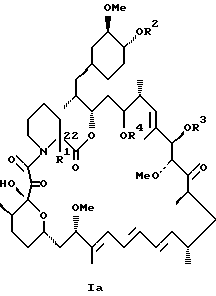
или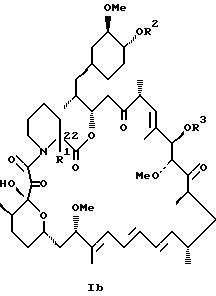
R1 обозначает C1-C6 алкил, C2-C7 ацил, C1-C6 алкилтио, C7-C16 арилалкил, C6-C10 арилтио, C7-C16 арилалкилтио, циано, C1-C6 фторалкил или C6-C10 арил необязательно замещенный одним или более галогенами, C1-C6 алкокси, гидрокси, C2-C7 алканоил или циано, и
R2, R3 и R4 каждый независимо представляют:
а) водород, или
b) SiEt3, или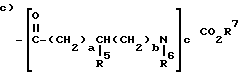
в которой R5 обозначает водород, алкил из 1-6 углеродных атомов, аминоалкил из 1-4 углеродных атомов, аралкил из 7-10 углеродных атомов, -(CH2)dCO2R8, -(CH2)eNR9CO2R10, карбамилалкил из 2-3 углеродных атомов, гидроксиалкил из 1-4 углеродных атомов, гуанилалкил из 2-4 углеродных атомов, меркаптоалкил из 1-4 углеродных атомов, алкилтиоалкил из 2-6 углеродных атомов, индолилметил, гидроксифенилметил, имидазоилметил или фенил необязательно моно-, ди- или три- замещенный заместителем выбранным из: алкила из 1-6 углеродных атомов, алкокси из 1-6 углеродных атомов, гидрокси, циано, галоида, нитро, карбалкокси из 2-7 углеродных атомов, трифторметила амино или карбоновой кислоты.
R6 и R9 каждый независимо представляют водород, алкил из 1-6 углеродных атомов или аралкил из 7-10 углеродных атомов.
R7, R8 и R10 каждый независимо представляют алкил из 1-6 углеродных атомов, аралкил из 7-10 углеродных атомов, флуоренил- метил или фенил необязательно моно-, ди- или три- замещенный заместителем, выбранным из: алкила из 1-6 углеродных атомов, алкокси из 1-6 углеродных атомов, гидрокси, циано, галоида, нитро, карбалкокси из 2-7 углеродных атомов, трифторметила амино или карбоновой кислоты,
a равно 0-4,
b равно 0-4,
c равно 1-2,
d равно 0-4,
e равно 0-4,
где R5, R6, a и b - независимы в каждой из субъединиц:
когда c = 2, или
d) -CONH(CR11R12)f - X
где R11 и R12 каждый независимо представляют водород, алкил из 1-6 углеродных атомов, аралкил из 7-10 углеродных атомов, циклоалкил из 3-8 углеродных атомов, галоген или трифторметил,
X представляет водород, низший алкил из 1-6 углеродных атомов, циклоалкил из 3-8 углеродных атомов, трифторметил, нитро, алкокси из 1-6 углеродных атомов, карбоалкокси из 2-7 углеродных атомов, аралкил из 7-10 углеродных атомов, галоид, диалкиламино из 1-6 углеродных атомов на алкил группу, тиоалкил из 1-6 углеродных атомов или Y,
Y обозначает фенил группу, которая может быть необязательно моно-, ди- или три- замещенной группой, выбранной из: алкила из 1-6 углеродных атомов, аралкила из 7-10 углеродных атомов, алкокси из 1-6 углеродных атомов, циано, галоида нитро, карбалкокси из 2-7 углеродных атомов, трифторметила, диалкиламино из 1-6 углеродных атомов в алкильной группе, алкилтио из 1-6 углеродных атомов, SO3H, PO3H или CO2H,
f = 0-5,
при условии, что R2, R3 и R4 не все обозначают водород и когда f = 0, X представляет низший алкил из 1-6 углеродных атомов, циклоалкил из 3-8 углеродных атомов, аралкил из 7-10 углеродных атомов или Y,
или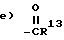
где R13 обозначает моно-, ди-, поли- или перфторированную алкил группу из 1-10 углеродных атомов, при условии, что R2, R3 и R4 не все являются водородом, алкил из 1-10 углеродных атомов или арил, в котором арил группа необязательно может быть моно-, ди- или три- замещенной группой, выбранной из: алкила из 1-6 углеродных атомов, арилалкила из 7-10 углеродных атомов, алкокси из 1-6 углеродных атомов, циано, галоида, нитро, карбалкокси из 2-7 углеродных атомов, трифторметил, амино, диалкиламино из 1-6 углеродных атомов в алкильной группе, алкилтио из 1-6 углеродных атомов, - SO3H, PO2H и CO2H, или
где X обозначает -(CH2)g- или -Ar.
R14 и R15 каждый независимо обозначают водород, алкил из 1-12 углеродных атомов, -(CH2)h-Ar, (CH2)i -NR16R17 или -(CH2)i -N+R16R17R18 Y-,
R16 и R17 каждый независимо обозначают водород, алкил из 1-12 углеродных атомов или -(CH2)h -Ar,
Ar является необязательно моно- или ди- замещенным группой, выбранной из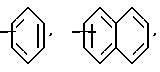
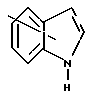
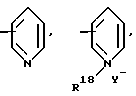
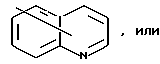
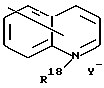
в которых необязательные заместители выбирают из группы, содержащей алкил из 1-6 углеродных атомов, аралкил из 7-10 углеродных атомов, алкокси из 1-6 углеродных атомов, циано, галоид, нитро, карбалкокси из 2-7 углеродных атомов или перфторалкил из 1-6 углеродных атомов,
R18 обозначает алкил из 1-6 углеродных атомов,
Y обозначает галид, сульфат, фосфат или п-толуолсульфонат анион,
g = 1-6,
h = 1-6,
i = 1 - 6 или
g) -CONHSO2-Ar
где Ar обозначает фенил, нафтил, пиридил, хинолил, изохинолил, хиноксалил, тиенил, тионафтил, фурил, бензофурил, бензодиоксил, бензоксазолил, бензоизоксазолил или бензодиоксолил; где Ar группа может быть необязательно моно-, ди- или три- замещенной группой, выбранной из: алкила из 1-6 углеродных атомов, арилалкила из 7-10 углеродных атомов, алкокси из 1-6 углеродных атомов, циано, галоида, нитро, карбалкокси из 2-7 углеродных атомов, трифторметила, амино, диалкиламино из 1-6 углеродных атомов на алкил группу, алкилтио из 1-6 углеродных атомов, - SO3H, -PO3H или CO2H, при условии, что R2, R3 и R4 не все обозначают водород, или его фармацевтически приемлемая соль, когда Ar группа содержит основной азот или когда Ar группа замещена диалкиламино из 1-6 углеродных атомов на алкил группу, - SO3H, -PO3H или CO2H,
h) -SO2R19
где R19 - алкил, алкенил или алкинил, содержащий 1-6 углеродных атомов, или ароматический радикал, выбранный из группы, содержащей фенил или нафтил, или гетероциклический радикал, выбранный из группы, содержащей тиофенил или хинолил или NHCO2R20, в котором R20 представляет низший алкил, содержащий от 1 до 6 углеродных атомов,
где R21 и R22 каждый представляют водород или алкил из 1-3 углеродных атомов или R21 и R22 вместе с азотом, к которому они присоединены, образуют насыщенный гетероцикл, имеющий 4-5 углеродных атомов, и j = 1-3, или R2 и R3, взятые вместе также представляют бирадикал формулы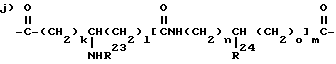
в которой R24 обозначает водород, алкил из 1-6 углеродных атомов, арилалкил из 7-10 углеродных атомов, (CH2)pNR25R26, аминоалкил из 1-4 углеродных атомов, гидроксиалкил из 1-4 углеродных атомов, гуанилалкил из 2-4 углеродных атомов, меркаптоалкил из 1-4 углеродных атомов, алкилтиоалкил из 2-6 углеродных атомов, индолметил, гидроксифенилметил, имидазолилметил или фенил, который необязательно моно-, ди- или три- замещен заместителем, выбранным из: алкила из 1-6 углеродных атомов, алкокси из 1-6 углеродных атомов, гидрокси, циано, галоида, нитро, карбалкокси из 2-7 углеродных атомов, трифторметила, амино или карбоновой кислоты,
R25 обозначает водород, алкил из 1-6 углеродных атомов или аралкил из 7-10 углеродных атомов,
R23 и R26 каждый независимо обозначают водород, формил, алканоил из 2-7 углеродных атомов, арилалканоил из 8-11 углеродных атомов, арилоил или CO2R27,
R27 обозначает алкил из 1-6 углеродных атомов, арилалкил из 7-10 углеродных атомов, аллил, флуоренилметил, или фенил, который необязательно моно-, ди- или три- замещен заместителем, выбранным из: алкила из 1-6 углеродных атомов, алкокси из 1-6 углеродных атомов, гидрокси, циано, галоида, нитро, карбалкокси из 2-7 углеродных атомов, трифторметила, амино или карбоновой кислоты,
k равно 0-4,
l равно 0-4,
m равно 0-1,
n равно 0-4,
о равно 0-4 и
p равно 0-4
или их фармацевтически приемлемая соль, или их пролиновый аналог, или фармацевтически приемлемая соль этого пролинового аналога.
Из заместителей, перечисленных выше, для R1 более предпочтительны C1-C6 низшие алкильные группы. Из них наиболее желательны метил и этил заместители. Специалисту в соответствующей технике должно быть ясно, что R1 заместители, перечисленные выше, могут быть линейными или разветвленными и могут включать заместители с дополнительными замещениями. Заместители этих R1 групп могут включать, но не ограничиваются, группами: гидрокси, кето, циано и галоид. В случае сложных аминоэфиров, описанных выше в разделе с), предпочтительными являются те соединения, в которых a = 0, d = 0, и C = 1; а = 0, b = 0, и c = 2; b = 0, и R5 обозначает -(CH2)d CO2R7; a = 0, b = 0 и R5 обозначает -(CH2)e NR9CO2R10; a = 0, b = 0 и R5 обозначает водород. Из C1-C6 фторалкил соединений, перечисленных для R1, наиболее предпочтительными заместителями являются трифторметил и трифторэтил.
В рамках данного описания термин "низший алкил", когда он используется отдельно или в комбинации, относится к радикалам, имеющим 1-6 углеродных атомов в углеродной цепи. Термин "ацил" относится к заместителям общей структуры 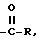 где R обозначает углеводород из 1-6 углеродных атомов, например, ацетил или пропионил.
где R обозначает углеводород из 1-6 углеродных атомов, например, ацетил или пропионил.
Под термином "арил", используемым здесь для обозначения группы или части группы, таким как арилалкил или арилокси, подразумевается (i) любой одновалентный карбоциклический радикал, обладающий ароматическим характером и включающий группы, содержащие от 6 до 10 углеродных атомов, такие как фенил или нафтил и (ii) гетероциклические группы, обладающие ароматическим характером с 5-10 атомами в цикл и одним или более (например, 2 или 3) гетероатомами, выбранными из кислорода, азота и серы. Примерами последних являются фурил, тиенил, пирролил, пиридинил, пиримидинил, хинолил, бензимидазолил, тиазолил и имидазолил. Эти группы могут необязательно быть замещены, под чем подразумевается необязательное замещение одним или более заместителями. Эти заместители могут быть обычно используемыми в фармацевтической химии, такими как галоген (например, Cl, Br, F), C1-C6 алкил, C1-C6 алкокси, галоид (C1-C6)-алкил (например, CF3, CF3CH2-) или галоид (C1-C6) алкокси (например, CHF2О, CF3CH2O-), NO2, NH2, CN, C1-C6 алкиламино, ди(C1-C6)алкил)амино, карбокси, (C1-C6 алкокси) карбонил, ацил (например (C1-C6 алкил) карбонил, арилкарбонил), ациламино, например, (C1-C6 алкил) CONH, арил (например, фенил) или амино (C1-C6)алкил.
Термин "арилалкил" относится к ароматическим заместителям, имеющим приблизительно от 7 до 16 углеродных атомов, с единичной валентностью, локализованной в боковой цепи. Примеры таких арилалкил заместителей включают бензил, толил, бензетил, фенетил и стирил группы. Термины "алкилтио", "арилтио" и "арилалкилтио" относятся к заместителям, имеющим структуру -SR, где R относится соответственно к только что упомянутым "низший алкил", "арил" и "арилалкил" группам. Термин "галоид" относится к фтору, хлору, брому или иоду.
Фармацевтически приемлемые соли этих соединений, которые содержат основные функциональные группы, такие как амино группа, могут быть получены из неорганических катионов, таких как натрий, калий и тому подобных и органических кислот, таких как уксусная, молочная, лимонная, винная, янтарная, малеиновая, малоновая, глюконовая и тому подобные.
Такие сложные аминоэфиры могут быть получены ацилированием соединения формулы 1a или 1b, приведенной выше, где один или более R2-4 представляет водород, ацилирующим агентом, имеющим общую структуру: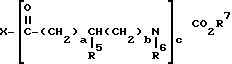
где X обозначает ОН в присутствии реагента конденсации, такого как дициклогексилкарбодиимид. Сложные аминоэфиры могут также быть получены использованием смешанного ангидрида вышеописанной карбоновой кислоты в качестве ацилирующего агента. Или же иначе, ацилирующим агентом может быть галоид ангидрид, в котором X является C1, Br или I. Группы ацилирующих соединений, используемые для получения соединений данного изобретения, выпускаются промышленностью или могут быть получены способами, описанными в литературе.
Из карбамилированных соединений, раскрытых выше в разделе d), предпочтительными членами являются те, в которых R3 представляет водород; те, в которых R2 представляет водород, те, в которых R4 представляет водород; те, в которых f равно 0 и X представляет Y; те, в которых R3 - водород, f = 0 и X обозначает Y; и те, в которых f равно 0, X является Y и Y представляет фенил, 4-фторфенил, 2,4-дифторфенил, 4-нитрофенил или 4-метилфенил.
В качестве одного из примеров этих соединений 31-карбамилированные соединения данного изобретения могут быть получены защитой 42-спиртовой группы рапамицина защитной группой, такой как трет-бутилдиметилсилил группа, с последующим карбамилированием 31-положения изоцианатом общей структуры:
OCN-C(R11R12)n X.
Удаление защитной группы дает 31-карбамилированные соединения. В случае трет-бутил диметилсилил защитной группы снятие защиты производится в умеренно кислой среде.
При использовании соединений с 31-карбамилированным положением и 42-положением со снятой защитой 42-положение может взаимодействовать с иным изоцианатом, чем тот, с которым взаимодействовала 31-спиртовая группа, что дает соединения, имеющие различные карбаматы в 31- и 42-положениях. Или же иначе, 42-карбамилированные соединения, полученные как описано выше, могут взаимодействовать с другим изоцианатом, что дает соединения, имеющие различные карбаматы в 31- и 42-положениях.
Изоцианаты, используемые для получения этих карбамилированных соединений, выпускаются промышленностью или могут быть получены по опубликованным методикам.
Фармацевтически приемлемыми солями этих карбамилированных рапамицин соединений являются соли, полученные из таких органических и неорганических кислот, как; уксусная, молочная, лимонная, винная, янтарная, малеиновая, малоновая, глюконовая, хлористоводородная, бромистоводородная, фосфорная, азотная, серная метансульфоновая и аналогичные известные приемлемые кислоты.
Предпочтительными фторированными сложными эфирами группы, описанной выше в разделе e), являются те, в которых R3 представляет водород; те, в которых R13 представляет моно-, ди-, поли- или пер-фторированную алкил группу из 1-6 углеродных атомов; и те, в которых R3 представляет водород и R13 обозначает моно-, ди-, поли- или пер-фторированную алкил группу из 1-6 углеродных атомов.
Когда другие ацилированные соединения из разделов c) - j) содержат арил или арилалкил радикалы, предпочтительно, чтобы арил радикал представлял фенил, нафтил, пиридил, хинолил, изохинолил, хиноксалил, тиенил, тионафтил, фурил, бензофурил, бензодиоксил, бензоксозолил, бензоизоксазолил или бензодиоксолил группу, которая может быть необязательно моно-, ди- или три- замещенной группой заместителей, выбранных из: алкил из 1-6 углеродных атомов, арилалкил из 7-10 углеродных атомов, алкокси из 1-6 углеродных атомов, циано, галоид, нитро, карбалкокси из 2-7 углеродных атомов, трифторметил, амино, диалкиламино из 1-6 углеродных атомов на алкил группу, алкилтио из 1-6 углеродных атомов, - SO3H, -PO3H и -CO2H. Более предпочтительно, чтобы арил радикал представлял фенильную группу, необязательно моно-, ди- или три-замещенную группой, выбранной из: алкил из 1-6 углеродных атомов, арилалкил из 7-10 углеродных атомов, алкокси из 1-6 углеродных атомов, циано, галоид, нитро, карбалкокси из 2-7 углеродных атомов, трифторметил, амино, диалкиламино из 1-6 углеродных атомов на алкил группу, алкилтио из 1-6 углеродных атомов, - SO3H, -PO3H и -CO2H.
Ацилированные соединения данного изобретения могут быть получены ацилированием рапамицина с помощью ацилирующего агента, имеющего общую структуру
в которой X представляет OH, в присутствии реагента сочетания, такого как CMC (1-циклогексил-3-(2-морфолиноэтил) карбодиимид мето-пара-толуолсульфонат). Соединения данного изобретения могут также быть получены использованием ангидрида вышеописанной карбоновой кислоты в качестве ацилирующего агента. Вдобавок ацилирующим агентом может быть галоидангидрид, в котором X может быть Cl, Br или I. Или же иначе, такие реагенты как Реагент Ishikawa (N, N-диэтил-1,1-2,3,3,3-гексафторпропиламин), могут быть использованы в качестве ацилирующего агента для получения соединений данного изобретения.
Соединения данного изобретения, ацилированные в каждом из 27-, 31- и 42- положений, могут быть получены способами, описанными выше, путем увеличения таких переменных, как реакционное время, температура и количество ацилирующего агента.
Например, 31-ацилированные соединения данного изобретения могут быть получены защитой 27- и 42-спиртовых групп рапамицина защитными группами, такими как трет-бутил диметилсилил группа, в присутствии основания, такого как имидазол, с последующим ацилированием 31-положения ацилирующим агентом, имеющим вышеприведенную общую структуру. Удаление защитных групп дает 31-ацилированные соединения. В случае трет-бутилдиметилсилил защитной группы удаление защиты выполняется в условиях умеренной кислотности, таких как смесь водной уксусной кислоты и ТГФ.
При использовании 31-ацилированного положения и 42-положения со снятой защитой, 42-положение может взаимодействовать с заданным ацилирующим агентом, что дает соединения, имеющие различные ацил составляющие в 31- и 42-положениях. Аналогично, 42-ацил соединения, полученные как описано выше, могут взаимодействовать с ацилирующим агентом, имеющим различную структуру для введения различных ацильных групп в 31-положение. Ацилирующие агенты, используемые для получения соединений данного изобретения, выпускаются промышленностью или могут быть получены описанными в литературе способами.
Предпочтительными амидо эфирами рапамицина из описанных выше эфиров в разделе f) являются те, в которых X представляет -(CH2)g-; те, в которых X представляет -(CH2)g и R14 и R15 обозначают алкил из 1-6 углеродных атомов; и те, в которых X обозначает -(CH2)-, R14 обозначает водород и R15 обозначает Ar обозначает -(CH2)h -Ar.
Фармацевтически приемлемые соли этих соединений могут быть получены, когда R14 или R15 представляют -(CH2)i -NR16R17 или когда Ar обозначает необязательно моно- или ди- замещенную пиридил или хинолил группу. Фармацевтически приемлемые соли являются производными таких органических или неорганических кислот, как уксусная, молочная, лимонная, винная, янтарная, малеиновая, малоновая, глюконовая, хлористоводородная, бромистоводородная, фосфорная, азотная серная, метансульфоновая и тому подобные.
Получение амидо эфиров рапамицина приведено в патенте США N 5.118.677, который приводится здесь в качестве ссылки.
Фармацевтически приемлемыми солями сульфонил карбамат рапамициновых соединений, описанных выше в разделе g), являются соли, полученные из таких неорганических катионов, как натрий, калий и тому подобные; органических оснований, таких как моно-, ди- и триалкил амины из 1-6 углеродных атомов на алкил группу, и моно-, ди- и тригидроксиалкил амины из 1-6 углеродных атомов на алкил группу и тому подобные, и органических и неорганических кислот, таких как уксусная, молочная, лимонная, винная, янтарная, малеиновая, малоновая, глюконовая, хлористоводородная, бромистоводородная, фосфорная, азотная, серная, метансульфоновая и подобные известные приемлемые кислоты.
Из этих соединений предпочтительными являются те, в которых Ar является необязательно моно-, ди- или три- замещенным фенилом. Когда Ar замещен арилалкилом из 7-10 углеродных атомов, предпочтительно, чтобы эта арил составляющая арилалкильного радикала являлась фенильной группой.
Соединения данного изобретения, карбамилированные по одному из 27-, 31- или 42-спиртовых положений или по всем этим положениям, могут быть получены взаимодействием рапамицина с изоцианатом, имеющим общую структуру
O = C = N- SO2-Ar,
в нейтральной среде или в присутствии основания, такого как пиридин.
Карбамилированные соединения данного изобретения могут быть получены путем защиты спиртовых групп, которые не должны взаимодействовать, защитной группой, такой как трет-бутил диметилсилил группа, с последующим карбамилированием незащищенного положения изоцианатом, имеющим вышеуказанную общую структуру. Удаление защитной группы дает заданные карбамилированные соединения. В случае трет-бутил диметилсилил защитной группы снятие защиты может выполняться в умеренно кислой среде.
Имея одно или два положения карбамилированных и другое положение со снятой защитой, можно осуществить взаимодействие незащищенного положения с изоцианатом, отличным от того, который взаимодействовал с первой спиртовой группой, что дает соединения, имеющие различные карбаматы в рассматриваемых позициях.
Изоцианаты, используемые для получения соединений данного изобретения, выпускаются промышленностью или могут быть получены способами, раскрытыми в литературе. March J. [Advanced Organic Chemistry, 3d ed., p.p. 391, 452 и 479 (1985)] раскрывает общий способ получения арилсульфонил изоцианатов, которые могут быть использованы, когда отсутствует промышленный арилсульфонилизоцианат. Следующая схема иллюстрирует один способ, исходящий из арил-соединения. Другие способы получения арилсульфонил изоцианатов известны по литературе: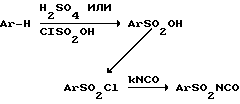
Рапамицин 27-, 31- и 42-сульфонаты данного изобретения, приведенные выше в разделе h), могут быть получены по стандартным литературным методикам, как описано в приведенной ниже общей реакционной схеме: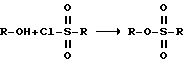
Образование сульфоната между спиртом и сульфонил галогенидом описано [Jerry March, Advanced Organic Chemistry, 3rd edition, punlished in 1985, p. 444] . Специфические условия реакции, используемые в данном изобретении, разработаны S. Rakhit of Ayerst Laboratories и приведены в патенте США N 4.316.885 (Февр. 23, 1982).
27-, 31- и 42-(N-карбоалкокси)сульфаматы данного изобретения могут также быть получены по реакции рапамицина с внутренней солью гидроокиси алкил (карбоксисульфамоил)триэтиламмония (Соли Бургесса, см. J.M. Atkins Jr. and E. M. Burgess, J.Am. Chem. Soc., 90, 4744, 1968, Е.M. Burgess, H.R. Penton Ir. and E.A. Faylor, J. Orh. Chem. 38, 26, 1978).
где R1 и R2 - как определено выше.
Амино алкил сложноэфирные соединения раздела i), приведенного выше, могут быть получены ацилированием заданных положений(ия) подходящим ацилирующим агентом, как описано в патенте США N 4.650.803, который включен в описание в качестве ссылки, в то время как оставшиеся из положений 27-, 31- или 42- защищены такой группой, как триэтилсилил.
Сложные аминодиэфиры, включающие соединения данного изобретения, приведенные выше в разделе i), могут быть получены ацилированием рапамицина ацилирующим агентом, имеющим общую структуру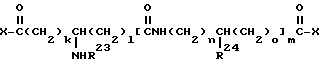
в которой R23, R24, k, l, m, n и o - как описано выше, и X обозначает ОН,
в присутствии агента сочетания (конденсации), такого как дициклогексилкарбодиимид или водорастворимый карбодиимид. Соединения данного изобретения могут также быть получены применением смешанного ангидрида или активированного сложного эфира вышеуказанной карбоновой кислоты (такого как эфир с п-нитрофенолом, пентахлор или пентафторфенолом, 2,4,5-нитрофенолом, N-гидроксисукцинимидом, N-гидроксифтальимидом или 1-гидрокси-1Н-бензотриазолом) в качестве ацилирующих агентов. Или же иначе, ацилирующими агентами могут быть галоидангидриды, в которых X может быть Cl, Br или F (за исключением, когда R23 или R27 = т-Bu), азид или имидазол производное названной кислоты.
Ацилирующие агенты, используемые для получения соединений данного изобретения, выпускаются промышленностью или могут быть получены описанными в литературе способами. Аминокислоты, используемые для получения соединений данного изобретения, могут быть либо R, либо S конфигурации, и оптически активный углерод сохраняет свою конфигурацию при превращении в соединение данного изобретения. В случаях, когда m равно 1, ацилирующие агенты могут обычно быть получены конденсацией двух аминокислот с образованием дипептида, который превращают в приведенный выше ацилирующий агент по стандартным химическим методикам.
Из этих аминодиэфирных соединений, предпочтительны те, в которых W равно 0, и те, в которых m равно 0 и l равно 1-2. Одним из наиболее предпочтительных сложных аминодиэфиров настоящего изобретения является C-22-метил-42-диметилглицин рапамицин. Данные по синтезу этого соединения приведены ниже в примере 7.
Каждому специалисту в соответствующей области техники должно быть ясно, что C-22 замещенные рапамицин производные данного изобретения могут включать те, в которых каждый из R2, R3 и R4 представляют водород или в котором один или два из R2, R3 и R4 представляют водород, а другая(ие) группы являются перечисленными выше в разделах b) - j). Данное изобретение включает также те соединения C-22 замещенного рапамицина, в которых каждый из R2, R3 и R4 представляет группы, выбранные из разделов a) - j), приведенных выше, включая производные, в которых R2, R3, R4 не являются выбранными из одних и тех же разделов, представленных выше в b) - j). Должно быть также понятно, что настоящее изобретение включает все фармацевтически приемлемые соли, аналоги, рацематы и индивидуальные энантиомеры таких соединений.
C-22 замещенные соединения данного изобретения могут быть получены сначала защитой 31- и 42- гидроксил положений и затем выполнением замещения по положению C-22. Пример такого замещения демонстрируется C-22 метилированием по схеме 1, приведенной ниже: 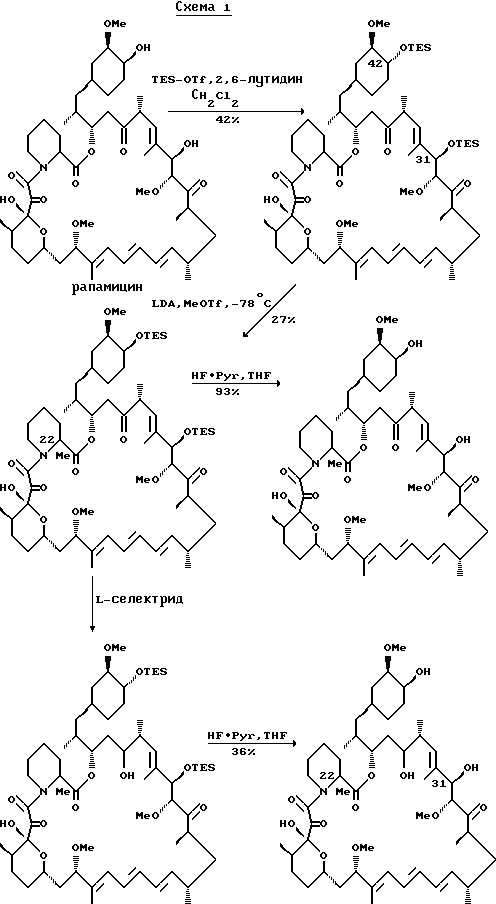
В схеме 1, которая иллюстрирует синтез альфа-метил соединений, положения 31- и 42- гидроксильных групп сначала защищают с помощью подходящей защитной группы, такой как три-этилсилил (TES) защитная группа. Затем бис-TES рапамицин обрабатывают литий диизопропил амидом (LDA) с последующим алкилированием метилтрифторметансульфонатом (MeOTf). В соответствии с этой схемой ацилирование протекает исключительно по C-22 положению. TES защитные группы могут затем быть удалены обработкой HF-Pyr-комплексом. 31- и 42- гидроксил положения могут затем быть замещены, как описано выше. Или же иначе, C-22 замещенное, TES - защищенное соединение может быть восстановлено L - селектридом (L - selectride) с получением C-27-гидрокси-C-22-метил защищенного производного. После чего могут быть введены функциональные группы по 31 и 42 положениям, как описано ранее.
Аналогичным образом могут быть приготовлены другие продукты алкилирования. Приведенная ниже схема 2 иллюстрирует C-22 этилирование рапамицина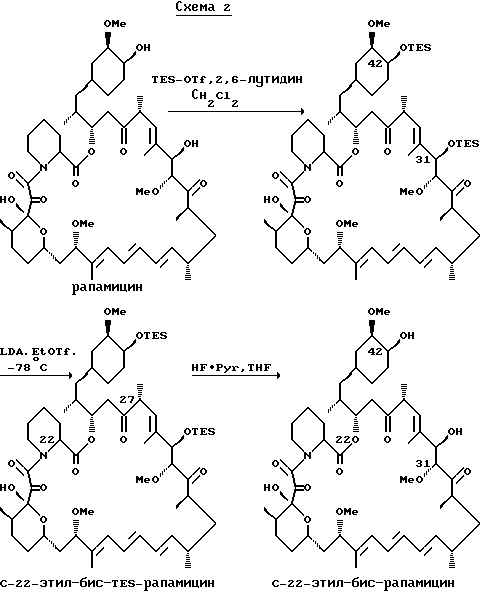
Каждому специалисту в соответствующей области техники должно быть понятно, что данные соединения легко могут быть приготовлены. Например, каждый специалист может приготовить C-42-ацилированные производные, такие как C-42-глицин-C-22-метил-рапамицин. Аналогичным образом могут быть введены функциональные группы по C-27 и C-31 положениям. Кроме того, поскольку пролиновые аналоги рапамицина известны, можно приготовить соответствующие алкилированные пролиновые аналоги, как иллюстрируется ниже схемой 3: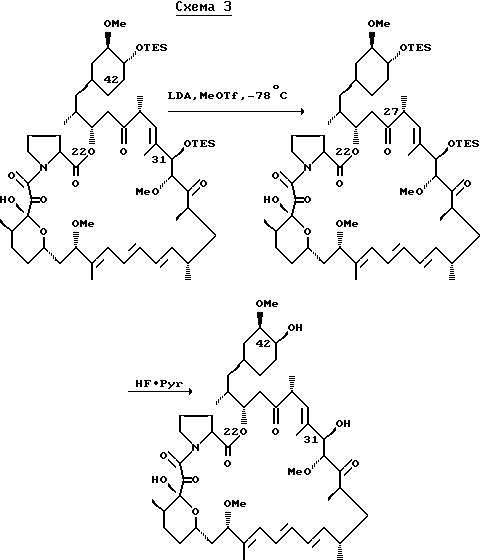
где заданная алкильная группа иллюстрируется замещенной метил группой. Могут быть также получены C-42 и C-31 функциональные производные C-22 алкилированного пролинового аналога. Аналогично, может быть получен C-27 восстановленный пролиновый аналог, по способу приведенному в схеме 1. В это положение может также быть введена функциональная группа, как описано.
Любому специалисту в соответствующей технике совершенно очевидно, что алкилирования по C-22 положению могут быть выполнены хорошо известными в литературе способами.
Такие алкилирования должны включать способы, использующие альдегиды типа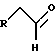
или хлорангидриды типа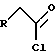
или алкилгалогениды типа
где X означает C1, бром или иод. В каждой из формул альдегида, хлорангидрида и алкилгалогенида R обозначает часть C-22 замещенного R1 заместителя указанного выше.
Понятно также, что алкилирование по положению C-22 может выполняться с использованием сульфонилхлорида, RSCl, такого, в котором R = алкил или фенил, или тозил цианида (TsCN). Понятно также, что предполагаемое тиоалкилирование может выполняться аналогичными способами. Примером может служить защита гидроксильной группы TES, обработка LDA (лаурилдиэтаноламидом) и тиоалкилирование диметилсульфидом или фенил-S-бромидом.
Иммуносупрессивные активности представленных соединений данного изобретения оценивают in vitro стандартными фармакологическими испытаниями для оценки пролиферации лимфацитов (LAF) и in vivo стандартными фармакологическими испытаниями для оценки времени выживания отщипнутого полнослойного кожного лоскута. Для in vitro измерения иммуносупрессивных воздействий представленных соединений используют методику комитогениндуцируемой тимоцит пролиферации (LAF). Кратко: клетки вилочковой железы нормального BALB/c мыши культивируют в течение 72 часов с РНА (ФГА) и IL-1 и вызывают пульсацию тритированным тимидином в течение шести часов. Клетки культивируют в присутствии или отсутствии различных концентраций рапамицина, циклоспорина A или испытуемого соединения. Собирают выращенные в культуре клетки и определяют содержащуюся в них радиоактивность. Ингибирование лимфопролиферации оценивают в процентах как изменение в счете импульсов в минуту по отношению к необработанным лекарственными средствами контролям. Результаты выражают в IC50.
Представленные соединения изобретения оценивают также in vivo испытанием, предназначенным для определения времени выживания отщипнутого полнослойного кожного лоскута от мужских DBA/2 доноров, трансплантированного мужским BALB/c реципиентам. Способ адаптирован из Billingham R.E. and Medawar P.B. J. Exp. Biol. , 28:385-402, (1951). Кратко, отщипнутый полнослойный кожный лоскут у донора пересаживают на спину реципиента, как гомотрансплантант и аутотрансплантант используют в качестве контроля в той же области. Реципиенты обрабатываются внутрибрюшинно либо различными концентрациями циклоспорина A как контроля для испытания, либо испытуемых соединений. Необработанные реципиенты служат как контроль отторжения. Трансплантат подвергают ежедневно дозиметрическому контролю и записывают результаты до тех пор, пока трансплантат не станет сухим и не образуется почерневшая корка. Это считают днем отторжения. Среднее время жизни трансплантата (число дней ± стандартное отклонение (S.D.)) группы, обработанной лекарственным препаратом, сравнивают с группой контроля.
Кроме того, исследуют стабильность представленных соединений путем ВДЖХ в 0,1 М фосфатном буфере (pH 7.4, 37oC).
Результаты каждого из этих испытаний (LAF, полнослойного кожного лоскута и стабильности) приведены в таблице.
Результаты этих стандартных фармакологических испытаний иллюстрируют как in vivo, так и in vitro иммуносупрессивную активность соединений настоящего изобретения. Результаты LAF испытания показывают, что стабилизированные рапамицин производные индуцируют супрессию T-клеточной пролиферации. Поскольку трансплантированные отщипнутые полнослойные кожные лоскуты обычно отторгаются на 6-7 дни без использования иммуносупрессивного агента, увеличенное время жизни, наблюдаемое при использовании 22-метил рапамицина дополнительно демонстрирует применимость соединений данного изобретения для иммуносупресcии.
Поскольку соединения данного изобретения структурно аналогичны рапамицину и имеют аналогичный рапамицину профиль активности, они также должны обладать противоопухолевой, противогрибковой и антипролиферативной активностями. В качестве таковых соединения данного изобретения полезны при лечении отторжения трансплантанта, такого как сердце, почки, печень, костный мозг и кожные трансплантанты; аутоиммунных заболеваний, таких как волчанка, ревматоидный артрит, сахарный диабет, миастения gravis и рассеянный склероз; воспалительных заболеваний, таких как псориаз, дерматит, экзема, себоррея; воспалительных заболеваний желудочно-кишечного тракта и увеита; твердых опухолей, грибковых инфекций и гиперпролиферативных сосудистых заболеваний, таких как рестеноз. Кроме того, данное изобретение дает также способ индуцирования иммуносупрессии у нуждающихся в этом млекопитающих, включающий введение названному млекопитающему иммуносупрессивного количества одного или более описанных здесь соединений.
Результаты испытания стабильности в 0.1 М фосфатном буфере показывают, что соединения данного изобретения обладают повышенной стабильностью по сравнению с рапамицином. Как показано выше, найдено, что 22-метил-рапамицин и 22-метил-27-гидрокси-рапамицин, соответственно, имеют периоды полураспада 38 и 21,8 часа. Эти результаты сопоставляются с 12-13 часами периода полураспада, определенного для рапамицина в тех же условиях.
Соединения данного изобретения могут быть составлены и назначаться неразбавленными или с фармацевтическими носителями для нуждающихся в них млекопитающих. Фармацевтический носитель может быть твердым или жидким.
Твердый носитель может включать одно или более веществ, которые могут действовать как корригенты, смазывающие вещества, растворители, суспендирующие средства, наполнители, вещества, обеспечивающие скольжение, уплотняющие добавки, связывающие вещества или разъединяющие вещества; это может быть также материал, образующий желатиновую капсулу. В порошках - носитель представляет собой тонко измельченное твердое вещество, смешанное с тонко измельченным активным ингредиентом. В таблетках - активный ингредиент смешан с носителем, обладающим необходимыми уплотняющими свойствами в подходящих пропорциях, и сжат в заданную форму с заданным размером. Порошки и таблетки обычно содержат до 99% активного ингредиента. Подходящие твердые носители включают, например, фосфат кальция, стеарат магния, тальк, сахар, лактозу, декстрин, крахмал, желатин, целлюлозу, метил целлюлозу, натрий карбоксиметил целлюлозу, поливинилпирролидин, низко плавящиеся парафины и ионообменные смолы.
Жидкие носители используются в приготовлении растворов, суспензий, эмульсий, сиропов, эликсиров и прессованных композиций. Активный ингредиент может быть растворен или суспендирован в фармацевтически приемлемом жидком носителе, таком как вода, органический растворитель, смеси того и другого или в фармацевтически приемлемых маслах и жирах. Жидкий носитель может содержать другие подходящие фармацевтические добавки, такие как солюбилизаторы, эмульгаторы, буферы, консерванты, вкусовые добавки, корригенты, суспендирующие вещества, загустители, красители, регуляторы вязкости или осмо-регуляторы. Подходящие примеры жидких носителей для перорального или парентерального введения включают воду (частично содержащие вышеуказанные добавки, например, производные целлюлозы, предпочтительно, раствор натрий карбоксиметил целлюлозы), спирты (включая одноатомные спирты и многоатомные спирты, например, гликоли) и их производные, и масла (например, фракционированные кокосовое масло и арахисовое масло). Для парентерального введения носитель может быть также жирным сложным эфиром, таким как этил олеат и изопропил миристат. Стерильные жидкие носители полезны для стерильных композиций в жидкой форме для парентерального введения. Жидкие носители для прессованных композиций могут быть галогенизированными углеводородами или другими фармацевтически приемлемыми обеспечивающими передвижение веществами. Жидкие фармацевтические композиции, представляющие стерильные растворы или суспензии, могут применяться, например, путем внутримышечной, внутрибрюшинной или подкожной инъекции. Стерильные растворы могут также вводиться внутривенно. Соединения данного изобретения могут также вводиться перорально либо в жидкой, либо в твердой композиционной форме.
Соединения данного изобретения могут вводиться ректально, например, в форме обычного суппозитория или мази.
Для введения путем внутриносовой или внутрибронхиальной ингаляции или инсуффляции соединения данного изобретения могут быть составлены в водных, частично водных растворах, которые могут быть затем переведены в форму аэрозоля. Соединения данного изобретения могут также вводиться трансдермально путем использования трансдермального пластыря, содержащего активное соединение и носитель, являющийся инертным по отношению к активному соединению, не токсичным по отношению к коже и обеспечивать доставку лекарственного средства для системной абсорбции через кожу в кровоток. Носитель может принимать любые формы, такие как кремы и мази, пасты, гели, используются также обтурирующие устройства. Кремы и мази могут быть вязкими жидкостями или полутвердыми эмульсиями либо типа масло-в-воде, либо типа вода-в-масле. Пасты включают абсорбирующие порошки, диспергированные в нефти или гидрофильной нефти, содержащей активный ингредиент, который также должен быть подходящим. Для высвобождения активного ингредиента в кровоток могут использоваться различные обтурирующие устройства, такие как полупроницаемое мембранное покрытие резервуара, содержащего активный ингредиент с носителем или без носителя, или матрица, содержащая активный ингредиент. Другие обтурирующие устройства известны из литературы.
Кроме того, соединения настоящего изобретения могут применяться в виде раствора, крема или лосьона при составлении с фармацевтически приемлемым растворителем, содержащим 0,1-5%, предпочтительно 2%, активного соединения, которое должно быть нанесено на пораженную грибком поверхность.
Требуемые дозы изменяются в зависимости от конкретных используемых композиций, способа введения, тяжести симптомов и конкретного пациента, нуждающегося в лечении. Основанные на результатах, полученных в стандартных фармакологических испытаниях для других соединений рапамицина, предлагаемые ежедневные дозы для внутривенного введения соединений данного изобретения должны составлять 0,001-25 мг/кг, предпочтительно между 0,005-5 мг/кг, и наиболее желательно между 0,01-0,5 мг/кг. Предлагаемые ежедневные пероральные дозы соединений данного изобретения должны составлять 0,005-75 мг/кг, предпочтительно между 0,01-50 мг/кг и наиболее желательно между 0,05-10 мг/кг.
Лечение обычно начинается с минимальных доз, меньших чем оптимальная доза соединения. После чего доза увеличивается до достижения оптимального эффекта при данных обстоятельствах; точная доза для перорального, парентерального, интраназального, внутрибронхиального, трансдермального или ректального введения будет определяться лечащим врачом опытным путем для конкретного больного. В основном соединения данного изобретения наиболее желательно вводить в концентрациях, которые дают наиболее эффективные результаты, не оказывая никаких пагубных или разрушительных побочных действий.
Предполагается, что когда соединения данного изобретения используются как иммуносупрессорные и противогрибковые средства, они могут вводиться в сочетании с одним или более другими иммунорегуляторами. Такие другие препятствующие отторжению химиотерапевтические средства включают, но не ограничиваются: азатиоприн, кортикостероиды, такие как преднизон и метилпреднизолон, циклофосфамид, рапамицин, циклоспорин A, FK-506, ОКТ-3 и ATG (антитимоцитарный глобулин). При объединении одного или более лекарственных средств данного изобретения с такими другими лекарственными средствами или агентами для индуцирования иммуносупрессии или лечения воспалительных заболеваний, меньшее количество каждого из веществ требуется для достижения заданного эффекта. Основы такой объединенной терапии разработаны Stepkowski, чьи результаты показывают, что применение комбинации рапамицина и циклоспорина A при меньших терапевтических дозах существенно увеличивает время жизни сердечного аллотрансплантата [Transplantation Proc. 23:507 (1991)].
Данное изобретение дает также способы получения соединений изобретения, которые включают один из следующих:
(i) алкилирование или ацилирование по C-22 положению соединения формулы:
или его пролинового аналога,
где R2' и R3' каждый обозначают триэтилсилил, с ацилирующим агентом формулы RaCHO или RbCOhal или Rc-hal или RdS-hal или тозил цианид или ReS - SRe, где Ra обозначает алкил из 1-5 углеродных атомов, арил или фторарил из 1-5 углеродных атомов, Rb - алкил из 1-6 углеродных атомов, Rc - алкил из 1-6 углеродных атомов, арилалкил из 7-16 углеродных атомов или фторалкил из 1-6 углеродных атомов, Rd - алкил из 1-6 углеродных атомов, арилалкил или арил, Re - алкил из 1-6 углеродных атомов, арил, или арилалкил и галоид (hal) является хлором или бромом; что дает соединение приведенной выше формулы, где R2-3 обозначают триэтилсилил и, при желании, удаление одной или более защитных групп,
(ii) восстановление соединения формулы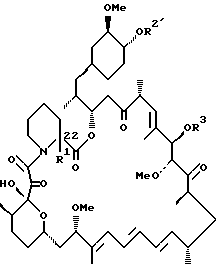
или его пролинового аналога, где R2' и R3' - как описано здесь выше, до получения соединения формулы 1a, в которой R2 и R3 обозначают триэтилсилил и R4 является водородом и, при желании, удаление одной или более защитных групп, или
(iii) ацилирование соединения вышеприведенной формулы, в которой R2-3 - такие, как определено ранее, при условии, что по крайней мере один из R2-3 является водородом, соединением формулы HOA
или его реакционноспособным производным, где A - одно из определений c), e), f), i) или j), как определено выше для R2-4, что дает соответствующее соединение формулы 1a или 1b, в котором один или более заместителей имеют значение от c) до j), приведенное выше, и, при желании, удаление любой из присутствующих триэтилсилил защитной группы, или
(iv) карбамилирование соединения приведенной выше формулы, в которой R2-4 - как определено выше, при условии, что по крайней мере один из R2-4 является водородом, соединением формулы:
OCN- SO2Ar или OCN-(CR11R12)nX
что дает соответствующее соединение формулы 1a или 1b и, при желании, удаление любой присутствующей триэтилсилил группы, или
(v) сульфирование соединения формулы, приведенной выше, в которой R2-4 - такие, как определено выше, при условии, что по крайней мере один из R2-4 является водородом, соединением формулы R19SO2hal или (R19SO2)2O или R20OCONSO2N+(aлкил)3, где hal является галогеном, таким как хлор или бром, и алкил обозначает алкил группу, например, алкил из 1-6 углеродных атомов и R19 и R20 - такие, как определено выше, и при желании, удаление присутствующей триэтилсилил группы, или
(vi) превращение основного соединения приведенной выше формулы в фармацевтически приемлемую соль и наоборот.
Относительно способа i), алкилирование (включая тиоалкилирование) или ацилирование может обычно выполняться в присутствии сильного основания, например литий диизопропиламида (LDA). Когда агент имеет формулу R2CHO, продукт формулы 1c имеет R1, обозначающую алкил или фторалкил из 1-6 углеродных атомов, замещенный гидрокси в положении -1. Относительно способа ii) продукт имеет алкил заместитель из 1-6 углеродных атомов, замещенный кето в положении -1; продукт способа iii) обычно замещен алкилом из 1-6 углеродных атомов, арилалкилом или фторалкилом из 1-6 углеродных атомов; продукты способов iv) и v) имеют алкил заместители из 1-6 углеродных атомов, арилалкил или арил с положением 1, представляющим "S".
Следующие синтетические методики приведены для демонстрации способов получения, полезных для получения соединений данного изобретения. Эти примеры исключительно иллюстративного характера и не ограничивают рамки объема данного соединения.
В приведенных далее ПМР-экспериментальных данных приняты следующие обозначения;
m - мультиплет,
s - синглет,
d - дублет,
t - триплет,
dd - двойной дублет.
Пример 1
C-22-метил-бис-TES-рапамицин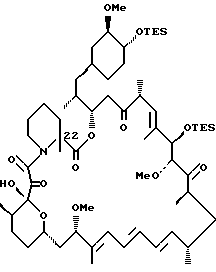
Бис-TES рапамицин (0,50 г, 0,483 ммоль) растворяют в ТГФ (4,4 мл, 0,1М) и охлаждают до -78oC. Раствор перемешивают 10 мин. Добавляют по каплям к раствору LDA. ТГФ комплекс (1,5М в циклогексане, 0,73 мл, 1,1 ммоль) и перемешивают 45 мин. К реакционной смеси добавляют метил трифторметансульфонат (0,18 г, 1,1 ммоль) и перемешивают 1,5 ч при -78oC. Реакцию прерывают, добавляя охлажденный до -78oC NaHCO3 (5 мл). Затем реакционной смеси дают нагреться до комнатной температуры. Смесь экстрагируют этилацетатом, промывают раствором соли, сушат над Na2SO4 и концентрируют в вакууме, получая желтый пенообразный продукт. Остаток хроматографируют, используя гексан-этилацетат (9:1 затем 4:1) в качестве элюента, что дает 22-метилированный Bil - TES рапамицин (0,138 г, 27% выход) в виде белой пены. ПМР (400 MHz, CDCl3) δ 0.50 (соед. m, 9Н), 0.71 (m, 1Н), 0.82-1.59 (соед. m, 38H), 1.65 (s, 3H), 1.69 (s, 3H), 1.78 (m, 2Н), 1.81 (s, 3Н), 1.85-1.95 (соед. m, 4Н), 2.05 (m, 2Н), 2.32 (m, 3Н), 2.36 (s, 1Н), 2.39 (s, 1Н), 2.61 (m, 2Н), 2.89 (m, 1H), 3.14 (s, наложен. на m, 1Н), 3.14 (m, 1Н), 3.28 (s, 3Н), 3.38 (m, 1Н), 3.40 (s, наложен. на m, 3Н), 3.40 (m, 2Н), 3.51 (m, 1Н), 3.53 (m, 1Н), 3.67 (dd, J= 5.71, 9.47 Hz, 1H), 3.79 (d, J = 5.57 Hz, 1H), 3.82 (m, 1H), 4.17 (d, J=5.35 Hz, 1H), 4.84 (m, 1H), 5.15 (d, J = 10.2 Hz, 1H), 5.42 (dd, J = 9.01, 14.2 Hz, 1H), 6.08 (d, J = 10.9 Hz, 1H), 6.20 (m, 1H), 6.23 (m, 1H), 6.42 (dd, J = 11.01, 14.01 Hz, 1H), масс-спектр высокого разрешения (отрицательный ион FAB) m/z 1155.7 [(М-H), рассчитано для C64H108NO13Si2: 1155.7].
Пример 2
C-22-метил-рапамицин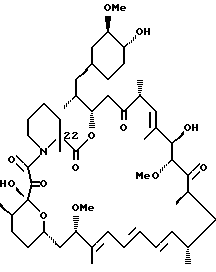
C-22-метил-бис-TES-рапамицин (0,489 г, 0,42 ммоль) растворяют в ТГФ (4 мл) и переносят в nalgene пробирку, содержащую некоторое количество 4  молекулярных сит. В разделительную пробирку halgene добавляют HF /пиридин комплекс (1 мл) к сухому пиридину (1 мл) при 0oC. К субстрату при 0oC добавляют 1,8 мл вышеупомянутого раствора. Реакционную смесь выдерживают 15 мин при 0oC, затем дают нагреться до комнатной температуры и перемешивают 2 ч. Затем реакционную смесь охлаждают до 0oC и реакцию прерывают медленным добавлением N2HCO3. Затем смесь экстрагируют EtOAc, после чего промывают 0,1 н. HCl, NaHCO3 и раствором соли. Органическую фазу сушат над Na2SO4 и упаривают в вакууме. Смесь хроматографируют, используя 2.5%, затем 5% MeOH/CH2Cl2 в качестве элюента, что дает C-22-метил-рапамицин (0,36 г, 93%). ИК (KBr) 3440 (s), 2940 (s), 1725 (s), 1650 (m), 1455 (w), 1380 (w), 1240 (w), 1195 (w), 1090 (s), 995 (m), 915 (w), 735 (w), ПМР (400 MHz, CDCl3) δ 0.67 (m, 1H), 0.90 (d, J = 6.38 Hz, 3H), 0.92 (d, J = 6.32 Hz, 3H), 1.01 (d, J = 6.56 Hz, 3H), 1.05 d, J = 7.24 Hz, 3H), 1.07 (d, J = 6.95 Hz, 3H), 1.11-1.48 (соед. m, 15H), 1.63 (s, 3H), 1.65 (s, 3H), 1.75 (s, 3H), 1.58-2.06 (соед. m, 10Н), 2.63 (s, 1Н), 2.65 (m, 3Н), 3.00 (m, 2Н), 3.14 (s, 3Н), 3.34 (m, 1Н), 3.36 (m, 3Н), 3.38 (s, 3Н), 3.50 (m, 2Н), 3.63 (m, 1H), 3.76 (m, 3Н), 4.21 (d, J = 4.47 Hz, 1Н), 4.52 (s, 1Н), 5.04 (m, 1H), 5,43 (m, 2Н), 6.05 (d, J = 10.1 Hz, 1Н), 6.13 (m, 1H), 6.25 (m, 1H), 6.41 (m, 1H), масс-спектр высокого разрешения (отрицательн. ион FAB) m/z 927 [(M-H), рассчитано для C52H80NO13:927].
молекулярных сит. В разделительную пробирку halgene добавляют HF /пиридин комплекс (1 мл) к сухому пиридину (1 мл) при 0oC. К субстрату при 0oC добавляют 1,8 мл вышеупомянутого раствора. Реакционную смесь выдерживают 15 мин при 0oC, затем дают нагреться до комнатной температуры и перемешивают 2 ч. Затем реакционную смесь охлаждают до 0oC и реакцию прерывают медленным добавлением N2HCO3. Затем смесь экстрагируют EtOAc, после чего промывают 0,1 н. HCl, NaHCO3 и раствором соли. Органическую фазу сушат над Na2SO4 и упаривают в вакууме. Смесь хроматографируют, используя 2.5%, затем 5% MeOH/CH2Cl2 в качестве элюента, что дает C-22-метил-рапамицин (0,36 г, 93%). ИК (KBr) 3440 (s), 2940 (s), 1725 (s), 1650 (m), 1455 (w), 1380 (w), 1240 (w), 1195 (w), 1090 (s), 995 (m), 915 (w), 735 (w), ПМР (400 MHz, CDCl3) δ 0.67 (m, 1H), 0.90 (d, J = 6.38 Hz, 3H), 0.92 (d, J = 6.32 Hz, 3H), 1.01 (d, J = 6.56 Hz, 3H), 1.05 d, J = 7.24 Hz, 3H), 1.07 (d, J = 6.95 Hz, 3H), 1.11-1.48 (соед. m, 15H), 1.63 (s, 3H), 1.65 (s, 3H), 1.75 (s, 3H), 1.58-2.06 (соед. m, 10Н), 2.63 (s, 1Н), 2.65 (m, 3Н), 3.00 (m, 2Н), 3.14 (s, 3Н), 3.34 (m, 1Н), 3.36 (m, 3Н), 3.38 (s, 3Н), 3.50 (m, 2Н), 3.63 (m, 1H), 3.76 (m, 3Н), 4.21 (d, J = 4.47 Hz, 1Н), 4.52 (s, 1Н), 5.04 (m, 1H), 5,43 (m, 2Н), 6.05 (d, J = 10.1 Hz, 1Н), 6.13 (m, 1H), 6.25 (m, 1H), 6.41 (m, 1H), масс-спектр высокого разрешения (отрицательн. ион FAB) m/z 927 [(M-H), рассчитано для C52H80NO13:927].
Пример 3
C-22-метил-C-27-гидрокси-бис-TES-рапамицин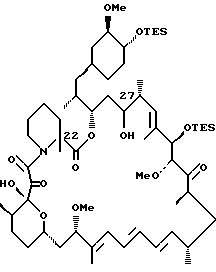
C-22-метил-бис-TES-рапамицин (0,27 г, 0,23 ммоль) растворяют в ТГФ (1.5 мл) и охлаждают до -78oC. Добавляют быстро L-селектрид (1,0 М в ТГФ, 0,17 мл). Реакционную смесь перемешивают 30 мин, затем добавляют 0,5 эквивалента L-селектрида (0,05 мл). Реакционную смесь медленно нагревают до комнатной температуры около 2 ч. Реакцию прерывают смешением с холодной водой, экстрагируют EtOAc, промывают раствором соли, сушат над Na2SO4 и упаривают в вакууме. Смесь хроматографируют, используя 20%, затем 40% EtOAc, гексан в качестве элюента. Затем продукт чистят ВДЖХ, 20% EtOAc, гексан, 21 мм - силикагелевая колонка, получая C-22-метил-C-27-гидрокси-бис-TES-рапамицин (0,08 г, 30% выход). ПМР (400 MHz, CDCl3) δ 0.0-0.3 (соед. m, 18Н), 0.40 (m, 1Н), 0.89-0.94 (соед. m, 36Н), 1.55 (s, 3Н), 1,62(s, 3Н), 1.10-2.10 (соед. m, 13Н), 1.93-2.02 (соед. m, 6Н), 2.65 (m, 3Н), 2.88 (m, 1Н), 3.12 (s, 3Н), 3.24 (m, 1Н), 3.26 (s, наложен. на m, 3Н), 3.26 (m, 1Н), 3,40 (s, наложен. на m, 3Н), 3,40 (m, 1Н), 3.51 (m, 1Н), 3.53 (m, 2Н), 3.63 (кажущийся t, J = 7.16 Hz, 1Н), 3.77 (d, J = 5.19 Hz, 1Н), 4.17 (d, J = 0.62 Hz, 1Н), 4.82 (m, 2Н), 5.37 (m, 1Н), 5.48 (m, 1Н), 5.98 (кажущийся t, J = 0.62 Hz, 1Н), 6.15 (m, 2Н), 6.38 (m, 1Н), масс-спектр высокого разрешения (отрицательный ион FAB) m/z 1157 [(M-H), рассчитано для C64H110NO13Si2: 157].
Пример 4
C-22-метил-C-27-гидрокси-рапамицин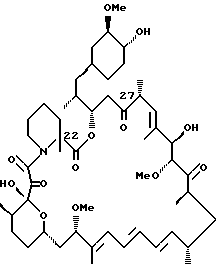
Восстановленный метилированный BiS-TES рапамицин (0,055 г, 0.047 ммоль) растворяют в ТГФ (0.5 мл) и переносят в halgene пробирку, содержащую некоторое количество 4  молекулярных сит. В отдельную halgene пробирку к пиридину (1 мл) при 0oC добавляют HF/пиридин комплекс (1 мл). К субстрату при 0oC добавляют 1 мл вышеуказанного раствора. Реакционную смесь перемешивают 15 мин при 0oC, затем оставляют перемешиваться при комнатной температуре в течение 2 ч. Затем реакционную смесь охлаждают до 0oC и реакцию прерывают медленным добавлением NaHCO3. Смесь экстрагируют EtOAc, промывают 0,1 н. HCl, NaHCO3 раствором соли. Органическую фазу сушат над Na2SO4 упаривают в вакууме. Смесь хроматографируют на флэш силикагеле и элюируют 2,5%, затем 5% MeOH/CH2Cl2, получая C-22-метил-C-27-гидрокси-рапамицин (0,016 г, 36% выход).
молекулярных сит. В отдельную halgene пробирку к пиридину (1 мл) при 0oC добавляют HF/пиридин комплекс (1 мл). К субстрату при 0oC добавляют 1 мл вышеуказанного раствора. Реакционную смесь перемешивают 15 мин при 0oC, затем оставляют перемешиваться при комнатной температуре в течение 2 ч. Затем реакционную смесь охлаждают до 0oC и реакцию прерывают медленным добавлением NaHCO3. Смесь экстрагируют EtOAc, промывают 0,1 н. HCl, NaHCO3 раствором соли. Органическую фазу сушат над Na2SO4 упаривают в вакууме. Смесь хроматографируют на флэш силикагеле и элюируют 2,5%, затем 5% MeOH/CH2Cl2, получая C-22-метил-C-27-гидрокси-рапамицин (0,016 г, 36% выход).
ИК (KBr) 3440 (s), 2920 (s), 1720 (s), 1645 (m), 1455 (m), 1375 (w), 1235 (w), 1190 (w), 1085 (s), 995 (m), 915 (wo), 735 (w), ПМР (400 МНz, CDCl3) δ 0.77 (m, 1H), 0.87 (d, J = 6.64 Hz, 3H), 0.90 (d, J = 6.85 Hz, 3H), 0.93 (d, J = 6.64 Hz, 3H), 0.97 (d, J = 6.64 Hz, 3H), 1.02 (d, J = 6.423 Hz, 3H), 1.04 (m, 1H), 1.18-1.42 (соед. m, 15Н), 1.61 (s, 3Н), 1.63 (s, 3H), 1.65 (s, 3Н), 1.53-2.06 (соед. m, 10Н), 2.28 (m, 2Н), 2.65 (m, 2Н), 2.92 (m, 2Н), 3.10 (m, 1Н), 3.13 (s, 3Н), 3.32 (s, 3Н), 3.36 (m, 2Н), 3.38 (s, 3Н), 3.51 (m, 2Н), 3.61 (кажущийся t, J = 7.37 Hz, 1H), 3.78 (d, J = 5.19 Hz, 1Н), 3.87 (m, 1H), 4.29 (m, 1H), 4.30 (m, 1H), 4.81 (m, 1H), 5.40 (m, 2H), 6.00 (d, J = 10.2, 1H), 6.09 (m, 1H), 6.22 (dd, J = 10.4, 14.7 Hz, 1H), 6.37 (dd, J = 11.0, 14.73 Hz, 1H), масс-спектр высокого разрешения (отрицательный ион FAB) m/z 929 [(M-H), рассчитано для C52H82NO13:929].
Пример 5
C-22-этил-бис-TES-рапамицин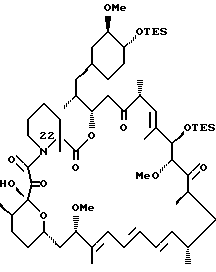
Бис-TES-рапамицин (2,0 г, 1,75 ммоль) растворяют в ТГФ (17.6 мл) и охлаждают до -78oC. LDA ТГФ комплекс (1,5 М в циклогексане, 2,9 мл, 4.38 ммоль) добавляют по каплям к раствору и перемешивают 45 мин. К реакционной смеси добавляют этил трифторметансульфонат (0,78 г, 4,38 ммоль) и перемешивают 2 ч при -78oC. Реакцию обрывают при -78oC насыщенным NaHCO3 (5 мл) и дают нагреться реакционной смеси до комнатной температуры. Смесь экстрагируют этилацетатом, промывают раствором соли, сушат над Na2SO4 и упаривают в вакууме, получая желтый пенообразный продукт. Смесь хроматографируют, используя 10% и 20% этилацетат/гексан в качестве элюента. Затем продукт чистят ВДЖХ, 20% EtOAc/гексан, на 41 мм колонке с двуокисью кремния, получая C-22-этил-бис-TES-рапамицин (0.25 г, 13%). ПМР (400 МНz, CDCl3) δ 0.0-0.64 (соед. m, 18Н), 0.73 (m, 1Н), 0.85-1.50 (соед. m, 46Н), 1.54-1.78 (соед. m, 3Н), 1.67 (s, 3H), 1.80 (s, 3H), 1.85-2.05 (соед. m, 4H), 2.32 (m, 3H), 2.37 (s, 1H), 2.41 (s, 1H), 2.56 (m, 2H), 2.89 (m, 1H), 3.14 (m, 1H), 3.15 (s, 3H), 3.26 (s, 3H), 3.40 (s, 3H), 3.42 (m, 1H), 3.48 (s, 1H), 3.50 (s, 1H), 3.74 (m, 1H), 3.89 (m, 1H), 4.14 (m, 1H), 4.88 (m, 2H), 5.16 (d, J = 10.3 Hz, 1H), 5.40 (dd, J = 9.28, 14.21 Hz, 1H), 6.05 (d, J = 11.1 Hz, 1H), 6.16 (m, 1H), 6.25 (m, 1H), 6.43 (dd, J = 11.1, 13.9 Hz, 1H), 13C ЯМР (100 MHz, CDCl3) δ 3.74, 4.37, 4.50, 4.76, 6.31, 6.42, 6.50, 8.33, 9.98, 12.79, 13.18, 13.83, 14.80, 15.25, 15.49, 16.39, 20.71, 21.81, 26.26, 26.71, 29.83, 30.61, 31.31, 31.37, 33.00, 33.94, 34.64, 35.99, 39.23, 39.47, 39.75, 41.34, 41.75, 42.52, 46.06, 48.37, 49.22, 55.56, 56.41, 57.78, 60.40, 64.02, 66.76, 73.62, 75.53, 75.56, 78.85, 83.37, 83.99, 84.16, 85.60, 98.77, 126.54, 126.94, 128.63, 130.87, 132.61, 136.50, 137.14, 139.55, 167.70, 171.56, 172.01, 207.99, 211.38, масс-спектр высокого разрешения (отрицательный ион FAB) m/z 1169.7 [(M-H), рассчитано для C65H111NO13Si2:1169.7].
Пример 6
C-22-этил-рапамицин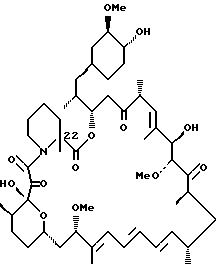
C-22-этил-бис-TES-рапамицин (0.25 г, 0.21 ммоль) растворяют в ТГФ (2 мл) и переносят в halgene пробирку, содержащую некоторое количество 4  молекулярных сит. В отдельной пробирке к сухому пиридину (1 мл) при 0oC добавляют HF/пиридин комплекс (1 мл). К субстрату при 0oC добавляют вышеупомянутый раствор (1,8 мл). Реакционную смесь перемешивают 15 мин при 0oC, затем оставляют перемешиваться при комнатной температуре 2 ч. После чего реакционную смесь охлаждают до 0oC и медленно добавляют NaHCO3 для того, чтобы прервать реакцию. Смесь экстрагируют EtOAc, промывают 0,1 н. HCl, NaHCO3 и раствором соли. Органическую фазу сушат над Na2SO4 и упаривают в вакууме. Смесь хроматографируют, используя 2,5%, затем 5% MeOH/CH2Cl2 в качестве элюента, что дает C-22-этил-рапамицин (0,13 г, 64% выход). ИК (KBr) 3440 (s), 2940 (s), 1725 (s), 1650 (m), 1460 (w), 1380 (w), 1235 (w), 1190 (w), 1090 (s), 995 (m), 915 (w), 735 (w), ПМР (400 MHz, CDCl3) δ 0.68 (m, 1H), 0.87-1.06 (соед. m, 15H), 1.06-1.45 (соед. m, 15H), 1.57 (m, 3Н), 1.66 (m, 3Н), 1.75 (m, 3Н), 1.90-2.08 (соед. m, 10Н), 2.31 (m, 3Н), 2.64 (m, 3Н), 2.89 (s, 1Н), 2.90 (m, 2Н), 3.13 (s, 3Н), 3.33 (m, 1Н), 3.38 (s, 3Н), 3.47 (m, 2Н), 3.66 (кажущийся t, J = 7.24 Hz, 1H), 3.82 (d, J = 5,47 Hz, 1H), 3,95 (m, 1H), 4.21 (d, J = 5,54 Hz, 1H), 4.43 (s, 1H), 5.09 (m, 1H), 5.37 (d, J = 9.62 Hz, 1H), 5.42 (d, J = 9.49 Hz, 1H), 6.05 (d, J = 10.93 Hz, 1H), 6.13 (dd, J = 10.5, 14.8 Hz, 1H), 6.24 (m, 1H), 6.39 (m, 1H), 13C ЯМР (100 MHz, CDCl3) δ 8.53, 10.19, 13.44, 13.98, 15.66, 15.91, 15.96, 16.53, 21.83, 21.97, 26.47, 26.93, 29.74, 30.79, 31.23, 31.44, 32.79, 33.27, 34.10, 34.59, 35.97, 39.12, 39.89, 40.04, 40.66, 41.36, 42.76, 46.20, 55.85, 56.58, 58.94, 64.17, 67.41, 73.97, 74.97, 74.96, 76.81, 83.37, 84.42, 85.16, 98.82, 126.84, 127.03, 128.66, 130.78, 132.82, 135.61, 136.99, 139.66, 167.40, 172.03, 194.05, 207.88, 213.99, масс-спектр высокого разрешения (отрицательный ион FAB) m/z 941 [(M-H), рассчитано для C53H82NO13: 941].
молекулярных сит. В отдельной пробирке к сухому пиридину (1 мл) при 0oC добавляют HF/пиридин комплекс (1 мл). К субстрату при 0oC добавляют вышеупомянутый раствор (1,8 мл). Реакционную смесь перемешивают 15 мин при 0oC, затем оставляют перемешиваться при комнатной температуре 2 ч. После чего реакционную смесь охлаждают до 0oC и медленно добавляют NaHCO3 для того, чтобы прервать реакцию. Смесь экстрагируют EtOAc, промывают 0,1 н. HCl, NaHCO3 и раствором соли. Органическую фазу сушат над Na2SO4 и упаривают в вакууме. Смесь хроматографируют, используя 2,5%, затем 5% MeOH/CH2Cl2 в качестве элюента, что дает C-22-этил-рапамицин (0,13 г, 64% выход). ИК (KBr) 3440 (s), 2940 (s), 1725 (s), 1650 (m), 1460 (w), 1380 (w), 1235 (w), 1190 (w), 1090 (s), 995 (m), 915 (w), 735 (w), ПМР (400 MHz, CDCl3) δ 0.68 (m, 1H), 0.87-1.06 (соед. m, 15H), 1.06-1.45 (соед. m, 15H), 1.57 (m, 3Н), 1.66 (m, 3Н), 1.75 (m, 3Н), 1.90-2.08 (соед. m, 10Н), 2.31 (m, 3Н), 2.64 (m, 3Н), 2.89 (s, 1Н), 2.90 (m, 2Н), 3.13 (s, 3Н), 3.33 (m, 1Н), 3.38 (s, 3Н), 3.47 (m, 2Н), 3.66 (кажущийся t, J = 7.24 Hz, 1H), 3.82 (d, J = 5,47 Hz, 1H), 3,95 (m, 1H), 4.21 (d, J = 5,54 Hz, 1H), 4.43 (s, 1H), 5.09 (m, 1H), 5.37 (d, J = 9.62 Hz, 1H), 5.42 (d, J = 9.49 Hz, 1H), 6.05 (d, J = 10.93 Hz, 1H), 6.13 (dd, J = 10.5, 14.8 Hz, 1H), 6.24 (m, 1H), 6.39 (m, 1H), 13C ЯМР (100 MHz, CDCl3) δ 8.53, 10.19, 13.44, 13.98, 15.66, 15.91, 15.96, 16.53, 21.83, 21.97, 26.47, 26.93, 29.74, 30.79, 31.23, 31.44, 32.79, 33.27, 34.10, 34.59, 35.97, 39.12, 39.89, 40.04, 40.66, 41.36, 42.76, 46.20, 55.85, 56.58, 58.94, 64.17, 67.41, 73.97, 74.97, 74.96, 76.81, 83.37, 84.42, 85.16, 98.82, 126.84, 127.03, 128.66, 130.78, 132.82, 135.61, 136.99, 139.66, 167.40, 172.03, 194.05, 207.88, 213.99, масс-спектр высокого разрешения (отрицательный ион FAB) m/z 941 [(M-H), рассчитано для C53H82NO13: 941].
Пример 7
Синтез C-22-метил-42-диметилглицин рапамицин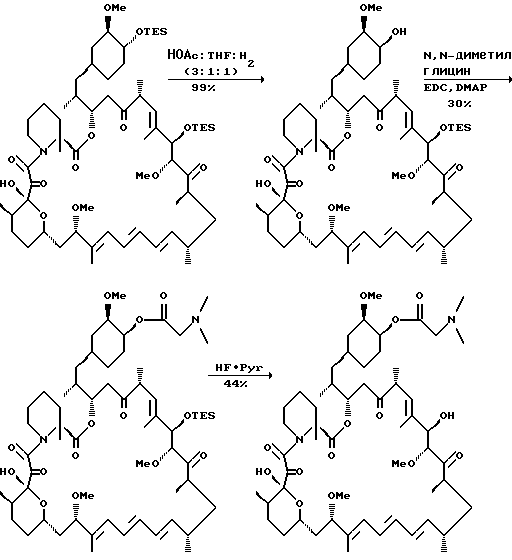
а) снятие одной защиты в 22-метилированном бис-TES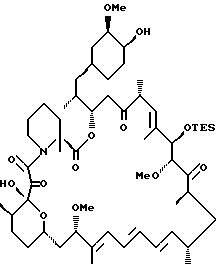
Метилированный бис-TES рапамицин (0,42 г, 0,36 ммоль) растворяют в 3:1:1 растворе НОАс: ТГФ:H2O. Реакция по данным ТСХ проходит за 10 минут, ее прерывают добавлением NaHCO3 и перемешивают 10 мин. Смесь экстрагируют EtOAc, сушат над Na2SO4, фильтруют и упаривают в вакууме. Остаток хроматографируют на силикагеле, элюируя 2,5% MeOH/CH2Cl2, что дает моно-TES метилированный гар (рапамицин) (0.37 г, 99% выход). ПМР (300 MHz, CDCl3) δ 0.47 (m, 6Н), 0.68 (m, 1H), 0.79-1.0 (соед. m, 30Н), 1.01-1.83 (соед. m, 13Н), 1.60 (s, 3Н), 1.63 (s, 3Н), 1.75 (s, 3Н), 1.92 (m, 2Н), 2.10 (m, 3Н), 2.31 (m, 2Н), 2.65 (m, 2H), 2.91 (m, 1H), 3.16 (s, 3Н), 3.21 (m, 2Н), 3.29 (s, 3Н), 3.36 (s, 3Н), 3.53 (m, 2Н), 3.64 (m, 1H), 3.78 (m, 1H), 3.81 (m, 1H), 4.14 (d, 1H), 4.75 (s, 1H), 4.85 (m, 1H), 5.13 (m, 1H), 5.4 (m, 1H), 6.07 (m, 1H), 6.2 (m, 2H), 6.4 (m, 1H)
b) ацилирование 22-метилированного моно-TES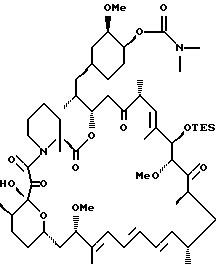
Метилированный моно-TES рапамицин (0,36 г, 0.35 ммоль) растворяют в метиленхлориде (8 мл). К реакционной смеси добавляют последовательно диметилглицин (3 экв. , 0.11 г), (3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (4 эквивал. , 0,27 г) и DМАР на кончике шпателя, после чего реакционную смесь оставляют перемешиваться в течение ночи. Затем реакционную смесь разбавляют метиленхлоридом и промывают водой. Водную фазу экстрагируют метиленхлоридом. Объединенные органические фазы промывают еще раз водой, сушат над Na2SO4, фильтруют и упаривают в вакууме. Смесь хроматографируют на силикагеле, элюируя 1%, затем 2,5% MeOH/CH2Cl2, что дает 42-глицинат моно-TES гар (0,12 г, 30% выход). ПМР (400 МНz, CDCl3) δ 0.73-1.06 (соед. m, 30Н), 1.18-1.81 (соед. m, 23Н), 1.62 (s, 3Н), 1.67 (s, 3Н), 1.82 (s, 3Н), 1.87-2.1 (соед. m, 4Н), 2.41 (s, 6Н), 2.63 (m, 2Н), 3.16 (m, 2Н), 3.29 (s, 3H), 3.35 (s, 3H), 3.36 (s, 2Н), 3.21-3.37 (соед. m, 2Н), 3.53 (m, 2Н), 3.68 (кажущийся t, J = 7.41 Hz, 1H), 3.89 (m, 2Н), 4.18 (d, J = 4.1 Hz, 1H), 4.74 (m, 1H), 4.80 (s, 1H), 4.92 (m, 1H), 5.20 (d, J = 10.4 Hz, 1H), 5.40 (dd, J = 7.0, 7.1 Hz, 1H), 6.09 (d, J = 10.7 Hz, 1H), 6.22 (m, 2Н), 6.42 (dd, J = 7.1, 7.2 Hz, 1H), масс спектр высокого разрешения (отрицательный ион FAB) m/z 1126.5 [(М-*), рассчитано для C62H102NO14Si:1126.5].
c) Снятие защиты 42-глицинат моно-TES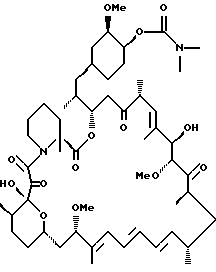
42-глицинат моно-TES рапамицин (0.11 г, 0.1 ммоль) растворяют в ТГФ (2 мл) и переносят в halgene пробирку, содержащую некоторое количество молекулярных сит. В отдельной halgene пробирке к 1 мл сухого пиридина при 0oC добавляют HF/пиридин комплекс (1 мл). 1 мл этого раствора добавляют в пробирку, содержащую субстрат при 0oC. Реакционную смесь перемешивают 15 мин, затем убирают ледяную баню и реакционную смесь оставляют перемешиваться при комнатной температуре 2 ч. Затем реакционную смесь охлаждают до 0oC и медленно добавляют NaHCO3, чтобы прервать реакцию. Смесь экстрагируют ЕtOAc, промывают 0,1н. HCl, NaHCO3 и раствором соли, последовательно. Органическую фазу сушат над Na2SO4 и упаривают в вакууме. Смесь хроматографируют на силикагеле, элюируя 2,5%, затем 5% MeOH/CH2Cl2, что дает C-22-метил-42-глицинат гар (0,043 г, 44% выход). ИК (KBr) 3440 (s), 2940 (s), 1730 (s), 1650 (m), 1460 (m), 1375 (w), 1290 (w), 1240 (w), 1190 (w), 1135 (w), 1100 (w), 990 (w), 730 (w), ПМР (400 MHz, CDCl3) δ 0.84 (dd, J = 12.1, 12.2 Hz, 1Н), 0.89 (d, J = 5.26 Hz, 3Н), 0.91 (d, J = 5.32 Hz, 3H), 1.00 (d, J = 6.56 Hz, 3Н), 1.05 (d, J = 1.7 Hz, 3Н), 1.07 (d, J = 1.73 Hz, 3Н), 0.87-1.45 (соед. m, 15H), 1.46-2.08 (соед. m, 10Н), 1.61 (s, 3Н), 1.65 (s, 3Н), 1.75 (s, 3Н), 2.35 (s, 6H), 2.65 (m, 2Н), 2.95 (s, 1Н), 3.14 (s, 3Н), 3.15 (m, 1Н), 3.17 (s, 2Н), 3.32 (m, 1H), 3.34 (s, 3Н), 3.36 (s, 3Н), 3.37 (m, 1Н), 3.50 (m, 2Н), 3.64 (кажущийся t, J = 7.4 Hz, 1H), 3.79 (m, 2Н), 4.22 (d, J = 5.7 Hz, 1H), 4.46 (s, 1H), 4.59 (m, 1H), 5.04 (m, 1H), 5.41 (d, J = 9.92 Hz, 1H), 5.44 (dd, J = 4.2, 7.1 Hz, 1H), 6.05 (d, J = 10.4 Hz, 1H), 6.13 (dd, J = 10.4, 12.6 Hz, 1H), 6.26 (dd, J = 12.6, 12.7 Hz, 1H), 6.38 (dd, J = 7.2, 7.3 Hz, 1H), 13C ЯМР (100 MHz, CDCl3) δ 10.15, 13.31, 14.02, 15.72, 16.01, 16.08, 17.17. 19.67, 21.81, 22.50, 26.96, 29.77, 30.91, 31.10, 32.88, 32.96, 33.71, 33.77, 35.80, 36.28, 38.84, 39.66, 39.98, 40.64, 41.32, 42.44, 45.21, 46.25, 55.93, 57.21, 59.09, 60.85, 67.32, 75.14, 76.33, 76.69, 77.00, 77.21, 77.32, 80.79, 83.68, 85.43, 98.66, 126.85, 128.93. 130.59, 133.03, 135.75, 136.49, 139.94, 167.86, 170.19, 172.36, 194.84, 207.97, 214.39, масс-спектр высокого разрешения (отрицательный ион FAB) m/z 1012.6 [(М-*), рассчитано для C62H102NO14Si:1012.6].
Описываются новые соединения общей формулы Iа и Ib, где R1 - С1-C6алкил; R2 - водород; SiEt3 или радикал формулы С(=O)-(СН2)yNR21R22, где j - целое число 1-3; R21 и R22 - С1-C3алкил; R3 - водород или SiEt3; R4 - водород или SiEt3, или их пролиновые аналоги, или их фармацевтически приемлемые соли, которые полезны для индуцирования иммуносупрессии и лечения отторжения трансплантата, гомологичной болезни, аутоиммунных заболеваний, воспалительных заболеваний, твердых опухолей, грибковых инфекций, лейкоза/лимфом дифференцированных Т-клеток и гиперпролиферативных васкулярных нарушений. Описываются также способ индуцирования иммуносупрессии, способ лечения или профилактики отторжения трансплантата или гомологичной болезни, способ лечения или профилактики гиперпролиферативных сосудистых нарушений. 4 с. и 18 з.п.ф-лы, 1 табл.
или
или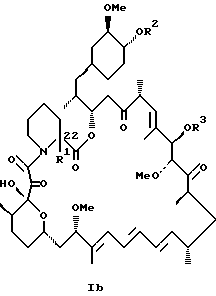
где R1 - C1 - C6 алкил,
R2 - водород, SiEt3 или радикал формулы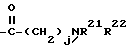
где j - целое число 1 - 3;
R21 и R22 - C1 - C3 алкил;
R3 - водород или SiEt3;
R4 - водород или SiEt3,
или их пролиновые аналоги, или их фармацевтически приемлемые соли.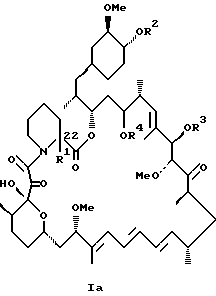
или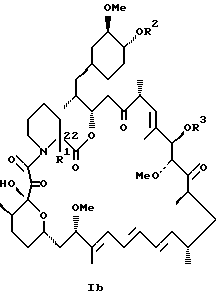
где R1 - C1 - C6 алкил,
R2 - водород, SiEt3 или радикал формулы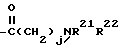
где j - целое число 1 - 3;
R21 и R22 - C1 - C3 алкил;
R3 - водород или SiEt3;
R4 - водород или SiEt3,
или его пролиновых аналогов или его фармацевтически приемлемой соли.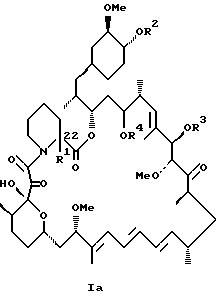
или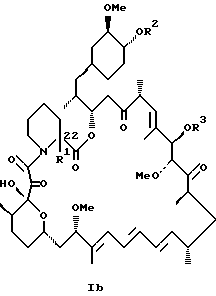
где R1 - C1 - C6 алкил,
R2 - водород, SiEt3 или радикал формулы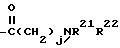
где j - целое число 1 - 3;
R21 и R22 - C1 - C3 алкил;
R3 - водород или SiEt3;
R4 - водород или SiEt3,
или его пролиновых аналогов, или его фармацевтически приемлемой соли.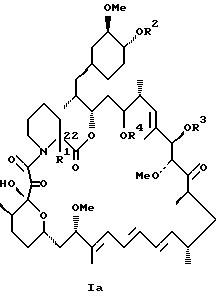
или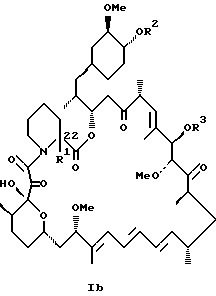
где R1 - C1 - C6 алкил,
R2 - водород, SiEt3 или радикал формулы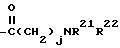
где j - целое число 1 - 3;
R21 и R22 - C1 - C3 алкил;
R3 - водород или SiEt3;
R4 - водород или SiEt3,
или его пролиновых аналогов, или его фармацевтически приемлемой соли.
| ^^СШОЮОНАЯ j'''' ^i''>&,v-n*C^-''?^t?u^|керамики | 0 |
|
SU378321A1 |
| US 5162333, 10.11.1992 | |||
| Способ получения производных 4(3Н)-оксо-5,6,7,8-тетрагидропиридо(2,3- @ )пиримидина или их таутомерных форм | 1987 |
|
SU1581222A3 |
| 0 |
|
SU401747A1 | |
| US 5665728, 09.09.1997. | |||

