Настоящее изобретение относится к соединениям формулы I
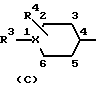
где R1 обозначает группу формулы (А), (В) или (С)
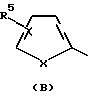

где Х в каждом случае независимо друг от друга обозначает S, О или N;
R2 и R4 каждый независимо друг от друга обозначает
(1) водород,
(2) алкоксигруппу или
(3) галоген;
R3 в каждом случае независимо друг от друга обозначает
(1) алкил,
(2) циклоалкил,
(3) галоген,
(4) гетероциклил,
(5) -NR8R9,
(6) -(CH2)mCONR8R9, где m обозначает целое число 0-3,
(7) -(CH2)mSO2NR8R9, где m обозначает целое число 0-3,
(8) -(CH2)mNR7COR9, где m обозначает целое число 0-3,
(9) -(CH2)mNR7SO2R9, где m обозначает целое число 0-3,
(10) -(СН2)mNR7C(V)NR8R9, где V обозначает S или О, а m обозначает целое число 0-3,
(11) -(CH2)mOY, где m обозначает целое число 0-3, a Y обозначает водород, алкил, алкилоксиалкил, циклоалкил, галоидалкил, гидроксиалкил, гетероциклил или карбоксиалкил, или
(12) -O(CH2)nZ, где n обозначает целое число 1-4, a Z обозначает циклоалкил, гидроксиалкил, циклоалкоксигруппу, гетероциклил, арилоксигруппу, гетероарил, -COR9, -CONR8R9, -SO2R9, -SO2NR8R9, -NR7SO2R9, незамещенный арил, моно-, ди- или тризамещенный арил, причем заместители независимо друг от друга выбраны из алкила, галогенов и алкилоксигруппы;
R5 в каждом случае независимо друг от друга обозначает
(1) -(CH2)mOY, где m обозначает целое число 0-3, а Y обозначает водород, алкил, алкилоксиалкил, циклоалкил, галоидалкил, гидроксиалкил, гетероциклил или карбоксиалкил, или
(2) -O(CH2)nZ, где n обозначает целое число 1-4, а Z обозначает циклоалкил, гидроксиалкил, циклоалкилоксигруппу, гетероциклил, арилоксигруппу, гетероарил, -COR9, -CONR8R9, -SO2R9, -SO2NR8R9, -NR7SO2R9, незамещенный арил, моно-, ди- или тризамещенный арил, причем заместители независимо друг от друга выбраны из алкила, галогенов и алкилоксигруппы;
R6 в каждом случае независимо друг от друга обозначает
(1) водород,
(2) -COR9,
(3) -CONR8R9,
(4) -С(V)NR8R9, где V обозначает О или S,
(5) -SO2R9 или
(6) -SO2NR8R9,
R7 и R8 каждый независимо друг от друга обозначает
(1) водород,
(2) алкил или
(3) гидроксиалкил;
R9 в каждом случае независимо друг от друга обозначает
(1) алкил,
(2) циклоалкил,
(3) арилалкил,
(4) гидроксиалкил,
(5) галоидалкил,
(6) гетероциклил,
(7) незамещенный арил, моно-, ди- или тризамещенный арил, причем заместители независимо друг от друга выбраны из алкила, галогенов и алкилоксигруппы, или
(8) гетероарил, или
R8 и R9 вместе с атомом азота, с которым они связаны, образуют 5- или 6-членное моноциклическое насыщенное или ненасыщенное кольцо, причем это кольцо необязательно замещено оксогруппой, или
R7 и R9 вместе с атомом азота, с которым они связаны, образуют 5- или 6-членное моноциклическое насыщенное или ненасыщенное кольцо, причем это кольцо необязательно замещено оксогруппой;
а также к их фармацевтически приемлемым солям или их кристаллическим формам.
По настоящему изобретению предлагаются далее фармацевтические композиции, содержащие в качестве компонента терапевтически эффективное количество соединения формулы I, его фармацевтически приемлемой соли или его кристаллической формы в смеси с одним или несколькими приемлемыми носителями.
Далее по настоящему изобретению предлагается способ лечения болевых состояний, вызванных самыми разнообразными причинами, включая, но не ограничиваясь ими, боли при воспалительных процессах, боли после хирургического вмешательства, висцеральные боли, зубную боль, предменструальную боль, головную боль, боль от ожога, мигрень или гистаминовые головные боли, боли при травмировании нервов, невритах, невралгиях, боли при отравлениях, ишемические боли, интерстициальный цистит, боли при раковых заболеваниях, болевые ощущения при заражении вирусами, паразитами и бактериями, посттравматические боли (включая болевые ощущения после переломов и спортивных травм); способ лечения воспалений, вызванных различными причинами, включая (но не ограничиваясь ими) бактериальную, грибковую и вирусную инфекцию, ревматоидный артрит, остеоартрит, хирургическое вмешательство, инфекции мочевого пузыря и идиопатическое воспаление мочевого пузыря, злоупотребления, пожилой возраст, недостаточность питания, простатит, конъюнктивит, боль, связанная с расстройствами пищеварительного тракта, такая, как синдром раздраженной толстой кишки, и, кроме того, способ лечения расстройств работы мочевого пузыря, связанных с синдромом инфравезикальной обструкции, и недержания мочи (включая острое недержание, недержание мочи при напряжении и повышенную реактивность мочевого пузыря); способ лечения астмы и септического шока у млекопитающих, включающий введение нуждающимся млекопитающим терапевтически эффективного количества соединения формулы I, его фармацевтически приемлемой соли или его кристаллической формы.
Простагландины или простаноиды (PG) составляют группу биологически активных соединений, выделенных из мембранных фосфолидов, и их получают из некоторых полиненасыщенных жирных кислот. Они относятся к нескольким основным классам, обозначенным литерами, включая D, Е, F, G, Н и I (простациклин). Эти основные классы далее подразделяются на подклассы, обозначенные подстрочными цифрами 1, 2 или 3, которые соответствуют предшествующей жирной кислоте, например PGE1 или PGE2. Простаноиды продуцируются убиквитарно, причем скорость их продуцирования обычно возрастает в ответ на действие различных раздражителей. Они, таким образом, проявляют самые различные фармакологические свойства.
Разнообразие действия простаноидов можно объяснить существованием ряда определенных рецепторов, которые медиируют их действие. Рецепторы для природного простагландина, к которому они обладают наибольшим сродством, получили названия и их разделили на пять основных типов, обозначенных как DP (PGD2), FP (PG2α), IP (PGI2), TP (ТХА2) и ЕР (РСЕ2). Дополнительная информация, касающаяся простагландинов и их рецепторов, приведена в работе Goodman & Gillman's, The Pharmacological Basis of Therapeutics, 9-е изд., McGraw-Hill, Нью-Йорк, 1996, глава 26, стр. 601-616.
Простаноиды продуцируются большинством клеток в ответ на механическую, термическую или химическую травму и воспалительный процесс и обусловливают сенсибилизацию или прямую активацию ближних чувствительных нервных окончаний. Для нескольких моделей ноцицептивных воспалительных процессов имеются сообщения о гиперальгетическом действии (повышенной чувствительности к раздражителю, которым обычно является болезненное ощущение). Даже несмотря на то, что PGE2 получил широкое признание как первичный медиатор гиперальгезии, под влиянием боли или воспалительного процесса выделяются значительные количества других простаноидов, включая PGI2. В самом деле, при прямом сравнении действия PGE2 и PGI2 на афферентный нейрон в качестве гиперальгетических или сенсибилизирующих агентов PGI2 как in vivo, так и in vitro оказывается равным или более эффективным. Однако до сих пор не существует каких-либо селективных рецепторных веществ-антагонистов, которые позволили бы с определенностью охарактеризовать подтип (подтипы) простаноидных рецепторов, медиирующих сенсибилизирующее действие PGE2 или PGI2.
Если принять во внимание характеристическую нестабильность и фармакокинетические свойства PGI2, то результаты большинства исследований анальгезии in vivo на грызунах позволяют предположить, что PGI2 играет основную роль в индукции гиперальгезии. Подобным же образом результаты исследований in vitro дают по существу явное основание полагать, что рецепторы IP действуют как важные модуляторы сенсорной функции нейронов. Поскольку рецепторы IP у сенсорных нейронов сообща активируют как аденилилциклазу, так и фосфолипазу С и, следовательно, кАМФ-зависимую протеинкиназу и протеинкиназу С, эти рецепторы способны оказывать мощное воздействие на активность ионных каналов и, таким образом, секретировать нейромедиаторы.
Последнее неоспоримое доказательство выдающейся роли рецепторов IP (предпочтительно PGI2) в болевых ощущениях при воспалительных процессах получено в недавних исследованиях на мышах, выведенных внутригенной мутацией, лишенных рецепторов IP (Т. Murata и др., Nature 1997, 388, 678-682). У этих животных вызываемые уксусной кислотой судороги или вызываемый каррагенаном отек лап ослабляли до уровней, аналогичных тем, который проявлялся у мышей диких разновидностей при введении в организм индометацина. В противоположность этому спинальные ноцицептивные рефлексы, которые определяли по ударам хвостом и испытанием с горячей плитой, были нормальными. Умеренные судороги как реакция, вызванная PGE2, у животных, выведенных внутригенной мутацией, оставались неизменными.
Основываясь на этих наблюдениях, можно полагать, что соединения по настоящему изобретению являются эффективными антиноцицептивными средствами.
Помимо того, что простаноиды являются медиаторами гиперальгезии, они, как известно, локально продуцируются в мочевом пузыре в ответ на действие физиологических раздражителей, таких, как растягивание детрузорной гладкой мышцы, травмы везикальной слизистой оболочки и стимуляции нервов [К. Anderson, Pharmacological Reviews, 1993, 45(3), 253-308]. PGI2 является основным простаноидом, выделяемым мочевым пузырем человека. Основываясь на нескольких сериях доказательств, полагают, что простаноиды могут служить связующим звеном между растягиванием детрузорной мышцы, происходящим при наполнении мочевого пузыря, и активацией С-волокнистых афферентов вследствие растяжения мочевого пузыря. Было высказано предположение о том, что простаноиды могут участвовать в патофизиологии расстройств работы мочевого пузыря, например в синдроме инфравезикальной обструкции, и состояниях, связанных с недержанием мочи, таких, как острое недержание, недержание мочи при напряжении и повышенная реактивность мочевого пузыря. Таким образом, полагают, что при лечении таких состояний окажутся полезными вещества-антагонисты простаноидных рецепторов IP.
В патентной литературе в качестве примеров упомянуты некоторые 2-(замещенный фенил) аминоимидазолиновые соединения. Так, в частности, в европейской заявке ЕР 0017484 В1 (поданной фирмой Fujisawa Pharmaceutical) описаны соединения, которые могут быть использованы для лечения гипертензивных, воспалительных и желудочно-кишечных заболеваний и для ослабления болей различной природы; в патенте США 4287201 (выдан на имя Olson и др.) описаны соединения, которые можно применять при задержке начала яйценоскости у молодых кур, при прерывании яйценоскости у взрослых кур и при инициировании искусственной линьки; в патенте США 4396617 (выдан на имя Dolman и Kuipers) описаны фунгициды, действующие против ржавчины бобов, бурой ржавчины пшеницы и мучнистой росы зерновых; в патенте США 4889868 (выдан на имя Huang) описаны ингибиторы липоксигеназы и фосфолипазы С и вещества-антагонисты рецептора фактора активации тромбоцитов, которые могут быть использованы для лечения воспалительных или аллергических состояний и инфаркта миокарда; в патенте США 5326776 (выдан на имя Winn и др.) описаны соединения, которые являются веществами-антагонистами рецептора ангиотензина II; в заявке на патент Великобритании GB 2038305 (поданной фирмой Duphar International Research) описаны соединения, которые можно применять для ингибирования роста боковых побегов растений табака или томатов, или ингибирования роста растительности на газонах, или для выращивания низкорослых декоративных растений; а в заявке WO 96/30350 (поданной фирмой Fujisawa Pharmaceutical) описаны соединения, которые могут быть использованы в качестве медикаментозных средств для профилактики и терапевтического лечения заболеваний, медиированных оксидазотной синтазой.
На фиг. 1 представлена рентгенограмма кристаллической формы I сульфата 2-[4-(4-изопропоксибензил)фенил]аминоимидазолина.
На фиг. 2 представлена рентгенограмма кристаллической формы II сульфата 2-[4-(4-изопропоксибензил)фенил]аминоимидазолина.
Используемые в данном описании и формуле изобретения следующие термины во всех случаях, если не указано иное, имеют приведенные ниже значения.
"Алкил" обозначает одновалентную разветвленную или неразветвленную насыщенную углеводородную группу, содержащую от одного до восьми углеродных атомов включительно, такую, как метил, этил, пропил, изопропил, изобутил, втор-бутил, трет-бутил, пентил, н-гексил и т.п.
"Циклоалкил" означает одновалентную насыщенную карбоциклическую группу, содержащую от трех до четырнадцати углеродных атомов включительно, такую, как циклопропилметил, циклопропилэтил, циклопропил, циклобутил, 3-этилциклобутил, циклопентил, циклогексил, циклогептил и т.п.
"Алкилоксигруппа" означает радикал -OR, где R обозначает алкил, который описан выше, необязательно замещенный одной или несколькими алкилоксигруппами. Примеры включают (хотя не ограничиваются ими) метокси-, этокси-, изопропокси-, втор-бутокси-, изобутокси-, 2-этокси-1-(этоксиметил)этоксигруппы и т.п.
"Циклоалкилоксигруппа" означает радикал -OR, где R обозначает циклоалкил, который описан выше, например циклопентилокси-, циклогексилоксигруппу и т.п.
"Гидроксиалкил" означает линейный одновалентный углеводородный радикал, содержащий от одного до четырех углеродных атомов, или разветвленный одновалентный углеводородный радикал, содержащий три или четыре углеродных атома, замещенный одной или двумя гидроксильными группами, при условии, что в случае наличия двух гидроксильных групп они не связаны с одним и тем же углеродным атомом. Примеры гидроксиалкильных радикалов включают, но не ограничены ими, гидроксиметил, 1-гидроксиметилэтил, 2-гидроксипропил, 3-гидроксипропил, 2-гидроксибутил, 3-гидроксибутил, 4-гидроксибутил, 2,3-дигидроксипропил, 1-(гидроксиметил)-2-гидроксиэтил, 2,3-гидроксибутил, 3,4-дигидроксибутил, 2-(гидроксиметил)-3-гидроксипропил и т.п., предпочтительно 2-гидроксиэтил и 1-(гидроксиметил)-2-гидроксиэтил.
"Алкилоксиалкил" означает гидроксиалкил, в котором водородный атом (водородные атомы) или одна или обе гидроксильные группы замещены С1-4алкилом, например 2-метоксиэтил, 3-метоксибутил, 2-метоксиметил, 2-изопропоксиэтил, 2-этокси-1-(этоксиметил)этил и т.п.
"Карбоксиалкил" означает радикал -RCOOH, где R обозначает алкил, который описан выше, например остаток 2-пропионовой кислоты, 3-бутановой кислоты и т.п.
"Арил" означает моноциклический ароматический углеводородный радикал, содержащий пять или шесть кольцевых атомов, или 9-14-членную бициклическую или трициклическую кольцевую систему, в которой по меньшей мере одно кольцо по своей природе является ароматическим. Примеры арильных радикалов включают, но не ограничены ими, бензил, фенил, нафтил и т.п.
"Арилоксигруппа" означает радикал -OR, где R обозначает арил, который описан выше, например феноксигруппу и т.п.
"Арилалкил" означает радикал RаRb-, где Ra обозначает арил, который описан выше, a Rb обозначает алкил, который описан выше, например бензил, фенетил, 3-фенилпропил и т.п.
"Гетероарил" означает моноциклическое ароматическое кольцо или 9-14-членную бициклическую кольцевую систему, в которой по меньшей мере одно кольцо по своей природе является ароматическим; это понятие охватывает гетероциклы, содержащие в кольце один, два или три гетероатома, выбранных из азота, кислорода и серы. Примеры гетероарильных радикалов включают, но не ограничены ими, тиенил, имидазолил, пиридинил, пиразинил и т.п.
"Гетероциклил" означает одновалентный насыщенный карбоциклический радикал, содержащий пять, шесть или семь кольцевых атомов, из которых один или два выбирают из азота, кислорода и серы. Примеры гетероциклильных радикалов включают, но не ограничены ими, тетрагидрофуранил, тетрагидропиранил, [1,3] диоксан-5-ил, 5-метил[1,3]диоксан-5-ил, морфолин, имидазолинил, пиперидинил, пирролидинил, пирролидин-2-он, пирролидин-2,3-дион и т.п., наиболее предпочтителен тетрагидропиранил.
"Галоген" означает атом фтора, брома, хлора или иода, предпочтительно фтора или хлора.
"Галоидалкил" означает алкил, замещенный одним-тремя атомами фтора или хлора, например хлорметил, трифторметил, 2,2,2-трифторэтил, 2,2,2-трихлорэтил и т.п.
"Аминозащитной группой" называют защитную группу, защищающую реакционноспособную аминогруппу, которая в противном случае в ходе проведения некоторых химических реакций подверглась бы превращению. Обычно используемые аминозащитные группы включают те, которые хорошо известны в данной области техники, например бензилоксикарбонил (карбобензилоксигруппа, КБЗ), пара-метоксибензилоксикарбонил, триалкилсилилкарбоксил, трифторметилкарбонил, пара-нитробензилоксикарбонил, N-трет-бутоксикарбонил (ВОС) и т.п. Некоторые аминозащитные группы предпочтительнее других благодаря относительной простоте удаления.
Термины "необязательный" и "необязательно" означают, что описываемое событие или обстоятельство может иметь место или его может не быть, и описание включает случаи, когда такие события или обстоятельства имеют место, и случаи, когда они отсутствуют. Так, например, "необязательно связанный" означает, что связь может содержаться или отсутствовать и что описание охватывает как одинарные, так и двойные связи.
"Инертный органический растворитель" или "инертный растворитель" означает растворитель, инертный в указанных в настоящем описании условиях проведения с его использованием в реакции, включая, например, бензол, толуол, ацетонитрил, тетрагидрофуран, N,N-диметилформамид, хлороформ (СНС13), метиленхлорид и дихлорметан (CH2Cl2), дихлорэтан, диэтиловый эфир, этилацетат, ацетон, метилэтилкетон, метанол, этанол, пропанол, изопропанол, трет-бутанол, диоксан, пиридин и т.п. Во всех случаях, если не указано иное, в качестве растворителей для проведения реакций по настоящему изобретению применяют инертные растворители.
Термин "фармацевтически приемлемый носитель" означает носитель, который может быть использован для приготовления фармацевтической композиции, который обычно совместим с другими компонентами композиции, не оказывает нежелательного действия на реципиента, не является нежелательным ни биологически, ни как-либо иначе и охватывает носители, которые приемлемы для ветеринарного применения, а также для фармацевтического применения при лечении человека. Термин "фармацевтически приемлемый носитель" в настоящем описании и формуле изобретения использован в качестве охватывающего как один, так и более чем один такой носитель.
Термин "фармацевтически приемлемая соль" соединения означает соль, которая фармацевтически приемлема и которая обладает требуемым фармакологическим действием исходного соединения. Такие соли включают
(1) кислотно-аддитивные соли, полученные с использованием минеральных кислот, таких, как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или полученные с использованием органических кислот, таких, как уксусная кислота, пропионовая кислота, гексановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфокислота, этансульфокислота, 1,2-этандисульфокислота, 2-гидроксиэтансульфокислота, бензолсульфокислота, 2-нафталинсульфокислота, 4-метилбицикло[2.2.2]окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 4,4'-метиленбис(3-гидрокси-2-ен-1-карбоновая кислота), 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и т.п., и
(2) соли, образующиеся, когда кислотный протон, содержащийся в исходном соединении, либо замещается ионом металла, например ионом щелочного металла, ионом щелочно-земельного металла или ионом алюминия, либо образует координационную связь с органическим основанием, таким, как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п.
Термин "кристаллическая форма" относится к различным твердым формам одного и того же соединения, например, к полиморфам, сольватам и аморфным формам.
(а) Полиморфы представляют собой кристаллические структуры, которые могут образовываться при кристаллизации соединения в виде различных кристаллических упаковок, причем все они характеризуются одинаковым элементным составом. Различным полиморфам обычно свойственны различные рентгенограммы, инфракрасные спектрограммы, точки плавления, значения плотности, твердости, кристаллические формы, оптические и электрические свойства, стабильность и растворимость. Конкретно используемый для перекристаллизации растворитель, скорость кристаллизации, температура хранения и другие факторы могут обусловить преобладание какой-либо одной кристаллической формы.
(б) Сольваты представляют собой в общем кристаллическую форму, которая включает либо стехиометрические, либо нестехиометрические количества растворителя. В процессе кристаллизации некоторые соединения в твердом кристаллическом состоянии часто проявляют тенденцию захватывать определенную молярную долю молекул растворителя, образуя таким образом сольват. Когда растворителем служит вода, могут образовываться гидраты.
(в) Аморфные формы представляют собой некристаллические материалы, не имеющие сколько-нибудь значительной упорядоченной зоны, которым обычно не свойственна четкая порошковая рентгенограмма.
Кристаллические формы по настоящему изобретению получены для сульфата 2-[4-(4-изопропоксибензил)фенил] аминоимидазолина, они обозначены как кристаллические формы I и II. Кристаллические формы I и II получали методами, описанными в примерах соответственно 1 и 22, они более подробно описаны в примерах 21-23. В целом кристаллические формы далее описаны в работе Вуrn и др. в Pharmaceutical Research, 1995, том 12(7), 945-954, и в Remington: The Science and Practice of Pharmacy, 1995, под редакцией E.W.Martin, Mack Publisching Company, 19-е изд. , Истон, шт. Пенсильвания, том 2, глава 83, 1447-1462.
"Лечение" заболевания включает
(1) профилактику заболевания, то есть предотвращение развития клинических симптомов заболевания у млекопитающих, которые могут подвергаться воздействию или оказаться предрасположенными к заболеванию, но еще не подвержены и не проявляют симптомов заболевания;
(2) ингибирование заболевания, т.е. задержку развития заболевания или его клинических симптомов, или
(3) ослабление заболевания, т.е. обеспечение регресса заболевания или его клинических симптомов.
"Терапевтически эффективное количество" означает то количество соединения, которое при введении в организм млекопитающего для лечения заболевания является достаточным для эффекта такого лечения заболевания. "Терапевтически эффективное количество" обычно варьируется в зависимости от соединения, болезненного состояния, которое необходимо устранить лечением, серьезности заболевания, которое лечат, возраста и относительного состояния здоровья пациента, пути и формы введения в организм, мнения соответствующего лечащего врача и других факторов.
Как хорошо известно в данной области техники, имидазолин-2-иламиновые группы в соединениях, таких, как соединения формулы I, находятся в таутомерном равновесии с имидазолин-2-илиденаминогруппами
Для удобства все соединения формулы I представлены как обладающие имидазолин-2-иламиновой структурой, но необходимо иметь в виду, что в объем изобретения включены обе таутомерные формы.
Наименование и нумерация положений в соединениях по настоящему изобретению проиллюстрированы ниже.

Боковые цепи заместителя R1 пронумерованы так, как это показано ниже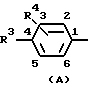

Соединения по изобретению называют имидазолиновыми производными, а номенклатура, используемая в настоящей заявке, основана в общем на рекомендациях ИЮПАК. Однако, поскольку жесткая привязка к этим рекомендациям приводила бы к существенному изменению названий при замене всего лишь одного заместителя, форма наименований соединений сохраняет соответствие номенклатуре в отношении базовой структуры молекулы.
Так, например, соединение формулы I, у которого R1 обозначает группу формулы (А), R2 и R4 обозначают водородные атомы, а R3 обозначает втор-бутоксигруппу, называют 2-[4-(4-втор-бутоксибензил)фенил]аминоимидазолином.
В частности, соединение формулы I, у которого R1 обозначает группу формулы (С), где Х обозначает S, R2 и R4 обозначают водородные атомы, а R5 обозначает метоксигруппу, называют 2-[4-(5-метокситиенил-2-илметил)фенил] аминоимидазолином.
Например, соединение формулы I, у которого R1 обозначает группу формулы (С), где Х обозначает N, R2 и R4 обозначают водородные атомы, а R3 обозначает этиламинокарбонил, называют 2-[4-(1-этиламинокарбонилпиперидин-4-илметил)фенил] аминоимидазолином.
В группе соединений по настоящему изобретению, которые представлены в кратком изложении сущности изобретения, предпочтительная категория включает соединения формулы I, у которых R2 и R4 каждый независимо друг от друга в каждом случае обозначает водород или галоген, предпочтительно водород, фтор или хлор.
В рамках этой категории одна предпочтительная подгруппа включает соединения формулы I, у которых R1 обозначает группу формулы (А), где
(1) R3 обозначает -(СН2)mОY, где m обозначает целое число 0-3, а предпочтительный Y обозначает
(а) алкил, предпочтительно метил, изопропил, втор-бутил, изобутил или трет-бутил;
(б) алкилоксиалкил, предпочтительно 2-этокси-1-(этоксиметил)этил;
(в) циклоалкил, предпочтительно циклопентил или циклогексил, или
(г) гетероциклил, предпочтительно тетрагидропиран-2-ил или тетрагидропиран-4-ил;
(2) R3 обозначает -O(CH2)nZ, где n обозначает целое число 1-4, a Z предпочтительно обозначает
(а) циклоалкил, предпочтительно циклопентил или циклогексил;
(б) гетероциклил, предпочтительно тетрагидропиран-2-ил или тетрагидропиран-4-ил;
(в) гидроксиалкил, предпочтительно 1-гидроксиметил;
(3) R3 обозначает -(CH2)mSO2NR8R9 или -(СН2)mСОNR8R9, где m обозначает целое число 0-3, а
(а) R8 обозначает водород или алкил, предпочтительно водород, метил, этил или изопропил;
(б) R9 обозначает
(I) алкил, предпочтительно метил, этил, пропил, изопропил, втор-бутил, н-бутил, изобутил, втор-бутил или трет-бутил, или
(II) арилалкил, предпочтительно бензил, или
(4) R3 обозначает -(CH2)mNR7SO2R9 или -(CH2)mNR7COR9, где m обозначает целое число 0-3, а
(а) R7 обозначает водород или алкил, предпочтительно водород, метил, этил или пропил;
(б) R9 обозначает
(I) алкил, предпочтительно метил, этил, пропил или изопропил;
(II) арил, предпочтительно фенил, или
(III) арилалкил, предпочтительно бензил.
В рамках этой категории другая предпочтительная подгруппа включает соединения формулы I, у которых R1 обозначает группу формулы (В), где Х обозначает S, а
(1) R3 обозначает -(СН2)mОY, предпочтительно где m обозначает целое число 0 или 1, a Y обозначает
(а) алкил, предпочтительно метил, изопропил, изобутил, втор-бутил или трет-бутил;
(б) алкилоксиалкил, предпочтительно 2-этокси-1-(этоксиметил)этил;
(в) циклоалкил, предпочтительно циклопентил или циклогексил, или
(г) гетероциклил, предпочтительно тетрагидропиран-2-ил или тетрагидропиран-4-ил, или
(2) R3 обозначает -O(CH2)nZ, где n обозначает целое число 1-4, a Z имеет значение, указанное для группы формулы (А).
В рамках этой категории другая предпочтительная подгруппа включает соединения формулы I, у которых R1 обозначает группу формулы (С), в которой Х обозначает N.
Примерами особенно предпочтительных соединений являются
2-[4-(4-изопропоксибензил)фенил]аминоимидазолин;
2-{4-[4-(втор-бутокси)бензил]фенил}аминоимидазолин;
2-{4-[4-(циклопентилокси)бензил]фенил}аминоимидазолин;
2-{4-[4-(тетрагидропиран-4-илокси)бензил]фенил}аминоимидазолин;
2-{4-[4-(тетрагидропиран-4-илметокси)бензил]фенил}аминоимидазолин;
2-{4-[4-(тетрагидропиран-2-илметокси)бензил]фенил}аминоимидазолин;
2-{ 4-[2-фтор-4-(тетрагидропиран-4-илметокси)бензил] фенил} аминоимидазолин;
2-{4-[4-(2-этокси-1-(этоксиметил)этокси)бензил]фенил}аминоимидазолин;
2-[4-(4-циклопентилокситиенил-2-илметил)фенил]аминоимидазолин;
2-{4-[4-(4-метоксифенил)сульфонилметиламиноэтокси)бензил]фенил}аминоимидазолин;
2-{4-[4-(1-гидроксиметилэтокси)бензил]фенил}аминоимидазолин;
2-[4-(5-метокситиенил-2-илметил)фенил]аминоимидазолин;
2-[4-(4-бутиламиносульфонилбензил)фенил]аминоимидазолин;
2-[4-(4-изопропоксиметилбензил)фенил]аминоимидазолин;
2-[4-(4-втор-бутоксиметилбензил)фенил]аминоимидазолин;
2-{4-[4-(изобутиламиносульфонил)бензил]фенил}аминоимидазолин;
2-[4-(4-бензиламинокарбонилбензил)фенил]аминоимидазолин;
2-[4-(4-изопропиламиносульфонилбензил)фенил]аминоимидазолин;
2-[4-(4-изобутиламинокарбонилбензил)фенил]аминоимидазолин и
2-[4-(4-трет-бутиламиносульфонилбензил)фенил]аминоимидазолин.
Соединения по настоящему изобретению могут быть получены методами, проиллюстрированными реакционными схемами, приведенными ниже.
Исходные материалы и реагенты, которые использованы при получении этих соединения, могут быть либо приобретены у промышленных поставщиков, таких, как фирма Aldrich Chemical Co., либо получены методами, которые известны специалистам в данной области техники, в ходе проведения процессов, описанных в такой литературе, как Fieser and Fieser's Reagents for Organic Synthesis, Wiley & Sons: Нью-Йорк, 1991, тома 1-15; Rodd's Chemistry of Carbon Compounds, Elsevier Science Publishers, 1989, тома 1-5 и дополнения, и Organic Reactions, Wiley & Sons: Нью-Йорк, 1991, тома 1-40. Эти схемы являются лишь иллюстрациями некоторых методов, в соответствии с которыми могут быть синтезированы соединения по настоящему изобретению, поэтому такие схемы можно подвергать различным модификациям, которые обычно может предположить любой специалист в данной области техники, ссылаясь на настоящее описание. Если необходимо, исходные материалы и промежуточные продукты такой реакции можно выделять и очищать в соответствии с обычными техническими приемами, включая, но не ограничиваясь ими, фильтрование, перегонку, кристаллизацию, хроматографию и т. п. Такие материалы можно охарактеризовать с применением обычных средств, включая физические константы и спектрографические данные.
Во всех случаях, если не указано иное, представляемые в настоящем описании реакции проводят под атмосферным давлением в температурном интервале от примерно -78 до примерно 150oС, более предпочтительно от примерно 0 до примерно 125oС, а наиболее предпочтительно при примерно комнатной температуре (или температуре окружающей среды), например при приблизительно 20oС.
Обычно соединения формулы I получают проведением взаимодействия промежуточных фениламиновых соединений формул Ia-II с имидазолиновым соединением 40 в форме кислотно-аддитивной соли или свободного основания. Схемы А-Ж иллюстрируют методы получения промежуточных фениламиновых соединений, у которых R1 обозначает группу формулы (А); схемы З и И иллюстрируют методы получения промежуточных фениламиновых соединений, у которых R1 обозначает группу формулы (В); схемы К-М иллюстрируют методы получения промежуточных фениламиновых соединений, у которых R1 обозначает группу формулы (С). Схема М иллюстрирует метод получения соединений формулы I.
На схеме А проиллюстрирован способ получения соединений формулы I, у которых R1 обозначает группу формулы (А), а R3 обозначает -O(CH2)nZ или -(СН2)mОY, из соответствующего промежуточного соединения формулы Iа.
Обычно исходные соединения формул 1а, 1b, 1с, 2а, 2с и 3а технически доступны, например, на фирме Aldrich Chemical Company, или либо известны, либо могут быть легко синтезированы любыми специалистами в данной области техники. Так, например, метоксибензоилнитробензол 3а можно получить по методу, который описан в работе Shani, Jashovam и др., опубликованной в J. Med. Chem., 1985, 28, 1504.
Метод (а) иллюстрирует получение соединения формулы Iа, в которой R3 имеет указанные выше значения, в частности где m обозначает 0.
На стадии 1 метоксибензоилнитробензол 3а получают ацилированием метоксибензола 1а ацилирующим агентом 2а, где L обозначает уходящую группу, такую, как хлор, в условиях ацилирования по Фриделю-Крафтсу. Эту реакцию проводят в инертной атмосфере в присутствии кислоты Льюиса, такой, как хлорид алюминия, трифторид бора и т.п. Приемлемые для проведения этой реакции инертные органические растворители включают галоидированные углеводороды, такие, как дихлорметан, дихлорэтан, сероуглерод и т.п., предпочтительно сероуглерод.
На стадии 2 гидроксибензоилнитробензол 4а получают обработкой соединения 3а сильной кислотой, такой, как смесь бромистоводородной кислоты с ледяной уксусной кислотой. Реакция деметилирования протекает с выдержкой при высокой температуре или при температуре кипения с обратным холодильником.
На стадии 3 соединение 5а получают прямым алкилированием соединения 4а алкилирующим агентом, таким, как алкилгалогенид, или ацилированием ацилирующим агентом, таким, как эфир галоидкарбоновой кислоты. Реакция протекает в инертной атмосфере в присутствии иодидного катализатора, такого, как иодид натрия или калия, и основания, такого, как карбонат калия, карбонат натрия или карбонат цезия. Приемлемые для проведения такой реакции растворители включают апротонные органические растворители, например ацетон, ацетонитрил, N, N-диметилформамид, N-метилпирролидон, тетрагидрофуран и т.п., предпочтительно тетрагидрофуран.
По другому варианту соединение 5а получают взаимодействием соединения 4а с органическим фосфином, таким, как трифенилфосфин, в сочетании с диалкилазодикарбоксилатом, таким, как диэтилазодикарбоксилат, в условиях реакции Мицунобу. Приемлемые для проведения этой реакции растворители включают инертные органические растворители, такие, как N,N-диметилформамид, N-метилпирролидон, этилацетат, тетрагидрофуран и т.п., предпочтительно тетрагидрофуран.
На стадии 4 фениламиновое соединение формулы Iа получают восстановлением кетогруппы и нитрогруппы соединения 5а. Приемлемые для восстановления кето- и нитрогрупп условия включают наличие борида никеля в подкисленном метаноле или каталитическую гидрогенизацию с использованием платинового или палладиевого катализатора (например, РtO2 или Pd/C, предпочтительно 10%-ного Pd/C) в протонном органическом растворителе, таком, как подкисленный метанол или подкисленный этанол, предпочтительно подкисленный этанол.
Метод (б) иллюстрирует другой вариант получения соединения формулы Iа, в которой R3 имеет указанные выше значения, в частности где m обозначает 0.
На стадии 1 замещенный бромбензол 2b получают обработкой бромфенола 1b алкилирующим агентом, таким, как алкилгалогенид. Эта реакция протекает в присутствии основания, такого, как карбонат калия, карбонат натрия или карбонат цезия, и катализатора, такого, как иодид натрия. Приемлемые для проведения этой реакции растворители включают апротонные растворители, такие, как N,N-диметилформамид, тетрагидрофуран, ацетонитрил и т.п.
На стадии 2 металлорганическое соединение 3b, где М+Br- обозначает металлорганический реагент, может быть легко синтезировано обычными специалистами в данной области техники, например, путем обработки соединения 2b подходящим металлом в условиях реакции Гриньяра. Реакция протекает в инертной атмосфере в среде апротонного органического растворителя, такого, как тетрагидрофуран.
На стадии 3 бензоилнитробензол 5b получают взаимодействием соединения 3b с ацилирующим агентом, таким, как ацилгалогенид, где L обозначает уходящую группу, такую, как хлор, в присутствии катализатора, такого, как тетракис(трифенилфосфин) палладий. Реакция протекает в инертной атмосфере в среде апротонного органического растворителя, такого, как тетрагидрофуран.
В дальнейшем фениламиновое соединение формулы Iа получают в соответствии с методами, представленными выше на схеме А, метод (а), стадия 4.
Метод (в) иллюстрирует другой вариант получения соединения формулы Iа, в которой R3 имеет указанные выше значения, в частности где m обозначает 1.
Бензилбромид 1d получают взаимодействием бромбензилбромида 1с с целевым спиртом в присутствии сильного основания, такого, как гидрид натрия. Реакция протекает в инертной атмосфере в среде апротонного растворителя, такого, как N,N-диметилформамид, тетрагидрофуран, ацетонитрил и т.п.
Хлорбензилфениламин 2d с защищенным амином, у которого Р обозначает аминозащитную группу, получают взаимодействием хлорбензилизоцианата 2с с аминозащитным реагентом, таким, как триалкилсилилэтиловый спирт. Реакция протекает в инертной атмосфере в среде апротонного растворителя, такого, как N, N-диметилформамид, тетрагидрофуран, ацетонитрил и т.п.
На стадии 1 бензилфенил 3d с защищенным амином получают реакцией сочетания соединения 1d с соединением 2d в условиях реакции Стилла. Эта реакция протекает, например, в присутствии литиированных соединений, таких, как трет-бутиллитий, соединений олова, таких, как трибутилоловогалогенид, и катализатора, такого, как тетракис(трифенилфосфин)палладий. Реакция протекает в инертной атмосфере в среде апротонного растворителя, такого, как гексаметилфосфорамид, N, N-диметилформамид, тетрагидрофуран, ацетонитрил, диметилсульфоксид и т.п.
На стадии 2 фениламиновое соединение формулы Iа получают удалением аминозащитной группы из соединения 3d путем его обработки специальным отщепляющим реагентом, например тетра-н-бутиламмонийфторидом. Эту реакцию проводят в инертной атмосфере в среде апротонного растворителя, такого, как N,N-диметилформамид, тетрагидрофуран, ацетонитрил, диметилсульфоксид и т.п.
Процессы получения соединений формулы I по такому методу из соответствующих соединений формулы Iа подробно описаны в примерах 1-4. Процессы получения соединений формул 1d и 2d подробно представлены в примерах получения соответственно 1 и 2.
На схеме Б проиллюстрирован другой способ получения соединений формулы I, в которой R1 обозначает группу формулы (A), a R3 обозначает алкил, циклоалкил, галоид, гетероциклил или -NR8R9, из соответствующего промежуточного соединения формулы Ib.
Обычно исходные соединения le, 2e, 1f и 2f либо коммерчески доступны, например поставляются фирмой Aldrich Chemical Company, либо известны специалистам в данной области техники, либо могут быть легко синтезированы. Метод (а) иллюстрирует получение соединения формулы Ib, R3 которой указаны выше, с использованием в качестве исходного продукта фторбензола le.
На стадии 1 фторбензоилнитробензол 3е получают ацилированием фторбензола le ацилирующим агентом 2e, где L обозначает уходящую группу, такую, как хлор. Приемлемые для проведения этой реакции растворители включают галоидированные углеводороды, такие, как дихлорметан, дихлорэтан, сероуглерод и т.п., предпочтительно сероуглерод.
На стадии 2 R3-замещенный бензоилнитробензол 6 получают замещением атома фтора в соединении формулы 3е первичным или вторичным амином, таким, как диметиламин, морфолин и т.п. Эту реакцию проводят в присутствии основания, например карбоната калия, карбоната натрия, карбоната цезия и т.п., в среде апротонного органического растворителя, такого, как тетрагидрофуран, N,N-диметилформамид, диметилсульфоксид и т.п., предпочтительно в диметилсульфоксиде.
На стадии 3 фениламиновое соединение формулы Ib получают восстановлением кетогруппы и нитрогруппы соединения формулы 6 в реакционных условиях, описанных в отношении схемы А, метод (а), стадия 4.
Метод (б) иллюстрирует другой вариант получения соединения формулы Ib, в которой R3 обозначает, в частности, алкил или циклоалкил.
На стадии 1 алкилбензоилнитробензол 6 получают проведением взаимодействия алкилбензола 1f с ацилирующим агентом 2f, где L обозначает уходящую группу, такую, как хлор, в условиях ацилирования по Фриделю-Крафтсу. Эту реакцию проводят в присутствии кислоты Льюиса, такой, как хлорид алюминия, трифторид бора и т.п. Приемлемые для проведения этой реакции растворители включают галоидированные углеводороды, такие, как дихлорметан, дихлорэтан, сероуглерод и т.п., предпочтительно сероуглерод.
На стадии 2 фениламиновое соединение формулы Ib получают восстановлением кетогруппы и нитрогруппы соединения формулы 6 в реакционных условиях, описанных в отношении схемы А, метод (а), стадия 4.
Процесс получения соединения формулы I по такому методу из соответствующего соединения формулы Ib подробно описан в примере 5.
На схеме В проиллюстрирован альтернативный способ получения соединений формулы I, R1 которой обозначает группу формулы (А), а R3 обозначает -(CH2)mNR7SO2R9, из соответствующих промежуточных соединений формулы Ic.
Метод (а) иллюстрирует получение соединения формулы Iс, в которой R3 обозначает сульфонамидную группу, в частности, где m обозначает 1.
На стадии 1 метилбензоилнитробензол 3g получают ацилированием метилбензола lg ацилирующим агентом 2g, где L обозначает уходящую группу, такую, как хлор, в условиях ацилирования по Фриделю-Крафтсу. Эту реакцию проводят в инертной атмосфере в присутствии кислоты Льюиса, такой, как хлорид алюминия, трифторид бора и т.п. Приемлемые для проведения этой реакции инертные органические растворители включают галоидированные углеводороды, такие, как дихлорметан, дихлорэтан, сероуглерод и т.п., предпочтительно сероуглерод.
На стадии 2 бромбензоилнитробензол 7 получают бензилбромированием соединения формулы 3g подходящим бромирующим агентом, таким, как N-бромсукцинимид. Бромирование протекает при повышенной температуре в присутствии инициатора свободнорадикальной полимеризации, такого, как перекись бензоила, в инертной атмосфере (например, в аргоне или азоте, предпочтительно в аргоне). Приемлемыми для проведения этой реакции неполярными растворителями являются хлорированные и ароматические углеводороды, такие, как тетрахлорид углерода и бензол.
На стадии 3 бромбензилнитробензол 8 получают восстановлением кетогруппы соединения формулы 7 обработкой восстановителем, селективным в отношении кетогруппы, таким, как триэтилсилан. Эта реакция протекает в инертной атмосфере в присутствии сильной кислоты, такой, как трифторметансульфокислота. Приемлемые для проведения этой реакции растворители включают галоидированные углеводороды, такие, как дихлорметан и дихлорэтан.
На стадии 4 азидобензилнитробензол 9 получают замещением бензилбромида в соединении 8 нуклеофильным азидным анионом. Приемлемыми для проведения этой реакции растворителями являются апротонные органические растворители, такие, как N,N-диметилформамид, N-метилпирролидон, тетрагидрофуран и т.п.
На стадии 5 аминобензилнитробензол 10 получают восстановлением азида до первичного амина проведением взаимодействия соединения 9 с приемлемым восстанавливающим азид агентом, таким, как трифенилфосфин и вода. Приемлемыми для проведения этой реакции растворителями являются органические растворители, такие, как диэтиловый эфир, 1,4-диоксан, тетрагидрофуран и т.п., предпочтительно тетрагидрофуран.
На стадии 6 сульфониламинобензилнитробензол 11 получают проведением взаимодействия соединения 10 с сульфонилирующим агентом R9SO2L, где L обозначает уходящую группу, в частности хлор, в присутствии основания, например триэтиламина. Сульфонилгалогениды технически доступны или могут быть получены по таким методам, которые описаны в работах Langer R.F., Can. J. Chem., 1983, 61, 1583-1592; Aveta R. и др., Gazetta Chimica Italiana, 1986, 116, 649-652, и King J.F. и Hillhouse J.H., Can. J. Chem., 1976, 54, 498. Приемлемыми для проведения этой реакции растворителями являются галоидированные углеводороды, такие, как дихлорметан, или двухфазные системы, включающие воду и этилацетат (например, реакция по методике Шоттен-Баумана).
На стадии 7 фениламиновое соединение Iс получают восстановлением нитрогруппы соединения 11 до аминогруппы. Приемлемые восстанавливающие нитрогруппу средства включают борид никеля в подкисленном метаноле и каталитическую гидрогенизацию с использованием платинового или палладиевого катализатора (например, PtO2 или Pd/C) в органическом растворителе, таком, как этанол или этилацетат.
В другом варианте метод (б) иллюстрирует получение соединения формулы Iс, в которой R3 обозначает сульфонамидную группу, в частности где m обозначает 0.
Соединение формулы Iс' получают проведением взаимодействия 4,4'-метилендианилина 12 с сульфонилирующим агентом, таким, как сульфонилгалогенид, в реакционных условиях, указанных выше в описании стадии 6, и с осуществлением кислотно-щелочной экстракции с помощью гидроксида и минеральной кислоты.
Соединение формулы Iс может быть (но необязательно) получено дальнейшим алкилированием соединения формулы Iс' приемлемым алкилирующим агентом в присутствии сильного основания, такого, как трет-бутоксид калия. Подходящие растворители включают апротонные органические растворители, такие, как ацетонитрил, N,N-диметилформамид, диметилсульфоксид и т.п., предпочтительно диметилсульфоксид.
Процессы получения соединений формулы I по такому методу из соответствующих соединений формул Iс' и Iс подробно описаны в примерах 6-9.
На схеме Г проиллюстрирован альтернативный способ получения соединений формулы I, в которой R1 обозначает группу формулы (А), а R3 обозначает -(CH2)mNR7COR9, из соответствующих промежуточных соединений формулы Id.
Метод (а) иллюстрирует получение соединения формулы Id, в которой R3 обозначает карбоксамидную группу, в частности где m обозначает 1.
В целом соединение формулы Id получают в реакционных условиях, которые ранее указаны в отношении схемы В, метод (а), но на стадии 6 проводят взаимодействие аминобензилнитробензола 10 с ацилирующим агентом R9COL, где L обозначает уходящую группу, такую, как хлор, с получением карбоксамида 13. Далее фениламиновое соединение формулы Id получают проведением соответствующего процесса так, как указано в описании стадии 7. В другом варианте метод (б) иллюстрирует получение соединения формулы Id, R3 которой обозначает сульфонамидную группу, в частности где m обозначает 0.
В целом соединение формулы Id' получают в реакционных условиях, которые ранее указаны в отношении схемы В, метод (б), но проводят взаимодействие 4,4'-метилендианилина 12 с ацилирующим агентом, таким, как ацилгалогенид, с получением соединения формулы Id'. Затем можно (но необязательно) соединение формулы Id получать дальнейшим алкилированием соединения формулы Id' подходящим алкилирующим агентом в присутствии сильного основания, такого, как трет-бутоксид калия. Подходящие растворители включают апротонные органические растворители, такие, как тетрагидрофуран, N,N-диметилформамид, диметилсульфоксид и т.п., предпочтительно диметилсульфоксид.
Процесс получения соединения формулы I по этому методу из соответствующего соединения формулы Id описан в примере 7.
На схеме Д проиллюстрирован альтернативный способ получения соединений формулы I, в которой R1 обозначает группу формулы (А), а R3 обозначает -(CH2)mNR7C(V)NR8R9, где V обозначает S или О, из соответствующих промежуточных соединений формулы Iе.
Метод (а) иллюстрирует получение соединения формулы Iе, в которой R3 обозначает мочевиновую/тиомочевиновую группу, где m, в частности, обозначает 1.
В целом соединение формулы Iе получают в реакционных условиях, которые ранее указаны в отношении схемы В, метод (а), но на стадии 6 проводят взаимодействие аминобензилнитробензола 10 с изоциана-том/изотиоцианатом в апротонном органическом растворителе с получением мочевинового/тиомочевинового соединения 14. Далее фениламиновое соединение формулы Iе получают проведением соответствующего процесса, как это указано в описании стадии 7.
В другом варианте метод (б) иллюстрирует получение соединения формулы Iе, в которой R3 обозначает мочевиновую/тиомочевиновую группу, в частности где m обозначает 0.
В целом соединение формулы Iе' получают в реакционных условиях, которые ранее указаны в отношении схемы В, метод (б), но проводят взаимодействие 4,4'-метилендианилина 12 с изоцианатом/изотиоцианатом в апротонном органическом растворителе, таком, как дихлорметан, тетрагидрофуран, N,N-диметилформамид и т.п. Затем дальнейшим алкилированием соединения формулы Iе' необязательно можно получить соединение формулы Iе пригодным алкилирующим агентом в присутствии сильного основания, такого, как трет-бутоксид калия. Пригодные растворители включают апротонные органические растворители, такие, как тетрагидрофуран, диметилсульфоксид, N,N-диметилформамид и т.п., предпочтительно диметилсульфоксид.
В другом варианте метод (в) иллюстрирует получение соединения формулы I, в которой R3 обозначает мочевиновую/тиомочевиновую группу, в частности где m обозначает 0.
На стадии 1 соединение 15, у которого Р обозначает аминозащитную группу, получают присоединением приемлемой аминозащитной группы, такой, как бензил, трет-бутоксикарбонил (ВОС) или карбобензилоксигруппа (КБЗ), к соединению 12 методами, которые обычному специалисту в данной области техники известны, например, в условиях реакции Шоттен-Баумана.
На стадии 2 мочевиновое/тиомочевиновое соединение 16 получают проведением взаимодействия соединения 15 с изоцианатом/изотиомочевиной в органическом растворителе, включая дихлорметан, дихлорэтан или тетрагидрофуран.
На стадии 3 соединение формулы Iе получают удалением аминозащитной группы из соединения 16 в условиях гидрогенизации с использованием катализатора, такого, как палладиевые и платиновые катализаторы. Приемлемые для проведения этой реакции растворители включают протонные и апротонные органические растворители, такие, как метанол, этанол, этилацетат и т.п.
Процесс получения соединения формулы I по такому методу из соответствующего соединения формулы Iе подробно описан в примере 10.
На схеме Е проиллюстрирован альтернативный способ получения соединений формулы I, R1 которой обозначает группу формулы (A), a R3 обозначает группу -(CH2)mSO2NR8R9, из соответствующих промежуточных соединений формулы If.
Метод (а) иллюстрирует получение соединения формулы If, в которой R3 обозначает сульфонамидную группу, в частности где m обозначает 0.
На стадии 1 хлорсульфонилбензилнитробензол 18 получают взаимодействием бензилнитробензола 17 с хлорсульфирующим агентом, таким, как хлорсульфокислота. Эта реакция протекает при температуре от примерно -50 до 10oС в инертном органическом растворителе, таком, как дихлорметан или дихлорэтан.
На стадии 2 аминосульфонилбензилнитробензол 20 получают проведением взаимодействия соединения 18 с первичным или вторичным амином. Приемлемые для проведения этой реакции растворители включают инертные органические растворители, такие, как дихлорметан, дихлорэтан или тетрагидрофуран.
На стадии 3 фениламиновое соединение It получают восстановлением нитрогруппы соединения 37 до аминогруппы. Приемлемые для восстановления нитрогруппы условия включают каталитическую гидрогенизацию с использованием платинового или палладиевого катализатора в протонном органическом растворителе, таком, как метанол, этанол или этилацетат.
Метод (б) иллюстрирует другой вариант получения соединения формулы If, в которой R3 обозначает сульфонамидную группу, в частности где m обозначает 1.
Бромбензилнитробензол 7 получают аналогично тому, как это описано выше в отношении схемы В.
На осуществляемой по другому варианту стадии 1а соединение 19 получают взаимодействием соединения 7 с солью сернистой кислоты, такой, как водный сульфит натрия или сульфит калия. Реакция протекает при температуре кипения с обратным холодильником в воде или смеси ацетонитрила с водой.
Далее на осуществляемой по другому варианту стадии 1б соединение 18 получают обработкой соединения 19 хлорирующим агентом, таким, как пентахлорид фосфора. Реакцию можно проводить с использованием только реагентов или в присутствии оксихлорида фосфора.
Далее фениламиновое соединение формулы If получают соответствующим осуществлением стадий, аналогичных стадиям 2 и 3 в методе (а) на схеме Е.
Процесс получения соединений формулы I по этому методу из соответствующих соединений формулы If подробно описан в примерах 11-12.
На схеме Ж проиллюстрирован альтернативный способ получения соединений формулы I, в которой R1 обозначает группу формулы (A), a R3 обозначает -(CH2)mCONR8R9, из соответствующих промежуточных соединений формулы Ig.
На стадии 1 бензилбензойную кислоту 22 получают восстановлением кетоновой группы бензоилбензойной кислоты 21 с помощью восстановителя, селективного в отношении кетоновой группы, в частности в гидрогенизационных условиях с использованием палладиевого или платинового катализатора. Реакция протекает при комнатной температуре в присутствии сильной кислоты, такой, как перхлорная кислота. Приемлемыми для проведения этой реакции растворителями являются протонные и апротонные растворители, такие, как метанол, этанол, этилацетат и т.п.
На стадии 2 нитробензилбензойную кислоту 23 получают по методу, который описан в химической литературе, например в работе Coon и др. в J. Org. Chem. , 1973, 38, 4243. В целом соединение 22 нитруют с получением нитрониевых солей таким путем, как взаимодействие с трифторметансульфоновой кислотой и азотной кислотой. Приемлемые для проведения этой реакции растворители включают инертные органические растворители, такие, как галоидированные углеводороды, например дихлорметан или дихлорэтан.
На стадии 3 нитробензилбензоилхлорид 24 получают обработкой соединения 23 хлорирующим агентом, таким, как фосген и эквиваленты фосгена, оксихлорид фосфора и оксалилхлорид, в среде N,N-диметилформамида (условия реакции Вильсмайера). Приемлемые для проведения этой реакции растворители включают инертные органические растворители, такие, как галоидированные углеводороды, например дихлорметан и дихлорэтан.
На стадии 4 аминокарбонилбензилнитробензол 25 получают проведением взаимодействия соединения 24 с первичным или вторичным амином. Реакция протекает в присутствии основания, такого, как пиридин, в среде инертного органического растворителя, такого, как дихлорметан, дихлорэтан и тетрагидрофуран.
На стадии 5 фениламиновое соединение Ig получают восстановлением нитрогруппы соединения 25 до аминогруппы. Приемлемые для восстановления нитрогруппы условия включают гидрогенизацию с использованием платинового или палладиевого катализатора в спиртовом растворителе, таком, как метанол и этанол.
Процесс получения соединений формулы I по этому методу из соответствующего соединения формулы Ig подробно описан в примере 13.
На схеме 3 проиллюстрирован альтернативный способ получения соединений формулы I, в которой R1 обозначает группу формулы (В), где Х обозначает S, а R5 обозначает либо -O(CH2)nZ, либо -(СН2)mОY, в частности где m обозначает 0, из соответствующих промежуточных соединений формулы Ih.
На стадии 1 тиениловое соединение 27b получают обработкой бромтиенила 26а алкоксидным анионом, например анионом метоксида натрия, в присутствии солей меди, таких, как иодид одновалентной меди. Реакция протекает в инертной атмосфере в приемлемом апротонном органическом растворителе, таком, как N,N-диметилформамид, N-метилпирролидон, тетрагидрофуран и т.п.
На стадии 2 орто-хлорированное тиениловое соединение 28 может быть получено методами, описанными в химической литературе, например в работе Stanetty и др. в Monatshefte Chemie, 1989, 120, 65. В целом соединение 27b в инертной атмосфере обрабатывают галоидирующим агентом, таким, как хлористый сульфурил. Приемлемые для проведения этой реакции растворители включают гексан, дихлорметан и дихлорэтан.
На стадии 3 тиенилгидроксиметилнитробензол 29 получают обработкой соединения 28 сильным основанием, таким, как н-бутиллитий, а затем бензальдегидом. Реакцию проводят с охлаждением в инертной атмосфере. Приемлемые для проведения этой реакции растворители включают апротонные органические растворители, такие, как тетрагидрофуран, диэтиловый эфир и т.п., предпочтительно тетрагидрофуран.
На стадии 4 тиенилметилнитробензоловое соединение 30 получают восстановлением гидроксиметильной группы соединения 29 алкилированным галоидсиланом, таким, как триметилсилилхлорид, в присутствии галогенидной соли, например иодида натрия. Приемлемые для проведения этой реакции растворители включают апротонные растворители, например ацетонитрил, N,N-диметилформамид и т.п.
На стадии 5 фениламиновое соединение формулы Ih получают восстановлением нитрогруппы соединения 30 до аминогруппы. Приемлемые для восстановления нитрогруппы условия включают наличие борида никеля в подкисленном метаноле, хлорида олова(II) в этаноле или каталитическую гидрогенизацию с использованием платинового или палладиевого катализатора (например, РtO2 или Pd/C) в органическом растворителе, таком, как этанол, изопропанол и этилацетат.
Процесс получения соединений формулы I по этому методу из соответствующего соединения формулы Ih подробно описан в примере 14.
На схеме И проиллюстрирован альтернативный способ получения соединений формулы I, в которой R1 обозначает группу формулы (В), где Х обозначает S, а R5 обозначает либо -O(CH2)nZ, либо -(СН2)mОY, в частности где m обозначает 0, из соответствующего промежуточного соединения формулы Ii.
На стадии 1 тиениловое соединение 27b можно получать по методу, который описан в химической литературе, например в работе М.А. Keeystra и др. в Tetrahedron, 1992, 48, 3633. В целом бромтиениловое соединение 26b обрабатывают алкоксидным анионом, например анионом метоксида или циклопентоксида, в присутствии сильного основания, такого, как гидрид натрия. Реакция протекает при повышенной температуре в инертной атмосфере, после чего следует добавление солей меди, таких, как бромид одновалентной меди или иодид одновалентной меди. Приемлемые для проведения этой реакции растворители включают инертные органические растворители, такие, как метанол, этанол, диоксан и тетрагидрофуран.
На стадии 2 алкилстаннан 31 получают станнилированием соединения формулы 27b, его обработкой галоидалкилстаннаном, таким, как хлорид (три-н-бутил)олова, в присутствии литийсодержащего реагента, например н-бутиллития. Реакция протекает в инертной атмосфере в среде апротонного органического растворителя, такого, как тетрагидрофуран или диэтиловый эфир.
На стадии 3 Р-защищенное соединение 32, где Р обозначает аминозащитную группу, получают проведением взаимодействия соединения 31 с бензилхлоридным реагентом с защищенной аминогруппой, в частности триметилсилилалкилкарбонильной группой. Реакция протекает в присутствии подходящего катализатора, такого, как платиновый или палладиевый катализатор, например тетракис(трифенилфосфин)палладий, в сорастворителе, таком, как гексаметилфосфорамид.
На стадии 4 фениламиновое соединение формулы Ii получают удалением из соединения 32 аминозащитной группы путем обработки нуклеофилом, таким, как источник фторидного иона, например фторидом (тетра-н-бутил)аммония, в инертном органическом растворителе, таком, как диоксан, тетрагидрофуран, диэтиловый эфир и т.п.
В другом варианте другие фениламиновые соединения формулы Ii могут быть получены заменой группы -O(CH2)nZ или -(СН2)mОY в соединениях формулы Ii, где Y или Z обозначает алкил или циклоалкил, другими алкильными группами в присутствии кислоты, такой, как п-толуолсульфокислота. Эта реакция протекает в инертной атмосфере при кипячении с обратным холодильником. Приемлемые для проведения реакции растворители включают спиртовые растворители, такие, как метанол, этанол и изопропанол.
Процесс получения соединений формулы I по этому методу из соответствующих соединений формулы Ii подробно описан в примерах 15-17.
На схеме К проиллюстрирован альтернативный способ получения соединений формулы I, в которой R1 обозначает группу формулы (С), где Х обозначает N, а R6 обозначает группу -C(V)NR8R9, где V обозначает О или S, из соответствующего промежуточного соединения формулы Ij.
На стадии 1 гетероциклилметилфениламин 34 получают восстановлением ароматических и нитрогрупп гетероарилметилнитробензола 33 в условиях
каталитической гидрогенизации, например, с использованием платинового или палладиевого катализатора (в частности, PtO2 или Pd/C, предпочтительно 10%-ного Pd/C) в протонном органическом растворителе, таком, как подкисленный метанол или подкисленный этанол, предпочтительно подкисленный этанол. Реакция протекает при температуре от примерно 20 до 100oС и под давлением 20-100 фунтов/кв.дюйм.
На стадии 2 фениламиновое соединение формулы Ij получают проведением взаимодействия соединения 34 с изоцианатом/тиоизоцианатом в инертном органическом растворителе, таком, как дихлорметан, диэтиламин или тетрагидрофуран. Реакция протекает в инертной атмосфере при температуре от примерно -10 до 30oС.
Процесс получения соединения формулы I по этому методу из соответствующего соединения формулы Ij подробно описан в примере 18.
На схеме Л проиллюстрирован альтернативный способ получения соединений формулы I, R1 которой обозначает группу формулы (С), где Х обозначает N, a R6 обозначает группу -COR9 или -SO2R9, из соответствующего промежуточного соединения формулы Ik.
Гетероциклилметилфениламин 34 получают аналогично тому, как это представлено выше на схеме К.
На стадии 1 Р1-защищенное соединение 35, где Р1 обозначает аминозащитную группу, получают присоединением к соединению 34 приемлемой аминозащитной группы, такой, как трифторацетил, бензил, трет-бутоксикарбонил (ВОС) или карбобензилоксигруппы (КБЗ), предпочтительно ВОС, методами, известными обычному специалисту в данной области техники. В целом соединение 34 обрабатывают ди-трет-бутилдикарбонатом в апротонном органическом растворителе, таком, как тетрагидрофуран.
На стадии 2 Р1- и Р2-защищенное соединение 36, где Р2 также обозначает аминозащитную группу, получают присоединением соответствующей аминозащитной группы, такой, как трифторацетил, бензил, трет-бутоксикарбонил (ВОС) или карбобензилоксигруппы (КБЗ), предпочтительно трифторацетила, к фениламиногруппе соединения 35 методами, известными специалисту в данной области техники. В целом соединение 35 обрабатывают трифторуксусным ангидридом в присутствии основания, такого, как триэтиламин. Реакция протекает в инертной атмосфере в среде инертного органического растворителя, такого, как дихлорметан, дихлорэтан, тетрагидрофуран и т.п.
На стадии 3 Р2-защищенное соединение 37 получают удалением аминозащитной Р1-группы в соединении 36 его обработкой сильной органической кислотой, такой, как трифторуксусная кислота, в инертном органическом растворителе, таком, как галоид ированные углеводороды, например дихлорметан или дихлорэтан.
На стадии 4 соединение 38 получают в результате взаимодействия соединения 37 с сульфирующим агентом, таким, как сульфонилгалогенид, или с ацилирующим агентом, таким, как ацилгалогенид. Реакция протекает в инертной атмосфере в присутствии основания, такого, как триэтиламин, в галоидированном органическом растворителе, таком, как дихлорметан или дихлорэтан.
На стадии 5 фениламиновое соединение формулы Ik получают удалением аминозащитной Р2-группы у соединения 38 его обработкой основанием, таким, как гидроксид лития. Приемлемые для проведения этой реакции растворители включают спиртовые и протонные растворители, такие, как метанол, этанол и вода.
Процесс получения соединения формулы I по этому методу из соответствующего соединения формулы Ik подробно описан в примере 19.
На схеме М проиллюстрирован альтернативный способ получения соединений формулы I, в которой R1 обозначает группу формулы (С), где Х обозначает N, а R6 обозначает группу -CONR8R9 или -SO2NR8R9, из соответствующих промежуточных соединений формулы Il.
Р2-защищенное соединение 37 получают аналогично тому, как это проиллюстрировано на приведенной выше схеме Л.
Соединение 39 получают в результате взаимодействия соединения 37 с карбамоилгалогенидом или сульфамоилгалогенидом. Реакция протекает в инертной атмосфере в присутствии основания, такого, как триэтиламин, в галоидированном органическом растворителе, таком, как дихлорметан или дихлорэтан.
На последующей стадии фениламиновое соединение формулы Il получают удалением защищающей Р2-группы у соединения 39 его обработкой основанием, таким, как гидроксид лития. Приемлемые для проведения этой реакции растворители включают спиртовые и протонные растворители, такие, как метанол, этанол и вода.
Процесс получения соединения формулы I по этому методу из соответствующего соединения формулы Il подробно описан в примере 20.
Схема Н в общем иллюстрирует получение соединений формулы I, где значения R1 и R2 указаны при кратком изложении сущности изобретения, из соответствующих промежуточных соединений формул Iа-II.
2-Имидазолиновые соединения формулы 40 известны или они могут быть легко синтезированы обычными специалистами в данной области техники. Так, например, синтез сульфатной соли формулы 40, где L обозначает хлор, описан A. Trani и Е. Bellasio, J. Het. Chem., 1974, 11, 257.
В целом имидазолиновые соединения формулы I могут быть получены в результате взаимодействия соответствующих промежуточных соединений формул Ia-II с 2-имидазолиновым соединением 40 в виде кислотно-аддитивной соли или свободного основания. Реакция протекает при кипячении с обратным холодильником, как правило, в инертной атмосфере. Приемлемыми для проведения этой реакции растворителями являются инертные органические растворители, такие, как метанол, этанол, изопропанол, дихлорметан, ацетонитрил, тетрагидрофуран, диоксан и т. п. Выбор растворителя обычно зависит от цели применения кислотно-аддитивной соли или свободного основания.
Общие показания по применению
Вещества-антагонисты рецептора IP, такие, как предлагаемые по настоящему изобретению, проявляют как противовоспалительные, так и болеутоляющие свойства in vivo. Таким образом, эти соединения могут быть использованы в качестве противовоспалительных и болеутоляющих средств для млекопитающих, прежде всего для человека. Они находят применение при болевых ощущениях, вызванных самыми разнообразными причинами, включая, но не ограничиваясь ими, боли при воспалительных процессах, боли после хирургического вмешательства, висцеральные боли, зубную боль, предменструальную боль, головную боль, боль от ожогов, мигрень или гистаминовые головные боли, боли при травмировании нервов, невритах, невралгии, боли при отравлениях, ишемические боли, боли при интерстициальном цистите, боли при раковых заболеваниях, болевые ощущения при заражении вирусами, паразитами и бактериями, посттравматические боли (включая болевые ощущения после переломов и спортивных травм) и боль, связанную с расстройствами пищеварительного тракта, такую, как синдром раздраженной толстой кишки.
Эти соединения находят также применение при лечении воспалительных процессов, вызванных различными причинами, включая, но не ограничиваясь ими, бактериальную, грибковую и вирусную инфекцию, ревматоидный артрит, остеоартрит, хирургическое вмешательство, инфекцию мочевого пузыря и идиопатическое воспаление мочевого пузыря, злоупотребления, пожилой возраст, недостаточность питания, простатит, конъюнктивит.
Такие соединения находят также применение при лечении расстройств работы мочевого пузыря, связанных с синдромом инфравезикальной обструкции, и недержания мочи, таком, как самопроизвольное мочеиспускание, недержание мочи при напряжении и повышенной реактивности мочевого пузыря.
Кроме того, эти соединения находят применение при лечении респираторных заболеваний, таких, как астма, при которой С-волокна в легких проявляют сверхчувствительность к ряду окружающих раздражителей, включая холодный воздух, пыль, цветочную пыльцу и другие антигены. Поскольку эти С-волокна выражают простаноидные рецепторы IP, активация таких рецепторов посредством PGI2 и последующее выделение нейрокининов могут способствовать сокращению ткани гладкой мускулатуры легких, отеку и секреции слизи. Таким образом, соединения по настоящему изобретению, используемые либо системно, либо в виде аэрозольных препаратов, приемлемы для эффективного терапевтического лечения астмы.
Более того, эти соединения находят также применение при лечении септического шока.
Испытание
Противовоспалительное/болеутоляющее действие соединений по настоящему изобретению можно оценить проведением in vivo испытаний, таких, как оценка по вызванной у крыс каррагенаном механической гиперальгезии лап и оценка по симптому Фройнда вызванной адъювантом механической гиперальгезии у крыс, которые более подробно описаны в примерах соответственно 30 и 31. Эффективность ингибирования сокращений можно оценить проведением in vitro испытаний, таких, как ингибирование сокращений мочевого пузыря, вызванных испытанием с изоволюметрическим растяжением мочевого пузыря, которое более подробно описано в примерах 32 и 33. Эффективность ингибирования септического шока можно оценить in vivo испытаниями, такими, как испытание с устранением у крыс вызванной действием эндотоксинов гипотензии, которое более подробно описано в примере 34.
Методы введения и фармацевтическая композиция
По изобретению предлагается фармацевтическая композиция, включающая соединение по настоящему изобретению, его фармацевтически приемлемую соль или его кристаллическую форму совместно с одним или несколькими фармацевтически приемлемыми носителями и необязательными другими терапевтическими и/или профилактическими компонентами.
Обычно соединения по настоящему изобретению вводят в организм в терапевтически эффективном количестве любым приемлемым путем введения лекарственных средств, которые служат такому назначению. В зависимости от ряда факторов, таких, как серьезность заболевания, от которого необходимо вылечить, возраст и относительное состояние здоровья пациента, активность используемого соединения, путь и форма введения в организм, показание, в отношении которого предусмотрено введение, выбор и опыт лечащего врача, приемлемые интервалы доз составляют 1-500 мг ежедневно, предпочтительно 1-100 мг ежедневно, а наиболее предпочтительно 1-30 мг ежедневно. Терапевтически эффективное количество соединений по настоящему изобретению в случае конкретного заболевания в состоянии установить обычный специалист в области лечения таких заболеваний без проведения чрезмерного количества экспериментов, опираясь на собственные познания и исходя из описания к настоящей заявке.
Соединения по настоящему изобретению обычно вводят в виде фармацевтических композиций, включая композиции, приемлемые для применения перорально (включая трансбуккальный и подъязычный методы), ректально, интраназально, локально, через легкие, вагинально и парентерально (включая внутримышечный, внутриартериальный, подоболочечный, подкожный и внутривенный методы), или в препаративной форме, приемлемой для введения ингаляционно или инсуффляционно. Предпочтительным методом введения является пероральный, в котором используют удобную схему ежедневного приема лекарственного средства, в которую можно вносить коррективы в соответствии с серьезностью заболевания.
Соединениям по изобретению совместно с обычными адъювантами, носителями или разбавителями можно придавать форму фармацевтических композиций и их одноразовых доз. Фармацевтические композиции и одноразовые дозированные формы могут включать обычные компоненты в обычных пропорциях вместе с дополнительными действующими соединениями или действующими началами или без них, и в такой одноразовой дозированной форме могут содержать любое приемлемое эффективное количество действующего компонента, соответствующее предусмотренному диапазону ежедневно вводимых доз. Такую фармацевтическую композицию можно применять в виде твердых препаратов, таких, как таблетки и наполненные капсулы, полутвердых препаратов, порошков, препаратов с выделением в постоянной концентрации или жидкостей, таких, как растворы, суспензии, эмульсии, эликсиры или наполненные ими капсулы, для перорального введения, в форме суппозиториев для ректального или вагинального введения или в форме стерильных инъекционных растворов для парентерального введения. Следовательно, типичным примером приемлемых препаративных форм в дозах на один прием служат композиции, содержащие 1 мг действующего вещества или, если брать шире, от 0,01 до 100 мг на таблетку.
На основе соединений по настоящему изобретению можно готовить самые разнообразные лекарственные формы для перорального введения. В качестве действующего компонента фармацевтические композиции и лекарственные формы могут включать соединения по изобретению, их фармацевтически приемлемые соли и кристаллические формы. Фармацевтически приемлемые носители могут находиться либо в твердой, либо в жидкой форме. Твердые препаративные формы включают порошки, таблетки, пилюли, капсулы, крахмальные облатки, суппозитории и диспергируемые гранулы. Твердый носитель может включать одно или несколько веществ, которые могут также выполнять функции разбавителей, вкусовых и ароматизирующих добавок, солюбилизаторов, смазывающих добавок, суспендирующих веществ, связующих веществ, консервантов, добавок, способствующих механическому разрушению таблеток, или инкапсулирующего материала. В порошках носитель находится в виде тонкоизмельченного твердого вещества, которое представляет собой смесь с тонкоизмельченным действующим компонентом. В таблетках действующий компонент смешан с носителем, обладающим необходимой связывающей способностью, в приемлемых пропорциях и спрессован с приданием желаемых формы и размера. Предпочтительные порошки и таблетки содержат от одного до примерно семидесяти процентов действующего вещества. Приемлемые носители включают карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатину, трагант, метилцеллюлозу, натрийкарбоксиметилцеллюлозу, низкоплавкий воск, масло какао и т.п. Термин "препарат" включает сочетание действующего вещества с инкапсулирующим материалом как носителем, образующим капсулу, в которой действующий компонент, совместно с носителями или без них, окружен носителем, связанным, таким образом, с ним. Подобным же образом этим термином охватываются крахмальные облатки и лепешки. Таблетки, порошки, капсулы, пилюли, крахмальные облатки и лепешки могут находиться в твердой форме, приемлемой для перорального введения.
Другие формы, приемлемые для перорального введения, включают препараты в жидком виде, к которым относятся эмульсии, сиропы, эликсиры, водные растворы, водные суспензии, и препараты в твердом виде, которые предназначены для перевода в форму жидких препаратов незадолго до применения. Эмульсии могут быть приготовлены в виде растворов в водных пропиленгликолиевых растворах или могут содержать эмульгаторы, такие, как лецитин, моноолеат сорбитана и аравийская камедь. Водные растворы можно готовить растворением действующего компонента в воде и добавлением приемлемых красителей, ароматизирующих и вкусовых добавок, стабилизаторов и загустителей. Водные суспензии можно готовить диспергированием тонкоизмельченного действующего компонента в воде с вязким материалом, таким, как природные и синтетические камеди, смолы, метилцеллюлоза, натрийкарбоксиметилцеллюлоза и другие хорошо известные суспендирующие агенты. Препараты в жидкой форме включают растворы, суспензии и эмульсии, они могут включать, помимо действующего компонента, красители, ароматизирующие и вкусовые добавки, стабилизаторы, буферные добавки, искусственные и природные подслащивающие вещества, диспергаторы, загустители, солюбилизирующие агенты и т.п.
С использованием соединений по настоящему изобретению можно готовить препараты для парентерального введения (например, путем инъекций, в частности для инъекции ударной дозы вещества или постоянного вливания), они могут содержаться в лекарственном средстве в дозах на один прием в виде ампул, предварительно наполненных шприцев, контейнеров малого объема для вливания и контейнеров для нескольких доз с добавленным консервантом. Композициям можно придавать такие формы, как суспензии, растворы или эмульсии в маслянистых или водных наполнителях, например растворы в водном полиэтиленгликоле. Примеры маслянистых или неводных носителей, разбавителей, растворителей или наполнителей включают пропиленгликоль, полиэтиленгликоль, растительные масла (например, оливковое масло) и органические сложные эфиры для инъекций (например, этилолеат), они могут содержать используемые для приготовления препаратов добавки, такие, как консерванты, смачивающие вещества, эмульгаторы, суспендирующие агенты, стабилизаторы и/или диспергаторы. По другому варианту активный компонент может находиться в виде порошка, полученного выделением в асептических условиях стерильного твердого вещества или лиофилизацией из раствора, и перед использованием его совмещают с приемлемым наполнителем, например со стерильной апирогенной водой.
Для местного введения в эпидермис на основе соединений по настоящему изобретению можно готовить мази, кремы и лосьоны или пластыри для введения через кожу. Мази и кремы можно готовить, например, с использованием водной или маслянистой основы с добавлением приемлемых загущающих и/или желатинизирующих добавок. Лосьоны можно готовить с использованием водной или маслянистой основы, обычно они содержат также один или несколько эмульгаторов, стабилизирующих агентов, диспергаторов, суспендирующих добавок, загущающих веществ и красителей. Препараты, приемлемые для местного введения в полость рта, включают лепешки, содержащие действующие вещества в ароматической и вкусовой добавке как в основе, обычно в сахарозе и аравийской камеди или траганте, пастилки, содержащие действующее вещество в инертной основе, такой, как желатин, глицерин или сахароза и аравийская камедь, и жидкости для полоскания рта, содержащие действующее вещество в приемлемом жидком носителе.
С использованием соединений по настоящему изобретению можно готовить препараты для введения в виде суппозиториев. Низкоплавкий воск, такой, как смесь глицеридов жирных кислот, или масло какао, вначале плавят, затем действующий компонент гомогенно диспергируют, например, перемешиванием. Далее расплавленную гомогенную смесь разливают по формам обычного размера, дают остыть и, следовательно, затвердеть.
С использованием соединений по настоящему изобретению можно готовить препараты для вагинального введения. Приемлемыми можно считать вагинальные суппозитории, тампоны, кремы, гели, пасты, губки или спреи, содержащие, помимо действующего вещества, такие носители, которые известны в данной области техники.
С использованием соединений по настоящему изобретению можно готовить препараты для интраназального введения. Растворы и суспензии вводят непосредственно в носовую полость с помощью обычных средств, например капельницы, пипетки или распылительного устройства. Препараты могут быть приготовлены в виде одноразовой или многоразовой дозированной формы. В этом последнем случае при наличии капельницы или пипетки это может быть достигнуто введением пациенту соответствующего предопределенного объема раствора или суспензии. В случае спрея этого можно добиться, например, с помощью атомизационного распылителя с дозирующим насосом.
С использованием соединений по настоящему изобретению можно готовить препараты для аэрозольного введения, в частности для введения в дыхательные пути, включая интраназальное введение. При этом вещество обычно характеризуется малым размером частиц, в частности примерно 5 мкм или меньше. Такие размеры частиц могут быть достигнуты с помощью средств, известных в данной области техники, например обработкой в микронной коллоидной мельнице. Действующее вещество находится в упаковке под давлением приемлемого пропеллента, такого, как хлорфторуглерод (ХФУ), например дихлордифторметан, трихлорфторметан или дихлортетрафторметан, диоксид углерода или другой приемлемый газ. Аэрозольный препарат обычно может также включать поверхностно-активное вещество, такое, как лецитин. Дозу лекарственного средства можно регулировать с помощью дозирующего клапана. В другом варианте с использованием действующих веществ могут быть приготовлены сухие порошки, например порошкообразная смесь такого вещества с приемлемой порошкообразной основой, такой, как лактоза, крахмал, производные крахмала, такие, как гидроксипропилметилцеллюлоза, и поливинилпирролидон (ПВП). Порошкообразный носитель в носовой полости обычно образует гель. Порошкообразную композицию можно готовить в форме лекарственного средства в дозах на один прием, например капсулы или облатки, в частности желатиновые и пузырьковые упаковки, из которых порошки можно вводить с помощью ингалятора.
При необходимости препараты могут быть снабжены покрытием для тонкой кишки, приспособленном для постоянного или регулируемого выделения в организм действующего компонента.
В предпочтительном варианте фармацевтические композиции готовят в форме лекарственного средства в дозах на один прием. В такой форме препарат разделен на унифицированные дозы, содержащие соответствующие количества действующего компонента. Унифицированная дозированная форма может представлять собой упакованный препарат, причем эта упаковка содержит раздельные количества препарата, в частности упакованные таблетки, капсулы, порошки в пузырьках или ампулы. Лекарственный препарат в дозах на один прием может представлять собой капсулу, таблетку, крахмальную облатку или лепешку или включать соответствующее число доз в любой из таких упакованных форм.
Другие пригодные фармацевтические носители и приготовленные с их применением препараты описаны в Remington: The Science and Practice of Pharmacy, 1995, под ред. E.W. Martin, Mack Publishing Company, 19-е изд., Истон, шт. Пенсильвания. Типичные фармацевтические композиции, содержащие соединения по настоящему изобретению, описаны в примерах 24-29.
Описанные ниже композиции и примеры приведены с целью дать возможность специалистам в данной области техники более четко понять сущность настоящего изобретения и пути его практического выполнения. Их следует рассматривать не как ограничивающие объем изобретения, а как примеры, приведенные лишь с иллюстративной целью.
ПРИМЕР ПОЛУЧЕНИЯ 1
1-Бром-4-изопропоксиметилбензол.
0,96 г гидрида натрия (24 ммоля 60%-ного) в минеральном масле при 0-5oС в атмосфере аргона добавляли в раствор 7,59 мл (36 ммолей) изопропанола в 30 мл сухого N, N-диметилформамида. После перемешивания смеси в течение приблизительно 25 мин добавляли 4-бромбензилбромид и смесь перемешивали при 20oС в течение еще 1 ч. Раствор распределяли между 50 мл насыщенного раствора хлорида аммония и 50 мл диэтилового эфира. Водную фазу экстрагировали 3 порциями по 20 мл диэтилового эфира, а объединенные органические слои промывали водой, сушили (над Na2SO4) и упаривали в вакууме. Сырой продукт фильтровали через силикагель с получением в виде прозрачного масла 2,33 г (85%) чистого 1-бром-4-изопропоксиметилбензола.
ПРИМЕР ПОЛУЧЕНИЯ 2
2-Триметилсиланилэтиловый эфир 4-хлорметилфенилкарбаминовой кислоты
В смесь 1,07 г (6,4 ммолей) 4-хлорметилфенилизоцианата в 22 мл тетрагидрофурана при 20-25oС в токе аргона добавляли 0,91 мл (6,4 ммолей) 2-триметилсилилэтанола. Смесь перемешивали в течение 4 ч при 20-25oС. Растворитель выпаривали в вакууме. Добавляли насыщенный раствор бикарбоната натрия и продукт экстрагировали этилацетатом. Экстракт промывали водой, рассолом, сушили (над Na2SO4) и концентрировали досуха. В результате очистки экспресс-хроматографией на диоксиде кремния и элюирования гексаном/этилацетатом в виде белого твердого кристаллического вещества получили 1,24 г (68%-ный выход) 2-триметилсиланилэтилового эфира 4-хлорметилфенилкарбаминовой кислоты с tпл 55-56oС.
1H-ЯМР: 7,38 (d, J=8,6, 2Н), 7,32 (d, J=8,6, 2Н), 6,60 (bs, 1H), 4,55 (s, 2H), 4,26 (m, 2H), 1,05 (m, 2H), 0,06 (s, 9H).
ПРИМЕР 1.
2-[4-(4-Изопропоксибензил)фенил]аминоимидазолин.
Ниже описано получение соединения формулы I из соответствующего соединения формулы Iа, в которой R1 обозначает группу формулы (А), R2 и R4 каждый обозначает водород, Y обозначает изопропил, а m обозначает 0.
Стадия 1.
Соединение формулы 3а получали в соответствии с методом Shani J. и др., J. Med. Chem., 1985, 28, 1504. Так, например, смесь 90 г (0,48 молей) 4-нитробензоилхлорида с 57,24 г (0,53 моля) анизола в 450 мл сероуглерода перемешивали в трехгорлой круглодонной колбе, снабженной приспособлением для подачи азота, холодильником и механической мешалкой, причем эту колбу охлаждали на ледяной бане. Отдельными порциями добавляли 84,0 г (0,63 моля) хлорида алюминия и перемешивание продолжали при температуре ледяной бани в течение 30 мин, а затем при комнатной температуре в течение еще 1 ч. Реакционную смесь охлаждали, обрабатывали 150 мл концентрированной соляной кислоты и разбавляли 250 мл холодной воды. Продукт собирали, отфильтровывали, промывали, сушили и кристаллизовали из этилацетата с получением в виде не совсем белого твердого вещества 4-(4-метоксибензоил)нитробензола с tпл 120-122oC. Данные элементного анализа для C14H11NО4, рассчитано: С 65,3, Н 4,31, N 5,44; обнаружено: С 65,22, Н 4,16, N 5,69.
Стадия 2.
Раствор 150 г (0,58 моля) 4-(4-метоксибензоил)нитробензола, 500 мл ледяной уксусной кислоты и 400 мл бромистоводородной кислоты концентрацией 48 мас. % кипятили с обратным холодильником в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и выливали в измельченный лед. Сырой продукт отфильтровывали, несколько раз промывали водой и сушили в высоком вакууме при приблизительно 50oС. В результате кристаллизации из этилацетата/гексана получали 114 г (81%) 4-(4-гидроксибензоил)нитробензола с tпл 190-193oС. Данные элементного анализа для С13Н9NO4, рассчитано: С 64,20, Н 3,73, N 5,76; обнаружено: С 63,95, Н 3,65, N 5,85. МС m/e (%): 243 (М+; 45).
Стадия 3.
Реакционную смесь, содержавшую 48,63 г (0,2 моля) 4-(4-гидроксибензоил)нитробензола, 98,4 г (0,8 моля) 2-бромпропана, 1,5 г иодида натрия и 27,6 г (0,2 моля) безводного карбоната калия в 200 мл N,N-диметилформамида, выдерживали при приблизительно 60-70oС в токе азота в течение 18 ч. Реакционную смесь концентрировали, а остаток перемешивали с водой и фильтровали. Сырой продукт несколько раз промывали водой и сушили с получением 54,8 г (96%) окрашенного в кремовый цвет продукта, который далее кристаллизовали из этилацетата с получением 4-(4-изопропоксибензоил)нитробензола с tпл 138oС. Данные элементного анализа для C16H18NO4: рассчитано: С 67,36, Н 5,30, N 4,91; обнаружено: С 67,39, Н 5,28, N 5,07.
Стадия 4.
Смесь 14,0 г (49,09 ммоля) 4-(4-изопропоксибензоил)нитробензола с 2,0 г 10%-ного палладия на угле в растворе 250 мл этанола и 30 мл концентрированной соляной кислоты под давлением 50 фунтов/кв.дюйм в течение 16 ч гидрировали в аппарате Парра. Катализатор удаляли фильтрованием через тампон из целита (броунмиллерита), а фильтрат концентрировали в вакууме. Остаток разбавляли охлажденной льдом водой, подщелачивали концентрированным раствором гидроксида аммония и экстрагировали этилацетатом. Органические экстракты промывали водой и рассолом и сушили (над Na2SО4). В результате удаления растворителя получали густое масло, которое подвергали кристаллизации из этилацетата/гексана с получением в виде белого твердого продукта 10,4 г (87%) 4-(4-изопропоксибензил)фениламина с tпл 92-93oC; данные элементного анализа для C16H19NO, рассчитано: С 79,63, Н 7,94, N 5,80; обнаружено: С 79,51, Н 7,92, N 5,96. МС m/e (%): 241 (М+; 83).
Заключительная стадия.
2-Хлор-2-имидазолинсульфат получали в соответствии с методами, описанными в работе A. Trani и Е. Bellasio в J. Het. Chem., 1974, 11, 257.
Смесь 24,36 г (120 ммолей) сульфата 2-хлор-2-имидазолина с 24,1 г (100 ммолей) 4-(4-изопропоксибензил)фениламина в 300 мл изопропанола в течение 1-2 ч кипятили с обратным холодильником в инертной атмосфере. Реакционную смесь концентрировали в вакууме и остаток разбавляли охлажденной льдом водой. Смесь подщелачивали 10%-ным гидроксидом натрия и тщательно экстрагировали дихлорметаном. Объединенные органические экстракты промывали холодной водой и рассолом, сушили (над К2СО3) и концентрировали. В результате кристаллизации из диэтилового эфира/гексана в виде не совсем белого твердого вещества получали 9,94 г (96%) 2-[4-(4-изопропоксибензил)фенил]аминоимидазолина с tпл 103-104oС. Данные элементного анализа для С19Н22N3O, рассчитано: С 73,76, Н 7,49, N 13,58; обнаружено: С 73,51, Н 7,42, N 13,57. МС m/e (%): 309 (М+; 100).
В смесь 0,5 г 2-[4-(4-изопропоксибензил)фенил]аминоимидазолина с 14 мл ацетона добавляли раствор 0,08 г серной кислоты в 1 мл ацетона. Смесь нагревали, перемешивали в течение 15 мин и фильтровали, получая в виде белого твердого вещества 0,56 г 2-[4-(4-изопропоксибензил)фенил]аминоимидазолинсульфата с tпл 215-216oС. Данные элементного анализа для С38Н48N6O6S, рассчитано: С 63,66, Н 6,75, N 11,72; обнаружено: С 63,50, Н 6,64, N 11,72.
В результате проведения процесса аналогично стадии 1 в примере 1 и проведения процесса непосредственно до стадии 4 и заключительной стадии в примере 1 получали 2-[4-(4-метоксибензил)фенил]аминоимидазолин с tпл 114-116oС.
В результате проведения процесса аналогично стадии 1 в примере 1, но с заменой 4-нитробензоилхлорида 3-метокси-4-нитробензоилхлоридом, и проведения процесса непосредственно до стадии 4 и заключительной стадии в примере 1 получали 2-[4-(4-метоксибензил)-3-метоксифенил] аминоимидазолин с tпл 127-128oС.
В результате проведения процесса аналогично стадии 3 в примере 1, но с заменой 2-бромпропана этил-2-бромпропионатом, а затем соответственно аналогично последующим стадиям в примере 1, получали 2-[4-[4-(4,5-дигидро-Н-имидазол-2-иламино)бензил] фенокси} пропионовую кислоту с tпл >300oС. Данные элементного анализа для C19H21N3О3: рассчитано: С 67,24, Н 6,24, N 12,38; обнаружено: С 66,90, Н 6,23, N 12,31.
В результате проведения процесса аналогично стадии 3 в примере 1, но с заменой 2-бромпропана другими алкилгалогенидами, а затем соответственно аналогично последующим стадиям в примере 1, получали следующие соединения формулы I:
2-[4-(4-этоксибензил)фенил]аминоимидазолин с tпл 152-153oС;
гидрохлорид 2-{4-[4-(2,2,2-трифторэтокси)бензил]фенил}аминоимидазолина с tпл 75-78oС;
оксалат 2-[4-(4-пропоксибензил)фенил]аминоимидазолина с tпл 146-147oС;
гидрохлорид 2-[4-(4-бутоксибензил)фенил]аминоимидазолина с tпл 97-100oС;
оксалат 2-[4-(4-бутоксибензил)фенил]аминоимидазолина с tпл 172-174oC;
гидрохлорид 2-[4-(4-изобутоксибензил)фенил]аминоимидазолина с tпл 127-129oС;
оксалат 2-[4-(4-пентилоксибензил)фенил]аминоимидазолина с tпл 163-166oС;
2-{4-[4-(1-метилбутокси)бензил]фенил}аминоимидазолин с tпл 99-112oC;
гидрохлорид 2-{4-[4-(4-гидроксипропокси)бензил]фенил}аминоимидазолина с tпл 129-133oС;
малеат 2-{ 4-[4-(3-гидрокси-2-гидроксиметилпропокси)бензил]фенил}аминоимидазолина с tпл 70-75oС;
гидрохлорид 2-[4-(4-бензилоксибензил)фенил] аминоимидазолина; данные элементного анализа для С23Н24N3OCl, рассчитано: С 70,13, Н 6,14, N 10,67; обнаружено: С 69,79, Н 6,10, N 10,74;
гидрохлорид 2-[4-(4-циклопентилоксибензил)фенил] аминоимидазолина с tпл 116-119oС;
гидрохлорид 2-[4-(4-циклогексоксибензил)фенил] аминоимидазолина с tпл 108-110oС;
гидрохлорид 2-[4-(4-циклогексилметоксибензил)фенил] аминоимидазолина с tпл 95-100oС;
оксалат 2-[4-(4-тетрагидропиран-2-илоксибензил)фенил]аминоимидазолина с tпл 168-170oС;
2-{ 4-[2-(4-метоксифенил)этоксибензил] фенил}аминоимидазолин с tпл 122-124oС;
гидрохлорид 2-[4-(4-бензоилметоксибензил)фенил]аминоимидазолина; данные элементного анализа для С24Н24N3O2Сl, рассчитано: С 67,28, Н 5,86, N 9,81; обнаружено: С 67,27, Н 5,76, N 9,62;
гидрохлорид 2-{ 4-[4-(циклопентиламинокарбонил)метоксибензил]фенил}аминоимидазолина с tпл 78-81oС;
гидрохлорид 2-[4-[4-(1-пиперидинкарбонил)метоксибензил] фенил]аминоимидазолина с tпл 65-67oС;
гидрохлорид 2-{ 4-[4-(фениламинокарбонил)метоксибензил] фенил}аминоимидазолина с tпл 186-187oС;
гидрохлорид 2-{ 4-[4-(диизопропиламинокарбонил)метоксибензил]фенил}аминоимидазолина с tпл 62-65oС;
гидрохлорид 2-{ 4-[4-(диэтиламинокарбонил)метоксибензил]фенил}аминоимидазолина; данные элементного анализа для C22H29N4O2C1, рассчитано: С 60,75, Н 7,18, N 12,88; обнаружено: С 60,91, Н 7,04, N 12,95;
гидрохлорид 2-[4-[4-(изопропиламинокарбонил)метоксибензил]фенил}аминоимидазолина с tпл 66-78oС;
гидрохлорид 2-{4-[4-(N-изопропил-N-метиламинокарбонил)метоксибензил]фенил}аминоимидазолина с tпл 77-81oС;
гидрохлорид 2-{ 4-[4-(4-метоксифенил)аминокарбонилметоксибензил] фенил} аминоимидазолина; данные элементного анализа для C25H27N4O3C1, рассчитано: С 63,33, Н 5,91, N 11,82; обнаружено: С 63,34, Н 5,78, N 11,67;
оксалат 2-[4-(2-фтор-4-пропоксибензил)фенил] аминоимидазолина с tпл 130-133oС;
оксалат 2-[4-(3-фтор-4-изопропоксибензил)фенил] аминоимидазолина с tпл 120-121oC;
малеат 2-[4-(2-фтор-4-тетрагидропиран-2-илметоксибензил)фенил] аминоимидазолина с tпл 138-141oС;
гидрохлорид 2-[4-(3-хлор-4-изопропоксибензил)фенил] аминоимидазолина с tпл 118-120oС;
гидрохлорид 2-[4-(2-хлор-4-метоксибензил)фенил] аминоимидазолина с tпл 126-129oC;
гидрохлорид 2-[4-(3-фтор-4-метоксибензил)фенил] аминоимидазолина с tпл 138-140oC;
гидрохлорид 2-[4-(4-фтор-2-метоксибензил)фенил] аминоимидазолина с tпл 230-233oС;
гидрохлорид 2-[4-(2,4-диметоксибензил)фенил] аминоимидазолина с tпл 137-143oС;
гидрохлорид 2-[4-(3,4-диметоксибензил)фенил] аминоимидазолина с tпл 127-128oС и
гидрохлорид 2-[4-(3-хлор-4-метоксибензил)фенил] аминоимидазолина с tпл 169-172oС.
ПРИМЕР 2
2-[4-(4-Тетрагидропиран-4-илоксибензил)фенил]аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Iа, в которой R1 обозначает группу формулы (А), R2 и R4 каждый обозначает водород, Z обозначает 4-тетрагидропиран-4-ил, а n обозначает 0.
2-[4-(4-Тетрагидропиран-4-илоксибензил)фенил] аминоимидазолин получали в результате проведения процесса аналогично стадиям 1 и 2 в примере 1, а затем осуществлением альтернативной стадии 3.
1,47 г (8,4 ммоля) диэтилазодикарбоксилата медленно, по каплям, добавляли в инертной атмосфере с одновременным перемешиванием при комнатной температуре в раствор 1,7 г (7 ммолей) 4-(4-гидроксибензоил) нитробензола (полученного аналогично описанным в примере 1 стадиям), 0,78 г (7,7 ммоля) 4-гидрокситетрагидропирана и 2,2 г (8,4 ммоля) трифенилфосфина в 20 мл сухого тетрагидрофурана. Реакционную смесь перемешивали в течение еще одного часа, а затем реакцию прекращали добавлением 1 мл воды и массу концентрировали в вакууме. Остаток разбавляли водой и экстрагировали этилацетатом. Объединенные органические экстракты промывали водой и рассолом, сушили (над Na2SO4) и концентрировали в вакууме. Сырой продукт хроматографировали на силикагеле (25% этилацетата/гексаны) и кристаллизовали из гексанов с получением в виде белого твердого вещества 1,4 г (61%) 4-(4-тетрагидропиран-4-илоксибензоил)нитробензола с tпл 105-106oС; данные элементного анализа для C18H17NO5, рассчитано: С 66,05, Н 5,23, N 4,28; обнаружено: С 65,95, Н 5,14, N 4,38. МС m/e (%): 283 (М+; 100).
В результате проведения процесса аналогично стадии 4 и заключительной стадии в примере 1, но с заменой 4-(4-изопропоксибензоил)нитробензола 4-(4-тетрагидропиран-4-илоксибензоил)нитробензолом получали 2-[4-(4-тетрагидропиран-4-илоксибензил)фенил]аминоимидазолин с tпл 169-170oС.
В результате проведения процесса аналогично примеру 2, но с заменой 4-гидрокситетрагидропирана другими гидроксисоединениями, получали следующие соединения формулы I:
2-{4-[4-(1-этилпропокси)бензил]фенил}аминоимидазолин; данные элементного анализа для С21Н27N3О, рассчитано: С 74,74, Н 8,06, N 12,45; обнаружено %, С 74,62, Н 7,90, N 12,33;
гидрохлорид 2-{ 4-[4-(втор-бутокси)бензил] фенил}аминоимидазолина с tпл 118-119oC;
малеат (R)-2-{ 4-[4-(втор-бутокси)бензил] фенил} аминоимидазолина с tпл 163-164oС;
малеат (S)-2-{ 4-[4-(втор-бутокси)бензил] фенил} аминоимидазолина с tпл 163oС;
гидрохлорид (S)-2-{4-[4-(2-метилбутокси)бензил]фенил}аминоимидазолина с tпл 119-122oC;
оксалат 2-[4-(4-гексилоксибензил)фенил]аминоимидазолина с tпл 150-161oС;
2-{4-[4-(2-метоксиэтокси)бензил]фенил]аминоимидазолин с tпл 110-112oС;
2-[4-(4-гидроксибензил)фенил]аминоимидазолин с tпл 170-177oC;
2-{4-[4-(2-гидроксиэтокси)бензил]фенил}аминоимидазолин с tпл 164-165oС;
гидрохлорид 2-{ 4-[4-(3-этоксипропокси)бензил] фенил}аминоимидазолина с tпл 91-92oС;
2-{ 4-[4-(хлорбутокси)бензил] фенил} аминоимидазолин; данные элементного анализа для C20H24N30OCl, рассчитано: С 67,12, Н 6,76, N 11,74; обнаружено: С 66,84, Н 6,79, N 11,80;
гидрохлорид 2-{ 4-[4-(2-метокси-1-метилэтокси)бензил] фенил} аминоимидазолина с tпл 71-74oC;
гидрохлорид 2-{ 4-[4-(3-метоксибутокси)бензил] фенил]аминоимидазолина с tпл 71-76oC;
гидрохлорид 2-{ 4-[4-(1-гидроксиметилэтокси)бензил] фенил}аминоимидазолина, m/s 326 (М+1);
гидрохлорид 2-{4-[4-(2-гидрокси-1-гидроксиметилэтокси)бензил]фенил}аминоимидазолина с tпл 50-55oС;
гидрохлорид 2-{4-[4-(2-этокси-1-этоксиметил)этоксибензил]фенил}аминоимидазолина, смолоподобная масса;
гидрохлорид 2-{ 4-[4-(2,3-дигидроксипропокси)бензил]фенил}аминоимидазолина с tпл 55-60oС;
гидрохлорид 2-{4-[4-(2-фенилэтокси)бензил]фенил}аминоимидазолина; данные элементного анализа для C24H26N3OCl, рассчитано: С 70,66, Н 6,42, N 10,30; обнаружено: С 70,42, Н 6,37, N 10,42;
гидрохлорид 2-{4-[4-(2-феноксиэтокси)бензил]фенил}аминоимидазолина c tпл 140-141oC;
гидрохлорид 2-{4-[4-(3-фенилпропокси)бензил]фенил}аминоимидазолина c tпл 101-104oC;
гидрохлорид 2-[4-(4-циклопропилметоксибензил)фенил] аминоимидазолина с tпл 121-122oС;
2-[4-(4-циклобутилметоксибензил)фенил]аминоимидазолин; данные элементного анализа для С21Н25N3О, рассчитано: С 75,19, Н 7,51, N 12,53; обнаружено: С 74,69, Н 7,32, N 11,96;
оксалат 2-{4-[4-(2-циклопентилэтокси)бензил]фенил}аминоимидазолина c tпл 152-153oС;
малеат 2-{4-[4-(2-циклогексилэтокси)бензил]фенил}аминоимидазолина с tпл 144-147oC;
оксалат 2-{4-[4-(2-циклогексилоксиэтокси)бензил]фенил}аминоимидазолина с tпл 120-127oС;
гидрохлорид 2-{4-[4-(2-изопропоксиэтокси)бензил]фенил}аминоимидазолина с tпл 76-80oС;
гидрохлорид 2-{ 4-[4-(2-(2-оксопирролидин-1-ил)этокси)бензил]фенил}аминоимидазолина с tпл 143-145oС;
гидрохлорид 2-{ 4-[4-(2-(2-оксоимидазолин-1-ил)этокси)бензил]фенил}аминоимидазолина с tпл 85-88oС;
2-[4-(4-тетрагидропиран-4-илметоксибензил)фенил] аминоимидазолин с tпл 159-160oC;
2-[4-(4-тетрагидрофуран-3-илметоксибензил)фенил] аминоимидазолин с tпл 147-149oC;
2-[4-(4-тетрагидрофуран-3-илоксибензил)фенил] аминоимидазолин с tпл 149-150oС;
2-{ 4-[4-(4-метилциклогексилокси)бензил]фенил}аминоимидазолин с tпл 80-85oС;
гидрохлорид 2-{ 4-[4-(5-метил[1,3]диоксан-5-илметокси)бензил]фенил}аминоимидазолина с tпл 85-90oC;
гидрохлорид 2-{4-[4-(3-хлор-2-гидроксиметил-2-метилпропокси) бензил]фенил}аминоимидазолина с tпл 65-70oС;
гидрохлорид 2-{ 4-[4-(2-тиен-2-илэтокси)бензил] фенил}аминоимидазолина; данные элементного анализа для С22Н24N3ОClS, рассчитано: С 63,83, Н 5,84, N 10,15; обнаружено: С 63,85, Н 5,80, N 10,14;
гидрохлорид 2-{ 4-[4-(2-тиен-3-илэтокси)бензил] фенил}аминоимидазолина; данные элементного анализа для С22Н24ОClS, рассчитано: С 63,14, Н 5,90, N 10,04; обнаружено: С 63,30, Н 5,81, N 10,11;
гидрохлорид 2-{ 4-[4-(2-метансульфонилэтокси)бензил]фенил}аминоимидазолина с tпл 172-175oC;
гидрохлорид 2-{ 4-[4-(4-метоксифенил)сульфониламиноэтоксибензил] фенил} аминоимидазолина; данные элементного анализа для С26Н31N4O6СlS, рассчитано: С 58,08, Н 5,95, N 10,41; обнаружено: С 57,97, Н 9,94, N 10,58;
оксалат 2-[4-(3-фтор-4-изобутоксибензил)фенил] аминоимидазолина с tпл 134-135oС;
гидрохлорид 2-[4-(2-фтор-4-изобутоксибензил)фенил]аминоимидазолина c tпл 134-135oC;
гидрохлорид 2-{ 4-[3-фтор-4-(тетрагидропиран-4-илокси)бензил]фенил}аминоимидазолина с tпл 149-151oС;
2-{ 4-[2-фтор-4-(тетрагидропиран-4-илокси)бензил]фенил}аминоимидазолин с tпл 169-170oC;
2-{ 4-[4-(тетрагидропиран-4-илокси)бензил] фенил} аминоимидазолин с tпл 124-127oС;
2-{4-[3-фтор-4-(тетрагидропиран-4-илметокси)бензил]фенил}аминоимидазолин с tпл 154-155oС;
малеат 2-{4-[2-фтор-(4-тетрагидропиран-4-илметокси)бензил]фенил}аминоимидазолина с tпл 134-135oС;
гидрохлорид 2-[4-(2-фтор-4-пентилоксибензил)фенил] аминоимидазолина, плавится со сморщиванием при 72oС;
оксалат 2-{4-[4-(2-изопропоксиэтокси)бензил]фенил}аминоимидазолина c tпл 134-137oC;
2-[4-(3-хлор-4-изобутоксибензил)фенил]аминоимидазолин с tпл 126-128oС и
гидрохлорид 2-{ 4-[3-хлор-4-(тетрагидропиран-4-илокси)бензил]фенил}аминоимидазолина с tпл 128-130oС.
ПРИМЕР 3.
2-[4-(4-Изопропоксибензил)фенил]аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Iа, в которой R1 обозначает группу формулы (A), R2 и R4 каждый обозначает водород, Y обозначает изопропоксигруппу, a m обозначает 0.
Стадия 1.
Смесь 30,0 г (173 ммоля) 4-бромфенола, 26,3 г (190 ммолей) карбоната калия, 0,60 г (4 ммоля) иодида натрия, 85,1 г (0,692 ммоля) 2-бромпропана и 173 мл N, N-диметилформамида выдерживали при 60oС в течение 17 ч. Раствор охлаждали до комнатной температуры и добавляли 300 мл воды. Раствор экстрагировали диэтиловым эфиром. Экстракт промывали водным гидроксидом натрия, водой и водным хлоридом натрия, сушили (над Na2SO4), фильтровали и концентрировали. Продукт очищали вакуумной перегонкой с получением в виде бесцветной жидкости 25,3 г (118 ммолей) 4-бромизопропоксибензола.
Стадия 2.
Смесь 0,534 г (22,0 ммоля) магния и 20 мл тетрагидрофурана в токе азота доводили до кипения с обратным холодильником. В раствор медленно добавляли 3,10 г (14,4 ммолей) 4-бромизопропоксибензола и для гарантии инициирования реакции 0,74 г (3,93 ммоля) 1,2-дибромэтана. После завершения реакции Гриньяра раствор охлаждали до комнатной температуры и добавляли 14,5 мл (7,3 ммолей) 0,5 М раствора хлорида цинка в тетрагидрофуране. Раствор выдерживали при температуре кипения с обратным холодильником в течение 30 мин, а затем охлаждали до комнатной температуры.
Стадия 3.
Смесь 1,47 г (7,92 ммоля) 4-нитробензоилхлорида, 0,46 г (0,40 ммолей) тетракис(трифенилфосфин)палладия и 10 мл тетрагидрофурана в токе азота охлаждали на бане из воды со льдом. В раствор добавляли порцию (3,5 ммоля) раствора диарилцинка. Раствор перемешивали в течение 1 ч на бане из воды со льдом, а затем в течение ночи при комнатной температуре. Раствор разбавляли 15 мл воды и концентрировали. Концентрат экстрагировали дихлорметаном. Экстракт сушили (над MgSO4), фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле, получая 1,0 г (3,5 ммоля) 4-изопропоксибензоил-4-нитробензола с tпл 135,6-136,9oС.
Заключительная стадия.
В ходе проведения процесса аналогично тому, как это указано в описании заключительной стадии примера 1, в качестве продукта, идентичного тому, который получали в примере 1, образовывался 2-[4-(4-изопропоксибензил)фенил] аминоимидазолин.
ПРИМЕР 4.
2-[4-(4-Изопропоксиметилбензил)фенил] аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Iа, в которой R1 обозначает группу формулы (A), R2 и R4 каждый обозначает водород, Y обозначает изопропил, a m обозначает 1.
Стадия 1.
В перемешиваемый раствор 300 мг (13,1 ммолей) 1-бром-4-изопропоксиметилбензола (получен аналогично примеру получения 1) в 1,4 мл сухого тетрагидрофурана при -78oС в атмосфере аргона по каплям добавляли 1,62 мл (2,75 ммолей) 1,7 М трет-бутиллития в пентане. После перемешивания смеси в течение приблизительно 20 мин добавляли 0,35 мл (1,31 ммоля) хлорида три-н-бутилолова. Смеси давали нагреться до 0-5oС и перемешивали в течение примерно 1 ч. При 0-5oС добавляли смесь 374 мг (13,1 ммолей) 2-триметилсиланилэтилового эфира (4-хлорметилфенил)карбаминовой кислоты (полученного так, как изложено в примере получения 2), 4,4 мл гексаметилфосфорамида и 29,3 мг (0,025 ммолей) тетракис(трифенилфосфин)палладия(0) и затем выдерживали при 65oС в течение 20 ч. Раствор распределяли между 15 мл воды и 15 мл диэтилового эфира. Водную фазу экстрагировали диэтиловым эфиром и объединенные органические растворы промывали водой и рассолом, сушили (над Na2SO4) и выпаривали в вакууме. Остаток растворяли в ацетонитриле, промывали 2 порциями по 20 мл гексана, сушили (над Na2SO4), концентрировали в вакууме и очищали экспресс-хроматографией с получением в виде прозрачного масла 204 мг (26%) чистого 2-триметилсиланилэтилового эфира 4-(4-изопропоксиметилбензил)фенилкарбаминовой кислоты.
Стадия 2.
В раствор 734 мг (1,84 ммолей) 2-триметилсиланилэтилового эфира 4-(4-изопропоксиметилбензил)фенилкарбаминовой кислоты в сухом диметилсульфоксиде при 20oС в атмосфере аргона добавляли 5,52 мл (5,52 ммоля) фторида тетра-н-бутиламмония в тетрагидрофуране. После перемешивания смеси в течение 1 ч раствор распределяли между 50 мл воды и 50 мл диэтилового эфира. Водную фазу экстрагировали 2 порциями по 20 мл диэтилового эфира и объединенные органические слои промывали водой и рассолом, сушили (над MgSO4) и выпаривали в вакууме. Сырой продукт очищали экспресс-хроматографией, получая в виде прозрачного масла 405 мг (86%) чистого 4-(4-изопропоксиметилбензил)фениламина.
Заключительная стадия.
В раствор 400 мг (1,57 ммолей) 4-(4-изопропоксиметилбензил) фениламина в 4,8 мл изопропанола при 20oС в атмосфере аргона добавляли 382мг (1,88 ммолей) 2-хлор-2-имидазолинсульфата. Смесь выдерживали при 80oС с перемешиванием в течение 3 ч. Растворитель выпаривали в вакууме, добавляли воду и 10%-ный гидроксид натрия до достижения рН 11-12. Основную водную фазу экстрагировали 3 порциями по 20 мл дихлорметана и объединенные органические слои промывали водой, сушили (над Na2SO4) и выпаривали в вакууме. Сырой продукт очищали экспресс-хроматографией, промывали 0,07 М карбонатом калия и концентрировали с получением в виде прозрачного масла 463 мг (91%) чистого 2-[4-(4-изопропоксиметилбензил)фенил]имидазолина.
К 2-[4-(4-изопропоксиметилбензил)фенил] имидазолину добавляли 129 мг (1,43 ммоля) щавелевой кислоты и в результате перекристаллизации из ацетона в виде белого кристаллического твердого вещества получали 507 мг оксалата 2-[4-(4-изопропоксиметилбензил)фенил]имидазолина с tпл 156,3-156,7oС; данные элементного анализа для С20Н25N3О•С2Н2O4, рассчитано: С 63,91, Н 6,58, N 10,16; обнаружено: С 63,98, Н 6,53, N 10,24.
В ходе проведения процесса аналогично тому, как это указано в описании стадии 1 примера 4, но с заменой 1-бром-4-изопропоксиметилбензола 1-бром-4-втор-бутоксиметилбензолом, а затем в соответствии с последующими стадиями получали оксалат 2-[4-(4-втор-бутоксиметилбензил)фенил] имидазолина с tпл 145,0-145,3oС; данные элементного анализа для C21H27N3O•C2H2O4, рассчитано: С 64,62, Н 6,84, N 9,83; обнаружено: С 64,81, Н 6,82, N 9,98.
ПРИМЕР 5.
2-[4-(4-Морфолинобензил)фенил]аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Ib, в которой R1 обозначает группу формулы (А), R2 и R4 каждый обозначает водород, а R3 обозначает морфолин.
Стадия 1.
26,0 г (195 ммолей) хлорида алюминия отдельными порциями добавляли в раствор 27,8 г (150 мл) 4-нитробензоилохлорида и 15,8 г (165 ммолей) 4-фторбензола в 100 мл сероуглерода. Через 1 ч образовавшуюся желтую смесь осторожно обрабатывали 60 мл концентрированной соляной кислоты и перемешивали в течение 30 мин. Далее смесь разбавляли водой и экстрагировали этилацетатом. Органический слой промывали разбавленным раствором гидроксида натрия, водой и рассолом, сушили (над MgSO4) и в вакууме удаляли растворитель. В результате кристаллизации из диэтилового эфира/гексанов в виде белого твердого вещества получали 10,6 г (82%) 4-(4-фторбензоил) нитробензола с tпл 87-88oС; данные элементного анализа для C13H8NO3F, рассчитано: С 63,69, Н 3,26, N 5,71; обнаружено: С 63,89, Н 3,28, N 5,78.
Стадия 2.
Смесь, содержавшую 1,96 г (8 ммолей) 4-(4-фторбензоил)нитробензола, 0,84 г (9,6 ммолей) морфолина, 1,33 г (9,6 ммолей) карбоната калия и 15 мл диметилсульфоксида, выдерживали при 100-110oС в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли холодной водой и фильтровали. Сырой продукт несколько раз промывали водой и сушили, получая 2,28 г (91%) 4-(4-морфолинобензоил)нитробензола с tпл 173-175oС, который использовали на последующей стадии без дальнейшей очистки.
Стадия 3.
1,0 г 4-(4-морфолинобензоил)нитробензола гидрировали под давлением 50 фунтов/кв. дюйм с использованием 10%-ного палладия на угле в этаноле и минеральной кислоте, как указано в вышеприведенном описании стадии 5 в примере 1. 0,66 г (77%) 4-(4-морфолинобензил)фениламина, полученного таким образом в качестве продукта, использовали на последующей стадии без дальнейшей очистки.
Заключительная стадия.
В ходе проведения процесса аналогично заключительной стадии в примере 1, но с заменой 4-(4-изопропоксибензил)фениламина 4-(4-морфолинобензил)фениламином (0,63 г) получали 0,51 г (63%) 2-[4-(4-морфолинобензил)фенил] аминоимидазолина с tпл 177-179oС; данные элементного анализа для C20H24N4О, рассчитано: С 71,44, Н 7,14, N 16,66; обнаружено: С 71,62, Н 7,24, N 16,41.
В ходе проведения процесса аналогично стадии 1 в примере 5 а затем соответственно аналогично стадии 3 и заключительной стадии в примере 5, получали 2-[4-(4-фторбензил)фенил]аминоимидазолин с tпл 110-112oС.
В ходе проведения процесса аналогично стадии 2 в примере 5, но с заменой морфолина N,N-(2-гидроксиэтил)амином, а затем соответственно аналогично последующим стадиям в примере 5 получали 2- { 4- [4-(N,N-(2-гидроксиэтил)амино)бензил]фенил}аминоимидазолин с tпл 150-152oC.
В ходе проведения процесса аналогично стадии 1 в примере 5, но с заменой 4-фторбензола 2,4-дифторбензолом, а затем соответственно аналогично стадии 3 и заключительной стадии в примере 5 получали гидрохлорид 2- [4-(2,4-дифторбензил)фенил] аминоимидазолина с tпл 150-152oС.
В ходе проведения процесса аналогично стадии 1 в примере 5, но с заменой 4-фторбензола алкилированными бензолами, а затем соответственно аналогично последующим стадиям в примере 5 получали следующие соединения формулы I:
гидрохлорид 2-[4-(4-этилбензил)фенил]аминоимидазолина с tпл 72-74oС;
фумарат 2-[4-(4-изопропилбензил)фенил]аминоимидазолина; данные элементного анализа для С19Н23N3О•0,75С4Н4О2, рассчитано: С 62,60, Н 6,28, N 9,78; обнаружено: С 62,61, Н 6,44, N 10,00;
фумарат 2-[4-(4-изобутилбензил)фенил] аминоимидазолина с tпл 182-184oС;
гидрохлорид 2-{4-[4-(3-метилбутил)бензил]фенил}аминоимидазолина; данные элементного анализа для C21H28N3Cl, рассчитано: С 69,94, Н 7,91, N 7,76; обнаружено: С 69,84, Н 11,65, N 11,75;
гидрохлорид 2-[4-(4-пропилбензил)фенил] аминоимидазолина; данные элементного анализа для C19H24N3O, рассчитано: С 68,07, Н 7,39, N 12,53; обнаружено: С 68,05, Н 7,21, N 12,70;
фумарат 2-{ 4-[4-(циклопентил)бензил] фенил} аминоимидазолина; данные элементного анализа для С25Н29N3O4, рассчитано: С 62,20, Н 6,19, N 8,43; обнаружено: С 62,42, Н 6,23, N 8,63, и
фумарат 2-{ 4-[4-(циклогексил)бензил] фенил} аминоимидазолина; данные элементного анализа для С22Н28N3Сl, рассчитано: С 68,43, Н 7,78, N 10,88; обнаружено: С 68,36, Н 7,45, N 11,23.
ПРИМЕР 6.
2-{4-[4-(4-метоксифенил)сульфониламинометилбензил]фенил}аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Iс, в которой R1 обозначает группу формулы (А), R2, R4 и R7 каждый обозначает водород, R9 обозначает 4-метоксифенил, a m обозначает 1.
Стадия 1.
9,3 г хлорида алюминия в виде одной порции вводили в смесь 10 г 4-нитробензоилхлорида и 6,3 мл толуола, растворенных в 35 мл сероуглерода. Смесь нагревали от комнатной температуры до температуры кипения и кипятили с обратным холодильником в течение 3 ч. Медленно добавляли 19 мл концентрированной соляной кислоты и смесь перемешивали в течение еще 30 мин, быстро выливали в воду и 3 раза экстрагировали дихлорметаном. Экстракт промывали разбавленным гидроксидом аммония и водой, сушили (над Na2SO4) и выпаривали. Перекристаллизацией из этилацетата в виде бледно-желтого твердого вещества получали 10,6 г (82%) 4-(4 -метилбензоил)нитробензола с tпл 122,4-123,1oС.
Стадия 2.
В смесь 1,20 г 4-(4-метилбензоил) нитробензола и 0,89 г N-бромсукцинимида, суспендированную в 63 мл тетрахлорида углерода, добавляли 0,012 г перекиси бензоила. Реакционную смесь кипятили в токе аргона с обратным холодильником в течение 4 ч, одновременно облучая лампой накаливания. Смесь фильтровали, а желтый раствор, содержавший сырой продукт, выпаривали и хроматографировали на силикагеле, элюируя гексаном/этилацетатом, с получением в виде белого твердого вещества 1,79 г (81%) 4-(4-бромметилбензоил)нитробензола с tпл 112,7-113,1oС.
Стадия 3.
500 мг 4-(4-бромметилбензоил)нитробензола растворяли в 5 мл дихлорметана и перемешивали в токе аргона. По каплям добавляли раствор 0,27 мл трифторметансульфокислоты в 2 мл дихлорметана, а затем раствор 0,37 мл триэтилсилана в 2 мл дихлорметана. По прошествии 5 мин добавляли вторую порцию трифторметансульфокислоты и триэтилсилана (в тех же самых пропорциях). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, выливали в избыток водного бикарбоната натрия и 3 раза экстрагировали дихлорметаном. Раствор, содержавший сырой продукт, выпаривали, хроматографировали на силикагеле, элюируя продукт гексаном/этилацетатом, и выпаривали досуха, получая 270 мг (56%) 4-(4-бромметилбензил)нитробензола.
Стадия 4.
210 мг (1,15 экв.) азида натрия вводили в раствор 901 мг 4-(4-бромметилбензил)нитробензола в 10 мл N,N-диметилформамида. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, выливали в воду и 3 раза экстрагировали диэтиловым эфиром. Органическую фазу 3 раза промывали водой, сушили (над MgSO4) и выпаривали досуха. Хроматографией на силикагеле, элюируя гексаном/этилацетатом, в виде желтого масла получали 492 мг (62%) 4-(4-азидометилбензил)нитробензола.
Стадия 5.
Раствор 5,835 г 4-(4-азидометилбензил)нитробензола в 175 мл тетрагидрофурана обрабатывали 0,43 мл воды и 6,45 г (1,1 экв.) трифенилфосфина. Смесь перемешивали при комнатной температуре в течение 18 ч и выпаривали растворитель. Остаток суспендировали в воде, суспензию подкисляли добавлением по каплям соляной кислоты до значения рН приблизительно 1, а затем 3 раза экстрагировали диэтиловым эфиром. Водную фазу подщелачивали 50%-ным водным гидроксидом натрия и 3 раза экстрагировали дихлорметаном с получением в виде коричневого масла 5,66 г сырого продукта. Хроматографией на силикагеле, элюируя дихлорметаном/метанолом/гидроксидом аммония, в виде желтого твердого вещества получали 3,91 г (79%) 4-(4-аминометилбензил)нитробензола.
Стадия 6.
500 мг 4-(4-аминометилбензил)нитробензола растворяли в 7 мл дихлорметана и нагревали с 426 мг 4-метоксибензолсульфонилхлорида и 0,3 мл триэтиламина. Смесь перемешивали при комнатной температуре в течение 15 ч, выливали в разбавленный водный раствор соляной кислоты и 3 раза экстрагировали дихлорметаном. Растворитель выпаривали, получая в виде желтого твердого продукта 851 мг (~100%) 4-[4-(4-метоксифенил)сульфониламинометилбензил] нитробензола, который без дальнейшей очистки использовали на последующей стадии.
Стадия 7.
791 мг 4-[4-(4-метоксифенил)сульфониламинометилбензил] нитробензола растворяли в 15 мл этилацетата и гидрировали при комнатной температуре над 5%-ным палладием на угле. Реакцию проводили в течение 6 ч, смесь фильтровали и выпаривали досуха. Полученный таким образом сырой продукт хроматографировали в короткой колонке с диоксидом кремния, элюируя гексаном/этилацетатом в соотношении 1: 1, с получением в виде желтого твердого вещества 645 мг (87%) 4-[4-(4-метоксифенил)сульфониламинометилбензил]фениламина.
Заключительная стадия.
В ходе проведения процесса аналогично заключительной стадии в примере 1, но с заменой 4-(4-изопропоксибензил)фениламина 645 мг 4-[4-(4-метоксифенил)сульфониламинометилбензил]фениламина концентрированием путем выпаривания растворителей и хроматографией на силикагеле, элюируя дихлорметаном/метанолом/гидроксидом аммония в соотношении 60:10:1, и перекристаллизацией из ацетона со 134 мг (1 экв.) щавелевой кислоты в виде белого твердого вещества получали 685 мг (75%) оксалата 2-{4-[4-(4-метоксифенил)сульфониламинометилбензил] фенил} аминоимидазолина с tпл 167,0-167,5oС; данные элементного анализа для C26H28N4О7S, рассчитано: С 57,89, Н 5,11, N 10,47; обнаружено: С 57,77, Н 5,22, N 10,36.
В ходе проведения процесса аналогично стадии 6 в примере 6, но с заменой 4-метоксибензолсульфонилхлорида другими сульфонилхлоридами, а затем соответственно аналогично последующим стадиям в примере 6 получали следующие соединения формулы I:
гидрохлорид 2-[4-(4-бензолсульфониламинометилбензил)фенил]аминоимидазолина с tпл 228,2-229,2oС;
оксалат 2-{4-[4-(4-фторфенил)сульфониламинометилбензил]фенил}аминоимидазолина с tпл 170,0-171,2oС;
оксалат 2-{4-[4-(2-фторфенил)сульфониламинометилбензил]фенил}аминоимидазолина с tпл 94,4-95,6oС;
оксалат 2-[4-(4-изопропилсульфониламинометилбензил)фенил]аминоимидазолина с tпл 128,5-129,5oC и
оксалат 2-[4-(4-пропилсульфониламинометилбензил)фенил]аминоимидазолина с tпл 122,8-123,5oС.
ПРИМЕР 7.
2-[4-(4-Этансульфониламинобензил)фенил]аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Ic', в которой R1 обозначает группу формулы (А), R2 и R4 каждый обозначает водород, R9 обозначает этил, a m обозначает 0.
Стадия 1.
1,1 мл (10 ммолей) этансульфонилхлорида в виде одной порции вводили в раствор 1,98 г (10 ммолей) 4,4'-метилендианилина в 25 мл дихлорметана. Реакционную смесь перемешивали в течение 1 ч и выпаривали с получением твердого вещества, которое растворяли в 50 мл дихлорметана и выливали в смесь диэтилового эфира/2%-ного водного раствора карбоната калия в соотношении 1:1. После экстракции водный слой удаляли и отбрасывали. Органический слой экстрагировали 2 порциями по 100 мл 1%-ного водного раствора гидроксида калия, водный слой обрабатывали избыточным количеством диоксида углерода и экстрагировали 3 порциями по 25 мл дихлорметана. Дихлорметановый слой разбавляли 125 мл диэтилового эфира и экстрагировали 2 порциями по 100 мл 1%-ного водного раствора соляной кислоты. Слои вновь разделяли, водную фазу экстрагировали 50 мл диэтилового эфира, а органические фазы сбрасывали в отход. Водную фазу, которая содержала продукт, нейтрализовали твердым карбонатом калия, экстрагировали 4 порциями по 20 мл дихлорметана и выпаривали досуха. Перекристаллизацией из ацетона/гексанов в виде белых игольчатых кристаллов получали 940 мг (32%) 4-[4-(этансульфонил)аминобензил]фениламина с tпл 108-109oC.
Заключительная стадия.
В ходе проведения процесса аналогично заключительной стадии в примере 1, но с заменой 4-(4-изопропоксибензил)фениламина 290 мг (1 ммоль) 4-[4-(этансульфонил)аминобензил]фениламина и кристаллизацией из этилацета получали 319 мг (89%) 2-[4-(4-этансульфониламинобензил)фенил]аминоимидазолина.
Гидрохлорид получали суспендированием свободного основания в 10 мл метанола и добавлением этанольного раствора соляной кислоты до достижения кислой реакции. Растворители выпаривали и продукт кипятили с обратным холодильником при перемешивании в 5 мл этилацетата. Фильтрованием и сушкой гидрохлорида 2-[4-(4-этансульфониламинобензил)фенил] аминоимидазолина получали продукт с tпл 178-178,5oС; данные элементного анализа для C18H23ClN4O2S, рассчитано %: С 54,74, Н 5,87, N 14,19; обнаружено %: С 54,65, Н 5,79, N 14,21.
В ходе проведения процесса аналогично стадии 1 в примере 7, но с заменой этансульфонилхлорида другими сульфонилхлоридами, а затем соответственно аналогично последующим стадиям в примере 7 получали следующие соединения формулы I:
гидрохлорид 2-[4-(4-бензолсульфониламинобензил)фенил] аминоимидазолина; данные элементного анализа для С22Н23СlN4O2S, рассчитано: С 57,89, Н 5,41, N 12,27; обнаружено: С 57,66, Н 5,17, N 11,95;
гидрохлорид 2-{ 4-[4-(4-метилфенил)сульфониламинобензил]фенил}аминоимидазолина; данные элементного анализа для С23Н25СlN4О2S, рассчитано: С 59,40, Н 5,61, N 12,07; обнаружено: С 59,59, Н 5,64, N 11,66;
гидрохлорид 2-[4-(4-изопропилсульфониламинобензил)фенил]аминоимидазолина с tпл 206,6-207oС;
гемиоксалат 2-[4-(4-метансульфониламинобензил)фенил] аминоимидазолина с tпл 254,2-254,5oC;
гидрохлорид 2-[4-(4-бензилсульфониламинобензил)фенил] аминоимидазолина; данные элементного анализа для C23H25ClN4O2S, рассчитано: С 60,45, Н 5,51, N 12,26; обнаружено: С 60,33, Н 5,67, N 12,39;
гидрохлорид 2-{4-[4-(2,2,2-трифторэтил)сульфониламинобензил] фенил}аминоимидазолина; данные элементного анализа для С18Н20С1F3N4O2S, рассчитано: С 48,16, Н 4,49, N 12,48; обнаружено: С 47,89, Н 4,47, N 12,33;
гидрохлорид 2-[4-(4-пропилсульфониламинобензил)фенил] аминоимидазолина; данные элементного анализа для C19H25ClN4O2S, рассчитано: С 55,56, Н 6,18, N 13,64; обнаружено: С 55,34, Н 6,17, N 13,44;
гидрохлорид 2-[4-(4-бутилсульфониламинобензил)фенил] аминоимидазолина с tпл 157-160oС;
гидрохлорид 2-{4-[4-(4-метоксифенил)сульфониламинобензил]фенил}аминоимидазолина; данные элементного анализа для С23Н25СlN4О3S, рассчитано: С 57,96, Н 5,35, N 11,76; обнаружено: С 57,81, Н 5,35, N 11,58;
гидрохлорид 2-{ 4-[4-(тиен-2-илсульфонил)аминобензил] фенил}аминоимидазолина с tпл 109,5-110oС и
гидрохлорид 2-[4-(4-диметиламиносульфониламинобензил)фенил] аминоимидазолина с tпл 198,5-201oС.
В ходе проведения процесса аналогично стадии 1 в примере 7, но с заменой этансульфонилхлорида карбонилсульфонилхлоридами, а затем соответственно аналогично последующим стадиям в примере 7 из соответствующего соединения формулы Id получали следующие соединения формулы I:
2-{ 4-[4-(тетрагидропиран-4-илкарбонил)аминобензил]фенил}аминоимидазолин с tпл 225-227oС и
хлорид 2-{ 4-[4-(изопропилкарбонил)аминобензил] фенил}аминоимидазолина, смолоподобная масса.
ПРИМЕР 8.
2-{4-[4-(Этансульфонил)метиламинобензил]фенил}аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Iс, в которой R1 обозначает группу формулы (А), R2 и R4 каждый обозначает водород, R7 обозначает метил, R9 обозначает этил, a m обозначает 0.
113 мг (1 ммоль) твердого трет-бутоксида калия вводили в раствор 290 мг 4-(4-этансульфониламинобензил)фениламина (полученного по вышеизложенному в примере 7) в 2 мл диметилсульфоксида. Смесь перемешивали и добавляли 0,1 мл (1,5 ммоля) метилиодида. По прошествии 1 ч реакционную смесь выливали в воду и 2 раза экстрагировали диэтиловым эфиром. Органическую фазу сушили над карбонатом калия, выпаривали и хроматографировали на силикагеле, элюируя дихлорметаном/ацетоном, с получением в виде желтого твердого продукта 200 мг (66%) 4-[4-(этансульфонил)метиламинобензил]фениламина.
В ходе проведения процесса аналогично заключительной стадии в примере 1, но с заменой 4-(4-изопропоксибензил)фениламина 300 мг (0,99 ммолей) 4-[4-(этансульфонил)метиламинобензил] фениламина, кипячением реакционной смеси с обратным холодильником в изопропаноле в течение 16 ч и кристаллизацией из этилацетата/гексана получали 318 мг (89%) 2-{4-[4-(этансульфонил)метиламинобензил]фенил}аминоимидазолина. Далее этот свободный амин превращали в гидрохлоридную соль с tпл 178-178,5oС; данные элементного анализа для C19H23ClN4O2S, рассчитано: С 53,68, Н 6,35, N 13,18; обнаружено: С 53,72, Н 6,01, N 13,09.
В ходе проведения процесса так, как в примере 8, но с заменой метилиодида другими алкилиодидами и затем соответственно так, как в примере 8, получали следующие соединения формулы I:
гидрохлорид 2-{4-[4-(метансульфонил)бензиламинобензил]фенил}аминоимидазолина; данные элементного анализа для C24H2ClN4O2S, рассчитано: С 58,08, Н 5,95, N 10,41; обнаружено: С 57,97, Н 5,94, N 10,58;
гидрохлорид 2-{4-[4-(изопропилсульфонил)метиламинобензил]фенил}аминоимидазолина; данные элементного анализа для С20Н2СlN4О2S, рассчитано: С 55,15, Н 6,57, N 12,86; обнаружено: С 55,11, Н 6,39, N 12,76;
гидрохлорид 2-{4-[4-(пропилсульфонил)метиламинобензил]фенил}аминоимидазолина; данные элементного анализа для C20H2ClN4O2S, рассчитано: С 56,31, Н 6,47, N 13,13; обнаружено: С 56,10, Н 6,34, N 13,04;
гидрохлорид 2-{4-[4-(этансульфонил)этиламинобензил]фенил)аминоимидазолина; данные элементного анализа для С21Н29СlN4О2S, рассчитано: С 56,55, Н 6,78, N 12,56; обнаружено: С 56,51, Н 6,61, N 12,51, и
гидрохлорид 2-{4-[4-(этансульфонил)пропиламинобензил]фенил}аминоимидазолина; данные элементного анализа для С22Н31ClN4O2S, рассчитано: С 54,47, Н 6,62, N 12,70; обнаружено: С 54,40, Н 6,44, N 12,59.
ПРИМЕР 9.
2-{4-[4-(1,1-Диоксоизотиазолидин-1-ил)бензил]фенил}аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Iс, в которой R1 обозначает группу формулы (А), R2 и R4 каждый обозначает водород, а R3 обозначает 1,1-диоксоизотиазолидин-1-ил.
1,40 г (3,9 ммолей) 4-[4-(3-хлорпропил)сульфониламинобензил]фениламина (полученного аналогично примеру 7) перемешивали в растворе в 25 мл тетрагидрофурана, содержавшего 180 мг гидрида натрия (4,5 ммолей, 60%-ного). Смесь кипятили с обратным холодильником в течение 16 ч, выливали в воду и 3 раза экстрагировали дихлорметаном. Выпариванием и перекристаллизацией в виде не совсем белого твердого продукта получали 1,09 г (86%) 4-[4-(1,1-диоксоизотиазолидин)бензил]фениламина с tпл 134,5-135,5oС.
В ходе проведения процесса аналогично заключительной стадии в примере 1, но с заменой 4-(4-изопропоксибензил)фениламина 4-[4-(1,1-диоксоизотиазолидин-1-ил)бензил] фениламином, получали 2-{4-[4-(1,1-диоксоизотиазолидин-1-ил)бензил] фенил} аминоимидазолин с tпл 197,2-198,5oС; данные элементного анализа для C21H24N4O6S, рассчитано: С 54,74, Н 5,25, N 12,17; обнаружено: С 54,63, Н 5,28, N 12,11.
ПРИМЕР 10.
2-{4-[4-(3-Фенилуреидо)бензил]фенил}аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Iе, в которой R1 обозначает группу формулы (A), R2, R4, R7 и R8 каждый обозначает водород, R9 обозначает фенил, V обозначает О, a m обозначает 0.
Стадия 1.
Смесь 19,8 г 4,4'-метилендианилина с 20 г карбоната калия в 300 мл этилацетата и 200 мл воды перемешивали на ледяной бане. В смесь медленно добавляли 15 мл бензилхлорформиата. Образовавшуюся смесь перемешивали в течение 1 ч, затем отделяли органический слой, а водный слой экстрагировали дополнительным количеством этилацетата. Объединенные органические экстракты промывали водой и рассолом, сушили (над Na2SO4) и в вакууме удаляли растворитель. Хроматографией в колонке с силикагелем выделяли сырую смесь, элюируя смесью 30% этилацетата/гексан, с получением в виде твердого вещества бензилового эфира 4-(4-аминобензил)фенилкарбаминовой кислоты.
Стадия 2.
0,997 г (3 ммоля) бензилового эфира 4-(4-аминобензил)фенилкарбаминовой кислоты вводили в раствор 0,393 г (3,3 ммоля) фенилизоцианата в 20 мл дихлорметана. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре в токе азота. Затем реакцию в реакционной смеси прекращали смешением с водой и смесь концентрировали в вакууме. Остаток суспендировали в воде, фильтровали, промывали водой и сушили с получением в виде белого твердого продукта 1,38 г бензилового эфира {4-[4-(3-фенилуреидо)бензил]фенил}карбаминовой кислоты.
Стадия 3.
Смесь 1,3 г бензилового эфира {4-[4-(3-фенилуреидо)бензил]фенил}карбаминовой кислоты с 0,35 г 10%-ного палладия на угле в 150 мл этанола гидрировали под давлением 50 фунтов/кв.дюйм в аппарате Парра в течение 12 ч. Реакционную смесь фильтровали через тампон из целита (броунмиллерита) с целью удалить катализатор. При концентрировании фильтрата в виде белого твердого вещества получали 0,76 г 4-[4-(3-фенилуреидо)бензил]фениламина.
Заключительная стадия.
Смесь 0,7 г (2,21 ммоля) 4-[4-(3-фенилуреидо)бензил]фениламина с 0,673 г (3,32 ммоля) 2-хлор-2-имидазолинсульфата в 20 мл 2-пропанола кипятили с обратным холодильником в течение 1 ч. Реакционную смесь концентрировали, разбавляли дихлорметаном и подщелачивали 10%-ным раствором гидроксида натрия. Органический слой отделяли, а водный слой экстрагировали дополнительным количеством дихлорметана. Объединенные органические экстракты промывали водой и рассолом, сушили (над К2СО3) и в вакууме удаляли растворитель. Остаток хроматографировали на оксиде алюминия (нейтральном активностью I, 15% метанола/дихлорметан) с получением в виде белого твердого вещества 2-{4-[4-(3-фенилуреидо)бензил] фенил}аминоимидазолина с tпл 167-170oС; данные элементного анализа для C23H23N5O, рассчитано: С 71,70, Н 5,97, N 18,18; обнаружено: С 71,34, Н 5,98, N 17,91.
В ходе проведения процесса аналогично тому, как это указано в описании стадии 6 в примере 6, но с заменой 4-метоксибензолсульфонилхлорида фенилизоцианатом, а затем соответственно аналогично примеру 6 получали фумарат 2-{ 4-[4-(3-фенилуреидо)метилбензил]фенил}аминоимидазолина с tпл 207-208,5oС.
ПРИМЕР 11.
2-[4-(4-Диметиламиносульфонилбензил)фенил]аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы If, в которой R1 обозначает группу формулы (А), R2, R4, R8 и R9 каждый обозначает метил, a m обозначает 0.
Стадия 1.
Раствор 4,26 г (70 ммолей) 4-бензилнитробензола в 25 мл дихлорметана при -30oС по каплям вводили в раствор 6 мл хлорсульфоновой кислоты в 25 мл дихлорметана. Смесь перемешивали в течение 10 мин при 0oС, а затем выливали в лед и встряхивали. Органическую фазу отделяли и выпаривали растворители. Остаток перекристаллизовывали из дихлорметана/гексана с получением в виде дихлорметановых гемисольватных кристаллов 5,52 г (78%) 4-(4-хлорсульфонилбензил)нитробензола.
Стадия 2.
354 мг (1 ммоль) дихлорметанового гемисольвата 4-(4-хлорсульфонилбензил) нитробензола, растворенного в 5 мл тетрагидрофурана, добавляли в 2 М раствор (избыточное количество) диметиламина в тетрагидрофуране. Смесь перемешивали в течение 1 ч, экстрагировали раствором диэтилового эфира и разбавленным карбонатом калия. Органический слой отделяли и выпаривали растворители. Остаток перекристаллизовывали из ацетона/гексана с получением 311 мг (98%) 4-(4-диметиламиносульфонилбензил)нитробензола.
Стадия 3.
311 мг 4-(4-диметиламиносульфонилбензил)нитробензола растворяли в этилацетате и гидрировали под давлением 40 фунтов/кв.дюйм с использованием 10%-ного палладия на угле в качестве катализатора в течение 2 ч. Смесь фильтровали и выпаривали растворители с получением в виде белого твердого вещества 288 мг (99%) 4-(4-диметиламиносульфонилбензил)фениламина.
Заключительная стадия.
Смесь 288 мг 4-(4-диметиламиносульфонилбензил)фениламина и 110 мг сульфата 2-хлоримидазолина в 2-пропаноле кипятили с обратным холодильником в течение 16 ч. В эту смесь быстро выливали разбавленный раствор карбоната калия и экстрагировали 4 порциями по 15 мл дихлорметана. Растворители выпаривали, получая в виде твердого вещества 320 мг 2-[4-(4-диметиламиносульфонилбензил)фенил]аминоимидазолина.
Продукт растворяли в этилацетате и обрабатывали избыточным количеством метанольного раствора хлорида водорода с получением сырого продукта. Растворители выпаривали, а остаток перекристаллизовывали из 2-пропанола/этилацетата с получением в виде белого твердого вещества 292 мг (76%) гидрохлорида 2-[4-(4-диметиламиносульфонилбензил)фенил] аминоимидазолина с tпл 194,1-195,3oС; данные элементного анализа для C18H23ClN4О2S, рассчитано: С 54,99, Н 5,87, N 14,19; обнаружено: С 54,74, Н 5,87, N 13,96.
В ходе проведения процесса аналогично стадии 2 в примере 11, но с заменой диметиламина другими аминами, а затем соответственно аналогично последующим стадиям в примере 11, получали другие соединения формулы I:
гидрохлорид 2-[4-(4-бензиламиносульфонилбензил)фенил] аминоимидазолина, плавившийся подобно стеклообразной массе; данные элементного анализа для C23H23ClN4O2S•0,7H2O, рассчитано %: С 59,04, Н 5,45, N 11,97; обнаружено %: С 59,05, Н 5,42, N 11,90;
гидрохлорид 2-[4-(4-изобутиламиносульфонилбензил)фенил]аминоимидазолина, плавившийся подобно стеклообразной массе; данные элементного анализа для C20H27ClN4O2S•0,7H2O, рассчитано %: С 55,36, Н 6,34, N 12,91; обнаружено %: С 55,38, Н 6,21, N 12,66;
гидрохлорид 2-[4-(4-пирролидин-1-илсульфонилбензил)фенил] аминоимидазолина с tпл 190,0-191,2oС; данные элементного анализа для C20H25ClN4O2S, рассчитано %: С 57,06, Н 5, 99, N 13,31; обнаружено %: С 56,97, Н 5,93, N 13,15;
оксалат 2-[4-(4-изопропиламиносульфонилбензил)фенил] аминоимидазолина с tпл 138,0-140,5oС; данные элементного анализа для C21H26N4O6S, рассчитано: С 54,53, Н 5,67, N 12,11; обнаружено: С 54,39, Н 5,58, N 12,02;
гидрохлорид 2-[4-(4-диизопропиламиносульфонилбензил)фенил]аминоимидазолина, плавившийся подобно стеклообразной массе; данные элементного анализа для C22H31ClN4O2S•0,5H2O, рассчитано: С 57,57, Н 6,81, N 12,20; обнаружено: С 57,67, Н 6,85, N 11,81;
оксалат 2-[4-(4-трет-бутиламиносульфонилбензил)фенил] аминоимидазолина, плавившийся подобно стеклообразной массе; данные элементного анализа для С22Н28N4O6S•Н2O, рассчитано: С 53,43, Н 6,11, N 11,33; обнаружено: С 53,67, Н 6,30, N 11,03, и
оксалат 2-[4-(4-бутиламиносульфонилбензил)фенил] аминоимидазолина с tпл 153,6-154,4oС; данные элементного анализа для C22H28N4O6S, рассчитано: С 55,45, Н 5,92, N 11,76; обнаружено: С 55,23, Н 5,80, N 11,67.
ПРИМЕР 12.
2-[4-(4-Бензиламиносульфонилметилбензил)фенил]аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы If, в которой R1 обозначает группу формулы (А), R2, R4 и R8 каждый обозначает водород, R9 обозначает бензил, а m обозначает 1.
Альтернативная стадия 1а.
Раствор 0,73 г (7 ммолей) сульфита натрия в 10 мл воды добавляли в раствор 1,41 г (5,6 ммолей) 4-(4-бромметилбензил) нитробензола в 10 мл ацетонитрила. Смесь перемешивали и кипятили с обратным холодильником в течение 2 ч. Выпаривали растворители и сушили с получением в виде белого порошка 2,29 г натриевой соли 4-(4-нитробензил)фенилметансульфокислоты.
Альтернативная стадия 1б.
2,29 г натриевой соли 4-(4-нитробензил)фенилметансульфокислоты объединяли с 1,45 г пентахлорида фосфора. Смесь выдерживали при 90oС в течение 5 мин, выливали в воду и экстрагировали 3 порциями по 20 мл дихлорметана. Выпаривали растворители с получением в виде неочищенного желтого твердого вещества 0,63 г 4-(4-хлорсульфонилметилбензил)нитробензола. Этот продукт использовали непосредственно на последующей стадии.
Стадия 2.
0,3 мл бензиламина добавляли в раствор 100 мг 4-(4-хлорсульфонилметилбензил)нитробензола в 4 мл тетрагидрофурана. Смесь перемешивали в течение 2 ч, выливали в разбавленный водный раствор карбоната калия и экстрагировали 3 порциями по 15 мл дихлорметана. Выпаривали растворители и остаток хроматографировали на силикагеле, элюируя 2%-ным раствором ацетона в дихлорметане, с получением в виде твердого вещества 44 мг 4-(4-бензиламиносульфонилметилбензил)нитробензола.
Стадия 3.
Раствор 44 мг 4-(4-бензиламиносульфонилметилбензил)нитробензола растворяли в этилацетате и гидрировали под давлением 40 фунтов/кв.дюйм с 10%-ным палладием на угле в качестве катализатора в течение 2 ч. Смесь фильтровали и растворитель выпаривали с получением в виде не совсем белого твердого продукта 39 мг 4-(4-бензиламиносульфонилметилбензил)фениламина.
Заключительная стадия.
Смесь 39 мг 4-(4-бензиламиносульфонилметилбензил)фениламина с 1 экв. 2-хлоримидазолинового основания в 2-пропаноле кипятили с обратным холодильником в течение 16 ч. В смесь приливали разбавленный раствор карбоната калия и экстрагировали 4 порциями по 15 мл дихлорметана. Выпаривали растворители с получением в виде твердого продукта 41,7 мг 2-[4-(4-бензиламиносульфонилметилбензил)фенил] аминоимидазолина с tпл 115-118oС; данные элементного анализа для С24Н25СlN4O2S•Н2О, рассчитано: С 58,94, Н 5,98, N 11,46; обнаружено: С 59,01, Н 5,91, N 11,30.
В ходе проведения процесса аналогично тому, как это указано в описании стадии 2 в примере 12, но с заменой бензиламина другими аминами, а затем соответственно аналогично последующим стадиям в примере 12, получали другие соединения формулы I:
гидрохлорид 2-[4-(4-изобутиламиносульфонилметилбензил)фенил]аминоимидазолина (51,5 мг в виде твердого вещества) с tпл 113,2-114,6oС; данные элементного анализа для C21H29ClN4O2S•0,5H2O, рассчитано: С 56,55, Н 6,78, N 12,56; обнаружено: С 56,68, Н 6,67, N 12,40.
оксалат 2-[4-(4-диметиламиносульфонилметилбензил)фенил]аминоимидазолина (73 мг, 58%) с tпл 154,4-154,8oС; данные элементного анализа для C23H28N4O6S, рассчитано: С 56,54, Н 5,78, N 11,42; обнаружено: С 56,56, Н 5,67, N 11,46, и
оксалат 2-[4-(4-пирролидин-1-илсульфонилметилбензил)фенил] аминоимидазолина (105 мг, 63%) с tпл 160-161oС; данные элементного анализа для C21H26N4O6S, рассчитано: С 54,53, Н 5,67, N 12,11; обнаружено: С 54,48, Н 5,58, N 12,13.
ПРИМЕР 13.
2-[4-(4-Пирролидин-1-иламинокарбонилбензил)фенил]аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Ig, в которой R1 обозначает группу формулы (А), R2 и R4 каждый обозначает водород, R8 и R9, взятые вместе с азотным атомом, с которым они связаны, образуют пирролидин, a m обозначает 0.
Стадия 1.
0,5 г 10%-ного палладия на угле добавляли в раствор 11,31 г (50 ммолей) 4-бензоилбензойной кислоты в 250 мл этанола и 10 мл 70%-ной перхлорной кислоты. Суспензию гидрировали при комнатной температуре под давлением 40 фунтов/кв.дюйм в течение 8 ч. Фильтрованием удаляли катализатор и фильтрат нейтрализовали водным раствором бикарбоната натрия. Выпаривали растворители и остаток распределяли между этилацетатом и разбавленным водным раствором гидроксида калия. Водную фазу подкисляли соляной кислотой. Выпадавшую в осадок кислоту отфильтровывали, промывали и сушили с получением 10,74 г (~100%) 4-бензилбензойной кислоты.
Стадия 2.
4-(4-Нитробензил)бензойную кислоту получали в соответствии с методами, описанными в работе Coon и др. в J. Org. Chem., 1973, 38, 4243.
3,16 мл 70%-ной азотной кислоты по каплям добавляли в суспензию 9,34 мл (105,6 ммолей) трифторметансульфокислоты в 250 мл дихлорметана. Эту суспензию охлаждали на бане из сухого льда с ацетоном и по каплям добавляли раствор 10,19 г (48 ммолей) 4-бензилбензойной кислоты в 50 мл дихлорметана. Смесь перемешивали в течение приблизительно 2 ч при -78oС и дополнительно 2 ч при комнатной температуре. Реакционную смесь выливали в измельченный лед. Выделенный органический слой 2 раза промывали дихлорметаном и объединенные органические слои сушили (над Na2SO4) и выпаривали растворители. Перекристаллизацией сырого продукта из метанола/этилацетата в виде желтого твердого продукта получали 9,27 г (58%) 4-(4-нитробензил)бензойной кислоты.
Стадия 3.
1,03 г (4 ммоля) 4-(4-нитробензил)бензойной кислоты растворяли в 40 мл дихлорметана. В смесь добавляли 0,42 мл (1,2 экв.) оксалилхлорида, а затем 1 каплю N, N-диметилформамида. Смесь перемешивали в течение 1 ч при комнатной температуре и выпаривали растворители с получением в виде бледно-желтого твердого продукта 1,10 г 4-(4-хлоркарбонилбензил)нитробензола.
Стадия 4.
4-(4-Хлоркарбонилбензил)нитробензол растворяли в 40 мл дихлорметана и добавляли раствор 64 мг (1 экв.) пирролидина в 0,2 мл пиридина. Смесь перемешивали в течение 2 ч при комнатной температуре, промывали разбавленным раствором гидроксида калия и выпаривали растворители с получением остатка в виде желтого масла. Остаток хроматографировали на силикагеле, элюируя дихлорметаном/метанолом, с получением 299 мг (99%) 4-(4-пирролидин-1-илкарбонилбензил)нитробензола.
Стадия 5.
Смесь 202 мг (0,65 ммоля) 4-(4-пирролидин-1-илкарбонилбензил)нитробензола со 110 мг 10%-ного палладия на угле и 20 мл этанола гидрировали под давлением 40 фунтов/кв.дюйм в течение 16 ч. Смесь фильтровали через тампон из целита (броунмиллерита) и растворители выпаривали с получением в виде белого твердого продукта 187 мг (99%) 4-[4-(1-пирролидинкарбонил)бензил]фениламина.
Заключительная стадия.
Смесь 182 мг (0,64 ммоля) 4-[4-(1-пирролидинкарбонил)бензил]фениламина со 131 мг (1 экв.) бисульфата 2-хлор-2-имидазолина в 30 мл 2-пропанола перемешивали при 60oС в течение 60 ч. Выпаривали растворители, а остаток суспендировали в разбавленном растворе гидроксида калия. Суспензию экстрагировали дихлорметаном и хроматографировали на силикагеле, элюируя метанолом/гидроксидом аммония, с получением 205 мг белого твердого вещества. Это белое твердое вещество экстрагировали этилацетатом, промывали разбавленным гидроксидом калия и обрабатывали избыточным количеством соляной кислоты в диэтиловом эфире с получением 193 мг (77%) гидрохлорида 2-{4-[4-(1-пирролидинкарбонил)бензил] фенил} аминоимидазолина, который при 46oС сморщивался; данные элементного анализа для C21H25ClN4O•0,7H2O, рассчитано: С 63,71, Н 6,69, N 14,09; обнаружено: С 63,44, Н 6,38, N 13,81.
В ходе проведения процесса аналогично тому, как это указано в описании стадии 4 в примере 13, но с заменой пирролидина другими аминами, а затем соответственно аналогично последующим стадиям в примере 13, получали другие соединения формулы I:
оксалат 2-[4-(4-изобутиламинокарбонилбензил)фенил]аминоимидазолина с tпл 100-144oС; данные элементного анализа для C23H28N4O5, рассчитано: С 62,71, Н 6,41, N 12,72; обнаружено: С 62,44, Н 6,36, N 12,72, и
оксалат 2-[4-(4-бензиламинокарбонилбензил)фенил] аминоимидазолина с tпл 188,5-195,0oС; данные элементного анализа для C24H24N4O•0,85C2H2O4, рассчитано: С 66,95, Н 5,62, N 12,15; обнаружено: С 67,05, Н 5,55, N 12,26.
ПРИМЕР 14.
2-[4-(4-Циклопентилокситиен-2-илметил)фенил]аминоимидазолин.
Ниже описано получение соединения формулы I из соответствующего соединения формулы Ih, в которой R1 обозначает группу формулы (В), где Х обозначает S, R2 обозначает водород, Y обозначает циклопентил, a m обозначает 0.
Стадия 1.
В раствор 8,75 мл (95,8 ммолей) циклопентанола в 250 мл N,N-диметилформамида при 0-5oС в токе азота в виде 60%-ной дисперсии в минеральном масле вводили 3,84 г (95,8 ммолей) гидрида натрия. По прошествии 10 мин смеси позволяли достичь комнатной температуры и перемешивали в течение 40 мин. Добавляли 3,59 мл (38,3 ммоля) 3-бромтиофена, а затем 14,63 г (76,8 ммоля) иодида одновалентной меди. Смесь выдерживали при 120oC в течение 22 ч. После охлаждения до примерно 10oС при интенсивном перемешивании добавляли раствор 12,1 г (0,25 моля) цианида натрия в 200 мл воды. Смесь перемешивали еще в течение 10 мин, а затем фильтровали. Фильтрат экстрагировали гексаном. Экстракт промывали водой, сушили (над Na2SO4) и концентрировали досуха. Перегонкой (100oС, остаточное давление 8 мм рт.ст.) в виде слегка бледно-желтого масла получали 4,52 г (70%) 3-циклопентилокситиофена.
1H-ЯМР (300 МГц, CDCl3, δ): 7,15 (dd, J=5,2, 3,1 Гц, 1Н), 6,72 (dd, J= 5,2, 15 Гц, 1Н), 6,19 (dd, J=3,1, 1,5 Гц, 1H), 4,65 (квинтет, J=4,2 Гц, 1H), 1,55-1,95 (m, 8H).
MC m/e (%): 168 (M+; 17).
Стадия 2.
2-Хлор-3-циклопентилокситиофен получали в соответствии с методами, описанными Р. Stanetty и Е. Puschautz в Monatsheefte Chemie, 1989, 120, 65. Так, например, в раствор 3,98 г (23,7 ммоля) 3-циклопентилокситиофена в 35 мл дихлорметана при 15oС в токе аргона вводили 2 мл (24,9 ммоля) сульфурилхлорида. Смесь перемешивали в течение 1 ч, а затем концентрировали досуха. В результате очистки экспресс-хроматографией (диоксид кремния, 100%-ный гексан) в виде бледно-желтого масла получали 2,75 г (59%) 2-хлор-3-циклопентилокситиофена.
1H-ЯМР (300 МГц, CDCl3, δ): 6,99 (d, J=6,0 Гц, 1H), 6,75 (d, J=6,0 Гц, 1H), 4,69-4,74 (m, 1H), 1,7-1,93 (m, 6H), 1,5-1,7 (m, 2H).
MC m/e (%): 202 (М+; 6).
Стадия 3.
В раствор 2,15 г (10,6 ммоля) 2-хлор-3-циклопентилокситиофена в 27 мл диэтилового эфира по каплям при -78oС в токе аргона вводили 4,4 мл 2,5 н. (11 ммолей) н-бутиллития в гексане. Смеси позволяли достичь 20-25oС и перемешивали в течение 4 ч. После повторного охлаждения до -78oС по каплям добавляли раствор 1,56 г (10,3 ммоля) п-нитробензальдегида в тетрагидрофуране. Смесь перемешивали при -78oС в течение 1 ч. При -78oС добавляли насыщенного хлорида аммония и смеси позволяли достичь приблизительно 10oС. Этилацетатом экстрагировали сырой продукт, промывали водой и рассолом, сушили (над Na2SO4) и концентрировали досуха с получением 3,6 г 4-(5-хлор-4-циклопентилокситиен-2-ил)-(4-нитрофенил)метанола, который использовали непосредственно на последующей стадии.
Стадия 4.
4-(4-Циклопентилокситиен-2-илметил)нитробензол может быть получен по методам, которые описаны E.J.Stoner и др. в Tetrahedron, 1995, 51, 11043. Так, например, в суспензию 6,64 г (44,3 ммоля) иодида натрия в 10 мл ацетонитрила в токе аргона при 20-25oС вводили 5,6 мл (44,3 ммоля) триметилсилилхлорида. После перемешивания в течение 15 мин при 20-25oС смесь охлаждали до 0-5oС и медленно добавляли 3,6 г 4-(5-хлор-4-циклопентилокситиен-2-ил)-(4-нитрофенил)метанола в 10 мл ацетонитрила. Добавляли 11,5 мл 10%-ного водного раствора гидроксида натрия, а затем избыточное количество воды. Продукт экстрагировали этилацетатом, промывали раствором 4,83 г тиосульфата натрия в 10 мл воды, водой и рассолом, сушили (над Na2SO4) и концентрировали досуха. В результате очистки экспресс-хроматографией (диоксид кремния, гексан/этилацетат в соотношении 98,5:1,5) в виде бледно-желтого масла получали 1,25 г (40%) 4-(4-циклопентилокситиен-2-илметил) нитробензола.
1Н-ЯМР (300 МГц, CDCl3, δ): 11,97 (d, J=8,8 Гц, 2Н), 7,40 (d, J=8,8 Гц, 2Н), 6,45 (m, 1H), 6,03 (d, J=1,7 Гц, 1Н), 4,60 (квинтет, J=4,3 Гц, 1H), 4,13 (s, 2Н), 1,5-1,89 (m, 8H).
МС m/e (%): 303 (M+; 15).
Стадия 5.
В раствор 1,28 г (4,2 ммоля) 4-(4-циклопентилокситиен-2-илметил)нитробензола в 34 мл абсолютного этанола в токе азота при 20-25oС добавляли 4,76 г (21,2 ммоля) гидрата дихлорида олова. Смесь выдерживали при 75oС в течение 2,5 ч и охлаждали до 0-5oС. До достижения рН 8 добавляли насыщенного раствора бикарбоната натрия. Добавляли этилацетата и смесь фильтровали. Происходило расслоение, водную фазу экстрагировали дополнительным количеством этилацетата. Объединенные органические фазы промывали рассолом, сушили (над Na2SO4) и концентрировали досуха. В результате очистки экспресс-хроматографией (диоксид кремния, гексан/этилацетат) в виде бледно-желтого масла получали 0,47 г (41%) 4-(4-циклопентилокситиен-2-илметил)фениламина.
1Н-ЯМР (300 МГц, CDCl3, δ): 7,03 (d, J=8,5 Гц, 2Н), 6,63 (d, J=8,5 Гц, 2Н), 6,41 (m, 1H), 5,96 (d, J=1,7 Гц, 1H), 4,58 (квинтет, J=4,3 Гц, 1H), 3,91 (s, 2H), 3,59 (bs, 2H), 1,67-1,88 (m, 6H), 1,46-1,67 (m, 2H).
Заключительная стадия.
В раствор 463 мг (1,69 ммоля) 4-(4-циклопентилокситиен-2-илметил)фениламина в 7 мл изопропилового спирта вводили раствор 293 мг (2,8 ммоля) 2-хлор-2-имидазолина в 7 мл изопропилового спирта. Смесь кипятили с обратным холодильником в течение ночи и в вакууме удаляли изопропиловый спирт. Добавляли 10%-ного гидроксида натрия и продукт экстрагировали дихлорметаном. Экстракт промывали водой, сушили (над Nа2SO4) и концентрировали досуха с получением сырого продукта.
578 мг этого сырого продукта растворяли в 20 мл толуола с последующим добавлением 4 мл циклопентанола и 674 мг гидрата п-толуолсульфокислоты. Смесь выдерживали при 100-110oС в течение 2 ч и охлаждали до комнатной температуры. Добавляли 10%-ного раствора гидроксида натрия. Конечный продукт 3 раза экстрагировали дихлорметаном, промывали водой, сушили (над Na2SO4) и концентрировали досуха. В результате очистки препаративной тонкослойной хроматографией с элюированием этилацетатом/метиловым спиртом/изопропиламином в виде бледно-желтого масла получали 290 мг (50%) 2-[4-(4-циклопентилокситиен-2-илметил)фенил]аминоимидазолина.
1Н-ЯМР (300 МГц, CDCl3, δ): 7,13 (d, J=8,2 Гц, 2Н), 6,93 (d, J=8,2 Гц, 2Н), 6,42 (m, 1H), 5,97 (d, J=1,7 Гц, 1H), 4,59 (квинтет, J=4,3 Гц, 1H), 3,96 (s, 2H), 3,8-4,1 (широкий s, 2H), 3,52 (s, 4H), 1,67-1,89 (m, 6H), 1,51-1,66 (m, 2H).
Оксалат 2-[4-(4-циклопентилокситиен-2-илметил)фенил] аминоимидазолина имеет tпл 142,4-143,3oС.
В ходе проведения процесса аналогично стадии 1 в примере 14, но с заменой циклопентанола изопропанолом, а затем соответственно аналогично последующим стадиям в примере 14 получали оксалат 2-[4-(4-изопропокситиен-2-илметил)фенил]аминоимидазолина с tпл 151,3-151,8oС.
ПРИМЕР 15.
2-[4-(5-Метокситиен-2-илметил)фенил]аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Ii, в которой R1 обозначает группу формулы (В), где Х обозначает S, R2 обозначает водород, Y обозначает метил, а m обозначает 0.
Стадия 1.
2-Метокситиофен получали в соответствии с методами, описанными Н.А. Keeystra и др. в Tetrahedron, 1992, 48, 3633. Так, например, раствор метоксида натрия в метаноле готовили добавлением 2,12 г (92,2 ммоля) натрия в 14 мл метанола. Поддерживая кипячение с обратным холодильником, добавляли 10 г (61,3 ммоля) 2-бромтиофена. Вводили 0,88 г (6,1 ммоля) бромида одновалентной меди, поддерживая кипячение смеси с обратным холодильником в течение 5,5 ч. При интенсивном перемешивании при 20-25oС добавляли раствор 3 г (61,3 ммоля) цианида натрия в 30 мл воды. Смесь перемешивали до полного растворения твердых частиц, экстрагировали гексаном, сушили (над Na2SO4) и концентрировали досуха. В результате перегонки (90oС под остаточным давлением 80 мм рт.ст.) в виде бесцветного масла получали 5,35 г (76%) 2-метокситиофена.
Стадия 2.
5-Метокситиен-2-ил(три-н-бутил)станнан получали добавлением в токе аргона при -78oС 3,81 мл 1,98 М (7,54 ммоля) н-бутиллития в гексане в раствор 860 мг (7,54 ммоля) 2-метокситиофена в 4,3 мл тетрагидрофурана. Смеси давали достичь 0-5oС и перемешивали в течение 2 ч, вновь охлаждали до -78oС и добавляли 2,05 мл (7,54 ммоля) хлорида трибутилолова. Смеси давали достичь 0-5oС и перемешивали в течение 1 ч. Продукт, 5-метокситиен-2-ил(три-н-бутил)станнан, использовали непосредственно на последующей стадии.
Стадия 3.
В 2,15 г (7,54 ммоля) 2-триметилсиланилэтилового эфира 4-хлорметилфенилкарбаминовой кислоты (описанного в примере получения 2) при 20-25oС добавляли 5-метокситиен-2-ил(три-н-бутил)станнан, а затем 11 мл гексаметилфосфорамида и 174,2 мг (0,146 ммоля) тетракис(трифенилфосфин) палладия. Смесь выдерживали при 65oС в течение 4,5 ч. Добавляли воды и продукт экстрагировали диэтиловым эфиром. Экстракт промывали водой, сушили (над Na2SO4) и концентрировали досуха. Остаток растворяли в ацетонитриле и дважды промывали гексаном. Ацетонитрильную фазу концентрировали досуха и очищали экспресс-хроматографией на диоксиде кремния, после чего, элюируя гексаном/этилацетатом, в виде желтой жидкости получали 690 мг (25%) 2-триметилсиланилэтилового эфира [4-(5-метокситиен-2-илметил)фенил]карбаминовой кислоты.
1Н-ЯМР (300 МГц, CDCl3, δ): 7,25 (d, J=8,5 Гц, 2Н), 7,16 (d, J=8,5 Гц, 2Н), 6,49 (bs, 1H), 6,37 (dt, J=3,7, 1,0 Гц, 1H), 5,98 (d, J=3,7 Гц, 1H), 4,25 (m, 2H), 3,94 (bs, 2H), 3,82 (s, 3H), 1,04 (m, 2H), 0,06 (s, 9H).
Стадия 4.
В раствор 684 мг (1,88 ммоля) 2-триметилсиланилэтилового эфира [4-(5-метокситиен-2-илметил)фенил] карбаминовой кислоты в 24 мл диметилсульфоксида при 20-25oС в токе аргона добавляли 5,6 мл 1 М раствора тетра-н-бутиламмонийфторида в тетрагидрофуране. Смесь перемешивали в течение 1 ч. Добавляли диэтилового эфира и раствор промывали водой и рассолом, сушили (над Na2SО4) и концентрировали досуха. Очисткой экспресс-хроматографией на диоксиде кремния, элюируя гексаном/этилацетатом, в виде густого желтого масла получали 372 мг (90%) 4-(5-метокситиен-2-илметил)фениламина.
1Н-ЯМР (300 МГц, CDCl3, δ): 7,02 (d, J=8,5 Гц, 2H), 6,63 (d, J=8,5 Гц, 2H), 6,36 (dt, J=3,7, 1,1 Гц, 1H), 5,97 (d, J=3,7 Гц, 1H), 3,87 (bs, 2H), 3,81 (s, 3H), 3,4-3,7 (широкий s, 1H).
MC m/e (%): 219 (М+; 100).
Заключительная стадия.
В раствор 145 мг (0,бб ммоля) 4-(5-метокситиен-2-илметил)фениламина в 10 мл ацетонитрила при 20-25oС в токе азота вводили 155 мг (0,76 ммоля) сульфата 2-хлор-2-имидазолина. Образовавшуюся суспензию выдерживали при 80oС в течение 1,5 ч. Смесь разбавляли дихлорметаном и промывали 10%-ным раствором гидроксида натрия и водой, сушили (над Na2SO4) и концентрировали досуха. В результате очистки препаративной тонкослойной хроматографией с элюированием этилацетатом/метиловым спиртом/изопропиламином в виде желтого масла получали 132 мг (70%) 2-[4-(5-метокситиен-2-илметил)фенил]аминоимидазолина.
1H-ЯМР (300 МГц, CDCl3, δ): 7,12 (d, J=8,4 Гц, 2Н), 6,93 (d, J=8,4 Гц, 2Н), 6,39 (dt, J=3,7, 1,1 Гц, 1Н), 5,98 (d, J=3,7 Гц, 1Н), 3,92 (bs, 2H), 3,82 (s, 3H), 3,52 (s, 4H), 3,15-3,35 (широкий s, 2H).
Оксалат 2-[4-(5-метокситиен-2-илметил)фенил] аминоимидазолина имеет tпл 121,8-122,8oС.
ПРИМЕР 16.
2-[4-(5-Циклопентилокситиен-2-илметил)фенил]аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Ii, в которой R1 обозначает группу формулы (В), где Х обозначает S, R2 обозначает водород, Y обозначает циклопентил, а m обозначает 0.
В смесь 51,1 мл (0,56 моля) циклопентанола с 50 мл диоксана при 0-5oС в токе аргона вводили 60%-ный гидрид натрия (4,91 г, 0,12 моля) в минеральном масле. Смесь выдерживали при 80oС до образования гомогенного раствора. При 80oС добавляли 10 г (5,9 мл, 0,061 моля) 2-бромтиофена, а затем 11,7 г (0,061 моля) иодида одновалентной меди. Смесь выдерживали при 120oС в течение 6 ч. После охлаждения до 20-25oС добавляли 30 г (0,61 моля) цианида натрия в 200 мл воды. Смесь интенсивно перемешивали в течение 20 мин, фильтровали и экстрагировали гексаном. Гексановый экстракт промывали водой, сушили (над Na2SO4) и концентрировали досуха. Фильтрованием через колонку с диоксидом кремния (с элюированием 100%-ным гексаном) в виде бесцветного масла получали 2,6 г (25,2%) 2-циклопентилокситиофена.
1H-ЯМР (300 МГц, CDCl3, δ): 6,7 (dd, J=5,7, 3,7 Гц, 1Н), 6,54 (dd, J= 5,7, 1,5 Гц, 1Н), 6,18 (dd, J=3,7, 1,5 Гц, 1Н), 4,66 (септет, J=2,7 Гц, 1Н), 1,5-2,0 (m, 8H).
В ходе проведения процесса аналогично стадии 2 из примера 15, но с заменой 2-метокситиофена 2-циклопентилокситиофеном, а затем соответственно аналогично последующим стадиям в примере 14 получали оксалат 2-[4-(5-циклопентилокситиен-2-илметил)фенил]аминоимидазолина с tпл 71,2-75,5oС.
ПРИМЕР 17.
2-[4-(5-Изопропокситиен-2-илметил)фенил]аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Ii, в которой R1 обозначает группу формулы (В), где Х обозначает S, R2 обозначает водород, Y обозначает изопропил, а m обозначает 0.
В раствор 210 мг (0,96 ммоля) 4-(5-метокситиен-2-илметил)фениламина в 20 мл изопропилового спирта в токе азота добавляли 460 мг (2,4 ммоля) гидрата п-толуолсульфокислоты. Смесь кипятили с обратным холодильником в течение 24 ч и охлаждали до 20-25oС. Добавляли 5%-ного раствора гидроксида натрия и продукт экстрагировали дихлорметаном. Экстракт сушили (над Na2SO4) и концентрировали досуха. В результате очистки препаративной тонкослойной хроматографией с элюированием гексаном/этилацетатом в виде бледно-желтого масла получали 135 мг (57%) 4-(5-изопропокситиен-2-илметил)фениламина.
1H-ЯМР (300 МГц, CDCl3, δ): 7,02 (d, J=8,5 Гц, 2Н), 6,63 (d, J=8,5 Гц, 2Н), 6,34 (dt, J=3,7, 1,1 Гц, 1Н), 6,01 (d, J=3,7 Гц, 1Н), 4,26 (квинтет, J= 6,1 Гц, 1Н), 3,88 (bs, 2Н), 1,31 (d, J=6,1 Гц, 6Н).
MС m/e (%): 247 (M+; 58).
Заключительная стадия.
В раствор 131 мг (0,53 ммоля) 4-(5-изопропокситиен-2-илметил)фениламина в 8 мл ацетонитрила при 20-25oС в токе аргона добавляли 121 мг (0,59 ммоля) сульфата 2-хлор-2-имидазолина. Смесь выдерживали при 80oС в течение 1,5 ч. Смесь разбавляли дихлорметаном и промывали гидроксидом натрия и водой, сушили (над Na2SO4) и концентрировали досуха. В результате очистки препаративной тонкослойной хроматографией с элюированием этилацетатом/метиловым спиртом/изопропиламином в виде густого желтого масла получали 150 мг (90%) 2-[4-(5-изопропокситиен-2илметил)фенил]аминоимидазолина.
1H-ЯМР (300 МГц, CDCl3, δ): 7,13 (d, J=8,4 Гц, 2Н), 6,93 (d, J=8,4 Гц, 2Н), 6,37 (dt, J=3,7, 1,0 Гц, 1Н), 6,02 (d, J=3,7 Гц, 1Н), 4,28 (квинтет, J= 6,2 Гц, 1Н), 3,93 (bs, 2Н), 3,52 (s, 4H), 3,24-3,5 (широкий s, 2H), 1,32 (d, J=6,2 Гц, 6Н).
МС m/е (%): 316 (М+; 100%).
Оксалат 2-[4-(5-изопропокситиен-2-илметил)фенил] аминоимидазолина имеет tпл 134,4-135oC.
ПРИМЕР 18.
2-[4-(1-Изопропиламинокарбонилпиперидин-4-илметил)фенил]аминоимидазолин.
Ниже описан процесс получения соединения формулы I из соответствующего соединения формулы Ij, в которой R1 обозначает группу формулы (С), где Х обозначает N, R2, R4 и R8 каждый обозначает водород, R9 обозначает изопропил, V обозначает О, a m обозначает 0.
Стадия 1.
Смесь 12,85 г (60 ммолей) 4-(4-нитробензил)пиридина с 1,0 г оксида платины (IV), 5 мл (60 ммолей) 12 н. соляной кислоты и 5 мл воды в 200 мл этанола гидрировали под давлением 40 фунтов/кв.дюйм в аппарате Парра в течение 12 ч. Реакционную смесь концентрировали в вакууме, остаток разбавляли холодной водой и подщелачивали 10%-ным раствором гидроксида натрия. Образовывавшуюся смесь экстрагировали этилацетатом. Объединенные органические экстракты промывали водой и рассолом, сушили (над Na2SО4) и концентрировали в вакууме. В результате кристаллизации остатка из этилацетата/гексана в виде белого кристаллического твердого вещества получали 9,85 г (86%) 4-(пиперидин-4-илметил)фениламина с tпл 110-113oC.
Стадия 2.
Раствор 0,57 г (3 ммоля) 4-(пиперидин-4-илметил)фениламина в 20 мл дихлорметана в атмосфере азота охлаждали на ледяной бане. По каплям в раствор добавляли 0,28 г (3,3 ммоля) изопропилизоцианата и перемешивали при температуре ледяной бани в течение 30 мин. Реакцию в реакционной смеси прекращали смешением с водой и смесь экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой и рассолом, сушили и в вакууме удаляли растворители. Сырой продукт хроматографировали на силикагеле, элюируя системой 2% метанола/дихлорметан, содержавшей 0,01% гидроксида аммония, с получением в виде смолистого продукта 0,66 г (80%) 4-(1-изопропиламинокарбонилпиперидин-4-илметил)фениламина.
Заключительная стадия.
Смесь 0,64 г (2,31 ммоля) 4-(1-изопропиламинокарбонилпиперидин-4-илметил)фениламина с 0,70 г (3,47 ммолей) сульфата 2-хлор-2-имидазолина в 20 мл 2-пропанола кипятили с обратным холодильником в течение 30 мин. Реакционную смесь концентрировали в вакууме. Остаток разбавляли водой, подщелачивали 10%-ным раствором гидроксида натрия и экстрагировали дихлорметаном. Органический слой промывали водой и рассолом, сушили (над Nа2SO4) и концентрировали в вакууме. Сырой продукт хроматографировали на нейтральном оксиде алюминия, элюируя системой 5% метанола/дихлорметан, и кристаллизовали из этилацетата, содержавшего некоторое количество этанола, с получением в виде белого твердого продукта 0,47 г (59%) 2- [4-(1-изопропиламинокарбонилпиперидин-4-илметил)фенил]аминоимидазолина с tпл 191-192oC.
В ходе проведения процесса аналогично стадии 1 в примере 18 и при проведении процесса непосредственно до заключительной стадии в виде пены получали гидрохлорид 2-[4-(пиперидин-4-илметил)фенил]аминоимидазолина.
В ходе проведения процесса аналогично тому, как это указано в описании стадии 2 в примере 18, но с заменой изопропилизоцианата другими изоцианатами, а затем соответственно аналогично заключительной стадии в примере 15, получали другие соединения формулы I:
гидрохлорид 2-[4-(1-фениламинокарбонилпиперидин-4-илметил)фенил]аминоимидазолина, который при 99oС сморщивался (высокогигроскопичный), С22Н28N5ОCl, и
гидрохлорид 2-[4-(1-этиламинокарбонилпиперидин-4-илметил)фенил] аминоимидазолина, который при 97oС сморщивался (очень гигроскопичный), C18N28N5OCl.
ПРИМЕР 19.
2-[4-(1-Бензолсульфонилпиперидин-4-илметил)фенил]аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Ik, в которой R1 обозначает группу формулы (С), где Х обозначает N, R2 и R4 каждый обозначает водород, R9 обозначает бензол.
Стадия 1.
Раствор 7,5 г (39,44 ммолей) 4-(пиперидин-4-илметил)фениламина в 200 мл сухого тетрагидрофурана в атмосфере азота охлаждали на ледяной бане. В этот раствор отдельными порциями добавляли 9,76 г ди-трет-бутилдикарбоната и перемешивали в течение 30 мин. Реакцию в образовавшейся смеси прекращали смешением с водой, смесь концентрировали в вакууме и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили и в вакууме удаляли растворители. Сырой продукт хроматографировали в короткой колонке с силикагелем, элюируя системой 30% этилацетата/гексан, получали в виде масла, которое затвердевало, 9,25 г (81%) 4-[1-(N-трет-бутоксикарбонил)пиперидин-4-илметил]фениламина с tпл 91-92oС.
Стадия 2.
Раствор 3,55 г (12,24 ммоля) 4-[1-(N-трет-бутоксикарбонил)пиперидин-4-илметил] фениламина и 10,2 мл (73,4 ммоля) триэтиламина в 70 мл дихлорметана охлаждали в атмосфере азота на ледяной бане. По каплям в раствор вводили 5,2 мл (36,7 ммолей) трифторуксусного ангидрида. Образовавшуюся смесь перемешивали в течение 30 мин, реакцию прекращали смешением с 100 мл фосфатного буфера (рН 7,0) и 150 мл метанола и перемешивали при комнатной температуре в течение 15 мин. Реакционную смесь концентрировали в вакууме и остаток экстрагировали этилацетатом. Органический слой промывали холодной водой и рассолом, сушили и в вакууме удаляли растворители. Сырой продукт хроматографировали на силикагеле, элюируя системой 30% этилацетата/гексан, получали в виде твердого продукта 4,43 г (94%) 2,2,2-трифтор-N-{4-[1-(N-трет-бутоксикарбонил)пиперидин-4-илметил] фенил}ацетамида с tпл 145-146oС; данные элементного анализа для С19Н25N2О3F3, рассчитано: С 59,06, Н 6,52, N 7,25; обнаружено: С 59,40, Н 6,54, N 7,42.
Стадия 3.
Смесь 3,3 г 2,2,2-трифтор-N-{4-[1-(N-трет-бутоксикарбонил)пиперидин-4-илметил] фенил}ацетамида с 5 мл трифторуксусной кислоты в 30 мл дихлорметана перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли дихлорметаном и холодной водой и нейтрализовали раствором бикарбоната натрия. Органический слой отделяли, промывали водой и рассолом, сушили и в вакууме удаляли растворители с получением в виде пены 1,5 г 2,2,2-трифтор-N-[4-(пиперидин-4-илметил)фенил]ацетамида.
Стадия 4.
0,5 г (1,75 ммолей) 2,2,2-трифтор-N-[4-(пиперидин-4-илметил)фенил]ацетамида и 0,23 г (2,1 ммоля) триэтиламина в 10 мл дихлорметана в атмосфере азота охлаждали на ледяной бане. В эту смесь при ее перемешивании добавляли раствор 0,37 г (2,1 ммоля) бензолсульфонилхлорида в 1 мл дихлорметана. Через 2 ч реакцию в реакционной смеси прекращали смешением с водой. Выделенный органический слой промывали холодной водой и рассолом, сушили и в вакууме удаляли растворители. Остаток кристаллизовали из этилацетата/гексана с получением 0,43 г (57%) 2,2,2-трифтор-N-[4-(1-бензолсульфонилпиперидин-4-илметил)фенил] ацетамида с tпл 194-195oС; данные элементного анализа для C20H21N2O3SF3, рассчитано: С 56,32, Н 4,96, N 6,57; обнаружено: С 56,54, Н 4,99, N 6,68.
Стадия 5.
Смесь 0,45 г 2,2,2-трифтор-N-[4-(1-бензолсульфонилпиперидин-4-илметил)фенил] ацетамида с 0,23 г гидроксида лития в 10 мл метанола и 1 мл воды перемешивали в течение приблизительно 48 ч. Реакционную смесь концентрировали в вакууме, разбавляли холодной водой и экстрагировали дихлорметаном. Органический слой промывали холодной водой и рассолом, сушили и в вакууме удаляли растворители. Остаток кристаллизовали из этилацетата/гексана с получением 0,29 г (83%) 4-(1-бензолсульфонилпиперидин-4-илметил)фениламина с tпл 158oС; данные элементного анализа для C18H22N2O2S, рассчитано: С 65,43, Н 6,71, N 8,48; обнаружено: С 65,59, Н 6,61, N 8,66.
Заключительная стадия.
Смесь 0,28 г (0,83 ммоля) 4-(1-бензолсульфонилпиперидин-4-илметил)фениламина с 0,25 г (1,25 ммолей) сульфата 2-хлор-2-имидазолина в 20 мл 2-пропанола кипятили с обратным холодильником в атмосфере азота в течение 30 мин. Реакционную смесь концентрировали в вакууме. Остаток подщелачивали 10%-ным раствором гидроксида натрия и экстрагировали дихлорметаном. Органический слой промывали холодной водой и рассолом, сушили и в вакууме удаляли растворители. Остаток хроматографировали на нейтральном оксиде алюминия, элюируя системой 1% метанола/дихлорметан, с получением в виде пены 0,29 г (89%) 2-[4-(1-бензолсульфонилпиперидин-4-илметил)фенил] аминоимидазолина. Данные элементного анализа для C21H26N4O2S•5H2O, рассчитано: С 61,89, Н 6,68, N 13,75; обнаружено: С 62,00, Н 6,52, N 13,85.
В ходе проведения процесса аналогично тому, как это указано в описании стадии 4 в примере 19, но с заменой бензолсульфонилхлорида другими сульфонилхлоридами или карбонилхлоридами, а затем соответственно аналогично последующим стадиям в примере 19, получали другие соединения формулы I:
гидрохлорид 2-[4-(1-метансульфонилпиперидин-4-илметил)фенил]аминоимидазолина; данные элементного анализа для C16H25N4O2ClS;
2-[4-(1-изопропилсульфонилпиперидин-4-илметил)фенил] аминоимидазолин с tпл 193-194oС; данные элементного анализа для C18H28N4O2S•4H2O, рассчитано: С 58,16, Н 7,81, N 15,07; обнаружено: С 58,26, Н 7,52, N 14,96;
2-[4-(1-изопропилкарбонилпиперидин-4-илметил)фенил]аминоимидазолин; данные элементного анализа для C19H28N4O, рассчитано: С 69,48, Н 8,59, N 17,06; обнаружено: С 69,41, Н 8,59, N 16,95;
2-[4-(1-изобутилкарбонилпиперидин-4-илметил)фенил] аминоимидазолин с tпл 122-125oС; МС m/z 343 (М+1), и
гидрохлорид 2-{4-[1-(3-метилбутилкарбонил)пиперидин-4-илметил]фенил}аминоимидазолина с tпл 155-157oС; данные элементного анализа для С21Н33N4OСl, рассчитано: С 64,19, Н 8,46, N 14,26; обнаружено: С 64,05, Н 8,39, N 14,27.
ПРИМЕР 20.
2-{4-[1-(1-Пиперидинсульфонил)пиперидин-4-илметил]фенил}аминоимидазолин.
Ниже описан другой вариант процесса получения соединения формулы I из соответствующего соединения формулы Il, в которой R1 обозначает группу формулы (С), где Х обозначает N, R2 и R4 каждый обозначает водород, R8 и R9 вместе с атомом азота, с которым они связаны, образуют пиперидин.
Стадия 4.
Раствор 0,5 г (1,75 ммолей) 2,2,2-трифтор-N-[4-(пиперидин-4-илметил)фенил]ацетамида (полученного так, как указано в вышеприведенном описании стадий 1-3 в примере 19) и триэтиламина в 10 мл дихлорметана в атмосфере азота охлаждали на ледяной бане. Затем смесь обрабатывали раствором 0,39 г (2,09 ммолей) 1-пиперидинсульфонилхлорида в 1 мл дихлорметана. Реакционную смесь перемешивали в течение 1,5 ч при 0-5oС и реакцию прекращали смешением с водой. Органический слой отделяли и водный слой экстрагировали дихлорметаном. Объединенные органические экстракты промывали водой и рассолом, сушили и в вакууме удаляли растворители. Остаток хроматографировали на силикагеле, элюируя системой 30% этилацетата/гексан, с получением в виде белого твердого продукта 0,48 г 2,2,2-трифтор-N-[4-(1-пиперидинсульфонилпиперидин-4-илметил)фенил] ацетамида с tпл 156-157oС; данные элементного анализа для С19Н26 N3О3SF3, рассчитано: С 52,64, Н 6,05, N 9,69; обнаружено: С 52,84, Н 6,00, N 9,79.
Стадия 5.
Смесь 0,48 г (1,11 ммолей) 2,2,2-трифтор-N-[4-(1-пиперидинсульфонилпиперидин-4-илметил)фенил]ацетамида с 0,23 г (5,54 ммолей) гидроксида лития в 10 мл метанола и 1 мл воды выдерживали при 60oС в течение примерно 2 ч. Реакционную смесь концентрировали в вакууме, разбавляли водой и экстрагировали дихлорметаном. Органические экстракты промывали водой и рассолом и сушили (над Na2SО4). Остаток кристаллизовали из этилацетата/гексана с получением в виде белого твердого вещества 0,30 г 4-(1-пиперидинсульфонилпиперидин-4-илметил)фениламина с tпл 144-145oC; данные элементного анализа для C17H27N3О2S, рассчитано: С 60,50, Н 8,06, N 12,45; обнаружено: С 60,76, Н 8,07, N 12,56.
Заключительная стадия
В ходе проведения процесса аналогично тому, как это указано в вышеприведенном описании заключительной стадии в примере 19, в виде пены получали 0,29 г (89%) 2-[4-(1-пиперидинсульфонилпиперидин-4-илметил)фенил]аминоимидазолина. Данные элементного анализа для C20H31N5O2S, рассчитано: С 59,23, Н 7,70, N 17,27; обнаружено: С 59,13, Н 7,56, N 17,13.
В ходе проведения процесса аналогично стадии 4 в примере 20, но с заменой 1-пиперидинсульфонилхлорида 1-пирролидинсульфонилхлоридом и соответственно аналогично последующим стадиям в примере 20 в виде пены получали 2-[4-(1-пирролидинсульфонилпиперидин-4-илметил)фенил] аминоимидазолин; данные элементного анализа для С19H29N5O2S, рассчитано: С 57,23, Н 7,53, N 17,56; обнаружено: С 57,27, Н 7,24, N 17,40.
ПРИМЕР 21.
Рентгенограммы кристаллической формы I.
Кристаллическую форму I сульфата 2-[4-(4-изопропоксибензил)фенил]аминоимидазолина готовили методами, описанными выше в примере 1. Рентгенограмму, представленную на фиг. 1, получали с помощью дифрактометра для порошковой рентгенографии Scintag X1, снабженного медным источником излучения Kα1. Числа, приведенные на фиг.2 в верхней и нижней частях оси абсцисс, указывают соответственно постоянную "d" и 2θ,, а с правой и левой сторон оси ординат указывают соответственно относительную интенсивность в % и количество импульсов в секунду (имп./с).
Приведенные ниже данные порошковых рентгенограмм представлены в единицах постоянной d и относительной интенсивности (ОИ) свыше 3%. Средневзвешенное значение длины волны рентгеновского излучения, которое применяли для расчетов, составляло 1,5406710-10 см (см. табл. 1).
ПРИМЕР 22.
Получение кристаллической формы II.
194 мг сульфата 2-[4-(4-изопропоксибензил)фенил]аминоимидазолина растворяли при 60oС в 1 мл воды, прозрачный верхний слой переносили в трубку Крейга и охлаждали на бане из воды со льдом. Центрифугированием собирали кристаллы и сушили в вакууме при комнатной температуре с получением 138 мг кристаллической формы II сульфата 2-[4-(4-изопропоксибензил)фенил]аминоимидазолина с tпл 217-218oС.
В другом варианте 38 г сульфата 2-[4-(4-изопропоксибензил)фенил]аминоимидазолина растворяли при 80oС в 500 мл воды. После горячего фильтрования раствор охлаждали до комнатной температуры и выдерживали при 4oС в течение 5 ч. Фильтрованием собирали кристаллы и сушили при комнатной температуре с получением 33,6 г кристаллической формы II сульфата 2-[4-(4-изопропоксибензил)фенил]аминоимидазолина с tпл 216-217oC.
ПРИМЕР 23.
Рентгенограммы кристаллической формы II.
Рентгенограмму кристаллической формы II, представленную на фиг.2, получали с помощью дифрактометра для порошковой рентгенографии Scintag X1, снабженного медным источником излучения Kα1. Числа, приведенные на фиг.1 в верхней и нижней частях оси абсцисс, указывают соответственно постоянную d и 2θ, а с правой и левой сторон оси ординат указывают соответственно относительную интенсивность (в %) и количество импульсов в секунду (имп./с).
Приведенные ниже данные порошковых рентгенограмм представлены в единицах постоянной d и относительной интенсивности (ОИ) свыше 3%. Средневзвешенное значение длины волны рентгеновского излучения, которое применяли для расчетов, составляло 1,5406•10-10 см (см. табл.2).
ПРИМЕР 24.
Композиция для перорального введения, мас.%:
Действующее вещество - 20,0
Лактоза - 79,5
Стеарат магния - 0,5
Оба компонента смешивают и распределяют по капсулам, каждая из которых содержит по 100 мг продукта; одна капсула в целом приблизительно соответствует одной суточной дозе.
ПРИМЕР 25.
Композиция для перорального введения, мас.%:
Действующее вещество - 20,0
Стеарат магния - 0,5
Натрийкросскармеллоза - 2,0
Лактоза - 76,5
ПВП (поливинилпирролидон) - 1,0
Вышеперечисленные компоненты смешивают и гранулируют с использованием метанола в качестве растворителя. Далее композицию сушат и с помощью соответствующей таблетирующей машины формуют с получением таблеток, каждая из которых содержит по 20 мг действующего вещества.
ПРИМЕР 26.
Композиция для парентерального введения (IV):
Действующее вещество, г - 0,25
Хлорид натрия - В количестве, необходимом для изотонического раствора
Вода для инъекций, мл - До 100
Действующее вещество растворяют в части воды для инъекции. Далее с перемешиванием добавляют хлорид натрия в количестве, необходимом для получения изотонического раствора. Оставшейся водой для инъекции массу раствора доводят до необходимой, фильтруют через 0,2-микрометрический мембранный фильтр и расфасовывают в стерильных условиях.
ПРИМЕР 27.
Композиция для суппозиториев, мас.%:
Действующее вещество - 1,0
Полиэтиленгликоль 1000 - 74,5
Полиэтиленгликоль 4000 - 24,5
Компоненты сплавляют между собой, смешивают на паровой бане и разливают в формы, содержащие в общем по 2,5 г продукта.
ПРИМЕР 28.
Композиция для местного применения, г:
Действующее вещество - 0,2-2
Спан 60 - 2
Твин 60 - 2
Минеральное масло - 5
Вазелин - 10
Метилпарабен - 0,15
Пропилпарабен - 0,05
Бутилированный гидроксианизол (БГА) - 0,01
Вода - До 100
Все вышеперечисленные компоненты, кроме воды, смешивают и с перемешиванием нагревают до 60oС. Далее при интенсивном перемешивании добавляют воды, нагретой до 60oС, в достаточном количестве для эмульгирования компонентов и затем добавляют воду в количестве, не достающем до общей массы 100 г.
ПРИМЕР 29.
Композиции для интраназального спрея.
В качестве композиций для интраназального спрея готовят несколько водных суспензий, содержащих 0,025-0,5% действующего вещества. Эти композиции необязательно включают инертные компоненты, такие, как микрокристаллическая целлюлоза, натрийкарбоксиметилцеллюлоза, декстроза и т.п. Для регулирования значений рН можно добавить соляной кислоты. Композиции интраназального спрея можно дозировать с помощью дозирующего насоса для интраназального спрея, как правило, подающего за каждый цикл по 50-100 мкл препарата. В типичном режиме дозирования предусмотрено по 2-4 цикла пульверизаций каждые 4-12 ч.
ПРИМЕР 30.
Оценка механической гиперальгезии, вызванной каррагинаном.
Противовоспалительное/болеутоляющее действие соединений по настоящему изобретению определяли оценкой по вызванной каррагинаном механической гиперальгезии, устанавливая степень ингибирования вызванной каррагинаном у крыс гиперальгезии лап с использованием модификации метода, описанного L.O. Randall и J.J. Selitto в Archives of International Pharmacodynamics, 1957, 11, 409-419, и Vinegar и др. в Journal of Pharmacology and Experimental Therapeutics, 1969, 166, 96-103.
Самцов крыс Sprague-Dawley (130-150 г) взвешивали и бессистемно распределяли по группам (численностью 10) подопытных животных. С целью вызвать механическую гиперальгезию животных подвергали легкому наркозу галотаном и через кожу подошвы левой задней лапы вводили 1%-ный каррагинан или наполнитель (по 100 мкл). За час до начала испытаний крысам вводили наполнитель (по 10 мл/кг перорально или по 1 мл/кг внутривенно) или соединения по настоящему изобретению (в дозе 1, 3, 10, 30 и 100 мг/кг перорально или 0,3, 1,0, 3,0 и 10 мг/кг внутривенно). Механическую гиперальгезию определяли с помощью анальгезиметра (UGO BASILE фирмы Biological Research Apparatus, Комерио, Италия). Обработанную наполнителем или каррагинаном заднюю лапу помещали на арку прибора таким образом, чтобы поверхность подошвы была обращена вниз. Далее на дорсальную поверхность лапы воздействовали постоянно возрастающим усилием. За конечную точку принимали усилие, при котором животное отдергивало лапу, начинало борьбу или издавало звуки.
Подопытные группы сравнивали по результатам одностороннего анализа, основанного на различии усилия, вызывавшего отдергивание лапы (PEAK). Попарные сравнения животных обработанных лекарственными средствами групп с животными группы, которых обрабатывали наполнителем, используя методологию НЗР (наименьшей значимой разности) Фишера и метод Данна. Для каждого животного рассчитывали степень ингибирования механической гиперальгезии в процентах, а среднее значение ИД50 вычисляли с помощью следующей сигмоидальной модели:
% ингибирования = 100/(1 + эксп.[(ИД50 - доза)/К)],
где ИД50 обозначает дозу соединения, необходимую для ингибирования наполовину максимальной реакции (т.е. 100% в этой модели), а К обозначает параметр кривизны.
В этом испытании соединения по настоящему изобретению проявляли активность.
ПРИМЕР 31.
Оценка по симптому Фройнда механической гиперальгезии, вызванной адъювантом.
Противовоспалительное/болеутоляющее действие соединений по настоящему изобретению можно также определить с помощью модели вызванных адъювантом у крыс болей при артрите, когда болевые ощущения оценивают по реакции животных на сдавливание воспаленной ступни в соответствии с вариантом методики, описанной J. Hylden и др. в Pain, 1989, 37, 229-243. Этот вариант включает оценку гиперальгезии, а не изменений активности нейронов спинного мозга.
В целом животных взвешивали и бессистемно распределяли по группам подопытных животных. С целью вызвать механическую гиперальгезию животных подвергали легкому наркозу галотаном и через кожу подошвы левой задней лапы вводили 100 мкл адъюванта для симптома Фройнда или солевого раствора. Спустя двадцать четыре часа крысам за один час до начала испытаний перорально вводили воду (наполнитель) или соединения по настоящему изобретению. Механическую гиперальгезию определяли с помощью анальгезиметра (UGO BASILE фирмы Biological Research Apparatus, Комерио, Италия). Обработанную солевым раствором или каррагинаном заднюю лапу помещали на арку прибора таким образом, чтобы поверхность подошвы была обращена вниз. Далее на дорсальную поверхность лапы воздействовали постоянно возрастающим усилием и за конечную точку принимали усилие, при котором животное отдергивало лапу, начинало борьбу или издавало звуки. Подопытные группы сравнивали по результатам одностороннего анализа, основанного на различии усилия, вызывавшего отдергивание лапы. Для каждого животного степень ингибирования в процентах рассчитывали в форме
100 x [(к/л - к/н)÷(с/н - к/н)],
где к/л обозначает усилие, при котором животное, которому вводили лекарственное средство, отдергивало обработанную каррагинаном лапу; к/н обозначает усилие, при котором животное, которому вводили наполнитель, отдергивало обработанную каррагинаном лапу; а с/н обозначает усилие, при котором животное, которому вводили наполнитель, отдергивало обработанную солевым раствором лапу. Значимость определяли с помощью критерия Стьюдента.
В этом испытании соединения по настоящему изобретению проявляли активность.
ПРИМЕР 32.
Ингибирование у крыс сокращений мочевого пузыря, вызванных изоволюметрическим растяжением мочевого пузыря.
Степень ингибирования сокращений мочевого пузыря определяли проведением испытания в соответствии с вариантом метода, описанного С.А. Maggi и др. в J. Pharm. and Exper. Therapeutics, 1984, 230, 500-513.
В целом самцов крыс Sprague-Dawley (200-250 г) взвешивали и бессистемно распределяли по группам подопытных животных. С целью вызвать сокращения мочевого пузыря через мочеиспускательный канал в мочевой пузырь вводили катетер и осуществляли вливание 5 мл теплого солевого раствора. У примерно 30% животных это вызывало ритмические сокращения. В начале регулярных ритмических сокращений внутривенно вводили соединения по изобретению (0,1, 0,3 или 1 мг/кг). Затем определяли влияние на ритмические сокращения.
В этом испытании соединения по настоящему изобретению проявляли активность.
ПРИМЕР 33.
Ингибирование обусловленных объемом сокращений у крыс.
Степень ингибирования сокращений мочевого пузыря определяли испытанием в соответствии с вариантом метода, описанного S.S. Hegde и др. в Proceedings of the 26th Annual Meeting of the International Continence Society, 27-30 августа 1996, реферат 126.
Самцов крыс Sprague-Dawley подвергали наркозу уретаном и к ним подключали приборы для внутривенного введения лекарственных средств, а в некоторых случаях для измерения артериального давления, частоты сердечных сокращений и давления внутри мочевого пузыря. На отдельной группе животных определяли влияние испытываемых соединений на обусловленные объемом сокращения мочевого пузыря. Обусловленные объемом рефлекторные сокращения мочевого пузыря вызывали наполнением мочевого пузыря солевым раствором. Испытываемые соединения вводили внутривенно в кумулятивном режиме через 10-минутные интервалы. По завершении исследований для позитивного контрольного эффекта внутривенно вводили 0,3 мг/кг атропина.
В этом испытании соединения по настоящему изобретению проявляли активность.
ПРИМЕР 34.
Устранение у крыс гипотензии, вызванной действием эндотоксинов.
Септический шок, иногда называемый эндотоксиновым бактериально-токсическим шоком, индуцируется присутствием в токах крови инфекционных агентов, в частности бактериальных эндотоксинов, и характеризуется гипотензией и дисфункцией органов. Многие симптомы септического шока, в частности гипотензию, вызывают у крыс введением бактериальных эндотоксинов. Таким образом, способность соединения ингибировать вызванную эндотоксинами гипотензию является показателем полезности соединения при лечении септического или эндотоксинового бактериально-токсического шока.
Действие соединений по изобретению при лечении септического или эндотоксинового бактериально-токсического шока оценивали определением степени устранения вызванной у крыс эндотоксинами гипотензии в соответствии с вариантом метода, описанного М. Giral и др. в British Journal of Pharmacology, 1969, 118,1223-1231.
В целом взрослых особей крыс (> 200 г) подвергали наркозу путем ингаляции анестезирующего средства и для ввода датчиков кровяного давления и размещения линий введения лекарственных средств канюлировали соответственно бедренные артерии и вены. Пока животные находились под влиянием анестезирующего средства, их помещали в приспособление Мэйо для фиксирования конечностей. После завершения действия наркоза и стабилизации частоты сердечных сокращений и кровяного давления (на что обычно требовалось примерно 30 мин) внутривенно вводили эндотоксин (50 мг/кг E.coli и 25 мг/кг Salmonella). Следили за изменениями кровяного давления и частоты сердечных сокращений. По прошествии одного часа также внутривенно вводили соединения по настоящему изобретению или наполнитель и в течении последующих трех часов постоянно следили за сердечно-сосудистыми показателями. Реакцию оценивали в процентах возврата к первоначальному диастолическому кровяному давлению. Значение определяли с помощью критерия Стьюдента.
В этом испытании соединения по настоящему изобретению проявляли активность.
Хотя настоящее изобретение описано со ссылкой на конкретные варианты его выполнения, специалистам в данной области техники следует иметь в виду, что в них можно вносить самые разнообразные изменения и их заменять эквивалентами, не меняя при этом истинной сущности и не выходя за рамки изобретения. Кроме того, для достижения соответствия конкретных ситуации, материала, рассматриваемой композиции, способа и стадии или стадий способа целевой сущности и рамкам настоящего изобретения можно прибегнуть ко многим модификациям. Все эти модификации необходимо рассматривать как подпадающие под объем прилагаемой формулы изобретения.
2. Данные по активности.
Сродство к IP-рецептору выражено как рКi, значение которого находится в пределах от 8,00-9,06 для соединений по настоящему изобретению.
Значения рКi (рКi=-1оgКi) определялись по следующей формуле:
при том, что значения IС50 являются такими концентрациями испытуемых соединений в nМ, при которых замещается 50% лигандов, связывающихся с рецепторами. [L] обозначает концентрацию лиганда, а значение КD является константой диссоциации лиганда. Данные таблицы показывают, что соединения настоящего изобретения обладают желаемой активностью.
Коренной проблемой, лежащей в основе настоящего изобретения, является создание новых соединений, которые являлись бы антагонистами IP-рецепторов и которые, следовательно, могли бы быть использованы для лечения заболеваний, таких как болевой или септический шок, воспалительные заболевания, недержание мочи, астма.
Из приведенной табл.4 можно видеть, что соединения, у которых фенильное кольцо присоединено к аминогруппе в 4 положении, оказались совершенно неожиданно более активными в примерно от 100 до 1000 раз, чем соединения, у которых фенильное кольцо замещено в положении 2.
Более того, соединения со связующим мостиком, описанным в ЕР 017484 (предпочтительно А= -О- или -S-), являются менее активными в части IP-рецептора по сравнению с соединениями, описанными в настоящем изобретении. Степень сродства измерена в показателях значений рКi, которая является логарифмической величиной, соответственно единичная величина увеличивается (или возрастает) на 10 в части сродства. В общем, более высокое значение рКi означает более высокое сродство связывания между потенциальным лекарством и его мишенью.
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕНЗОЛСУЛЬФОНОВЫЕ ПРОИЗВОДНЫЕ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1999 |
|
RU2201922C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1997 |
|
RU2184112C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНДИОНА, ТРИАЗИНДИОНА, ТЕТРАГИДРОХИНАЗОЛИНДИОНА В КАЧЕСТВЕ АНТАГОНИСТОВ α-АДРЕНЕРГИЧЕСКИХ РЕЦЕПТОРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2175322C2 |
| ПРОИЗВОДНЫЕ КАРБАМИНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1999 |
|
RU2179969C2 |
| ФЕНИЛГЛИЦИНОВЫЕ ПРОИЗВОДНЫЕ | 1999 |
|
RU2198871C1 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 2-ФЕНИЛАМИНОИМИДАЗОЛИНФЕНИЛКЕТОНА В КАЧЕСТВЕ АНТАГОНИСТОВ IP | 2001 |
|
RU2284995C2 |
| ПРОИЗВОДНЫЕ ТИАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1999 |
|
RU2166503C2 |
| ПРОИЗВОДНЫЕ 5H-ТИАЗОЛ[3,2-А]ПИРИМИДИНА, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1998 |
|
RU2197493C2 |
| ПРОИЗВОДНЫЕ ОКСАЗОЛИДИН-2-ОНА И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1994 |
|
RU2133743C1 |
| ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРАЗОЛА | 1995 |
|
RU2152388C1 |







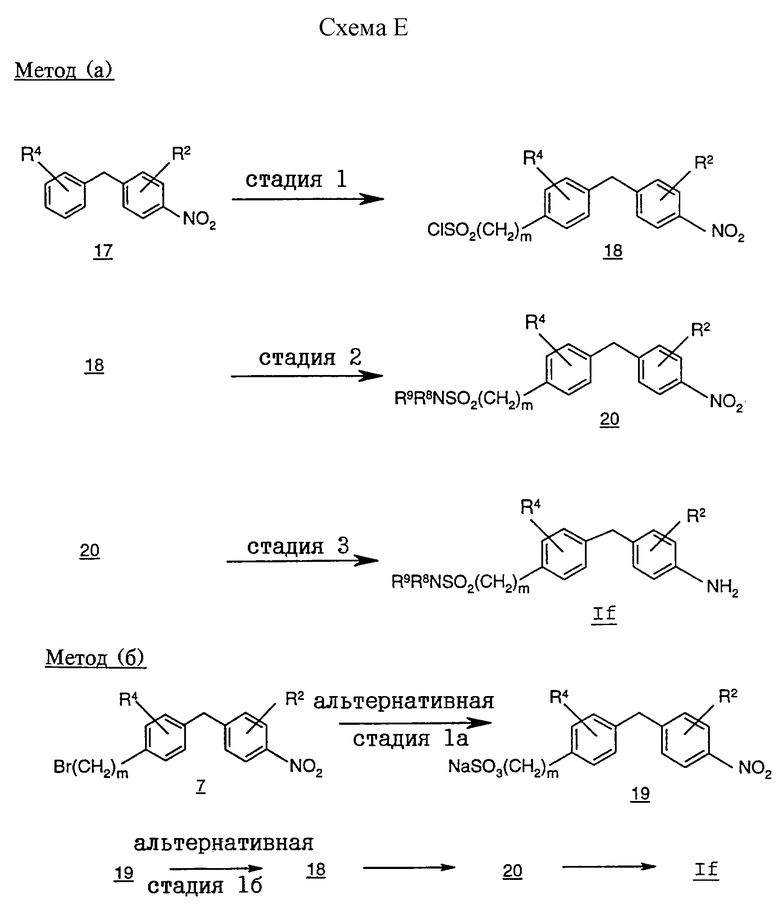
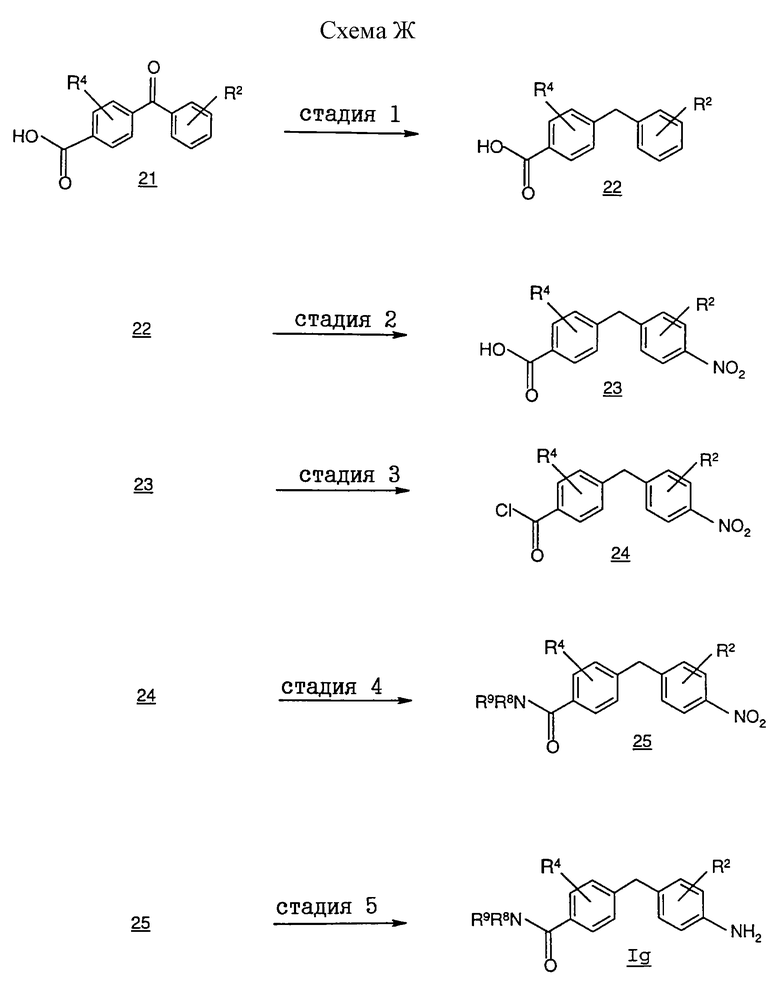
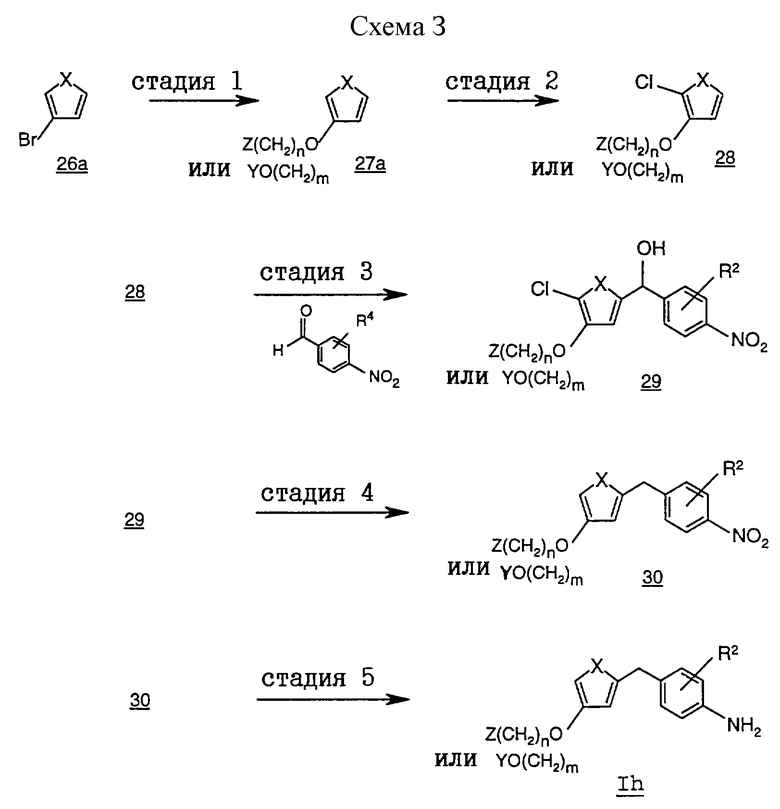

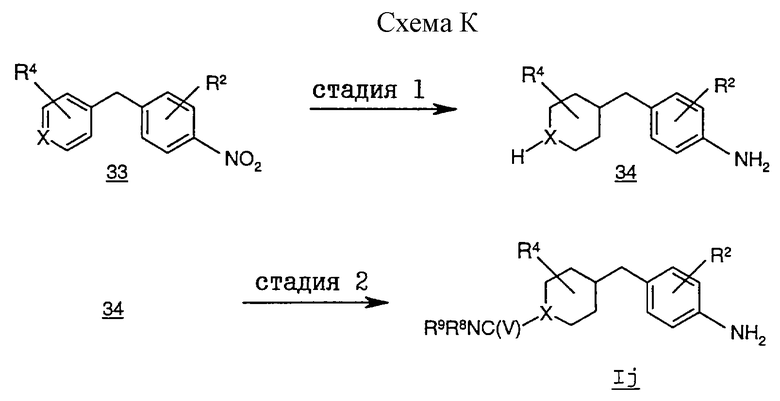





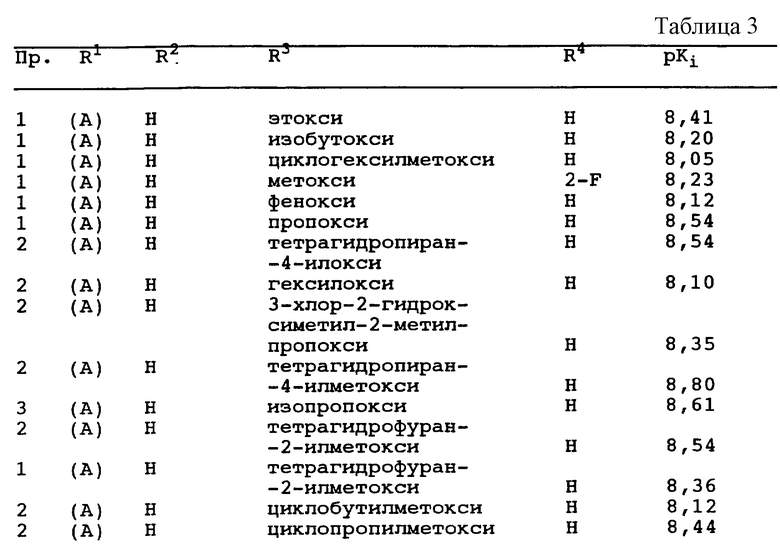

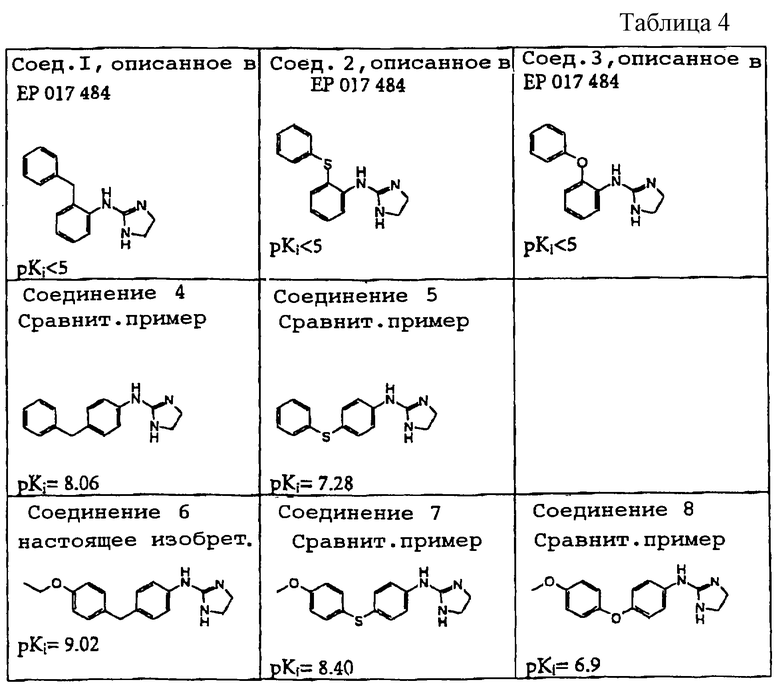

Изобретение относится к производным 2-(арилфенил)аминоимидазолина формулы I, где R1 обозначает группу формулы (А), (В) или (С), a R2, R3, R4, R5, R6 и Х такие, как определено в формуле изобретения. Также описана фармацевтическая композиция на основе этих соединений. Технический результат: получены новые соединения, обладающие ценными биологическими свойствами. Изобретение может быть использовано в области медицины в качестве лекарственного средства. 2 с. и 18 з.п.ф-лы, 2 ил., 4 табл.





где R1 обозначает группу формулы (А), (В) или (С)

где Х независимо в каждом случае обозначает S или N;
R2 и R4 каждый независимо друг от друга в каждом случае обозначает (1) водород, (2) алкоксигруппу или (3) галоген;
R3 независимо в каждом случае обозначает (1) алкил, (2) циклоалкил, (3) галоген, (4) морфолинил, 1,1-диоксоизотиазолидин-1-ил, (5) -NR8R9, (6) -(CH2)mCONR8R9, где m= 0, (7) -(CH2)mSO2NR8R9, где m= 0-3, целое число, (8) -(CH2)mNR7OR9, где m= 0, (9) -(CH2)mNR7SO2R9, где m= 0-3, целое число, (10) -(CH2)mNR7C(V)NR8R9, где V обозначает O, a m= 0-3, целое число, (11) -(СН2)mОY, где m= 0-3, целое число, Y - водород, алкил, алкилоксиалкил, циклоалкил, галоидалкил, гидроксиалкил, тетрагидропиранил, тетрагидрофурил, 5-метил-[1,3] -диоксан-5-ил или карбоксиалкил, или (12) -O(CH2)nZ, где n= 1-4, целое число, a Z - циклоалкил, гидроксиалкил, циклоалкилоксигруппу, тетрагидропиранил, тетрагидрофурил, феноксигруппа, тиенил, -COR9, -CONR8R9, -SO2R9, -NR7SO2R9, незамещенный фенил;
R5 независимо в каждом случае обозначает -(СН2)mОY, где m= 0, а Y - водород, алкил, циклоалкил;
R6 независимо в каждом случае обозначает: (1) водород, (2) -OR9, (3) -CONR8R9, (4) -С(V)NR8R9, где V обозначает О, 2 (5) -SO2R9 или (6) -SO2NR8R9, R7 и R8 каждый независимо друг от друга в каждом случае обозначает (1) водород, (2) алкил или (3) гидроксиалкил;
R9 независимо в каждом случае обозначает (1) алкил, (2) циклоалкил, (3) бензил, (4) гидроксиалкил, (5) галоидалкил, (6) пиперидинил, тетрагидропиранил, пирролидинил, (7) незамещенный или монозамещенный фенил, причем заместители независимо друг от друга выбраны из алкила, галогенов и алкилоксигруппы, или (8) тиенил; или
R8 и R9 вместе с атомом азота, с которым они связаны, образуют 5- или 6-членное моноциклическое насыщенное кольцо,
а также его фармацевтически приемлемая соль или его кристаллическая форма.
2-[4-(4-изопропоксибензил)фенил] аминоимидазолин;
2-[4-[4-(втор-бутокси)бензил] фенил} аминоимидазолин;
2-{ 4-[4-(циклопентилокси)бензил] фенил} аминоимидазолин;
2-{ 4-[4-(тетрагидропиран-4-илокси)бензил] фенил] аминоимидазолин;
2-{ 4-[4-(тетрагидропиран-4-илметокси)бензил] фенил} аминоимидазолин;
2-{ 4-[2-фтор-4-(тетрагидропиран-4-илметокси)бензил] фенил)-аминоимидазолин;
2-{ 4-[4-(2-этокси-1-(этоксиметил)этокси)бензил] фенил} аминоимидазолин;
2-[4-(4-циклопентилокситиенил-2-илметил)фенил] аминоимидазолин;
2-{ 4-[4-(1-гидроксиметилэтокси)бензил] фенил} аминоимидазолин;
2-[4-(5-метокситиен-2-илметил)фенил] аминоимидазолин;
2-[4-(4-бутиламиносульфонилбензил)фенил] аминоимидазолин;
2-[4-(4-изопропоксиметилбензил)фенил] аминоимидазолин;
2-[4-(4-втор-бутоксиметилбензил)фенил] аминоимидазолин;
2-{ 4-[4-(изобутиламиносульфонил)бензил] фенил] аминоимидазолин;
2-[4-(4-бензиламинокарбонилбензил) фенил] аминоимидазолин;
2-[4-(4-изопропиламиносульфонилбензил)фенил] аминоимидазолин;
2-[4-(4-изобутиламинокарбонилбензил)фенил] аминоимидазолин и
2-[4-(4-трет-бутиламиносульфонилбензил)фенил] аминоимидазолин.
Приоритет по пунктам:
19.06.1998 по пп. 1,4-12 и 14-19;
04.09.1977 по пп. 2,3,13 и 20.
| ЭЛЕКТРИЧЕСКАЯ МАШИНА С ПЕРЕМЕННО-ВОЗВРАТНЫМ ДВИЖЕНИЕМ СЕРДЕЧНИКА | 1928 |
|
SU17484A1 |
| US 4889868 А, 26.12.1989 | |||
| US 4396617 А, 02.08.1983 | |||
| WO 9842679 A1, 01.10.1998 | |||
| СПОСОБ ПОЛУЧЕНИЯ ИМИДАЗОЛОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ СОЛИ | 1989 |
|
RU2028293C1 |

