Данная заявка является частичным продолжением совместно поданной заявки США №09/074251, поданной 7 мая 1998 г. под заглавием "Способ и аппаратура для получения очищенной терефталевой кислоты", которая является выделенной заявкой совместно поданной заявки США №08/477898, поданной 7 июня 1995 г. под заглавием "Способ и аппаратура для получения очищенной терефталевой кислоты", ныне патента США №5767311, а также частичным продолжением заявки США №08/962030, поданной 31 октября 1997 г. под заглавием "Способ и аппаратура для получения очищенной терефталевой кислоты", которая является частичным продолжением совместно поданной заявки США №08/760890, поданной 6 декабря 1996 г. под заглавием "Способ и аппаратура для получения очищенной терефталевой кислоты", которая, в свою очередь, является частичным продолжением совместно поданной заявки США №08/477898, поданной 7 июня 1995 г. под заглавием "Способ и аппаратура для получения очищенной терефталевой кислоты", ныне патента США №5767311, все четыре из которых поданы тем же заявителем, что и данная заявка, и описание которых во всей полноте включено сюда настоящим в качестве ссылки для всех целей.
Настоящее изобретение относится к способам снижения содержания 4-карбоксибензальдегида (4-КБА) и 3-карбоксибензальдегида (3-КБА) и, более конкретно, к способу снижения содержания 4-КБА в сырой терефталевой кислоте (ТФК) и 3-карбоксибензальдегида (3-КБА) в сырой изофталевой кислоте (ИФК).
Последние достижения в производстве терефталевой кислоты (ТФК) требуют сравнительно высокой чистоты п-ксилола (99,7+%) для того, чтобы улучшить качество продукта и снизить производственные затраты. Это обусловлено тем, что в процессе в качестве основного способа очистки сырой терефталевой кислоты, получаемой в окислительной части процесса, используют гидрирование. Хотя способ гидрирования является весьма эффективным для исключения основной примеси, 4-карбоксибензальдегида (4-КБА), путем превращения ее в п-толуиловую кислоту, такие способы могут быть применены лишь для очень малых количеств 4-КБА (предпочтительно, менее 3000 ppm).
Малое количество 4-КБА (или 3-КБА в производстве изофталевой кислоты (ИФК)) не может быть окислено в ТФК (или ИФК) в реакторе окисления, так как используемый в реакторе растворитель, уксусная кислота, является слабым растворителем для значительного растворения ТФК (или ИФК) и 4-КБА (или 3-КБА). В действительности в традиционном процессе почти все полученное количество ТФК (или ИФК) и 4-КБА (или 3-КБА) выпадает в реакторе окисления в осадок, образуя суспензию. Поэтому небольшое количество 4-КБА (или 3-КБА) инкапсулируется внутри твердой фазы ТФК (или ИФК) и не может быть далее окислено воздухом в ТФК (или ИФК) в реакторе окисления. Следует отметить, что несмотря на то, что 4-КБА (или 3-КБА) представляет собой альдегид, который может быть легко окислен воздухом в ТФК (или ИФК) при рабочих условиях реактора окисления, он трудно окисляется, поскольку находится в твердой фазе. Таким образом, сохраняется потребность в таком способе снижения содержания 4-КБА или 3-КБА в сырой ТФК или в сырой ИФК, соответственно, который не имел бы вышеописанных недостатков.
В соответствии с настоящим изобретением предложен способ снижения содержания изомеров карбоксибензальдегида (4-КБА или 3-КБА) в сырой ТФК/ИФК, в котором сырую ТФК или сырую ИФК растворяют в N-метилпирролидоне (НМЛ) и затем вводят во взаимодействие с существенно безводной перекисью водорода (содержащей оптимизированное количество воды от 0 до 5%) или с воздухом для превращения 4-КБА в ТФК (или 3-КБА в ИФК) со степенью конверсии от 40 до 50% за проход при умеренных температуре и давлении, а именно в пределах от 0 до 150°С и давлении от 1 до 20 атм.
Задачей настоящего изобретения является разработка патентованных растворителей, которые могли бы полностью растворить и ТФК (или ИФК), и 4-КБА (или 3-КБА) из твердых осадков сырой ТФК (или ИФК), полученных в реакторе окисления. Другой задачей настоящего изобретения является разработка способов и условий для реакции растворенного 4-КБА (или 3-КБА) с окислителями, такими как перекись водорода, чистый кислород, воздух или другие окислители, таким образом, чтобы окислитель оказывал минимальное вредное влияние на растворитель в растворе. При использовании настоящего изобретения основные примеси (т.е. 4-КБА или 3-КБА) могут быть окислены до целевых продуктов (т.е. ТФК или ИПК), которые, в свою очередь, могут быть выделены в процессе, увеличив тем самым выход на п-ксилол (или м-ксилол). Окислители КБА, используемые в настоящем изобретении, включают окислители, которые: (а) не вносят в процесс других примесей или побочных продуктов кроме воды; (в) могут быть концентрированы в стабильной форме в предпочтительном растворителе, таком как N-метилпирролидон (N-МП) (или метанол при очистке МФК); (с) с которыми может быть достигнута относительно высокая конверсия КБА при низком мольном соотношении окислитель/КБА; и (d) с которыми продуктами окисления должны быть преимущественно ТФК (или ИФК) и вода в качестве побочного продукта. Следует понимать, что термин "окислители КБА" в данной заявке предназначен для того, чтобы охватить все окислители, которые используются для окисления 4-КБА и 3-КБА в ТФК и ИФК, соответственно, как описано здесь. В соответствии с изобретением одним из предпочтительных окислителей КБА является преимущественно безводная перекись водорода, концентрированная в предпочтительном растворителе, с небольшим количеством добавленной воды (до 5%). Этот окислитель КБА является предпочтительным для окисления небольших количеств 4-КБА или 3-КБА, содержащихся в растворе. Безводная перекись водорода является чрезвычайно реакционноспособной и выпускается в промышленном масштабе и продукты ее разложения являются экологически безопасными. Перекись водорода может быть подвергнута концентрированию в стабильной форме в N-МП (в одном из предпочтительных растворителей для окисления КБА).
В соответствии с изобретением другим окислителем предпочтительным для окисления небольших количеств 4-КБА или 3-КБА, содержащихся в растворе, является воздух (или чистый кислород). Хотя предпочтительными окислителями КБА являются практически безводная перекись водорода (содержание воды в перекиси водорода находится в пределах от 0 до 5 маc.%) и воздух (или чистый кислород), следует понимать, что в соответствии с данным изобретением окислители КБА, используемые для конверсии изомеров КБА в соответствии с данным изобретением, могут быть выбраны из различных окислителей, которые способны окислять альдегиды в растворе, включая, не ограничиваясь, перекись водорода в воде, озон, четыреххлористый углерод, трихлорацетальдегид, гексамин, ацетон, циклогексанон, бензофенон, циннамальдегид, диметилсульфоксид, сульфиды, хиральные окислители, глиоксали и органические надкислоты, которые включают надмуравьиную кислоту, надуксусную кислоту, пербензойную кислоту, надпропионовую кислоту, надмалеиновую кислоту и надфталевую кислоту.
Предпочтительно использовать неочищенную терефталевую кислоту (ТФК), содержащую, по меньшей мере, 80-99% терефталевой кислоты и 0-20% изофталевой кислоты.
Предпочтительна неочищенная изофталевая кислота (ИФК), содержащая, по меньшей мере, 95% изофталевой кислоты и 0-5% карбоксибензальдегида (КБА).
Предпочтительно окисление осуществляют при температуре в пределах от 0 до 150°С и давлении от 1 до 20 атм, при мольном отношении перекиси водорода к карбоксибензальдегиду (КБА) от 0,5 до 15.
Более предпочтительно окисление осуществляют при температуре в пределах от 20 до 100°С и при мольном отношении перекиси водорода к карбоксибензальдегиду (КБА) от 2 до 5. Кроме того, окисление осуществляют при мольном отношении воды к карбоксибензальдегиду (КБА), равном от 0 до 5, более предпочтительно от 1 до 5.
Предпочтительное время пребывания в указанном реакторе окисления КБА составляет от 0,1 до 6 часов, более предпочтительно от 1 до 5 минут.
В случае, когда окисление осуществляют воздухом, предпочтительное мольное отношение воздуха к карбоксибензальдегиду (КБА) составляет от 1 до 100, более предпочтительно от 1 до 20.
Аппарат для растворения также может служить в качестве реактора окисления КБА и работает при мольном отношении перекиси водорода к карбоксибензальдегиду (КБА) от 2 до 5, а в случае использования в качестве окислителя воздуха отношение его к КБА составляет от 1 до 20. Причем при использовании в качестве окислителя перекиси водорода, стабилизированной NMP, мольное отношение воды к карбоксибензальдегиду (КБА) составляет от 1 до 5. Время пребывания в нем реакционной смеси составляет от 1 до 5 минут.
Предпочтительное время пребывания в указанном реакторе окисления КБА составляет от 0,1 до 6 часов.
Более полное понимание способов по настоящему изобретению может быть достигнуто при обращении к нижеследующему подробному описанию, которое рассматривается вместе с прилагаемыми чертежами, на которых на фиг.1 представлена схема проведения способа получения ТФК и МФК из смеси ксилолов и на фиг.2 - схема проведения способа усовершенствованного снижения содержания изомеров карбоксибензальдегида по настоящему изобретению.
Подробное описание вариантов способа
Настоящее изобретение относится к способу снижения концентрации изомеров карбоксибензальдегида в сырой ТФК или ИФК. Для иллюстрации осуществление способа по настоящему изобретению будет рассмотрено в связи с недавно разработанным процессом получения ТФК и ИФК из смеси ксилолов.
На фиг.1 дано схематическое изображение нового способа получения ТФК и ИФК из смеси ксилолов. Этот новый способ более полно описан в совместно поданной заявке США №09/097930, заявленной 16 июня 1998 г. также заявителем описанного здесь изобретения, содержание которой включено сюда в качестве ссылки.
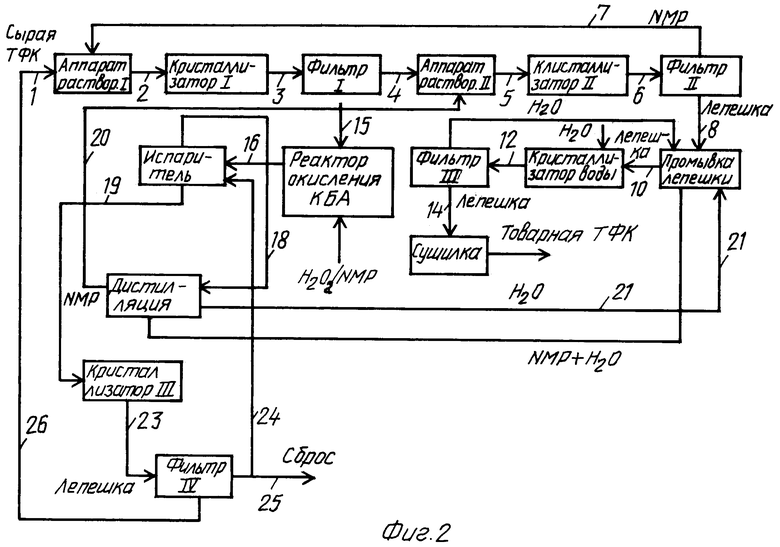
Сырую терефталевую кислоту (фиг.1) с участка окисления реактора (не показано), содержащую приблизительно 95% терефталевой кислоты, 5% изофталевой кислоты и небольшое количество других примесей (4-карбоксибензальдегид, 3-карбоксибензальдегид, п-толуиловую кислоту, м-толуиловую кислоту и т.д.) подают в первый чан для растворения 100 по трубопроводу 1 для смешивания с маточным раствором из фильтра П 105 (по трубопроводу 7) и шламом из фильтра IV 113 (по трубопроводу 19). Температуру в первом чане для растворения 100 поддерживают на уровне от 160 до 180°С для полного растворения твердых веществ и выпаривания по существу всего метанола, поступающего по трубопроводу 19. Затем насыщенный раствор из первого чана для растворения 100 непрерывно подают в первый охлаждающий кристаллизатор 101 по трубопроводу 2 для получения кристаллов соли терефталевой кислоты при температуре от 30 до 50°С. Шлам, содержащий кристаллы соли терефталевой кислоты, покидает первый охлаждающий кристаллизатор 101 по трубопроводу 3 в фильтр I 102, где сырой кристаллический осадок удаляют и подают во второй чан для растворения 103 по трубопроводу 4. Во втором чане для растворения 103 осадок вновь растворяют в чистом N-метилпирролидоне, рециркулированном по трубопроводу 35 из системы по восстановлению растворителя. Температуру во втором чане для растворения 103 вновь поддерживают на уровне от 160 до 180°С, чтобы полностью растворить кристаллы соли терефталевой кислоты. Насыщенный раствор из второго чана для растворения 103 непрерывно подают по трубопроводу 5 во второй флэш-кристаллизатор 104, где температуру поддерживают на уровне как минимум 60°С, чтобы предотвратить образование кристаллов соли терефталевой кислоты. Степень снижения температуры в кристаллизаторе контролируют количеством N-метилпирролидона, быстро испаряемого из кристаллизатора в результате снижения давления. Выпаренный N-метилпирролидон рециркулируют в первый чан для растворения 100 по трубопроводу 36.
Шлам из второго кристаллизатора 104 подают в фильтр П 105 по трубопроводу 6, где очищенный твердый осадок терефталевой кислоты восстанавливают и подают на установку для промывания осадка 106, в то время как маточный раствор рециркулируют в первый чан для растворения 100 по трубопроводу 7. В установке для промывания осадка 106 общую массу остаточного N-метилпирролидона удаляют противоточным промыванием водой, а промытый осадок подают по трубопроводу 10 в реакционную камеру 107 для удаления окончательных следов N-метилпирролидона в твердых веществах терефталевой кислоты вымачиванием водой при температуре от 160 до 280°С. Осадок, не содержащий N-метилпирролидона, фильтруют в фильтре III 108 и сушат в сушилке I 109, получая конечную терефталевую кислоту.
Маточный раствор из фильтра I 102 подают по трубопроводу 15 в аппарат для осаждения 112, при этом маточный раствор проходит через окислительный аппарат 111. Метанол подают в аппарат для осаждения по трубопроводу 16, вызывая полное осаждение (или кристаллизацию) терефталевой кислоты и незначительное осаждение изофталевой кислоты из маточного раствора. Шлам из аппарата для осаждения 112 подают в фильтр IV 113 по трубопроводу 18 для удаления основной части маточного раствора из шлама перед его рециркулированием в первый чан для растворения 100 по трубопроводу 19. Маточный раствор из фильтра IV 113 подают в выпарной аппарат 114 для удаления N-метилпирролидона и метанола путем теплового и вакуумного выпаривания таким образом, что концентрированный маточный раствор становится насыщенным раствором изофталевой кислоты, который подают в первый кристаллизатор 115 изофталевой кислоты для ее кристаллизации при температуре от 30 до 50°С в результате охлаждения или быстрого испарения. Испаренный N-метилпирролидон и метанол из выпарного аппарата 114 подают в дистилляционную колонну 110, получая N-метилпирролидон из нижней части, а метанол - из верхней части колонны. Поток метанола рециркулируют в аппарат для осаждения 112 по трубопроводу 16, в то время как поток N-метилпирролидона подают во второй чан для растворения 103 по трубопроводу 35. Шлам из первого кристаллизатора 115 изофталевой кислоты подают в фильтр V 116, получая сырой осадок изофталевой кислоты и маточный раствор. Маточный раствор подают в аппарат для осаждения 112 по трубопроводу 17, однако часть потока 17 продувают через трубопровод 37 для предотвращения накопления примесей и красящих веществ. Осадок из фильтра V 116 затем подают по трубопроводу 25 в чан для растворения 117 изофталевой кислоты, где ее сырой осадок растворяют метанолом при подходящих температуре и давлении. Насыщенный раствор изофталевой кислоты фильтруют в фильтре VI 118, чтобы удалить следы нерастворимых веществ для продувания трубопровода 28. Свободный от твердых веществ раствор подают по трубопроводу 29 во второй кристаллизатор 119 для изофталевой кислоты, получая ее кристаллы путем быстрого испарения метанола из кристаллизатора в результате снижения давления. Шлам из второго кристаллизатора 119 изофталевой кислоты подают по трубопроводу 30 в фильтр VII 120, чтобы восстановить и промыть кристаллы очищенной изофталевой кислоты для окончательной сушки в сушилке II 121, получая конечную изофталевую кислоту, в то время как маточный раствор из фильтра VII 120 рециркулируют в выпарной аппарат 114 по трубопроводу 31.
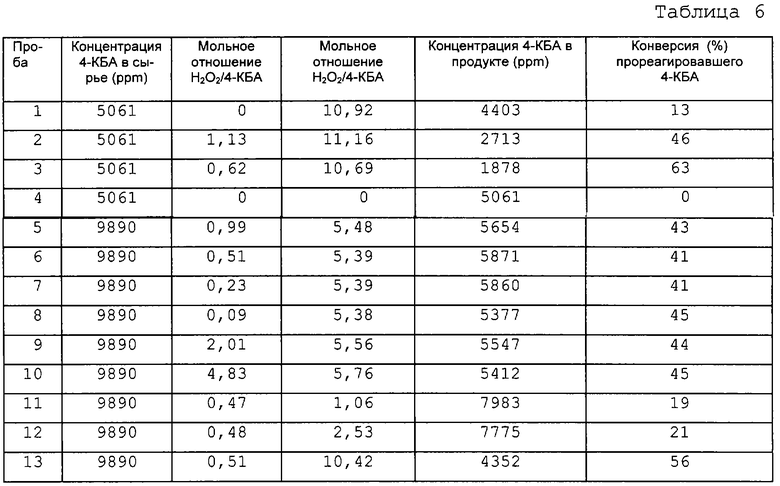
Сырую ТФК из секции окисления (содержащую главным образом ТФК и небольшие примеси 4-КБА и п-толуиловой кислоты) вводят в аппарат для растворения I по линии 1 (фиг.2) для смешения с маточным раствором из фильтра II (по линии 7) и суспензией из фильтра IV (по линии 26). Температуру в аппарате для растворения I поддерживают на уровне от 160 до 180°С для того, чтобы полностью растворить твердые вещества.
Затем насыщенный раствор из аппарата для растворения I по линии 2 непрерывно подают в первый охлаждаемый кристаллизатор (кристаллизатор I) для образования кристаллов соли ТФК при температуре от 30 до 50°С. Суспензия, содержащая указанные кристаллы соли ТФК, выходит из кристаллизатора I по линии 3 в фильтр I, где сырая лепешка удаляется и по линии 4 вводится в аппарат для растворения II. В аппарате для растворения II лепешку заново растворяют в чистом N-МП, подаваемом в рецикл по линии 20 из колонны дистилляции смеси N-МП/вода (дистилляция). Температуру в аппарате для растворения II опять-таки поддерживают на уровне от 160 до 180°С для того, чтобы полностью растворить кристаллы соли ТФК. Насыщенный раствор из аппарата для растворения II по линии 5 непрерывно подают в кристаллизатор II, где поддерживают температуру минимум 60°С, чтобы предотвратить образование кристаллов соли ТФК. Степень понижения температуры в кристаллизаторе II регулируют количеством N-МП, испаряемым из кристаллизатора II за счет снижения давления. Испаренный N-МП возвращают в рецикл в аппарат для растворения I по линии 27.
Суспензию из кристаллизатора II по линии 6 подают на питание фильтра 11, где лепешку твердой очищенной ТФК извлекают и направляют на промывку лепешки по линии 8, в то время как маточный раствор возвращают в рецикл в аппарат для растворения I по линии 7. При промывке лепешки основную часть остаточного N-МП из лепешки удаляют методом противоточной промывки, используя воду, и после этого промытую лепешку подают по линии 10 в Сокинг-камеру (кристаллизатор воды) для удаления любых следов N-МП из лепешки ТФК путем промывки водой при температурах между 160 и 280°С. Затем очищенную от N-МП лепешку ТФК подают по линии 12 в фильтр III, где ее отфильтровывают перед подачей по линии 14 в сушилку I для сушки и получения конечной товарной ТФК.
Маточный раствор из фильтра I (содержащий растворитель (например, N-МП), ТФК, 4-КБА, п-толуиловую кислоту и другие незначительные примеси) по линии 15 передают в реактор окисления КБА для взаимодействия с перекисью водорода в N-МП (с нужным количеством воды). Работу реактора окисления КБА предпочтительно ведут при температуре от 25 до 50°С и под давлением между примерно 1 и примерно 5 атм. В описанном выше процессе примерно 40-60% 4-КБА, содержащегося в маточном растворе из фильтра I, превращается в ТФК. Выходящий поток из реактора окисления КБА по линии 16 передают в испаритель для отгонки N-МП и воды от смеси. Полученный концентрированный раствор выводят из куба испарителя и по линии 19 подают в кристаллизатор III. В кристаллизаторе III добиваются максимального извлечения путем работы кристаллизатора III при температуре между 0 и 60°С и под давлением. Далее суспензию, полученную в кристаллизаторе III, по линии 23 подают на фильтр IV, откуда лепешку по линии 26 возвращают в рецикл в аппарат для растворения I, а основную часть маточного раствора возвращают в испаритель по линии 24. Небольшую оставшуюся часть маточного раствора выводят из системы по линии 25.
Смесь N-МП и воды из испарителя по линии 18 подают в колонну дистилляции (Дистилляция), где воду выделяют в качестве верхнего потока и направляют на промывку лепешки по линии 21. Растворитель (например, N-МП) извлекают в виде кубового продукта дистилляции и направляют в рецикл в аппарат для растворения II по линии 20.
Следующие примеры показывают эффективность окислителя КБА при превращении 4-КБА в ТФК в органическом растворе, таком как N-МП, что является основным и отличительным признаком данного изобретения.
Пример 1
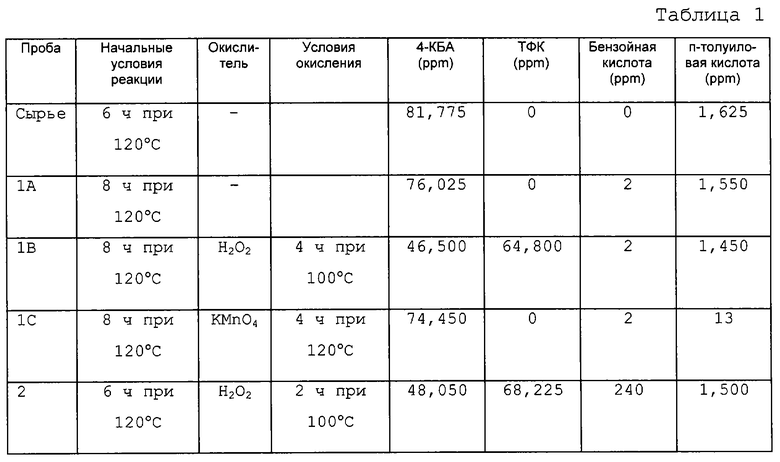
В данном примере восстановление 4-КБА в ТФК в присутствии перекиси водорода с использованием способа по настоящему изобретению описано по данным газовой хроматографии. Готовили раствор 4-КБА в N-МП. Этот раствор нагревали до 120°С в течение примерно 6 часов. Затем раствор делили на 4 пробы по 30 мл. К одной пробе в качестве окислителя добавляли имеющуюся в продаже 30% перекись водорода. Три другие пробы нагревали дополнительно в течение 2 часов при 120°С перед тем, как к двум из них добавили окислитель. Использованными окислителями являлись 30% перекись водорода и перманганат калия (KMnO4). После добавления окислителя пробы нагревали в течение еще 2 часов. Пробы, содержавшие перекись водорода, нагревали при 100°С, в то время как пробу, содержавшую КМnO4, выдерживали при 120°С. В таблице 1 представлены полученные результаты. Как можно видеть, перекись водорода оказалась эффективным окислителем 4-КБА в ТФК (пробы 1В и 2). Другой сильный окислитель, КМnO4, не окислял 4-КБА в ТФК (проба 1С). В контрольной пробе (проба 1А) также не наблюдалось окисление 4-КБА в ТФК.
Пример 2
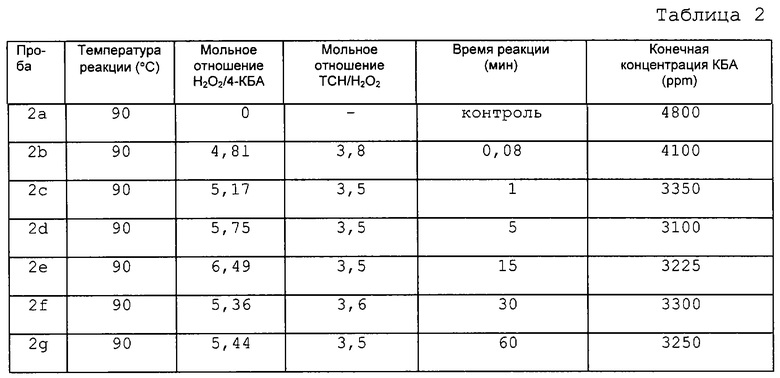
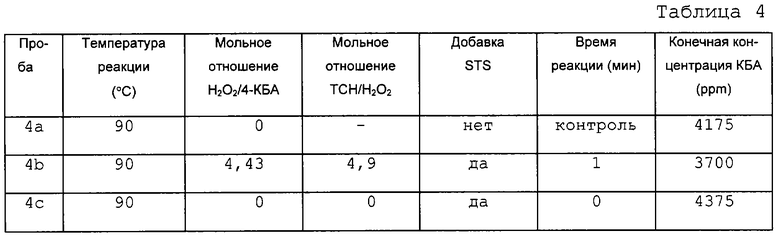
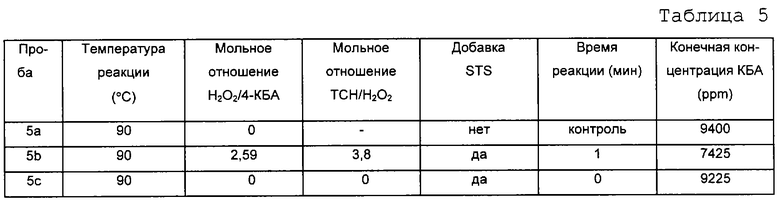
В данном примере показано влияние времени реакции на количество ТФК, получаемое из 4-КБА по способу настоящего изобретения. Окисление происходит быстро, так что после первой минуты реакции нельзя видеть существенного изменения концентрации 4-КБА. Этот пример показывает также, что температура реакции мало влияет на ее скорость. В данном эксперименте исходный раствор делили на равные части, каждая из которых весила примерно 25 г. Эти пробы нагревали до температуры, показанной ниже в таблице, используя масляную баню. Одну пробу из каждого опыта использовали в качестве контрольной без добавления окислителя. Раствор безводной перекиси водорода готовили путем добавления имеющейся в продаже 30% перекиси водорода в воде к N-МП и отгонки воды, что давало практически безводный раствор перекиси водорода, стабилизированной в N-МП. Определенные количества безводной перекиси водорода добавляли к остальным пробам. Раствор 40% тиосульфата натрия (ТСН) готовили в HPLC воде. После заданного периода реакции добавляли избыток ТСН для того, чтобы оборвать реакцию. Каждую пробу вынимали из масляной бани и охлаждали до комнатной температуры (где требовалось). Затем пробы отфильтровывали и фильтрат анализировали методом газовой хроматографии. Результаты приведены в таблицах 2-5.

Пример 3
В данном примере показано влияние отношения количеств перекиси водорода и 4-КБА на превращение 4-КБА в ТФК при использовании способа по настоящему изобретению. Этот пример показывает также необходимость добавления небольшого количества воды для улучшения реакции окисления. Готовили два раствора 4-КБА в N-МП и грели их при 165°С в течение приблизительно 10 минут для того, чтобы быть уверенными в полном растворении. Концентрация ТФК в этих растворах была в четыре раза больше концентрации 4-КБА. Растворы охлаждали до 23°С и делили на пробы. К каждой пробе добавляли разное количество воды и безводной перекиси водорода в N-МП. Пробы включали контрольную пробу, к которой не добавляли ни воду, ни перекись водорода. Затем пробы анализировали на содержание 4-КБА методом газовой хроматографии. Результаты анализа компенсировали для устранения эффектов разведения. Результаты суммированы в таблице 6.
Из результатов для проб 1, 3 и 13 ясно, что для данного мольного соотношения Н2O2/4-КБА присутствие воды существенно увеличивает конверсию 4-КБА в ТФК.
Хотя предпочтительное осуществление изобретения иллюстрируется на прилагаемых чертежах и в таблицах и описано в предшествующем подробном описании, следует понимать, что изобретение не ограничивается описанными вариантами, но может быть подвергнуто многочисленным перестановкам, модификациям и заменам без отступления от сути изобретения, которая представлена и определена нижеследующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОЧИСТКИ ИЗОФТАЛЕВОЙ КИСЛОТЫ ПУТЕМ КРИСТАЛЛИЗАЦИИ | 1999 |
|
RU2208606C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЧИЩЕННОЙ ТЕРЕФТАЛЕВОЙ И ИЗОФТАЛЕВОЙ КИСЛОТЫ ИЗ СМЕСИ КСИЛОЛОВ | 1999 |
|
RU2214391C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЧИСТОЙ ИЗОФТАЛЕВОЙ КИСЛОТЫ И СОПУТСТВУЮЩИХ ПРОДУКТОВ ИЗ КСИЛОЛЬНЫХ ФРАКЦИЙ | 2009 |
|
RU2430911C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОФТАЛЕВОЙ КИСЛОТЫ С ВЫСОКОЙ СТЕПЕНЬЮ ЧИСТОТЫ | 2004 |
|
RU2266277C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЧИСТОЙ ИЗОФТАЛЕВОЙ КИСЛОТЫ И СОПУТСТВУЮЩИХ ПРОДУКТОВ ИЗ ИЗОМЕРОВ ЦИМОЛА И ДИИЗОПРОПИЛБЕНЗОЛА | 2009 |
|
RU2415836C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОЧИСТОЙ ИЗОФТАЛЕВОЙ КИСЛОТЫ | 1997 |
|
RU2137753C1 |
| НЕПРЕРЫВНЫЙ СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОЧИСТОЙ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ | 2003 |
|
RU2254324C2 |
| СИСТЕМА ПОЛУЧЕНИЯ ПОЛИКАРБОНОВОЙ КИСЛОТЫ, ИСПОЛЬЗУЮЩАЯ ОХЛАЖДЕННЫЙ МАТОЧНЫЙ РАСТВОР ИЗ ОКИСЛИТЕЛЬНОГО СЖИГАНИЯ В КАЧЕСТВЕ ЗАГРУЗКИ СИСТЕМЫ ОЧИСТКИ ОТ ЗАГРЯЗНЕНИЙ | 2007 |
|
RU2458907C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2388743C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЧИЩЕННОЙ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ | 2002 |
|
RU2292332C2 |
Изобретение относится к усовершенствованному способу снижения содержания 4-карбоксибензальдегида в производстве терефталевой или 3-карбоксибензальдегида в производстве изофталевой кислоты, включающему: (а) растворение сырой терефталевой кислоты или сырой изофталевой кислоты в растворителе при температуре от 50 до 250°С с получением раствора; (b) кристаллизацию очищенной кислоты из указанного раствора путем снижения его температуры и/или давления; (с) отделение указанной кристаллизованной терефталевой кислоты или изофталевой кислоты от указанного раствора; (d) добавление окислителя в реактор окисления карбоксибензальдегида для окисления указанного отфильтрованного раствора на стадии (с), приводящее к превращению 4-карбоксибензальдегида или 3-карбоксибензальдегида в терефталевую кислоту или изофталевую кислоту; (е) выпаривание растворителя из указанного раствора со стадии (d); (f) охлаждение концентрированного раствора со стадии (е) для кристаллизации дополнительного количества очищенной терефталевой кислоты или изофталевой кислоты и фильтрацию указанной суспензии, а также рециркуляцию большей части маточного раствора со стадии (f) в аппарат для растворения его на стадии (а) и подачу меньшей части маточного раствора со стадии (f) на сброс. Возможно также добавление окислителя к раствору сырой терефталевой кислоты или сырой изофталевой кислоты после стадии (а), в этом случае стадия (d) отсутствует. Способ позволяет получить хорошо очищенный целевой продукт при умеренных условиях температуры и давления. 2 н. и 38 з.п. ф-лы, 6 табл., 2 ил.
| СПОСОБ ПОЛУЧЕНИЯ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ | 1992 |
|
RU2083550C1 |
| Нефтяной конвертер | 1922 |
|
SU64A1 |
| WO 9824749 A, 11.06.1998 | |||
| WO 9509143 A, 06.04.1995 | |||
| WO 9640612 A, 19.12.1996. | |||

