Изобретение относится к промышленному органическому синтезу, конкретно к технологическому процессу получения чистой изофталевой кислоты (ЧИФК) - мономеру и полупродукту для синтеза широкого ассортимента полимерных материалов.
Чистая ИФК применяется для получения полиэфиров, которые, в свою очередь, используют для производства покрытий, красок, армированных пластиков, упаковок и бутылок (из ПЭТФ) пищевого и технического назначения. Кроме того, ИФК как сомономер или полупродукт идет для изготовления прочных и малотоксичных электроизоляционных лаков, волокон с повышенной химической и термической стойкостью, цветообразующих компонентов для кино- и фотопленок, синтетической кожи, полимербетонов и многих других изделий.
Современные промышленные синтезы изофталевой кислоты основаны на двухстадийных химических процессах, включающих обычно так называемый SD-способ - жидкофазное окисление метаксилола кислородсодержащим газом в уксуснокислой среде в присутствии металлбромидного катализатора с получением изофталевой кислоты - сырца (ИФК) с последующей ее очисткой методом гидрирования нежелательных примесей (в основном м-карбоксибензальдегида (м-КБА) молекулярным водородом в водной среде в присутствии палладия и других металлов, нанесенных на углеродный носитель (Pd/C) с получением высокочистой изофталевой кислоты, пригодной в качестве мономерного сырья для изготовления качественных полимерных материалов и изделий на их основе [1, 2].
Основным недостатком двухстадийных процессов получения ЧИФК является необходимость использования разных растворителей, катализаторов и параметров на стадиях окисления м-ксилола до ИФК и очистки ИФК до ЧИФК методом гидрирования примесей, что по существу обуславливает неизбежность применения двух независимых установок, принципиально различных по химической технологии и аппаратурному оформлению.
Для преодоления этих недостатков были предложены другие методы, исключающие вторую химическую стадию очистки ИФК методом гидрирования. Так, например, в известных способах [3, 4] очистку ИФК и других ароматических кислот осуществляют путем их растворения к уксусной кислоте и других растворителях с последующим доокислением примесей О2-газом. Эти способы характеризуются многостадийностью и сложностью аппаратурного оформления. Для растворения ИФК в уксусной кислоте необходимы высокие температурные условия - 230-280°С. Сам процесс перекристаллизации для очистки указанных ароматических кислот, содержащих от 0,1 до 0,35% карбоксибензальдегида, неэффективный, а в случае доокисления примесей в указанном температурном интервале в процесс окислительного декарбоксилирования вовлекается уксусная кислота. Это снижает экономическую эффективность указанных процессов. Кроме того, многостадийность и относительная сложность аппаратурного оформления обуславливают необходимость повышенных капитальных затрат при создании производственных установок.
Согласно другому известному непрерывному способу получения изофталевой кислоты окисление м-ксилола осуществляют при температуре 190-205°С, давлении 1,9-1,7 МПа кислородом воздуха в растворе уксусной кислоты в присутствии ацетатов кобальта и марганца, а также при эквимолярном и суммарном содержании ацетатов металлов, бромида натрия [4, 5]. Окисление проводят в одну ступень при температуре 190-205°С, давлении 1,9-2,7 МПа в течение 30-40 мин [5].
Смесь ацетатов кобальта и марганца берут в молярном соотношении 2-5:1, а мольное соотношение м-ксилол:вода:суммарное содержание ацетатов кобальта и марганца в подаваемой реакционной смеси поддерживают в интервале 1,0-1,75:1,0-2,5:1,0-1,2·10-2. Расход воздуха составляет 360-320 нл/моль м-ксилола.
Содержание основной примеси, м-карбоксибензальдегида, в выделенной из оксидата ИФК в зависимости от предельных значений концентраций катализатора, м-ксилола в ИРС, а также времени пребывания находится в пределах 0,003-0,03, показатель цветности 19-30°Н.
Указанный способ обладает рядом недостатков, а именно:
1) повышенное качество ИФК ([м-КБА]=0,003÷0,005%, цветность 19,0-19,2°Н) достигается при малых концентрациях м-ксилола в реакционной смеси 0,75-1,0 моль/л, что указывает на относительно низкую производительность реакционного объема реактора. Но даже и в этих условиях качество ИФК не соответствует требованиям качества высокочистого продукта, основные показатели которого должны соответствовать следующим значениям:
[м-КБА]≤0,0025%, цветность ≤10°Н;
2) с повышением концентрации м-ксилола в исходной реакционной смеси (1,5-1,8 моль/л) качество ИФК по показателю цветности находится в пределах 30-105°Н, что не соответствует требованиям к чистой ИФК, используемой для получения высококачественных полимерных материалов;
3) проведение реакции непрерывного окисления в одном аппарате в одну стадию с высокой степенью превращения м-ксилола в ИФК до остаточной ничтожно малой концентрации промежуточных продуктов (концентрации м-толуиловой кислоты 0,004-0,005%, м-карбоксибензальдегида 0,003-0,005%) неизбежно приводит к резкому возрастанию потерь уксусной кислоты за счет ее окислительного каталитического карбоксилирования. Это происходит в результате «высвобождения» значительной доли катализатора от участия в основной радикально-цепной реакции окисления м-ксилола и его промежуточных соединений из-за ничтожно малой их стационарной концентрации в зоне реакции, и катализ окисления смещается в сторону побочной сопряженной реакции «сгорания» СН3СООН.
Промышленная практика показывает, что «сгорание» СН3СООН в аналогичных случаях составляет 120-220 кг/т целевого продукта.
Существенным недостатком способа является относительно низкий выход целевого продукта 86,7-93,4%.
Известен также способ получения изофталевой и терефталевой кислоты [6] повышенного качества путем жидкофазного окисления м-ксилола или п-ксилола в две ступени в присутствии кобальт-марганец-бромидного катализатора. На первой ступени окисление осуществляют при 200-215°С и давлении 2,0-2,6 МПа, на второй ступени при температуре 180-200°С реакционную массу обрабатывают одновременно кислородсодержащей парогазовой смесью, выходящей из первой ступени окисления, и флегмой, взятой из зоны ступенчатой кристаллизации и имеющей в своем составе 80-88% СН3СООН, 10-16% Н2О, 1,5-4,0% алифатических соединений общей формулой
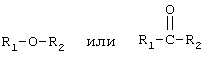
R1, R2-СН3 или СН3О. Массовое соотношение флегма:реакционная смесь составляет 0,3-0,6:1. Содержание основной примеси изомеров КБА в получаемых ТФК и ИФК находится в пределах 0,006-0,018%.
Этот способ получения изофталевой и терефталевой кислот позволяет в процессе двухступенчатого окисления м-ксилола или п-ксилола повысить качество ИФК и ТФК, однако, как другие, ранее описанные методы, он обладает рядом недостатков. Наиболее существенным недостатком этого способа является то, что с повышением качества ИФК или ТФК возрастают потери уксусной кислоты из-за необходимости повышения температуры, концентрации катализатора и/или времени окисления [6]. Так, при различном, остаточном содержании п-КБА в ТФК или м-КБА в ИФК удельные потери СН3ООН за счет сгорания составляют, кг/т ТФК, ИФК
Таким образом, указанный способ экономически оправдан при получении ИФК и ТФК средней степени чистоты, а при производстве высокочистых мономеров он существенно уступает лучшим современным способам, в том числе двухстадийным процессам. Близким по технической сущности является способ получения высокочистой изофталевой кислоты в три стадии [7].
На первой стадии процесса окисление м-ксилола осуществляют в температурном интервале 180-200°С O2-газом в среде уксусной кислоты в присутствии Со-Mn-Br катализатора при суммарной концентрации Со и Mn 300-1500 ppm и соотношениях Mn:Со=0,5-5. Br:[Со+Mn]=0,5-1,5 до достижения содержания в продуктах м-КБА 500-10000 ppm (0,05-1,0%), на второй стадии проводят обработку оксидата первой стадии O2-газом до достижения остаточного содержания в продуктах окисления м-КБА 100-800 ppm (0,01-0,08%), после чего из реакционной массы выделяют ИФК, смешивают ее с уксусной кислотой и подают на третью стадию. На третьей стадии суспензию нагревают до температуры выше 100°С и экстрагируют примеси, содержащиеся в ИФК до достижения качества продукта по содержанию м-КБА, удовлетворяющем требованиям ЧИФК.
Осуществление процесса получения ЧИФК в три стадии (2 стадии окисления и 1 стадия экстракции) с использованием известных Со-Mn-Br-катализатора, растворителя (СН3СООН) и O2-газа при температуре от 180 до 210°С принципиально позволяет достигнуть ЧИФК по содержанию м-КБА, не уступающему по данным патента качественным показателям ЧИФК, получаемой по двухстадийным процессам (синтез - окислением, очистка - гидрированием). В то же время этот способ обладает рядом существенных недостатков, среди которых наиболее существенным являются следующие:
1) в предложенном способе отсутствует стадия удаления из ЧИФК остаточного (после выделения и сушки) количества уксусной кислоты, которая является, как и м-КБА, нежелательной примесью в конечном продукте. Известно, что обычный метод сушки не позволяет количественно удалить СН3СООН, находящуюся внутри кристаллической решетки ИФК (т.н. межкристаллитный захват с образованием водородной связи). Для удаления СН3СООН из кристаллической ИФК требуется метод возгонки или применения других способов очистки;
2) проведение процесса окисления в две ступени до остаточного содержания в ИФК м-КБА 0,01-0,08%, которое может быть снижено до требуемой нормы (0,0025%) на третьей, конечной, стадии экстракции уксусной кислотой при Т>100°С, неизбежно приводит к необходимости использования повышенного температурного режима (до 210°С - верхний предел) и, как следствие, к увеличению скорости сопряженной реакции окислительного декарбоксилирования, т.е. к ее необратимым потерям;
3) метод экстракции м-КБА из кристаллической ИФК в среде СН3СООН неэффективный, т.к. в условиях неполного растворения скорость диффузионного процесса перехода м-КБА в раствор из твердой кристаллической ИФК ничтожно мал, а по мере перехода м-КБА в раствор градиент концентраций примеси в твердой и жидкой фазах снижается, что дополнительно замедляет и до того низкую скорость процесса очистки этим способом;
4) между 2-ой стадией окисления и 3-ей стадий экстракции требуется замена растворителя (СН3СООН), что усложнит схему процесса необходимостью применения стадий охлаждения (кристаллизации), выделения кристаллической ИФК из оксидата методом фильтрации, суспендирования ИФК в чистой СН3СООН и подачи суспензии на третью ступень.
Наиболее близким по технической сущности и достигаемым результатом является способ получения ИФК и других изомеров бензолдикарбоновых кислот путем окисления м-ксилола О2-газом в три стадии [8]. На первой стадии исходную реакционную смесь, содержащую изомер ксилола, низкую алифатическую кислоту и катализатор подают в реакционную зону первой ступени с начальной скоростью 6-30 м/с противотоком по отношению к направлению реакционной смеси и полученный продукт реакции окисляют на второй стадии O2-газом и обрабатывают флегмой из кристаллизатора 2-ой стадии с последующей кристаллизацией и выделением продуктов окисления, которую после отжатия обрабатывают возвращаемым в цикл растворителем - уксусной кислотой, экстрагируют в раствор примеси из твердого вещества при 200-250°С в течение 5-60 мин и проводят третью стадию окисления. В качестве катализатора используют соли Со и Mn и соединения Br с добавлением солей никеля, хрома, циркония или церия. Соотношение концентраций модифицированного катализатора на первой, второй и третьей стадиях окисления составляет 1:0,5-0,9:0,01-0,2, соответственно. Согласно этому способу получают целевой продукт, содержащий КБА в изомерах фталевых кислот ≤25 ppm.
К недостаткам этого способа следует отнести сложность схемы процесса, требующей после 2-й ступени окисления включение стадий кристаллизации и выделения сырой ИФК, экстрагирования примесей в растворе СН3СООН при температуре до 250°С и последующего доокисления на 3-ей ступени. Сам процесс экстрагирования примесей из кристаллов ИФК в условиях неполного ее растворения является медленным процессом.
Кроме того, выделенная чистая ароматическая кислота (ИФК) после сушки содержит остатки СН3СООН, что недопустимо при использовании ИФК для получения высококачественного ПЭТФ. В этом случае необходима 4-я стадия процесса - перекристаллизация ИФК в воде.
Целью настоящего изобретения является упрощение процесса и улучшение качества ИФК.
Поставленная цель достигается тем, что окисление м-ксилола проводят в три ступени при дискретном изменении (колебании) температуры по ступеням в сторону понижения с последующим повышением или повышения с последующим понижением по схеме Т1>Т2<Т3 или Т1<Т2>Т3 в температурном интервале 180-200°С в присутствии марганец-кобальт-бромидного катализатора, модифицированного добавками солей цинка и/или никеля в соотношении металлов Mn:Co:Zn:Ni=1:(0,5-2):(0,005-0,01):(0,005-0,01) при суммарной концентрации 490 ppm в реакционной массе, эквимолярном по отношению к металлам количества брома и при времени смешения вводимых в зону реакции реагентов <10 сек, после чего оксидат с 3й ступени подвергают охлаждению, выделяют кристаллическую ИФК и последовательно обрабатывают (промывают) уксусной кислотой при температуре 80-100°С в массовом соотношении ИФК:СН3СООН=1:(2-2,5) для удаления катализатора, затем водой при повышенной температуре 150-230°С в массовом соотношении ИФК:Н2О=1:(2-3) для удаления уксусной кислоты, после чего промытый продукт выделяют и сушат известными приемами с получением высокочистой ИФК.
Новым в предложенном способе являются проведение процесса окисления в трехступенчатом режиме с дискретным изменением (колебанием) температуры на каждой ступени в присутствии модифицированного Mn-Co-Zn-Ni-Br катализатора с достижением найденных пределов степени конверсии м-ксилола на каждой ступени в ИФК, оцениваемой содержанием в ИФК основного промежуточного продукта - м-КБА (нежелательная примесь, лимитирующая качество конечного продукта), и по суммарному содержанию побочных высококипящих продуктов, оцениваемых показателем цветности, а также осуществление процесса на каждой ступени в условиях практически мгновенного смешения реагентов (≤10 сек), обеспечивающего максимально возможный тепло- и массоперенос в трехфазной системе газ-жидкость-твердое тело, максимально возможную скорость растворения кислорода в жидкой фазе реакционной зоны и, как следствие, селективное превращение м-ксилола в целевой продукт, не требующий сложных химических или физико-химических стадий очистки. Проведение реакций в узком температурном диапазоне с колебанием температуры на каждой ступени в присутствии модифицированного катализатора при быстром смешении привело к неожиданным результатам по качеству продукта и позволило упростить технологический процесс, исключив из схемы стадии выделения продуктов (кристаллизация, фильтрация, экстракция) после 2-ой ступени окисления. Как показали эксперименты, колебания температурного режима при выбранном катализаторе обусловлены необходимостью обеспечения разных темпратурных условий окисления для исходного субстрата (м-ксилола) и промежуточных продуктов (м-толуиловый альдегид, м-толуиловая кислота, м-карбоксибензальдегид), имеющих различную реакционную способность к окислению, растворимость и термостабильность, а также уровнем активности и модифицированного Mn-Co-Zn-Ni-Br катализатора.
Ниже приводятся блок-схема установки окисления м-ксилола и получения чистой ИФК (см. чертеж) и ее описание, а также примеры, характеризующие сущность изобретения.
Установка включает сборник раствора катализатора 1, смеситель исходной реакционной смеси (ИРС) 2, реактор I ступени 3, реактор II ступени 4, реактор III ступени 5, кристаллизаторы 6, центрифуги 7, 11, сушилку 8, 12, суспензатор ИФК в воде 9, кристаллизаторы 10, колонну регенерации СН3СООН 13, узел регенерации катализатора.
Реакторы I, II и III ступени объемом 1 л снабжены турбинными мешалками и обратными холодильниками, элементами обогрева, патрубками для ввода и вывода реагентов, отбора проб, а также приборами КИПиА для измерения температуры, давления и непрерывного измерения и регистрации концентраций О2, CO2, CO в газовых потоках, выходящих из реакторов.
Исходные реагенты, содержащие м-ксилол, уксусную кислоту и катализатор, приготавливают в смесителе ИРС 2, подают в реактор I ступени 3 дозировочным насосом. Воздух от компрессора подводят в каждый реактор (3, 4, 5). Время смешения реагентов в реакторах окисления устанавливают изменением числа оборотов турбинной мешалки, время пребывания - количеством ИРС, подаваемой в процесс окисления с коррекцией реакционного объема на величину газонаполнения. Температурный режим в автотермическом режиме устанавливается установкой регулятора давления на линии отходящих из реакторов отработанных газов (т.н. абгазов). На II и III ступенях окисления предусмотрен подвод тепла с помощью электронагревателя. Подачу воздуха в реакторы устанавливают регулятором расхода на линии абгазов с замером объемного расхода ротаметрами.
Продукты окисления после реактора окисления III ступени 5 охлаждают до 60°С в кристаллизаторе 6. Суспензию ИФК в растворе СН3СООН фильтруют на центрифуге или вакуумном фильтре 7. Выделенную ИФК промывают уксусной кислотой непосредственно на фильтре (7) в массовом соотношении ИФК:СН3СООН=1:2-2,5. После промывки осадка ИФК уксусной кислотой до полного удаления остатков катализатора влажный продукт подвергают сушке до постоянного веса, после чего его репульпируют в чистой химочищенной воде в суспензаторе 9 в массовом соотношении ИФК:Н2О=1:2-3 при температуре 150-230°С. Для выделения растворенной части ИФК в воде суспензию охлаждают в кристаллизаторах 10 до 100-150°С, далее фильтруют на центрифуге или фильтре (11). Выделенную ИФК сушат в сушилке 12 до постоянного веса с получением высокочистой ИФК. Отработанный растворитель - СН3СООН, содержащий катализатор и примеси, подвергают регенерации в узлах регенерации СН3СООН 13 и катализатора 14. Очистку отработанных газов после реакторов окисления проводят в узле абсорбции 15. Очищенные абгазы сбрасывают в атмосферу, а регенерированный растворитель и катализатор возвращают в процесс окисления (рецикл).
Оценка качества ИФК осуществлялась хроматографическим, полярографическим и спектральным методами анализа.
Предлагаемое изобретение иллюстрируется следующими примерами.
Пример 1.
В сборник исходной реакционной смеси 2 загружают 513 мл (442 г) м-ксилола, 40 мл (40 г) Н2О и заранее приготовленный в сборнике 1 уксуснокислый раствор катализатора в следующем количественном соотношении:
СН3СООН - 1872 мл (1964 г.)
тетрагидраты ацетатов
До пуска реактор заполняют на 50% раствором катализатора, состав которого соответствует содержанию компонентов в ИРС (без м-ксилола), нагревают до 200°С в атмосфере азота. С момента достижения указанной температуры подают исходную реакционную смесь и воздух. Температурный режим по ступеням окисления принят по схеме Т1>Т2<Т3 (200°С-185°C-200°C). Стационарный режим на I ступени при времени пребывания 52 мин достигают после 3-х кратного обмена реакционного продукта в зоне реакции, достижения постоянного состава продуктов окисления по содержанию м-КБА не более 1,35%, по цветности не более 135°Н и стационарного состава газа, выходящего из реактора ([О2]≤5% об., [СО2]-const, [СО]-const).
Контроль осуществляют по показаниям непрерывно действующих газоанализаторов.
После окончания окисления первой ступени оксидат направляют в реактор второй ступени, в котором температуру снижают до 185°С и продолжают обработку реакционной массы О2-газом в течение 20 минут до достижения [м-КБА] не более 0,19% и показателя цветности не более 35°Н. Затем суспензию после второй ступени нагревают до температуры 200°С и в реакторе III ступени обрабатывают О2-газом в течение 10 минут до достижения остаточной концентрации м-КБА в продуктах окисления не более 0,01% и показателя цветности не более 16°Н.
Содержимое реактора III ступени охлаждают до 80°С, выделяют из оксидата твердую фазу, которую далее последовательно обрабатывают уксусной кислотой при 80°С и водой при 227°С и после выделения ИФК сушат до постоянного веса. Условия и результаты опыта, приведенные в таблице 1, показывают, что в принятых условиях окисления качественные показатели ИФК соответствуют требованиям высокочистого мономера: [м-КБА] <0,001%, (<10 ppm), показатель цветности 5°Н. Сгорание СН3СООН=41 кг/т ИФК.
Пример 2.
Опыт проводят в условиях примера 1 с той лишь разницей, что дискретное изменение температурного режима окисления по ступеням осуществлено по схеме Т1<Т2>Т3, а именно 190°С-200°С-190°С при времени пребывания на II ступени 5 мин, а на III ступени 20 мин.
Получены результаты: [м-КБА]<0,001%, цветность 6,0°Н, сгорание СН3СООН=35 кг/т ИФК. Качественные показатели ИФК удовлетворяют требованиям высокочистой ИФК. Сгорание снизилось на 6 кг/т ИФК.
Пример 3.
Опыт проводят в условиях примера 2 с той лишь разницей, что температуру на I и III ступенях снизили на 10°С.
Получены результаты: [м-КБА]=0,0022%. Цветность=7,0°Н. Сгорание СН3СООН=39 кг/т ИФК. Качество ЧИФК соответствует предельным значениям высокочистого мономера. Потери СН3СООН за счет сгорания снизились на 6 кг/т ИФК.
Пример 4 (сравнительный).
Опыт проводят в условиях примера 3 с той лишь разницей, что температуру на II ступени снизили на 30°С, а время пребывания увеличили в 4 раза.
Результат: [м-КБА]=0,005%, цветность 12°Н, т.е. качество ИФК не достигает требуемых значений.
Пример 5.
Опыт проводят в условиях примера 1 с той лишь разницей, что соотношение компонентов катализатора изменили. Вместо Mn:Со=2:1 приняли Mn:Со=1:2 при сохранении общей концентрации металлов.
Результат: [м-КБА]<0,001%, цветность 9. Сгорание СН3СООН=51 кг/т ИФК. В приведенных условиях качество ИФК соответствует требованиям высокочистого мономера. Сгорание СН3СООН увеличилось на 10,5 кг/т ИФК.
Пример 6 (сравнительный).
Опыт проводят в условиях примера 5 с той лишь разницей, что из состава катализатора исключают Zn и Ni.
Результат: [м-КБА]=0,0015%, цветность 11°Н. Сгорание СН3СООН=50,9 кг/т ИФК. Исключение модифицирующих добавок (Zn и Ni) приводит к ухудшению показателей цветности.
Пример 7 (сравнительный).
Опыт проводят в условиях примера 3 с той лишь разницей, что суммарную концентрацию Mn-Co-Zn-Ni-Br катализатора снизили на 30%.
Результат: [м-КБА]=0,0045%, цветность 12°Н, т.е. снижение суммарной концентрации катализатора не обеспечивает достижения требуемого качества конечного продукта.
Пример 8 (сравнительный).
Опыт проводят в условиях примера 3 с той лишь разницей, что соотношение Mn:Со увеличили с 1:0,5 до 1:0,35. Результат: [м-КБА]=0,0045%, цветность 11°Н. При увеличении соотношения Mn:Со≥1:0,35 качество продукта ухудшается и не достигает требуемых показателей.
Пример 9 (сравнительный).
Опыт проводят в условиях примера 3 с той лишь разницей, что время смешения реагентов увеличили до 30 сек.
Результат: [м-КБА]=0,0029%, цветность 16°Н.
Пример 10 (прототип).
Опыт проводят в условиях прототипа (патент RU 2047594).
Результат: [[м-КБА]=0,003%, цветность 10°Н, сгорание=82 кг/т ИФК.
В условиях прототипа сгорание СН3СООН возросло в ˜ 2 раза, качественные показатели - на пределе граничных значений. В ИФК присутствует 0,006% СН3СООН.
Преимуществом заявленного изобретения по сравнению с прототипом является упрощение процесса, позволяющего исключить из технологической схемы после 2-ой ступени окисления стадию кристаллизации с выделением сырой ИФК и малоэффективную стадию экстрагирования примесей в растворе СН3СООН при температуре до 250°С с последующим доокислением на 3-ей ступени.
Кроме того, заявляемый процесс обеспечивает получение ИФК более высокого качества, а именно:
- очистка ИФК от СН3СООН обеспечивает полное удаление этой нежелательной примеси (в прототипе удаление СН3СООН из кристаллической ИФК не предусматривается и остаточное содержание уксусной кислоты 0,1-0,01%);
- обеспечивается снижение содержания основной примеси - м-КБА с 25 ppm до 10 ppm и ниже (примеры 1, 2, 5);
- улучшается показатель качества ИФК по цветности с 10°Н до 5-7°Н (примеры 1, 2, 3).
°С
°Н
°Н
°С
°Н
°Н
°Н
Источники литературы
1. Патент США №5110984, 1992.
2. Заявка РФ №95117934, 1997.
3. Патент США №3708532, 1975.
4. Авт. свид. СССР №313829, 1971.
5. Патент РФ №1031120, 1980.
6. Патент РФ №117452, 1983.
7. Патент Японии №454149, 1992.
8. Патент РФ №2047594, 1995.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ИЗОФТАЛЕВОЙ КИСЛОТЫ | 1998 |
|
RU2163592C2 |
| НЕПРЕРЫВНЫЙ СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОЧИСТОЙ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ | 2003 |
|
RU2254324C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЧИСТОЙ ИЗОФТАЛЕВОЙ КИСЛОТЫ И СОПУТСТВУЮЩИХ ПРОДУКТОВ ИЗ КСИЛОЛЬНЫХ ФРАКЦИЙ | 2009 |
|
RU2430911C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЧИСТОЙ ИЗОФТАЛЕВОЙ КИСЛОТЫ И СОПУТСТВУЮЩИХ ПРОДУКТОВ ИЗ ИЗОМЕРОВ ЦИМОЛА И ДИИЗОПРОПИЛБЕНЗОЛА | 2009 |
|
RU2415836C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОЧИСТОЙ ИЗОФТАЛЕВОЙ КИСЛОТЫ | 1997 |
|
RU2137753C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОФТАЛЕВОЙ И МУРАВЬИНОЙ КИСЛОТ ОКИСЛЕНИЕМ м-ДИИЗОПРОПИЛБЕНЗОЛА И м-ЭТИЛ-ИЗОПРОПИЛБЕНЗОЛА | 2011 |
|
RU2485091C2 |
| СПОСОБ ПОЛУЧЕНИЯ И ОЧИСТКИ ИЗОФТАЛЕВОЙ КИСЛОТЫ | 2010 |
|
RU2458042C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВНУТРИМОЛЕКУЛЯРНЫХ АНГИДРИДОВ БЕНЗОЛПОЛИКАРБОНОВЫХ КИСЛОТ | 2009 |
|
RU2412178C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВНУТРИМОЛЕКУЛЯРНОГО АНГИДРИДА ТРИМЕЛЛИТОВОЙ КИСЛОТЫ | 2003 |
|
RU2266276C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОМЕРОВ БЕНЗОЛДИКАРБОНОВЫХ КИСЛОТ С ВЫСОКОЙ СТЕПЕНЬЮ ОЧИСТКИ | 1993 |
|
RU2047594C1 |
Настоящее изобретение относится к способу получения чистой изофталевой кислоты путем ступенчатого окисления м-ксилола кислородсодержащим газом в среде уксусной кислоты в присутствии катализатора, включающего соли тяжелых металлов и галоидных соединений при повышенных давлении и температуре до определенной степени конверсии м-ксилола в изофталевую кислоту с последующим выделением целевого продукта. При этомокисление м-ксилола проводят в три ступени при дискретном изменении (колебании) температуры по ступеням в сторону понижения с последующим повышением или повышения с последующим понижением по схеме Т1 >Т2 <Т3 или Т1 <Т2 >Т3 в температурном интервале 180-200 0С в присутствии марганец-кобальт-бромидного катализатора, модифицированного добавками солей цинка и/или никеля в соотношении металлов Mn:Co:Zn:Ni = 1:(0,5-2):(0,005-0,01):(0,005-0,01) при суммарной концентрации металлов 490 ppm в реакционной массе, эквимолярном по отношению к металлам количестве брома и при времени смешения вводимых в зону реакции реагентов < 10 сек, после чего оксидат с 3й ступени подвергают охлаждению, выделяют кристаллическую ИФК и последовательно обрабатывают (промывают) уксусной кислотой при температуре 80-100 0С в массовом соотношении ИФК:СН3СООН=1:(2-2,5) для удаления катализатора, затем водой при повышенной температуре 150-230 0С в соотношении ИФК:Н2О=1:(2-3) для удаления уксусной кислоты, после чего промытый продукт выделяют и сушат известными приемами с получением высокочистой ИФК. Способ позволяет упростить процесс и улучшить качество ИФК. 2 табл., 1 ил.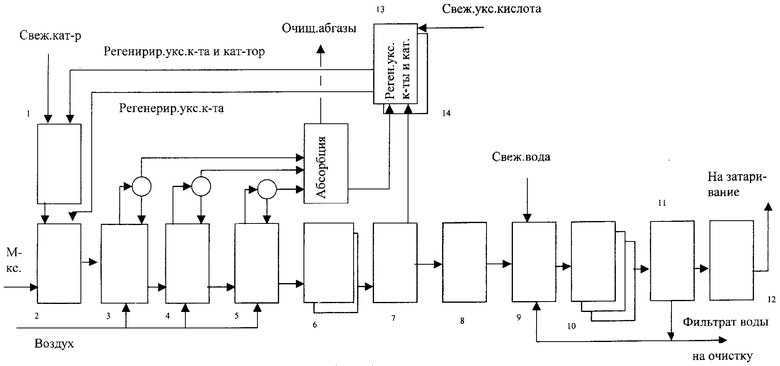
Способ получения чистой изофталевой кислоты путем ступенчатого окисления м-ксилола кислородсодержащим газом в среде уксусной кислоты в присутствии катализатора, включающего соли тяжелых металлов и галоидных соединений при повышенных давлении и температуре до определенной степени конверсии м-ксилола в изофталевую кислоту с последующим выделением целевого продукта, отличающийся тем, что окисление м-ксилола проводят в три ступени при дискретном изменении (колебании) температуры по ступеням в сторону понижения с последующим повышением или повышения с последующим понижением по схеме Т1 >Т2 <Т3 или Т1 <Т2 >Т3 в температурном интервале 180-2000С в присутствии марганец-кобальт-бромидного катализатора, модифицированного добавками солей цинка и/или никеля в соотношении металлов Mn:Co:Zn:Ni = 1:(0,5-2):(0,005-0,01):(0,005-0,01) при суммарной концентрации металлов 490 ppm в реакционной массе, эквимолярном по отношению к металлам количестве брома и при времени смешения вводимых в зону реакции реагентов < 10 с, после чего оксидат с 3-ей ступени подвергают охлаждению, выделяют кристаллическую ИФК и последовательно обрабатывают (промывают) уксусной кислотой при температуре 80-1000С в массовом соотношении ИФК : СН3СООН=1:(2÷2,5) для удаления катализатора, затем водой при повышенной температуре 150-2300С в соотношении ИФК : Н2О=1:(2÷3) для удаления уксусной кислоты, после чего промытый продукт выделяют и сушат известными приемами с получением высокочистой ИФК.
| СПОСОБ ПОЛУЧЕНИЯ ИЗОМЕРОВ БЕНЗОЛДИКАРБОНОВЫХ КИСЛОТ С ВЫСОКОЙ СТЕПЕНЬЮ ОЧИСТКИ | 1993 |
|
RU2047594C1 |

