ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА СМЕЖНЫЕ ЗАЯВКИ
Эта заявка претендует на приоритет временного заявочного серийного № 60/606619, зарегистрированного 2 сентября 2004 года, и № 60/631345, зарегистрированного 29 ноября 2004 года, описания которых включены в этот документ в виде ссылки на их сущность в такой степени, которая не противоречит нижеприведенным заявлениям.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Это изобретение вообще относится к процессу для жидкофазного каталитического окисления ароматического соединения. Один аспект изобретения касается частичного окисления диалкилового ароматического соединения (например, параксилола) для получения неочищенной ароматической дикарбоновой кислоты (например, неочищенной терефталевой кислоты), которая может затем подвергаться очистке и разделению. Другой аспект изобретения касается усовершенствованного реактора в виде барботажной колонны, которая обеспечивает более эффективный и экономичный процесс жидкофазного окисления.
ТЕХНИЧЕСКИЙ УРОВЕНЬ ИЗОБРЕТЕНИЯ
Реакции жидкофазного окисления используются в самых различных коммерческих процессах. Например, жидкофазное окисление в настоящее время используется для окисления альдегидов до кислот (например, пропиональдегида до пропионовой кислоты), окисление циклогексана до адипиновой кислоты и окисление алкиловых ароматических соединений до спиртов, кислот или дикарбоновых кислот. Особенно важным коммерческим процессом окисления в последней категории (окисление алкиловых ароматических соединений) является жидкофазное каталитическое частичное окисление параксилола до терефталевой кислоты. Терефталевая кислота является важным соединением, применяемым в самых различных областях. Терефталевая кислота главным образом используется в качестве сырья для получения полиэтилентерефталата (ПЭТ). ПЭТ является хорошо известной пластмассой, используемой в больших количествах во всем мире для изготовления таких продуктов как бутылки, волокна и упаковочные материалы.
В типовом процессе жидкофазного окисления, включающем в себя частичное окисление параксилола до терефталевой кислоты, поток жидкофазного сырья и поток газообразного окислителя вводятся в реактор и образуют в реакторе многофазную реакционную среду. Поток жидкофазного сырья, введенный в реактор, содержит по меньшей мере одно окисляемое органическое соединение (например, параксилол), а поток газообразного окислителя содержит молекулярный кислород. По меньшей мере часть молекулярного кислорода, введенная в реактор в виде газа, растворяется в жидкофазной реакционной среде, обеспечивая тем самым доступность кислорода для реакции в жидкой фазе. Если жидкая фаза многофазной реакционной среды содержит молекулярный кислород в недостаточной концентрации (т.е., если часть реакционной среды испытывают «кислородное голодание»), то нежелательные побочные реакции могут давать примеси и/или заданные реакции могут протекать более медленно. Если жидкая фаза реакционной среды содержит слишком мало окисляемого соединения, то скорость реакции может быть медленной, что нежелательно. Кроме того, если жидкая фаза реакционной среды содержит избыточную концентрацию окисляемого соединения, то побочные реакции могут образовывать нежелательные дополнительные примеси.
Обычные реакторы для жидкофазного окисления снабжены средствами перемешивания для размешивания многофазной реакционной среды, содержащейся в них. Перемешивание реакционной среды предназначено для улучшения растворения молекулярного кислорода в жидкой фазе реакционной среды, поддержания сравнительно равномерной концентрации растворенного кислорода в жидкой фазе реакционной среды и для поддержания сравнительно равномерной концентрации окисляемого органического соединения в жидкой фазе реакционной среды.
Перемешивание реакционной среды, подвергаемой жидкофазному окислению, часто обеспечивается механическими средствами перемешивания в таких резервуарах, как, например, у непрерывно действующих с мешалкой реакторов (CSTR). Хотя CSTR обеспечивают тщательное перемешивание реакционной среды, но они имеют ряд недостатков. Например, CSTR имеют сравнительно высокую стоимость из-за того, что для них требуются дорогие моторы, герметизированные подшипники и приводные оси и/или сложные механизмы для перемешивания. Кроме того, вращающиеся и/или вибрирующие механические части CSTR требуют регулярного технического обслуживания. Время, затраченное на обслуживание и отключение в связи с техническим обслуживанием, увеличивает стоимость эксплуатации CSTR. Однако даже при регулярном техническом обслуживании системы механического перемешивания, применяемые в CSTR, склонны к механическим отказам и могут требовать замены через сравнительно короткое время.
Реакторы типа барботажных колонн (барботажные реакторы колонного типа) являются привлекательной альтернативой CSTR и другим реакторам окисления с механическим перемешиванием. Барботажные реакторы колонного типа обеспечивают перемешивание реакционной среды без использования дорогого и ненадежного механического оборудования. Барботажные реакторы колонного типа обычно включают в себя удлиненную вертикальную реакционную зону, внутри которой содержится реакционная среда. Перемешивание реакционной среды в реакционной зоне обеспечивается преимущественно за счет естественного всплывания газовых пузырьков, поднимающихся через жидкую фазу реакционной среды. Это перемешивание за счет естественного всплывания пузырьков, обеспеченное в барботажных реакторах колонного типа, уменьшает общие затраты и затраты на обслуживание по сравнению с соответствующими затратами для реакторов с механическим перемешиванием. Кроме того, по существу отсутствие движущихся механических частей в таких реакторах обеспечивает систему окисления, которая менее склонна к механическим отказам, чем реакторы с механическим перемешиванием.
Когда жидкофазное частичное окисление параксилола проводится в обычном реакторе окисления (в CSTR или в колонне с колпачковыми тарелками), продукт, выводимый из реактора, является обычно суспензией, содержащей неочищенную терефталевую кислоту (НТК) и маточный раствор. НТК содержит сравнительно высокие количества примесей (например, 4-карбоксибензальдегид, паратолуиловую кислоту, флуореноны и другие окрашенные вещества), что делает ее неподходящей в качестве сырья для получения ПЭТ. Таким образом НТК, полученную в обычных реакторах окисления, обычно подвергают процессу очистки, превращающему НТК в очищенную терефталевую кислоту (ОТК), подходящую для получения ПЭТ.
Один типовой процесс очистки для превращения НТК в ОТК включает в себя следующие стадии: (1) замену маточного раствора водой в шламе, содержащем НТК, (2) нагревание водного шлама НТК для растворения НТК в воде, (3) каталитическую гидрогенизацию водного раствора НТК для превращения примесей в более подходящие и/или легко разделяемые соединения, (4) осаждение полученной ОТК из гидрогенизированного раствора посредством многих ступеней кристаллизации, и (5) отделение кристаллизованной ОТК от оставшихся жидкостей. Хотя этот тип общепринятого способа очистки является эффективным, но он может быть очень дорогим. Отдельные факторы, обуславливающие высокую стоимость обычных способов очистки НТК, включают в себя, например, тепловую энергию, необходимую для улучшения растворения НТК в воде, катализатор, требующийся для гидрогенизации, поток водорода, необходимый для гидрогенизации, потери выхода, вызванные гидрогенизацией некоторой части терефталевой кислоты, и несколько емкостей, требующихся для многоступенчатой кристаллизации. Таким образом было бы желательно получить НТК продукт, который мог бы быть очищен без необходимости использования тепла для улучшения растворения в воде, гидрогенизации и/или многоступенчатой кристаллизации.
ЦЕЛИ ИЗОБРЕТЕНИЯ
Поэтому целью настоящего изобретения является обеспечение более эффективных и экономичных реактора и способа для жидкофазного окисления.
Другой целью изобретения является обеспечение более эффективных и экономичных реактора и способа для жидкофазного каталитического частичного окисления параксилола до терефталевой кислоты.
Еще одной целью изобретения является обеспечение барботажного реактора колонного типа, который облегчает улучшение проведения жидкофазных реакций окисления при пониженном образовании примесей.
Еще одной целью изобретения является обеспечение более эффективной и экономичной системы для получения чистой терефталевой кислоты (ЧТК) жидкофазным окислением параксилола для получения неочищенной терефталевой кислоты (НТК) и последующей очистки НТК до ЧТК.
Кроме того, целью изобретения является обеспечение барботажного реактора колонного типа для окисления параксилола и получения НТК продукта, который можно очистить без необходимости использования тепла для улучшения растворения НТК в воде, без гидрогенизации растворенной НТК и/или многоступенчатой кристаллизации гидрогенизированной ЧТК.
Следует отметить, что объем настоящего изобретения, как он определен в приложенной формуле изобретения, не ограничен способами и аппаратами, которые могут реализовать все вышеперечисленные цели. Более того, объем заявленного изобретения может охватывать самые разные системы, которые не осуществляют все или какие-либо из вышеперечисленных целей. Дополнительные цели и преимущества настоящего изобретения станут вполне очевидными специалисту в этой области при рассмотрении им следующего подробного описания и соответствующих чертежей.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Одно воплощение настоящего изобретения относится к способу окисления, включающему в себя следующие операции: (a) ввод потока сырья, содержащего параксилол, в реакционную зону барботажного реактора колонного типа, в котором по меньшей мере часть реакционной зоны ограничена одной или больше вертикальными боковыми стенками реактора, в котором по меньшей мере около 25 мас.% параксилола поступает в реакционную зону на один или более участков, отстоящих от боковой стенки по меньшей мере на 0,05D, в котором реакционная зона имеет максимальный диаметр (D); и (b) окисление по меньшей мере части параксилола в жидкой фазе трехфазной реакционной среде, содержащейся в реакционной зоне, в которой образуется в результате окисления неочищенная терефталевая кислота, в которой количество твердых веществ, присутствующих в реакции, поддерживается в диапазоне от около 5 до около 40 мас.%, усредненных по времени и по объему.
Другое воплощение настоящего изобретения относится к способу, включающему в себя следующие стадии: (a) ввод потока сырья, содержащего параксилол, в реакционную зону барботажного реактора колонного типа, в котором по меньшей мере часть реакционной зоны ограничена одной или более вертикальными боковыми стенками реактора, в котором по меньшей мере около 25 мас.% параксилола поступает в реакционную зону на один или более участков, отстоящих от боковой стенки по меньшей мере на 0,05D, в котором реакционная зона имеет максимальный диаметр (D); (b) окисление по меньшей мере части параксилола в жидкой фазе трехфазной реакционной среды, содержащейся в реакционной зоне, для образования тем самым частиц твердой неочищенной терефталевой кислоты в реакционной среде, при этом количество твердых веществ, присутствующих в реакционной среде, поддерживается в диапазоне от около 5 до около 40 мас.%, усредненных по времени и по объему; и (с) окисление по меньшей мере части частиц твердой неочищенной терефталевой кислоты во втором реакторе окисления для получения тем самым более чистой терефталевой кислоты.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Ниже подробно описаны предпочтительные воплощения со ссылками на приложенные чертежи, на которых:
фиг.1 является видом сбоку реактора окисления, сконструированного в соответствии с одним из воплощений настоящего изобретения, в особенности показывающим подачу сырья, окислителя и возвратных потоков в реактор, наличие многофазной реакционной среды в реакторе и отвод газа и суспензии соответственно из верхней и нижней частей реактора;
фиг.2 является увеличенным изображением нижней части барботажного реактора колонного типа на сечении по линии 2-2 на фиг.3, в особенности показывающим расположение и форму распределителя окислителя, используемого для ввода его потока в реактор;
фиг.3 является видом сверху распределителя окислителя по фиг.2, в особенности показывающим отверстия в верхней части распределителя для окислителя;
фиг.4 является видом снизу распределителя на фиг.2, в особенности показывающим отверстия для окислителя в нижней части распределителя для окислителя;
фиг.5 является изображением распределителя на сечении по линии 5-5 на фиг.3, в особенности показывающим ориентацию отверстий для окислителя в верхней и нижней частях распределителя для окислителя;
фиг.6 является увеличенным видом сбоку нижней части барботажного реактора колонного типа, в особенности показывающим систему для ввода потока сырья в реактор во многих вертикально распределенных местах;
фиг.7 является видом сверху нижней части реактора на сечении по линии 7-7 на фиг.6, в особенности показывающим, как система ввода сырья, показанная на фиг.6, распределяет поток сырья в предпочтительную радиальную зону подачи (ЗП) и в более чем одну из равных зон (Q1,Q2,Q3,Q4).
фиг.8 является видом сверху нижней части реактора, аналогичным виду на фиг.7, но показывающим альтернативное средство для подачи потока сырья в реактор с использованием соединительных трубопроводов, каждый из которых имеет большое число небольших подающих отверстий;
фиг.9 является изометрическим изображением альтернативной системы для ввода потока сырья в реакционную зону через многие точки ввода, расположенные вертикально, в особенности показывающим, что система распределения исходных материалов может по меньшей мере частично опираться на распределитель для окислителя;
фиг.10 является видом сбоку системы для распределения исходных материалов и распределителя окислителя, показанных на фиг.9;
фиг.11 является видом сверху на сечении по линии 11-11 на фиг.10, также показывающим систему распределения исходных материалов с одним вводом, опирающуюся на распределитель окислителя;
фиг.12 является изометрическим изображением альтернативного распределителя для окислителя, имеющего все отверстия для окислителя, расположенные в нижней части кольцевого элемента;
фиг.13 является видом сверху альтернативного распределителя окислителя на фиг.12;
фиг.14 является видом снизу альтернативного распределителя окислителя на фиг.12, в особенности показывающим расположение отверстий в нижней части для ввода потока окислителя в реакционную зону;
фиг.15 является видом сбоку сечения по линии 15-15 распределителя окислителя на фиг.13, в особенности показывающим ориентацию нижних отверстий;
фиг.16 является видом сбоку барботажного реактора колонного типа, снабженного внутренней камерой для деаэрации рядом с выходом из нижней части реактора;
фиг.17 является увеличенным видом сбоку нижней части барботажного реактора колонного типа на фиг.16 на сечении по линии 17-17 на фиг.18, в особенности показывающим конфигурацию внутренней камеры для деаэрации, расположенной у выхода из нижней части барботажного реактора колонного типа;
фиг.18 является видом сверху на сечении по линии 18-18 на фиг.16, в особенности показывающим вихревой разбиватель потока, расположенный в камере для деаэрации;
фиг.19 является видом сбоку барботажного реактора колонного типа, снабженного внешней камерой для деаэрации, и показывающим, каким образом часть деаэрированного шлама, выходящая из нижней части камеры для деаэрации, может быть использована для промывки непроизводственной линии трубопроводов, связанной с нижней частью реактора;
фиг.20 является видом сбоку барботажного реактора колонного типа, снабженного гибридной внутренней/внешней камерой деаэрации для отделения газовой фазы от реакционной среды, выводимой из приподнятого бокового расположения в реакторе;
фиг.21 является видом сбоку барботажного реактора колонного типа, снабженного альтернативной гибридной камерой для деаэрации нижней части реактора;
фиг.22 является увеличенным видом сбоку в разрезе нижней части барботажного реактора колонного типа на фиг.21, в особенности показывающим использование альтернативного распределителя окислителя, в котором используются входные штуцера для подачи потока окислителя через кубовую часть реактора;
фиг.23 является увеличенным видом сбоку в разрезе, аналогичным виду на фиг.22, в особенности показывающим альтернативное устройство ввода потока окислителя в реактор через большое число отверстий в кубовой части реактора и необязательно использующее отбойные тарелки для более равномерного распределения потока в реакторе;
фиг.24 является видом сбоку барботажного реактора колонного типа, использующего внутренний трубопровод для вводимого потока, способствующий улучшению дисперсии окисляемого соединения за счет рециркуляции части реакционной среды от верхнего участка реактора до нижнего;
фиг.25 является видом сбоку барботажного колонного реактора, использующего внешний трубопровод для вводимого потока, способствующий улучшению дисперсии окисляемого соединения благодаря рециркуляции части реакционной среды от верхнего участка реактора до нижнего;
фиг.26 является видом сбоку в разрезе горизонтального эжектора, который может быть использован для улучшения дисперсии окисляемого соединения в реакторе окисления, в особенности показывающим эжектор, в котором используется поступающее жидкое сырье для втягивания реакционной среды в эжектор и для подачи с высокой скоростью смеси сырья и реакционной среды в реакционную зону;
фиг.27 является видом сбоку в разрезе вертикального эжектора, который может быть использован для улучшения дисперсии окисляемого соединения в реакторе окисления, в особенности показывающим эжектор, который соединяет жидкое сырье и поступающий газ и использует соединенную двухфазную жидкость для втягивания реакционной среды в эжектор и подачи с высокой скоростью смеси жидкого сырья, поступающего газа и реакционной среды в реакционную зону;
фиг.28 является видом сбоку барботажного реактора колонного типа, содержащего многофазную реакционную среду, особенно показывающим теоретическое разделение реакционной среды на 30 горизонтальных слоев равного объема для количественной оценки перепада некоторых показателей в реакционной среде;
фиг.29 является видом сбоку барботажного реактора колонного типа, содержащего многофазную реакционную среду, особенно показывающим первый и второй дискретные 20%-е сплошные объемы реакционной среды, которые имеют существенно отличающиеся концентрации кислорода и/или скорости его расхода;
фиг.30 является видом сбоку двух расположенных один на другом реакторов с возможным механическим перемешиванием или без него, содержащих многофазную реакционную среду, особенно показывающим, что резервуары, представляющие собой дискретные 20% сплошные объемы реакционной среды, имеющие существенно отличающиеся концентрации кислорода и/или скорости его расходования;
фиг.31 является видом сбоку трех рядом расположенных реакторов с возможным механическим перемешиванием или без него, содержащих многофазную реакционную среду, особенно показывающим, что резервуары представляют собой дискретные 20% сплошные объемы реакционной среды, имеющие существенно отличающиеся концентрации кислорода и/или скорости его расходования;
фиг.32А и 32В являются увеличенными изображениями частиц неочищенной терефталевой кислоты (НТК), полученных в соответствии с одним воплощением настоящего изобретения, особенно показывающими, что каждая частица НТК имеет низкую плотность, при этом частица с высокой площадью поверхности состояла из большого числа свободно связанных субчастиц НТК;
фиг.33А и 33В являются увеличенными изображениями обычно получаемых частиц НТК, особенно показывающими, что частица обычной НТК имеет больший размер, меньшую плотность и меньшую площадь поверхности, чем частицы НТК в соответствии с изобретением на фиг.32А и 32В;
фиг.34 является упрощенной поточной блок-схемой процесса известного уровня техники для получения очищенной терефталевой кислоты (ОТК);
фиг.35 является упрощенной поточной блок-схемой процесса получения ОТК в соответствии с одним воплощением настоящего изобретения; и
фиг.36 является графиком, показывающим, что результаты испытаний твердой фракции, описанные в разделе "Примеры", в частности иллюстрируют, что получение 4-4'-дикарбоксистильбена существенно возрастает, когда используется большее содержание твердой фазы в реакционной среде.
ПОДРОБНОЕ ОПИСАНИЕ
Одно воплощение настоящего изобретения относится к жидкофазному частичному окислению окисляемого соединения. Такое окисление предпочтительно проводится в жидкой фазе многофазной реакционной среды, содержащейся в одном или большем количестве реакторов смешения. Подходящие реакторы смешения включают в себя, например, реакторы с барботажным перемешиванием (например, барботажные реакторы колонного типа), реакторы смешения (например, проточные реакторы смешения) и реакторы, перемешиваемые потоками (например, реакторы со струйным перемешиванием). В одном воплощении изобретения жидкофазное окисление проводится в одном барботажном реакторе колонного типа.
Используемый здесь термин «барботажный реактор колонного типа» означает реактор для облегчения химических реакций в многофазной реакционной среде, в котором перемешивание реакционной среды осуществляется преимущественно движением вверх пузырьков газа через реакционную среду. Используемый здесь термин «перемешивание» означает работу, затраченную в реакционной среде, вызывающую образование потока и/или перемешивание. Используемые здесь термины «большинство», «преимущественно» и «в основном» означает больше 50%. Используемый здесь термин «механическое перемешивание» означает перемешивание реакционной среды, вызванное физическим движением твердого или подвижного элемента (элементов) против реакционной среды или внутри нее. Например, механическое перемешивание может осуществляться вращением, колебанием и/или колебанием внутренних мешалок, лопаток, вибраторов или акустических диафрагм, расположенных в реакционной среде. Используемый здесь термин «перемешивание потоками» означает перемешивание реакционной среды, вызываемое высокоскоростной инжекцией и/или рециркуляцией одного или больше потоков в реакционной среде. Например, перемешивание потоками может обеспечиваться соплами, эжекторами и/или эжекционными устройствами.
В предпочтительном воплощении настоящего изобретения менее около 40% перемешивания реакционной среды в барботажном реакторе колонного типа во время окисления осуществляется механическим и/или перемешиванием потоком, более предпочтительно, чтобы примерно менее 20% перемешивания обеспечивалось механическим и/или перемешиванием потоком, и наиболее предпочтительно, чтобы менее около 5% перемешивания обеспечивалось механическим и/или перемешиванием потоком. Предпочтительно, чтобы величина механического и/или перемешивания потоком, осуществляемая для многофазной реакционной среды во время окисления, была меньше около 3 кВт на кубический метр реакционной среды, более предпочтительно меньше около 2 кВт на кубический метр и наиболее предпочтительно меньше 1 кВт на кубический метр.
Показанный на фиг.1 предпочтительный барботажный реактор колонного типа 20, содержит корпус 22 резервуара, имеющего реакционную секцию 24 и отделенную секцию 26. Реакционная секция 24 ограничивает внутреннюю реакционную зону 28, а отделенная секция 26 ограничивает внутреннюю отделенную зону 30. Преимущественно жидкофазный поток сырья вводится в реакционную зону 28 через входы 32a,b,c,d для сырья. Преимущественно газофазный поток окислителя вводится в реакционную зону 28 через распределитель 34, расположенный в нижней части реакционной зоны 28. Жидкофазный поток сырья и газофазный поток окислителя совместно образуют многофазную реакционную среду 36 в реакционной зоне 28. Многофазная реакционная среда 36 содержит жидкую и газовую фазы. Более предпочтительно, чтобы многофазная реакционная среда 36 представляла собой трехфазную среду, имеющую компоненты в твердой фазе, в жидкой фазе и в газовой фазе. Компонент твердой фазы реакционной среды 36 предпочтительно осаждается в реакционной зоне 28 в результате реакции окисления, проводимой в жидкой фазе реакционной среды 36. Барботажный реактор колонного типа 20 включает в себя выход 38 для шлама, расположенный близко к кубовой части реакционной зоны 28, и выход 40 для газа, расположенный рядом с верхней частью отделенной зоны 30. Выходящий поток шлама, содержащий компоненты жидкой фазы и твердой фазы реакционной среды 36, отводится из реакционной зоны 28 через выход 38 для шлама, а выходящий преимущественно газообразный поток выводится из отделенной зоны 30 через выход 40 для газа.
Поток жидкофазного сырья, вводимый в барботажный реактор колонного типа 20 через входы 32a,b,c,d для сырья, предпочтительно содержит окисляемое соединение, растворитель и каталитическую систему.
Окисляемое соединение, присутствующее в потоке жидкофазного сырья, предпочтительно содержит по меньшей мере одну углеводородную группу. Более предпочтительно, чтобы окисляемое соединение было ароматическим соединением. Еще более предпочтительно, чтобы окисляемое соединение было ароматическим соединением по меньшей мере с одной присоединенной замещенной углеводородной группой или по меньшей мере с одним присоединенным гетероатомом или по меньшей с одной присоединенной функциональной группой карбоновой кислоты (-COOH). Даже еще более предпочтительно, чтобы окисляемое соединение было ароматическим соединением по меньшей мере с одной присоединенной углеводородной группой или по меньшей мере с одной присоединенной замещенной углеводородной группой, при этом каждая присоединенная группа содержит от 1 до 5 атомов углерода. Еще более предпочтительно, чтобы окисляемое соединение было ароматическим соединением, имеющим только две присоединенные группы, при этом каждая присоединенная группа содержит только один атом углерода и состоит из метильных групп и/или замещенных метильных групп и/или максимально одной карбоксильной группы. Даже еще более предпочтительно, чтобы окисляемое соединение было параксилолом, метаксилолом, паратолуиловым альдегидом, метатолуиловым альдегидом, паратолуиловой кислотой, метатолуиловой кислотой и/или ацетальдегидом. Наиболее предпочтительно, чтобы окисляемое соединение было параксилолом.
«Углеводородная группа», как здесь определено, является по меньшей мере одним атомом углерода, который связан только с атомами водорода или с другими атомами углерода. «Замещенная углеводородная группа», как здесь определено, является по меньшей мере одним атомом углерода, связанным по меньшей мере с одним гетероатомом и по меньшей мере с одним атомом водорода. «Гетероатомами», как здесь определено, являются все атомы, кроме атомов углерода и водорода. Ароматические соединения, как здесь определено, содержат ароматическое кольцо, предпочтительно имеющее по меньшей мере 6 атомов углерода, даже более предпочтительно, имеющее только атомы углерода, как часть кольца. Подходящие примеры таких ароматических колец включают, но не ограничиваются только такими, как бензол, бифенил, трифенил и другие конденсированные ароматические кольца на основе углерода.
Подходящие примеры окисляемого соединения включают алифатические углеводороды (например, алканы, разветвленные алканы, циклические алканы, алифатические алкены, разветвленные алкены и циклические алкены); алифатические альдегиды (например, ацетальдегид, пропиональдегид, изомасляный альдегид и n-изомасляный альдегид); алифатические спирты (например, этанол, изопропанол, n-пропанол, n-бутанол и изобутанол); алифатические кетоны (например, диметилкетон, этилметилкетон, диэтилкетон и изопропилметилкетон); алифатические сложные эфиры (например, метилформиат, метилацетат, этилацетат); алифатические перекиси, перкислоты и гидроперекиси (например, трет-бутилгидроперекись, надуксусная кислота и ди-трет-бутилперекись); алифатические соединения с группами, которые являются комбинациями из вышеупомянутых алифатических соединений с другими гетероатомами (например, алифатические соединения, содержащие один или больше молекулярных частей из углеводородов, альдегидов, кетонов, сложных эфиров, перекисей, перкислот и/или гидроперекисей в комбинации с натрием, бромом, кобальтом, марганцем и цирконием); различные бензольные кольца, нафталиновые кольца бифенилы, трифенилы и другие ароматические группы с одной или большим количеством присоединенных углеводородных групп (например, толуол, этилбензол, изопропилбензол, n-пропилбензол, неопентилбензол, параксилол, метаксилол, ортоксилол, все изомеры триметилбензолов, все изомеры тетраметилбензолов, пентаметилбензол, гексаметилбензол, все изомеры этилметилбензолов, все изомеры диэтилбензолов, все изомеры этилметилнафталинов, все изомеры диэтилнаталинов, все изомеры диметилбифенилов, все изомеры этилметилбифенилов и все изомеры диэтилбифенилов, стильбен и с одной или большим количеством присоединенных углеводородных групп, флуорен и с одной или большим количеством присоединенных углеводородных групп, антрацен и с одной или большим количеством присоединенных углеводородными групп, и дифенилэтан и с одной или большим количеством присоединенных углеводородных групп); различные бензольные кольца, нафталиновые кольца, бифенилы, трифенилы и другие ароматические группы с одной или большим количеством присоединенных углеводородных групп и/или с одним или большим количеством присоединенных гетероатомов, которые могут быть соединены с другими атомами или группами атомов (например, фенол, все изомеры метилфенолов, все изомеры диметилфенолов, все изомеры нафтолов, бензилметиловый эфир, все изомеры бромфенолов, бромбензол, все изомеры бромтолуолов, включая альфа-бромтолуол, дибромбензол, нафтенат кобальта и все изомеры бромбифенилов); различные бензольные кольца, нафталиновые кольца, бифенилы, трифенилы и другие ароматические группы с одной или большим количеством присоединенных углеводородных групп и/или с одним или большим количеством присоединенных гетероатомов и/или с одной или большим количеством присоединенных замещенных углеводородных групп (например, бензальдегид, все изомеры бромбензальдегидов, все изомеры бромированных толуиловых альдегидов, включая все изомеры альфа-бромтолуиловых альдегидов, все изомеры гидроксибензальдегидов, все изомеры бромгидроксибензальдегидов, все изомеры бензолдикарбоновых альдегидов, все изомеры бензолтрикарбоновых альдегидов, пара-толуиловый альдегид, мета-толуиловый альдегид, орто-толуиловый альдегид, все изомеры толуолдикарбоновых альдегидов, все изомеры трикарбоновых альдегидов, все изомеры толуолтетракарбоновых альдегидов, все изомеры диметилбензолдикарбоновых альдегидов, все изомеры диметилбензолтрикарбоновых альдегидов, все изомеры диметилбензолтетракарбоновых альдегидов, все изомеры триметилбензолтрикарбоновых альдегидов, все изомеры этилтолуиловых альдегидов, все изомеры триметилбензолдикарбоновых альдегидов, тетраметилбензолдикарбоновый альдегид, оксиметилбензол, все изомеры оксиметилтолуолов, все изомеры оксиметилбромтолуолов, все изомеры оксиметилтолуиловых альдегидов, все изомеры оксиметилбромтолуиловых альдегидов, гидроперекись бензила, гидроперекись бензоила, все изомеры толилметилгидроперекисей и все изомеры метилфенолметилгидроперекисей); различные бензольные кольца, нафталиновые кольца, бифенилы, трифенилы и другие ароматические группы с одной или большим количеством присоединенных выбранных групп, при этом выбранные группы представляют собой углеводородные группы и/или присоединенные гетероатомы, и/или замещенные углеводородные группы, и/или карбоксильные группы, и/или перкислотные группы (например, бензойная кислота, пара-толуиловая кислота, мета-толуиловая кислота, орто-толуиловая кислота, все изомеры этилбензойных кислот, все изомеры пропилбензойных кислот, все изомеры бутилбензойных кислот, все изомеры пентилбензойных кислот, все изомеры диметилбензойных кислот, все изомеры триметилбензойных кислот, все изомеры тетраметилбензойных кислот, пентаметилбензойная кислота, все изомеры диэтилбензойных кислот, все изомеры бензолдикарбоновых кислот, все изомеры бензолтрикарбоновых кислот, все изомеры метилбензолдикарбоновых кислот, все изомеры диметилбензолдикарбоновых кислот, все изомеры метилбензолтрикарбоновых кислот, все изомеры бромбензойных кислот, все изомеры дибромбензойных кислот, все изомеры бромтолуиловых кислот, включая альфа-бромтолуиловые кислоты, толилуксусная кислота, все изомеры оксибензойных кислот, все изомеры оксиметилбензойных кислот, все изомеры окситолуиловых кислот, все изомеры оксиметилтолуиловых кислот, все изомеры оксиметилбензолдикарбоновых кислот, все изомеры оксибромбензойных кислот, все изомеры оксибромтолуиловых кислот, все изомеры оксиметилбромбензойных кислот, все изомеры карбоксибензальдегидов, все изомеры дикарбоксибензальдегидов, пербензойная кислота, все изомеры гидропероксиметилбензойных кислот, все изомеры гидропероксиметилоксибензойных кислот, все изомеры гидропероксикарбонилбензойных кислот, все изомеры гидропероксикарбонилтолуолов, все изомеры метилбифенилкарбоновых кислот, все изомеры диметилбифенилкарбоновых кислот, все изомеры метилбифенилдикарбоновых кислот, все изомеры бифенилтрикарбоновых кислот, все изомеры стильбена с одной или большим количеством присоединенных выбранных групп, все изомеры флуоренона с одной или большим количеством присоединенных выбранных групп, все изомеры нафталина с одной или большим количеством присоединенных выбранных групп, бензил, все изомеры бензила с одной или большим количеством присоединенных выбранных групп, бензофенон, все изомеры бензофенона с одной или большим количеством присоединенных выбранных групп, антрахинон, все изомеры антрахинона с одной или большим количеством присоединенных выбранных групп, все изомеры дифенилэтана с одной или большим количеством присоединенных выбранных групп, бензокумарин и все изомеры бензокумарина с одной или большим количеством присоединенных выбранных групп).
Если окисляемое соединение, присутствующее в потоке жидкофазного сырья, является обычно твердым соединением (т.е. является твердым при стандартных температуре и давлении), то предпочтительно растворять окисляемое соединение в растворителе перед вводом в реакционную зону 28. Предпочтительно, чтобы температура кипения окисляемого соединения при атмосферном давлении была по меньшей мере около 50°С. Более предпочтительно, чтобы температура кипения окисляемого соединения была в диапазоне от около 80 до около 400°С, и наиболее предпочтительно в диапазоне от 125 до 155°С. Количество окисляемого соединения, присутствующего в жидкофазном вводимом сырье предпочтительно в диапазоне от около 2 до около 40 мас.%, более предпочтительно в диапазоне от около 4 до около 20 мас.%, и наиболее предпочтительно в диапазоне от 6 до 15 мас.%.
Следует отметить, что окисляемое соединение, присутствующее в вводимом жидкофазном сырье, может представлять комбинацию двух или более различных окисляемых химических веществ. Эти два или больше различных химических веществ могут быть введены в виде смеси в поток подаваемого жидкофазного сырья или могут вводиться по отдельности в виде нескольких подаваемых потоков сырья. Например, окисляемое соединение, содержащее параксилол, метаксилол, паратолуиловый альдегид, паратолуиловую кислоту и ацетальдегид, может подаваться в реактор через один вход или через несколько отдельных входов.
Растворитель, присутствующий во вводимом потоке сырья, предпочтительно содержит кислотный компонент и воду. Растворитель предпочтительно присутствует в потоке жидкофазного сырья в концентрации от около 60 до около 98 мас.%, более предпочтительно от около 80 до около 96 мас.% и наиболее предпочтительно от 85 до 94 мас.%. Кислотный компонент растворителя является предпочтительно преимущественно органической низкомолекулярновой монокарбоновой кислотой, имеющей 1-6 атомов углерода, более предпочтительно 2 атома углерода. Наиболее предпочтительно, чтобы кислотный компонент растворителя был в основном уксусной кислотой. Предпочтительно, чтобы кислотный компонент составлял по меньшей мере около 75 мас.% растворителя, более предпочтительно по меньшей мере около 80 мас.% растворителя и наиболее предпочтительно от 85 до 98 мас.% растворителя, а остальное является в основном водой. Растворитель, вводимый в барботажный реактор колонного типа 20, может содержать малые количества таких примесей, как, например, паратолуиловый альдегид, терефталевый альдегид, 4-карбоксибензальдегид (4-КБА), бензойная кислота, паратолуиловая кислота, паратолуиловый альдегид, альфа-бромпаратолуиловая кислота, изофталевая кислота, фталевая кислота, тримеллитовая кислота, полиароматические соединения и/или взвешенные дисперсные вещества. Предпочтительно, чтобы общее количество примесей в растворителе, вводимом в барботажный реактор колонного типа 20, было меньше, чем около 3 мас.%.
Каталитическая система, присутствующая в подаваемом потоке жидкофазного сырья, является предпочтительно однородной жидкофазной каталитической системой, которая может ускорить окисление (включая частичное окисление) окисляемого соединения. Более предпочтительно, чтобы каталитическая система содержала по меньшей мере один многовалентный переходный металл. Еще более предпочтительно, чтобы каталитическая система содержала кобальт и бром. Наиболее предпочтительно, чтобы каталитическая система содержала кобальт, бром и марганец.
Когда кобальт присутствует в каталитической системе, то предпочтительно, чтобы его количество, присутствующее в потоке жидкофазного сырья, было таким, при котором концентрация кобальта в жидкой фазе реакционной среды 36 поддерживалась бы в интервале от около 300 до около 6000 мас.ч. на миллион (частей на миллион), более предпочтительно в интервале от около 700 до около 4200 частей на миллион и наиболее предпочтительно в интервале от 1200 до 3000 частей на миллион. Когда бром присутствует в каталитической системе, то предпочтительно, чтобы его количество, присутствующее в потоке жидкофазного сырья, было таким, при котором его концентрация в жидкой фазе реакционной среды 36 поддерживалась бы в интервале от около 300 до около 5000 частей на миллион, более предпочтительно в интервале от около 600 до около 4000 частей на миллион и наиболее предпочтительно в интервале от 900 до 3000 частей на миллион. Когда марганец присутствует в каталитической системе, то предпочтительно, чтобы его количество, присутствующее в потоке жидкофазного сырья, было таким, при котором его концентрация в жидкой фазе реакционной среды 36 поддерживалась бы в интервале от около 20 до около 1000 частей на миллион, более предпочтительно в интервале от около 40 до около 500 частей на миллион, наиболее предпочтительно в интервале от 50 до 200 частей на миллион.
Концентрации кобальта, брома и/или марганца в жидкой фазе реакционной среды 36, приведенные выше, выражается в усредненных по времени и по объему величинах. Используемый здесь термин «усредненный по времени» означает среднюю величину по меньшей мере из 10 измерений, сделанных через равные промежутки времени в течение непрерывного периода по меньшей мере в 100 секунд. Используемый здесь термин «усредненный по объему» означает среднюю величину по меньшей мере из 10 замеров, сделанных в равномерных 3-мерных местах, в некотором объеме.
Отношение массы кобальта к массе брома (Co:Br) в системе катализаторов, вводимых в реакционную зону 28 находится предпочтительно в интервале от около 0,25:1 до около 4:1, более предпочтительно в интервале от около 0,5:1 до около 3:1, и наиболее предпочтительно в интервале от 0,75:1 до 2:1. Отношение массы кобальта к массе марганца (Co:Mn) в системе катализаторов, вводимых в реакционную зону 28, предпочтительно в интервале от около 0,3:1 до около 40:1, более предпочтительно в интервале от около 5:1 до около 30:1, и наиболее предпочтительно в интервале от 10:1 до 25:1.
Поток жидкофазного сырья, вводимый в барботажный реактор колонного типа 20, может включать в себя малые количества таких примесей, как, например, толуол, этилбензол, паратолуиловый альдегид, терефталевый альдегид, 4-карбоксибензальдегид (4-КБА), бензойная кислота, паратолуиловая кислота, паратолуиловый альдегид, альфа-бромпаратолуиловая кислота, изофталевая кислота, фталевая кислота, тримеллитовая кислота, полиароматические соединения и/или взвешенные микрочастицы. Когда используется барботажный реактор колонного типа 20 для получения терефталевой кислоты, метаксилол, и ортоксилол также считаются примесями. Предпочтительно, чтобы общее количество примеси в потоке жидкофазного сырья, вводимом в барботажный реактор колонного типа 20, было меньше примерно 3 мас.%.
Хотя на фиг.1 показано воплощение изобретения, где окисляемое соединение, растворитель и система катализаторов смешиваются и вводятся в барботажный реактор колонного типа 20 в виде одного потока сырья, в альтернативном воплощении настоящего изобретения окисляемое соединение, растворитель и катализатор могут по отдельности вводиться в барботажный реактор колонного типа 20. Например, возможна подача потока чистого параксилола в барботажный реактор колонного типа 20 через вход, отделенный от входа (входов) для растворителя и катализатора.
Газофазный поток окислителя, вводимый в барботажный реактор колонного типа 20 через распределитель 34 для окислителя, содержит преимущественно молекулярный кислород (О2). Предпочтительно, чтобы поток окислителя содержал молекулярный кислород в интервале от около 5 до около 40% мольных, более предпочтительно в интервале от около 15 до около 30% мольных, и наиболее предпочтительно в интервале от 18 до 24% мольных. Предпочтительно, чтобы остальным газом или газами в окисляющем потоке был преимущественно газ или газы, такой как азот, являющийся инертным при окислении. Более предпочтительно, чтобы поток окислителя состоял по существу из молекулярного кислорода и азота. Наиболее предпочтительно, чтобы поток окислителя был сухим воздухом, состоящим из 21% мольных молекулярного кислорода и из примерно 78-81% мольных азота. В альтернативном воплощении настоящего изобретения поток окислителя может содержать по существу чистый кислород.
Как снова показано на фиг.1, барботажный реактор колонного типа 20 предпочтительно снабжен обратным распределителем 42, расположенным выше уровня верхней поверхности 44 реакционной среды 36. Обратный распределитель 42 предназначен для ввода капель преимущественно жидкофазного обратного потока в отделенную зону 30 посредством любого средства для образования капель, известного в этой технике. Более предпочтительно, чтобы обратный распределитель образовывал брызги капель, направленные вниз к уровню верхней поверхности 44 реакционной среды 36. Предпочтительно, чтобы эти направленные вниз брызги из капель воздействовали (т.е. касались и влияли) по меньшей мере примерно на 50% максимальной горизонтальной площади сечения отделенной зоны 30. Более предпочтительно, чтобы брызги из капель воздействовали по меньшей мере на 75% максимальной горизонтальной площади сечения отделенной зоны 30. Наиболее предпочтительно, чтобы брызги из капель воздействовали по меньшей мере на 90% максимальной горизонтальной площади сечения отделенной зоны 30. Эти направленные вниз жидкие обратные брызги могут способствовать предотвращению вспенивания на уровне верхней поверхности 44 реакционной среды 36 или выше этого уровня поверхности и могут также способствовать отсоединению капель любой жидкости или шлама, унесенных восходящим газом, который направляется к выходу 40 для газа. Кроме того, обратный поток жидкости может служить для уменьшения количества (микро)частиц и возможно осаждающихся соединений (например, растворенной бензойной кислоты, паратолуиловой кислоты, 4-КБА, терефталевой кислоты и солей металлического катализатора), выходящих с газообразным потоком, отводимым от отделенной зоны 30 через выход 40 для газа. Помимо этого ввод обратных капель в отделенную зону 30 посредством дистилляции может использоваться для корректировки состава удаляемого газа, отводимого через выход 40.
Жидкий обратный поток, вводимый в барботажный реактор колонного типа 20 через обратный распределитель 42, предпочтительно имеет примерно такой же состав, как и растворитель для жидкофазного потока сырья, вводимого в барботажный реактор колонного типа 20 через входы 32a,b,c,d для сырья. Таким образом предпочтительно, чтобы жидкий обратный поток содержал кислотный компонент и воду. Кислотный компонент обратного потока предпочтительно является низкомолекулярной органической монокарбоновой кислотой, имеющей 1-6 атомов углерода, более предпочтительно имеющей 2 атома углерода. Наиболее предпочтительно, чтобы кислотный компонент обратного потока был уксусной кислотой. Предпочтительно, чтобы кислотный компонент состоял по меньшей мере на около 75 мас.% из обратного потока, более предпочтительно по меньшей мере на около 80 мас.% из обратного потока, и наиболее предпочтительно от 85 до 98 мас.% из обратного потока, а остальное вода. Так как обратный поток обычно имеет такой же состав, как и растворитель в жидкофазном потоке сырья, то когда это описание относится ко «всему растворителю», введенному в реактор, такой «весь растворитель» должен включать как обратный поток, так и часть растворителя во входном потоке сырья.
Во время жидкофазного окисления в барботажном реакторе колонного типа 20 предпочтительно, чтобы потоки сырья, окислителя и обратный поток по существу непрерывно вводились в реакционную зону 28, а выходящие потоки газа и шлама по существу непрерывно отводились из реакционной зоны 28. Используемый здесь термин «по существу непрерывно» означает промежуток времени по меньшей мере 10 часов с перерывом менее 10 минут. Во время окисления предпочтительно, чтобы окисляемое соединение (например, параксилол) по существу непрерывно вводилось в реакционную зону 28 со скоростью по меньшей мере около 8000 килограммов в час, более предпочтительно со скоростью в интервале от около 13000 до около 80000 килограммов в час, еще более предпочтительно в интервале от около 18000 до около 50000 килограммов в час, и наиболее предпочтительно в интервале от 22000 до 30000 килограммов в час. Хотя обычно предпочтительно, чтобы скорости вводимых потоков сырья, окислителя и обратного потока были по существу постоянны, но следует отметить, что в одном воплощении настоящего изобретение предусмотрен импульсный ввод потоков сырья, окислителя и обратного потока для улучшения перемешивания и массопередачи. Когда подаваемые потоки сырья, окислителя и обратный поток вводятся импульсно, то предпочтительно, чтобы их скорости потоков изменялись в пределах от около 0 до около 500% от скоростей потоков в установившемся режиме, указанных здесь, более предпочтительно в пределах от около 30 до около 200% от скоростей потоков в установившемся режиме, указанных здесь, и наиболее предпочтительно в пределах от 80 до 120% от скоростей потоков в установившемся режиме, указанных здесь.
Средняя объемно-временная скорость реакции (ОВС) в барботажном реакторе колонного типа 20 окисления определяется как масса подаваемого окисляемого соединения, приходящаяся на единицу объема реакционной среды 36, в единицу времени (например, подаваемые килограммы параксилола, приходящиеся на кубический метр, в час. При обычном использовании количество окисляемого соединения, не превращенного в продукт, обычно будет вычитаться из количества окисляемого соединения в потоке сырья перед расчетом ОВС. Однако превращения и выходы являются обычно высокими для многих окисляемых соединений, предпочитаемых здесь (например, для параксилола), и удобно здесь определять этот термин так, как это сделано выше. По причинам капитальных и эксплуатационных затрат среди прочего обычно предпочтительно, чтобы реакция проводилась с высокими ОВС. Однако проведение реакции при постоянно возрастающей ОВС может повлиять на качество или выход при частичном окислении. Барботажный реактор колонного типа 20 особенно полезен, когда ОВС окисляемого соединения (например, параксилола) находится в интервале от около 25 до около 400 килограммов на кубический метр в час, более предпочтительно в интервале от около 30 до около 250 килограммов на кубический метр в час, еще более предпочтительно в интервале от около 35 до около 150 килограммов на кубический метр в час, и наиболее предпочтительно в интервале от 40 до 100 килограммов на кубический метр в час.
Кислородная ОВС в барботажном реакторе колонного типа 20 окисления определяется как масса молекулярного кислорода, расходуемая на единицу объема реакционной среды 36 в единицу времени (например, килограммов молекулярного кислорода, расходуемых на кубический метр в час). По причинам капитальных затрат и расхода растворителя при окислении среди прочих обычно предпочтительно, чтобы реакция проводилась с высокой кислородной ОВС. Однако проведение реакции при постоянно возрастающей кислородной ОВС в конце концов приводит к снижению качества или выхода при частичном окислении. Не прибегая к теории, это, по-видимому, связано со скоростью переноса молекулярного кислорода из газовой фазы в жидкую на поверхности раздела между ними и затем в объем жидкости. Слишком высокая кислородная ОВС возможно приводит к слишком низкому содержанию растворенного кислорода в объеме жидкой фазе реакционной среды.
Общая средняя кислородная ОВС определяется здесь как масса всего кислорода, расходуемая во всем объеме реакционной среды 36 в единицу времени (например, килограммов молекулярного кислорода, расходуемых на кубический метр в час). Барботажный реактор колонного типа 20 особенно полезен, когда общая средняя кислородная ОВС находится в интервале от около 25 до около 400 килограммов на кубический метр в час, более предпочтительно в интервале от около 30 до около 250 килограммов на кубический метр в час, еще более предпочтительно от около 35 до около 150 килограммов на кубический метр в час и наиболее предпочтительно от 40 до 100 килограммов на кубический метр в час.
Во время окисления в барботажном реакторе колонного типа 20 предпочтительно, чтобы отношение скорости потока массы всего растворителя (как в сырьевом потоке, так и обратном потоке) к скорости потока массы окисляемого соединения, поступающего в реакционную зону 28, поддерживалось в интервале от около 2:1 до около 50:1, более предпочтительно в интервале от около 5:1 до около 40:1 и наиболее предпочтительно в интервале от 7,5:1 до 25:1. Предпочтительно, чтобы отношение скорости потока массы растворителя, вводимого, в виде части потока сырья, к скорости потока массы растворителя, вводимого, в виде части обратного потока, поддерживалось в интервале от около 0,5:1 до полного отсутствия обратного потока, более предпочтительно в интервале от около 0,5:1 до около 4:1, еще более предпочтительно в интервале от около 1:1 до около 2:1 и наиболее предпочтительно в интервале от 1,25 до 1,5:1.
Во время жидкофазного окисления в барботажном реакторе колонного типа 20 предпочтительно, чтобы поток окислителя, вводимый в барботажный реактор колонного типа 20, был в количестве, которое обеспечивало бы некоторое превышение содержания кислорода над его стехиометрическим содержанием в молекулярном кислороде. Количество избыточного молекулярного кислорода, требуемое для наилучших результатов для конкретного окисляемого соединения, влияет на общую экономику жидкофазного окисления. Во время жидкофазного окисления в барботажном реакторе колонного типа 20 предпочтительно, чтобы отношение скорости потока массы окислителя к скорости потока массы окисляемого органического соединения (например, параксилола), поступающих в реактор 20, поддерживалось в интервале от около 0,5:1 до около 20:1, более предпочтительно в интервале от около 1:1 до около 10:1 и наиболее предпочтительно в интервале от 2:1 до 6:1.
Как снова видно на фиг.1, потоки сырья, окислителя и обратный поток, вводимые совместно в барботажный реактор колонного типа 20, образуют по меньшей мере часть многофазной реакционной среды 36. Реакционная среда 36 является предпочтительно трехфазной средой, содержащей твердую, жидкую и газообразную фазы. Как упомянуто выше, окисление окисляемого соединения (например, параксилола) происходит преимущественно в жидкой фазе реакционной среды 36. Таким образом жидкая фаза реакционной среды 36 содержит растворенный кислород и окисляемое соединение. Экзотермический характер реакции окисления, которая происходит в барботажном реакторе колонного типа 20, вызывает кипение/испарение части растворителя (например, уксусной кислоты и воды), введенного через входы 32a,b,c,d. Таким образом газообразная фаза реакционной среды 36 в реакторе 20 образуется преимущественно из испаряющегося растворителя и нерастворенной непрореагировавшей части потока окислителя. В некоторых реакторах окисления известного уровня техники используются теплообменные трубки/радиаторы для нагревания или охлаждения реакционной среды. Однако такие теплообменные структуры могут быть нежелательны в описываемых здесь реакторе и способе в соответствии с настоящим изобретением. Таким образом предпочтительно, чтобы барботажный реактор колонного типа 20 не включал в себя по существу поверхностей, контактирующих с реакционной средой 36, и не имел усредненный по времени тепловой поток, превышающий 30000 Вт на квадратный метр.
Концентрация растворенного кислорода в жидкой фазе реакционной среды 36 находится в динамическом равновесии между скоростью массопередачи от газовой фазы и скоростью его расходования в жидкофазной реакции (т.е. не задается просто парциальным давлением молекулярного кислорода в подаваемой газовой фазе, хотя это является одним фактором в скорости подачи растворенного кислорода, и он все же влияет на предельное верхнее значение концентрации растворенного кислорода). Количество растворенного кислорода локально изменяется, являясь более высоким у поверхностей раздела пузырьков. Вообще количество растворенного кислорода зависит от соотношения между факторами накопления и расходования в различных областях реакционной среды 36. Во времени количество растворенного кислорода зависит от равномерности перемешивания газа и жидкости, имеющей отношение к скоростям расхода химических веществ. При расчете подходящего соответствия между накоплением и расходованием растворенного кислорода в жидкой фазе реакционной среды 36, предпочтительно, чтобы усредненная по времени и по объемам концентрация кислорода в жидкой фазе реакционной среды 36 поддерживалась выше приблизительно 1 частей на миллион мольных, более предпочтительно в интервале от около 4 до около 1000 частей на миллион мольных, еще более предпочтительно в интервале от около 8 до около 500 частей на миллион мольных и наиболее предпочтительно в интервале от 12 до 120 частей на миллион мольных.
Реакция жидкофазного окисления, проводимая в барботажном реакторе колонного типа 20, предпочтительно является реакцией осаждения с образованием твердых веществ. Более предпочтительно, чтобы жидкофазное окисление, проводимое в барботажном реакторе колонного типа 20, вводимых в реакционную зону 28 по меньшей мере около 10 мас.% окисляемого соединения, вело к образованию твердого соединения (например, частиц неочищенной терефталевой кислоты) в реакционной среде 36. Еще более предпочтительно, чтобы жидкофазное окисление по меньшей мере около 50 мас.% окисляемого соединения вело к образованию твердого соединения в реакционной среде 36. Наиболее предпочтительно, чтобы жидкофазное окисление по меньшей мере 90 мас.% окисляемого соединения вело к образованию твердого соединения в реакционной среде 36. Предпочтительно, чтобы общее количество твердых веществ в реакционной среде 36 было приблизительно больше 3 мас.% усредненных по времени и по объемам. Более предпочтительно, чтобы общее количество твердых веществ в реакционной среде 36 поддерживалось в интервале от около 5 до около 40 мас.%, еще более предпочтительно в интервале от около 10 до около 35 мас.%, и наиболее предпочтительно в интервале от 15 до 30 мас.%. Предпочтительно, чтобы значительная часть продукта окисления (например, терефталевой кислоты), получаемого в барботажном реакторе колонного типа 20, присутствовала в реакционной среде 36, в виде твердого вещества в противоположность оставшейся части, растворенной в жидкой фазе реакционной среды 36. Количество твердого продукта окисления, присутствующего в реакционной среде, предпочтительно составляет по меньшей мере около 25 мас.% от всего продукта окисления (в твердой и жидкой фазах) в реакционной среде 36, более предпочтительно по меньшей мере около 75 мас.% от всего продукта окисления в реакционной среде 36 и наиболее предпочтительно по меньшей мере 95 мас.% от всего продукта окисления в реакционной среде 36. Численные интервалы, приведенные выше для количества твердых веществ в реакционной среде 36, применимы по существу к работе в установившемся режиме барботажной колонны 20 в течение значительного непрерывного промежутка времени, но не при пуске, остановке или не установившейся работе барботажного реактора колонного типа 20. Количество твердых веществ в реакционной среде 36 определяется гравиметрическим способом. В этом гравиметрическом способе представительную часть шлама удаляют из реакционной среды и взвешивают. В условиях, которые эффективно поддерживают общее разделение между твердой и жидкой фазами в реакционной среде, свободная жидкость удаляется из части твердых веществ осаждением или фильтрацией эффективно без потерь осажденных твердых веществ и при этом менее 10% исходной массы жидкости остается с частью твердых веществ. Оставшаяся жидкость на твердых веществах выпаривается до сухости эффективно без сублимации твердых веществ. Отношение массы части твердых веществ к массе исходной части шлама является долей твердых веществ, обычно выражаемой в процентах.
Реакция осаждения, проводимая в барботажном реакторе колонного типа 20, может вызвать загрязнения (т.е. наслаивания твердых веществ) на поверхности некоторых жестких структур, с которыми контактирует реакционная среда 36. Таким образом в одном воплощении настоящего изобретения предпочтительно, чтобы барботажный реактор колонного типа 20 не содержал никаких внутренних теплообменных, перемешивающих структур или внутренних перегородок в реакционной зоне 28 в связи с тем, что такие структуры имеют склонность к загрязнению. Если внутренние структуры присутствуют в реакционной зоне 28, то желательно их избегать, т.к. они имеют наружные поверхности со значительным количеством обращенных вверх плоских участков, которые будут очень сильно загрязняться. Таким образом, если какие-либо внутренние структуры присутствуют в реакционной зоне 28, то предпочтительно, чтобы меньше приблизительно 20% всех обращенных вверх открытых внешних поверхностных участков таких внутренних структур были образованы существенно плоскими поверхностями, имеющими наклон менее 15° по отношению к горизонтали.
Как снова показано на фиг.1, физическая конфигурация барботажного реактора колонного типа 20 помогает обеспечить оптимальное окисление окисляемого соединения (например, параксилола) с минимальным образованием примесей. Предпочтительно, чтобы удлиненная реакционная секция 24 в объеме корпуса 22 включала в себя по существу цилиндрическую основу 46 и нижнюю часть 48. Верх реакционной зоны 28 ограничен горизонтальной плоскостью 50, расположенной поперек верхней части цилиндрической основы 46. Низ 52 реакционной зоны 28 ограничен самой нижней внутренней поверхностью нижней части 48. Обычно низ 52 реакционной зоны 28 расположен рядом с отверстием 38 для выхода шлама. Таким образом удлиненная реакционная зона 28 внутри барботажного реактора колонного типа 20 имеет максимальную длину «L», измеренную от верхнего конца 50 до нижнего конца 52 реакционной зоны 28 вдоль продольной оси цилиндрической основы 46. Длина L реакционной зоны 28 предпочтительно находится в интервале от около 10 до около 100 метров, более предпочтительно в интервале от около 20 до около 75 метров и наиболее предпочтительно в интервале от 25 до 50 метров. Реакционная зона 28 имеет максимальный диаметр (ширину) «D», обычно равный максимальному внутреннему диаметру цилиндрического основного тела 46. Максимальный диаметр D реакционной зоны 28 предпочтительно находится в интервале от около 1 до около 12 метров, более предпочтительно в интервале от около 2 до около 10 метров, еще более предпочтительно в интервале от около 3,1 до около 9 метров и наиболее предпочтительно в интервале от 4 до 8 метров. В предпочтительном воплощении настоящего изобретения реакционная зона 28 имеет отношение длины к диаметру «L:D» в интервале от около 6:1 до около 30:1. Еще более предпочтительно, чтобы реакционная зона 28 имела отношение L:D в интервале от около 8:1 до около 20:1. Наиболее предпочтительно, чтобы реакционная зона 28 имела отношение L:D в интервале от 9:1 до 15:1.
Как описано выше, в реакционную зону барботажного реактора колонного типа 20 поступает многофазная реакционная среда 36. Реакционная среда 36 имеет нижний уровень, совпадающий с низом 52 реакционной зоны 28, и верхний уровень, расположенный на верхней поверхности 44. Верхняя поверхность 44 реакционной среды 36 расположена вдоль горизонтальной плоскости, которая проходит через реакционную зону 28, где содержимое реакционной зоны 28 непрерывно переходит из газофазного состояния в жидкофазное сплошное состояние. Верхняя поверхность 44 предпочтительно расположена, где усредненная по локальному времени задержка газа тонким горизонтальным слоем содержимого реакционной зоны 28 составляет 0,9.
Реакционная среда 36 имеет максимальную высоту «Н», измеренную между ее верхним и нижним уровнями. Максимальная ширина «W» реакционной среды 36 обычно равна максимальному диаметру «D» цилиндрической основы 46. Во время жидкофазного окисления в барботажном реакторе колонного типа 20 предпочтительно, чтобы Н поддерживалась в интервале от около 60 до около 120% от L, более предпочтительно от около 80 до около 110% L и наиболее предпочтительно от 85 до 100% L. В предпочтительном воплощении настоящего изобретения реакционная среда 36 имеет отношение высоты к ширине «Н:W» больше приблизительно 3:1. Более предпочтительно, чтобы реакционная среда 36 имела отношение «Н:W» в интервале от около 7:1 до около 25:1. Еще более предпочтительно, чтобы реакционная среда 36 имела отношение «Н:W» в интервале от около 8:1 до около 20:1. Наиболее предпочтительно, чтобы реакционная среда 36 имела отношение «Н:W» в интервале от 9:1 до 15:1. В одном воплощении изобретения L=H и D=W, так что различные размеры или отношения, представленные здесь для L и W, также относятся к H и W, и наоборот.
Относительно высокие отношения L:D и H:W, обеспеченные в соответствии с воплощением изобретения, могут способствовать нескольким важным преимуществам системы по изобретению. Как рассмотрено более подробно ниже, оказалось, что более высокие отношения L:D и H:W, а также некоторые другие отличительные особенности, рассмотренные ниже, могут способствовать полезным вертикальным градиентам концентраций молекулярного кислорода и/или окисляемого соединения (например, параксилола) в реакционной среде 36. Вопреки общепринятым взглядам, которые позитивно относятся к хорошо перемешанной реакционной среде со сравнительно равномерным распределением концентраций, было обнаружено, что вертикальное распределение концентраций кислорода и/или концентраций окисляемого соединения способствует более эффективной и экономичной реакции окисления. Минимизация концентраций кислорода и окисляемого соединения вблизи верхней части реакционной среды 36 может помочь избавиться от таких потерь, как непрореагировавший кислород или непрореагировавшее окисляемое соединение, выводимые через верхний выход 40 для газов. Однако если концентрации окисляемого соединения и непрореагировавшего кислорода являются низкими во всей реакционной среде 36, то скорость и/или избирательность окисления уменьшаются. Таким образом предпочтительно, чтобы концентрации молекулярного кислорода и/или окисляемого соединения были значительно выше в нижней части реакционной среды 36, чем в верхней части реакционной среды 36.
Кроме того, высокие отношения L:D и H:W вызывают давление на нижнюю часть реакционной среды, которое будет значительно выше давления на верхнюю часть реакционной среды 36. Этот градиент давления по высоте является результатом высоты и плотности реакционной среды 36. Одним преимуществом этого градиента давления по высоте является то, что повышенное давление на дно резервуара обеспечивает большую растворимость кислорода и лучшую массопередачу, чем было бы иначе достижимо при сравнительных температурах и верхних давлениях в малого объема реакторах. Таким образом реакцию окисления можно было проводить при более низких температурах, чем потребовались бы в более малого объема резервуарах. Когда используется барботажный реактор колонного типа 20 для частичного окисления параксилола до неочищенной терефталевой кислоты (НТК), то способность работать при более низких температурах для проведения реакции с такими же или лучшими скоростями массопередачи имеет целый ряд преимуществ. Например, низкотемпературное окисление параксилола уменьшает количество растворителя, сжигаемого во время реакции. Как подробно рассмотрено ниже, низкотемпературное окисление также благоприятно влияет на образование малых, с высокой площадью поверхности, свободно связанных легко растворимых частиц НТК, которые можно подвергнуть более экономичным способам очистки, чем большие, с малой площадью поверхности, плотные частицы НТК, полученные в обычных процессах высокотемпературного окисления.
Во время окисления в реакторе 20 предпочтительно, чтобы усредненная по времени и по объемам температура реакционной среды 36 поддерживалась в интервале от около 125 до около 200°С, более предпочтительно в интервале от около 140 до около 180°С, и наиболее предпочтительно в интервале от 150 до 170°С. Давление в верхней части над реакционной средой 36 предпочтительно поддерживается в интервале от около 1 до около 20 бар (избыточного давления), более предпочтительно в интервале от около 2 до около 12 бар и наиболее предпочтительно в интервале от 4 до 8 бар. Предпочтительно, чтобы разность давлений между верхней частью реакционной среды 36 и нижней частью реакционной среды 36 была в интервале от около 0,4 до около 5 бар, более предпочтительно в интервале от около 0,7 до около 3 бар и наиболее предпочтительно в интервале от 1 до 2 бар. Хотя обычно предпочитается, чтобы давление верха над реакционной средой поддерживалось относительно постоянным, в одном воплощении настоящего изобретения предусматривается импульсный режим для давления верха с целью облегчения улучшенного перемешивания и/или массопередачи в реакционной среде 36. Когда давление верха меняется импульсно, предпочтительно, чтобы величина таких пульсирующих давлений была в интервале от около 60 до около 140% давления верха в установившемся режиме, указанного здесь, более предпочтительно от около 85 до около 115% давления верха в установившемся режиме, указанного здесь, и наиболее предпочтительно от 95 до 105% давления верха в установившемся режиме, указанного здесь.
Другим преимуществом высокого отношения L:D реакционной зоны 28 является то, что оно может способствовать увеличению средней поверхностной скорости реакционной среды 36. Термин «поверхностная скорость» или «поверхностная скорость газа», как он здесь используется со ссылкой на реакционную среду 36, означает объемную скорость восходящего потока газовой фазы реакционной среды 36, разделенного горизонтальной площадью поперечного сечения реактора в конкретном месте. Повышенная поверхностная скорость, обеспеченная высоким отношением L:D реакционной зоны 28, может улучшить локальное перемешивание и увеличить удерживание газа реакционной средой 36. Усредненные по времени поверхностные скорости реакционной среды 36 при одной четверти высоты, при половине высоты и/или при трех четвертях высоты реакционной среды 36 предпочтительно больше приблизительно 0,3 м/с, более предпочтительно находятся в интервале от около 0,8 до около 5 м/с, еще более предпочтительно в интервале от около 0,9 до около 4 м/с и наиболее предпочтительно в интервале от 1 до 3 м/с.
Как показано на фиг.1, отделенная секция 26 барботажного реактора колонного типа 20 является просто расширенной частью корпуса 22, расположенной сразу над реакционной секцией 24. Отделенная секция 26 уменьшает скорость восходящей газовой фазы в барботажном реакторе колонного типа 20, когда газовая фаза поднимается над верхнем уровнем поверхности 44 реакционной среды 36 и приближается к выходу 40 для газа. Это уменьшение скорости движения вверх газовой фазы способствует облегчению удаления захваченных жидкостей и/или твердых веществ восходящей газовой фазой и тем самым уменьшает нежелательные потери некоторых компонентов, присутствующих в жидкой фазе реакционной среды 36.
Отделенная секция 26 предпочтительно включает в себя переходную стенку 54 обычно в виде усеченного конуса, обычно цилиндрическую широкую боковую стенку 56 и верх 58. Узкий низ переходной стенки 54 соединен с верхней частью цилиндрической основы 46 реакционной секции 24. Широкий верх переходной стенки 54 соединен с нижней частью широкой боковой стенки 56. Предпочтительно, чтобы переходная стенка 54 была направлена вверх и во внешнюю сторону от ее узкого низа под углом в интервале от около 10 до около 70° от вертикали, более предпочтительно в интервале от около 15 до около 50° от вертикали и наиболее предпочтительно в интервале от 15 до 45° от вертикали. Широкая боковая стенка 56 имеет максимальный диаметр «Х», который обычно больше максимального диаметра «D» реакционной секции 24, хотя, если верхняя часть реакционной секции 24 имеет меньший диаметр, чем максимальный диаметр реакционной секции 24, то Х может действительно быть меньше D. В предпочтительном воплощении настоящего изобретения отношение диаметра широкой боковой стенки 56 к максимальному диаметру реакционной секции 24 «Х:D» находится в интервале от около 0,8:1 до около 4:1, наиболее предпочтительно в интервале от 1,1:1 до 2:1. Верх 58 соединен с верхней частью широкой боковой стенки 56. Верх 58 является предпочтительно вообще эллиптическим, ограничивающий центральное отверстие, позволяющее газу выйти из отдельной зоны 30 через выход 40 для газа. Альтернативно, верх 58 может иметь любую форму, включая коническую. Отделенная зона 30 имеет максимальную высоту «Y», измеренную от верхней части 50 реакционной зоны 28 до самой верхней части отделенной зоны 30. Отношение длины реакционной зоны 28 к высоте отсоединяющей зоны 30 «L:Y» предпочтительно находится в интервале от около 2:1 до около 24:1, более предпочтительно в интервале от около 3:1 до около 20:1 и наиболее предпочтительно в интервале от 4:1 до 16:1.
Теперь со ссылкой на фиг.1-5, будет более подробно рассмотрено расположение и конфигурация распределителя 34 окислителя. На фиг.2 и 3 показано, что распределитель 34 окислителя может включать в себя кольцевой элемент 60, поперечный элемент 62 и два входных трубопровода 64a,b для окислителя. Для удобства эти входные трубопроводы для окислителя могут входить в резервуар при некотором возвышении над кольцевым элементом 60 и затем поворачивать вниз, как показано на фиг.2 и 3. Альтернативно входные трубопроводы 64a,b могут входить в резервуар ниже кольцевого элемента 60 или примерно в той же горизонтальной плоскости, как и кольцевой элемент 60. Каждый входной трубопровод 64a,b включает в себя первый фланец, соединенный с соответствующим входом 66a,b для окислителя, смонтированным в корпусе 22 резервуара, а второй фланец входного трубопровода подвижно соединен с кольцевым элементом 60. Кольцевой элемент 60 предпочтительно образован трубопроводами, более предпочтительно большим числом прямых трубопроводных секций и наиболее предпочтительно большим числом прямых трубчатых секций, жестко соединенных друг с другом для образования трубчатого многоугольника. Предпочтительно, чтобы кольцевой элемент 60 был смонтирован по меньшей мере из 3 прямых трубчатых секций, более предпочтительно из 6-10 трубчатых секций и наиболее предпочтительно из 8 трубчатых секций. Поэтому, когда кольцевой элемент 60 сформирован из 8 трубчатых секций, он имеет вообще восьмиугольную форму. Поперечный элемент 62 предпочтительно сформирован по существу из прямых трубчатых секций, которые подвижно соединены с противоположными трубчатыми секциями кольцевого элемента 60 и расположены по диагонали между ними. Трубчатая секция, используемая для поперечного элемента 62, предпочтительно имеет такой же диаметр, как и у трубчатых секций, используемых для формирования кольцевого элемента 60. Предпочтительно, чтобы трубчатые секции, которые образуют входные трубопроводы 64a,b для окислителя, кольцевой элемент 60 и поперечный элемент 62, имели номинальный диаметр больше приблизительно 0,1 м, более предпочтительно в интервале от около 0,2 до около 2 м и наиболее предпочтительно в интервале от 0,25 до 1 м. Как возможно лучше всего показано на фиг.3, кольцевой элемент 60 и поперечный элемент 62, каждый из них, имеют большое число верхних отверстий 68 для окислителя, выводящих поток окислителя вверх в реакционную зону 28. Как возможно лучше всего показано на фиг.4, кольцевой элемент 60 и поперечный элемент 62 могут иметь одно или больше нижних отверстий 70 для окислителя, выводящих поток окислителя вниз в реакционную зону 28. Нижние отверстия 70 для окислителя могут также использоваться для вывода жидкостей и/или твердых веществ, которые могут проникать в кольцевой элемент 60 и/или в поперечный элемент 62. Чтобы предотвратить осаждение твердых веществ внутри распределителя окислителя 34, поток жидкости может непрерывно или периодически пропускаться через распределитель 34 для вымывания любых накопившихся твердых веществ.
Снова со ссылкой на фиг.1-4, во время окисления в барботажном реакторе колонного типа потоки окислителя принудительно поступают через входы 66a,b соответственно во входные трубопроводы 64a,b для окислителя. Потоки окислителя затем транспортируются по входным трубопроводам 64a,b для окислителя в кольцевой элемент 60. После того как поток окислителя вошел в кольцевой элемент 60, он распределяется по внутренним объемам кольцевого элемента 60 и поперечного элемента 62. Поток окислителя затем принудительно выводится из распределителя окислителя 34 в реакционную зону 28 через верхние и нижние отверстия 68, 70 в кольцевом элементе 60 и поперечном элементе 62.
Выходы верхних отверстий 68 окислителя распределены одно от другого в поперечном направлении и размещены на одинаковом приподнятом уровне в реакционной зоне 28. Таким образом выходы верхних отверстий 68 окислителя обычно расположены в основном вдоль горизонтальной плоскости, определяемой верхней частью распределителя окислителя 34. Выходы нижних отверстий 70 для окислителя распределены одно от другого в поперечном направлении и размещены на одинаковом существенно приподнятом уровне в реакционной зоне 28. Таким образом выходы нижних отверстий 70 для окислителя обычно расположены в основном вдоль горизонтальной плоскости, определяемой нижней частью распределителя окислителя 34.
В одном воплощении настоящего изобретения распределитель окислителя 34 имеет по меньшей мере около 20 верхних отверстий 68. Более предпочтительно распределитель окислителя 34 имеет от около 40 до около 800 верхних отверстий. Наиболее предпочтительно распределитель окислителя 34 имеет от 60 до 400 верхних отверстий 68. Распределитель окислителя 34 предпочтительно имеет по меньшей мере примерно 1 нижнее отверстие 70 окислителя, сформированное в нем. Более предпочтительно распределитель окислителя 34 имеет от около 2 до около 40 нижних отверстий 70. Отношение числа верхних отверстий 68 для окислителя к числу нижних отверстий 70 для окислителя в распределителе окислителя 34 предпочтительно находится в интервале от около 2:1 до около 100:1, более предпочтительно в интервале от около 5:1 до около 25:1 и наиболее предпочтительно в интервале от 8:1 до 15:1. Диаметры по существу всех верхних и нижних отверстий 68, 70 для окислителя предпочтительно по существу одинаковы, поэтому отношения объемных скоростей потока окислителя из верхних и нижних отверстий 68, 70 для окислителя по существу такие же, как и отношения, приведенные выше для сравнения значения верхних и нижних отверстий 68, 70 для окислителя.
На фиг.5 показано направление выхода окислителя из верхних и нижних отверстий 68, 70 для окислителя. Что касается верхних отверстий 68 для окислителя, то предпочтительно, чтобы по меньшей мере часть верхних отверстий 68 для окислителя выводила поток под углом «А» от вертикали. Предпочтительно, чтобы процент верхних отверстий 68 для окислителя, отклоняющихся от вертикали на угол «А», был в интервале от около 30 до около 90%, более предпочтительно в интервале от около 50 до около 80%, еще более предпочтительно в интервале от около 60 до 75% и наиболее предпочтительно около 67%. Угол «А» находится предпочтительно в интервале от около 5 до около 60°, более предпочтительно в интервале от около 10 до около 45° и наиболее предпочтительно в интервале от 15 до 30°. Что касается нижних отверстий 70 для окислителя, то предпочтительно, чтобы существенно все нижние отверстия 70 для окислителя были расположены рядом с самой нижней частью кольцевого элемента 60 и/или поперечного элемента 62. Таким образом любые жидкости и/или твердые вещества, которые могут быть ненамеренно введены в распределитель окислителя 34, могут быть легко выведены из него через нижние отверстия 70 для окислителя. Предпочтительно, чтобы нижние отверстия 70 для окислителя выводили его поток вниз под существенно вертикальным углом. Для целей этого описания верхнее отверстие для окислителя может быть любым отверстием, которое выводит поток окислителя вообще в направлении вверх (т.е. под углом выше горизонтали) и нижнее отверстие для окислителя может быть любым отверстием, которое выводит поток окислителя вообще в направлении вниз (т.е. под углом ниже горизонтали).
Во многих обычных барботажных реакторах колонного типа, содержащих многофазную реакционную среду, по существу вся реакционная среда, расположенная ниже распределителя окислителя (или другого механизма для ввода потока окислителя в реакционную зону), имеет очень низкую величину задержки газа. Как известно в этой технике, «задержка газа» означает просто долю объема многофазной среды, которая находится в газообразном состоянии. Зоны низкой задержки газа в среде можно также называть «неаэрированными зонами». Во многих общеизвестных барботажных реакторах колонного типа для шлама значительная часть общего объема реакционной среды распложена ниже распределителя окислителя (или другого механизма для ввода потока окислителя в реакционную зону). Таким образом, значительная часть реакционной среды, присутствующая в нижней части общеизвестных барботажных реакторов колонного типа, является неаэрированной.
Оказалось, что минимизация неаэрированных зон в реакционной среде, подвергаемой окислению в барботажном реакторе колонного типа, может минимизировать образование некоторых типов нежелательных примесей. Неаэрированные зоны реакционной среды содержат сравнительно мало пузырьков окислителя. Этот малый объем пузырьков окислителя уменьшает количество молекулярного кислорода, доступного для растворения в жидкой фазе реакционной среды. Таким образом, жидкая фаза в неаэрированной зоне реакционной среды имеет сравнительно низкую концентрацию молекулярного кислорода. Эти испытывающие нехватку кислорода неаэрированные зоны реакционной среды имеют склонность ускорять нежелательные побочные реакции вместо требуемой реакции окисления. Например, когда параксилол частично окисляется с образованием терефталевой кислоты, недостаток кислорода в жидкой фазе реакционной среды может вызвать образование нежелательных больших количеств бензойной кислоты и связанных ароматических колец, явно включающих в себя очень нежелательные окрашенные молекулы, известные как флуореноны и антрахиноны.
В соответствии с одним воплощением настоящего изобретения жидкофазное окисление проводится в барботажном реакторе колонного типа с такой конфигурацией и режимом работы, при которых минимизируется объемная доля реакционной среды с низкими величинами задержки газа. Эта минимизация неаэрированных зон может быть определена количественно теоретическим разделением всего объема реакционной среды на 2000 дискретных горизонтальных слоев одинакового объема. При исключении верхнего и нижнего из горизонтальных слоев, каждый из оставшихся горизонтальных слоев является дискретным объемом, ограниченным с боковых сторон боковыми стенками реактора и с верхней и нижней сторон воображаемыми горизонтальными плоскостями. Самый верхний горизонтальный слой ограничен с нижней стороны воображаемой горизонтальной плоскостью и с верхней стороны верхним уровнем поверхности реакционной среды. Самый нижний горизонтальный слой ограничен с его верхней стороны воображаемой горизонтальной плоскостью и с его нижней стороны - нижним краем резервуара. После теоретического разделения реакционной среды на 2000 горизонтальных слоев одинакового объема можно будет определить усредненную по времени и по объемам задержку газа каждого горизонтального слоя. Когда используется этот способ количественного определения величины неаэрированных зон, то предпочтительно, чтобы число горизонтальных слоев, имеющих усредненную по времени и по объемам задержку газа меньше 0,1, было меньше 30, более предпочтительно меньше 15, еще более предпочтительно меньше 6 и даже более предпочтительно меньше 4, и наиболее предпочтительно меньше 2. Предпочтительно, чтобы число горизонтальных слоев, имеющих задержку газа меньше 0,2, было меньше 80, более предпочтительно меньше 40, еще более предпочтительно меньше 20, даже более предпочтительно меньше 12 и наиболее предпочтительно меньше 5. Предпочтительно, чтобы число горизонтальных слоев, имеющих задержку газа меньше 0,3, было меньше 120, более предпочтительно меньше 80, еще более предпочтительно меньше 40, даже более предпочтительно меньше 20 и наиболее предпочтительно меньше 15.
Снова со ссылкой на фиг.1 и 2, было обнаружено, что размещение распределителя окислителя 34 в более низком положении в реакционной зоне 28 обеспечивает несколько преимуществ, включая уменьшение количества неаэрированных зон в реакционной среде 36. При заданных высоте «Н» реакционной среды 36, длине «L» реакционной зоны 28 и максимальном диаметре «D» реакционной зоны 28 предпочтительно, чтобы большая часть (т.е.,>50 мас.%) потока окислителя вводилась в реакционную зону 28 в пределах приблизительно 0,025Н, 0,022L и/или 0,25D от нижнего штуцера 52 реакционной зоны 28. Более предпочтительно, чтобы большая часть потока окислителя вводилась в реакционную зону 28 в пределах около 0,02Н, 0,01L и/или 0,2D от нижнего штуцера 52 реакционной зоны 28. Наиболее предпочтительно, чтобы большая часть потока окислителя вводилась в реакционную зону 28 в пределах около 0,015Н, 0,013L и/или 0,15D от нижнего штуцера 52 реакционной зоны 28.
В воплощении, показанном на фиг.2, расстояние по высоте «Y1» между нижним штуцером 52 реакционной зоны 28 и выходом верхних отверстий 68 в распределителе окислителя 34 меньше приблизительно 0,25Н, 0,022L и/или 0,25D, поэтому по существу весь поток окислителя поступает в реакционную зону 28 в пределах около 0,25Н, 0,022L и/или 0,25D от нижнего штуцера 52 реакционной зоны 28. Более предпочтительно, чтобы Y1 было меньше около 0,02Н, 0,018L и/или 0,2D. Наиболее предпочтительно, чтобы Y1 было меньше 0,015Н, 0,013L и/или 0,15D, но больше 0,005Н, 0,004L и/или 0,06D. На фиг.2 показана касательная линия 72 на участке, где нижний край цилиндрической основы 46 корпуса 22 с резервуаром соединяется с верхним краем эллиптического низа 48 корпуса 22 с резервуаром. Альтернативно низ 48 может быть любой формы, включая коническую, и касательная линия все еще будет определять нижний край цилиндрической основы 46. Вертикальное расстояние «Y2» между касательной линией 72 и верхней частью распределителя окислителя 34 предпочтительно составляет по меньшей мере около 0,0012Н, 0,001L и/или 0,01D; более предпочтительно по меньшей мере около 0,005Н, 0,004L и/или 0,05D, и наиболее предпочтительно по меньшей мере 0,01Н, 0,008L и/или 0,1D. Вертикальное расстояние «Y3» между нижним штуцером 52 реакционной зоны 28 и выходом нижних отверстий 70 для окислителя в распределителе окислителя 34 предпочтительно меньше около 0,015Н, 0,013L и/или 0,15D; более предпочтительно меньше около 0,012Н, 0,01L и/или 0,075D, но больше 0,003Н, 0,002L и/или 0,025D.
В предпочтительном воплощении настоящего изобретения отверстия, выводящие поток окислителя и поток сырья в реакционную зону, имеют такую форму, при которой количество (масса) потока окислителя или потока сырья, выводимое из отверстия, прямо пропорционально открытой площади сечения отверстия. Таким образом, например, если 50% общей открытой площади сечения, определяемой всеми отверстиями для окислителя, размещено в пределах 0,15D нижней части реакционной зоны, то 50 мас.% потока окислителя поступает в реакционную зону в пределах 0,15D нижней части реакционной зоны, и наоборот.
Помимо преимуществ, обеспеченных минимизацией неаэрированных зон (т.е. зон с низкой задержкой газа) в реакционной среде 36, было обнаружено, что окисление может быть ускорено максимизацией задержки газа всей реакционной средой 36. Реакционная среда 36 предпочтительно имеет усредненную по времени и объемам задержку газа по меньшей мере около 0,4, более предпочтительно в интервале от около 0,6 до около 0,9 и наиболее предпочтительно в интервале от 0,65 до 0,85. Несколько физических и рабочих характеристик барботажного реактора колонного типа 20 способствуют высокой задержке газа, приведенной выше. Например, для реактора данного размера и для величины потока окислителя в нем высокое отношение L:D реакционной зоны 28 дает меньший диаметр, который увеличивает поверхностную скорость в реакционной среде 36, что в свою очередь увеличивает задержку газа. Кроме того, действительный диаметр барботажной колонны и отношение L:D, как известно, влияют на среднюю задержку газа даже при заданной постоянной поверхностной скорости. Помимо этого, минимизация неаэрированных зон, особенно в нижней части реакционной зоны 28 способствует увеличению величины задержки газа. Также давление верха и механическая конфигурация барботажного реактора колонного типа могут влиять на стабильность его работы при высоких поверхностных скоростях и величинах задержек газа, рассмотренных здесь.
Кроме того, изобретатели открыли важность работы при оптимизированном давлении верха для получения повышенной задержки газа и повышенной массопередачи. Может казаться, что работа при более низком давлении верха, которое уменьшает растворимость молекулярного кислорода в соответствии с законом Генри, будет снижать скорость массопередачи молекулярного кислорода от газа к жидкости. В резервуаре с механическим перемешиванием, здесь именно такой случай, так как уровни аэрации и скорости массопередачи определяются главным образом типом мешалки и давлением верха. Однако в барботажном реакторе колонного типа в соответствии с предпочтительным воплощением настоящего изобретения было выявлено, как использовать более низкое давление верха для того, чтобы вызвать увеличение объема заданной массы газофазного потока окислителя, повышением поверхностной скорости в реакционной среде 36, что в свою очередь вызывает увеличение задержки газа и скорости массопередачи молекулярного кислорода.
Баланс между слиянием пузырьков и их разделением является чрезвычайно сложным явлением, приводящим, с одной стороны, к склонности к пенообразованию, понижающему скорость внутренней циркуляции жидкой фазы, и к возможной потребности в чрезвычайно больших отделенных зонах, а, с другой стороны, к склонности образовывать меньшее количество, но очень больших пузырьков, что дает меньшую задержку газа и более низкую скорость массопередачи от потока окислителя к жидкой фазе. Что касается жидкой фазы, то ее состав, плотность, вязкость и поверхностное натяжение среди прочих факторов, как известно, взаимодействуют очень сложным образом и приводят к очень сложным результатам, даже при отсутствии твердой фазы. Например, лабораторные исследователи показали, что полезно определять, является ли «вода» водопроводной водой, дистиллированной водой или деионизованной водой, когда сообщали о наблюдениях или оценивали их, даже при использовании простых водно-воздушных систем в барботажных колоннах. Для сложных смесей в жидкой фазе и при добавлении твердой фазы степень сложности повышается. Поверхностные неровности на отдельных частицах твердых веществ, средний размер твердых частиц, распределение частиц по размерам относительно жидкой фазы и способность жидкости смачивать поверхность твердого вещества среди прочего - все эти факторы являются очень важными при взаимодействии частиц с жидкой фазой и с потоком окислителя и при установлении, какие конфигурации потоков возникнут из-за влияния характера поведения пузырьков и природной конвекции.
Таким образом, способность барботажного реактора колонного типа полезно работать при высоких поверхностных скоростях и большой задержке газа, раскрытая здесь, зависит, например, от соответствующего выбора: (1)состава жидкой фазы реакционной среды; (2) количества и типа осажденных твердых веществ, которыми можно управлять (и то и другое) условиями реакции; (3) величины потока окислителя, подаваемого в реактор; (4) давления верха, которое влияет на объемный расход потока окислителя, на стабильность пузырьков и из-за энергетического баланса на температуру реакции; (5) температуры реакции, которая влияет на свойства жидкости, свойства осажденных твердых веществ и на удельный объем потока окислителя; и (6) геометрии и механических элементов резервуара для проведения реакции, включая отношение L:D.
Еще раз обращаясь к фиг.1, было обнаружено, что улучшенное распределение окисляемого соединения (например, параксилола) в реакционной среде 36 может быть осуществлено введением жидкофазного потока сырья в реакционную зону 28 на многих участках, распределенных по высоте. Предпочтительно, чтобы жидкофазный поток сырья вводился в реакционную зону 28 через по меньшей мере 3 входных отверстия, более предпочтительно через по меньшей мере 4 входных отверстия. Используемый здесь термин «входные отверстия» означает отверстия, которые выводят жидкофазный поток сырья в реакционную зону 28 для смешивания с реакционной средой 36. Предпочтительно, чтобы по меньшей мере 2 из входных отверстий были распределены по высоте одно от другого по меньшей мере на около 0,5D, более предпочтительно по меньшей мере на около 1,5D и наиболее предпочтительно по меньшей мере на 3D. Однако предпочтительно, чтобы самое высокорасположенное входное отверстие, удаленное по высоте от самого низко расположенного отверстия, было удалено от последнего не более чем на около 0,75Н, 0,65L и/или 8D; более предпочтительно не более чем на около 0,5Н, 0,4L и/или 5D; и наиболее предпочтительно не более чем на 0,4Н, 0,35L и/или 4D.
Хотя желательно вводить жидкофазный поток сырья на многих распределенных по высоте участках, было также обнаружено, что улучшенное распределение окисляемого соединения в реакционной среде 36 обеспечивается, если большая часть жидкофазного потока сырья вводится в нижнюю половину реакционной среды 36 и/или реакционной зоны 28. Предпочтительно, чтобы по меньшей мере около 75 мас.% жидкофазного потока сырья вводилась в нижнюю половину реакционной среды 36 и/или реакционной зоны 28. Более предпочтительно, чтобы по меньшей мере 90 мас.% жидкофазного потока сырья вводилось в нижнюю половину реакционной среды 36 и/или реакционной зоны 28. Кроме того, предпочтительно, чтобы по меньшей мере около 30 мас.% жидкофазного потока сырья вводилось в реакционную зону в пределах около 1,5D самого нижнего участка по высоте, где вводится поток окислителя в реакционную зону 28. Этот самый нижний вертикальный участок, где поток окислителя вводится в реакционную зону 28, обычно находится в нижней части распределителя окислителя; однако в предпочтительном воплощении настоящего изобретения предусмотрены различные альтернативные конфигурации для ввода потока окислителя в реакционную зону 28. Предпочтительно, чтобы по меньшей мере около 50 мас.% жидкофазного сырья вводилось в пределах около 2,5D самого нижнего участка по высоте, где поток окислителя вводится в реакционную зону 28. Предпочтительно, чтобы по меньшей мере около 75 мас.% жидкофазного потока сырья вводилось в пределах около 5D самого нижнего участка по высоте, где поток окислителя вводится в реакционную зону 28.
Каждое входное отверстие определяет открытую площадь, через которую выводится сырье. Предпочтительно, чтобы по меньшей мере около 30% от общей открытой площади сечения всех подающих входов было расположено в пределах около 1,5D самого нижнего участка по высоте, где поток окислителя вводится в реакционную зону 28. Предпочтительно, чтобы по меньшей мере около 50% от общей открытой площади сечения всех входов было расположено в пределах около 2,5D самого нижнего участка по высоте, где поток окислителя вводится в реакционную зону 28. Предпочтительно, чтобы по меньшей мере около 75% от общей открытой площади всех входов было расположено в пределах около 5D самого нижнего участка по высоте, где поток окислителя вводится в реакционную зону 28.
Как снова показано на фиг.1, в одном воплощении настоящего изобретения входы 32a,b,c являются просто группой вертикально распределенных отверстий, расположенных вдоль одной стороны корпуса 22 с резервуаром. Эти отверстия предпочтительно имеют в основном одинаковые диаметры меньше около 7 см, более предпочтительно в интервале от около 0,25 до около 5 см, и наиболее предпочтительно в интервале от 0,4 до 2 см. Барботажный реактор колонного типа 20 предпочтительно снабжен системой управления скоростью потока жидкофазного сырья из каждого входного отверстия. Такая система управления потоком предпочтительно включает в себя отдельный вентиль управления потоком 74a,b,c,d для каждого соответствующего входа 32a,b,c,d. Кроме того, предпочтительно, чтобы барботажный реактор колонного типа 20 был снабжен системой управления потоком, которая позволяет вводить по меньшей мере часть жидкофазного потока сырья в реакционную зону 28 с повышенной входной поверхностной скоростью по меньшей мере около 2 метров в секунду, более предпочтительно со скоростью по меньшей мере около 5 метров в секунду, еще более предпочтительно со скоростью по меньшей мере около 6 метров в секунду и наиболее предпочтительно со скоростью в интервале от 8 до 20 метров в секунду. Используемый здесь термин «входная поверхностная скорость» означает усредненную по времени объемную скорость подаваемого потока из входного отверстия, деленную на площадь входного отверстия. Предпочтительно, чтобы по меньшей мере около 50 мас.% подаваемого потока сырья вводилось в реакционную зону 28 с повышенной входной поверхностной скоростью. Наиболее предпочтительно, чтобы по существу весь подаваемый поток сырья вводился в реакционную зону с повышенной входной поверхностной скоростью.
На фиг.6, 7 показана альтернативная система для ввода жидкофазного потока сырья в реакционную зону 28. В этом воплощении входной поток сырья вводится в реакционную зону в четырех различных по высоте участках. Каждый участок по высоте снабжен соответствующей системой 76a,b,c,d распределения подаваемого потока. Каждая система 76 распределения подаваемого потока включает в себя основной подающий трубопровод 78 и коллектор 80. Каждый коллектор 80 снабжен по меньшей мере двумя выходами 82, 84, соединенными с соответствующими вставными трубопроводами 86, 88, которые входят в реакционную зону 28 резервуара в корпусе 22. Каждый вставной трубопровод 86, 88 имеет соответствующее входное отверстие 87, 89 для вывода подаваемого потока в реакционную зону 28. Входные отверстия 87, 89 предпочтительно имеют по существу одинаковые диаметры менее около 7 см, более предпочтительно в интервале от около 0,25 до около 5 см и наиболее предпочтительно в интервале от 0,4 до 2 см. Предпочтительно, чтобы входные отверстия 87, 89 каждой системы 76a,b,c,d распределения подаваемого потока были расположены диаметрально противоположно, чтобы вводить подаваемый поток в реакционную зону с противоположных направлений. Также предпочтительно, чтобы диаметрально противоположные входные отверстия 86, 88 соседних систем 76 распределения подаваемых потоков были ориентированы с поворотом на угол в 90° по отношению друг к другу. При работе жидкофазный подаваемый поток сырья вводится в основной входной трубопровод 78 и затем поступает в коллектор 80. Коллектор 80 равномерно распределяет подаваемый поток для его одновременного ввода с противоположных сторон реактора 20 через входные отверстия 87, 89.
На фиг.8 показана альтернативная конфигурация, в которой каждая система 76 для распределения подаваемого потока снабжена съемными трубами 90, 92, а не вставными трубопроводами 86, 88 (показанными на фиг.7). Съемные трубы 90, 92 входят в реакционную зону 28 и имеют большое число малых подающих отверстий 94, 96 для вывода жидкофазного подаваемого сырья в реакционную зону 28. Предпочтительно, чтобы малые подающие отверстия 94, 96 в съемных трубах 90, 92 имели по существу одинаковые диаметры менее около 50 мм, более предпочтительно от около 2 до около 25 мм и наиболее предпочтительно от 4 до 15 мм.
На фиг.9-11 показана альтернативная система 100 распределения входного потока. Система 100 распределения входного потока вводит жидкофазный поток сырья во многих участках, расположенных по высоте и горизонтально, не требующих многих входов через боковую стенку барботажного реактора колонного типа 20. Система 100 распределения входного потока вообще включает в себя один входной трубопровод 102, распределитель 104, большое число вертикальных распределительных трубопроводов 106, горизонтальное поддерживающее устройство 108 и вертикальное поддерживающее устройство 110. Входной трубопровод 102 проходит через боковую стенку основы 46 корпуса 22 с резервуаром. Входной трубопровод 102 подвижно соединен с распределителем 104. Распределитель 104 равномерно распределяет входной поток, выходящий из входного трубопровода 102, между отвесными распределительными трубопроводами 106. Каждый распределительный трубопровод 106 имеет большое число входных отверстий, расположенных по высоте 112a,b,c,d, для вывода подаваемого потока сырья в реакционную зону 28. Горизонтальное поддерживающее устройство 108 соединено с каждым распределительным трубопроводом 106 и препятствует относительному горизонтальному смещению распределительных трубопроводов 106. Вертикальное поддерживающее устройство 110 предпочтительно соединено с горизонтальным поддерживающим устройством 108 и с верхней частью распределителя 34 для окислителя. Вертикальное поддерживающее устройство 110 в основном препятствует вертикальному перемещению распределительных трубопроводов 106 в реакционной зоне 28. Предпочтительно, чтобы входные отверстия 112 имели в основном одинаковый диаметр менее около 50 мм, более предпочтительно от около 2 до около 25 мм и наиболее предпочтительно от 4 до 15 мм. Вертикальные разделительные промежутки между входными отверстиями 112 системы 100 распределения входного потока, как показано на фиг.9-11, могут быть одинаковыми, как описано выше со ссылкой на систему распределения входного потока на фиг.1.
Было обнаружено, что конфигурации потоков реакционной среды во многих барботажных реакторах колонного типа могут дать неравномерное азимутальное распределение окисляемого соединения в реакционной среде, особенно когда окисляемое соединение преимущественно вводится вдоль одной стороны реакционной среды. Используемый здесь термин «азимутальный» означает угол или промежуток вокруг отвесной продольной оси реакционной зоны. Используемый здесь термин «отвесный» означает расположение в пределах 45° от вертикали. В одном воплощении настоящего изобретения входной поток сырья, содержащий окисляемое соединение (например, параксилол), вводится в реакционную зону через большое число азимутально распределенных входных отверстий. Эти азимутально распределенные входные отверстия могут помочь исключить области с избыточно высокой или со слишком низкой концентрацией окисляемого соединения в реакционной среде. Различные системы распределения входного потока сырья, показанные на фиг.6-11, являются примерами систем, которые обеспечивают правильное азимутальное распределение входных отверстий.
Как показано на фиг.7, чтобы количественно определить азимутально распределенный ввод жидкофазного потока сырья в реакционную среду, реакционная среда может быть теоретически разделена на четыре равные зоны «Q1,Q2,Q3,Q4» приблизительно одинакового объема. Эти равные зоны «Q1,Q2,Q3,Q4» ограничены двумя воображаемыми пересекающимися перпендикулярными вертикальными плоскостями «Р1,Р2», выходящими за пределы максимального вертикального размера и максимального радиального размера реакционной среды. Когда реакционная среда содержится в цилиндрическом резервуаре, то линия пересечения воображаемых пересекающихся вертикальных плоскостей Р1,Р2 будет приблизительно совпадать с вертикальной центральной осью цилиндра, и каждая равная зона Q1,Q2,Q3,Q4 будет вообще клиновидным вертикальным объемом, имеющим высоту, равную высоте реакционной среды. Предпочтительно, чтобы значительная часть окисляемого соединения выводилась в реакционную среду через входные отверстия, расположенные по меньшей мере в двух различных равных зонах.
В предпочтительном воплощении настоящего изобретения не более чем около 80 мас.% окисляемого соединения выводятся в реакционную среду через входные отверстия, которые могут быть расположены в одной зоне. Более предпочтительно, чтобы не более чем около 60 мас.% окисляемого соединения выводилось в реакционную среду через входные отверстия, которые могут быть расположены в одной зоне. Наиболее предпочтительно, чтобы не более 40 мас.% окисляемого соединения выводилось в реакционную среду через входные отверстия, которые могут быть расположены в одной зоне. Эти параметры распределения окисляемого соединения по зонам измеряются, когда зоны ориентированы таким образом, чтобы максимально возможное количество окисляемого соединения подавалось в одну из равных зон. Например, если весь входной поток сырья выводится в реакционную среду через два входных отверстия, которые распределены одно от другого на 89°, то для целей определения азимутального распределения в четырех равных зонах, 100 мас.% подаваемого потока выводятся в реакционную среду в одной зоне, так как равные зоны ориентированы таким образом, чтобы оба эти входные отверстия были расположены в одной из равных зон.
Кроме преимуществ, связанных с правильным распределением входных отверстий, также было обнаружено, что правильное радиальное распределение входных отверстий в барботажном реакторе колонного типа также может быть важным. Предпочтительно, чтобы значительная часть окисляемого соединения вводилась в реакционную среду для подачи ее через входные отверстия, которые радиально распределены в направлении внутрь от боковой стенки резервуара. Таким образом в одном воплощении настоящего изобретения значительная часть окисляемого соединения поступает в реакционную зону через входные отверстия, расположенные в «предпочтительной радиальной зоне входа», которая отделена во внутреннем направлении от боковых образующих стенок, ограничивающих реакционную зону.
Как снова показано на фиг.7, предпочтительная радиальная зона подачи «FZ» может принять форму теоретического отвесного цилиндра, центрированного в реакционной зоне 28 и имеющего внешний диаметр «D0», равный 0,9D, где «D» является диаметром реакционной зоны 28. Таким образом образуется внешнее кольцо «ОА» толщиной 0,05D между предпочтительной радиальной зоной входа FZ и внутренней частью боковой стенки, ограничивающей реакционную зону 28. Предпочтительно, чтобы малое количество окисляемого соединения поступало в реакционную зону 28 или оно совсем туда не поступало через входные отверстия, расположенные в этом внешнем кольце ОА.
В другом воплощении предпочтительно, чтобы малое количество окисляемого соединения вводилось в центр реакционной зоны 28 или оно туда совсем не поступало. Таким образом, как показано на фиг.8, предпочтительная радиальная зона входа FZ может принять форму теоретического отвесного кольца, центрированного в реакционной зоне 28 и имеющего внешний диаметр D0, равный 0,9D, и внутренний диаметр D1, равный 0,2D. Таким образом в этом воплощении внутренний цилиндр IC, имеющий диаметр 0,2D, «вырезается» из центра предпочтительной радиальной зоны подачи FZ. Предпочтительно, чтобы малое количество окисляемого соединения вводилось или вообще оно не вводилось в реакционную зону 28 через входные отверстия, расположенные в этом внутреннем цилиндре IC.
В предпочтительном воплощении настоящего изобретения значительная часть окисляемого соединения вводится в реакционную среду 36 через входные отверстия, расположенные в предпочтительной радиальной зоне подачи, независимо от того, имеет ли предпочтительная радиальная зона подачи цилиндрическую или кольцевую форму, описанную выше. Более предпочтительно, чтобы по меньшей мере около 25 мас.% окисляемого соединения вводились в реакционную среду 36 через входные отверстия, расположенные в предпочтительной радиальной зоне подачи. Еще более предпочтительно, чтобы по меньшей мере около 50 мас.% окисляемого соединения вводилось в реакционную среду 36 через входные отверстия, расположенные в предпочтительной радиальной зоне подачи. Наиболее предпочтительно, чтобы по меньшей мере 75 мас.% окисляемого соединения вводилось в реакционную среду 36 через входные отверстия, расположенные в предпочтительной радиальной зоне подачи.
Хотя теоретические равные зоны и теоретическая предпочтительная радиальная зона подачи, показанные на фиг.7 и 8, описаны со ссылкой на распределение подаваемого жидкофазного потока сырья, было обнаружено, что правильное распределение по зонам и радиальное распределение газофазного потока окислителя также может дать некоторые преимущества. Таким образом в одном воплощении настоящего изобретения описание распределения по зонам и радиального распределения подаваемого жидкофазного потока сырья, приведенное выше, также относится и к тому, каким образом газофазный поток окислителя вводится в реакционную среду 36.
Теперь, как показано на фиг.12-15, альтернативный распределитель 200 окислителя вообще включает в себя кольцевой элемент 202 и два входных трубопровода 204 и 206. Распределитель окислителя 200 на фиг.12-15 является таким же, как и распределитель окислителя 34 на фиг.1-11, за исключением следующих трех основных отличий: (1) распределитель окислителя 200 не имеет диагонального поперечного элемента; (2) верхняя часть кольцевого элемента 202 не имеет отверстий для подачи окислителя по высоте; и (3) распределитель окислителя 200 имеет гораздо больше отверстий в нижней части кольцевого элемента 202.
Как возможно лучше всего показано на фиг.14 и 15, нижняя часть кольцевого распределителя окислителя 202 имеет большое число отверстий 208. Отверстия 208 предпочтительно имеют форму, при которой по меньшей мере около 1% общей открытой площади сечения, определяемой отверстиями 208 для окислителя, расположено ниже осевой линии 210 (фиг.15), при этом осевая линия 210 расположена выше объемной центроиды кольцевого элемента 202. Более предпочтительно, чтобы по меньшей мере около 5% общей открытой площади сечения, определяемой всеми отверстиями окислителя 208, было расположено ниже осевой линии 210, при этом по меньшей мере около 2% общей открытой площади, определяемой отверстиями 208, выводили поток окислителя в общем нижнем направлении в пределах около 30° от вертикали. Еще более предпочтительно, чтобы по меньшей мере около 20% общей открытой площади сечения, определяемой всеми отверстиями окислителя 208, были расположены ниже осевой линии 210, при этом по меньшей мере около 10% общей открытой площади сечения, определяемой отверстиями 208, выводили поток окислителя в общем нижнем направлении в пределах 30° от вертикали. Наиболее предпочтительно, чтобы по меньшей мере около 75% общей открытой площади сечения, определяемой всеми отверстиями окислителя 208, были расположены ниже осевой линии 210, при этом по меньшей мере около 40% общей открытой площади сечения, определяемой отверстиями 208, выводили поток окислителя в общем нижнем направлении в пределах 30° от вертикали. Доля общей открытой площади сечения, определяемой всеми отверстиями 208 для окислителя, которые расположены выше осевой линии 210, составляет предпочтительно меньше около 75%, более предпочтительно меньше около 50%, еще более предпочтительно меньше около 25% и наиболее предпочтительно меньше 5%.
Как показано на фиг.14 и 15, отверстия 208 для окислителя включают в себя направленные вниз отверстия 208a и скошенные отверстия 208b. Направленные вниз отверстия 208a конфигурированы для вывода потока окислителя в общем нижнем направлении под углом в пределах около 30° от вертикали, более предпочтительно в пределах около 15° от вертикали и наиболее предпочтительно в пределах 5° от вертикали. Скошенные отверстия 208b конфигурированы для вывода потока окислителя в общем направлении в сторону и вниз под углом «А», который находится в интервале от около 15 до около 75° от вертикали, более предпочтительно угол А находится в интервале от около 30 до около 60° от вертикали и наиболее предпочтительно угол А находится в интервале от 40 до 50° от вертикали.
Предпочтительно, чтобы по существу все отверстия окислителя 208 имели приблизительно одинаковый диаметр. Диаметр отверстий окислителя 208 предпочтительно находится в интервале от около 2 до около 300 мм, более предпочтительно в интервале от около 4 до около 120 мм и наиболее предпочтительно в интервале от 8 до 60 мм. Общее число отверстий окислителя 208 в кольцевом элементе 202 выбирается для того, чтобы обеспечивалось соответствие критериям низкого перепада давления, указанным ниже. Предпочтительно, чтобы общее число отверстий окислителя 208, смонтированных в кольцевом элементе 202, было по меньшей мере около 10, более предпочтительно, чтобы общее число отверстий окислителя 208 было в интервале от около 20 до около 200 и наиболее предпочтительно, чтобы общее число отверстий 208 было в интервале от 40 до 100.
Хотя на фиг.12-15 показана очень специальная конфигурация распределителя окислителя 200, но следует отметить, что могут быть использованы самые различные конфигурации распределителя окислителя для получения описанных здесь преимуществ. Например, распределитель окислителя не обязательно должен иметь конфигурацию восьмиугольного кольцевого элемента, показанную на фиг.12, 13. Скорее распределитель для окислителя возможно будет смонтирован из любой конфигурации трубопровода (трубопроводов) для потока, в котором используется большое число раздельных отверстий для выхода потока окислителя. Размер, число и направление отверстий окислителя в трубопроводе для потока предпочтительно находятся в пределах интервалов, указанных выше. Кроме того распределитель окислителя предпочтительно имеет конфигурацию, обеспечивающую продольное и радиальное распределение молекулярного кислорода, описанное выше.
Независимо от конкретной конфигурации распределителя окислителя, предпочтительно, чтобы распределитель окислителя имел физическую конфигурацию и работал таким образом, чтобы при этом минимизировалось падение давления, связанное с выводом потока окислителя из входного трубопровода (трубопроводов) через отверстия для окислителя в реакционную зону. Такое падение давления рассчитывается как усредненное по времени статическое давление потока окислителя внутри входного трубопровода у входов 66a,b распределителя окислителя минус усредненное по времени статическое давление в реакционной зоне на высоте, где половина потока окислителя вводится выше этого места по высоте и другая половина потока окислителя вводится ниже этого места по высоте. В предпочтительном воплощении настоящего изобретения усредненное по времени падение давления, связанное с выводом потока окислителя из распределителя, составляет менее чем около 0,3 МПа, более предпочтительно менее чем около 0,2 МПа, еще более предпочтительно менее чем около 0,1 МПа и наиболее предпочтительно менее 0,05 МПа. В предпочтительных рабочих условиях барботажного реактора колонного типа, описанного здесь, давление потока окислителя внутри входного трубопровода (трубопроводов) распределителя окислителя предпочтительно находится в интервале от около 0,35 до около 1 МПа, более предпочтительно в интервале от около 0,45 до около 0,85 МПа и наиболее предпочтительно в интервале от 0,5 до 0,7 МПа.
Как уже было показано со ссылкой на конфигурацию распределителя окислителя на фиг. 2-5, может быть желательно непрерывно или периодически промывать распределитель окислителя жидкостью (например, уксусной кислотой, водой и/или параксилолом) для предотвращения загрязнения распределителя окислителя твердыми веществами. Когда используется такое промывание жидкостями, предпочтительно, чтобы эффективное количество жидкости (т.е. не только малое количество капель жидкости, которые могут естественно присутствовать в потоке окислителя) пропускалось через распределитель окислителя и через отверстия для окислителя в течение по меньшей мере одного промежутка времени больше одной минуты ежедневно. Когда жидкость непрерывно или периодически выводится из распределителя окислителя, то предпочтительно, чтобы усредненное по времени отношение скорости потока массы жидкости через распределитель окислителя к скорости потока массы молекулярного кислорода, проходящего через распределитель окислителя было в интервале от около 0,05:1 до около 30:1, или в интервале от около 0,1:1 до около 2:1, или даже в интервале от 0,2:1 до 1:1.
В одном воплощении настоящего изобретения значительная часть окисляемого соединения (например, параксилола) может быть введена в реакционную зону через распределитель окислителя. В такой конфигурации предпочтительно, чтобы окисляемое соединение и молекулярный кислород выводились из распределителя окислителя через те же самые отверстия в распределителе окислителя. Как было отмечено выше, окисляемое соединение обычно является жидкостью при нормальных температуре и давлении. Поэтому в этом воплощении двухфазный поток может выводится из распределителя окислителя с жидкой фазой, содержащей окисляемое соединение, и с газообразной фазой, содержащей молекулярный кислород. Однако следует признать, что по меньшей мере часть окисляемого соединения может быть в газообразном состоянии, когда оно выводится из распределителя окислителя. В одном воплощении жидкая фаза, выводимая из распределителя окислителя, сформирована преимущественно из окисляемого соединения. В другом воплощении жидкая фаза, выводимая из распределителя окислителя, имеет по существу тот же состав, как и подаваемый поток сырья, описанный выше. Когда жидкая фаза, выводимая из распределителя окислителя, имеет тот же состав, как и подаваемый поток сырья, такая жидкая фаза может содержать растворитель и/или систему катализаторов в количествах и с соотношениями, приведенными выше со ссылкой на состав вводимого потока сырья.
В одном воплощении настоящего изобретения предпочтительно, чтобы по меньшей мере около 10 мас.% всего окисляемого соединения, вводимого в реакционную зону, вводилось через распределитель окислителя, более предпочтительно, чтобы по меньшей мере около 40 мас.% окисляемого соединения вводилось в реакционную зону через распределитель окислителя и наиболее предпочтительно, чтобы по меньшей мере 80 мас.% окисляемого соединения вводилось в реакционную зону через распределитель окислителя. Когда все окисляемое соединение или его часть вводится в реакционную зону через распределитель окислителя, то предпочтительно, чтобы по меньшей мере около 10 мас.% всего молекулярного кислорода, вводимого в реакционную зону, вводилось через тот же самый распределитель окислителя, более предпочтительно, чтобы по меньшей мере около 40 мас.% окисляемого соединения вводилось в реакционную зону через тот же самый распределитель окислителя и наиболее предпочтительно, чтобы по меньшей мере 80 мас.% окисляемого соединения вводилось в реакционную зону через тот же самый распределитель окислителя. Когда значительная часть окисляемого соединения вводится в реакционную зону через распределитель окислителя, то предпочтительно, чтобы один или несколько датчиков температуры (например, термопар) были расположены в распределителе окислителя. Эти датчики температуры могут быть использованы для того, чтобы удостовериться, что температура в распределителе окислителя не становится опасно высокой.
Теперь, как показано на фиг. 16-18, барботажный реактор колонного типа 20 включает в себя внутренний резервуар 300 для деаэрации, расположенный в нижней части реакционной зоны 28 рядом с выходом 38 для шлама. Было обнаружено, что побочные реакции, образующие примеси, происходят со сравнительно высокой скоростью во время деаэрации реакционной среды 36. Используемый здесь термин «деаэрация» означает отделение газовой фазы от многофазной реакционной среды. Когда реакционная среда 36 сильно аэрирована (задержка газа >0,3), образование примесей минимально. Когда реакционная среда 36 сильно деаэрирована (задержка газа <0,01), образование примесей также минимально. Однако, когда реакционная среда частично аэрирована (задержка газа 0,01-0,3), ускоряются побочные реакции и увеличивается образование примесей. Резервуар 300 для деаэрации позволяет решить эту и другие проблемы за счет минимизации объема реакционной среды 36 в частично аэрированном состоянии и за счет минимизации времени, требующегося для ее деаэрации. Из нижней части резервуара 300 для деаэрации получают по существу деаэрированный шлам и он выводится из реактора 20 через выход 38 для шлама. Существенно деаэрированный шлам предпочтительно содержит менее чем около 5% объемных газовой фазы, более предпочтительно менее чем около 2% объемных газовой фазы и наиболее предпочтительно менее 1% объемного газовой фазы.
На фиг.16 показан барботажный реактор колонного типа 20, включающий в себя регулятор 302 уровня и регулятор 304 потока. Регулятор 302 уровня и регулятор 304 потока взаимодействуют для поддержания реакционной среды 36 в основном на постоянном уровне в реакционной зоне 28. Регулятор 302 уровня срабатывает для определения (например, считыванием величины избыточного давления или ядерного импульса) поднятия верхней поверхности 44 реакционной среды 36 и формирования управляющего сигнала 306, соответствующего поднятию реакционной среды 36. Регулятор 304 потока принимает управляющий сигнал 306 и регулирует скорость потока шлама через выходной трубопровод 308 для шлама. Таким образом скорость потока шлама на выходе 38 для шлама может изменяться в интервале от максимальной объемной скорости потока шлама (Fmax), когда подъем уровня реакционной среды 36 слишком большой, и до минимальной объемной скорости потока шлама (Fmax), когда подъем реакционной среды 36 слишком мал.
Чтобы удалить твердофазный продукт окисления из реакционной зоны 28, его часть должна сначала пройти через резервуар 300 для деаэрации. Резервуар 300 для деаэрации обеспечивает слаботурбулентный внутренний объем, позволяющий газовой фазе реакционной среды 36 естественно выделиться из жидкой и твердой фаз реакционной среды, когда поток жидкости и твердых веществ стекает вниз к выходу 38 для шлама. Выделение газовой фазы из жидкой и твердой фаз вызван естественным всплытием вверх газовой фазы в жидкой и твердой фазах. Когда используется резервуар 300 для деаэрации, превращение реакционной среды 36 из полностью аэрированной трехфазной среды в полностью деаэрированный двухфазный шлам является быстрым и эффективным.
Как показано теперь на фиг. 17 и 18, резервуар 300 для деаэрации включает в себя в общем образующую боковую стенку 308, ограничивающую зону 312 деаэрации. Предпочтительно, чтобы боковая стенка 308 была направлена вверх под углом в пределах около 30° от вертикали, более предпочтительно в пределах около 10° от вертикали. Наиболее предпочтительно, чтобы боковая стенка 308 была по существу вертикальной. Зона 312 деаэрации отделена от реакционной зоны 28 и имеет высоту «h» и диаметр «d». Верх 310 боковой стенки 308 открыт для приема реакционной среды из реакционной зоны 28 во внутренний объем 312. Низ боковой стенки 308 подвижно соединен с выходом 38 для шлама переходной секцией 314. В некоторых случаях, когда отверстие выхода 38 для шлама является большим или когда диаметр «d» боковой стенки 308 мал, переходную секцию 314 можно исключить. Как возможно лучше всего показано на фиг.18, резервуар 300 для деаэрации может включать в себя гаситель 316 вихрей, расположенный в зоне 312 деаэрации. Гаситель 316 вихрей может быть любой структурой, предназначенной для предотвращения образования вихрей, когда твердая и жидкая фазы стекают вниз к выходу 38 для шлама.
Чтобы обеспечивалось должное отделение газовой фазы от твердой и жидкой фаз в резервуаре 300 для деаэрации, высота «h» и горизонтальная площадь поперечного сечения внутренней зоны 312 деаэрации должны тщательно подбираться. Высота «h» и горизонтальная площадь поперечного сечения внутренней зоны 312 деаэрации должны предоставлять достаточное расстояние и время для того, чтобы даже при выведении максимального количества шлама (т.е., когда шлам выводится с Fmax), по существу весь объем пузырьков газа мог быть выделен из твердой и жидкой фаз, перед тем как пузырьки газа достигнут нижнего выхода из резервуара 300 для деаэрации. Таким образом предпочтительно, чтобы площадь поперечного сечения зоны 312 деаэрации была такой, чтобы максимальная скорость в нижнем направлении (Vdmax) жидкой и твердой фаз через зону 312 деаэрации была меньше скорости естественного выделения (Vu) пузырьков газа из жидкой и твердой фаз. Максимальная скорость в нижнем направлении (Vdmax) жидкой и твердой фаз через зону 312 деаэрации возникает при максимальной объемной скорости потока шлама (Fmax), рассмотренной выше. Естественная скорость выделения (Vu) пузырьков газа через жидкую и твердую фазы изменяется в зависимости от размера пузырьков; однако естественная скорость выделения (Vu0,5) пузырьков газа диаметром 0,5 см из жидкой и твердой фаз может быть использована как предельная величина, так как по существу весь исходный объем пузырьков в реакционной среде 36 будет больше 0,5 см. Предпочтительно, чтобы площадь поперечного сечения зоны 312 деаэрации была такой, при которой
Vdmax меньше 75% Vu0,5, более предпочтительно, чтобы Vdmax была менее около 40%
Vu0,5, наиболее предпочтительно, чтобы Vdmax была менее 20% Vu0,5.
Скорость стекания вниз жидкой и твердой фаз в зоне 312 деаэрации резервуара 300 для деаэрации рассчитывается как объемная скорость потока деаэрированного шлама через выход 38 для шлама, деленная на минимальную площадь поперечного сечения зоны 312 деаэрации. Скорость стекания вниз жидкой и твердой фаз в зоне 312 деаэрации резервуара 300 для деаэрации предпочтительно меньше чем около 50 см в секунду, более предпочтительно меньше чем около 30 см в секунду и наиболее предпочтительно меньше 10 см в секунду.
Следует отметить, что хотя образующая боковая стенка 308 резервуара 300 для деаэрации, как показано, имеет цилиндрическую форму, боковая стенка 308 могла бы включать в себя большое число стенок с самыми различными конфигурациями (например, треугольной, квадратной или овальной), пока стенки ограничивают внутренний объем, имеющий подходящий объем, площадь поперечного сечения, ширину «d» и высоту «h». В предпочтительном воплощении настоящего изобретения «d» находится в интервале от около 0,2 до около 2 м, более предпочтительно в интервале от около 0,3 до около 1,5 м и наиболее предпочтительно в интервале от 0,4 до 1,2 м. В предпочтительном воплощении настоящего изобретения «h» находится в интервале от около 0,3 до около 5 м, более предпочтительно в интервале от около 0,5 до около 3 м и наиболее предпочтительно в интервале от 0,75 до 2 м.
В предпочтительном воплощении настоящего изобретения боковая стенка 308 является по существу вертикальной, так что площадь поперечного сечения зоны 312 деаэрации по существу постоянна вдоль всей высоты «h» зоны 312 деаэрации. Предпочтительно, чтобы максимальная горизонтальная площадь сечения зоны 312 деаэрации была менее около 25% от максимальной поперечной площади горизонтального сечения реакционной зоны 28. Более предпочтительно, чтобы максимальная площадь поперечного сечения зоны 312 деаэрации была в интервале от около 0,1 до около 10% максимальной площади поперечного сечения реакционной зоны 28. Наиболее предпочтительно, чтобы максимальная площадь поперечного сечения зоны 312 деаэрации была в интервале о 0,25 до 4% максимальной горизонтальной площади сечения реакционной зоны 28. Предпочтительно, чтобы максимальная площадь поперечного сечения зоны 312 деаэрации была в интервале от около 0,02 до около 3 м2, более предпочтительно от около 0,05 до около 2 м2 и наиболее предпочтительно в интервале от 0,1 до 1,2 м2. Объем зоны 312 деаэрации предпочтительно меньше около 5% от общего объема реакционной среды 36 или реакционной зоны 28. Более предпочтительно, чтобы объем зоны 312 деаэрации находился в интервале от около 0,01 до около 2% общего объема реакционной среды 36 или реакционной зоны 28. Наиболее предпочтительно, чтобы объем зоны 312 деаэрации был в интервале от 0,05 до около 1% общего объема реакционной среды 36 или реакционной зоны 28. Объем зоны 312 деаэрации предпочтительно менее около 2 м3, более предпочтительно находился в интервале от около 0,01 до около 1 м3 и наиболее предпочтительно в интервале от 0,05 до 0,5 м3.
Теперь, как показано на фиг.19, барботажный реактор колонного типа 20 включает в себя внешний резервуар 400 для деаэрации. В этой конфигурации аэрированная реакционная среда 36 выводится из реакционной зоны 28 через приподнятое по высоте отверстие в корпусе 22 c резервуаром. Выведенная аэрированная среда перемещается по выходному трубопроводу 402 во внешний резервуар 400 для деаэрации для удаления газовой фазы из твердой и жидкой фаз. Удаленная газовая фаза отводится из резервуара 400 для деаэрации через трубопровод 404, а существенно деаэрированный шлам выводится из резервуара 400 для деаэрации через трубопровод 406.
Как показано на фиг.19, выходной трубопровод 402 является приблизительно прямым, горизонтальным и ортогональным по отношению к корпусу 22 с резервуаром. Это просто одна удобная конфигурация; и выходной трубопровод 402 может быть другим в любом отношении, при условии, что он эффективно соединяет барботажный реактор колонного типа 20 с внешним резервуаром 400 для деаэрации. Что касается трубопровода 404, то для него выгодно, что он присоединен рядом или почти к верхней части резервуара 400 для деаэрации, что позволяет контролировать проблемы безопасности, связанные с застойной газовой зоной, содержащей окисляемое соединение и окислитель.
Кроме того трубопроводы 402 и 404 могут содержать такие средства регулирования потока, как вентили.
Когда реакционная среда 36 выводится из реактора 20 через приподнятый по высоте выход, как показано на фиг.19, предпочтительно, чтобы барботажный реактор колонного типа 20 был снабжен нижним выходом 408 рядом с нижней частью 52 реакционной зоны 28. Нижний выход 408 и нижний трубопровод 410, присоединенный к упомянутому выходу, могут использоваться для удаления реакционной среды из реактора 20 во время остановок реактора. Предпочтительно, чтобы обеспечивался один или больше нижних выходов 308 в нижней трети по высоте реакционной среды 36, более предпочтительно в нижней четверти по высоте реакционной среды 36 и наиболее предпочтительно в самой нижней точке реакционной зоны 28.
При приподнятом по высоте выводе шлама и при системе деаэрации, показанных на фиг.19, нижние трубопровод 410 и выход 408 не используются для вывода шлама из реакционной зоны 28 во время окисления. В данной технике известно, что твердые вещества склонны к осаждению под действием силы тяжести в неаэрированных и других неперемешиваемых частях шлама, включая застойные трубопроводы для потоков. Кроме того, осажденные твердые вещества, например терефталевая кислота, могут иметь тенденцию отверждаться в виде больших агломератов при непрерывном осаждении и/или кристаллизации. Таким образом, чтобы избежать забивания нижнего трубопровода 410 для потока, может быть использована часть деаэрированного шлама из нижней части резервуара 400 для деаэрации для непрерывной или периодической промывки нижнего трубопровода 410 во время нормальной работы реактора 20. Предпочтительным средством для обеспечения такой промывки шламом трубопровода 410 является периодическое открывание вентиля 412 в трубопроводе 410 и обеспечение прохождения части деаэрированного шлама через трубопровод 410 в реакционную зону 28 через нижнее отверстие 408. Даже когда вентиль 412 полностью или частично открыт, только часть деаэрированного шлама протекает через нижний трубопровод 410 и возвращается обратно в реакционную зону 28. Оставшаяся часть деаэрированного шлама, не использованная для промывки нижнего трубопровода 410, выводится через трубопровод 414 из реактора 20 для дальнейшей обработки вниз по потоку (например, для очистки).
Во время нормальной работы барботажного реактора колонного типа 20 в течение значительного промежутка времени (например, >100 часов) предпочтительно, чтобы количество деаэрированного шлама, используемого для промывки нижнего трубопровода 410, было менее 50 мас.% от общего количества деаэрированного шлама, полученного из нижней части резервуара 400 для деаэрации, более предпочтительно менее около 20 мас.% и наиболее предпочтительно менее 5 мас.%. Кроме того предпочтительно, чтобы в течение значительного промежутка времени средняя скорость потока массы деаэрированного шлама, использованного для промывки нижнего трубопровода 410, была примерно в 4 раза меньше средней скорости потока массы окисляемого соединения в реакционную зону 28, более предпочтительно около 2 раз меньше средней скорости потока массы окисляемого соединения в реакционную зоны 28, еще более предпочтительно менее средней скорости потока массы окисляемого соединения в реакционную зону 28 и наиболее предпочтительно менее 0,5 средней скорости потока массы окисляемого соединения в реакционную зону 28.
Как снова показано на фиг.19, резервуар 400 для деаэрации включает в себя существенно отвесную предпочтительно цилиндрическую боковую стенку 416, ограничивающую зону 418 деаэрации. Зона 318 деаэрации имеет диаметр «d» и высоту «h». Высота «h» определяется как вертикальное расстояние между местом, где аэрированная реакционная среда входит в резервуар 400 для деаэрации, и нижней частью боковой стенки 416. Высота «h», диаметр «d», площадь и объем зоны 418 деаэрации предпочтительно существенно такие же, как описано выше со ссылкой на зону 312 деаэрации резервуара 300, показанных на фиг. 16-18. Кроме того, резервуар 400 для деаэрации включает в себя верхнюю секцию 420, сформированную боковой стенкой 416 выше зоны 418 деаэрации. Верхняя секция 420 резервуара 400 для деаэрации может быть любой высоты, хотя она предпочтительно вытянута вверх до уровня реакционной среды 36 в реакционной зоне 28 или выше упомянутого уровня. Верхняя секция 420 обеспечивает пространство для газовой фазы, чтобы она могла должным образом отделиться от жидкой и твердой фаз перед выходом ее из резервуара 400 для деаэрации через трубопровод 404. Теперь следует отметить, что хотя трубопровод 404, как показано, возвращает отделившуюся газовую фазу в зону отделения реактора 20, трубопровод 404 может быть альтернативно подсоединен к корпусу 22 резервуара на любом возвышении по высоте над выходным трубопроводом 402. Возможно, но не обязательно, трубопровод 404 может быть соединен с выходным трубопроводом 40 для газа для того, чтобы отделившаяся газовая фаза из резервуара 40 для деаэрации могла быть соединена с удаленным верхним потоком пара в трубопроводе 40 и направлена вниз для дальнейшей обработки.
Теперь, как показано на фиг.20, барботажный реактор колонного типа 20 включает в себя гибридный внутренне-внешний резервуар 500 для деаэрации. В этой конфигурации часть реакционной среды 36 выводится из реакционной зоны 28 через сравнительно большое расположенное выше по высоте отверстие 502 в боковую стенку резервуарного корпуса 22. Выведенная реакционная среда 36 затем переносится по коленчатому трубопроводу 504 сравнительно большого диаметра и поступает в верхнюю часть резервуара 500 для деаэрации. На фиг.20 коленчатый трубопровод 504, как показано, соединен ортогонально с боковой стенкой резервуарного корпуса 22 и имеет плавный изгиб с углом около 90°. Это просто одна удобная конфигурация; и коленчатый трубопровод 504 может быть другим в любом отношении, при условии, что он эффективно соединяет барботажный реактор колонного типа 20 с внешним резервуаром 500 для деаэрации, как описано. Кроме того, коленчатый трубопровод 504 может с пользой включать в себя такие средства для регулирования потоков, как вентили.
В резервуаре 500 для деаэрации газовая фаза перемещается вверх, а твердая и жидкая фазы двигаются вниз. Перемещающаяся вверх газовая фаза может снова войти в коленчатый трубопровод 504 и затем выйти через отверстие 502 обратно в реакционную зону 28. Таким образом противоток поступающей реакционной среды 36 и выходящего отделившегося газа может возникнуть у отверстия 502. Деаэрированный шлам выводится из резервуара 500 для деаэрации через трубопровод 506. Резервуар 500 для деаэрации включает в себя существенно отвесную, предпочтительно цилиндрическую боковую стенку 508, ограничивающую зону 510 деаэрации. Зона 510 деаэрации имеет высоту «h» и диаметр «d». Предпочтительно, чтобы приподнятое отверстие 502 и коленчатый трубопровод 504 имели такой же диаметр, как и диаметр «d» зоны 510 деаэрации, или больше этого диаметра. Высота «h», диаметр «d», площадь и объем зоны 510 деаэрации предпочтительно существенно такие же, как описано выше со ссылкой на зону 312 деаэрации резервуара 300 для деаэрации, показанных на фиг. 16-18.
На фиг. 19 и 20 показано воплощение барботажного реактора колонного типа 20, где твердый продукт (например, неочищенная терефталевая кислота), полученный в реакционной зоне 28, выводится из реакционной зоны 28 через приподнятый по высоте выход. Вывод аэрированной реакционной среды 36 через приподнятое место выше нижней части барботажного реактора колонного типа 20 может помочь избежать накопления и застаивания плохо аэрированной реакционной среды 36 в нижней части 52 реакционной зоны 28. В соответствии с другими аспектами настоящего изобретения концентрация кислорода и окисляемого соединения (например, параксилола) в реакционной среде 36 рядом с верхней частью реакционной среды 36 предпочтительно ниже, чем рядом с ее нижней частью. Таким образом, отвод реакционной среды 36 у приподнятого по высоте участка может повысить выход за счет уменьшения количества непрореагировавших реагентов, отводимых из реактора 20. Температура реакционной среды 36 также значительно изменяется в продольном (по высоте) направлении, когда барботажный реактор колонного типа 20 работает при высоких нормальных температуре и давлении и при градиентах химического состава, приведенных здесь. При таких условиях температура реакционной среды будет обычно иметь локальные минимумы около низа и верха реакционной зоны 28. Минимум около низа связан с выпариванием растворителя вблизи того места, куда вводится весь окислитель или его часть. Минимум около верха снова обусловлен выпариванием растворителя, хотя здесь это обусловлено падающим давлением внутри реакционной среды. Кроме того, могут возникать другие локальные минимумы между верхом и низом каждый раз, когда в реакционную среду вводится дополнительно сырье или окислитель. Таким образом, существует один или больше температурных максимумов, вызванных экзотермической теплотой реакций окисления, между низом и верхом реакционной зоны 28. Отвод реакционной среды 36 у приподнятого по высоте места и при более высокой температуре может быть особенно выгодным, когда обработка нижнего потока происходит при более высоких температурах, так как уменьшаются затраты на энергию, связанные с нагреванием отводимой среды для обработки направленного вниз потока.
Таким образом, в предпочтительном воплощении настоящего изобретения и особенно, когда происходит обработка направленного вниз потока при более высоких температурах, реакционная среда 36 отводится из барботажного реактора колонного типа 20 через приподнятый по высоте выход (выходы), расположенные над участком (участками), через который по меньшей мере 50 мас.% жидкофазного потока сырья поступает в реакционную зону 28. Более предпочтительно, чтобы реакционная среда 36 отводилась из барботажного реактора колонного типа 20 через приподнятый выход (выходы), расположенный над участком (участками), через который по существу весь жидкофазный поток сырья и/или газофазный поток окислителя поступают в реакционную зону 28. Предпочтительно, чтобы по меньшей мере 50 мас.% компонентов твердой и жидкой фаз, выведенных из барботажного реактора колонного типа 20, были выведены через приподнятый по высоте выход (выходы). Более предпочтительно, чтобы по существу все компоненты твердой и жидкой фаз, выведенные из барботажного реактора колонного типа 20, были выведены через приподнятый по высоте выход (выходы). Предпочтительно, чтобы приподнятый по высоте выход (выходы) был расположен по меньшей мере примерно на 1D выше нижнего штуцера 52 реакционной зоны 28. Более предпочтительно, чтобы приподнятый по высоте выход (выходы) был расположен по меньшей мере на около 2D выше нижнего штуцера 52 реакционной зоны 28. Наиболее предпочтительно, чтобы приподнятый по высоте выход (выходы) был расположен по меньшей мере на 3D выше нижнего штуцера 52 реакционной зоны 28. При заданной высоте «H» реакционной среды 36 предпочтительно, чтобы приподнятый по высоте выход (выходы) был вертикально размещен между около 0,2H и около 0,8H, более предпочтительно между около 0,3H и около 0,7H и наиболее предпочтительно между 0,4H и 0,6H. Кроме того, предпочтительно, чтобы температура реакционной среды 36 при приподнятом по высоте выходе из реакционной зоны 28, была по меньшей мере на 1°С выше температуры реакционной среды у нижнего штуцера 52 реакционной зоны 28. Более предпочтительно, чтобы температура реакционной среды 36 у приподнятого по высоте выхода из реакционной зоны 28 была в интервале от около 1,5 до около 16°С выше температуры реакционной среды 36 у нижнего штуцера 52 реакционной зоны 28. Наиболее предпочтительно, чтобы температура реакционной среды 36 у приподнятого по высоте выхода из реакционной зоны 28 была в интервале от 2 до 12°С выше температуры реакционной среды 36 у нижнего штуцера 52 реакционной зоны 28.
Теперь, как показано на фиг.21, барботажный реактор колонного типа 20 включает в себя альтернативный гибридный резервуар 600 для деаэрации, расположенный в нижней части реактора 20. В этой конфигурации аэрированная реакционная среда 36 выводится из реакционной зоны 28 через относительно большое отверстие 602 в нижнем штуцере 52 резервуарного корпуса 22. Отверстие 602 ограничивает открытый верх резервуара 600 для деаэрации. В резервуаре 600 для деаэрации газовая фаза перемещается вверх, а твердая и жидкая фазы стекают вниз. Перемещающаяся вверх газовая фаза может повторно войти в реакционную зону 28 через отверстие 602. Таким образом у отверстия 602 может возникнуть противоток входящей реакционной среды 36 и выходящего отделившегося газа. Деаэрированный шлам выходит из резервуара 600 для деаэрации через трубопровод 604. Резервуар 600 для деаэрации включает в себя по существу отвесную, предпочтительно цилиндрическую боковую стенку 606, ограничивающую зону 608 деаэрации. Зона 608 деаэрации имеет высоту «h» и диаметр «d». Предпочтительно, чтобы отверстие 602 имело такой же диаметр, как и диаметр зоны 608 деаэрации, или больше последнего. Высота «h», диаметр «d», площадь и объем зоны 608 деаэрации были предпочтительно по существу такими же, как описано выше со ссылкой на зону 312 деаэрации резервуара 300 для деаэрации, показанных на фиг. 16-18.
Теперь, как показано на фиг.22, барботажный реактор колонного типа 20 включает в себя альтернативный распределитель окислителя 620. Распределитель окислителя 620 включает в себя кольцевой элемент 622 и два входных трубопровода 624, 626. Кольцевой элемент 622 предпочтительно имеет существенно такую же конфигурацию, как и кольцевой элемент 202, описанный выше со ссылкой на фиг. 12-15. Входные трубопроводы 624, 626 проходят отвесно через отверстия внизу 48 резервуарного корпуса 22 и обеспечивают вход потока окислителя в кольцевой элемент 622.
Теперь, как показано на фиг.23, барботажный реактор колонного типа 20, показанный на фиг.21, включает в себя не распределяющее средство для ввода потока окислителя в реакционную зону 28. В конфигурации на фиг.23 поток окислителя подается в реактор 20 через трубопроводы 630, 632 для окислителя. Трубопроводы 630, 632 для окислителя соединены с соответствующими отверстиями 634, 636 в низу 48 резервуарного корпуса 22. Поток окислителя непосредственно вводится в реакционную зону 28 через отверстия 634, 636 для окислителя. Возможно, но не обязательно, могут быть предусмотрены отбойники 638, 640 для отклонения потока окислителя, после того как он первоначально вошел в реакционную зону 28.
Как упомянуто выше, предпочтительно, чтобы реактор окисления имел конфигурацию и работал таким образом, чтобы устранялись зоны высокой концентрации окисляемого соединения в реакционной среде, так как такие зоны могут привести к образованию примесей. Одним способом улучшения исходной дисперсии окисляемого соединения (например, параксилола) в реакционной среде является разбавление окисляемого соединения жидкостью. Жидкость, используемая для разбавления окисляемого соединения, может быть получена из части реакционной среды, расположенной на значительном расстоянии от того места (мест), где окисляемое соединение вводится в реакционную зону. Эта жидкость из отдаленной части реакционной среды может циркулироваться к месту, близко расположенному к месту ввода окисляемого соединения через впускной трубопровод, который расположен внутри и/или с внешней стороны по отношению к основному резервуару для проведения реакций.
На фиг. 24 и 25 показаны два предпочтительных способа циркуляции жидкости из отдаленной части реакционной среды до места около входа окисляемого соединения с использованием внутреннего (фиг.24) или внешнего (фиг.25) трубопровода. Предпочтительно, чтобы длина подающего трубопровода от его входа (т.е. отверстия (отверстий), где жидкость поступает в трубопровод) до его выхода (т.е. отверстия (отверстий), где жидкость выводится из трубопровода) была больше около 1 м, более предпочтительно больше около 3 м и наиболее предпочтительно больше 9 м. Однако действительная длина трубопровода становится менее важной, если жидкость поступает из отдельного резервуара, вероятно расположенного непосредственно над резервуаром, в который первоначально выпускается окисляемое соединение, или рядом с этим резервуаром. Жидкость из любого отдельного резервуара, содержащая по меньшей мере некоторое количество реакционной среды, является предпочтительным источником для первоначального разбавления окисляемого соединения.
Предпочтительно, чтобы жидкость, стекающая по трубопроводу, какой бы не был ее источник, имела более низкую постоянную концентрацию окисляемого соединения, чем у реакционной среды, непосредственно примыкающей по меньшей мере к одному выходу трубопровода. Кроме того, предпочтительно, чтобы жидкость, протекающая через трубопровод, имела концентрацию окисляемого соединения в жидкой фазе меньше около 100000 частей на миллион, более предпочтительно меньше около 10000 частей на миллион, еще более предпочтительно меньше около 1000 частей на миллион и наиболее предпочтительно меньше 100 частей на миллион, в котором концентрации измеряются перед добавлением в трубопровод небольших количеств окисляемого соединения и любого возможного отдельного растворителя. При измерении после добавления дополнительного количества вводимого окисляемого соединения и возможного вводимого растворителя предпочтительно, чтобы объединенный поток жидкости, поступающий в реакционную среду, имел концентрацию окисляемого соединения в жидкой фазе меньше около 300000 частей на миллион, более предпочтительно меньше около 50000 частей на миллион и наиболее предпочтительно меньше 10000 частей на миллион.
Желательно поддерживать поток через трубопровод на достаточно низкой скорости так, чтобы циркулирующая жидкость подавляла требующийся общий градиент окисляемого соединения внутри реакционной среды. В этой связи предпочтительно, чтобы отношение массы жидкой фазы в реакционной зоне, в которую первоначально вводилась добавка окисляемого соединения, к скорости потока массы жидкости, протекающей через трубопровод, было больше около 0,3 минуты, более предпочтительно больше около 1 минуты, еще более предпочтительно от около 2 до около 120 минут и наиболее предпочтительно от 3 до 60 минут.
Имеется много средств заставить жидкость протекать через трубопровод. Предпочтительными средствами являются сила тяжести, эжекторные устройства всех типов, использующие или газ или жидкость, или то и другое, в качестве движущей среды, и механические насосы всех типов. Когда используется эжектор, то в одном воплощении изобретения применяется в качестве движущей среды по меньшей мере одна среда, выбранная из группы, состоящей из потока сырья окисляемого соединения (жидкости или газа), потока окислителя (газа), потока растворителя (жидкости), прокаченной реакционной среды (шлама). В другом воплощении используется в качестве движущей среды по меньшей мере две жидкости, выбранные из группы, состоящей из потока сырья окисляемого соединения, потока окислителя и потока растворителя. В другом воплощении еще используется в качестве движущей среды комбинация потока сырья окисляемого соединения, потока окислителя и потока растворителя.
Подходящий диаметр или диаметры циркуляционного трубопровода могут меняться в соответствии с количеством и свойствами переносимого материала, энергии, доступной для принудительной перекачки потока, и соображений, касающихся капитальных затрат. Предпочтительно, чтобы минимальный диаметр такого трубопровода был больше около 0,02 м, более предпочтительно между около 0,06 и около 2 м и наиболее предпочтительно между 0,12 и 0,8 м.
Как отмечалось выше, желательно регулировать поток по трубопроводу в некоторых предпочтительных интервалах. Имеется много средств, известных в этой технике, воздействия на это регулирование посредством задания подходящей фиксированной геометрии во время изготовления трубопровода для потока. В другом предпочтительном воплощении используются конфигурации, которые меняются во время работы, преимущественно включающие в себя вентили всех видов и описаний как с ручным, так и с механическим управлением любыми средствами, включая замкнутые системы автоматического регулирования с обратной связью от датчиков или без них. Другим предпочтительным средством регулирования потока разбавленной жидкости является изменение подвода энергии между входом и выходом трубопровода. Предпочтительными средствами являются изменение скорости потока одной или больше движущих жидкостей к эжектору, изменение ввода энергии в двигатель насоса и изменение разности в плотностях или в высоте размещения при использовании силы тяжести. Эти предпочтительные средства могут использоваться также во всех комбинациях.
Трубопровод, используемый для циркуляции жидкости из реакционной среды, может быть любого типа, известного в этой технике. В одном воплощении используется трубопровод, целиком или частично изготовленный с применением обычных материалов для труб. В другом воплощении применяется трубопровод, сконструированный полностью или частично с использованием стенки реактора, как одной части трубопровода. Трубопровод может быть сконструирован полностью заключенным в реактор (фиг.24) или он может быть сконструирован полностью расположенным вне его (фиг.25), или он может включать в себя секции, размещенные как внутри, так и снаружи реактора.
Изобретатели предусматривают, что особенно в более крупногабаритных реакторах может быть желательно иметь много трубопроводов различных конструкций для перемещения по ним жидкости. Кроме того, может быть желательно предусмотреть много выходов во многих позициях на одном или на всех трубопроводах. Особенности конструкции будут уравновешивать желательный общий градиент в постоянных концентрациях окисляемого соединения посредством желательного начального разбавления подаваемого окисляемого соединения в соответствии с другими аспектами данного изобретения.
На фиг. 24 и 25 показаны конструкции, в которых используются резервуар для деаэрации, соединенный с трубопроводом. Этот резервуар для деаэрации дает возможность части реакционной среды, используемой для разбавления поступающего окисляемого соединения, быть по существу частью деаэрированного шлама. Однако теперь следует отметить, что жидкость или шлам, используемый для разбавления поступающего окисляемого соедиенения, может также находится как в аэрированном виде, так и в деаэрированном виде.
Использование жидкости, протекающей по трубопроводу, для обеспечения разбавления подаваемого окисляемого соединения особенно применимо в барботажных реакторах колонного типа, кроме того в барботажных реакторах колонного типа большая выгода от начального разбавления подаваемого окисляемого соединения может быть достигнута даже без добавления подаваемого окисляемого соединения непосредственно в трубопровод при условии, что выход трубопровода расположен достаточно близко к месту добавления окисляемого соединения. В таком воплощении предпочтительно, чтобы выход трубопровода был расположен в пределах около 27 выходных диаметров трубопровода от ближайшего места добавления окисляемого соединения, более предпочтительно в пределах около 9 выходных диаметров трубопровода, еще более предпочтительно в пределах около 3 выходных диаметров трубопровода и наиболее предпочтительно в пределах 1 выходного диаметра трубопровода.
Также было обнаружено, что эжекторы потока могут быть пригодны для начального разбавления подаваемого окисляемого соединения в барботажных колоннах окисления в соответствии с одним воплощением настоящего изобретения даже без использования трубопроводов для получения разбавляющей жидкости из удаленной части реакционной среды. В таких случаях эжектор расположен внутри реакционной среды и имеет открытый проток из реакционной среды во вход эжектора, соседняя реакционная среда втягивается за счет пониженного давления. Примеры двух возможных конфигураций эжектора приведены на фиг. 26 и 27. В предпочтительном воплощении этих эжекторов самое ближайшее место расположения подаваемого окисляемого соединения находится в пределах около 4 м, более предпочтительно в пределах около 1 м и наиболее предпочтительно в 0,3 м от входа эжектора. В другом воплощении окисляемое соединение подается под давлением в качестве движущей жидкости. Еще в одном воплощении или растворитель или окислитель подается под давлением в качестве дополнительной движущей среды вместе с окисляемым соединением. Еще в одном воплощении как растворитель, так и окислитель подаются под давлением в качестве дополнительной движущей среды вместе с окисляемым соединением.
Изобретатели предусматривают, что особенно в более крупногабаритных реакторах может быть желательно иметь много эжекторов и различных конструкций, расположенных в различных местах внутри реакционной среды. Особенности конструкций сбалансируют желательный общий градиент постоянных концентраций окисляемого соединения посредством желательного начального разбавления подаваемого окисляемого соединения в соответствии с другими аспектами данного изобретения. Кроме того, изобретатели предусматривают, что распределение выходного потока из эжектора может быть ориентировано в любом направлении. Когда используются много эжекторов, каждый эжектор может быть ориентирован по отдельности и опять-таки в любом направлении.
Как упомянуто выше, некоторые физические и эксплуатационные особенности барботажного реактора колонного типа 20, описанные выше со ссылкой на фиг. 1-27, обеспечивают градиенты давления по высоте, температуре и концентрациям реагентов (т.е. кислорода и окисляемого соединения) в реакционной среде 36. Как рассмотрено выше, эти градиенты по высоте могут обеспечить более эффективный и экономичный процесс окисления по сравнению с общеизвестными процессами окисления, для которых предпочтительна хорошо перемешанная реакционная среда со сравнительно равномерными давлением, температурой и концентрациями реагентов по всему объему. Градиенты по высоте для кислорода, окисляемого соединения (например, параксилола) и температуры стали возможны благодаря использованию системы окисления в соответствии с воплощением настоящего изобретения, которое теперь будет рассмотрено более подробно.
Теперь, как показано на фиг.28, чтобы количественно оценить градиенты концентраций реагентов, имеющие место в реакционной среде 36 во время окисления в барботажном реакторе колонного типа 20, весь объем реакционной среды 36 теоретически может быть разделен на 30 дискретных горизонтальных слоев равного объема. На фиг.28 представлена эта концепция разделения реакционной среды на 30 дискретных горизонтальных слоев одинакового объема. За исключением верхнего и нижнего горизонтальных слоев, каждый горизонтальный слой является дискретным объемом, ограниченным сверху и снизу воображаемыми горизонтальными плоскостями и с боковых сторон стенкой реактора 20. Самый высокий горизонтальный слой ограничен со своей нижней стороны воображаемой горизонтальной плоскостью, а со своей верхней стороны - верхней поверхностью реакционной среды 36. Самый нижний горизонтальный слой ограничен со своей верхней стороны воображаемой горизонтальной плоскостью, а со своей нижней стороны - нижней частью корпуса резервуара. После того как реакционная среда 36 была теоретически разделена на 30 дискретных горизонтальных слоев равного объема, может быть определена усредненная по времени и по объему концентрация реагентов в каждом горизонтальном слое. Отдельный горизонтальный слой, имеющий максимальную концентрацию из всех 30 горизонтальных слоев, может быть идентифицирован как «горизонтальный слой с максимальной концентрацией, С-макс». Отдельный горизонтальный слой, расположенный выше горизонтального слоя с максимальной концентрацией и имеющий минимальную концентрацию из всех горизонтальных слоев, расположенных выше горизонтального слоя с максимальной концентрацией, может быть идентифицирован как «горизонтальный слой с минимальной концентрацией, С-мин». После этого может быть рассчитан градиент концентрации по высоте как отношение концентрации в горизонтальном слое с С-макс к концентрации в горизонтальном слое с С-мин.
По отношению к количественной оценке градиента концентрации кислорода, когда реакционная среда 36 теоретически делится на 30 дискретных горизонтальных слоев равного объема, горизонтальный слой с O2-макс идентифицируется как имеющий максимальную концентрацию кислорода из всех 30 горизонтальных слоев, и горизонтальный слой с O2-мин идентифицируется как имеющий минимальную концентрацию кислорода из всех горизонтальных слоев, расположенных выше горизонтального слоя с O2-макс. Концентрации кислорода в горизонтальных слоях измеряются в газовой фазе реакционной среды 36 в пересчете на усредненное по времени и по объемам влажное вещество в молях. Предпочтительно, чтобы отношение концентрации кислорода горизонтального слоя с O2-макс к концентрации кислорода горизонтального слоя с O2-мин было в интервале от около 2:1 до около 25:1, более предпочтительно в интервале от около 3:1 до около 15:1 и наиболее предпочтительно в интервале от 4:1 до 10:1.
Обычно горизонтальный слой с O2-макс будет расположен около нижней части реакционной среды 36, а горизонтальный слой с O2-мин будет расположен около верхней части реакционной среды 36. Предпочтительно, чтобы горизонтальный слой с O2-мин был одним из 5 самых верхних горизонтальных слоев из 30 дискретных горизонтальных слоев. Наиболее предпочтительно, чтобы горизонтальный слой с O2-мин был самым верхним из 30 дискретных горизонтальных слоев, как показано на фиг.28. Предпочтительно, чтобы горизонтальный слой с O2-макс был одним из 10 самых нижних горизонтальных слоев из 30 дискретных горизонтальных слоев. Наиболее предпочтительно, чтобы горизонтальный слой с O2-макс был одним из 5 самых нижних горизонтальных слоев из 30 дискретных горизонтальных слоев. Например, на фиг.28 показан горизонтальный слой с O2-макс как третий горизонтальный слой из нижней части реактора 20. Предпочтительно, чтобы вертикальный промежуток между горизонтальными слоями с O2-мин и с O2-макс был по меньшей мере около 2W, более предпочтительно по меньшей мере около 4W и наиболее предпочтительно по меньшей мере 6W. Предпочтительно, чтобы вертикальный промежуток между горизонтальными слоями с O2-мин и c O2-макс был по меньшей мере около 0,2Н, более предпочтительно по меньшей мере около 0,4Н и наиболее предпочтительно по меньшей мере 0,6Н.
Усредненная по времени и по объему концентрация кислорода в пересчете на влажное вещество горизонтального слоя с O2-мин предпочтительно находится в интервале от около 0,1 до около 3% мольных, более предпочтительно в интервале от около 0,3 до около 2% мольных и наиболее предпочтительно в интервале от 0,5 до 1,5% мольных. Усредненная по времени и усредненная по объему концентрация кислорода горизонтального слоя с O2-макс предпочтительно находится в интервале от около 4 до около 20% мольных, более предпочтительно в интервале от около 5 до около 15% мольных и наиболее предпочтительно в интервале от 6 до 2% мольных. Усредненная по времени концентрация кислорода в пересчете на сухое вещество в газообразном выходящем потоке, выводимом из реактора 20 через выход 40 для газа, предпочтительно находится в интервале от около 0,5 до около 9% мольных, более предпочтительно в интервале от около 1 до около 7% мольных и наиболее предпочтительно в интервале от 1,5 до 5% мольных.
В связи с тем, что концентрация кислорода так заметно уменьшается к верхней части реакционной среды 36, желательно, чтобы потребность в кислороде уменьшалась в верхней части реакционной среды 36. Эта сниженная потребность в кислороде около верхней части реакционной среды 36 может быть достигнута созданием градиента концентрации по высоте окисляемого соединения (например, параксилола) там, где минимальная концентрация окисляемого соединения расположена около верхней части реакционной среды 36.
Что касается количественной оценки градиента концентрации окисляемого соединения (например, параксилола), когда реакционная среда 36 теоретически делится на 30 дискретных горизонтальных слоев, то горизонтальный слой с ОС-макс идентифицируется как слой, имеющий максимальную концентрацию окисляемого соединения из всех 30 горизонтальных слоев, и горизонтальный слой с ОС-мин идентифицируется как слой, имеющий минимальную концентрацию окисляемого соединения из всех слоев, расположенных над горизонтальным слоем с ОС-макс. Концентрации окисляемого соединения в горизонтальных слоях измеряются в жидкой фазе в пересчете на усредненную по времени и усредненную по объему массовую фракцию. Предпочтительно, чтобы отношение концентрации окисляемого соединения в горизонтальном слое с ОС-макс к концентрации окисляемого соединения в горизонтальном слое с ОС-мин было больше около 2:1, более предпочтительно больше около 10:1, еще более предпочтительно больше около 20:1 и наиболее предпочтительно в интервале от 40:1 до 1000:1.
Обычно горизонтальный слой с ОС-макс будет расположен около нижней части реакционной среды 36, а горизонтальный слой с ОС-мин будет расположен около верхней части реакционной среды 36. Предпочтительно, чтобы горизонтальный слой с ОС-мин был одним из 5 самых верхних горизонтальных слоев из 30 дискретных горизонтальных слоев. Наиболее предпочтительно, чтобы горизонтальный слой с ОС-мин был самым верхним из 30 дискетных горизонтальных слоев, как показано на фиг.28. Предпочтительно, чтобы горизонтальный слой с ОС-макс был одним из 10 самых нижних горизонтальных слоев из 30 дискретных горизонтальных слоев. Наиболее предпочтительно, чтобы горизонтальный слой с ОС-макс был одним из 5 самых нижних горизонтальных слоев из 30 дискретных горизонтальных слоев. Например, как показано на фиг.28, горизонтальный слой с ОС-макс является пятым горизонтальным слоем от нижней части реактора 20. Предпочтительно, чтобы вертикальный промежуток между горизонтальными слоями с ОС-мин и ОС-макс был по меньшей мере около 2W, где «W» является максимальной шириной реакционной среды 36. Более предпочтительно, чтобы вертикальный промежуток между горизонтальными слоями с ОС-мин и ОС-макс был по меньшей мере около 4W и наиболее предпочтительно по меньшей мере 6W. При заданной высоте «Н» реакционной среды 36, предпочтительно, чтобы вертикальный промежуток между горизонтальными слоями с ОС-мин и ОС-макс был по меньшей мере около 0,2Н, более предпочтительно по меньшей мере около 0,4Н и наиболее предпочтительно по меньшей мере 0,6Н.
Усредненная по времени и усредненная по объему концентрация окисляемого соединения (например, параксилола) в жидкой фазе горизонтального слоя с ОС-мин предпочтительно меньше около 5000 частей на миллион, более предпочтительно меньше около 2000 частей на миллион, еще более предпочтительно меньше около 400 частей на миллион и наиболее предпочтительно в интервале от 1 до 100 частей на миллион. Усредненная по времени и усредненная по объему концентрация окисляемого соединения в жидкой фазе горизонтального слоя с ОС-макс предпочтительно находится в интервале от около 100 до около 10000 частей на миллион, более предпочтительно в интервале от около 200 до около 5000 частей на миллион и наиболее предпочтительно в интервале от 500 до 3000 частей на миллион.
Хотя предпочтительно, чтобы барботажный реактор колонного типа 20 обеспечивал градиенты по высоте концентрации окисляемого соединения, также предпочтительно, чтобы минимизировался объемный процент реакционный среды 36, имеющей концентрацию окисляемого соединения в жидкой фазе выше 1000 частей на миллион. Предпочтительно, чтобы усредненный по времени объемный процент реакционной среды 36, имеющей концентрацию окисляемого соединение в жидкой фазе выше 1000 частей на миллион, был меньше чем около 9%, более предпочтительно меньше чем около 6% и наиболее предпочтительно меньше 3%. Предпочтительно, чтобы усредненный по времени объемный процент реакционной среды 36, имеющей концентрацию окисляемого соединения в жидкой фазе выше 2500 частей на миллион, был меньше чем около 1,5%, более предпочтительно меньше чем около 1% и наиболее предпочтительно меньше 0,5%. Предпочтительно, чтобы усредненный по времени объемный процент реакционной среды 36, имеющей концентрацию окисляемого соединения в жидкой фазе выше 10000 частей на миллион, был меньше чем около 0,3, более предпочтительно меньше чем 0,1% и наиболее предпочтительно меньше 0,03%. Предпочтительно, чтобы усредненный по времени объемный процент реакционной среды 36, имеющей концентрацию окисляемого соединения в жидкой фазе выше 25000 частей на миллион, был меньше чем около 0,03%, более предпочтительно меньше чем около 0,015% и наиболее предпочтительно меньше 0,007%. Изобретатели отмечают, что объем реакционной среды 36, имеющий повышенные уровни окисляемого соединения, не должен находится в одном непрерывном объеме. В различное время хаотичные конфигурации потока в резервуаре барботажного реактора колонного типа дают одновременно две или больше смежных, но отделенных частей реакционной среды 36, имеющих повышенные уровни окисляемого соединения. В каждом промежутке времени, используемом для усреднения по времени, все такие смежные, но отделенные объемы больше 0,0001 объемного процента всей реакционной среды суммируются для определения общего объема, имеющего повышенные уровни концентраций окисляемого соединения в жидкой фазе.
Кроме градиентов концентрации кислорода и окисляемого соединения, рассмотренных выше, предпочтительно, чтобы существовал температурный градиент в реакционной среде 36. Как опять показано на фиг.28, этот температурный градиент может быть количественно оценен таким же образом, как и градиенты концентрации, теоретическим разделением реакционной среды 36 на 30 дискретных горизонтальных слоев равного объема и измерением усредненной по времени и усредненной по объему температуры каждого слоя. Горизонтальный слой с самой низкой температурой из самых низко расположенных 15 горизонтальных слоев после этого может быть идентифицирован как «горизонтальный слой с Т-мин», а горизонтальный слой, расположенный выше горизонтального слоя с Т-мин и имеющий максимальную температуру из всех слоев выше горизонтального слоя с Т-мин, может быть после этого идентифицирован как «горизонтальный слой с Т-макс». Предпочтительно, чтобы температура горизонтального слоя с Т-макс была по меньшей мере на около 1°С выше температуры горизонтального слоя с Т-мин. Более предпочтительно, чтобы температура горизонтального слоя с Т-макс была выше в интервале от около 1,25 до около 12°С температуры горизонтального слоя с Т-мин. Наиболее предпочтительно, чтобы температура горизонтального слоя с Т-макс была выше в интервале от 2 до 8°С температуры горизонтального слоя с Т-мин. Температура горизонтального слоя с Т-макс предпочтительно находится в интервале от около 125 до около 200°С, более предпочтительно в интервале от около 140 до около 180°С и наиболее предпочтительно в интервале от 150 до 170°С.
Обычно горизонтальный слой с Т-макс будет расположен около центра реакционной среды 36, а горизонтальный слой с Т-мин будет расположен около нижней части реакционной среды 36. Предпочтительно, чтобы горизонтальный слой с Т-мин был одним из 10 наиболее низких горизонтальных слоев из 15 самых низких горизонтальных слоев. Наиболее предпочтительно, чтобы горизонтальный слой с Т-мин был одним из 5 наиболее низких горизонтальных слоев из 15 самых низких горизонтальных слоев. Например, на фиг.28 показан горизонтальный слой с Т-мин как второй горизонтальный слой от нижней части реактора 20. Предпочтительно, чтобы горизонтальный слой с Т-макс был одним из 20 средних горизонтальных слоев из 30 дискретных горизонтальных слоев. Наиболее предпочтительно, чтобы горизонтальный слой с Т-мин был одним из 14 средних горизонтальных слоев из 30 дискретных слоев. Например, на фиг.28 показан горизонтальный слой с Т-макс как двадцатый горизонтальный слой от нижней части реактора 20 (т.е. один из средних 10 горизонтальных слоев). Предпочтительно, чтобы промежуток по высоте между горизонтальным слоем с Т-мин и горизонтальным слоем с Т-макс был по меньшей мере около 2W, более предпочтительно по меньшей мере около 4W и наиболее предпочтительно по меньшей мере 6W. Предпочтительно, чтобы вертикальный промежуток между горизонтальными слоями с Т-мин и Т-макс был по меньшей мере около 0,2Н, более предпочтительно по меньшей мере около 0,4Н и наиболее предпочтительно по меньшей мере 0,6Н.
Как рассмотрено выше, когда градиент температуры по высоте существует в реакционной среде 36, то может быть выгодно выводить реакционную среду 36 у приподнятого по высоте места, где температура реакционной среды самая высокая, особенно когда выведенный продукт подвергается дальнейшей обработке по ходу потока при более высоких температурах. Таким образом, когда реакционная среда 36 выводится из реакционной зоны 28 через один или больше приподнятых по высоте выходов, как показано на фиг. 19 и 20, то предпочтительно, чтобы приподнятый по высоте выход (выходы) был расположен близко к горизонтальному слою с Т-макс. Предпочтительно, чтобы приподнятый по высоте выход был расположен в пределах 10 горизонтальных слоев от горизонтального слоя с Т-макс, более предпочтительно в пределах 5 горизонтальных слоев от горизонтального слоя с Т-макс и наиболее предпочтительно в пределах 2 горизонтальных слоев от горизонтального слоя с Т-макс.
Теперь следует отметить, что многие из отличительных особенностей изобретения, описанных здесь, могут быть использованы во многих системах реакторов окисления - не только в системах, использующих один реактор окисления. Кроме того, некоторые отличительные особенности изобретения, рассмотренные здесь, могут быть использованы в реакторах окисления с механическим перемешиванием или с перемешиванием протоками - не только в реакторах с барботажным перемешиванием (т.е. в барботажных реакторах колонного типа). Например, изобретатели обнаружили некоторые преимущества, связанные со ступенчатой/изменяющейся концентрацией кислорода и/или скоростью расходования кислорода в объеме реакционной среды. Преимущества, полученные при ступенчатом изменении концентрации/расходования кислорода в реакционной среде, могут быть реализованы независимо от того, содержится ли весь объем реакционной среды в одном резервуаре или в нескольких резервуарах. Кроме того, преимущества, полученные при ступенчатом изменении концентрации/расходования кислорода в реакционной среде, могут быть реализованы независимо от того, имеет ли реактор (реакторы) механическое перемешивание, перемешивание потоками и/или барботажное перемешивание.
Одним способом количественной оценки степени ступенчатого изменения концентрации кислорода и/или скорости его расходования в реакционной среде является сравнение двух или нескольких отчетливых 20% непрерывных объемов реакционной среды. Эти 20% непрерывные объемы не должны определяться какой-либо определенной формой. Однако каждый 20% непрерывный объем должен быть сформирован из непрерывного объема реакционной среды (т.е. каждый объем непрерывен), и 20% непрерывные объемы не должны перекрывать друг друга (т.е. объемы «отчетливые»). На фиг. 29-31 показано, что эти отчетливые 20% непрерывные объемы могут быть расположены в одном и том же реакторе (фиг.29) или в разных реакторах (фиг. 30 и 31). Следует отметить, что реакторы, показанные на фиг. 29-31, могут быть реакторами с механическим перемешиванием, с перемешиванием потоками и/или барботажным перемешиванием. В одном воплощении предпочтительно, чтобы реакторы, показанные на фиг. 29-31, были реакторами с барботажным перемешиванием (т.е. барботажными реакторами колонного типа).
Теперь, как показано на фиг.29, реактор 20 содержит реакционную среду 36. Реакционная среда 36 включает в себя первый четкий 20% непрерывный объем 37 и второй четкий 20% непрерывный объем 39.
Теперь, как показано на фиг.30, система с несколькими реакторами включает в себя первый реактор 720a и второй реактор 720b. Реакторы 720a,b совместно содержат общий объем реакционной среды 736. Первый реактор 720a содержит первую часть 736a реакционной среды, а второй реактор 720b содержит вторую часть 736b реакционной среды. Первый четкий 20% непрерывный объем 737 реакционной среды 736, как показано, определен внутри первого реактора 720a, а второй четкий 20% непрерывный объем 739 реакционной среды 736, как показано, определен внутри второго реактора 720b.
Теперь, как показано на фиг.31, система с несколькими реакторами включает в себя первый реактор 820a, второй реактор 820b и третий реактор 820с. Реакторы 820a,b,c совместно содержат общий объем реакционной среды 836. Первый реактор 820a содержит первую часть 836a реакционной среды, второй реактор 820b содержит вторую часть 836b реакционной среды и третий реактор 820с содержит третью часть 836с реакционной среды. Первый четкий 20% непрерывный объем 837 реакционной среды 836, как показано, определен внутри первого реактора 820a; второй четкий 20% непрерывный объем реакционной среды 836, как показано, определен внутри второго реактора 820b; и третий четкий 20% непрерывный объем 841 реакционной среды 836, как показано, определен внутри третьего реактора 820с.
Ступенчатое распределение содержания кислорода в реакционной среде может быть количественно представлено со ссылкой на 20% непрерывный объем реакционной среды, имеющей наиболее обогащенную мольную фракцию кислорода в газовой фазе, и со ссылкой на 20% непрерывный объем реакционной среды, имеющей наиболее обедненную мольную фракцию кислорода в газовой фазе. В газовой фазе четкого 20% непрерывного объема реакционной среды, содержащей самую высокую концентрацию кислорода в газовой фазе, усредненная по времени и усредненная по объему концентрация кислорода в пересчете на влажное вещество находится предпочтительно в интервале от около 3 до около 18 мольных %, более предпочтительно в интервале от около 3,5 до около 14 мольных % и наиболее предпочтительно в интервале от 4 до 10 мольных %. В газовой фазе четкого 20% непрерывного объема реакционной среды, содержащей самую низкую концентрацию кислорода в газовой фазе, усредненная по времени и усредненная по объему концентрация кислорода в пересчете на влажное вещество находится предпочтительно в интервале от около 0,3 до около 5 мольных %, более предпочтительно в интервале от около 0,6 до около 4 мольных % и наиболее предпочтительно в интервале от 0,9 до 3 мольных %. Более того, отношение усредненной по времени и усредненной по объему концентрации кислорода в пересчете на влажное вещество в наиболее обогащенном 20% непрерывном объеме реакционной среды к усредненной по времени и по объему концентрации кислорода в пересчете на влажное вещество в наиболее обедненном 20% непрерывном объеме реакционной среды находится предпочтительно в интервале от около 1,5:1 до около 20:1, более предпочтительно в интервале от около 2:1 до около 12:1 и наиболее предпочтительно в интервале от 3:1 до 9:1.
Ступенчатое распределение скорости расходования кислорода в реакционной среде может быть количественно оценено в виде объемно-временной скорости расхода кислорода в реакции, первоначально рассмотренной выше (кислородной ОВС). Кислородная ОВС ранее рассматривалась в общем смысле (т.е. с точки зрения средней кислородной ОВС всей реакционной среды), однако кислородная ОВС может также рассматриваться в локальном смысле (т.е. части реакционной среды), чтобы количественно представить ступенчатое распределение скорости расходования кислорода в объеме реакционной среды.
Изобретатели выявили, что очень полезно вызвать изменение кислородной ОВС во всем объеме реакционной среды в полном соответствии с желательными градиентами, рассмотренными здесь в связи с давлением в реакционной среде и с мольной фракцией молекулярного кислорода в газовой фазе реакционной среды. Таким образом, предпочтительно, чтобы отношения кислородной ОВС первого четкого 20% непрерывного объема реакционной среды к кислородной ОВС второго четкого 20% непрерывного объема реакционной среды было в интервале от около 1,5:1 до около 20:1, более предпочтительно в интервале от около 2:1 до около 12:1 и наиболее предпочтительно в интервале от 3:1 до 9:1. В одном воплощении «первый четкий 20% непрерывный объем» расположен ближе «второго четкого 20% непрерывного объема» к месту, где молекулярный кислород первоначально вводится в реакционную среду. Эти большие градиенты кислородной ОВС желательны, независимо от того, проводится ли частичное окисление реакционной среды в барботажном реакторе колонного типа окисления или в любом другом типе реактора, в котором имеют место градиенты давления и/или мольной фракции молекулярного кислорода в газовой фазе реакционной среды (например, в реакторе с механическим перемешиванием, имеющим несколько вертикально расположенных зон перемешивания, достигаемых с помощью нескольких вращающихся крыльчаток с сильным радиальным потоком, возможно увеличенным преимущественно горизонтальными сборками отражательных перегородок, при этом поток окислителя поднимается обычно вверх от места ввода рядом с нижней частью реактора, несмотря на это может произойти значительное обратное перемешивание потока окислителя внутри каждой вертикально расположенной зоны перемешивания и некоторое обратное перемешивание потока окислителя может произойти между соседними вертикально расположенными зонами перемешивания). То есть, когда существует градиент давления и/или мольной фракции молекулярного кислорода в газовой фазе реакционной среды, то изобретатели обнаружили, что желательно создавать аналогичный градиент химической потребности в растворенном кислороде такими средствами, как описаны здесь.
Предпочтительным средством для создания изменения локальной кислородной ОВС является регулирование мест ввода окисляемого соединения и управление смешиванием жидкой фазы реакционной среды, чтобы управлять градиентами концентрации окисляемого соединения в соответствии с другими описаниями настоящего изобретения. Другие полезные средства, способные вызвать локальное изменение кислородной ОВС, включают в себя изменение активности реакции благодаря изменению локальной температуры и изменению локальной смеси компонентов катализатора и растворителя (например, за счет ввода дополнительных количеств газа, чтобы вызвать охлаждение испарением в определенной части реакционной среды, и за счет добавления потока растворителя, содержащего большее количество воды, чтобы уменьшить активность в определенной части реакционной среды).
Как рассмотрено выше со ссылкой на фиг.30 и 31, реакция частичного окисления может с пользой проводиться в нескольких реакторах, в которых по меньшей мере часть, предпочтительно по меньшей мере 25%, более предпочтительно по меньшей мере 50% и наиболее предпочтительно по меньшей мере 75% молекулярного кислорода, выходящего из первого реактора, отводится к одному или нескольким последующим реакторам для потребления дополнительных добавок, предпочтительно больше 10%, более предпочтительно больше 20% и наиболее предпочтительно больше 40% молекулярного кислорода, выходящего из первого по направлению потока реактора. При использовании такой последовательности потока молекулярного кислорода из одного реактора в другой желательно, чтобы первый реактор работал при более высокой интенсивности реакции, чем по меньшей мере один из последующих реакторов, предпочтительно при отношении усредненной по реактору кислородной ОВС внутри первого реактора к усредненной по реактору кислородной ОВС внутри следующего реактора в интервале от около 1,5:1 до около 20:1, более предпочтительно в интервале от около 2:1 до около 12:1 и наиболее предпочтительно в интервале от 3:1 до 9:1.
Как рассмотрено выше, все типы первого реактора (например, барботажная колонна, с механическим перемешиванием, с обратным смешиванием, с внутренним ступенчатым распределением, с потоком вытеснения и так далее) и все типы последующих реакторов, которые могут быть других типов, а могут и не быть другими типами, отличающимися от первого реактора, пригодны для последовательностей потока молекулярного кислорода в последующие реакторы в соответствии с настоящим изобретением. Средства, вызывающие уменьшение усредненной по резервуару кислородной ОВС внутри последующих реакторов, полезно включают в себя понижение температуры, уменьшение концентрации окисляемого соединения и уменьшение реакционной активности конкретной смеси каталитических компонентов и растворителя (например, уменьшение концентрации кобальта, повышение концентрации воды и добавление каталитического замедлителя, такого как небольшие количества ионов меди).
При протекании от первого реактора к следующему реактору поток окислителя может быть обработан любым средством, известным в этой технике, таким как сжатие или уменьшение давления, охлаждение или нагревание, и удаление массы или добавление массы в любом количестве или любого типа. Однако использование уменьшающейся усредненной по резервуару кислородной ОВС в последующих реакторах особенно полезно, когда абсолютное давление в верхней части первого реактора меньше чем около 2,0 МПа, более предпочтительно меньше чем около 1,6 МПа и наиболее предпочтительно меньше 1,2 МПа. Кроме того, использование уменьшающейся усредненной по резервуару кислородной ОВС в последующих реакторах особенно полезно, когда отношение абсолютного давления в верхней части первого реактора к абсолютному давлению в верхней части по меньшей мере одного следующего реактора находится в интервале от около 0,5:1 до 6:1, более предпочтительно в интервале от около 0,6:1 до около 4:1 и наиболее предпочтительно в интервале от 0,7:1 до 2:1. Уменьшение давления в последующих реакторах ниже этих нижних границ слишком снижает доступность молекулярного кислорода, и повышение давления выше этих верхних границ обходится чрезвычайно дорого по сравнению с использованием свежего окислителя.
При использовании последовательного потока молекулярного кислорода в следующие реакторы, имеющие уменьшающиеся усредненные по резервуару кислородные ОВС, могут подаваться свежие входные потоки окисляемого соединения, растворителя и окислителя в последующие реакторы и/или в первый реактор. Потоки жидкой фазы и твердой фазы реакционной среды, если они присутствуют, могут протекать в любом направлении между реакторами. Вся газовая фаза или часть ее, выходящая из первого реактора и поступающая в следующий реактор, может вытекать из первого реактора отдельно от частей жидкой фазы или твердой фазы реакционной среды, если они присутствуют, или в виде смеси с последними. Поток продукта, содержащего жидкую фазу и твердую фазу, если присутствует, может отводиться из реакционной среды в любой реактор в системе.
Как показано снова на фиг. 1-29, окисление предпочтительно проводится в барботажном реакторе колонного типа 20 в условиях, которые заметно отличаются, в соответствии с предпочтительными воплощениями, раскрытыми здесь, от условий в известных реакторах окисления. Когда используется барботажный реактор колонного типа 20 для проведения жидкофазного частичного окисления параксилола до неочищенной терефталевой кислоты (НТК) в соответствии с предпочтительными воплощениями, раскрытыми здесь, пространственные профили локальной интенсивности реакции, локальной интенсивности выпаривания и локальной температуры наряду с видами течения жидкости в реакционной среде и предпочтительные сравнительно низкие температуры окисления способствуют формированию частиц НТК, имеющих уникальные и полезные свойства.
На фиг. 32A и 32B показаны исходные частицы НТК, полученной в соответствии с одним воплощением настоящего изобретения. На фиг.32А показаны исходные частицы НТК под 500-кратным увеличением, а на фиг.32В - изображение одной из частиц НТК, увеличенной в 2000 раз. Как вероятно лучше всего показано на фиг.32В, каждая исходная частица НТК обычно образована из большого числа малых агломерированных субчастиц НТК, обеспечивающих тем самым исходную частицу НТК со сравнительно большой площадью поверхности, высокой пористостью, низкой плотностью и хорошей растворимостью. Исходные частицы НТК обычно имеют средний размер в интервале от около 20 до около 150 мк, более предпочтительно в интервале от около 30 до около 120 мк и наиболее предпочтительно в интервале от 40 до 90 мк. Субчастицы НТК обычно имеют средний размер в интервале от около 0,5 до около 30 мк, более предпочтительно от около 1 до около 15 мк и наиболее предпочтительно в интервале от 2 до 5 мк. Относительно высокая площадь поверхности исходных частиц НТК, показанных на фиг. 32А и 32В, количественно определена с использованием способа измерения площади поверхности Браунауэра-Эмметта-Теллера (способ БЭТ). Предпочтительно, чтобы исходные частицы НТК имели среднюю площадь поверхности (по БЭТ) по меньшей мере около 0,6 кв.м/г. Более предпочтительно, чтобы исходные частицы НТК имели среднюю площадь поверхности (по БЭТ) в интервале от около 0,8 до около 4 кв.м/г. Наиболее предпочтительно, чтобы исходные частицы НТК имели среднюю площадь поверхности (по БЭТ) в интервале от 0,9 до 2 кв.м/г. Физические свойства (например, размер частиц, площадь поверхности по БЭТ, пористость и растворимость) исходных частиц НТК, полученных по оптимизированному процессу окисления в соответствии с предпочтительным воплощением настоящего изобретения, позволяют очистить частицы НТК более эффективными и/или экономичными способами, как более подробно описано ниже со ссылкой на фиг.35.
Средние размеры частиц, представленные выше, были определены с использованием микроскопии в поляризованном свете и анализа изображений. Оборудование, используемое при анализе размера частиц, включало в себя оптический микроскоп Е800 фирмы Nikon c объективом 4× Plan Flour N.A. 0.13, цифровую фотокамеру Spot RTTM и персональный компьютер с программой для анализа изображений Image Pro PlusTM V4.5.0.19. Способ анализа размера частиц включал в себя следующие основные операции: (1) диспергирование порошка НТК в минеральном масле; (2) приготовление предметных стекол с дисперсией для микроскопа; (3) изучение предметных стекол с помощью микроскопии в поляризованном свете (условие скрещенных поляр - частицы видны как яркие объекты на черном фоне); (4) получение различных изображений для каждого приготовленного образца (размер поля = 3×2,25 мм, размер пикселя = 1,84 мк/пиксель); (5) проведение анализа с помощью программы Image Pro Plus; (6) перенос размеров частиц в сводную таблицу и (7) осуществление статистической обработки данных в сводной таблице. Операция (5) «проведение анализа изображений с помощью программы Image Pro Plus» включала в себя операции: (а) задание оптического порога для детектирования белых частиц на темном фоне; (b) создание бинарного изображения; (с) использование однопроходного открытого фильтра для отфильтровывания шумовых пикселей; (d) измерение всех частиц в изображении и (е) сообщение о среднем измеренном диаметре для каждой частицы. Программа Image Pro Plus определяет средний диаметр отдельных частиц как числовую среднюю длину диаметров частицы, измеренных с интервалами во 2-й степени и проходящих через центроиду частицы. Операция 7 «осуществление статистической обработки данных в сводной таблице» включает в себя расчет объемно-взвешенного среднего размера частиц следующим образом. Объем каждой из n частиц в образце рассчитывается так, как будто он имеет сферическую форму, используя pi/6*di^3; объем каждой частицы умножается на ее диаметр для нахождения pi/6*di^4; для всех частиц в образце суммируются величины pi/6*di^4; суммируются объемы всех частиц в образце; и рассчитывается объемно-взвешенный диаметр частицы как сумма для всех n частиц в образце (pi/6*di^4), деленная на сумму для всех n частиц в образце (pi/6*di^3). Используемый здесь термин «средний размер частицы» относится к объемно-взвешенному среднему размеру частицы, определенному в соответствии с вышеприведенным способом испытания; и на него также дается ссылка как на D(4,3), рассчитываемому по формуле
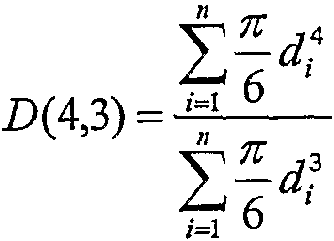
Кроме того, операция 7 включает в себя определение размеров частиц, для которых различные фракции во всем объеме образца меньше. Например, D(v,0,1) является размером частиц, для которых 10% общего объема образца меньше и 90% больше; D(v,0,5) является размером частиц, для которых одна половина объема образца больше, а другая половина меньше; D(v,0,9) является размером частиц, для которых 90% общего объема образца меньше, и так далее. Кроме того, операция 7 включает в себя расчет величины D(v,0,9) минус D(v,0,1), которая здесь определена как «разброс размеров частиц», и операция 7 включает в себя расчет величины разброса размера частиц, деленного на D(4,3), которая здесь определена как «относительный разброс размера частиц».
Помимо этого предпочтительно, чтобы D(v,0,1) частиц НТК, измеренная выше, была в интервале от около 5 до около 65 мк, более предпочтительно в интервале от около 15 до около 55 мк и наиболее предпочтительно в интервале от 25 до 45 мк. Предпочтительно, чтобы D(v,0,5) частиц НТК, измеренная выше, была в интервале от около 10 до около 90 мк, более предпочтительно в интервале от около 20 до около 80 мк и наиболее предпочтительно в интервале от 30 до 70 мк. Предпочтительно, чтобы D(v,0,9) частиц НТК, измеренная выше, была в интервале от около 30 до около 150 мк, более предпочтительно в интервале от около 40 до около 130 мк и наиболее предпочтительно в интервале от 50 до 110 мк. Предпочтительно, чтобы относительный разброс размера частиц был в интервале от около 0,5 до около 2,0, более предпочтительно в интервале от около 0,6 до около 1,5 и наиболее предпочтительно в интервале от 0,7 до 1,3.
Площади поверхности частиц по способу БЭТ, приведенные выше, были измерены на приборе Micromeritics ASAP2000 (поставляемом фирмой Micromeritics Instrument Corp., из Норкросса, шт. Джорджия). В первой операции в процессе измерения взвешивалось от 2 до 4 г частиц для образца и образец высушивался в вакууме при 50°С. Образец затем помещался в аналитический газовый коллектор и охлаждался до 77К. Затем измерялась изотерма поглощения азота при по меньшей мере 5 равновесных давлениях, при этом образец подвергался воздействию известных объемов газообразного азота и измерялось падение давления. Равновесные давления были соответственно в интервале Р/Р0=0,01-0,20, где Р равновесное давление, а Р0 давление паров жидкого азота при 77К. Полученная изотерма затем была изображена в виде графика в соответствии со следующим уравнением БЭТ:

где Va является объемом газа, адсорбированного образцом при Р, Vm является объемом газа, требующимся для покрытия всей поверхности образца монослоем газа, и С является константой. Из этого графика определялись Vm и С. Vm затем преобразовывалась в площадь поверхности с использованием площади сечения азота при 77К по формуле
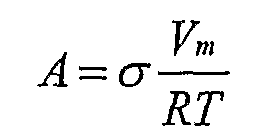
где σ - площадь поперечного сечения азота при 77К, Т является 77К и R является газовой константой.
Как уже было рассмотрено выше, НТК, полученная в соответствии с одним воплощением настоящего изобретения, проявляет очень высокую растворимость по сравнению с обычной НТК, полученной по другим способам. Эта повышенная скорость растворения позволяет более эффективно очищать и/или использовать более эффективные способы для очистки НТК, полученной по изобретению. Следующее описание относится к тому, как может быть количественно оценена скорость растворения НТК.
Скорость растворения известного количества твердых веществ в известном количестве растворителя в перемешиваемой смеси может быть измерена по различным методикам. Используемый здесь способ измерения, называемый «испытание на растворение во времени» выполняется следующим образом. В течение всего времени испытания на растворение используется давление окружающей среды около 0,1 МПа. Окружающая температура, используемая в течение всего временного испытания на растворение, была около 22°С. Кроме того, твердые вещества, растворитель и весь аппарат для растворения при этой температуре находятся в полном равновесном состоянии перед началом испытания и не происходит никакого заметного нагревания или охлаждения химического стакана или его содержимого во время периода растворения. Порция растворителя из свежего аналитического качества (способ ВЭЖХ) тетрагидрофурана (чистотой выше 99,9) в количестве 250 г помещалась в чистый (KIMAX) высокий стеклянный 400 мл химический стакан (инвентарный номер 14020 фирма Kimble/Kontes, Вайнленд, Нью-Йорк), который является неизолированным гладкостенным и преимущественно цилиндрическим по форме. Магнитная мешалка с покрытием из тефлона (инвентарный номер VWR 58948-230, длина около 1 дюйма, диаметр 3/8 дюйма восьмиугольное поперечное сечение, фирма VWR International, Вест Честер, Пенсильвания) помещается в стакан, где она естественно оседает на дно. Образец размешивается с помощью магнитной мешалки Variomag multipoint 15 (фирма H@P Labortechnik AG, Обершлайсхайм, Германия), установленной на 800 оборотов в минуту. Размешивание начинается не более чем за 5 минут до добавления твердых веществ и продолжается непрерывно в течение по меньшей мере 30 минут после добавления твердых веществ. Твердый образец из частиц неочищенной или очищенной ТФК массой до 250 миллиграммов, взвешивается в неклейкой чашке весов для образцов. В начальный момент времени, обозначенный как t=0, взвешенные твердые вещества высыпаются все сразу в размешиваемый тетрагидрофуран (далее ТГФ) и одновременно запускается таймер. При правильном проведении испытания ТГФ очень быстро смачивает твердые вещества и образует разбавленный хорошо размешанный шлам в течение 5 секунд. Образцы этой смеси последовательно получают в течение следующих промежутков времени, измеряемых в минутах от t=0: 0,08, 0,25, 0,50, 0,75, 1,00, 1,50, 2,00, 2,50, 3,00, 4,00, 5,00, 6,00, 8,00, 10,00, 15,00 и 30,00. Каждый малый образец удаляется из разбавленной хорошо размешанной смеси с использованием нового одноразового шприца (фирма Becton, Dickinson and Co, 5 мл, инвентарный номер 30163, Франклин Лейкс, Нью-Йорк 07417). Непосредственно после удаления из химического стакана образец из приблизительно 2 миллилитров прозрачной жидкости быстро выводится через новый неиспользованный шприцевой фильтр (диаметр 25 мм, 0,45 мка, Gelman GHP Acrodisk GF, фирма Pall Corp., Ист Хилз, Нью-Йорк 11548) в новую помеченную стеклянную пробирку для образцов. Продолжительность наполнения каждого шприца, ввода в фильтр и вывода в пробирку для образцов составляет обычно менее чем около 5 секунд, и этот промежуток времени соответствующим образом начинается и завершается в пределах около 3 секунд с каждой стороны от каждого заданного времени отбора. В течение около 5 минут каждого заполнения пробирки с образцами плотно закрывались и выдерживались при приблизительно постоянной температуре до проведения следующего химического анализа. После отбора последнего образца через 30 минут после t=0 все шестнадцать образцов анализировались для определения количества растворенной ТФК с использованием способа ВЭЖХ-ДМД, вообще рассмотренного в другом месте этого описания. Однако в настоящем испытании стандарты калибровки и сообщаемые результаты основаны на миллиграммах растворенной ТФК на грамм растворителя из ТГФ (в дальнейшем «частей на миллион в ТГФ»). Например, если все 250 миллиграммов твердых веществ были из очень чистой ТФК и если все это количество полностью растворялось в 250 граммах ТГФ растворителе, перед тем как был отобран определенный образец, то правильно измеренная концентрация будет около 1000 частей на миллион в ТГФ.
Когда НТК в соответствии с настоящим изобретением подвергается временному испытанию на растворение, описанному выше, то предпочтительно, чтобы образец, взятый через одну минуту после t=0, растворялся до концентрации по меньшей мере около 500 частей на миллион в ТГФ, более предпочтительно до по меньшей мере 600 частей на миллион в ТГФ. Для образца, взятого через две минуты после t=0, предпочтительно, чтобы НТК в соответствии с настоящим изобретением растворялась в ТГФ до концентрации по меньшей мере около 700 частей на миллион, более предпочтительно до по меньшей мере 750 частей на миллион в ТГФ. Для образца, взятого через 4 минуты после t=0, предпочтительно, чтобы НТК в соответствии с настоящим изобретением растворялась в ТГФ до концентрации по меньшей мере около 840 частей на миллион, более предпочтительно до концентрации по меньшей мере 880 частей на миллион в ТГФ.
Изобретатели обнаружили, что модель относительно простого экспоненциального роста приемлема для описания временной зависимости всего набора данных от полного временного испытания на растворимость, несмотря на сложность дисперсных образцов и процесса растворения. Форма уравнения здесь в соответствии с «моделью временного растворения» является следующей:
S=A+B*(1-exp(-C*t)),
где t=время в минутах;
S=растворимость в ТГФ в частях на миллион за время t;
exp=экспоненциальная функция натурального логарифма с основанием 2;
A, B = постоянные регрессии в частях на миллион в ТГФ, где A относится преимущественно к быстрому растворению более мелких частиц за очень короткое время и где сумма A+B относится преимущественно к общей величине растворения в конце заданного периода испытания; и
С = временная константа регрессии в обратных минутах.
Регрессивные константы подобраны для уменьшения суммы квадратов расхождений между точками действительных данных и соответствующими модельными величинами, и этот способ обычно называется подбором по методу «наименьших квадратов». Предпочтительным пакетом программ для выполнения этой регрессии данных является JMP Realease 5.1.2 (SAS Institute Inc., JMP Software, SAS Campus Drive, Cary, NC 27513).
Когда НТК в соответствии с настоящим изобретением испытывается способом временного испытания на растворение и подгоняется к модели растворения во времени, описанной выше, то предпочтительно, чтобы НТК имела временную константу «С» больше чем около 0,5 обратных минут, более предпочтительно больше чем около 0,6 обратных минут и наиболее предпочтительно больше 0,7 обратных минут.
На фиг. 33А м 33В показана частица обычной НТК, полученной по обычному высокотемпературному способу окисления в реакторе с непрерывным перемешиванием (РРНП). На фиг.33А показана частица обычной НТК с 500-кратным увеличением, а на фиг.33В выделена и показана эта частица НТК с увеличением в 2000 раз. Визуальное сравнение частиц НТК по изобретению, показанных на фиг. 32А и 32В, и частицы обычной НТК, показанной на фиг. 33А и 33В, свидетельствует, что частица обычной НТК имеет более высокую плотность, меньшую площадь поверхности, меньшую пористость и больший размер, чем частицы НТК по изобретению. Действительно обычная НТК, представленная на фиг. 33А и 33В, имеет средний размер частиц около 205 мк и площадь поверхности по способу БЭТ около 0,57 кв.м/г.
На фиг.34 показан обычный способ получения очищенной терефталевой кислоты (ОТК). В обычном способе для получения ОТК параксилол частично окисляется в реакторе 700 для высокотемпературного окисления с механическим перемешиванием. Шлам, содержащий НТК, выводится из реактора 700 и затем очищается в системе 702 для очистки. ОТК продукт системы 702 очистки вводится в систему 706 разделения для отделения и сушки частиц ОТК. Системы 702 очистки представляют собой большую часть расходов, связанных с получением частиц ОТК обычными способами. Система 702 очистки обычно включает в себя систему 708 добавления/обмена воды, систему 710 растворения, систему 712 гидрогенизации и три отдельных резервуара 704a,b,c для кристаллизации. В системе 708 добавления/обмена воды значительная часть материнского раствора вытесняется водой. После добавления воды шлам вода/НТК вводится в систему 710 растворения, где смесь вода/НТК нагревается, пока частицы НТК полностью не растворятся в воде. После растворения НТК водный раствор НТК подвергается гидрогенизации в системе 712 гидрогенизации. Гидрогенизированный выходящий поток из системы 712 гидрогенизации затем подвергается трем операциям кристаллизации в резервуарах 704a,b,c для кристаллизации и после этого подвергается отделению ОТК в разделяющей системе 706.
На фиг.35 показан усовершенствованный способ получения ОТК с использованием барботажного реактора колонного типа 800 для окисления, имеющего конфигурацию в соответствии с одним воплощением настоящего изобретения. Исходный шлам, содержащий частицы твердой НТК и жидкий материнский раствор, выводится из реактора 800. Обычно исходный шлам может содержать частицы твердой НТК размером в интервале от около 10 до около 50 мас.%, а оставшаяся часть является жидким материнским раствором. Частицы твердой НТК, присутствующие в исходном шламе, обычно содержат по меньшей мере 400 частей на миллион (ppmw) 4-карбоксибензальдегида (4-КБА), более обычно по меньшей мере около 800 частей на миллион и наиболее обычно в интервале от 1000 до 15000 частей на миллион 4-КБА. Исходный шлам, выведенный из реактора 800, вводится в систему 802 очистки для уменьшения концентрации 4-КБА и других примесей, присутствующих в НТК. Более чистый/очищенный шлам получают от системы 802 очистки и подвергают его разделению и сушке в разделяющей системе 804, чтобы тем самым получить частицы более чистой твердой терефталевой кислоты, содержащей меньше около 400 частей на миллион 4-КБА, более предпочтительно меньше около 250 частей на миллион 4-КБА и наиболее предпочтительно в интервале от 10 до 200 частей на миллион 4-КБА.
Система 802 очистки в системе для получения ОТК, показанная на фиг.35, дает целый ряд преимуществ над системой 802 очистки в системе известного уровня техники, показанной на фиг.34. Предпочтительно, чтобы система 802 очистки обычно включала в себя систему 806 для обмена раствора, автоклав 808 и один кристаллизатор 810. В системе 806 для обмена раствора по меньшей мере около 50 мас.% материнского раствора, присутствующего в исходном шламе, заменяется свежим заменяющим растворителем, чтобы тем самым обеспечить шлам с замененным растворителем, содержащий частицы НТК и заменяющий растворитель. Шлам с замененным растворителем, выходящий из системы 806 для замены раствора, вводится в автоклав (или второй реактор окисления) 808. В автоклаве 808 проводится вторая реакция окисления при слегка более высоких температурах, чем использовались при исходной/первичной реакции окисления, проводимой в барботажном реакторе колонного типа 800. Как было рассмотрено выше, большая площадь поверхности, малый размер и низкая плотность частиц НТК, полученной в реакторе 800, вызывают захват некоторых примесей частицами НТК, что дает возможность проводить окисление в автоклаве 808 без необходимости полного растворения частиц НТК. Таким образом температура в автоклаве 808 может быть ниже чем во многих аналогичных процессах известного уровня техники. Вторичное окисление, проводимое в автоклаве 808, предпочтительно снижает концентрацию 4-КБА в НТК по меньшей мере на около 200 частей на миллион, более предпочтительно по меньшей мере на около 400 частей на миллион и наиболее предпочтительно в интервале от 600 до 6000 частей на миллион. Предпочтительно, чтобы температура вторичного окисления в автоклаве 808 была по меньшей мере на около 10°С выше температуры первичного окисления в барботажном реакторе колонного типа 800, более предпочтительно на около 20-80°С выше температуры первичного окисления в реакторе 800 и наиболее предпочтительно на 30-50°С выше температуры первичного окисления в реакторе 800. Температура вторичного окисления предпочтительно находится в интервале от около 160 до около 240°С, более предпочтительно в интервале от около 180 до около 220°С и наиболее предпочтительно в интервале от 190 до 210°С. Очищенный продукт из автоклава 808 требует только одной операции кристаллизации в кристаллизаторе 810 перед его отделением в системе 804 разделения. Подходящие способы вторичного окисления/варки подробно рассматриваются в опубликованной патентной заявке США № 2005/0065373, содержание которой здесь представлено ссылкой.
Терефталевая кислота (например, ОТК), полученная в системе, показанной на фиг.35, предпочтительно образована из частиц ОТК, имеющих средний размер по меньшей мере около 40 мк, более предпочтительно от около 50 до около 2000 мк и наиболее предпочтительно в интервале от 60 до 200 мк. Частицы ОТК предпочтительно имеют среднюю площадь поверхности по БЭТ меньше чем около 0,25 кв.м/г, более предпочтительно в интервале от около 0,005 до около 0,2 кв.м/г и наиболее предпочтительно в интервале от 0,01 до 0,18 кв.м/г. ОТК, полученная посредством системы, показанной на фиг.35, является подходящей для использования в качестве сырья для получения ПЭТ. Обычно ПЭТ получают этерификацией терефталевой кислоты этиленгликолем с последующей поликонденсацией. Предпочтительно, чтобы терефталевая кислота, полученная в соответствии с одним каким-то воплощением настоящего изобретения, была использована как сырье для процесса с трубчатым реактором для получения ПЭТ, описанного в патентной заявке США с серийным № 10/013318, зарегистрированной 7 декабря 2001 года и приведенной здесь в ссылке.
Частицы НТК с раскрытой здесь морфологией особенно пригодны для использования в вышеприведенном способе варки с окислением для уменьшения содержания 4-КБА. Кроме того, эти предпочтительные частицы НТК дают преимущества в самых различных других последующих способах, включающих в себя растворение и/или химическую реакцию частиц. Эти дополнительные последующие способы включают в себя и также без ограничения реакцию по меньшей мере с одним гидроксилсодержащим соединением для получения эфирных соединений, особенно реакцию НТК с метанолом с образованием диметилтерефталата и сложных эфиров, в качестве примесей; реакцию по меньшей мере с одним диолом с образованием эфирного мономера и/или полимерных соединений, особенно реакцию НТК с этиленгликолем с образованием полиэтилентерефталата (ПЭТ); и полное или частичное растворение в растворителях, включая, но также без ограничений, воду, уксусную кислоту и N-метил-2-пирролидон, которые могут подвергаться дальнейшей обработке, включающей, но также без ограничения, повторное осаждение более чистой терефталевой кислоты и/или избирательное химическое восстановление карбонильных групп, но не групп карбоновых кислот. Также сюда относится хорошее растворение НТК в растворителе, содержащем воду, с частичной гидрогенизацией, которая уменьшает количество альдегидов, особенно 4-КБА, флуоренонов, фенонов и/или антрахинонов.
Изобретатели также полагают, что частицы НТК, имеющие предпочтительные рассмотренные здесь свойства, могут быть получены из частиц НТК, не имеющих предпочтительных свойств, описанных здесь (неподходящие частицы НТК) посредством введения, но не только, механического измельчения неподходящих частиц НТК и полного или частичного их растворения не подходящих частиц НТК, после чего следует полное или частичное повторное осаждение.
В соответствии с одним воплощением настоящего изобретения предлагается способ для частичного окисления окисляемого ароматического соединения до одного или больше типов ароматической карбоновой кислоты, в котором чистота растворяющей части подаваемого вещества (т.е. «подаваемого растворителя») и чистота части из окисляемого соединения в подаваемом веществе (т.е. «подаваемого окисляемого соединения») подбираются в пределах определенных интервалов, приведенных ниже. Вместе с другими воплощениями настоящего изобретения это дает возможность управлять степенью чистоты жидкой фазы и твердой фазы, если последняя присутствует, и объединенной шламовой фазой (т.е. твердое вещество плюс жидкость) реакционной среды в некоторых предпочтительных интервалах, приведенных ниже.
Что касается подаваемого растворителя, то известно, что он окисляет окисляемое ароматическое соединение (соединения) для получения ароматической карбоновой кислоты, в котором подаваемый растворитель, вводимый в реакционную среду, является смесью чистой для анализа уксусной кислоты и воды, которая часто используется в лабораторной практике и в опытном производстве. Также известно, что он требуется для проведения окисления окисляемого ароматического соединения до ароматической карбоновой кислоты, при этом растворитель, выводимый из реакционной среды, отделяется от полученной ароматической карбоновой кислоты и затем обратно рециркулирует в реакционную среду как вводимый растворитель преимущественно для уменьшения производственных затрат. Эта рециркуляция растворителя вызывает накопление с течением времени некоторых примесей и побочных продуктов процесса в рециркулирующем растворителе. В этой технике известны различные средства, помогающие очищать рециркулирующий растворитель перед его повторным вводом в реакционную среду. Обычно более высокая степень очистки рециркулирующего растворителя приводит к значительно более высоким производственным затратам, чем при более низкой степени очистки подобными средствами. Одно воплощение настоящего изобретения относится к пониманию и определению предпочтительных интервалов содержания большого числа примесей в подаваемом растворителе, многие из которых раньше считались в основном не вредными, чтобы найти оптимальный баланс между общими производственными затратами и общей чистотой продукта.
Термин «подаваемый рециркулирующий растворитель» определяется здесь как растворитель, содержащий по меньшей мере около 5 мас.%, которая раньше проходила через реакционную среду, содержащую одно или больше окисляемых ароматических соединений, подвергаемых частичному окислению. Из-за производственного запаса растворителя и рабочего цикла аппарата предпочтительно, чтобы части рециркулирующего растворителя пропускались через реакционную среду по меньшей мере один раз в рабочий день, более предпочтительно по меньшей мере раз в день в течение по меньшей мере семи последовательных рабочих дней и наиболее предпочтительно по меньшей мере раз в день в течение по меньшей мере 30 последовательных рабочих дней. По экономическим соображениям предпочтительно, чтобы по меньшей мере около 20 мас.% растворителя, подаваемого в реакционную среду по настоящему изобретению, составлял рециркулирующий растворитель, более предпочтительно, чтобы по меньшей мере около 40 мас.%, еще более предпочтительно по меньшей мере около 80 мас.% и наиболее предпочтительно по меньшей мере 90 мас.% составлял рециркулирующий растворитель.
Изобретатели обнаружили, что из-за реакционной активности и из-за металлических примесей, оставшихся в продукте окисления, концентрации выбранных переменной валентности металлов во вводимом рециркулирующем растворителе предпочтительно находятся в интервалах, приведенных здесь же. Концентрация железа во вводимом рециркулирующем растворителе предпочтительно ниже около 150 частей на миллион, более предпочтительно ниже около 40 частей на миллион и наиболее предпочтительно от 0 до 8 частей на миллион. Концентрация никеля в рециркулирующем растворителе предпочтительно ниже около 150 частей на миллион, более предпочтительно ниже около 40 частей на миллион и наиболее предпочтительно от 0 до 8 частей на миллион. Концентрация хрома в рециркулирующем растворителе предпочтительно ниже около 150 частей на миллион, более предпочтительно ниже около 40 частей на миллион и наиболее предпочтительно от 0 до 8 частей на миллион. Концентрация молибдена в рециркулирующем растворителе предпочтительно ниже около 75 частей на миллион, более предпочтительно ниже около 20 частей на миллион и наиболее предпочтительно от 0 до 4 частей на миллион. Концентрация титана в рециркулирующем растворителе предпочтительно ниже около 75 частей на миллион, более предпочтительно ниже около 20 частей на миллион и наиболее предпочтительно от 0 до 4 частей на миллион. Концентрация меди в рециркулирующем растворителе предпочтительно ниже около 20 частей на миллион, более предпочтительно ниже около 4 частей на миллион и наиболее предпочтительно от 0 до 1 частей на миллион. Другие металлические примеси также обычно присутствуют в рециркулирующем растворителе, и они обычно изменяются при более низких уровнях пропорционально присутствию одного или нескольких вышеперечисленных металлов. Управление концентрациями вышеперечисленных металлов в предпочтительных интервалах будет удерживать другие металлические примеси в подходящих пределах.
Эти металлы могут возникать как примеси в любом из входящих потоков в процессе (например, в поступающем окисляемом соединении, растворителе, окислителе и каталитических соединениях). Альтернативно, металлы могут образовываться как продукты коррозии любого из производственных аппаратов в способе, контактирующих с реакционной средой и/или контактирующих с рециркулирующим растворителем. Средства для управления концентрациями металлов в раскрытых интервалах их концентраций включают в себя соответствующие спецификации и контроль за чистотой различных подаваемых потоков и использование соответствующих конструктивных материалов, включая, но без ограничения, многие коммерческие марки титана и нержавеющую сталь таких сортов, как известная дуплексные нержавеющие стали и высокомолибденистые нержавеющие стали.
Изобретатели также выявили предпочтительные интервалы содержания выбранных ароматических соединений в рециркулирующем растворителе. К ним относятся как осажденные, так и растворенные ароматические соединения в рециркулирующем растворителе.
Удивительно, что даже осажденный продукт (например, ТФК) частичного окисления параксилола является загрязнителем, который следует контролировать в рециркулирующем растворителе. В связи с тем, что имеются удивительно предпочтительные интервалы для уровней твердых веществ в реакционной среде, любой осажденный продукт в подаваемом растворителе непосредственно вычитается из количества окисляемого соединения, которое может быть введено совместно с другими. Помимо этого подача осажденных твердых веществ из ТФК в рециркулирующем растворителе при повышенных их уровнях, как оказалось, неблагоприятно влияет на свойства частиц, образующихся в осаждающейся среде окисления, что приводит к нежелательным проявлениям при последующих обработках потока (например, при фильтрации продукта, промывке растворителем, оксидирующей варке неочищенного продукта, растворении неочищенного продукта для дальнейшей обработки и т.д.). Другой нежелательной особенностью осажденных твердых веществ в подаваемом рециркулирующем растворителе является то, что они часто содержат очень большие количества осажденных примесей по сравнению с концентрациями этих примесей в объеме твердых веществ внутри ТФК шлама, из которого получают значительную часть рециркулирующего растворителя. Возможно, что повышенные уровни примесей, наблюдаемые в твердых веществах, взвешенных в рециркулирующем фильтрате, могут быть связаны со временем нуклеации для осаждения некоторых примесей из рециркулирующего растворителя и/или с охлаждением рециркулирующего растворителя, являющимся намеренным, или обусловлены потерями в окружающую среду. Например, концентрации сильно окрашенного и нежелательного 2,6-дикарбоксифлуоренона наблюдались гораздо в более высоких уровнях в твердых веществах, присутствующих в рециркулирующем растворителе при 80°С, чем они наблюдались в ТФК твердых веществах, отделенных от рециркулирующего растворителя при 160°С. Аналогичным образом концентрации изофталевой кислоты наблюдались гораздо более высокие в твердых веществах, присутствующих в рециркулирующем растворителе, по сравнению с их уровнями, наблюдаемыми в ТФК твердых веществах из реакционной среды. Как именно будут вести себя конкретные осажденные примеси, захваченные рециркулирующим растворителем, когда их повторно вводят в реакционную среду, по-видимому, будет по-разному. Это зависит вероятно от относительной растворимости примеси в жидкой фазе реакционной среды, от того, как осажденная примесь наслаивается в осажденные твердые вещества, и вероятно от локальной скорости осаждения ТФК в том месте, где твердое вещество первое вторично поступает в реакционную среду. Таким образом изобретатели выяснили, что полезно управлять уровнем некоторых примесей в рециркулирующем растворителе, как рассмотрено ниже, не принимая во внимание, присутствуют ли эти примеси в рециркулирующем растворителе в растворенном виде или в виде захваченных частиц.
Количество осажденных твердых веществ, присутствующих в рециркулирующем фильтрате, определяется гравиметрическим способом следующим образом. Представительный образец отводится из подаваемого растворителя в реакционную среду, когда растворитель протекает по трубопроводу в сторону реакционной среды. Требуемое количество образца около 100 г помещается в стеклянный сосуд с внутренним объемом около 250 мл. Перед сбросом давления до атмосферного, но при непрерывном отборе в сосуд для образца, рециркулирующий фильтрат охлаждается до менее 100°С; это охлаждение производится для ограничения испарения растворителя в течение короткого промежутка времени перед его отбором в стеклянный сосуд и его запайкой. После того как образец был отобран при атмосферном давлении, стеклянный сосуд сразу же запаивается. Затем образцу дают охладиться до около 20°С при температуре окружающей среды около 20°С без принудительной конвекции. После достижения около 20°С образец выдерживается в этом состоянии в течение по меньшей мере около 2 часов. Затем запаянный сосуд энергично встряхивается пока не будет достигнуто видимое равномерное распределение твердых веществ. Сразу же после этого подводится магнитная мешалка к сосуду с образцом и она вращается с достаточной скоростью для эффективного поддержания равномерного распределения твердых веществ в сосуде. Затем пипеткой отбирается 10 миллилитровая часть перемешанной жидкости с взвесью твердых веществ и взвешивается. Затем масса жидкой фазы из этой части эффективно отделяется фильтрацией под вакуумом все еще при около 20°С без потери твердых веществ. Влажные твердые вещества, отфильтрованные от этой части, затем сушатся без сублимации твердых веществ и эти высушенные твердые вещества взвешиваются. Отношение массы высушенных твердых веществ к массе исходной части шлама является долей твердых веществ, выраженной в процентах, и обозначающей здесь содержание осажденных твердых веществ в рециркулирующем фильтрате при 20°С.
Изобретатели обнаружили, что ароматические соединения, растворенные в жидкой фазе реакционной среды и содержащие ароматические карбоновые кислоты без неароматических углеводородных групп (например, изофталевую кислоту, бензойную кислоту, фталевую кислоту, 2,5,4'-трикарбоксибифенил) являются чрезвычайно вредными компонентами. Хотя эти соединения имеют очень пониженную химическую активность в описанной реакционной среде по сравнению с окисляемыми соединениями, имеющими неароматические углеводородные группы, изобретатели выявили, что эти соединения тем не менее подвержены многочисленным вредным реакциям. Таким образом выгодно управлять составом этих соединений в предпочтительных интервалах в жидкой фазе реакционной среды. Это приводит к предпочтительным интервалам выбранных соединений в подаваемом рециркулирующем растворителе и также к предпочтительным интервалам выбранных исходных веществ в подаваемом окисляемом ароматическом соединении.
Например, при жидкофазном частичном окислении параксилола до терефталевой кислоты (ТФК) изобретатели выявили, что сильно окрашенную и нежелательную примесь 2,7-дикарбоксифлуоренона (2,7-ДКФ) фактически нельзя обнаружить в реакционной среде и в отбираемом продукте, если метазамещенные ароматические соединения находятся в очень малых количествах в реакционной среде. Изобретатели выявили, что когда примесь изофталевой кислоты присутствует во все возрастающих количествах в подаваемом растворителе, образование 2,7-ДКФ возрастает почти прямо пропорционально. Изобретатели также обнаружили, что когда примесь метаксилола присутствует в подаваемом параксилоле, образование 2,7-ДКФ снова увеличивается почти прямо пропорционально. Более того, даже если подаваемые растворитель и окисляемое соединение не содержат метазамещенных органических соединений, изобретатели обнаружили, что образуется некоторое количество изофталевой кислоты во время обычного частичного окисления очень чистого параксилола, особенно когда присутствует бензойная кислота в жидкой фазе реакционной среды. Эта самообразованная изофталевая кислота из-за своей большей растворимости чем у ТФК в растворителе, содержащем уксусную кислоту и воду, осаждается со временем в коммерческих аппаратах, в которых используется рециркулирующий растворитель. Таким образом количество изофталевой кислоты в подаваемом растворителе, количество метаксилола в подаваемом окисляемом ароматическом соединении и скорость самообразования изофталевой кислоты в реакционной среде соответствующим образом все рассматриваются в балансе друг с другом и в балансе со всеми реакциями, по которым расходуется изофталевая кислота. Изофталевая кислота, как было обнаружено, расходуется по дополнительным реакциям с образованием 2,7-ДКФ, как рассмотрено ниже. Кроме того, изобретатели выявили, что имеются другие проблемы для рассмотрения, когда устанавливаются соответствующие интервалы для метазамещенных ароматических соединений при частичном окислении параксилола до ТФК. Другие сильно окрашенные и нежелательные примеси, такая как 2,6-дикарбоксифлуоренон (2,6-ДКФ), по-видимому, в значительной степени связаны с растворенными паразамещенными ароматическими соединениями, которые всегда присутствуют при подаче параксилола для жидкофазного окисления. Таким образом подавление 2,7-ДКФ является наилучшим решением в перспективе для регулирования уровня других образующихся окрашенных примесей.
Например, в жидкофазном частичном окислении параксилола до ТФК изобретатели выявили, что образование тримеллитовой кислоты возрастает по мере возрастания уровней изофталевой и фталевой кислот в реакционной среде. Тримеллитовая кислота является трехфункциональной карбоновой кислотой, приводящей к разветвлению полимерных цепей во время получения ПЭТ из ТФК. Во многих применениях ПЭТ степени разветвления должны доводиться до низких уровней и, следовательно, уровень тримеллитовой кислоты должен доводиться до низкой величины в очищенной ТФК. Помимо того, что это приводит к тримеллитовой кислоте, присутствие метазамещенных и ортозамещенных соединений в реакционной среде также способствует образованию других трикарбоновых кислот (например, 1,3,5-трикарбоксибензола). Более того, повышенное присутствие трикарбоновых кислот в реакционной среде увеличивает количество образующейся тетракарбоновой кислоты (например, 1,2,4,5-тетракарбоксибензола). Сдерживание общего образования всех ароматических карбоновых кислот, имеющий больше двух групп карбоновой кислоты, является одним фактором при установлении предпочтительных уровней метазамещенных и ортозамещенных соединений в подаваемом рециркулирующем растворителе, в подаваемом окисляемом соединении и в реакционной среде в соответствии с настоящим изобретением.
Например, в жидкофазном частичном окислении параксилола до ТФК изобретатели открыли, что повышенные уровни в жидкой фазе реакционной среды нескольких растворенных ароматических карбоновых кислот без неароматических углеводородных групп приводят непосредственно к повышенному образованию окиси и двуокиси углерода. Это повышенное образование оксидов углерода приводит к потере выхода, как со стороны окислителя, так и со стороны окисляемого соединения на более поздней стадии процесса, так как многие из совместно получаемых ароматических карбоновых кислот, которые, с одной стороны, могут рассматриваться как примеси, с другой стороны, также имеют коммерческую ценность. Таким образом соответствующее удаление относительно растворимых карбоновых кислот, лишенных неароматических углеводородных групп, из рециркулирующего растворителя, имеет экономическую ценность в том, что это предотвращает снижение выхода окисляемого ароматического соединения и окислителя помимо вклада в подавление образования очень нежелательных примесей, таких как различные флуореноны и тримеллитовая кислота.
Например, в жидкофазном частичном окислении параксилола до ТФК изобретатели открыли, что образование 2,5,4'-трикарбоксибифенила, по-видимому, является неизбежным. 2,5,4'-трикарбоксибифенил является ароматической трикарбоновой кислотой, полученной связыванием двух ароматических колец с ариловым радикалом, вероятно образованным декарбоксилированием или декарбонилированием паразамещенных ароматических соединений. К счастью 2,5,4'-трикарбоксибифенил обычно образуется в меньших количествах, чем тримеллитовая кислота и обычно не увеличивает трудности при разветвлении полимерных молекул при производстве ПЭТ. Однако изобретатели открыли, что повышенные уровни 2,5,4'-трикарбоксибифенила в реакционной среде, содержащей окисленные алкилароматические соединения, в соответствии с предпочтительными воплощениями настоящего изобретения приводят к повышенным уровням сильно окрашенного и нежелательного 2,6-ДКФ. Повышенный уровень 2,6-ДКФ возможно образовался из 2,5,4'-трикарбоксибифенила посредством связывания колец с потерей одной молекулы воды, хотя точный механизм реакции до конца не известен. Если 2,5,4'-трикарбоксибифенил, который более растворим в растворителе, содержащем уксусную кислоту и воду, чем ТФК, будет беспрепятственно накапливаться в рециркулирующем растворителе, то степень превращения в 2,6-ДКФ может стать недопустимо большой.
Например, в жидкофазном частичном окислении параксилола до ТФК изобретатели открыли, что ароматические дикарбоновые кислоты без неароматических углеводородных групп (например, изофталевой кислоты) обычно приводят к мягкому подавлению химической активности реакционной среды, когда присутствуют в жидкой фазе при достаточной концентрации.
Например, в жидкофазном частичном окислении параксилола до ТФК изобретатели открыли, что осаждение очень часто не является идеальным (т.е. не равновесным) по отношению к относительным концентрациям различных веществ в твердой и в жидкой фазах. Вероятно это вызвано тем, что скорость осаждения очень велика при объемно-временных скоростях реакций, предпочитаемых здесь, что приводит к неидеальному совместному осаждению примесей или даже к поглощению. Таким образом, когда желательно ограничить концентрацию некоторых примесей (например, тримеллитовой кислоты и 2,6-ДКФ) в неочищенной ТФК, посредством конфигурации аппаратов для последующих операций, то предпочтительно управлять их концентрацией в подаваемом растворителе, а также скоростью их образования в реакционной среде.
Например, изобретатели открыли, что соединения бензофенона (например, 4,4'-дикарбоксибензофенон и 2,5,4'-трикарбоксибензофенон), полученные во время частичного окисления параксилола, имеют нежелательное влияние на реакционную среду для ПЭТ, даже если соединения бензофенона не являются такими сильно окрашенными в ТФК, как флуореноны и антрахиноны. Поэтому желательно ограничивать присутствие бензофенонов и подбирать исходные вещества в рециркулирующем растворителе и в подаваемом окисляемом соединении. Более того, изобретатели открыли, что присутствие повышенных уровней бензойной кислоты, введенной в рециркулирующий растворитель или образовавшейся в реакционной среде, приводит к повышенным скоростям образования 4,4'-дикарбоксибензофенона.
Просматривая свою работу, изобретатели открыли и количественно представили в достаточной степени удивительный набор реакций для ароматических соединений без неароматических углеводородных групп, которые присутствуют при жидкофазном частичном окислении параксилола до ТФК. Возвращаясь к тому единственному случаю с бензойной кислотой, изобретатели открыли, что повышенные уровни бензойной кислоты в реакционной среде по некоторым воплощениям настоящего изобретения, приводят к чрезвычайно повышенному образованию сильно окрашенной и нежелательной 9-флуоренон-2-карбоновой кислоты, к чрезвычайно повышенным уровням 4,5'-дикарбоксифенила, к повышенным уровням 4,4'-дикарбоксибензофенона, к мягкому подавлению химической активности проводимого окисления параксилола, к повышенным уровням оксидов углерода и к сопутствующим потерям выхода продукта. Изобретатели открыли, что повышенные уровни бензойной кислоты в реакционной среде также приводят к повышенному образованию изофталевой и фталевой кислот, уровни которых желательно удерживать в малых интервалах в соответствии с аналогичными аспектами настоящего изобретения. Число и важность реакций с участием бензойной кислоты являются даже более удивительными, так как некоторые изобретатели в последнее время рассматривают использование бензойной кислоты вместо уксусной кислоты в качестве основного компонента растворителя (см., например, патент США № 6562997). Кроме того, эти изобретатели заметили, что бензойная кислота образуется сама во время окисления параксилола со скоростями, которые очень важны в связи с ее образованием из примесей, таких как толуол и этилбензол, обычно обнаруживаемых в подаваемом окисляемом соединении, содержащем параксилол коммерческой чистоты.
С другой стороны, изобретатели обнаружили мало пользы в дополнительном регулировании состава рециркулирующего растворителя в отношении присутствия окисляемого соединения и в отношении промежуточных продуктов реакций с ароматическими соединениями, которые сохраняют неароматические углеводородные группы и также сравнительно растворимы в рециркулирующем растворителе. Вообще эти соединения или подаются в реакционную среду или образуются в последней со скоростями, значительно более высокими, чем соответствуют их присутствию в рециркулирующем растворителе; и скорость расхода этих соединений в реакционной среде достаточно высока; сохранение одной или больше неароматических углеводородных групп позволяет подходящим образом ограничить их накопление в рециркулирующем растворителе. Например, во время частичного окисления параксилола во многофазной реакционной среде параксилол испаряется в ограниченной степени вместе с большим количеством растворителя. Когда этот испарившийся растворитель выводится из реактора как часть отходящего газа и конденсируется для возвращения в качестве рециркулирующего растворителя, значительная часть испарившегося параксилола также конденсируется в него. Не нужно ограничивать концентрацию этого параксилола в рециркулирующем растворителе. Если растворитель отделяется от твердых веществ, когда шлам выводится из реакционной среды после окисления параксилола, этот возвращенный растворитель будет содержать такую же концентрацию растворенной паратолуиловой кислоты, как и растворитель, присутствующий в точке удаления из реакционной среды. Хотя может быть важно ограничивать постоянную концентрацию паратолуиловой кислоты в жидкой фазе реакционной среды, как показано ниже, не нужно по отдельности регулировать концентрацию паратолуиловой кислоты в этой части рециркулирующего растворителя благодаря ее сравнительно хорошей растворимости и ее низкой скорости потока массы по отношению к образованию паратолуиловой кислоты в реакционной среде. Аналогичным образом изобретатели обнаружили мало смысла в ограничении концентраций в рециркулированном растворителе ароматических соединений с метиловыми заместителями (например, толуиловых кислот), ароматических альдегидов (например, терефталевого альдегида), ароматических соединений с оксиметиловыми заместителями (например,4-оксиметилбензойной кислоты) и бромированных ароматических соединений, сохраняющих по меньшей мере одну неароматическую углеводородную группу (например, альфа-бромпаратолуиновой кислоты) ниже тех концентраций, которые по существу присутствуют в жидкой фазе, выходящей из реакционной среды, возникающей при частичном окислении ксилола в соответствии с предпочтительными воплощениями настоящего изобретения. Удивительно, что изобретатели также обнаружили, что не нужно также регулировать в рециркулирующем растворителе концентрацию выбранных фенолов, по существу получаемых во время частичного окисления ксилола, так как эти соединения образуются и распадаются в реакционной среде со скоростями, которые гораздо более высокие, чем соответствуют их присутствию в рециркулирующем растворителе. Например, изобретатели обнаружили, что 4-оксибензойная кислота имеет относительно малое влияние на химическую активность в предпочтительных воплощениях настоящего изобретения, когда совместно вводится со скоростью выше 2 г 4-оксибензойной кислоты на 1 кг параксилола, что гораздо выше ее естественного уровня в рециркулирующем растворителе, несмотря на то, что другие сообщают о ее ингибирующем действии в подобной реакционной среде (См., например, W. Partenheimer, Catalysis Today 23 (1995), p.81).
Таким образом, имеется много реакций и много соображений для установления предпочтительных интервалов различных ароматических примесей в подаваемом растворителе, как теперь раскрыто. Эти раскрытия изложены в виде агрегированного средневзвешенного состава всех потоков растворителя, вводимых в реакционную среду в течение заданного промежутка времени, предпочтительно одного дня, более предпочтительно одного часа и наиболее предпочтительно одной минуты. Например, если один подаваемый поток растворителя течет по существу непрерывно при составе 40 частей на миллион изофталевой кислоты и со скоростью 7 кг в минуту, второй подаваемый поток растворителя течет по существу непрерывно при составе 2000 частей на миллион изофталевой кислоты со скоростью 10 кг в минуту и не имеется никаких других потоков растворителя, вводимых в реакционную среду, то агрегированный средневзвешенный состав подаваемого растворителя рассчитывается как (40*7+2000*10)/(7+10)=1193 частей на миллион изофталевой кислоты. Примечательно, что масса любого подаваемого окисляемого соединения или любого вводимого окислителя, которые вероятно смешиваются с подаваемым растворителем перед поступлением в реакционную среду, не учитываются при расчете агрегированного средневзвешенного состава подаваемого растворителя.
В таблице 1, приведенной ниже, перечислены предпочтительные величины некоторых компонентов в подаваемом растворителе, вводимом в реакционную среду. Компоненты подаваемого растворителя, перечисленные в таблице 1, следующие: 4-карбоксибензальдегид (4-КБА), 4,4'-дикарбоксистильбен (4,4'-ДКС), 2,6-дикарбоксиантрахинон (2,6-ДКА), 2,6-дикарбоксифлуоренон (2,6-ДКФ), 2,7-дикарбоксифлуоренон (2,7-ДКФ), 3,5-дикарбоксифлуоренон (3,5-ДКФ), 9-флуоренон-2-карбоновая кислота (9Ф-2КК), 9-флуоренон-4-карбоновая кислота (9Ф-4КК), все флуореноны, включая другие флуореноны, отдельно не приведенные (все флуореноны), 4,4'-дикарбоксибифенил (4,4'-ДКБ), 2,5,4'-трикарбоксибифенил (2,5,4'-ТКБ), фталевая кислота (ФК), изофталевая кислота (ИФК), бензойная кислота (БК), тримеллитовая кислота (ТМК), 2,6-дикарбоксибензокумарин (2,6-ДКБК), 4,4'-дикарбоксибензил (4,4'-ДКБЗ), 4,4'-дикарбоксибензофенон (4,4'-ДКБФ), 2,5,4'-трикарбоксибензофенон (2,5,4'-ТКБФ), терефталевая кислота (ТФК), осажденные твердые вещества при 20°С и все ароматические карбоновые кислоты без неароматических углеводородных групп. В нижеприведенной таблице 1 даются предпочтительные количества этих примесей в НТК, полученной в соответствии с воплощениями настоящего изобретения.
Многие другие ароматические примеси также обычно присутствуют в рециркулирующем растворителе, обычно изменяющиеся даже при более низких уровнях и/или пропорционально одному или нескольким приведенных ароматических соединений. Способы регулирования приведенных ароматических соединений в предпочтительных интервалах обычно удерживают другие ароматические примеси на подходящих уровнях.
Когда в реакционной среде используется бром, в динамическом равновесии, как известно, существует большое число ионных и органических форм брома. Эти различные формы брома имеют разные характеристики растворимости, после того как они покидают реакционную среду и проходят через различные операции в аппаратах, относящиеся к рециркулирующему растворителю. Например, альфа-бромпаратолуиловая кислота может сохраняться, как таковая, при некоторых условиях или может быстро подвергнуться гидролизу при других условиях с образованием 4-оксиметилбензойной кислоты и бромистого водорода. В настоящем изобретении предпочтительно, чтобы по меньшей мере около 40 мас.%, более предпочтительно по меньшей мере около 60 мас.% и наиболее предпочтительно по меньшей мере около 80 мас.% общей массы брома, присутствующей в агрегированном растворителе, подаваемом в реакционную среду, было в виде одной или больше следующих химических форм: ионов брома, альфа-бромтолуиловой кислоты и бромуксусной кислоты.
Хотя важность и ценность управления агрегированной средневзвешенной чистотой подаваемого растворителя в пределах приведенных желательных интервалов в соответствии с настоящим изобретением не были пока что выявлены и/или раскрыты, подходящие средства для управления чистотой подаваемого растворителя отобраны из уже известных в этой области различных способов. Во-первых, любой растворитель, испаряющийся из реакционной среды, имеет обычно подходящую чистоту при условии, если вместе с испаряющимся растворителем из реакционной среды не будут уноситься жидкие или твердые вещества. Подача капель возвратного растворителя в пространство для отходящих газов над реакционной средой, как здесь рассмотрено, подходящим образом ограничивает такой унос; и рециркулирующий растворитель подходящий чистоты по отношению к ароматическому соединению может быть сконденсирован из таких отходящих газов. Во-вторых, более трудная и дорогая очистка подаваемого рециркулирующего растворителя обычно относится к растворителю, отобранному из реакционной среды в жидком виде, и к растворителю, который последовательно контактирует с жидкой и/или твердой фазами реакционной среды, выводимой из реактора (например, рециркулирующий растворитель, полученный из фильтра, в котором концентрируются твердые вещества, и/или промытый рециркулирующий растворитель из центрифуги, в которой концентрируются твердые вещества, и/или промытый рециркулирующий растворитель, взятый от операции кристаллизации и т.д.). Однако также известны средства в этой области для проведения необходимой очистки этих потоков рециркулирующего растворителя с использованием одного или больше средств известной техники. Что касается удерживания осажденных твердых веществ в рециркулирующем растворителе в пределах заданных интервалов, то подходящие для этого средства включают в себя, но без ограничения, гравиметрическое осаждение, механическую фильтрацию с использованием тканевого фильтра на поворотных ленточных или вращающихся барабанных фильтрах, механическую фильтрацию с использованием стационарной фильтрующей среды в резервуарах высокого давления, гидроциклонов и центрифуг. Что касается удерживания растворенных ароматических соединений в рециркулирующем растворителе в пределах заданных интервалов, средства для этого включают в себя, но без ограничения, средства, раскрытые в патенте США № 4939297 и патентной заявке США № 2005-0038288, приведенных здесь в качестве ссылок. Однако ни одно из этих уже известных изобретений не открыло и не раскрыло предпочтительные уровни чистоты подаваемого агрегированного растворителя, как это сделано здесь. Скорее эти предшествующие изобретения просто предлагали средства для очистки выборочных и частичных потоков рециркулирующего растворителя без вывода инновационных оптимальных величин по составу агрегированного средневзвешенного растворителя, подаваемого в реакционную среду.
Обращаясь теперь к чистоте подаваемого окисляемого соединения, известно, что некоторые уровни изофталевой кислоты, фталевой кислоты и бензойной кислоты присутствуют и допустимы при их низком содержании в очищенной ТФК, используемой для производства полимеров. Более того, известно, что эти соединения относительно более растворимы во многих растворителях и могут быть просто удалены из очищенной ТФК кристаллизацией. Однако из раскрытого здесь воплощения этого изобретения теперь известно, что управление уровнем нескольких сравнительно растворимых ароматических соединений, явно включающих в себя изофталевую кислоту, фталевую кислоту и бензойную кислоту, в жидкой фазе реакционной среды, является чрезвычайно важным для сдерживания уровня полициклических и окрашенных ароматических соединений, образовавшихся в реакционной среде, для контроля соединений с более чем 2 карбоновыми кислотами на молекулу, для управления активностью реакций в реакционной среде для частичного окисления и для управления потерями выхода окислителя и ароматического соединения.
В этой области техники известно, что изофталевая кислота, фталевая кислота и бензойная кислота образуются в реакционной среде следующим образом. Вводимая примесь метаксилола окисляется при высокой степени превращения и выходе до ИФК. Вводимая примесь ортоксилола окисляется при высокой степени превращения и выходе до ИФК. Вводимые примеси этилбензола и толуола окисляются при высокой степени превращения и выходе до бензойной кислоты. Однако изобретатели обнаружили, что значительные количества изофталевой кислоты, фталевой кислоты и бензойной кислоты также образуются в реакционной среде, содержащей параксилол, посредством неокисления метаксилола, ортоксилола, этилбензола и толуола, а других реакций. Эти существенно другие химические пути возможно включают в себя декарбонилирование, декарбоксилирование, реорганизацию переходных состояний и добавление метиловых и карбониловых радикалов к ароматическим кольцам.
При определении предпочтительных интервалов содержания примесей в подаваемом окисляемом соединении важны многие факторы. Любая примесь в подаваемом потоке является вероятной причиной прямой потери выхода и затрат на очистку продукта, если требования к чистоте являются достаточно строгими (например, в реакционной среде для частичного окисления параксилола, толуол и этилбензол, обычно обнаруживаемые в параксилоле коммерческой чистоты, приводят к бензойной кислоте и эта бензойная кислота в основном удаляется из многих коммерческих ТФК). Когда продукт частичного окисления вводимой примеси участвует в дополнительных реакциях, другие факторы помимо простой потери выхода и удаления становятся важными при расчете, во сколько обойдется очистка вводимого потока сырья (например, в реакционной среде для частичного окисления параксилола этилбензол приводит к бензойной кислоте, а бензойная кислота последовательно приводит к сильно окрашенной 9-флуоренон-2-карбоновой кислоте, к изофталевой кислоте, к фталевой кислоте и среди прочего к повышенному образованию оксидов углерода). Когда в реакционной среде образуются сами по себе дополнительные количества примеси по химическим механизмам, непосредственно не связанным с вводимыми примесями, анализ становится еще более сложным (например, в реакционной среде для частичного окисления параксилола бензойная кислота также образуется сама по себе в самом параксилоле). Кроме того, обработка по потоку неочищенного продукта окисления может повлиять на соображения относительно предпочтительной чистоты вводимого сырья. Например, стоимость удаления до подходящих уровней прямой примеси (бензойной кислоты) и последующих примесей (изофталевой кислоты, фталевой кислоты, 9-флуоренон-2-карбоновой кислоты и др.) может быть одной и той же, может отличаться одна от другой и может отличаться от требований удаления в основном не значимой примеси (например, продукта 4-КБК неполного окисления при окислении параксилола до ТФК).
Следующие приведенные интервалы чистоты для параксилола являются предпочтительными, когда параксилол с растворителем и окислителем вводится в реакционную среду для частичного окисления для получения ТФК. Эти интервалы более предпочтительны в процессе получения ТФК, включающем в себя операции после окисления для удаления из реакционной среды примесей, отличающихся от окислителя и растворителя (например, металлических катализаторов). Эти интервалы еще более предпочтительны в процессах получения ТФК, в которых удаляется дополнительная 4-КБК из НТК (например, превращением НТК в диметилтерефталат плюс примесные эфиры и последующим отделением метилового эфира от 4-КБК дистилляцией, способами окислительного дегирирования для превращения 4-КБК в ТФК, способами гидрогенизации для превращения 4-КБК в паратолуиловую кислоту, которая затем отделяется частичной кристаллизацией). Эти интервалы наиболее предпочтительны в процессах получения ТФК, в которых удаляется дополнительная 4-КБК из НТК способами окислительного дегирирования для превращения 4-КБК в ТФК.
С использованием вновь узнанных предпочтительных интервалов рециркулирующих ароматических соединений и относительных количеств ароматических соединений, полученных непосредственно при окислении вводимых примесей, по сравнению с другими по существу химическими путями, были установлены улучшенные интервалы для примесей при вводе нечистого параксилола в процесс частичного окисления для получения ТФК. В нижеприведенной Таблице 2 даны предпочтительные значения для количеств метаксилола, ортоксилола и этилбензола+толуола во вводимом параксилоле.
Специалисты в этой области теперь признают, что вышеприведенные примеси в нечистом параксилоле могут оказывать существенное влияние на реакционную среду, после того как продукты их частичного окисления накопились в рециркулирующем растворителе. Например, ввод верхнего количества в наиболее предпочтительном интервале содержания метаксилола, т.е. 400 частей на миллион, сразу же приведет к образованию около 200 частей на миллион изофталевой кислоты в жидкой фазе реакционной среды, когда работают с около 33 мас.% твердых веществ в реакционной среде. Это сравнимо с вводом верхнего количества из наиболее предпочтительного интервала для изофталевой кислоты 400 частей на миллион в рециркулирующий растворитель, которое после обеспечения типичного охлаждения испарившегося растворителя реакционной средой, доходит до около 1200 частей на миллион изофталевой кислоты в жидкой фазе реакционной среды. Таким образом, именно накопление продуктов частичного окисления в рециркулирующем растворителе с течением времени будет представлять собой самое большое возможное воздействие примесей метаксилола, ортоксилола, этилбензола и толуола на подаваемый поток нечистого параксилола. Поэтому предпочтительно, чтобы вышеприведенные интервалы содержания примесей в подаваемом нечистом параксилоле поддерживались в течение по меньшей мере половины каждого дня проведения любого частичного окисления реакционной среды в конкретном производственном аппарате, более предпочтительно в течение по меньшей мере трех четвертей каждого дня в течение по меньшей мере семи последовательных рабочих дней и наиболее предпочтительно, когда средневзвешенные массы подаваемой композиции нечистого параксилола находятся в пределах предпочтительных интервалов в течение по меньшей мере 30 последовательных рабочих дней.
Средства для получения нечистого параксилола предпочтительной чистоты, уже известные в этой области техники, включают в себя, но без ограничения, дистилляцию, способы частичной кристаллизации при температуре ниже окружающей, и способы молекулярных сит с использованием адсорбции при избирательном размере пор. Однако предпочтительные интервалы чистоты параксилола, заданные здесь, верхние пределы являются более труднодостижимыми и дорогими для получения, чем обычно используемые коммерческими поставщиками параксилола; и тем не менее на нижнем пределе устраняется слишком дорогая очистка параксилола для подачи в реакционную среду для частичного окисления благодаря открытию и определению, где совместное влияние самообразования примеси из самого параксилола и реакций ее расходования в реакционной среде становятся более важными, чем скорости ввода примесей в нечистый параксилол.
Когда вводимый поток, содержащий ксилол, содержит выбранные примеси, такие как этилбензол и/или толуол, при окислении этих примесей может образоваться бензойная кислота. Используемый здесь термин «образованная примесью бензойная кислота» обозначает бензойную кислоту, полученную из любого источника, за исключением ксилола, во время его окисления.
Как здесь рассмотрено, часть бензойной кислоты, произведенной во время окисления ксилола, получена из самого ксилола. Это получение бензойной кислоты из ксилола является по существу добавкой к любой части полученной бензойной кислоты, которая может образоваться из примеси. Если не связывать себя теорией, то можно предположить, что бензойная кислота образовалась из ксилола в реакционной среде, когда различные промежуточные продукты окисления ксилола непроизвольно декарбонилируются (теряют моноокись углерода) или декарбоксилируются (теряют двуокись углерода), образуя арильные радикалы. Эти арильные радикалы могут затем отнять атом водорода у одного или у многих источников в реакционной среде и образовать самовозникшую бензойную кислоту. Какой бы не был химический механизм для этого, используемый здесь термин «самовозникшая бензойная кислота» обозначает бензойную кислоту, образовавшуюся из ксилола при его окислении.
Как здесь также раскрыто, когда параксилол окисляется для производства терефталевой кислоты (ТФК), получение самовозникшей бензойной кислоты приводит к потере выхода параксилола и потерю выхода окислителя. Кроме того, присутствие самовозникшей бензойной кислоты в жидкой фазе реакционной среды связано с возрастанием многих нежелательных побочных реакций, включающих в себя преимущественно образование сильно окрашенных соединений, так называемых монокарбоксифлуоренами. Самовозникшая бензойная кислота также способствует нежелательному накоплению бензойной кислоты в рециркулирующем фильтрате, который затем повышает концентрацию бензойной кислоты в жидкой фазе реакционной среды. Таким образом, образование самовозникшей бензойной кислоты желательно минимизировать, но это также рассматривается соответствующим образом одновременно с факторами, влияющими на расходование бензойной кислоты, с факторами, относящимися к другим проблемам избирательности реакций и с общими экономическими соображениями.
Изобретатели открыли, что самовозникновение бензойной кислоты может удерживаться на низких уровнях соответствующим подбором, например, температуры, распределения ксилола и доступности кислорода в реакционной среде во время окисления. Не желая быть связанным теорией, но более низкие температуры и улучшенная доступность кислорода по-видимому подавляют скорости декарбонилования и/или декарбоксилирования, что позволяет избежать потерь выхода, вызванных самовозникшей бензойной кислотой. Достаточная доступность кислорода по-видимому направляет арильные радикалы в другие более желательные продукты, особенно оксибензойные кислоты. Распределение ксилола в реакционной среде может также влиять на баланс между превращением арильных радикалов в бензойную кислоту или в оксибензойные кислоты. Какие бы не были химические механизмы, изобретатели открыли условия реакции, которые хотя достаточно мягкие, чтобы снизить образование бензойной кислоты, являются достаточно жесткими для окисления высокой доли производимых оксибензойных кислот до моноокиси углерода и/или двуокиси углерода, которые легко удаляются из продукта окисления.
В предпочтительном воплощении настоящего изобретения реактор окисления конфигурируется и работает таким образом, чтобы образование самовозникающей бензойной кислоты было минимизировано и было максимизировано окисление оксибензойных кислот до моноокиси углерода и двуокиси углерода. Когда реактор окисления используется для окисления параксилола до терефталевой кислоты, предпочтительно, чтобы параксилол составлял по меньшей мере около 50 мас.% всего ксилола в потоке сырья, вводимого в реактор. Более предпочтительно, чтобы параксилол составлял по меньшей мере до около 75 мас.% всего ксилола во вводимом потоке. Еще более предпочтительно, чтобы параксилол составлял по меньшей мере до 95 мас.% всего ксилола во вводимом потоке. Наиболее предпочтительно, чтобы параксилол составлял по существу весь ксилол во вводимом потоке.
Когда используется реактор окисления параксилола до терефталевой кислоты, предпочтительно, чтобы скорость получения терефталевой кислоты была максимальна, а скорость получения самовозникающей бензойной кислоты была минимизирована. Предпочтительно, чтобы отношение скорости получения (по массе) терефталевой кислоты к скорости получения (по массе) самовозникающей бензойной кислоты было по меньшей мере около 500:1, более предпочтительно по меньшей мере около 1000:1 и наиболее предпочтительно по меньшей мере 1500:1. Как можно видеть ниже, скорость получения самовозникающей бензойной кислоты предпочтительно измеряется, когда концентрация бензойной кислоты в жидкой фазе реакционной среды ниже 2000 частей на миллион, более предпочтительно ниже 1000 частей на миллион и наиболее предпочтительно ниже 500 частей на миллион, так как эти низкие концентрации подавляют до достаточно низких скоростей реакции, которые превращают бензойную кислоту в другие соединения.
Объединяя самовозникающую бензойную кислоту и бензойную кислоту, образующуюся из примеси, получают предпочтительную величину отношения скорости образования (по массе) терефталевой кислоты к суммарной скорости получения (по массе) бензойной кислоты по меньшей мере около 400:1, более предпочтительно по меньшей мере около 700:1 и наиболее предпочтительно 1100:1. Как можно видеть ниже, суммарная скорость получения самовозникающей бензойной кислоты плюс бензойной кислоты, образующейся из примеси, предпочтительно измеряется, когда концентрация бензойной кислоты в жидкой фазе реакционной среды ниже 2000 частей на миллион, более предпочтительно, когда она ниже 1000 частей на миллион и наиболее предпочтительно, когда она ниже 500 частей на миллион, так как эти низкие концентрации подавляют до подходящих низких скоростей те реакции, которые превращают бензойную кислоту в другие соединения.
Как здесь раскрыто, повышенные концентрации бензойной кислоты в жидкой фазе реакционной среды приводят к возрастающему образованию многих других ароматических соединений, некоторые из которых являются вредными примесями в ТФК; и, как здесь раскрыто, повышенные концентрации бензойной кислоты в жидкой фазе реакционной среды приводят к возрастающему образованию газообразных окислов углерода, что приводит к потере выхода для окислителя и ароматических соединений и/или растворителя. Более того, теперь раскрыто, что изобретатели выяснили, что значительная часть этого повышенного образования других ароматических соединений и окислов углерода возникает в результате реакций превращения в них некоторого количества молекул самой бензойной кислоты, что контрастирует с каталитическим действием бензойной кислоты на другие реакции, при которых она не расходует себя. Поэтому термин «суммарное образование бензойной кислоты» здесь определяется как усредненная по времени масса всей бензойной кислоты, выходящая из реакционной среды, минус усредненная по времени масса всей бензойной кислоты, поступающей в реакционную среду в течение такого же промежутка времени. Это суммарное образование бензойной кислоты часто является положительной величиной, обусловленной скоростями образования бензойной кислоты, из примеси, и самообразующейся бензойной кислоты. Однако изобретатели открыли, что скорость превращения бензойной кислоты в окислы углерода и в несколько других соединений по-видимому увеличивается приблизительно линейно, когда возрастает концентрация бензойной кислоты в жидкой фазе реакционной среды, измеряемая, когда условия других реакций, включающие температуру, доступность кислорода, ОВС и активность реакций, поддерживаются постоянными. Таким образом, когда концентрация бензойной кислоты в жидкой фазе реакционной среды довольно велика, вероятно из-за повышенной концентрации бензойной кислоты в рециркулирующем растворителе, то превращение молекул бензойной кислоты в другие соединения, включая окислы углерода, может стать равным или большим, чем химическое образование новых молекул бензойной кислоты. В этом случае суммарное образование бензойной кислоты может оказаться сбалансированным (быть около нуля) или даже стать отрицательным. Изобретатели открыли, что при положительном суммарном получении бензойной кислоты отношение скорости образования (по массе) терефталевой кислоты в реакционной среде к скорости суммарного образования бензойной кислоты в реакционной среде предпочтительно выше, чем около 700:1, более предпочтительно выше, чем около 1100:1, и наиболее предпочтительно выше 4000:1. Изобретатели открыли, что когда суммарное образование бензойной кислоты является отрицательным, отношение скорости получения (по массе) терефталевой кислоты в реакционной среде к скорости суммарного образования бензойной кислоты в реакционной среде предпочтительно выше, чем около 200:(-1), более предпочтительно выше, чем около 1000:(-1), и наиболее предпочтительно выше 5000:(-1).
Изобретатели также открыли предпочтительные интервалы для состава шлама (жидкость + твердое вещество), выводимого из реакционной среды, и твердой НТК части шлама. Предпочтительный состав шлама и предпочтительный состав НТК являются удивительно превосходными и полезными. Например, очищенная ТФК, полученная из этой предпочтительной НТК окислительным дигирированием, имеет довольно низкий уровень всех примесей и окрашенных примесей, так что очищенная ТФК является подходящей без гидрогенизации дополнительной 4-КБК и/или окрашенных примесей для широкого применения в производстве ПЭТ волокон и ПЭТ упаковки. Например, предпочтительный состав шлама обеспечивает жидкую фазу реакционной среды, которая имеет относительно низкую концентрацию важных примесей и это значительно снижает образование других даже более нежелательных примесей, как показано здесь. Кроме того, предпочтительный состав шлама существенно способствует последующей обработке жидкости из шлама, чтобы она стала приемлемо чистым рециркулирующим раствором в соответствии с другими воплощениями настоящего изобретения.
НТК, полученная в соответствии с одним воплощением настоящего изобретения, содержит меньше различных примесей типов, чем НТК, полученная по известным способам и аппаратам, особенно в тех, где используется рециркулирующий растворитель. Примесями, которые могут присутствовать в НТК, являются следующие: 4-карбоксибензальдегид (4-КБА), 4,4'-дикарбоксистильбен (4,4'-ДКС), 2,6-дикарбоксиантрахинон (2,6-ДКА), 2,6-дикарбоксифлуоренон (2,6-ДКФ), 2,7-дикарбоксифлуоренон (2,7-ДКФ), 3,5-дикарбоксифлуоренон (3,5-ДКФ), 9-флуоренон-2-карбоновая кислота (9Ф-2КК), 9-флуоренон-4-карбоновая кислота (9Ф-4КК), 4,4'-дикарбоксибифенил (4,4'-ДКБ), 2,5,4'-трикарбоксибифенил (4,4'-ТКБ), фталевая кислота (ФК), изофталевая кислота (ИФК), бензойная кислота (БК), тримеллитовая кислота (ТМК), паратолуиловая кислота (ПТК), 2,6-дикарбоксибензокумарин (2,6-ДКБК), 4,4'-дикарбоксибензил (4,4'-ДКБЗ), 4,4'-дикарбоксибензофенон (4,4'-ДКБФ), 2,5,4'-трикарбоксибензофенон (2,5,4'-ТКБФ). В нижеприведенной Таблице 3 приведены предпочтительные количества этих примесей в НТК, полученной в соответствии с одним воплощением настоящего изобретения.
Кроме того, предпочтительно, чтобы НТК, получаемая в соответствии с настоящим изобретением, имела пониженное содержание окрашенных соединений по отношению к их содержанию в НТК, полученной по обычным способам и аппаратам, преимущественно таких, в которых используется рециркулирующий растворитель. Таким образом предпочтительно, чтобы НТК, полученная в соответствии с одним воплощением настоящего изобретения, имела прозрачность в процентах при длине волны 340 нанометрах (нм) по меньшей мере около 25%, более предпочтительно по меньшей мере около 50% и наиболее предпочтительно по меньшей мере 60%. Также предпочтительно, чтобы НТК, полученная в соответствии с одним воплощением настоящего изобретения, имела прозрачность в процентах при 400 нм по меньшей мере около 88%, более предпочтительно по меньшей мере около 90% и наиболее предпочтительно по меньшей мере 92%.
Испытание на прозрачность в процентах дает возможность измерить окрашенные поглощающие свет примеси, присутствующие в ТФК или НТК. Используемое здесь испытание относится к измерениям, выполненным над частью раствора, приготовленного растворением 2,00 г сухой твердой ТФК или НТК в 20,0 миллилитрах диметилсульфоксида (ДМСО) качества чистый для анализа или лучше. Часть этого раствора затем помещается в полумикропроточную кювету PN 176700 фирмы Hellma, которая сделана из кварца и имеет толщину 1,0 см и объем 0,39 миллилитра (Hellma, USA, 80 Skyline Drive, Plainview, NY 11803). Для измерения прозрачности при различных длинах волн света через эту наполненную проточную кювету используется спектрофотометр с диодной матрицей Agilent 8453 (фирма Agilent Technologies, 395 Page Mill Road, Palo Alto, CA 94303). После соответствующей коррекции для учета спектральной поглощающей способности фона, включая, но без ограничения, в отношении кюветы и используемого растворителя, получают непосредственно на приборе прозрачность в процентах, характеризующую долю падающего света, которая проходит через растворитель. Процентные величины прозрачности для света с длиной волны 340 нм и 400 нм особенно полезны, чтобы отличить чистую ТФК от многих примесей обычно обнаруживаемых в ней.
Предпочтительные интервалы различных ароматических примесей в шламе (твердое вещество+жидкость) реакционной среды представлены в нижеприведенной таблице 4.
Эти предпочтительные составы шлама дают предпочтительный состав жидкой фазы реакционной среды и в то же время эффективно устраняют экспериментальные трудности, связанные с осаждением дополнительных компонентов жидкой фазы из реакционной среды в твердофазные компоненты во время отбора образцов из реакционной среды, разделения жидкостей и твердых веществ и перехода к условиям для анализа.
Много других ароматических примесей, также обычно присутствующих в шламовой фазе реакционной среды и в НТК из реакционной среды, обычно изменяются при даже более низких уровнях и/или пропорционально одному или нескольким раскрытым ароматическим соединениям. Удерживание раскрытых ароматических соединений в предпочтительных интервалах будет удерживать другие ароматические примеси на требуемых уровнях. Эти предпочтительные составы для шламовой фазы в реакционной среде и для твердой НТК, отобранной непосредственно из шлама, достигаются в соответствии с воплощениями изобретения, раскрытыми здесь для частичного окисления параксилола до ТФК.
Измерение концентрации компонентов с низкими уровнями содержания в растворителе, рециркулирующем растворителе, НТК, шламе из реакционной среды и в ОТК обычно осуществляется методами жидкостной хроматографии. Теперь будут описаны два взаимозаменяемых воплощения.
Способ, называемый здесь ЖХВД-ДМД, представляет собой жидкостную хроматографию высокого давления (ЖХВД), связанную с диодным матричным детектором (ДМД) для обеспечения разделения и количественного определения различных химических молекул в данном образце. Прибор, используемый для этого измерения, является моделью 1100 HPLC, снабженную ДМД, предлагаемой фирмой Agilent Technologies (Palo Alto, CA), хотя имеются и другие подходящие приборы, коммерчески доступные и у других поставщиков. Как известно, и время удержания, и чувствительность детектора, калибруются с использованием известных соединений, присутствующих в известных количествах, которые соответствуют присутствующим в действительных неизвестных образцах.
Способ, называемый здесь как ЖХВД-МС, представляет собой жидкостный хроматограф высокого давления, связанный с масс-спектрометром (МС), для обеспечения разделения, идентификации и количественного определения различных молекул соединений в данном образце. Приборами, используемыми в этом измерении, являются ЖХВД Alliance HPLC и МС ZQ MS, предлагаемые фирмой Waters Corp. (Milford,MA), хотя коммерчески доступны и другие подходящие приборы от других поставщиков. Как известно в этой технике, и время удержания, и чувствительность масс-спектрометра калибруются с использованием известных соединений, присутствующих в известных количествах, которые присутствуют в действительных неизвестных образцах.
Другое воплощение настоящего изобретения относится к частичному окислению ароматического окисляемого соединения с соответствующим уравновешиванием подавления вредных ароматических примесей, с одной стороны, и получения двуокиси углерода и моноокиси углерода, т.е. суммарных окислов углерода (СОх), с другой стороны. Эти окислы углерода обычно выводятся из реактора вместе с отходящим газом, и они приводят к значительной потере растворителя и окисляемого соединения, включая в конечном счете и предпочтительные окисленные производные (например, уксусной кислоты, параксилола и ТФК). Изобретатели обнаружили более низкие пределы для образования окислов углерода, ниже которых, по-видимому, высокое образование вредных ароматических примесей, как описано ниже, и низкий общий уровень превращения неизбежно слишком малы, чтобы быть экономически выгодными. Изобретатели также выявили верхние пределы для окислов углерода, выше которых образование окислов углерода продолжает увеличиваться с незначительным дальнейшим приростом, обеспечиваемым снижением образования вредных ароматических примесей.
Изобретатели открыли, что уменьшение концентраций в жидкой фазе подаваемого ароматического окисляемого соединения и ароматических промежуточных соединений в реакционной среде приводит к более низким скоростям образования вредных примесей во время частичного окисления ароматического окисляемого соединения. Эти вредные примеси включают в себя связанные ароматические кольца и/или ароматические молекулы, содержащие больше требуемого числа групп карбоновой кислоты (например, при окислении параксилола вредными примесями являются 2,6-дикарбоксиантрахинон, 2,6-дикарбоксифлуоренон, тримеллитовая кислота, 2,5,4'-трикарбоксибифенил, и 2,5,4'-бензофенон). Ароматические промежуточные соединения включают в себя ароматические соединения, образовавшиеся из подаваемого окисляемого ароматического соединения и все еще сохраняющие неароматические углеводородные группы (например, при окислении параксилола ароматическими промежуточными соединениями являются паратолуиловый альдегид, терефталевый альдегид, паратолуиловая кислота, 4-КБК, 4-оксиметилбензойная кислота и альфа-бромпаратолуиловая кислота). Подаваемое ароматическое окисляемое соединение и ароматические промежуточные соединения, сохраняющие неароматические углеводородные группы, когда присутствуют в жидкой фазе реакционной среды, по-видимому, приводят к вредным примесям таким же образом, как уже раскрыто здесь для растворенных ароматических соединений без неароматических углеводородных групп (например, изофталевой кислоты).
Пытаясь исключить эту необходимость в более высокой активности реакций для подавления образования вредных ароматических примесей во время частичного окисления окисляемого ароматического соединения, изобретатели открыли, что нежелательным побочным результатом этого является повышенное образование окислов углерода. Важно оценить, что эти окислы углерода приводят к снижению выхода окисляемого соединения и окислителя, а не только растворителя. Ясно, что значительная и иногда основная доля окислов азота возникает из окисляемого соединения и его производных соединений, а не из растворителя; и часто окисляемое соединение обходится дороже на единицу углерода, чем растворитель. Более того, важно оценить, что желательный продукт в виде карбоновой кислоты (например, ТФК) также подвержен дальнейшему окислению до окислов углерода, когда присутствует в жидкой фазе реакционной среды.
Важно также принять во внимание, что настоящее изобретение относится к реакциям в жидкой фазе реакционной среды и концентрациям в ней реагентов. Это контрастирует с некоторыми прежними изобретениями, которые относятся непосредственно к образованию в осажденном твердом веществе ароматического соединения, сохраняющего неароматические углеводородные группы. В частности, для частичного окисления параксилола до ТФК, в некоторых прежних изобретениях внимание обращается на количество 4-КБА, осажденного в твердой фазе НТК. Однако изобретатели настоящего изобретения обнаружили разницу больше чем два к одному для отношения 4-КБА в твердой фазе к 4-КБА в жидкой фазе, используя те же условия по температуре, давлению, катализу, составу растворителя и объемно-временной скорости реакции параксилола, в зависимости от которых проводится частичное окисление в хорошо перемешиваемом автоклаве или в реакционной среде с кислородом и параксилолом, выполняемое постадийно в соответствии с настоящим изобретением. Кроме того, изобретатели обнаружили, что отношение 4-КБА в твердой фазе к 4-КБА в жидкой фазе также может изменяться от более чем двух к одному в хорошо перемешиваемой или постадийно изменяемой реакционной среде, в зависимости от объемно-временной скорости реакции параксилола при других, но аналогичных условиях температуры, давления, катализа и состава растворителя. Кроме того, 4-КБА в твердой фазе НТК, по-видимому, не способствует образованию вредных примесей, и 4-КБА в твердой фазе может быть регенерирован и окислен до ТФК просто и с высоким выходом (например, окислительным дегирированием НТК шлама так, как здесь описано); в то время как удаление вредных примесей гораздо более трудное и дорогое, чем удаление твердофазной 4-КБА, и образование окислов углерода приводит к постоянной потере выхода. Таким образом важно представлять, что этот аспект настоящего изобретения относится к жидкофазным составам в реакционной среде.
Независимо от того, образуются ли окислы углерода из растворителя или окисляемого соединения, изобретатели обнаружили, что при экономически оправданных превращениях образование окислов углерода сильно связано с уровнем общей реакционной активности, несмотря на широкое различие конкретных сочетаний значений температуры, металлов, галогенов, кислотности реакционной среды, измеряемой в рН, содержания воды, используемой для получения уровня общей реакционной активности. Изобретатели нашли, что полезно проводить частичное окисление ксилола для оценки уровня общей реакционной активности с использованием концентрации толуиловых кислот в жидкой фазе на половине высоты столба реакционной среды, в нижней части реакционной среды и в верхней части реакционной среды.
Таким образом возникает важное одновременное уравновешивание для минимизации образования вредных примесей благодаря повышению реакционной активности и в то же время уменьшения до минимума образования окислов углерода благодаря снижению реакционной активности. То есть, если общее образование окислов углерода подавляется до слишком низкой величины, то образуются слишком большие уровни вредных примесей, и наоборот.
Кроме того, изобретатели обнаружили, что растворимость и относительная реакционная способность заданной карбоновой кислоты (например, ТФК) и присутствие других растворенных ароматических соединений без неароматических углеводородных групп вводят очень важное средство в этом равновесии между окислами углерода и вредными примесями. Требующаяся карбоновая кислота в качестве продукта обычно растворима в жидкой фазе реакционной среды, даже когда она также присутствует в твердом виде. Например, при температурах в предпочтительных диапазонах ТФК растворима в реакционной среде, содержащей уксусную кислоту и воду, в уровнях от около одной тысячи частей на миллион до более 1 мас.%, по мере повышения температуры растворимость растет. Несмотря на это имеются различия в скоростях реакций при образовании различных вредных примесей из подаваемого окисляемого ароматического соединения (например, параксилола), из ароматических образовавшихся промежуточных соединений (например, паратолуиловой кислоты), из заданной ароматической карбоной кислоты (например, ТФК) и из ароматических соединений без неароматических углеводородных групп (например, изофталевой кислоты), при этом присутствие и реакционная способность последних двух групп определяет степень уменьшения возврата по отношению к дальнейшему подавлению первых двух групп, подаваемого окисляемого ароматического соединения и ароматических промежуточных соединений. Например, при частичном окислении параксилола до ТФК, если растворенная ТФК достигает концентрации 7000 частей на миллион в жидкой фазе реакционной среды при заданных условиях, растворенная бензойная кислота достигает 8000 частей на миллион, растворенная изофталевая кислота достигает 6000 частей на миллион и растворенная фталевая кислота достигает 2000 частей на миллион, то дальнейшее снижение общего содержания вредных соединений начинает снижаться по мере повышения реакционной активности при подавлении жидкофазной концентрации паратолуиловой кислоты и 4-КБА ниже аналогичных уровней. То есть присутствие и концентрация в жидкой фазе реакционной среды ароматических соединений без неароматических углеводородных групп очень мало изменяются при повышении реакционной активности, и их присутствие служит для увеличения области уменьшающихся возвратов для снижения концентрации образовавшихся промежуточных соединений, чтобы подавить образование вредных примесей.
Таким образом, в одном воплощении настоящего изобретения обеспечиваются предпочтительные интервалы для окислов углерода, ограниченные у нижнего предела интервала низкой реакционной активностью и чрезмерным образованием вредных примесей, а у верхнего предела интервала избыточными потерями углерода, но с уровнями, которые ниже ранее описанных и раскрытых как экономически оправданные. Поэтому образование окислов углерода предпочтительно сдерживается следующим образом. Отношение молей всех полученных окислов углерода к молям вводимого окисляемого ароматического соединения предпочтительно больше чем около 0,02:1, более предпочтительно больше чем около 0,04:1, еще более предпочтительно больше чем около 0,05:1 и наиболее предпочтительно больше 0,06:1. В то же время отношение молей всех полученных окислов углерода к молям вводимого окисляемого ароматического соединения предпочтительно меньше чем около 0,24:1, более предпочтительно меньше чем около 0,22:1, еще более предпочтительно меньше чем около 0,19:1 и наиболее предпочтительно меньше 0,15:1. Отношение молей полученной двуокиси углерода к молям вводимого окисляемого ароматического соединения предпочтительно больше чем около 0,01:1, более предпочтительно больше чем около 0,03:1, еще более предпочтительно больше чем около 0,04:1 и наиболее предпочтительно больше 0,05:1. В то же время отношение молей полученной двуокиси углерода к молям вводимого окисляемого ароматического соединения предпочтительно меньше чем около 0,21:1, более предпочтительно меньше чем около 0,19:1, еще более предпочтительно меньше чем около 0,16:1 и наиболее предпочтительно меньше 0,11:1. Отношение молей полученной окиси углерода к молям вводимого окисляемого ароматического соединения предпочтительно больше чем около 0,005:1, более предпочтительно больше чем около 0,010:1, еще более предпочтительно больше чем около 0,015:1, и наиболее предпочтительно больше 0,020:1. В то же время отношение молей полученной окиси углерода к молям вводимого окисляемого ароматического соединения предпочтительно меньше чем около 0,09:1, более предпочтительно меньше чем около 0,07:1, еще более предпочтительно меньше чем около 0,05:1 и наиболее предпочтительно меньше 0,04:1.
Содержание двуокиси углерода в сухом отходящем газе реактора окисления предпочтительно больше чем около 0,10% мольных, более предпочтительно больше чем около 0,20% мольных, еще более предпочтительно больше чем около 0,25% мольных и наиболее предпочтительно больше 0,30% мольных. В то же время содержание двуокиси углерода в сухом отходящем газе из реактора окисления предпочтительно меньше чем около 1,5% мольных, более предпочтительно меньше чем около 1,2% мольных, еще более предпочтительно менее чем около 0,9% мольных и наиболее предпочтительно меньше чем около 0,8% мольных. Содержание окиси углерода в сухом отходящем газе из реактора окисления предпочтительно больше чем около 0,05% мольных, более предпочтительно больше чем около 0,10% мольных, еще более предпочтительно больше 0,15 и наиболее предпочтительно больше 0,18% мольных. В то же время содержание окиси углерода в сухом отходящем газе из реактора окисления предпочтительно меньше чем около 0,60% мольных, более предпочтительно меньше чем около 0,50% мольных, еще более предпочтительно меньше чем около 0,35% мольных и наиболее предпочтительно меньше 0,28% мольных.
Изобретатели открыли, что важным фактором для уменьшения получения окислов углерода в этих предпочтительных интервалах является повышение чистоты рециркулирующего фильтрата и вводимого окисляемого соединения для снижения концентрации ароматических соединений без неароматических углеводородных групп в соответствии с описаниями настоящего изобретения - это одновременно снижает образование окислов углерода и вредных примесей. Другим фактором является улучшение распределения параксилола и окислителя в реакторе в соответствии с описанием настоящего изобретения. Другими факторами, обеспечивающими вышеприведенные предпочтительные уровни окислов углерода, являются градиенты давления, температуры, концентрации окисляемого соединения в жидкой фазе и для окислителя в газовой фазе. Другими факторами, обеспечивающими вышеприведенные предпочтительные уровни окислов углерода, являются предпочтительные величины объемно-временной скорости реакции, давления, температуры, состава растворителя, состава катализатора и конструкция реактора.
Важным преимуществом работы в предпочтительных интервалах образования окислов углерода является возможность уменьшения расхода молекулярного кислорода, хотя не до стехиометрических значений. Несмотря на хорошее распределение окислителя и окисляемого соединения в соответствии с настоящим изобретением, избыток кислорода может удерживаться выше его стехиометрического значения, рассчитанного только для подачи окисляемого соединения, для обеспечения некоторых потерь с окислами углерода и для создания избытка молекулярного кислорода для управления образованием вредных примесей. В частности для случая, в котором ксилол является подаваемым окисляемым соединением, отношение массы подаваемого молекулярного кислорода к массе подаваемого ксилола предпочтительно больше чем около 0,91:1,00, более предпочтительно больше чем около 0,95:1,00 и наиболее предпочтительно больше 0,99:1,00. В то же время отношение массы подаваемого кислорода к массе подаваемого ксилола предпочтительно меньше чем около 1,20:1,00, более предпочтительно меньше чем около 1,22:1,00 и наиболее предпочтительно меньше 1,06:1,00. В частности, для подаваемого ксилола усредненное по времени содержание молекулярного кислорода в сухом отходящем газе из реактора окисления предпочтительно больше чем около 0,1% мольного, более предпочтительно больше чем около 1% мольный и наиболее предпочтительно больше 1,5% мольного. В то же время усредненное по времени содержание молекулярного кислорода в сухом отходящем газе из реактора окисления предпочтительно меньше чем около 6% мольных, более предпочтительно меньше чем около 4% мольных и наиболее предпочтительно меньше 3% мольных.
Другим важным преимуществом работы в предпочтительных интервалах образования окислов углерода является то, что меньшее количество ароматического соединения превращается в окислы углерода и другие менее ценные продукты. Это преимущество оценивается с использованием суммы молей всех ароматических соединений, выходящих из реакционной среды, деленной на сумму молей всех ароматических соединений, поступающих в реакционную среду в течение непрерывного промежутка времени, предпочтительно одного часа, более предпочтительно в течение одного дня и наиболее предпочтительно в течение 30 дней подряд. Это отношение здесь называется «мольным отношением сохранения» для ароматических соединений в реакционной среде и выражается в процентах. Если все вводимые ароматические соединения выходят из реакционной среды в виде ароматических соединений, хотя преимущественно в виде окисленных вводимых ароматических соединений, то мольное отношение сохранения имеет максимальную величину 100%. Если точно 1 из каждых 100 вводимых ароматических молекул превращается в окислы углерода и/или другие неароматические молекулы (например, уксусной кислоты) при прохождении через реакционную среду, то мольное отношение сохранения составляет 99%. В частности для случая, когда ксилол является основным сырьем из окисляемого ароматического соединения, мольное отношение сохранения этого соединения, проходящего через реакционную среду, предпочтительно больше чем около 98%, более предпочтительно больше чем около 98,5% и наиболее предпочтительно меньше 99,0%. В то же время, чтобы была достаточная общая реакционная активность, мольное отношение сохранения для ароматических соединений, проходящих через реакционную среду, составляет предпочтительно меньше чем около 99,9%, более предпочтительно меньше чем около 99,8% и наиболее предпочтительно меньше 99,7%, когда ксилол является основным подаваемым окисляемым ароматическим соединением.
Другой аспект настоящего изобретения относится к получению метилацетата в реакционной среде, содержащей уксусную кислоту и одно или несколько окисляемых ароматических соединений. Метилацетат является относительно летучим соединением по сравнению с водой и уксусной кислотой и таким образом имеет склонность к уносу вместе с отходящим газом, если не будет использовано дополнительное охлаждение или другие операции в аппаратах для его регенерации и/или утилизации перед выпуском вместе с отходящим газом в окружающую среду. Образование метилацетата таким образом представляет собой дополнительные эксплуатационные и также капитальные затраты. Вероятно метилацетат образуется благодаря первичному соединению метильного радикала, вероятно образующегося при разложении уксусной кислоты, с кислородом с образованием метилгидроперекиси, затем разлагающейся с образованием метанола, и наконец метилацетата при реакции метанола с оставшейся уксусной кислотой. Какой бы не была химическая последовательность реакций, изобретатели открыли, что каждый раз, когда получение метилацетата находится на слишком низком уровне, образование окислов углерода также слишком мало и получение вредных ароматических примесей находится на слишком высоком уровне. Если получение метилацетата находится на слишком высоком уровне, то образование окислов углерода также является недопустимо высоким, что приводит к потерям выхода растворителя, окисляемого соединения и окислителя. При использовании предпочтительных воплощений, раскрытых здесь, производственное отношение молей получаемого метилацетата к молям вводимого окисляемого ароматического соединения предпочтительно больше чем около 0,005:1, более предпочтительно больше чем около 0,010:1 и наиболее предпочтительно больше 0,020:1. В то же время производственное отношение молей получаемого метилацетата к молям вводимого окисляемого ароматического соединения предпочтительно меньше чем около 0,09:1, более предпочтительно меньше чем около 0,07:1, еще более предпочтительно меньше чем около 0,05:1 и наиболее предпочтительно меньше 0,04:1.
Некоторые воплощения этого изобретения могут быть также проиллюстрированы следующими примерами, хотя следует понимать, что эти примеры приведены просто для иллюстрации и не предназначены для ограничения объема изобретения, если на это конкретно не указано.
ПРИМЕРЫ 1 и 2
В примерах 1 и 2 промышленный барботажный реактор колонного типа использовался для окисления параксилола до неочищенной терефталевой кислоты (НТК). В примере 1 шлам в реакционной среде содержал около 28 мас.% твердых веществ. В примере 2 шлам в реакционной среде содержал около 38 мас.% твердых веществ. Эти примеры демонстрируют, что количество твердых веществ в реакционной среде влияют на процесс в отношении образования нежелательных примесей.
В примерах 1 и 2 использовался промышленный барботажный реактор колонного типа с резервуаром, имеющим почти вертикальную по существу цилиндрическую основу с внутренним диаметром около 2,44 м. Высота цилиндрической основы была около 32 м от нижней тангенциальной линии (ТЛ) до верхней тангенциальной линии цилиндрической основы. Резервуар снабжен с 2:1 эллиптическими днищем на крышкой на цилиндрической основе. Общая высота реактора была около 33,2 м. Масса шлама внутри вертикального цилиндра реактора поддерживалась приблизительно постоянной около 63500 кг. Скорость подачи параксилола коммерческой чистоты эффективно постоянна с величиной около 101 килограммов в минуту через круглое отверстие, расположенное в стенке основного тела на высоте около 4,35 м выше нижней ТЛ резервуара. Подавался фильтрат растворителя, хорошо смешанный с параксилолом, с эффективно стабильной скоростью. Фильтрат растворителя подавался из аппарата системы рециркуляции и содержал приблизительно выше 97 мас.% уксусной кислоты и воды. Скорость потока фильтрата растворителя и концентрация каталитических компонентов в фильтрате растворителя были такими, что состав жидкой фазы реакционной среды включал около 2100 частей на миллион кобальта, около 1900 частей на миллион брома и около 125 частей на миллион марганца. Отдельный поток возвратного растворителя подавался в виде капель в зону отсоединения газа выше рабочего уровня реакционной среды при эффективно постоянной скорости. Этот обратный поток растворителя содержал приблизительно выше 99 мас.% уксусной кислоты и воды; и обратный поток растворителя был из отдельного аппарата системы рециркуляции и не имел существенных уровней каталитических компонентов. Окислителем был сжатый воздух, подаваемый с эффективно постоянной скоростью около 450 килограммов в минуту через распределитель окислителя, подобный показанному на фиг. 2-5. Этот распределитель окислителя включал в себя скошенный трубопровод для потока, который имел форму приблизительно равностороннего восьмиугольника с поперечным элементом, соединяющим одну сторону с противоположной стороной и пересекающим вертикальную ось симметрии реактора. Скошенный трубопровод для потока был изготовлен из номинальных 12 дюймовых трубчатых компонентов Schedule 10S. Ширина восьмиугольника от центроиды одной стороны трубопровода для потока до центроиды противоположной стороны была около 1,83 м. Восьмиугольник расположен приблизительно горизонтально и средняя высота размещения ортогонального трубопровода была около 0,11 м над нижней ТЛ реактора. Распределитель окислителя имел около 75 круглых отверстий диаметром около 0,025 м. Указанные отверстия были расположены приблизительно равномерно вокруг указанного восьмиугольника, и поперечный элемент был расположен около верхней части указанных 12 дюймовых труб. Имелось одно круглое отверстие диаметром около 0,012 м около нижней части только одной стороны восьмиугольного трубопровода. Рабочее давление газов в верхней части реактора было стабильным около 0,46 МПа. Реактор работал по существу в адиабатическом режиме, так как теплота реакции повышала температуру входящих потоков и испаряла большое количество поступающего растворителя. Выходящий шлам, содержащий НТК, выводился вблизи нижней части эллиптического днища реактора с существенно стабильной скоростью потока НТК, хотя количество жидкости в шламе изменялось, как описано ниже.
Некоторые воплощения этого изобретения могут быть также проиллюстрированы следующими примерами, хотя следует понимать, что эти примеры приведены только для иллюстрации и не имеют цели ограничить объем изобретения, если на это не будет конкретно указано.
ПРИМЕР 1
В этом примере состав шлама удерживался при содержании в нем около 28 мас.% твердых веществ. Это осуществлялось подстройкой скорости подачи фильтрата растворителя примерно на уровне около 1421 килограмм в минуту и скорости подачи возвратного растворителя примерно на уровне 374 килограмма в минуту. Общее содержание воды в подаваемых фильтрате растворителя и в возвратном растворителе было таково, что концентрация воды в жидкой фазе реакционной среды составляла около 5,7 мас.%. Рабочая температура, измеренная около среднего возвышения реакционной среды, была около 157°С.
ПРИМЕР 2
В этом примере состав шлама удерживался при содержании в нем около 38 мас.% твердых веществ. Это осуществлялось подстройкой скорости подачи фильтрата растворителя примерно на уровне около 775 килограммов в минуту и скорости подачи возвратного растворителя примерно на уровне около 791 килограммов в минуту. Общее содержание воды в подаваемых фильтрате растворителя и в возвратном растворителе было таково, что концентрация воды в жидкой фазе реакционной среды составляла около 6,2 мас.%. Рабочая температура, измеренная примерно посередине высоты столба реакционной среды, была около 159°С. Вопреки большей доли твердых веществ в шламе примера 2, никаких трудностей не наблюдалось при эксплуатации барботажной колонны со шламом или при переносе шлама для последующих операций.
Имеется много примесей, которые обычно образуются при связывании ароматических колец во время частичного окисления параксилола. Одной из примесей является 4-4'-дикарбоксистильбен. Это соединение имеет гораздо более высокое поглощение света, чем терефталевая кислота, и таким образом оно снижает оптическую прозрачность заданного продукта. Кроме того, 4-4'-дикарбоксистильбен является удобной примесью для использования при контроле качества окисления, так как она разделяется избирательно по отношению к твердой фазе реакционной среды, в частности фактически 4-4'-дикарбоксистильбен не присутствовал в потоках рециркулирующего растворителя, вводимых в промышленные барботажные реакторы колонного типа, рассмотренные в примерах 1 и 2.
В примерах 1 и 2 концентрация 4-4'-дикарбоксистильбена измерялась аналитическим способом с использованием ЖХВД-ДМД, калиброванных соответствующей контрольной смесью, содержащей растворитель и известные количества нескольких стандартов, в частности содержащих известное количество 4-4'-дикарбоксистильбена. Аналитический способ с использованием ЖХВД-ДМД рассмотрен в вышеприведенном разделе Подробное описание.
Результаты анализов 4-4'-дикарбоксистильбена (ДКС) с использованием аналитических образцов для примеров 1 и 2 показаны на фиг.36. Эти данные включают в себя данные по около 40 образцам НТК шлама из нижнего потока окислителя, взятых в течение приблизительно 5 дней. Каждый из образцов НТК шлама перед анализом разделялся на жидкую и твердую фазы. Кроме того, анализировалось большое число образцов подаваемого фильтрата и был проведен расчет материального баланса (т.е. суммарная масса соединений Х, созданных в реакторе= массе выводимых соединений Х минус вводимая масса соединений. Из этого материального баланса рассчитывалось отношение скорости образования ДКС к скорости образования НТК. Все данные отдельных анализов и отношений по отдельным материальным балансам были отложены на левой оси в частях на миллион массовых (частей на миллион). Кроме того, на фиг.36 показаны на правой оси массовые проценты твердых веществ в шламе, измеренные не автономным прибором, который был откалиброван с использованием автономных гравиметрических образцов для анализа.
Эти данные показывают, что получение 4-4'-дикарбоксистильбена статистически и сильно возрастает, когда в примере 2 использовалось большее содержание твердых веществ. При содержании 28 мас.% твердых веществ в шламе, как в примере 1, средний уровень образования 4-4'-дикабоксистильбена был 5,7 частей на миллион. При содержании 38 мас.% твердых веществ в шламе, как в примере 2, средний уровень образования 4-4'-дикарбоксистильбена был 8,2 частей на миллион. Это повышение на 44%. Кроме того, следует отметить, что образование 4-4'-дикарбоксистильбена быстро уменьшается на правой оси фиг.36, когда этот эксперимент был завершен и уровень твердых веществ снизился до 33 мас.%. Более того, было отмечено увеличение многих других нежелательных окрашенных примесей в такой же степени, как и 4-4'-декарбоксистильбена, когда массовая доля твердых веществ в шламе была на более высоком уровне, как в примере 2. Таким образом в реакторе окисления, содержащем твердые вещества, слишком высокая доля твердых веществ вызывает существенное вредное воздействие на избирательность окисления. То есть имеется предпочтительный верхний предел в отношении доли твердых веществ в окислителе, который неожиданно оказался меньше, чем было указано при рассмотрении поддержания гидравлической стабильности шлама в реакторе и в последующих после реактора операциях. Кроме того, имеется нижний предел концентрации твердых веществ, ниже которого целесообразность избирательно окисления уменьшается; и объемы оборудования и реальное потребление энергии установками преобладает над расчетами и эксплуатацией в этом режиме.
Изобретение было подробно описано с конкретными ссылками на его предпочтительные воплощения, но следует понимать, что изменения и модификации могут быть осуществлены в пределах его сущности и объема.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2388738C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2363535C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2381212C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2393146C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2384563C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2006 |
|
RU2435753C2 |
| СОСТАВ СЫРОЙ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2005 |
|
RU2388744C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2388745C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ | 2005 |
|
RU2382758C2 |
| ОПТИМИЗИРОВАННОЕ ЖИДКОФАЗНОЕ ОКИСЛЕНИЕ В БАРБОТАЖНОЙ КОЛОННЕ РЕАКТОРНОГО ТИПА | 2005 |
|
RU2381211C2 |
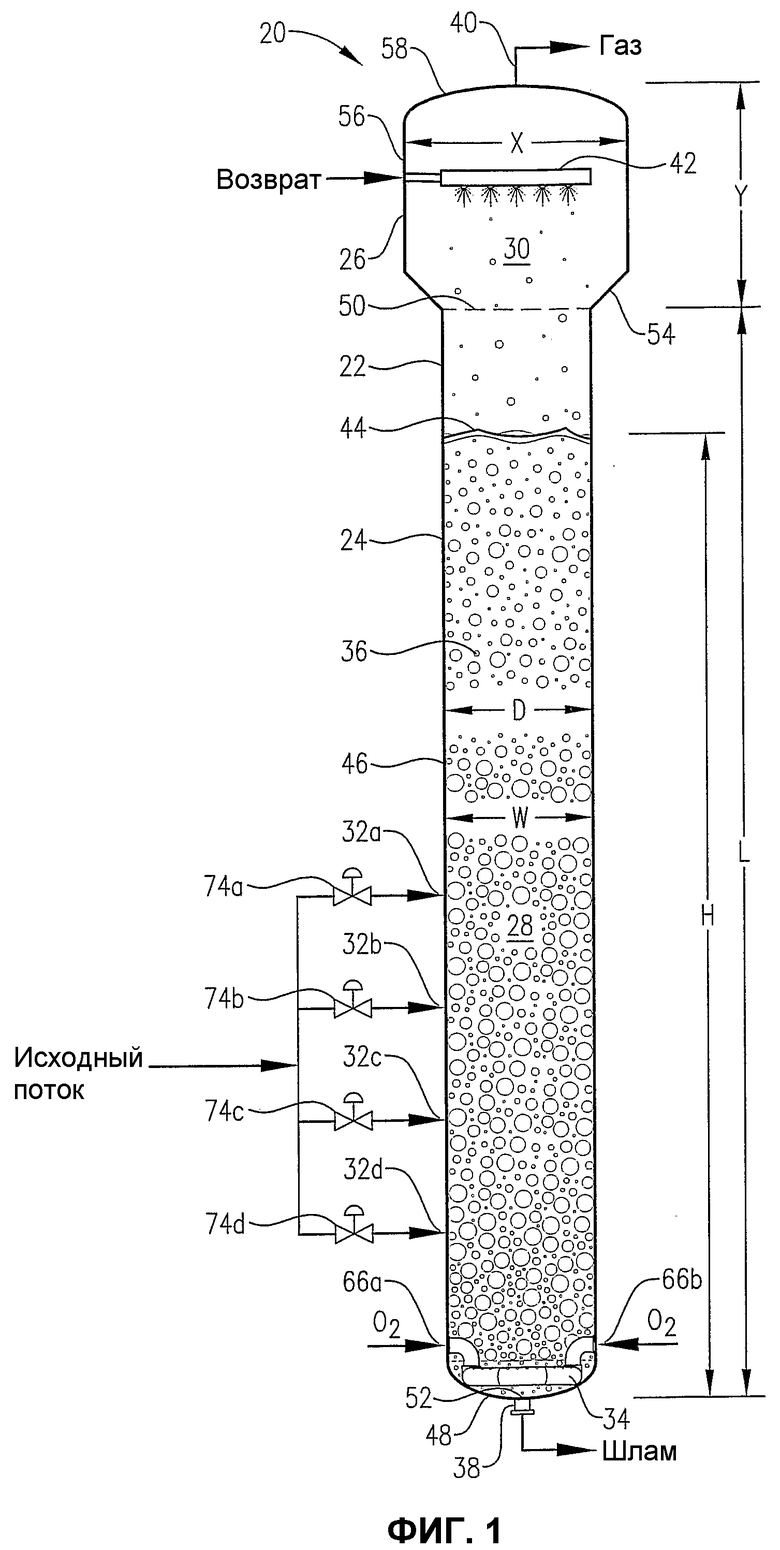
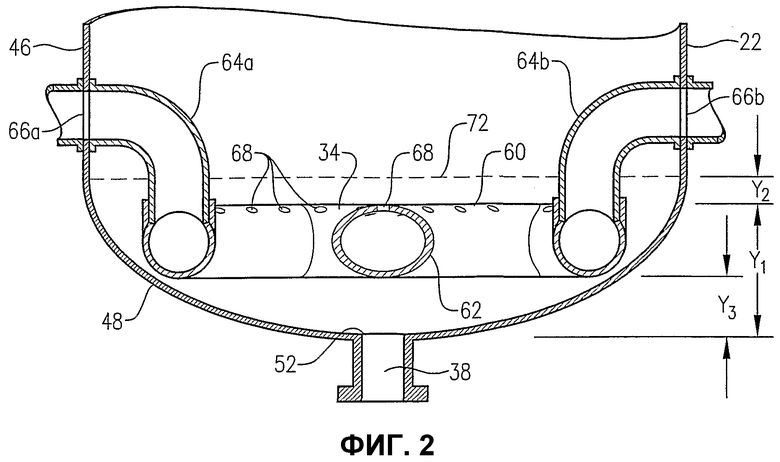


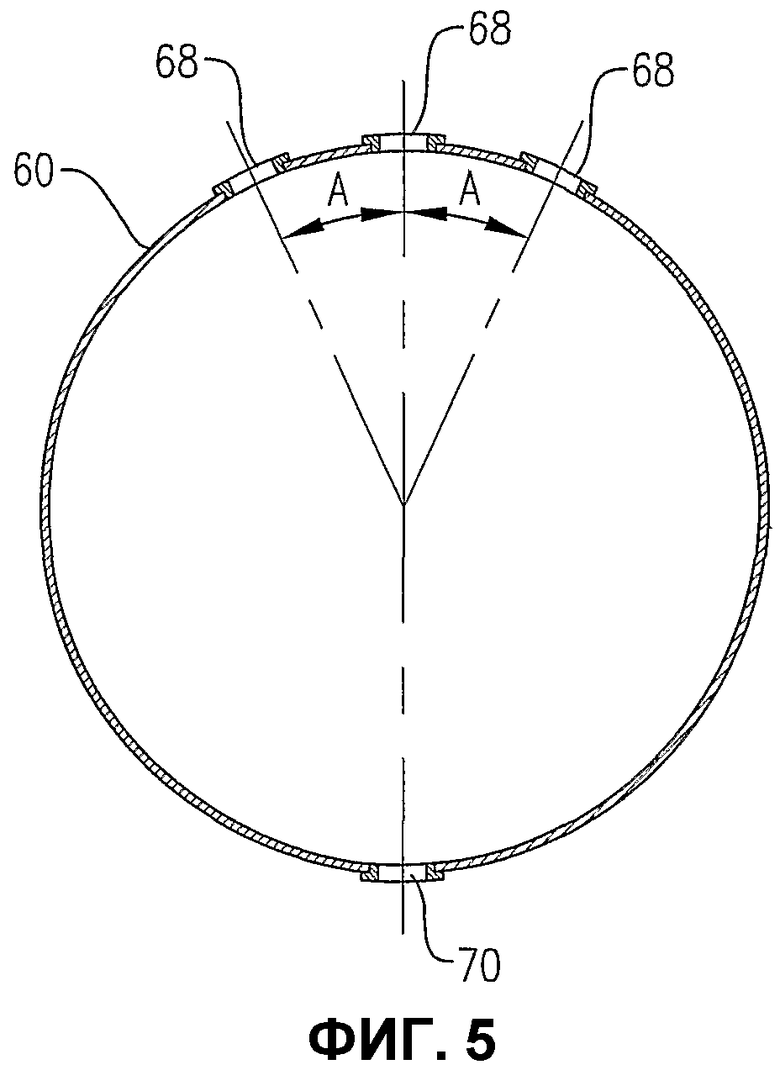
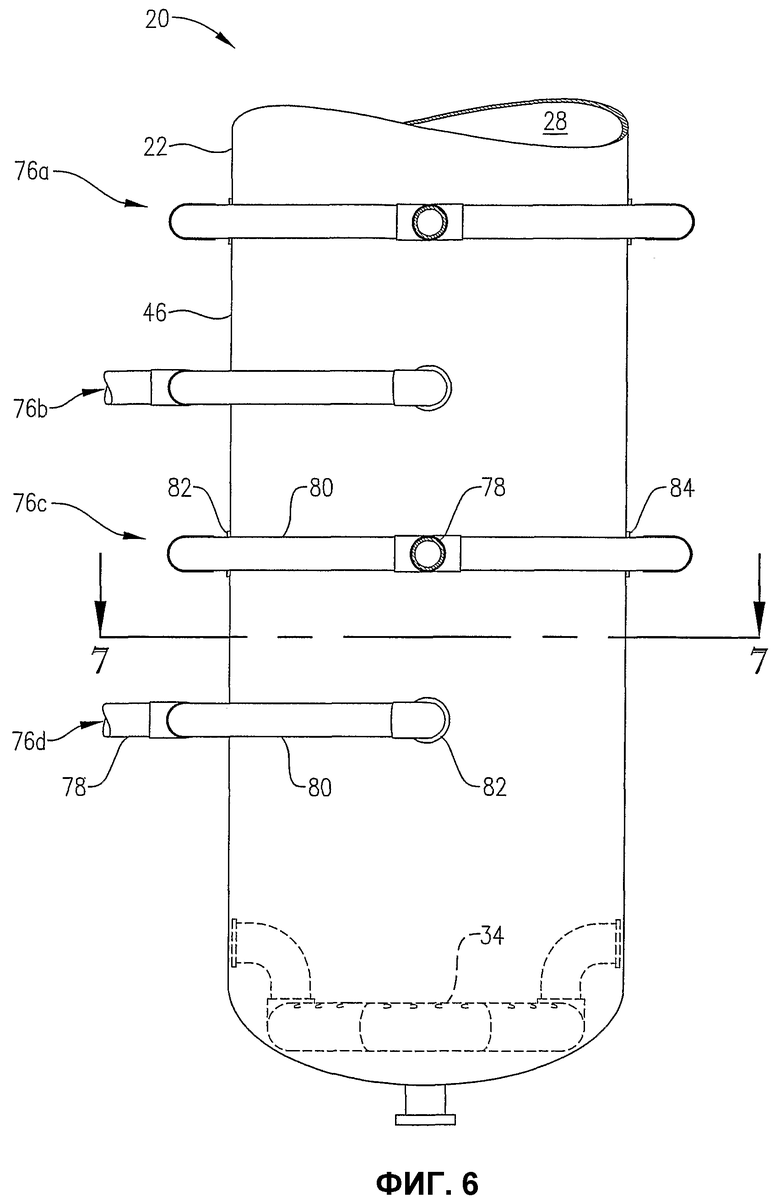
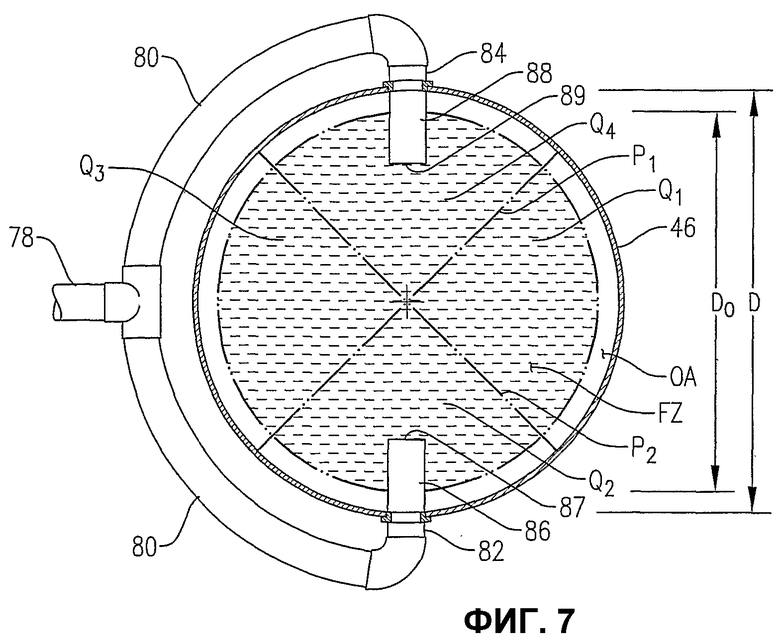

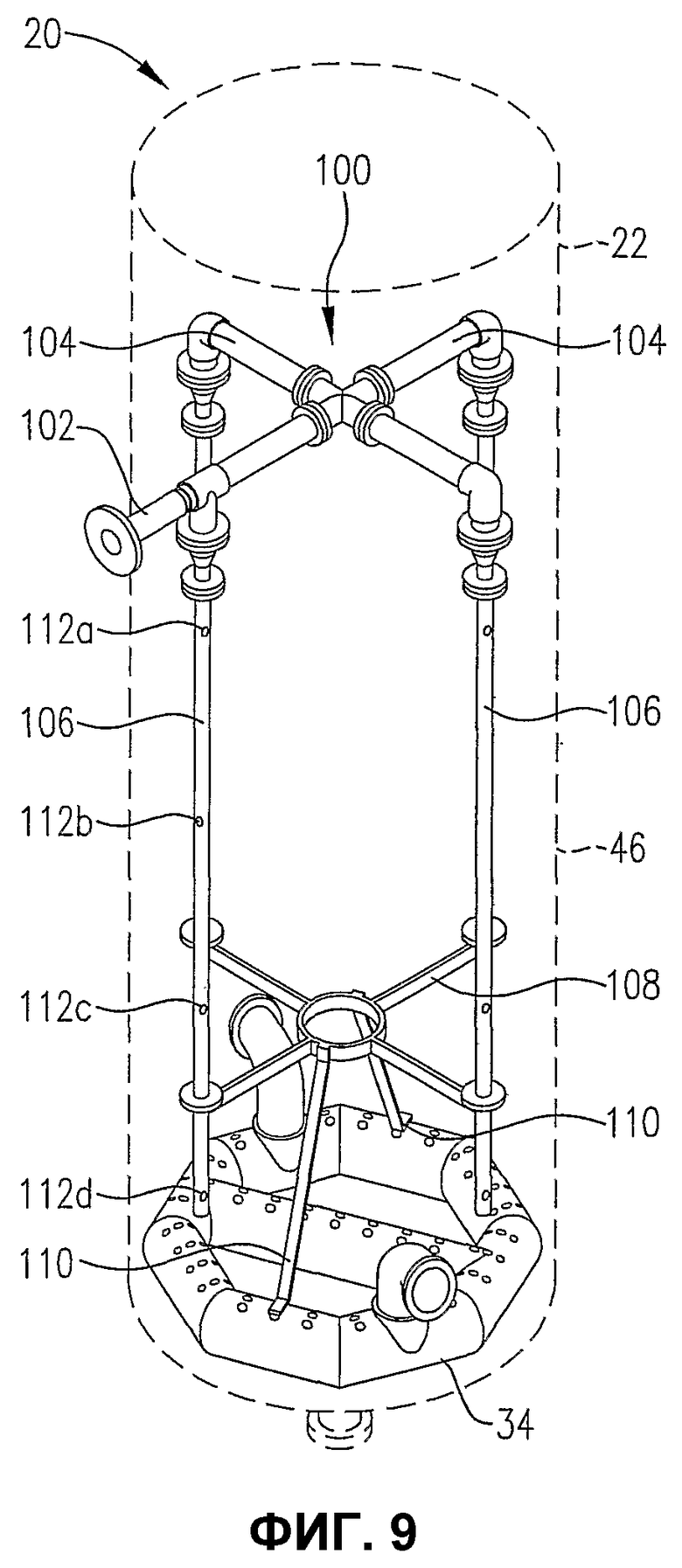
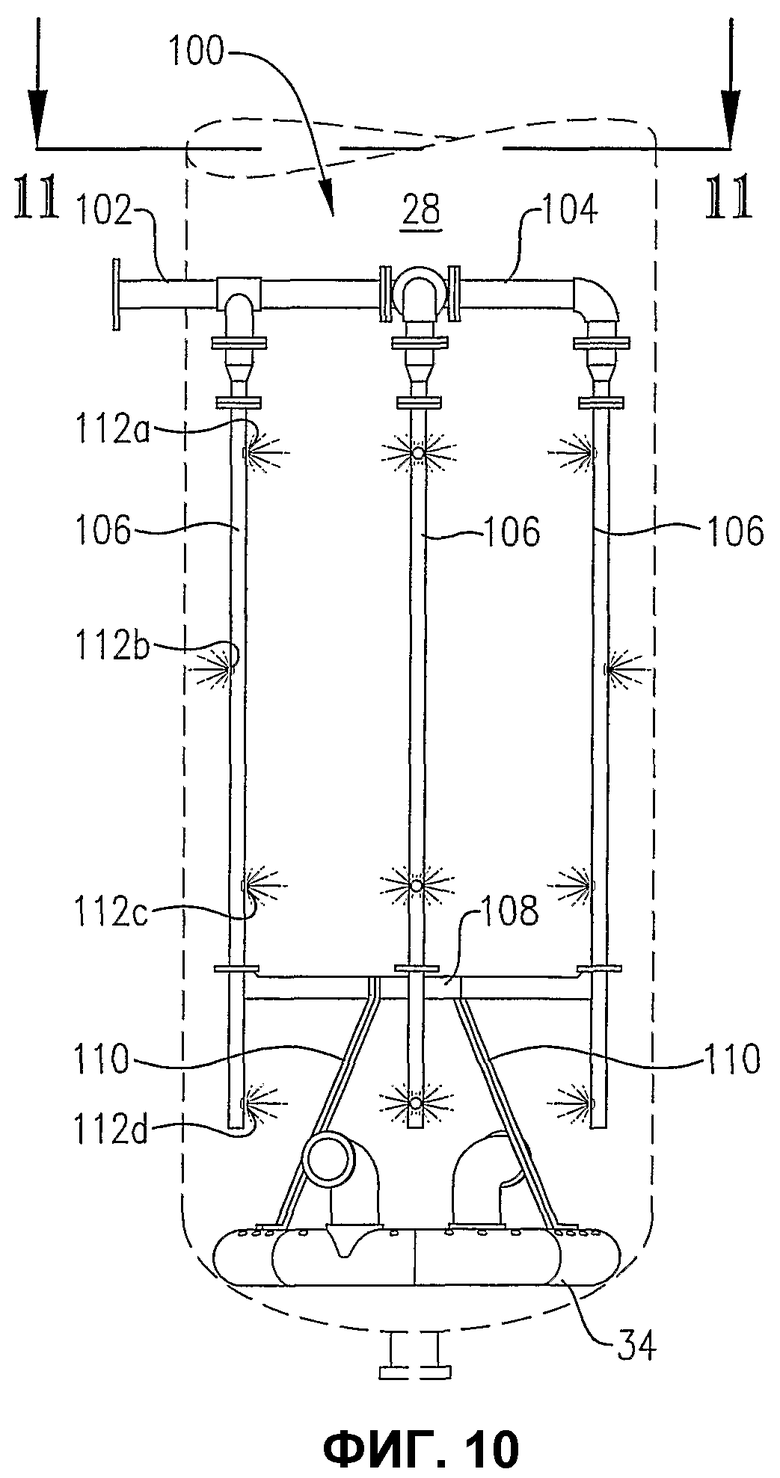

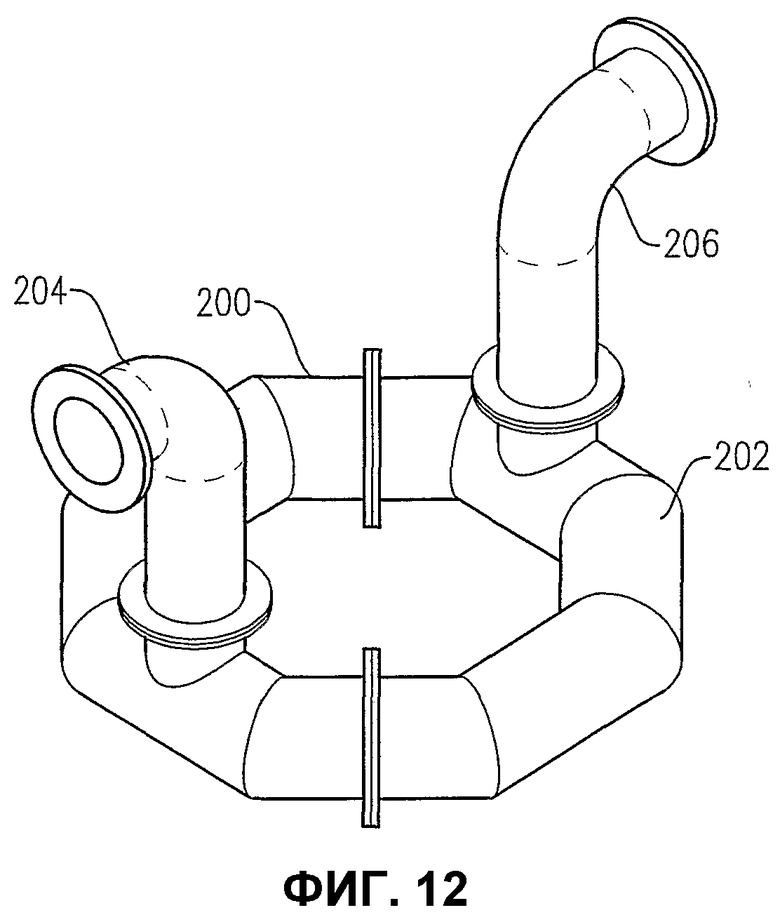
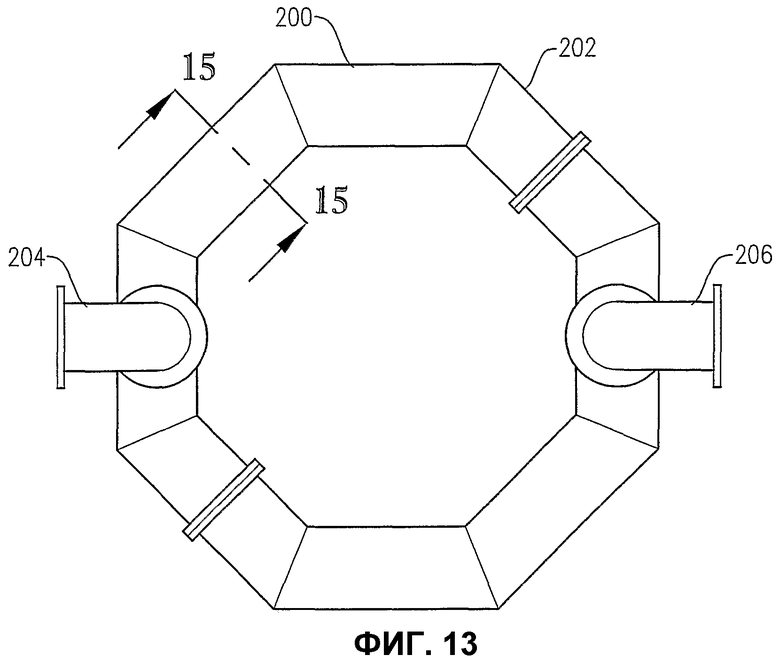

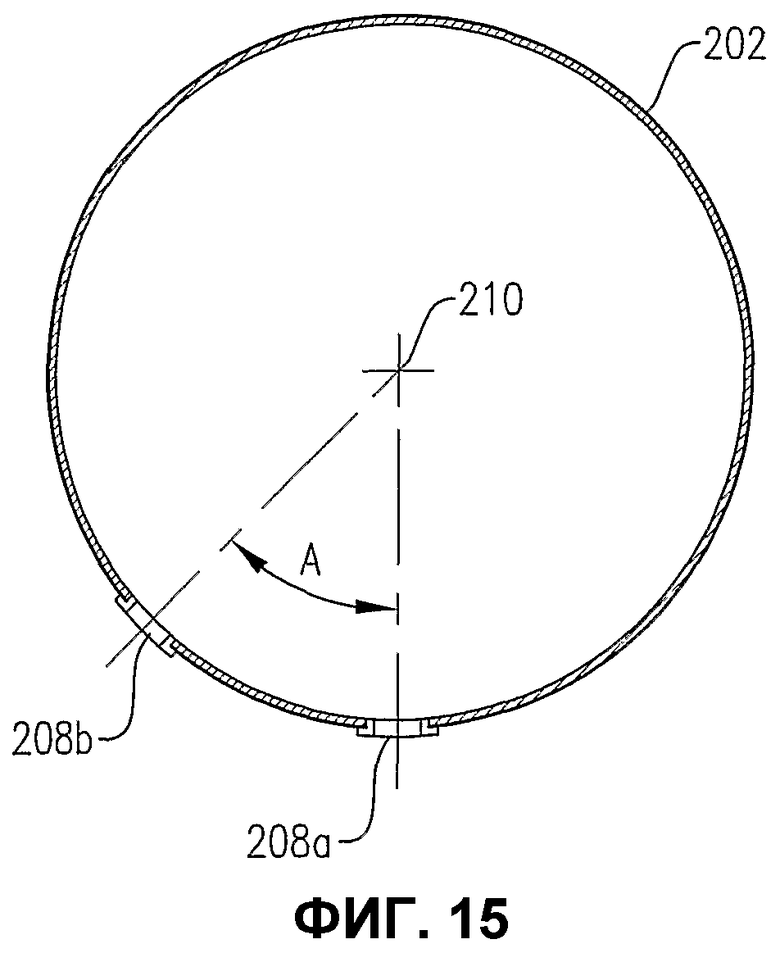

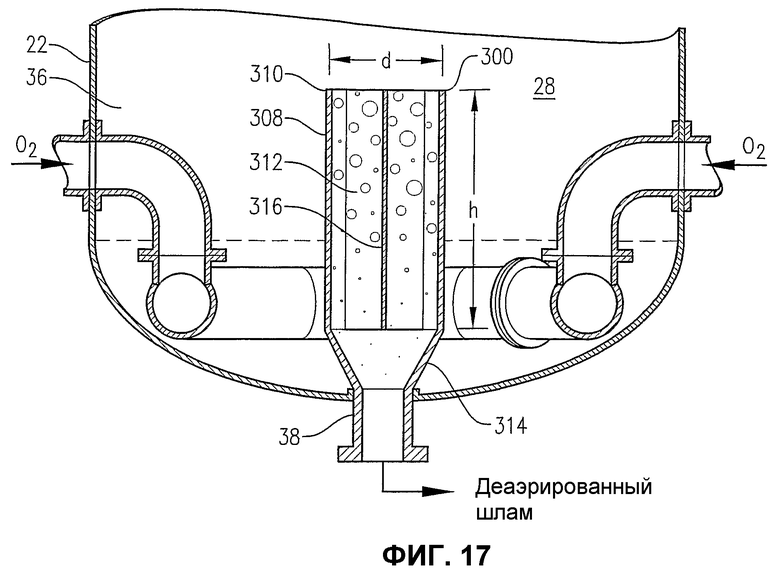
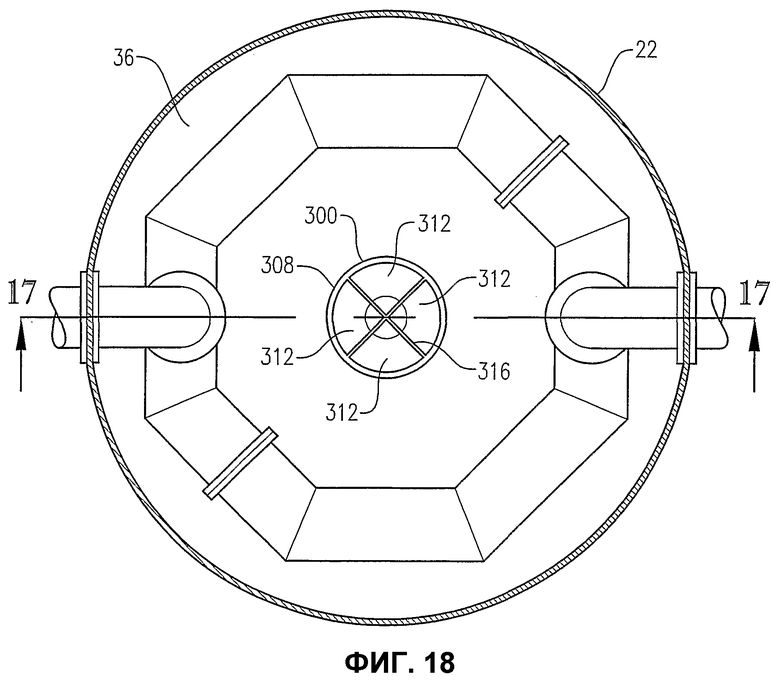
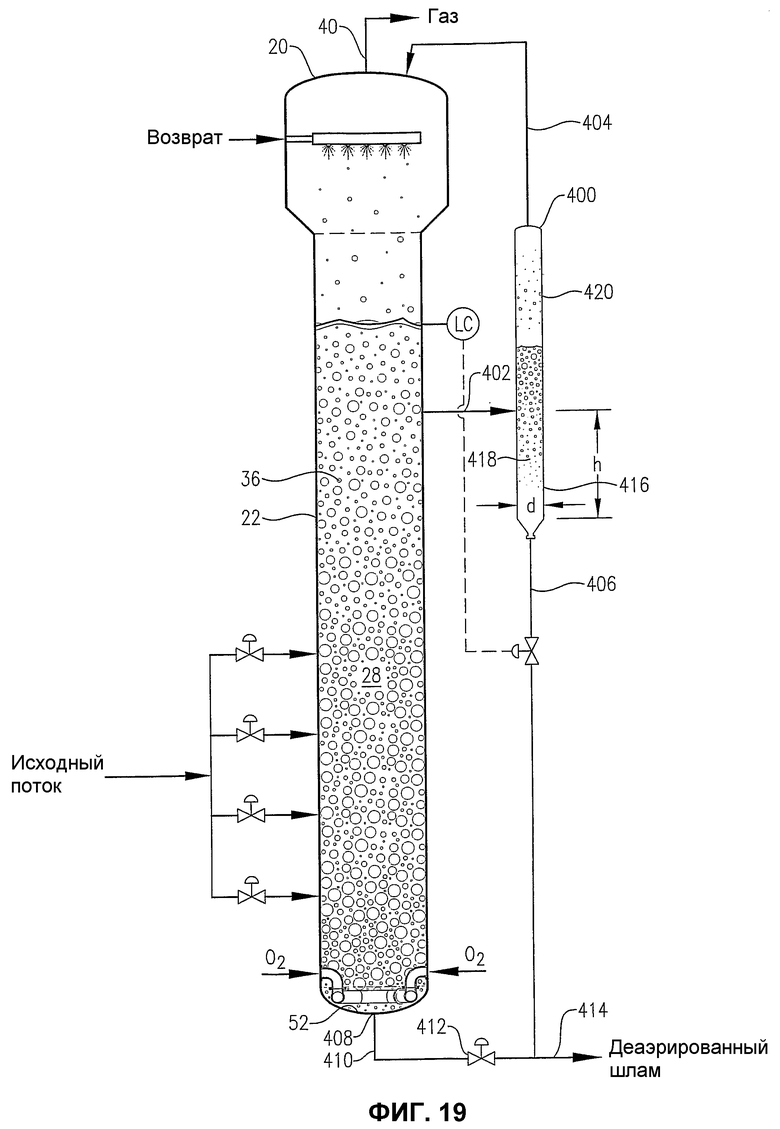
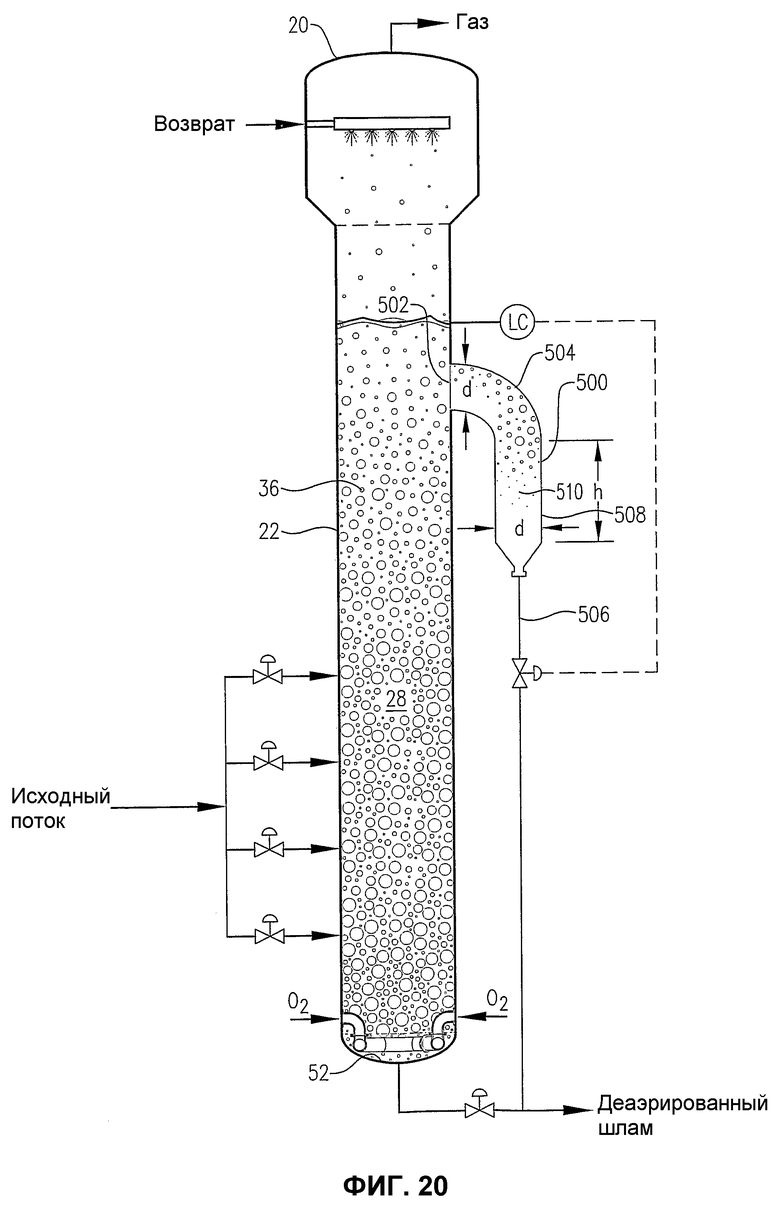
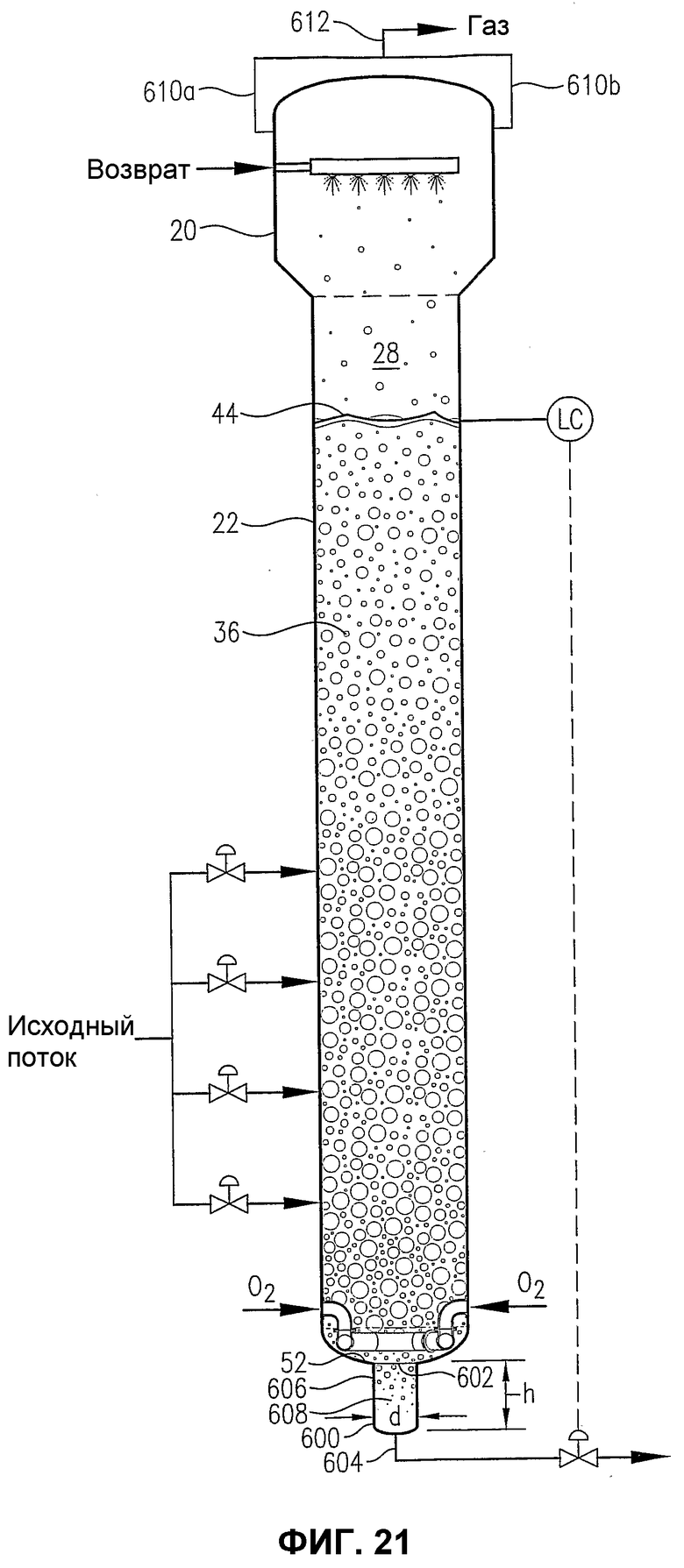


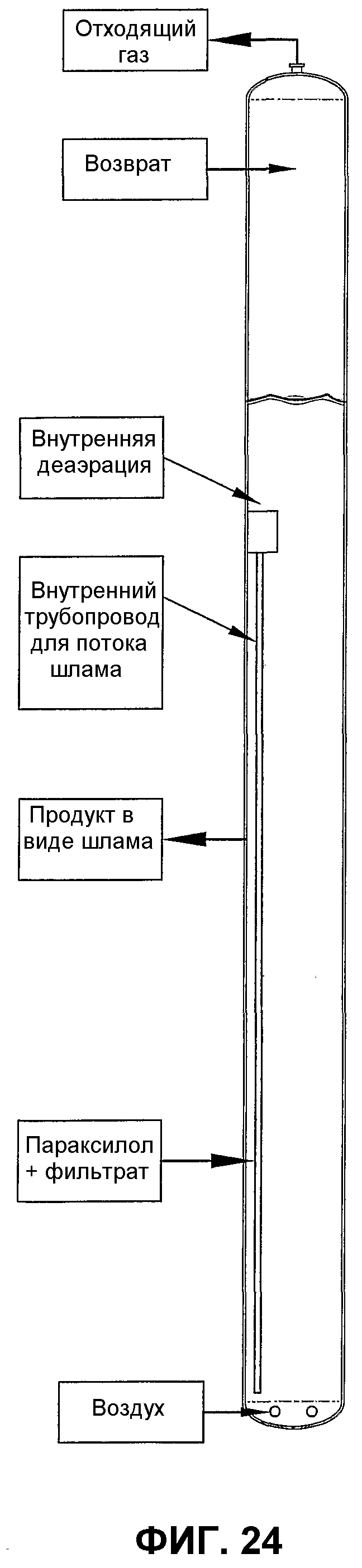
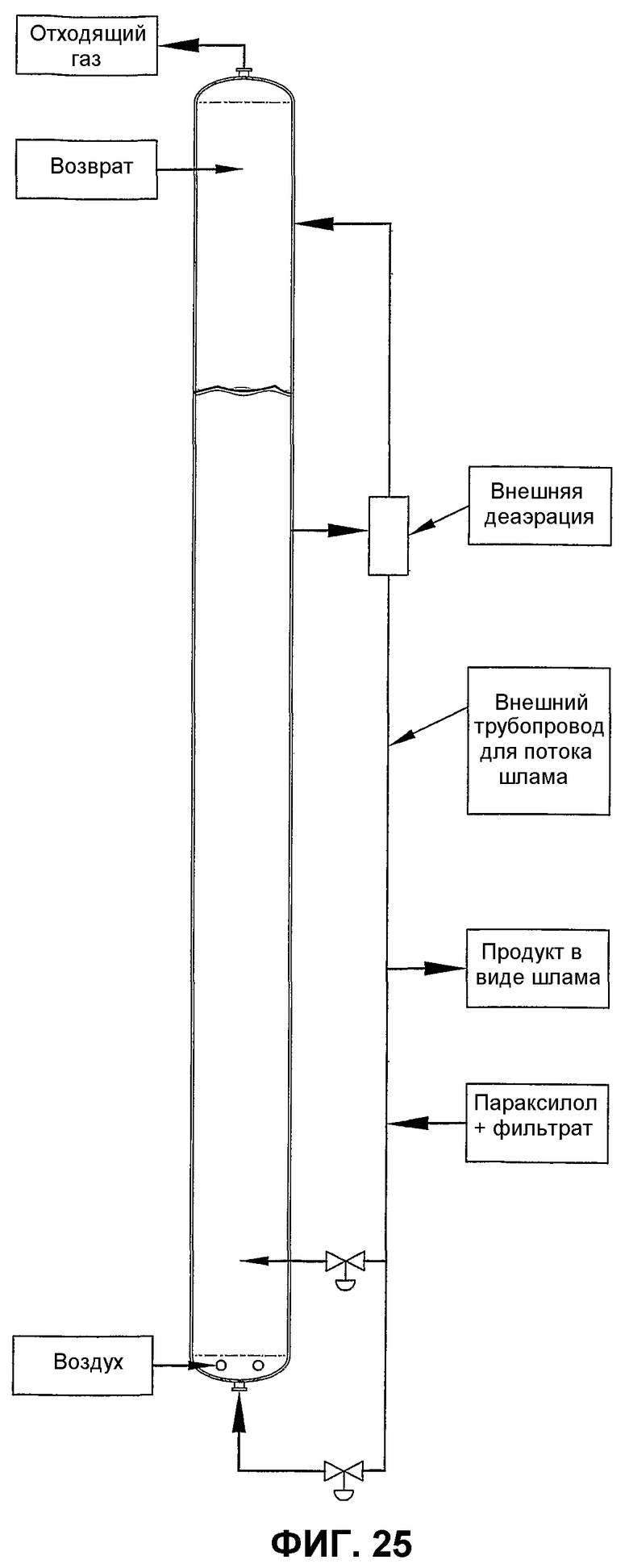

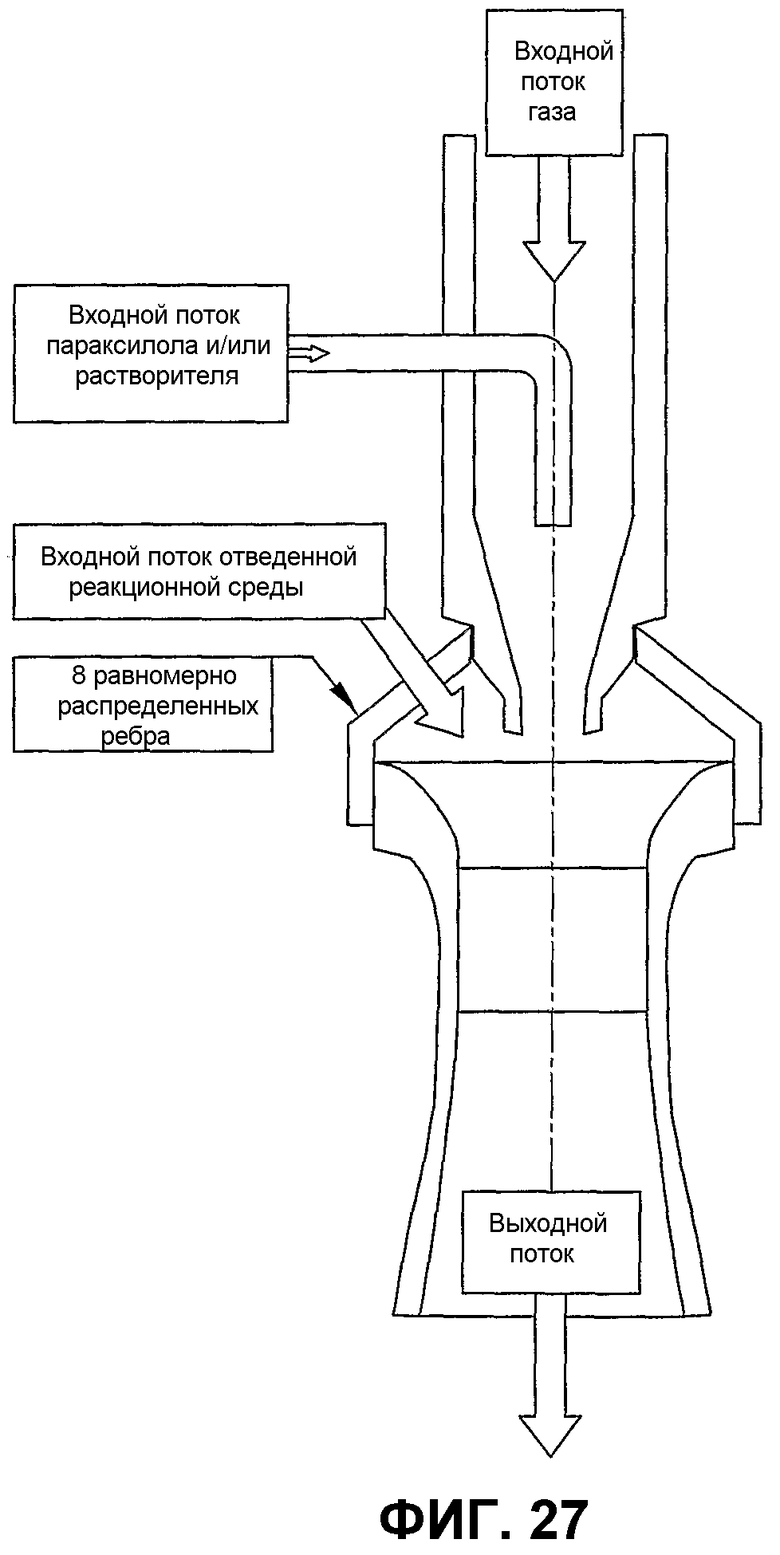
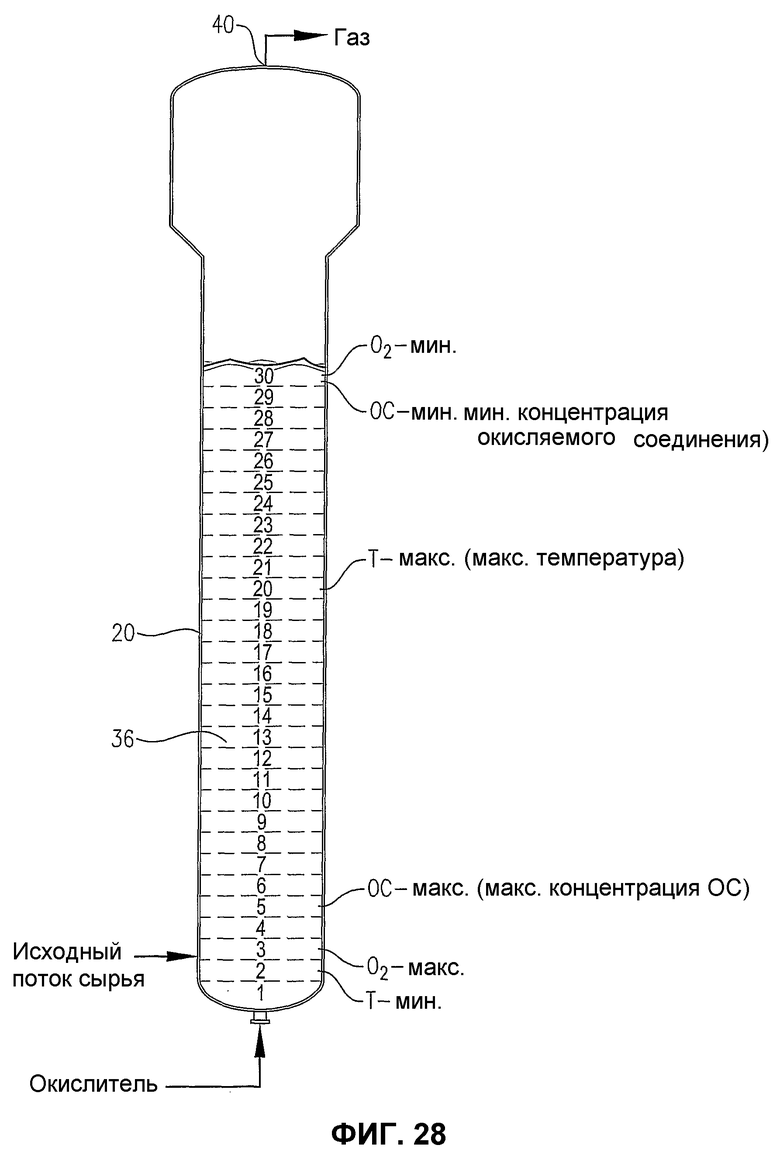
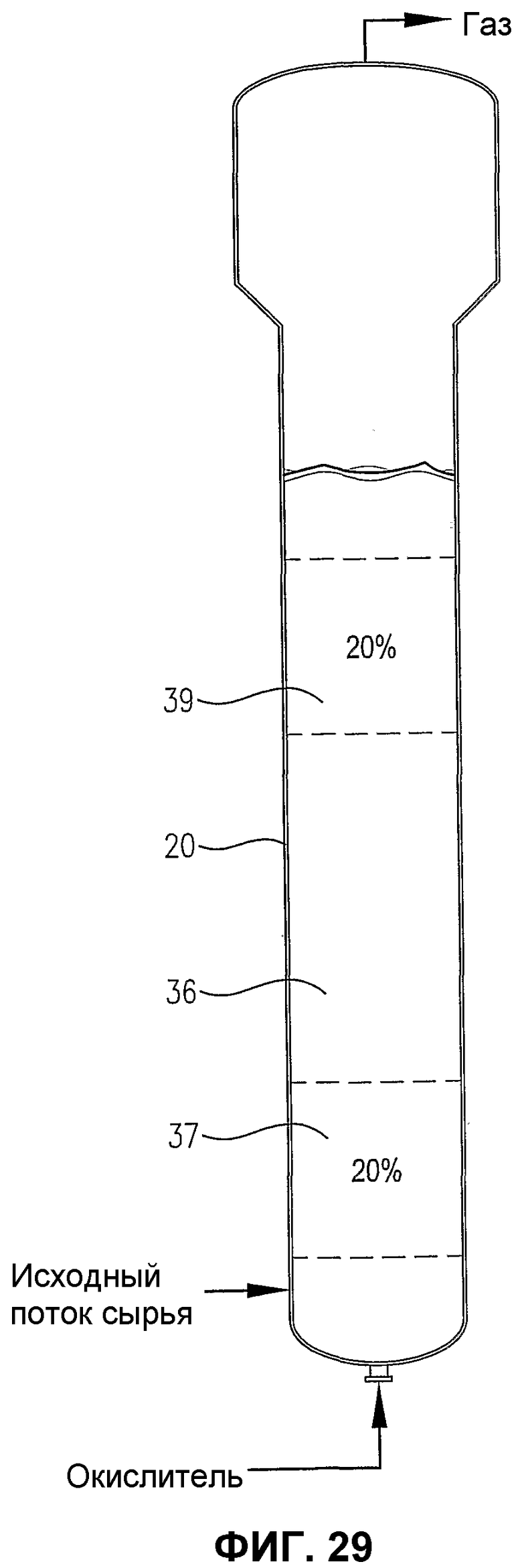
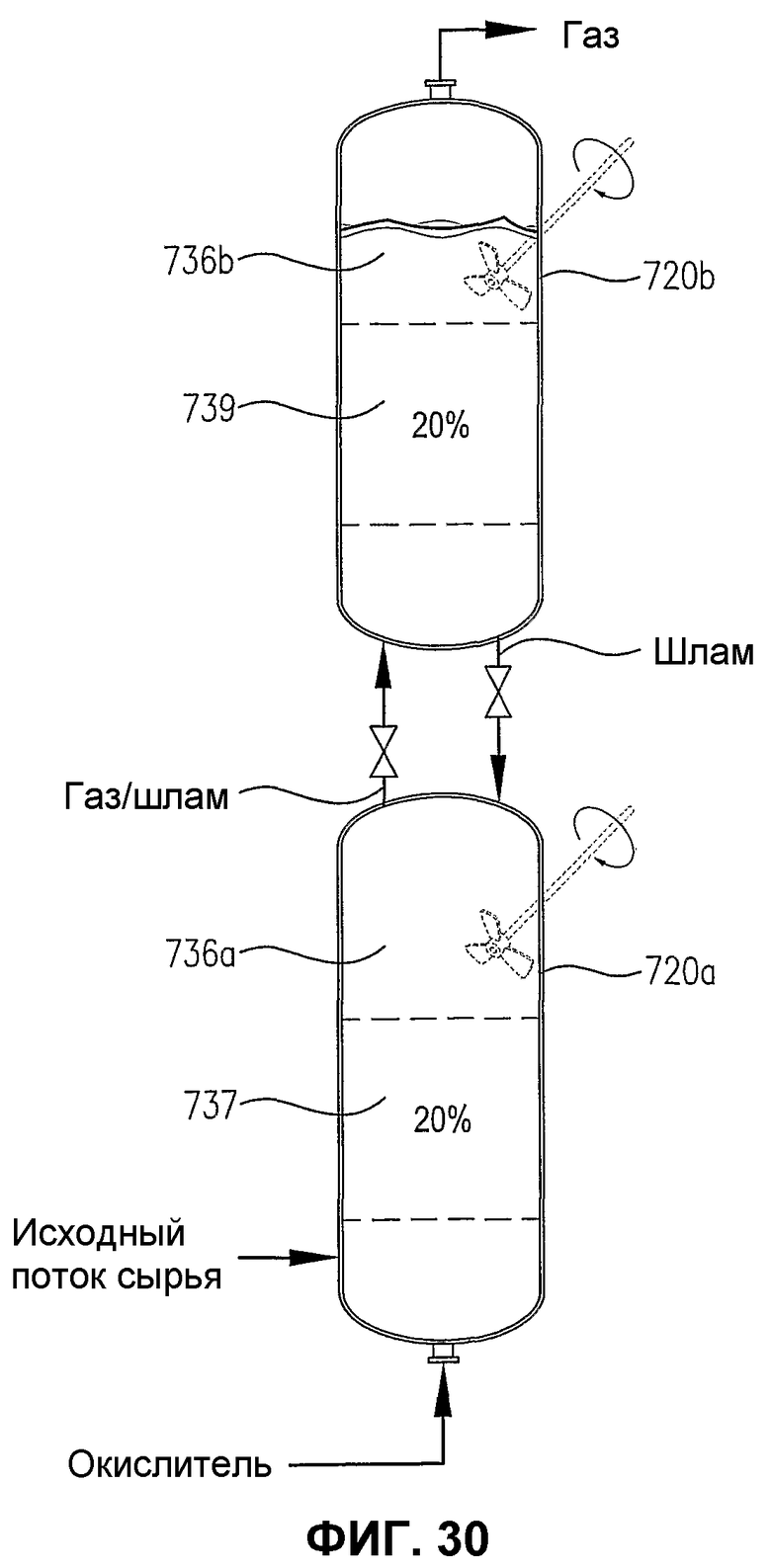
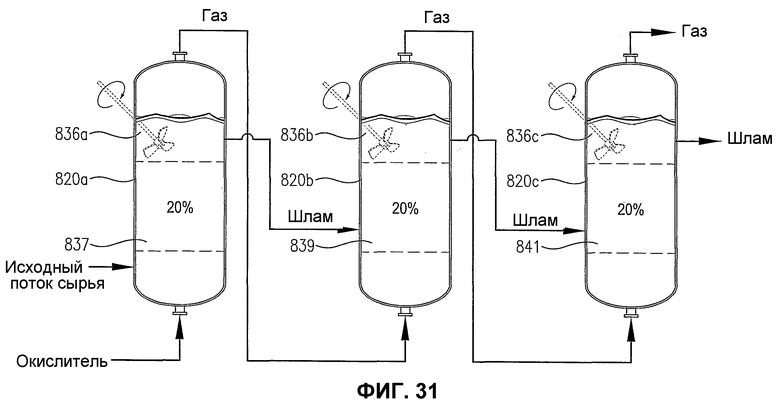





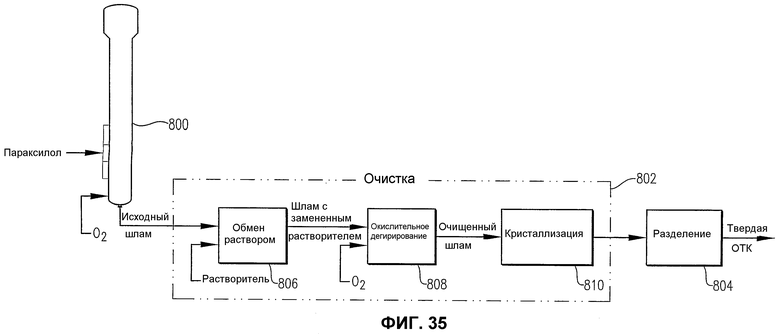
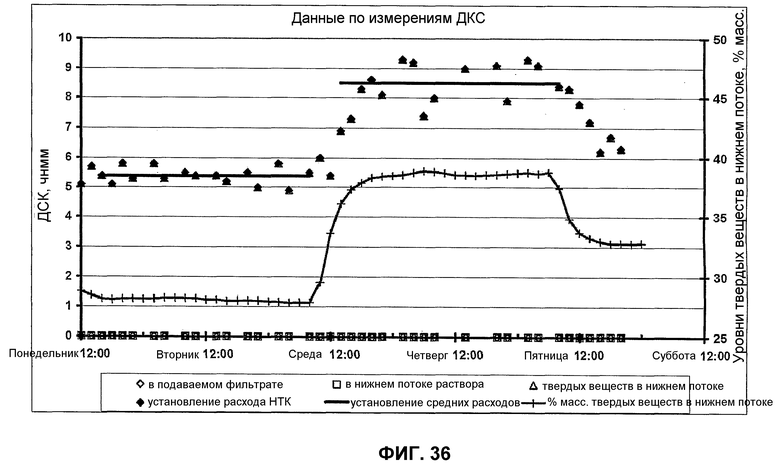
Изобретение относится к процессу жидкофазного каталитического окисления параксилола с получением терефталевой кислоты. Сырье, содержащее параксилол, вводят в реакционную зону барботажного реактора колонного типа. По меньшей мере, 25 мас.% параксилола поступает в реакционную зону на один или более участков, отстоящих от боковой стенки, по меньшей мере, на 0,05D, где D - максимальный диаметр реакционной зоны. Окисление параксилола с образованием неочищенной терефталевой кислоты происходит в жидкой фазе трехфазной реакционной среды, в которой количество твердых веществ поддерживается в интервале от 5 до 40 мас.%, усредненных по времени и объему. Во втором реакторе осуществляют окисление неочищенной терефталевой кислоты для получения более чистой терефталевой кислоты. Обеспечивается повышение эффективности и экономичности процесса. 2 н. и 31 з.п. ф-лы, 36 ил., 4 табл.
1. Способ получения терефталевой кислоты, включающий:
а) подачу исходного потока, содержащего параксилол, в реакционную зону барботажного реактора колонного типа, причем, по меньшей мере, часть реакционной зоны ограничена одной или более вертикальными боковыми стенками реактора, в котором, по меньшей мере, 25 мас.% параксилола подают в реакционную зону на один или более участков, отстоящих от боковой стенки, по меньшей мере, на 0,05D, где D - максимальный диаметр реакционной зоны; и
(b) окисление, по меньшей мере, части параксилола в жидкой фазе трехфазной реакционной среды, размещенной в реакционной зоне, с образованием неочищенной терефталевой кислоты в реакционной среде, при этом количество твердых веществ, присутствующих в реакционной среде, поддерживают в интервале от около 5 до около 40 мас.%, усредненных по времени и по объему.
2. Способ по п.1, в котором окисление проводят таким образом, что при теоретическом разделении реакционной зоны на 2000 дискретных горизонтальных слоев равного объема менее 40 горизонтальных слоев имеют среднюю задержку газа менее 0,3, а задержку газа реакционной среды поддерживают выше около 0,4, усредненных по времени и по объему.
3. Способ по п.1, в котором количество твердых веществ, присутствующих в реакционной среде, поддерживают в интервале от 10 до 35 мас.%, усредненных по времени и по объему, причем задержку газа реакционной среды поддерживают в интервале от около 0,6 до около 0,9, усредненных по времени и по объему.
4. Способ по п.1, в котором количество твердых веществ, присутствующих в реакционной среде, поддерживают в интервале от 15 до 30 мас.%, усредненных по времени и по объему, причем задержку газа реакционной среды поддерживают в интервале от 0,65 до 0,85, усредненных по времени и по объему, и окисление проводят таким образом, что при теоретическом разделении указанной реакционной зоны на 2000 дискретных горизонтальных слоев равного объема менее 20 указанных горизонтальных слоев имеют задержку газа менее 0,3, усредненных по времени и по объему.
5. Способ по п.1, в котором окисление вызывает, по меньшей мере, из около 10 мас.% параксилола образование частиц твердой неочищенной терефталевой кислоты в реакционной среде.
6. Способ по п.1, в котором окисление проводят в присутствии каталитической системы, содержащей кобальт.
7. Способ по п.1, в котором каталитическая система также содержит бром и марганец.
8. Способ по п.1, в котором реакционная среда имеет максимальную высоту (Н), максимальную ширину (W) и отношение H:W, по меньшей мере, около 3:1.
9. Способ по п.8, в котором отношение H:W находится в интервале от около 8:1 до около 20:1.
10. Способ по п.8, который включает ввод в реакционную зону потока преимущественно газофазного окислителя, содержащего молекулярный кислород.
11. Способ по п.10, в котором большую часть молекулярного кислорода вводят в реакционную зону в место, находящееся в пределах около 0,25W от низа реакционной зоны.
12. Способ по п.10, в котором вводимый поток содержит уксусную кислоту.
13. Способ по п.12, в котором, по меньшей мере, около 50 мас.% параксилола поступает в реакционную зону в место, находящееся в пределах около 2,5 W от самого низкого места, в котором молекулярный кислород поступает в реакционную зону.
14. Способ по п.12, в котором подаваемый поток вводят в реакционную зону через большое число подающих отверстий, причем, по меньшей мере, два из указанных подающих отверстия вертикально отделены одно от другого промежутком, составляющим, по меньшей мере, около 0,5W.
15. Способ по п.1, в котором параксилол вводят в реакционную зону со скоростью потока массы, по меньшей мере, около 8000 кг в час.
16. Способ по п.1, в котором параксилол вводят в реакционную зону с объемно-временной скоростью потока в интервале от приблизительно 25 до приблизительно 40 кг/м3 в час.
17. Способ по п.1, в котором параксилол вводят в барботажный реактор колонного типа со скоростью потока массы в интервале от около 18000 до около 50000 кг в час, при этом параксилол вводят в реакционную зону с объемно-временной скоростью в интервале от около 35 до около 150 кг/м3 в час.
18. Способ получения терефталевой кислоты, включающий:
а) подачу исходного потока, содержащего параксилол, в реакционную зону барботажного реактора колонного типа, причем, по меньшей мере, часть реакционной зоны ограничена одной или более вертикальными боковыми стенками реактора, при этом, по меньшей мере, 25 мас.% параксилола подают в реакционную зону на один или более участков, отстоящих от боковой стенки, по меньшей мере, на 0,05D, где D - максимальный диаметр реакционной зоны;
(b) окисление, по меньшей мере, части параксилола в жидкой фазе трехфазной реакционной среды, размещенной в реакционной зоне, с образованием неочищенной терефталевой кислоты в реакционной среде, при этом количество твердых веществ, присутствующих в реакционной среде, поддерживают в интервале от около 5 до около 40 мас.%, усредненных по времени и по объему, и
с) окисление, по меньшей мере, части частиц твердой неочищенной терефталевой кислоты во втором реакторе окисления для получения более чистой терефталевой кислоты.
19. Способ по п.18, в котором окисление проводят таким образом, что при теоретическом разделении реакционной зоны на 2000 дискретных горизонтальных слоев равного объема менее 40 горизонтальных слоев имеют среднюю задержку газа менее 0,3, а задержку газа реакционной среды поддерживают выше около 0,4, усредненных по времени и по объему.
20. Способ по п.18, в котором количество твердых веществ, присутствующих в реакционной среде, поддерживают в интервале от 10 до 35 мас.%, усредненных по времени и по объему.
21. Способ по п.18, в котором задержку газа реакционной среды поддерживают в интервале от около 0,6 до около 0,9, усредненных по времени и по объему.
22. Способ по п.18, в котором количество твердых веществ, присутствующих в реакционной среде, поддерживают в интервале от 15 до 30 мас.%, усредненных по времени и по объему, причем задержку газа реакционной среды поддерживают в интервале от 0,65 до 0,85, усредненных по времени и по объему, при этом окисление проводят таким образом, что при теоретическом разделении реакционной зоны на 2000 дискретных горизонтальных слоев равного объема менее 20 указанных горизонтальных слоев имеют задержку газа менее 0,3, усредненных по времени и по объему.
23. Способ по п.18, в котором окисление во втором реакторе окисления снижает среднюю концентрацию 4-КБА, присутствующую в твердых частицах неочищенной терефталевой кислоты, до 200 мас. ч. на миллион с получением более чистой терефталевой кислоты.
24. Способ по п.18, в котором твердые частицы неочищенной терефталевой кислоты имеют среднюю концентрацию 4-КБФ, по меньшей мере, 400 мас. ч. на миллион и более чистая терефталевая кислота имеет среднюю концентрацию 4-КБА менее 400 мас. ч. на миллион.
25. Способ по п.18, в котором окисление во втором реакторе окисления снижает среднюю концентрацию 4-КБА, присутствующую в твердых частицах неочищенной терефталевой кислоты, до 400 мас. ч. на миллион с получением более чистой терефталевой кислоты, причем твердые частицы неочищенной терефталевой кислоты имеют среднюю концентрацию 4-КБА, по меньшей мере, 800 мас. ч. на миллион и более чистая терефталевая кислота имеет среднюю концентрацию 4-КБА менее 250 мас. ч. на миллион.
26. Способ по п.18, в котором окисление во втором реакторе окисления проводят при средней температуре, которая, по меньшей мере, на около 10°С выше средней температуры окисления в исходном реакторе окисления.
27. Способ по п.18, в котором окисление в реакторе колонного типа проводят при средней температуре в интервале от около 125 до около 200°С, причем окисление во втором реакторе проводят при средней температуре в интервале от около 160 до около 240°С.
28. Способ по п.18, в котором окисление во втором реакторе окисления проводят при средней температуре, которая на около 20 до около 80°С выше средней температуры исходного реактора окисления, в котором окисление проводят при средней температуре в интервале от около 140 до около 180°С, при этом окисление в указанном втором реакторе окисления проводят при средней температуре в интервале от около 180 до около 220°С.
29. Способ по п.18, в котором частицы неочищенной терефталевой кислоты имеют площадь поверхности по БЭТ больше, чем около 0,6 м2/г.
30. Способ по п.18, в котором частицы неочищенной терефталевой кислоты имеют средний размер в интервале от около 20 до около 150 мкм.
31. Способ по п.18, в котором существенная часть твердых частиц неочищенной терефталевой кислоты содержит агломерированные субчастицы, имеющие средний размер в интервале от около 0,5 мкм до около 30 мкм.
32. Способ по п.18, в котором частицы неочищенной терефталевой кислоты имеют средний размер в интервале от около 30 до около 120 мкм, причем указанные субчастицы имеют средний размер в интервале от около 1 мкм до около 15 мкм.
33. Способ по п.18, который дополнительно включает извлечение исходной взвеси, содержащей маточную жидкость и твердые частицы неочищенной терефталевой кислоты, из барботажного реактора колонного типа, причем указанный процесс включает замену, по меньшей мере, 50 мас.% маточной жидкости в начальной смеси заменяющим растворителем с получением взвеси, содержащей заменяющий растворитель и твердые частицы неочищенной терефталевой кислоты, причем полученную взвесь вводят во второй реактор окисления.
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИГИДРОКСИБЕНЗОЛОВ | 2002 |
|
RU2228326C2 |
| US 3240803 A, 15.03.1966 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |

