Настоящее изобретение относится к новым фармацевтическим применениям соединений, обладающим антагонистической активностью в отношении метаботропных глутаматных рецепторов (mGluR).
Глутамат является основным медиатором возбуждения в центральной нервной системе, осуществляющим свое действие посредством ионотропных глутаматных рецепторов. Он также играет основную роль в активации путей модуляции метаболизма посредством mGluR.
На основе гомологии их аминокислотных последовательностей, фармакологических характеристик агонистов и участия в механизмах трансдукции 8 известных в настоящее время подтипов mGluR подразделяют на три группы. Было установлено, что рецепторы группы I (mGluR1 и mGluR5) участвуют в стимуляции фосфолипазы С (PLC), вызывающей гидролиз фосфоинозитида и повышение уровней Са++, и в некоторых экспрессионных системах участвуют в модуляции ионных каналов, таких как К+-каналы, Са++-каналы, катионные каналы неизбирательного действия или NMDA-рецепторы. Рецепторы группы II (mGluR2 и mGluR3) и рецепторы группы III (mGluR4, mGluR6, mGluR7 и mGluR8) взаимодействуют с аденилилциклазой по типу отрицательной связи, и было установлено, что они принимают участие в ингибировании образования цАМФ при гетерологичной экспрессии в клетках млекопитающих и во внутренней очистке активированных протеином G калиевых каналов в ооцитах Xenopus и в однополярных щеточных клетках мозжечка. За исключением mGluR6, который экспрессируется практически только в сетчатке, mGluR, по-видимому, широко экспрессируются в центральной нервной системе (ЦНС).
Указанные mGluR представляют собой потенциальные важные терапевтические мишени при многочисленных неврологических и психических нарушениях, это мнение в значительной степени основано на исследованиях соединений, которые не обладают избирательностью в отношении подтипов mGluR (см. обзор Knopfel и др., J. Med. Chem. 38, 1417-1426, 1995; Conn и Pin, Annu. Rev. Pharmacol. Toxicol. 37, 205-237, 1997). В частности, для группы I mGluR выяснение роли отдельных подтипов рецепторов в значительной степени сдерживается отсутствием соединений, обладающих выраженной системной активностью и избирательным действием в отношении отдельных подтипов.
При создании настоящего изобретения неожиданно было установлено, что избирательные антагонисты mGluR5 обладают высокой эффективностью в отношении лечения боли и состояния тревоги.
Эти данные основаны на результатах экспериментов, проведенных с новым классом соединений, обладающих высокой избирательностью и афинностью в качестве антагонистов рецептора mGluR5 человека и крысы (избирательные антагонисты mGluR5). Избирательные антагонисты mGluR5 в контексте настоящего описания, как правило, обладают приблизительно в 100 раз большей активностью в отношении рецептора mGluR5, чем в отношении рецептора mGluR1, предпочтительно приблизительно в 200 раз большей активностью и наиболее предпочтительно приблизительно в 400 раз большей активностью. Эти избирательные антагонисты mGluR представляют собой 2-арилалкенил-, 2-гетероарилалкенил-, 2-арилалкинил-, 2-гетероарилалкинил-, 2-арилазо- и 2-гетероарилазопиридины, более конкретно 6-метил-2-(фенилазо)-3-пиридинол, (Е)-2-метил-6-стирилпиридин и соединения формулы I
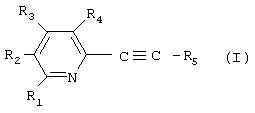
где
R1 обозначает водород, С1-С4алкил, С1-С4алкокси, циано, этинил или диС1-С4алкиламино,
R2 обозначает водород, гидрокси, карбокси, С1-С4алкоксикарбонил, диС1-С4алкиламинометил, 4-(4-фторбензоил)пиперидин-1-илкарбокси, 4-трет-бутилоксикарбонилпиперазин-1-илкарбокси, 4-(4-азидо-2-гидроксибензо-ил)пиперазин-1-илкарбокси или 4-(4-азидо-2-гидрокси-3-йодбензоил)пиперазин-1-илкарбокси,
R3 обозначает водород, С1-С4алкил, карбокси, С1-С4алкоксикарбонил, С1-С4алкилкарбамоил, гидроксиС1-C4алкил, диС1-С4алкиламинометил, морфолинокарбонил или 4-(4-фторбензоил)пиперидин-1-илкарбокси,
R4 обозначает водород, гидрокси, карбокси, С2-С5алканоилокси, С1-С4алкоксикарбонил, аминоС1-С4алкокси, диС1-С4алкиламиноС1-С4алкокси, диС1-С4алкиламиноС1-С4алкил или гидроксиС1-С4алкил и
R5 обозначает группу формулы
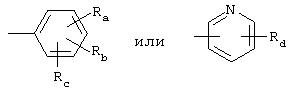
где Ra и Rb независимо друг от друга обозначают водород, галоген, нитро, циано, С1-С4алкил, С1-C4алкокси, трифторметил, трифторметокси или С2-С5алкинил и Rc обозначает водород, фтор, хлор, бром, гидрокси, С1-С4алкил, С2-С5алканоилокси, С1-С4алкокси или циано и
Rd обозначает водород, галоген или С1-С4алкил, в свободной форме или в форме фармацевтически приемлемых солей.
Более конкретно данные основаны на экспериментах, проведенных с соединениями, включающими 2-[2-(пиридин-3-ил)этинил]-6-метилпиридин, 3-метокси-6-метил-2-мета-толилэтинилпиридин, 2-метил-6-(2,3,5-трихлорфенилэтинил)пиридин, 2-метил-6-(фенилэтинил)пиридин и 2-(3-фторфенилэтинил)-6-метилпиридин (используемыми в форме свободных оснований).
Соединения формулы I могут быть получены путем взаимодействия соединения формулы II
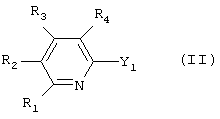
с соединением формулы III

где один из Y1 и Y2 обозначает реакционноспособную этерифицированную гидроксигруппу или галоген, такой как бром или йод, а другой обозначает группу Y3-C≡C-, где Y3 обозначает водород или металл, и R1, R2, R3, R4 и R5 имеют указанные выше значения и функциональные группы R1, R2, R3 и R4, а также функциональные заместители R5 могут быть временно защищены.
Реакция может быть проведена обычными методами, например, в условиях реакции сочетания Хека и Соногашира или Гриньяра. Исходные продукты являются известными или могут быть получены из известных продуктов общепринятыми методами.
Установлено, что соединения формулы I могут применяться в качестве модуляторов mGluR, прежде всего в качестве избирательных антагонистов mGluR5.
Возможность модулирования mGluR может быть продемонстрирована различными путями, в том числе с помощью анализов связывания и функциональных анализов, таких как анализы вторичных мессенджеров, или измерений изменений концентраций внутриклеточного кальция. Например, измерения превращения инозитолфосфата в рекомбинантных линиях клеток, экспрессирующих hmGluR5a, дают значения IС50 для соединений формулы I примерно от 1 нМ до приблизительно 50 нМ.
В частности, соединения формулы I обладают выраженным и избирательно модулирующим, прежде всего антагонистическим, действием в отношении человеческих mGluR, прежде всего mGluR5. Это может быть установлено в опытах in vitro, например, на рекомбинантных человеческих метаботропных глутаматных рецепторах, прежде всего на их подтипах, связанных с PLC, таких как mGluR5, с использованием различных методов, например, путем измерения ингибирования индуцированного агонистом увеличения внутриклеточной концентрации Са2+ согласно методу, описанному у L.P.Daggett и др., Neuropharm. том 34, стр. 871-886 (1995), P.J.Flor и др., J.Neurochem., том 67, стр. 58-63 (1996), или путем определения степени, в которой ингибируется индуцированное агонистом увеличение метаболизма инозитолфосфата, согласно методу, описанному у Knoepfel и др., Eur. J.Pharmacol. том. 288, стр. 389-392 (1994), L.P.Daggett и др., Neuropharm., том 67, стр. 58-63 (1996) и в цитированных там ссылках. Выделение и экспрессия человеческих подтипов mGluR описаны в патенте US 5521297. Значения IС50 соединений имели в отношении ингибирования индуцированного квискуалатом метаболизма инозитолфосфата, измеренные с использованием рекомбинантных клеток, экспрессирующих hmGluR5a, находятся в диапазоне от приблизительно 1 нМ до приблизительно 50 нМ.
Активность антагонистов mGluR5 в качестве аналгетиков по изобретению может быть продемонстрирована в опытах на моделях постоянной воспалительной боли и невропатической боли, проводимых, как описано ниже.
Введение пероральным путем избирательных антагонистов mGluR5, например, описанных выше изменяет в зависимости от дозы механическую гипералгезию при моделировании на крысах воспалительной боли с использованием полного адъюванта Фрейнда (Bartho и др., Naunyn Schmiedebergers Arch. Pharmacol. 342, 666-670, 1970). При использовании этой модели пероральное введение, например, антагониста формулы I приводит к максимальной реверсии на уровне 60-95% воспалительной гипералгезии, при этом значение ED50 находится в диапазоне от 4 до 25 мг/кг. Антигипералгезивное действие продолжается достаточно долго (более 5 ч) и начинается очень быстро.
Эти результаты свидетельствуют о том, что избирательные антагонисты mGluR5 могут применяться для лечения боли.
Введение в подошву определенных антагонистов mGluR5, например указанных выше, вызывает зависящую от дозы реверсию вызванной механическим путем гипералгезии при моделировании на мышах невропатической боли с помощью наложения частичной лигатуры на седалищный нерв (в соответствии с модификациями, описанными у Seltzer и др., Pain 43:205-218, 1990). При использовании этой модели введение в подошву, например, антагонистов формулы I в дозах приблизительно от 1 до приблизительно 100 мг/кг вызывает значительную реверсию вызванной механическим путем гипералгезии.
Эти данные указывают на то, что применение антагонистов для лечения боли не ограничено лечением воспалительной боли.
Таким образом, анестезирующее действие, достигаемое согласно изобретению, может применяться для лечения боли различного происхождения или этиологии, в частности для лечения воспалительной боли и связанной с ней гипералгезии, невропатической боли и связанной с ней гипералгезии, хронической боли, например серьезной хронической боли, послеоперационной боли и боли, связанной с различными состояниями, включая рак, стенокардию, почечные или желчные колики, менструацию, мигрень и подагру.
Воспалительная боль может иметь различное происхождение, включая артрит и ревматоидное заболевание, тендосиновит и васкулит. Невропатическая боль включает тригеминальную невралгию или невралгию при опоясывающем лишае, диабетическую невропатическую боль, каузалгию, боль в нижней части спины и синдромы деафферентации, такие как авульсия плечевого сплетения.
Активность антагонистов mGluR5 по изобретению в отношении состояния страха может быть продемонстрирована на стандартных моделях, таких как тест с использованием каскадного лабиринта на мышах, тест с индуцированной стрессом гипотермией у мышей и тест по оценке социальных исследовательских способностей на крысах, как это описано ниже.
В тесте с использованием каскадного лабиринта, проводимом на мышах линии OF1 [R.G. Lister, Psychopharmacology-Berl. 92, 180-185 (1987)], соединения формулы I, например, при введении в дозах от приблизительно 0,1 до приблизительно 100 мг/кг приводят к увеличению случаев входа в ответвления (открытые плечи) лабиринта и времени, проведенного в открытых плечах каскадного лабиринта. Тест также может быть проведен на самцах мышей линии C57/BL6 согласно методу Razo и др. [Naunyn-Schmiedeberg's Arch. Pharmacol. 337. 675-678, 1988].
В тесте с индуцированной стрессом гипотермией у мышей [В. Oliver и др., Euro. Neuropsychopharmacol. 4, 93-102 (1994)], например, соединения формулы I в дозах от приблизительно 0,1 до приблизительно 100 мг/кг уменьшают индуцированную стрессом гипотермию у мышей линии OF1.
В тесте по оценке социальных исследовательских способностей на крысах, например, соединения формулы I в дозах от приблизительно 0,03 до приблизительно 3 мг/кг приводят к увеличению количества социальных контактов с животными-резидентами. Тест проводят следующим образом.
В тесте используют взрослых самцов крыс линии Sprague Dawley ("резиденты") и молодых самцов листерных крыс линии Hooded ("гости"), "Гостей" содержат парами, а "резидентов" содержат по одиночке в пластиковых клетках (Macrolon, 42×27×15 см) в течение двух недель до начала теста. Все обработки проводят только с использованием крыс-"гостей". Тестируемое соединение вводят перорально (2 мл/кг). В тест включают две дополнительные группы: крысам контрольной группы вводят 0,5%-ный метоцел, а дополнительную группу обрабатывают хлордиазэпоксидом бензодиазепина, который служит в качестве позитивного стандарта. В каждую группу включают по двенадцать крыс. Пары, состоящие из одной крысы-"гостя" и одной крысы-"резидента", причисляют случайным образом к одной из экспериментальных или контрольных групп. В каждой паре только "гостя" обрабатывают пероральным путем перед тем, как его помещают в клетку-дом животного-"резидента". Продолжительность активных типов поведения крысы-"гостя", направленных на сближение (фыркание, исследование аногенитальной области, обнюхивание, ухаживание, лизание, игра) с "резидентом" регистрируют вручную и записывают суммарные данные в течение 5-минутного периода. Все наблюдения проводят во время светлой фазы (с 11 ч утра до 4 ч дня) в клетке-доме "резидента".
В соответствии с вышеизложенным в настоящем изобретении предложены:
а) применение антагониста mGluR5 для лечения боли и состояния страха,
б) применение антагониста mGluR5 для приготовления фармацевтической композиции, предназначенной для лечения боли и состояния страха,
в) фармацевтическая композиция, включающая в качестве действующего вещества антагонист mGluR5, предназначенная для применения при лечения боли и состояния страха,
г) способ лечения боли и состояния страха у пациента, нуждающегося в таком лечении, предусматривающий введение такому пациенту терапевтически эффективного количества антагониста mGluR5.
Для новых применений по изобретению соответствующая доза должна, конечно, варьироваться в зависимости, например, от используемого соединения, хозяина, пути введения и природы и серьезности состояния, подлежащего лечению. Однако в целом установлено, что удовлетворительные результаты на животных могут быть получены при использовании суточной дозы от приблизительно 0,1 до приблизительно 100 мг/кг веса тела. Для крупных млекопитающих, например людей, показанная суточная доза находится в диапазоне от приблизительно 5 до приблизительно 1000 мг соединения при использовании согласно изобретению, вводимая обычным путем, например в виде разделенных доз до пяти раз в день.
Антагонист mGluR5 можно вводить перорально, например, в форме таблеток или капсул, или парентерально, например, с помощью внутривенной, внутрибрюшинной, внутримышечной, подкожной, интраназальной или внутрикожной инъекции, а также путем трансдермального введения (например вместе с жирорастворимым носителем в форме кожного пластыря, помещаемого на кожу), или путем введения в желудочно-кишечный тракт (например, с помощью капсулы или таблетки). Предпочтительные терапевтические композиции для инокулята и доза варьируются в зависимости от клинических показаний. Инокулят обычно приготавливают из высушенного препарата антагониста mGluR5 (например, из лиофилизированного порошка) путем суспендирования препарата в физиологически приемлемом разбавителе, таком как вода, физиологический раствор или забуференный фосфатом физиологический раствор.
Фармацевтические композиции, содержащие в качестве действующего вещества антагонист mGluR5, вводят индивидуально или в сочетании с фармацевтически приемлемыми носителями в виде однократной дозы или в виде нескольких доз. Пригодные фармацевтические носители включают инертные твердые разбавители или наполнители, стерильные водные растворы и различные нетоксичные органические растворители. Фармацевтические композиции, полученные путем объединения антагониста mGluR5 с фармацевтически приемлемым носителем, затем могут быть легко введены в различных дозируемых формах, таких как таблетки, лепешки, сиропы, инъецируемые растворы и т.п. Такие фармацевтические носители могут при необходимости содержать дополнительные ингредиенты, такие как корригенты, связующие вещества, эксципиенты и т.п. Так, для целей перорального введения применяют таблетки, содержащие различные эксципиенты, такие как цитрат натрия, карбонат кальция и фосфат кальция, наряду с различными разрыхлителями, такими как крахмал, и предпочтительно картофельный крахмал или крахмал из тапиоки, альгиновая кислота и определенные комплексы силикатов, наряду со связующими агентами, такими как поливинилпирролидон, сахароза, желатин и камедь акации. Для целей таблетирования часто дополнительно применяют замасливатели, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции этого типа также могут применяться в качестве наполнителей желатиновых капсул с солевым и твердым наполнителем. Предпочтительные вещества для этой цели включают лактозу или молочный сахар и полиэтиленгликоли с высокой молекулярной массой. Если для перорального введения требуются водные суспензии эликсиров, то действующее вещество антагонист mGluR5 объединяют с различными подслащивающими веществами или корригентами, красителями и при необходимости с эмульгаторами или суспендирующими агентами, а также с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин и их комбинации. Для парентерального введения применяют растворы антагониста mGluR5 в кунжутном или арахисовом масле или в водном полипропиленгликоле, а также в стерильных водных физиологических растворах, содержащих соответствующие водорастворимые фармацевтически приемлемые соли металлов. Такой водный раствор должен быть при необходимости соответствующим образом забуферен, и жидкий разбавитель сначала делают изотоничным с помощью достаточного количества физиологического раствора или глюкозы. Такие специальные водные растворы особенно пригодны для внутривенной, внутримышечной, подкожной и внутрибрюшинной инъекции. Все используемые стерильные водные среды легко могут быть получены стандартными методами, хорошо известными специалистам в данной области. Кроме того, можно вводить указанные выше соединения местно (например, через введенный катетер), используя соответствующий раствор, пригодный для данной цели.
Следующие варианты осуществления изобретения представляют собой виды продукта, содержащие вкладыш для упаковки с инструкциями по терапевтическому применению, упаковочный материал и фармацевтические композиции, включающие препаративную форму одного или нескольких антагонистов mGluR5. Инструкции по применению в целом должны содержать особенности применения антагониста mGluR5 для облегчения одного или нескольких симптомов дисфункций, сопровождающихся, в частости, болью и/или состоянием страха. Определенный вид продукта также обычно содержит этикетку, на которой приводятся данные о соединении или композиции и особенностях ее применения для облегчения одного или нескольких симптомов, связанных с указанной дисфункцией.
Способ лечения боли и состояния страха по изобретению в контексте настоящего описания означает способ введения в организм пациента, нуждающегося в этом, фармацевтической композиции, содержащей антагонист mGluR5, с целью лечения или предупреждения одного или нескольких симптомов дисфункций, сопровождающихся, в частости, болью и/или состоянием страха. Указанный способ предусматривает введение композиции в организм пациента I) до того, как дисфункция диагностирована, например, согласно профилактическим протоколам введения с целью предупреждения развития дисфункции, а также II) после того, как дисфункция диагностирована, например, согласно терапевтическим протоколам.
Согласно указанному способу лечения боли и состояния страха антагонист mGluR5 вводят в состав любой медицинской формы или композиции. Его применяют в качестве единственного агента лекарственной терапии или в сочетании с другими медицинскими препаратами. Поскольку фармакокинетика и фармакодинамика антагониста mGluR5 варьируется для различных пациентов, то наиболее предпочтительный метод достижения терапевтически эффективной концентрации в ткани заключается в постепенном увеличении дозы и наблюдении за клиническими воздействиями. Начальная доза для такого режима терапии с возрастающими дозами зависит от пути введения.
Помимо обнаружения выраженной активности избирательных антагонистов mGluR5 в отношении лечения боли неожиданно было установлено, что действия антагонистов mGluR в отношении гипералгезии в основном опосредуются mGluR, экпрессируемыми в периферической нервной системе (периферическими mGluR), прежде всего mGluR5. Этот результат является полностью неожиданным, принимая во внимание известные данные, основанные на следующих исследованиях.
Электрофизиологические исследования mGluR продемонстрировали, что их активация оказывает сильное влияние на синаптическую модуляцию в центральной нервной системе (ЦНС). Результаты фармакологических и физиологических исследований рефлекса спинного мозга позволили предположить, что mGluR могут как ослаблять, так и усиливать моторную реакцию спинного мозга (см. Boxall и др., Neuroscience, 82:591-602, 1998). Внутриклеточные исследования выявили, что активация mGluR также оказывает непосредственное воздействие на свойства мембран интернейронов с широким моторным спектром и нейронов переднего рога спинного мозга (Morisset и Nagy, J.Neurophysiol. 76:2794-2798, 1996; Liu и King, Br. J.Pharmacol. 116, стр. 105, 1995).
Молекулярные биологические исследования подтвердили наличие экспрессии РНК mGluR в ЦНС млекопитающих. Также были описаны протеины рецепторов подтипов mGluR1-5, mGluR7 и mGluR8 в головном мозге млекопитающих. Эти подтипы рецепторов, по-видимому, локализованы на нейронах как пре-, так и постсинаптически, а также присутствуют в глиальных клетках. С помощью методов гибридизации in situ было продемонстрировано наличие мРНК mGluR в спинном мозге взрослых крыс (Boxall и др., 1998, см. выше). В спинном мозге крыс экпрессируются мРНК подтипов 1, 3-5 и 7 mGluR. Кроме того, иммуногистохимическими методами была выявлена экспрессия протеина mGluR5 в спинном мозге человека и крысы и в клетках спинно-мозговых узлов крысы (Valerio и др., Neuroscience Research. 28:49-57, 1997).
Электрофизиологическими исследованиями in vivo было выявлено, что активация mGluR спинного мозга оказывает влияние на развитие гипервозбудимости спинного мозга (обзор см. у Boxall и др., 1998).
Поведенческие фармакологические исследования на крысах показали, что вводимый внутриоболочечно агонист 3,5-дигидроксифенилглицин (ДГФГ) группы I mGluR индуцирует увеличение спонтанных ноцицептивных реакций у крыс (Fisher и Coderre, Neuroreport. 9:1169-1172, 1998). Другим доказательством роли рецепторов группы I mGluR в процессинге болезненных реакций спинного мозга являются данные о антиноцицептивном воздействии вводимых внутриоболочечно антител к крысиным mGluR1 и mGluR5. Оба эти антитела приводят к реверсии спонтанных ноцицептивных реакций, вызываемых внутриоболочечным введением ДГФГ (Fundytus и др., Neuroreport. 9:731-735, 1998). Кроме того, они оба приводят к реверсии холодной аллодинии, развивающейся после провреждения седалищного нерва у крысы, свидетельствуя о том, что рецепторы mGluR1 и mGluR5 спинного мозга могут играть роль в опосредовании невропатической боли.
Все известные данные, основанные на вышеуказанных исследованиях, свидетельствуют о том, что участие mGluR в процессинге ноцицептивной реакции ограничено ЦНС. Поэтому следует ожидать, что терапия с использованием антагонистов mGluR для того, чтобы она обладала аналгезирующей активностью, должна требовать доступа к ЦНС, например, введения в ЦНС, или способности антагониста проникать через гематоэнцефалический барьер.
Результат, полученный при создании изобретения, заключающийся в том, что гипералгезивные действия антагонистов mGluR в основном медиируются mGluR, экспрессируемыми в периферической нервной системе, прежде всего mGluR5, может быть продемонстрирован на стандартных моделях, описанных ниже.
После интрацеребровентрикулярного или внутриоболочечного введения антагонистов mGluR, способных проникать через гематоэнцефалический барьер, например, определенных антагонистов mGluR5, при моделировании на крысах с использованием полного адъюванта Фрейнда (Bartho и др., 1990, выше) антагонисты mGluR вызывают лишь слабые антигипералгезивные действия.
Поэтому маловероятно, что сайты головного мозга и спинного мозга являются основными сайтами действия при оральном введении.
В тесте с механической гипералгезией задней лапы необработанных крыс (Randall и Selitto, Arch. Int. Pharmacodyn. Ther. 111:409-419, 1957) агонисты глутаматных рецепторов по их эффективности могут быть расположены в следующум порядке: глутамат ~ 2-хлор-3-гидроксифенилглицин (ХГФГ) > ДГФГ > NMDA > АМРА > (±)-2-аминобицикло [3.1.0] гексан-2,6-дикарбоновая кислота (LY 314582) > L-4-фосфоно-2-аминомасляная кислота (L-AP-4). Среди тестированных обладающих избирательным действием в отношении рецептра соединений наиболее сильными гипералгезивными агентами являются те, которые действуют на mGluR группы I.
Эти результаты свидетельствуют о том, что рецепторы mGluR группы I, в частности, участвуют в передаче ноницептивного импульса и что они экспрессируются в периферической нервной системе.
При совместном введении агониста mGluR группы I ДГФГ в заднюю лапу крысы при использовании той же модели, которая описана у Randall и Selitto, антагонисты mGluR5 ингибируют в зависимости от дозы индуцируемую ДГФГ гипералгезию, в то время как антагонист mGluR группы I (S)-4-карбоксифенил-глицин [(S)-4-C-PG], обладающий избирательным действием в отношении mGluRl по сравнению с рецепторами mGluR5, оказывает ограниченное действие.
Эти результаты свидетельствуют о том, что рецептор mGluR5, в частности, участвует в ноницептивной передаче и подтверждают, что он экспрессируется в периферической нервной системе.
Приведенные выше данные свидетельствуют о том, что гипералгезию, связанную с воспалительной болью, можно лечить с помощью антагонистов mGluR, например, антагонистов mGluR, обладающих, в частности, антагонистическим действием в отношении mGluR5. Кроме того, они свидетельствуют о том, что антагонист mGluR, который практически не действует (или вводится таким образом, что он практически не действует) на рецепторы mGluR центральной нервной системы, практически не действуя на центральную нервную систему, не является менее активным в отношении его антигипералгезивной активности, чем антагонист mGluR, проникающий в ЦНС.
В соответствии с вышеизложенным в настоящем изобретении также предложены:
а) Применение антагониста mGluR для лечения боли, где аналгезирующее действие достигается в результате взаимодействия указанного антагониста прежде всего или в основном с периферическими рецепторами mGluR.
б) Применение антагониста mGluR для приготовления фармацевтической композиции, предназначенной для лечения боли путем взаимодействия указанного антагониста прежде всего или в основном с периферическими рецепторами mGluR.
в) Фармацевтическая композиция, содержащая в качестве действующего вещества антагонист mGluR, предназначенная для лечения боли, где аналгезирующее действие достигается в результате взаимодействия указанного антагониста прежде всего или в основном с периферическими рецепторами mGluR.
г) Способ лечения боли у пациента, нуждающегося в таком лечении, предусматривающий введение антагониста mGluR, где аналгезирующее действие достигается в результате взаимодействия указанного антагониста прежде всего или в основном с периферическими рецепторами mGluR.
Предпочтительно указанное аналгезирующее действие достигается исключительно или практически исключительно в периферических рецепторах mGluR.
Основное взаимодействие с периферическими рецепторами mGluR предпочтительно достигается за счет выбора действующего вещества, которое практически не проникает в ЦНС или вводится таким образом, что оно практически не проникает в ЦНС.
Пути введения, при которых вводимый антагонист mGluR практически не проникает в ЦНС, включают местное, прежде всего трансдермальное введение.
Для осуществления трансдермального введения антагонист mGluR может вводиться в составе любой общепринятой жидкой или твердой трансдермальной фармацевтической композиции, например, из числа таких, которые описаны в Remington's Pharmaceutical Sciences, 16-е изд., изд-во Mack; Sucker, Fuchs и Spieser, Pharmaceutische Technologie, 1-е изд., изд-во Springer и в GB 2098865 A или DOS 3212053, содержание которых включено в настоящее описание в качестве ссылки.
Дозы антагониста mGluR, требующиеся для оказания указанного аналгезирующего действия, соответствуют таковым, описанным выше для применения антагонистов mGluR5 для лечения боли и состояния страха.
Предпочтительные антагонисты mGluR5 для применения согласно изобретению включают указанные выше соединения формулы I в свободной форме или в форме фармацевтически приемлемых солей. Репрезентативные соединения формулы I включают 2-метил-6-(фенилэтинил) пиридин, 2-[(пиридин-3-ил)этинил]-6-метилпиридин, 2-(3-фторфенилэтинил)-6-метилпиридин и (3-{2-[2-транс-(3,5-дихлорфенил)винил]-6-метилпиридин-3-илокси} пропил) диметиламин. Эти и другие соединения и группы соединений формулы I, пригодные для применения согласно изобретению, а также их получение описаны, например, в WO 99/02497, включенном в настоящее описание в качестве ссылки.
Переносимость антагонистов mGluR5 формулы I может быть определена общепринятым методом. При введении в дозах, указанных в описанных выше тестах, не обнаружено никакого существенного токсикологического действия.
Также и в стандартных анализах мутагенности, например в скрининге Эймса, у соединений не обнаружено наличия потенциальнй мутагенной активности.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТАГОНИСТИЧЕСКИМ ДЕЙСТВИЕМ НА MGLU R ЧЕЛОВЕКА | 1998 |
|
RU2203889C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ВАНИЛОИДНЫХ АНТАГОНИСТОВ | 2005 |
|
RU2449995C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ВАНИЛОИДНЫХ АНТАГОНИСТОВ | 2005 |
|
RU2396261C2 |
| 7-ХЛОР-4-ГИДРОКСИ-2-(2-ХЛОР-4-МЕТИЛФЕНИЛ)-1,2,5,10-ТЕТРАГИДРОПИРИДАЗИНО[4, 5В]ХИНОЛИН-1,10-ДИОН, СПОСОБЫ ЛЕЧЕНИЯ БОЛИ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2000 |
|
RU2234507C2 |
| ЛЕЧЕНИЕ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2006 |
|
RU2424795C2 |
| ПРИМЕНЕНИЕ ЗОЛЕДРОНОВОЙ КИСЛОТЫ, ЕЕ СОЛЕЙ, ГИДРАТОВ И СПОСОБ АНТИНОЦИЦЕПТИВНОГО ИЛИ АНТИАЛЛОДИНИЧЕСКОГО ЛЕЧЕНИЯ БОЛИ, СПОСОБ ЛЕЧЕНИЯ НЕВРОПАТИЧЕСКОЙ БОЛИ | 2001 |
|
RU2325913C2 |
| ЭТИНИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2712633C1 |
| ИНГИБИТОРЫ FAAH ПЕРИФЕРИЧЕСКИ-ОГРАНИЧЕННОГО ДЕЙСТВИЯ | 2011 |
|
RU2583435C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ NEP-ИНГИБИТОРЫ, ИНГИБИТОРЫ ЭНДОГЕННОЙ ЭНДОТЕЛИНПРОДУЦИРУЮЩЕЙ СИСТЕМЫ И АНТАГОНИСТЫ AT-РЕЦЕПТОРА | 2005 |
|
RU2384346C2 |
| ПРОИЗВОДНЫЕ 1-АЗАБИЦИКЛО[3.3.1]НОНАНОВ | 2005 |
|
RU2445313C2 |
Изобретение относится к фармакологии и может быть использовано в неврологии и психиатрии. Используют антагонисты рецепторов mGluR5 2-арилалкенил-2-гетероарилалкенил-, 2 арилалкинил-, 2-гетероарилалкинил-, 2-арилазо- и - 2-гетероарилазо, более конкретно 6-метил-2-(фенилазо)-3-пиридинол, (Е)-2-метил-6-стирилпиридин для лечения боли и состояния страха путем введения указанных средств в свободной форме, в форме фармацевтически приемлемых солей или в составе фармацевтической композиции. Изобретение расширяет арсенал лекарственных средств. 6 с. и 3 з.п. ф-лы.
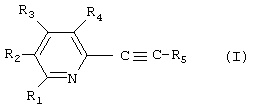
где R1 обозначает водород, С1-С4алкил, С1-С4алкокси, циано, этинил или диС1-С4алкиламино;
R2 обозначает водород, гидрокси, карбокси, С1-С4алкоксикарбонил, диС1-С4алкиламинометил, 4-(4-фторбензоил)пиперидин-1-илкарбокси, 4-трет-бутилоксикарбонилпиперазин-1-илкарбокси, 4-(4-азидо-2-гидроксибензоил)пиперазин-1-илкарбокси или 4-(4-азидо-2-гидрокси-3-йодбензоил)пиперазин-1-илкарбокси;
R3 обозначает водород, С1-С4алкил, карбокси, С1-С4алкоксикарбонил, С1-С4алкилкарбамоил, гидроксиС1-С4алкил, диС1-С4алкиламинометил, морфолинокарбонил или 4-(4-фторбензоил)пиперидин-1-илкарбокси;
R4 обозначает водород, гидрокси, карбокси, С2-С5алканоилокси, С1-С4алкоксикарбонил, аминоС1-С4алкокси, диС1-С4алкиламиноС1-С4алкокси, диС1-С4алкиламиноС1-С4алкил или гидроксиС1-С4алкил;
R5 обозначает группу формулы
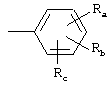
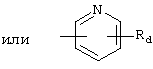
где Ra и Rb независимо друг от друга обозначают водород. галоген, нитро, циано, С1-С4алкил, С1-С4алкокси, трифторметил, трифторметокси или С2-С5алкинил;
Rc обозначает водород, фтор, хлор, бром, гидрокси, С1-С4алкил, С2-С5алканоилокси, С1-С4алкокси или циано;
Rd обозначает водород, галоген или С1-С4алкил,
в свободной форме или в форме фармацевтически приемлемых солей вместе с фармацевтически приемлемыми носителями или разбавителями, предназначенная для лечения состояния страха.
Приоритет по пунктам:
| WO 97051109 А, 13.02.1997 | |||
| Способ получения производных пиридина | 1985 |
|
SU1424731A3 |
| ПРИСПОСОБЛЕНИЕ ДЛЯ ЗАГЛУБЛЕНИЯ ЖЕЛЕЗОБЕТОННОГО ЯКОРЯ | 0 |
|
SU334119A1 |

