Настоящее изобретение относится к лечению или предупреждению боли или ноцицепции.
Уровень техники
Боль представляет собой сенсорное ощущение, отличное от восприятий прикосновения, давления, тепла и холода. Она часто описывается пострадавшими такими терминами как яркая, тупая, ноющая, колющая, режущая или обжигающая и, как обычно считают, включает в себя как первоначальное восприятие, так и реакцию на это восприятие. Этот диапазон восприятий, а также изменчивость в восприятии боли различными индивидуумами затрудняет точное определение боли, однако многие индивидуумы страдают от сильной и постоянной боли.
Боль, которая вызвана повреждением нервных структур, часто проявляется как нервная сверхчувствительность или гипералгезия, и ее называют “невропатическая” боль. Боль также может быть “вызвана” стимуляцией ноцицептивных рецепторов и может передаваться через интактные нервные пути, причем такую боль называют “ноцицептивной” болью.
Уровень стимуляции, при котором боль становится заметна, называется “порог болевой чувствительности”. Аналгетики представляют собой фармацевтические агенты, которые облегчают боль путем повышения порога болевой чувствительности без потери сознания. После введения аналгезирующего лекарства для ощущения боли требуется стимул большей интенсивности или большей продолжительности. У индивидуума, страдающего от гипералгезии, аналгезирующее лекарство может оказывать анти-гипералгезирующий эффект. В отличие от аналгетиков, агенты, такие как местные анестетики, блокируют передачу в периферических нервных волокнах, таким образом блокируя осознание боли. Анестетики общего действия, с другой стороны, уменьшают осознание боли путем потери сознания.
Сообщалось, что антонтагонисты тахикинина индуцируют антиноцицепцию у животных, которая, как считается, является аналогичной аналгезии у человека (Maggi et al, J. Auton. Pharmacol. (1993) 13, 23-93). В частности, было показано, что непептидные антагонисты NK-1 рецептора обеспечивают такую аналгезию. Например, антагонист NK-1 рецептора RP 67,580 обеспечивает аналгезию, эффективность которой сравнима с эффективностью морфина (Garret et al, Proc. Natl. Acad. Sci. USA (1993) 88, 10208-10212).
Опиоидные аналгетики представляют собой прочно установившийся класс аналгезирующих агентов с морфиноподобным действием. Синтетические и полусинтетические опиоидные аналгетики представляют собой производные пяти химических классов соединений: фенантренов, фенилгептиламинов, фенилпиперидинов, морфинанов и бензоморфанов. Фармакологически эти соединения имеют различные активности, таким образом, некоторые являются сильными агонистами опиоидных рецепторов (например морфин), другие представляют собой от умеренных до мягких агонистов (например кодеин), еще одни проявляют смешанную активность агониста-антагониста (например налбуфин), и еще одни являются частичными агонистами (например налорфин). Несмотря на то, что опиоидный частичный агонист, такой как налорфин (N-алкильный аналог морфина), будет антагонизировать аналгезирующие эффекты морфина, при приеме отдельно он может быть сильнодействующим аналгетиком сам по себе.
Из всех опиоидных аналгетиков морфин остается наиболее широко используемым, но, в дополнение к его терапевтическим свойствам, он имеет и ряд недостатков, включая угнетение дыхания, пониженную моторику желудочно-кишечного тракта (приводящую к запору), тошноту и рвоту. Переносимость и физическая зависимость также ограничивают клинические применения опиоидных соединений.
Аспирин и другие салицилатные соединения часто используют в лечении для прерывания усиления воспалительного процесса при ревматоидных заболеваниях и артрите и временного облегчения боли. Другие лекарственные соединения, используемые для этих целей, включают в себя производные фенилпропионовой кислоты, такие как Ибупрофен и Напроксен, Сулиндак, фенилбутазон, кортикостероиды, противомалярийные средства, такие как хлорохин и гидроксихлорохина сульфат, и фенематы (J. Hosp. Pharm., 36:622 (May 1979)). Однако эти соединения неэффективны при невропатической боли.
Доступные терапии боли также имеют недостатки. Некоторые терапевтические агенты требуют пролонгированного применения перед тем как пациент ощутит эффект. Другие существующие лекарства вызывают серьезные побочные эффекты у некоторых пациентов, и за субъектами нужно внимательно следить, чтобы гарантировать, что какие-либо побочные эффекты не являются чрезмерно угрожающими. Большинство существующих лекарств дают только временное облегчение боли и должны приниматься последовательно на суточной или недельной основе. С развитием заболевания количество лекарственного средства, необходимого для облегчения боли, часто увеличивается, таким образом увеличивая возможность вредных побочных эффектов.
Рецепторы NMDA определяют путем связывания N-метил-D-аспартат (NMDA) содержащего комплекса рецептора/ионного канала с несколькими различными идентифицированными доменами связывания. NMDA сам по себе представляет собой молекулу, структурно сходную с глутаматом (Glu), которая связывается на глутаматсвязывающем сайте и является высокоселективной и сильнодействующей в активации рецептора NMDA (Watkins (1987); OIney (1989)).
Известны многие соединения, которые связываются на NMDA/Glu связывающем сайте (например СРР, DCPP-ene, CGP 40116, CGP 37849, CGS 19755, NPC 12626, NPC 17742, D-AP5, D-AP7, CGP 39551, CGP-43487, MDL-100,452, LY-274614, LY-233536 и LY-233053). Другие соединения, которые называют неконкурентными антагонистами NMDA, связываются на других сайтах в NMDA рецепторном комплексе (примерами являются фенциклидин, дизоцилпин, кетамин, тилетамин, CNS 1102, декстрометорфан, мемантин, кинуреновая кислота, CNQX, DNQX, 6,7-DCQX, 6,7-DCHQC, R(+)-HA-966, 7-хлор-кинуреновая кислота, 5,7-DCKA, 5-иод-7-хлор-кинуреновая кислота, MDL-28,469, MDL-100,748, MDL-29,951, L-689,560, L-687,414, ACPC, ACPCM, ACPCE, аркаин, диэтилентриамин, 1,10-диаминодекан, 1,12-диаминододекан, ифенпродил и SL-82.0715). Эти соединения подробно рассмотрены Rogawski (1992) и Massieu et al., (1993) и в статьях, перечисленных там.
В дополнение к его физиологической функции, глутамат (Glu) может быть нейротоксичным. Нейротоксичность Glu называют “эксайтотоксичностью”, поскольку нейротоксическое действие Glu, подобно его полезным свойствам, опосредовано процессом возбуждения (OIney (1990); Choi (1992)). В норме, когда Glu высвобождается у синаптического рецептора, он связывается только временно, а затем быстро удаляется от рецептора с помощью процесса, который транспортирует его обратно в клетку. В некоторых аномальных условиях, включая удар, эпилепсию и травму ЦНС, захвата Glu не происходит, и Glu накапливается около рецептора, приводя к устойчивому возбуждению электрохимической активности, что приводит к гибели нейронов, которые имеют рецепторы Glu. Многие нейроны в ЦНС имеют рецепторы Glu, таким образом, эксайтотоксичность может вызывать огромное количество повреждений ЦНС.
Острое эксайтотоксическое повреждение может происходить как результат ишемических событий, гипоксических событий, травмы головного или спинного мозга, некоторых типов пищевого отравления, в которое вовлечен эксайтотоксический яд, такой как домоевая кислота, и опосредованной припадком нейрональной дегенерации, которая может являться результатом устойчивой эпилептической активности (эпилептический статус). Большое количество доказательств указывает на рецептор NMDA как на один из подтипов рецепторов, через которые Glu опосредует существенное количество повреждений ЦНС, и хорошо установлено, что антагонисты NMDA эффективны в защите нейронов ЦНС от эксайтотоксической дегенерации при этих синдромах острого повреждения ЦНС (Choi (1988); OIney (1990)).
В дополнение к нейрональному повреждению, вызванному острыми инсультами, избыточная активация рецепторов Glu может также вносить вклад в постепенно развивающиеся нейродегенеративные процессы, приводящие к гибели клеток при различных хронических нейродегенеративных заболеваниях, включая болезнь Альцгеймера, боковой амиотрофический склероз, деменцию, связанную со СПИДом, болезнь Паркинсона и болезнь Хантигтона (OIney (1990)). Обычно считают, что антагонисты NMDA могут быть полезны при терапевтическом лечении таких хронических заболеваний.
В 1980-е гг. обнаружили, что РСР (фенциклидин) (также известный как "ангельская пыль") действует как "сайт распознавания РСР" в ионном канале NMDA Glu рецептора. РСР действует как неконкурентный антагонист, который блокирует поток ионов через ионный канал NMDA. Совсем недавно стало очевидно, что лекарства, которые действуют на РСР сайт в качестве неконкуретных антагонистов NMDA, вероятно, обладают психотомиметическими побочными эффектами. Более того, в настоящее время известно, что некоторые конкурентные и неконкуретные антагонисты NMDA могут вызывать подобные патоморфологические эффекты в головном мозге крыс (OIney et at., (1991); Hargreaves et ai., (1993)). Такие соединения также обладают психотомиметическими эффектами у людей (Kristensen et al., (1992); Herrling (1994); Grotta (1994)).
Сайт связывания глицина NMDA рецепторного комплекса отличается от сайтов связывания Glu и РСР. Также недавно было обнаружено, что NMDA рецепторы существуют в качестве нескольких подтипов, которые характеризуются различными свойствами глицинсвязывающего сайта рецептора. Многие соединения, которые связываются на сайте глицина NMDA рецептора, полезные для лечения удара и нейродегенеративных состояний, описаны в патентах США 5604227, 5733910, 5599814, 5593133, 5744471, 5837705 и 6103721.
Сущность изобретения
В настоящее время обнаружено, что некоторые соединения, которые проявляют свойство связывания с сайтом глицина NMDA рецептора, полезны для уменьшения боли и особенно для уменьшения невропатической боли.
Поэтому, в одном аспекте, в данном изобретении предложено соединение, 7-хлор-4-гидрокси-2-(2-хлор-4-метилфенил)-1,2,5,10-тетрагидропиридазино[4,5-b]хинолин-1,10-дион, в соответствии со структурной формулой I
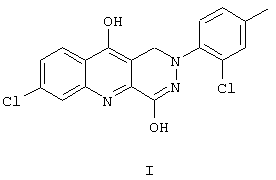
В другом аспекте в изобретении предложен способ лечения боли с использованием соединения в соответствии со структурной формулой I, при котором вводят уменьшающее боль эффективное количество данного соединения.
В еще одном воплощении данный способ включает в себя введение уменьшающего боль эффективного количества соединения в соответствии со структурной формулой I в форме фармацевтической композиции, содержащей соединение в соответствии со структурной формулой 1 в качестве активного ингредиента вместе с одной или более чем одной фармацевтически приемлемой добавкой.
В дополнительном воплощении данный способ включает в себя связывание соединения по изобретению с сайтом глицина NMDA рецептора теплокровного животного, такого как человек, так, чтобы полезным образом ингибировать активность NMDA рецептора.
Другой аспект изобретения представляет собой способ приготовления соединения в соответствии со структурной формулой I.
Еще одни аспекты изобретения представляют собой фармацевтические композиции, которые содержат соединение в соответствии со структурной формулой I, и применение соединения в соответствии со структурной формулой I для получения лекарственных средств и фармацевтических композиций.
Подробное описание изобретения
В данном изобретении предложено соединение, 7-хлор-4-гидрокси-2-(2-хлор-4-метилфенил)-1,2,5,10-тетрагидропиридазино[4,5-b]хинолин-1,10-дион, его фармацевтически приемлемые соли, способы приготовления соединения и его солей, фармацевтические композиции, содержащие соединение или его соли, и способы применения соединения, солей и фармацевтических композиций.
Подходящие фармацевтически приемлемые соли соединений по изобретению включают в себя соли присоединения кислоты, такие как метансульфонат, фумарат, гидрохлорид, гидробромид, цитрат, трис(гидроксиметил)аминометан, малеат, и соли, образованные с фосфорной и серной кислотой. В других воплощениях подходящие соли представляют собой основные соли, такие как соли щелочных металлов, например натрия, соли щелочноземельных металлов, например кальция или магния, соли органических аминов, например триэтиламина, морфолина, N-метилпиперидина, N-этилпиперидина, прокаина, дибензиламина, холина, N,N-дибензилэтиламина, или аминокислот, таких как лизин.
Для применения соединения по изобретению или его фармацевтически приемлемой соли для терапевтического лечения, которое может включать в себя профилактическое лечение, боли у млекопитающих, которые могут являться людьми, соединение может быть приготовлено в соответствии со стандартной фармацевтической практикой в виде фармацевтической композиции.
Подходящие фармацевтические композиции, которые содержат соединение по изобретению, можно вводить обычными путями, например путем перорального, местного, парентерального, трансбуккального, назального, вагинального или ректального введения или путем ингаляции. Для этих целей соединение по изобретению может быть изготовлено в форме, например, таблеток, капсул, водных или масляных растворов, суспензий, эмульсий, кремов, мазей, гелей, назальных спреев, суппозиториев, тонкоизмельченных порошков или аэрозолей для ингаляции и стерильных водных или масляных растворов или суспензий или стерильных эмульсий для парентерального применения (включая внутривенное, внутримышечное или инфузию) способами, известными в данной области техники. Предпочтительным путем введения является пероральный путь с помощью таблетки или капсулы.
Кроме соединения по настоящему изобретению, фармацевтическая композиция по настоящему изобретению может также содержать один или более чем один другой фармакологически активный агент, либо такая фармацевтическая композиция может быть одновременно или последовательно введена совместно с одним или более чем одним другим фармакологически активным агентом.
Фармацевтические композиции по настоящему изобретению должны быть в норме введены так, чтобы субъект получал уменьшающую боль эффективную суточную дозу. Суточная доза может быть дана в разделенных дозах, как необходимо, при этом точное количество получаемого соединения и путь введения зависят от массы, возраста и пола пациента, которого лечат, и от конкретного заболевания, которое лечат, в соответствии с правилами, известными в данной области техники. Предпочтительный режим дозирования составляет один раз в сутки.
В дополнительном воплощении изобретения предложена фармацевтическая композиция, которая содержит соединение по изобретению, как оно определено здесь, или его фармацевтически приемлемую соль вместе с фармацевтически приемлемой добавкой, такой как эксципиент или носитель.
В еще одном воплощении изобретения предложено применение соединения по изобретению или его фармацевтически приемлемой соли, в производстве лекарственного средства, полезного для связывания с сайтом глицина NMDA рецептора у теплокровного животного, такого как человек.
В еще одном воплощении изобретения предложен способ связывания соединения по изобретению с сайтом глицина NMDA рецептора теплокровного животного, такого как человек, нуждающегося в лечении боли, при котором указанному животному вводят эффективное количество соединения структурной формулы 1 или его фармацевтически приемлемой соли.
Определения:
Обычно в способах, процессах и примерах, описанных здесь:
концентрирования осуществляли путем выпаривания с помощью роторного испарителя in vacuo;
операции осуществляли при температуре окружающей среды, то есть в диапазоне 18-26°С, и в атмосфере азота;
колоночную хроматографию (с помощью флэш-процедуры) осуществляли на кремнеземе Merck Kieselgel (Art. 9385), если не указано иначе;
выходы приведены только для иллюстрации и не обязательно являются максимально достижимыми;
структуру конечных продуктов формулы I обычно подтверждали с помощью ЯМР и масс-спектральных методик, спектры протонного магнитного резонанса определяли в DMSO-d6, если не указано иначе, с использованием спектрометра Varian Gemini 2000, работающего при напряженности поля 300 МГц; химические сдвиги приведены в частях на миллион по отношению к тетраметилсилану в качестве внутреннего стандарта (8 шкала), и пиковые мультиплетности показаны таким образом: s, синглет; bs, широкий синглет; d, дублет; АВ или dd, дублет дублетов; t, триплет, dt, дублет триплетов; m, мультиплет; bm, широкий мультиплет; масс-спектральные данные бомбардировки быстрыми атомами (FAB) получали, используя спектрометр Platform (снабженный Micromass), работающий в электроспрее, и, как подходит, собирали данные либо положительных ионов, либо отрицательных ионов, в данной заявке приведены (М+Н)+.
промежуточные соединения обычно полностью не охарактеризовывали, и чистоту обычно оценивали масс-спектральным (МС) или ЯМР анализом.
Использовали следующие обозначения и определения, имеющие следующие значения:
CDCl3 обозначает дейтерированный хлороформ;
CMC обозначает 1-циклогексил-3-(2-морфолиноэтил)карбодиимида мето-р-толуолсульфонат;
DCM обозначает дихлорметан;
DCU обозначает дициклогексилмочевину;
DHC обозначает 1,3-дициклогексилкарбодиимид;
DMAP обозначает 4-(диметиламино)пиридин;
DMF обозначает N,N-диметилформамид;
DMSO обозначает диметилсульфоксид;
м/с обозначает масс-спектроскопию;
NMP обозначает N-метилпирролидинон;
ЯМР обозначает ядерный магнитный резонанс;
р.о. обозначает per os (перорально);
THF обозначает тетрагидрофуран;
t.i.d. обозначает три раза в сутки.
Примеры и тесты, описанные здесь, предназначены для иллюстрации, но не для ограничения изобретения.
Примеры
Пример 1
Соединение по изобретению 7-хлор-4-гидрокси-2-(2-хлор-4-метилфенил)-1,2,5,10-тетрагидропиридазино[4,5-b]хинолин-1,10-дион может быть получено с помощью следующей процедуры:
2-Хлор-4-метил-сренилгидразина гидрохлорид
Суспензию 2-хлор-4-метил-анилина (10,1 мл, 11,63 г, 82,1 ммоль) в 64 мл воды и 60 мл 12 н HCI охлаждали до -5°С (внутренняя температура) и перемешивали с помощью механической мешалки. Раствор нитрита натрия (8,26 г, 119,7 ммоль) в 56 мл воды добавляли в течение 30 минут. Раствор становился более прозрачным, однако оставалось некоторое количество твердого вещества. Смесь перемешивали при -5°С в течение 20 минут, а затем охлаждали до -10°С. Раствор дигидрата хлорида олова (II) (53,60 г, 237,6 ммоль) в 36 мл 12 н HCI добавляли по каплям в течение 30 минут, поддерживая внутреннюю температуру от -5 до -10°С. Полученную розовато-коричневую смесь перемешивали при температуре от -5 до -10°С в течение 2 часов, а затем фильтровали холодной через предварительно охлажденную фриттованную стеклянную воронку. Собранные твердые вещества промывали холодным 1% этанолом в эфире (100 мл), затем холодным эфиром (500 мл) и сушили на воздухе в течение 30 минут. После сушки в вакууме желаемый продукт получали в виде бледно-желтого кристаллического твердого вещества (7,76 г, 49%).
1H-ЯМР δ (300 МГц, CDCl3) δ 10.09 (bs, 2Н), 7.89 (s, 1H), 7.25 (d, 1H, Jm=1,2 Гц), 7.13 (dd, 1H, Jo=8,4 Гц, Jm=1,2 Гц), 7.02 (d, 1H, Jo=8,4 Гц), 2.24 (s, 3Н); MC(Cl) m/z 157/159.
(трет-Бутокси)-N-[(2-хлор-4-метил-фенил)амино]карбоксамид
Суспензию 2-хлор-4-метилфенилгидразина гидрохлорида (7,74 г, 40,09 ммоль) в 95 мл насыщенного водного NaHCO3 перемешивали в течение 10 минут, а затем обрабатывали твердым К2СО3 (9,45 г, 68,37 ммоль). Полученную высокодисперсную светло-желтую суспензию перемешивали в течение 10 минут. Раствор ди-трет-бутилдикарбоната (12,97 г, 46,12 ммоль) в 195 мл THF добавляли в течение 5 минут, и полученную двухфазную смесь энергично перемешивали в течение 3 часов. Реакционную смесь разделяли, и водный слой экстрагировали эфиром (5×25 мл). Объединенные органические слои промывали дистиллированной водой (2×75 мл), сушили над МgSO4 и концентрировали при пониженном давлении. После высушивания в вакууме получили светло-оранжевое масло (14,07 г). Материал очищали с помощью флэш-хроматографии на силикагеле, используя смесь эфира и гексанов 10:90 в качестве элюента. Продукт получали в виде светло-желтого масла, которое затвердевало при стоянии (9,92 г, 96%).
1H-ЯМР (300 МГц, CDCl3) δ 8.88 (s, 1H), 7.15 (s, 1Н), 7.09 (d, 1H, Jm=1,2 Гц), 6.97 (d, 1H, Jo=8,1 Гц), 6.64 (d, 1H, Jo=8,1 Гц), 2.18 (s, 3Н), 1.41 (s, 9H); МС (Cl) m/z 279/281.
Диметил-7-хлор-4-гидроксихинолин-2,3-дикарбоксилат
Перемешиваемую смесь метил-2-амино-4-хлорбензоата (2,50 г, 13,5 ммоль) и диметил-ацетилендикарбоксилата (2,05 г, 14,4 ммоль) в трет-бутаноле (22 мл) кипятили с обратным холодильником в течение 7 часов в атмосфере азота. После добавления дополнительного количества диметил-ацетилендикарбоксилата (1,16 г, 8,13 ммоль) и кипячения с обратным холодильником в течение еще 2,5 часов, реакционной смеси давали охладиться до комнатной температуры, и добавляли одной порцией трет-бутоксид калия (1,56 г, 13,9 ммоль). Образовывался осадок, и полученную смесь кипятили с обратным холодильником в течение 1,5 часов. Данную смесь охлаждали до комнатной температуры и фильтровали для отделения твердых веществ, которые промывали mpem-бутанолом и эфиром. Твердые вещества растворяли в воде и подкисляли 1 н серной кислотой с образованием осадка. Полученную смесь экстрагировали метиленхлоридом, и объединенные экстракты промывали солевым раствором и водой, сушили над MgSO4, фильтровали и концентрировали с получением зеленого твердого вещества. Перекристаллизацией этого материала из метанола получили соединение, указанное в заголовке (1,15 г, 47%), в виде кремового твердого вещества, т.пл. 232-233°С; МС (Сl):296 (М+Н). Анализ для C13H10ClNO5: вычислено: С, 52,81; Н, 3,41; N, 4,74; найдено: С, 52,75; Н, 3,47; N, 4,69.
3-Карбометокси-7-хлор-4-гидроксихинолин-2-карбоновая кислота
К перемешиваемой суспензии диметил-7-хлор-4-гидроксихинолин-2,3-дикарбоксилата (1,0 г, 3,38 ммоль) в воде (20 мл) добавляли водный раствор гидроксида натрия (0,27 г, 6,75 ммоль), при этом суспензия растворялась. Реакционную смесь нагревали до 60°С в течение 1 часа. После этого времени реакционную смесь охлаждали до комнатной температуры и подкисляли концентрированной соляной кислотой. Продукт затем экстрагировали в диэтиловый эфир и этилацетат. Органические экстракты сушили над MgSO4, фильтровали и концентрировали в вакууме с получением соединения, указанного в заголовке, в виде твердого вещества (900 мг). Этот материал очищали с помощью перекристаллизации, используя систему сорастворителей этилацетат/гексан с получением соединения, указанного в заголовке (571 мг, 60%), в виде белого твердого вещества, т.пл. 296°С (разлагается). МС (Cl)=238 (М+Н). Анализ для C12H8NO5Cl·0,45 СН3СО2СН2СН3·0,10Н2О: вычислено: С, 51,30; Н, 3,68; N, 4,34; найдено: С, 51,28; Н, 3,62; N, 3,97. 1H-ЯМР 8.22 (d, J=8,7 Гц, 1Н), 7.92 (d, J=1,8 Гц, 1Н), 7.28 (dd, J=8,7; 1,8 Гц, 1Н), 3.90 (s, 3H).
3-Карбометокси-2-пирролидинокарбамид-7-хлор-4-гидроксихинолин
К суспензии 3-карбометокси-7-хлор-4-гидроксихинолин-2-карбоновой кислоты (2,25 г, 8,0 ммоль) в ТHF (20 мл) при температуре окружающей среды в атмосфере азота добавляли дициклогексилкарбодиимид (1,65 г, 8,0 ммоль) и пирролидин (0,596 г, 8,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, после этого времени побочный продукт мочевину удаляли путем фильтрования. Желаемый продукт очищали с помощью флэш-хроматографии на колонке, используя 5% метанол в хлороформе, с получением соединения, указанного в заголовке (2,52 г, 94,3%), в виде твердого вещества коричнево-рыжего цвета, т.пл.=215°С; МС (Cl): 335 (М+Н). 300 МГц 1H-ЯМР (DMSO-d6): 8.12 (d, J=8,7 Гц, 1Н), 7.60 (d, 1H, J=1,8 Гц), 7.47 (dd, 1H, J=8,8; 2,0 Гц), 3.69 (s, 3H), 3.40-3.49 (m, 2H), 3.27-3.33 (m, 2H), 1.80-1.96(m,4H).
7-Хлор-4-оксо-2-(пирролидинилкарбонил)гидрохинолин-3-карбоновая кислота
К суспензии 3-карбометокси-2-пирролидинокарбамид-7-хлор-4-гидроксихинолина (2,52 г, 7,5 ммоль) в деионизированной воде (40 мл) добавляли по каплям раствор (20 мл) водного гидроксида калия (882 мг, 15,75 ммоль). При завершении добавления реакционную смесь нагревали до 60°С. Через 3 часа реакционную смесь фильтровали для удаления небольшого количества нерастворимого материала. Фильтрат затем подкисляли до рН 1, что давало белый осадок. Твердое вещество выделяли путем фильтрования под вакуумом, промывали водой и сушили при 30°С в вакууме в течение 16 часов. Это давало соединение, указанное в заголовке (1,5 г, 64%), в виде белого твердого вещества, т.пл.=225-8°С; МС (Сl): 321 (М+Н). 300 МГц 1H-
ЯМР (DMSO-d6): 8.28 (d, J=8,8 Гц, 1Н), 7.77 (s, 1H), 7.64 (d, 1H, J=8,7), 3.52-3.57 (m, 2H), 3.17-3.19 (m, 2H), 1.83-1.98 (m, 4H).
N-[(трет-бутокси)карбониламино][7-хлор-4-оксо-2-(пирролидинилкарбонил)(3-гидрохинолил)]-N-(2-хлор-4-метилфенил)карбоксамид
К перемешиваемой суспензии 7-хлор-4-оксо-2-(пирролидинилкарбонил)гидрохинолин-3-карбоновой кислоты (14,57 г, 45,43 ммоль) в безводном THF (300 мл) в атмосфере азота добавляли 1-циклогексил-3-(2-морфолиноэтил)карбодиимида мето-пара-толуолсульфонат (CMC, 34,89 г, 82,37 ммоль). Белая суспензия сразу же становилась яркожелтой. Добавляли (трет-бутокси)-N-[(2-хлор-4-метилфенил)амино]карбоксамид (13,89 г, 54,10 ммоль) в виде твердого вещества, затем 50 мл безводного THF. Ярко желтую реакционную смесь перемешивали при комнатной температуре в течение 22 часов. Добавляли вторую порцию CMC (16,77 г, 39,59 ммоль) к реакционной смеси. После 2,5 часов при комнатной температуре реакционную смесь нагревали при 60°С в течение 5,5 часов. После охлаждения до комнатной температуры, реакционную смесь фильтровали, и собранные твердые вещества промывали THF. Фильтрат и промывные воды концентрировали и сушили в вакууме с получением светло-желтой пены. Материал растворяли в метиленхлориде (400 мл), промывали дистиллированной водой (2×150 мл) и экстрагировали 10% NаНСО3 (2×500 мл). Органический слой сушили над Na2SO4, концентрировали и сушили в вакууме с получением светло-коричневой пены. Материал очищали с помощью флэш-хроматографии на силикагеле, используя градиент от 95:5 до 85:15 смеси хлороформа и метанола в качестве элюента, с получением 15,42 г (61%) желаемого продукта в виде белого твердого вещества. 1H-ЯМР (300 МГц, DMSO-d6) δ 13.03 (bs, 1H), 9.19 (bs, 1H), 8.25 (d, 1H, Jo=8,7 Гц), 7.68 (d, 1H, Jm=1,8 Гц), 7.54 (dd, 1H, Jo=8,7 Гц, Jm=1,8 Гц), 7.50 (d, 1H, Jm=1,8 Гц), 7.45 (d, 1H, Jo=7,8 Гц), 6.81 (d, 1H, Jo=7,8 Гц), 3.47 (m, 4H), 2.34 (s, 3Н), 1.90 (m, 4H), 1.40 (s, 9Н); МС (-Cl) m/z 559/561.
7-хлор-4-гидрокси-2-(2-хлор-4-метилфенил)-1,2,5,10-тетрагидропиридазино[4,5-b]хинолин-1,10-дион
К перемешиваемой суспензии N-[(трет-бутокси)карбониламино][7-хлор-4-оксо-2-(пирролидинилкарбонил)(3-гидрохинолил)]-N-(2-хлор-4-метилфенил)карбоксамида (21,16 г, 37,82 ммоль) в 900 мл безводного THF в атмосфере азота медленно добавляли метансульфоновую кислоту (120,0 мл, 184,9 ммоль). Полученный темно-желтый раствор перемешивали при комнатной температуре в течение 18 часов. Данный раствор выливали в 7 л воды, перемешивали в течение 3 часов и фильтровали с получением светло-желтого твердого вещества. Твердое вещество обрабатывали ультразвуком в метаноле, выделяли путем фильтрования и сушили в вакууме (30 мм) при 40°С с получением продукта в виде белого твердого вещества (12,93 г, 88%). 1H-ЯМР (300 МГц, DMSO-d6) δ 12.90 (bs, 1H), 12.10 (bs, 1H), 8.16 (d, 1H, Jo=8,7 Гц), 8.07 (d, 1H, Jm=1,8 Гц), 7.47 (dd, 1H, Jo=8,7 Гц, Jm=1,8 Гц), 7.47 (d, 1H, Jm=1,2 Гц), 7.42 (d, 1H, Jo=8,1 Гц), 7.29 (dd, 1H, Jo=8,1 Гц, Jm=1,2 Гц), 2.38 (s, 3Н); МС (Cl) m/z 388/390/392. Вычислено для С18Н11Сl2N3О3: С, 55,69; Н, 2,86; N, 10,82; найдено: С, 55,78; Н, 2,89; N, 10,79.
Пример 2
7-хлор-4-гидрокси-2-(2-хлор-4-метилфенил)-1,2,5,10-тетрагидропиридазино[4,5-b]хинолин-1,10-диона холиновая соль
Суспензию 7-хлор-4-гидрокси-2-(2-хлор-4-метилфенил)-1,2,5,10-тетрагидропиридазин[4,5-b]хинолин-1,10-диона (753 мг, 1,94 ммоль) в метаноле (50 мл) обрабатывали гидроксидом холина (550 мл 45% раствора в метаноле, 1,94 ммоль). Большая часть твердых веществ немедленно растворилась, и данную смесь обрабатывали ультразвуком в течение 10 минут для растворения оставшихся веществ. Раствор фильтровали через 0,2 микронный нейлоновый фильтрующий шприц. Раствор концентрировали путем упаривания с помощью роторного испарителя до 1,01 г (>100%) желтого твердого вещества. Твердое вещество перекристаллизовывали из кипящего с обратным холодильником этанола (25 мл), и раствору давали кристаллизоваться медленно и без перемешивания. Примерно через 2 часа кристаллы собирали путем вакуумного фильтрования. Желтое твердое вещество сушили на воздухе с получением (696 мг, 73%) соединения, указанного в заголовке, которое перекристаллизовывали из кипящего с обратным холодильником этанола (20 мл). Твердым веществам давали образоваться в течение 16 часов и аккуратно соскребали с колбы, собирали путем вакуумного фильтрования и промывали этанолом (2×3 мл) с получением 500 мг соединения, указанного в заголовке, которое при высушивании при 100 мТор (13,33·10-3 КПа) при 30°С в течение трех дней давало 480 мг соединения, указанного в заголовке (50%). Т.пл. 239,5-240,5°С (разлагается); 1H-ЯМР (300 МГц, DMSO-d6) δ 8.12-8.09 (2Н, m), 7.34-7.17 (4Н, m), 3.86-3.80 (2Н, m); 3.39 (2Н, t, J=5,25 Гц), 3.09 (9Н, s); 2.35 (3Н, s); Вычислено для C18H10N303Cl2·1,0 C5H14NO·0,6 H2O: С, 55,01; Н, 5,06; N, 11,16; найдено: С, 55,04; 54,75; Н, 4,86; 4,86; N, 11,05; 11,07.
Тесты биологической функции
Тест А: ингибирование связывания [3H]-MDL105,519
Мембраны головного мозга крыс: мембраны головного мозга крыс, использованные в этих экспериментах, получали от Analytical Biological Services Inc., и готовили по существу в соответствии со способом В.М. Baron et al., J. Pharmacol. Exp. Ther. 250, 162 (1989). Кратко, свежую ткань головного мозга, включая кору головного мозга и гиппокамп, от самцов крыс линии Sprague Dawley гомогенизировали в 0,32 М сахарозе и центрифугировали при низкой скорости для отделения клеточных мембран от других клеточных компонентов. Мембраны затем промывали 3 раза с использованием деионизированной воды, за чем следовала обработка 0,04% Triton X-100. Наконец, мембраны промывали шесть раз в 50 мМ Трис-цитратном буфере, рН 7,4, и замораживали при -80°С до использования.
[3H]-MDL105,519 (72 Ки/ммоль) закупали у Amersham. Холодный MDL.105,519 закупали у Sigma/RBI. Анализы связывания осуществляли по существу в соответствии с протоколом В.М. Baron et al., J. Pharmacol. Exp. Ther. 279, 62 (1996), следующим образом. В день эксперимента мембраны головного мозга размораживали при комнатной температуре и суспендировали в 50 мМ трис-ацетатном буфере, рН 7,4 ("TAB"). Семьдесят пять микрограмм на миллилитр белка (путем использования красителя BioRad) использовали для конкурентного связывания. Эксперименты осуществляли, используя 96-луночные планшеты. Мембраны инкубировали с 20 мкл соединений в различных концентрациях и 1,2 нМ [3H]-MDL 105,519 в течение 30 минут при комнатной температуре в общем объеме 250 мкл. Неспецифическое связывание определяли путем использования 100 мкМ немеченного MDL 105,519. Немеченный MDL 105,519 и соединения растворяли в виде 12,5 мМ маточных растворов в DMSO. Конечную концентрацию DMSO в каждой лунке поддерживали ниже 1%, причем было обнаружено, что эта концентрация не изменяет результатов связывания. После инкубации несвязавшийся [3Н]-MDL 105,519 удаляли путем фильтрования на планшеты GF/B Unifilter, используя харвестер Packard. Фильтры промывали четыре раза охлажденным на льду TAB (в сумме 1,2 мл буфера). Планшеты сушили в течение ночи при комнатной температуре, и связанную радиоактивность измеряли на Packard TopCount после добавления 45 мкл на лунку MICROSCINT О. Эффективность соединений выражали как Ki, и данные анализировали, используя таблицу Microsoft Excel и программное обеспечение GraphPad Prizm.
Мембраны головного мозга человека: мембраны головного мозга человека получали от Analytical Biological Services Inc., и анализы осуществляли, как описано для мембран крыс.
Тест Б: тест с формалином
Тест с формалином позволяет оценить способность перорально вводимого соединения ингибировать формалин-индуцированные ноцицептивные поведения у крыс (D. Dubuisson, et at., Pain 4, 161-174 (1977); Н. Wheeler-Aceto et al., Psychopharmacology 104, 35-44 (1991); T.J. Coderre, et al., Pain 54, 43-50 (1993)). В данном тесте наблюдают две различные фазы формалин-индуцированных поведений. Первая фаза ответа, вызванная острой ноцицепцией на химическое вещество (формалин), инъецированное в лапу, имеет место между нулем и пятью минутами. Следует период покоя между 5 - 15 минутами после инъекции. После периода покоя вторая фаза ответа, вызванная сенсибилизацией центральных нейронов в заднем роге, имеет место через 15 минут и продолжается вплоть до 60 минут. Сенсибилизация центральных нейронов в позвоночном столбе увеличивает вредный афферентный вход и вызывает более сильную блокаду боли, которая должна передаваться в головной мозг. Ингибирование второй фазы ответа указывает на спинномозговой механизм действия лекарства.
Процедура для теста с формалином следующая: самцов крыс помещают в камеру из органического стекла и наблюдают в течение 30-45 минут за их исходной активностью. Животных предварительно обрабатывают либо носителем, либо различными дозами тестируемого соединения. Животным вводят дозы 0,05 мл стерильного 1% формалина в заднюю лапу (под дорсальную кожу) за 3 часа до инъекции формалина. Число отдергиваний лапы (ответов) во время первой фазы (0-5 минут) и второй фазы (20-35 минут) подсчитывают и записывают. Ответное отдергивание вычисляют как процент ингибирования по сравнению со средней оценкой группы с физиологическим раствором в качестве контроля. ЭД50 представляет собой дозу соединения, которая дает 50% ингибирования ноцицептивного ответа.
% ингибирования ноцицептивного ответа=
100 × (число ответов в группе носителя - число ответов в группе соединения)/(число ответов в группе носителя)
Т-критерий Стьюдента использовали для статистического анализа, чтобы определить значимость эффектов соединений. Соединения считаются активными в зависимости от их способности ингибировать ответное отдергивание.
Тест В: модель невропатической боли (хроническое повреждение сокращения):
Тест хронического повреждения сокращения (Chronic Constriction injury, CCl) представляет собой модель невропатической боли, связанной с повреждениями нервов, которые могут возникать непосредственно от травмы и сдавления, либо опосредованы широким рядом заболеваний, таких как инфекция, рак, метаболические состояния, токсины, дефициты питательных веществ, иммунологическая дисфункция и мышечно-скелетные изменения. В данной модели одностороннюю периферическую гипералгезию продуцируют у крыс путем наложения лигатуры на нерв (G.J. Bennett, et al., Pain 33, 87-107 (1988)).
Методически, крыс линии Sprague-Dawley (250-350 г) анестезировали пентобарбиталом натрия, и общий седалищный нерв экспонировали на уровне середины бедра путем тупого рассечения через двуглавую мышцу бедра. Срез нерва (примерно 7 мм), проксимальный к трифуркации седалищного нерва, освобождали от ткани, и накладывали лигатуру в четырех положениях с помощью хромированного кетгута. Шов завязывали примерно с 1 мм расстоянием между лигатурами. Разрез закрывали слоями, и животным давали оправиться. Термальную гипералгезию измеряли, используя тест отдергивания лапы (К. Hargreaves, et al., Pain 32, 77-88 (1988)). Чтобы осуществить данный тест, животных приучали к стеклянному полу с повышенной температурой. Нагреватель, выделяющий тепловую энергию за счет излучения, был нацелен на середину подошвы задней лапы (территория седалищного нерва) через стеклянный пол с 20 сек перерывами, используемыми для предотвращения повреждения кожи. Записывали время латентного состояния для рефлекса отдергивания в обеих задних лапах.
Поврежденные лапы с лигированными нервами показали более короткие периоды латентного состояния для отдергивания лапы по сравнению с неповрежденными или симулированно оперированными лапами. Ответы на тестируемые соединения оценивали в различные моменты времени после перорального введения для определения начала и продолжительности эффекта соединения. Исследования ответной реакции на дозу проводили с многочисленными группами ССl крыс, которые получали либо носитель, либо тестируемое соединение перорально три раза в сутки в течение 5 дней. Периоды латентного состояния для отдергивания лапы измеряли каждые сутки за 10 минут до и через 2 или 3 часа после первой суточной дозы. Эффективность подсчитывали как средний процент уменьшения гипералгезии в течение 5 дней дозирования по сравнению с таковым у животных, которым давали носитель. Эффективности соединений выражали в виде минимальной эффективной дозы (МЭД) в мг/кг/сутки, которая дает процент уменьшения гипералгезии, который статистически значим, причем анти-гипералгезический эффект вычисляли следующим образом:
% анти-гипералгезии=(среднее значение группы носителя - среднее значение группы соединения)х100/(среднее значение группы носителя)
Анализ данных осуществляли с помощью теста сравнения множества значений (тест Дьюннета (Dunnett)).
В таблице показаны результаты тестов А, Б и В для соединений по изобретению.
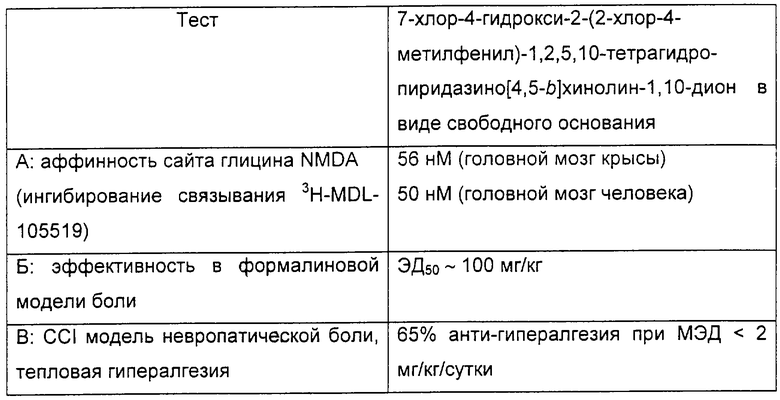
В формалиновой модели боли эффективная доза соединения по изобретению, которая вызывает 50% уменьшение чувствительности к болевому стимулу, составляла примерно 100 мг/кг, доза, которая сравнима с дозой габапентина, необходимой для достижения подобного результата. В CCl модели невропатической боли, однако минимальная эффективная доза соединения по изобретению составляла менее 2 мг/кг/сутки для достижения 65% анти-гипералгезии. Для сравнения, для достижения примерно 46% анти-гипералгезии требуется примерно 90 мг/кг/сутки габапентина.
При введении путем интратекальной инъекции соединение по изобретению ингибировало развитие NMDA индуцированного поведения/припадка с ЭД50 110 нмоль.
Соединение по изобретению также тестировали на связывание с панелью из более чем 80 не-NMDA рецепторов. Соединение показало отсутствие значительного взаимодействия с любым протестированным рецептором, отличным от NMDA рецептора.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРИДАЗИНОХИНОЛИНА | 1994 |
|
RU2168511C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНОХИНОЛИНА | 1994 |
|
RU2279432C2 |
| АНАЛЬГЕЗИРУЮЩЕЕ СРЕДСТВО | 2018 |
|
RU2711968C1 |
| НОВЫЙ ХИМИЧЕСКИЙ СПОСОБ ПОЛУЧЕНИЯ 6-ХЛОР-4-(4-ФТОР-2-МЕТИЛФЕНИЛ)ПИРИДИН-3-АМИНА, КЛЮЧЕВОГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПРОИЗВОДСТВА NT-814 | 2024 |
|
RU2840774C2 |
| БЕНЗО[d] ИЗОКСАЗОЛ-3-ОЛЬНЫЕ ИНГИБИТОРЫ DAAO | 2004 |
|
RU2384574C2 |
| Соединения 6, 7-дигидро-5H-пиразоло[5,1-b][1,3]оксазин-2-карбоксамида | 2017 |
|
RU2719599C2 |
| АНАЛЬГЕЗИРУЮЩЕЕ СРЕДСТВО | 2016 |
|
RU2634618C1 |
| 3R, 4S -3-[4-(4-ФТОРФЕНИЛ-4-ГИДРОКСИПИПЕРИДИН-1-ИЛ] ХРОМАН- 4,7-ДИОЛ, ЕГО ОПТИЧЕСКИЕ ИЗОМЕРЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ | 1994 |
|
RU2126404C1 |
| (2R,4R)-5-(5′-ХЛОР-2′-ФТОРБИФЕНИЛ-4-ИЛ)-2-ГИДРОКСИ-4-[(5-МЕТИЛОКСАЗОЛ-2-КАРБОНИЛ)АМИНО]ПЕНТАНОВАЯ КИСЛОТА | 2016 |
|
RU2715241C2 |
| СОЕДИНЕНИЯ ((ФЕНИЛ)ИМИДАЗОЛИЛ)МЕТИЛГЕТЕРОАРИЛА, ИХ ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ УКАЗАННЫЕ СОЕДИНЕНИЯ, И НАБОР, ВКЛЮЧАЮЩИЙ ТАКУЮ КОМПОЗИЦИЮ | 2008 |
|
RU2478631C2 |
Изобретение относится к 7-хлор-4-гидрокси-2(2-хлор-4-метилфенил)-1,2,5,10-тетрагидропиридазино[4,5-b]хинолин-1,10-диону, его фармацевтически приемлемым солям, способам лечения боли, при которых вводят уменьшающее боль эффективное количество данного соединения, и фармацевтические композиции, содержащие данное соединение. 5 н. и 1 з.п. ф-лы, 1 табл.
Приоритет по пунктам
| WO 9511244, 27.04.1995. |

