Перекрестные ссылки на родственные заявки
Для настоящего изобретения испрашивается приоритет по Предварительной Заявке США No. 61/368500 от 28 июля 2010 года, содержание которой в полном объеме и для всех целей включено в настоящее описание посредством ссылки.
Это изобретение было создано при поддержке правительства США по грантам No. DA012413, DA012447 и АА017538, полученным национальным Институтом Здравоохранения США. Правительство США обладает определенными правами на настоящее изобретение.
Уровень техники
Периферические каннабиноидные рецепторы осуществляют мощный ингибирующий контроль над возникновением болевых ощущений, однако конкретный эндогенный каннабиноидный сигнал, который обычно запускает этот внутренний анальгетический механизм, все еще неизвестен. Чтобы ответить на этот вопрос, авторы изобретения разработали периферический ингибитор гидролазы амидов жирных кислот (FAAH), фермента, ответственного за деградацию эндоканнабиноида анандамида. Соединение под названием URB937 подавляет активность FAAH и повышает уровень анандамида за пределами центральной нервной системы (ЦНС). Несмотря на неспособность URB937 попасть в мозг и спинной мозг, это соединение ослабляет поведенческую реакцию, характерную для постоянной боли, в моделях воспаления и поражения периферических нервов на грызунах, а также подавляет вызванную вредоносным стимулом нейронную активацию в областях спинного мозга, вовлеченных в обработку болевого сигнала. Блокада CB1-рецептора нивелирует эти эффекты. Эти результаты позволяют предположить, что анандамид-опосредованная передача сигналов на периферических CB1-рецепторах контролирует передачу информации о боли в ЦНС. Неспособные проникать в мозг ингибиторы FAAH, усиливающие данный механизм, представляют собой новый способ терапии боли.
Анандамид, природный амид арахидоновой кислоты и этаноламина, обладает всеми ключевыми свойствами эндогенного каннабиноидного вещества (Devane, W.A. et al. Science, 258, 1946-1949 (1992)): он высвобождается стимулированными нейронами при необходимости (Di Marzo, V. et al., Nature, 372, 686-691 (1994); Giuffrida, A. et al., Nat. Neurosci., 2, 358-363 (1999)); он активирует каннабиноидные рецепторы с высокой аффинностью (Devane, W.A. et al. Science, 258, 1946-1949 (1992)) и он быстро выводится посредством двухстадийного процесса, состоящего из транспорта, опосредуемого переносчиком, и последующего внутриклеточного гидролиза (Di Marzo, V. et al., Nature, 372, 686-691 (1994); Beltramo, M. et al., FEBS Lett, 403, 263-267 (1997)). Гидролиз анандамида катализируется ферментом гидролазой амидов жирных кислот (FAAH), мембранно-связанной сериновой гидролазой (Cravatt, B.F. et al., Nature, 384, 83-87 (1996); Patricelli, M.P. et al., Biochemistry, 38, 9804-9812 (1999)) (WO 98/20119) (Патент США No. 6271015), которая также расщепляет другие биоактивные жирные этаноламиды, такие как олеоилэтаноламид (цис-9-октадеценамид)) (Rodriguez de Fonseca, F. et al. Nature, 414, 209-212 (2001)) и палмитоилэтаноламид (Calignano, A. et al., Nature, 394, 277-281 (1998)). Мутантные мыши, не имеющие гена, кодирующего FAAH, не могут метаболизировать анандамид (Cravatt, B.F. et al., Proc. Natl. Acad. Sci. U.S.A., 98, 9371-9376 (2001)) и, несмотря на фертильность и общую нормальность, демонстрируют признаки повышенной активности анандамида на каннабиноидных рецепторах, такие как пониженная чувствительность к боли (Cravatt, B.F. et al., Proc. Natl. Acad. Sci. U.S.A., 98, 9371-9376 (2001)). Это позволяет предположить возможность того, что лекарственные средства, нацеленные на FAAH, могут усиливать местное действие анандамида, и при этом, возможно, позволяют избежать множества зачастую нежелательных эффектов, сопровождающих использование Δ9-ТНС и других антагонистов каннабиноидов прямого действия (Hall, W., et al., Lancet, 352, 1611-1616 (1998); Chaperon, F., et al., Crit. Rev. Neurobiol, 13, 243-281 (1999)).
Ощущение боли может эффективно контролироваться нейромедиаторами, которые функционируют внутри ЦНС. Эта модуляция была хорошо охарактеризована в заднем роге спинного мозга, где обрабатываются импульсы, переносимые ноцицептивными (болечувствительными) волокнами перед тем, как они передаются в мозг. В дополнение к этим основным механизмам, может осуществляться внутренний контроль передачи боли на концах афферентного нервного волокна вне ЦНС.Один из известных примеров периферической регуляции представлен эндогенными опиоидами, которые высвобождаются из активированных иммунных клеток во время воспаления и ингибируют инициацию боли путем взаимодействия с опиоидными рецепторами, локализованными на чувствительных нервных окончаниях1,2.
Предполагается, что эндоканнабиноидные медиаторы могут выполнять функцию, аналогичную функции опиоидов, так как фармакологическая активация периферических каннабиноидных рецепторов CB1 и CB2 ингибирует связанную с болью поведенческую реакцию3-7, в то время как генетическое нарушение экспрессии рецептора CB1 в первичных ноцицептивных нейронах обостряет такую реакцию8. Кроме того, есть данные, из которых следует, что клинические состояния, ассоциированные с невропатической болью или воспалением, такой как комплексный регионарный болевой синдром и артрит, могут сопровождаться периферическими повышениями уровня эндоканнабиноида анандамида9,10. Полагают, что другой важный эндоканнабиноид-лиганд, 2-арахидоноилглицерин (2-AG), также вовлечен в ноцицептивную передачу сигналов вне ЦНС8,11.
Огромное внимание уделяется роли анандамида в болевых ощущениях. Методы лечения боли посредством введения анандамида и палмитоиланандамида раскрыты в публикации патентной заявки США No.: 20020173550. Методы лечения боли путем введения ингибиторов FAAH раскрыты в публикациях патентных заявок США No. 20040127518 и 20030134894. Способы лечения боли путем введения ингибиторов транспорта анандамида раскрыты в публикации патентной заявки США No. 20030149082.
Хотя эти сведения намекают на то, что эндоканнабиноидная система выполняет важную функцию в периферической регуляции ноцицепции, они не дают однозначного представления о природе эндогенного лиганда или лигандов, вовлеченных в данную функцию. При этом устранение этого пробела является необходимым для получения представления о внутренних механизмах, которые контролируют возникновение боли на молекулярном уровне, а также для обнаружения новых обезболивающих средств, лишенных основных побочных эффектов. В настоящем исследовании для более подробного изучения функций периферического анандамида и раскрытия его возможной роли в контроле появления болевых сигналов авторами был идентифицирован и охарактеризован не проникающий в мозг ингибитор фермента деградации анандамида, FAAH12. Особо важной при разработке терапевтического применения ингибиторов FAAH является их способность модулировать действие эндогенных каннабиноидных систем внутри ЦНС, вызывающих нежелательные психотропные или седативные эффекты.
Настоящее изобретение направлено на решение этих и других потребностей, для чего предлагаются ингибиторы FAAH периферически-ограниченного действия, а также способ их применения для лечения разнообразных состояний, включающих боль и/или воспаление.
Раскрытие изобретения
В первом аспекте изобретения описываются соединения и фармацевтические композиции, имеющие следующую формулу:
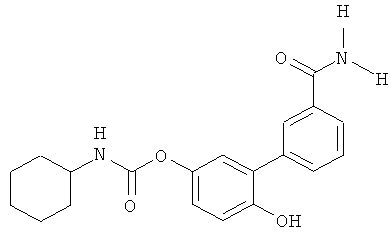 ,
,
в которой R1 выбирают из группы, состоящей из гидроксигруппы и ее физиологически гидролизуемых сложных эфиров, -SH, -o-карбоксамидо, -OC(O)R7, -О-CO-NR8R9 и -NR8R9, где R7 представляет собой замещенный или незамещенный гидрокарбил, a R8 и R9 независимо выбирают из группы, состоящей из водорода и (C1-C3)алкила; R2 и R3 независимо выбирают из группы, состоящей из водорода и гидрокарбила; каждый R4 независимо выбирают из группы, состоящей из галогена и гидрокарбила, а n представляет собой целое число от 0 до 4; каждый R5 независимо выбирают из группы, состоящей из галогена или гидрокарбила, a m представляет собой целое число от 0 до 3; и R6 представляет собой замещенный или незамещенный циклогексил; а также их фармацевтически приемлемые соли. В предпочтительных воплощениях распределение этих соединений в реципиенте ограничено периферическими областями.
Во втором аспекте изобретения описываются фармацевтические композиции, включающие терапевтически эффективное количество соединений по изобретению. Композиции могут быть включены в состав, предназначенный для введения любым подходящим образом, включая пероральный и парентеральный пути введения. Кроме того, композиции могут иметь вид единичной лекарственной дозировки.
В третьем аспекте изобретения описывается способ лечения пациента, нуждающегося в ингибиторе FAAH периферически-ограниченного действия (например, соединении-ингибиторе FAAH по изобретению). В предпочтительных воплощениях пациентом является человек. В некоторых воплощениях необходимость связана с лечением боли, воспаления или иммунного расстройства у пациента. В некоторых воплощениях боль может быть ноцицептивной, воспалительной или невропатической болью. В предпочтительном варианте соединение-ингибир FAAH периферически-ограниченного действия представляет собой соединение по изобретению.
В четвертом аспекте изобретения описывается способ повышения периферической активности эндогенно вырабатываемого (т.е. эндоканнабиноида, такого как анандамид, N-арахидоноил допамин) или поступающего извне каннабиноидного амида жирной кислоты у пациента путем введения соединения по изобретению. В предпочтительном варианте амидом жирной кислоты является анандамин, N-арахидоноил допамин, олеоилэтаноламид, стеароилэтаноламид или палмитоилэтаноламид. В случае, когда этаноламид жирной кислоты поступает извне, он может вводиться пациенту перед, после или одновременно с введением соединения по изобретению. В некоторых воплощениях пациент нуждается в лечении боли, воспаления или иммунного расстройства. В предпочтительных воплощениях боль может быть ноцицептивной, воспалительной или невропатической болью.
В пятом аспекте изобретения описываются способы скрининга соединений на предмет их способности к вытеснению из мозга транспортной системой белка устойчивости к раку молочной железы (BCRP). Структуры, аналогичные URB937, и соединения по изобретению могут служить в качестве субстратов для BCRP. Соответственно, в некоторых воплощениях в изобретении описываются способы анализа аналога URB937 и/или соединений по изобретению на основе их способности транспортироваться транспортной системой BCRP in vitro.
Краткое описание чертежей
Фиг.1 URB937 представляет собой ингибитор FAAH периферически-ограниченного действия, (a) Активность FAAH в печени (закрашенные круги) и в мозге (закрашенные квадраты) через 1 ч после введения различных доз URB937 (0,03-100 мг-кг-1, подкожно) у мышей Swiss, (b) Распределение URB937 в печени (закрашенные круги), в мозге (закрашенные квадраты) и в сыворотке (вставка) после однократной инъекции мышам (1 мг-кг-1, внутрибрюшинно). (с) Зависимость ингибирования активности FAAH в печени (закрашенные круги) или в мозге (закрашенные квадраты) от времени после введения URB937 (1 мг-кг-1, внутрибрюшинно). (d) Воздействие URB937 (1 мг-кг-1, внутрибрюшинно, закрашенные столбцы) или пустого носителя (незакрашенные столбцы) на уровень анандамида и пальмитоилэтаноламида (PEA) в печени, в переднем мозге и в гипоталамусе мышей Swiss, (e) Воздействие URB937 на уровень анандамида и PEA в печени мышей С57 В1/6 дикого типа (+/+) и FAAH-дефицитных мышей (-/-). (f) Отсутствие влияния URB937 (1 мг-кг-1, внутрибрюшинно, закрашенные столбцы) на уровень 2-арахидоноилглицерина (2-AG) в мышах Swiss. Результаты выражены как среднее значение±стандартная погрешность среднего; n=3; *P<0,05; ***P<0,001 по сравнению с пустым носителем.
Фиг.2 URB937 ингибирует поведенческие реакции на вредоносные химические вещества у мышей и крыс, (a-d) Болевое поведение мышей, индуцированное уксусной кислотой (НАс). (a) Болевое поведение (количество эпизодов скорчивания) оценивали через 1 ч после введения пустого носителя (V), URB937 (URB, 1 мг-кг-1, внутрибрюшинно) или индометацина (IDM, 1 мг-кг-1, внутрибрюшинно). Также показан эффект от пустого носителя и URB937, вводимых без уксусной кислоты. (b) Статистическая корреляция между антиноцицепцией и ингибированием активности FAAH в печени, вызванным URB937 (1 мг-кг-1, внутрибрюшинно). (с) Влияние URB937 (1 мг-кг-1, внутрибрюшинно) на скорчивания, индуцированные уксусной кислотой у мышей дикого типа C57B 1/6 (+/+) и у FAAH-дефицитных мышей (-/-). (d) Антагонист CB1 римонабант (Rim, 1 мг-кг-1, подкожно), в отличие от антагониста CB2 АМ630 (1 мг-кг-1, подкожно), нивелирует антиноцицептивное действие URB937. Результаты выражены в виде средних значений±стандартная погрешность среднего; n=5-17. *P<0,05 по сравнению с пустым носителем; **P<0,01 по сравнению с пустым носителем; и ***P<0,001 по сравнению с пустым носителем; ##P<0,01 по сравнению с URB937; ###P<0,001 по сравнению с URB937. (e-g) Болевое поведение крыс, индуцированное формалином, (e) URB937 (1 мг-кг-1, внутрибрюшинно) демонстрировал зависимое от времени изменение суммарного болевого индекса по отношению к пустому носителю, римонабанту (2 мг-кг-1, внутрибрюшинно) или комбинации URB937 и римонабанта (F14,22=1.86, P=0.039). Формалин инъецировали в момент времени=0. (f) URB937 (1 мг-кг-1, внутрибрюшинно) уменьшал площадь под кривой (AUC) болевого поведения во время полного цикла реакции на формалин (F3,22=3.32, P=0.039). (g) Антиноцицептивный эффект URB937 был ограничен Фазой 2 реакции на формалин (10-60 мин; F1,3=3.05, P=0,050), тогда как достоверных изменений болевого поведения во время Фазы 1 (0-10 мин) не наблюдалось (F1,3=2.22, P=0.115). Результаты выражены в виде средних значений±стандартная погрешность среднего; n=5-7. *P<0,05, все группы по сравнению с URB937; #P<0,05, URB937 или URB937 вместе с римонабантом по сравнению с пустым носителем.
Фиг.3 URB937 подавляет индуцированную формалином экспрессию белка Fos в поясничной области (L4) спинного мозга крыс, (a-c) Репрезентативные срезы, демонстрирующие индуцированные формалином Fos-положительные клетки в поясничных сегментах после инъекции (a) пустого носителя; (b) URB937 (1 мг-кг-1, внутрибрюшинно); или (c) URB937 вместе с римонабантом (2 мг-кг-1, внутрибрюшинно). Калибровочная шкала, 1 мм. (d) Количественный анализ влияния пустого носителя (незакрашенные столбцы), URB937 (закрашенные столбцы), римонабанта, и URB937 вместе с римонабантом на количество Fos-положительных клеток в поверхностном слое заднего рога (пластинка I, II), nucleus proprius (пластинка III, IV), в шейной области заднего рога (пластинка V, VI), и в переднем роге. Данные о поведении тех же объектов исследования представлены на Фиг.2. Результаты выражены в виде средних значений±стандартная погрешность среднего; n=5-7. *P<0,05, все группы по сравнению с URB937; #P<0,05, URB937 вместе с римонабантом, или римонабант отдельно по сравнению с URB937; &P<0,05, пустой носитель, римонабант по сравнению с URB937.
Фиг.4 URB937 ослабляет болевую поведенческую реакцию, вызванную периферическим воспалением у мышей. Влияние URB937 (1 мг-кг-1, внутрибрюшинно), вводимого отдельно или в комбинации с римонабантом (R, 1 мг-кг-1, внутрибрюшинно) или АМ630 (AM, 1 мг-кг-1, внутрибрюшинно), на (a) индуцированную каррагенаном механическую гипералгезию; (b) термическую гипералгезию; (с) механическую аллодинию; и (d) отек лапы. Механическую и термическую гипералгезию измеряли сразу перед инъекцией каррагенана (0 ч) или через 4 ч и 24 ч после инъекции. Механическую аллодинию измеряли в момент Очи через 24 ч после каррагенана. Результаты выражены в виде средних значений±стандартная погрешность среднего; n=6. *p<0,05 по сравнению с пустым носителем; **P<0,01 по сравнению с пустым носителем; ***P<0,001 по сравнению с пустым носителем; #P<0,05 по сравнению с URB937; ###P<0,01 по сравнению с URB937.
Фиг.5 URB937 подавляет болевую поведенческую реакцию, вызванную периферическим невральным поражением у мышей, (a-c) Влияние однократного введения носителя (заштрихованные столбцы) или URB937 (закрашенные столбцы; 1 мг-кг-1, внутрибрюшинно) на (a) механическую гипералгезию, (b) термическую гипералгезию и (c) механическую аллодинию, индуцированную перевязыванием седалищного нерва, (d-f) Влияние повторных инъекций URB937 (1 мг-кг-1, внутрибрюшинно, один раз ежедневно в течение 4 следующих друг за другом дней) на (d) механическую гипералгезию, (е) термическую гипералгезию и (f) механическую аллодинию. BL - исходный уровень (измеренный перед перевязыванием); IL - ипсилатеральная лапа (с перевязкой); CL - контралатеральная лапа (без перевязки). Результаты выражены в виде средних значений±стандартная погрешность среднего; n=6. ***P<0,001 по сравнению с исходным уровнем; #P<0,05 по сравнению с пустым носителем; ##P<0,01 по сравнению с пустым носителем; ###P<0,001 по сравнению с пустым носителем.
Фиг.6 Влияние URB937 (1 мг-кг-1, внутрибрюшинно) на уровень анандамида и палмитоилэтаноламида (PEA) в тканях мышей Swiss. Незакрашенные столбцы - пустой носитель; закрашенные столбцы - URB937. Результаты выражены в виде средних значений±стандартная погрешность среднего; n=4-6.*, P<0,05, **, P<0,01, ***, P<0,001 по сравнению с пустым носителем;
Фиг.7 Внутриподошвенная инъекция каррагенана не влияет на механическую (a) и термическую гипералгезию (b) или механическую аллодинию (c) в контралатеральных (без инъекции) лапах мышей Swiss. Римонабант (R, 1 мг-кг-1, внутрибрюшинно) и АМ630 (1 мг-кг-1, внутрибрюшинно) не имели эффекта. Результаты выражены в виде средних значений±стандартная погрешность среднего.
Подробное описание изобретения
После возникновения боли периферические каннабиноидные рецепторы оказывают мощный ингибирующий контроль, однако эндогенный каннабиноидный сигнал, который обычно запускает этот внутренний анальгетический механизм, все еще неизвестен. Авторы настоящего изобретения разработали периферический ингибитор гидролазы амидов жирных кислот (FAAH), фермента, ответственного за деградацию эндоканнабиноида анандамида. Соединение URB937 подавляет активность FAAH и повышает уровень анандамида за пределами центральной нервной системы (ЦНС). Несмотря на удивительную относительную неспособность проникать в мозг и в спинной мозг (соединение кроме того удивительно чувствительно к системе транспорта, опосредующей вытеснение из мозга), URB937 ослабляет поведенческую реакцию, характерную для постоянной боли в моделях воспаления и поражения периферических нервов на грызунах, а также подавляет вызванную вредоносным стимулом нейронную активацию в областях спинного мозга, вовлеченных в обработку болевого сигнала. Блокада CBi-рецептора предотвращает эти эффекты. Эти результаты позволяют предположить, что анандамид-опосредованная передача сигналов на периферических CB1-рецепторах контролирует передачу информации о боли в ЦНС. Относительно неспособные проникнуть в мозг ингибиторы FAAH, усиливающие данный механизм, представляют собой новый способ терапии боли.
Ощущение боли может эффективно контролироваться нейромедиаторами внутри ЦНС. Эта модуляция была хорошо изучена в заднем роге спинного мозга, где импульсы, переносимые ноцицептивными (болечувствительными) волокнами, обрабатываются перед тем, как они передаются в мозг. Дополнительно к этим основным механизмам, внутренний контроль передачи боли может осуществляться на концах афферентных нервных волокон вне ЦНС. Одним из известных примеров периферической регуляции являются эндогенные опиоиды, которые высвобождаются из активированных иммунных клеток во время воспаления и ингибируют инициацию боли путем взаимодействия с опиоидными рецепторами, локализованными на чувствительных нервных окончаниях1,2.
Соединение URB937 представляет собой мощный ингибитор FAAH, который практически не проникает в ЦНС и, таким образом, в основном препятствует деактивации анандамида только в периферических тканях. Несмотря на ограниченный спектр действия URB937 оказывает значительное антиноцицептивное влияние в моделях острой и постоянной боли на грызунах, которое нивелируется блокадой каннабиноидного рецептора CB1. Из этих наблюдений можно предположить, что ингибирование периферической активности FAAH усиливает эндогенный анальгетический механизм, регулирующий передачу возникающих ноцицептивных входных сигналов в спинной мозг и в мозг. Этот механизм, вероятно, опосредован анандамидом или другим эндогенным каннабиноидным амидом жирной кислоты.
Не желая связывать себя какой-либо теорией, авторы предполагают, что периферическая передача сигналов с помощью анандамида служит в качестве диффузной паракринной системы, которая модулирует интенсивность болевых стимулов по мере их нарастания в поврежденных тканях. Две линии рассуждений подтверждают эту мысль. Во-первых, сигналы, вызываемые воспалением и невральным поражением, могут запускать локальное высвобождение анадамида. Например, деполяризация мембраны и активация каналов TRPV-1 стимулируют выработку анандамида в культурах сенсорных нейронов25, в то время как активация провоспалительного рецептора, Toll-подобного рецептора 4, вызывает аналогичный эффект в макрофагах26. Эти сигналы, а также, возможно, другие, которые еще не идентифицированы, могут вносить вклад в повышение уровня периферического анандамида, обнаруженное в животных моделях поражения спинного нерва и воспаления8,11, а также при болезненных состояниях человека, таких как комплексный регионарный болевой синдром9 и артрит10. Во-вторых, хотя рецепторы CBi особенно широко представлены в мозге, они также повсеместно распространены в тканях и органах млекопитающих. Более конкретно, они экспрессируются в крупных первичных сенсорных нейронах и транспортируются в периферические нервные окончания27,28, где их присутствие может быть как необходимым для поддержания обычных болевых порогов8, и так и достаточным для того, чтобы оказывать значительный ноцицептивный эффект'. Рецепторы CB1 на болечувствительных окончаниях могут опосредовать анальгетическое действие местно вырабатываемого анандамида и могут также быть вовлечены в противовоспалительную активность этого липидного медиатора через его ингибирующее влияние на высвобождение возбуждающих нейропептидов29. Тем не менее, разумно предполагать, что другие каннабиноидные и каннабиноид-подобные рецепторы также прямо или косвенно вносят свой вклад в передачу сигналов анандамида в ответ на поражение. Двумя вероятными кандидатами являются рецепторы CB2, которые могут активироваться или с помощью анандамида, или с помощью 2-AG30, а также рецепторы, активируемые пероксисомными пролифераторами α-типа, которые активируются с помощью PEA и других медиаторов, имеющих липидное происхождение7,20,21. Эти рецепторы и их эндогенные лиганды присутствуют в периферических сенсорных нейронах и иммунных клетках, и вовлечены в модуляцию ноцицепции и воспаления21,31,32.
Мутантные мыши, в которых FAAH селективно делетирован во всех клетках, кроме нервных, и сохранен в периферических и центральных нейронах, демонстрируют поразительный фенотип, в котором нормальная ноцицептивная передача сопровождается уменьшением чувствительности к провоспалительным триггерам33. Возможное объяснение этого открытия, которое согласуется с настоящими результатами, заключается в том, что активность сигналов анадамида в периферических ноцицепторах регулируется FAAH, локализованным на самих ноцицепторах, а не на других соседних клетках, отличных от нервных. Это согласуется с тем наблюдением, что периферическая аксотомия индуцирует экспрессию FAAH в крупных сенсорных нейронах, реакцию, которая, как ожидается, расширяет совместную локализацию FAAH и рецепторов 
Агонисты опиоидных рецепторов прямого действия оказывают значительное анальгетическое воздействие в животных и человеческих экспериментальных моделях боли2,35. Авторы настоящего изобретения показали, что достижение значительной аналгезии возможно также путем усиления активности механизма на основе анандамида, вовлеченного в сохранение ноцицептивного гомеостаза. Эти факты дают новый взгляд на внутренний контроль боли и могут использоваться в терапевтических целях для разработки эффективных анальгетиков, лишенных основных побочных эффектов.
Определения
Следует отметить, что использованные в данном описании и в последующей формуле изобретения существительные в единственном числе включают также и множественное число, если в контексте явно не указано иное.
«FAAH» служит для обозначения гидролазы амидов жирных кислот млекопитающих и включает человеческую, крысиную и мышиную форму этого фермента, однако не огранивается только ими. Патент США No. 6,271,015 раскрывает выделенные и очищенные формы FAAH. В ряде воплощений IC50 FAAH рассматриваемых соединений определяют по ингибированию крысиной формы фермента в физиологически приемлемых условиях. Гидролазы амидов жирных кислот (FAAH) (Deutsch, D.G., et al., Prostaglandins Leukot. Essent. Fatty Acid, 66, 201-210 (2002)) - это ферменты, ответственные за деградацию липидных эталонамидов (Fowler, С.J., et al., Biochem. Pharmacol. 62, 517-526 (2001); Patricelli, M.P., et al. Vitam. Horm., 62, 663-674 (2001)), например анандамида (AEA, 1, Фиг.1), (Devane, W.A., et al., Science 258, 1946-1949 (1992)) олеоилэтаноламида (Rodriguez de Fonseca, F., et al. Nature (London) 414, 209-212 (2001); Fu, J., et al., Nature (London) 425, 90-93 (2003)) и палмитоилэтаноламида (Calignano, A., et al. Nature (London) 394, 277-281 (1998); Lambert, D.M., et al., Curr. Med. Chem. 9, 663-674 (2002)), биохимического процесса, который наряду с селективным транспортом в клетки в случае AEA (Di Marzo, V., Nature (London) 372, 686-691 (1994); Beltrama, M., et al., Science 277, 1094-1097 (1997); Piomelli, D., et al., Proc. Natl. Acad. Sci. U.S.A. (2002)), приводит к прекращению клеточных эффектов данных физиологически активных веществ. Вследствие различных важных физиологических ролей этаноламидов жирных кислот классы низкомолекулярных соединений, которые способны блокировать гидролазу FAAH или гидролазы FAAH, но не связываются с другими ферментами, участвующими в метаболизме эндоканнабиноидов, например с моноглицерид-липазой (MGL) (Dinh, Т.Р., et al., Proc. Natl. Acad. Sci. U.S.A. 99, 10819-10824 (2002)), или с каннабиноидными рецепторами, могут быть полезны как в качестве фармакологических средств, так и в качестве прототипов для разработки новых лекарственных средств (Piomelli, D., et al. Trends Pharmacol. Sci. 21, 218-224 (2000); Bisogno, Т., et al., Curr. Pharm. Des. 8, 533-547 (2002); Yarnell, A., Chem. Eng. News 80(49), 32 (2002); Smith, A., Nat. Rev. Drug Discov. 2, 92 (2003); Wendeler, M., et al. Angew. Chem. Int. Ed. 42, 2938-2941 (2003)).
Термин «фармацевтически приемлемый носитель» охватывает все стандартные фармацевтические носители, буферы и наполнители, включая фосфатно-солевой буферный раствор, воду и эмульсии (такие как масло-вода или вода-масло), а также различные типы увлажняющих агентов и/или адъювантов. Подходящие фармацевтические носители и их составы описаны в «Remington's Pharmaceutical Sciences» (Mack Publishing Co., Easton, 19th ed. 1995). Предпочтительные фармацевтические носители зависят от предполагаемого способа введения активного агента. Типичные способы введения описаны ниже.
Термин «эффективное количество» обозначает дозировку, достаточную для получения искомого результата в отношении выявленного расстройства, состояния или психического состояния. Искомый результат может включать субъективное или объективное улучшение у реципиента такой дозировки. В отношении боли улучшение может представлять собой уменьшение признаков или симптомов боли.
Термины «лечение», «терапия» и им подобные включают, в частности, способы и операции, осуществляемые для достижения полезных изменений в состоянии здоровья реципиента. Изменения могут быть или субъективными или объективными и могут относиться к таким характеристикам, как симптомы или признаки заболевания, расстройства или состояния, которое необходимо лечить. Например, если пациент замечает уменьшение боли, то было проведено успешное лечение. Например, если наблюдается уменьшение отека, то было проведено полезное лечение воспаления. Аналогичным образом, если врач замечает объективные изменения, такие как улучшенный диапазон движения, то лечение боли или воспаления, которые ухудшали это движение, оказалось полезным. Предотвращение ухудшения состояния реципиента также охватывается этим термином.
Терапевтическая польза включает любой из ряда субъективных или объективных факторов, выявляющих благотворную реакцию или улучшение состояния, которое необходимо лечить, как обсуждается в настоящем документе.
«Фармацевтически приемлемый» или «терапевтически приемлемый» относится к веществу, которое не препятствует эффективности или биологической активности активных ингредиентов и которое не токсично по отношению к хозяину в используемых количествах, причем хозяева могут быть людьми или животными, которым вводится это вещество.
«Терапевтически эффективное количество» обозначает количество активного агента, достаточное для получения искомого биологического или клинического результата. Этот результат может представлять собой ослабление признаков, симптомов или причин заболевания или может быть другим целевым изменением биологической системы. Термин «терапевтически эффективное количество» используется в настоящем изобретении для обозначения любого количества состава, которое вызывает существенное улучшение заболевания, расстройства или состояния при введении объекту. Это количество будет меняться в зависимости от состояния, которое необходимо лечить, стадии прогрессии состояния, а также типа и концентрации применяемого состава. Подходящие количества в любом случае будут очевидны специалистам в данной области техники или их можно определить с помощью стандартного эксперимента.
«Профилактическое лечение» представляет собой лечение пациента, не имеющего явных признаков неврологического или психологического расстройства или состояния или же имеющего только ранние или слабые признаки такого расстройства или состояния, где лечение вводят с целью уменьшения риска развития патологии или ухудшения расстройства или состояния. Соединения по изобретению могут применяться в качестве профилактического лечения для предотвращения ненужных или нежелательных страхов или панических атак или для уменьшения уровня страхов на тот случай, если произойдет ухудшение состояния.
При использовании в данном документе термин «пациент» или «объект» включает любое подвергающееся лечению животное, включая, без ограничения перечисленным, млекопитающих (например, крыс, мышей, кошек, собак), в том числе человека.
При использовании в настоящем изобретении термин «гидрокарбил» означает (C1-С8) углеводородный остаток, который является (C1-С8)алкильным остатком, (C1-C8)алкенильным, (C3-С8)циклоалкильным, (C3-С8)циклоалкельным, (C1-С8)гетероалкильным, (C3-C8)гетероалкенильным, (C3-С8)гетероциклоалкильным, или (C3-C8)гетероциклоалкенильным остатком. В предпочтительном варианте гидрокарбил в каждом случае представляет собой замещенный или незамещенный (C1-C6), (С1-C3) или (C1-С2)гидрокарбил и более предпочтительно незамещенный (С1-C3)алкил. В еще более предпочтительном варианте гидрокарбил в каждом случае представляет собой метил или этил, или трифторметил. Термин «гидрокарбил» также включает группы, содержащие до 1, 2, или 3 атомов гидрокарбильных групп, как они описаны выше, замещенных гетероатомом при условии, что гетероатомы гидрокарбила не располагаются смежно друг с другом и гидрокарбил не присоединен к оставшемуся соединению с помощью гетероатома.
В настоящем изобретении термин «алкил», сам по себе или в виде части другого заместителя, означает, если явно не указано иное, прямую или разветвленную цепь насыщенного углеводородного остатка, содержащего указанное число углеродных атомов (т.е. (C1-C6) означает от одного до шести атомов углерода). Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, н-пентил, н-гексил, н-гептил, н-октил, и так далее.
В настоящем изобретении термин «алкокси» означает алкильный остаток, присоединенный к остальной молекуле через атом кислорода алкокси-группы. Соответственно, примеры алкоксигрупп включают, в частности, метокси, этокси, пропокси и так далее.
Термин «алкенил» берет свое название от соответствующей алкильной группы, но отличается от нее наличием одной или нескольких двойных связей. Аналогично, «алкинильные» группы названы вслед за соответствующими алкильными группами, но отличаются наличием одной или нескольких тройных связей. Неограниченные примеры таких ненасыщенных алкенильных и алкинильных групп включают винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил, а также высшие гомологи и изомеры.
В настоящем изобретении термин «гетероалкил» берет свое название от соответствующей алкильной группы, но отличается тем, что содержит один, два или три гетероатома, независимо выбранных из N, O, и S и замещающих атом углерода алкильной группы. Гетероатомы азота и серы необязательно окислены, а атомы азота необязательно кватернизированы. Гетероалкильная группа присоединена к остатку молекулы через атом углерода гетероалкильной группы, а гетероатомы гетероалкила не располагаются смежно с другими гетероатомами.
Термин «гетероалкенил» берет свое название от соответствующей алкенильной группы, но отличается тем, что имеет 1, 2, или 3 гетероатома, замещающих атом углерода алкенильной группы. Гетероатомы азота и серы необязательно окислены, а атомы азота необязательно кватернизированы. Гетероатом может образовывать двойную связь с углеродным атомом. Гетероалкенильная группа присоединена к остатку молекулы через атом углерода алкила, а гетероатомы алкила не располагаются смежно с другими гетероатомами.
В настоящем изобретении термин «циклоалкил» означает насыщенный моноциклический углеводородный радикал, включающий от около 3 до около 8 атомов углерода, а более предпочтительно - 3-6 атомов углерода. Термин «циклоалкенил» означает моноциклический неароматический углеводородный радикал, включающий от около 5 до около 6 атомов углерода и имеющий по меньшей мере одну двойную связь. Типичные циклоалкильные группы и циклоалкенильные группы включают циклопропил, циклобутил, циклопнтил, циклогексил, циклогексенил, циклогепта-1,3-диенил и так далее.
В настоящем изобретении термин «гетероциклоалкил» означает насыщенный или частично насыщенный углеводородный остаток, включающий от около 3 до около 8 атомов углерода, а более предпочтительно - 3-6 атомов углерода, в которых 1, 2 или 3 из атомов углерода независимо замещены гетероатомом, выбранным из O, N, или S. Атомы азота и серы необязательно окислены, а атом(ы) азота необязательно кватернизированы. Окисленная сера может находиться в форме тио-, сульфинил- или сульфонилгрупп. Термин «гетероциклоалкенил» означает гетероциклоалкильную группу, имеющую по меньшей мере одну двойную связь. Гетероциклоалкильная или гетероциклоалкенильная группа присоединена к остатку молекулы через атом углерода гетероциклоалкильной или гетероциклоалкенильной группы, соответственно; и гетероатомы гетероциклоалкила или гетероциклоалкенила не располагаются смежно с другими гетероатомами гетероциклоалкила или гетероциклоалкенила.
В настоящем изобретении подразумевается, что термин «гетероатом» включает кислород (O), азот (N) и серу (S)).
В настоящем изобретении термин «галоген» или «гало» означает иод (I), бром (Br), хлор (Cl) и/или фтор (F).
Вышеописанный гидрокарбильцый, алкильный, алкенильный, циклоалкильный, циклоалкенильный, гетероалкильный, гетероалкенильный, циклогетероалкильный и циклогетероалкенильный остатки могут быть замещены одним, двумя или тремя заместителями, независимо выбранными из незамещенного (C1-С6) или (С1-C3)алкила, незамещенной (C1-C6) или (С1-C3)алкоксигруппы, незамещенной аминогруппы, незамещенной (C1-С6) или (C1-С3) алкиламиногруппы, ди-(незамещенный (C1-C6) или (C1-C3)алкил)аминогруппы, гидроксигруппы, галогена, незамещенной карбоксамидогруппы, незамещенной (C1-C6) или (С1-C3)алкилкарбоксамидогруппы, оксогруппы и нитрогруппы. Неограничивающие примеры алкоксигрупп включают метокси-, этокси-, трет-бутокси, циклопентилокси-, трифторметокси- и так далее. В настоящем изобретении термин «оксо» означает=0. В настоящем изобретении термин «амино» означает -NH2. В некоторых воплощениях каждая из гидрокарбильных групп является незамещенной. В некоторых воплощениях каждая из углеводородной, алкильной, алкенильной, циклоалкильной, циклоалкенильной, гетероалкильной, гетероалкенильной, циклогетероалкильной и циклогетероалкенильной групп является незамещенной.
Соединение периферически-ограниченного действия представляет собой соединение, которое плохо проникает через гематоэнцефалический барьер или вытесняется из мозга быстрее, чем проникает в него. Соответственно, соединение периферически-ограниченного действия по изобретению может вводиться в дозировках, которые ингибируют активность FAAH в периферической нервной системе в гораздо большей степени, чем в центральной нервной системе (например, в мозге). В некоторых воплощениях ингибитор FAAH по изобретению вводят подкожно, внутривенно или перорально для ингибирования периферической активности FAAH (например, в печени) в концентрации ED50, которая составляет не более чем ¼, 1/8, или 1/10 от ED50 для ингибирования активности FAAH в мозге мыши. В предпочтительном варианте ингибитор FAAH периферически-ограниченного действия уменьшает активность FAAH в периферической нервной системе по меньшей мере в 3, 4, 5, 7, 8 или 10 раз сильнее, чем в центральной нервной системе (например, в мозге) млекопитающего. Например, активность FAAH на периферии может ингибироваться на 80% (т.е. остается 20% от исходного неингибированного уровня активности FAAH), в то время как активность FAAH в центральной нервной системе будет ингибироваться на 10% (остается 90% от исходного не ингибированного уровня активности FAAH), обеспечивая 80%/10% или 8-кратное различие в ингибировании FAAH.
Физиологически расщепляемый сложный эфир представляет собой сложный эфир, который является субстратом для карбоксиэстераз in vivo. Физиологически расщепляемые сложные эфиры, как правило, быстро гидролизуются, так что концентрация соответствующего спирта превышает концентрацию эфира в крови или плазме. Например, физиологически расщепляемый сложный эфир представляет собой эфир, который быстро гидролизуется до соответствующего спирта и кислоты in vivo за полупериод, составляющий менее чем ¼, 1,2 или 3 часа в терапевтически разумных дозировках.
Соединения по изобретению
Соединения по изобретению соответствуют формуле:
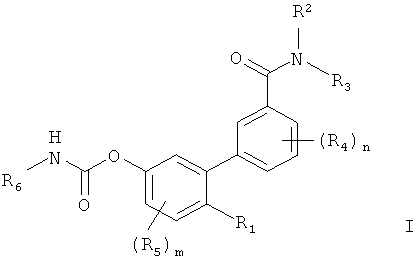
в которой R1 представляет собой полярную группу. В некоторых воплощениях R1 выбирают из группы, состоящей из гидроксигруппы и ее физиологически гидролизуемых сложных эфиров, -SH, -О-карбоксамидо, -OC(O)R7, -O-CO-NR8R9 и -NR8R9, где R7 представляет собой замещенный или незамещенный гидрокарбил, a R8 и R9 независимо выбирают из группы, состоящей из водорода и замещенного или незамещенного гидрокарбила; R2 и R3 независимо выбирают из группы, состоящей из водорода и замещенного или незамещенного гидрокарбила; каждый R4 независимо выбирают из группы, состоящей из галогена и замещенного или незамещенного гидрокарбила, а n представляет собой целое число от 0 до 4; каждый R5 независимо выбирают из группы, состоящей из галогена или замещенного или незамещенного гидрокарбила, a m представляет собой целое число от 0 до 3; и R6 представляет собой замещенный или незамещенный циклогексил; а также их фармацевтически приемлемые соли. В некоторых воплощениях каждый из R2, R3, R7, R8 и R9 независимо выбирают из водорода и незамещенного гидрокарбила. В некоторых других воплощениях каждый из R2, R3, R7, R8, и R9 независимо представляет собой водород или незамещенный C1-С3 гидрокарбил. В других воплощениях каждый из R4 и R5 независимо представляет собой галоген или C1-С3 гидрокарбил. В предпочтительном варианте действие вышеописанных соединений по изобретению ограничено периферической нервной системой.
В некоторых воплощениях, которые применимы ко всему вышесказанному, m равно 0, а n равно 0, 1, 2, 3 или 4. В других воплощениях m равно 1, а n равно 0, 1, 2, 3 или 4. В еще одних воплощениях m равно 2, а n равно 0, 1, 2, 3 или 4. Еще в одних воплощениях m равно 3, а n равно 0, 1, 2, 3 или 4. В некоторых воплощениях сумма шип равна 0, 1, 2 или 3. В других воплощениях каждый гидрокарбил является незамещенным.
В предпочтительном варианте R1 представляет собой гидроксигруппу или ее физиологически гидролизуемый сложный эфир. Такие сложные эфиры включают эфиры формулы -OC(O)R7, где R7 представляет собой замещенный или незамещенный гидрокарбил, более предпочтительно замещенный или незамещенный алкил, алкенил, циклоалкил, гетероалкил, гетероциклоалкил, гетероалкенил, гетероциклоалкенил и циклоалкенил, а еще более предпочтительно замещенный или незамещенный (С1-C3)алкил (например, метил, этил, пропил, трифторметил) или замещенный или незамещенный (C1-С3) гидрокарбил, выбранный из алкенила, циклоалкила, гетероалкила, гетероциклоалкила, гетероалкенила, гетероциклоалкенила и циклоалкенила. В некоторых из этих воплощений m равно 0, а n равно 0, 1 или 2; m равно 1, а n равно 0, 1 или 2; или m равно 2, a n равно 0,1 или 2.
В других воплощениях, которые применимы ко всему вышесказанному, R2 и R3 представляют собой водород или замещенный или незамещенный (С1-C3) гидрокарбил, выбранный из алкила, алкенила, циклоалкила, гетероалкила, гетероциклоалкила, гетероалкенила, гетероциклоалкенила и циклоалкенила. В еще одних таких воплощениях m равно 0, а n равно 0, 1 или 2; m равно 1, а n равно 0, 1 или 2; или m равно 2, a n равно 0, 1 или 2. В некоторых воплощениях по меньшей мере один или оба из R2 и R3 представляют собой водород. В некоторых из этих воплощений m равно 0, a n равно 0, 1 или 2; m равно 1, a n равно 0, 1 или 2; или m равно 2, а n равно 0, 1 или 2. В других воплощениях гидрокарбил из R2 и/или R3 является незамещенным.
В еще одних воплощениях, которые применимы ко всему вышеуказанному, R1 представляет собой гидрокси и по меньшей мере один из R2 и R3 представляет собой водород. В некоторых из этих воплощений и R2, и R3 представляют собой водород. В других воплощениях, в которых R1 представляет собой водород, R2 и R3 независимо выбирают из замещенного или незамещенного (C1-C3)алкила (например, метила, этила, пропила) и H. В еще одних из таких воплощений m равно 0, а п равно 0, 1 или 2; m равно 1, а n равно 0, 1 или 2; или m равно 2, а n равно 0, 1 или 2.
В других воплощениях, которые применимы ко всему вышесказанному, R6 представляет собой замещенный или незамещенный циклогексил. Заместители для циклогексила включают алкил (например, метил, этил), галоген (F, Cl, I, Br и предпочтительно F или Cl) и трифторметил. В некоторых из этих воплощений m равно 0, а n равно 0, 1 или 2; m равно 1, а n равно 0, 1 или 2; или m равно 2, a n равно 0, 1 или 2.
В других воплощениях, которые применимы ко всему вышесказанному, R4 представляет собой замещенный или незамещенный алкил, алкенил, циклоалкил, гетероалкил, гетероциклоалкил, гетероалкенил, гетероциклоалкенил или циклоалкенил, а более предпочтительно - замещенный или незамещенный (С1-C3) алкил (например, метил, этил, пропил, трифторметил) или замещенный или незамещенный (C1-С3) гидрокарбил, выбранный из алкенила, циклоалкила, гетероалкила, гетероциклоалкила, гетероалкенила, гетероциклоалкенила и циклоалкенила. В других воплощениях R4 выбирают из (C1-С3) алкила (например, метила, этила, пропила), а n равно 0, 1, 2 или 3. В других воплощениях каждый из R4 представляет собой галоген или алкилгалогенид (например, трифторметил). В других воплощениях каждый из R4 представляет собой галоген или незамещенный (C1-C3)алкил (например, метил, этил, пропил). В других таких воплощениях m равно 0 или 1.
В других воплощениях, которые применимы ко всему вышесказанному, R5 представляет собой замещенный или незамещенный алкил, алкенил, циклоалкил, гетероалкил, гетероциклоалкил, гетероалкенил, гетероциклоалкенил или циклоалкенил, а более предпочтительно - замещенный или незамещенный (C1-C3)алкил (например, метил, этил, пропил, трифторметил) или замещенный или незамещенный (C1-С3) гидрокарбил, выбранный из алкенила, циклоалкила, гетероалкила, гетероциклоалкила, гетероалкенила, гетероциклоалкенила и циклоалкенила. В других воплощениях любого из вышеуказанного R5 выбирают из (С1-C3)алкила (например, метила, этила, пропила), a m равно 1, 2 или 3. В других воплощениях каждый из R5 представляет собой галоген или алкилгалогенид (например, трифторметил). В еще одних воплощениях каждый из R5 представляет собой галоген или незамещенный (C1-C3)алкил (например, метил, этил, пропил). В еще одних воплощениях n равно 0 или 1.
В особенно предпочтительных вариантах осуществления изобретения Ri представляет собой гидроксигруппу или ее физиологически гидролизуемый сложный эфир, где гидролиз высвобождает соответствующее соединение, где R1 представляет собой гидрокси, R6 представляет собой незамещенный циклогексил, m равно 0, a n равно 90, 1 или 2; или m равно 1, an равно 0, 1 или 2, или m равно 2, a n равно 0, 1 или 2. В некоторых воплощениях сложный эфир имеет формулу -OC(O)R7, где R7 представляет собой замещенный или незамещенный гидрокарбил, более предпочтительно замещенный или незамещенный алкил, алкенил, циклоалкил, гетероалкил, гетероциклоалкил, гетероалкенил, гетероциклоалкенил или циклоалкенил, а более предпочтительно-замещенный или незамещенный (C1-C3) алкил (например, метил, этил, пропил, трифторметил) или замещенный или незамещенный (C1-C3) гидрокарбил, выбранный из алкенила, циклоалкила, гетероалкила, гетероциклоалкила, гетероалкенила, гетероциклоалкенила и циклоалкенила. В некоторых других воплощениях R7 представляет собой незамещенный гидрокарбил, незамещенный алкил, незамещенный алкенил, незамещенный циклоалкил, незамещенный гетероалкил, незамещенный гетероциклоалкил, незамещенный гетероалкенил, незамещенный гетероциклоалкенил или незамещенный циклоалкенил; или незамещенный (C1-С3) алкил (например, метил, этил, пропил, трифторметил) или незамещенный (C1-C3) гидрокарбил, выбранный из алкенила, циклоалкила, гетероалкила, гетероциклоалкила, гетероалкенила, гетероциклоалкенила и циклоалкенила.
В особенно предпочтительном воплощении соединение имеет формулу:

3'-карбамоил-6-гидроксибифенил-3-ил циклогексилкарбамат
или представляет собой физиологически гидролизуемый эфир этого соединения, из которого при гидролизе высвобождается 3'-карбамоил-6-гидроксибифенил-3-ил циклогексилкарбамат, а также его фармацевтически приемлемые соли.
В предпочтительных воплощениях действие всех вышеописанных соединений ограничено периферической нервной системой.
Соединения по изобретению могут содержать один или несколько асимметричных центров и таким образом могут существовать в виде рацемических смесей, индивидуальных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. Подразумевается, что настоящее изобретение включает все такие изомерные формы соединений по изобретению.
Соединения по изобретению включают любые диастереомеры или пары любых энантиомеров. Диастереомеры, например, могут быть получены с помощью фракционной кристаллизации из подходящего растворителя, например, метанола или этилацетата или их смеси. Полученная таким образом пара энантиомеров может быть разделена на индивидуальные стереоизомеры с помощью традиционных способов, например с использованием оптически активной кислоты в качестве растворяющего агента.
Также любой энантиомер такого соединения по изобретению может быть получен с помощью стереоспецифического синтеза с использованием оптически чистых реагентов известной конфигурации.
Соединения по настоящему изобретению могут иметь не встречающиеся в природе соотношения атомных изотопов для одного или нескольких из атомов. Например, соединения могут быть радиоактивно-меченными изотопами, как тритий или углерод-14. Все изотопные вариации соединений по настоящему изобретению, как радиоактивные, так и нет, находятся в рамках настоящего изобретения.
Соединения по настоящему изобретению могут быть выделены в форме их фармацевтически приемлемых аддитивных солей с кислотами, например в форме солей, полученных с использованием неорганических или органических кислот. Такие кислоты могут включать соляную, азотную, серную, фосфорную, муравьиную, уксусную, трифторуксусную, пропионовую, малеиновую, янтарную, малоновую и т.п. Кроме того, некоторые соединения, имеющие кислотную функциональность, могут быть представлены в форме их неорганической соли, в которой противоион может быть выбран из натрия, калия, лития, кальция, магния и так далее, а также из органических оснований. Термин «фармацевтически приемлемые соли» означает соли, полученные из фармацевтически приемлемых нетоксичных оснований или кислот, включающих неорганические основания или кислоты и органические основания или кислоты.
Изобретение также охватывает пролекарственные формы настоящих соединений, которые при введении, перед тем как стать активными фармакологическими агентами подвергаются химическому превращению в метаболических процессах. Вообще говоря, такие пролекарственные средства могут представлять собой производные соединений по настоящему изобретению, которые легко превращаются in vivo в функциональное соединение по изобретению. Стандартные процедуры для выбора и получения подходящих пролекарственных производных описаны, например, в «Design of Prodrugs)), ed. H. Bundgaard, Elsevier, 1985. Настоящее изобретение также охватывает активные метаболиты соединений по настоящему изобретению.
Некоторые из соединений, описанных в данном изобретении, содержат олефиновые двойные связи и до тех пор, пока не указано иное, подразумевается, что они включают оба геометрических изомера, E и Z.
Некоторые из соединений, описанных в данном изобретении, могут существовать в формах с различными местами присоединения водорода, что обозначается термином «таутомеры». Таким примером может быть кетон и его енольная форма, которые известны как кето-енольные таутомеры. Индивидуальные таутомеры, а также их смеси охватываются формулами соединений по изобретению.
Высокопроизводительный анализ ингибирования FAAH
Исследования соединений, описанные в данном документе, пригодны для высокопроизводительного скрининга. Таким образом, предпочтительные анализы отслеживают связывание ингибитора с FAAH или высвобождение продукта реакции (например, амида жирной кислоты или этаноламина), получаемого в ходе гидролиза субстрата, такого как олеоил этанол амид или анандамид. Субстрат может быть помечен для облегчения обнаружения высвобождаемых продуктов реакции. Высокопроизводительные анализы на присутствие, отсутствие или для количественной оценки конкретных продуктов реакции хорошо известны специалистам в данной области техники. Так, например, патент США No. 5,559,410 раскрывает высокопроизводительные методы скрининга белков, а Патенты США No. 5,576,220 и No. 5,541,061 раскрывают высокопроизводительные методы скрининга связывания лиганд/антитело.
Кроме того, коммерчески доступны высокопроизводительные системы скрининга (см., например, «Zymark Согр.», Хопкинтон, Массачусетс; «Air Technical Industries)), Ментор, Огайо; «Весктап Instruments, Inc.» Фуллертон, Калифорния; «Precision Systems, Inc.», Натик, Массачусетс, и т.п.). В этих системам, как правило, все процессы, включая забор образцов и реактивов, диспергирование в жидкости, инкубацию по времени и финальное считывание микропланшета на детекторе, соответствующем осуществляемому анализу, автоматизированы. Эти конфигурируемые системы позволяют осуществлять высокопроизводительный и быстрый запуск, а также обеспечивают высокую степень гибкости и настройки. Производители таких систем предоставляют подробные протоколы для различных анализов. Так, например, «Zymark Согр.» предоставляет технические бюллетени, описывающие системы скрининга для детекции модуляции транскрипции гена, связывания лиганда и т.п.
Способы скрининга соединений на антиноцицептивную активность.
Способы скрининга ингибиторов FAAH на антиноцицептивный эффект хорошо известны специалистам в данной области. Например, тестируемые соединения могут быть введены объектам-животным в тесте горячей пластинки для мышей, в тесте с формалином для мышей и затем могут быть измерены ноцицептивные реакции на термическое или химическое поражение тканей. См. также патент США No. 6,326,156, в котором раскрыты способы скрининга антиноцицептивной активности. См. Cravatt et al. Proc. Natl. Acad. Sci. U.S.A. 98:9371-9376 (2001).
Фармацевтические композиции
В настоящем изобретении также описываются фармацевтические композиции вышеописанных соединений-ингибиров FAAH периферически-ограниченного действия. Термин «композиция», как он используется в фармацевтической композиции, предназначен для описания продукта, включающего активный ингредиент(ы) и любой инертный ингредиент(ы), который представляет собой носитель, а также любого продукта, получаемого прямо или косвенно в результате объединения, комплексообразования или агрегации любых двух или нескольких ингредиентов, или в результате диссоциации одного или нескольких ингредиентов, или в результате реакций других типов, или в результате взаимодействий одного или нескольких ингредиентов. Соответственно, фармацевтические композиции по настоящему изобретению охватывают любую композицию, полученную путем смешивания соединения по настоящему изобретению и фармацевтически приемлемого носителя. Термин «фармацевтическая композиция» служит для обозначения композиции, подходящей для фармацевтического использования для пациента, включая животное или человека. Фармацевтическая композиция, как правило, включает эффективное количество активного агента и фармацевтически приемлемый носитель.
Композиция включает композиции, подходящие для перорального, ректального, местного, парентерального (включая подкожное введение, внутримышечное и внутривенное), окулярное (офтальмическое), пульмональное (назальную или буккальную ингаляцию) и назальное введение, хотя наиболее подходящий путь в любом конкретном случае будет зависеть от характера и тяжести состояний, подвергаемых лечению, а также от свойств активного ингредиента. Типичным путем введения является пероральный. Композиции по изобретению могут быть традиционно иметь вид единичной лекарственной дозировки и быть получены способами, хорошо известными в области фармацевтики.
Для практического применения соединения по изобретению могут быть объединены, как активный ингредиент, с фармацевтическим носителем согласно стандартным фармацевтическим способам составления однородных смесей. Носитель может иметь широкий спектр форм в зависимости от формы препарата, целесообразной для введения, например перорального или парентерального (включая внутривенное). В получении композиций для пероральной лекарственной формы в случае пероральных жидких препаратов, таких как, например, суспензии, эликсиры и растворы может применяться любая из обычных фармацевтических сред, такая как, например, вода, гликоли, масла, спирты, отдушки, консерванты, красители и т.п.; а в случае пероральных твердых препаратов, таких как, например, порошки, твердые и мягкие капсулы и таблетки - могут применяться обычные фармацевтические носители, такие как крахмал, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазки, связующие агенты, разрыхлители и так далее, причем твердые пероральные препараты являются предпочтительными по отношению к жидким препаратам.
Ввиду простоты введения таблетки и капсулы представляют собой наиболее предпочтительные пероральные лекарственные формы. В таком случае обычно применяются твердые фармацевтические носители. При необходимости таблетки могут быть покрыты стандартными водными или безводными методами. Такие композиции и препараты могут содержать по меньшей мере 0,1 процент активного соединения. Процент активного соединения этих композиций может, конечно, варьироваться и обычно находится в интервале от около 2 процентов до около 60 процентов в пересчете на единицу массы лекарственной формы. Количество активного соединения в таких терапевтических композициях должно быть таким, чтобы получаемая дозировка была терапевтически эффективной. Активные соединения также могут вводиться интраназально, например в виде жидких капель или спрея.
Таблетки, пилюли, капсулы и т.п. также могут содержать связующий агент, такой как трагакантовая камедь, аравийская камедь, кукурузный крахмал или желатин; наполнители, такие как дикальция фосфат; разрыхлитель, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота; смазывающее вещество, такое как стеарат магния; и подсластитель, такой как сахароза, лактоза или сахарин. Когда единичная лекарственная форма представляет собой капсулу, она может дополнительно к материалам, перечисленным выше, содержать жидкий носитель, такой как жирное масло.
Различные другие материалы могут присутствовать в качестве покрытий или для модификации физической формы лекарственной формы. Например, таблетки могут быть покрыты шеллаком, сахаром или и тем, и другим. Сироп или эликсир может содержать дополнительно к активному ингредиенту сахарозу в качестве подсластителя, метил- и пропилпарабены в качестве консервантов, краситель и ароматизатор, такой как вишневый или апельсиновый коррегент. Для предотвращения распада в процессе прохождение через верхние пути ЖК-тракта, композиция может представлять собой состав, покрытый кишечнорастворимой оболочкой.
Что касается составов и разнообразия путей введения, то способы и составы для введения лекарственных средств раскрыты в Remington's Pharmaceutical Sciences, 17th Edition, (Gennaro et al. Eds., Mack Publishing Co., 1985). Remington's Pharmaceutical Sciences, Gennaro AR ed. 20th edition, 2000: Williams & Wilkins PA, USA.
Введение
Соединения по изобретению также могут вводиться парентерально. Растворы и суспензии этих активных соединений могут быть получены в воде, подходящим образом смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии также могут быть получены в глицерине, жидких полиэтиленгликолях, и их смесях в масле. В обычных условиях хранения и применения эти препараты могут содержать консервант для предотвращения роста микроорганизмов.
Фармацевтические формы, подходящие для инъекционного введения, включают стерильные водные растворы или дисперсии, а также стерильные порошки для изготовления стерильных инъецируемых растворов или дисперсий по мере надобности. Во всех случаях форма должна быть стерильной и должна быть настолько жидкой, насколько это необходимо для введения шприцом. Она должна быть стабильной в условиях изготовления и хранения и должна быть ограждена от загрязняющего воздействия микроорганизмов, таких как бактерии и грибы. Носитель может быть растворителем или дисперсионной средой, содержащей, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль и т.п.) и их подходящие смеси, а также растительными маслами.
Соединения по изобретению могут быть эффективны в широком спектре дозировок. Например, при лечении взрослого человека, могут быть необходимы дозировки от около 10 до около 1000 мг, от около 100 до около 500 мг или от около 1 до около 100 мг. Могут использоваться суточные дозы от 0,05 до около 100 мг, а более предпочтительно - от около 0,1 до около 100 мг. Наиболее предпочтительная дозировка составляет от около 0,1 мг до около 70 мг в день. При выборе подходящего для пациента режима часто может быть необходимо начинать с дозировки от около 2 до около 70 мг в день, и когда состояние находится под контролем - уменьшать дозировку до уровня от около 0,1 до около 10 мг в день. Например, при лечении взрослого человека могут использоваться дозировки от около 0,05 до около 100 мг, предпочтительно, от около 0,1 до около 100 мг в день. Точная дозировка будет зависеть от способа введения, целей терапии, формы введения, пациента, подвергаемого лечению, и от массы тела пациента, подвергаемого лечению, а также от предпочтений и опыта компетентного врача или ветеринара.
Как правило, соединения по настоящему изобретению могут быть разделены на единичные лекарственные формы, содержащие предпочтительно от около 0,1 до около 100 мг активного ингредиента вместе с фармацевтически приемлемым носителем на единицу дозировки. Обычно лекарственные формы, подходящие для перорального, назального, пульмонального или трансдермального введения, включают от около 0,001 мг до около 100 мг, предпочтительно от около 0,01 мг до около 50 мг соединений по изобретению, смешанных с фармацевтически приемлемым носителем или разбавителем. Для хранения и применения эти препараты предпочтительно содержат консервант, предотвращающий рост микроорганизмов.
Введение соответствующего количества соединения-кандидата может осуществляться с помощью любых способов, известных в данной области техники, например перорально, ректально, парентерально, внутрибрюшинно, внутривенно, подкожно, субдермально, интраназально или внутримышечно. В некоторых воплощениях используется трансдермальное введение. Подходящее количество соединения-кандидата или его доза могут определяться эмпирически, как известно в данной области техники. Подходящее терапевтическое количество - это количество, достаточное для достижения целевого терапевтического эффекта (например, лечения или ослабления боли или лечения или уменьшения воспаления). Соединение-кандидат может вводиться так часто, как это требуется для облегчения боли или для уменьшения воспаления, например раз в час, каждые шесть часов, каждые восемь, двенадцать или восемнадцать часов, ежедневно или раз в неделю.
Составы, подходящие для перорального введения, могут состоять из (a) жидких растворов, таких как эффективное количество упакованной нуклеиновой кислоты, суспендированной в разбавителях, таких как вода, солевой раствор или PEG 400; (b) капсул, саше или таблеток, каждая из которых содержит определенное количество каждого ингредиента, в виде жидкостей, твердых веществ, гранул или желатина; (c) суспензий в соответствующей жидкости; и (d) подходящих эмульсий. Таблетированные формы могут включать один или несколько компонентов из числа лактозы, сахарозы, маннита, сорбита, фосфата кальция, кукурузного крахмала, картофельного крахмала, микрокристаллической целлюлозы, желатина, коллоидного диоксида кремния, талька, стеарата магния, стеариновой кислоты и других вспомогательных веществ, красителей, наполнителей, связующих агентов, разбавителей, буферных агентов, увлажнителей, консервантов, отдушек, красителей, разрыхлителей и фармацевтически приемлемых носителей. Лекарственные формы в виде пастилок могут включать активный ингредиент во вкусовом агенте, например в сахарозе, а также пастилки могут включать активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и эмульсии аравийской камеди, гели и так далее, содержащие дополнительно к активному ингредиенту носители, известные в данной области техники.
Инъекционные растворы и суспензии могут быть получены из стерильных порошков, гранул и таблеток, описанного выше типа. Составы, подходящие для парентерального введения, например с помощью внутрисуставного (в сустав), внутривенного, внутримышечного, интрадермального, внутрибрюшинного и подкожного пути, включают водные и неводные, изотонические стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатики и растворы, придающие составу изотоничность к крови предполагаемого реципиента, а также водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты, солюбилизаторы, загустители, стабилизаторы и консерванты.
Что касается трансдермального пути введения, то способы трансдермального введения лекарственных средств раскрыты в Remington's Pharmaceutical Sciences, Gennaro AR ed. 20th edition, 2000: Williams & Wilkins PA, USA. Дермальные или кожные пластыри представляют собой предпочтительные средства для чрескожной доставки соединений по изобретению. Пластыри предпочтительно обеспечивают усилитель абсорбции, такой как ДМСО, для усиления абсорбции соединений. Другие методы чрескожной доставки лекарственных средств раскрыты в патентах США No. 5,962,012, 6,261,595, и 6,261,595. Каждая из этих публикаций в полном объеме включена в данный документ посредством ссылки.
Предпочтительные пластыри включают такие пластыри, которые контролируют скорость доставки лекарственного средства в кожу. Пластыри могут обеспечивать разнообразные системы дозирования, включающие «резервуарную» систему или «монолитную» систему, соответственно. Резервуарная модель может, например, иметь четыре слоя: адгезионный слой, который непосредственно контактирует с кожей, контрольная мембрана, которая контролирует диффузию молекул лекарственного средства, резервуар молекул лекарственного средства и водостойкое покрытие.
Такая модель поставляет одинаковые количества лекарственного средства в течение определенного периода времени, причем скорость доставки должна быть меньше чем предел насыщения различных типов кожи.
Монолитная модель, как правило, например, имеет только три слоя: адгезионный слой, полимерная матрица, содержащая соединение, и водостойкое покрытие. Эта модель несет насыщающее кожу количество лекарственного средства. Таким образом, скорость доставки контролируется кожей. При уменьшении количества лекарственного средства в пластыре до количества ниже уровня предела насыщения скорость доставки падает.
Соединения по изобретению могут использоваться в комбинации с другими соединениями по изобретению или с другими лекарственными средствами, которые также могут быть полезны для лечения, предотвращения или в подавления боли, воспаления или иммунных расстройств. В одном воплощении второе лекарственное средство не является ингибитором FAAH, но его действие направлено на то же расстройство, что и ингибитор FAAH. Такие лекарственные средства могут вводиться с помощью того пути введения и в таком количестве, какие стандартно используются в их случае, одновременно или последовательно с соединением по изобретению. Когда соединение по изобретению используется одновременно с одним или несколькими другими лекарственными средствами, то предпочтительной является фармацевтическая композиция в единичной лекарственной дозировке, содержащая как соединение по изобретению, так и эти другие лекарственные средства. При использовании в комбинации с одним или несколькими активными ингредиентами соединение по настоящему изобретению и другие активные ингредиенты могут использоваться в более низких дозах, чем когда каждый из них используется отдельно. Соответственно, фармацевтические композиции по настоящему изобретению включают такие композиции, которые содержат один или более других активных ингредиентов в дополнение к соединениям, раскрытым выше.
В фармацевтических композициях по настоящему изобретению для перорального, подъязычного, подкожного, внутримышечного, внутривенного, трансдермального, местного или ректального введения активное начало само по себе или в ассоциации с другим активным началом может вводиться животным и людям в единичных дозированных формах, смешанных со стандартными фармацевтическими носителями. Подходящие единичные дозированные формы включают формы для перорального введения, такие как таблетки, желатиновые капсулы, порошки, гранулы и пероральные суспензии или растворы, подъязычные и буккальные формы, аэрозоли, импланты, подкожные, внутримышечные, внутривенные, интраназальные или интраокулярные и ректальные формы.
В другом воплощении фармацевтические композиции по настоящему изобретению, активное начало или активные начала, как правило, включены в состав стандартных лекарственных форм. Стандартная лекарственная форма содержит от 0.5 до 1000 мг, предпочтительно от 1 до 500 мг, и более предпочтительно от 2 до 200 мг ингибитора FAAH на единичную лекарственную форму для ежедневного введения.
Способы лечения
Контроль боли
В некоторых воплощениях соединения Формулы I и II могут вводиться для облегчения или для лечения боли у пациента. Лечение может быть профилактическим или терапевтическим. Пациент, получающий лечение может быть человеком. Соединения и композиции по изобретению могут вводиться индивидуально в целях уменьшения тяжести или частоты или степени боли. Лечение может вводиться в комбинации с другим обезболивающим средством или противовоспалительным агентом. В некоторых воплощениях боль может быть невропатической болью, выбранной из группы, состоящей из посттригеминальной невралгии, невропатической боли в нижней части спины, периферической или полиневропатической боли, комплексного регионарного болевого синдрома (каузалгии и симпатической рефлекторной дистрофии), диабетической невропатии, токсической невропатии и хронической невропатии, вызванной химиотерапевтическими агентами. В других воплощениях боль является почечными или печеночными коликами или фибромиалгией. В некоторых воплощениях, связанных с невропатической болью, первичное поражение или дисфункция нервной системы вызывается механическим повреждением нерва пациента. В некоторых из таких воплощений механическое повреждение обусловлено компрессией нерва, рассечением нерва, каузалгией, повреждением спинного мозга, постхирургической болью, фантомной болью или образованием рубца у пациента.
В других воплощениях боль вызвана воспалением или повреждением ткани. Воспалительная боль развивается в ответ на поражение ткани, вызванное вредоносными стимулами. В ответ на повреждение ткани высвобождаются цитокины и другие медиаторы, что усиливает ноцицепцию. Как результат, в месте повреждения возникает первичная гипералгезия (повышенная чувствительность к боли), а в ткани, окружающей участок повреждения, возникает вторичная гипералгезия. Гипералгезия убывает вместе с воспалением при заживлении ткани. В некоторых других воплощениях воспаление ассоциировано с отеком легких, почечными камнями, незначительными травмами, заживлением ран, заживление ран на коже, вагинитом, кандидозом, поясничным спондилоартрозом, сосудистыми заболеваниями, головными болями при мигрени, синусовой головной болью, тензионной головной болью, зубной болью, узелковым периартеритом, тиреоидит, апластической анемией, заболеванием Ходжкина, болезнью Рейно, ревматическим полиартритом, диабетом I типа, диабетом II типа, миастенией гравис, рассеянным склерозом, саркоидозом, нефротическим синдромом, синдромом Бехчета, полиомиозитом, гингивитом, гиперчувствительностью, посттравматическим отеком или ишемией миокарда или остеоартритом.
Контроль воспаления
В некоторых воплощениях соединения формул I и II могут использоваться для облегчения воспаления у объекта. Лечение может быть профилактическим или терапевтическим. Пациентом, получающим лечение, может быть человек. Соединения и композиции по изобретению могут вводиться индивидуально в целях уменьшения тяжести или частоты или степени воспаления. Лечение может проводиться в комбинации с другим обезболивающим средством или с противовоспалительным агентом.
Контроль иммунологических нарушений
Следующие примеры представлены в иллюстративных целях и не предназначены для ограничения объема притязаний изобретения, раскрытого в данном документе. Любые вариации иллюстративных примеров изделий и/или способов, которые могут прийти на ум специалисту в данной области техники, также подпадают под объем притязаний настоящего изобретения.
Примеры
Материалы и методы
Коэффициенты распределений лекарственных средств. Авторы при комнатной температуре (25±1°C) определяли значения log D7,4,oct, отражающие разделение растворенных веществ между н-октанолом и водным раствором, содержащим буфер с pH 7,436.
Анализ ферментов. Авторы проводили стандартные анализы FAAH и моноацилглицерин-липазы, как описано в 15 и 31, с использованием в качестве субстратов [3H]-анандамида (подарок Национального института по проблемам злоупотребления наркотиками) и 2-олеоил-sn-глицерина («Nu-Check Ргер», Элизиан, Миннесота), соответственно.
Анализ транспорта лекарственных средств. Анализы были проведены в «Cerep Inc.» (Редмонд, Вашингтон), согласно протоколам, изложенным на веб-сайте компании (http://wvvw.cerep.fr).
Анализ тканей. Авторы проводили экстракцию из тканей и анализ эндоканабиноидов методом жидкостной хроматографии/масс-спектрометрии, как описано в 38. Похожие процедуры с ЖХ/МС использовались для экстракции из тканей и количественной оценки URB937, как подробно описано ниже.
Экспрессия Fos. Авторы измеряли уровень белка Fos методом количественной иммуноцитохимии5 на срезах поясничного (L4/L5) спинного мозга самцов крыс Sprague-Dawley.
Хирургическое вмешательство. Авторы проводили перевязывание седалищных нервов у самцов мышей Swiss, как было описано для крыс24 с минимальными модификациями39.
Поведенческие тесты. Авторы измеряли защитные реакции, вызванные в.б. инъекцией уксусной кислоты у самцов мышей Swiss и C57B 1/6 (дикого типа или дефектных по FAAH)40, внутриподошвенной инъекцией каррагинана у самцов мышей Swiss20, внутриподошвенной инъекцией формалина у самцов крыс Sprague-Dawley41 и перевязыванием седалищного нерва у самцов мышей Swiss39.
Синтез ингибиторов FAAH URB937 синтезировали в основном следуя опубликованной процедуре36. Соединение получали в пять стадий начиная с 3-бромо-4-гидроксибензальдегида, который бензилировали (BzCl, DMF, CsCO3, КТ, 3 ч), затем окисляли и гидролизовали (m-CPBA, CH2Cl2, 40°C, 72 ч; NaOMe, EtOH, КТ, 1 ч) до 4-бензилокси-3-бромфенола; последний дорабатывали сочетанием по Сузуки [3-карабмоилфенилбориновая кислота, толуол, Pd(PPh3)4, Na2CO3/H2O, дефлегмация, 2 ч], карбамированием (с-C6H11CNO), Et3N, толуол/CH3CN 1:1, дефлегмация, 18 ч) и гидрогенизационным снятием защиты с получением искомого соединения. N-циклогексил-O-бифенил-3-ил карбаматы 1c-e синтезировали реакцией подходящего 3'-карбамоил-4-замещенного фенола с циклогексил изоцианатом, тогда как 1f получали катализированной Pd/C гидрогенизацией соответствующего предшественника, который был получен из подходящего фенольного производного. Все бифенолы были синтезированы реакцией перекрестного сочетания по Сузуки между 3-карбамоил фенил бориновой кислотой и соответствующими 3-бромо-4-замещенными фенолами (в случае предшественников 1c,d) или 3-хлор-4-фторфенол (для 1e). Подробные методики синтеза всех соединений будут представлены далее.
Синтез 3'-карбамоил-6-гидроксибифенил-3-илового эфира циклогексилкарбаминовой кислоты (URB937). К перемешиваемой суспензии 3'-карбамоил-6-бензилоксилбифенил-3-илового эфира циклогексилкарбаминовой кислоты (222 мг; 0,5 ммоль) в EtOAc (2.5 мл) и EtOH (2.5 мл) добавляли 10% Pd/C (22 мг). Смесь гидрогенировали при 4 атм и 50°C в течение 4 часов, охлаждали, фильтровали на целите и концентрировали. Очистка остатка колоночной хроматографией (циклогексан/EtOAc 1:9) и рекристаллизация дали URB937 в виде белого твердого вещества. Выход: 92% (0,163 г). Тп: 128-130°C (CH2Cl2/н-гексан). MS (ESI) m/z: 355,2 (М+Н+). 1H ЯМР (200 МГц, CDCl3) δ:=1,13-2,02 (m, 10H), 3,55 (m, 1Н), 5,13 (br d, 1H), 5,85 (br s, 1H), 6,59 (br s, 1H), 6,74-6,95 (m, 3H), 7,07 (s, 1H), 7,34-7,41 (m, 1H), 7,56 (m, 1H), 7,68-7,75 (m, 2H) ppm. ИК (Nujol)nmax: 3333, 1701, 1655 см-1.
Другие химические вещества. [3H]-Анандамид был приобретен у «American Radiolabeled Chemicals, Inc.» (Сент-Луис, Миссури). 2-[2H8]-AG и АМ630 - у «Cayman Chemical» (Энн-Арбор, Мичиган). Анандамид, [2Н4]-анандамид и PEA были синтезированы в лаборатории42. Римонабант и N-циклогексил бифенил-3-илацетамид являются подарками Национального института по проблемам злоупотребления наркотиками и «Kadmus Pharmaceuticals Inc.», соответственно.
Животные Авторы использовали самцов мышей Swiss Webster («Charles River», 20-30 г), самцов C57B 1/6 («Jackson Laboratory)), 20-25 г), самцов мышей с дефектом FAAH (25-35 г) обратноскрещенных более 10 раз с исходными C57B 1/6, самцов крыс Wistar («Charles River», 250-300 г) и самцов крыс Sprague-Dawley (SD) («Harlan Laboratories)), 275-350 г). Мышей и крыс Wistar рассаживали по группам в стандартные клетки при комнатной температуре с циклом дня и ночи 12:12 ч с неограниченным доступом к воде и стандартным кормовым гранулам. Крыс Wistar, как правило, использовали для исследований FAAH. Все эксперименты соответствовали указаниям Национальных Институтов Здоровья по уходу и использованию лабораторных животных, и были разрешены институциональным комитетом по содержанию и использованию животных Университета Калифорнии, Ирвин, и Университета Джорджии, Афины, и соответствовали Директиве Совета ЕС 86 (609) EEC, а экспериментальный протокол был проведен в соответствии с итальянскими нормативно-правовыми актами (DL 116/92).
Извлечение тканей. Мышей умерщвляли изофлураном, сразу же собирали и замораживали ткани в жидком азоте. Замороженные ткани взвешивали и гомогенизировали в метаноле (1 мл, содержащем [2H4]-анандамид, [2H4]-PEA, [2H8]-2-AG, и N-циклогексил бифенил-3-илацетамид в качестве внутренних стандартов. Аналиты экстрагировали хлороформом (2 об.) и отмывали водой (1 об.). Органические фазы объединяли и высушивали под азотом. Нефракционированный органический экстракт использовали для количественной оценки URB937. Для других анализов органический экстракт фракционировали хроматографией в открытой колонке со слоем силикагеля, как описано в 43. Вкратце, экстракт растворяли в хлороформе и загружали в маленькие стеклянные колонки заполненные силикагелем G (60~A 230-400 Mesh ASTM; «Whatman», Клифтон, Нью-Джерси). Анандамид, PEA и 2-AG элюировали хлороформом/метанолом (9:1, об./об.).
Получение сыворотки. Кровь, слитую из туловища, собирали из декапитированных мышей, давали ей свернуться и помещали на лед. Свернувшуюся кровь центрифугировали при 18,000 x g в течение 10 мин при 4°C, переносили сыворотку в стеклянные сосуды и разводили дистиллированной водой до 1 мл. Белки осаждали ледяным ацетоном (1 мл), содержащим N-циклогексил бифенил-3-илацетамид в качестве внутреннего стандарта, и преципитат удаляли центрифугированием при 3000 xg в течение 10 мин при 4°C. Образцы высушивали под азотом для удаления ацетона, и экстрагировали хлороформом/метанолом как описано выше.
Жидкостная хроматография/масс-спектрометрия (ЖХ/МС). Тканевый уровень анандамида, PEA, 2-AG и URB937 определяли с помощью системы «1100-LC», соединенной с детектором «1946A-MS)) («Agilent Technologies, Inc.», Пало-Алто, Калифорния) и снабженной интерфейсом для ионизации электрораспылением. URB937 и N-циклогексил бифенил-3-илацетамид (m/z=294) элюировали на колонке «XDB Eclipse С18» (50×4,6-мм внутренний диаметр, 1,8 µм, «Zorbax») линейным градиентом 60%-100% A в B в течение 3 мин. при скорости потока 1,0 мл-мин-1. Мобильная фаза A состояла из метанола, содержащего 0,25% уксусную кислоту, 5 мМ ацетат аммония; мобильная фаза B состояла из воды, содержащей 0,25% уксусную кислоту, 5 мМ ацетат аммония. Анандамид, 2-AG и PEA элюировали градиентом метанола в воде (от 85% до 90% метанола в течение 2,5 мин) при скорости потока 1,0 мл-мин-1. Температуру колонки поддерживали при 40°C. Детекцию МС осуществляли в режиме положительной ионизации, капиллярное напряжение было установлено равным 3 кВ, напряжение фрагментора варьировало от 120 до 140 B. Азот использовали как сушильный газ при скорости потока 13 л-мин-1 и при температуре 350°C. Давление небулайзера было установлено равным 60 psi. Na+ аддукты ([M+Na+]) аналитов и внутренние стандарты отслеживали в селективном режиме отслеживания ионов. Предел количественной оценки составил 0,4 пмоль.
Анализ транспорта лекарственных веществ. Анализ транспорта проводили в «Cerep Inc.» (Редмонд, Вашингтон). Клеточную проницаемость определяли в направлении апикальное (A) - базальное (B) и в направлении B-A в присутствии 10 мкМ соединения в растворе Хенкса, содержащем 1% диметилсульфоксид (ДМСО). Соотношения потоков рассчитывали как отношение проницаемостей B-A к A-B.
Иммуногистохимия Fos. Через два часа после инъекции формалина в дорсальную часть лапы крысы получали летальную дозу нембутала (50 мг-мл-1). Животных перфузировали через сердце 100 мл 1% гепаринизированного фосфатно-солевого буфера (PBS) с последующей перфузией 300 мл ледяного 4% параформальдегида. Забирали пояснично-крестцовую область спинного мозга каждой крысы. Спинной мозг постфиксировали в 4% параформальдегиде при 4°C в течение 24 часов и затем осуществляли защиту от замораживания в 30% сахарозе при 4°C в течение 24-48 часов. Спинной мозг нарезали криостатом на 40 мкм поперечные срезы на уровне поясничного утолщения (L4/L5). Свободно плавающие альтернативные срезы держали в сериях лунок, заполненных PBS. Каждый четвертый срез обрабатывали на предмет иммунореактивности Fos для гарантии того, что одна и та же клетка не будет подсчитана дважды. Эндогенные пероксидазы инактивировали инкубацией в пероксиде водорода. Срезы блокировали козьей сывороткой для предотвращения неспецифического связывания и инкубировали в присутствии первичного антитела Fos (1:20,000, «Аbсаm», Кембридж, Массачусетс, США), разведенного в PBS, содержащем 0,4% Triton (в течение 1 ч при 37°C, а затем 48 ч при 4°С). Ткань инкубировали в присутствии биотинилированных вторичных козьих анти-кроличьих IgG (1:600, «Vector Laboratories)), Берлингейм, Калифорния, США) в течение 1 ч при 37°C. Затем, срезы инкубировали с реактивом «Vectastain Elite АВС» (1:200, «Vector Laboratories))) в течение 1 ч, с последующей инкубацией в течение 5 мин в 2% усиленном никелем диаминобензидине. Срезы помещали на предметные стекла, сушили на воздухе, обезвоживали в повышающихся концентрациях этанола, обесцвечивали в ксилоле, и закрывали покровным стеклом, прикрепленным «Permount». Все данные в одном и том же эксперименте обрабатывались одновременно для контроля разброса иммуноокрашивания разных прогонов. Специфичность иммуноокрашивания доказывали исключением первичного антитела из протокола иммуноокрашивания и демонстрацией того, что предварительная адсорбция пептидной антисыворотки блокировала специфическое окрашивание5.
Количественная оценка иммунореактивности. С помощью микроскопа для количественной оценки были выбраны три среза L4, которые демонстрировали наибольшее количество Fos-положительных клеток. Отбор срезов и оценка количества Fos-положительных клеток осуществлялись наблюдателем, неосведомленным об условиях эксперимента. Срезы регистрировали при 5х увеличении с помощью светового микроскопа «DMLB» и 1300 цифровой камеры в схожих условиях яркости/контраста для демонстрации сравнимого окрашивания фона. Пластинчатые подотделы выделяли на всех срезах с помощью программного обеспечения «Image J» (Национальные Институты здоровья США, Бетезда, Мэриленд, США). Подотделы серого вещества спинного мозга определяли как поверхностные пластинки (пластинки I и II), nucleus proprius (пластинки III и IV), шейку заднего рога (пластинки V и VI), и передний рог (пластинки VII, VIII, IX и X)44. С помощью «Image J» в каждом подотделе экспрессирующие Fos клетки, вне зависимости от интенсивности окрашивания, подсчитывались наблюдателем, неосведомленным об условиях эксперимента. Внутриэкспертная надежность находилась в диапазоне от 93% для поверхностной пластинки до 81% для всех подотделов пластинок.
Получение лекарственных средств для in vivo экспериментов. Лекарственные средства растворяли в полиэтиленгликоле 400/Tween-80/физрастворе (1/1/18; по объему) и вводили в.б. (5-10 мл-кг-1) или п.к. инъекцией (10 мл-кг-1). Для инъекций в латеральные желудочки мозга URB937 растворяли в 100% ДМСО и инъецировали в объеме 5 мкл.
Хирургическое вмешательство. Все хирургические вмешательства проводили в асептических условиях. Имплантация канюли для интрацеребровентрикулярного (Lev.) введения лекарственного средства. Крыс SD анастезировали смесью кетамина (70 мг-кг-1, в.б.) и ксилазина (9,33 мг-кг-1, в.б.). Животных помещали в стереотаксическую рамку и стабилизировали ушными фиксаторами («David Kopf Instruments)), Туюнга, Калифорния, США). Направляющую канюлю 22 калибра из нержавеющей стали имплантировали в правый латеральный желудочек мозга за 7 дней до проведения экспериментов. Координаты для имплантации (-0,9 мм в антеропостериорном направлении и -1,5 мм в медиолатеральном направлении относительно брегмы, и 3,8 мм ниже поверхности черепа) были определены с помощью атласа мозга крысы Paxinos и Watson45. Направляющие канюли прикрепляли к черепу с помощью 3 шурупов из нержавеющей стали и стоматологического цемента и держали открытыми до инъекции вставкой предохранительного стилета. Для инъекций стилет удаляли и лекарственное средство или носитель инфузировали в объеме 5 мкл с помощью 10 мкл микрошприца Гамильтона, соединенного с инъектором 28 калибра из нержавеющей стали, который выдавался на 1 мм за пределы направляющей канюли, посредством катетера PTFE 24G («Small parts Inc», Логанспорт, Индиана) заполненного PBS. Маленький пузырек воздуха (3 мкл) вводится в дистальный конец катетера PTFE 24G для отделения инъецируемого раствора от PBS и для визуального отслеживания инъекции. Инъекции осуществляли в течение периода 1 мин и инъектор оставляли на месте в течение еще 1 дополнительной минуты для предотвращения выброса обратным потоком. Размещение канюль проверяли в конце экспериментов инъекцией трипана синего (5 мкл) перед эвтаназией крыс. Только животных с правильным расположение включали в данное исследование. Перед проведением эксперимента крысам позволяли восстановиться в течение 7-10 дней. Перевязку седалищного нерва проводили на мышах Swiss, с помощью адаптации способа Bennett и Xie24. Мышей анастизировали ксилазином (10 мг-кг-1, в.б.) и кетамином (100 мг-кг-1, в.б.), левое бедро брили и очищали «Betadine®», делали небольшой разрез (2 см в длину) в середине левого бедра для открытия седалищного нерва. Нерв несильно связывали в двух различных участках (расположенных с 2 мм интервалом) вокруг всего нерва, с помощью шелковых нитей (7-0). Хирургическую область посыпали порошком стрептомицина и закрывали одиночным мышечным швом и двумя кожными скобами и в конце очищали «Betadine®». В ложнооперированных животных нервы обнажали, но не перевязывали. Животных помещали под лампу для обогрева до их пробуждения.
Поведенческие тесты. Корчи, вызванные уксусной кислотой, измеряли в мышах Swiss или в мышах C57B 1/6 (дикого типа и дефектных по FAAH), как описано в 40 с незначительными модификациями. Вкратце, мышей акклиматизировали в экспериментальном помещении в течение 2 ч. Каждому животному инъецировали уксусную кислоту (150 мкл, 0,6% в физрастворе) и помещали в стеклянный цилиндр. Абдоминальные растяжки (удлинения тела и задних конечностей), подсчитывали в течение 20 минут, начиная с 5 мин после инъекции уксусной кислоты. URB937, римонабант и АМ630 вводили п.к. инъекцией за 1 ч до уксусной кислоты. Поведение по баллам оценивал наблюдатель, не осведомленный об условиях обработки. Индуцированную формалином ноцицепцию оценивали в крысах Sprague-Dawley, как описано в 41. Крыс рассаживали отдельно и содержали в комнате для содержания общего пользования с циклом свет:темнота 12:12 ч. Животные получали свободный доступ к пище и воде, и им давали возможность акклиматизироваться в помещении перед проведением эксперимента. За один час до введения формалина крысы получали в.б. инъекции носителя, URB937 (1 мг-кг-1, в.б.), римонабанта (2 мг-кг-1 в.б.) или комбинации URB937 и римонабанта. Животных акклиматизировали в контейнере для наблюдения (прозрачный плексигласовый ящик, 29×29×25 см) за 15 мин. до введения инъекции формалина (50 мкл, 5% в физрастворе) в дорсальную поверхность правой задней лапы. Сразу после инъекции формалина крыс возвращали в контейнер для наблюдения и записывали с помощью видеокамеры защитное поведение в течение 60 мин. Записи анализировались наблюдателями, не осведомленными об условиях обработки. Защитное поведение измеряли непрерывно в течение 60 мин41. Общее время, потраченное животными в трех различных поведенческих категориях (0, 1, 2) было зафиксировано в 5 мин интервалах, где: (0) крыса демонстрирует нормальную позу, (1) поднимает инъецированную лапу, или (2) лижет, трясет или кусает инъецированную лапу. В каждом 5-мин интервале был проанализировано время, потраченное на (1) подъем и (2) лизание или кусание инъецированной лапы. Защитное поведение было проанализировано с помощью метода взвешенной оценки суммарных баллов боли (CPS-WST1,2) рассчитанных для всего периода наблюдения (0-60 мин), и, отдельно, для первой (0-10 мин) и второй фазы (10-60 мин) поведенческого ответа46. Площадь под кривой (AUC), соответствующую CPS-WST1,2, рассчитывали с помощью правила трапеций. Отек лапы индуцировали у мышей инъекцией в правые задние лапы 50 мкл стерильного физраствора, содержащего 1% λ-каррагинана. Объем лапы измеряли с помощью плетизмометра («Ugo Basile», Милан, Италия). Носитель или URB937 (1 мг-кг-1, в.б.) инъецировали непосредственно перед каррагинаном. Римонабант и АМ630 (1 мг кг-1, в.б.) инъецировали за 30 минут перед каррагинаном. Механическую гипералгезию оценивали измерением задержки (c) отдергивания лапы от постоянного механического давления, оказываемого на ее дорсальную поверхность. 15-г калиброванный стеклянный цилиндрический стержень (диаметр=10 мм,) скошенный до конической точки (диаметр=3 мм), использовался для приложения механического воздействия. Груз был вертикально подвешен между двумя кольцами, прикрепленными к подставке, и свободно перемещался вертикально. Использовали предельное время 3 мин. Термическую гипералгезию оценивали, как описано в 47, измеряли задержку отдергивания задней лапы от фокусированного луча источника лучистого тепла (термическая интенсивность: инфракрасное излучение 3,0), воздействующего на поверхность подошвы, с помощью коммерческого устройства («Ugo Basile», Варезе, Италия). Предельное время было равно 30 сек. Механическую аллодинию оценивали путем приложения постепенно изменяющейся силы на поверхность подошвы задней ноги с помощью нити Вон Фрея, при использовании динамического подошвенного анастезиометра («Ugo Basile»). Предельная сила была установлена на уровне 50 г.
Статистический анализ Результаты выражены как среднее±стандартная ошибка среднего. Статистическую значимость определяли по t-критерию Стьюдента, однофакторному или двухфакторному дисперсионному анализу (ANOVA) с последующим post hoc использованием критерия Бонферрони в случае необходимости. Отдельный одномерный дисперсионный анализ осуществляли для определения эффектов экспериментального лечения индуцированного формалином защитного поведения, измеренного по площади под кривой. Повторные измерения (Обработка×Время [повторный фактор]) дисперсионного анализа осуществляли в системе измерения суммарных баллов индуцированной формалином боли. Поправку Гринхауза-Гессера применяли ко всем повторяющимся факторам. Для каждого пластинчатого подраздела при L4/L5 спинного мозга одномерный дисперсионный анализ осуществляли для определения воздействия экспериментальной обработки на количество экспрессирующих Fos клеток. Post hoc критерии наименьшей значимой разности Фишера и Тьюки применяли к поведенческим данным и данным иммуноокрашивания Fos, соответственно. Post hoc сравнения, которые не соответствуют условию равной дисперсии, исправляли дробной корректировкой степеней свободы. Анализы осуществляли с помощью статистического программного обеспечения «SPSS» (версия 17.0; «SPSS Incorporated)), Чикаго, Иллинойс, США).
Результаты
Обнаружение ингибитора FAAH периферически-ограниченного действия
Известные в настоящее время ингибиторы FAAH легко пересекают гематоэнцефалический барьер12. Для получения ингибиторов с ограниченным доступом к ЦНС авторы настоящего изобретения добавили химические группы различной полярности к проксимальному фенильному кольцу Оарилкарбамата URB59713,14 (таблица 1, 1a), в положении, в котором, как ожидалось, малые гидрофильные заместители не ухудшат биологическую активность15. Новые соединения имеют сравнимую эффективность при тестировании FAAH в препаратах мембран мозга крыс и были равно эффективны при блокировании активности FAAH в печени при систематическом введении мышам (1 мг-кг-1, внутрибрюшинно, в.б.) (таблица 1, 1b-1f). Они значительно отличались, однако, по способности проникать в ЦНС. В частности, p-нитрофенильное производное URB937 (таблица 1, 1b) супрессирует активность FAAH в периферических тканях мышей и крыс, но не оказывает влияния на активность FAAH в мозге (таблица 1, таблица 2). Исследование дозировок в мышах показало, что средняя эффективная доза (ED50) URB937 для ингибирования FAAH в мозге была в 200 раз выше чем ED50 для ингибирования FAAH в печени (фиг.1а). Более того, после системного введения URB937 (1 мг-кг-1, в.б.) быстро распределился в сыворотке или печени, но не детектировался в ткани мозга (фиг.lb). Как и в случае других O-арилкарбаматов, известных как взаимодействующие с FAAH через ковалентный механизм13,16, URB937 ингибирует периферическую активность FAAH in vivo быстро и надолго (фиг.1c).
Механизм периферической сегрегации Анализы взаимодействий структура-активность указывают на то, что полярность p-гидроксифенильной составляющей вносит существенный вклад в периферическую сегрегацию URB937. В таблице 1 показано, что аналоги, в которых R-заместитель был слабополярным или аполярным - соединения 1c, 1d и 1e - легко проникали в мозг после системного введения мышам, тогда как аналог, в котором R состоит из полярной аминогруппы, соединение 1f, в основном не проникал в мозг. Тем не менее, относительно высокая липофильность URB937 (коэффициент распределения, LogDoct, pH7,4: URB937, 3.03±0,01; URB597, 3.71±0,01; среднее±стандартная ошибка среднего, п=3) позволяет молекуле пассивно диффундировать в ЦНС, если активно не противодействовать этому процессу. Для первой проверки данной идеи авторы определили соотношения проницаемости и оттока URB937 через поляризованные монослои человеческих эпителиальных клеток ТС7, которые экспрессируют различные белки-транспортеры, вовлеченные в вытеснение лекарственных средств из мозга17. URB937 не распределен равномерно по апикальному (A) и базальному (B) компартментам монослоев ТС7, как ожидалось бы от липофильной молекулы, переносимой пассивной диффузией. Скорее, соединение аккумулировалось в компартменте A [проницаемость, в нм-с-1 (% выделения) A-B, 38 (83%); B-A, 371 (95%); соотношение выделения, 9,8; среднее 2 независимых экспериментов], через механизм, который был нечувствителен к верапамилу, ингибитору Р-гликопротеина (Pgp) (100 мкМ) [B-A проницаемость в нм-с-1 (% выделения): 322 (94%)]. Эти сведения подтверждают, что URB937 вытесняется из ЦНС транспортной системой, которая фармакологически отличается от Pgp. В соответствии с этой интерпретацией, инъекция субмаксимальной дозы URB937 (0,1 мг-кг-1) в латеральные желудочки мозга крыс, дают в течение 1 ч почти полное ингибирование FAAH печени (остаточная активность: 11,3±1,9% контроля; среднее±стандартная ошибка среднего, n=3). Напротив, системное введение в 10 раз более высокой дозы лекарственного средства (1 мг-кг-1, в.б.) не оказало детектируемого воздействия на FAAH из мозга крысы (таблица 2).
Для исследования идентичности транспортной системы, вовлеченной в вытеснение URB937 из ЦНС, мы ввели в крыс различные фармакологические ингибиторы транспортеров гематоэнцефалического барьера вместе с наибольшей системной дозой URB937, которая не проникает в мозг (25 мг-кг-1, в.б.) (Фиг.1a). Совместное введение соединения Ко-143, ингибитора белка устойчивости к раку молочной железы (BCRP, ABCG1, вызвало дозозависимое повышение доступа URB937 в мозг. Напротив, совместное введение верапамила, ингибитора P-гликопротеина (Pgp), или пробенецида, ингибитора транспортера органических ионов, не дало такого эффекта. Результаты подтверждают, что URB937 вытесняется из ЦНС белком-транспортером, который фармакологически неотличим от BCRP.
Усиление периферической передачи сигналов анандамида Введение URB937 (1 мг-кг-1, в.б.) в мышах повышает уровни анандамида на периферии, но не в переднем мозге или гипоталамусе (фиг.1d, фиг.6). Этот эффект вызван селективным ингибированием активности FAAH из-за того, что (i) сопровождается повышениями эндогенных субстратов FAAH, таких как пальмитоилэтаноламид (PEA) (фиг.1d); и (ii) не наблюдается в мутантных мышах в которых экспрессия гена faah разрушена гомологичной рекомбинацией18 (фиг.1e). Важно отметить, что URB937 не влияет на активность моноацилглицерин-липазы in vitro (медианная ингибирующая концентрация, IC50, >100 мкМ; n=3) и не изменяет в тканях уровни ее эндоканабиноидного субстрата, 2-AG, in vivo (фиг.1f).
Модуляция висцеральной боли Проникающие в мозг ингибиторы FAAH ослабляют поведенческие ответы на болевые стимулы в грызунах, свойство, которое в целом приписывается их способности усиливать проведение сигнала анандамида в мозге и спинном мозге12,13. Для проверки вклада периферического анандамида в эти процессы, мы проверили воздействие URB937 на защитный ответ (избегание боли), вызванный инъекцией уксусной кислоты в перитонеальную полость мышей. Подкожное введение URB937 уменьшает корчи, вызванные уксусной кислотой, с ED50 0,1 мг-кг-1 (фиг.2a и данные не представлены). Этот эффект (i) был сравним по эффективности с тем, что вызывается мощным нестероидным анальгетиком индометацином (1 мг-кг-1, п.к.) (фиг.2a); (ii) коррелировал со степенью периферического ингибирования FAAH (коэффициент корреляции Пирсона, r=0,96, фиг.2b); и (iii) отсутствовал у мутантных мышей с дефектом FAAH (фиг.2 с). Антиноцицептивные эффекты URB937 нивелировались CB1-антагонистом римонабантом, но не CB1-антагонистом АМ630 (каждый по 1 мг-кг-1, п.к.) (фиг.2d).
Хотя анандамид является агонистом каналов ваниллоидного рецептора 1 типа (vanilloid type-1 transient receptor potential (TRPV-1) channels)19, URB937 вызывал недетектируемое защитное поведение при индивидуальном введении (1 мг-кг-1) (фиг.2a), что позволяет предположить, что тканевые концентрации, достигаемые эндогенным анандамидом после ингибирования периферических FAAH, не способны активировать каналы TRPV-1. Более того, стимуляция рецепторов активируемых пероксисомными пролифераторами а типа, которые могут быть индуцированы PEA20,21 (фиг.1d), вряд ли объясняет антиноцицептивные эффекты URB937, из-за того, что эти эффекты нивелируются блокадой CB1-рецепторов (фиг.2d).
Модуляция боли, индуцированной повреждением ткани В других сериях экспериментов мы оценивали влияние URB937 (1 мг-кг-1, в.б.) в модели постоянной боли при повреждении ткани, вызванном введением формалина в дорсальную часть задней лапы крысы. Инъекция формалина вызывает заметный защитный ответ, который времязависимо ослаблялся URB937 при сравнении с носителем, римонабантом (2 мг-кг-1, в.б.) или комбинацией римонабанта и URB937 (фиг.2е). Дополнительные анализы показали, что (i) URB937 уменьшает площадь под кривой формалин-индуцированного болевого поведения относительно всех других групп обработки (фиг.2f); и (ii) этот эффект главным образом был вызван уменьшением в последней фазе (Фазе 2) ответа на формалин (фиг.2g), при котором непрерывная активность первичных афферентных волокон сопровождалась воспалением и центральной сенсибилизацией спинномозговых ноцицептивных цепей 22,23.
Для выяснения, будет ли усиление периферической активности анандамида изменять обработку ноцицептивных входящих сигналов в спинном мозге, авторы измерили формалин-индуцируемую экспрессию Fos в тех же животных, которые были подвергнуты поведенческому тестированию. URB937 понижал ответ Fos на формалин (фиг.3a, b), уменьшая количество Fos-положительных клеток в поверхностном слое заднего рога (пластинка I, II), nucleus proprius (пластинка III, IV), шейной области заднего рога (пластинка V, VI), и переднего рога (фиг.3d). Этот эффект нивелировался CB1-антагонистом римонабантом (2 мг-кг-1, в.б.), который значимо изменял уровни Fos при введении без URB937 (фиг.3c, d). Способность URB937 подавлять обработку болевых сигналов спинным мозгом, несмотря на утрату проникновения в ЦНС, позволяет предположить, что периферический анандамид модулирует болевые входящие сигналы, перед тем как они войдут в спинной мозг.
Модуляция воспалительной и нейропатической боли Авторы также интересовались, может ли периферическое ингибирование активности FAAH оказать влияние на устойчивые болевые ответы, вызванные воспалением или повреждением нерва. Авторы индуцировали воспалительную реакцию в мышах инъекцией полисахарида каррагинана в одну из задних лап. Это привело к развитию механической и термической гипералгезии (повышенной чувствительности к вредоносным стимулам) а также к местному отеку (фиг.4). Одиночная системная инъекция URB937 (1 мг-кг-1, в.б.), введенная одновременно с каррагинаном, вызывает значимое уменьшение механической и термической гиперплазии, оцениваемое через 4 ч и 24 ч после обработки каррагинаном (фиг.4a, b). Лекарственное средство также подавляет механическую аллодинию (боль вследствие воздействия стимулов, обычно ее не вызывающих), измеренную через 24 ч после каррагинана (фиг.4c). Эти действия были ограничены воспаленными лапами (фиг.7) и нивелировались CBi-антагонистом римонабантом, но не CB2-антагонистом АМ630 (каждый по 1 мг-кг-1, в.б.) (фиг.4a-c). URB937 не оказывает влияния на отек лапы через 4 часа после инъекции каррагинана, но вызывает обратное развитие отека через 24 часа после инъекции посредством механизма, который был чувствителен блокировке как CB1-так и CB2-рецепторов (фиг.4d).
У другой группы мышей повреждали периферический нерв, слабо перевязывая левый седалищный нерв24. Одиночная доза URB937 (1 мг-кг-1, в.б., за 2 ч перед тестированием), вводимая через одну неделю после проведения хирургического вмешательства, ослабляла термическую гипералгезию и подавляла механическую гипералгезию и механическую аллодинию на оперированных лапах (фиг.5a-c). В частности, эффект сопровождался изменениями в реактивности неоперированных лап, указывая на то, что URB937 селективно нормализует механические и термические пороги, измененные повреждением нерва (фиг.5a-c). И наконец, авторы проверили эффект повторных инъекций URB937 (1 мг-кг-1, в.б., за 2 ч перед тестированием) один раз в день в течение 4 дней, начиная с 3 дня после перевязки нерва. Обработка вызвала антиноцицептивный эффект, который был неотличим от эффекта, вызванного однократной дозой лекарственного средства (фиг.5d-f), что наводит на мысль, что URB937 облегчает устоявшуюся нейропатическую боль без возникновения толерантности.

***P<0,001; **P<0,01; n=3
Список литературы
1. Stein, С., Schafer, М., & Machelska, Н., Attacking pain at its source: new perspectives on opioids. Nat Med 9 (8), 1003-1008 (2003).
2. Stein, С. & Zollner, С, Opioids and sensory nerves. Handb Exp Pharmacol (194), 495-518 (2009).
3. Calignano, A., La Rana, G., Giuffrida, A., & Piomelli, D., Control of pain initiation by endogenous cannabinoids. Nature 394 (6690), 277-281 (1998).
4. Jaggar, S.I., Sellaturay, S., & Rice, A.S., The endogenous cannabinoid anandamide, but not the CB2 ligand palmitoylethanolamide, prevents the viscero-visceral hyper-reflexia associated with inflammation of the rat urinary bladder. Neurosci Lett 253 (2), 123-126 (1998).
5. Nackley, A.G., Suplita, R.L., 2nd, & Hohmann, A.G., A peripheral cannabinoid mechanism suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience 117 (3), 659-670 (2003).
6. Dziadulewicz, E.K. et al., Naphthalen-1-y1-(4-pentyloxynaphthalen-1-y1)methanone: a potent, orally bioavailable human CB1/CB2 dual agonist with antihyperalgesic properties and restricted central nervous system penetration. J Med Chem 50 (16), 3851-3856 (2007).
7. Anand, P., Whiteside, G., Fowler, C.J., & Hohmann, A.G., Targeting CB2 receptors and the endocannabinoid system for the treatment of pain. Brain Res Rev 60 (1), 255-266 (2009).
8. Agarwal, N. et al., Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci 10 (7), 870-879 (2007).
9. Kaufmann, I. et al., Enhanced anandamide plasma levels in patients with complex regional pain syndrome following traumatic injury: a preliminary report. Eur Surg Res 43 (4), 325-329 (2009).
10. Richardson, D. et al., Characterisation of the cannabinoid receptor system in synovial tissue and fluid in patients with osteoarthritis and rheumatoid arthritis. Arthritis Res Ther 10 (2), R43 (2008).
11. Mitrirattanakul, S. et al., Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain 126 (1-3), 102-114 (2006).
12. Schlosburg, J.E., Kinsey, S.G., & Lichtman, A.H., Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J 11 (1), 39-44 (2009).
13. Kathuria, S. et al., Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med 9 (1), 76-81 (2003).
14. Piomelli, D. et al., Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev 12 (1), 21-38 (2006).
15. Clapper, J.R. et al., A second generation of carbamate-based fatty acid amide hydrolase inhibitors with improved activity in vivo. ChemMedChem 4 (9), 1505-1513 (2009).
16. Alexander, J.P. & Cravatt, B.F., Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem Biol 12 (11), 1179-1187 (2005).
17. Loscher, W. & Potschka, H., Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2 (1), 86-98 (2005).
18. Cravatt, B.F. et al., Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A 98 (16), 9371-9376 (2001).
19. Starowicz, K., Nigam, S., & Di Marzo, V., Biochemistry and pharmacology of endovanilloids. Pharmacol Ther 114 (1), 13-33 (2007).
20. LoVerme, J., La Rana, G., Russo, R., Calignano, A., & Piomelli, D., The search for the palmitoylethanolamide receptor. Life Sci 77 (14), 1685-1698 (2005).
21. Sagar, D.R., Kendall, D.A., & Chapman, V., Inhibition of fatty acid amide hydrolase produces PPAR-alpha-mediated analgesia in a rat model of inflammatory pain. Br J Pharmacol 155 (8), 1297-1306 (2008).
22. Coderre, T.J. & Melzack, R., The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury. J Neurosci 12 (9), 3665-3670 (1992).
23. Puig, S. & Sorkin, L.S., Formalin-evoked activity in identified primary afferent fibers: systemic lidocaine suppresses phase-2 activity. Pain 64 (2), 345-355 (1996).
24. Bennett, G.J. & Xie, Y.K., A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33 (1), 87-107 (1988).
25. Ahluwalia, J., Yaqoob, M., Urban, L., Bevan, S., & Nagy, I., Activation of capsaicin-sensitive primary sensory neurones induces anandamide production and release. J Neurochem 84 (3), 585-591 (2003).
26. Liu, J. et al., A biosynthetic pathway for anandamide. Proc Natl Acad Sci U S A 103 (36), 13345-13350(2006).
27. Hohmann, A.G. & Herkenham, M., Localization of central cannabinoid CBI receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: a double-label in situ hybridization study. Neuroscience 90 (3), 923-931 (1999).
28. Hohmann, A.G. & Herkenham, M., Cannabinoid receptors undergo axonal flow in sensory nerves. Neuroscience 92 (4), 1171-1175 (1999).
29. Richardson, J.D., Kilo, S., & Hargreaves, K.M., Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain 75 (1), 111-119 (1998).
30. Mackie, K., Cannabinoid receptors as therapeutic targets. Annu Rev Pharmacol Toxicol 46, 101-122 (2006).
31. LoVerme, J. et al., Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther 319 (3), 1051-1061 (2006).
32. Guindon, J. & Hohmann, A.G., Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol 153 (2), 319-334 (2008).
33. Cravatt, B.F. et al., Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proc Natl Acad Sci U S A 101 (29), 10821-10826 (2004).
34. Lever, I.J. et al., Localization of the endocannabinoid-degrading enzyme fatty acid amide hydrolase in rat dorsal root ganglion cells and its regulation after peripheral nerve injury. J Neurosci 29 (12), 3766-3780 (2009).
35. Tegeder, I. et al., Peripheral opioid analgesia in experimental human pain models. Brain 126 (Pt 5), 1092-1102 (2003).
36. Моr, M. et al., Cyclohexylcarbamic acid 3'- or 4'-substituted biphenyl-3-y1 esters as fatty acid amide hydrolase inhibitors: synthesis, quantitative structure-activity relationships, and molecular modeling studies. J Med Chem 47 (21), 4998-5008 (2004).
37. King, A.R. et al., URB602 inhibits monoacylglycerol lipase and selectively blocks 2-arachidonoylglycerol degradation in intact brain slices. Chem Biol 14 (12), 1357-1365 (2007).
38. Astarita, G., Ahmed, F., & Piomelli, D., Identification of biosynthetic precursors for the endocannabinoid anandamide in the rat brain. J Lipid Res 49 (1), 48-57 (2008).
39. Russo, R. et al., The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3'-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther 322 (1), 236-242 (2007).
40. Calignano, A., La Rana, G., & Piomelli, D., Antinociceptive activity of the endogenous fatty acid amide, palmitylethanolamide. Eur J Pharmacol 419 (2-3), 191-198 (2001).
41. Tjolsen, A., Berge, O.G., Hunskaar, S., Rosland, J.H., & Hole, K., The formalin test: an evaluation of the method. Pain 51 (1), 5-17 (1992).
42. Fegley, D. et al., Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3'-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther 313 (1), 352-358 (2005).
43. Cadas, H., di Tomaso, E., & Piomelli, D., Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidylethanolamine, in rat brain. J Neurosci 17 (4), 1226-1242 (1997).
44. Presley, R.W., Menetrey, D., Levine, J.D., & Basbaum, A.I., Systemic morphine suppresses noxious stimulus-evoked Fos protein-like immunoreactivity in the rat spinal cord. J Neurosci 10 (1), 323-335 (1990).
45. Paxinos, G. & Watson, C, The rat brain in stereotaxic coordinates, 6th ed. (Academic Press/Elsevier, Amsterdam; Boston; 2007).
46. Ruda, M.A., Ling, Q.D., Hohmann, A.G., Peng, Y.B., & Tachibana, Т., Altered nociceptive neuronal circuits after neonatal peripheral inflammation. Science 289 (5479), 628-631 (2000).
47. Hargreaves, K., Dubner, R., Brown, F., Flores, C, & Joris, J., A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32 (1), 77-88 (1988).
48. Greenhouse and Geisser, 1959. Psychometrika. 24:95-112.





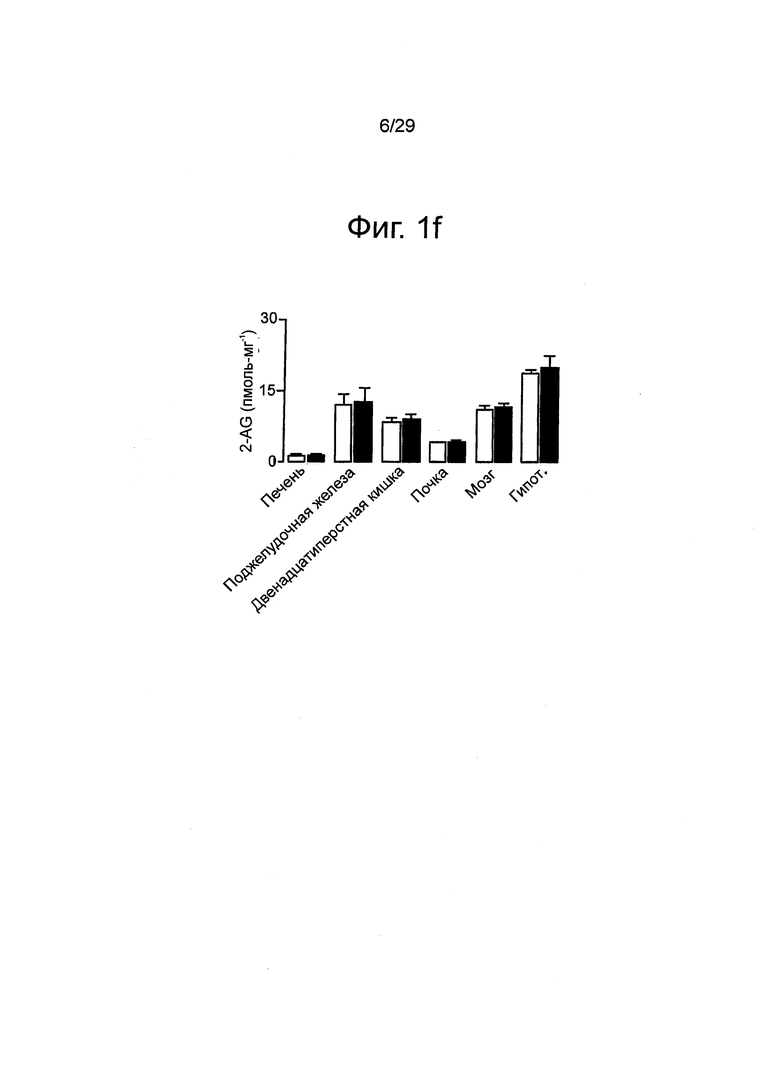
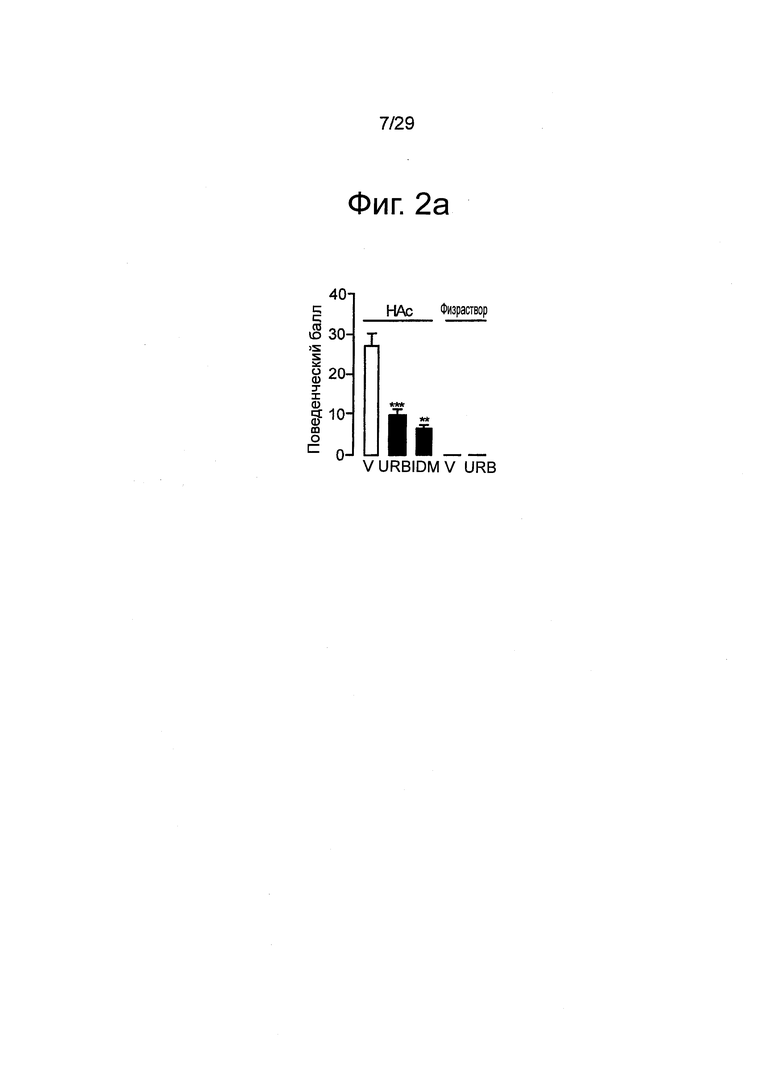




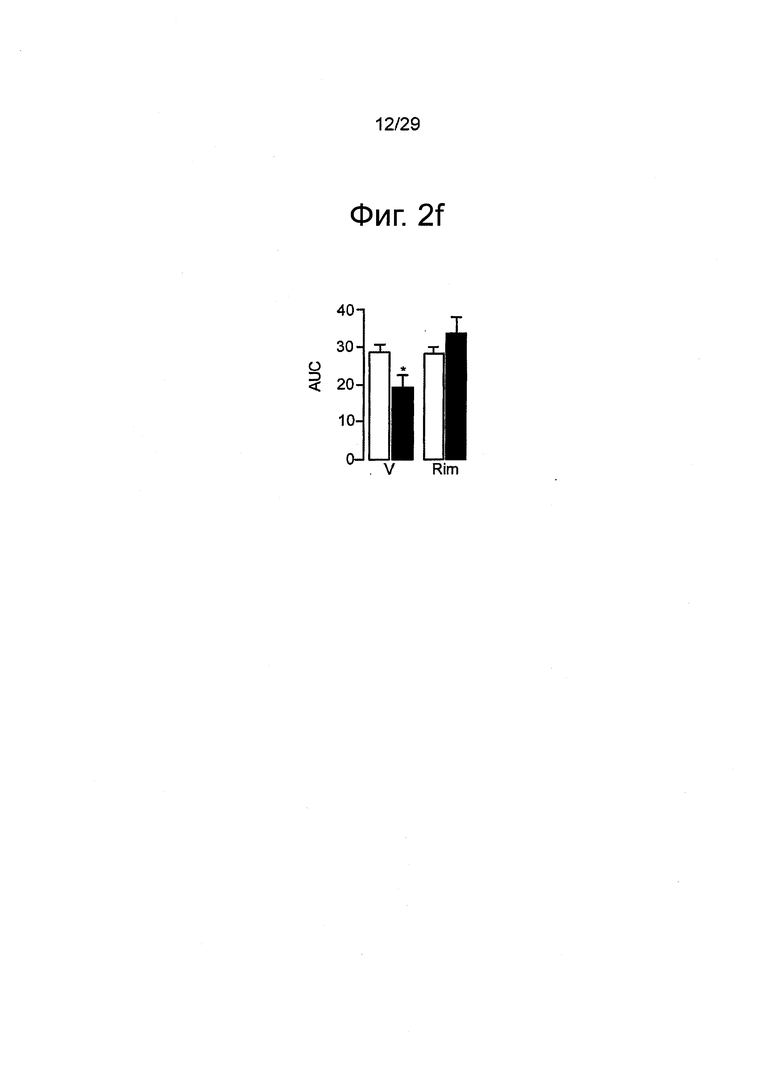


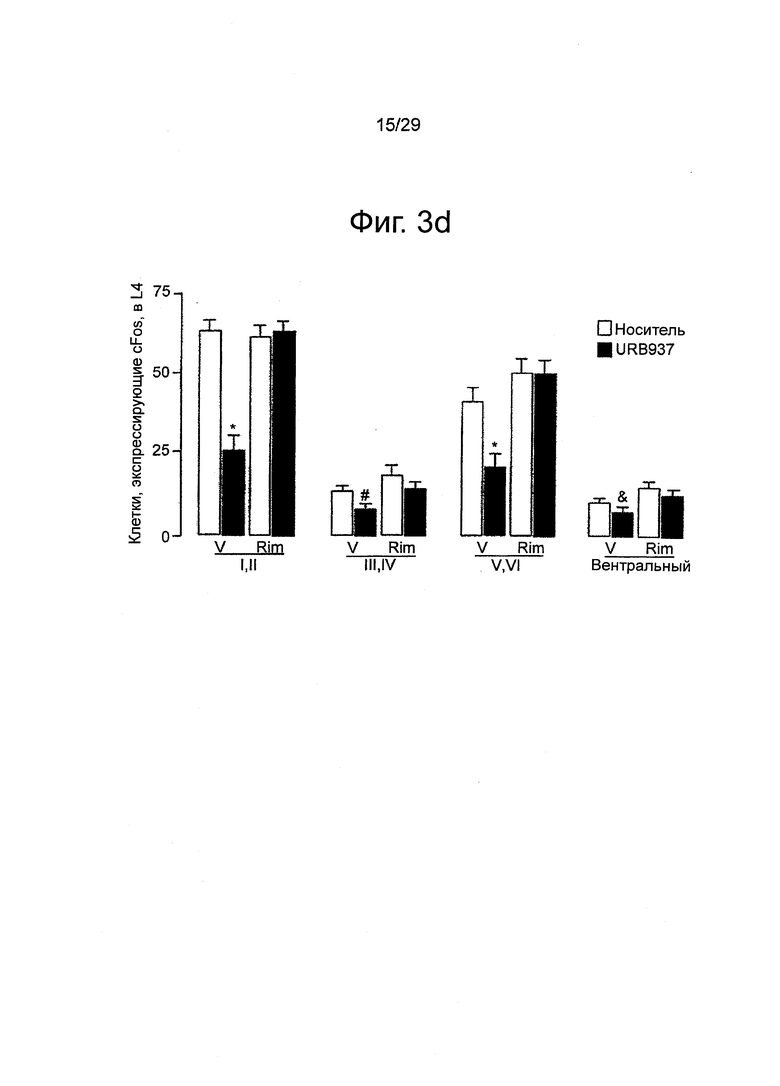
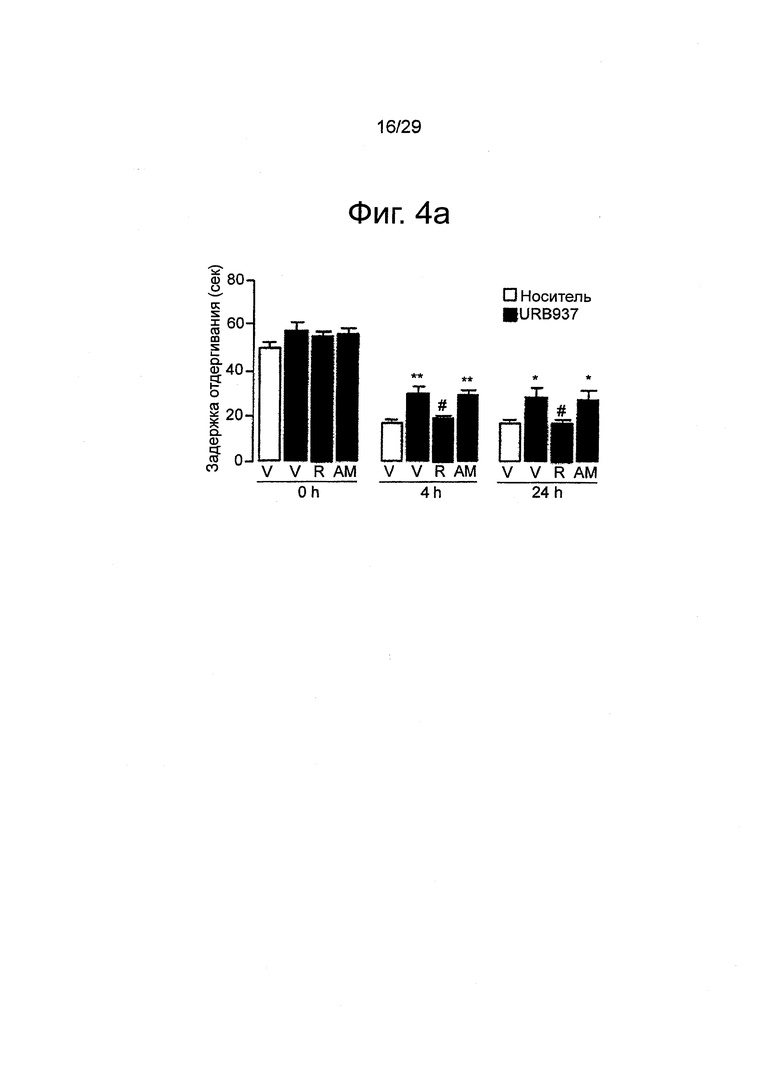


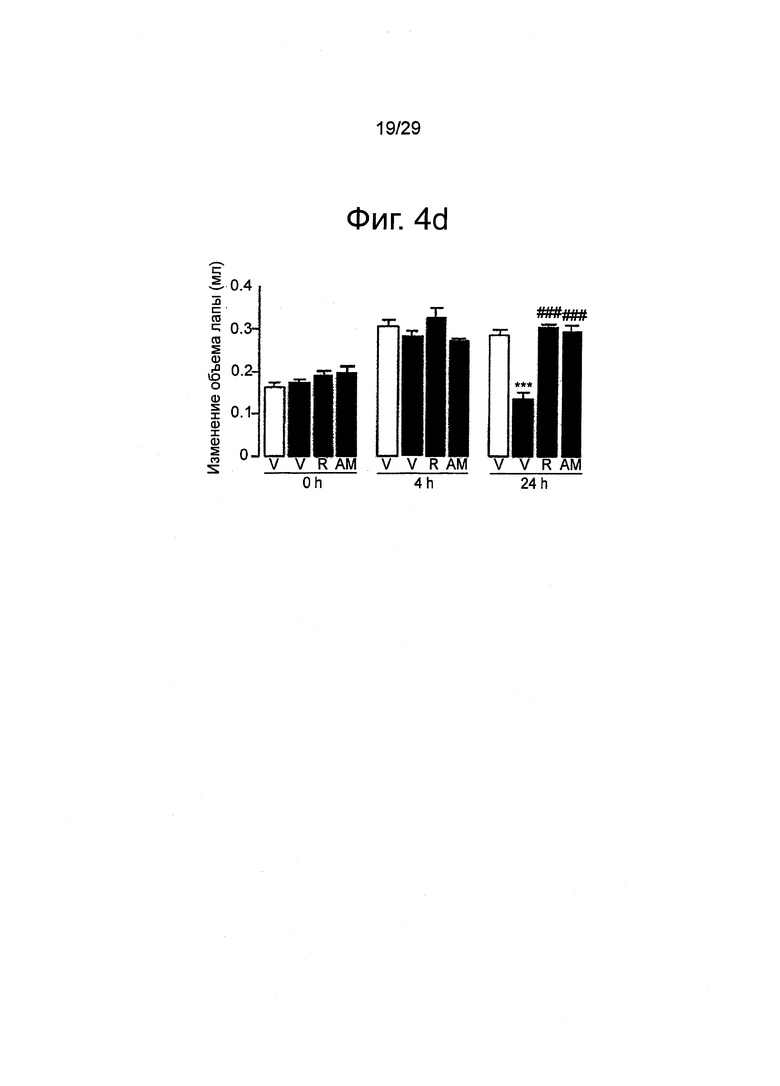


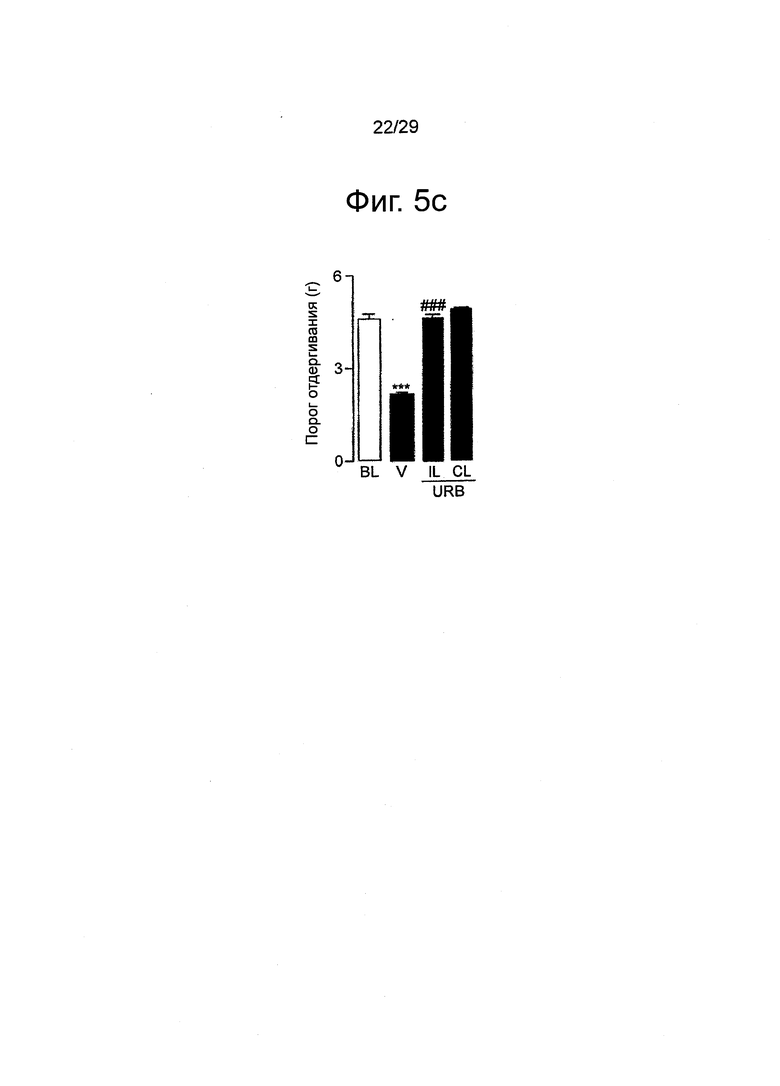



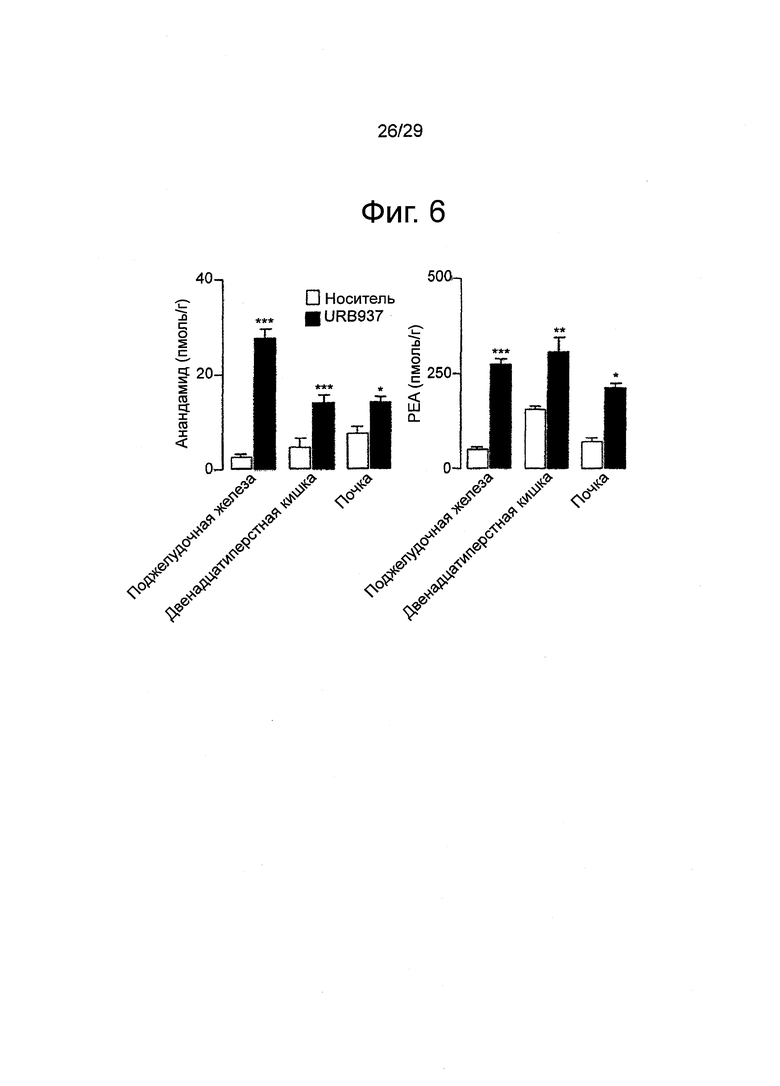
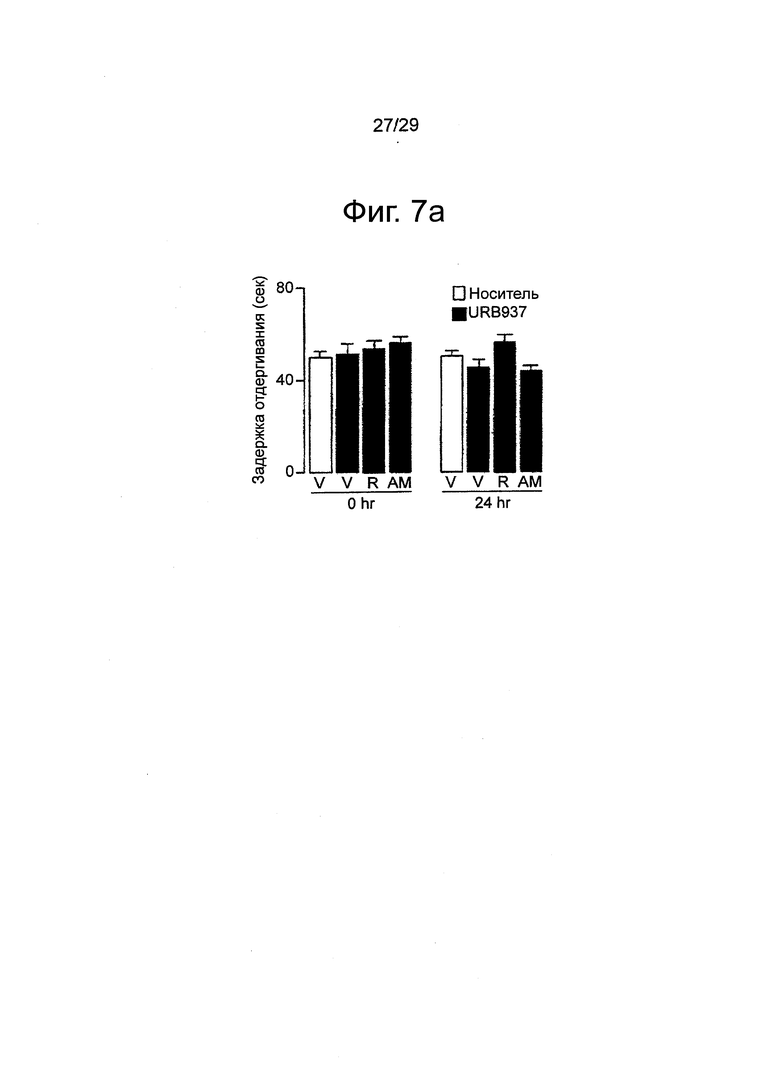

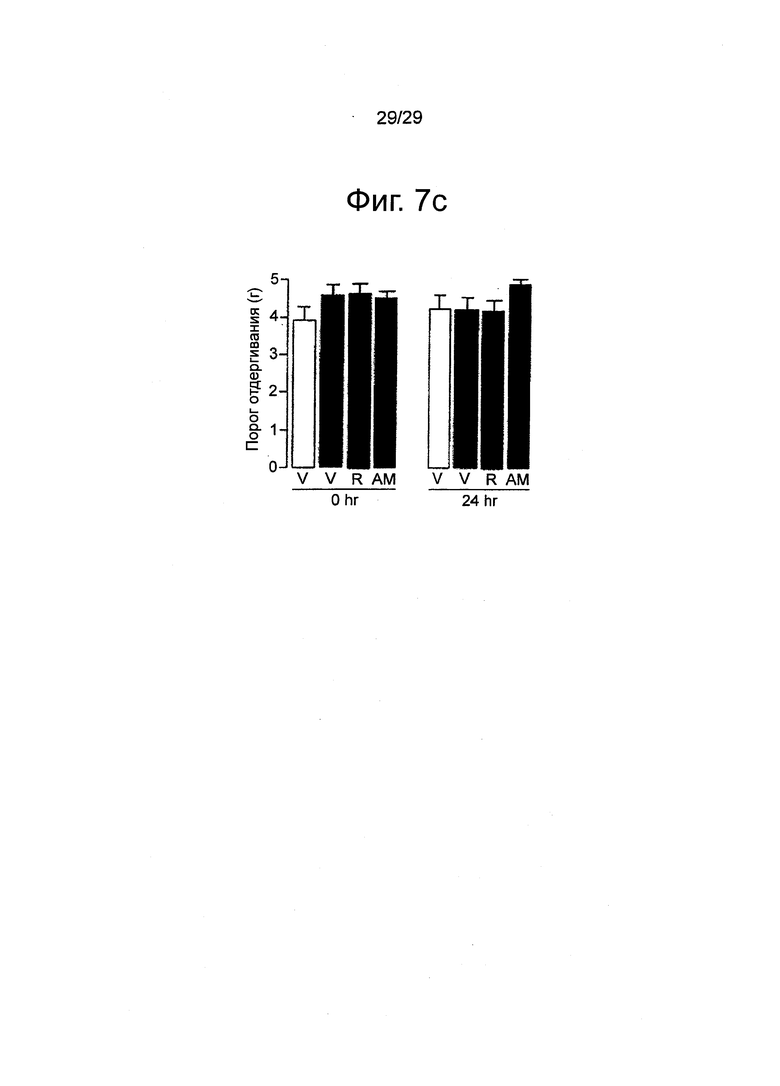
Изобретение относится к соединению формулы (1), где R1 выбирают из группы, состоящей из гидроксигруппы и ее физиологически гидролизуемых сложных эфиров и -NR8R9, где R8 и R9 независимо выбирают из группы, состоящей из водорода и незамещенного (С1-С3)алкила; R2 и R3 независимо выбирают из группы, состоящей из водорода или незамещенного (С1-С3)алкила; каждый из R4 независимо выбирают из группы, состоящей из галогена и незамещенного (С1-С3)алкила, а n представляет собой целое число от 0 до 4; каждый из R5 независимо выбирают из группы, состоящей из галогена и незамещенного (С1-С3)алкила, a m представляет собой целое число от 0 до 3; R6 представляет собой незамещенный циклогексил или циклогексил, замещенный (С1-С6)алкилом, галогеном или трифторметилом; и его фармацевтически приемлемые соли. Соединения предназначены для изготовления фармацевтической композиции или лекарственного средства для ингибирования FAAH у нуждающегося в этом млекопитающего, где млекопитающее нуждается в лечении боли, воспалительного расстройства или нуждается в лечении для ускоренного заживления ран. Соединения также предназначены для повышения периферического уровня анандамида, олеоилэтаноламида (OEA), палмитилэтаноламида (PEA) или стеароилэтаноламида (SEA) у нуждающегося в этом млекопитающего. Технический результат - ингибиторы FAAH периферически-ограниченного действия. 8 н. и 20 з.п. ф-лы, 2 табл., 29 ил.
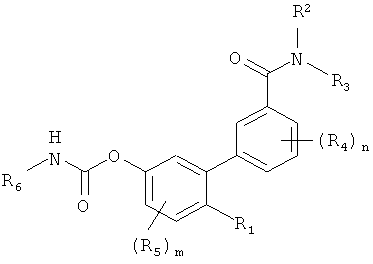 (1)
(1)
1. Соединение формулы:

где R1 выбирают из группы, состоящей из гидроксигруппы и ее физиологически гидролизуемых сложных эфиров и -NR8R9, где R8 и R9 независимо выбирают из группы, состоящей из водорода и незамещенного (С1-С3)алкила;
R2 и R3 независимо выбирают из группы, состоящей из водорода или незамещенного (С1-С3)алкила;
каждый из R4 независимо выбирают из группы, состоящей из галогена и незамещенного (С1-С3)алкила, а n представляет собой целое число от 0 до 4;
каждый из R5 независимо выбирают из группы, состоящей из галогена и незамещенного (С1-С3)алкила, a m представляет собой целое число от 0 до 3;
R6 представляет собой незамещенный циклогексил или циклогексил, замещенный (С1-С6)алкилом, галогеном или трифторметилом;
и его фармацевтически приемлемые соли.
2. Соединение по п.1, в котором R1 представляет собой физиологически гидролизуемый сложный эфир.
3. Соединение по п.1 или 2, в котором физиологически гидролизуемый сложный эфир представляет собой -OC(O)R7, где R7 представляет собой незамещенный (С1-С3)алкил.
4. Соединение по п.1, в котором R1 представляет собой гидроксигруппу.
5. Соединение по п.1, в котором R1 представляет собой гидроксигруппу и по меньшей мере один из R2 и R3 представляет собой водород.
6. Соединение по п.1, в котором R1 представляет собой гидроксигруппу и оба из R2 и R3 представляют собой водород.
7. Соединение по любому из пп.1-2, 4-6, в котором R6 представляет собой незамещенный циклогексил.
8. Соединение по любому из пп.1-2, 4-6, в котором каждый из R4 и R5 независимо выбирают из галогена и незамещенного (С1-С3)алкила.
9. Соединение по любому из пп.1-2, 4-6, в котором m равно 0.
10. Соединение по любому из пп.1-2, 4-6, в котором n равно 0.
11. Соединение по любому из пп.1-2, 4-6, в котором сумма m и n равна 1, 2 или 3.
12. Соединение по п.1, имеющее формулу:
,
и его фармацевтически приемлемые соли.
13. Фармацевтическая композиция для ингибирования гидролазы амидов жирных кислот (FAAH), включающая эффективное количество соединения по любому из пп.1-12.
14. Применение соединения по любому из пп.1-12 при изготовлении лекарственного средства для ингибирования FAAH у нуждающегося в этом млекопитающего.
15. Применение по п.14, где млекопитающее нуждается в лечении боли, воспалительного расстройства или нуждается в лечении для ускоренного заживления ран.
16. Применение по п.15, где боль является ноцицептивной, воспалительной или невропатической болью.
17. Применение по п.15, где млекопитающее имеет воспалительное расстройство или нуждается в лечении для ускоренного заживления ран.
18. Фармацевтическая композиция для селективного ингибирования периферической гидролазы амидов жирных кислот (FAAH), включающая терапевтически эффективное количество соединения по любому из пп.1-12 и фармацевтически приемлемое вспомогательное вещество.
19. Фармацевтическая композиция для лечения состояния, выбранного из боли, воспаления и медленного заживления ран, включающая терапевтически эффективное количество соединения по любому из пп.1-12 и фармацевтически приемлемое вспомогательное вещество.
20. Способ лечения боли и/или воспаления путем введения нуждающемуся в этом млекопитающему терапевтически эффективного количества соединения по любому из пп.1-12.
21. Способ по п.20, где лечат воспаление.
22. Способ по п.20, где лечат боль.
23. Способ лечения состояния, выбранного из группы, состоящей из боли, воспаления и медленного заживления ран, путем введения нуждающемуся в этом млекопитающему терапевтически эффективного количества соединения по любому из пп.1-12.
24. Способ повышения периферического уровня анандамида, олеоилэтаноламида (OEA), палмитилэтаноламида (PEA) или стеароилэтаноламида (SEA) у нуждающегося в этом млекопитающего путем введения соединения по любому из пп.1-12.
25. Способ по п.24, где OEA, PEA, SEA или анандамид является эндогенным для млекопитающего.
26. Способ по п.24, где OEA, PEA, SEA или анадамид вводят млекопитающему перед, после или одновременно с введением соединения.
27. Способ по п.24, где введение ОЕА, PEA, SEA или анандамида происходит одновременно с введением соединения.
28. Способ по любому из пп.24-27, где млекопитающее нуждается в лечении боли, воспаления или нуждается в лечении для ускоренного заживления ран.
| Горелка для кухни типа "Примус" | 1928 |
|
SU10267A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| FEGLEY DARREN et al: "Characterization of the fatty acid amide | |||

