Изобретение относится к анальгезирующим фармацевтическим композициям, содержащим опиоидное анальгезирующее средство и ингибитор циклооксигеназы-2 (СОХ-2). Изобретение также относится к способам лечения боли, заключающим в себе введение подобных фармацевтических композиций больным.
Существует неослабеваемая потребность в анальгезирующих средствах, способных ослаблять боль с высокой эффективностью и в то же время снижать возможность нежелательного действия. Нестероидные противовоспалительные средства ("NSAID"), включающие в себя соединения, такие как ибупрофен, кетопрофен и диклофенак, проявляют противовоспалительное действие и являются эффективными при боли, связанной с высвобождением простагландинов и других медиаторов воспаления. Например, диклофенак рассматривают как чрезвычайно сильнодействующее и эффективное средство, применяемое в качестве анальгезирующего и противовоспалительного агента. Диклофенак утвержден в США для продолжительного симптоматического лечения ревматоидного артрита, остеоартрита и анкилозирующего спондилоартрита. Кроме того, считают, что он пригоден для кратковременного лечения острого скелетно-мышечного повреждения, острого плечекистевого синдрома, послеоперационной боли и дисменореи. Однако такие NSAID как диклофенак приблизительно у 20% больных вызывают побочное действие, что требует прекращения лекарственной терапии. Побочные эффекты включают в себя, например, желудочно-кишечное кровотечение и аномальное повышение уровня ферментов печени.
Опиоиды представляют собой группу лекарственных средств как природного, так и синтетического происхождения, которые применяют главным образом в качестве действующих на центральную нервную систему анальгезирующих средств и которые обладают свойствами, подобными опиуму и морфину (Gilman et al., 1980, Goodman and Oilman’s, The Pharmacological basis of Theurapeutics, Chapter 24:494-534, Pub. Pergamon Press; публикация включена в описание в виде ссылки). Опиоды включают в себя морфин и морфиноподобные гомологи, содержащие полусинтетические производные кодеин (метилморфин) и гидрокодон (дигидрокодеинон), наряду со многими другими подобными производными. Морфин и родственные опиоиды проявляют активность агонистов в центральной нервной системе или (мю) μ-опиоидных рецепторов ЦНС (относится к головному и спинному мозгу), а также проявляют сродство к (дельта) δ- и (каппа) к-опиоидным рецепторам, чтобы осуществлять ряд эффектов, включающих в себя обезболивание, сонливость, изменения настроения и психическое помрачение сознания. Кроме сильнодействующего анальгезирующего действия, родственные морфину опиоиды могут вызывать ряд нежелательных эффектов, включающих в себя, например, угнетение дыхания, тошноту, рвоту, головокружение, психическое помрачение сознания, дисфорию, зуд, запор, увеличение давления в желчных путях, задержку мочи и гипотензию. Развитие толерантности к опиоидным лекарственным средствам и риск химической зависимости и злоупотребление этими лекарственными средствами представляет другой нежелательный эффект.
Морфин, который рассматривается как прототип опиоидного анальгетика, имеется в различных лекарственных формах, включающих в себя пероральные лекарственные формы немедленного высвобождения, а позднее стал производиться в виде композиций с 12-часовым контролируемым высвобождением (например, MS Contin® таблетки, коммерчески доступные из Purdue Frederick Company). Другие опиоидные анальгетики имеются в виде пероральных лекарственных форм немедленного высвобождения, такие как гидроморфин (например, Dilaudid®, коммерчески доступный из Knoll Pharmaceuticals). Позднее другой опиоидный анальгетик с контролируемым высвобождением, оксикодон, стал доступным (OxyContin, коммерчески доступный из Purdue Pharma). Конечно, существует множество других пероральных композиций немедленного высвобождения и отсроченного высвобождения опиоидов, которые являются коммерчески доступными во всем мире.
В предшествующих публикациях сообщалось, что анальгезирующее действие может быть улучшено путем снижения нежелательных эффектов благодаря комбинации опиоида с NSAID или анальгезирующими средствами, такими как ацетилсалициловая кислота или ацетаминофен, таким образом, чтобы получить синергическое анальгезирующее действие, что позволяет снизить общую дозу как NSAID, так и анальгетика. Например, в патенте США №4569937, выданном Baker et al. 11 февраля 1986, описана комбинация оксикодона с ибупрофеном в соотношении оксикодон/ибупрофен, составляющем от 1:6 до приблизительно 1:400. В патенте США №4690927, выданном Voss et al. 1 сентября 1987, описана комбинация представителя NSAID диклофенака и кодеина в весовом отношении диклофенака к кодеину, составляющем приблизительно от 1:1 до 3:1. В патенте США №5190947, выданном Riess et al. 2 марта 1993, описана диклофенак-соль кодеина ([2-[2,6-дихлорфенил,)-амино]-фенил]-уксусная кислота). В патенте США №4844907, полученном Elger et al. 4 июля 1989, описана многофазная таблетка, представляющая собой комбинацию наркотической анальгезирующей фазы и фазы NSAID в раздельных слоях. В патенте США №4587252, полученном Arnold et al. 6 мая 1986, описан способ лечения боли с использованием комбинации гидрокодона и ибупрофена.
Нестероидные противовоспалительные средства (NSAID) проявляют большую часть их противовоспалительной, анальгезирующей и жаропонижающей активности и ингибируют вызванные гормонами сокращения матки и рост определенных типов раковых опухолей посредством ингибирования простагландин G/H синтазы, известной также как циклооксигеназа.
Циклооксигеназа жирных кислот (СОХ) была описана как источник простагландинов, тромбоксанов и ряда других биологически активных гидроксилированных метаболитов, произведенных из арахидоновой кислоты, и более ненасыщенных жирных кислот. Начиная с конца 1960-х годов В.Samnuelsson, S.Bergstrom и их коллеги обнаружили биологическую активность продуктов циклооксигеназы и объяснили их структуру. В конце 1960-х и в начале 1970-х годов J.Vane обнаружил, что аспирин и другие NSAIDs проявляют их основные биологические активности путем ингибирования циклооксигеназы. СОХ непосредственно отвечает за образование PGG и PGH, и они служат в качестве промежуточных соединений в синтезе PGD, PGE, PGF, PGI и ТХА. В конце 1970-х и начале 1980-х годов было показано, что многие гормоны и другие биологически активные агенты могут регулировать клеточную активность СОХ. Во-первых, было предположено, что индукция СОХ является простым результатом окислительной инактивации СОХ, которая происходит только после нескольких оборотов субстрата. Этот факт является общим среди ферментов, которые включают молекулярный кислород в их субстраты - кислород способствует быстрой деградации фермента. Иногда эти ферменты называют ферментами-самоубийцами. В ответ на быструю (в течение секунд) инактивацию циклооксигеназы ее матричная РНК транскрибируется, и фермент быстро индуцируется, чтобы восстановить эту потерю посредством катализа. Несколькими группами исследователей было замечено, что циклооксигеназа индуцируется в значительно большей степени, чем это необходимо для восстановления потерянной активности. Используя олигонуклеотид, направленный на клонированный фермент СОХ-1, была идентифицирована вторая полоса при назернблоттинге с низкой достоверностью. Этот ген был клонирован и идентифицирован как второй фермент СОХ, названный СОХ-2, и было найдено, что он, при нормальных условиях, главным образом отсутствует во многих клетках, но быстро индуцируется некоторыми цитокинами и медиаторами. Было обнаружено, что экспрессия этого фермента в значительной степени ответственна за отмеченный ранее избыток активности СОХ в активированных клетках. Гены для СОХ-1 и СОХ-2 отличаются, и ген для СОХ-1 составляет 22 т.н. и размер матричной РНК 2,8 т.н., в то время как ген для СОХ-2 составляет 8,3 т.н. и размер матричной РНК 4,1 т.н. В то время как промотор СОХ-1 не содержит связывающих сайтов, распознающих фактор транскрипции, промотор СОХ-2 содержит участки для NF-kB, АР-2, NF-IL-6 и глюкокортикоидов (Н.R.Herschman, Cane. Metas. Rev. 13:256, 1994). Имеются некоторые различия в активных сайтах ферментов. Аспирин ингибирует активность циклооксигеназы СОХ-1, но оставляет интактной ее пероксидазную активность, в то время как аспирин превращает СОХ-2 из циклооксигеназы в 15-липоксигеназу (E.A.Meade et al., J. Biol. Chem. 268:6610, 1993).
Было высказано предположение, что во многих клетках СОХ-1 является ответственной за эндогенное базальное высвобождение простагландинов и представляет важность в физиологических функциях простагландинов, которые включают в себя сохранение желудочно-кишечной целостности и почечного кровотока. Ингибирование СОХ-1 вызывает ряд побочных эффектов, включая ингибирование агрегации тромбоцитов, связанной с нарушениями коагуляции, и желудочно-кишечную токсичность с вероятностью образования язв и кровотечения. Полагают, что желудочно-кишечная токсичность является следствием уменьшения биосинтеза простагландинов, которые проявляют цитозащитное действие по отношению к слизистой желудка.
Большая вероятность побочных эффектов исторически связана с постоянным использованием классических ингибиторов циклооксигеназы, из которых все являются приблизительно равными по силе действия в отношении СОХ-1 или СОХ-2 или которые являются избирательными к СОХ-1. Хотя почечная токсичность имеет место, она обычно явно проявляется у больных, которые уже страдают от почечной недостаточности (D.Kleinknecht, Sem. Nephrol. 15:228, 1995). Самой распространенной и болезненной токсичностью является желудочно-кишечная токсичность. Даже относительно нетоксичные лекарственные средства, такие как пироксикам, вызывают у вплоть до 4% больных сильное кровотечение и образование язв (M.J.S.Langman et al., Lancet 343:1075, 1994). В США подсчитали, что около 2000 больных с ревматоидным артритом и 20000 больных с остеоартритом умирает каждый год вследствие желудочно-кишечных побочных эффектов, связанных с применением ингибиторов СОХ. В Великобритании приблизительно 30% из ежегодных 4000 смертей, связанных с язвой желудка, приписывают ингибиторам СОХ (Scrip 2162, р.17). Ингибиторы СОХ вызывают желудочно-кишечную и почечную токсичность вследствие ингибирования синтеза гомеостатических простагландинов, ответственных за образование эпителиальной слизи и за почечный кровоток соответственно.
Вторая форма циклооксигеназы, СОХ-2, легко и быстро индуцируется рядом агентов, включая митогены, эндотоксины, гормоны, цитокины и факторы роста.
Предполагают, что СОХ-2 ответственна главным образом за патологические эффекты простагландинов, которые возникают в случае, когда имеет место быстрая индукция СОХ-2 в ответ на такие агенты, как воспалительные агенты, гормоны, факторы роста и цитокины. Поэтому избирательный ингибитор СОХ-2 должен обладать противовоспалительными, жаропонижающими и анальгезирующими свойствами, подобными свойствам стандартных нестероидных противовоспалительных лекарственных средств (NSAID). Кроме того, ингибитор СОХ-2 должен ингибировать индуцированные гормонами сокращения матки и обладать потенциалом противоракового действия. Ингибитор СОХ-2 должен иметь преимущества над NSAID, такие как снижение способности индуцировать некоторые из основанных на механизме действия побочных эффектов. Кроме того, полагают, что ингибиторы СОХ-2 имеют пониженный потенциал токсичности в отношении желудочно-кишечного тракта, сниженный потенциал в отношении почечных побочных эффектов, сниженный эффект в отношении времени кровотечения и уменьшенную способность вызывать приступы астмы у астматических субъектов, чувствительных к аспирину.
Таким образом, соединения с высокой специфичностью по отношению к СОХ-2 в сравнении с СОХ-1 могут применяться как средства, альтернативные стандартным NSAID. Это имеет место особенно в случаях, когда применение NSAID противопоказано, как, например, больным с пептическими язвами, гастритом, региональным энтеритом, язвенным колитом, дивертикулитом или с рецидивами желудочно-кишечных патологических изменений; с желудочно-кишечным кровотечением, нарушениями свертывания, включая анемию, гипопротромбинемию, гемофилию или другие случаи кровотечения; с заболеванием почек и для больных, готовящихся к операции или к принятию антикоагулянтов.
Как только стало очевидным, что СОХ-1, а не СОХ-2, является ответственной за продукцию простагландинов желудочно-кишечным эпителием и вносит основной вклад в синтез простагландинов почек, поиск избирательных ингибиторов СОХ-2 стал чрезвычайно активным. Это привело очень быстро к признанию того, что несколько ингибиторов СОХ, включая нимесалид и Dup-697, которые, как известно, вызывают незначительное или совсем не вызывают желудочно-кишечное раздражение, являются избирательными по отношению к СОХ-2.
В патенте США №5409944 (Black et al.) описаны некоторые производные алкансульфонамидоинданона, применяемые для лечения боли, лихорадочного состояния, воспаления, артрита, рака и других болезненных состояний. В патенте также обсуждаются композиции для лечения заболеваний, опосредованных циклооксигеназой-2, содержащие описанные новые производные алкансульфонамидоинданона вместе с успокаивающими боль средствами, включая ацетаминофен или фенацетин; потенцирующее средство, включая кофеин; антагонист Н-2, гидроокись алюминия или магния, симетисон, противоотечное средство, включая фенилэфрин, фенилпропаноламин, псевдофедрин, оксиме-тазолин, эпинефрин, нафазолин, ксилонетазолин, пропилгекседрин или лево-дезокси-эфедрин; противокашлевое средства, включая кодеин, гидрокодон, карамифен, карбетапентан или декстраметорфан; мочегонное средство и/или седативное средство или неседативный антигистамин. Хотя Black et al. ссылаются на применение противокашлевой дозы двух опиоидных анальгетиков (кодеин и гидрокодон), они не описывают и не предполагают использовать их ингибиторы СОХ-2 с эффективными количествами для обезболивания любых опиоидных анальгетиков.
Целью настоящего изобретения является представление способа и фармацевтической композиции (лекарственного средства), которые позволяют снизить концентрации опиоидных анальгетиков в плазме, в то же время сохраняя способность эффективного устранения боли.
Кроме того, целью настоящего изобретения является обеспечение способа и фармацевтической композиции (лекарственного средства) для эффективного лечения боли у больных посредством опиоидных анальгетиков, которые обеспечивают продолжительное и эффективное устранение боли, в то же время обеспечивая возможность снижения побочных эффектов, зависимости и толерантности, которые больные могут испытывать при продолжительном лечении опиоидом.
Далее, целью является обеспечение способа и фармацевтической композиции (лекарственного средства) для эффективного лечения боли у больных путем усиления анальгезирующего действия ингибитора СОХ-2.
Изобретение относится к удивительному синергическому действию, полученному посредством введения опиоидного анальгетика вместе с ингибитором СОХ-2.
Настоящее изобретение относится отчасти к анальгезирующим фармацевтическим композициям, содержащим ингибитор СОХ-2 вместе с опиоидными анальгетиками. Опиоидный анальгетик и ингибитор СОХ-2 могут быть введены перорально, посредством имплантата, парентерально, подъязычно, ректально, местно, посредством ингаляции и т.д. В других аспектах изобретения ингибитор СОХ-2 может быть введен отдельно от опиоидного анальгетика, как изложено более детально ниже.
Изобретение позволяет применять более низкие дозы опиоидного анальгетика или ингибитора СОХ-2 (что упоминается здесь как "кажущийся односторонний синергизм") или более низкие дозы обоих лекарственных средств (в тексте упоминается как "двусторонний синергизм"), чем обычно требуется, когда применяется одно любое лекарственное средство. Благодаря применению более низких количеств любого или обоих лекарственных средств побочные эффекты, связанные с эффективным устранением боли у человека, значительно снижаются.
В некоторых предпочтительных аспектах изобретение относится отчасти к синергическим комбинациям ингибитора СОХ-2 в достаточном для оказания терапевтического действия количестве вместе с опиоидным анальгетиком, так, что достигаемое анальгезирующее действие, по меньшей мере, приблизительно в 5 (и предпочтительно, по меньшей мере, приблизительно в 10) раз выше, чем действие, достигаемое при отдельном применении опиоидного анальгетика, за исключением комбинаций ингибитора СОХ-2 с противокашлевыми дозами гидрокодона или кодеина. В некоторых аспектах синергическая комбинация обеспечивает анальгезирующее действие, приблизительно до 30-40 раз превышающее действие, достигаемое опиоидным анальгетиком в отдельности.
В таких аспектах синергические комбинации проявляют так называемый "кажущийся односторонний синергизм", означая, что доза ингибитора СОХ-2 синергически усиливает действие опиоидного анальгетика, но доза опиоидного анальгетика, по-видимому, не усиливает значительно действие ингибитора СОХ-2. В некоторых аспектах комбинацию вводят в единой лекарственной форме. В других аспектах комбинацию вводят отдельно, предпочтительно одновременно. В некоторых предпочтительных аспектах проявляемый между ингибитором СОХ-2 и опиоидным анальгетиком синергизм является таковым, что дозировка опиоидного анальгетика должна быть субтерапевтической, если ее вводят без ингибитора СОХ-2. В других предпочтительных аспектах настоящее изобретение относится к фармацевтической композиции, содержащей эффективную для обезболивания дозу опиоидного анальгетика вместе с дозой ингибитора СОХ-2, эффективной для усиления анальгезирующего действия опиоидного анальгетика.
Хотя некоторые аспекты изобретения относятся к синергическим комбинациям ингибитора СОХ-2 вместе с опиоидным анальгетиком, где имеется "кажущийся односторонний синергизм", полагают, что в действительности эти комбинации проявляют двусторонний синергизм, подразумевая, что ингибитор СОХ-2 усиливает действие опиоидного анальгетика, а другой опиоидный анальгетик усиливает действие ингибитора СОХ-2. Таким образом, другие аспекты изобретения относятся к комбинациям ингибитора СОХ-2 и опиоидного анальгетика, в которых доза каждого лекарственного средства снижается благодаря обнаруженному между лекарственными препаратами синергизму, и обезболивание, являющееся результатом комбинации лекарственных средств в сниженных дозах, удивительным образом усиливается. Двусторонний синергизм не всегда проявляется в действующих дозировках из-за того, что существует эффективное отношение опиоидного анальгетика к ингибитору СОХ-2 (означая, что опиоид обычно проявляет намного более высокую относительную анальгезирующую активность).
В некоторых предпочтительных аспектах изобретение относится к фармацевтическим композициям, содержащим ингибитор СОХ-2 в достаточном для оказания терапевтического действия количестве вместе с терапевтически эффективным или субтерапевтическим количеством опиоидного анальгетика, выбранного из группы, состоящей из альфентанила, аллилпродина, альфапродина, анилеридина, бензилморфина, безитрамида, бупренорфина, буторфанола, клонитазена, циклазоцина, дезоморфина, декстроморамида, дезоцина, диампромида, диаморфона, дигидрокодеина, дигидроморфина, дименоксадола, димефептанола, диметилтиамбутена, диоксафетилбутирата, дипипанона, эптазоцина, этогептазина, этилметилтиамбутена, этилморфина, этонитазена фентанила, героина, гидроморфона, гидроксипетидина, изометадона, кетобемидона, леваллорфана, леворфанола, левофенацилморфана, лофентанила, меперидина, мептазинола, метазоцина, метадона, метопона, морфина, мирофина, налбуфина, нарцеина, никоморфина, норлеворфанола, норметадона, налорфина, норморфина, норпипанона, опиума, оксикодона, оксиморфона, папаверитума, пентазоцина, фенадоксона, феноморфана, феназоцина, феноперидина, пиминодина, пиритрамида, профептазина, промедола, проперидина, пропирама, пропоксифена, суфентанила, тилидина, трамадола, их солей, их комплексов; смеси любого из вышеприведенных средств, смешанных мю-агонистов/антагонистов, комбинаций мю-антагонистов, их солей или комплексов и тому подобное. В некоторых предпочтительных аспектах опиоидный анальгетик является мю- или каппа-опиоидным агонистом. В некоторых предпочтительных аспектах изобретение относится к фармацевтическим композициям, содержащим ингибитор СОХ-2 в достаточном для оказания терапевтического действия количестве вместе с терапевтически эффективным или субтерапевтическим количеством опиоидного анальгетика, выбранного из группы, состоящей из морфина, дигидрокодеина, гидроморфона, оксикодона, оксиморфона, их солей и смесей любых из перечисленных средств.
В некоторых предпочтительных аспектах изобретение относится к фармацевтическим композициям, содержащим ингибитор СОХ-2 в достаточном для оказания терапевтического действия количестве вместе с дозой кодеина, которая является анальгезирующей, если вводится без ингибитора СОХ-2. Такая доза кодеина предпочтительно составляет приблизительно от 30 до 400 мг.
В некоторых предпочтительных аспектах изобретение относится к фармацевтическим композициям, содержащим ингибитор СОХ-2 в достаточном для оказания терапевтического действия количестве вместе с дозой гидрокодона, которая является анальгезирующей, если вводится без ингибитора СОХ-2. Такая доза гидрокодона предпочтительно составляет приблизительно от 5 до 2000 мг и предпочтительно, по меньшей мере, приблизительно 15 мг гидрокодона.
Кроме того, изобретение относится к способу эффективного лечения боли у человека, включающему в себя введение больному терапевтически эффективного количества ингибитора СОХ-2 вместе с дозой опиоидного анальгетика таким образом, что комбинация оказывает анальгезирующее действие, которое, по меньшей мере, приблизительно в 5 (и предпочтительно, по меньшей мере, в 10) раз выше, чем действие, производимое дозой опиоидного анальгетика в отдельности. В некоторых аспектах, синергическая комбинация оказывает анальгезирующее действие, которое вплоть до 30-40 раз выше действия, производимого дозой опиоидного анальгетика в отдельности. В некоторых предпочтительных аспектах дозы ингибитора СОХ-2 и опиоидного анальгетика вводят перорально. В дальнейших предпочтительных аспектах ингибитор СОХ-2 и опиоидный анальгетик вводят в форме единичных пероральных доз. В некоторых предпочтительных аспектах доза опиоидного анальгетика должна быть субтерапевтической, если ее вводят без ингибитора СОХ-2. В других предпочтительных аспектах доза опиоидного анальгетика является эффективной для обеспечения только обезболивания, но доза опиоида обеспечивает анальгезирующее действие, по меньшей мере, в 5 раз выше, чем обычно достигаемое дозой опиоида, применяемого в отдельности.
Кроме того, изобретение относится к применению фармацевтической комбинации ингибитора СОХ-2 с опиоидным анальгетиком для эффективного устранения боли у человека.
Далее, изобретение относится к использованию ингибитора СОХ-2 в производстве фармацевтического препарата, содержащего ингибитор СОХ-2 и опиоидный анальгетик для лечения боли.
Кроме того, изобретение относится к использованию опиоидного анальгетика в производстве фармацевтического препарата, содержащего ингибитор СОХ-2 и опиоидный анальгетик для лечения боли.
Изобретение относится также к способу эффективного устранения боли у человека, заключающемуся во введении анальгезирующего эффективного или субтерапевтического количества опиоидного анальгетика; и введении эффективного количества ингибитора СОХ-2 в количестве, эффективном для усиления анальгезирующего действия, оказываемого вышеупомянутым опиоидным анальгетиком. Ингибитор СОХ-2 может быть введен до, одновременно или после введения опиоидного анальгетика, пока интервал действия дозировки ингибитора СОХ-2 накладывается на интервал действия опиоидного анальгетика (или его анальгезирующего действия). Другими словами, согласно способу настоящего изобретения, в некоторых предпочтительных аспектах ингибитор СОХ-2 не нужно вводить в такой же дозировке или даже тем же путем, что и опиоидный анальгетик. Скорее способ относится к удивительному синергическому и/или аддитивному действию, достигаемому у человека, когда ему вводят анальгезирующие эффективные уровни опиоидного анальгетика и, до или в течение интервала действия дозировки опиоидного анальгетика, или во время действия обезболивания, вводят эффективное количество ингибитора СОХ-2 для усиления анальгезирующего действия опиоидного анальгетика. Если СОХ-2 вводят до введения опиоидного анальгетика, предпочтительно, чтобы интервалы действия доз для двух лекарственных средств накладывались, т.е. таким образом, чтобы анальгезирующая активность, превышающая, по крайней мере, частично действие в интервале доз опиоидного анальгетика, по крайней мере, отчасти приписывалась ингибитору СОХ-2.
В дополнительном способе согласно изобретению удивительное синергическое и/или аддитивное действие, достигаемое у человека, когда анальгезирующие эффективные уровни ингибитора СОХ-2 вводят человеку, и в интервале действия ингибитора СОХ-2 или во время действия обезболивания благодаря введению ингибитора СОХ-2, вводят эффективное количество опиоидного анальгетика для усиления анальгезирующего действия ингибитора СОХ-2.
В дальнейшем аспекте настоящее изобретение связано с пероральной твердой лекарственной формой, содержащей анальгезирующее эффективное количество опиоидного анальгетика вместе с таким количеством ингибитора СОХ-2 или его фармацевтически приемлемой соли, которое усиливает активность опиоидного анальгетика.
Необязательно, пероральная твердая лекарственная форма включат в себя носитель отсроченного высвобождения, который вызывает отсроченное высвобождение опиоидного анальгетика или как опиоидного анальгетика, так и ингибитора СОХ-2, когда лекарственная форма соприкасается с желудочно-кишечной жидкостью. Лекарственная форма отсроченного высвобождения может содержать множество субстратов, которые включают в себя лекарственные средства. Субстраты могут содержать сфероиды матрикса или инертные фармацевтически приемлемые шарики, которые покрыты лекарственными препаратами. Покрытые шарики затем предпочтительно покрывают сверху оболочкой отсроченного высвобождения, содержащей носитель отсроченного высвобождения. Матриксный сфероид может включать в себя носитель отсроченного высвобождения в самом матриксе; или матрикс может представлять собой матрикс нормального высвобождения, содержащий лекарства, матрикс, имеющий оболочку, которая содержит носитель отсроченного высвобождения. В других аспектах пероральная твердая лекарственная форма включает в себя ядро таблетки, содержащее лекарственные средства в матриксе нормального высвобождения, с ядром таблетки, покрытым оболочкой, включающей в себя носитель отсроченного высвобождения. В других аспектах таблетка содержит лекарственные средства в матриксе отсроченного высвобождения, содержащем носитель отсроченного высвобождения. В дальнейших аспектах таблетка содержит опиоидный анальгетик в матриксе отсроченного высвобождения и ингибитор СОХ-2, покрытый оболочкой, в таблетке в слое немедленного высвобождения.
Во многих предпочтительных аспектах изобретения композиции, содержащие перечисленные в описании ингибиторы СОХ-2 и опиоидные лекарственные средства, вводят перорально. Подобные пероральные лекарственные формы могут содержать одно или оба лекарственных средств в форме немедленного или отсроченного высвобождения. Для облегчения введения предпочтительно, чтобы пероральная лекарственная форма содержала оба лекарственных препарата. Пероральные лекарственные формы могут быть в виде таблеток, пастилок, лепешек, водных или масляных эмульсий, диспергирующих порошков или гранул, эмульсий, многокомпонентных композиций, сиропов, эликсиров и тому подобного.
Фармацевтические композиции, содержащие СОХ-2 и/или опиоидные лекарственные средства, перечисленные в описании, также могут быть в виде микрочастиц, например микрокапсул, микросфер и тому подобного, которые могут быть инъецированы или имплантированы больному человеку, или других имплантируемых лекарственных форм, известных специалистам в области фармацевтических композиций. Для облегчения введения предпочтительно, чтобы подобные лекарственные формы содержали оба лекарственных препарата.
Дополнительные фармацевтические композиции, рассматриваемые далее в изобретении, включают в себя трансдермальные лекарственные формы, суппозитории, порошки и аэрозоли для ингаляции и трансбуккальные таблетки.
Для комбинации ингибитора СОХ-2 и опиоидного анальгетика к тому же существуют разные способы введения.
Следует иметь в виду, что в рамках настоящего изобретения вводят названия, имеющие следующие значения:
В настоящем изобретении название "эффективное обезболивание" определяется как удовлетворительное снижение или снятие боли наряду с допустимым уровнем побочных эффектов, определяемых больным человеком.
Согласно изобретению, название "эффективное устранение боли" означает объективную оценку ответа больного человека (испытываемая боль и наличие побочных эффектов) на лечение анальгезирующим средством, а также субъективную оценку терапевтического лечения больным, подвергающимся такому лечению. Специалисты в данной области понимают, что эффективное обезболивание будет варьировать, в зависимости от многих факторов, включая индивидуальные особенности больного.
В настоящем изобретении название "опиоидный анальгетик" определяется как лекарственное средство в его основной форме, или его фармацевтически приемлемая соль, или комплекс.
В настоящем изобретении название "ингибитор СОХ-2" определяется как лекарственное средство в его основной форме, или его фармацевтически приемлемая соль, или комплекс.
В настоящем изобретении название "непрерывное (отсроченное) высвобождение" определяется как высвобождение лекарственного средства (опиоидного анальгетика) из трансдермальной композиции с такой скоростью, что концентрации (уровни) в крови (например, в плазме) поддерживаются в пределах терапевтического диапазона (выше минимальной эффективной анальгезирующей концентрации или "МЕАС"), но ниже токсических уровней, в течение приблизительно 12 часов или дольше.
Название "устойчивое состояние" означает, что кривая концентрации в плазме крови данного лекарственного средства в значительной степени воспроизводится от дозы к дозе.
Название "минимальная эффективная анальгезирующая концентрация" определяют согласно изобретению как минимальная эффективная терапевтическая концентрация лекарственного средства в плазме крови, при которой достигается, по крайней мере, некоторое ослабление боли у данного больного. Специалисты в данной области медицины поймут, что измерение боли является весьма субъективным фактором, и среди больных могут наблюдаться значительные индивидуальные различия.
Применяемые в настоящем изобретении ингибиторы СОХ-2 будут иметь противовоспалительные, жаропонижающие и анальгезирующие свойства, подобные стандартным нестероидным противовоспалительным лекарственным средствам, и, кроме того, будут ингибировать индуцируемые гормонами сокращения матки и обладать потенциальным противораковым действием, но при этом будут обладать пониженной способностью индуцировать некоторые связанные с механизмом действия побочные эффекты. В частности, такие ингибиторы СОХ-2 должны обладать сниженным потенциалом токсичности в отношении желудочно-кишечного тракта, сниженным потенциалом в отношении индукции почечных побочных эффектов, сниженным по отношению ко времени кровотечения действием и уменьшенной способностью индуцировать приступы астмы у чувствительных к аспирину субъектов-астматиков. Об ингибиторах СОХ-2 сообщалось в данной области, и было показано, что многие известные химические структуры производят ингибирование циклооксигеназы-2. В целях настоящего изобретения название "ингибитор СОХ-2" определяют как все соединения, которые обладают ингибиторной по отношению к СОХ-2 активностью и которые предпочтительно проявляют специфичность, по меньшей мере, в 9 раз выше для СОХ-2 по сравнению с СОХ-1, либо in vitro (как определено, например, посредством измерений IC50), либо in vivo (как определено, например, посредством измерений ED50). Подобные ингибиторы СОХ-2 будут применяться в связи с настоящим изобретением и рассматриваются в прилагаемой формуле изобретения. Предпочтительно, чтобы применяемые в настоящем изобретении ингибиторы СОХ-2 имели такие значения IC50 in vitro и/или ED50 in vivo, чтобы отношение СОХ-1 к СОХ-2 было выше приблизительно в 20 раз или более, предпочтительно в 100 раз или более и самое предпочтительное - в 1000 раз или более.
Некоторые предпочтительные ингибиторы СОХ-2 включают в себя целекоксиб (SC-58635), DUP-697, флосулид (CGP-28238), мелоксикам, 6-метокси-2-нафтилуксусную кислоту (6-MNA), Vioхх (МК-966), набуметон (пролекарство для 6-MNA), нимесулид, NS-398, SC-5766, SC-58215, Т-614 или их комбинации. Существует ряд ингибиторов СОХ-2, находящихся в стадии разработки по состоянию на середину 1998 г. Они включают в себя мелоксикам (коммерчески доступный в Великобритании по состоянию на 1996 г. Boerhinger-Ingelheim); нимесулид (выпущенный в 1985 в Европе из Hesinn); набуметон (6-MNA является активным метаболитом) (коммерчески доступный в США как Relafin™); целекоксиб (SC-58635) (NDA регистрация посредством Searle, оценена в сентябре 1998); Vioxx (MK-966, L745337) (NDA регистрация посредством Merck, оценена в ноябре 1998); D-1367 (Chiroscience; в фазе I в Великобритании); Т-614 (Toyama; в фазе II в Японии и в фазе I в Великобритании); SC-57666 (Monsanto; в фазе I в США).
В опытах, обсуждаемых в 1996 на ежегодной конференции American College of Rheumatology, продемонстрировали, что целекоксиб является эффективным для больных и лишен желудочно-кишечных побочных эффектов у нормальных добровольцев (Scrip 2175, октябрь 1996, р. 15). В исследованиях на нормальных добровольцах 128 субъектов получали целекоксиб, 100 мг или 200 мг дважды в день, или напроксен, или плацебо в течение одной недели. В группе, получавшей целекоксиб, и у субъектов, получавших плацебо, не наблюдали желудочно-кишечных признаков болезни или симптомов, в то время как в группе, получавшей напроксен, 20% субъектов испытывали желудочно-кишечные признаки болезни и симптомы. Кроме того, у нормальных добровольцев целекоксиб не вызывал изменений функции тромбоцитов. 293 исследуемых больных с остеоартритом получали целекоксиб 40 мг, 100 мг или 200 мг или плацебо дважды в день в течение четырех недель. Целекоксиб снижал симптомы в значительной степени, и скорости исчезновения симптомов в группе, получавшей целекоксиб в высоких дозах, были ниже, чем в группе, получавшей плацебо. Больные с ревматоидным артритом получали целекоксиб в дозах 100 мг, 200 мг или 400 мг или плацебо дважды в день в течение четырех недель. Что касается больных остеоартритом, показатели симптомов улучшались у больных, получавших целекоксиб, по сравнению с принимающими плацебо, а скорость исчезновения симптомов была ниже у больных, принимавших целекоксиб. Об ингибиторах СОХ-2 сообщалось в данной области, и было показано, что многие известные химические структуры производят ингибирование циклооксигеназы-2.
Ингибиторы СОХ-2 описаны в патентах США №№5616601; 5604260; 5593994; 5550142; 5536752; 5521213; 5639780; 5604253; 5552422; 5510368; 5436265; 5409944 и 5130311, все из которых включены в данный текст в виде ссылки. Многие ингибиторы СОХ-2 могут быть описаны химически как арилсульфонамиды. Действительно, как целекоксиб, так и Vioxx, которые рассматривают как "сверхизбирательные", являются арилсульфонамидами, и, более специфично, бензолсульфонамидами. Эти соединения будут применяться в способах и композициях настоящего изобретения. Однако специалисты в данной области оценивают, что многие дополнительные ингибиторы СОХ-2 идентифицированы в данной области и используются в связи со способами и композициями настоящего изобретения.
Использование связи между структурой и функцией для оценки ингибиторов СОХ-2 является проблематичным, поскольку эти ингибиторы СОХ являются ферментами-самоубийцами. Таким образом, при анализе in vitro показано, что величина IC50 изменяется с течением времени. По этой причине опубликованные величины IC50, полученные в разных лабораториях, для обычных ингибиторов СОХ представлены как величины, отличающиеся более чем на два порядка. Это затрудняет сравнение величины ингибирования СОХ-1, полученной в одной лаборатории, с величиной ингибирования СОХ-2, полученной в другой лаборатории (см., например, D.E.Griswold and J.L.Adams, Med. Res, Rev. 16:181-206). Таким образом, предпочтительно, чтобы при исследовании ингибиторов СОХ для сравнения их относительных активностей сравнивались только результаты одного и того же анализа, проводимого в одно и то же время. При использовании ранее полученных данных предпочтительно брать данные только из перечня некоторых соединений, которые получены одной группой исследователей, так, чтобы можно было определить относительные активности. Таблица 1, представленная ниже, показывает характерные данные для характерных NSAID и некоторых ингибирующих СОХ-2 соединений. Данные собраны из ряда разных источников и выбраны из доступных лабораторий с использованием ссылок, в которых приводятся некоторые соединения из одной и той же статьи и которые содержат данные, относительно сравнимые с данными, полученными из некоторых других лабораторий (т.е. в пределах разумного диапазона вариаций, понимая, что результаты из разных лабораторий могут варьировать вплоть до трех порядков величин для агентов, действующих как ферменты-самоубийцы). Следует учитывать, что большинство приведенных в таблице 1 величин являются результатом анализа in vitro (за исключением тех случаев, где активность выражают в мг/кг). Литературные данные подтверждают, что отношения активности СОХ-1/СОХ-2 обычно сохраняются in vivo, но это не всегда справедливо. Например, индометацин является всегда избирательным в отношении COX-1 in vitro и in vivo, а напроксен, который является избирательным в отношении COX-1 in vitro, часто (но не всегда) является избирательным к COX-2 In vivo. Отчасти это происходит вследствие весьма искусственных условий анализа in vitro. Первых два структурных ряда были признаны в качестве ингибиторов СОХ-2, проявляющих мало заметную ульцерогенную активность. Эти ранние соединения включают в себя арилсульфонамиды нимесулид, NS-398, и CGP 23238 и 1,2-диарилгетероциклы Dup-697 и SC-58125. Griswold and Adams описывают связь структуры и активности в некоторых деталях (Med. Res. Rev. 16:282-206, 1996).
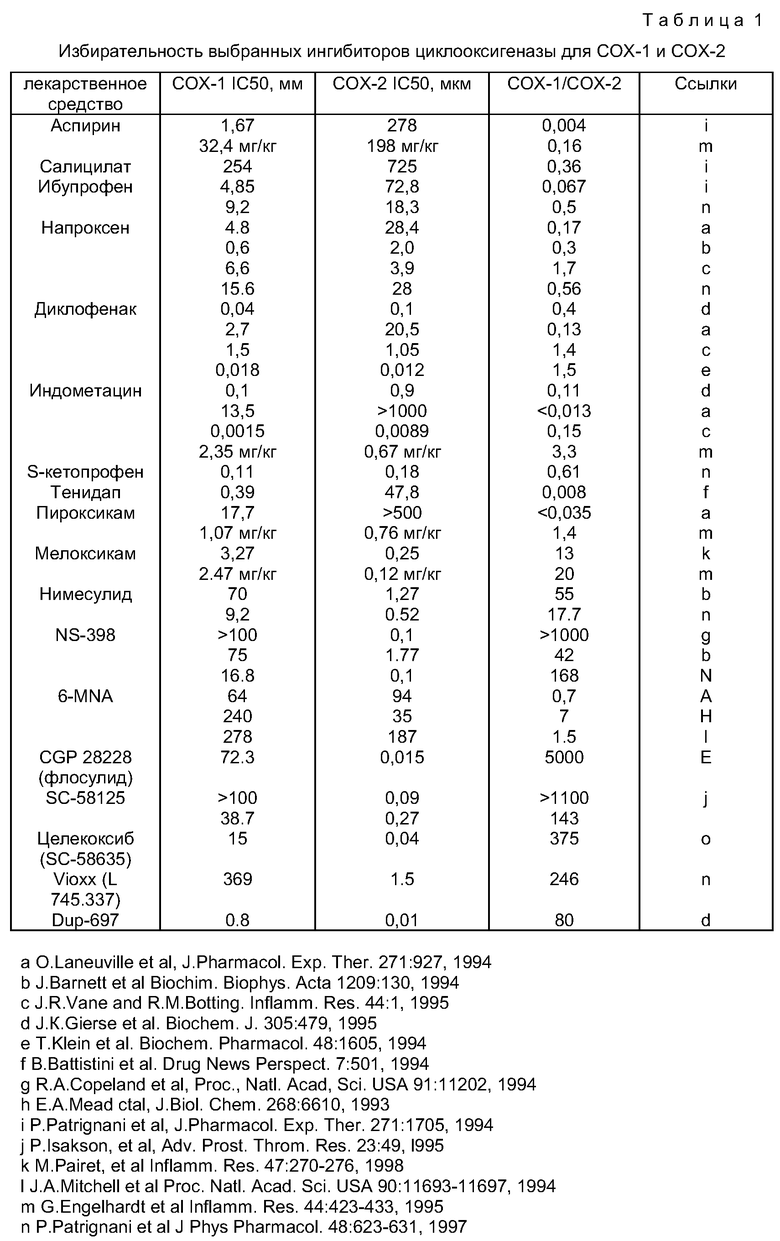
Например, как сообщил Famaey J.P., Inflamm. Res. 1997 Nov. 46 (11):437-446, нимесулид, сульфонанилидное соединение с противовоспалительными свойствами, обладает фармакологическим действием, что предполагает, что оно может быть избирательным ингибитором СОХ-2. В некоторых анализах in vitro, в которых использованы либо очищенные препараты СОХ-2 и СОХ-1, либо клеточные препараты, исследователи десяти из одиннадцати различных групп показали, что нимесулид избирательно ингибирует СОХ-2. Сообщалось, что отношение ингибиторов СОХ-2/СОХ-1 изменяется соответственно анализируемому препарату, приблизительно от 0,76 до 0,0004, т.е. от 1,3 до 2512 раз избирательность в отношении СОХ-2 выше, чем в отношении COX-1. Кроме того, проводимый на здоровых добровольцах анализ цельной крови in vivo показал, что имеет место значительное снижение продукции COX-2 PGE2, при этом не наблюдается какого-либо влияния на продукцию СОХ-1 ТХВ2 (субъекты, проходившие лечение нимесулидом по 100 мг дважды в день в течение 2 недель) в сравнении с субъектами, проходившими лечение аспирином (300 мг три раза в день в течение 2 недель), у которых не наблюдали влияния на продукцию СОХ-2 PGE2 и отмечали почти полное подавление продукции СОХ-1 ТХВ2. Таким образом, нимесулид можно рассматривать как относительно избирательный ингибитор СОХ-2. При рекомендуемой дозировке 100 мг дважды в день он является таким же эффективным анальгезирующим и противовоспалительным агентом, как и классические NSAID, и хорошо переносимым лекарственным средством с немногими побочными эффектами согласно результатам открытых исследований крупного масштаба и глобальной оценке большого ряда контролируемых и неконтролируемых сравнительных опытов.
Неограничивающий перечень опиоидных анальгезирующих лекарственных средств, которые могут быть использованы в настоящем изобретении, включает в себя альфентанил, аллилпродин, альфапродин, анилеридин, бензилморфин, безитрамид, бупренорфин, буторфанол, клонитазен, кодеин, циклазоцин, десоморфин, декстроморамид, дезоцин, диампромид, диаморфон, дигидрокодеин, дигидроморфин, дименоксадол, димефептанол, диметилтиамбутен, диоксафетилбутират, дипипанон, эптазоцин, этогептазин, этилметилтиамбутен, этилморфин, этонитазен, фентанил, героин, гидрокодон, гидроморфон, гидроксипетидин, изометадон, кетобемидон, леваллорфан, леворфанол, левофенацилморфан, лофентанил, меперидин, мептазинол, метазоцин, метадон, метопон, морфин, мирофин, налбуфин, нарцеин, никоморфин, норлеворфанол, норметадон, налорфин, норморфин, норпипанон, опиум" оксикодон, оксиморфон, папаверетум, пентазоцин, фенадоксон, феноморфан, феназоцин, феноперидин, пиминодин, пиритрамид, профептазин, промедол, проперидин, пропирам, пропоксифен, суфентанил, тилидин, трамадол, их соли, их комплексы; смеси любых из перечисленных средств, смешанные мю-агонисты/антагонисты, комбинации мю-антагонистов, их соли или комплексы и тому подобное. В некоторых предпочтительных аспектах опиоидным анальгетиком является мю- или каппа-опиоидный агонист. В дополнительных предпочтительных аспектах опиоидным анальгетиком является избирательный каппа-агонист.
В некоторых предпочтительных аспектах опиоидный анальгетик выбирается из кодеина, гидроморфона, гидрокодона, оксикодона, дигидрокодеина, дигидроморфина, диаморфона, морфина, трамадола, оксиморфона, их солей или смесей.
Настоящее изобретение относится к анальгезирующим препаратам для перорального введения, которые представляют собой комбинацию ингибитора СОХ-2 или его фармацевтически приемлемой соли и опиоидного анальгетика или его фармацевтически приемлемой соли. Комбинация предпочтительно проявляет синергическое или, по крайней мере, аддитивное действие при анальгезирующих дозировках.
Дозированные уровни ингибитора СОХ-2 приблизительно от 0,005 мг до 140 мг на киллограмм веса тела в день становятся на порядок терапевтически более эфективными в комбинации с опиоидным анальгетиком. Или же ингибитор СОХ-2, приблизительно от 0,25 мг до 7 г на больного в день, вводится в комбинации с опиоидным анальгетиком. Например, воспаление можно лечить эффективно путем введения приблизительно от 0,005 до 50 мг ингибитора СОХ-2 на кг веса тела в день или иначе приблизительно от 0,25 мг до 3,5 г на больного в день.
Количество ингибитора СОХ-2, которое можно комбинировать с носителями с получением единичной лекарственной формы, содержащей комбинацию ингибитора СОХ-2 и опиоидного анальгетика, будет зависеть от конкретного больного и способа введения. Например, предназначенная для перорального введения композиция может содержать от 0,25 мг до 5 г ингибитора СОХ-2, смешанного с соответствующим и подходящим количеством носителя, которое может колебаться приблизительно от 5 до 95% тотальной композиции. Стандартные лекарственные формы будут содержать в основном приблизительно от 0,5 мг до 1500 мг ингибитора СОХ-2 и обычно 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, 600 мг, 800 мг или 1000 мг и так далее, вплоть до 1500 мг.
В одном из вариантов ингибитор СОХ-2 находится в пероральной лекарственной форме отсроченного высвобождения с гидроморфоном в качестве терапевтически активного опиоида в количестве приблизительно от 2 мг до 64 мг хлоргидрата гидроморфона. Или же лекарственная форма может содержать молярные эквивалентные количества других солей гидроморфона или основания гидроморфона. В других аспектах опиоидный анальгетик представляет собой морфин, и пероральные лекарственные формы отсроченного высвобождения настоящего изобретения по весу включают в себя приблизительно от 2,5 мг до 800 мг морфина. В другом варианте опиоидный анальгетик представляет собой оксикодон, и пероральные лекарственные формы отсроченного высвобождения включают в себя приблизительно от 2,5 мг до 800 мг оксикодона. Опиоидный анальгетик может представлять собой гидрокодон, и пероральные дозированные формы отсроченного высвобождения могут включать в себя анальгезирующие дозы приблизительно от 8 мг до 50 мг гидрокодона на стандартную лекарственную форму. Опиоидный анальгетик может представлять собой трамадол, и пероральные лекарственные формы отсроченного высвобождения могут включать в себя приблизительно от 25 мг до 800 мг трамадола на стандартную лекарственную форму. Лекарственная форма может содержать более одного опиоидного анальгетика для достижения, по существу, эквивалентного терапевтического действия.
Предпочтительные комбинации согласно изобретению содержат эффективное количество ингибитора СОХ-2, выбранного из группы, состоящей из нимесулида, мелорикана и флосулида, и эффективное количество опиоидного анальгетика, выбранного из группы, состоящей из трамадола, гидроморфона, морфина, оксикодона, гидрокодона и дигидрокодеина в отношениях, представленных в таблице 2. В некоторых предпочтительных аспектах отношение вышеупомянутых опиоидов к вышеупомянутым ингибиторам СОХ-2 представлены в таблице 2.
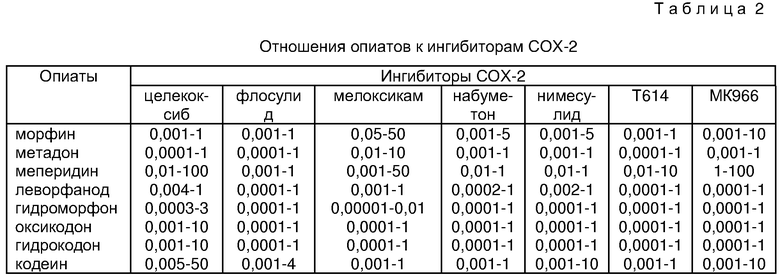
Другими словами, в таблице II приведены отношения морфина к целекоксибу приблизительно от 0,001:1 до 1:1; отношение метадона к флосулиду отношение составляет приблизительно от 0,0001:1 до 1:1 и так далее.
В некоторых предпочтительных вариантах согласно настоящему изобретению предпочтительной является такая пероральная лекарственная форма, которая включает в себя следующие комбинации опиоида и ингибитора СОХ-2: морфина 40 мг плюс 40 мг флосулида; морфина 40 мг плюс 6 мг нимесулида; оксикодона 20 мг плюс 20 мг флосулида; оксикодона 40 мг плюс 4 мг нимесулида; гидроморфона 5 мг плюс 20 мг флосулида или гидроморфона 5 мг плюс 4 мг нимесулида.
Вводимая дозировка будет, конечно, изменяться в зависимости от известных факторов, таких как фармакодинамические характеристики каждого агента комбинации и ее способ введения или применения, а также возраст, здоровье и вес больного. Дозировка также зависит от природы и выраженности симптомов, конкурентного лечения, если оно имеется, частоты лечения и желаемого результата. Композиция, содержащая любую из вышеперечисленных комбинаций опиоидных анальгетиков и ингибиторов СОХ-2, может быть введена дробными дозами в пределах от 2 до 6 раз в день или в форме отсроченного высвобождения, которая обеспечивает скорость высвобождения, эффективную для достижения желаемых результатов.
Оптимальные отношения ингибитора СОХ-2 и опиоидного анальгетика определяют стандартными анализами, хорошо известными в данной области, для определения опиоидной и анальгезирующей активности. Например, фенил-п-бензохиноновый тест можно использовать для установления анальгезирующей активности. Фенил-п-бензохиноновый тест, индуцирующий корчи у мышей (H.Bluroberg et al., 1965, Proc. Spc. Exp. Med. 118:763-766, включен в данный текст в виде ссылки, также как и его известные модификации), является стандартной процедурой, которая используется для определения и сравнения анальгезирующей активности различных классов анальгезирующих лекарственных средств с хорошей корреляцией с анальгезирующей активностью у человека. Представленные на изоболограмме данные для мыши могут быть перенесены на другие виды, у которых пероральные анальгезирующие дозы индивидуальных соединений являются известными или могут быть установлены. Способ состоит из определения процента дозы ED50 для каждого соотношения доз по оптимальной кривой, полученной с помощью регрессионного анализа, из изобалограммы мыши, умножения каждого компонента на его эффективную дозу для данного вида (животного) и затем получения соотношения количества ингибитора СОХ-2 и опиоидного анальгетика. Эта основная корреляция для анальгезирующих свойств дает возможность установить диапазон эффективности и для человека (E.W.Pelikan, 1959, The Pharmacologist 1:73; включено в данный текст в виде ссылки).
Применение равноэффективной дозовой заместительной модели и анализ криволинейной регрессии, использующий все данные для индивидуальных соединений и различных дозовых соотношений для комбинаций, указывает на существование неожиданно повышенной анальгезирующей активности комбинаций ингибитора СОХ-2 и опиоидного анальгетика, т.е. полученная активность выше, чем активность, ожидаемая от суммы активностей индивидуальных компонентов.
Настоящее изобретение относится к лекарственным формам немедленного высвобождения эффективного анальгезирующего количества комбинации ингибитора СОХ-2 и опиоидного анальгетика. Лекарственная форма немедленного высвобождения может быть получена в виде таблетки или мультичастицы, которую можно инкапсулировать. Можно использовать также и другие лекарственные формы немедленного высвобождения, известные в данной области.
Композиции согласно изобретению представляют собой благоприятную возможность для достижения ослабления боли от умеренной до сильной при наличии воспаления или без него. Благодаря синергическому и/или аддитивному действию, производимому полученной комбинацией опиоидного анальгетика и ингибитора СОХ-2, можно применять сниженные дозировки каждого из препаратов - ингибитора СОХ-2 и опиоидного анальгетика. Используя меньшие количества одного или обоих лекарственных средств, можно снизить количество и степень выраженности связанных с каждым компонентом побочных эффектов. Кроме того, изобретенная комбинация лишена побочных эффектов, к которым некоторые больные особенно чувствительны.
Настоящее изобретение относится к способу ингибирования СОХ-2 и лечения заболеваний, опосредованных СОХ-2, заключающему в себе введение больному, нуждающемуся в подобном лечении, нетоксического терапевтически эффективного количества комбинации ингибитора СОХ-2 и опиоидного анальгетика настоящего изобретения. Эти заболевания включают в себя боль от умеренной до сильной, имеющую самые различные этиологии, включая, но не ограничиваясь ими, раковую боль и послеоперационную боль, лихорадку и воспаление целого ряда состояний, включая ревматическую атаку, симптомы, связанные с гриппом или другими вирусными инфекциями, насморк, боли в задненижнем и шейном отделе, дисменорею, головную боль, зубную боль, растяжения и деформации, миозит, невралгию, синовит, артрит, включая ревматоидный артрит, дегенеративные заболевания суставов (остеоартрит), подагру и анкилозирующий спондилоартрит, бурсит, ожоги и раны. Кроме того, комбинацию ингибитора СОХ-2 и опиоидного анальгетика применяют в качестве альтернативных обычным нестероидным противовоспалительным лекарственным средствам или комбинациям NSAID’s с другими лекарственными препаратами, особенно когда подобные нестероидные противовоспалительные лекарственные средства могут быть противопоказаны, как, например, больным с пептической язвой, гастритами, регионарным энтеритом, язвенным колитом, дивертикулитом или с повторяющимися случаями желудочно-кишечных поражений; GI-кровотечением, расстройствами свертывания, включая анемию, как, например, гипопротромбинемию, гемофилию или другими проблемами кровотечения; заболеванием почек; больным до операции или принимающим антикоагулянты.
Лекарственные формы отсроченного высвобождения согласно изобретению обычно позволяют достичь и поддерживать терапевтические уровни, в основном, без значительного увеличения в интенсивности и/или степени выраженности сопутствующих побочных эффектов, как, например, тошнота, рвота или сонливость, которые часто связаны с высокими уровнями в крови опиоидных анальгетиков. Имеются данные, позволяющие предполагать, что применение настоящих лекарственных форм приводит к снижению риска привыкания к чрезмерному употреблению лекарственных средств.
Может быть получена комбинация ингибитора СОХ-2 и опиоидного анальгетика для увеличения продолжительности анальгезирующего действия, допускающая однократную суточную дозировку. Эти композиции, при сравнимых суточных дозировках подходящего лекарственного средства немедленного высвобождения, ассоциированы со снижением частоты случаев с тяжелыми неблагоприятными реакциями от лекарственных средств и могут вводиться в более низкой суточной дозе, чем общепринятые пероральные лекарственные препараты, при этом сохраняя контроль над болью.
Комбинацию ингибитора СОХ-2 и опиоидного анальгетика можно использовать в смеси со стандартными наполнителями, т.е. фармацевтически приемлемыми органическими и неорганическими веществами в качестве носителей, подходящими для перорального, парентерального, назального, внутривенного, подкожного, тонкокишечного или любого другого подходящего способа введения, известного в данной области. Подходящие фармацевтически приемлемые носители включают в себя, не ограничиваясь ими, воду, солевые растворы, спирты, аравийскую камедь, растительные масла, бензиловые спирты, полиэтиленгликоли, желатин, углеводы, как, например, лактозу, амилозу или крахмал, стеарат магния, тальк, кремневую кислоту, вязкий парафин, парфюмерное масло, моноглицериды и диглицериды жирных кислот, эфиры жирных кислот и пентаэритрита, гидроксиметилцеллюлозу, поливинилпирролидон и т.д. Фармацевтические препараты можно стерилизовать и, если желательно, смешивать с добавочными агентами, например смазочными маслами, консервантами, стабилизаторами, смачивателями, эмульгаторами, солями для поддержания осмотического давления, буферами, красителями, вкусовыми и/или ароматическими веществами и тому подобное. Их также можно комбинировать, если желательно, с другими активными агентами, например другими анальгезирующими агентами. Для парентерального применения особенно подходящими являются масляные или водные растворы, а также суспензии, эмульсии или имплантаты, включая суппозитории. Ампулы являются подходящими стандартными лекарственными формами. Для перорального применения особенно подходящими являются таблетки, драже, жидкости, капли, суппозитории или капсулы, микрокапсулы (caplets) и желатиновые капсулы (gelcaps). Композиции, предназначенные для перорального применения, можно приготовить согласно любому способу, известному в данной области, и такие композиции могут содержать один или более агентов, выбранных из группы, состоящей из инертных, нетоксических фармацевтических наполнителей, которые являются подходящими для производства таблеток. Такие наполнители включают в себя, например, инертный разбавитель, как, например, лактозу; гранулирующие и дезинтегрирующие агенты, как, например, кукурузный крахмал; связующие агенты, как, например, крахмал и смазочные агенты, как, например, стеарат магния. Таблетки могут быть не покрыты оболочкой или их покрывают оболочкой по известной технологии для изящества или чтобы задержать высвобождение активных ингредиентов. Композиции для перорального применения могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешивают с инертным разбавителем.
Водные суспензии содержат вышеупомянутую комбинацию лекарственных средств, и такая смесь имеет один или более наполнителей, подходящих в качестве суспендирующих агентов, например фармацевтически приемлемых синтетических смол, как, например, гидроксипропилметилцеллюлозы, или природных смол. Масляные суспензии могут быть приготовлены путем суспендирования вышеуказанной комбинации лекарственных средств в растительном масле или минеральном масле. Масляные суспензии могут содержать загуститель, как, например, пчелиный воск или цетиловый спирт. Сироп, эликсир или тому подобное можно использовать, когда добавляют подсластитель. Можно приготовить также инъецируемые суспензии, в которых употребляют соответствующие жидкие носители, суспендирующие агенты и тому подобное. Также можно высушить активные соединения при температуре ниже 0°С и применять полученные лиофилизированные соединения, например, для приготовления продуктов для инъекции.
Кроме того, способ лечения и фармацевтические композиции настоящего изобретения могут включать в себя одно или более лекарственных средств, кроме ингибитора СОХ-2 и опиоидного анальгетика, где дополнительное лекарство (лекарства) может или не может действовать с ними синергическим образом. Примеры подобных дополнительных лекарственных средств включают в себя нестероидные противовоспалительные агенты, включая ибупрофен, диклофенак, напрофен, беноксапрофен, флурбипрофен, фенопрофен, флубуфен, кетопрофен, индопрофен, пиропрофен, карпрофен, оксапрозин, прамопрофен, муропрофен, триоксапрофен, супрофен, аминопрофен, тиапрофеновую кислоту, флупрофен, буклоксовую кислоту, индометацин, сулиндак, толметин, зомепирак, тиопинак, зидометацин, ацетемацин, фентиазак, клиданак, окспинак, мефенамовую кислоту, меклофенамовую кислоту, флуфенамовую кислоту, нифлумовую кислоту, толфенамовую кислоту, дифлурисал, флуфенисал, пироксикам, судоксикам или изоксикам и тому подобное. Другие подходящие дополнительные лекарственные средства, которые могут быть включены в лекарственные формы настоящего изобретения, включают в себя ацетаминофен, аспирин и другие неопиоидные анальгетики.
Лекарственные формы контролируемого высвобождения
Комбинацию ингибитора СОХ-2 и опиоидного анальгетика можно получить в качестве пероральной композиции контролируемого или отсроченного высвобождения в любой подходящей таблетке, покрытой оболочкой таблетке или многокомпонентной композиции, известной специалистам в данной области. Лекарственная форма отсроченного высвобождения может необязательно включать в себя носитель отсроченного высвобождения, который заключается в матрикс наряду с опиоидом или который применяется в качестве покрытия отсроченного высвобождения.
Дозированная форма отсроченного высвобождения может включать в себя опиоидный анальгетик в форме отсроченного высвобождения и ингибитор СОХ-2 в форме отсроченного высвобождения или в форме немедленного высвобождения. Ингибитор СОХ-2 может быть заключен в матрикс отсроченного высвобождения наряду с опиоидом; заключен в покрытие отсроченного высвобождения; заключен как отдельный слой отсроченного высвобождения или слой немедленного высвобождения; или может быть заключен как порошок, гранулы и т.д. в желатиновую капсулу с субстратами настоящего изобретения. Или же дозированная форма отсроченного высвобождения может иметь ингибитор СОХ-2 р форме отсроченного высвобождения и опиоидный анальгетик в форме отсроченного высвобождения или в форме немедленного высвобождения.
Пероральная лекарственная форма согласно изобретению может быть получена в виде, например, гранул, сфероидов, шариков, пилюль (далее совокупно называются "мультичастицами") и/или частиц. Количество мультичастиц, которое является эффективным для достижения желаемой дозы опиоида во времени, может быть помещено в капсулу или включено в любую другую подходящую пероральную твердую форму.
В одном предпочтительном аспекте настоящего изобретения лекарственная форма отсроченного высвобождения включает в себя подобные частицы, содержащие или включающие в себя активный ингредиент, где частицы имеют диаметр приблизительно от 0,1 мм до 2,5 мм, предпочтительно от 0,5 мм до 2 мм.
В некоторых аспектах частицы включают в себя матриксы нормального высвобождения, содержащие опиоидный анальгетик с ингибитором СОХ-2 или без него. Эти частицы затем покрывают оболочкой с носителем отсроченного высвобождения в тех аспектах, где ингибитор СОХ-2 немедленно высвобождается, ингибитор СОХ-2 может быть включен в отдельные матриксные частицы нормального высвобождения или может быть введен совместно в отличную композицию немедленного высвобождения, которая либо заключена в желатиновую капсулу, либо вводится отдельно. В других аспектах частицы включают в себя инертные шарики, которые покрывают оболочкой с опиоидным анальгетиком с ингибитором СОХ-2 или без него. Согласно этому оболочку, включающую в себя носитель отсроченного высвобождения, применяют к шарикам в качестве покрытия.
Частицы предпочтительно покрывают тонкой пленкой из материала, который позволяет высвобождение опиоида (или соли) и, если желательно, ингибитора СОХ-2, с замедленной скоростью в водной среде. Покрытие из тонкой пленки выбирается таким, чтобы достигалась в комбинации с другими установленными свойствами желаемая скорость высвобождения in vitro. Покрытия композиций отсроченного высвобождения настоящего изобретения должны быть способными производить крепкую сплошную пленку, которая является гладкой и изящной, способной поддерживать пигменты и другие покрывающие добавки, нетоксические, инертные и без отлипа.
ПОКРЫТИЯ
Лекарственные формы настоящего изобретения необязательно покрывают оболочкой из одного или более материалов, способствующих регуляции высвобождения или для защиты композиции. В одном аспекте покрытия делают для того, чтобы облегчить либо рН-зависимое либо рН-независимое высвобождение, например, в случаях, когда они подвергаются действию жидкостей желудочно-кишечного тракта. рН-зависимое покрытие служит для того, чтобы обеспечить высвобождение опиоида в желаемые области желудочно-кишечного (GI) тракта, например желудок или тонкий кишечник, таким образом, что происходит всасывание, способное поддержать анальгезирующее действие у больного, по крайней мере, приблизительно 12 часов и предпочтительно вплоть до 24 часов. В случаях, когда желательно рН-независимое покрытие, то это покрытие программируется таковым, чтобы достичь оптимального высвобождения независимо от изменений рН окружающей жидкости, например желудочно-кишечного тракта (ЖКТ). Также можно получить композиции, которые высвобождают часть дозы в одном желаемом месте ЖКТ, например желудке, и высвобождают остальную часть дозы в другом месте ЖКТ, например тонком кишечнике.
Композиции согласно изобретению, для получения которых используют рН-зависимые покрытия, могут обладать эффектом повторного действия таким образом, что незащищенное лекарственное средство покрывают энтеросолюбильным покрытием, и оно высвобождается в желудок, в то время как остальная часть, защищенная энтеросолюбильным покрытием, высвобождается в нижнем отделе желудочно-кишечного тракта. Покрытия, которые являются рН-зависимыми и могут быть использованы в соответствии с настоящим изобретением, включают в себя шеллак, ацетатфталат целлюлозы (CAP), ацетатфталат поливинила (PVAP), фталат гидроксипропилметилцеллюлозы и сополимеры эфира метакриловой кислоты, зеин и тому подобное.
В некоторых предпочтительных аспектах субстрат (например, шарик, представляющий собой ядро таблетки, матриксная частица), содержащий опиоидный анальгетик (с или без ингибитора СОХ-2), покрывают гидрофобным материалом, выбранным из (i) алкилцеллюлозы; (ii) акрилового полимера или (iii) из их смесей. Покрытие может находиться в виде органического или водного раствора или дисперсии. Покрытие можно использовать для получения привеса субстрата приблизительно от 2 до 25%, чтобы достичь желаемой характеристики отсроченного высвобождения. Подобные композиции описаны, например, детально в патентах США №№5273760 и 5286493, права на которые переданы владельцу настоящего изобретения и включены в данный текст посредством ссылки.
Другие примеры композиций отсроченного высвобождения и покрытий, которые могут быть использованы в соответствии с настоящим изобретением, включены в патенты США №№5324351; 5356467 и 5472712, в которых они зарегистрированы со ссылкой на универсальность.
Алкилцеллюлозные полимеры
Целлюлозные материалы и полимеры, включая алкилцеллюлозы, представляют собой гидрофобные материалы, которые хорошо подходят для покрытия шариков согласно изобретению. Просто в качестве примера, одним предпочтительным алкилцеллюлозным полимером является этилцеллюлоза, хотя специалисты отдают должное тому, что другие полимеры целлюлозы и/или алкилцеллюлозы можно легко использовать, раздельно или в любой комбинации, в качестве всего или части гидрофобного покрытия согласно изобретению.
Одной коммерчески доступной водной суспензией этилцеллюлозы является Aquacoat® (FMC Corp., Philadelphia, Pennsylvania, U.S.A.). Aquacoat® получают путем растворения этилцеллюлозы в не смешивающемся с водой органическом растворителе и затем эмульгирования в воде в присутствии поверхностно-активного вещества и стабилизатора. После гомогенизации, в результате которой образуются субмикронные капли, органический растворитель упаривают под вакуумом с получением псевдолатекса. Пластификатор не включают в псевдолатекс в процессе производственного цикла. Таким образом, до применения его в качестве покрытая необходимо перед использованием тщательно смешать Aquacoat® с подходящим пластификатором.
Другая водная дисперсия этилцеллюлозы коммерчески доступна как Surelease18 (Colorcon, Inc., West Point, Pennsylvania, U.S.A.). Этот продукт получают путем включения пластификатора в дисперсию в течение производственного процесса. Горячий плав полимера, пластификатор (дибутиловый эфир себациновой кислоты) и стабилизатор (олеиновая кислота) получают в виде гомогенной смеси, которую затем разбавляют щелочным раствором с получением водной суспензии, которую можно наносить непосредственно на субстраты.
Акриловые полимеры
В других предпочтительных аспектах настоящего изобретения гидрофобный материал, заключающий в себе покрытие контролируемого высвобождения, представляет собой фармацевтически приемлемый акриловый полимер, включая, но не ограничиваясь, сополимеры акриловой кислоты и метакриловой кислоты, сополимеры метилметакрилата, этоксиэтилметакрилаты, цианоэтилметакрилат, поли(акриловую кислоту), поли(метакриловую кислоту), сополимер алкиламида метакриловой кислоты, поли(метилметакрилат), полиметакрилат, сополимер поли(метилметакрилата), полиакриламид, сополимер аминоалкилметакрилата, поли(ангидрид метакриловой кислоты) и сополимеры глицидилметакрилата.
В некоторых предпочтительных аспектах акриловый полимер состоит из одного или более аммониевых сополимеров метакрилата. Аммониевые сополимеры метакрилата хорошо известны в данной области и описаны в NF XVII как полностью полимеризованные сополимеры эфиров акриловой и метакриловой кислот с низким содержанием четвертичных аммониевых групп.
Для того чтобы получить желаемую степень растворения, необходимо включить два или более аммониевых сополимеров метакрилата, обладающих различными физическими свойствами, как, например, различными молярными отношениями четвертичных аммониевых групп к нейтральным эфирам (мет)акриловой кислоты.
Некоторые полимеры метакриловой кислоты в виде эфиров используют для получения рН-зависимых покрытий, которые можно применять в соответствии с настоящим изобретением. Например, существует семейство сополимеров, синтезированных из диэтиламиноэтилметакрилата и других нейтральных эфиров метакриловой кислоты, также известных как сополимер метакриловой кислоты или полимерные метакрилаты, коммерчески доступные как Eudragit® из Rohm Tech, Inc. Существует несколько различных типов Eudragit®. Например, Eudragit® является примером сополимера метакриловой кислоты, который набухает и растворяется в кислой среде. Eudragit® является сополимером метакриловой кислоты, который не набухает приблизительно при рН<5,7 и растворяется при рН>6. Eudragit® не набухает приблизительно при рН<6,5 и растворяется при рН>7. Eudragit® RL и Eudragit® RS набухают в воде, и количество воды, адсорбированной этими полимерами, зависит от рН, однако дозированные формы, покрытые Eudragit® RL и RS, являются рН-независимыми.
В некоторых предпочтительных аспектах акриловое покрытие содержит смесь двух лаков из акриловой смолы, коммерчески доступных из Rohm Pharma under the Tradenames Eudragit® RL30D and Eudragit® RS30D соответственно. Eudragit® RL30D и Eudragit® RS30D являются сополимерами эфиров акриловой кислоты и метакриловой кислоты с низким содержанием четвертичных аммониевых групп, молярное отношение аммониевых групп к остающимся нейтральным эфирам (мет)акриловой кислоты составляет 1:20 в Eudragit® RL30D и 1:40 в Eudragit® RS30D. Средний молекулярный вес составляет приблизительно 150000. Кодовые обозначения RL (высокая проницаемость) и RS (низкая проницаемость) относятся к свойствам проницаемости этих агентов. Смеси Eudragit® RL/RS не растворяются в воде и в жидкостях желудочно-кишечного тракта. Однако покрытия, образованные из этих агентов, проявляют способность к набуханию и проницаемости в водных растворах и в жидкостях желудочно-кишечного тракта.
Дисперсии Eudragit® RL/RS настоящего изобретения можно смешивать вместе в любом желаемом соотношении, для того чтобы в конечном счете получить композицию отсроченного высвобождения, имеющую желаемую степень растворения. Желаемые композиции отсроченного высвобождения можно получить, например, из задерживающего покрытия, произведенного из 100% Eudragit® RL, 50% Eudragit® RL и 50% Eudragit® RS и 10% Eudragit® RL:Eudragit® 90% RS. Конечно, специалисты в данной области признают, что другие акриловые полимеры также можно использовать, как, например, Eudragit® L.
Пластификаторы
В тех аспектах настоящего изобретения, где покрытие содержит водную дисперсию гидрофобного материала, включение эффективного количества пластификатора в водную дисперсию гидрофобного материала в дальнейшем улучшит физические свойства покрытия отсроченного высвобождения. Например, так как этилцеллюлоза имеет относительно высокую температуру стеклования и не образует эластичные пленки в нормальных условиях покрытия, предпочтительно включить пластификатор в этилцеллюлозное покрытие, содержащее покрытие отсроченного высвобождения, до его использования в качестве покрывающего материала. Обычно количество пластификатора, включенного в раствор для покрытия, основано на концентрации пленкообразователя, например, наиболее часто приблизительно от 1 до 50% по весу пленкообразователя. Однако концентрацию пластификатора можно правильно определить после тщательного экспериментирования с раствором данного покрытия и способом применения.
Примеры подходящих для этилцеллюлозы пластификаторов включают в себя водонерастворимые пластификаторы, как, например, дибутилсебацинат, диэтилфталат, триэтилцитрат, трибутилцитрат и триацетин, хотя можно применять и другие водонерастворимые пластификаторы (такие как ацетилированные моноглицериды, эфиры фталевой кислоты, касторовое масло и т.д.). Триэтилцитрат является особенно предпочтительным пластификатором для водных дисперсий этилцеллюлозы настоящего изобретения.
Примеры подходящих пластификаторов для акриловых полимеров настоящего изобретения включают в себя, не ограничиваясь ими, эфиры лимонной кислоты, как, например, триэтилцитрат NF XVI, трибутилцитрат, дибутилфталат и, возможно, 1,2-пропиленгликоль. Другие пластификаторы, которые являются подходящими для увеличения эластичности пленок, образованных из акриловых пленок, таких как лаковые растворы Eudragit® RL/RS, включают в себя полиэтиленгликоли, пропиленгликоль, диэтилфталат, касторовое масло и триацетин. Триэтилцитрат является особенно предпочтительным пластификатором для водных дисперсий этилцеллюлозы настоящего изобретения.
Кроме того, было найдено, что добавление малого количества талька снижает стремление водной дисперсии к слипанию в процессе производства и действует как полирующий агент.
Способы получения покрытых оболочкой шариков
При использовании водной дисперсии гидрофобного материала для покрытия оболочкой инертных фармацевтических шариков, таких как голые полые шарики 18/20, множество полученных стабилизированных твердых шариков контролируемого высвобождения затем может быть помещено в желатиновую капсулу в количестве, достаточном для обеспечения эффективной дозы контролируемого высвобождения при проглатывании и контактировании с окружающей жидкостью, например желудочным соком или средой растворения.
Из стабилизированных шариковых композиций контролируемого высвобождения настоящего изобретения медленно высвобождается терапевтически активный агент, например, при проглатывании и взаимодействии с желудочной жидкостью, а затем с жидкостью в кишечнике. Характер контролируемого высвобождения композиций изобретения можно изменить, например, путем изменения количества покрытия из водной дисперсии гидрофобного материала, изменения способа, с помощью которого добавляют пластификатор к гидрофобному материалу, путем включения дополнительных ингредиентов или наполнителей, путем изменения способа производства и т.д. Характер растворения конечного продукта можно изменить, например, путем увеличения или уменьшения толщины задерживающего покрытия.
Сфероиды или шарики, покрытые терапевтически активным агентом, получают, например, путем растворения терапевтически активного агента в воде и затем разбрызгивания раствора на субстрат, например голые полые шарики 18/20, при использовании вставки Wuster. Необязательно, также добавляют дополнительные ингредиенты до покрытия шариков, чтобы содействовать связыванию опиоидов с шариками и/или чтобы подкрасить раствор и т.д. Например, продукт, который включает в себя гидроксипропилметилцеллюлозу и т.д., в присутствии или в отсутствии красителя (например, Opadry®, коммерчески доступный из Colorcon, Inc.), может быть добавлен к раствору, и раствор перемешивают (например, в течение приблизительно 1 часа) до использования этого раствора для покрытия шариков. Полученный покрытый оболочкой субстрат, в данном случае шарики, может затем необязательно быть покрыт мембраной, для того чтобы отделить терапевтически активный агент от гидрофобного покрытия контролируемого высвобождения. Примером подходящей мембраны является мембрана, которая содержит гидроксипропилметилцеллюлозу. Однако может быть использован любой пленкообразователь, известный в данной области. Предпочтительным является то, что мембрана не воздействует на скорость растворения конечного продукта.
Далее шарики могут быть покрыты водной дисперсией гидрофобного материала. К тому же водная дисперсия гидрофобного материала предпочтительно включает в себя эффективное количество пластификатора, например триэтилцитрата. Предварительно созданные водные дисперсии этилцеллюлозы, такие как Aquacoat® и Surelease®, могут быть использованы. Если используют Surelease®, то необходимо отдельно добавить пластификатор. Или же предварительно созданные водные дисперсии акриловых полимеров, такие как Eudragit®, могут быть использованы.
Кроме пленкообразователя, пластификатора и системы растворителя (т.е воды) растворы для покрытия настоящего изобретения предпочтительно содержат краситель, чтобы придать продукту изящество и отличительную особенность. Краситель может быть добавлен к раствору терапевтически активного агента вместо или кроме водной дисперсии гидрофобного материала. Например, краситель добавляют к Aquacoat®, используя основанные на спирте и пропиленгликоле окрашенные дисперсии, измельченные алюминиевые красочные лаки и глушители, такие как двуокись титана, путем добавления красителя со сдвигом в сторону раствора водорастворимого полимера и затем использования незначительного сдвига в сторону пластифицированного Aquacoat®. Или же любой подходящий способ окрашивания композиций настоящего изобретения может быть использован. Подходящие ингредиенты для окрашивания композиции при использовании водной дисперсии акрилового полимера включают в себя двуокись титана и цветные пигменты, такие как пигменты оксида железа. Однако включение пигментов может увеличить задерживающее действие покрытия.
Пластифицированная водная дисперсия гидрофобного материала может быть нанесена на субстрат, содержащий терапевтически активный агент, путем распыления при использовании любого подходящего устройства для распыления, известного в данной области. В предпочтительном способе используется система псевдожидкого слоя Wurster, в которой воздушная струя, впущенная снизу, разжижает основу материала и осуществляет высушивание, тогда как покрытие из акрилового полимера распыляется. Достаточное количество водной дисперсии гидрофобного материала предпочтительно используют для того, чтобы достичь предопределенного контролируемого высвобождения вышеуказанного терапевтически активного агента, когда вышеуказанный покрытый оболочкой субстрат подвергается действию водных растворов, например, желудочного сока, принимая во внимание физические характеристики терапевтически активного агента, способ введения пластификатора и т.д. После покрытия гидрофобным материалом дальнейшее покрытие пленкообразователем, как, например, Opadry®, необязательно применяют к шарикам. Это покрытие применяют, если оно вообще требуется, чтобы существенно снизить агломерацию шариков.
На высвобождение терапевтически активного агента из композиции контролируемого высвобождения настоящего изобретения может влиять, т.е. регулировать до желаемой скорости, добавление одного или более модифицирующих высвобождение агентов или образование одного или более проходов через покрытие. Отношение гидрофобного материала к водорастворимому материалу определяют, наряду с другими факторами, с помощью характеристик требуемой скорости высвобождения и растворимости выбранных материалов.
Модифицирующие высвобождение агенты, которые функционируют как порообразователи, могут быть органическими или неорганическими и включать в себя материалы, которые могут быть растворены, экстрагированы или выщелочены из покрытия в среду потребления. Порообразователи могут представлять собой один или более гидрофильных материалов, как, например, гидроксипропилметилцеллюлозу.
Покрытия отсроченного высвобождения настоящего изобретения также могут включать в себя материалы, применяемые для производства микропористой пластинки в среде потребления, как, например, поликарбонаты, состоящие из линейных полиэфиров угольной кислоты, у которых карбонатные группы повторяются в полимерной цепи.
Модифицирующий высвобождение агент может также представлять собой полупроницаемый полимер.
В некоторых предпочтительных аспектах модифицирующий высвобождение агент выбирают из гидроксипропилметилцеллюлозы, лактозы, стеаратов металлов и смесей любых из перечисленных компонентов.
Покрытия отсроченного высвобождения настоящего изобретения также могут включать в себя средства выхода, представляющие собой, по крайней мере, один проход, отверстие или тому подобное. Проход может быть образован с помощью методов, которые представлены в патенте США №№3845770; 3916889; 4063064 и 4088864 (все из которых цитируются в данном тексте). Проход может иметь любую форму, как, например, круглую, треугольную, квадратную, эллиптическую, неравномерную и т.д.
Композиции на основе матриксных шариков
В других аспектах настоящего изобретения композицию контролируемого высвобождения получают посредством матрикса, имеющего упомянутое выше покрытие контролируемого высвобождения. Настоящее изобретение также может применять матрикс контролируемого высвобождения, который обусловливает in vitro скорости растворения опиоида в предпочтительных диапазонах и который высвобождает опиоид рН-зависимым и рН-независимым образом. Материалы, подходящие для включения в матрикс контролируемого высвобождения, зависят от используемого для образования матрикса способа.
Например, матрикс, кроме опиоидного анальгетика и (необязательно) СОХ-2, может включать в себя:
гидрофильные или гидрофобные материалы, как например смолы, эфиры целлюлозы, акриловые смолы, произведенные из белка материалы; перечень не является исключительным, и любой фармацевтически приемлемый гидрофобный материал или гидрофильный материал, который способствует контролируемому высвобождению активного агента и который плавится (или смягчается до степени, необходимой для экструзии), может быть использован в соответствии с настоящим изобретением;
перевариваемые длинноцепочечные (С8-С50, особенно С12-С40), замещенные или незамещенные углеводороды, такие как жирные кислоты, спирты жирного ряда, сложные эфиры глицерина и жирных кислот, минеральные и растительные масла и воска и стеариловый спирт; и полиалкиленгликоли.
Из этих полимеров акриловые полимеры, особенно Еudragit® RSPO эфиры целлюлозы, особенно гидроксиалкилцеллюлозы и карбоксиалкилцеллюлозы, являются предпочтительными. Пероральные дозированные формы могут содержать между 1% и 80% (по весу), по крайней мере, одного гидрофильного или гидрофобного материала.
Когда гидрофобным материалом является углеводород, то углеводород предпочтительно имеет точку плавления между 25° и 90°С. Из длинноцепочечных углеводородных материалов предпочтительными являются спирты жирного (алифатического) ряда. Пероральная дозированная форма может содержать вплоть до 60% (по весу), по крайней мере, одного перевариваемого, длинноцепочечного углеводорода.
Предпочтительно, пероральная дозированная форма содержит вплоть до 60% (по весу), по крайней мере, одного полиалкиленгликоля.
Гидрофобный материал предпочтительно выбирают из группы, состоящей из алкилцеллюлоз, полимеров и сополимеров акриловой и метакриловой кислот, шеллака, зеина, гидрированного касторового масла, гидрированного растительного масла или из их смесей. В некоторых предпочтительных аспектах настоящего изобретения гидрофобным материалом является фармацевтически приемлемый акриловый полимер, включая, но не ограничивась, сополимеры акриловой кислоты и метакриловой кислоты, метилметакрилат, сополимеры метилметакрилата, этоксиэтилметакрилаты, цианоэтилметакрилаты, сополимер аминоалкилметакрилата, поли(акриловую кислоту), поли(метакриловую кислоту), сополимер алкиламина метакриловой кислоты, поли(метилметакрилат), поли(метакриловую кислоту) (ангидрид), полиметакрилат, полиакриламид, поли(ангидрид метакриловой кислоты) и сополимеры глицидилметакрилата. В других аспектах гидрофобный материал выбирают из материалов, как, например, гидроксиалкилцеллюлоза, таких как гидроксипропилметилцеллюлозы, и смесей из вышеперечисленных компонентов.
Предпочтительными гидрофобными материалами являются водонерастворимые материалы с более или менее выраженными гидрофильными и/или гидрофобными свойствами. Предпочтительно, используемые в изобретении гидрофобные материалы имеют точку плавления приблизительно от 30° до 200°С, предпочтительно приблизительно от 45° до 90°С. Особенно, гидрофобный материал может включать в себя природные или синтетические воски, спирты жирного ряда (как например, лауриловый, миристиловый, стеариловый, цетиловый или предпочтительно цетостеариловый спирт), жирные кислоты, включая, но не ограничивась, эфиры жирных кислот, глицериды жирных кислот (моно-, ди- и триглицериды), гидрированные жиры, углеводороды, нормальные воски, стеариновую кислоту, стеариловый спирт и гидрофобные и гидрофильные материалы, имеющие углеводородные скелеты. Подходящие воски включают в себя, например, пчелиный воск, гликовоск (glycowax), касторовый воск и карнаубский воск. В описании настоящего изобретения воскоподобное вещество определяют как любой материал, который обычно является твердым при комнатной температуре и имеет точку плавления приблизительно от 30° до 100°С.
Подходящие гидрофобные материалы, которые могут быть использованы в соответствии с настоящим изобретением, включают в себя перевариваемые, длинноцепочечные (С8-С50, особенно C12-C40), замещенные или незамещенные углеводороды, такие как жирные кислоты, спирты жирного ряда, сложные эфиры глицерина и жирных кислот, минеральные и растительные масла и воска и природные и синтетические воски. Предпочтительными являются углеводороды, имеющие точку плавления между 25° и 90°С. В некоторых аспектах, из длинноцепочечных углеводородных материалов предпочтительными являются спирты жирного (алифатического) ряда. Пероральная дозированная форма может содержать вплоть до 60% (по весу), по крайней мере, одного перевариваемого, длинноцепочечного углеводорода.
Предпочтительно комбинации двух или более гидрофобных материалов включают в матриксные композиции. Если включают дополнительный гидрофобный материал, то его предпочтительно выбирают из природных и синтетических восков, жирных кислот, спиртов жирного ряда и из их смесей. Примеры включают в себя пчелиный воск, карнаубский воск, стеариновую кислоту и стеариловый спирт. Этот перечень не является исключительным.
Один особенно подходящий матрикс включает в себя, по крайней мере, одну водорастворимую гидроксиалкилцеллюлозу, по крайней мере, один С12-С36, предпочтительно C14-C22 алифатический спирт и, необязательно, по крайней мере, один полиалкиленгликоль. По крайней мере, одной гидроксиалкилцеллюлозой предпочтительно является гидрокси(от C1 до С6)алкилцеллюлоза, как, например, гидроксипропилцеллкотоза, гидроксипролилметилцеллюлоза и, особенно, гидроксиэтилцеллюлоза. Количество, по крайней мере, одной гидроксиалкилцеллюлозы в настоящей пероральной дозированной форме определяется, между прочим, с помощью определенной скорости требуемого высвобождения опиоида. По крайней мере, одним алифатическим спиртом может быть, например, лауриловый спирт, миристиловый спирт или стеариловый спирт, в особенно предпочтительных аспектах настоящей пероральной дозированной формы, однако, по крайней мере, один алифатический спирт представляет собой цетиловый спирт или цетостеариловый спирт. Количество, по крайней мере, одного алифатического спирта в настоящей пероральной дозированной форме определяется, как и выше, с помощью определенной скорости требуемого высвобождения опиоида. Оно будет зависеть от того, присутствует, по крайней мере, один полиалкиленгликоль или отсутствует в пероральной дозированной форме. В отсутствие, по крайней мере, одного полиалкиленгликоля пероральная дозированная форма содержит между 20% и 50% (по весу), по крайней мере, одного алифатического спирта. Когда, по крайней мере, один полиалкиленгликоль присутствует в пероральной дозированной форме, то объединенный вес, по крайней мере, одного алифатического спирта и, по крайней мере, одного полиалкиленгликоля предпочтительно составляет между 20% и 50% (по весу) тотальной дозировки.
В одном аспекте соотношение, например, по крайней мере, одной гидроксиалкилцеллюлозы или акрилового полимера, по крайней мере, к одному алифатическому спирту/полиалкиленгликолю определяет в значительной степени скорость высвобождения опиоида из композиции. Соотношение, по крайней мере, одной гидроксиалкилцеллюлозы, по крайней мере, к одному алифатическому спирту/полиалкиленгликолю между 1:2 и 1:4 является предпочтительным, а соотношение между 1:3 и 1:4 является особенно предпочтительным.
По крайней мере, один полиалкиленгликоль может быть представлен, например, полипропиленгликолем или полиэтиленгликолем, который является предпочтительным. Средний молекулярный вес, по крайней мере, одного полиалкиленгликоля составляет предпочтительно между 1000 и 15000, особенно между 1500 и 12000.
Другой подходящий матрикс контролируемого высвобождения включает в себя алкилцеллюлозу (особенно этилцеллюлозу), от C12 до С36 алифатический спирт и, необязательно, полиалкиленгликоль.
В другом предпочтительном аспекте матрикс включает в себя фармацевтически приемлемую комбинацию, по крайней мере, из двух гидрофобных материалов.
Кроме вышеуказанных ингредиентов, матрикс контролируемого высвобождения также может содержать подходящие количества других материалов, например разбавителей, загустителей, связующих агентов, гранулирующих добавок, красителей, ароматизаторов и способствующих скольжению агентов, которые являются обычными в фармацевтической области.
Способы приготовления основанных на матриксе шариков
Для того чтобы облегчить приготовление твердой, пероральной дозированной формы контролируемого высвобождения согласно изобретению, можно использовать любой способ получения матриксной композиции, известный специалистам в данной области. Например, включение в матрикс может быть осуществлено, например, путем (а) образования гранул, содержащих, по крайней мере, одну водорастворимую гидроксиалкилцеллюлозу и опиоид или соль опиоида; (b) смешивания содержащей гранулы гидроксиалкилцеллюлозы, по крайней мере, с одним С12-С36 алифатическим спиртом; и (с) необязательно, прессования и формования гранул. Предпочтительно, гранулы образуются путем влажного измельчения гидроксиалкилцеллюлозы/опиоида с водой. В особенно предпочтительном аспекте этого способа количество добавленной воды на стадии влажного измельчения составляет предпочтительно между 1,5 и 5 раз, особенно между 1,75 и 3,5 раз выше сухого веса опиоида.
В других альтернативных аспектах сфероидизирующий агент вместе с активным ингредиентом может быть сфероидизирован с образованием сфероидов. Микрокристаллическая целлюлоза является предпочтительной. Подходящей микрокристаллической целлюлозой является, например, продаваемая как Avicel РН 101 (Trade Mark, FMC corporation). В подобных аспектах кроме активного ингредиента и сфероидизирующего агента сфероиды могут также содержать связующий агент. Подходящие связующие агенты, такие как водорастворимые полимеры низкой вязкости, хорошо известны специалистам в фармацевтической области. Однако водорастворимая гидроксиалкилцеллюлоза с низшим алкилом, как, например, гидроксипропилцеллюлоза, является предпочтительной. Дополнительно (или альтернативно) сфероиды могут содержать водонерастворимый полимер, особенно акриловый полимер, акриловый сополимер, такой как сополимер метакриловой кислоты и этилакрилата, или этилцеллюлозу. В таких аспектах покрытие отсроченного высвобождения включает в себя, в основном, гидрофобный материал, как, например, (а) воск, либо только в смеси со спиртом жирного ряда; или (b) шеллак или зеин.
Матрикс, полученный расплавом-экструзией
Матрикс отсроченного высвобождения может быть получен посредством техники расплавления-грануляции или расплавления-экструзии. Обычно техника расплавления-грануляции включает в себя расплавление твердого гидрофобного материала, например воска, и включение в него порошкообразного лекарственного средства. Для того, чтобы получить лекарственную форму отсроченного высвобождения, необходимо включить дополнительное гидрофобное вещество, например этилцеллюлозу или водонерастворимый акриловый полимер, в расплавленный восковой гидрофобный материал. Примеры композиций отсроченного высвобождения, полученные посредством техники расплавления-грануляции, приведены в патенте США №4861598, права на которые переданы владельцу настоящего изобретения, где они зарегистрированы со ссылкой на универсальность.
Дополнительный гидрофобный материал может включать в себя одно или более водонерастворимых воскоподобных термопластичных веществ, возможно, смешанных с одним или более воскоподобных термопластичных веществ, которые являются менее гидрофобными, чем одно или более водонерастворимое воскоподобное вещество. Для того чтобы достичь постоянного высвобождения, индивидуальные воскоподобные вещества в композиции должны быть, в основном, неперевариваемыми и нерастворимыми в жидкостях желудочно-кишечного тракта в течение первоначальных стадий высвобождения. Применяемые водонерастворимые воскоподобные вещества могут быть веществами с растворимостью в воде, которая ниже, чем приблизительно 1:5000 (вес к весу).
Кроме вышеуказанных ингредиентов, матрикс отсроченного высвобождения может также содержать соответствующие количества других материалов, например разбавителей, смазок, связывающих веществ, гранулирующих добавок, красителей, ароматизаторов и способствующих скольжению агентов, которые являются стандартными в фармацевтической области. Количество этих дополнительных материалов является достаточным для достижения желаемого действия, проявляемого желаемой композицией.
Кроме вышеуказанных ингредиентов, матрикс отсроченного высвобождения с включенными в него мультичастицами, полученными расплавом-экструзией, также может содержать соответствующие количества других материалов, например разбавителей, смазок, связывающих веществ, гранулирующих добавок, красителей, ароматизаторов и способствующих скольжению агентов, которые являются стандартными в фармацевтической области в количествах вплоть до приблизительно 50% по весу частицы, если желательно.
Специфические примеры фармацевтически приемлемых носителей и наполнителей, которые могут быть использованы для приготовления пероральных дозированных форм, описаны в Handbook of Pharmaceutical Excipients, American Pharmaceutical Association (1986), где они зарегистрированы посредством ссылки.
Мультичастицы, полученные расплавом-экструзией
Получение подходящего матрикса расплавом-экструзией согласно настоящему изобретению может, например, включать в себя стадии смешивания опиоидного анальгетика вместе, по крайней мере, с одним гидрофобным материалом и предпочтительно дополнительным гидрофобным материалом для получения гомогенной смеси. Затем гомогенную смесь нагревают до температуры, достаточной, по крайней мере, для размягчения смеси, достаточного для того, чтобы экструдировать эту смесь. Затем полученную гомогенную смесь экструдируют с образованием нитей. Экструдат предпочтительно охлаждают и размельчают до мультичастиц с помощью средств, известных в данной области. Нити охлаждают и размельчают до мультичастиц. Затем мультичастицы делят на стандартные лекарственные формы. Экструдат предпочтительно имеет диаметр приблизительно от 0,1 до 5 мм и способствует отсроченному высвобождению терапевтически активного агента в течение периода времени приблизительно от 8 до 24 часов.
Необязательный способ приготовления расплавленных экструдатов настоящего изобретения включает в себя непосредственное дозирование в экструдер гидрофобного материала, терапевтически активного агента и необязательного связующего вещества; нагревание гомогенной смеси; экструзию гомогенной смеси с образованием нитей; охлаждение содержащих гомогенную смесь нитей; размельчение нитей на частицы, имеющие размер приблизительно от 0,1 мм до 12 мм; разделение вышеуказанных частиц на стандартные лекарственные формы. В этом аспекте изобретения осуществляется относительно непрерывный производственный процесс.
Диаметр отверстия экструдера или выходного отверстия можно регулировать, чтобы изменить толщину выдавленных нитей. Кроме того, выходная часть экструдера не должна быть круглой; она может быть вытянутой, прямоугольной и т.д. Выходящие нити могут быть измельчены до частиц, используя горячие кусачки, гильотину и т.д.
Система мультичастиц, полученная расплавом-экструзией, могжет быть, например, в виде гранул, сфероидов или окатышей в зависимости от выходного отверстия экструдера. В описании настоящего изобретения названия "расплавленная и экструдтрованная мультичастица(ы)" и "система(ы) расплавленной и экструдированной мультичастицы" и "расплавленные и экструдированные частицы" относятся к множеству единиц, предпочтительно в области подобного размера и/или формы, содержащих один или более активный агент и один или более наполнитель, предпочтительно включая гидрофобный материал, который приведен в описании. В этом отношении, расплавленные и экструдированные мультичастицы имеют размер в области приблизительно от 0,1 до 12 мм в длину и диаметр приблизительно от 0,1 до 5 мм. Кроме того, следует понимать, что расплавленные и экструдированные мультичастицы могут быть любой геометрической формы в пределах этого диапазона размера. Или же экструдат может быть просто разрезан на частицы желаемой длины и разделен на единичные дозы терапевтически активного агента без стадии сфероидизации.
В одном предпочтительном аспекте пероральные дозированные формы приготовляют для того, чтобы включить эффективное количество расплавленных и формованных мультичастиц в капсулу. Например, множество расплавленных и формованных мультичастиц можно поместить в желатиновую капсулу в количестве, достаточном для получения эффективной дозы отсроченного высвобождения при проглатывании и контактировании с желудочным соком.
В другом предпочтительном аспекте подходящее количество мультичастиц из экструдата прессуют в пероральные таблетки, используя соответствующее оборудование для таблетирования с применением стандартной технологии. Технологии и композиции для приготовления таблеток (прессованные и формованные), капсулы (твердые и мягкие желатиновые) и пилюли также описаны в Remingtons Pharmaceutical Sciences (Arthur Osol, editor), 1553-1593 (1980), и включены в данный текст посредством ссылки.
В другом предпочтительном аспекте экструдат может быть формован в таблетки, как изложено в патенте США №4957681 (Klimesch, et. al.), описанном подробно выше, и включен в данный текст посредством ссылки.
Необязательно, системы мультичастиц, полученные расплавом-экструзией, отсроченного высвобождения или таблетки могут быть покрыты, или желатиновая капсула может быть покрыта, покрытием отсроченного высвобождения, как, например, описанные выше покрытия отсроченного высвобождения. Подобные покрытия предпочтительно включают в себя достаточное количество гидрофобного материала для достижения уровня привеса приблизительно от 2 до 30%, хотя покрытие может быть больше в зависимости от физических свойств применяемого отдельного опиоидного анальгезирующего соединения и желаемой скорости высвобождения, наряду с другими факторами.
Стандартные лекарственные формы настоящего изобретения, полученные посредством расплава и экструзии, могут также включать в себя расплавленные и экструдированные мультичастицы, содержащие один или более терапевтически активный агент, представленные выше, до инкапсулирования. Кроме того, стандартные лекарственные формы могут содержать количество терапевтически активного агента немедленного высвобождения для быстрого терапевтического эффекта. Терапевтически активный агент немедленного высвобождения может быть включен, например, как отдельные окатыши в желатиновую капсулу, или может быть покрыт на поверхности мультичастиц после приготовления дозированных форм (например, покрытие контролируемого высвобождения или основанное на матриксе). Стандартные лекарственные формы настоящего изобретения также могут содержать комбинацию контролируемого высвобождения из шариков и матриксных мультичастиц для достижения желаемого эффекта.
Композиции отсроченного высвобождения настоящего изобретения предпочтительно медленно высвобождают терапевтически активный агент, например, при проглатывании и контактировании с желудочным соком и затем жидкостями кишечника. Характеристика композиций отсроченного высвобождения изобретения, полученных расплавом и экструзией, может быть изменена, например, путем изменения количества замедляющего агента, т.е. гидрофобного материала, путем изменения количества пластификатора относительно гидрофобного материала, путем включения дополнительных ингредиентов или наполнителей, путем изменения способа приготовления и т.д.
В других аспектах изобретения расплавленный и экструдированный материал получают без включения терапевтически активного агента, который добавляют к экструдату. Подобные композиции обычно имеют терапевтически активный агент, смешанный вместе с экструдированным матриксным материалом, и затем смесь таблетируют для того, чтобы получить композицию медленного высвобождения. Подобные композиции могут иметь преимущества, например, когда терапевтически активный агент, включенный в композицию, чувствителен к температурам, которые необходимы для размягчения гидрофобного материала и/или замедляющего материала.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ АСПЕКТОВ
Следующие примеры иллюстрируют различные аспекты настоящего изобретения. Они не должны истолковываться как ограничивающие область притязания изобретения.
ПРИМЕРЫ 1-2
Оценка комбинации морфина и набуметона (пример 1) и морфина и мелоксикама (пример 2)
В примерах 1-2 проверяют комбинацию, обладающую синергическим действием, ингибитора СОХ-2 и опиата путем проверки набуметона (пример 1) и мелоксикама (пример 2) посредством теста на вытягивание (коруа), индуцируемого фенилхиноном (PPQ).
Набуметон не является собственно избирательным для COX-2, но его оценивают здесь, поскольку его применение связано с крайне низким уровнем образования язв. Набуметон является пролекарством, приводящим к действительному ингибитору COX-2, 6-метокси-2-нафтилуксусной кислоте (6-MNA) (см. таблицу 1). Низкая способность набуметона вызывать язву может быть следствием рН-зависимого образования 6-MNA. Этого не происходит при низких величинах рН, таких, которые найдены в слизистой оболочке желудка. Таким образом, СОХ-2 является, по-видимому, избирательно функциональным. При клинических испытаниях найдено, что набуметон является вполне эффективным, с крайне незначительной способностью, к образованию язв. При испытаниях на больных с остеоартритом набуметон сравнивают с диклофенаком. Было найдено, что он является таким же эффективным, как диклофенак (он является крайне слабым, требуя 1500 мг ежедневно), однако ни у одного из 382 больных, леченых набуметоном, не проявлялась желудочно-кишечная токсичность (S.H.Roth et al., J. Rheumatol. 21:1118, 1994). В результате наблюдений за больными, принимающими лечение набуметоном, в течение года, показано, что случаи язв составляют только 0,5% (PDR 1995, р.2396).
Методы: Изоболографический анализ взаимодействия лекарственного средства выполняют на мышах-самцах ICR. Во время=0 вводят перорально мелоксикам или набуметон или наполнитель. Во время (Т)=9 минутам перорально вводят морфин или наполнитель. Во время Т=29 минутам PPQ (фенил-п-бензилхинон), 2 мг/кг, инъецируют внутрибрюшинно. Во время Т=36 минутам число брюшных вытягивании подсчитывают для каждой мыши в течение 1 минуты. Во время Т=40 минутам вытягивания вновь подсчитывают в течение 1 минуты. Исследовано 6-8 мышей на дозу.
Концентрации морфина, используемые для ответа на его дозу, составляют 0,5, 1, 2 и 5 мг/кг. Концентрации набуметона, используемые для ответа на его дозу, являются 20, 50, 100 и 300 мг/кг. Концентрации мелоксикама, используемые для ответа на его дозу, являются 1, 3, 10 и 50 мг/кг.
Процент ингибирования индуцируемого посредством PPQ вытягивания (корчи) рассчитывают следующим образом:
=1 - {[полное # вытягивания при двух подсчетах с лекарственным средством]/[полное # вытягивания при двух подсчетах с наполнителем]}×100.
ED50 (доза лекарственного средства, вызывающая 50% ингибирования) определяют путем нелинейной регрессии. Когда вводят комбинации морфина и мелоксикама, соотношение всегда соответствует 1:10 или 1:1000 соответственно. Для изучения комбинаций следующие концентрации используют: для морфина/набуметона составляет 0,036/36, 0,072/72, 0,1/100 и 0,144/144 мг/кг, для морфина/мелоксикама составляет 0,18/1,8, 0,36/3,6, 0,72/7,2 и 1,44/14,4 мг/кг. ED50 для каждого лекарственного средства в комбинации определяют путем простого расчета количества каждого в комбинации при ED50 дозе комбинации. Результаты ED50 для примера 1 (набуметон) против морфина изложены ниже:
мелоксикам: морфин ED50=1,86 мг/кг перорально
мелоксикам ED50=15,2 мг/кг перорально (слабая экстраполяция) с комбинацией доза-ответ, используя морфин:
мелоксикам 1:10
ED50 морфин = 0,62
ED50 мелоксикам = 6,22.
Как видно из данных ED50, мелоксикам в значительной степени увеличивает активность морфина, в то время как морфин не воздействует на активность мелоксикама. Однако морфин помогает мелоксикаму достичь большей эффективности - 72% против 45% ингибирования.
Данные, представленные из примеров 1-2, в дальнейшем представлены на фиг.1, которая представляет собой график, изображающий процент ингибирования (ED50) против дозы (мг/кг). На фиг.1 изображены кривые доза-ответ для набуметона, мелоксикама и морфина только и для комбинаций набуметона + морфина и мелоксикама + морфина. Как видно из представленных на фиг.1 результатов, морфин не изменяет дозу-ответ для набуметона или мелоксикама. Однако набуметон и мелоксикам оба изменяют дозу-ответ для морфина (указано стрелками).
Взаимодействие морфина и флусолида может быть продемонстрировано с помощью изоболограммы (См., например, S.Loewe, Pharm. Rev., 9; 237 (1957) относительно построения и базы изоболограммы; включено в данный текст посредством ссылки).
На фиг.2 изображена изоболограмма для набуметона при взаимодействии с морфином (включенными являются 95% уверенных интервалов). Диагональ, соединяющая величины ED50 двух данных раздельно лекарственных средств, представляет собой простую аддитивность эффектов при различных соотношениях компонентов. Величины ED50, находящиеся ниже кривой (между линией и началом), указывают на сверхаддитивность. Как видно из фиг.2, комбинация набуметона и морфина проявляет синергическое действие, поддерживаемое соотношениями комбинаций этих лекарственных средств, перечисленных в таблице 2.
На фиг.3 изображена изоболограмма для мелоксикама при взаимодействии с морфином (включенными являются 95% уверенных интервалов). Как видно из фиг.3, комбинация набуметона и морфина проявляет синергическое действие, поддерживаемое соотношениями комбинаций этих лекарственных средств, перечисленных в таблице II.
Известно в данной области, что данные для мыши, представленные на изоболограмме, могут быть перенесены на другие виды, для которых пероральные эффективные анальгезирующие дозы индивидуальных соединений известны или могут быть определены. Поэтому специалисты в данной области оценивают, что эта основная корреляция для анальгезирующих свойств дает возможность определить диапазон эффективностей для человека.
Заключение
В то время как изобретение описывает и иллюстрирует данные со ссылкой на определенные предпочтительные аспекты изобретения, специалисты в данной области оценивают, что очевидные модификации могут быть сделаны без отступления от характера и области притязания изобретения. Например, эффективные дозировки и специфические фармакологические ответы могут изменяться в зависимости от соотношений данного опиоида и данного используемого ингибитора СОХ-2, а также от композиции и способа введения. Подобные варианты рассматривают как находящиеся в области притязания изобретения.
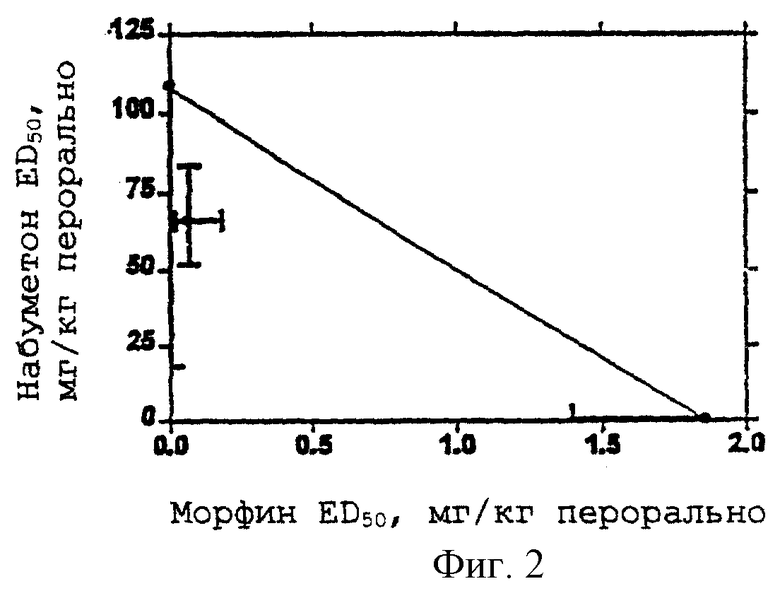
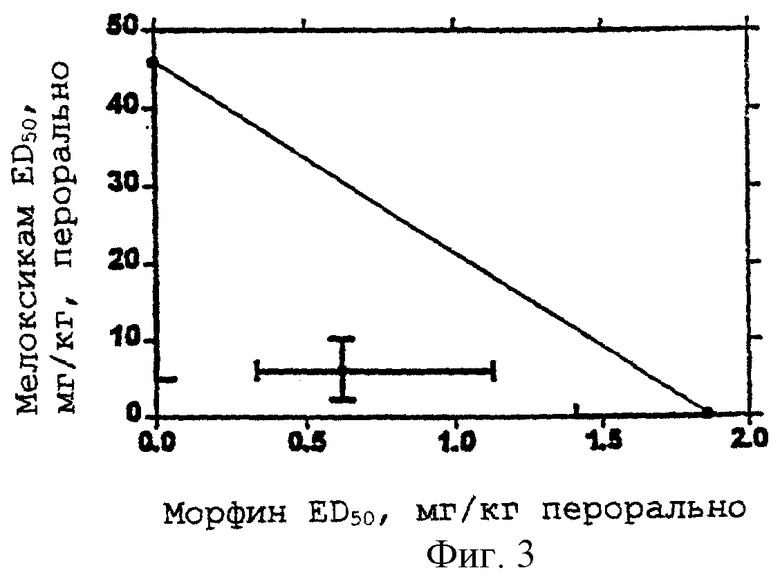
Изобретение относится к медицине, в частности к лечению боли. Для этого предлагается фармацевтическая композиция, содержащую ингибитор циклооксигеназы-2 и опиоидный анальгетик. Это позволяет снизить концентрацию опиоидных анальгетиков в плазме при сохранении эффективного устранения боли и уменьшения побочных эффектов лечения. 11 з.п. ф-лы, 3 ил., 2 табл.
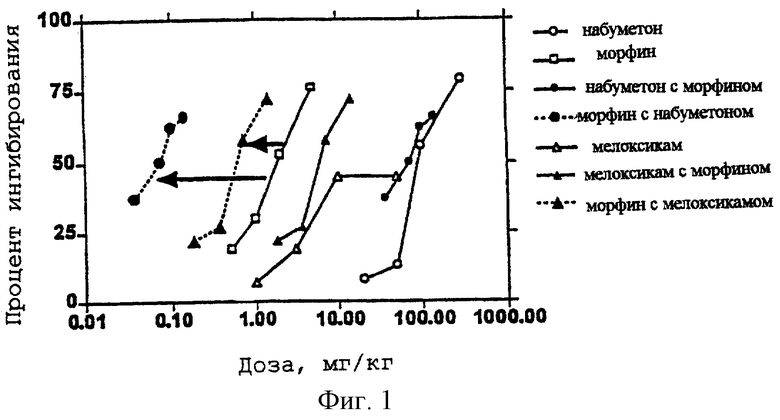
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| TWYCROSS R.G | |||
| et al | |||
| Monitoring drug use in palliative care | |||
| Palliat | |||
| Med | |||
| Прибор для охлаждения жидкостей в зимнее время | 1921 |
|
SU1994A1 |
| ГЛЕЗЕР М.Г | |||
| и др | |||
| Справочник по фармакотерапии сердечно-сосудистых заболеваний | |||
| - М.: Авиценна, 1996, с.262 и 263 | |||
| ЛОУРЕНС Д.Р | |||
| и др | |||
| Клиническая фармакология | |||
| - М..: Медицина, 1993, т.2, с.16 и 17. | |||

