2. ИнгибированиеДанное изобретение относится к замещенным производным пропаноламина и их фармацевтически приемлемым солям и физиологически функциональным производным.
Уже описано несколько классов биологически активных веществ для лечения ожирения (болезни Деркума) и нарушений липидного обмена:
- полимерные адсорбенты, такие как, например, холестирамин,
- бензотиазепины (WO 93/16055),
- димеры и конъюгаты желчных кислот (ЕР 0489423),
- амиды 4-амино-2-уреидопиримидин-5-карбоновых кислот (ЕР 0557879).
Задачей изобретения являются соединения, проявляющие терапевтически применимое гиполипидемическое (понижающее содержание липидов в крови) действие.
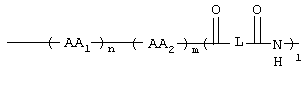
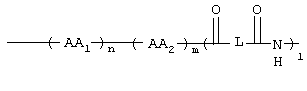
Таким образом, данное изобретение относится к соединениям формулы I
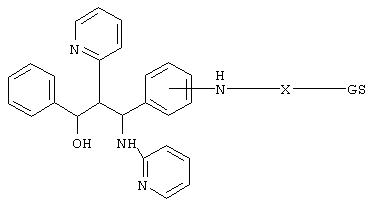
где GS обозначает группу желчной кислоты формулы
R1 обозначает связь с X, ОН;
R2 обозначает связь с X, ОН, О-(С1-С6)-алкил, NН-(С2-С6)-алкил-SО3Н, N(СН3)-СН2-СН2-SО3Н, NH(C1-С6)-алкил-СООН, N (СН3)-(C1-C6)-алкил-СООН;
при условии, что R1 и R2 не имеют одновременно следующее значение
R1 связь с Х и
R2 связь с X;
R3, R4 обозначают независимо друг от друга Н, ОН;
Х обозначает
или связь;
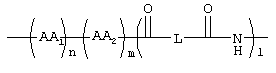
1, n, m независимо друг от друга равны 0 или 1;
L обозначает (C1-C6)-алкил, фенил;
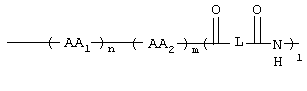
AA1, AA2 независимо друг от друга обозначают аминокислотный остаток или аминокислотный остаток, который одно- или многократно замещен аминозащитной группой;
а также их фармацевтически приемлемым солям и физиологически функциональным производным.
Предпочтительными являются соединения формулы I, в которых один или несколько остатков имеют следующее значение:
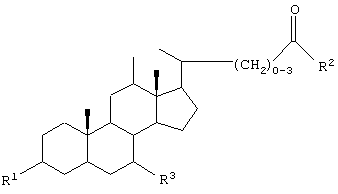
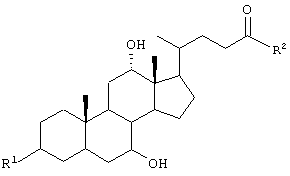
GS обозначает группу желчной кислоты формулы
R обозначает связь с X, ОН;
R2 обозначает связь с X, ОН, О-(C1-C6)-алкил, NH-(C2-C6)- алкил-SО3Н, N(СН3)-CH2-CH2-SO3H, NH(C1-C6)-алкил-СООН, N(СН3)-(C1-С6)-алкил-СООН;
при условии, что R1 и R2 не имеют одновременно следующее значение
R1 связь с Х и
R2 связь с X;
Х обозначает
или связь;
1, m, n независимо друг от друга равны 0 или 1;
L обозначает (C1-C6)-алкил, фенил;
AA1, AA2 независимо друг от друга обозначают аминокислотный остаток или аминокислотный остаток, который одно- или многократно замещен аминозащитной группой;
а также их фармацевтически приемлемые соли и физиологически функциональные производные.
Особенно предпочтительными являются соединения формулы 1, в которых один или несколько остатков имеют следующее значение:
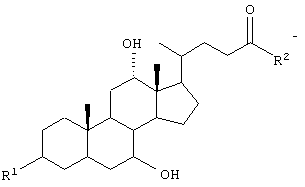
GS обозначает группу желчной кислоты формулы
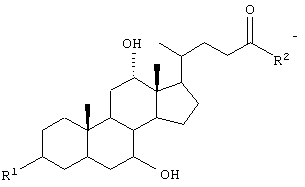
R1 обозначает связь с X, ОН;
R2 обозначает связь с X, ОН, О-(C1-С6)-алкил, NH-(C2-C6)-алкил-SО3Н, NH(C1-C6)-алкил-СООН;
при условии, что R1 и R2 не имеют одновременно следующее значение
R1 связь с Х и
R2 связь с X;
Х обозначает
или связь;
1, m, n независимо друг от друга равны 0 или 1;
L обозначает (C1-С6)-алкил;
AA1, AA2 независимо друг от друга обозначают аминокислотный остаток или аминокислотный остаток, который одно- или многократно замещен аминозащитной группой;
а также их фармацевтически приемлемые соли.
Под термином ″алкил" имеют в виду линейные или разветвленные углеводородные цепи.
Под ″аминокислотами" или "аминокислотными остатками" имеют в виду стереоизомерные формы, т.е. D- или L-формы следующих соединений:

Сокращенные обозначения аминокислот использовали согласно общепринятому способу обозначений (срав. Schroder, Lubke, The Peptides, Band I, New York 1965, Seiten XXII-XXIII; Houben-Weyl, Methoden der Organischen Chemie, Band XV/1 und 2, Stuttgart 1974). Аминокислота D-Asp обозначает D-форму аспарагиновой кислоты. Пептиды являются по их химической природе амидами кислот и при гидролизе разлагаются до аминокислот.
Под аминокислотными защитными группами следует понимать подходящие группы, которыми могут быть защищены функциональные группы боковых цепей аминокислотных остатков (см., например, T.W. Greene, P.G.M. Wuts, Protective Groups in Organic Synthesis, 2nd Edition, John Wiley and Sons, New York 1991). Предпочтительными аминозащитными группами являются третбутилоксикарбонил (ВОС), 9-флуоренилметоксикарбонил (Fmoc), бензилоксикарбонил (Z), 2-(3,5-диметоксифенил)проп-2-илокси-карбонил (Ddz), метил, трет-бутил, тритил, S-трет-бутил, трет-бутиламинокарбонил.
Данное изобретение относится далее к способам получения соединений формулы 1, которые отличаются следующими схемами реакции (Схемы 1-4):

Соединения типа IV получают, подвергая о-, м- или п-замещенные имины типа II реакции с кетоном III. Эту реакцию можно проводить, например, смешиванием обоих соединений, без растворителя и последующего нагревания, или в подходящем растворителе, таком как этанол, тетрагидрофуран (ТГФ), толуол, диглим или тетрадекан, при температурах от 20°С до 150°С.
Кетосоединения типа IV восстанавливают в подходящем растворителе, таком как, например, метанол, ТГФ или ТГФ/вода с помощью NaBH4 или другого подходящего восстановителя при температурах между -30°С и +40°С до гидроксисоединений типа V. При восстановлении в качестве основных продуктов реакции образуются в большинстве случаев две смеси изомеров (рацематы). Различные рацематы могут быть отделены друг от друга фракционированной кристаллизацией или хроматографией на силикагеле. Нитрогруппа в соединениях типа V может быть восстановлена известными способами, такими как, например, каталитическое гидрирование с Pd или Pd на угле и H2 в метаноле.
Полученные таким образом рацемические соединения типа VI могут быть далее разделены на их энантиомеры. Разделение рацематов VI на энантиомеры типа VII может проводиться хроматографией через хиральный колоночный материал или при помощи известных в литературе способов с оптически активными вспомогательными реагентами (срав. J. Org. Chem. 44, 1979, 4891).
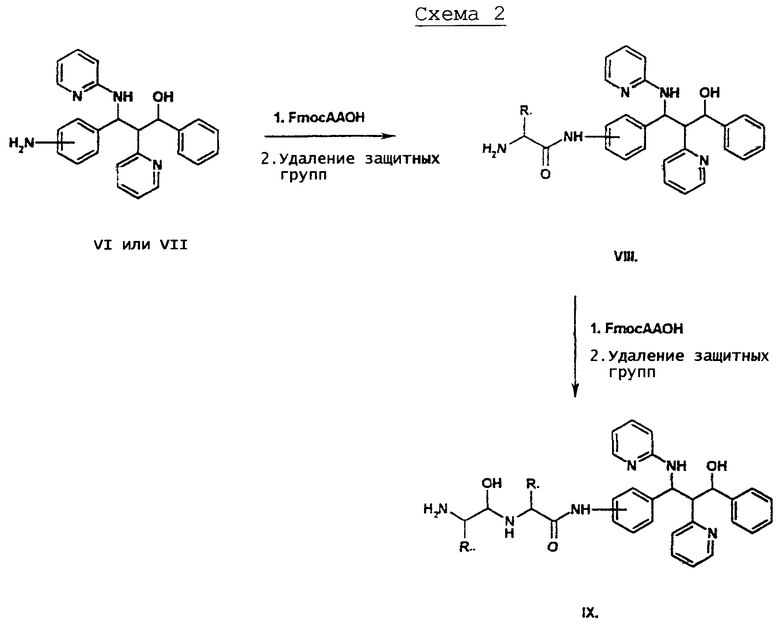
Согласно схеме 2 ароматические амины типа VI или VII (рацемат или чистый энантиомер) могут быть преобразованы аминокислотой по известному стандартному способу сочетания пептидов в производные VIII. В качестве такого способа пригодно, например, сочетание с TOTU и триэтиламином в ДМФ (см. литературу: G. Breipohl, W. Konig EP 0460446; W. Konig, G. Breipohl, P. Pokorny, M. Birkner in E. Girat and D. Andreu (Eds.) Peptides 1990, Escom, Leiden, 1991, 143-145). Остатки АА1 и АА2 имеют приведенное для формулы I значение. Аминогруппу аминокислоты обеспечивают защитной группой, например, Fmoc, карбоксильная группа является незащищенной.
У аминокислот с функциональными группами в боковой цепи эти группы являются соответственно защищенными, либо временно во время синтеза, либо с сохранением защитных групп в соединениях согласно изобретению.
Чтобы получить производное VIII, защитную группу аминогруппы отщепляют, например, в случае Fmoc в смеси из ДМФ и пиперидина. Получают дипептидные конъюгаты IX, если исходя из соединений типа VIII повторяют последовательность реакций (а) сочетания аминокислоты, (b) удаления защитной группы.
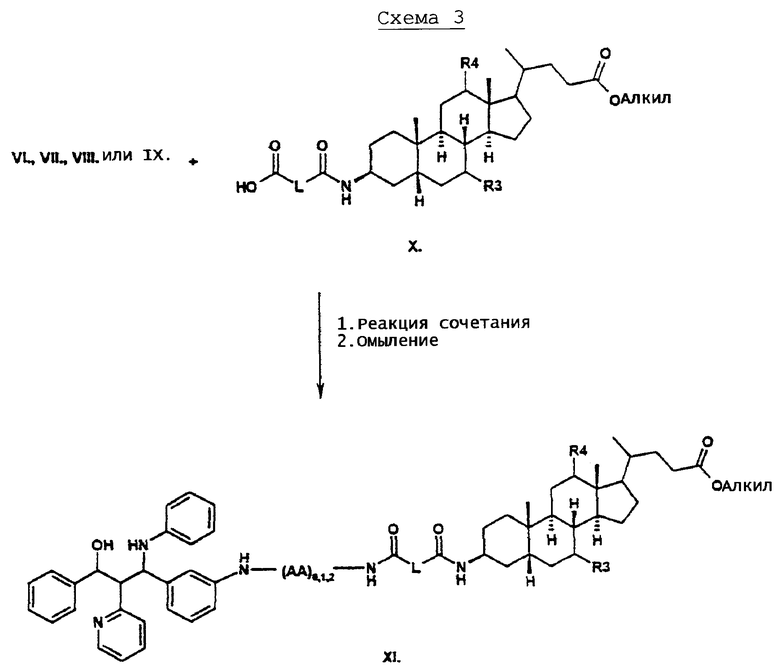
Производные желчных кислот типа Х могут быть получены из эфиров 3-аминожелчной кислоты путем связывания с алкил или арилкарбоновыми кислотами или их производными, такими как, например, янтарный ангидрид, согласно известным способам (например, ЕР 0614908, ЕР 0489423). Эти соединения (X) подвергают реакции с аминосоединениями типа VI, VII, VIII или IX согласно стандартному способу сочетания пептидов. После реакции сочетания посредством омыления алкиловой эфирной группы части желчной кислоты получают соединения типа XI.
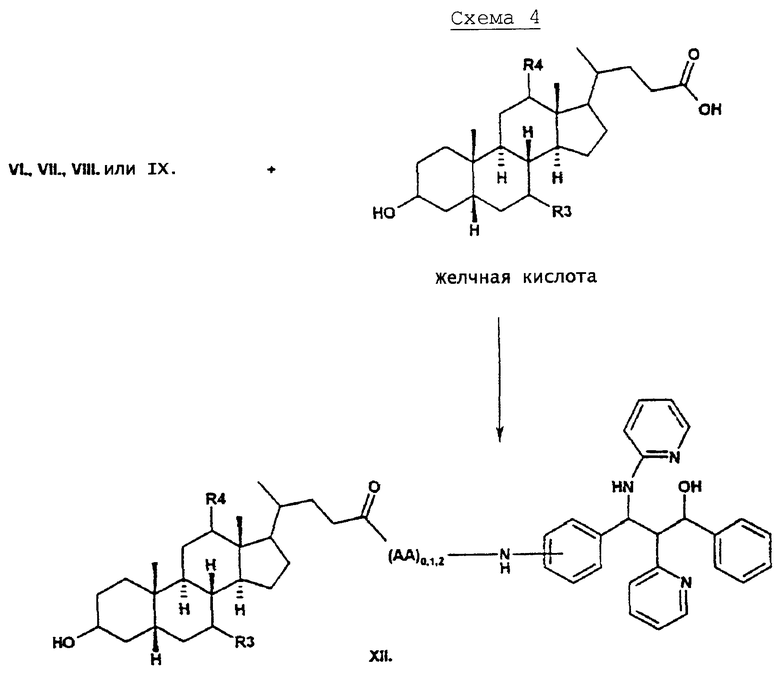
Аминосоединения типа VI, VII, VIII или XI могут быть подвергнуты реакции с карбоксильной группой желчных кислот. Здесь также находит применение известный способ сочетания пептидов, например, реакции сочетания в присутствии TOTU и триэтиламина или с дициклогексилкарбодиимидом, гидроксибензотриазолом и триэтиламином в ТГФ. Согласно этим способам могут быть получены соединения типа XII.
Фармацевтически приемлемые соли благодаря их более высокой водорастворимости по сравнению с исходными или основными соединениями являются особенно подходящими для медицинского применения. Эти соли должны содержать фармацевтически приемлемый анион или катион. Пригодными фармацевтически приемлемыми аддитивными солями кислот соединений данного изобретения являются соли неорганических кислот, таких как соляная кислота, бромистоводородная кислота, фосфорная, метафосфорная, азотная, сульфоновая и серная кислоты, а также органических кислот, таких как, например, уксусная кислота, бензолсульфоновая, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, изотионовая, молочная, лактобионовая, малеиновая, яблочная, метансульфоновая, янтарная, п-толуолсульфоновая, винная и трифторуксусная кислоты. Для медицинских целей особенно предпочтительно применяют хлоридную соль. Пригодными фармацевтически приемлемыми основными солями являются соли аммония, соли щелочных металлов (такие как соли натрия и калия) и соли щелочно-земельных металлов (такие как соли магния и кальция).
Соли с фармацевтически неприемлемым анионом также включены в объем данного изобретения в качестве полезных промежуточных продуктов для получения или очистки фармацевтически приемлемых солей и/или для применения в нетерапевтических, например, в in vitro-применениях.
Применяемое здесь понятие “физиологически функциональное производное” обозначает любое физиологически приемлемое производное соединения согласно изобретению формулы I, например эфир, которое при введении млекопитающему, такому как, например, человек, способно (прямо или косвенно) образовывать соединение формулы I или один из его активных метаболитов.
К физиологически функциональным производным причисляются также пролекарства соединений данного изобретения. Такие пролекарства могут метаболизироваться in vivo до соединения данного изобретения. Эти пролекарства сами по себе могут быть или могут не быть активными.
Соединения согласно данному изобретению могут также существовать в различных полиморфных формах, например в виде аморфной и кристаллической полиморфных форм. Все полиморфные формы соединений данного изобретения входят в объем данного изобретения и являются объектом изобретения.
Далее все ссылки на "соединение (соединения) согласно формуле (I)" относятся к соединению (соединениям) формулы (I), описанным выше, а также к их солям, сольватам и описанным физиологически функциональным производным.
Количество соединения формулы (I), которое требуется для достижения желательного биологического эффекта, зависит от ряда факторов, например выбранного специфического соединения, предполагаемого применения, способа введения и клинического состояния пациента. В общем, суточная доза находится в диапазоне от 0,3 мг до 100 мг (обычно от 3 мг до 50 мг) в сутки на килограмм веса тела, например 3-10 мг/кг/сутки. Внутривенная доза находится, например, в диапазоне от 0,3 мг до 1,0 мг/кг/сутки, которая подходящим образом в виде инфузии может вводиться от 10 нг до 100 нг на килограмм в минуту. Пригодные инфузионные растворы для этих целей могут, например, содержать от 0,1 нг до 10 мг, обычно от 1 нг до 10 мг на миллилитр. Разовые дозы могут, например, содержать от 1 мг до 10 мг активного вещества. Таким образом, ампулы для инъекций могут содержать, например, от 1 мг до 100 мг, а перорально вводимые разовые готовые к употреблению формы, такие как, например, таблетки или капсулы, могут содержать от 1,0 до 1000 мг, обычно от 10 до 600 мг. В случае фармацевтически приемлемых солей вышеуказанные весовые данные относятся к весу бензотиазепиновых ионов солей. Для профилактики или терапии вышеуказанных состояний соединения формулы (I) могут применяться в виде индивидуального соединения, но предпочтительно они находятся с приемлемым носителем в виде фармацевтической композиции. Носитель, конечно, должен быть приемлемым, в том смысле, что он совместим с другими компонентами композиции и не является вредным для здоровья пациентов. Носитель может быть твердым веществом или жидкостью или и тем и другим, и его готовят предпочтительно с соединением в виде разовой дозы, например, в виде таблетки, которая может содержать от 0,05% до 95% активного вещества. Могут присутствовать также дополнительные фармацевтически активные вещества, в том числе дополнительные соединения формулы (I). Фармацевтические композиции согласно данному изобретению могут быть приготовлены в соответствии с известными фармацевтическими способами, которые по существу состоят в том, что компоненты смешивают с фармакологически приемлемыми носителями и/или вспомогательными веществами (добавками).
Фармацевтическими композициями согласно данному изобретению являются такие композиции, которые пригодны для приема внутрь, ректального, местного, перорального (например, подъязычного) и парентерального (например, подкожного, внутримышечного, внутрикожного или внутривенного) введения, хотя самый пригодный способ введения в каждом отдельном случае зависит от типа и тяжести подлежащего лечению состояния и от типа применяемого в каждом отдельном случае соединения формулы (I). В объем данного изобретения входят также готовые формы в виде драже и дюрантные формы в виде драже (формы пролонгированного действия, депо-формы). Предпочтительными являются формы, устойчивые к действию кислот и желудочного сока. Пригодные устойчивые к желудочному соку покрытия включают в себя ацетат-фталат целлюлозы, поливинилацетат-фталат, фталат гидроксипропилметилцеллюлозы и анионные полимеры метакриловой кислоты и метилметакрилата метакриловой кислоты.
Пригодные фармацевтические соединения для орального введения могут существовать в виде отдельных единиц, таких как, например, капсулы, капсулы с облатками, таблетки для сосания или таблетки, которые в каждом отдельном случае содержат определенное количество соединения формулы (I); в виде порошков или гранулятов; в виде раствора или суспензии в водной или неводной жидкости; или в виде эмульсии типа масло в воде или вода в масле. Эти композиции могут быть приготовлены, как уже упоминалось, согласно любому подходящему фармацевтическому способу, который включает в себя стадию, при которой активное вещество и носитель (который может состоять из одного или нескольких дополнительных компонентов) приводят в контакт. В общем, эти композиции готовят посредством равномерного и гомогенного смешивания активного вещества с жидким и/или мелкоизмельченным твердым носителем, после чего этот продукт, если необходимо, формуют. Так, например, таблетка может быть изготовлена прессованием или формованием порошка или гранулята соединения, в случае необходимости с одним или несколькими дополнительными компонентами. Прессованные таблетки могут быть изготовлены посредством таблетирования соединения в свободно-текучей форме, такой как, например, порошок или гранулят, в случае необходимости, смешанные со связующим, мягчителем, инертным разбавителем и/или одним или несколькими поверхностно-активными / диспергирующими веществами, в подходящем аппарате. Формованные таблетки могут быть изготовлены формованием порошкообразного увлажненного инертным жидким разбавителем соединения в подходящем аппарате.
Фармацевтические композиции, пригодные для перорального (подъязычного) введения, включают в себя таблетки для сосания, которые содержат соединение формулы (I) с вкусовым веществом, обычно сахарозой и гуммиарабиком или трагантом, и пастилки, которые содержат соединение в инертной основе, такое как желатин и глицерин или сахароза и гуммиарабик.
Пригодные фармацевтические композиции для парентерального введения включают в себя предпочтительно стерильные водные композиции соединения формулы (I), которые предпочтительно являются изотоничными с кровью предполагаемого реципиента. Эти композиции предпочтительно вводят внутривенно, хотя введение может производиться также подкожно, внутримышечно или внутрикожно в виде инъекции. Эти композиции могут быть предпочтительно приготовлены смешиванием соединения с водой и стерилизацией полученного раствора и приданием ему изотоничности с кровью. Инъекционные композиции данного изобретения содержат обычно от 0,1 до 5 мас.% активного соединения.
Пригодные фармацевтические композиции для ректального введения предпочтительно готовят в виде свечей с разовой дозой. Они могут быть приготовлены смешиванием соединения формулы (I) с одним или несколькими обычными твердыми носителями, например какао-маслом, и помещением полученной смеси в форму.
Пригодные фармацевтические композиции для местного (локального) применения на коже существуют в виде мази, крема, лосьона, пасты, спрея, аэрозоля или масла. В качестве носителя могут применяться вазелины, ланолин, полиэтиленгликоли, спирты и комбинации из двух или нескольких из этих веществ. Активное вещество обычно присутствует в концентрации от 0,1 до 15 мас.% композиции, например от 0,5 до 2%.
Возможным является также чрескожное введение. Пригодные фармацевтические композиции для чрескожных применений могут быть в виде отдельных пластырей, которые пригодны для продолжительного тесного контакта с эпидермисом пациента. Такие пластыри содержат активное вещество в забуференном в случае необходимости водном растворе, растворенное и/или диспергированное в повышающем адгезию средстве или диспергированное в полимере. Подходящая концентрация активного вещества составляет приблизительно 1-35%, предпочтительно приблизительно 3-15%. В качестве особой возможности активное вещество, как, например, описано в Pharmaceutical Research, 2(6):318 (1986), может высвобождаться посредством электротранспорта или ионофореза.
Данное изобретение относится к соединениям формулы I в виде их рацематов, рацемических смесей и чистых энантиомеров, а также их диастереомеров и их смесей.
Соединения формулы I и их фармацевтически приемлемые соли и физиологически функциональные производные отличаются благоприятным действием на липидный обмен. Эти соединения могут применяться отдельно или в комбинации с дополнительными активными веществами, понижающими содержание липидов в крови. Эти соединения пригодны для профилактики, а также, в частности, для лечения нарушений липидного обмена, в частности гиперлипидемии. Соединения формулы I пригодны также для воздействия на уровень холестерина в сыворотке, а также для предупреждения и лечения артериосклеротических явлений.
Следующие данные подтверждают фармакологическую эффективность рассматриваемых в данном изобретении соединений.
Биологическое испытание соединений данного изобретения проводили посредством исследования ингибирования поглощения [3H]-таурохолата в мембранных везикулах щеточной каймы подвздошной кишки кролика. Тест ингибирования проводили следующим образом:
1. Получение мембранных везикул щеточной каймы подвздошной железы кролика.
Получение мембранных везикул щеточной каймы из кишечных клеток тонкой кишки проводили с использованием так называемого способа Мg2+-преципитации. Самцов Ново-Зеландских кроликов (вес тела 2-2,5 кг) умерщвляли внутривенной инъекцией 0,5 мл Т61®, водного раствора 2,5 мг тетракаин-HCl, 100 мг эмбутрамида и 25 мг мебезонийиодида. Тонкую кишку извлекали и промывали охлажденным на льду физиологическим раствором поваренной соли. Концевые 7/10 тонкой кишки (отмеренные в орально-ректальном направлении, т.е. концевую подвздошную кишку, которая содержит активную Nа+-зависимую систему транспорта желчных кислот) применяли для получения мембранных везикул щеточной каймы. Кишки замораживали в пластиковых мешках под азотом при -80°С. Для получения мембранных везикул замороженные кишки оттаивали при 30°С в водяной бане. Слизистую оболочку соскребали и суспендировали в 60 мл охлажденной льдом смеси 12 мМ Трис/НСl-буфер (рН 7,1)/300 мМ маннит, 5 мМ ЭГТА/10 мг/л фенилметилсульфонилфторида/1 мг/л ингибитора трипсина, полученного из сои (32 Е/мг)/0,5 мг/л ингибитора трипсина, полученного из бычьего легкого (193 Е/мг)/5 мг/л бацитрацина. После разбавления до 300 мл охлажденной на льду дистиллированной водой гомогенизировали при помощи Ultraturrax (18-Stab, IKA Werk Staufen, Deutschland) 3 минуты при 75% максимальной мощности при охлаждении льдом. После добавления 3 мл 1 М раствора МgС12 (конечная концентрация 10 мМ) выдерживали точно 1 минуту при 0°С. Посредством добавления Мg2+ клеточные мембраны агрегировали и осаждали за исключением мембран щеточной каймы. После 15-минутного центрифугирования при 3000×g (5000 об/мин, ротор SS-34) осадок выбрасывали, а супернатант, который содержит мембраны щеточной каймы, центрифугировали 30 минут при 48000×g (20000 об/мин, ротор SS-34). Супернатант выбрасывали, осадок повторно гомогенизировали в 60 мл смеси 12 мМ Трис/НС1-буфер (рН 7,1)/60 мМ маннит, 5 мМ ЭГТА с использованием гомогенизатора Potter Elvejhem (Braun, Melsungen, 900 об/мин, 10 тактов). После добавления 0,1 мл раствора MgCl2 и 15-минутного инкубационного периода при 0°С снова центрифугировали 15 минут при 3000×g. Супернатант центрифугировали затем еще раз 30 минут при 48000×g (20000 об/мин, ротор SS-34). Осадок помещали в 30 мл смеси 10 мМ Трис/Нереs-буфер (рН 7,4)/300 мМ маннит и повторно гомогенно суспендировали посредством 20 тактов в гомогенизаторе Potter Elvejhem при 1000 об/мин. После 30-минутного центрифугирования при 48000×g (20000 об/мин, ротор SS-34) осадок помещали в 0/5-2 мл смеси Трис/Нереs-буфер (рН 7,4)/280 мМ маннит (конечная концентрация 20 мг/мл) и ресуспендировали при помощи туберкулинового шприца с иглой 27 Gauge. Везикулы либо использовали непосредственно после получения для исследований транспорта, либо хранили при -196°С в порциях по 4 мг в жидком азоте.
2. Ингибирование поглощения [3Н]-таурохолата в мембранных везикулах щеточной каймы подвздошной кишки.
Поглощение субстратов в вышеописанные мембранные везикулы щеточной каймы определяли с использованием так называемого способа мембранной фильтрации. 10 мкл суспензии везикул (100 мкг белка) пипетировали в виде капель на стенку полистирольной инкубационной пробирки (11×70 мм), которая содержала инкубационную среду с соответствующими лигандами (90 мкл). Эта инкубационная среда содержит 0,75 мкл=0,75 мкКи [3H(G)]-таурохолата (удельная активность: 2,1 Ки/ммоль)/0,5 мкл 10 мМ таурохолата/8,75 мкл буфера для транспорта натрия (10 мМ Трис/Hepes (рН 7,4)/100 мМ маннит/100 мМ NaCl) (Na-T-Б) или 8,75 мкл буфера для транспорта калия (10 мМ Трис/Hepes (рН 7,4)/100 мМ маннит/100 мМ КСl) (К-Т-Б) и 80 мкл раствора соответствующего ингибитора, в зависимости от эксперимента растворенного в Na-T-буфере или К-Т-буфере. Эту инкубационную среду фильтровали через поливинилиденфторидный мембранный фильтр (SYHV LO 4NS, 0,45 мкм, 4 мм диаметр, Millipore, Eschborn/ Deutschland). Посредством перемешивания везикул с инкубационной средой начинали измерение транспорта. Концентрация таурохолата в инкубационной исходной смеси составляла 50 мкМ. После желательного инкубационного периода (обычно 1 минуты) транспорт останавливали добавлением 1 мл охлажденного льдом стопраствора (10 мМ Трис/Hepes (рН 7,4)/150 мМ КСl). Полученную смесь тотчас же отсасывали под вакуумом 25-35 мбар через мембранный фильтр из нитрата целлюлозы (ME 25, 0,45 мкм, 25 мм диаметр, Schleicher (Schuell, Dassel, Deutschland). Фильтр промывали 5 мл охлажденного льдом стоп-раствора.
Для измерения поглощения радиоактивно меченного таурохолата этот мембранный фильтр суспендировали с 4 мл сцинтиллятора Quickszint 361 (Zinsser Analytik GmbH, Frankfurt, Deutschland) и радиоактивность измеряли в измерительном приборе TriCarb 2500 (Canberra Packard GmbH, Frankfurt, Deutschland). Измеренные показатели после калибровки прибора с использованием стандартных проб и после корректировки возможной присутствующей хемилюминесценции получали в виде расп/мин (распадов в минуту).
Контрольные показатели определяли в каждом случае в Na-T-Б и К-Т-Б. Разность между поглощением в Na-T-Б и К-Т-Б давала Na+-зависимую долю транспорта. Концентрацию ингибитора, при которой Nа+-зависимая доля транспорта ингибировалась на 50% в расчете на контроль, обозначали как IC50 Na+.
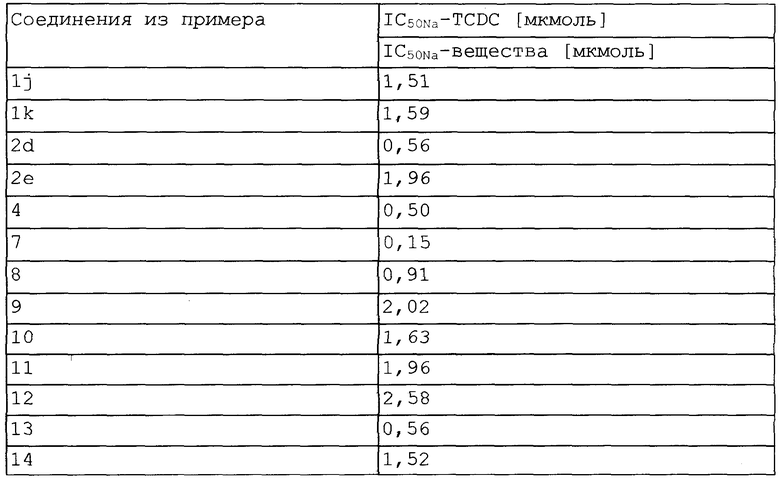
Фармакологические данные включают в себя серию тестов, в которых исследовали взаимодействие соединений данного изобретения с кишечной системой транспорта желчных кислот в концевой тонкой кишке. Результаты приведены в таблице.
Таблица показывает результаты измерений ингибирования поглощения [3Н]-таурохолата в мембранные везикулы щеточной каймы подвздошной кишки кроликов. Приведены отношения IC50Na-показателей сравнительного вещества в виде таурохенодезоксихолата (TCDC) и тестируемого в каждом случае вещества.
Нижеследующие примеры более подробно поясняют данное изобретение, не ограничивая изобретение описанными в этих примерах продуктами и вариантами осуществления.
Пример 1
а.
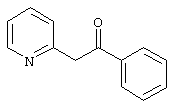
К 50 г (0,54 моль) пиколина в 770 мл тетрагидрофурана добавляли по каплям при -55°С 366 мл 15% н-бутиллития в н-гексане. Нагревали до комнатной температуры и снова охлаждали до -55°С. Медленно добавляли по каплям 77 г N,N-диметилбензамида (0,52 моль) в 570 мл тетрагидрофурана, затем нагревали до комнатной температуры и перемешивали еще в течение 1 часа. После добавления 550 мл 1 н. соляной кислоты экстрагировали этилацетатом (3х), органические фазы сушили MgSO4, и упаривали. Посредством перегонки остатка получали 47,5 г (47%) продукта. Точка кипения 134-136°С/0,28 мбар.
b.
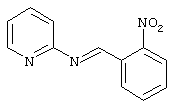
20,0 г (0,13 моль) б-нитробензальдегида, 12,5 г (0,13 моль) 2-аминопиридина и 0,3 г п-толуолсульфоновой кислоты в 150 мл толуола нагревали в течение 2,5 часов с обратным холодильником. Раствор охлаждали, образующийся осадок отсасывали и сушили.
Выход: 18,1 г (60%) продукта
Точка плавления: 93-95°C
С12Н9N3О2; (227) MS (FAB) 228 М+H+
с.
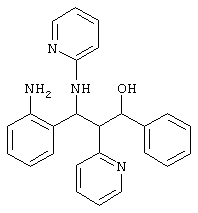
12,0 г (61 ммоль) кетона из примера la и 15,0 г (66 ммоль) имина из примера 1b нагревали в течение 45 минут на паровой бане. Реакционную смесь растворяли в этаноле при нагревании. После охлаждения осадок отсасывали и перекристаллизовывали из этанола.
Выход: 11,8 г (46%) продукта
C25H20N403 (424,2) MS (FAB) 425 М+H+
d.

8,0 г (18,8 ммоль) кетосоединения из примера 1с растворяли в 300 мл смеси тетрагидрофуран/вода 10:1, соединяли с 4,67 г боргидрида натрия и перемешивали при комнатной температуре в течение 2 часов. Затем раствор упаривали, остаток соединяли со 100 мл 2 н. соляной кислоты и нагревали на паровой бане до полного растворения. После охлаждения доводили до щелочной реакции 4 н. раствором NaOH и экстрагировали этилацетатом (2х). Органические фазы сушили MgSO4 и упаривали. Остаток хроматографировали на силикагеле (гептан/этилацетат 1:1). Получали два рацемических соединения в качестве продукта.
1-я фракция: 3,9 г (48%) неполярного рацемата (пример 1 d/1)
С25Н22N403 (426,2) MS (FAB) 427 М+Н+
2-я фракция: 2,5 г (31%) полярного рацемата (пример 1 d/2)
С25Н22N403 (426,2) MS (FAB) 427 М+Н+
е.
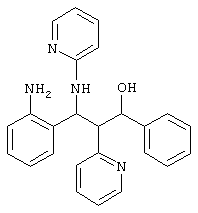
2,5 г (5,86 ммоль) неполярного рацемата из примера 1 d/1 растворяли в 300 мл метанола, соединяли с приблизительно 20 мг Pd/C (10%) и гидрировали в атмосфере Н2 при комнатной температуре. Катализатор отфильтровывали и раствор упаривали. Остаток хроматографировали через силикагель (н-гептан/этилацетат 7:13).
Выход: 1/9 г (82%) продукта
С25Н24N40 (396,22) MS (FAB) 397 М+Н+
f.
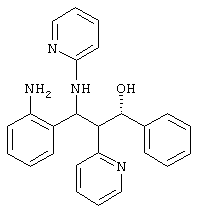
(+)-энантиомер (пример 1 f/2)
100 мг рацемического соединения из примера 1е разделяли при помощи препаративной ВЖХ на энантиомеры. Разделение проводили с использованием колонки CSP-Chiralpak (Fa. Daicel, Dusseldorf) со смесью н/гексан/этанол 4:1.
В качестве первой фракции получали 40 мг (-)-энантиомера (пример 1 f/1), в качестве 2-й фракции получали 40 мг (+)-энантиомера (пример 1 f/2).
g.
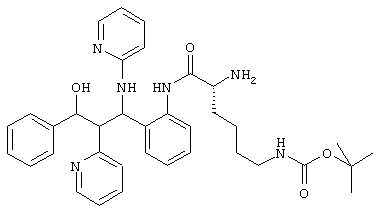
4,0 г (10,1 ммоль) аминосоединения из примера 1е (неполярного рацемата), 4,85 г (10,3 ммоль) N-Fmoc-D-Lys(BOC)ОН, 4/0 г (12,2 ммоль) TOTU и 2,7 мл триэтиламина растворяли в 300 мл диметилформамида и перемешивали 2 часа при комнатной температуре. Реакционную смесь выливали на воду и экстрагировали этилацетатом (2х). Органические фазы сушили (МgSО4) и упаривали. Остаток растворяли в 150 мл смеси диметилформамид/пиперидин 2:1 для отщепления Fmoc-группы и перемешивали 1 час при комнатной температуре. Этот раствор выливали на воду, экстрагировали этилацетатом (3х). Органические фазы сушили (MgSO4) и упаривали. Хроматографией на силикагеле (дихлорметан/метанол 9:1) получено 4,0 г (63,5%) продукта.
C36H44N604 (624,3) MS (FAB) 625 М+Н+
h.
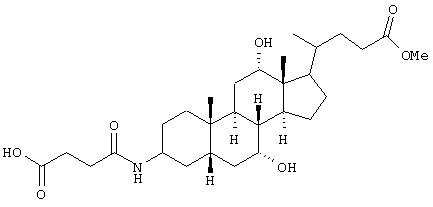
5,0 г (11,86 ммоль) метилового эфира 3β-аминохолевой кислоты (Европейская патентная заявка ЕР 0614908), 1,3 г (13 ммоль) янтарного ангидрида и 16,5 мл триэтиламина растворяли в 75 мл тетрагидрофурана и перемешивали 1 час при комнатной температуре. Раствор упаривали. Остаток растворяли в воде, подкисляли соляной кислотой, экстрагировали этилацетатом (3х).
Органические фазы сушили (MgSO4) и упаривали.
Выход: 5,8 г (94%)
C29H47NО7 (521,3) MS (FAB) 528 M+Li+.
i.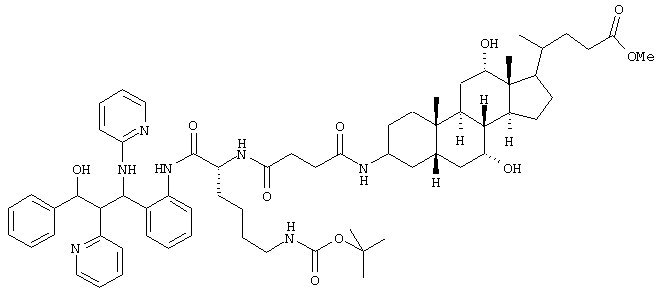
4,0 г (6,4 ммоль) соединения из примера 1g, 3,45 г (6,6 ммоль) производного желчной кислоты из примера 1h, 1,2 мл триэтиламина, 2,16 г (16 ммоль) гидроксибензотриазола и 2,56 г дициклогексилкарбодиимида (12,4 ммоль) растворяли в 250 мл тетрагидрофурана и 5 часов перемешивали при комнатной температуре. Смесь упаривали, остаток растворяли в этилацетате и промывали раствором NаНСО3. Органические фазы сушили (МgSО4) и упаривали. Хроматографией на силикагеле (дихлорметан/метанол 19:1, затем 9:1) получено 3,1 г (43%) продукта.
С65Н89N7О10 (1127,7) MS (FAB) 397,2 M+Li+
J.
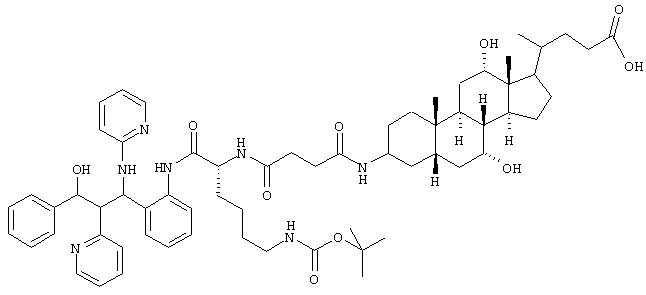
3,1 г (2,75 ммоль) метилового эфира из примера li растворяли в 200 мл этанола, соединяли с 31 мл 1 н. раствора NaOH и перемешивали 5 часов при комнатной температуре. Смесь упаривали, остаток растворяли в воде и соединяли с насыщенным раствором NaH2PO4. Экстрагировали этилацетатом (2х), органические фазы сушили над MgSO4 и упаривали. Неочищенный продукт хроматографировали на силикагеле (дихлорметан/метанол 4:1).
Выход: 2,25 г (73%)
C64H87N7O10 (1113,7) MS (FAB) 1120,7 M+Li+
k.
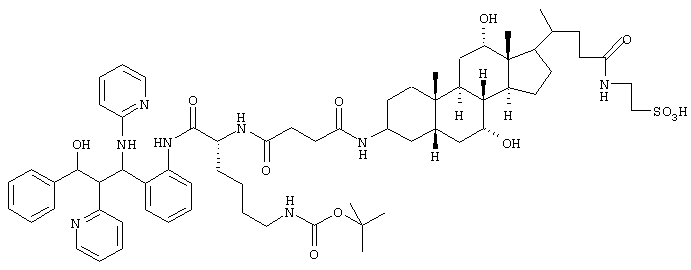
1,5 г (1,35 ммоль) соединения из примера lj и 0,81 мл триэтиламина соединяли при 0°С с 0,48 мл этилового эфира хлор-муравьиной кислоты и перемешивали в течение 10 минут. После этого добавляли 0,6 г таурина, растворенного в 30 мл 0,1 н. раствора NaOH, и перемешивали в течение 24 часов при комнатной температуре. Смесь упаривали, остаток растворяли в небольшом количестве воды и выливали в насыщенный раствор Na2HPO4. Эту смесь экстрагировали этилацетатом (3х), органические фазы сушили MgSO4 и упаривали. После хроматографии на силикагеле (дихлорметан/метанол 4:1, затем метанол) получали 0,98 г (60%) конъюгата таурина.
С66H92N8O12S (1270,7) MS (FAB) 1243,6 M+Na+
Пример 2
а.
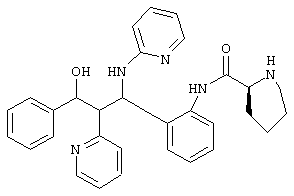
2,5 г (6,31 ммоль) аминосоединения из примера 1е (неполярного рацемата), 2/2 г (6,52 ммоль) Fmoc-L-пролина, 2,5 г (7,62 ммоль) TOTU и 1,7 мл триэтиламина растворяли в 100 мл диметилформамида и перемешивали 3 часа при комнатной температуре. Реакционную смесь упаривали наполовину, соединяли с водой и экстрагировали этилацетатом (3х). Органические фазы сушили МgS04 и упаривали. После хроматографии на силикагеле (этилацетат/н-гептан 7:3) получали 3,85 г (85%) продукта.
Этот Fmoс-защищенный промежуточный продукт (3,6 г) растворяли в 110 мл смеси пиперидин/ДМФ 1:10 и перемешивали в течение 1 часа при комнатной температуре. Смесь упаривали и хроматографировали на силикагеле (дихлорметан/метанол 19:1, затем 9:1).
Выход: 1,8 г (72/5%)
С30Н31N502 (493,2) MS (FAB) 494 М+H+
b.
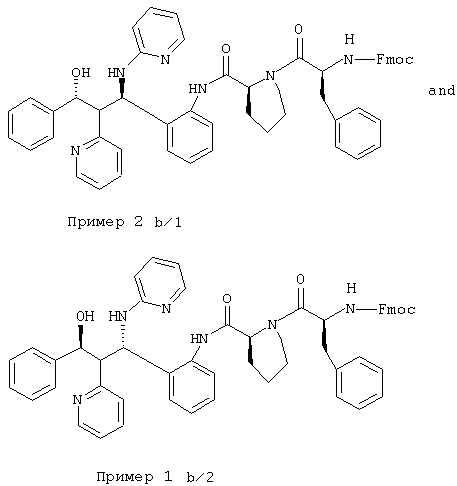
1,7 г (3,44 ммоль) соединения из примера 2а перемешивали с 1,4 г (3,61 ммоль) Finoc-L-фенилаланина, 1,9 г (5,80 ммоль) TOTU и 1,0 мл триэтиламина в 150 мл ДМФ 4 часа при комнатной температуре. Реакционную смесь упаривали и остаток хроматографировали на силикагеле (этилацетат/н-гептан 4:1). Получали две фракции:
1-я фракция: 1,28 г (43%) неполярного диастереомера (пример 1 b/1)
С54Н50N6О5 (862,4) MS (FAB) 863,4 М+Н+
2-я фракция: 0,82 г (28%) полярного диастереомера (пример 2 b/1)
С54Н50N6О5 (862,4) MS (FAB) 863,4 М+Н+
с.
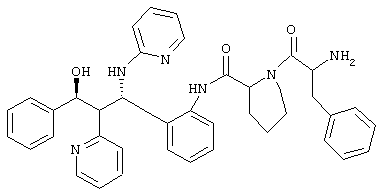
0,8 г (0,93 ммоль) соединения из примера 2 b/2 растворяли в 33 мл смеси ДМФ/пиперидин 10:1 и перемешивали 1 час при комнатной температуре. После упаривания остаток хроматографировали на силикагеле (дихлорметан/метанол 19:1, затем 9:1).
Выход: 0,35 г (59%)
С39Н40N6О3 (640,3) MS (FAB) 641,3 M+H+
d.
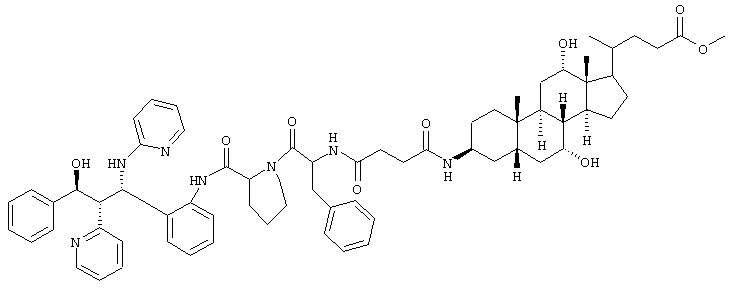
0,5 г (0,78 ммоль) соединения из примера 2с, 0,45 г (0,86 ммоль) производного желчной кислоты из примера lh подвергали реакции согласно описанному в примере li способу. Получали 0,38 г (43%) продукта.
С68Н85N7О9 (1143,6) MS (FAB) 1144,6 М+Н+
е.
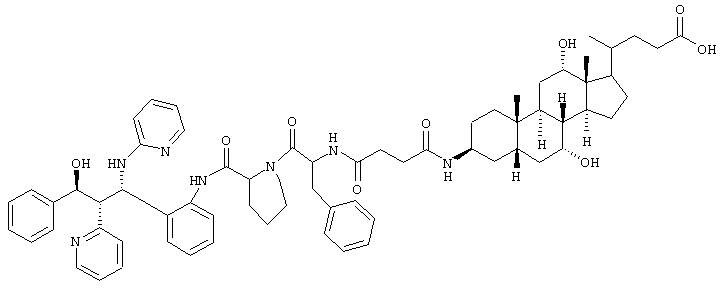
0,31 г (0,27 ммоль) метилового эфира из примера 1d растворяли в 30 мл этанола, соединяли с 3,0 мл 1 н. раствора NaOH и перемешивали 12 часов при комнатной температуре. Реакционную смесь упаривали, и остаток хроматографировали на силикагеле (дихлорметан/метанол 4:1).
Выход: 220 мг (72%)
C67H83N7O9 (1129,6) MS (FAB) 1130,6 M+H+
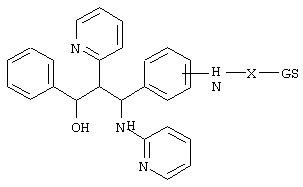
Пример 3
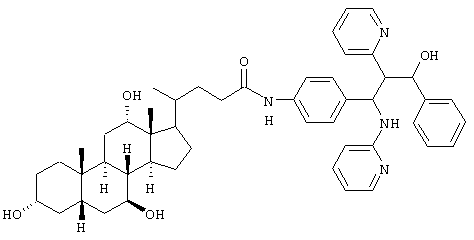
0,3 г (0,78 ммоль) 3-(4-аминофенил)-1-фенил-2-пиридин-2-ил-3-(пиридин-2-иламино)пропан-1-ола (получение аналогично примеру le), 0,34 г (0,83 ммоль) урсохолевой кислоты, 0,34 г (2,52 ммоль) гидроксибензотриазола, 0,41 г (2 ммоль) дициклогексилкарбодиимида и 0,15 мл триэтиламина перемешивали в 50 мл тетрагидрофурана в течение 2 дней при комнатной температуре. После завершения реакции твердые вещества отфильтровывали. Раствор упаривали, и остаток хроматографировали на силикагеле (дихлорметан/метанол 9:1, затем 17:3). Получали 0,33 г (55%) продукта.
C49H62N4O5 (786,5) MS (FAB) 787,5 M+H+
С использованием соответствующих исходных соединений аналогично примерам 1-3 получали примеры 4-14.
Пример 4
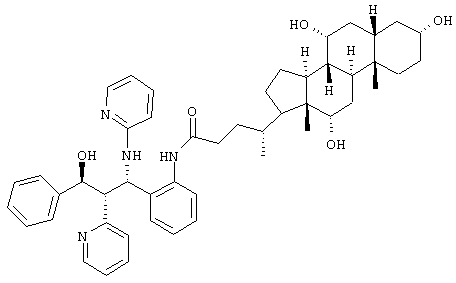
C49H62N4O5 (787,1) MS (FAB) 788,1 M+H+
Пример 5 (неполярный диастереомер)
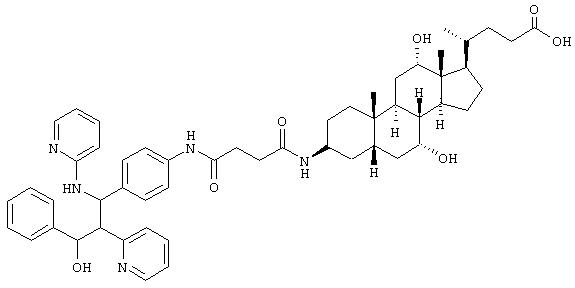
C53H67N5O7 (886,2) MS (FAB) 887,2 M+H+
Пример 6
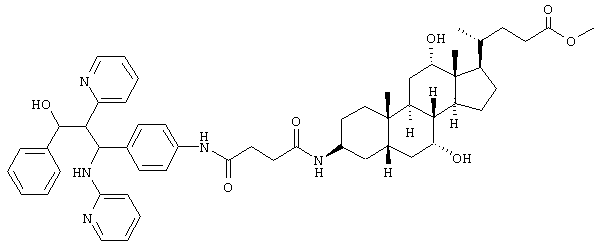
C54H69N5O7 (900,2) MS (FAB) 901,2 M+H+
Пример 7 (полярный диастереомер)
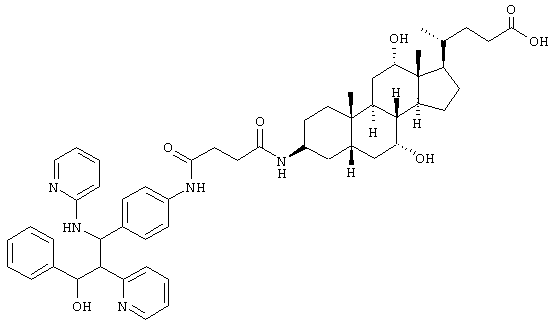
C53H67N5O7 (886,2) MS (FAB) 887,2 M+H+
Пример 8
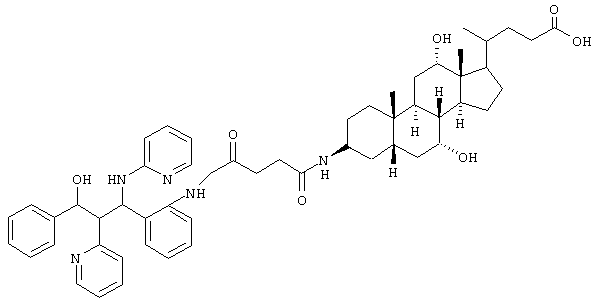
C53H67N5O7 (886,2) MS (FAB) 887,2 M+H+
Пример 9
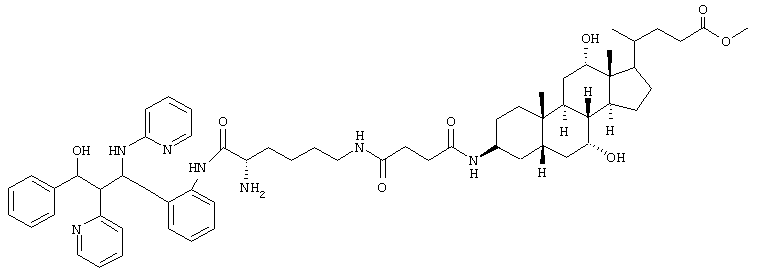
C60H81N7O8 (1028,4) MS (FAB) 1029,4 M+H+
Пример 10
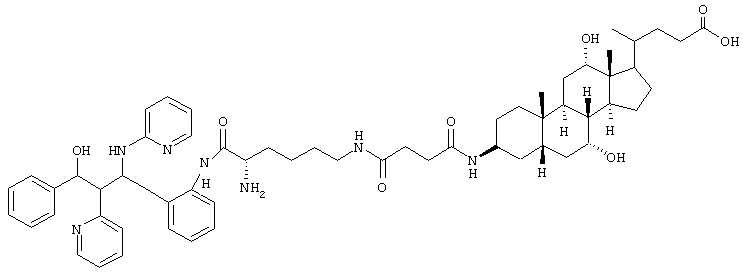
C59H79N7O8 (1014,3) MS (FAB) 1015,3 M+H+
Пример 11
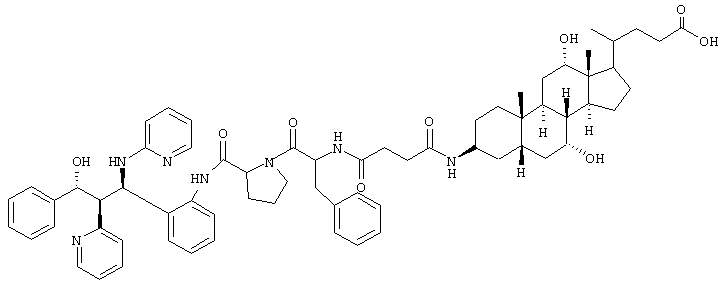
C67H83N7O9 (1130,5) MS (FAB) 1031,5 M+H+
Пример 12
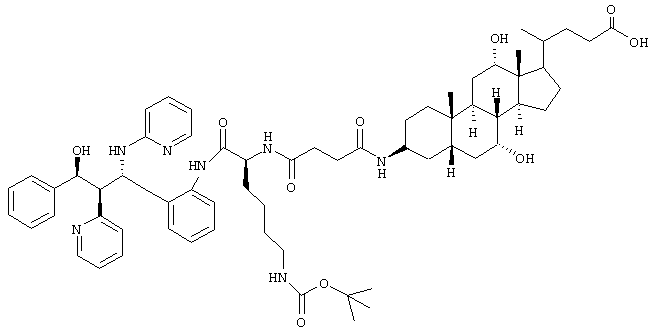
C64H87N7O10 (1114,4) MS (FAB) 1115,4 M+H+
Пример 13
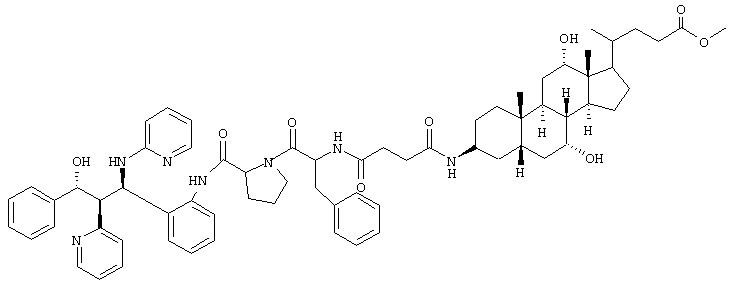
C68H85N7O9 (1144,5) MS (FAB) 1145,5 M+H+
Пример 14
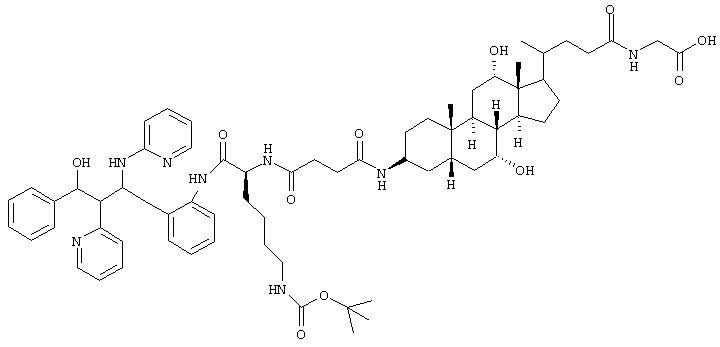
C66H90N8O11 (1171,5) MS (FAB) 1172,5 M+H+.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПРОПАНОЛАМИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СРЕДСТВО НА ИХ ОСНОВЕ | 1998 |
|
RU2198876C2 |
| ЗАМЕЩЕННЫЕ ЖЕЛЧНЫМИ КИСЛОТАМИ ФЕНИЛАЛКЕНОИЛГУАНИДИНЫ, ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1999 |
|
RU2232769C2 |
| НОВЫЕ АРОМАТИЧЕСКИЕ ФТОРГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ ЛЕКАРСТВЕННЫЕ СРЕДСТВА, И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2340622C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ФТОРГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ ЛЕКАРСТВЕННЫЕ СРЕДСТВА И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2339641C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРОВ ЭНДОТЕЛИНА | 1994 |
|
RU2126418C1 |
| ПРОИЗВОДНЫЕ ЖЕЛЧНОЙ КИСЛОТЫ В КАЧЕСТВЕ АГОНИСТОВ FXR/TGR5 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2707280C2 |
| ФТАЛИМИДОПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ МОНОАМИНООКСИДАЗЫ В | 2003 |
|
RU2317289C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ КИСЛОТНОЙ ПОМПЫ | 2007 |
|
RU2412188C2 |
| ПРОТИВОВИРУСНЫЕ ЭФИРЫ ИЗОСТЕРОВ СУБСТРАТОВ АСПАРТАТПРОТЕАЗЫ ИЛИ ИХ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И КОМПОЗИЦИЯ | 1995 |
|
RU2164229C2 |
| НОВЫЕ ФТОРГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ ПИРАЗОЛОВ, СОДЕРЖАЩЕЕ ИХ ЛЕКАРСТВЕННОЕ СРЕДСТВО И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2370499C2 |
Изобретение относится к замещенным производным пропаноламина с желчными кислотами формулы I и их фармацевтически приемлемым солям и физиологически функциональным производным, где GS - группа желчной кислоты формулы II, R1 – связь с X, ОН, R2 – связь с X, ОН, -О-(C1-С6)алкил, -NH-(С2-С6)-алкил-SO3Н, -NH-(С1-С6)-алкил-СООН, R1 и R2 одновременно не означают связь с Х, Х –
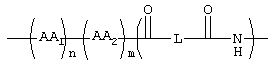
l, m, n – 0,1; L – (C1-C6)-алкил, AA1, АА2 независимо аминокислотный остаток, возможно одно- или многократно замещенный аминогруппой. Соединения I пригодны в качестве гиполипидемических средств. 2 н. и 7 з.п. ф-лы, 1 табл.
 I
I
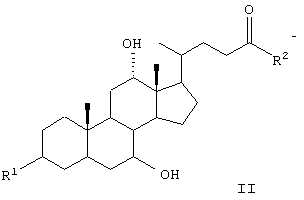
где GS обозначает группу желчной кислоты формулы
R1 обозначает связь с X, ОН;
R2 обозначает связь с X, ОН, О-(C1-С6)-алкил, NH-(С2-С6)-алкил-SО3Н, NH-(С1-С6)-алкил-СООН;
при условии, что R1 и R2 не имеют одновременно следующее значение: R1 связь с Х и R2 связь с X;
Х обозначает
или связь;
l, m, n независимо друг от друга обозначают 0 или 1;
L обозначает (C1-C6)-алкил;
AA1, АА2 независимо друг от друга обозначают аминокислотный остаток или аминокислотный остаток, который одно- или многократно замещен аминозащитной группой, а также их фармацевтически приемлемые соли.
| US 5610151 A, 11.03.1997.EP 0624593 A2, 17.11.1994.EP 0489423 A2, 10.06.1992.EP 0869121 A1, 07.10.1998. |

