Настоящее изобретение касается генного кластера, точнее - генов из генного кластера.
В частности, изобретение касается полинуклеотидов, таких как ДНК, ускоряющих биосинтез ML-236B - ингибитора HMG-CoA-редуктазы - в микроорганизмах, вырабатывающих ML-236B, при введении в эти микроорганизмы. Кроме того, изобретение касается векторов, в которые встраиваются эти полинуклеотиды, клетки-хозяина, трансформируемой этими векторами, белков, экспрессируемых этими векторами, способа получения ML-236B с помощью этих полинуклеотидов и/или белков, включающего получение ML-236B из культуры клетки-хозяина, а также касается других связанных с этим вопросов.
Правастатин является ингибитором HMG-CoA-редуктазы. Правастатин натрия применялся при лечении гиперлипидемии, он обладает полезным фармакологическим свойством - способностью снижать уровень холестерина в крови. Правастатин можно получить с помощью Streptomyces carbophilus путем микробиологического превращения ML-236B, вырабатываемого Penicillium citrinum (как описано в Endo A. et al., J. Antibiot., 29, 1346 (1976); Matsuoka T. et al., Eur. J. Biochem., 184, 707 (1989), и в японской патентной заявке №57-2240).
Было показано, что ML-236B - предшественник правастатина и ловастатин - ингибитор HMG-CoA-редуктазы обладают одинаковой частичной структурой. Они синтезируются биологическим путем через поликетиды (как описано в Moore R.N. et al., J. Am. Chem. Soc., 107, 3694 (1985); Shiao M. and Don H.S., Proc. Natl. Sci. Counc. Repub. China.B, 11, 223 (1987)).
Поликетиды - это соединения, происходящие из β-кето углеродных цепей; они образуются в ходе реакции конденсации низкомолекулярных карбоксикислот, таких как уксусная кислота, пропионовая кислота, масляная кислота и т.п. В зависимости от способа конденсации или восстановления каждой из карбонильных β-кетогрупп могут образовываться различные структуры (как описано в Hopwood D.A. and Sherman D.H., Annu. Rev. Genet., 24, 37-66 (1990); Hutchinson C.R. and Fujii I., Annu. Rev. Microbiol., 49, 201-238 (1995)).
Известно, что ферменты поликетидсинтетазы (в дальнейшем именуемые ПКС), участвующие в синтезе поликетидов, имеются у нитчатых грибов-гифомицетов и бактерий. Ферменты нитчатых грибов исследовались методами молекулярной биологии (как описано в Feng G.H. and Leonard T.J., J. BacterioL, 177, 6246 (1995); Takano Y. et al., Mol. Gen. Genet., 249, 162 (1995)). У микроорганизма Aspergillus terreus, вырабатывающего ловастатин, ген ПКС, связанный с биосинтезом ловастатина, подвергался анализу (как описано в открытой международной заявке в Японии (KOHYO) №9-504436, а также в соответствующем патенте WO 9512661 на ДНК, кодирующую триол-поликетидсинтетазу).
Гены, связанные с биосинтезом вторичных метаболитов у нитчатых грибов, часто образуют кластеры в геноме. Известно, что существуют кластеры генов, участвующих в путях биосинтеза поликетидов. Гены, кодирующие белки ферментов (типа ПКС), участвующих в биосинтезе афлатоксина - поликетида, вырабатываемого Aspergillus flavus и Aspergillus parasiticus, как известно, образуют кластерную структуру. Проводился геномный анализ и сравнение генов, участвующих в биосинтезе афлатоксина в этих микроорганизмах (Yu J. et al., Appl. Environ. Microbiol., 61, 2365 (1995)). Сообщали, что гены, участвующие в биосинтезе стеригматоцистина у Aspergillus nidulans, образуют кластерную структуру, занимающую непрерывный участок размером около 60 kb в геноме (как описано в Brown D.W. et al., Proc. Natl. Acad. Sci. USA, 93, 1418 (1996)).
Исследовалась модуляция поликетидсинтетазной активности вспомогательными белками при синтезе ловастатина (Kennedy J. et al., Science, vol. 284, 1368 (1999)).
В настоящее время, однако, молекулярно-биологический анализ биосинтеза ML-236В и факторов его регуляции нельзя считать достаточным.
Настоящее изобретение обеспечивает полинуклеотид, который может применяться для ускорения биосинтеза ML-236B.
Типичным полинуклеотидом является полинуклеотид, кодирующий белок, включающий или состоящий из аминокислотной последовательности SEQ ID No. 38, 42, 44, 46, 48 или 50. Также представлены варианты этого полинуклеотида, кодирующие модифицированную аминокислотную последовательность, имеющую по меньшей мере одну делецию, вставку, замену или изменение.
Перечень последовательностей в виде таблицы
Перечень последовательностей входит в состав описания настоящего патента. Для облегчения понимания он представлен в виде следующей таблицы А.



Полинуклеотиды, кодирующие аминокислотные последовательности SEQ ID No. 38, 42, 44, 46, 48 или 50, могут представлять собой кДНК, геномную ДНК или мРНК. Геномная ДНК, кодирующая эти 6 последовательностей, называется mlcE, mlcR, mlcA, mlcB, mlcC и mlcD соответственно. Независимо от этих обозначений мы полагаем, что эти структурные гены кодируют белки со следующими функциями:
mlcA Поликетидсинтетаза
mlcB Поликетидсинтетаза
mlcC Р450-Монооксигеназа
mlcD HMG-CoA-Редуктаза
mlcE Выводная помпа
mlcR Транскрипционный фактор
Мы открыли, что введение mlcE или кДНК, соответствующей mlcE, ускоряет биосинтез ML-236B, и введение mlcR или кДНК, соответствующей mlcR, тоже ускоряет биосинтез ML-236B. Кроме того, mlcR стимулирует транскрипционную экспрессию генов от mlcA до mIcD. Как показали опыты по инактивации генов, mlcA, В, С и D участвуют в продукции ML-236B независимо или в сочетании.
Варианты mlcA, В и/или С, которые можно получить природным или искусственным способом, используются для получения производных ML-236B, включая такие статины, как правастатин и ловастатин. В этом плане становится возможным получение правастатина непосредственно с помощью таких вариантов, при единственной стадии ферментации и без микробиологического превращения ML-236B в правастатин, которое сейчас проводится с помощью Streptomyces carbophilus.
Предпочтительным полинуклеотидом является последовательность, включающая SEQ ID No. 37, или мутант, или вариант таковой, способный ускорить биосинтез ML-236B. Такой ДНК-полинуклеотид можно получить из Escherichia coli, трансформированных pSAKexpE SANK 72499 (FERM BP-7005).
Другим предпочтительным полинуклеотидом является последовательность, включающая SEQ ID No. 41, или мутант, или вариант таковой, способный ускорить биосинтез ML-236B. Такой ДНК-полинуклеотид можно получить из Escherichia coli, трансформированных pSAKexpR SANK 72599 (FERM BP-7006).
Полинуклеотиды настоящего изобретения могут применяться в рабочем сочетании с одним или несколькими полинуклеотидами. Предпочтительны те комбинации, которые способны повысить продукцию ML-236B в микроорганизмах-продуцентах.
Примеры таких комбинаций включают полинуклеотид SEQ ID No. 37 или его вариант с подобной функцией в сочетании с одной или несколькими последовательностями, выбранными из числа самой SEQ ID No. 37 и 41, 43, 45, 47 и 49, а также полинуклеотид SEQ ID No. 41 или его вариант с подобной функцией в сочетании с одной или несколькими последовательностями, выбранными из числа самой SEQ ID No. 41, 37, 43, 45,47 и 49.
В одном воплощении полинуклеотидом предпочтительно является полинуклеотид, кодирующий белок, включающий или состоящий из аминокислотной последовательности SEQ ID No. 38, 42, 44, 46, 48 или 50 и способный ускорить биосинтез ML-236B сам по себе или вместе с полинуклеотидом SEQ ID No. 37, SEQ ID No. 41 или их вариантов, обладающих подобной функцией.
Настоящее изобретение также распространяется на полинуклеотиды, способные к гибридизации в строгих условиях с полинуклеотидом настоящего изобретения. К ним относятся полинуклеотиды, способные ускорить биосинтез ML-236B в продуцирующем ML-236B микроорганизме при введении в этот микроорганизм.
В типичном случае полинуклеотидом служит ДНК, кДНК или геномная ДНК, или РНК, и они могут быть смысловыми или антисмысловыми. Полинуклеотид обычно является очищенным, например очищенным от других клеточных компонентов.
Настоящее изобретение распространяется на варианты полинуклеотидов, кодирующих аминокислотные последовательности SEQ ID No. 38, 42, 44, 46, 48 или 50, в которых имеются замены одного или нескольких нуклеотидов. Эти замены могут произойти естественным путем или могут быть сделаны в пределах вырожденности триплетов генетического кода. Таким образом, эти вырожденные полинуклеотиды кодируют все ту же аминокислотную последовательность. К этим вариантам полинуклеотидов мы относим геномную ДНК с экзонами и интронами, а не только простые последовательности кДНК.
Настоящее изобретение также распространяется на варианты полинуклеотидов, кодирующих аминокислотные последовательности SEQ ID No. 38, 42, 44, 46, 48 или 50, в которых имеется по меньшей мере одна модификация типа делеции, вставки или замены. Таким образом, настоящее изобретение распространяется на полинуклеотидные варианты, кодирующие аминокислотные последовательности, укороченные, удлиненные или неизмененные по размеру относительно указанных последовательностей. Эти варианты полинуклеотидов сохраняют способность ускорять синтез ML-236B и обладают активностью, сравнимой или большей, чем у исходной последовательности, из которой происходит данный вариант.
Полинуклеотидные варианты в определенной степени идентичны исходной последовательности. Степень идентичности должна составлять не менее 60%, не менее 80%, не менее 90% или не менее 95% или 100%. Степень идентичности варианта предпочтительно оценивают с помощью компьютерных программ типа программы BLAST, в которой применяется алгоритм проведения поиска гомологий.
В одном из воплощений предпочтительным полинуклеотидом настоящего изобретения является ДНК, выбранная из группы, состоящей из:
(а) ДНК, включающей одну или несколько нуклеотидных последовательностей, представленных в нуклеотидах №1-1662 SEQ ID No. 37 из Перечня последовательностей, и отличающейся тем, что она ускоряет биосинтез ML-236B в продуцирующем ML-236B микроорганизме при введении в этот микроорганизм;
(б) ДНК, гибридизирующейся в строгих условиях с ДНК, описанной в пункте (а), и отличающейся тем, что она ускоряет биосинтез ML-236B в продуцирующем ML-236B микроорганизме при введении в этот микроорганизм;
(в) ДНК, включающей одну или несколько нуклеотидных последовательностей, представленных в нуклеотидах №1-1380 SEQ ID No. 41 из Перечня последовательностей, и отличающейся тем, что она ускоряет биосинтез ML-236B в продуцирующем ML-236B микроорганизме при введении в этот микроорганизм;
(г) ДНК, гибридизирующейся в строгих условиях с ДНК, описанной в пункте (в), и отличающейся тем, что она ускоряет биосинтез ML-236B в продуцирующем ML-236B микроорганизме при введении в этот микроорганизм.
Полинуклеотиды настоящего изобретения ускоряют биосинтез ML-236B в микроорганизме-продуценте. Примерами микроорганизмов, продуцирующих ML-236B, являются такие виды Penicillium, как Penicillium citrinum, Penicillium brevicompactum (описано в Brown A.G. et al., J. Chem. Soc. Perkin-1, 1165 (1976)), Penicillium cyclopium (описано в Doss S.L. et al., J. Natl. Prod., 49, 357 (1986)) и т.п. Другими примерами являются Eupenicillium sp. M6603 (описано в Endo A. et al., J. Antibiot. - Tokyo, 39, 1609 (1986)), Paecilomyces viridis FERM P-6236 (описано в японской патентной заявке №58-98092), Paecilomyces sp. M2016 (описано в Endo A. et al., J. Antibiot. -Tokyo, 39, 1609 (1986)), Trichoderma longibrachiatum M6735 (описано в Endo A. et al., J. Antibiot. - Tokyo, 39, 1609 (1986)), Hypomyces chrysospermus IFO 7798 9описано в Endo A. et al., J. Antibiot. - Tokyo, 39, 1609 (1986)), Gliocladium sp. YJ-9515 (описано в WO 9806867), Trichoderma viride IFO 5836 (описано в японской патентной заявке №62-19159), Eupenicillium reticulisporium IFO 9022 (описано в японской патентной заявке №62-19159) или любой другой подходящий организм.
Из этих продуцирующих ML-236B микроорганизмов предпочтителен Penicillium citrinum и более предпочтителен штамм Penicillium citrinum SANK 13380. Штамм Penicillium citrinum SANK 13380 депонирован в Research Institute of Life Science and Technology, Agency of Industrial Science and Technology, 22 декабря 1992 г. под кодовым номером PERM BP-4129 в соответствии с Будапештским договором о депонировании микроорганизмов. Примерами продуцирующих ML-236B микроорганизмов также являются те, которые выделены из природных источников, и те, которые мутировали естественным или искусственным путем.
Изобретение также обеспечивает векторы, включающие полинуклеотид настоящего изобретения, такие как вектор, получаемый из Escherichia coli pSAKexpE SANK 72499 (PERM BP-7005) или Escherichia coli pSAKexpR SANK 72599 (PERM BP-7006). К таким векторам настоящего изобретения относятся экспрессионные векторы.
Также обеспечиваются клетки-хозяев, трансформированные вектором настоящего изобретения, включая микроорганизмы, продуцирующие ML-236B. Клетки-хозяева по настоящему изобретению включают Penicillium citrinum и Escherichia coli, например, Escherichia coli pSAKexpE SANK 72499 (PERM BP-7005) или Escherichia coli pSAKexpR SANK 72599 (PERM BP-7006).
Кроме того, изобретение распространяется на полипептиды, кодируемые полинуклеотидом настоящего изобретения. Примерами полипептидов настоящего изобретения служат последовательности SEQ ID No. 38 или 42 или варианты таковых, имеющие определенную степень идентичности с SEQ ID No. 38 или 42 и способные к ускорению продукции ML-236B в микроорганизмах-продуцентах. Другие полипептиды - те, что кодируются другими полинуклеотидными последовательностями настоящего изобретения и их вариантами, сохраняющими определенную степень идентичности.
Степень идентичности полипептидных вариантов с SEQ ID No. 38 или 42 должна составлять не менее 80%, не менее 90%, или не менее 95%, или 100%. Степень идентичности варианта предпочтительно оценивают с помощью компьютерных программ типа программы BLAST, в которой применяется алгоритм проведения поиска гомологий.
К полипептидам настоящего изобретения относятся последовательности, укороченные или удлиненные по сравнению с SEQ ID No. 38 или 42 или их вариантами. Укороченные полипептиды включают частичные аминокислотные последовательности SEQ ID No. 38, 42 или их вариантов и сохраняют способность ускорять биосинтез ML-236B. Удлиненные полипептиды включают полные или частичные аминокислотные последовательности SEQ ID No. 38, 42 или их вариантов и сохраняют способность ускорять биосинтех ML-236B. Удлиненные полипептиды включают слитые (fusion) белки типа Fc-слитого белка.
К полипептидам настоящего изобретения относятся полипептиды, имеющие последовательность SEQ ID No. 38, SEQ ID No. 42, SEQ ID No. 44, SEQ ID No. 46, SEQ ID No. 48, или варианты таковых с одинаковой функцией. Антитела к полипептидам настоящего изобретения также включены. Как поликлональные, так и моноклональные антитела включены в настоящее изобретение. Они могут применяться для регуляции продукции ML-236B и для получения таких производных ML-236B, как статины, включая правастатин и ловастатин. Кроме того, антитела могут предпочтительно использоваться для анализа биосинтеза ML-236B и механизмов его регуляции. Такой анализ может применяться для модуляции продукции ML-236B и для получения производных ML-236B.
Клетки-хозяева по настоящему изобретению, содержащие вектор настоящего изобретения, могут применяться в способе получения ML-236B, включающем выращивание таких клеток и затем выделение ML-236B из культуры. В одном из способов вектор содержит m1сЕ или mlcR, но не содержит других генов, таких как mlcA, mlcB, mlcC или mlcD.
Продукция способом настоящего изобретения может происходить в отсутствие рекомбинантных полипептидов mlcA, mlcB, mlcC и/или mlcD, которые соответствуют SEQ ID No. 44, SEQ ID No. 46, SEQ ID No. 48 и SEQ ID No. 50.
Далее настоящее изобретение будет описано более подробно.
Авторы настоящего изобретения клонировали геномную ДНК, содержащую гены, участвующие в биосинтезе ML-236B в Penicillium citrinum. Эта геномная ДНК, в дальнейшем именуемая связанной с биосинтезом ML-236B, клонирована из геномной библиотеки ДНК микроорганизма, продуцирующего ML-236B. Геномную ДНК анализировали с целью выявления находящихся там структурных генов, а затем получали соответствующие этим структурным генам кДНК с помощью обратной транскрипции-полимеразной цепной реакции (в дальнейшем именуемой "ОТ-ПЦР"), используя тотальную РНК, содержащую мРНК Penicillium citrinum в качестве матрицы. Было обнаружено, что биосинтез ML-236B в микроорганизме-продуценте ускоряется, когда этот микрорганизм трансформирован вектором, несущим рекомбинантные кДНК.
Настоящее изобретение особенно касается кДНК (в дальнейшем именуемых ускоряющими биосинтез ML-236B), ускоряющих биосинтез ML-236B в продуцирующем ML-236B микроорганизме при введении в этот микроорганизм.
К ускоряющим биосинтез ML-236B полинуклеотидам настоящего изобретения, таким как ускоряющие биосинтез ML-236B кДНК, относятся, к примеру:
(I) ДНК, получаемая путем синтеза с применением в качестве матрицы продукта транскрипции (матричной РНК, в дальнейшем именуемой мРНК) структурного гена, участвующего в биосинтезе ML-236B и существующего в геномной ДНК продуцирующего ML-236B микроорганизма;
(II) двухцепочечная ДНК, полученная в результате ассоциации ДНК (I) и второй цепи ДНК, синтезированной с помощью ДНК (I) в качестве первой цепи;
(III) двухцепочечная ДНК, полученная путем репликации или амплификации двухцепочечной ДНК (II), например, методом клонирования или ему подобным;
(IV) ДНК, способная к гибридизации с одной из указанных выше ДНК или мРНК в строгих условиях.
В качестве ДНК (IV) могут служить любые из приведенных последовательностей структурных генов, например нуклеотиды от №1 до 1662 в SEQ ID No. 37 из Перечня последовательностей или нуклеотиды от №1 до 1380 в SEQ ID No. 41, в которых необязательно имеется замена, делеция и/или добавление одного или нескольких нуклеотидов и которые способны ускорять биосинтез ML-236B в продуцирующем ML-236B микроорганизме при введении в этот микроорганизм.
При гибридизации двух одноцепочечных нуклеиновых кислот образуется двухцепочечная молекула в том участке, где они комплементарны или сильно комплементарны друг другу, а "строгие условия" в данном случае означает, что гибридизационным раствором является 6×SSC (1×SSC состоит из 150 мМ NaCl, 15 мМ цитрата натрия), а температура при гибридизации равна 55°С.
Ускоряющую биосинтез ML-236B кДНК можно получить, например, путем выделения содержащего кДНК клона из библиотеки кДНК микроорганизма, продуцирующего ML-236B. В качестве альтернативы можно использовать ОТ-ПЦР с помощью пары праймеров, составленных на основе нуклеотидной последовательности связанной с биосинтезом ML-236B геномной ДНК, вместе с мРНК или тотальной РНК из микроорганизма, продуцирующего ML-236B.
Продуцирующим ML-236B микроорганизмом является любой микроорганизм, обладающий способностью продуцировать ML-236B. Как указано ранее, примерами микроорганизмов, продуцирующих ML-236B, являются такие виды Penicillium, как Penicillium citrinum, Penicillium brevicompactum, Penicillium cyclopium и т.д., а другими примерами являются Eupenicillium sp. M6603, Paecilomyces viridis FERM P-6236, Paecilomyces sp. M2016, Trichoderma longibrachiatum M6735, Hypomyces chrysospermus IFO 7798, Gliocladium sp. YJ-9515, Trichoderma viride IFO, Eupenicillium reticulisporium IFO 9022 и любые другие подходящие организмы.
Из этих продуцирующих ML-236B микроорганизмов предпочтителен Penicillium citrinum и более предпочтителен штамм Penicillium citrinum SANK 13380. Штамм Penicillium citrinum SANK 13380 депонирован в Research Institute of Life Science and Technology, Agency of Industrial Science and Technology, 22 декабря 1992 г. под кодовым номером PERM BP-4129 в соответствии с Будапештским договором о депонировании микроорганизмов. Примерами продуцирующих ML-236B микроорганизмов также являются те, которые выделены из природных источников, и те, которые мутировали естественным или искусственным путем.
Связанную с биосинтезом ML-236B геномную ДНК можно получить скринированием геномной библиотеки ДНК продуцирующего ML-236B микроорганизма с помощью соответствующего зонда. Зонд разрабатывают на основе последовательности ДНК, предположительно играющей роль в биосинтезе ML-236B, предпочтительно из нитчатого гриба.
Выбор методов создания геномной библиотеки ДНК не ограничен - можно применять любые подходящие методы, предпочтительно общие методы создания геномных библиотек ДНК эукариотических организмов. Примеры таковых включают метод Maniatis et al. (Maniatis T. et al., Molecular cloning, a laboratory manual, 2nd ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. (1989)). Известны и другие методы в этой области.
Вкратце, геномную ДНК из микроорганизма, продуцирующего ML-236B, можно получить путем сбора клеток из культуры этого микроорганизма, физического разрушения клеток, экстракции ДНК, находящейся в ядрах, и очистки этой ДНК.
Выращивать микроорганизм, продуцирующий ML-236B, можно в условиях, подходящих для данного микроорганизма. Например, Penicillium citrinum, предпочтительный микроорганизм, продуцирующий ML-236B, можно выращивать, инокулируя клетки в среду MBG3-8 (состав (вес/об.): 7% глицерина, 3% глюкозы, 1% соевого порошка, 1% пептона (фирмы Kyokuto Seiyaku Kogyo), 1% кукурузного экстракта (фирмы Honen), 0,5% нитрата натрия, 0,1% сульфата магния семиводного (рН 6,5)) и инкубируя со встряхиванием при 22-28°С в течение 3-7 дней. Косяки для хранения можно приготовить, заливая расплавленную агарную среду PGA (состав: 200 г/л картофельного экстракта, 15% глицерина, 2% агара) в пробирку и удерживая пробирку под углом до затвердения агара. Затем можно инокулировать Penicillium citrinum на косяк с помощью платиновой иглы с последующей инкубацией при 22-28°С в течение 7-15 дней. Выращенные таким образом микроорганизмы или бактерии можно долгое время хранить на косяке при 0-4°С.
Клетки микроорганизма, продуцирующего ML-236B, выращенные в жидкой культуре, можно собрать центрифугированием, а те, что выращены на твердой среде, можно собрать скребком или чем-то вроде этого.
Разрушить клетки можно путем растирания клеток в ступке с пестиком после замораживания их жидким азотом. Ядерную ДНК из разрушенных клеток можно экстрагировать с помощью детергента, например додецилсульфата натрия (в дальнейшем именуемого ДСН) или другого подходящего детергента. Экстрагированную геномную ДНК желательно обработать фенол-хлороформной смесью для удаления белка, а затем высадить в осадок этанолом.
Полученную геномную ДНК расщепляют на фрагменты соответствующим рестрикционным ферментом. Не имеется ограничений на применение рестрикционных ферментов для расщепления ДНК; предпочтительны общедоступные рестрикционные ферменты. К таковым относится Sau3AI. Другие ферменты известны в этой области. После расщепления ДНК подвергают гель-электрофорезу, затем из геля выделяют геномную ДНК требуемого размера. Нет особых ограничений на размер фрагмента ДНК, но предпочтительно он составляет 20 kb и больше.
Также нет ограничений на выбор ДНК-вектора, применяемого при построении геномной библиотеки ДНК, лишь бы этот вектор имел последовательность ДНК, необходимую для репликации в клетке-хозяине, которая будет им трансформирована. К числу таких векторов относятся плазмидные векторы, фаговые векторы, космидные векторы, ВАС-векторы и т.п., но предпочтительны космидные векторы. ДНК-векторы предпочтительно являются экспрессионными векторами. Более предпочтительно, когда ДНК-вектор включает ДНК или нуклеотидную последовательность, придающую селективный фенотип клеткам, трансформированным этим вектором.
Желательно, чтобы ДНК-вектор подходил как для клонировании, так и для экспрессии. Предпочтительным вектором является челночный вектор, который может применятся для трансформации более чем одного микроорганизма. Желательно, чтобы челночный вектор имел последовательность ДНК, позволяющую его репликацию в клетке-хозяине, и предпочтительно такую последовательность или последовательности, которые позволяют его репликацию в нескольких различных клетках из разных групп микроорганизмов, таких как бактерии и грибы. Кроме того, челночный вектор предпочтительно должен содержать последовательность ДНК, придающую селективный фенотип целому ряда различных клеток-хозяев, например, клеток из разных групп микроорганизмов.
Нет особых ограничений на выбор комбинации групп микроорганизмов и клеток, трансформируемых челночным вектором, при условии, что одна из групп микроорганизмов подходит для клонирования, а другая обладает способностью вырабатывать ML-236B. Такой комбинацией, например, может быть комбинация из бактерий и нитчатых грибов, комбинация из дрожжей и нитчатых грибов, причем комбинация из бактерий и нитчатых грибов предпочтительна. Нет особых ограничений на выбор бактерии, лишь бы она широко применялась в биотехнологии, как Escherichia coli, Bacillus subtilis и им подобные. Предпочтительна Escherichia coli, и более предпочтительна Escherichia coli XLl-Blue MR. Также нет ограничений на выбор дрожжей, лишь бы они широко применялись в биотехнологии, как Saccharomyces cerevisiae и им подобные. Примерами нитчатых грибов являются продуцирующие ML-236B микроорганизмы, описанные выше. Примеры других подходящих микроорганизмов известны в этой области.
В настоящем изобретении микроорганизм можно выбрать из таких групп, как бактерии, нитчатые грибы и дрожжи.
Примером вышеупомянутых челночных векторов является космидный вектор, несущий маркерный ген, пригодный для селекции фенотипа, и cos-участок. Другие подходящие векторы известны в этой области. Предпочтительным вектором является pSAKcos1, построенный путем вставки cos-участка из космидного вектора pWE15 (фирмы Stratagene) в плазмиду pSAK.333, включающую последовательность гена гигромицин В-фосфотрансферазы Escherichia coli (описано в японской патентной заявке №3-262486). Метод построения pSAKcos1 показан на Фигуре 1. Настоящее изобретение не ограничивается этим вектором.
Геномную библиотеку ДНК можно построить путем введения челночного вектора в клетку-хозяин с тем, чтобы вектор содержал фрагмент геномной ДНК из продуцирующего ML-236B микроорганизма. В качестве клетки-хозяина предпочтительно используют Escherichia coli, более предпочтительно Escherichia coli XLl-Blue MR. Когда клеткой-хозяином является Escherichia coli, введение может проводиться путем упаковки in vitro. В настоящем изобретении трансформация также включает введение чужеродной ДНК методом упаковки in vitro, а понятие трансформированной клетки охватывает также клетки, в которые чужеродная ДНК вводится методом упаковки in vitro.
Для идентификации требуемого клона геномную библиотеку можно скринировать с помощью антитела или зонда из нуклеиновой кислоты, причем зонды из нуклеиновой кислоты предпочтительны. Зонд из нуклеиновой кислоты предпочтительно получают на основе нуклеотидной последовательности гена или ДНК, связанной с биосинтезом поликетида, предпочтительно последовательности, происходящей из нитчатого гриба. Нет ограничений на выбор конкретного гена, лищь бы он участвовал в биосинтезе поликетидов и его нуклеотидная последовательность была известна. Примерами таких генов являются гены ПКС афлатоксина Aspergillus flavus и Aspergillus parasiticus, ген ПКС стеригматоцистина Aspergillus nidulans и им подобные.
Подходящие зонды из нуклеиновой кислоты можно получить, например, путем синтеза олигонуклеотидного зонда, включающего часть известной последовательности геномной ДНК, как описано выше, или получения олигонуклеотидных праймеров и амплификации требуемой ДНК с помощью полимеразной цепной реакции (в дальнейшей именуемой ПЦР, как описано в Saiki R.K. et al; Science, 239, 487 (1988)) и геномной ДНК в качестве матрицы. Другие методы, пригодные для получения таких зондов, хорошо известны в этой области.
Зонд из нуклеиновой кислоты можно получить из продуцирующего ML-236B микроорганизма, например, с помощью ПЦР или ОТ-ПЦР. Праймеры для ПЦР или ОТ-ПЦР (в дальнейшем именуемые "праймер для ПЦР") предпочтительно составляют на основе нуклеотидной последовательности того гена, связанного с биосинтезом поликетида, нуклеотидная последовательность которого известна. Предпочтительно этим геном является ген ПКС афлатоксина Aspergillus flavus, Aspergillus parasiticus или ген ПКС стеригматоцистина Aspergillus nidulans.
Праймеры для ПЦР желательно проектировать так, чтобы они содержали нуклеотидные последовательности, кодирующие сильно консервативные аминокислотные последовательности в генах ПКС. Методы определения нуклеотидных последовательностей, соответствующих данной аминокислотной последовательности, включают дедукцию на основе употребимости кодонов клетки-хозяина и методы создания смешанных олигонуклеотидных последовательностей с помощью множественных кодонов (в дальнейшем именуемых "вырожденные олигонуклеотиды"). В последнем случае множественность олигонуклеотидов можно снизить введением гипоксантина в их нуклеотидные последовательности.
Праймер для ПЦР может содержать нуклеотидную последовательность, которая при отжиге будет гибридизироваться с матричной цепью, причем праймер присоединяется к дополнительной 5'-последовательности. Нет особых ограничений на выбор такой дополнительной 5'-нуклеотидной последовательности, лишь бы этот праймер мог служить для ПЦР или ОТ-ПЦР. Такой дополнительной 5'-последовательностью, например, может быть нуклеотидная последовательность, удобная для клонирования продукта ПЦР. Такой нуклеотидной последовательностью, например, может быть сайт расщепления для рестрикционного фермента или нуклеотидная последовательность, содержащая такой сайт.
Кроме того, при проектировании праймера для ПЦР предпочтительно, чтобы сумма гуаниновых (G) и цитозиновых (С) оснований равнялась 40-60% от общего числа оснований. Более того, предпочтительно, чтобы почти или совсем не было самогибридизации данного праймера и, в случае пары праймеров, чтобы почти или совсем не было гибридизации между праймерами.
Нет особых ограничений на число нуклеотидов, входящих в состав праймера для ПЦР, лишь бы он подходил для ПЦР. Нижний предел этого числа обычно равен 10-14 нуклеотидам, а верхний - 40-60 нуклеотидам. Предпочтительная длина праймера от 14 до 40 нуклеотидов.
Праймер для ПЦР предпочтительно состоит из ДНК. Нуклеозидами в праймере могут служить дезоксиаденозин, дезоксицитидин, дезокситимидин и дезоксигуанозин, кроме того, также дезоксиинозин. Желательно, чтобы в 5'-позиции нуклеозида, находящегося на 5'-конце праймера для ПЦР, была гидроксильная группа или гидроксигруппа с одним остатком фосфорной кислоты, присоединенным с помощью эфирной связи.
Синтезировать праймер для ПЦР можно методами, которые обычно применяются для синтеза нуклеиновых кислот, например фосфоамидитным методом. В таком методе предпочтительно использовать автоматизованный синтезатор ДНК.
В качестве матрицы для ПЦР или ОТ-ПЦР можно использовать геномную ДНК или мРНК соответственно из продуцирующего ML-236B микроорганизма. Вместо мРНК можно также использовать тотальную РНК в качестве матрицы для ОТ-ПЦР.
Продукт ПЦР или ОТ-ПЦР можно клонировать, встроив его в соответствующий ДНК-вектор. В общем, на выбор ДНК-вектора для стадии клонирования не имеется ограничений. Имеются коммерческие наборы, позволяющие легко клонировать продукты ПЦР и ОТ-ПЦР. К примеру, для такого клонирования подходит набор Original ТА Cloning Kit (фирмы Invitrogen; в качестве ДНК-вектора применяется pCR2.1).
Для того чтобы получить клонированный продукт ПЦР, трансформированные клетки-хозяева, содержащие плазмиды, несущие требуемый продукт ПЦР, выращивают в культуре, а затем из клеток экстрагируют и очищают плазмиды. После этого выделяют встроенный фрагмент ДНК из этой плазмиды.
Трансформированные клетки желательно выращивать в условиях, подходящих именно для этих клеток. Предпочтительная клетка-хозяин - Escherichia coli, ее выращивают в среде LB (1% триптона, 0,5% дрожжевого экстракта, 0,5% хлористого натрия) со встряхиванием при 30-37°С от 18 часов до двух дней.
Выделить плазмиды из культуры трансформированных клеток можно путем сбора клеток и выделения плазмид, очищенных от других клеточных компонентов, таких как геномная ДНК или белки хозяина. Выделение плазмидной ДНК из культуры Escherichia coli можно проводить щелочным методом Маниатиса (описано в Maniatis Т. et al., Molecular cloning, a laboratory manual, 2nd ed.. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. (1989)). Имеются коммерческие наборы для выделения плазмид более высокой чистоты. Предпочтителен набор Plasmid Mini Kit (фирмы Qiagen AG). Кроме того, имеются коммерческие наборы для массовой продукции плазмид. Предпочтителен набор Plasmid Maxi Kit (фирмы Qiagen AG).
Концентрацию плазмидной ДНК в препарате можно определить, измеряя поглощение при длине волны 260 нм после соответствующего разбавления образца ДНК. Расчеты проводятся на основе того, что раствор со значением поглощения OD260, равном 1, содержит 50 мкг/мл ДНК (описано в Maniatis Т. et al., supra).
Чистоту ДНК можно вычислить из соотношения зачений поглощения при длинах волн 280 и 260 нм (описано в Maniatis Т. et al., supra).
Методы мечения зондов нуклеиновых кислот можно в общем разделить на радиоактивные и нерадиоактивные. В общем, нет ограничений на выбор радионуклеотида для метки, которым может служить, например, 32P, 35S, 14С и другие. Для метки предпочтительно применять 32Р. Выбор средства для нерадиоактивной метки тоже в общем не ограничен, лишь бы это было общеприменимое средство для мечения нуклеиновых кислот, которым может быть, например, дигоксигенин, биотин или им подобные, причем предпочтителен дигоксигенин.
В общем, на методы мечения зондов нуклеиновых кислот также не имеется ограничений. Предпочтительны общепринятые методы, например методы внедрения метки в продукт посредством ПЦР или ОТ-ПЦР с помощью меченых нуклеотидных субстратов, ник-трансляция, применение неупорядоченых праймеров, мечение по концам, и методы синтеза олигонуклеотидной ДНК с помощью меченых нуклеотидных субстратов. Из числа этих методов можно выбрать подходящий в зависимости от вида зонда из нуклеиновой кислоты.
Присутствие в геноме продуцирующего ML-236B микроорганизма нуклеотидной последовательности, тождественной нуклеотидной последовательности данного зонда нуклеиновой кислоты, можно подтвердить путем гибридизации по Сазерну с геномной ДНК этого микроорганизма.
Гибридизацию по Сазерну можно проводить согласно методу Маниатиса (описано в Maniatis Т. et al., supra).
Меченый зонд нуклеиновой кислоты, полученный как описано выше, можно применять для скринирования геномной библиотеки ДНК. Выбор метода скринирования не имеет особых ограничений, лишь бы он в общем подходил для клонирования генов, хотя предпочтителен метод гибридизации колоний Маниатиса (описано в Maniatis Т. et al., supra).
Колонии для гибридизации желательно выращивать в условиях, подходящих для клетки-хозяина. Предпочтительная клетка-хозяина - Escherichia coli, ее выращивают путем инкубации в агарной среде LB (1% триптона, 0,5% дрожжевого экстракта, 0,5% хлористого натрия, 1,5% агарозы) при 30-37°С от 18 часов до двух дней.
Выделение вектора, несущего рекомбинантную ДНК, из положительного клона, полученного при гибридизации колоний, обычно проводят путем экстракции плазмиды из культуры положительного клона и ее очистки.
Трансформированный штамм Escherichia coli - Escherichia coli pML48 SANK71199, представляющий собой положительный клон, полученный согласно настоящему изобретению, депонирован в Research Institute of Life Science and Technology, Agency of Industrial Science and Technology, 7 июля 1999 г. в соответствии с Будапештским договором о депонировании микроорганизмов и получил кодовый номер FERM ВР-6780.
Типичный ДНК-вектор, переносимый Escherichia coli pML48 SANK71199, был назван pML48.
Подтвердить то, что рекомбинантный ДНК-вектор, обнаруженный в положительном клоне, содержит геномную ДНК, связанную с биосинтезом ML-236B, можно путем определения нуклеотидной последовательности рекомбинатной вставки ДНК-вектора, гибридизации по Сазерну или экспрессии вставки для выявления функции.
Нуклеотидную последовательность ДНК можно определить методом химической модификации Максама и Гилберта (описано в Maxam A.M.M. and Gilbert W., Methods in Enzymology, 65, 499 (1980)) или методом дидезокси-терминации цепи (описано в Messing J. and Vieira J., Gene, 19, 269 (1982)). Другие методы хорошо известны в этой области. При определении нуклеотидной последовательности плазмидная ДНК должна быть высокой чистоты, как описано выше.
Нуклеотидная последовательность вставки pML48 показана в SEQ ID No. 1 из Перечня последовательностей. Нуклеотидная последовательность, показанная в SEQ ID No. 2 из Перечня последовательностей, полностью комплементарна нуклеотидной последовательности SEQ ID No. 1. В общем, нуклеотидная последовательность геномной ДНК может обладать генетическим полиморфизмом в пределах вида, иными словами, иметь аллогенные различия. Кроме того, известно, что в процессе клонирования и секвенирования с определенной частотой могут возникать замены нуклеотидов или другие изменения. Соответственно геномная ДНК настоящего изобретения, связанная с биосинтезом ML-236В, также охватывает геномную и другую ДНК, способную гибридизироваться с нуклеотидами №1-34203 SEQ IDО No. 1 или 2 из Перечня последовательностей. Предпочтительной является геномная или другая ДНК, которая может гибридизироваться в строгих условиях с ДНК, содержащей нуклеотиды №1-34203 SEQ ID No. 1 или 2 из Перечня последовательностей. Эти ДНК включают ДНК, содержащую нуклеотиды №1-34203 SEQ ID No. 1 или 2 из Перечня последовательностей, в которой имеется одна или несколько замен, делеций и/или вставок нуклеотидов. Кроме того, эти способные к гибридизации геномные или другие ДНК могут включать ДНК, происходящую не из Penicillium citrinum SANK 13380, а из других микроорганизмов, продуцирующих ML-236B, предпочтительно обладающую способностью к улучшению продукции ML-236B при введении в продуцирующий ML-236B микроорганизм.
Геномную ДНК, связанную с биосинтезом ML-236B, желательно анализировать в соответствии со следующими методами 1-3.
1) Анализ с помощью программ анализа генов
Гены из геномной ДНК можно определять с помощью программы для обнаружения генов (в дальнейшем именуемой "GRAIL") и программ поиска гомологичных последовательностей (BLASTN и BLASTX).
Программа GRAIL ищет структурные гены в геномной ДНК, разделяя геномную последовательность по 7 параметрам для выявления генной последовательности, а результаты обобщаются методом нейронной сети (описано в Uberbacher Е.С. & Mural R.J., Proc. Natl. Acad. Sci. USA, 88, 11261 (1991)). К примеру, можно использовать программный пакет ApoCom GRAIL Toolkit (фирмы Аросоm).
Программа BLAST использует алгоритм поиска гомологии в нуклеотидных и аминокислотных последовательностях (описано в Altschul S.F., Madden T.L. et al., Nucl. Acids Res., 25, 3389 (1997)).
Положение и направление структурного гена в образце геномной последовательности ДНК можно предсказать, разбивая последовательность ДНК на соответствующие отрезки и проводя поиск гомологии в генетической базе данных с помощью BLASTN. Положение и направление структурного гена в исследуемой последовательности ДНК можно предсказать, транслируя отрезки геномных последовательностей ДНК в шести рамках считывания (3 на смысловой цепи и еще 3 на антисмысловой цепи) и проводя поиск гомологии в полученных аминокислотных последовательностях в белковой базе данных с помощью BLASTX.
У эукариотических организмов участки, кодирующие структурные гены в геномной ДНК, зачастую разделены интронами. Для анализа структурных генов с интронами более эффективна программа BLAST, предназначенная для разделенных интронами генов, причем предпочтительна программа Gapped-BLAST (встроена в пакет BLAST2: WISCONSIN GCG версии 10.0).
2) Анализ методом гибридизации по Норзерну
Экспрессию структурного гена, который предсказан методами анализа, описанными в пункте 1, можно исследовать методом гибридизации по Норзерну.
Следует получить тотальную РНК из культуры продуцирующего ML-236B микроорганизма. Культуру предпочтительного микроорганизма, продуцирующего ML-236B, - Penicillium citrinum можно получить, инокулируя этот микроорганизм из косяка в среду MGB3-8 и инкубируя со встряхиванием при 22-28°С от одного до четырех дней.
Выбор метода экстракции РНК из продуцирующего ML-236B микроорганизма не ограничен, а предпочтительным является метод гуанидин тиоцианат-горячий фенол, метод гуанидин тиоцианат-гуанидин-соляная кислота и им подобные. Примером коммерческого набора для получения тотальной РНК высокой чистоты является RNeasy Plant Mini Kit (фирмы Qiagen AG). Кроме того, можно получить мРНК путем пропускания тотальной РНК через колонку с олиго(dТ) и сбора фракции, адсорбировавшейся на колонке.
Перенос РНК на мембрану, приготовление зонда, гибридизацию и обнаружение сигнала можно проводить тем же способом, что при рассмотренной выше гибридизации по Сазерну.
3) Анализ 5'- и 3'-концов транскрипта
Анализ 5'- и 3'-концов транскрипта можно проводить методом RACE (быстрой амплификации концов кДНК). Метод заключается в получении кДНК, включающей известный нуклеотидный участок и неизвестный участок на 5'- или 3'-конце гена с помощью ОТ-ПЦР, используя мРНК в качестве матрицы (описано в Frohman M.A., Methods Enzymol, 218, 340 (1998)).
5'-RACE проводится следующим образом. Первую цепь кДНК синтезируют с помощью обратной транскриптазы, используя мРНК в качестве матрицы. В качестве праймера применяются антисмысловые олигонуклеотиды (1), составленные на основе известной части нуклеотидной последовательности. К 3'-концу первой цепи кДНК присоединяют гомополимерную нуклеотидную цепь (состоящую из одного вида основания) с помощью терминальной дезоксинуклеотидилтрансферазы. Затем, используя первую цепь кДНК в качестве матрицы, амплифицируют двухцепочечную кДНК на 5'-конце методом ПЦР. Для этого используют 2 праймера: на смысловой цепи - ДНК-олигонуклеотид, содержащий последовательность, комплементарную гомополимерной последовательности, на антисмысловой цепи - олигонуклеотид (2), содержащий 3'-конец ДНК-олигонуклеотида (1) (описано в Frohman M.A., Methods in Enzymol., 218, 340 (1993)). Имеется коммерческий набор для 5'-RACE, а именно 5'-RACE System for Rapid Amplification of cDNA Ends, версия 2.0 (фирмы GIBCO).
В методе 3'-RACE используется участок полиА, существующий на 3'-конце мРНК. При этом первую цепь кДНК синтезируют с помощью обратной транскриптазы, используя мРНК в качестве матрицы и олиго(dТ)-адаптер в качестве праймера. Затем амплифицируют двухцепочечную кДНК на 3'-конце методом ПЦР, используя первую цепь кДНК в качестве матрицы. В качестве праймера на смысловой цепи используют ДНК-олигонуклеотид (3), составленный на основе известной части нуклеотидной последовательности смысловой цепи, а на антисмысловой цепи - oлигo(dT)-aдаптep. Имеется коммерческий набор для 3'-RACE, а именно Ready-To-Go T-primed First-Strand Kit (фирмы Pharmacia).
Результаты анализа методами 1 и 2 предпочтительно используются в методе RACE при составлении праймеров на основе известной части исследуемой нуклеотидной последовательности.
С помощью методов анализа, описанных выше в пунктах 1 и 2, можно определить направление структурного гена в последовательности ДНК, определить сайт инициации транскрипции в структурном гене, положение кодона инициации трансляции и кодона терминации инициации. На основе этой информации можно получить структурные гены и их кДНК, а именно кДНК, ускоряющие биосинтез ML-236B.
Предполагается, что в рекомбинантном ДНК-векторе pML48, полученном в соответствии с настоящим изобретением, имеется 6 структурных генов. Они названы mlcA, mlcB, mlcC, mlcD, mlcE и mlcR соответственно. Полагают, что кодирующие участки генов mlcA, mlcB, mlcE и mlcR находятся в нуклеотидной последовательности SEQ ID No. 2 из Перечня последовательностей, а кодирующие участки mlcC и mlcD находятся в нуклеотидной последовательности SEQ ID No. 1.
Примеры методов получения специфических кДНК, ускоряющих биосинтез ML-236В и соответствующих вышеуказанным структурным генам, включают клонирование методом ОТ-ПЦР с помощью праймеров, составленных на основе последовательностей этих структурных генов и обрамляющей их ДНК, и клонирование из библиотеки кДНК с помощью соответствующих ДНК-зондов по известным нуклеотидным последовательностям. Другие методы хорошо известны в этой области. Для функциональной экспрессии кДНК, полученной этими методами, предпочтительно следует получить полную кДНК.
Ниже излагается метод получения кДНК, ускоряющей биосинтез ML-236B, с помощью ОТ-ПЦР.
Для получения кДНК, ускоряющей биосинтез ML-236B, необходимо разработать пару праймеров для ОТ-ПЦР так, чтобы они избирательно гибридизировались с каждой из цепей матрицы, образуя кДНК. Однако праймеры для ОТ-ПЦР вовсе не должны быть полностью комплементарными к каждой части цепи матрицы, они лишь должны удовлетворять условиям, описанным выше. Праймеры для ОТ-ПЦР, которые гибридизируются с антисмысловой цепью (в дальнейшем именуемые "смысловыми праймерами"), могут быть полностью комплементарными к своей части антисмысловой цепи (в дальнейшем именуются "незамещенными смысловыми праймерами") или могут быть неполностью комплементарными (в дальнейшем именуются "частично замещенными смысловыми праймерами"). Те праймеры для ОТ-ПЦР, которые гибридизируются со смысловой цепью (в дальнейшем именуемые "антисмысловыми праймерами"), могут быть полностью комплементарными к своей части антисмысловой цепи (в дальнейшем именуются "незамещенными антисмысловыми праймерами") или могут быть неполностью комплементарными (в дальнейшем именуются "частично замещенными смысловыми праймерами").
Смысловые праймеры следует проектировать так, чтобы они давали продукт ОТ-ПЦР, содержащий кодон ATG в начальной позиции инициатора трансляции. Также желательно, чтобы продукт ОТ-ПЦР содержал правильный кодон терминации трансляции только в той рамке считывания, которая содержит исходный стартовый кодон ATG и не содержит других (лишних) сайтов остановки трансляции. Положение кодона инициации трансляции тех структурных генов, что представлены в настоящем изобретении, показано в таблице 5 для генов, входящих в SEQ ID No. 1 и SEQ ID No. 2 из Перечня последовательностей.
На 5'-конце незамещенного смыслового праймера должен находиться нуклеотид "А" из кодона инициации трансляции ATG или основание, примыкающее к нему с 5'-конца.
Частично замещенный смысловой праймер должен избирательно гибридизироваться со специфическим участком в SEQ ID No. 1 или SEQ ID No. 2 из Перечня последовательностей, причем нуклеотидная последовательность SEQ ID No. 2 полностью комплементарна SEQ ID No. 1.
Когда частично замещенный смысловой праймер содержит нуклеотидную последовательность, примыкающую с 3'-конца к кодону инициации трансляции ATG, он не должен содержать в этом месте нуклеотидных последовательностей, служащих кодонами терминации (ТАА, TAG или TGA) в той же рамке считывания, что ATG.
Частично замещенный смысловой праймер может содержать нуклеотид "А", последовательность "AT" или "ATG" (в дальнейшем именуемых "нуклеотид или нуклеотидная последовательность m'), которые соответствуют нуклеотиду "А", последовательности "AT" или "ATG" кодона инициации трансляции (в дальнейшем именуемых "нуклеотид или нуклеотидная последовательность m). Когда нуклеотидом m' является "А", который соответствует "А" последовательности m, то "А" в m' должен находиться на 3'-конце частично замещенного смыслового праймера. Точно так же, когда m' представляет собой "AT", то он должен находиться на 3'-конце частично замещенного смыслового праймера. Когда нуклеотидом или последовательностью m является "ATG", который соответствует "ATG" в m', то тринуклеотиды, находящиеся на 3'-стороне от ATG в праймере, не должны быть стоп-кодонами. Другими словами, тринуклеотиды, в которых 5'-концевым нуклеотидом является (3n+1)-й нуклеотид (n - целое число, равное или больше 1), считая от А из "ATG" в m' в направлении 3'-конца, не могут быть ни ТАА, ни TAG или TGA. Описанные выше праймеры можно применять для получения кДНК с кодоном метионина в положении, соответствующем кодону инициации трансляции мРНК, применяемой в качестве матрицы для ОТ-ПЦР.
Когда на 3'-конце частично замещенного смыслового праймера находится нуклеотид в положении (3n+1), то тринуклеотидом, начинающимся в этом положении, не может быть ТАА, TAG или TGA в продукте ОТ-ПЦР, полученном с применением этого праймера как одного из праймеров вместе с РНК или мРНК продуцирующего ML-236B микроорганизма в качестве матрицы, или в продукте ПЦР, полученном с применением геномной ДНК или кДНК в качестве матрицы. Положение нуклеотида отсчитывается от "А" из кодона инициации трансляции ATG в направлении 3'-конца, причем n - это целое число, равное или больше 1.
Когда на 3'-конце частично замещенного смыслового праймера находится нуклеотид в положении (3n+2), то триплетом, в центре которого находится нуклеотид в положении (3n+2), не может быть ТАА, TAG или TGA в продукте ПЦР или ОТ-ПЦР, полученном, как описано выше.
Когда на 3'-конце частично замещенного смыслового праймера находится нуклеотид в положении (3n+3), то триплетом, в котором положение (3n+3) занимает 3'-нуклеотид, не может быть ТАА, TAG или TGA.
Таковы требования к смысловому праймеру - как изложено выше.
Антисмысловой праймер проектируют так, чтобы в паре со смысловым праймером с помощью ОТ-ПЦР можно было амплифицировать кДНК, кодирующие каждый из структурных генов (mlcA, mlcB, mlcC, mlcD, mlcE и mlcR) в направлении, эквивалентном направлению от N-конца к С-концу соответствующих пептидов.
Выбор незамещенного антисмыслового праймера не ограничен, лишь бы его нуклеотидная последовательность была комплементарна последовательности, находящейся в районе сайта терминации трансляции кДНК. Однако предпочтителен праймер, имеющий на 5'-конце основание, комплементарное тому основанию, что находится на 3'-конце кодона терминации трансляции, или имеющий основание на 5'-стороне от этого основания праймера. Более предпочтителен праймер, имеющий 3 основания, комплементарных кодону терминации трансляции. В таблицах 8-10 показаны кодоны терминации трансляции всех структурных генов, комплементарные им последовательности, аминокислотные остатки на С-концах каждого из пептидов, кодируемых этими структурными генами, нуклеотидные последовательности, кодирующие эти аминокислотные остатки, и их расположение в SEQ ID No. 1 и SEQ ID No. 2.
Частично замещенные антисмысловые праймеры должны избирательно гибридизироваться со специфическим участком в нуклеотидной последовательности SEQ ID No. 1 или SEQ ID No. 2 из Перечня последовательностей.
Таковы требования к антисмысловому праймеру - как изложено выше.
Можно добавить соответствующие нуклеотидные последовательности к 5'-концам частично замещенных смысловых праймеров и частично замещенных антисмысловых праймеров, если при этом выполняются изложенные выше требования. Нет особых ограничений на выбор таких нуклеотидных последовательностей, лишь бы праймер подходил для ПЦР. Примеры таких последовательностей включают нуклеотидные последовательности, удобные для клонирования продуктов ПЦР, например сайты расщепления для рестрикционных ферментов и нуклеотидные последовательности, содержащие такие сайты.
Кроме того, смысловой и антисмысловой праймеры должны проектироваться согласно изложенному выше и в соответствии с общими правилами составления праймеров для ПЦР.
Как описано выше, в качестве матрицы для ОТ-ПЦР можно использовать мРНК или тотальную РНК из продуцирующего ML-236B микроорганизма. В настоящем изобретении кДНК, ускоряющую биосинтез ML-236B и соответствующую структурному гену mlсЕ, получали путем составления и синтеза пары праймеров, способных амплифицировать весь кодирующий участок структурного гена mlсЕ, находящегося во вставке плазмиды pML48, а затем проведения ОТ-ПЦР с применением тотальной РНК из SANK 13380 в качестве матрицы (праймеры соответствуют нуклеотидным последовательностям SEQ Ш No. 35 и 36 из Перечня последовательностей).
кДНК, ускоряющую биосинтез ML-236B и соответствующую структурному гену mlcR, получали таким же образом с помощью праймеров, соответствующих нуклеотидным последовательностям SEQ ID No. 39 и 40 из Перечня последовательностей.
Как описано выше, продукт ОТ-ПЦР можно клонировать путем введения его в соответствующий ДНК-вектор. Выбор ДНК-вектора для клонирования не ограничен, им может быть вектор, общеприменимый при клонировании фрагментов ДНК. Имеются коммерческие наборы, позволяющие легко клонировать продукты ОТ-ПЦР; предпочтителен набор Original ТА Cloning Kit (фирмы Invitrogen: в качестве ДНК-вектора служит pCR2.1).
Подтвердить функциональную экспрессию кДНК, ускоряющих биосинтез ML-236B и полученных вышеописанными способами, в микроорганизме, продуцирующем ML-236B, можно путем клонирования кДНК в ДНК-вектор, пригодный для функциональной экспрессии в этом микроорганизме. Рекомбинантным ДНК-вектором затем трансформируют соответствующие клетки и сравнивают трансформированные клетки с нетрансформированными по их способности к биосинтезу ML-236B. Если кДНК, ускоряющая биосинтез ML-236B, функционально экспрессируется в трансформированных клетках, то они будут обладать лучшей способностью к биосинтезу ML-236B, чем исходные клетки.
Нет особых ограничений на выбор ДНК-вектора для экспрессии в продуцирующем ML-236B микроорганизме (в дальнейшем именуется функциональным экспрессионным вектором), лишь бы он подходил для трансформации продуцирующего ML-236B микроорганизма и функциональной экспрессии полипептида, кодируемого кДНК, ускоряющей биосинтез ML-236B, в этом организме. Предпочтительно, чтобы вектор был стабильным в клетке-хозяине и имел нуклеотидную последовательность, позволяющую реплицироваться в клетке-хозяине.
Вектор для функциональной экспрессии может содержать одну или несколько кДНК, ускоряющих биосинтез ML-236B, например соответствующих структурным генам mlсЕ и/или mlcR.
Вектор для функциональной экспрессии может содержать один или несколько видов ДНК помимо той кДНК, что соответствует структурным генам mlсЕ и/или mlcR, ускоряющим биосинтез ML-236B при введении в продуцирующий ML-236B микроорганизм. Примерами таких ДНК являются: кДНК, соответствующие структурным генам mlcA, mlcB, mlcC или mlcD; геномная ДНК, связанная с синтезом ML-236B; ДНК, кодирующая факторы, регулирующие экспрессию ускоряющей биосинтез ML-236B кДНК настоящего изобретения, и им подобные.
Вектор для функциональной экспрессии предпочтительно содержит нуклеотидную последовательность, придающую селективный фенотип плазмиде в клетке-хозяине, и он предпочтительно является челночным вектором.
Кроме того, селективный фенотип может представлять собой лекарственную устойчивость и т.п., предпочтительно устойчивость к антибиотику и более предпочтительно устойчивость к ампициллину или гигромицину В.
В том случае когда экспрессионный вектор является челночным вектором, он должен содержать нуклеотидную последовательность, позволяющую ему реплицироваться в клетке-хозяине одной из групп микроорганизмов, и нуклеотидную последовательность, необходимую для экспрессии полипептида, кодируемого вставкой вектора в другом типе клетки-хозяина. Предпочтительно, чтобы вектор придавал различные селективные фенотипы трансформированным клеткам из разных групп микроорганизмов. Требования к сочетаниям групп микроорганизмов такие же, как требования к челночному вектору, применяемому для клонирования и экспрессии связанной с биосинтезом ML-236В геномной ДНК, описанные в настоящем описании.
В настоящем изобретении приемлемым челночным вектором является pSAK700, построенный сочетанием промотора 3-фосфоглицераткиназы (в дальнейшем именуемого "pgk"), происходящего из Aspergillus nidulans и встроенного в ДНК-вектор pSAK333 (описан в японской патентной заявке №3-262486), адаптера для встраивания чужеродных генов, и терминатора pgk, находящегося в ДНК, причем именно в таком порядке (см. Фигуру 4).
Полипептид можно экспрессировать в продуцирующем ML-236B микроорганизме путем встраивания кДНК, соответствующей описанному выше структурному гену mlсЕ, в экспрессионный вектор, описанный выше. В настоящем изобретении экспрессионный вектор для рекомбинантной кДНК pSAKexpE был получен встраиванием кДНК, соответствующей структурному гену mlсЕ, в адаптерный сайт pSAK700. Встроенная в pSAKexpE последовательность - нуклеотидая последовательность кДНК, соответствующей структурному гену mlcE, - показана в SEQ ID No. 37 из Перечня последовательностей. Таким же образом - встраиванием кДНК, соответствующей структурному гену mlcR, в адаптерный сайт pSAK700, - был получен экспрессионный вектор для рекомбинантной кДНК pSAKexpR. Встроенная в pSAKexpR последовательность - нуклеотидная последовательность кДНК, соответствующей структурному гену mlcR, - показана в SEQ ID No. 41 из Перечня последовательностей.
Escherichia coli pSAKexpE SANK 72499, то есть штамм Escherichia coli, трансформированный плазмидой pSAKexpE, депонирован в Research Institute of Life Science and Technology, Agency of Industrial Science and Technology, 25 января 2000 г. под кодовым номером PERM BP-7005 в соответствии с Будапештским договором о депонировании микроорганизмов. Escherichia coli pSAKexpR SANK 72599, то есть штамм Escherichia coli, трансформированный плазмидой pSAKexpR, депонирован в Research Institute of Life Science and Technology, Agency of Industrial Science and Technology, 25 января 2000 г. под кодовым номером PERM BP-7006 в соответствии с Будапештским договором о депонировании микроорганизмов.
Надлежащие методы трансформации выбирают в зависимости от клетки-хозяина, чтобы получить экспрессию ускоряющей биосинтез ML-236B кДНК, связанной с биосинтезом ML-236B геномной ДНК или их фрагментов. Трансформацию Penicillium citrinum - предпочтительного микроорганизма, продуцирующего ML-236B, - можно проводить путем введения рекомбинантного ДНК-вектора в протопласты, полученные из спор Penicillium citrinum (описано в Nara F. et al., Curr. Genet., 23, 28 (1993)).
Споры из косяка с культурой Penicillium citrinum инокулируют на чашку с агарной средой PGA и инкубируют при 22-28°С в течение 10-14 дней. Затем собирают споры из чашки и инокулируют 1×107-1×109 спор в 50-100 мл питательной среды YPL-20 (состав: 0,1% дрожжевого экстракта (фирмы Difco), 0,5% полипептона (фирмы Nihon Seiyaku), 20% лактозы, рН 5,0), после чего инкубируют при 22-28°С от 18 часов до двух дней. Собирают прорастающие споры из культуры и обрабатывают ферментами, разрушающими клеточную стенку, получая протопласты. Нет особых ограничений на выбор ферментов, разрушающих клеточную стенку, лишь бы они были способны разрушить клеточную стенку Penicillium citrinum и не оказывали повреждающего действия на этот микроорганизм. Примеры таковых включают зимолиазу, хитиназу и им подобные.
Смешивание рекомбинантного ДНК-вектора, несущего ускоряющую биосинтез ML-236B кДНК, с продуцирующим ML-236B микроорганизмом или его протопластом в соответствующих условиях дает возможность ввести рекомбинантный ДНК-вектор в этот протопласт и получить трансформант.
Трансформанты продуцирующего ML-236B микроорганизма следует выращивать в условиях, подходящих для данной клетки-хозяина. Трансформанты Penicillium citrinum - предпочтительного микроорганизма, продуцирующего ML-236B, - можно выращивать, проинкубировав предварительно трансформированные протопласты в условиях, способствующих регенерации клеточной стенки, а затем продолжая культуру. Для этого трансформированные протопласты можно внести в средний слой агарной среды VGS (состав: минимальная среда Вогеля, 2% глюкоза, 1 М глюцитол, 2% агар), наложенный на нижний слой агарной среды VGS (состав: минимальная среда Вогеля, 2% глюкоза, 1 М глюцитол, 2,7% агар) и покрытый верхним слоем агарной среды VGS (состав: минимальная среда Вогеля, 2% глюкоза, 1 М глюцитол, 1,5% агар), содержащей 800 мкг/мл гигромицина В, после чего инкубировать при 22-28°С в течение 7-15 дней. Полученный штамм подращивают в среде PGA, инкубируя при 22-28°С. Этот штамм инокулируют платиновой иглой в косяк, приготовленный на среде PGA, инкубируют при 22-28°С в течение 10-14 дней и хранят при 0-4°С.
Как описано выше, ML-236B эффективно вырабатывается при инокуляции трансформанта Penicillium citrinum с регенерированной клеточной стенкой из косяка, описанного выше, в среду MBG 3-8 с последующей инкубацией при 22-28°С в течение 7-12 дней со встряхиванием. Penicillium citrinum также можно выращивать в жидкой среде просто как клетку-хозяина и получать ML-236B.
ML-236B можно очистить из культуры трансформанта продуцирующего ML-236B микроорганизма сочетанием различных методов, широко применяемых для очистки натуральных продуктов. Выбор таких методов ничем особо не ограничен, например может применяться центрифугирование, отделение твердых веществ от жидкости фильтрованием, обработка щелочью или кислотой, экстракция органическими растворителями, растворение, хроматографические методы типа адсорбционной хроматографии, разделительной хроматографии и т.п., а также кристаллизация и другие. ML-236B может находиться в виде гидроксикислоты или лактона, которые взаимно превращаются. Гидроксикислоту можно превратить в ее соль, которая более стабильна. С учетом их физических свойств можно получить ML-236B в виде гидроксикислоты (в дальнейшем именуемой свободной гидроксикислотой), солей гидроксикислоты (в дальнейшем именуемых солями гидроксикислоты) или лактона ML-236B (в дальнейшем именуемого лактоном).
Культуру подвергают щелочному гидролизу при повышенной или комнатной температуре для раскрытия кольца и превращения в соль гидроксикислоты, затем раствор подкисляют и фильтруют. Фильтрат экстрагируют органическим растворителем, отделяя от воды требуемый продукт в виде свободной гидроксикислоты. Выбор органического растворителя ничем особо не ограничен. Примерами таковых являются алифатические углеводороды - гексан, гептан и другие; ароматические углеводороды - бензол, толуол и другие; галогенированные углеводороды - метиленхлорид, хлороформ и другие; эфиры - диэтиловый эфир и другие; сложные эфиры - этилформиат, этилацетат и другие; или смеси, состоящие из двух и более растворителей.
Требуемое соединение можно получить в виде соли гидроксикислоты путем растворения свободной гидроксикислоты в водном растворе соли щелочного металла типа гидроокиси натрия.
Кроме того, требуемое соединение можно получить в виде лактона посредством замыкания кольца при нагревании свободной гидроксикислоты в органическом растворителе, что вызывает дегидратацию, или другими методами.
Полученные таким образом свободную гидроксикислоту, гидроксикислоту или лактон можно очистить и выделить с помощью колоночной хроматографии. Носители для колонок, применяемых в хроматографии, ничем особо не ограничены. Примерами таковых являются Sephadex LH-20 (фирмы Pharmacia), Diaion HP-20 (фирмы Mitsubishi Kagaku), силикагель, обратнофазовые носители и другие, причем предпочтительны носители серии С 18.
Выбор метода количественного определения ML-236B ничем особо не ограничен; предпочтительны методы, общеприменимые при количественном определении органических соединений. Примерами таковых являются обратнофазовая высокоэффективная жидкостная хроматография (в дальнейшем именуемая "обратнофазовой ВЭЖХ") или подобные. Количественное определение методом обратнофазовой ВЭЖХ можно проводить, подвергая культуру продуцирующего ML-236B микроорганизма щелочному гидролизу, подвергая растворимую фракцию обратнофазовой ВЭЖХ на колонке с С18, измерения УФ-поглощения и перевода его в количество ML-236B. Выбор колонки с С18 ничем особо не ограничен; предпочтительны колонки, обычно применяемые для обратнофазовой ВЭЖХ. Примерами таковых являются SSC-ODS-262 (диаметр 6 мм, длина 100 мм, фирмы Senshu Kagaku) или подобные. Выбор растворителя для подвижной фазы ничем особо не ограничен, лишь бы это был растворитель, обычно применяемый для обратнофазовой ВЭЖХ. Таковым является, к примеру, 75% метанол - 0,1% триэтиламин - 0,1% уксусная кислота или подобные. Когда ML-236B пропускают при комнатной температуре через колонку SSC-ODS-262, в которой в качестве подвижной фазы применяется 75% метанол - 0,1% триэтиламин - 0,1% уксусная кислота со скоростью 2 мл/мин, ML-236B выходит через 4,0 минут. ML-236B можно обнаруживать с помощью УФ-детектора для ВЭЖХ. При УФ-детекции поглощение регистрируется при длине волны 220-280 нм, предпочтительно 220-260 нм и более предпочтительно 236 нм.
Предусматриваются фармацевтические композиции, содержащие ML-236B, полученный согласно настоящему изобретению, вместе с фармацевтическим носителем.
Также предусматриваются фармацевтические композиции, содержащие правастатин, приготовленный из ML-236B, полученного согласно настоящему изобретению, вместе с фармацевтическим носителем.
Фармацевтические композиции настоящего изобретения могут быть стандартными, похожими на те, что применяются в существующих формулах ML-236B или правастатина.
Методы лечения также являются составной частью настоящего изобретения, в них применяются эти соединения или композиции для лечения гиперлипидемии и других заболеваний.
Далее изобретение раскрывается более подробно в отношении фигур и примеров. Примеры представлены для иллюстрации, а не для ограничения настоящего изобретения.
Фигура 1. Диаграмма, изображающая конструкцию ДНК-вектора pSAKcosl.
Фигура 2. Результаты анализа структурных генов встроенной в pML48 последовательности.
Фигура 3. Гибридизация по Норзерну встроенной в pML48 последовательности.
Фигура 4. Диаграмма, изображающая конструкцию экспрессионного кДНК-вектора pSAK700.
Фигура 5. Анализ методом ОТ-ПЦР транскрипции mlсА-Е и R в трансформанте pSAKexpR.
Фигура 6. Анализ методом ОТ-ПЦР транскрипции т1сЕ в трансформанте pSAKexpE.
Пример 1. Построение вектора pSAKcos1
Плазмиду pSAK.333, содержащую ген гигромицин В-фосфотрансферазы (в дальнейшем именуемой "ГФТ") из Escherichia coli (японская патентная заявка №3-262486), расщепляли рестрикционным ферментом BamHI (фирмы Takara Shuzo Co., Ltd., Япония) и обрабатывали ДНК-полимеразой Т4 (фирмы Takara Shuzo Co., Ltd., Япония) для образования тупых концов.
Полученный фрагмент ДНК лигировали на себя в кольцевую форму с помощью набора для лигирования ДНК Ver.2 (фирмы Takara Shuzo Co., Ltd., Япония), а затем трансформировали им компетентные клетки Escherichia coli JM109 (фирмы Takara Shuzo Co., Ltd., Япония). Путем селекции трансформированных клеток Escherichia coli был получен штамм, содержащий плазмиду, названную pSAK360, у которой произошла делеция сайта BamHI.
pSAK360 расщепляли рестрикционным ферментом PvuII, затем обрабатывали щелочной фосфатазой и получали фрагмент, дефосфорилированный на 5'-конце. Из космидного вектора pWE15 (фирмы Stratagene) получали фрагмент SalI-ScaI (около 3 kb), содержащий cos-сайт, и обрабатывали ДНК-полимеразой Т4 для образования тупых концов. Затем этот фрагмент лигировали с pSAK360 по сайту PvuII. Этой ДНК трансформировали клетки JM109. Путем селекции трансформированных клеток Escherichia coli были получены штаммы, содержащие плазмиду, названную pSAKcos1, в которую встроился фрагмент SalI-ScaI (около 3 kb) по сайту PvuII. pSAKcos1 содержит сайты расщепления для рестрикционных ферментов ВаmHI, EcoRI и NotI, все они происходят из pWE15. pSAKcos1 содержит гены устойчивости к ампициллину и гигромицину в качестве маркеров для селекции.
В следующих примерах, когда в качестве клетки-хозяина использовали Escherichia coli, селекцию трансформантов pSAKcos1 или трансформантов pSAKcos1 со встроенным чужеродным геном проводили, добавляя 40 мкг/мл ампициллина (фирмы Sigma) в соответствующую среду. Когда в качестве клетки-хозяина использовали Penicillium citrinum SANK 13380, то селекцию трансформантов pSAKcos1 или трансформантов pSAKcos1 со встроенным чужеродным геном проводили, добавляя 200 мкг/мл гигромицина В (фирмы Sigma) в соответствующую среду.
Метод построения pSAKcos1 показан на Фигуре 1.
Пример 2. Выделение геномной ДНК Penicillium dtrinum SANK 13380
1) Выращивание Penicillium citrinum SANK 13380
Посевную культуру Penicillium dtrinum SANK 13380 делали на косяке из агарной среды PGA. Для этого агар инокулировали Penicillium citrinum SANK 13380 с помощью платиновой иглы и инкубировали при 26°С в течение 14 дней. Косяк хранили при 4°С.
Культуры выращивали в жидкой среде с аэрацией. Клетки с поверхности в 5 мм из указанного выше косяка инокулировали в 50 мл среды MBG3-8 в конической колбе на 500 мл и инкубировали при 26°С со встряхиванием при 210 об/мин в течение 5 дней.
2) Выделение геномной ДНК из Penicillium citrinum SANK 13380
Культуру, полученную на стадии 1, центрифугировали 10 минут при 1000×g при комнатной температуре и собирали клетки. Растирали 3 г (сырой вес) клеток в ступке, охлаждаемой сухим льдом, получая порошок. Разрушенные клетки помещали в центрифужную пробирку, заполненную 20 мл 62,5 мМ ЭДТА·2Na (фирмы Wako Pure Chemical Industries, Ltd.), 5% ДСН, 50 мМ трис-HCl (фирмы Wako Pure Chemical Industries, Ltd.) буфера (рН 8,0) и осторожно перемешивали, после чего оставляли при 0°С на 1 час. Добавляли туда 10 мл фенола, насыщенного 10 мМ трис·НСl,0,1 мМ ЭДТА·2Na (рН 8,0; в дальнейшем именуется "ТЕ"), и эту смесь осторожно перемешивали 1 час при 50°С.
После центрифугирования 10 минут при 10000×g при комнатной температуре отбирали 15 мл и переносили в другую центрифужную пробирку. Туда добавляли 1/2 объема насыщенного ТЕ фенола и 1/2 объема хлороформа. Смесь перемешивали 2 минуты и центрифугировали 10 минут при 10000×g при комнатной температуре (в дальнейшем именуется "экстракция фенол-хлороформом"). К 10 мл верхнего слоя (водная фаза) добавляли 10 мл 8 М ацетата аммония (рН 7,5) и 25 мл 2-пропанола (фирмы Wako Pure Chemical Industries, Ltd.), охлаждали 15 минут при -80°С и центрифугировали при 4°С при 1000×g в течение 10 минут.
После высаживания осадки растворяли в 5 мл ТЕ, после чего вносили 20 мкл рибонуклеазы А (10 мг/мл; фирмы Gibco) и инкубировали 20 минут при 37°С. Добавляли 20 мл 2-пропанола и осторожно перемешивали. Затем нити геномной ДНК наматывали на кончик пастеровской пипетки и растворяли в 2 мл ТЕ.
Далее в раствор ДНК добавляли 0,1 объем 3 М ацетата натрия (рН 6,5) и 2,5 объема этанола. Раствор охлаждали 15 минут при -80°С, а затем центрифугировали при 4°С при 1000×g в течение 5 минут (что в дальнейшем именуется "осаждение этанолом"). Полученный осадок растворяли в 200 мкл ТЕ - это и есть фракция геномной ДНК.
Пример 3. Получение геномной библиотеки ДНК Penicillium citrinum SANK 13380
1) Выделение фрагмента геномной ДНК
К 100 мкл водного раствора геномной ДНК (50 мкг) Penicillium citrinum SANK 13380, полученной в Примере 2, добавляли 0,25 единиц Sau3AI (Takara Shuzo Co., Ltd., Япония). Через 10, 30, 60, 90 и 120 секунд отбирали пробы по 20 мкл смеси и добавляли в них 0,5 М ЭДТА (рН 8,0) для остановки реакции рестрикционного фермента. Получали фрагменты частично расщепленной ДНК, которые разделяли методом гель-электрофореза в агарозе, и извлекали агарозный гель, содержащий фрагменты ДНК в 30 kb и больше.
Извлеченный гель измельчали и помещали в центрифужно-фильтровальный блок (фирмы Japan Millipore). Гель замораживали, охлаждая 15 минут при -80°С, а затем расплавляли 10 минут при 37°С. Из него экстрагировали ДНК, центрифугируя при 5000×g в течение 5 минут. ДНК подвергали экстракции фенол-хлороформом и осаждению этанолом. Полученные осадки растворяли в небольшом объеме ТЕ.
2) Предварительная обработка ДНК-вектора pSAKcos1
pSAKcos1 расщепляли рестрикционным ферментом ВаmHI (Takara Shuzo Co., Ltd., Япония), а затем обрабатывали щелочной фосфатазой (Takara Shuzo Co., Ltd., Япония) 30 минут при 65°С. Обработанный раствор подвергали экстракции фенол-хлороформом и осаждению этанолом. Полученный осадок растворяли в небольшом объеме ТЕ.
3) Лигирование и упаковка in vitro
Фрагмент геномной ДНК (2 мкг), описанный выше на стадии 1, и предварительно обработанную, как описано выше, плазмиду pSAKcos1 (1 мкг) смешивали вместе и лигировали при 16°С в течение 16 часов с помощью набора для лигирования ДНК Ver.2 (Takara Shuzo Co., Ltd., Япония). После обработки раствор подвергали экстракции фенол-хлороформом и осаждению этанолом. Полученные осадки растворяли в 5 мкл ТЕ. Продукт лигирования подвергали упаковке in vitro с помощью набора GIGAPAK П Gold (фирмы Stratagene), получая трансформанты Escherichia coli, содержащие рекомбинантный ДНК-вектор. На чашку с образовавшимися колониями трансформантов Escherichia coli наливали 3 мл среды LB, после чего колонии из чашки собирали с помощью "скребка" (что именуется "собранный раствор 1"). Чашку еще раз промывали 3 мл среды LB и собирали клетки (что именуется "собранный раствор 2"). К смеси собранных растворов 1 и 2 добавляли глицерин до конечной концентрации 18% (что именуется суспензией клеток Escherichia coli) и хранили при -80°С как геномную библиотеку ДНК Penicillium citrinum SANK 13380.
Пример 4. Амплификация фрагмента гена ПКС методом ПЦР с применением геномной ДНК Penicillium citrinum SANK 13380 в качестве матрицы
1) Разработка и синтез праймеров для ПЦР
На основе аминокислотной последовательности гена ПКС Aspergillus flavus (описано в Brown D.W. et aL, Proc. Natl. Acad. Sci. USA, 93, 1418 (1996)) были разработаны и синтезированы вырожденные праймеры, представленные в SEQ ID No. 3 и 4 из Перечня последовательностей. Синтез проводился фосфоамидитным методом.
SEQ ID No. 3 из Перечня последовательностей:
gayacngcntgyasttc
SEQ ID No. 4 из Перечня последовательностей:
tcnccnknrcwgtgncc
В нуклеотидных последовательностях SEQ ID No. 3 и 4 n означает инозин (гипоксантин), y означает t или с, s означает g или с, k означает g или t, r означает g или а, и w означает а или t.
2) Амплификация сегмента ДНК методом ПЦР
Приготовили 50 мкл реакционной смеси, содержащей праймеры для ПЦР, описанные выше на стадии 1 (по 100 пмоль), геномную ДНК Penicillium citrinum SANK 13380 из Примера 2 (500 нг), 0,2 мМ dATP, 0,2 мМ dCTP, 0,2 мМ dGTP, 0,2 мМ dTTP, 50 мМ хлористого калия, 2 мМ хлористого магния и 1,25 единиц ДНК полимеразы Ex. Tac (Takara Shuzo Co., Ltd., Япония). Смесь подвергали циклу реакций, состоящему из трех последовательных стадий, следующим образом: 1 минута при 94°С, 2 минуты при 58°С и 3 минуты при 70°С. Этот цикл повторяли 30 раз для амплификации фрагмента ДНК. ПЦР проводили в приборе TaKaRa PCR Thermal Cycler MP TP 3000 (фирмы Takara Shuzo Co., Ltd., Япония).
Амплифицированные фрагменты ДНК подвергали агарозному гель-электрофорезу, после чего извлекали агарозный гель, содержащий фрагменты ДНК размером от 1,0 до 2,0 kb. Из геля выделяли ДНК и подвергали экстракции фенол-хлороформом и осаждению этанолом. Полученный осадок растворяли в небольшом объеме ТЕ.
3) Лигирование и трансформация
Полученный на стадии 2 фрагмент ДНК лигировали с плазмидой pCR2.1 с помощью системы клонирования ТА Cloning System pCR2.1 (фирмы Invitrogen), в которой плазмида входит в состав набора. Плазмиду использовали для трансформации Escherichia coli JM109, чтобы получить трансформанты.
Выбирали несколько колоний из этих трансформантов и выращивали культуры согласно Maniatis et al. (описано в Maniatis T. et al., Molecular cloning, a laboratory manual, 2nd ed.. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. (1989)). Для этого каждую колонию высевали в пробирку на 24 мл, содержащую 2 мл среды LB, и инкубировали при 37°С в течение 18 часов со встряхиванием.
Из этой культуры выделяли рекомбинантный ДНК-вектор щелочным методом (описано в Maniatis Т. et al., supra). Для этого отбирали 1,5 мл культуры и центрифугировали при комнатной температуре 2 минуты при 1000×g. Клетки собирали из осадка. Их суспендировали, добавляя 100 мкл раствора 50 мМ глюкозы, 25 мМ трис·НСl, 10 мМ ЭДТА (рН 8,0). Туда же добавляли 200 мкл 0,2 N гидроокиси натрия - 1% ДСН. Суспензию осторожно перемешивали, лизируя микроорганизмы. Затем добавляли 150 мкл 3 М ацетата калия - 11,5% уксусной кислоты, чтобы денатурировать белки, и центрифугировали при комнатной температуре 10 минут при 1000×g. Собирали супернатант, который подвергали экстракции фенол-хлороформом и осаждению этанолом. Осадок растворяли в 50 мкл ТЕ, содержащего 40 мкг/мл рибонуклеазы А (фирмы Sigma).
Каждый из рекомбинантных ДНК-векторов расщепляли рестрикционными ферментами и подвергали электрофорезу. Во всех вставках из рекомбинантных ДНК-векторов, проявлявших отличия по профилю расщепления на электрофорезе, определяли нуклеотидные последовательности ДНК с помощью ДНК-секвенатора (модель 377 фирмы Perkin Elmer Japan).
Таким образом был идентифицирован штамм с рекомбинантным ДНК-вектором, содержащим фрагмент гена ПКС из Penicillium citrinum.
Пример 5. Гибридизация по Сазерну геномной ДНК Penicillium citrinum SAN К 13380
1) Электрофорез и перенос на мембрану
Геномную ДНК (10 мкг) Penicillium citrinum SANK 13380, полученную в Примере 2, расщепляли рестрикционными ферментами EcoRI, SalI, HindIII или SacI (все они фирмы Takara Shuzo Co., Ltd., Япония) и подвергали агарозному гель-электрофорезу. Для геля применяли агарозу L03 "TAKARA" (Takara Shuzo Co., Ltd., Япония). После электрофореза гель погружали в 0,25 М соляную кислоту (фирмы Wako Pure Chemical Industries, Ltd.) и инкубировали при комнатной температуре 10 минут с покачиванием. Гель переносили в 0,4 N гидроокись натрия (фирмы Wako Pure Chemical Industries, Ltd.) и инкубировали при комнатной температуре 30 минут. Методом щелочного переноса Maniatis et al. (supra) переносили ДНК из геля на нейлоновую мембрану Hybond™-N+ (фирмы Amersham) и фиксировали в ней. Мембрану промывали раствором 2×SSC (1×SSC содержит 150 мМ NaCl, 15 мМ цитрат натрия), а затем высушивали на воздухе.
2) Гибридизация и обнаружение сигнала
Полученную на стадии 1 мембрану гибридизировали, используя в качестве зонда фрагмент гена ПКС, полученный в Примере 4.
Для получения зонда 1 мкг ДНК вставки фрагмента гена ПКС, полученного в Примере 4, метили с помощью набора DIG DNA Labeling Kit (фирмы Boehringer-Mannheim), кипятили 10 минут, а затем быстро охлаждали непосредственно перед применением.
Описанную на стадии 1 мембрану погружали в гибридизационный раствор (DIG Easy Hyb фирмы Boehringer-Mannheim) и подвергали прегибридизации с покачиванием при 20 об/мин 2 часа при 42°С. Затем добавляли указанный выше зонд и проводили гибридизацию с покачиванием при 20 об/ми при 42°С в течение 18 часов в камере Multishaker Oven НВ (фирмы ТАГГЕС). После этого мембрану промывали 3 раза раствором 2×SSC при комнатной температуре в течение 20 минут и 2 раза раствором 0,1×SSC при 55°С в течение 30 минут.
Промытую мембрану обрабатывали с помощью набора для люминесцентной детекции DIG Luminescent Detection Kit for Nucleic Acids (фирмы Boehringer-Mannheim) и накрывали рентгеновской пленкой Lumifilm (фирмы Boehringer-Mannheim). Обработку проводили с помощью проявочной машины для медицины Fuji FPM 800A (фирмы Fuji Film Corporation).
В результате этого было подтверждено наличие гена ПКС, фрагмент которого был получен в Примере 4, в геноме Penicillium citrinum.
Пример 6. Скринирование геномной библиотеки ДНК Penicillium citrinum SANK 13380 с помощью зонда из фрагмента гена ПКС
Клонирование фрагмента геномной ДНК, содержащего ген ПКС, проводилось методом гибридизации колоний.
1) Подготовка мембраны
Клеточную суспензию Escherichia coli, служащую в качестве геномной библиотеки ДНК Penicillium citrinum SANK 13380 (описана в Примере 3), разводили и делали рассев на чашку с агарной средой LB так, чтобы на чашке выросло от 5000 до 10000 колоний. Чашку инкубировали при 26°С в течение 18 часов и охлаждали при 4°С в течение 1 часа. На чашку накладывали мембрану Hybond™-N+ (фирмы Amersham) и держали в контакте 1 минуту. Мембрану с налипшими колониями осторожно удаляли с чашки. Находившуюся в контакте с колониями поверхность переворачивали лицом вверх и погружали на 7 минут в 200 мл раствора 1,5 М хлористого натрия, 0,5 N гидроокиси натрия, а затем дважды погружали на 3 минуты в 200 мл раствора 1,5 М хлористого натрия, 0,5 М трис·НСl, 1 мМ ЭДТА (рН 7,5), после чего промывали 400 мл 2×SSC. Промытую мембрану подсушивали на воздухе в течение 30 минут.
2) Гибридизация
В качестве зонда использовали ДНК вставки гена ПКС, полученную в Примере 4 (1 мкг). ДНК метили с помощью набора DIG DNA Labeling Kit (фирмы Boehringer-Mannheim), кипятили 10 минут, а затем быстро охлаждали непосредственно перед применением.
Мембрану, описанную на стадии 1, погружали в гибридизационный раствор (DIG Easy Hyb фирмы Boehringer-Mannheim) и подвергали прегибридизации при 20 об/мин 2 часа при 42°С. Затем добавляли указанный выше зонд и проводили гибридизацию при 20 об/мин при 42°С в течение 18 часов в камере Multishaker Oven HB (фирмы TAITEC). После этого мембрану промывали 3 раза раствором 2×SSC при комнатной температуре в течение 20 минут и 2 раза раствором 0,1×SSC при 68°С в течение 30 минут.
Промытую мембрану обрабатывали с помощью набора для люминесцентной детекции DIG Luminescent Detection Kit for Nucleic Acids (фирмы Boehringer-Mannheim) и накрывали рентгеновской пленкой Lumifilm (фирмы Boehringer-Mannheim). Обработку проводили с помощью проявочной машины для медицины Fuji FPM 800A (фирмы Fuji Film Corporation).
Стадии 1 и 2 выше именуются скринингом.
Колонии на чашке, где был обнаружен положительный сигнал при первом скринировании, снимали и клетки суспендировали в среде LB. Затем клетки соответствующим образом разводили и наносили на чашку. После этого проводили второе скринирование для очистки положительного клона.
Положительный клон, полученный в настоящем примере, то есть трансформированный штамм Escherichia coli pML48 SANK 71199, депонирован в Research Institute of Life Science and Technology, Agency of Industrial Science and Technology, 7 июля 1999 г. под кодовым номером FERM BP-6780 в соответствии с Будапештским договором о депонировании микроорганизмов.
Пример 7. Анализ встроенной последовательности рекомбинантного ДНК-вектора pML48 (1)
Выращивание культуры штамма Escherichia coli pML48 SANK 71199, полученного в Примере 6, и выделение рекомбинантного ДНК-вектора из этой культуры проводили таким же образом, как описано в Примере 4.
Полученный ДНК-вектор назвали pML48. Вставку из pML48, которая является связанной с биосинтезом ML-236B геномной ДНК, расщепляли различными рестрикционными ферментами и образовавшиеся фрагменты субклонировали в плазмиду pUC119 (фирмы Takara Shuzo Co., Ltd., Япония). Для этого продукты расщепления pML48 различными рестрикционными ферментами подвергали электрофорезу, а затем ДНК переносили на мембрану и подвергали гибридизации. В результате этого получили рестрикционную карту встроенной последовательности pML48 стандартными для этой области методами.
Нуклеотидные последовательности всех встроенных в эти субклоны вставок определяли с помощью ДНК-секвенатора (модель 377 фирмы Perkin Elmer Japan Co. Ltd.), а затем определили всю нуклеотидную последовательность pML48.
Встроенная в pML48 вставка в целом состоит из 34203 оснований.
Нуклеотидная последовательность встроенной в pML48 вставки описана в SEQ ID No. 1 и 2 из Перечня последовательностей. Последовательности SEQ ID No. 1 и SEQ ID No. 2 полностью комплементарны друг другу.
Наличие структурных генов во встроенной в pML48 вставке анализировали с помощью программы поиска генов GRAIL (ApoCom GRAIL Toolkit фирмы Apocom Corporation) и программы поиска гомологии BLAST (Gapped-BLAST (BLAST2): встроена в пакет WISCONSIN GCG версии 10.0).
В результате этого было предсказано наличие 6 различных структурных генов во встроенной в pML48 вставке; они были названы mlcA, mlcB, mlcC, mlcD, mlcE и mlcR соответственно. Кроме того, было предсказано, что кодирующие участки mlcA, mlcB, mlcE и mlcR находятся в SEQ ID No. 2, а кодирующие участки mlcC и mlcD находятся в SEQ ID No. 1 из Перечня последовательностей. Также было предсказано относительное положение и длина каждого из предполагаемых структурных генов встроенной последовательности.
Результаты настоящего примера представлены на Фигуре 2. Стрелки обозначают локализацию, направление и относительный размер каждого структурного гена на вставке pML48. Направленная влево стрелка означает, что кодирующий участок структурного гена (mlcA, В, Е или R) находится в SEQ ID No. 2. Направленная вправо стрелка означает, что кодирующий участок структурного гена (mlcC или D) находится в SEQ ID No. 1.
Пример 8. Анализ встроенной последовательности рекомбинантного ДНК-вектора pML48 (2)
Анализ экспрессии структурных генов, существование которых было предсказано в Примере, проводили с посощью гибридизации по Норзерну и RACE. Анализировали 5'- и 3'-концевые участки.
1) Выделение тотальной РНК Penicillium citrinum SANK 13380
Клетки из площади в 5 мм косяка Penicillium citrinum SANK 13380 (описанного в Примере 2) инокулировали в 10 мл среды MGB3-8 в конической колбе на 100 мл и инкубировали при 26°С в течение 3 дней со встряхиванием.
Выделение тотальной РНК из этой культуры проводили с помощью набора RNeasy Plant Mini Kit (фирмы Qiagen AG), в котором применяется гуанидин-изотиоцианатный метод. Для этого культуру центрифугировали при комнатной температуре 10 минут при 5000xg и собирали клетки. После этого замораживали 2 г (сырой вес) клеток в жидком азоте и растирали в ступке, получая порошок. Разрушенные клетки суспендировали в 4 мл лизирующего буфера (из набора). По 450 мкл суспензии наносили на 10 центрифужных колонок QIAshredder, входящих в набор, и центрифугировали при комнатной температуре 10 минут при 1000×g. Собирали каждый из элюентов и добавляли в них по 225 мкл этанола, а затем переносили в центрифужные мини-колонки для РНК, входящие в набор. Колонки промывали промывочным буфером из набора, после чего адсорбировавшийся на колонках материал элюировали пропусканием 50 мкл свободной от рибонуклеазы дистиллированной воды. Этот элюент использовали в качестве тотальной фракции РНК.
2) Гибридизация по Норзерну
Образец РНК готовили, добавляя 2,25 мкл водного раствора, содержащего 20 мкг тотальной РНК Penicillium citrinum SANK 13380, к смеси из 1 мкл 10×MOPS (состав: 200 мМ 3-морфолинопропансульфоновой кислоты, 50 мМ ацетата натрия, 10 мМ ЭДТА·2Na, рН 7,0; стерилизован в автоклаве при 121°С в течение 20 минут; фирмы Dojinkagaku Laboratory Co. Ltd.), 1,75 мкл формальдегида и 5 мкл формамида, и перемешивая. Образец РНК держали 10 минут при 65°С, затем быстро охлаждали в ледяной воде и подвергали агарозному гель-электрофорезу. Гель для электрофореза получали, смешивая 10 мл 10×MOPS и 1 г агарозы L03 "TAKARA" (Takara Shuzo Co., Ltd., Япония) с 72 мл обработанной диэтилпирокарбонатом воды (фирмы Sigma) и нагревая до растворения агарозы, а затем охлаждали и добавляли 18 мл формальдегида. В качестве буфера для образца использовали 1×MOPS (полученный разбавлением 10xMOPS в 10 раз водой). РНК из геля переносили на мембрану Hybond™-N+ (фирмы Amersham) в растворе 10×SSC.
В качестве зондов использовали фрагменты ДНК а, b, с, d и е, полученные при расщеплении встроенной последовательности pML48 рестрикционными ферментами 1 и 2, показанными в нижеследующей Таблице 1. Локализация каждого зонда на вставке pML48 показана в верхней части Фигуры 3.
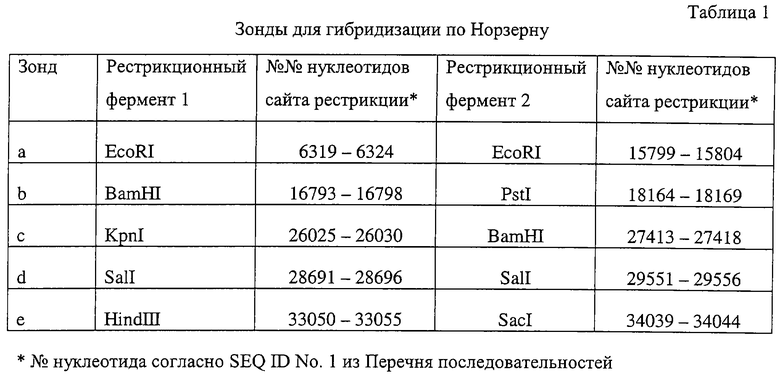
Мечение зондов, гибридизация и детекция сигнала проводились, как при гибридизации по Сазерну, описанной в Примере 5.
Результаты этого примера показаны в нижней части Фигуры 3. Каждый сигнал показывает присутствие продукта транскрипции, гомологичного нуклеотидной последовательности данного зонда.
Результаты показывают, что структурные гены, наличие которых во встроенной в pML48 последовательности было предсказано в настоящем примере, а именно - mlcA, mlcB, mlcC, mlcD, mlcE и mlcR, действительно транскрибируются в Penicillium citrinum SANK 13380.
Положение сигнала не соответствует относительному размеру продукта транскрипции.
3) Определение 5'-концевой последовательности методом 5'-RACE
кДНК, содержащие 5'-концевые участки структурных генов, получали с помощью системы для быстрой амплификации концов кДНК - 5'-RACE версии 2.0 (фирмы GDBCO).
Были получены 2 вида антисмысловых олигонуклеотидов. Их разработали на основе нуклеотидных последовательностей, находящихся в кодирующих участках и вблизи 5'-концов каждого из структурных генов во встроенной последовательности pML48, как предсказано результатами Примера 7 и пункта 2 настоящего примера.
Нуклеотидные последовательности антисмысловых олигонуклеотидов (1), составленных на основе последовательностей, находящихся на 3'-стороне от каждого из структурных генов, показаны в Таблице 2. Нуклеотидные последовательности антисмысловых олигонуклеотидов (2), составленных на основе последовательностей, находящихся на 5'-стороне от каждого из структурных генов, показаны в Таблице 3.


Первую цепь кДНК синтезировали по реакции обратной транскриптазы с помощью олигонуклеотида (1) в качестве праймера и тотальной РНК Penicillium citrinum SANK 13380 в качестве матрицы. Для этого 24 мкл реакционной смеси, содержащей 1 мкг тотальной РНК, 2,5 пмоль олигонуклеотида (1) и 1 мкл обратной транскриптазы SUPER SCRIPT™ (из набора), инкубировали при 16°С в течение 1 часа, а продукт реакции вносили в центрифужный картридж GLASSMAX (входящий в набор) и очищали первую цепь кДНК.
К 3'-концу первой цепи кДНК присоединяли цепочку полиС с помощью терминальной дезоксирибонуклеотидилтрансферазы, входящей в состав набора.
50 мкл реакционной смеси, включающей первую цепь кДНК с присоединенной цепочкой полиС, смешивали с 40 пмоль олигонуклеотида (2) и 40 пмоль праймера Abriged Anchor Primer (из набора) и инкубировали 2 минуты при 94°С. Затем повторяли 35 раз цикл инкубации, состоящий из 30 секунд при 94°С, 30 секунд при 55°С и 2 минут при 72°С, после чего инкубировали 5 минут при 72°С и 18 часов при 4°С. Продукт реакции подвергали агарозному гель-электрофорезу и выделяли ДНК из геля. Продукт очищали путем экстракции фенол-хлороформом и осаждения этанолом, а затем клонировали таким же образом, как описано в Примере 4, с помощью pCR2.1.
Описанная выше операция - это 5'-RACE.
Были определены нуклеотидные последовательности 5'-концевых фрагментов кДНК и предсказаны положения начала транскрипции и кодоны начала трансляции.
В таблице 4 показаны номера SEQ ID, в которых представлены нуклеотидные последовательности 5'-концевых фрагментов кДНК каждого из структурных генов, полученные методом 5'-RACE. В таблице 5 показаны номера SEQ ID, в которых находятся точки начала транскрипции и точки начала трансляции каждого из структурных генов и расположение этих точек.


4) Определение 3'-концевой последовательности методом 3'-RACE
кДНК, содержащие 3'-концевые участки структурных генов, получали с помощью набора Ready-To-Go T-Primed First-Strand Kit (фирмы Pharmacia).
Один из смысловых олигонуклеотидов (3) был разработан на основе нуклеотидных последовательностей, находящихся в кодирующих участках и вблизи 3'-концов каждого из структурных генов во встроенной последовательности pML48, как предсказано результатами Примера 7 и пункта 2 настоящего примера.
Нуклеотидные последовательности олигонуклеотидов (3), составленных для каждого из структурных генов, показаны в Таблице 6.

Первую цепь кДНК синтезировали по реакции обратной транскриптазы с помощью праймера NotI-d(T)18 (из набора) и тотальной РНК Penicillium citrinum SANK 13380 (1 мкг) в качестве матрицы.
100 мкл реакционной смеси, включающей первую цепь кДНК, 40 пмоль олигонуклеотида (3) и праймер NotI-d(T)18 (из набора) инкубировали 2 минуты при 94°С. Затем повторяли 35 раз цикл инкубации, состоящий из 30 секунд при 94°С, 30 секунд при 55°С и 2 минут при 72°С, после чего инкубировали 5 минут при 72°С и 18 часов при 4°С. Продукт реакции подвергали агарозному гель-электрофорезу и выделяли ДНК из геля. Продукт очищали путем экстракции фенол-хлороформом и осаждения этанолом, а затем клонировали таким же образом, как описано в Примере 4, с помощью pCR2.1.
Описанная выше операция - это 3'-RACE.
Были определены нуклеотидные последовательности на 3'-концах кДНК и предсказаны кодоны остановки трансляции.
В таблице 7 показаны номера SEQ ID, в которых представлены нуклеотидные последовательности 3'-концевых фрагментов кДНК каждого из структурных генов, полученные методом 3'-RACE. В таблице 8 показаны кодоны остановки трансляции и их положение согласно SEQ ID No. 1 и 2 из Перечня последовательностей.


В таблице 9 показаны предсказанные С-концевые аминокислотные остатки полипептидов, кодируемых каждым из структурных генов, последовательности тринуклеотидов, кодирующих эти аминокислотные остатки, и положение этих тринуклеотидов.

В таблице 10 представлены последовательности, комплементарные кодонам окончания трансляции, показанным в таблице 8, номера SEQ ID, где они находятся и положение комплементарных последовательностей.

Как описано выше, было установлено положение каждого из структурных генов, его направление и расположение. На основе этой информации можно получить продукты транскрипции и продукты трансляции каждого из структурных генов.
Пример 9. Получение кДНК, соответствующей структурному гену mlcE
1) Выделение тотальной РНК
Тотальную ДНК Penicillium citrinum. выделяли по методике Примера 8.
2) Составление праймеров
Для получения полной кДНК, соответствующей структурному гену mlcE, определенному в Примере 8, были разработаны и синтезированы следующие праймеры:
смысловой праймер - 5'-gttaacatgtcagaacctctaccccc-3' (SEQ ID No. 35) антисмысловой праймер - 5'-aatatttcaagcatcagtctcaggcac-3' (SEQ ID No. 36).
Праймеры происходят из последовательности, расположенной вверх от 5'-конца структурного гена mlcE, и из последовательности, расположенной вниз от 3'-конца, соответственно. Синтез проводили фосфоамидитным методом.
3) ОТ-ПЦР
Для получения полной кДНК, кодирующей продукт гена mlcE, использовали набор для ПЦР Takara RNA LA PCR Kit (AMV) версии 1.1.
Для этого 20 мкл реакционной смеси, содержащей 1 мкг тотальной РНК, 2,5 пмоль праймера Random 9mer (из набора) и 1 мкл обратной транскриптазы (из набора) инкубировали 30 минут при 42°С, получая первую цепь кДНК. Затем обратную транскриптазу инактивировали, нагревая при 99°С в течение 5 минут.
100 мкл второй реакционной смеси, включающей весь объем реакционной смеси для первой цепи кДНК (выше), 40 пмоль смыслового праймера и 40 пмоль антисмыслового праймера, инкубировали 2 минуты при 94°С. Затем повторяли 30 раз цикл инкубации, состоящий из 30 секунд при 94°С, 30 секунд при 60°С и 2 минут при 72°С, после чего инкубировали 5 минут при 72°С и 18 часов при 4°С. Продукт реакции подвергали агарозному гель-электрофорезу и выделяли ДНК из геля. Продукт очищали путем экстракции фенол-хлороформом и осаждения этанолом и использовали для трансформации компетентных клеток штамма Escherichia coli JM109 (фирмы Takara Shuzo Co., Ltd., Япония) таким же образом, как описано в примере 4 с помощью pCR2.1. Из трансформированных клеток Escherichia coli отбирали штамм, несущий плазмиду с этим фрагментом ДНК, которую назвали pCRexpE.
Была определена нуклеотидная последовательность ДНК, встроенной в рекомбинантный вектор pCRexpE. Вставка содержала полную кДНК, соответствующую структурному гену mlcE. Его нуклеотидная последовательность и аминокислотная последовательность пептида, предсказанного из нуклеотидной последовательности, показаны в SEQ ID No. 37 и/или SEQ ID No. 38 из Перечня последовательностей.
Из известных последовательностей наиболее близкой к mlcE (полипептиду) была ORF10 в генном кластере, связанном с биосинтезом ловастатина, на уровне идентичности в 70%.
Пример 10. Построение экспрессионного вектора рSАК700
Экспрессионный вектор pSAK700 для кДНК был построен с помощью векторов pSAK333 и pSAK360, описанных в примере 1.
pSAK333 расщепляли двумя рестрикционными ферментами ВаmHI и HindIII (фирмы Takara Shuzo Co., Ltd., Япония) и подвергали агарозному гель-электрофорезу. Из геля выделяли фрагмент ДНК в 4,1 kb и обрабатывали ДНК-полимеразой Т4 (фирмы Takara Shuzo Co., Ltd., Япония) для образования тупых концов.
К указанному выше фрагменту ДНК присоединяли адаптер EcoRI-NotI-BamHI (фирмы Takara Shuzo Co., Ltd., Япония) с помощью набора для лигирования ДНК Ver.2 (фирмы Takara Shuzo Co., Ltd., Япония). Этой лигированной ДНК трансформировали компетентные клетки штамма Escherichia coli JM109 (фирмы Takara Shuzo Co., Ltd., Япония). Из трансформированных клеток Escherichia coli отбирали штамм, несущий плазмиду с адаптером, которую назвали pSAK410.
pSAK360 расщепляли двумя рестрикционными ферментами PvuII и Sspl и подвергали электрофорезу. Из геля выделяли фрагмент ДНК (около 2,9 kb), содержащий промотор и терминатор гена 3-фосфоглицераткиназы (в дальнейшем именуемого "pgk") и ГФТ из Escherichia coli.
Выделенный фрагмент ДНК встраивали в pSAK410 по сайту PvuII с помощью набора для лигирования ДНК Ver.2 (фирмы Takara Shuzo Co., Ltd., Япония). Этой дотированной ДНК трансформировали компетентные клетки штамма Escherichia coli JM109 (фирмы Takara Shuzo Co., Ltd., Япония). Из трансформированных клеток Escherichia coli отбирали штамм, несущий плазмиду с этим фрагментом ДНК, которую назвали pSAK700.
Построение pSAK700 показано на Фигуре 4.
pSAK700 имеет сайты рестрикции для ферментов ВаmHI и NotI. pSAK700 также имеет ген устойчивости к ампициллину (в дальнейшем именуемый "Амп") и ген устойчивости к гигромицину ГТФ в качестве маркеров для селекции. В следующих примерах, когда в качестве клетки-хозяина использовали Escherichia coli, селекцию клеток, трансформированных pSAK700 или pSAK700 со встроенным чужеродным геном проводили, добавляя 40 мкг/мл ампициллина в соответствующую среду. Когда в качестве клетки-хозяина использовали Penicillium citrinum SANK 13380, то селекцию клеток, трансформированных pSAK700 или pSAK700 со встроенным чужеродным геном проводили, добавляя 200 мкг/мл гигромицина В в соответствующую среду.
Пример 11. Построение экспрессионного вектора pSAKexpE
Рекомбинантный ДНК-вектор pCRexpE, полученный в Примере 9, обрабатывали 2 часа при 37°С рестрикционными ферментами HpaI и SspI (фирмы Takara Shuzo Co., Ltd., Япония) и продукт реакции подвергали агарозному гель-электрофорезу. Из геля извлекали полосу, содержащую полную кДНК mlcE размером около 1,7 kb.
После обработки pSAK.700 рестрикционным ферментом NotI (фирмы Takara Shuzo Co., Ltd., Япония) 1 час при 37°С вектор обрабатывали ДНК-полимеразой Т4 (фирмы Takara Shuzo Co., Ltd., Япония) 5 минут при 37°С для образования тупых концов. Затем вектор подвергали экстракции фенол-хлороформом и осаждению этанолом. Осадок ДНК растворяли в небольшом объеме ТЕ. Туда добавляли щелочную фосфатазу и инкубировали 30 минут при 65°С. Обработанный pSAK700 лигировали с фрагментом ДНК в 1,7 kb, полученным на стадии 1, с помощью набора для лигирования ДНК Ver.2 (фирмы Takara Shuzo Co., Ltd., Япония). Этой лигированной ДНК трансформировали компетентные клетки штамма Escherichia coli JM109. Получали штамм Escherichia coli, трансформированный экспрессионным вектором с кДНК.
Полученный в настоящем примере трансформированный штамм Escherichia coli pSAKexpE SANK 72499 депонирован в Research Institute of Life Science and Technology, Agency of Industrial Science and Technology, 25 января 2000 г. под кодовым номером FERM ВР-7005 в соответствии с Будапештским договором о депонировании микроорганизмов.
Пример 12. Получение кДНК, соответствующей структурному гену mlcR
1) Выделение тотальной РНК
Тотальную РНК Penicillium citrinum выделяли по методике Примера 8.
2) Составление праймеров
Для получения полной кДНК, соответствующей структурному гену mlcR, определенному в Примере 8, были разработаны и синтезированы следующие праймеры:
смысловой праймер - 5'-ggatccatgtccctgccgcatgcaacgattc-3' (SEQ ID No. 39) антисмысловой праймер - 5'-ggatccctaagcaatattgtgtttcttcgc-3' (SEQ ID No. 40).
Праймеры составлены на основе последовательности, расположенной вверх от 5'-конца структурного гена mlcR, и из последовательности, расположенной вниз от 3'-конца соответственно. Синтез проводили фосфоамидитным методом.
3) ОТ-ПЦР
Для получения полной кДНК, кодирующей продукт гена mlcR, применяли набор для ПЦР Takara RNA LA PCR Kit (AMV) версии 1.1.
Для этого 20 мкл реакционной смеси, содержащей 1 мкг тотальной РНК, 2,5 пмоль праймера Random 9mer (из набора) и 1 мкл обратной транскриптазы (из набора) инкубировали 30 минут при 42°С, получая первую цепь кДНК. Затем обратную транскриптазу инактивировали, нагревая при 99°С в течение 5 минут.
100 мкл второй реакционной смеси, включающей весь объем реакционной смеси для первой цепи кДНК (выше), 40 пмоль смыслового праймера и 40 пмоль антисмыслового праймера, инкубировали 2 минуты при 94°С. Затем повторяли 30 раз цикл инкубации, состоящий из 30 секунд при 94°С, 30 секунд при 60°С и 2 минут при 72°С, после чего инкубировали 5 минут при 72°С и 18 часов при 4°С. Продукт реакции подвергали агарозному гель-электрофорезу и выделяли ДНК из геля. Продукт очищали путем экстракции фенол-хлороформом и осаждения этанолом и использовали для трансформации компетентных клеток штамма Escherichia coli JM109 (фирмы Takara Shuzo Co., Ltd., Япония) таким же образом, как описано в примере 4 с помощью pCR2.1. Из трансформированных клеток Escherichia coli отбирали штамм, несущий плазмиду с этим фрагментом ДНК, которую назвали pCRexpR.
Была определена нуклеотидная последовательность ДНК, встроенной в рекомбинантный вектор pCRexpR. Вставка содержала полную кДНК, соответствующую структурному гену mlcR. Его нуклеотидная последовательность и аминокислотная последовательность пептида, предсказанного из нуклеотидной последовательности, показаны в SEQ ID No. 41 и/или SEQ ID No. 42 из Перечня последовательностей.
Из известных последовательностей наиболее близким к mlcR (полипептиду) был lovE в генном кластере, связанном с биосинтезом ловастатина, на уровне идентичности в 34%.
Пример 13. Построение экспрессионного вектора pSAKexpR
Рекомбинантный ДНК-вектор pCRexpR, полученный в Примере 9, обрабатывали 2 часа при 37°С рестрикционным ферментом ВаmHI (фирмы Takara Shuzo Co., Ltd., Япония) и продукт реакции подвергали агарозному гель-электрофорезу. Из геля извлекали полосу, содержащую полную кДНК mlcR размером около 1,4 kb.
После обработки pSAK700 рестрикционным ферментом ВаmHI (фирмы Takara Shuzo Co., Ltd., Япония) 1 час при 37°С добавляли щелочную фосфатазу (фирмы Takara Shuzo Co., Ltd., Япония) и инкубировали 30 минут при 65°С. Расщепленный и обработанный таким образом pSAK700 лигировали с фрагментом ДНК в 1,4 kb, полученным на стадии 1, с помощью набора для дотирования ДНК Ver.2 (фирмы Takara Shuzo Co., Ltd., Япония). Этой лигированной ДНК трансформировали компетентные клетки штамма Escherichia coli JM109. Получали штамм Escherichia coli, трансформированный экспрессионным вектором с кДНК.
Полученный в настоящем примере трансформированный штамм Escherichia coli pSAKexpR SANK 72599 депонирован в Research Institute of Life Science and Technology, Agency of Industrial Science and Technology, 25 января 2000 г., под кодовым номером FERM ВР-7006 в соответствии с Будапештским договором о депонировании микроорганизмов.
Пример 14. Трансформация продуцирующих ML-236B микроорганизмов
1) Получение протопластов
Споры с косяка культуры штамма Penicillium citrinum SANK 13380 инокулировали на агарную среду PGA и инкубировали 14 дней при 26°С. Затем из этой культуры собирали споры Penicillium citrinum SANK 13380, инокулировали 1×108 спор в 80 мл питательной среды YPL-20 и инкубировали 1 день при 26°С. После подтверждения прорастания спор под микроскопом их собирали центрифугированием при комнатной температуре 10 минут при 5000×g в виде осадка.
Споры промывали 3 раза стерилизованной водой для получения протопластов. Для этого 200 мг зимолиазы 20Т (фирмы Seikagaku Kogyo) и 100 мг хитиназы (фирмы Sigma) растворяли в 10 мл 0,55 М раствора хлористого магния и центрифугировали при комнатной температуре 10 минут при 5000×g. Образовавшийся супернатант использовали в качестве ферментного раствора. В коническую колбу на 100 мл вносили 20 мл ферментного раствора и 0,5 г (сырой вес) проросших спор и инкубировали с покачиванием 60 минут при 30°С. После подтверждения с помощью микроскопа, что проросшие споры превратились в протопласты, обработанный раствор фильтровали через стеклянный фильтр 3G-2 (фирмы HARIO). Фильтрат центрифугировали при комнатной температуре 10 минут при 1000×g и собирали протопласты в виде осадка.
2) Трансформация
Полученные на стадии 1 протопласты дважды промывали 30 мл 0,55 М хлористого магния и один раз 30 мл раствора, состоящего из 0,55 М хлористого магния, 50 мМ хлористого кальция и 10 мМ 3-морфолинопропансульфоната (рН 6,3 или меньше; в дальнейшем именуется раствором МСМ). Затем протопласты суспендировали в 100 мкл раствора 4% полиэтиленгликоля 8000, 10 мМ 3-морфолинопропансульфоната, 0,0025% гепарина (фирмы Sigma), 50 мМ хлористого магния (рН 6,3 или меньше; в дальнейшем именуется раствором для трансформации).
Смешивали 96 мкл раствора для трансформации, содержащего около 5×107 протопластов, и 10 мкл ТЕ, содержащего 120 мкг pSAKexpE или pSAKexpR, и оставляли на льду на 30 минут. Затем добавляли 1,2 мл раствора 20% полиэтиленгликоля, 50 мМ хлористого магния, 10 мМ 3-морфолинопропансульфоновой кислоты (рН 6,3). Эту смесь осторожно перемешивали с помощью пипетки и оставляли при комнатной температуре на 20 минут. Затем добавляли 10 мл раствора МСМ, осторожно перемешивали и центрифугировали при комнатной температуре 10 минут при 1000×g. В осадке получали трансформированные протопласты.
3) Регенерация клеточной стенки трансформированных протопластов
Трансформированные протопласты, полученные на стадии 2, суспендировали в 5 мл жидкого среднего слоя агарной среды VGS и наносили на чашку, содержащую 10 мл твердого нижнего агара среды VGS. Чашку инкубировали при 26°С в течение 1 дня, после чего на поверхность наносили 10 мл жидкого верхнего агара среды VGS, содержащего 5 мг гигромицина В на чашку (конечная концентрация гигромицина 200 мкг/мл). После инкубации при 26°С в течение 14 дней оба штамма (то есть штаммы, происходящие из протопластов, трансформированных либо pSAKexpE, либо pSAKexpR) высевали на агарную среду PGA, содержащую 200 мкг/мл гигромицина В, а затем высевали на косяк из агарной среды PGA и инкубировали при 26°С в течение 14 дней.
Косяки хранили при 4°С.
Проверочный пример 1. Сравнение способности к биосинтезу ML-236B у трансформированных и исходных штаммов
Выращивали трансформированные штаммы, полученные в Примере 14, и Penicillium citrinum SANK 13380, а затем определяли количество ML-236B в каждой культуре.
Для инокуляции брали споры с поверхности в 5 мм из косяков, содержащих трансформированные штаммы, описанные в Примере 14, и косяка с Penicillium citrinum SANK 13380, описанного в Примере 2. Клетки инокулировали в 10 мл среды MBG3-8 в конической колбе на 100 мл и инкубировали при 24°С в течение 2 дней со встряхиванием, а затем добавляли 3,5 мл 50% раствора глицерина. После этого продолжали выращивать культуры при 24°С в течение 10 дней со встряхиванием.
К 10 мл культуры добавляли 50 мл 0,2 N гидроокиси натрия и инкубировали при 26°С в течение 1 часа со встряхиванием. Затем культуру центрифугировали при комнатной температуре 2 минуты при 3000×g. Отбирали 1 мл супернатанта, смешивали с 9 мл 75% метанола и подвергали ВЭЖХ. Для этого применяли колонку SSC-ODS-262 (диаметр 6 мм, длина 100 мм, фирмы Senshu Kagaku Co. Ltd.), а в качестве подвижной фазы систему 75% метанол - 0,1% триэтиламин - 0,1% уксусная кислота. Элюирование проводили при комнатной температуре со скоростью 2 мл/мин. В этих условиях ML-236B выходил из колонки через 4 минуты после нанесения. Детекция проводилась УФ-детектором при длине волны 236 нм.
Три из 8 трансформированных pSAKexpE штаммов обладали повышенной способностью к биосинтезу ML-236B. У этих штаммов способность к биосинтезу ML-236B была в среднем на 10% выше по сравнению с исходным штаммом. Способность к биосинтезу ML-236B у этих 3 штаммов также стабильно сохранялась после пересева, например, при моноспоровой обработке и т.п. Эти результаты показывают, что вставка в pSAKexpE является кДНК, ускоряющей биосинтез ML-236B.
Пять из числа трансформированных pSAKexpR штаммов обладали повышенной способностью к биосинтезу ML-236B. У этих штаммов способность к биосинтезу ML-236B была в среднем на 15% выше по сравнению с исходным штаммом. Способность к биосинтезу ML-236B у этих 5 штаммов также стабильно сохранялась после пересева, например, при моноспоровой обработке и т.п. Эти результаты показывают, что вставка в pSAKexpR является кДНК, ускоряющей биосинтез ML-236B.
Таким образом, ускоряющие биосинтез ML-236B кДНК, полученные из продуцирующего ML-236B микроорганизма согласно настоящему изобретению, ускоряют биосинтез ML-236B в продуцирующем ML-236B микроорганизме при введении в этот микроорганизм.
Пример 15. Определение последовательности кДНК, соответствующих структурным генам mlcA-D
Определяли последовательность кДНК, соответствующей структурному гену m1сА.
Первую цепь кДНК синтезировали с помощью набора TAKARA LA PCR Kit версии 1.1 (Takara Shuzo Co., Ltd.). Используя первую цепь в качестве матрицы и различные пары праймеров, проводили несколько реакций ПЦР для амплификации полной или частичной кДНК.
Повторяли 30 раз цикл, состоящий из 30 секунд при 94°С, 30 секунд при 60°С и 5 минут при 72°С, с помощью набора для ПЦР The Big Dye Primer/Terminator Cycle Sequencing Kit и прибора ABI Prism 377 (фирмы РЕ Applied Biosystems).
Каждый продукт реакции по отдельности встраивали в плазмиду pCR2.1.
Получали трансформанты Escherichia coli с каждой рекомбинантной плазмидой.
Определяли нуклеотидные последовательности каждой вставки из рекомбинантных плазмид.
Определяли последовательности экзонов и интронов путем сравнения нуклеотидных последовательностей указанных выше продуктов ОТ-ПЦР с последовательностью структурного гена mlcA.
После этого определяли последовательность кДНК, соответствующей структурному гену mlcA (SEQ ID No. 43). Вычисляли аминокислотную последовательность, соответствующую полипептиду, кодируемому этой кДНК (SEQ ID No. 44), и определяли его функцию на основе поиска гомологии по аминокислотной последовательности.
Из известных последовательностей наиболее близким к mlcA (полипептиду) был LNKS (lovB) в генном кластере, связанном с биосинтезом ловастатина, на уровне идентичности в 60%.
Таким же образом определяли последовательность кДНК, соответствующей структурному гену mlcB (SEQ ID No. 45). Вычисляли аминокислотную последовательность, соответствующую полипептиду, кодируемому этой кДНК (SEQ ID No. 46), и определяли его функцию на основе поиска гомологии по аминокислотной последовательности.
Из известных последовательностей наиболее близким к mlcB (полипептиду) был LDKS (lovB) в генном кластере, связанном с биосинтезом ловастатина, на уровне идентичности в 61%.
Подобным образом определяли последовательность кДНК, соответствующей структурному гену mlcC (SEQ ID No. 47). Вычисляли аминокислотную последовательность, соответствующую полипептиду, кодируемому этой кДНК (SEQ ID No. 48), и определяли его функцию на основе поиска гомологии по аминокислотной последовательности.
Из известных последовательностей наиболее близким к mlcC (полипептиду) был lovA в генном кластере, связанном с биосинтезом ловастатина, на уровне идентичности в 72%.
Кроме того, определяли последовательность кДНК, соответствующей структурному гену mlcD (SEQ ID No. 49). Вычисляли аминокислотную последовательность, соответствующую полипептиду, кодируемому этой кДНК (SEQ ID No. 50), и определяли его функцию на основе поиска гомологии по аминокислотной последовательности.
Из известных последовательностей наиболее близким к mlcD (полипептиду) был ORF8 в генном кластере, связанном с биосинтезом ловастатина, на уровне идентичности в 63%.
Было определено положение экзонов в каждом из структурных генов согласно SEQ ID No. 1 или SEQ ID No. 2, как показано ниже:

Было определено положение сайтов терминации трансляции в каждом из структурных генов согласно SEQ ID No. 1 или SEQ ID No. 2, как показано ниже:

Пример 16. Исследования инактивации генов
Структурные гены mlcA, В или D P. citrinum инактивировали посредством направленного мутагенеза с помощью гомологичной рекомбинации.
Рекомбинантную плазмиду для получения мутанта Р. citrinum с инактивированным структурным геном mlcA построили с помощью плазмиды pSAK333.
Выделяли внутренний фрагмент KpnI в 4,1 kb из локуса mlcA, встроенного в pML48, очищали, затупляли концы с помощью набора DNA Blunting Kit (фирмы Takara Shuzo Co., Ltd.) и лигировали с расщепленной PuuII плазмидой pSAK333. Получили плазмиду, которую назвали pdismlcA.
Плазмидой pdismlcA трансформировали Р. citrinum SANK 13380.
Геномную ДНК трансформанта pdismlcA гибридизировали по Сазерну для подтверждения инактивации структурного гена mlcA.
Этот мутант с инактивированным mlcA совсем не вырабатывал ML-236B или его предшественника.
Рекомбинантную плазмиду для получения мутанта Р. citrinum с инактивированным структурным геном m1сВ построили с помощью плазмиды pSAK333.
Выделяли фрагмент PstI-BamHI в 1,4 kb из локуса mZcB, встроенного в pML48, очищали, затупляли концы с помощью набора DNA Blunting Kit (Takara Shuzo Co., Ltd.) и лигировали с расщепленной РииП плазмидой pSAK333. Получили плазмиду, которую назвали pdismlcB.
Плазмидой pdismlcB трансформировали Р. citrinum SANK 13380.
Геномную ДНК трансформанта pdismlcB гибридизировали по Сазерну для подтверждения инактивации структурного гена mlсВ.
Этот мутант с инактивированным mlсВ не вырабатывал ML-236B, но вырабатывал его предшественник ML-236A.
Рекомбинантную плазмиду для получения мутанта Р. citrinum с инактивированным структурным геном mlcD построили с помощью плазмиды pSAK333.
Выделяли фрагмент PstI-ВаmHI в 1,4 kb из локуса mlcD, встроенного в pML48, очищали, затупляли концы с помощью набора DNA Blunting Kit (Takara Shuzo Co., Ltd.) и лигировали с расщепленной РuuII плазмидой pSAK333. Получили плазмиду, которую назвали pdismlcD.
Плазмидой pdismlcD трансформировали Р. citrinum SANK 13380.
Геномную ДНК трансформанта pdismlcD гибридизировали по Сазерну для подтверждения инактивации структурного гена mlcD.
Этот мутант с инактивированным mlcD вырабатывал ML-236B в количестве, равном 30% от того, что вырабатывал нетрансформированный контрольный штамм.
Пример 17. Функциональный анализ mlcR у трансформантов pSAKexpR
Два из трансформантов pSAKexpR, полученных в Примере 12 и названных TR1 и TR2 соответственно, а также нетрансформированные клетки Penicillium citrinum SANK 13380 инокулировали в среду MBG3-8 и инкубировали по отдельности, как описано в Примере 8.
Из каждой культуры экстрагировали тотальную РНК, как описано в Примере 8. Проводили ОТ-ПЦР, используя эту тотальную РНК в качестве матрицы, а в качестве праймеров - пару олигонуклеотидов, составленных на основе нуклеотидной последовательности структурных генов mlcA, В, С, D, Е или R.

Результаты анализа методом ОТ-ПЦР представлены на Фигуре 5 для нетрансформированных клеток Penicillium citrinum 13380 и для двух трансформантов TR1 и TR2.
Структурные гены mlсА, В, С, D и R экспрессировались на первый, второй и третий день роста культуры трансформантов pSAKexpR.
Напротив, все эти структурные гены экспрессировались только на третий день роста культуры нетрансформированных клеток.
В экспрессии структурного гена mlcE не наблюдалось отличий между трансформантами pSAKexpR и нетрансформированными клетками.
Результаты показывают, что белок, кодируемый кДНК, соответствующей структурному гену mlcR, индуцирует транскрипцию некоторых других структурных генов (например, mlсА, В, С, D) из генного кластера, связанного с биосинтезом ML-236B.
Пример 18. Функциональный анализ mlcE у трансформантов pSAKexpE
Полученный в Примере 12 трансформант pSAKexpE, названный ТЕ1, а также нетрансформированные клетки Penicillium citrinum SANK 13380 инокулировали в среду MBG3-8 и инкубировали по отдельности, как описано в Примере 8.
Из каждой культуры экстрагировали тотальную РНК, как описано в Примере 8.
Проводили ОТ-ПЦР, используя эту тотальную РНК в качестве матрицы, а в качестве праймеров - пару олигонуклеотидов, составленных на основе нуклеотидной последовательности структурных генов mlcA, В, С, D, E или R. Праймеры в настоящем примере были идентичны тем, что показаны в таблице из предшествующего примера.
Результаты анализа методом ОТ-ПЦР представлены на Фигуре 6 для нетрансформированных клеток Penicillium citrinum 13380 и для трансформанта ТЕ1.
Структурный ген mlсЕ экспрессировался на первый, второй и третий день роста культуры трансформантов pSAKexpE.
Напротив, структурный ген m1сЕ экспрессировался только на третий день роста культуры нетрансформированных клеток.
С другой стороны, в экспрессии структурных генов mlcA, В, С, D и R не наблюдалось отличий между трансформантом pSAKexpE и нетрансформированными клетками (данные не приводятся).
Результаты показывают, что белок, кодируемый кДНК, соответствующей структурному гену mlсЕ, ускоряет биосинтез ML-236B независимо от структурных генов mlсА, B,C,D и R.

























































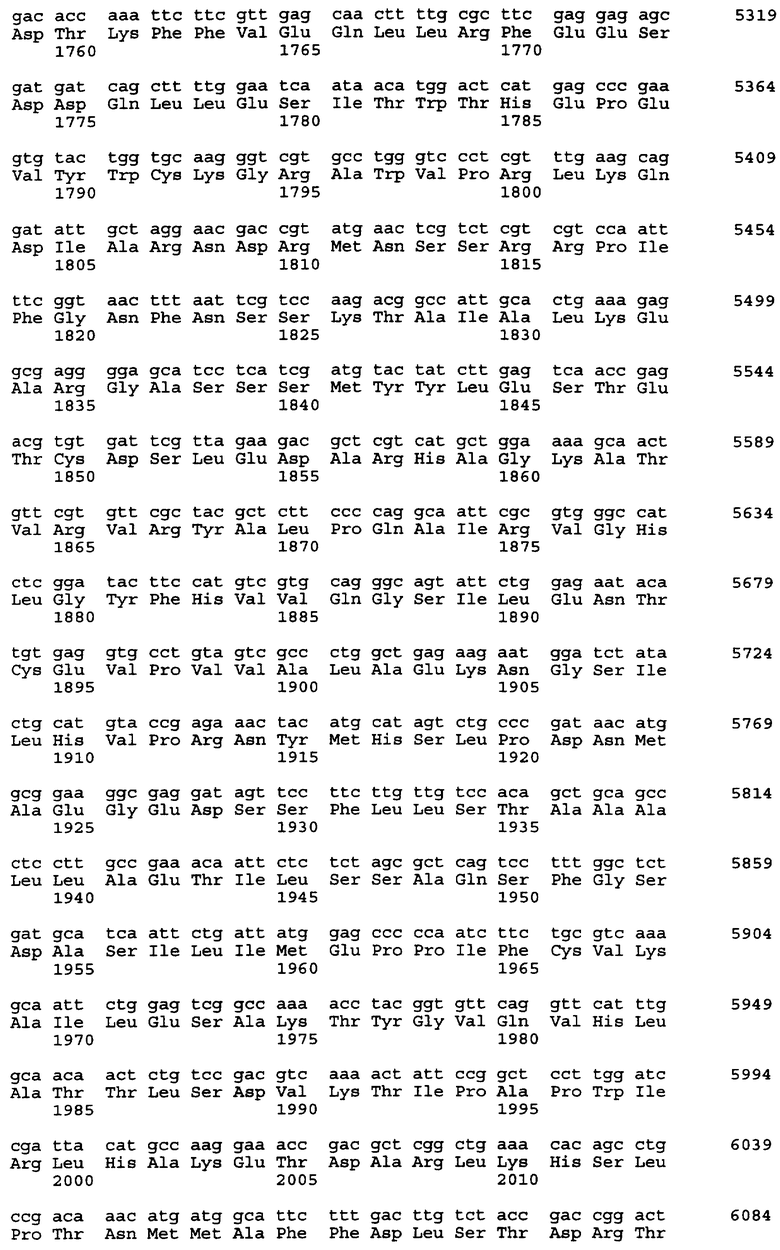
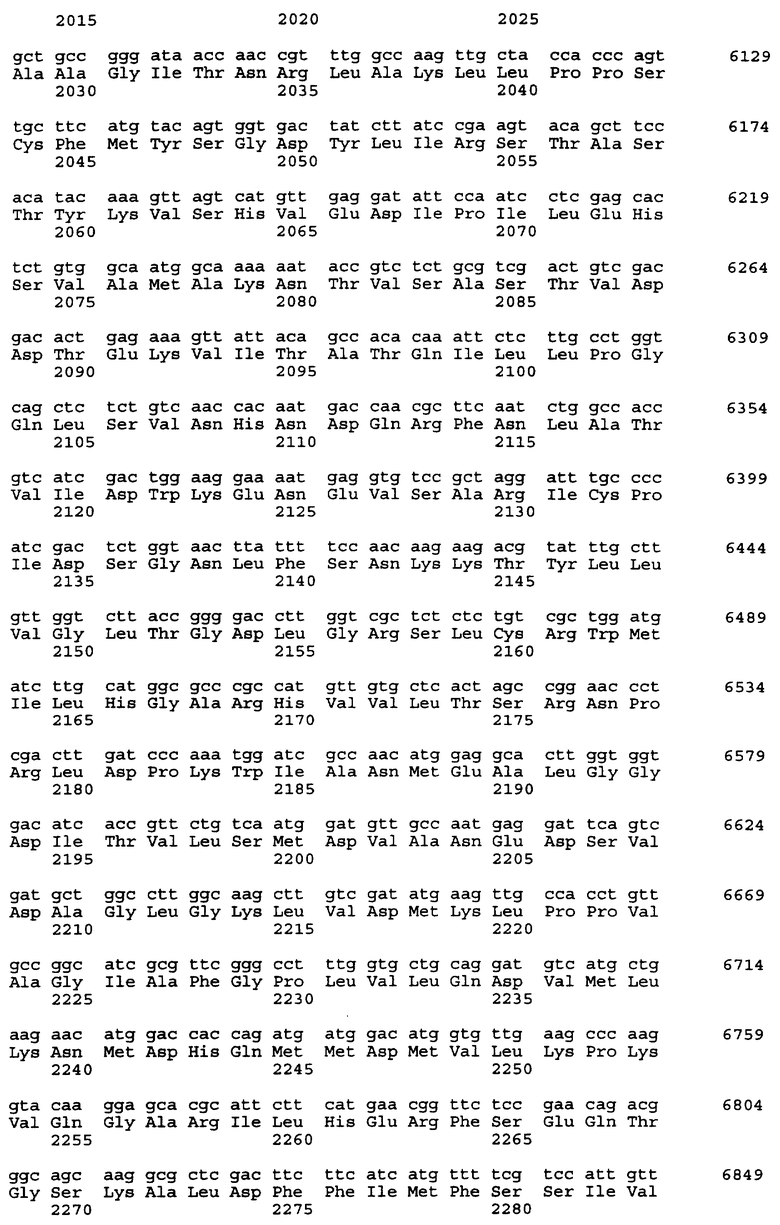

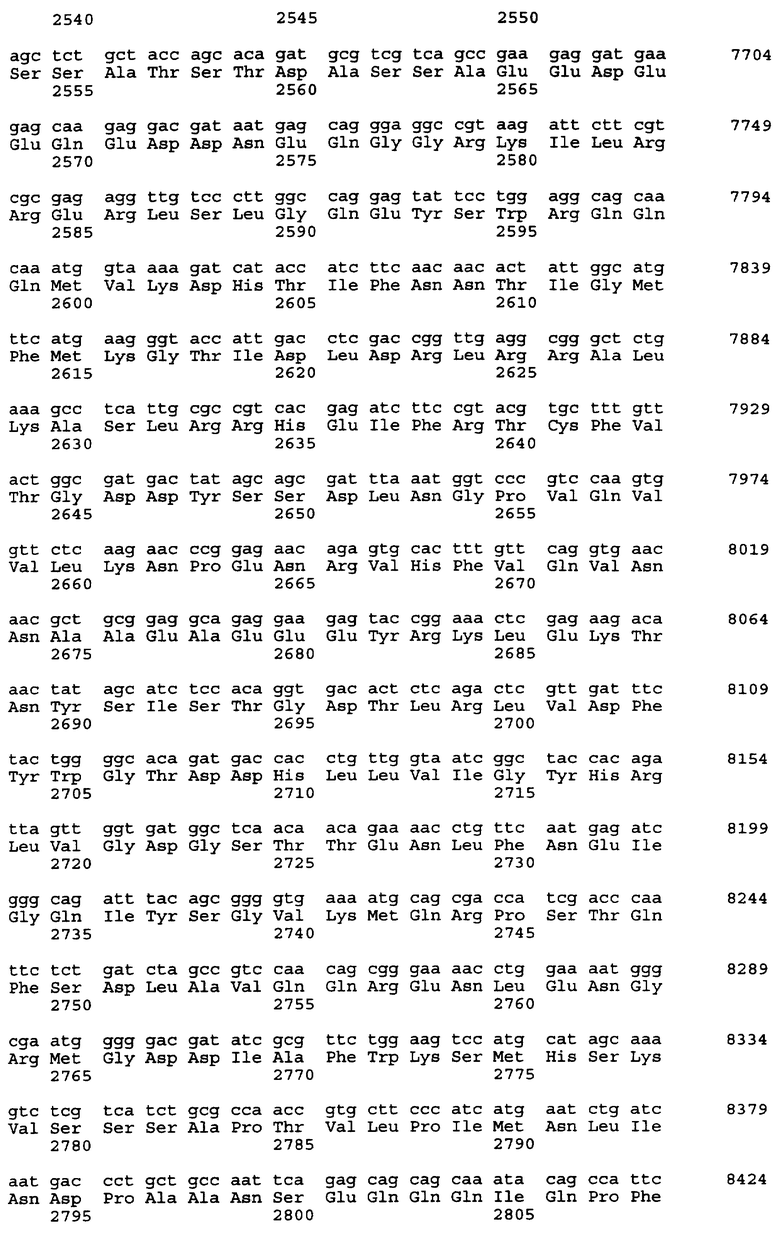









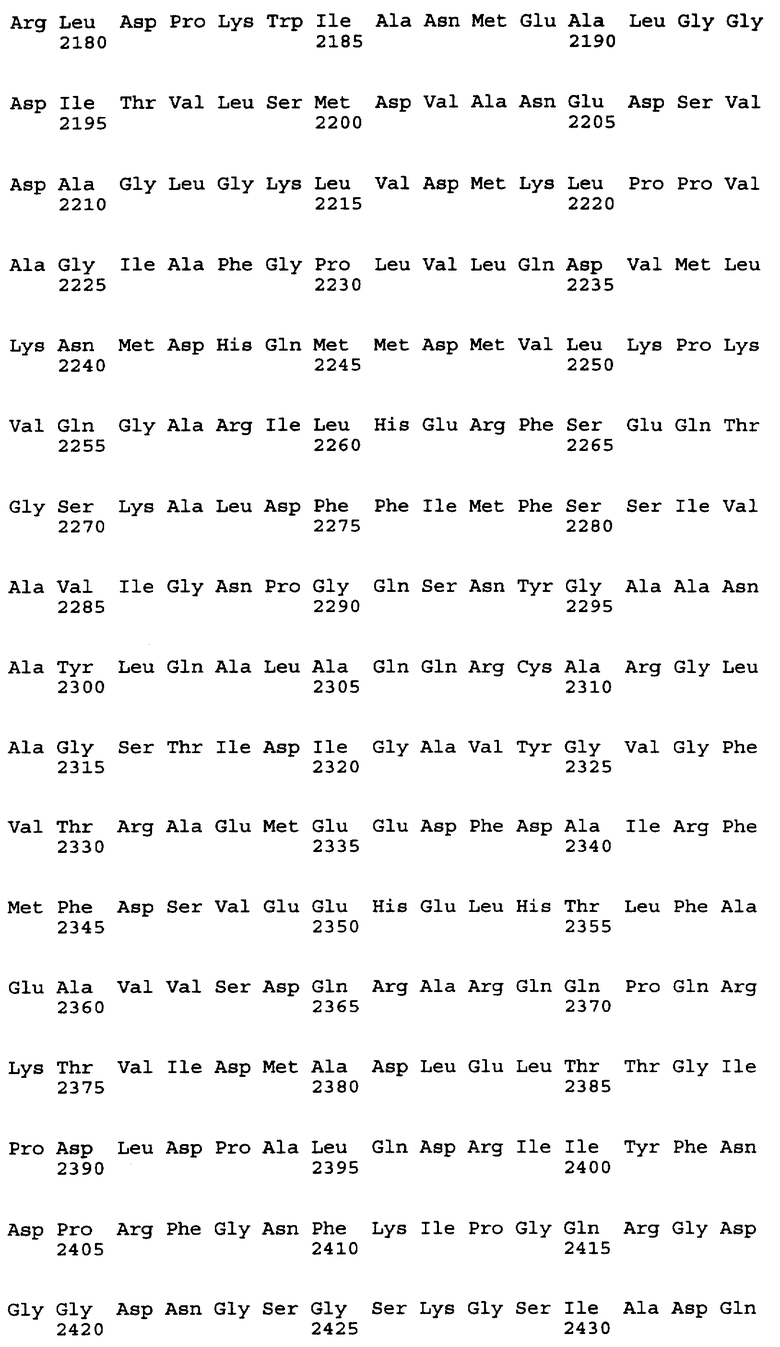





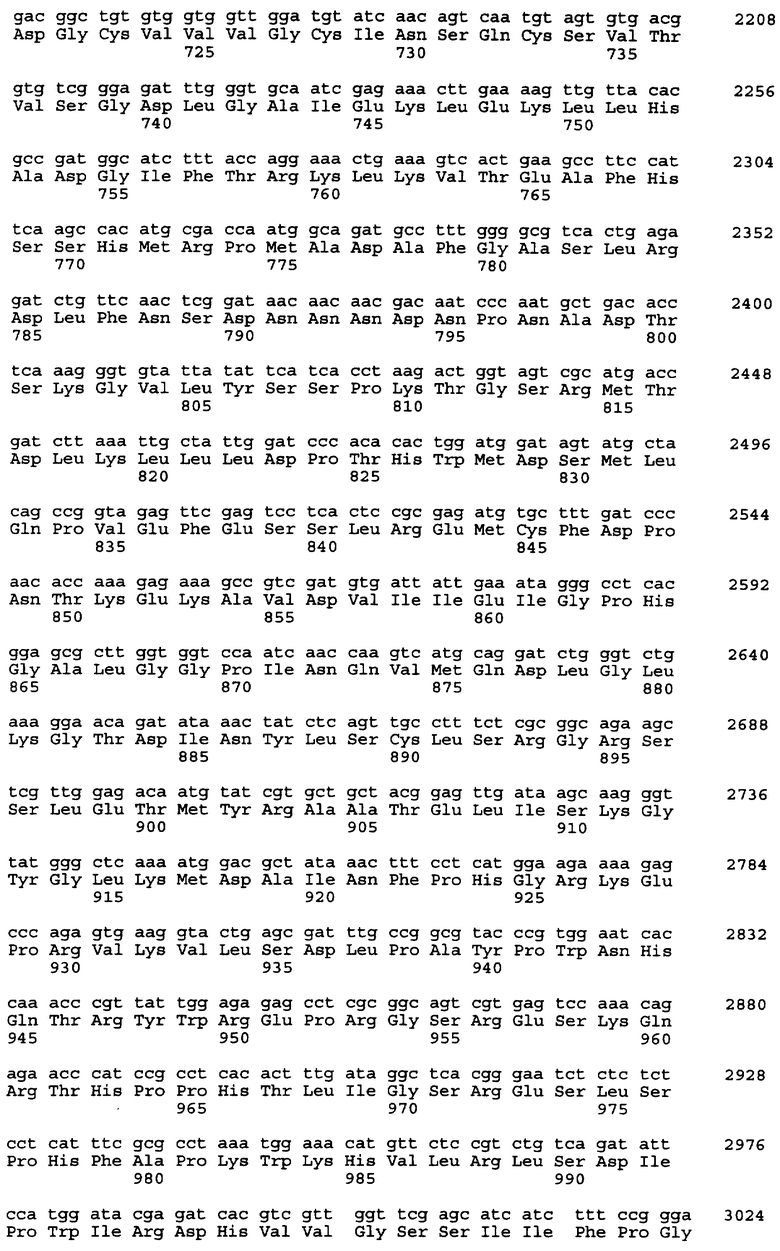


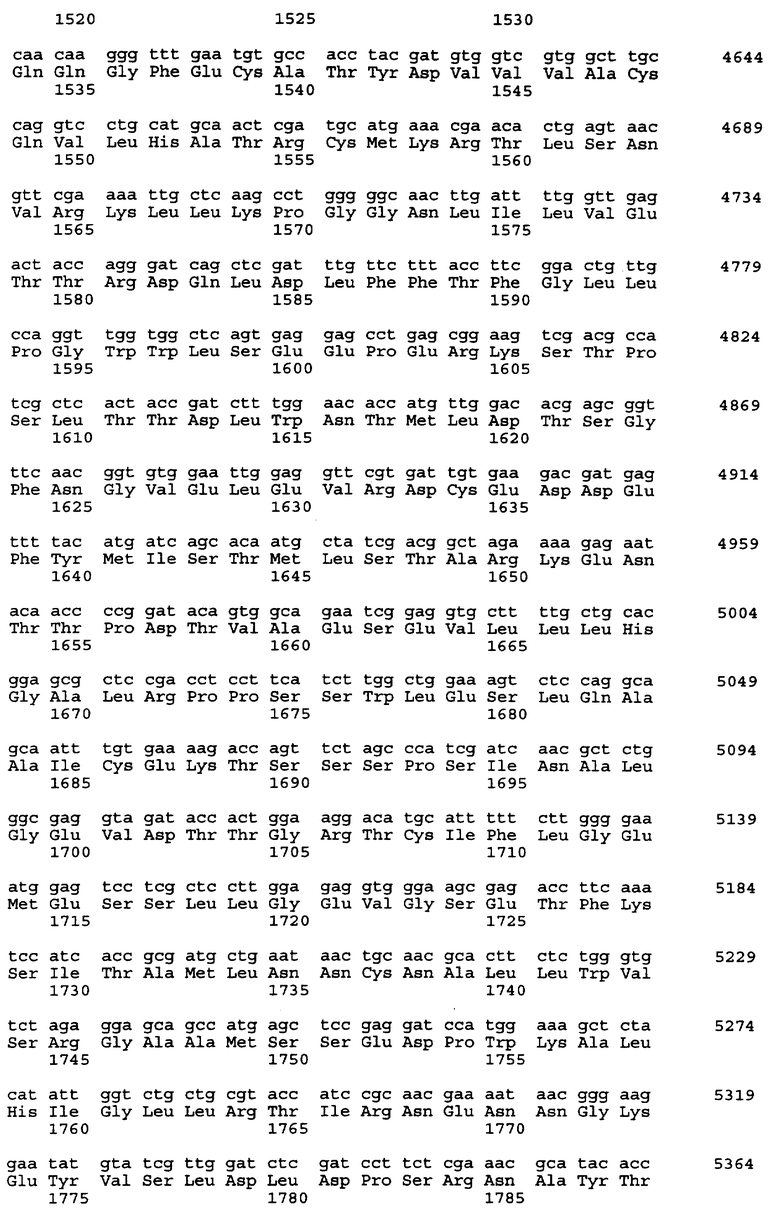

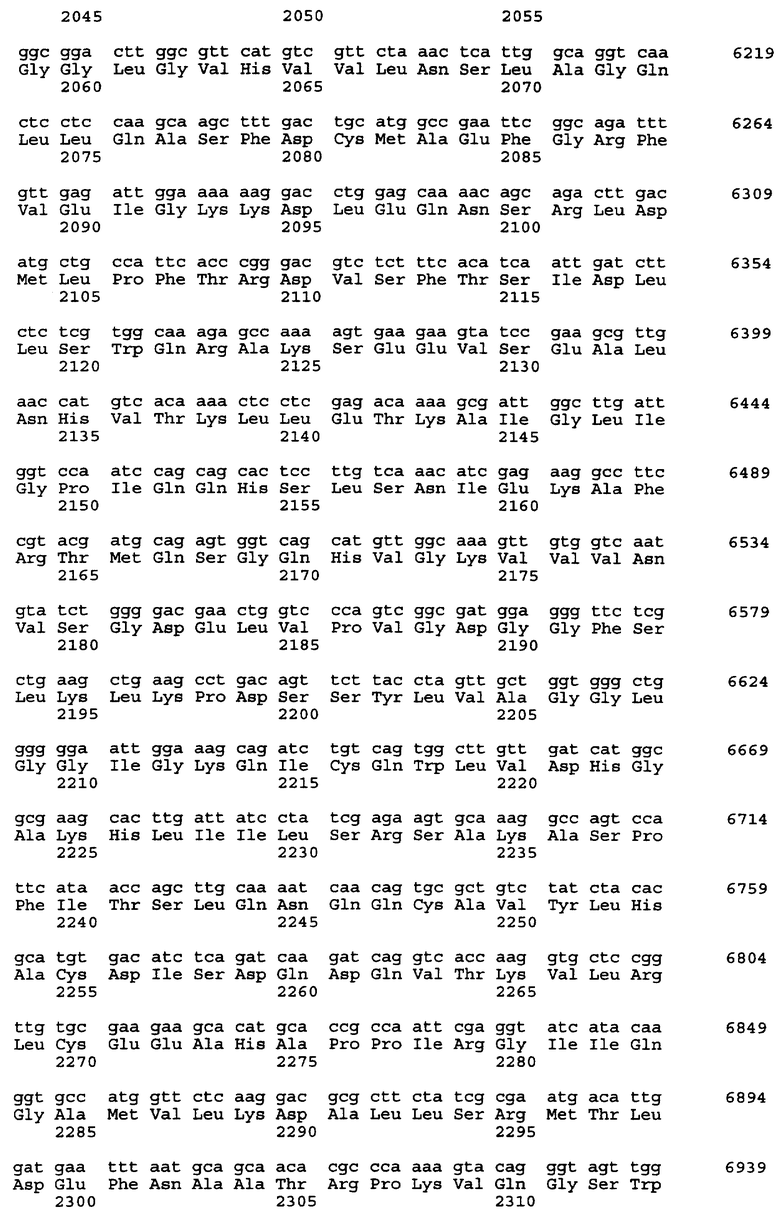































Изобретение относится к биотехнологии и представляет собой полинуклеотиды, обладающие способностью ускорять биоситнез предшественника правастатина ML-236В в микроорганизмах, вырабатывающих ML-236В, при введении в эти микроорганизмы. Данные полинуклеотиды встраиваются в вектора, которыми трансформируют клетки микроорганизмов - продуцентов предшественника правастатина, который является ингибитором HMG-CoA редуктазы. 20 c. и 26 з.п. ф-лы, 14 табл., 6 ил.
| WO 9512661 A, 11.05.1995.HENDRICKSON L | |||
| et al | |||
| Lovastatin biosynthesis in Aspergillus terreus: characterization ofblocked mutants, enzyme activities and a multifunctional polyketidesyntase gene | |||
| Current Biology | |||
| vol | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Et al | |||
| Modulation of polyketide syntuase activity by accessory proteins during lovastatin biosynthesis | |||
| Science | |||
| vol | |||
| СЧЕТНЫЙ ДИСК ДЛЯ РАСЧЕТА СОСТАВНЫХ ЧАСТЕЙ ПИЩИ | 1919 |
|
SU284A1 |

